

Summary
T cells are crucial to generate an effective response against numerous invading microbial pathogens and play a pivotal role in tumor surveillance and elimination. However, unwanted T cell activation can also lead to deleterious immune-mediated inflammation and tissue damage. To ensure that an optimal T cell response can be established, each step, beginning from T cell development in the thymus to their activation and function in the periphery, is tightly regulated by many transcription factors and epigenetic regulators including microRNAs (miRNAs). Here, we first summarize recent progress in identifying major immune regulatory miRNAs in controlling the differentiation and function of distinct T cell subsets. Moreover, as emerging evidence has demonstrated that miRNAs can impact T cell immunity through targeting both immune- and non-immune cell populations that T cells closely interact with, the T cell-extrinsic role of miRNAs in regulating different aspects of T cell biology is also addressed. Finally, we discuss the complex nature of miRNA-mediated control of T cell immunity and highlight important questions that remain to be further investigated.
Keywords: miRNA, T cell immunity, post-transcriptional regulation, immune regulation
1. INTRODUCTION
T cell-mediated immunity plays a central role in protecting hosts from the invasion of pathogenic microorganisms like viruses, bacteria, fungus, and parasites. T cells can directly eliminate pathogen-infected cells. They can also facilitate pathogen clearance through activating innate immune cells such as macrophages to enhance their phagocytic activities or helping B cells to generate memory B cells and plasma cells that produce high-affinity antibodies. On the other hand, in addition to mounting protective immune responses, T cells are also capable of controlling unwanted immune activation and preventing deleterious immune-mediated inflammation and tissue damage. One of the key T cell subsets involved in the latter process is regulatory T (Treg) cells, a dedicated immune population crucial for the negative regulation of immune responses. To date, many different transcriptional and epigenetic mechanisms that regulate the development and function of both conventional T (Tconv) cells and Treg cells have been identified. Among different epigenetic regulators, for almost two decades, microRNAs (miRNAs), a class of small non-coding RNA molecules, have been intensively investigated in the immune system. Unlike transcription factors, miRNAs are generally thought to fine-tune gene expression rather than enacting drastic gene changes. Nevertheless, many miRNAs exist in clusters and/or paralogs with high degrees of evolutionary conservation. These miRNA “families” exhibited elevated impact on gene regulation and resultant immunobiology.
The importance of miRNA-mediated gene regulation in T cell immunity was first revealed in the studies where Dicer, a key molecule required for the generation of mature miRNAs, was selectively deleted in T cells 1,2. In the thymus, while Dicer ablation in early T cell progenitors does not seem to lead to any substantial alterations in the development of Tconv cells except for reduced thymic cellularity 2, thymic Treg cell differentiation is significantly compromised 3. On the other hand, in the periphery, both Tconv and Treg cells are heavily impacted by the disruption of the entire miRNA network. Tconv cells with Dicer deficiency fail to differentiate into multiple helper T (Th) cell lineages and exhibit aberrant effector function 1. Similarly, deletion of Dicer or Drosha, another essential component in miRNA biogenesis specifically in Treg cells also severely impairs their homeostasis and suppressor capacity, leading to the development of fatal multi-organ autoimmunity 4,5. Together, these findings provided early evidence demonstrating an indispensable role of miRNA in controlling T cell immunity, a point that is further supported by subsequent studies in which distinct T cell subsets were shown to be regulated by individual miRNAs. Here, we will begin with reviewing the current knowledge of miRNA-mediated gene regulation in T cells with the focus on a select group of immune regulatory miRNAs and miRNA families that can either promote or inhibit a given type of T cell response, identified from our own work or reported in recent studies. Next, as differentiation, activation, and function of T cells rely on the close interaction with many different cell types, we will also address how miRNAs regulate T cell responses through targeting other immune and non-immune cell populations in health and disease. Finally, we will discuss the complex nature of miRNA-mediated control of T cell immunity and highlight important questions that remain to be further investigated.
2. CELL-INTRINSIC ROLE OF MIRNAS IN CONTROLLING T CELL IMMUNITY
2.1. Impact of miRNAs on different effector CD4 T cell subsets
Upon antigen recognition, depending on the presence of a particular cytokine milieu at the time, naïve CD4 T cells can differentiate into distinct Th cell subsets through acquiring corresponding lineage-specific transcription factors 6. These master regulators orchestrate a sophisticated transcriptional program involving other transcription factors to maintain Th cell lineage identity and to confer the effector function to different Th cell subsets via the production of unique sets of cytokines. Since the function of miRNAs in T cells was initially reported 1, over years, accumulating evidence has demonstrated a key role of miRNAs in regulating Th cell differentiation and function through targeting the corresponding lineage-specific transcription factors and/or effector cytokines and their receptors. Below we will discuss recent literature that provided examples of individual miRNAs or miRNA families controlling different types of CD4 T cell immunity (Fig. 1A).
FIGURE 1. miRNAs control the differentiation and function of distinct T cell subsets.
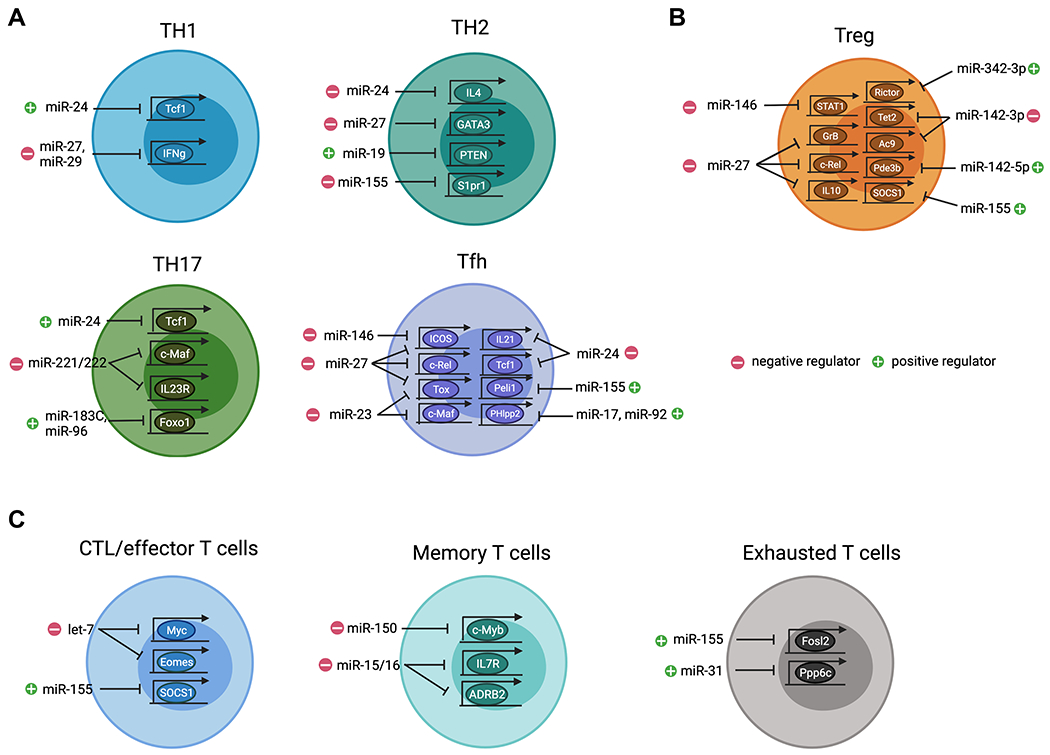
(A) Different miRNAs function either as positive or negative regulators in regulating CD4 Th cell responses. In Th1 cells, miR-24 promotes IFNγ responses through targeting Tcf7 whereas miR-27 and miR-29 inhibit IFNγ expression. In Th2 cells, miR-24, miR-27 and miR-155 serve as negative regulators by targeting, Il4, Gata3, and S1pr1, respectively, whereas miR-19 promotes Th2 cell response through targeting Pten. In Th17 cells, miR-24 and miR-183C promotes their function by inhibiting targeting Tcf7 and Foxo1, respectively, whereas miR-221/miR-222 negatively regulating Th17 cells by targeting Maf and Il23r. In Tfh cells, miR-23C (including miR-23, miR-24, and miR-27) and miR-146a/b coordinately restrict their responses by targeting, Maf, Tox, Il21, Tcf7, Rel, and Icos, respectively whereas miR-155 and miR-17/92 promote Tfh cells by targeting Peli1 and Phlpp2. (B) Development, maintenance, homeostasis, and function of Treg cells are regulated by different miRNAs. Specifically, miR-27, miR-142-3p, and miR-146a act as negative regulators in Treg cells by targeting Rel, Gzmb, Il10, Tet2, Ac9, and Stat1, respectively, whereas miR142-5p, miR-155, and miR-342-3p are essential to promote Treg cell homeostasis and function by targeting Pde3b, Socs1, and Rictor, respectively. (C) Distinct miRNAs regulate CD8 T cell immunity. In particular, let-7 and miR-155 serving as negative and positive regulators to control effector function by targeting Myc, Eomes and Socs1, respectively. Moreover, miR-150-mediated control of Myb and miR-15/16-mediated control of Il7r and Adrb2 respectively impacts memory T cell formation. Finally, both miR-31 and miR-155 promote T cell exhaustion by targeting Fosl2 and Ppp6c, respectively. All graphics are created with BioRender.com.
Th1 cells
IFNγ-secreting Th1 cells are crucial for eradicating intracellular pathogens but can also drive many autoimmune diseases when their responses are not properly regulated. IFNγ produced by Th1 cells is essential for their function. At the same time, it also serves as a key component in a positive feedback mechanism that ensures optimal Th1 differentiation through inducing T-bet, a Th1-specific master transcriptional regulator 7. The increased production of IFNγ observed in Dicer-deficient T cells under non-polarized or even Th2 conditions suggested that the combinatorial effect of miRNA-mediated gene regulation in T cells predominately plays a negative role in Th1 differentiation and/or IFNγ production 1. Subsequently, by taking a reconstitution approach, members of the miR-29 family were found capable of largely rescuing the aberrant IFNγ phenotype identified in miRNA-deficient T cells through targeting T-bet and a close relative molecule, Eomes 8. In addition to regulating master transcription factors, miR-29 is capable of directly regulating IFNγ expression. As such, mice devoid of miR-29-mediated gene regulation exhibit greater resistance to intracellular bacterial infection but also display more severe IFNγ-driven delayed-type hypersensitivity and autoimmunity 9,10. Like miR-29, miR-27 was also identified as a potent IFNγ suppressor. It was suggested that γ-herpesviruses-driven miR-27 degradation helps establish latent infection in T cells and facilitates a long-term sustained viral infection through down-regulating host IFNγ expression 11. Consistent with this finding, our previous analysis of T cells with miR-27 overexpression also pointed to miR-27 as a negative regulator in controlling Th1 differentiation and IFNγ production 12,13. Interestingly, even though miR-24 and miR-27 belong to the same evolutionarily conserved miR-23~27~24 clusters (miR-23Cs), mice with miR-24 overexpression in T cells exhibit enhanced Th1 and Th17 differentiation and heightened IFNγ and IL-17 responses 12,14. Further mechanistic studies identified TCF1, a high mobility group box transcription factor known for its role in restricting IFNγ and IL-17 expression 15,16, as a bona fide miR-24 target. These results supported a role of miR-24 as a positive regulator of both Th1 and Th17 immunity.
Th2 cells
Through the production of IL-4 and other type 2 cytokines, Th2 cells are pivotal in providing host defense against parasitic infections but also act as a key player in promoting asthma and allergic disease pathogenesis 17. Expression of GATA3, the Th2 master regulator, is essential for Th2 differentiation and cytokine expression. Like the aforementioned IFNγ-T-bet relationship, IL-4 is required for GATA3 expression and IL-4-mediated GATA3 induction also forms a positive feedback loop to secure Th2 lineage identity 18. In addition to miR-155 and miR-19 which have been shown to inhibit and promote Th2 responses, respectively 19–21, we and others have found that the aforementioned miR-23Cs also play a major role in regulating Th2 immunity through targeting not only IL-4 directly but also a network of regulators, including GATA3, upstream of IL-4 production and Th2 differentiation 12,22. Interestingly, unlike what was found in Th1 cells where miR-24 and miR-27 antagonize with each other in regulating Th1 responses, miR-24 and miR-27 collaboratively limit Th2 immunity 12. Consequently, despite that miR-23 seems to play a minimal role in Th2 cell differentiation and function, mice with T cell-specific deletion of the entire miR-23C exhibit exacerbated Th2 cell-driven airway inflammation and tissue pathology upon ovalbumin or house dust mite challenges 12.
Th17 cells
Despite that Th17 cells have been mostly studied for their role in mediating inflammatory pathology in numerous autoimmune disease settings, Th17 cells are also vital in conferring immunity to extracellular bacterial and fungal infections. Through the production of IL-17, Th17 cells can contribute to pathogen clearance by coordinating early neutrophil recruitment and augmenting their bactericidal activity 23. To date, multiple cytokines such as TGFβ, IL-1β, IL-6, IL-23 have all been shown to promote Th17 responses. Among them, TGFβ and IL-6 differentiated Th17 cells are considered to predominately play a protective role in promoting mucosal defense, barrier tissue integrity, and curtailing immune pathogenic responses. On the other hand, Th17 cells induced by IL-1β, IL-6, and IL-23 were shown to drive chronic tissue inflammation, granuloma formation and autoimmunity 24. Recently, it was demonstrated that a miRNA family, miR-221/miR-222, plays a negative role in controlling Th17 immunity 25. Loss of miR-221/miR-222 in T cells leads to increased IL-17 production as well as enhanced expression of RORγT, the Th17 cell master regulator. Mechanistically, IL-23R and c-Maf were suggested to be the direct targets of miR-221/miR-222. Previously, c-Maf has been shown to be induced in a STAT3-dependent manner and can directly activate RORγT expression in CD4 T cells leading to Th17 differentiation 26. In addition, c-Maf is also capable of promoting IL-23R expression and in turn, IL-23-mediated signaling further activates c-Maf forming a positive feedback loop. Considering the well-established role of IL-23 in promoting pathogenic Th17 cell differentiation 27, miR-221 and miR-222 were thus suggested to function as integral regulatory components of intestinal Th17 cell immunity by preventing it from shifting to the proinflammatory responses. As a consequence, while loss of miR-221/miR-222 does not impact intestinal Th17 cell homeostasis at steady state, mice with either global or T cell-specific miR-221/miR-222 ablation exhibit more severe DSS-induced mucosal damage 25.
In contrast to miR-221/miR-222, the miR-183-96-182 cluster (miR-183C) was reported to act as positive regulators of Th17 responses 28. It was shown that all individual miRNA members of miR-183C are highly expressed in Th17 cells. Specifically, elevated expression of miR-183C was found to be induced under IL-23-driven but not TGFβ-dependent Th17 differentiating condition. These results suggested that miR-183C is involved in regulating the pathogenicity of Th17 cells. Supporting this notion, loss of miR-183C in T cells led to reduced expression of not only IL-17 but also IL-22 and GM-CSF, two cytokines that are known to be produced by pathogenic Th17 cells 24. Further RNA-seq analysis has revealed many pathogenic signature genes, including Il23r and Il1r1, in Th17 cells were all downregulated in T cells devoid of miR-183C. Mechanistically, miR-183C was shown to promote the pathogenic function of Th17 cells in part via directly targeting Foxo1, a known negative regulator of Th17 cells through repressing the expression of both IL-1R and IL-23R 28–30. As a consequence, mice harboring miR-183C-deficient T cells showed reduced disease phenotype in experimental autoimmune encephalomyelitis (EAE) compared to the WT controls whereas mice having miR-183C-overexpressing T cells exhibited more severe disease 28.
Tfh cells
Follicular helper T (Tfh) cells are a specialized subset of CD4 T cells with a unique role in supporting B cell-mediated humoral immunity. Elevated expression of CXCR5 on Tfh cells allows them to respond to CXCL13 and migrate into B cell follicles. The interaction between Tfh and B cells within B cell follicles is crucial for the induction of the germinal center (GC) reaction, which is required for the generation of high-affinity antibody-producing plasma cells and long-lived memory B cells. The identification of BCL6 as a master regulator of Tfh cell differentiation has solidified Tfh cells as a distinct T helper cell lineage similar to Th1, Th2, Th17, and Treg cells 31,32. Previously, through studying mice with constitutive ablation or tamoxifen-inducible deletion of global miRNAs in T cells, miRNAs have been shown to be essential for not only the differentiation but also the maintenance of Tfh cells 33,34. Loss of miRNAs in T cells severely impaired Tfh cell phenotype and function, concomitant with reduced GC B cell responses. Subsequent studies have identified both miR-17~92 family and miR-155 to be the positive regulators of Tfh cells 33,35,36. While the former miRNA family controls the expression of BCL6 and CXCR5 required for their migration into B cell follicles, the latter is responsible for ensuring proper cellular proliferation during Tfh cell differentiation.
Nevertheless, not all miRNAs are involved in promoting Tfh cell responses. Recently, in our analysis of mice with T cell-specific deletion of miR-23C, in addition to unrestricted Th2-driven IgE responses as discussed above 12, we could also detect increased total serum IgG levels and considerably more and larger GCs accompanied with elevated Tfh cell numbers upon airway allergic reaction or during LCMV infection. In contrast, when the entire miR-23C or individual members in this miRNA family were overexpressed in T cells, reduced Tfh and GC B cell responses were observed 37. These results suggest that each member of miR-23C collaboratively limit Tfh cell responses and the resultant humoral immunity. Many previously known Tfh cell-associated molecules such as c-Maf, IL-21, and ICOS were found to be directly repressed by miR-23, miR-24, and miR-27, respectively. Moreover, our studies of miR-23C-mediated regulation of Tfh cell responses also offered the opportunity to identify TOX, a target of miR-23 and miR-27, as a new key regulator of Tfh cell responses 37. TOX, a transcription factor that was previously reported to be essential for CD4 T cell development in the thymus 38, was found to be highly up-regulated in Tfh cells in a BCL6-dependent manner in both mice and humans. Moreover, the Tfh cell-promoting activity of TOX was shown to be in part through driving the expression of many molecules such as TCF1, LEF1, and PD1 that are crucial for Tfh cell differentiation and function 39–41. It should be noted, TCF1 is also directly regulated by miR-24 as discussed above 14. Together with other Tfh-associated molecules like c-Rel that were previously shown to be the targets of this miRNA family 13, these studies established miR-23C as a central player that regulates multiple aspects of Tfh cell biology 37.
Like miR-23C, miR-146a was also previously reported to negatively control Tfh cells 42. By employing either adoptive T cell or mixed bone marrow stem cell transfers from miR-146a germline null mice, miR-146a has been demonstrated to play a T cell-intrinsic role in limiting Tfh cell responses 42. Interestingly, however, our recent work in mice with T cell-specific deletion of miR-146a showed no change in Tfh cells both at steady state and also after immunization 43. While the discrepancy between these two studies was initially puzzling, it is not entirely surprising. Impaired Treg cell-mediated regulation or the transient lymphopenia that resulted from the adoptive T cell transfer or mixed BM chimera approaches taken in the early study could both potentially cause accelerated and/or enhanced phenotypes that might not normally occur in unperturbed mice with T cell-specific miR-146a ablation. When miR-146b, the paralog of miR-146a which shares the same seed sequence and thus likely targets the same set of genes, was also deleted, spontaneous accumulation of Tfh and GC B cells could then be detected. Moreover, mice with T cell-specific ablation of both miR-146 paralogs harbored not only further increased numbers of Tfh and GC B cells but also elevated antigen-specific IgG levels upon immunization 43. Similarly, while ICOS was suggested to be the direct target of miR-146a and that dysregulated ICOS expression in the absence of miR-146a was responsible for the observed Tfh phenotype in the former study 42, the increase in ICOS expression only became significant when both miR-146a and miR-146b were simultaneously deleted 43. Collectively, these results demonstrated that albeit expressed at a lower level in T cells, miR-146b is still capable of regulating Tfh cell responses in the absence of miR-146a and that both members of this miRNA family are redundantly required to control Tfh cells, likely through coordinately targeting the same set of molecules.
2.2. Role of miRNAs in Treg cells
As discussed above, while Teff cell responses are required for long-term resistance and control of many types of infection, potent T cell immunity without the presence of appropriate regulations can also lead to deleterious T cell-mediated inflammation and tissue damage. The discovery of the transcription factor Foxp3 as a central molecular regulator of Treg cells has helped us gain a better understanding of the molecular mechanisms that control the development and function of Treg cells 44. Several thousand genes are differentially expressed in Treg cells, including miRNAs, and some of these are directly regulated by Foxp3. These findings suggest a role for miRNA-mediated regulation of gene expression in Treg cell differentiation, maintenance, or function 3,45,46. Previously, we have identified two key miRNAs, miR-155 and miR-146a that are pivotal for maintaining normal Treg cell homeostasis, and regulating their suppression of Th1 inflammation, respectively 47,48. Nevertheless, the complexity and the severity of the disease phenotypes observed in mice harboring miRNA-deficient Treg cells could not be attributed entirely to the loss of aforementioned miRNAs in Treg cells 4,5. It is clear that there are additional miRNAs that play crucial roles in controlling other important features of Treg cell biology. To this end, one hint of the involvement of another miRNA in regulating Treg cell responses came from our recent study of T cells with excessive miR-27-mediated gene regulation. As discussed above, we and others have found that miR-27 functions as a negative regulator in limiting Th1 responses at least in part through directly targeting IFNγ 11–13. However, mice with T cell-specific miR-27 overexpression were shown to harbor increased frequencies of IFNγ-producing Th1 cells and would eventually develop spontaneous lympho-hyperactivation diseases 13. Similarly, elevated expression of miR-27 detected in human patients with multiple sclerosis (MS), was also previously suggested to promote proinflammatory Th1 responses that aggravate autoimmunity 49. The seemingly contradicting results of elevated IFNγ responses in mice (or humans) that harbored T cells with a diminished capacity to produce IFNγ owing to miR-27-mediated repression raised a question as to whether the autoimmune pathology developed from a defect in the Treg cell compartment. After all, miR-27 was also overexpressed in Treg cells in mice in which excessive miR-27 expression was found in all T cells. Indeed, subsequent studies have shown that miR-27 not only impairs thymic Treg cell development and peripheral Treg cell homeostasis but also inhibits Treg cell suppressor function through targeting many key Treg cell-associated molecules including c-Rel, Granzyme B and IL-10. Consequently, Treg cells with miR-27 overexpression failed to maintain immunological tolerance even when Teff cell function was also impeded (Fig. 1B) 13.
Like miR-27, another miRNA, miR-142-3p, one of the two mature isoforms generated from the hairpin structure of the miR-142 duplex, has also been documented to act as a negative regulator in controlling Treg cell biology. As cAMP represents one of the key components of Treg cell-mediated immune regulation 50, it was shown that miR-142-3p can inhibit Treg cell function through repressing adenylyl cyclase 9 (AC9), an enzyme that is essential for the generation of cAMP. Moreover, Foxp3 enables AC9-dependent intracellular cAMP production by down-regulating miR-142-3p expression in Treg cells 51. Interestingly, while the level of miR-142-3p in Treg cells is low, the other isoform, miR-142-5p was previously shown to be highly up-regulated in Treg cells 52. When miR-142 (including both isoforms) was ablated in Treg cells, mice developed fatal multi-organ autoimmunity despite harboring Treg cells with seemingly unaltered development and homeostasis, suggesting a critical role of miR-142 in maintaining normal Treg cell suppressor function 53. Further studies have attributed the observed suppression defect in miR-142-deficient Treg cells to the loss of miR-142-5p-mediated regulation of Pde3b 53, a cAMP-degrading phosphodiesterase that was previously shown to be strongly repressed in Treg cells in a Foxp3-dependent manner 54. Together these results support a model whereby differential expression of miR-142 isoforms confers Treg cell suppression function through maintaining intracellular cAMP concentration. To this end, a low level of miR-142-3p in Treg cells ensures the optimal production of cAMP whereas a high level of miR-142-5p prevents its degradation (Fig. 1B) 53.
The miRNAs that have been discussed in Treg cells thus far are all involved in regulating either the entire Treg cell population or the ones that develop in the thymus (i.e. tTreg cells). As extrathymically generated Treg cells are equally important and play a non-redundant role in establishing immunological tolerance 44, several miRNAs have also been identified to exhibit regulatory function in those peripheral induced Treg (iTreg) cells. To this end, in addition to repressing AC9, miR-142-3p was also reported to target TET2, an enzyme that enables both passive and active demethylation through oxidizing the methyl group of 5-methylcytosine (5mC) to yield 5-hydroxymethylcytosine (5hmC) and other oxidized methylcytosines 55. Previously, TET2 has been shown to play an integral role in maintaining Foxp3 expression through regulating demethylation in two “conserved non-coding sequences” (CNS), CNS1 and CNS2, intronic cis-regulatory elements in the Foxp3 locus in addition to other Treg cell-specific hypomethylated regions 56. While the CpG sites in the Foxp3 CNS2 region are uniformly unmethylated in tTreg cells, they are primarily methylated in iTreg cells particularly in cells generated in the presence of TGFβ in vitro. During iTreg cell differentiation, by adding Vitamin C, a known co-activator for TET proteins, increased stability of Foxp3 expression was observed 56. Supporting this notion, during islet autoimmunity, aberrant miR-142-3p expression in T cells has been shown to impair iTreg cell differentiation and homeostasis through repressing TET2-dependent CNS2 demathylation, thereby contributing to autoimmune activation and progression 55. Analogous to miR-142-3p, miR-342-3p has also been previously shown to be expressed at a lower level in Treg cells compared to Tconv cells 57. Nevertheless, a recent study has demonstrated that glucocorticoids, a class of cholesterol-derived corticosteroid hormones with potent immunosuppressive and anti-inflammatory properties, were capable of inducing miR-342-3p expression in both tTreg and iTreg cells with a strong impact on the latter cell population 58. Induction of miR-342-3p in iTreg cells by synthetic glucocorticoids such as Dexamethasone (Dex) was shown to be necessary for glucocorticoid-mediated control of autoimmune inflammation as Dex administration exhibited no effect on attenuating the disease severity in mice receiving miR-342-3p inhibitor-treated iTreg cells. Further mechanistic studies have identified Rictor, a key adaptor protein of the mTORC2 complexes, as a direct target of miR-342-3p. Previously, it was reported that mTORC2 inactivation or Rictor ablation promotes the suppressor capacity of Treg cells in part through inhibiting glycolysis 59. Consistent with this notion, miR-342 inhibition was shown to reprogram Treg cells to glycolytic pathways, leading to impaired suppressor function regardless of Dex treatment. Altogether, this study demonstrated that glucocorticoid-induced miR-342-3p expression confers Treg cell suppressor function through targeting Rictor-mediated metabolic programming (Fig. 1B).
2.3. Function of miRNAs in CD8 T cells
Upon antigen stimulation, CD8 T cells, another arm of cell-mediated immunity, undergo massive clonal expansion and differentiate into Teff cells that can directly eliminate viral-infected or cancer cells. After clearance of pathogens as well as tumors, CD8 Teff cells rapidly die by apoptosis, whereas a small fraction of activated T cells differentiates into memory T cells to provide long-term protection. On the other hand, when CD8 T cells are exposed to persistent antigen and/or inflammatory signals during chronic infection or cancer, their function deteriorates. Those “exhausted” T cells exhibit dysregulated metabolism, poor homeostatic proliferation, and impaired memory recall responses 60. Like CD4 T cells, the function of miRNAs in controlling CD8 T cell immunity has also been long investigated. Deletion of Dicer resulted in a relatively small reduction in CD8 T cell frequencies in the periphery at steady state but a much more pronounced defect in CD8 Teff cell response upon viral infection 61. Interestingly, while the Dicer-dependent miRNA network was shown to be critical for CD8 T cell survival and migration, Dicer-deficient CD8 T cells respond more rapidly to TCR stimulation with enhanced activation and proliferation phenotype. They also express high levels of effector molecules such as perforin, granzymes, and cytokines, suggesting a selective role of miRNAs in restraining CD8 Teff cell biology 62. Supporting this notion, let-7 was recently identified as a key negative regulator of CD8 Teff cell differentiation and function 63. let-7 is expressed at a higher level in naïve CD8 T cells and its expression is down-regulated after TCR activation. Enforced expression of let-7 in CD8 T cells resulted in impaired clonal expansion and differentiation of effector cytotoxic T lymphocytes (CTLs), leading to diminished antiviral and antitumor immune responses. These results suggested that TCR-mediated downregulation of let-7 expression is needed for inducing proper CTL responses. Mechanistically, Myc and Eomes directly targeted by let-7 were shown to be respectively accounted for the observed compromised proliferation and cytotoxic function in CTLs with let-7 overexpression (Fig. 1C) 63. In addition to let-7, miR-150 is also highly expressed in naïve CD8 T cells 64. Upon LCMV infection, miR-150 overexpression resulted in a significant decrease in the memory precursor population during the effector phase, suggesting a negative role of miR-150 in establishing CD8 T cell memory 65. Consistent with this notion, significantly more miR-150-deficient CD8 T cells were recovered compared to WT counterparts at the memory phase. Moreover, memory CD8 T cells devoid of miR-150 also conferred enhanced protective immunity following recall infection 65. Further studies have identified c-Myb, a miR-150 target, as a key mediator of the negative effect of miR-150 in memory CD8 T cell formation. As the role of c-Myb in driving the expression of Bcl-2, an anti-apoptotic molecule with a direct connection to memory cell survival 66, has been long established 67, miR-150 was suggested to limit CD8 T cell memory responses at least in part by impacting c-Myb-dependent Bcl-2 expression. Analogous to miR-150, miR-15/16 family which is also downregulated upon T cell activation was recently shown to play a role in restraining memory CD8 T cell differentiation. While the absolute number of antigen-specific effector cells is unchanged in mice with T cell-specific deletion of miR-15/16 upon LCMV infection, memory CD8 T cell accumulation is preferentially affected 68 . It remains to be further investigated as to whether or not there is any specific target that could account for the effects of miR-15/16 on CD8 memory cell differentiation. Nevertheless, many memory-associated molecules such as IL-7R 69 and ADRB2 70 were found to be directly targeted by miR-15/16 (Fig. 1C).
Unlike the aforementioned let-7, miR-150, and miR-15/16, miR-31 is highly induced during T cell activation through the calcium-NFAT pathway 71. However, despite their different expression patterns, miR-31 was also suggested to act as a negative regulator in controlling CD8 T cell immunity. Previously, continued exposure to type I interferons was reported to contribute to CD8 T cell dysfunction in part through the induction of PD-1 72. To this end, the expression of miR-31 was shown to increase the sensitivity of T cells to type I interferons and promote T cell exhaustion during chronic infection through enhancing the expression of PD-1 and multiple other inhibitory molecules. Finally, considering that miR-31 is expressed only following TCR activation, it was suggested that miR-31 exhibits its regulatory function in response to secondary or chronic exposure to type I interferons. Supporting this notion, while miR-31 deficiency significantly enhances CD8 T cell responses leading to improved viral control during chronic viral infection, it has little or no effect on virus-specific CD8 T cells and viral burden during acute infection (Fig. 1C) 71. Similar to miR-31, miR-155 was also recently shown to promote CD8 T cell exhaustion during chronic viral infection despite the role of miR-155 in driving effector CD8 T cell responses against virus (and cancer) has being well documented 73,74,75. Specifically, overexpression of miR-155 increases the generation and/or survival of terminally differentiated exhausted T cells without altering the proliferation of the progenitor exhausted T cell pool. Moreover, by performing transcriptome analysis, it was found that several molecules involved in the AP-1 pathway including a direct miR-155 target, Fosl2, are all inhibited by miR-155 overexpression. Previously, it has been shown that while NFAT is generally considered to be a key factor in T cell activation, under conditions where it does not cooperate with AP-1, NFAT promotes CD8 T cell exhaustion by binding directly to regulatory regions of many exhaustion-associated genes, including PD-1 and Tim3 76. As such, miR-155 can drive the exhaustion program by downregulating Fosl2 and increasing the activity of “AP-1 less” NFAT functioning (Fig. 1C) 75.
3. CELL-EXTRINSIC ROLE OF MIRNAS IN CONTROLLING T CELL IMMUNITY
3.1. miRNAs control T cell development through targeting thymic epithelial cells
T cells originate from hematopoietic stem cells in the bone marrow. The lymphoid progenitors migrate to the thymus, where they go through a series of differentiation stages to generate self-tolerant, mature T lymphocytes with diverse specificities. Intra-thymic T cell development is a complex process that depends upon continuous guidance from the thymus stromal cell microenvironment. Thymic epithelial cells (TECs) make up the majority of thymic stromal cells and can be divided into two major populations, cortical thymic epithelial cells (cTECs) and medullary thymic epithelial cells (mTECs), based on their localization 77. cTECs are essential for the positive selection of developing T cells that are capable of recognizing and interacting with MHC molecules on their surface. On the other hand, mTECs play major roles in inducing the negative selection of highly self-reactive T cells as well as in promoting the generation of Treg cells needed for establishing immunological tolerance. Like T cell development, the establishment of an appropriate thymic microenvironment also requires proper differentiation of thymic stromal cell populations that involves multiple levels of signals including intracellular signaling events and dynamic gene regulatory networks. As important molecular regulators of gene expression, the role of miRNAs in TEC biology and, more importantly, its impact on thymic T cell development have been recently demonstrated.
Compared to the relatively minor effect of miRNA ablation in T cells during early thymic T cell development as discussed above, TEC-specific deletion of the miRNA network results in a dramatic disruption of thymic architecture with increased TEC apoptosis and a severe loss of thymic cellularity 78–80. Consequently, thymus with miRNA-deficient TECs exhibits notable defects in the positive selection, with reduction of CD4 and CD8 single-positive (SP) thymocytes, and the appearance of immature thymic B cells that resemble those observed in the bone marrow 78. Nevertheless, despite the reduced T cell cellularity both in the thymus and in the periphery, the compromised thymic microenvironment, particularly in the medullary compartment, as a result of miRNA deficiency in TECs leads to altered T cell phenotypes and increased autoimmunity susceptibility 78–80. Together, these studies demonstrated an essential role of miRNA-mediated gene regulation in establishing the thymic epithelia pivotal for appropriate thymocytes selection, T cell lineage commitment, and the generation of immunological tolerance.
To date, several studies were carried out to investigate individual miRNAs that regulate TEC differentiation and function. In one report, miRNA microarray profiling analysis of cTECs, immature mTEClow, and mature mTEChigh from mouse and human samples revealed evolutionarily conserved cell type- and differentiation-specific miRNA signatures. In particular, a mutual regulatory relationship between a subset of miRNAs and the expression of AIRE, a transcriptional regulator known to be involved in coordinating promiscuous expression of tissue-restricted antigens (TRAs) during mTEC maturation was identified 81. Further studies have shown that despite exhibiting no major impact on thymic architecture and the development of the cTEC and mTEC populations 80, TEC-specific deletion of miR-29a, one of the miRNAs highly expressed in AIREhi mTECs, led to reduced expression of AIRE, accompanied by down-regulation of AIRE-dependent TRA expression (Fig. 2A) 81.
FIGURE 2. T cell-extrinsic roles of miRNAs in regulating T cell immunity.
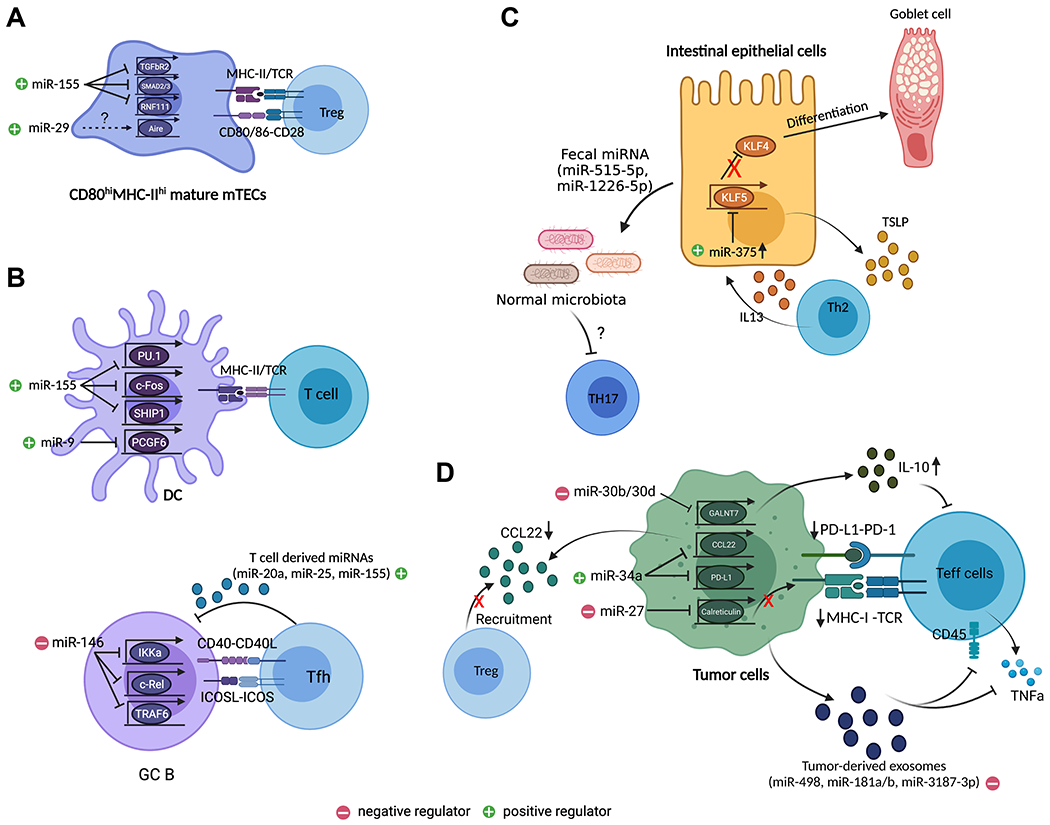
(A) miR-29 is required for the development of Aire+ mTEC while miR-155 safeguards mTEC maturation that is required for optimal thymic Treg cell development by targeting many components within the TGFβ signaling pathway including Tgfbr2, Smad2/3, and Rnf111. (B) In the periphery, miRNAs impact T cell responses through regulating their interacting partners. In DCs, miR-155 promotes their maturation and function through targeting Spi1 (encodes for PU.1), Fos, and Ship1. Similarly, miR-9 enhances function of DCs, cDC1s in particular, through targeting Pcgf6. In B cells, miR-146a limits GC B cell differentiation by targeting many signaling components downstream of CD40 signaling pathway including Ikka, Rel, and Traf6, which in turn impacts Tfh cell responses. On the other hand, exosomal miRNAs such as miR-20a, miR-25, and miR155, released from Tfh cells can promote GC B cell responses. (C) miRNAs can also affect T cell responses through shaping the tissue microenvironment. For example, in the intestine, miR-375 promotes IEC differentiation and function by targeting Klf5. Loss of miR-375 resulted in impaired TSLP secretion leading to defective intestinal Th2 immunity. Moreover, IEC-derived miRNAs, miR-515-5p and miR-1226-5p can regulate Th17 responses in the gut through targeting gut microbiota. (D) In tumor, downregulation of tumor suppressor miRNA, miR-34a led to enhanced Treg cell recruitment and impaired Teff cell activation due to the loss of miR-34a-mediated inhibition of CCL22 and PD-L1. On the other hand, miR-27 and miR-30b/d serve as oncomiRs by impairing MHCI-mediated antigen presentation and by promoting the production of immunosuppressive cytokine, IL-10 through targeting Calreticulin and Galnt7, respectively. Finally, tumor cell-derived exosomal miRNAs such as miR-181a/b, miR-498, miR3187-3p can also directly inhibit Teff cell function by targeting Cd45 and Tnf. All graphics are created with BioRender.com.
As discussed above, in addition to its role in mediating negative selection, the thymic medulla represents a specific site for establishing self tolerance via the generation of Treg cells. To this end, we have recently demonstrated that miR-155, a miRNA whose expression in T cells is critical for Treg cell development and homeostasis 48,82, also plays a pivotal role in driving thymic Treg cell differentiation through promoting mTEC maturation 82. To this end, miR-155 is preferentially expressed in mature CD80hiMHCIIhi mTECs in part via a RANK signaling-dependent manner. Elevated expression of miR-155 in mTECs is important for maintaining optimal mTEC maturation as TEC-specific deletion of miR-155 leads to a diminished mature mTEC population despite the overall thymic cellularity remaining unaltered. Consequently, the quantity of Treg cells particularly in the thymus was also impacted 82. Mechanistically, we have further demonstrated that miR-155 ensures proper mTEC maturation through targeting a network of molecules (including TGFβR2, SMAD2/3, and RNF111) within the TGFβ signaling pathway. As the TGFβ signaling pathway had been previously shown to play a key role in limiting the maturation and expansion of mTECs 83, our work reveals a previously unappreciated role of miR-155 in safeguarding mTEC maturation through counteracting the negative effects from the continuous presence of intrathymic TGFβ, thereby establishing an optimal thymic microenvironment favorable for thymic Treg development (Fig. 2A) 82. This study is the first demonstration of an individual miRNA with functional importance in driving Treg cell differentiation in both T cell-intrinsic and -extrinsic manners. It also suggests that additional attention should be paid to stromal cells residing in the same microenvironments to better understand the biological impact of a given miRNA on a selective T cell response or other immunological processes.
3.2. miRNAs impact T cell homeostasis and function through regulating different T cell-interacting immune cell partners
From going through their development in the thymus to being activated and mediating their effector functions in the periphery, interactions with other immune cell populations are crucial in conferring optimal T cell immunity. Below, we will review some key discoveries that described how miRNAs could impact T cell responses through regulating other immune cell types (Fig. 2B).
miRNAs in Dendritic cells
Dendritic cells, a type of professional antigen-presenting cells (APCs), play a vital role in connecting innate and adaptive immunity. Through presenting antigen peptides via MHC molecules along with the provision of co-stimulatory signals, DCs are not only important to help facilitate T cell development in the thymus but also critical in serving as a prominent player in the initiation and progression of adaptive immune response in the periphery. Thus, it is not surprising that miRNAs can impact T cell activation and proliferation through exerting their regulatory function to affect the development and function of DCs.
For example, miR-155, a miRNA that has already been discussed several times for its diverse function in different T cell subsets as well as in the thymic epithelium, has also been shown to control a variety of physiological and pathological processes in DCs 84–89. In myeloid DCs, miR-155 is upregulated upon LPS-induced DC maturation, which is characterized by a series of functional changes, including decreased pathogen binding/endocytic activity, increased cytokine production, and enhanced antigen presentation. Mechanistically, it was shown that through targeting PU.1, miR-155 decreases the pathogen binding capacity of DCs by downregulating DC-SIGN, thereby switching functions of DCs from endocytosis to promoting T cell activation 84. DCs with miR-155 deficiency exhibit reduced expression of MHCII and costimulatory molecules, such as CD40, CD80, and CD86, leading to impaired antigen presentation and costimulatory function that are necessary to activate T cells 85,86. In addition to the aforementioned PU.1, it was suggested that miR-155 can mediate its regulatory function in DCs through repressing c-Fos as all of the phenotypic and functional defects exhibited by miR155-deficient DCs could be reproduced by deregulated c-Fos expression 86. On the other hand, upregulation of miR-155 in DCs has been reported to promote autoimmune responses in many different disease settings. In one disease model in which the transfer of self-antigen–pulsed, TLR-matured DCs can induce a functional CD8 T cell response and autoimmune diabetes, DCs lacking miR-155 were found to exhibit an impaired ability to break immune tolerance 87. In contrast, transfer of self-antigen-pulsed DCs with miR-155 overexpression was sufficient to promote autoimmunity even in the absence of TLR stimuli. The autoimmune-driving effect of miR-155 is likely mediated through targeting SHIP1, an inositol polyphosphatase that is critical in regulating proinflammatory signaling pathways in DCs 88. Similarly, in experimental autoimmune encephalomyelitis (EAE), a murine model of multiple sclerosis (MS), elevated expression of miR-155 in DCs was shown to promote inflammatory T cell responses through the production of Th1- and Th17-polarizing cytokines, thus exacerbating the autoimmune conditions 89,90.
Recently, another miRNA, miR-9, was also demonstrated to facilitate the maturation of DCs and enhance their function to stimulate T cell activation 91. Interestingly, the positive effect of miR-9 on DCs was shown to be restricted to conventional DC1 (cDC1), a type of DCs that are particularly recognized for their role in eliciting anti-tumor immunity 92. Upon activation, the expression of mature miR-9 is specifically up-regulated in cDC1 but not cDC2. Consequently, PCGF6, a negative regulator of DC activation 93, is directly targeted by miR-9 in cCD1 leading to enhanced tumor-specific T cell responses and better control of tumor growth in vivo. It should be noted, the selective effect of miR-9 on promoting cDC1 function is not simply due to the difference in their miR-9 expression patterns. Overexpression of miR-9 in cDC2 does not lead to significant changes in the expression of costimulatory molecules and their capacity to activate T cells similar to what was observed in cDC1 with miR-9 overexpression 91. Collectively, these results suggest that the distinct expression patterns and functions of miR-9 in different DC subtypes allow this miRNA to coordinately control DC responses to elicit appropriate T cell immunity. This study also offers a great example for understanding context-dependent and cell-type-specific miRNA-mediated gene regulation within different immune cell subpopulations (Fig. 2B).
miRNAs in B cells
As discussed above, Tfh cells are pivotal in providing help to B cells in GCs, a specialized microstructure essential for the generation of B cell clones that produce high-affinity antibodies with different functional properties 94. Similarly, the differentiation and maintenance of Tfh cells also requires GC B cells and that the magnitude of the Tfh cell response was directly correlated with the magnitude of the GC B cell response 95. These two immune cell populations are tightly controlled by a variety of molecular regulators to ensure the generation of optimal humoral immunity without eliciting unwanted autoantibody responses. Interestingly, many of these molecular regulators are shared between GC B and Tfh cells. One perfect example is BCL6, the aforementioned Tfh cell master transcription factor that was originally identified as an essential regulator of GC B cell differentiation 96. The fact that BCL6 controls both GC B cells and Tfh cells suggests an intricate gene regulatory circuit is implemented in two interacting immune populations, thereby enabling them to generate a concerted response in the same tissue microenvironment. To this end, similar to what has been previously discussed in Tfh cells 33,34, by using a mouse model in which miRNAs were specifically deleted in B cells expressing AID, a DNA-editing enzyme expressed in GC B cells 97, miRNA-mediated gene regulation was also shown to be integral in GC B cell formation 98. Mice with AID-driven disruption of Dicer-dependent miRNA network fail to generate high-affinity class-switched antibodies and B cell memory in response to a T cell-dependent antigen. Together, the studies of global miRNA deletion in Tfh and GC B cells highlight a critical role of miRNAs in establishing protective humoral immunity.
One of the miRNAs that have been extensively studied in GC B cell responses is miR-155, which is upregulated upon B cell activation and highly expressed in GC B cells 19,52,99. Like the positive role that has already been described in Tfh cells 36, B cells devoid of miR-155 generate defective germinal center responses and fail to produce high-affinity isotype switched antibodies. Mechanistically, the observed impaired humoral immune responses are in part attributed to the loss of miR-155-mediated regulation of PU.1 100,101. In addition to the abovementioned role of PU.1 in regulating DC function, in B cells, PU.1 was shown to inhibit isotype switching and plasma cell differentiation by inducing the expression of PAX5 101, a transcription factor indispensable for B cell development but functioning as a negative regulator for plasma cell differentiation 102. Thus, the miR-155-PU.1 axis confers proper GC B cell responses through ensuring the effectiveness of terminal B cell differentiation as well as regulating other PU.1 targets that are directly involved in T-B cell interactions 101.
Reciprocal interactions between GC B and Tfh cells through many different ligand/receptor engagements are essential to support the formation of GCs 103. In addition to a sustained antigenic stimulation 95, two of the best characterized positive feedback signals between GC B cells and Tfh cells are through the CD40–CD40L and ICOSL–ICOS interactions 104. To this end, our recent study has shown that miR-146a, a miRNA highly expressed in both Tfh and GC B cells, is equally or perhaps even more important in controlling GC B cells through inhibiting multiple components (including TRAF6, RelB, c-Rel and IKKα) downstream of the CD40 signaling pathway than in limiting Tfh cell responses via targeting ICOS 43. Unlike what has been shown in Tfh cells, deletion of miR-146a alone in mature B cells is sufficient to lead to elevated GC reactions accompanied by increased Tfh cell frequencies and the development of spontaneous autoimmunity over time 43. It is thus not surprising that no further changes in humoral responses could be detected between mice with B cell-specific miR-146a ablation and those with miR-146a deletion in both B and T cells. These findings also suggest that the loss of a T cell-extrinsic, but B cell-intrinsic regulation exerted by miR-146a is primarily responsible for the dysregulated Tfh cell phenotype previously observed in mice with germline miR-146a deficiency (Fig. 2B) 42.
Finally, in addition to controlling GC B and Tfh cell responses by their respective miRNAs, it was recently demonstrated that miRNAs can also regulate GC T-B cell communication in a cell non-autonomous manner through the transfer of miRNAs via extracellular vesicles (EVs) during immune synapse formation 105. By using an in vitro experimental system in which antigen-specific T cells and Dicer-deficient B cells were co-cultured, it was shown that the formation of immune synapses facilitates the transfer of a defined set of T cell-derived miRNAs such as miR-20a, miR-25, and miR-155 to B cells to promote B cell homeostasis and function. Further analysis of mice harboring T cells incapable of releasing miRNA-containing EVs due to the deletion of Rab27a/b, two members of the Rab family that are required for exosome secretion 106, revealed dysfunctional GC responses and significantly reduced levels of serum IgM and class-switched IgG upon immunization (Fig. 2B) 105. These findings add another layer of complexity to miRNA-mediated regulation of T cell-dependent B cell-driven humoral immunity.
3.3. miRNAs regulate T cell immunity through shaping the tissue microenvironment in health and disease
In addition to interacting with different immune cell populations, it is now well recognized that T cell responses are heavily impacted by the environment they reside in. Many studies have demonstrated that T cells could acquire specific migratory, homeostatic, and functional features in response to different environmental stimuli, allowing them to exert their function in a given anatomical site 107. Similar to the aforementioned role of mTEC-derived miR-155 in regulating thymic Treg cell development, in the peripheral tissues, miRNAs can impact T cell responses by shaping the environmental cues supplied by nonimmune cells. Alternatively, miRNAs can also directly serve as molecular mediators (e.g. exosomal miRNAs) to facilitate the communication between the tissue-specific stromal cells and tissue-resident T cells. As discussed below, we will focus on the recent findings in miRNA-mediated control of local T cell responses in a T cell-extrinsic manner in two settings: the intestinal tract and tumors.
miRNAs in intestinal epithelial cells
Among different non-lymphoid tissues, the intestine represents the most unique tissue environment. It contains the largest surface of contact between the body and the external environment. Moreover, the intestine is also continuously exposed to vast amounts of harmless foreign antigenic materials including food proteins that are ingested daily in our diet as well as trillions of commensal microorganisms that colonize the intestinal lumen. Located between epithelial cells and in the lamina propria, different intestinal immune cell populations coordinate with each other to orchestrate protective immune responses against numerous gut-associated pathogens. In addition to providing a physical barrier between commensals and immune cells, intestinal epithelial cells (IECs) also exhibit immunological functions in the gut through secreting mucus, antimicrobial peptides, as well as cytokines and chemokines that regulate gut-resident T cells and other immune cell populations. To date, emerging evidence has highlighted the functional relevance of miRNAs within IECs to fine-tune host gene expression networks and signaling pathways that can modulate mucosal immunity in the intestine (Fig. 2C).
The critical role of miRNAs in the intestinal epithelium was first demonstrated by the specific ablation of Dicer within IECs in mice 108,109. Deletion of Dicer-dependent miRNA pathway in IECs results in disrupted intestinal architecture and defective intestinal epithelial cell differentiation. Consequently, mice with IEC-specific Dicer deficiency exhibit impaired growth, decreased water absorption in the colon, and are unable to efficiently absorb fat in their diet 108. Additionally, loss of the Dicer-dependent miRNA network also lead to significant loss of goblet cells due to impaired goblet cell differentiation and reduced production of a variety of Th2 cytokines, including IL-4, IL-5, IL-13, and RELMβ, a goblet cell-specific Th2 anti-parasitic effector molecule in the colon 109. As a result, mice with IEC devoid of miRNAs exhibit decreased mucus production and diminished functional Th2 immunity in the gut, accompanied by compromised resistance to parasitic infection. Subsequent studies have identified miR-375 as a key IEC-expressed miRNA that modulates goblet cell differentiation through targeting KLF5, a member of the KLF family of zinc-finger transcription factors that has been previously shown to be essential for goblet-cell differentiation and the maintenance of intestinal crypt architecture 110. Moreover, it was also shown that miR-375 functions as a positive regulator to promote the production of Thymic Stromal Lymphopoietin (TSLP), an epithelial cytokine known to promote Th2 immune responses 111. As expression of miR-375 in IECs was found to be induced by IL-13 109, these results suggest a Th2 feed-forward loop driven by intestinal epithelial miR-375 to maintain protective anti-parasitic immunity in the gut (Fig. 2C).
In addition to generating effective immune responses against gut-associated pathogens, one of the most important functions of the intestinal immune system is to establish tolerance to a vast amount of gut flora to prevent deleterious immune-mediated inflammation and tissue damage 112. It is thus not surprising that IEC-derived miRNAs could impact mucosal immune responses by influencing the gut microbiota. To this end, it was reported that IEC-derived miR-515-5p and miR-1226-5p promoted the growth of F. nucleatum and E. coli through directly regulating gene expression in the bacteria although it remains uncertain as to how host miRNAs are able to be transferred from IECs into these commensal microorganisms 113. Further studies have shown that loss of IEC-derived miRNAs in mice results in increased IL-17 responses and exacerbated DSS-induced colitis, which could be largely rescued by administration of WT fecal miRNAs 113. Considering the well-recognized role of gut microbiota in intestinal Th17 cell differentiation, these results strongly suggest that IEC-derived miRNAs can maintain intestinal homeostasis through regulating gut microbiota and intestinal T cell immunity (Fig. 2C).
Cancer-associated miRNAs regulate T cell function in the tumor microenvironment
Studies over the past 50 years have suggested a complex relationship between cancer and the immune system. T cells can exert their anti-tumor effects by either acting directly on tumor cells or enhancing immune responses against tumors through the stimulation of other immune cell populations. Nevertheless, tumors can also gain resistance by hijacking major components of immune regulatory function to inactivate anti-tumor immune responses 114,115. Since their initial discovery back in the late 90s, miRNAs have been intensively studied for their multifaceted roles in cancer 116. Recently, in addition to the cell-autonomous roles of miRNAs in either promoting (i.e. oncomiRs) or inhibiting (i.e. tumor suppressor miRNAs) tumorigenesis, emerging evidence has revealed non-cell-autonomous roles played by cancer–associated miRNAs to mediate tumor-T cell communications in the tumor microenvironment (Fig. 2D).
In hepatocellular carcinoma (HCC), infection of hepatitis B virus (HBV) has been well documented to play a key role in initiating the tumorigenic process and accounts for more than 60% of the total liver cancer cases in developing countries 117. It was shown that elevated TGF-β signaling activity caused by HBV infection could suppress the expression of miR-34a in HCC cells, resulting in enhanced production of the chemokine CCL22, a primary target of miR-34a 118. Through binding to its receptor CCR4 on the surface of Treg cells, HCC-derived CCL22 promotes the recruitment of Treg cells rendering the microenvironment of liver tissue around the portal venous system immunosuppressive to favor the colonization and expansion of HCC cells disseminated from the primary tumor site. In addition to driving Treg cell-mediated control of anti-tumor immunity, inhibition of miR-34a has also been previously demonstrated to negatively regulate T cell responses through inducing immune checkpoint molecule expression 119. Specifically, in Epstein-Barr virus (EBV)-associated B cell lymphomas, the co-localization of viral protein EBNA2 and B-cell-specific transcription factor EBF1 at the miR-34a promoter has been shown to repress miR-34a expression, leading to enhanced expression of PD-L1, a direct target of miR-34a 119. As the interaction between tumor-expressing PD-L1 and its receptor PD-1, which is highly expressed on tumor-infiltrating T cells, represents one of the major immune escape mechanisms in cancer 120, the miR-34a-PD-L1 axis in EBV-associated B cell lymphomas thus provides another example of the cell-extrinsic role of miR-34a in regulating T cell-mediated anti-tumor immunity (Fig. 2D).
Besides acting as tumor suppressors, many miRNAs have been functionally characterized as oncomiRs for their activities to inhibit anti-tumor immune responses. To this end, in human melanoma, elevated levels of miR-30b/30d have been reported to promote tumorigenesis through targeting GALNT7, a GalNAc transferase that modifies the O-linked glycosylation on target proteins 121. Loss of GALNT7 by excessive miR-30b/30d-mediated inhibition in human melanoma cells was shown to lead to increased production of immunosuppressive cytokine, IL-10. Consequently, the supernatants from miR-30d-overexpressing (or GALNT7-knockdown) melanoma cells display enhanced abilities to suppress T cell activation and to promote Treg cell differentiation. Moreover, mice harboring melanoma cells with aberrant miR-30d expression exhibit increased recruitment of Treg cells to the metastatic site 121. Another example is miR-27a, a miRNA with multiple functions in controlling T cell responses as discussed above. By targeting calreticulin, an endoplasmic reticulum (ER) chaperon protein responsible for the assembly and cell surface expression of MHC class I molecules 122, miR-27a was suggested to promote tumor progression at least in part through inhibiting tumor-infiltrating CD8 T cell activation, and cytotoxic activity in colorectal cancer (CRC) 123. Finally, in addition to modulating tumor’s activities to interact with T cells, tumor-derived miRNAs could also directly impact T cell function through exosome-mediated delivery similar to what was discussed above in B and T cell interaction. To this end, a recent study has found that melanoma cells can limit CD8 T cell activation and function through releasing melanoma-derived exosomes enriched with TNF- and CD45-targeting miRNAs such as miR-498, miR-181a/b, and miR-3187-3p (Fig. 2D) 124.
4. CONCLUSION, REMAINING QUESTIONS, AND FUTURE DIRECTIONS
Over the past two decades, thanks to the technological advancements in miRNA target identification and the establishment of a vast amount of genetic tools, our knowledge of the role of miRNAs in regulating T cell immunity has grown tremendously. However, with our understanding of miRNA-mediated control of T cell responses getting deeper, new questions have also arisen. For example, one of the most interesting findings from recent studies is the fact that several miRNAs that serve as important gene modulators in a given T cell population are also highly expressed and play key functions in other cell types those T cells closely interact with (e.g. miR-146a in Tfh and GC B cells or miR-155 in Treg cells and mTECs). These results suggest miRNAs can enforce their regulatory effects on T cells by targeting them in both cell-intrinsic and -extrinsic manners. Nevertheless, the detected similar expression patterns of miRNAs in T cells and their interacting partners could not be simply explained by the shared environmental signals they receive. These miRNAs also do not seem to target the same set of genes in the two interacting populations despite promoting concerted responses. It thus remains to be further investigated as to whether these are just some unique cases or represent a common mechanism by which miRNAs control T cell immunity. Moreover, the existence of evolutionarily conserved miRNA clusters has been generally thought to help ensure their biological impact through coordinately targeting one key gene or different components within the common biological pathway by different members in the same miRNA family (e.g. miR-23C in limiting Tfh cell responses). However, the observation that individual miRNAs from the same miRNA cluster can also antagonize each other in controlling a specific type of T cell immunity (e.g. miR-23C in regulating Th1 cell responses) also raises a question as to how such an unproductive feature in miRNA-mediated gene regulation could be retained evolutionarily.
Moving forward, to fully appreciate miRNA-mediated regulation of T cell immunity, besides continuing the current efforts to identify new miRNAs and their targets in different T cell subsets, additional attention should be paid to understand the molecular mechanisms underlying the regulation and function of miRNAs commonly expressed in T cells and their corresponding interacting partners as well as to further examine how the expression of individual miRNAs within the same cluster can be differently regulated under certain circumstances to optimally control a particular type of T cell response.
ACKNOWLEDGEMENT
This work was supported by National Institutes of Health grants AI108651, AI139753, and AI163813 (to L.-F. Lu). L.-F. Lu is a scientific advisor for Elixiron Immunotherapeutics. The authors declare no conflict of interest.
REFERENCES
- 1.Muljo SA, Ansel KM, Kanellopoulou C, Livingston DM, Rao A, Rajewsky K. Aberrant T cell differentiation in the absence of Dicer. J Exp Med. 2005;202(2):261–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cobb BS, Nesterova TB, Thompson E, et al. T cell lineage choice and differentiation in the absence of the RNase III enzyme Dicer. J Exp Med. 2005;201(9):1367–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cobb BS, Hertweck A, Smith J, et al. A role for Dicer in immune regulation. J Exp Med. 2006;203(11):2519–2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liston A, Lu LF, O’Carroll D, Tarakhovsky A, Rudensky AY. Dicer-dependent microRNA pathway safeguards regulatory T cell function. J Exp Med. 2008;205(9):1993–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhou X, Jeker LT, Fife BT, et al. Selective miRNA disruption in T reg cells leads to uncontrolled autoimmunity. J Exp Med. 2008;205(9):1983–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations (*). Annu Rev Immunol. 2010;28:445–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Afkarian M, Sedy JR, Yang J, et al. T-bet is a STAT1-induced regulator of IL-12R expression in naive CD4+ T cells. Nat Immunol. 2002;3(6):549–557. [DOI] [PubMed] [Google Scholar]
- 8.Steiner DF, Thomas MF, Hu JK, et al. MicroRNA-29 regulates T-box transcription factors and interferon-gamma production in helper T cells. Immunity. 2011;35(2):169–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ma F, Xu S, Liu X, et al. The microRNA miR-29 controls innate and adaptive immune responses to intracellular bacterial infection by targeting interferon-gamma. Nat Immunol. 2011;12(9):861–869. [DOI] [PubMed] [Google Scholar]
- 10.Smith KM, Guerau-de-Arellano M, Costinean S, et al. miR-29ab1 deficiency identifies a negative feedback loop controlling Th1 bias that is dysregulated in multiple sclerosis. J Immunol. 2012;189(4):1567–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guo YE, Riley KJ, Iwasaki A, Steitz JA. Alternative capture of noncoding RNAs or protein-coding genes by herpesviruses to alter host T cell function. Mol Cell. 2014;54(1):67–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cho S, Wu CJ, Yasuda T, et al. miR-23 approximately 27 approximately 24 clusters control effector T cell differentiation and function. J Exp Med. 2016;213(2):235–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cruz LO, Hashemifar SS, Wu CJ, et al. Excessive expression of miR-27 impairs Treg-mediated immunological tolerance. J Clin Invest. 2017;127(2):530–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cho S, Wu CJ, Nguyen DT, et al. A Novel miR-24-TCF1 Axis in Modulating Effector T Cell Responses. J Immunol. 2017;198(10):3919–3926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu Q, Sharma A, Oh SY, et al. T cell factor 1 initiates the T helper type 2 fate by inducing the transcription factor GATA-3 and repressing interferon-gamma. Nat Immunol. 2009;10(9):992–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu Q, Sharma A, Ghosh A, Sen JM. T cell factor-1 negatively regulates expression of IL-17 family of cytokines and protects mice from experimental autoimmune encephalomyelitis. J Immunol. 2011;186(7):3946–3952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fahy JV. Type 2 inflammation in asthma--present in most, absent in many. Nat Rev Immunol. 2015;15(1):57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang DH, Cohn L, Ray P, Bottomly K, Ray A. Transcription factor GATA-3 is differentially expressed in murine Th1 and Th2 cells and controls Th2-specific expression of the interleukin-5 gene. J Biol Chem. 1997;272(34):21597–21603. [DOI] [PubMed] [Google Scholar]
- 19.Thai TH, Calado DP, Casola S, et al. Regulation of the germinal center response by microRNA-155. Science. 2007;316(5824):604–608. [DOI] [PubMed] [Google Scholar]
- 20.Okoye IS, Czieso S, Ktistaki E, et al. Transcriptomics identified a critical role for Th2 cell-intrinsic miR-155 in mediating allergy and antihelminth immunity. Proc Natl Acad Sci U S A. 2014;111(30):E3081–3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Simpson LJ, Patel S, Bhakta NR, et al. A microRNA upregulated in asthma airway T cells promotes TH2 cytokine production. Nat Immunol. 2014;15(12):1162–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pua HH, Steiner DF, Patel S, et al. MicroRNAs 24 and 27 Suppress Allergic Inflammation and Target a Network of Regulators of T Helper 2 Cell-Associated Cytokine Production. Immunity. 2016;44(4):821–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lu YJ, Gross J, Bogaert D, et al. Interleukin-17A mediates acquired immunity to pneumococcal colonization. PLoS Pathog. 2008;4(9):e1000159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gaffen SL, Jain R, Garg AV, Cua DJ. The IL-23-IL-17 immune axis: from mechanisms to therapeutic testing. Nat Rev Immunol. 2014;14(9):585–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mikami Y, Philips RL, Sciume G, et al. MicroRNA-221 and -222 modulate intestinal inflammatory Th17 cell response as negative feedback regulators downstream of interleukin-23. Immunity. 2021;54(3):514–525 e516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tanaka S, Suto A, Iwamoto T, et al. Sox5 and c-Maf cooperatively induce Th17 cell differentiation via RORgammat induction as downstream targets of Stat3. J Exp Med. 2014;211(9):1857–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Langrish CL, Chen Y, Blumenschein WM, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201(2):233–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ichiyama K, Gonzalez-Martin A, Kim BS, et al. The MicroRNA-183-96-182 Cluster Promotes T Helper 17 Cell Pathogenicity by Negatively Regulating Transcription Factor Foxo1 Expression. Immunity. 2016;44(6):1284–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wan Q, Kozhaya L, ElHed A, et al. Cytokine signals through PI-3 kinase pathway modulate Th17 cytokine production by CCR6+ human memory T cells. J Exp Med. 2011;208(9):1875–1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu C, Yosef N, Thalhamer T, et al. Induction of pathogenic TH17 cells by inducible salt-sensing kinase SGK1. Nature. 2013;496(7446):513–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Crotty S T follicular helper cell differentiation, function, and roles in disease. Immunity. 2014;41(4):529–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vinuesa CG, Linterman MA, Yu D, MacLennan IC. Follicular Helper T Cells. Annu Rev Immunol. 2016;34:335–368. [DOI] [PubMed] [Google Scholar]
- 33.Baumjohann D, Kageyama R, Clingan JM, et al. The microRNA cluster miR-17 approximately 92 promotes TFH cell differentiation and represses subset-inappropriate gene expression. Nat Immunol. 2013;14(8):840–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zeitrag J, Dahlstrom F, Chang Y, et al. T cell-expressed microRNAs critically regulate germinal center T follicular helper cell function and maintenance in acute viral infection in mice. Eur J Immunol. 2021;51(2):408–413. [DOI] [PubMed] [Google Scholar]
- 35.Xiao C, Srinivasan L, Calado DP, et al. Lymphoproliferative disease and autoimmunity in mice with increased miR-17-92 expression in lymphocytes. Nat Immunol. 2008;9(4):405–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu WH, Kang SG, Huang Z, et al. A miR-155-Peli1-c-Rel pathway controls the generation and function of T follicular helper cells. J Exp Med. 2016;213(9):1901–1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu CJ, Cho S, Huang HY, et al. MiR-23~27~24-mediated control of humoral immunity reveals a TOX-driven regulatory circuit in follicular helper T cell differentiation. Sci Adv. 2019;5(12):eaaw1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aliahmad P, Seksenyan A, Kaye J. The many roles of TOX in the immune system. Curr Opin Immunol. 2012;24(2):173–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Choi YS, Gullicksrud JA, Xing S, et al. LEF-1 and TCF-1 orchestrate T(FH) differentiation by regulating differentiation circuits upstream of the transcriptional repressor Bcl6. Nat Immunol. 2015;16(9):980–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu L, Cao Y, Xie Z, et al. The transcription factor TCF-1 initiates the differentiation of T(FH) cells during acute viral infection. Nat Immunol. 2015;16(9):991–999. [DOI] [PubMed] [Google Scholar]
- 41.Shi J, Hou S, Fang Q, Liu X, Liu X, Qi H. PD-1 Controls Follicular T Helper Cell Positioning and Function. Immunity. 2018;49(2):264–274 e264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pratama A, Srivastava M, Williams NJ, et al. MicroRNA-146a regulates ICOS-ICOSL signalling to limit accumulation of T follicular helper cells and germinal centres. Nat Commun. 2015;6:6436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cho S, Lee HM, Yu IS, et al. Differential cell-intrinsic regulations of germinal center B and T cells by miR-146a and miR-146b. Nat Commun. 2018;9(1):2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol. 2012;30:531–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zheng Y, Josefowicz SZ, Kas A, Chu TT, Gavin MA, Rudensky AY. Genome-wide analysis of Foxp3 target genes in developing and mature regulatory T cells. Nature. 2007;445(7130):936–940. [DOI] [PubMed] [Google Scholar]
- 46.Marson A, Kretschmer K, Frampton GM, et al. Foxp3 occupancy and regulation of key target genes during T-cell stimulation. Nature. 2007;445(7130):931–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lu LF, Boldin MP, Chaudhry A, et al. Function of miR-146a in controlling Treg cell-mediated regulation of Th1 responses. Cell. 2010;142(6):914–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lu LF, Thai TH, Calado DP, et al. Foxp3-dependent microRNA155 confers competitive fitness to regulatory T cells by targeting SOCS1 protein. Immunity. 2009;30(1):80–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guerau-de-Arellano M, Smith KM, Godlewski J, et al. Micro-RNA dysregulation in multiple sclerosis favours pro-inflammatory T-cell-mediated autoimmunity. Brain. 2011;134(Pt 12):3578–3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bopp T, Becker C, Klein M, et al. Cyclic adenosine monophosphate is a key component of regulatory T cell-mediated suppression. J Exp Med. 2007;204(6):1303–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang B, Zhao J, Lei Z, et al. miR-142-3p restricts cAMP production in CD4+CD25-T cells and CD4+CD25+ TREG cells by targeting AC9 mRNA. EMBO Rep. 2009;10(2):180–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kuchen S, Resch W, Yamane A, et al. Regulation of microRNA expression and abundance during lymphopoiesis. Immunity. 2010;32(6):828–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Anandagoda N, Willis JC, Hertweck A, et al. microRNA-142-mediated repression of phosphodiesterase 3B critically regulates peripheral immune tolerance. J Clin Invest. 2019;129(3):1257–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gavin MA, Rasmussen JP, Fontenot JD, et al. Foxp3-dependent programme of regulatory T-cell differentiation. Nature. 2007;445(7129):771–775. [DOI] [PubMed] [Google Scholar]
- 55.Scherm MG, Serr I, Zahm AM, et al. miRNA142-3p targets Tet2 and impairs Treg differentiation and stability in models of type 1 diabetes. Nat Commun. 2019;10(1):5697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yue X, Trifari S, Aijo T, et al. Control of Foxp3 stability through modulation of TET activity. J Exp Med. 2016;213(3):377–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jeker LT, Zhou X, Gershberg K, et al. MicroRNA 10a marks regulatory T cells. PLoS One. 2012;7(5):e36684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim D, Nguyen QT, Lee J, et al. Anti-inflammatory Roles of Glucocorticoids Are Mediated by Foxp3(+) Regulatory T Cells via a miR-342-Dependent Mechanism. Immunity. 2020;53(3):581–596 e585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Charbonnier LM, Cui Y, Stephen-Victor E, et al. Functional reprogramming of regulatory T cells in the absence of Foxp3. Nat Immunol. 2019;20(9):1208–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.McLane LM, Abdel-Hakeem MS, Wherry EJ. CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annu Rev Immunol. 2019;37:457–495. [DOI] [PubMed] [Google Scholar]
- 61.Zhang N, Bevan MJ. Dicer controls CD8+ T-cell activation, migration, and survival. Proc Natl Acad Sci U S A. 2010;107(50):21629–21634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Trifari S, Pipkin ME, Bandukwala HS, et al. MicroRNA-directed program of cytotoxic CD8+ T-cell differentiation. Proc Natl Acad Sci U S A. 2013;110(46):18608–18613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wells AC, Daniels KA, Angelou CC, et al. Modulation of let-7 miRNAs controls the differentiation of effector CD8 T cells. Elife. 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wu H, Neilson JR, Kumar P, et al. miRNA profiling of naive, effector and memory CD8 T cells. PLoS One. 2007;2(10):e1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen Z, Stelekati E, Kurachi M, et al. miR-150 Regulates Memory CD8 T Cell Differentiation via c-Myb. Cell Rep. 2017;20(11):2584–2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kaech SM, Tan JT, Wherry EJ, Konieczny BT, Surh CD, Ahmed R. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat Immunol. 2003;4(12):1191–1198. [DOI] [PubMed] [Google Scholar]
- 67.Salomoni P, Perrotti D, Martinez R, Franceschi C, Calabretta B. Resistance to apoptosis in CTLL-2 cells constitutively expressing c-Myb is associated with induction of BCL-2 expression and Myb-dependent regulation of bcl-2 promoter activity. Proc Natl Acad Sci U S A. 1997;94(7):3296–3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gagnon JD, Kageyama R, Shehata HM, et al. miR-15/16 Restrain Memory T Cell Differentiation, Cell Cycle, and Survival. Cell Rep. 2019;28(8):2169–2181 e2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nanjappa SG, Walent JH, Morre M, Suresh M. Effects of IL-7 on memory CD8 T cell homeostasis are influenced by the timing of therapy in mice. J Clin Invest. 2008;118(3):1027–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Slota C, Shi A, Chen G, Bevans M, Weng NP. Norepinephrine preferentially modulates memory CD8 T cell function inducing inflammatory cytokine production and reducing proliferation in response to activation. Brain Behav Immun. 2015;46:168–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Moffett HF, Cartwright ANR, Kim HJ, et al. The microRNA miR-31 inhibits CD8(+) T cell function in chronic viral infection. Nat Immunol. 2017;18(7):791–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Terawaki S, Chikuma S, Shibayama S, et al. IFN-alpha directly promotes programmed cell death-1 transcription and limits the duration of T cell-mediated immunity. J Immunol. 2011;186(5):2772–2779. [DOI] [PubMed] [Google Scholar]
- 73.Dudda JC, Salaun B, Ji Y, et al. MicroRNA-155 is required for effector CD8+ T cell responses to virus infection and cancer. Immunity. 2013;38(4):742–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gracias DT, Stelekati E, Hope JL, et al. The microRNA miR-155 controls CD8(+) T cell responses by regulating interferon signaling. Nat Immunol. 2013;14(6):593–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Stelekati E, Chen Z, Manne S, et al. Long-Term Persistence of Exhausted CD8 T Cells in Chronic Infection Is Regulated by MicroRNA-155. Cell Rep. 2018;23(7):2142–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Martinez GJ, Pereira RM, Aijo T, et al. The transcription factor NFAT promotes exhaustion of activated CD8(+) T cells. Immunity. 2015;42(2):265–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Abramson J, Anderson G. Thymic Epithelial Cells. Annu Rev Immunol. 2017;35:85–118. [DOI] [PubMed] [Google Scholar]
- 78.Zuklys S, Mayer CE, Zhanybekova S, et al. MicroRNAs control the maintenance of thymic epithelia and their competence for T lineage commitment and thymocyte selection. J Immunol. 2012;189(8):3894–3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Khan IS, Taniguchi RT, Fasano KJ, Anderson MS, Jeker LT. Canonical microRNAs in thymic epithelial cells promote central tolerance. Eur J Immunol. 2014;44(5):1313–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Papadopoulou AS, Dooley J, Linterman MA, et al. The thymic epithelial microRNA network elevates the threshold for infection-associated thymic involution via miR-29a mediated suppression of the IFN-alpha receptor. Nat Immunol. 2011;13(2):181–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ucar O, Tykocinski LO, Dooley J, Liston A, Kyewski B. An evolutionarily conserved mutual interdependence between Aire and microRNAs in promiscuous gene expression. Eur J Immunol. 2013;43(7):1769–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dong J, Warner LM, Lin LL, Chen MC, O’Connell RM, Lu LF. miR-155 promotes T reg cell development by safeguarding medullary thymic epithelial cell maturation. J Exp Med. 2021;218(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hauri-Hohl M, Zuklys S, Hollander GA, Ziegler SF. A regulatory role for TGF-beta signaling in the establishment and function of the thymic medulla. Nat Immunol. 2014;15(6):554–561. [DOI] [PubMed] [Google Scholar]
- 84.Martinez-Nunez RT, Louafi F, Friedmann PS, Sanchez-Elsner T. MicroRNA-155 modulates the pathogen binding ability of dendritic cells (DCs) by down-regulation of DC-specific intercellular adhesion molecule-3 grabbing non-integrin (DC-SIGN). J Biol Chem. 2009;284(24):16334–16342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rodriguez A, Vigorito E, Clare S, et al. Requirement of bic/microRNA-155 for normal immune function. Science. 2007;316(5824):608–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dunand-Sauthier I, Santiago-Raber ML, Capponi L, et al. Silencing of c-Fos expression by microRNA-155 is critical for dendritic cell maturation and function. Blood. 2011;117(17):4490–4500. [DOI] [PubMed] [Google Scholar]
- 87.Lind EF, Millar DG, Dissanayake D, et al. miR-155 Upregulation in Dendritic Cells Is Sufficient To Break Tolerance In Vivo by Negatively Regulating SHIP1. J Immunol. 2015;195(10):4632–4640. [DOI] [PubMed] [Google Scholar]
- 88.Cekic C, Casella CR, Sag D, et al. MyD88-dependent SHIP1 regulates proinflammatory signaling pathways in dendritic cells after monophosphoryl lipid A stimulation of TLR4. J Immunol. 2011;186(7):3858–3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Murugaiyan G, Beynon V, Mittal A, Joller N, Weiner HL. Silencing microRNA-155 ameliorates experimental autoimmune encephalomyelitis. J Immunol. 2011;187(5):2213–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.O’Connell RM, Kahn D, Gibson WS, et al. MicroRNA-155 promotes autoimmune inflammation by enhancing inflammatory T cell development. Immunity. 2010;33(4):607–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cordeiro B, Jeon P, Boukhaled GM, et al. MicroRNA-9 Fine-Tunes Dendritic Cell Function by Suppressing Negative Regulators in a Cell-Type-Specific Manner. Cell Rep. 2020;31(5):107585. [DOI] [PubMed] [Google Scholar]
- 92.Bottcher JP, Reis e Sousa C. The Role of Type 1 Conventional Dendritic Cells in Cancer Immunity. Trends Cancer. 2018;4(11):784–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Boukhaled GM, Cordeiro B, Deblois G, et al. The Transcriptional Repressor Polycomb Group Factor 6, PCGF6, Negatively Regulates Dendritic Cell Activation and Promotes Quiescence. Cell Rep. 2016;16(7):1829–1837. [DOI] [PubMed] [Google Scholar]
- 94.Victora GD, Nussenzweig MC. Germinal centers. Annu Rev Immunol. 2012;30:429–457. [DOI] [PubMed] [Google Scholar]
- 95.Baumjohann D, Preite S, Reboldi A, et al. Persistent antigen and germinal center B cells sustain T follicular helper cell responses and phenotype. Immunity. 2013;38(3):596–605. [DOI] [PubMed] [Google Scholar]
- 96.Klein U, Dalla-Favera R. Germinal centres: role in B-cell physiology and malignancy. Nat Rev Immunol. 2008;8(1):22–33. [DOI] [PubMed] [Google Scholar]
- 97.Revy P, Muto T, Levy Y, et al. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the Hyper-IgM syndrome (HIGM2). Cell. 2000;102(5):565–575. [DOI] [PubMed] [Google Scholar]
- 98.Xu S, Guo K, Zeng Q, Huo J, Lam KP. The RNase III enzyme Dicer is essential for germinal center B-cell formation. Blood. 2012;119(3):767–776. [DOI] [PubMed] [Google Scholar]
- 99.Basso K, Sumazin P, Morozov P, et al. Identification of the human mature B cell miRNome. Immunity. 2009;30(5):744–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Vigorito E, Perks KL, Abreu-Goodger C, et al. microRNA-155 regulates the generation of immunoglobulin class-switched plasma cells. Immunity. 2007;27(6):847–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lu D, Nakagawa R, Lazzaro S, et al. The miR-155-PU.1 axis acts on Pax5 to enable efficient terminal B cell differentiation. J Exp Med. 2014;211(11):2183–2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Nera KP, Kohonen P, Narvi E, et al. Loss of Pax5 promotes plasma cell differentiation. Immunity. 2006;24(3):283–293. [DOI] [PubMed] [Google Scholar]
- 103.Crotty S Follicular helper CD4 T cells (TFH). Annu Rev Immunol. 2011;29:621–663. [DOI] [PubMed] [Google Scholar]
- 104.Liu D, Xu H, Shih C, et al. T-B-cell entanglement and ICOSL-driven feed-forward regulation of germinal centre reaction. Nature. 2015;517(7533):214–218. [DOI] [PubMed] [Google Scholar]
- 105.Fernandez-Messina L, Rodriguez-Galan A, de Yebenes VG, et al. Transfer of extracellular vesicle-microRNA controls germinal center reaction and antibody production. EMBO Rep. 2020;21(4):e48925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ostrowski M, Carmo NB, Krumeich S, et al. Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat Cell Biol. 2010;12(1):19–30; sup pp 11-13. [DOI] [PubMed] [Google Scholar]
- 107.Masopust D, Soerens AG. Tissue-Resident T Cells and Other Resident Leukocytes. Annu Rev Immunol. 2019;37:521–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.McKenna LB, Schug J, Vourekas A, et al. MicroRNAs control intestinal epithelial differentiation, architecture, and barrier function. Gastroenterology. 2010;139(5):1654–1664, 1664 e1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Biton M, Levin A, Slyper M, et al. Epithelial microRNAs regulate gut mucosal immunity via epithelium-T cell crosstalk. Nat Immunol. 2011;12(3):239–246. [DOI] [PubMed] [Google Scholar]
- 110.McConnell BB, Kim SS, Yu K, et al. Kruppel-like factor 5 is important for maintenance of crypt architecture and barrier function in mouse intestine. Gastroenterology. 2011;141(4):1302–1313, 1313 e1301-1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kitajima M, Lee HC, Nakayama T, Ziegler SF. TSLP enhances the function of helper type 2 cells. Eur J Immunol. 2011;41(7):1862–1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Izcue A, Coombes JL, Powrie F. Regulatory lymphocytes and intestinal inflammation. Annu Rev Immunol. 2009;27:313–338. [DOI] [PubMed] [Google Scholar]
- 113.Liu S, da Cunha AP, Rezende RM, et al. The Host Shapes the Gut Microbiota via Fecal MicroRNA. Cell Host Microbe. 2016;19(1):32–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Nishikawa H, Sakaguchi S. Regulatory T cells in tumor immunity. Int J Cancer. 2010;127(4):759–767. [DOI] [PubMed] [Google Scholar]
- 115.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Di Leva G, Garofalo M, Croce CM. MicroRNAs in cancer. Annu Rev Pathol. 2014;9:287–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69–90. [DOI] [PubMed] [Google Scholar]
- 118.Yang P, Li QJ, Feng Y, et al. TGF-beta-miR-34a-CCL22 signaling-induced Treg cell recruitment promotes venous metastases of HBV-positive hepatocellular carcinoma. Cancer Cell. 2012;22(3):291–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Anastasiadou E, Stroopinsky D, Alimperti S, et al. Epstein-Barr virus-encoded EBNA2 alters immune checkpoint PD-L1 expression by downregulating miR-34a in B-cell lymphomas. Leukemia. 2019;33(1):132–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Han Y, Liu D, Li L. PD-1/PD-L1 pathway: current researches in cancer. Am J Cancer Res. 2020;10(3):727–742. [PMC free article] [PubMed] [Google Scholar]
- 121.Gaziel-Sovran A, Segura MF, Di Micco R, et al. miR-30b/30d regulation of GalNAc transferases enhances invasion and immunosuppression during metastasis. Cancer Cell. 2011;20(1):104–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Raghavan M, Wijeyesakere SJ, Peters LR, Del Cid N. Calreticulin in the immune system: ins and outs. Trends Immunol. 2013;34(1):13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Colangelo T, Polcaro G, Ziccardi P, et al. Proteomic screening identifies calreticulin as a miR-27a direct target repressing MHC class I cell surface exposure in colorectal cancer. Cell Death Dis. 2016;7:e2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Vignard V, Labbe M, Marec N, et al. MicroRNAs in Tumor Exosomes Drive Immune Escape in Melanoma. Cancer Immunol Res. 2020;8(2):255–267. [DOI] [PubMed] [Google Scholar]