

Abstract
Cystic fibrosis (CF) is a monogenic disease caused by impaired production and/or function of the CF transmembrane conductance regulator (CFTR) protein. Although we have previously shown correction of the most common pathogenic mutation, there are many other pathogenic mutations throughout the CF gene. An autologous airway stem cell therapy in which the CFTR cDNA is precisely inserted into the CFTR locus may enable the development of a durable cure for almost all CF patients, irrespective of the causal mutation. Here, we use CRISPR-Cas9 and two adeno-associated viruses (AAVs) carrying the two halves of the CFTR cDNA to sequentially insert the full CFTR cDNA along with a truncated CD19 (tCD19) enrichment tag in upper airway basal stem cells (UABCs) and human bronchial epithelial cells (HBECs). The modified cells were enriched to obtain 60%–80% tCD19+ UABCs and HBECs from 11 different CF donors with a variety of mutations. Differentiated epithelial monolayers cultured at air-liquid interface showed restored CFTR function that was >70% of the CFTR function in non-CF controls. Thus, our study enables the development of a therapy for almost all CF patients, including patients who cannot be treated using recently approved modulator therapies.
Keywords: cystic fibrosis, airway stem cell therapy, genome editing for CF, CRISPR-Cas9, CFTR correction, CF, universal CFTR correction
Graphical abstract
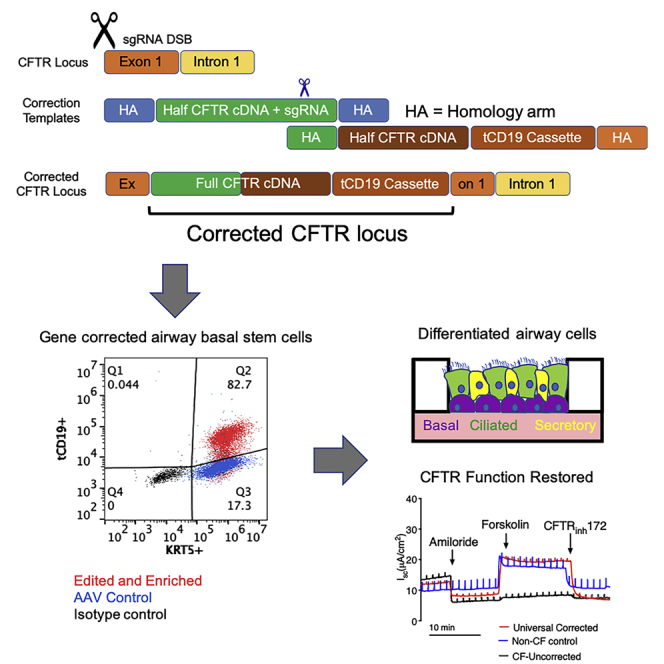
Cystic fibrosis (CF) is a life-threatening genetic disease caused by mutations in the CFTR gene. Porteus and coworkers use CRISPR-Cas9 and two adeno-associated viruses to insert the full CFTR cDNA into human airway stem cells in a “mutation-agnostic” manner, restoring CFTR function to levels seen in non-CF controls. These findings are an essential step toward autologous airway stem cell therapy for nearly all CF patients, including those with mutations not amenable to modulator therapy.
Introduction
Cystic fibrosis (CF), one of the most common life-shortening genetic diseases, can result from a variety of deleterious mutations spanning the CF transmembrane conductance regulator (CFTR) gene. Among the >1,700 known CFTR mutations that can cause CF, F508del affects most CF patients in Europe and North America. Small molecule CFTR modulators that aid CFTR folding or potentiation increase CFTR function and improve health outcomes in patients with at least one F508del allele or certain gating mutations, comprising ∼90% of the known CF population.1, 2, 3 However, significant interpatient heterogeneity of response has been observed in these modulators.1, 2, 3 In addition, modulators must be administered daily, are costly, only partially restore function, and do not cover the full spectrum of CFTR mutations.2,4,5 Thus, there is a continued need to develop a durable therapy that can treat 100% of CF patients.
Several gene-therapy trials for CF have been performed.6, 7, 8 Although CF is a systemic disease, past attempts to develop a gene therapy have focused on the respiratory tract because of the high morbidity and mortality associated with CF lung disease. These trials used viral or non-viral vectors to deliver a copy of the CFTR cDNA into the cells of the epithelia in the upper (nose, sinuses) and lower airways.6, 7, 8 These trials were unsuccessful in producing a durable improvement in pulmonary function in CF patients. Although in vivo genome editing is being attempted to treat other diseases (e.g., muscular dystrophy),9 immune responses against gene-editing enzymes such as Cas9 have been shown to limit the persistence of gene-corrected cells in mouse models.10 We pursued the development of an autologous gene-corrected airway stem cell therapy to treat CF in order to avoid anti-Cas9 adaptive immune responses, which have been shown to exist in humans.11,12
A major obstacle to the successful development of ex vivo gene therapy is the inability to gene correct CFTR mutations or add a functional copy of the CFTR cDNA safely with high efficiencies in airway basal stem cells. Several prior studies have attempted to insert the CFTR cDNA in airway basal stem cells and airway tissue using integrating viruses.13,14 In these studies, the expression of CFTR was driven by non-native promoters, and the integration of CFTR in the genome occurred in a semi-random manner. Nevertheless, these studies showed that the overexpression of CFTR using non-native promoters was sufficient to restore CFTR function, both in vitro and in vivo. In this study, we show the precise insertion of the CFTR cDNA in the endogenous CFTR locus, thereby preserving natural CFTR expression.
The discovery and development of zinc finger nucleases, transcription activator-like effective nucleases (TALEN), and Cas9 have enabled the precise insertion of exogenous DNA sequences into the genome.15,16 The ex vivo gene correction of F508del and other CFTR mutations using nuclease-mediated editing has been performed in induced pluripotent stem cells (iPSCs) and primary intestinal stem cells.17, 18, 19 Although the differentiation of iPSCs into CFTR-expressing airway epithelia consisting of cells resembling ciliated, secretory, and basal cells has been shown,20,21 long-term differentiation potential of the iPSC-derived basal cells still needs to be validated.22 As an alternative, we recently reported an ex vivo cell-therapy approach using airway stem cells derived from the nasal and sinus epithelia, termed upper airway basal stem cells (UABCs), as well as human bronchial epithelial cells (HBECs).23 With the use of Cas9 and adeno-associated virus (AAV), we achieved >40% correction of the F508del mutation in a selection-free manner and observed 30%–50% restoration of CFTR function relative to non-CF controls by Ussing chamber analysis.23 This approach can be adapted to correct other mutations individually. However, because numerous non-F508del mutations, each relatively rare, are responsible for CF in patients who are not helped by CFTR modulators, a more efficient gene-therapy strategy might be the insertion of the entire CFTR cDNA into the endogenous locus.
One challenge in using Cas9/AAV to insert the CFTR cDNA is that the CFTR cDNA (4,500 bp) is close to the packaging limit of AAV (4,800 bp). As a result, there is insufficient space for the inclusion of homology arms (HAs) and other components such as a poly(A) sequence or a detection tag to enrich for the edited basal stem cells. To overcome this challenge, we applied a previously reported strategy in which sequences >5,000 bp in length were inserted sequentially using two templates containing two halves of the homologous recombination (HR) template.24 In this study, we use this strategy to insert the CFTR cDNA and an enrichment cassette expressing truncated CD19 (tCD19) in the endogenous CFTR locus of airway basal stem cells.24 Thus, our strategy enables the development of a universal gene therapy (universal strategy) for virtually all CF patients and addresses a significant hurdle in the development of ex vivo gene therapies for CF.
Results
Optimized expansion of primary UABCs
We previously reported the expansion of UABCs as monolayers in media containing epidermal growth factor (EGF), the bone morphogenetic protein (BMP) antagonist noggin, and the transforming growth factor-β (TGF-β) inhibitor A83-01 (EN media) in plates coated with tissue-culture collagen/laminin basement membrane extract (BME).23 However, BME cannot be used to produce clinical-grade cell-therapy products because it is derived from mouse sarcoma cells. Hence, we sought to identify an alternative coating material that is clinically compatible.
Upper airway tissues were obtained from CF and non-CF patients undergoing functional endoscopic sinus surgeries (FESSs). UABCs were isolated by digestion with pronase, as previously reported, and expanded using EN media.23 We screened the use of amine, carboxyl, fibronectin-functionalized plates (data not shown), and a recombinant laminin 511 coating (iMatrix). The use of iMatrix coating improved the expansion of UABCs by a factor of 1.5 ± 0.4 with respect to UABCs cultured in plates coated with BME extract (Figure 1A). To test the impact of iMatrix coating on the expansion of genome-edited UABCs, we edited non-CF UABCs using our previously reported reagents specific to the F508del mutation and expanded the edited UABCs in plates coated with BME or iMatrix.23 Edited UABCs cultured in iMatrix-coated plates showed improved expansion consistent with our observations in experiments with unmanipulated UABCs (Figure 1B). All further experiments used UABCs plated on tissue-culture plates coated with iMatrix.
Figure 1.

Expansion and characterization of UABCs
(A and B) UABCs cultured in tissue-cultured plates coated with iMatrix (recombinant laminin 511) showed improved proliferation (A) immediately after plating from tissue and (B) during subsequent passages. Error bars indicate standard deviation. (C) Telomerase activity in the expanded UABCs was measured using telomeric repeat amplification protocol (TRAP), and telomerase activity was observed in UABCs across multiple passages. (D and E) Significant shortening of telomere lengths was also not observed in expanded UABCs when telomere lengths were probed using (D) Southern blot or (E) qPCR.
Since UABCs expanded significantly on iMatrix, we were concerned about the possibility of extensive telomere erosion, which could be detrimental to the long-term durability of a cell therapy. Hence, we measured telomerase expression and telomere lengths in UABCs cultured ex vivo. We observed telomerase activity in UABCs across multiple passages spanning 3 weeks in multiple donors (Figure 1C), and we did not observe a significant shortening of telomere lengths as measured by Southern blot and qPCR (Figures 1D and 1E).
Optimizing the insertion of the full-length CFTR cDNA and enrichment of corrected basal stem cells
We first evaluated the possibility of inserting the CFTR cDNA in the endogenous locus using a single AAV. Genome editing using Cas9 results in double-stranded breaks (DSBs) that are repaired either using non-homologous end joining (NHEJ) or HR. The insertion of the CFTR cDNA requires the HR pathway, which relies on correction templates containing the cDNA of interest flanked by HAs that resemble the DSB site. Previous studies reporting high levels of HR in primary cells using AAV reported HAs of ≥400 bp.25, 26, 27 Because the CFTR cDNA (∼4,500 bp) is close to the packaging capacity of AAV (4,800−4,900 bp), there is insufficient room to include HAs of 400 bp. However, the CFTR cDNA can be packaged into AAV with small HAs of ∼50 bp and a short poly(A) sequence (<100 bp).
To determine if efficient HR can be achieved with small HAs, we used HR templates coding for green fluorescent protein (GFP), but not CFTR, containing HAs of 50, 100, 150, and 400 bp. This GFP expression cassette utilized the spleen focus-forming virus (SFFV) promoter. In UABCs edited using these GFP-expressing templates, we observed that only 10% of UABCs treated with templates containing HA of 50 bp were GFP+ and only 15% GFP+ UABCs were produced when using HR templates with 100 bp HAs (Figure S1A). Since previous studies showed that allelic correction levels above 20% were most consistent in restoring CFTR function,23 we pursued an alternate strategy that would enable the enrichment of corrected UABCs.
To enable the inclusion of longer HAs and additional genetic sequences that allow the enrichment of corrected cells, we adapted a previously reported strategy in which two halves of the full template are packaged into two separate AAV vectors, which then undergo fusion at the target genomic site by HR following a DSB.24 In this strategy, two sequential HR events, as illustrated in Figure S2, will result in the insertion of the CFTR cDNA and an enrichment cassette containing tCD19 under the control of the phosphoglycerate kinase (PGK) promoter (Figure 2A). The use of truncated human cell-surface proteins such as CD19 and the nerve growth factor receptor (tNGFR) to enrich genetically modified cells for clinical applications has been previously reported.28, 29, 30, 31, 32 We used tCD19 in this study because airway basal stem cells are known to endogenously express NGFR. Notably, the tCD19 protein does not contain the intracellular signaling domains present in the endogenous protein and is not capable of affecting cell phenotype by intracellular signaling. We do note that the extracellular portion of CD19 could be a target for chimeric antigen receptor T cell (CAR-T) therapies (Yescarta and Kymriah) or bi-specific antibody therapy (blinatumomab) if it ever became necessary to eliminate the engineered cells and their progeny as a possible safety feature. Lastly, we used the PGK promoter for expression of the tCD19 tag because the PGK promoter has been shown to not exhibit strong enhancer effects in studies comparing the activity of different promoters.33 The CFTR cDNA was codon diverged to remove the sequence corresponding to the single-stranded guide RNA (sgRNA) in exon 1 and reduce homology to the endogenous sequence. The CFTR cDNA was split at 2,883 bp (amino acid 961, methionine) from the ATG site. The sgRNA sequence from exon 1 was engineered into the first half of the HR template, and this was followed by a 400-bp random stuffer sequence that does not bear homology to any human DNA sequence (Figure 2A). This stuffer serves as the right HA for the second HR template containing the remaining CFTR cDNA, a bovine growth hormone (BGH) poly(A) tail, a PGK promoter, tCD19, and an SV40 poly(A) tail.
Figure 2.

Insertion of the CFTR cDNA in UABCs and HBECs
(A) Schematic of the universal strategy with the two halves of the CFTR cDNA and the tCD19 enrichment cassette. (B) After editing, 2%–4% of edited UABCs were tCD19+. Edited UABCs were enriched by FACS to obtain >60% tCD19+ cells. A log base 2 scale has been used to make it easier to visualize the enrichment.Error bars indicate standard deviation. (C) UABCs and HBECs from CF donors were edited using the universal strategy. In this representative sample, 83% of the edited UABCs were positive for both tCD19 and KRT5. Controls treated with AAV but without Cas9 were KRT5+ and were negative for tCD19. (D) UABCs and HBECs from donors with different genotypes were corrected using the universal strategy. In most cases, >60% of enriched UABCs were tCD19+KRT5+. Error bars indicate standard deviation. (E–H) >90% of FACS-enriched UABCs were also positive for (E) p63, (F) KRT14, (G) ITGA6, and (H) NGFR.
Primary UABCs were electroporated with the Cas9/sgRNA complex and immediately incubated with two AAVs carrying the two halves of the HR template. We first tested this strategy in UABCs derived from donors without CF. We observed 3% ± 1% tCD19+ UABCs (Figure 2B). The edited UABCs were expanded for 10−14 days, and tCD19+ cells were enriched using fluorescence-activated cell sorting (FACS). We obtained 69% ± 19% tCD19+ cells after expansion of enriched cells (Figure 2B).
We extracted genomic DNA (gDNA) from the enriched tCD19+ cells to validate the proper insertion of the CFTR cDNA in the CFTR locus. The CFTR cDNA was codon diverged from the native sequence in order to prevent premature recombination crossovers and to distinguish it from the endogenous sequence. We analyzed the presence of this codon-diverged CFTR sequence by Sanger sequencing. The CFTR locus was amplified using junction-spanning primers at the 5′ insertion site, 3′ insertion site, and junction in which the two halves of the CFTR cDNA are joined. We confirmed that the two halves of the CFTR cDNA were joined correctly by sequential HR (Figures S1B and S1C). We also amplified the beginning and the end of the CFTR cDNA insert and the tCD19 enrichment cassette and confirmed the presence of the expected sequences.
To determine the number of alleles in the pool of cells with our CFTR cDNA insert, we optimized a droplet digital PCR (ddPCR) assay. Figure S1D plots the percentage of tCD19+ cells observed with respect to the percent of alleles positive for the tCD19 sequence. The percentage of alleles modified as measured by ddPCR is approximately one-half the percentage of tCD19+ cells detected by FACS. This ratio is consistent with the insertion of the full-length CFTR cDNA and tCD19 cassette into one allele per tCD19+ cell.
Insertion of the CFTR cDNA into UABCs and HBECs obtained from donors with CF
After optimizing the gene correction and enrichment strategy on healthy donor UABCs, we tested the sequential insertion strategy in UABCs and HBECs derived from 11 different CF patients. UABCs were derived from 4 different CF patients who were all homozygous or compound heterozygous for F508del. In these 4 samples, we observed 4% ± 2% tCD19+ UABCs (Figure S1E) after editing. We started by gene editing 500,000 to 1 million UABCs (passage 1) and obtained 10−46 million UABCs within two passages post-editing (passage 3) (Figure S1F). Only 3−5 million of the expanded UABCs were used for further FACS enrichment. On average, about 100,000 ± 50,000 tCD19+ cells were obtained after FACS. These were expanded to obtain 500,000−2 million cells after expansion for 1 passage. If all of the corrected cells had been FACS sorted, we estimate that up to 38 million cells could have been obtained after enrichment and expansion for one passage (Figure S1F). We obtained 68% ± 12% tCD19+ UABCs that were also cytokeratin (KRT)5+ after FACS enrichment and expansion in culture for 4 days (Figures 2C and 2D). On day 4 after FACS enrichment, >90% of the corrected UABCs were also positive for KRT5, P63, KRT14, integrin-alpha 6 (ITGA6), and NGFR (Figures 2E−2H).
In order to test the ability of our strategy to restore CFTR function in patients with other genotypes including class I nonsense mutations such as W1282X and R553X, we obtained HBECs derived from 7 different donors from 4 different sources. In HBECs derived from these 7 different donors with CF, we observed 10% ± 5% tCD19+ cells (Figure S1E) after editing and 72% ± 19% tCD19+ KRT5+ HBECs after enrichment (Figure 2D). Edited UABCs and HBECs stably express tCD19 only if both halves of the HR template are integrated into the genome. Episomal expression of tCD19 driven by the PGK promoter was measured using controls treated with only AAV, and significantly fewer cells (p < 0.05) in the control group expressed tCD19 compared to edited samples at the time of enrichment (Figure S1E). Thus, we successfully inserted the cDNA in UABCs and HBECs from 11 different donors with CF and enriched the cells containing the full CFTR cDNA using the tCD19 expression on the cell surface.
Restoration of CFTR function in CF patient samples
The restoration of CFTR function was tested on epithelial sheets generated by culturing the edited and enriched UABCs and HBECs at air-liquid interface (ALI).23 UABCs differentiated in ALI cultures for 28−35 days produced differentiated epithelial sheets containing ciliated cells expressing acetylated alpha-tubulin and secretory cells expressing MUC5AC oriented toward the apical surface (Figure 3A). Moreover, the transepithelial resistances of differentiated epithelial sheets produced by UABCs cultured on BME and iMatrix maintained high transepithelial resistances, indicative of the production of a well-differentiated epithelium (Figure 3B). The percent of tCD19+ alleles in the edited and enriched cells was not significantly different before and after differentiation (paired t test, p = 0.29) indicating that the corrected UABCs did not have a proliferative disadvantage (Figure 3C).
Figure 3.

Restoration of CFTR expression and function in differentiated epithelial sheets
(A) Epithelial sheets generated by differentiation of UABCs (cultured on iMatrix) in air-liquid interfaces (ALIs) contained cells positive for KRT5, acetylated tubulin, and mucin 5AC. Scale bar corresponds to 10 μm. (B) Epithelial sheets generated from UABCs cultured on iMatrix (unedited and universal edited) showed trans-epithelial resistances that were not significantly different from epithelial sheets generated from UABCs cultured on BME. (C) The percent alleles modified was quantified both right after sorting and after differentiation for 28−35 days on ALI cultures. The percent of tCD19+ alleles was not significantly different between these time points, indicating that the modified cells did not have any disadvantage in proliferation when compared to the unmodified tCD19− cells. (D) Western blot probing CFTR expression. CFTR expression in Calu-3 cells was used as a positive control (lane 1). Samples were diluted by 1/10 relative to other samples since Calu-3 cells are known to express high levels of CFTR. Lane 2 shows strong mature CFTR expression (band C) in a corrected CF sample, which is comparable to the mature CFTR expression (band C) observed in a non-CF control shown in lane 4. Uncorrected cells (F508del/3659delC) obtained from the same donor as the corrected cells used in lane 2 show reduced mature CFTR expression (band C, lane 3). (E) Representative traces obtained from epithelial sheets by Ussing chamber analysis in one non-CF control sample and one CF sample from donor 1 before and after correction. The traces from the corrected sample show similar forskolin and CFTRinh-172 responses as the non-CF control sample. (F) CFTRinh-172 short-circuit currents observed in corrected CF samples from 11 different donors as a percentage of the average CFTRinh-172 currents observed in non-CF controls. (G) CFTRinh-172 short-circuit currents observed in non-CF controls and epithelial monolayers derived from CF UABCs before and after correction. (H) CFTRinh-172 short-circuit currents observed in non-CF controls and epithelial monolayers derived from CF HBECs before and after correction.
We first measured CFTR expression in ALI cultures using immunoblotting. Figure 3D shows a western blot probing CFTR expression using anti-CFTR primary antibody Ab450 in non-CF, uncorrected, and corrected CF samples. The non-CF control shows one band corresponding to fully glycosylated mature CFTR (band C, 170−180 kDa) and incompletely glycosylated CFTR (band B) (lane 4). The CF sample shows a weak band C (lane 3) indicating impaired glycosylation of CFTR. The sample used in this study is a compound heterozygous sample (F508del/3659delC) with a frameshift present close to the end of the CFTR protein. The corrected sample showed an increase in band C intensity, demonstrating an increase in mature CFTR protein, relative to the uncorrected CF sample (lane 2).
CFTR function was quantified by measuring short-circuit currents from ALI-differentiated epithelial sheets. Representative traces obtained from an Ussing chamber assay for a CF sample before and after correction are shown in Figure 3E. Consistent with the observation of restored mature CFTR expression seen in the western blot, we observed restoration of CFTR function as indicated by the restoration of both the forskolin-stimulated short-circuit current and the inhibitable short-circuit current responsive to the selective CFTR inhibitor CFTRinh-172. The magnitudes of the responses to forskolin and CFTRinh-172 are similar to those observed in epithelial sheets derived from non-CF control donors. The CFTRinh-172 responses observed in corrected epithelial sheets derived from 11 different CF patients are presented in Table 1.
Table 1.
Summary of percent editing of CF upper airway basal cells (UABCs) and human bronchial epithelial cell (HBEC) samples and relative restoration of CFTR function as measured by responses to CFTRinh-172
| Patient | Genotype | Editing efficiency (% tCD19+ cells) | Raw-inhibitable CF current μA/cm2 | Percentage of non-CF-inhibitable current |
|---|---|---|---|---|
| UABCs | ||||
| WT average: −19 ± 11 μA/cm2 | ||||
| 1 | ΔF/ΔF | 67.7 | −13.1 | 69.81 |
| 1 | ΔF/ΔF | 67.7 | −12.78 | 68.10 |
| 1 | ΔF/ΔF | 67.7 | −8.96 | 47.75 |
| 1 | ΔF/ΔF | 67.7 | −7.8 | 41.57 |
| 2 | ΔF/3659delC | 56.8 | −9.15 | 48.76 |
| 2 | ΔF/3659delC | 56.8 | −24.8 | 132.16 |
| 2 | ΔF/3659delC | 56.8 | −28.74 | 153.15 |
| 3 | ΔF/ΔF | 68.5 | −8.79 | 46.84 |
| 3 | ΔF/ΔF | 68.5 | −6.14 | 32.72 |
| 4 | ΔF/ΔF | 82.3 | −25.59 | 136.37 |
| 4 | ΔF/ΔF | 82.3 | −10.75 | 57.29 |
| 4 | ΔF/ΔF | 82.3 | −9.04 | 48.17 |
| HBECs | ||||
| WT average: −18 ± 9 μA/cm2 | ||||
| 5 | ΔF/ΔF | 57.0 | −10.9 | 57.31 |
| 5 | ΔF/ΔF | 57.0 | −11.72 | 61.62 |
| 6 | ΔF/ΔF | 79.0 | −22.31 | 117.29 |
| 6 | ΔF/ΔF | 79.0 | −16.99 | 89.32 |
| 6 | ΔF/ΔF | 79.0 | −19.53 | 102.68 |
| 6 | ΔF/ΔF | 79.0 | −17.63 | 92.69 |
| 7 | ΔF/N1303K | 74.8 | −17.6 | 92.53 |
| 7 | ΔF/N1303K | 74.8 | −18.36 | 96.53 |
| 7 | ΔF/N1303K | 74.8 | −20.07 | 105.52 |
| 7 | ΔF/N1303K | 74.8 | −15.14 | 79.60 |
| 7 | ΔF/N1303K | 74.8 | −27.75 | 145.89 |
| 8 | 1717-1G/N1303K | 79.1 | −28.94 | 152.15 |
| 8 | 1717-1G/N1303K | 79.1 | −10.9 | 57.31 |
| 9 | ΔF/W1282X | 79.0 | −11.61 | 61.04 |
| 9 | ΔF/W1282X | 68.0 | −12.04 | 63.30 |
| 9 | ΔF/W1282X | 79.0 | −13.06 | 68.66 |
| 9 | ΔF/W1282X | 68.0 | −15.07 | 79.23 |
| 10 | W1282X/W1282X | 89.2 | −12.75 | 67.03 |
| 10 | W1282X/W1282X | 80.9 | −16.02 | 84.2239664 |
| 11 | W1282X/R553X | 73.1 | −16.1 | 84.6445604 |
CFTR contains 1,480 amino acids. The mutations presented here occur in the first 1/3 or the last 1/3 of the CFTR protein. ΔF refers to the F508del mutation.
Epithelial sheets derived from corrected UABCs and HBECs obtained from the 11 different donors with CF showed CFTRinh-172 responses that were 30%–150% of the CFTRinh-172 responses seen in non-CF controls (Table 1; Figure 3F). Corrected CF UABC cultures showed CFTRinh-172-sensitive short-circuit currents of 14 ± 8 μA/cm2 (74% ± 42% relative to non-CF), which was significantly higher than currents of 1 ± 2 μA/cm2 seen in uncorrected CF controls (adjusted p value = 0.0085, ANOVA followed by Tukey test). Non-CF UABC cultures showed CFTRinh-172-sensitive short-circuit currents of 19 ± 11 μA/cm2, which is not significantly different from the CFTRinh-172 responses seen in corrected UABC cultures (adjusted p value = 0.3540) (Figures 3F and 3G). Corrected CF HBEC cultures showed CFTRinh-172-sensitive short-circuit currents of 17 ± 5 μA/cm2 (88% ± 27% relative to non-CF), which was significantly higher than currents of 2 ± 1 μA/cm2 seen in uncorrected CF controls (adjusted p value < 0.0001, ANOVA followed by Tukey test). This indicates the ability of the universal strategy to restore CFTR function in samples with different genotypes (Table 1). Non-CF HBEC cultures showed CFTRinh-172-sensitive short-circuit currents of 18 ± 9 μA/cm2, which is not significantly different from the CFTRinh-172 responses seen in corrected UABC cultures (adjusted p value = 0.6989) (Figures 3F and 3H).
Off-target (OT) activity of sgRNA
The activity of Cas9 is directed to a target location in the genome by means of the sgRNA. However, Cas9 can tolerate mismatches between the sgRNA and the gDNA resulting in potential OT activity. We used in silico methods to identify over 50 possible OT sites and characterized the activity of the sgRNA at the top 47 predicted OT sites (Table S1) using both wild-type Cas9 (WT-Cas9) and high-fidelity Cas9 (HiFi Cas9), which was previously reported to be effective in maintaining high on-target activity while lowering OT activity34 (Figure S3; Table S1). We observed significant OT activity in OT-3. We observed insertions or deletions (indels) in 57% ± 3% alleles when we used WT-Cas9. However, this OT activity was reduced by >30-fold to 1.2% ± 0.4% indels by using HiFi Cas9. Consistent with previous reports, we did not observe any reduction in on-target activity as measured by indels (Figure S3).34 The OT site (OT-3) corresponds to an intergenic region that has no known function and is located 10 kbp away from the nearest gene, which codes for sideroflexin 1, which is a mitochondrial serine transporter. Table S1 presents the sequences and genes associated with each of the 47 OT sites.
No enrichment of edited cells with mutations in genes associated with solid tumors
Studies have reported that the ex vivo editing of cells may result in selection against cells with a functional p53 pathway and thus increase the risk of tumor formation if these cells were used for therapy.35,36 To characterize the enrichment of cells with increased tumor-forming potential, we used the Stanford Solid Tumor Actionable Mutation Panel (STAMP) to detect the enrichment of clinically actionable mutations in 130 genes associated with the formation of solid tumors in edited and enriched UABCs obtained from 1 donor with CF and HBECs obtained from 2 different donors.37, 38, 39, 40 No sample showed any mutation in TP53 with or without editing. Mutations were only observed in 1−3 genes out of the 130 genes listed in the panel (Table S2). However, these mutations were also present in the control unedited samples from the same donors, and the variable allele frequency (VAF) was not different between the control and edited samples. Because the identical mutations were present in both the edited and unedited samples, it means that the mutations must have been present in the cells originally (the shared common ancestor) and demonstrates that the engineering process did not generate them.
Discussion
Several previous studies have attempted to add a functional copy of the CFTR cDNA using viral and non-viral methods both in vitro and in vivo.6, 7, 8,13,14,41,42 In these reports, the expression of CFTR was driven by exogenous promoters, and the CFTR expression cassette was not inserted in the endogenous CFTR locus. Nevertheless, in vitro studies in human samples and an in vivo study in a CF pig model observed restoration of CFTR function following gene replacement.13,14 The development of CRISPR-Cas9-mediated genome editing enables the insertion of the CFTR cDNA in the endogenous locus and thus should enable the preservation of native CFTR expression, including in ciliated cells and ionocytes. However, it was unknown if the addition of the full-length CFTR cDNA in the endogenous locus under the control of the native promoter would be sufficient to restore CFTR function. Since the CFTR cDNA with the appropriate HAs cannot be packaged into one AAV capsid, insertion of partial CFTR cDNAs into introns 7 and 8 of the CFTR gene has been attempted, and this approach can potentially be used to treat ∼89% of CF patients.43 This approach, which did not enrich for corrected cells, was tested in four CF patient samples and resulted in CFTR function that was 30%–50% relative to non-CF controls. Our results show that the insertion of the full-length CFTR cDNA, which can treat >99.5% of CF patients, results in CFTR function similar to that seen in non-CF controls (Figures 3F−3H).
Concerns about the restoration of CFTR function using this universal strategy stem from previous studies that reported the importance of introns in regulating CFTR expression.44,45 Our results show that insertion of the CFTR cDNA in exon 1 of the endogenous locus can successfully restore CFTR function in airway epithelial sheets generated from the UABCs and HBECs of CF patients. The successful restoration of CFTR might be enabled by the fact that all of the exons and introns downstream of exon 1 are left intact when cells are corrected using the universal strategy. This result is also consistent with another study, which showed that the insertion of a partial CFTR cDNA construct into intron 8 did not disrupt the open chromatin profile of the CFTR locus.43 Future studies characterizing the open chromatin profile of corrected UABCs and HBECs will further inform us about the influence of our universal strategy on the regulation of both CFTR and neighboring genes.
Although our therapeutic approach is applicable to all CF patients harboring mutations in any combination of exons or introns, it is particularly relevant for patients with mutations that do not respond to recently developed CFTR modulators. These patients often have class I nonsense mutations such as W1282X and exhibit a very severe disease burden and would not be responsive to current CFTR modulator therapies. Significantly, our experiments demonstrate the feasibility of restoring CFTR function in the presence of undruggable nonsense mutations such as W1282X and R553X (Table 1). Our approach thus enables the development of an autologous airway stem cell therapy for all CF patients affected by mutations in any of the exons or introns of the CFTR gene including patients with nonsense mutations. Additional strategies may be needed to treat patients with biallelic mutations in the promoter, estimated to be <0.5% of the CF population.
Our experiments further show that the corrected UABCs and HBECs produce differentiated epithelial sheets with restored CFTR function (Figure 3). In epithelial sheets produced from corrected UABCs and HBECs obtained from 11 different CF patients, we measured restored CFTR-mediated chloride transport that was, on average, >70% of the levels seen in non-CF controls, with individual readings ranging from 32% to 150% of control values. Several previous studies have shown that when CF cells were either mixed with cells transduced with viruses inducing the overexpression of CFTR42,46, 47, 48 or WT cells,49,50 the presence of 10%–25% CFTR-expressing cells was sufficient to restore CFTR function close to levels in non-CF controls. Other studies have investigated CFTR function in patients with milder CF phenotypes associated with improved survival, and these studies also observed as little as 2%–15% CFTR function relative to WT.51,52 Our studies indicate that the universal strategy can restore CFTR function well beyond this 15% threshold and enables the restoration of CFTR function to levels seen in asymptomatic CF carriers.51,52
In addition to validating the universal CFTR correction strategy, our studies also advance our ability to generate a clinical cell-therapy product by replacing xenobiotic reagents such as BME with reagents compatible with good manufacturing practices. We previously reported the feeder-free culture of UABCs in a chemically defined media formulation.23 However, the UABCs were plated on tissue-culture-treated plates coated with BME derived from mouse sarcoma cells. Previous studies have reported the improved expansion of airway basal stem cells using laminin-enriched conditioned media along with dual-SMAD (small mothers against decapentaplegic) inhibition.53 Improving upon these reports, the results from this current study indicate that recombinant laminin 511 (iMatrix) is an effective replacement for BME that further improves our ability to expand UABCs using reagents compatible with the production of airway stem cells for clinical applications. Moreover, the primary UABCs cultured in this manner do not experience a significant shortening of their telomeres, presumably due to the retention of some telomerase activity (Figures 1C−1E). Telomerase activity has been reported to be associated with the self-renewal potential of hepatocytes and hematopoietic stem cells.54,55 In addition, telomerase activity has also been reported in airway cells obtained from freshly isolated nasal tissue.56 Thus, our results suggest that the durability of the primary UABCs used in our studies is unlikely to be limited by shortened telomeres. Lastly, previous studies in hematopoietic cells have reported that efficient HR is facilitated by improved cell proliferation,27 and these results were also consistent with our previously reported findings that improved culture conditions facilitate HR in airway stem cells.23 In this study, we did not evaluate if the modified culture conditions improved HR significantly.
In addition to assessing the potential durability of the gene-corrected stem cells, it is also important to test their safety. Previous studies that reported the enrichment of cells with a dysfunctional TP53 pathway used immortalized cells or primary cells from model organisms such as mice.35,36 Here, we have quantified the presence of mutations in TP53 and an additional 129 genes associated with solid tumors in gene-corrected primary UABCs and HBECs from humans. We did not observe any increase in the frequency of alleles with mutations in genes associated with solid tumors. These results are consistent with previous reports from our group, which did not observe any enrichment of alleles with mutations in TP53 in gene-edited hematopoietic stem and progenitor cells and iPSCs.37,57 Thus, our results indicate that our universal ex vivo strategy for CFTR correction may be safe for further clinical translation.
We previously reported our ability to embed corrected UABCs on a porcine small intestinal submucosal (pSIS) membrane that is US Food and Drug Administration (FDA) approved for use in facilitating sino-nasal repair after surgeries.23 In that previous study, we reported the need for 1.3 million UABCs in order to fully cover one maxillary sinus.23 The use of the optimized culture protocols reported here enables us to obtain this yield (Figure S1F). These studies thus build the foundation for the development of an autologous gene-corrected stem cell therapy for all CF patients. The transplantation of gene-corrected airway stem cells and the restoration of mucociliary transport in vivo are still significant hurdles that must be overcome for the clinical translation of this approach. Future studies will first investigate the transplantation of UABCs embedded on the pSIS membrane or other biomaterials into the sinuses of animal models to treat CF sinus disease, an important unmet need and common morbidity in nearly all CF patients. The knowledge developed in this process will be useful in optimizing the autologous transplantation of gene-corrected airway basal stem cells into the lower airways as a durable stem cell therapy for CF lung disease.
Conclusion
Our experiments show that the full-length CFTR cDNA along with an enrichment cassette expressing tCD19 can be inserted into the endogenous CFTR locus in airway basal stem cells using Cas9 and two AAV6 vectors containing two halves of the HR template. The corrected airway basal stem cells differentiate to produce epithelial sheets that exhibit restored CFTR-mediated chloride transport, which is, on average, 70%−80% of the levels seen in non-CF controls. Our findings enable the further development of a durable therapeutic option for all CF patients, especially those patients who cannot be treated using existing modulator therapies. In addition, the findings enable future studies to optimize the transplantation of gene-corrected UABCs into the upper airway epithelia of CF patients to treat CF sinus disease, as well as strategies to develop an autologous gene-corrected airway stem cell therapy for CF lung disease.
Materials and methods
Subject details
Upper airway tissue was obtained from adult patients undergoing FESS after obtaining informed consent. The protocol was approved by the Institutional Review Board at Stanford University. The CFTR mutations from each subject were recorded. HBECs were obtained from Lonza, the Primary Airway Cell Biobank at McGill University, the Cystic Fibrosis Foundation Cell Bank, or The University of North Carolina. CF bronchial epithelial cells were obtained from lungs explanted during transplantation under protocol #13-1396, approved by the Committee on the Protection of the Rights of Human Subjects at The University of North Carolina at Chapel Hill. All donors provided informed consent.
Method details
EN media
Advanced Dulbecco's Modified Essential Medium/Nutrient mixture F-12 (ADMEM/F12) was supplemented with B27 supplement, nicotinamide (10 mM), human EGF (50 ng/mL), human noggin (100 ng/mL), A83-01 (500 nM), N-acetylcysteine (1 mM), and HEPES (1 mM).
Cell culture of UABCs
Upper airway tissue pieces were cut into small pieces (1−2 mm2). Tissue pieces were washed with 10 mL sterile PBS with 2× antibiotic/antimycotic (penicillin, streptomycin, amphotericin B; Gibco; #15240062) on ice and digested with pronase (1.5 mg/mL; Sigma; #P5147) for 2 h at 37°C or at 4°C overnight. Digestion was stopped using 10% fetal bovine serum (FBS). Digested tissue was filtered through cell strainers (BD Falcon; #352350) into a sterile 50-mL conical tube. The mixture was centrifuged at 600 × g for 3 min at room temperature. Red blood cell (RBC) lysis was then performed using RBC lysis buffer (eBioscience) as per the manufacturer’s instructions. After RBC lysis, cells were suspended in 1 mL EN media and counted. A small sample was fixed using 2% paraformaldehyde and permeabilized using Tris-buffered saline with 0.1% Tween 20 (TBST). Cells were stained for KRT5 (Abcam; ab193895). An isotype control (Abcam; ab199093) was used to assess non-specific staining. KRT5+ cells were plated at a density of 10,000 cells per cm2 as monolayers in tissue-culture plates. At the time of plating, 4.5 μL of iMatrix per well of a 6-well plate was added immediately after the addition of EN media into the wells. Cells were incubated at 37°C in 5% O2 and 5% CO2 in EN media with 10 μM Rho-associated, coiled-coil containing protein kinase (ROCK) inhibitor (Y-27632; Santa Cruz; sc-281642A). Cells obtained from CF patients were grown in EN media supplemented with additional antimicrobials for 2 days (fluconazole, 2 μg/mL; amphotericin B, 1.25 μg/mL; imipenem, 12.5 μg/mL; ciprofloxacin, 40 μg/mL; azithromycin, 50 μg/mL; meropenem, 50 μg/mL). The concentration of antimicrobials was decreased by 50% after 2−3 days and then withdrawn after editing (days 5−6).
Cell culture of HBECs
HBECs were cultured in PneumaCult-Ex Plus at 3,000−10,000 cells/cm2 in tissue-culture flasks without any coating.
Gene editing of UABCs
UABCs and HBECs were edited as previously reported but with sgRNA and HR templates specific to the universal correction strategy.23 Briefly, cells were cultured in EN media with 10 μM ROCK inhibitor (Y-27632; Santa Cruz; sc-281642A). Gene correction was performed 5 days after plating. Cells were detached using TrypLE Express Enzyme (Gibco; 12605010). 5 × 105−1 × 106 cells were resuspended in 100 μL Opti-MEM (Gibco; 31985062) resulting in a density of 5−10 million cells/mL. Electroporation (nucleofection) was performed using Lonza 4D Nucleocuvette vessel (Lonza; V4XP-3024). 30 μg of WT-Cas9 (Integrated DNA Technologies, Coralville, IA, USA; cat. no. 1074182) or HiFi Cas9 (Integrated DNA Technologies, Coralville, IA, USA; cat. no. 1081061) and 16 μg of 2′-O-methyl (M), 2′-O-methyl 3′phosphorothioate modified sg-RNA (MS-sgRNA) (TriLink BioTechnologies, San Diego, CA, USA) (molar ratio = 1:2.5) were complexed at room temperature for 10 min, mixed with the cells suspended in 100 μL of Opti-MEM, and transferred to a Nucleocuvette vessel. Cells were electroporated using the program CA-137. 400 μL of Opti-MEM was added to each well after electroporation. Two AAVs carrying the two halves of the CFTR cDNA and the tCD19 enrichment cassette were added at a multiplicity of infection (MOI) of 105 genomes per cell (as determined by ddPCR). Our previous study reported an optimal MOI of 106 genomes per cell. However, those experiments used qPCR to quantify AAV genomes, which overestimates the number of AAV genomes. Functional titration experiments were used to determine the optimal AAV MOIs for this study. Cells, along with the AAVs, were transferred into multiple wells of a 6-well plate such that the cell density was ∼5,000 cells/cm2. 1.5 mL of EN media, followed by 4.5 μL of iMatrix, was added to each well. Media were replaced 48 h after electroporation.
UABCs were passaged 4−5 days after editing in order to expand the edited cells for FACS enrichment. Passaged UABCs were plated in tissue-culture-treated flasks with a surface area of 225 cm2. 100 μL of iMatrix was added to each flask in addition to 25 mL of EN media. UABCs were suspended by trypsinization 4−5 days after passaging and enriched for tCD19+ cells using FACS. tCD19 was detected using an anti-human CD19 antibody (BioLegend; 302206)
Gene editing of HBECs
The same protocol used for UABCs was used for HBECs except that the cells were cultured in PneumaCult-Ex Plus supplemented with 10 μM ROCK inhibitor.
FACS enrichment of edited UABCs and HBECs
Edited UABCs and HBECs were suspended by treatment with TrypLE Express Enzyme (Gibco; 12605010). UABCs were incubated with anti-human CD19 antibodies conjugated with FITC for 20−30 min. Cells were also incubated with 7-AAD for 10 min in order to label non-viable cells. The labeled cells were washed three times with FACS buffer (PBS with 1% BSA and EDTA) and resuspended in FACS buffer. The UABCs and HBECs were FACS sorted using FACSAria II SORP (BD Biosciences). For each experiment, cells that were unmodified and cells that were treated with only the AAVs but no Cas9 were used as controls in order to identify FACS settings that specifically detected genome-edited tCD19+ UABCs and HBECs.
Measuring gene correction using analytical flow cytometry
Cells were expanded for 4−5 days after FACS sorting. The percent of tCD19+ cells was quantified using a FACS analyzer (Beckman Coulter; CytoFLEX). Cells were also stained for other basal stem cell markers such as KRT14 (Abcam; ab181595), P63 (BioLegend; 687202), ITGA6 (BioLegend; 313608), and NGFR (BioLegend; 345106).
Measuring gene correction using ddPCR
Correction was also validated using ddPCR. gDNA was extracted from cells using Quick Extract (QE; Lucigen; QE09050). The cells were suspended by trypsinization and washed once with Opti-MEM or media. 50−100,000 cells were centrifuged, and the supernatant was discarded. 50 μL QE was added to the cell pellet and vortexed. The QE suspension was heated at 65°C for 6 min, vortexed, and then heated at 98°C for at least 10 min to inactivate QE. We observed that inactivation for less than 10 min often resulted in failed PCR. The modified CFTR locus was amplified using the primers forward: 5′-AGCATCACAAATTTCACAAATAAAGCA-3′ and reverse: 5′-ACCCCAAAATTTTTGTTGGCTGA-3′; and amplification was quantified using theprobe sequence: 5′-CACTGCATTCTAGTTGTGGTTTGTCCA-3′. A region in intron 1 of CFTR was used as a reference for allele quantification using ddPCR. The reference region was amplified using the primers forward: 5′-TGCTATGCCAGTACAAACCCA-3′ and reverse: 5′-GGAAACCATACTTCAGGAGCTG-3′; and amplification was quantified using the probe sequence: 5′-TTGTTTTTGTATCTCCACCCTG-3′.
Measuring OT activity
Primary UABCs from non-CF patients were electroporated with Cas9 and sgRNA without the HR template. gDNA was extracted 4−5 days after ribonucleoprotein (RNP) delivery. Potential OT sites were identified using the bioinformatic tool COSMID,58 allowing for 3 mismatches within the 19 protospacer adjacent motif (PAM) proximal bases. Predicted OT loci were initially enriched by locus-specific PCR, followed by a second round of PCR to introduce adaptor and index sequences for the Illumina MiSeq platform. All amplicons were normalized, pooled, and quantified using a Qubit (Thermo Fisher Scientific) and were sequenced using a MiSeq Illumina using 2 × 250 bp paired-end reads. Indels at potential OT sites were quantified as previously described.59
Briefly, paired-end reads from MiSeq were filtered by an average Phred quality (Qscore) greater than 20. Single reads were quality trimmed using cutadapt and trim galore prior to merging of paired-end reads using fast length adjustment of short reads (FLASH). Alignments to reference sequences were performed using Burrows-Wheeler aligner (BWA) for each barcode. Percentage of reads containing indels with a ±5-bp window of the predicted cut sites was quantified.
ALI culture of corrected UABCs and HBECs
Gene-corrected cells were plated either on the day of FACS sorting or after expansion for 4−5 days. 30,000 to 60,000 cells per well were plated in 6.5 mm Transwell plates with a 0.4-μm pore polyester membrane insert (Corning; 07-200-154). EN media were used to expand cells until they were confluent (3−7 days). Once cells were confluent in Transwell inserts and stopped translocating fluid between the upper and lower chambers, media in the bottom compartment were replaced with ALI media previously reported by Gentzsch et al obtained from the University of North Carolina (UNC).60
Immunoblot
Immunoblotting methods were used to compare CFTR protein expression pre/post-correction. Non-CF nasal cells and patient-derived CF cells pre/post-correction were plated and cultured according to the above methods. Lysis was performed by incubating cells for 15 min in ice-cold radioimmunoprecipitation assay (RIPA) buffer supplemented with EDTA-free protease inhibitor. Lysates were gathered and rotated at 4°C for 30 min and then spun at 10,000 × g at 4°C for 10 min to pellet insoluble genomic material. The supernatant was collected and mixed with 4× Laemmli sample buffer containing 100 mM DTT and subsequently heated at 37°C for 30 min. Lysate from ∼10,000 cells was loaded for the Calu-3 lane, and lysates from ∼100,000 cells were loaded for non-CF nasal, uncorrected, and corrected CF cells; fractionated by SDS-PAGE; and then transferred onto a polyvinylidene fluoride (PVDF) membrane. Blocking was performed with 5% nonfat milk in TBST (10 mM Tris, pH 8.0, 150 mM NaCl, 0.5% Tween 20) for 60 min. The membrane was probed with antibodies against CFTR (Ab450, 1:1,000) and histone H3 (1:10,000). Membranes were washed and incubated with a 1:10,000 dilution of horseradish peroxidase-conjugated anti-mouse for 1 h and then developed by SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific; 34095).
Ussing chamber functional assays
Ussing chamber measurements were performed 3−5 weeks after cells had stopped translocating fluid as described before.23 For chloride-secretion experiments with UABCs and HBECs, solutions were as follows (in mM): apical: Na(gluconate) 120, NaHCO3 25, KH2PO4 3.3, K2HPO4 0.8, Ca(gluconate)2 4, Mg(gluconate)2 1.2, and mannitol 10 and basolateral: NaCl 120, NaHCO3, 25, KH2PO4 3.3, K2HPO4 0.8, CaCl2 1.2, MgCl2 1.2, and glucose 10. The concentration of ion channel activators and inhibitors was as follows: amiloride (10 μM, apical), forskolin (10 μM, bilateral), VX-770 (10 μM, apical), CFTRinh-172 (20 μM, apical), and uridine triphosphate (UTP; 100 μM, apical).
Protein, RNA, and gDNA extractions from single UABC pellets for telomerase and telomere measurements
Frozen cell pellets containing ∼1 × 106 UABCs were resuspended in 100 μL of NP40 lysis buffer (25 mM HEPES, 150 mM KCl, 1.5 mM MgCl2, 0.5% NP-40, 10% glycerol, pH adjusted to 7.5 with KOH, with freshly added 1 mM DTT, 200 μM PMSF, and 1:1,000 protease inhibitor cocktail [Sigma; P8340]). Soluble supernatant was used for telomerase detection by TRAP (telomeric repeat amplification protocol), western blotting, as well as soluble RNA extraction; insoluble pellet is sufficient for gDNA extraction for telomere length measurement by Telo-qPCR and TRF (telomere/terminal restriction fragment) analyses.
For TRAP, 2 μg of protein extract per Bradford was programed into a 25-μL of “cold extension” reaction, 0.5 μL from which was subsequently PCR amplified in the presence of p32-labeled telomere strand (TS) primers, according to Chen et al.61 Total RNA was TRIzol extracted from the same soluble extract and subjected to qPCR analysis for telomerase reverse transcriptase (TERT) mRNA expression, which tightly correlates with TRAP activity (data not shown).
For telomere length measurements, gDNA from the insoluble pellet was resuspended in tail lysis buffer containing protease K and then extracted by phenol-chloroform. 10 ng of gDNA was used in Telo-qPCR reactions. Telomeric amplicon was PCR amplified with 5′-ACACTAAGGTTTGGGTTTGGGTTTGGGTTTGGGTTAGTGT-3′ and 5′-TGTTAGGTATCCCTATCCCTATCCCTATCCCTATCCCTAACA-3′; albumin housekeeping amplicon was amplified with 5′-CGGCGGCGGGCGGCGCGGGCTGGGCGGAAATGCTGCACAGAATCCTTG-3′ and 5′-GCCCGGCCCGCCGCGCCCGTCCCGCCGGAAAAGCATGGTCGCCTGTT-3′, according to Cawthon.62 For TRFs, 2 μg of gDNA was digested with a restriction enzyme cocktail, containing HinfI, RsaI, MspI, and AluI for 4 h at 37°C. In-gel Southern hybridization was performed with a 3× 5′-TTAGGG-3′ p32-labeled probe, according to Herbert et al.63
Stanford STAMP
UABCs and HBECs obtained from donors with CF were corrected and enriched using FACS. The FACS-enriched cells and control cells were then expanded for 1−2 passages to obtain ∼1 million cells per condition. The gDNA was extracted using the GeneJET gDNA Purification Kit (Thermo Fisher Scientific; cat. no. K0722). Next-generation sequencing of samples was performed at the Stanford Molecular Genetic Pathology Clinical Laboratory to quantify mutations in 130 genes (e.g., TP53, EGFR, KRAS, NRAS) identified in the Stanford STAMP.38, 39, 40 The STAMP assay is clinically validated and can detect variants with a variant allele fraction as low 5%.
The assay was performed as previously described to assess the enrichment of tumor-forming mutations in gene-edited hematopoietic cells using a similar panel.37 Briefly, the gDNA was sheared (M220-focused ultrasonicator; Covaris, Woburn, MA, USA), sequencing libraries were prepared (KK8232 KAPA LTP (low throughput) Library Preparation Kit Illumina Platforms; KAPABiosystems, Wilmington, MA, USA), and hybridization-based target enrichment was performed with custom-designed oligonucleotides (Roche NimbleGen, Madison, WI, USA). Illumina sequencing instruments were used to sequence pooled libraries (MiSeq or NextSeq 500 Systems; Illumina, San Diego, CA, USA). Sequencing results are analyzed with an in-house-developed bioinformatics pipeline. Sequence alignment against the human reference genome hg19 is performed with BWA in paired-end mode using the BWA-maximal exact match (MEM) algorithm and standard parameters. Variant calling was performed separately for single nucleotide variants (SNVs), indels <20 bp, and fusions. VarScan version (v.)2.3.6 was used for calling SNVs and indels, and FRACTERA v.1.4.4 was used for calling fusions. Variants are annotated using ANNOVAR and Ensembl reference transcripts.
Acknowledgments
We appreciate the efforts of Ivan T. Lee, Nicole Borchard, Sachi Dholakia, David Zarabanda, Phillip Gall, and Yasuyuki Noyama in prioritizing efforts at human tissue acquisition from the operating theater. We thank the CF Canada Primary Airway Cell Biobank at McGill University and the American Cystic Fibrosis Foundation Cell Bank for providing primary bronchial airway epithelial cells. CFTR antibodies were obtained from Dr. John Riordan at The University of North Carolina at Chapel Hill through the CF Foundation Antibody Distribution Program. The work was funded by grants from the California Institute of Regenerative Medicine (DISC2-09637), Stanford-SPARK MCHRI; NIH (K08DK124684 to Z.M.S, K08DE027730 to A.A.S., and U01DK085527, U01CA217851, U01CA176299, and U19AI116484 to C.J.K.); Cancer Prevention and Research Institute of Texas (RR14008 to G.B.); and Cystic Fibrosis Foundation (BAO19XX0). We thank the Crandall Foundation for a philanthropic gift that supported this work. S.V. and Z.M.S. were supported by a fellowship from the Cystic Fibrosis Foundation (VAIDYA19F0 and SELLER16L0). S.H.R. acknowledges funding from the NIH (DK065988) and CF Foundation (BOUCHE19RO).
Author contributions
Conceptualization, S.V. and M.H.P.; methodology, J.V.N., Z.M.S., T.J.D., L.C., D.T.B., J.J.W., S.E.A., C.J.K., G.B., C.M.L., S.H.R., and C.J.S.; investigation, S.V., R.B., L.C., Z.M.S., D.T.B., S.M.A., C.M.L., T.H.D., A.A.S., C.A.T., and A.M.D.; writing, S.V., R.B., L.C., Z.M.S., D.T.B., S.M.A., C.M.L., T.H.D., A.A.S., C.A.T., A.M.D., J.V.N., T.J.D., J.J.W., S.E.A., C.J.K., G.B., S.H.R., C.J.S., and M.H.P. All authors participated in the design and conception of experiments and provided editorial feedback on the manuscript.
Declaration of interests
M.H.P. has equity and serves on the Scientific Advisory Board of CRISPR Therapeutics and Graphite Bio. J.V.N. is a consultant with COOK Medical, which manufactures the pSIS graft. Neither company had any input on the design, execution, interpretation, or publication of the work in this manuscript.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2021.03.023.
Contributor Information
Sriram Vaidyanathan, Email: svaidy@stanford.edu.
Matthew H. Porteus, Email: mporteus@stanford.edu.
Supplemental information
References
- 1.Taylor-Cousar J.L., Munck A., McKone E.F., van der Ent C.K., Moeller A., Simard C., Wang L.T., Ingenito E.P., McKee C., Lu Y., et al. Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del. N. Engl. J. Med. 2017;377:2013–2023. doi: 10.1056/NEJMoa1709846. [DOI] [PubMed] [Google Scholar]
- 2.Heijerman H.G.M., McKone E.F., Downey D.G., Van Braeckel E., Rowe S.M., Tullis E., Mall M.A., Welter J.J., Ramsey B.W., McKee C.M., et al. VX17-445-103 Trial Group Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: a double-blind, randomised, phase 3 trial. Lancet. 2019;394:1940–1948. doi: 10.1016/S0140-6736(19)32597-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Keating D., Marigowda G., Burr L., Daines C., Mall M.A., McKone E.F., Ramsey B.W., Rowe S.M., Sass L.A., Tullis E., et al. VX16-445-001 Study Group VX-445-Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N. Engl. J. Med. 2018;379:1612–1620. doi: 10.1056/NEJMoa1807120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bose S.J., Krainer G., Ng D.R.S., Schenkel M., Shishido H., Yoon J.S., Haggie P.M., Schlierf M., Sheppard D.N., Skach W.R. Towards next generation therapies for cystic fibrosis: Folding, function and pharmacology of CFTR. J. Cyst. Fibros. 2020;19(Suppl 1):S25–S32. doi: 10.1016/j.jcf.2019.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Konstan M.W., Flume P.A. Clinical care for cystic fibrosis: preparing for the future now. Lancet Respir. Med. 2020;8:10–12. doi: 10.1016/S2213-2600(19)30334-0. [DOI] [PubMed] [Google Scholar]
- 6.Moss R.B., Milla C., Colombo J., Accurso F., Zeitlin P.L., Clancy J.P., Spencer L.T., Pilewski J., Waltz D.A., Dorkin H.L., et al. Repeated aerosolized AAV-CFTR for treatment of cystic fibrosis: a randomized placebo-controlled phase 2B trial. Hum. Gene Ther. 2007;18:726–732. doi: 10.1089/hum.2007.022. [DOI] [PubMed] [Google Scholar]
- 7.Alton E.W.F.W., Armstrong D.K., Ashby D., Bayfield K.J., Bilton D., Bloomfield E.V., Boyd A.C., Brand J., Buchan R., Calcedo R., et al. UK Cystic Fibrosis Gene Therapy Consortium Repeated nebulisation of non-viral CFTR gene therapy in patients with cystic fibrosis: a randomised, double-blind, placebo-controlled, phase 2b trial. Lancet Respir. Med. 2015;3:684–691. doi: 10.1016/S2213-2600(15)00245-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wagner J.A., Nepomuceno I.B., Messner A.H., Moran M.L., Batson E.P., Dimiceli S., Brown B.W., Desch J.K., Norbash A.M., Conrad C.K., et al. A phase II, double-blind, randomized, placebo-controlled clinical trial of tgAAVCF using maxillary sinus delivery in patients with cystic fibrosis with antrostomies. Hum. Gene Ther. 2002;13:1349–1359. doi: 10.1089/104303402760128577. [DOI] [PubMed] [Google Scholar]
- 9.Amoasii L., Long C., Li H., Mireault A.A., Shelton J.M., Sanchez-Ortiz E., McAnally J.R., Bhattacharyya S., Schmidt F., Grimm D., et al. Single-cut genome editing restores dystrophin expression in a new mouse model of muscular dystrophy. Sci. Transl. Med. 2017;9:eaan8081. doi: 10.1126/scitranslmed.aan8081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li A., Tanner M.R., Lee C.M., Hurley A.E., De Giorgi M., Jarrett K.E., Davis T.H., Doerfler A.M., Bao G., Beeton C., Lagor W.R. AAV-CRISPR Gene Editing Is Negated by Pre-existing Immunity to Cas9. Mol. Ther. 2020;28:1432–1441. doi: 10.1016/j.ymthe.2020.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Charlesworth C.T., Deshpande P.S., Dever D.P., Camarena J., Lemgart V.T., Cromer M.K., Vakulskas C.A., Collingwood M.A., Zhang L., Bode N.M., et al. Identification of preexisting adaptive immunity to Cas9 proteins in humans. Nat. Med. 2019;25:249–254. doi: 10.1038/s41591-018-0326-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wagner D.L., Amini L., Wendering D.J., Burkhardt L.-M., Akyüz L., Reinke P., Volk H.D., Schmueck-Henneresse M. High prevalence of Streptococcus pyogenes Cas9-reactive T cells within the adult human population. Nat. Med. 2019;25:242–248. doi: 10.1038/s41591-018-0204-6. [DOI] [PubMed] [Google Scholar]
- 13.Cooney A.L., Abou Alaiwa M.H., Shah V.S., Bouzek D.C., Stroik M.R., Powers L.S., Gansemer N.D., Meyerholz D.K., Welsh M.J., Stoltz D.A., et al. Lentiviral-mediated phenotypic correction of cystic fibrosis pigs. JCI Insight. 2016;1:e88730. doi: 10.1172/jci.insight.88730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ostedgaard L.S., Zabner J., Vermeer D.W., Rokhlina T., Karp P.H., Stecenko A.A., Randak C., Welsh M.J. CFTR with a partially deleted R domain corrects the cystic fibrosis chloride transport defect in human airway epithelia in vitro and in mouse nasal mucosa in vivo. Proc. Natl. Acad. Sci. USA. 2002;99:3093–3098. doi: 10.1073/pnas.261714599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Porteus M. Genome Editing: A New Approach to Human Therapeutics. Annu. Rev. Pharmacol. Toxicol. 2016;56:163–190. doi: 10.1146/annurev-pharmtox-010814-124454. [DOI] [PubMed] [Google Scholar]
- 16.Porteus M.H. A New Class of Medicines through DNA Editing. N. Engl. J. Med. 2019;380:947–959. doi: 10.1056/NEJMra1800729. [DOI] [PubMed] [Google Scholar]
- 17.Schwank G., Koo B.K., Sasselli V., Dekkers J.F., Heo I., Demircan T., Sasaki N., Boymans S., Cuppen E., van der Ent C.K., et al. Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell Stem Cell. 2013;13:653–658. doi: 10.1016/j.stem.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 18.Firth A.L., Menon T., Parker G.S., Qualls S.J., Lewis B.M., Ke E., Dargitz C.T., Wright R., Khanna A., Gage F.H., Verma I.M. Functional Gene Correction for Cystic Fibrosis in Lung Epithelial Cells Generated from Patient iPSCs. Cell Rep. 2015;12:1385–1390. doi: 10.1016/j.celrep.2015.07.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Crane A.M., Kramer P., Bui J.H., Chung W.J., Li X.S., Gonzalez-Garay M.L., Hawkins F., Liao W., Mora D., Choi S., et al. Targeted correction and restored function of the CFTR gene in cystic fibrosis induced pluripotent stem cells. Stem Cell Reports. 2015;4:569–577. doi: 10.1016/j.stemcr.2015.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McCauley K.B., Hawkins F., Serra M., Thomas D.C., Jacob A., Kotton D.N. Efficient Derivation of Functional Human Airway Epithelium from Pluripotent Stem Cells via Temporal Regulation of Wnt Signaling. Cell Stem Cell. 2017;20:844–857.e6. doi: 10.1016/j.stem.2017.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Firth A.L., Dargitz C.T., Qualls S.J., Menon T., Wright R., Singer O., Gage F.H., Khanna A., Verma I.M. Generation of multiciliated cells in functional airway epithelia from human induced pluripotent stem cells. Proc. Natl. Acad. Sci. USA. 2014;111:E1723–E1730. doi: 10.1073/pnas.1403470111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Berical A., Lee R.E., Randell S.H., Hawkins F. Challenges Facing Airway Epithelial Cell-Based Therapy for Cystic Fibrosis. Front. Pharmacol. 2019;10:74. doi: 10.3389/fphar.2019.00074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vaidyanathan S., Salahudeen A.A., Sellers Z.M., Bravo D.T., Choi S.S., Batish A., Le W., Baik R., de la O S., Kaushik M.P., et al. High-Efficiency, Selection-free Gene Repair in Airway Stem Cells from Cystic Fibrosis Patients Rescues CFTR Function in Differentiated Epithelia. Cell Stem Cell. 2020;26:161–171.e4. doi: 10.1016/j.stem.2019.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bak R.O., Porteus M.H. CRISPR-Mediated Integration of Large Gene Cassettes Using AAV Donor Vectors. Cell Rep. 2017;20:750–756. doi: 10.1016/j.celrep.2017.06.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dever D.P., Bak R.O., Reinisch A., Camarena J., Washington G., Nicolas C.E., Pavel-Dinu M., Saxena N., Wilkens A.B., Mantri S., et al. CRISPR/Cas9 β-globin gene targeting in human haematopoietic stem cells. Nature. 2016;539:384–389. doi: 10.1038/nature20134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bak R.O., Dever D.P., Porteus M.H. CRISPR/Cas9 genome editing in human hematopoietic stem cells. Nat. Protoc. 2018;13:358–376. doi: 10.1038/nprot.2017.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Charlesworth C.T., Camarena J., Cromer M.K., Vaidyanathan S., Bak R.O., Carte J.M., Potter J., Dever D.P., Porteus M.H. Priming Human Repopulating Hematopoietic Stem and Progenitor Cells for Cas9/sgRNA Gene Targeting. Mol. Ther. Nucleic Acids. 2018;12:89–104. doi: 10.1016/j.omtn.2018.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Scaife M., Pacienza N., Au B.C.Y., Wang J.C.M., Devine S., Scheid E., Lee C.J., Lopez-Perez O., Neschadim A., Fowler D.H., et al. Engineered human Tmpk fused with truncated cell-surface markers: versatile cell-fate control safety cassettes. Gene Ther. 2013;20:24–34. doi: 10.1038/gt.2011.210. [DOI] [PubMed] [Google Scholar]
- 29.Nakazawa Y., Huye L.E., Salsman V.S., Leen A.M., Ahmed N., Rollins L., Dotti G., Gottschalk S.M., Wilson M.H., Rooney C.M. PiggyBac-mediated cancer immunotherapy using EBV-specific cytotoxic T-cells expressing HER2-specific chimeric antigen receptor. Mol. Ther. 2011;19:2133–2143. doi: 10.1038/mt.2011.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tey S.-K., Dotti G., Rooney C.M., Heslop H.E., Brenner M.K. Inducible caspase 9 suicide gene to improve the safety of allodepleted T cells after haploidentical stem cell transplantation. Biol. Blood Marrow Transplant. 2007;13:913–924. doi: 10.1016/j.bbmt.2007.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Passerini L., Mel E.R., Sartirana C., Fousteri G., Bondanza A., Naldini L., Roncarolo M.G., Bacchetta R. CD4+ T Cells from IPEX Patients Convert into Functional and Stable Regulatory T Cells by FOXP3 Gene Transfer. Sci. Transl. Med. 2013;5:215ra174. doi: 10.1126/scitranslmed.3007320. [DOI] [PubMed] [Google Scholar]
- 32.Pavel-Dinu M., Wiebking V., Dejene B.T., Srifa W., Mantri S., Nicolas C.E., Lee C., Bao G., Kildebeck E.J., Punjya N., et al. Gene correction for SCID-X1 in long-term hematopoietic stem cells. Nat. Commun. 2019;10:1634. doi: 10.1038/s41467-019-09614-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lombardo A., Cesana D., Genovese P., Di Stefano B., Provasi E., Colombo D.F., Neri M., Magnani Z., Cantore A., Lo Riso P., et al. Site-specific integration and tailoring of cassette design for sustainable gene transfer. Nat. Methods. 2011;8:861–869. doi: 10.1038/nmeth.1674. [DOI] [PubMed] [Google Scholar]
- 34.Vakulskas C.A., Dever D.P., Rettig G.R., Turk R., Jacobi A.M., Collingwood M.A., Bode N.M., McNeill M.S., Yan S., Camarena J., et al. A high-fidelity Cas9 mutant delivered as a ribonucleoprotein complex enables efficient gene editing in human hematopoietic stem and progenitor cells. Nat. Med. 2018;24:1216–1224. doi: 10.1038/s41591-018-0137-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Haapaniemi E., Botla S., Persson J., Schmierer B., Taipale J. CRISPR-Cas9 genome editing induces a p53-mediated DNA damage response. Nat. Med. 2018;24:927–930. doi: 10.1038/s41591-018-0049-z. [DOI] [PubMed] [Google Scholar]
- 36.Ihry R.J., Worringer K.A., Salick M.R., Frias E., Ho D., Theriault K., Kommineni S., Chen J., Sondey M., Ye C., et al. p53 inhibits CRISPR-Cas9 engineering in human pluripotent stem cells. Nat. Med. 2018;24:939–946. doi: 10.1038/s41591-018-0050-6. [DOI] [PubMed] [Google Scholar]
- 37.Gomez-Ospina N., Scharenberg S.G., Mostrel N., Bak R.O., Mantri S., Quadros R.M., Gurumurthy C.B., Lee C., Bao G., Suarez C.J., et al. Human genome-edited hematopoietic stem cells phenotypically correct Mucopolysaccharidosis type I. Nat. Commun. 2019;10:4045. doi: 10.1038/s41467-019-11962-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Newman A.M., Bratman S.V., To J., Wynne J.F., Eclov N.C.W., Modlin L.A., Liu C.L., Neal J.W., Wakelee H.A., Merritt R.E., et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat. Med. 2014;20:548–554. doi: 10.1038/nm.3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hadd A.G., Houghton J., Choudhary A., Sah S., Chen L., Marko A.C., Sanford T., Buddavarapu K., Krosting J., Garmire L., et al. Targeted, high-depth, next-generation sequencing of cancer genes in formalin-fixed, paraffin-embedded and fine-needle aspiration tumor specimens. J. Mol. Diagn. 2013;15:234–247. doi: 10.1016/j.jmoldx.2012.11.006. [DOI] [PubMed] [Google Scholar]
- 40.Wong S.Q., Li J., Tan A.Y.-C., Vedururu R., Pang J.-M.B., Do H., Ellul J., Doig K., Bell A., MacArthur G.A., et al. CANCER 2015 Cohort Sequence artefacts in a prospective series of formalin-fixed tumours tested for mutations in hotspot regions by massively parallel sequencing. BMC Med. Genomics. 2014;7:23. doi: 10.1186/1755-8794-7-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Engelhardt J.F., Yang Y., Stratford-Perricaudet L.D., Allen E.D., Kozarsky K., Perricaudet M., Yankaskas J.R., Wilson J.M. Direct gene transfer of human CFTR into human bronchial epithelia of xenografts with E1-deleted adenoviruses. Nat. Genet. 1993;4:27–34. doi: 10.1038/ng0593-27. [DOI] [PubMed] [Google Scholar]
- 42.Johnson L.G., Olsen J.C., Sarkadi B., Moore K.L., Swanstrom R., Boucher R.C. Efficiency of gene transfer for restoration of normal airway epithelial function in cystic fibrosis. Nat. Genet. 1992;2:21–25. doi: 10.1038/ng0992-21. [DOI] [PubMed] [Google Scholar]
- 43.Suzuki S., Crane A.M., Anirudhan V., Barillà C., Matthias N., Randell S.H., Rab A., Sorscher E.J., Kerschner J.L., Yin S., et al. Highly Efficient Gene Editing of Cystic Fibrosis Patient-Derived Airway Basal Cells Results in Functional CFTR Correction. Mol. Ther. 2020;28:1684–1695. doi: 10.1016/j.ymthe.2020.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ott C.J., Blackledge N.P., Kerschner J.L., Leir S.-H., Crawford G.E., Cotton C.U., Harris A. Intronic enhancers coordinate epithelial-specific looping of the active CFTR locus. Proc. Natl. Acad. Sci. USA. 2009;106:19934–19939. doi: 10.1073/pnas.0900946106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rowntree R.K., Vassaux G., McDowell T.L., Howe S., McGuigan A., Phylactides M., Huxley C., Harris A. An element in intron 1 of the CFTR gene augments intestinal expression in vivo. Hum. Mol. Genet. 2001;10:1455–1464. doi: 10.1093/hmg/10.14.1455. [DOI] [PubMed] [Google Scholar]
- 46.Goldman M.J., Yang Y., Wilson J.M. Gene therapy in a xenograft model of cystic fibrosis lung corrects chloride transport more effectively than the sodium defect. Nat. Genet. 1995;9:126–131. doi: 10.1038/ng0295-126. [DOI] [PubMed] [Google Scholar]
- 47.Zhang L., Button B., Gabriel S.E., Burkett S., Yan Y., Skiadopoulos M.H., Dang Y.L., Vogel L.N., McKay T., Mengos A., et al. CFTR delivery to 25% of surface epithelial cells restores normal rates of mucus transport to human cystic fibrosis airway epithelium. PLoS Biol. 2009;7:e1000155. doi: 10.1371/journal.pbio.1000155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Farmen S.L., Karp P.H., Ng P., Palmer D.J., Koehler D.R., Hu J., Beaudet A.L., Zabner J., Welsh M.J. Gene transfer of CFTR to airway epithelia: low levels of expression are sufficient to correct Cl- transport and overexpression can generate basolateral CFTR. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005;289:L1123–L1130. doi: 10.1152/ajplung.00049.2005. [DOI] [PubMed] [Google Scholar]
- 49.Shah V.S., Ernst S., Tang X.X., Karp P.H., Parker C.P., Ostedgaard L.S., Welsh M.J. Relationships among CFTR expression, HCO3- secretion, and host defense may inform gene- and cell-based cystic fibrosis therapies. Proc. Natl. Acad. Sci. USA. 2016;113:5382–5387. doi: 10.1073/pnas.1604905113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dannhoffer L., Blouquit-Laye S., Regnier A., Chinet T. Functional properties of mixed cystic fibrosis and normal bronchial epithelial cell cultures. Am. J. Respir. Cell Mol. Biol. 2009;40:717–723. doi: 10.1165/rcmb.2008-0018OC. [DOI] [PubMed] [Google Scholar]
- 51.Char J.E., Wolfe M.H., Cho H.J., Park I.H., Jeong J.H., Frisbee E., Dunn C., Davies Z., Milla C., Moss R.B., et al. A little CFTR goes a long way: CFTR-dependent sweat secretion from G551D and R117H-5T cystic fibrosis subjects taking ivacaftor. PLoS ONE. 2014;9:e88564. doi: 10.1371/journal.pone.0088564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sheppard D.N., Rich D.P., Ostedgaard L.S., Gregory R.J., Smith A.E., Welsh M.J. Mutations in CFTR associated with mild-disease-form Cl- channels with altered pore properties. Nature. 1993;362:160–164. doi: 10.1038/362160a0. [DOI] [PubMed] [Google Scholar]
- 53.Mou H., Vinarsky V., Tata P.R., Brazauskas K., Choi S.H., Crooke A.K., Zhang B., Solomon G.M., Turner B., Bihler H., et al. Dual SMAD Signaling Inhibition Enables Long-Term Expansion of Diverse Epithelial Basal Cells. Cell Stem Cell. 2016;19:217–231. doi: 10.1016/j.stem.2016.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Morrison S.J., Prowse K.R., Ho P., Weissman I.L. Telomerase activity in hematopoietic cells is associated with self-renewal potential. Immunity. 1996;5:207–216. doi: 10.1016/s1074-7613(00)80316-7. [DOI] [PubMed] [Google Scholar]
- 55.Lin S., Nascimento E.M., Gajera C.R., Chen L., Neuhöfer P., Garbuzov A., Wang S., Artandi S.E. Distributed hepatocytes expressing telomerase repopulate the liver in homeostasis and injury. Nature. 2018;556:244–248. doi: 10.1038/s41586-018-0004-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hajj R., Baranek T., Le Naour R., Lesimple P., Puchelle E., Coraux C. Basal cells of the human adult airway surface epithelium retain transit-amplifying cell properties. Stem Cells. 2007;25:139–148. doi: 10.1634/stemcells.2006-0288. [DOI] [PubMed] [Google Scholar]
- 57.Martin R.M., Ikeda K., Cromer M.K., Uchida N., Nishimura T., Romano R., Tong A.J., Lemgart V.T., Camarena J., Pavel-Dinu M., et al. Highly Efficient and Marker-free Genome Editing of Human Pluripotent Stem Cells by CRISPR-Cas9 RNP and AAV6 Donor-Mediated Homologous Recombination. Cell Stem Cell. 2019;24:821–828.e5. doi: 10.1016/j.stem.2019.04.001. [DOI] [PubMed] [Google Scholar]
- 58.Cradick T.J., Qiu P., Lee C.M., Fine E.J., Bao G. COSMID: A web-based tool for identifying and validating CRISPR/Cas off-target sites. Mol. Ther. Nucleic Acids. 2014;3:e214. doi: 10.1038/mtna.2014.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lee C.M., Cradick T.J., Bao G. The Neisseria meningitidis CRISPR-Cas9 System Enables Specific Genome Editing in Mammalian Cells. Mol. Ther. 2016;24:645–654. doi: 10.1038/mt.2016.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gentzsch M., Boyles S.E., Cheluvaraju C., Chaudhry I.G., Quinney N.L., Cho C., Dang H., Liu X., Schlegel R., Randell S.H. Pharmacological rescue of conditionally reprogrammed cystic fibrosis bronchial epithelial cells. Am. J. Respir. Cell Mol. Biol. 2017;56:568–574. doi: 10.1165/rcmb.2016-0276MA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen L., Roake C.M., Freund A., Batista P.J., Tian S., Yin Y.A., Gajera C.R., Lin S., Lee B., Pech M.F., et al. An Activity Switch in Human Telomerase Based on RNA Conformation and Shaped by TCAB1. Cell. 2018;174:218–230.e13. doi: 10.1016/j.cell.2018.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cawthon R.M. Telomere length measurement by a novel monochrome multiplex quantitative PCR method. Nucleic Acids Res. 2009;37:e21. doi: 10.1093/nar/gkn1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Herbert B.-S., Shay J.W., Wright W.E. Analysis of Telomeres and Telomerase. Curr. Protoc. Cell Biol. 2003;20:18.6.1–18.6.20. doi: 10.1002/0471143030.cb1806s20. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.