

Abstract
Background:
Phthalates may disturb metabolic homeostasis in the liver by interfering with the peroxisome proliferator-activated receptors (PPARs). However, the role of hepatic macrophages in the lipid metabolic dysregulation induced by diethylhexyl phthalate (DEHP) remains unclear.
Objectives:
We aimed to evaluate the respective role of hepatocyte- and macrophage-specific in the hepatotoxicity induced by DEHP.
Methods:
Wild-type (WT), hepatocyte-specific knockout (Hep-KO), and macrophage-specific PPAR knockout (Mac-KO) mice were administered DEHP ( body weight) by daily gavage for 28 d, followed by hepatotoxicity examination and macrophage analysis. RNA sequencing and lipid metabolomic analysis were used to characterize the molecular changes in mouse liver. Mouse bone marrow-derived macrophages (BMDMs) and human monocytic THP-1 cell-derived macrophages were used to investigate the mechanistic regulation of macrophages’ polarization by DEHP and mono(2-ethylhexyl) phthalate (MEHP).
Results:
The levels of hepatic steatosis and triglyceride were significantly higher in the mice treated with DEHP compared with the control mice in the WT and Hep-KO model. Lipid accumulation induced by DEHP was notably attenuated in the Mac-KO mice, but M2-polarization of hepatic macrophages in the Mac-KO mice was significantly higher compared with the WT mice under DEHP treatment. The M2-polarization of BMDMs and human macrophages was suppressed by DEHP and MEHP. Transcriptomic and lipidomic data suggested lower levels of lipid biosynthesis, fatty acid oxidation, and oxidative phosphorylation in the Mac-KO mice compared with the WT and Hep-KO mice under DEHP treatment.
Conclusions:
Our data suggested that the orchestrated activation of and by MEHP may reprogram hepatic macrophages’ polarization, thereby affecting lipid homeostasis in the mouse liver. Although this conclusion was based on studies conducted in mice and in vitro, these findings may aid in elucidating the health effect of environmental phthalate exposure. https://doi.org/10.1289/EHP9373
Introduction
Phthalates are widely used as industrial plasticizers in the manufacture of plastics, resins, rubbers, paints, emulsifiers, medical devices, personal care products, and so on. Diethylhexyl phthalate (DEHP) is the most common member in the class of phthalates and noncovalently binds to plastics, resulting in ubiquitous human exposure to DEHP in the United States via consumer products containing plasticizers (Zota et al. 2014). In addition, the widespread use of plastics in food-packaging materials potentially increases the oral exposure to phthalates (EFSA CEP Panel et al. 2019). DEHP and some other phthalates have demonstrated endocrine-disrupting effects, which may disturb organism-wide metabolic homeostasis and lead to various adverse health outcomes, including nonalcoholic fatty liver disease (NAFLD) (Maradonna and Carnevali 2018; Mariana et al. 2016). Experimental studies have shown that chronic oral exposure to DEHP can induce liver tumors in rodents (NTP 1982). A recent 28-d repeated gavage study identified the liver and metabolic system as main targets of DEHP in rats (Tassinari et al. 2021). Currently, DEHP has been classified as a nongenotoxic substance “possibly carcinogenic to humans” by the International Agency for Research on Cancer (IARC 2013). Although there is insufficient data of carcinogenicity in humans, the health risks posed by DEHP and other phthalates have raised wide public concern owing to the increasing emergence of numerous plastic products in the environment globally (Deng et al. 2020).
As typical peroxisome proliferators, DEHP and its derivatives have been demonstrated to induce fatty liver in rodents by interfering with the peroxisome proliferator-activated receptor alpha () (Feige et al. 2010; Issemann and Green 1990). PPARs, consisting of , , and , are a group of steroid hormone receptors that provide fine modulation for glucose and fat metabolism, as well as regulate inflammatory cell activation and fibrotic processes (Wahli and Michalik 2012). Accumulating evidence has revealed the crucial role of PPARs in metabolic syndromes, including NAFLD (Francque et al. 2021). may serve as the central bridge connecting the lipid metabolic disorder in the liver with DEHP-induced hepatocarcinogenesis (Corton et al. 2014). Interspecies differences between murine and human hepatic energy metabolism were also documented for (Feige et al. 2010). However, DEHP may induce liver cancer in animals through signaling pathways (Takashima et al. 2008). In addition, a recent study has shown that mono(2-ethylhexyl) phthalate (MEHP), the main metabolite of DEHP, could induce abnormal lipid metabolism and the pro-inflammatory state in differentiated adipocytes through , thereby affecting the overall homeostasis of energy metabolism (Manteiga and Lee 2017). Although the hepatotoxicity mechanism of DEHP remains incompletely known at present, recent experimental studies with mouse (Li et al. 2020) and hepatic cellular models (Xu et al. 2020) has suggested disruption of lipid metabolic homeostasis as a hallmark response for the adverse outcome.
A challenge to elucidating the mechanistic process of NAFLD is attributed to the sophisticated context of the tissue microenvironment in the liver, which comprises multiple cell types and extracellular matrix (Hotamisligil 2017). In the induction of fatty liver by DEHP, hepatocytes are a major responsive cell type, but other cell types may also be involved. In particular, hepatic macrophages, including the resident Kupffer cells and recruited monocyte-derived macrophages, play a central role in maintaining metabolic homeostasis and in the pathogenesis of liver injury (Tacke 2017). Moreover, macrophages are highly plastic and produce diverse functional subtypes, which dynamically change to adapt to the tissue microenvironment. The activation of PPARs in macrophages has been shown to be critical in governing the phenotype switch (Chawla 2010). Together, these findings suggest that macrophages may be targeted by DEHP, resulting in fundamental effects on liver disease progression via altering cell–cell interactions. However, the interaction between hepatocytes and macrophages in DEHP-induced fatty liver remains unclear. Thus, the identification of regulatory mechanisms of PPARs in driving cell type-specific behaviors will help to employ their functions in protecting the liver from metabolic disorders induced by environmental pollutants (e.g., phthalates).
In the present study, we aimed to investigate the respective role of in hepatocytes and macrophages in the lipid metabolic dysregulation induced by DEHP. The wild-type (WT), knockout (Hep-KO), and knockout (Mac-KO) mice were used as animal models for the in vivo study of DEHP oral exposure. Human hepatic cells and monocyte–macrophage polarization cellular models were introduced to elucidate the cell-specific functions of and .
Methods
Reagents
Detailed information for the key reagents and other materials are supplied in Table S1. For mice experiments, DEHP was suspended in 0.5% (wt/vol in distilled water) sodium carboxymethylcellulose (CMC) and stored at 4°C. The DEHP suspension was freshly prepared every 7 d. For in vitro experiments, DEHP and MEHP were dissolved in dimethyl sulfoxide and stored at . The solutions were refreshed every 4 wk.
Animals
Animal study protocols complied with national guidelines for the care and use of laboratory animals and were approved by the Institutional Animal Care and Use Committee of China National Center for Food Safety Risk Assessment (No. 2019004). The experiments were reported following the Animal Research: Reporting In Vivo Experiments guidelines. Six- to 8-wk-old male WT and () mice on a C57BL/6J background were purchased from Vital River Laboratory Inc. and the Jackson Laboratories, respectively. The Hep-KO and Mac-KO mice were respectively generated by intercrossing male mice with female and mice on the C57BL/6J background. The mice were crossed at the Shanghai Model Organisms Center (Shanghai, China). Animals were maintained at 20°C, in a 12-h light/dark cycle with 50–60% relative humidity and fed standard chow diets. Because no significant sex difference was observed in the hepatotoxicity of DEHP in the long-term feeding study with mice (NTP 1982) and because the estrous cycle may interfere with the toxicological end points in females (Liang et al. 2018), only male mice were used in the entire study.
Animal Treatment
In the dose–response study, WT mice were administered DEHP at doses of 0, 625, 1,250, and body weight (BW) () by daily gavage for 28 d. The dosage design was based on the oral median lethal dose () of DEHP in mice () (NTP 1982). We used about 1/10 of the as the highest dose for the 28-d study and 2-fold intervals for the lower doses. In the study, the WT () and Hep-KO () mice were administered DEHP ( BW) or 0.5% (wt/vol) CMC by daily gavage for 28 d. In the study, the WT () and Mac-KO () mice were administered DEHP ( BW) or 0.5% (wt/vol) CMC by daily gavage for 28 d. Animals administered 0.5% (wt/vol) CMC served as the vehicle control (CT). At the end of experiments, mice were weighed and anesthetized by intraperitoneal injection with sodium pentobarbital ( BW), and blood was collected from the retro-orbital sinus via orbital puncture. Then, the mice were euthanized via cervical dislocation. The liver was isolated, weighed, and divided into three parts: One part was fixed with 10% neutral-buffered formalin for histopathology examination and lipid staining; one part was frozen in liquid nitrogen and stored at for the determination of DEHP, MEHP, cytokines, RNA sequencing, and lipidomic analysis; and the third part was used for flow cytometry.
Determination DEHP and MEHP in Plasma and Liver
DEHP and MEHP in mouse plasma and liver from the dose–response study were determined according to a method modified from Miao et al. (2018). Briefly, plasma samples (; plasma samples with hemolysis were not used) or liver tissue samples (; two mice in the same group were combined in a pooled sample) was homogenized in acetate buffer () with () in a Dounce homogenizer. After spiking with of the isotope internal standards solution () (Cat# ALR-138S-CN; AccuStandard), the mixture was shaken for 12 h at 37°C for the enzymatic hydrolysis of phthalate conjugates. The analytes were chromatographically resolved using an ACQUITY BEH Phenyl column (, ; Waters) with a linear solvent gradient from mobile phase A (acetonitrile) to mobile phase B (0.02% formic acid) at a flow rate of . The eluent was analyzed using a TSQ Altis triple quadrupole mass spectrometer (Thermo Scientific) equipped with an electrospray ionization (ESI) interface in the multiple reaction monitoring mode.
Blood Biochemistry
Blood samples collected from the mice were centrifuged at for 10 min to obtain plasma. Plasma samples with hemolysis from each group were not used and the sample sizes () can be found in the corresponding Supplemental Material summary tables and figure captions. The concentration of triglycerides (TGs), cholesterol (CHO), cholinesterase (CHE), high-density lipoprotein cholesterol (HDLC), low-density lipoprotein cholesterol (LDLC), aspartate aminotransferase (AST), and alkaline phosphatase (ALP) were determined using an automatic biochemistry analyzer Beckman Coulter AU680 with the following reagents: OSR60118 TG kit, OSR6116 CHO kit, OSR6114 CHE kit, OSR6187 HDLC kit, OSR6183 LDLC kit, OSR6109 AST kit, and OSR6104 ALP kit (Beckman Coulter, USA).
Liver Index, Histopathology, and Lipid Staining
Livers of mice in the three individual studies were isolated and weighed. The liver index was calculated as the ratio of liver weight to body weight in percentage. The liver was fixed in 10% neutral-buffered formalin for 48 h, followed by embedding with paraffin or optimal cutting temperature (OCT) compound for the immunohistochemistry (IHC) and Oil Red O staining, respectively. Paraffin-embedded tissue sections () were stained with hematoxylin and eosin for histopathological examination. The severity of hepatic steatosis was quantitatively evaluated by multiplying the grading scores of the pathological lesion area in the section (, 1; 5–25%, 2; 25–50%, 3; and , 4) and the size of cell vacuolation (−, 1; ; ; and ).
The OCT-embedded tissue was sectioned () in a cryostat (CryoStar NX50, Thermo Scientific). The lipid deposition in mouse liver was visualized by Oil Red O staining with frozen tissue sections (). Briefly, Oil Red O working solution was prepared by diluting stock solution ( Oil Red O in isopropanol) with distilled water at . Sections were incubated with freshly prepared Oil Red O working solution for 1 h at room temperature (RT), then washed with distilled water three times and allowed to air dry. Images were captured on a Zeiss Imager M2 with ZEN imaging software (version 2.5, Zeiss). The staining intensity was quantitatively evaluated by multiplying the grading scores of stain area in the section (, 1; 5–25%, 2; 25%–50%, 3; and , 4) and the general size of lipid stain (−, 1; ; ; and ).
Determination of Hepatic TG
The total protein was isolated from of mouse liver (; for groups that had more than five mice, five mice were randomly selected) by homogenization on ice with a lipid extraction kit (Cat# ab211044; Abcam). The TG level in the lipid extract was determined using a Triglyceride Colorimetric Assay Kit (Cat# 10010303; Cayman), according to the manufacturer’s instructions.
Cell Lines
Human monocytic THP-1 cells (TIB-202) were acquired from American Type Culture Collection (ATCC) and maintained in Roswell Park Memorial Institute (RPMI) 1640 (ATCC-modified) complete medium with 10% (vol/vol) fetal bovine serum (FBS), 1% (vol/vol) penicillin/streptomycin, and 2-mercaptoethanol. Human liver hepatocellular carcinoma HepG2 cells were obtained from the Cell Bank of Type Culture Collection Committee of the Chinese Academy of Sciences and maintained in minimal essential medium (MEM) supplemented with 10% (vol/vol) FBS, 1% (vol/vol) penicillin/streptomycin, 1% (vol/vol) nonessential amino acids, and 1% (vol/vol) sodium pyruvate. The peroxisome proliferator-activated receptor response element (PPRE)-eGEP-THP-1 cell line was established by GENECHEM Inc. (Shanghai, China) using the PPRE-H2B-eGFP plasmid, which was a gift from S. Degrelle and T. Fournier (Addgene plasmid #84393) (Degrelle et al. 2017). The PPRE-eGEP-THP-1 cell line was maintained in RPMI 1640 complete medium with 10% (vol/vol) FBS and 1% (vol/vol) penicillin/streptomycin. All cells were maintained in an atmosphere of 37°C, 5% carbon dioxide (), and 95% humidity.
Human Macrophage Polarization in Vitro
Macrophage polarization was induced in vitro as previously described (Yang et al. 2020). Briefly, THP-1 cells or PPRE-eGEP-THP-1 cells were seeded in black-wall 96-well plates (Cat# 3603; Corning) at 100,000 cells/well with culture medium containing phorbol 12-myristate 13-acetate (). After adhesion for 48 h, the cells were allowed to rest in fresh media for 24 h to acquire M0 macrophages. To induce the polarization of M1 or M2 macrophages, M0 macrophages were treated with lipopolysaccharide (LPS; ) and recombinant human interferon gamma (; ) or interleukin-4 (IL-4; ) and IL-13 (), respectively, for 24–48 h. Macrophage polarization was validated with high-content cellular imaging method using the specific markers CCR7 and CD209, see the section “High-Content Cellular Imaging.” The number of replicate wells for each outcome are available in the corresponding figure captions and summary tables in the Supplemental Material.
Polarization of Bone Marrow-Derived Macrophages
Femurs and tibias isolated from 12-wk-old WT () and Mac-KO () male mice were used for generating bone marrow-derived macrophages (BMDMs) as previously reported (Giannakis et al. 2019). Briefly, bone marrow cells were collected and cultured in Dulbecco’s Modified Eagle Medium (DMEM) complete medium supplemented with 10% FBS (vol/vol), 1% penicillin/streptomycin (vol/vol), and recombinant mouse macrophage colony-stimulating factor () at 37°C and 5% for 7 d with the addition of fresh medium every 3 d. For the induction of polarization, the adherent M0 macrophages were respectively treated (3 wells/treatment) with LPS () and murine () or murine () and () for 24 h to obtain M1 or M2 type macrophages.
HepG2-Macrophages Coculture
HepG2 cells were preattached in the 6- or 24-well Transwell plates (Cat# 3450, Cat# 3413; Corning) at for 18 h. THP-1–derived M1/M2 macrophages were harvested with Accutase cell detachment solution (Cat# 07922; Stemcell) and plated in the upper inserts at . After coculture for 24 h, the HepG2 cells were harvested for RNA sequencing () or mitochondrial function analysis ().
Cell Treatment
Human HepG2 cells (20,000 cells/well) and THP-1/PPRE-eGEP THP-1–derived M0 macrophages (30,000 cells/well) were seeded in black-wall 96-well plates (Cat# 3603; Corning) for 24 h and then treated with DEHP and MEHP alone or in combination with selective PPARs ligands in replicate wells () for 24 h. The treatment concentrations of each compound were as follows: DEHP (), MEHP (), Wy14643 (WY; agonist, ), GW7647 ( agonist, ), rosiglitazone (RGZ; agonist, ), GW6471 ( antagonist, ), GW9662 ( antagonist, ), and T0070907 ( antagonist, ). For the dose–responsiveness experiments, treatment doses are available in Tables S11–S13, S29, and S30. BMDMs (40,000 cells/well, wells/treatment) were treated with DEHP () or MEHP () during macrophage polarization for 24 h.
High-Content Cellular Imaging
Cells treated as described in the sections “Cell Treatment” and “Mitochondrial Oxidative Phosphorylation Analysis” were blocked with FcR blocking buffer (Biolegend) for 10 min at RT and incubated with the fluorescence-conjugated antibodies against CD36 (1:100), CD209 (1:50), CCR7 (1:50), CD206 (1:100), CD69 (1:100), or lipid probes (1:1,000) for 30 min at RT. After refreshing with FluoroBrite DMEM (Gibco), cell images were acquired using the ImageXpress Micro Confocal High Content Imaging System (version 6.5, Molecular Device). The fluorescent images from at least five sites per well were analyzed using CellReporterXpress Imaging and Analysis Software (version 6.5, Molecular Device). The mean stain area and percentage of positive-staining cells (%) were calculated by normalizing the overall fluorescent staining cells (area and counts) to the total cells in the same image.
Surface Plasmon Resonance Analysis
Recombinant human (), (), and () proteins were separately fixed on the carboxyl sensor chips (Cat# SEN-AU-100-10-COOH; Nicoya Lifesciences) by amine-coupling. Amine-coupling uses the N-terminus and groups of lysine residues of the ligand to immobilize a ligand via covalent links. For fixing, the sensor chips were first activated for 240 s with a mixture of 1-Ethyl-3-(3-dimethylaminopropyl)-carbodiimide (EDC) and N-Hydroxysuccinimide (NHS) at a flow rate of . Then the PPARs proteins were each diluted with immobilization buffer ( sodium acetate, ) and then injected into chips. Finally, the chips were deactivated by ethanolamine hydrochloride at a flow rate of for . DEHP (100 and ) or MEHP (100, 200, 400, and ) in a volume of was injected sequentially into the prepared sensor chips at for an interaction time of 400 s in phosphate-buffered saline (PBS; ) running buffer. The interaction of the proteins with the chemicals was detected using the OpenSPR system (Nicoya Lifesciences). The binding time was 240 s, and the disassociation time was 160 s. The chips were then regenerated with PBS running buffer at a flow rate of . A one-to-one diffusion-corrected model was fitted to the wavelength shifts of resonance units (RUs) corresponding to the varied chemical concentrations. The data were retrieved and analyzed using TraceDrawer software (version 1.9, Ridgeview Instruments ab).
Molecular Docking and Dynamics Simulation
The crystal structures of the ligand binding domain from human (ID: 1I7G) and (ID: 1I7I) were obtained from the Research Collaboratory for Structural Bioinformatics (RCSB) Protein Data Bank database (https://www.rcsb.org). The chemical structure of MEHP was retrieved from the PubChem database (http://pubchem.ncbi.nlm.nih.gov/). The protein–ligand docking studies between and MEHP were based on these compounds’ structures and carried out with AutoDock (version 4.2, Arthur Olson, Scripps Research Institute) packages. The docked protein–ligand complexes were subjected to molecular dynamics simulation by the Desmond module of Schrodinger software (version 4.2) (https://www.schrodinger.com/Desmond/). Briefly, the optimal complex was placed in a cubic water box with a minimum distance of between the protein surface and box edges. Sodium and chloride ions were added to achieve physiological salt conditions with an electroneutrality of concentration. Energy minimization was performed under an OPLS-2005 force field. Twenty equilibration simulations were run in the isothermal-isobaric (NpT) ensemble with the following parameters: temperature, 300 K; pressure, 1.0135 bar; and integration time step, 2 fs. Constraint of all bonds involving hydrogen atoms was achieved with the Secure Hash Algorithm Keccak (SHAKE) method. Root mean square deviation and root mean square fluctuation were monitored and average structures were extracted for interaction mode analysis.
RNA Sequencing
Total RNA was extracted from mouse livers or cultured cells with TRIzol reagent (Thermo Fisher Scientific) and quantified using an Agilent 2100 Bioanalyzer. Libraries were prepared with TruSeq Stranded mRNA Sample Prep Kit (Illumina) and sequenced with Illumina Hiseq 2500 (for mouse liver tissues) or Illumina Noveseq 6000 (for cultured human cells) in pair-end mode. Clean reads were mapped to the reference genome mm10 and hg38 with HISAT (Kim et al. 2015). Mapped reads were assembled with StringTie (version 1.2.1) and quantified using FeatureCounts (Liao et al. 2014). Gene expression was calculated using Bowtie2 (version 2.2.6, Ben Langmead, Johns Hopkins University) and RSEM (version 1.2.12). Differentially expressed genes (fold change and ) were identified using DESeq2 (version 1.10.1, Bo Li and Colin Dewey, University of Wisconsin-Madison). Gene set enrichment and pathway enrichment were analyzed according to the Kyoto Encyclopedia of Genes and Genomes (KEGG) database. Gene Ontology analysis was performed using the Database for Annotation, Visualization and Integrated Discovery (https://david.ncifcrf.gov) to identify enriched items ( and top 20). All raw data are available in Gene Expression Omnibus database (https://www.ncbi.nlm.nih.gov/geo/) with the accession numbers GSE159120, GSE160373, GSE160004, and GSE160826.
IHC and Multicolor Immunofluorescence
Liver sections () from the Hep-KO model ( group, group) and Mac-KO model () were de-waxed in xylene for 10 min three times and successively rehydrated in ethanol (100%, 95%, 70%; vol/vol). After antigen unmasking with microwave heating for 30 min at 95°C in sodium citrate (), the endogenous peroxidase was quenched by incubation with 3% (vol/vol) hydrogen peroxide in PBS. The sections were blocked with 5% normal goat serum and then incubated with primary antibodies overnight at 4°C. After incubation with the horseradish peroxidase-conjugated secondary antibody, the expression and localization of antigens was visualized with a 3,3′-diaminobenzidine substrate kit (Cat# 8095S; Cell Signaling Technology) or Opal fluorophores (Cat# NEL810001KT; PerkinElmer). IHC images of liver sections were captured on a Zeiss Imager M2 and analyzed using ZEN imaging software (version 2.5, Zeiss).
Flow Cytometry
The freshly isolated mouse livers () were minced with a syringe needle, and gently crushed in PBS with a cell pestle and cell strainers (BD Biosciences). The cell suspensions were filtered through cell strainers, followed by centrifugation at for 5 min. The isolated cells were resuspended and blocked with the Fc receptor blocking solution for 10 min, followed by incubation with fluorophore-conjugated antibodies against CD45, CD11b, F4/80, CD36, CD206, Ly6c, and CCR2 for 30 min at RT. Cells were acquired using a FACSCalibur (version 10.5, BD Biosciences) and analyzed using FlowJo software. The gating strategy for flow cytometry analysis is provided in Figure S1. Briefly, leukocytes were first distinguished from the total cells, then two populations of macrophages were gated and classified into M1 and M2 macrophages. Macrophages with high levels of CD36 and lipid droplets (LDs) were respectively gated from macrophages. Patrolling and inflammatory monocyte populations were separated with Ly6c from monocytes.
Determination of Cytokines
The total protein was isolated from of mouse liver by homogenization on ice with PBS containing ethylenediaminetetraacetic acid and protease inhibitor cocktail (Cat# 04693116001; Roche). The protein concentrations were determined using a Pierce bicinchoninic acid protein assay kit (Thermo). Homogenate proteins () were used for the detection of tissue level of cytokines with a Legendplex Mouse Macrophage/Microglia 13-plex panel kit (Biolegend).
Cell supernatants from THP-1–derived macrophages were collected and separated from cell debris by centrifugation at for 10 min. The cytokines were determined using a Legendplex Human Macrophage/Microglia 13-plex Panel (Biolegend) according to the manufacturer’s instructions. Data were acquired by FACSCalibur flow cytometry and analyzed using LEGENDplex software (version 8.0; Biolegend).
Untargeted Lipid Metabolomic Analysis
Freshly isolated livers were snap frozen and saved at until processing. Eight hundred microliters of dichloromethane/methanol (3:1, vol/vol) was added to the sample, followed by grinding and centrifugation at for 15 min at 4°C. The organic fraction was transferred to a new vial and dried under a nitrogen stream, followed by reconstitution in isopropanol/acetonitrile/water (2:1:1, vol/vol/vol). Reconstituted extracts were injected () into an ultra-performance liquid chromatography (UPLC) system coupled to a high-resolution tandem mass spectrometer Xevo G2 XS QTOF (Waters), as previously described (Liu et al. 2020). Lipids were separated using an ACQUITY UPLC CSH C18 column (, ; Waters) with the solvents A (, ammonium formate, 0.1% formic acid) and B (, ammonium formate, 0.1% formic acid) at a flow rate of . The gradient program was applied as follows: 0–2 min, 40–43% B; 2–7 min, 50–54% B; 7.1–13 min, 70–99% B; and 13.1–15 min, 40% B. The mass spectrometer was equipped with an ESI source and was performed in both positive ion (POS) and negative ion (NEG) mode. The capillary and sampling cone voltages were set as follows: 3,000 V and 40 V in POS mode; and 2,000 V and 40 V in NEG mode. Data with mass ranges of m/z 100–2,000 and m/z 50–2,000 were acquired at the POS and NEG modes with data-dependent tandem mass spectrometry acquisition. Data were analyzed according to accurate mass measurement and product ion spectrum matching with online database Lipid Maps.
Mitochondrial Oxidative Phosphorylation Analysis
HepG2 cells were seeded in black-wall 96-well plates (Cat# 3603; Corning) at 300,000 cells/mL. After adhesion for 24 h, the cells were treated with MEHP (M; ) alone or in combination with IACS-10759 (IACS; ), VLX600 (VLX; ), 3-nitropropanoic acid (3-NP; ), or () in replicate wells ( wells/group) for 24 h. For the determination of mitochondrial membrane potential (MMP), HepG2 cells were incubated with tetramethylrhodamine ethyl ester (TMRE; ) in MEM complete medium for 30 min at 37°C. For the detection of mitochondrial reactive oxygen species, HepG2 cells were incubated with MitoSOX Red Mitochondrial Superoxide Indicator (Invitrogen) in Hanks’ Balanced Salt Solution for 10 min at 37°C. After incubation, cells were washed twice with PBS containing 0.2% (vol/vol) bovine serum albumin and stained with Hoechst 33342 in FluoroBrite DMEM. Subsequently, the cells were analyzed using the ImageXpress High Content Imaging System, see the section “High-Content Cellular Imaging.”
Statistical Analysis
Graphpad Prism (version 6.0; GraphPad) was used for statistical analysis and graphing. Data were analyzed by one-way or two-way analysis of variance, as appropriate, and post hoc testing with correction for multiple comparisons was used to determine differences between specific groups. was considered statistically significant. All group numbers and detailed significant values are provided in the experiment methods and within the figure captions. No methods were used for sample size determination or randomization.
Results
Effects of DEHP on Hepatic Lipid Metabolism
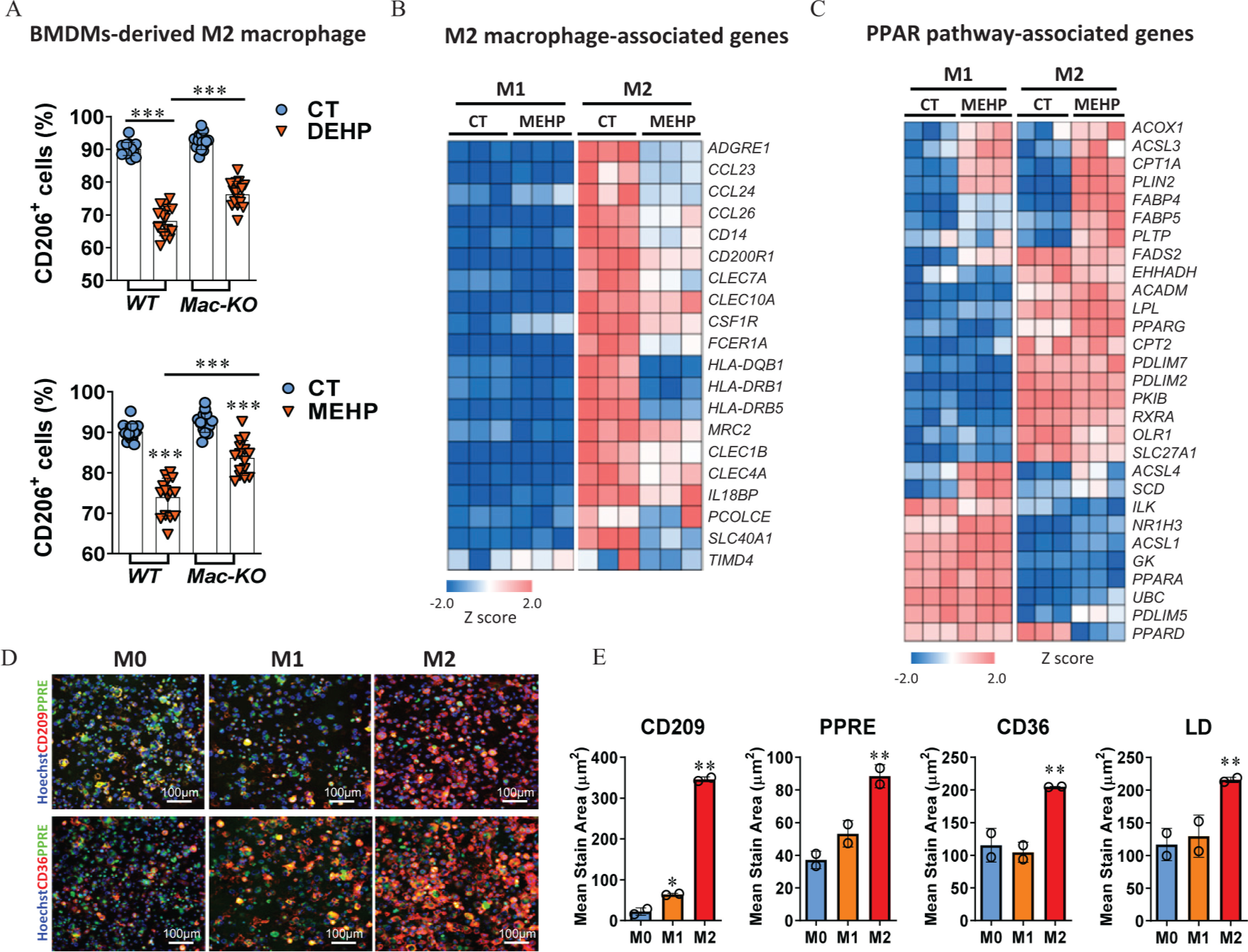
After repeated dosing for 28 d, both liver weight and liver index of mice treated with DEHP were significantly higher compared with control mice, and the response appeared to be dose dependent (Figure 1A; Table S2), whereas the body weight was not evidently different (Figure S2A, Table S44). The concentrations of the major metabolite MEHP in plasma (Figure 1B; Table S3) and liver tissue (Figure 1C; Table S3) of mice treated with DEHP were also significantly higher than for the control mice. Blood biochemistry analysis indicated that the plasma level of TGs in mice of the DEHP group was significantly lower than for the control group at the end of the treatment, whereas the plasma levels of CHO, LDLC, HDLC, and ALP were higher than for the control group (Figure 1D; Table S4). The hepatic TG level of mice treated with DEHP was markedly higher than for the control mice and the response was dose dependent (Figure 1E; Table S5). Furthermore, histology examination showed dose-related hepatic steatosis characterized by cytoplasmic vacuolization in the mouse liver of the DEHP group, and the quantitative severity of steatosis was in line with the intensity of Oil Red O staining (Figure 1F; Table S6). In human hepatic HepG2 cells, treatment with MEHP at the concentration of for 24 h resulted in higher intracellular levels of BODIPY FL and LDs compared with the control cells, and the response was concentration dependent (Figure 1G; Table S7). Thus, the in vitro data supported the effect of DEHP on hepatic lipid accumulation in the mice model. In addition, RNA sequencing with the mouse liver and human HepG2 cells indicated that the differently expressed genes (DEGs; fold change , ) between the DEHP and control group were closely associated with lipid metabolism (Figure S2B,C and Tables S45 and S46) and PPAR signaling pathway (Figure S2D,E and Tables S47 and S48).
Figure 1.
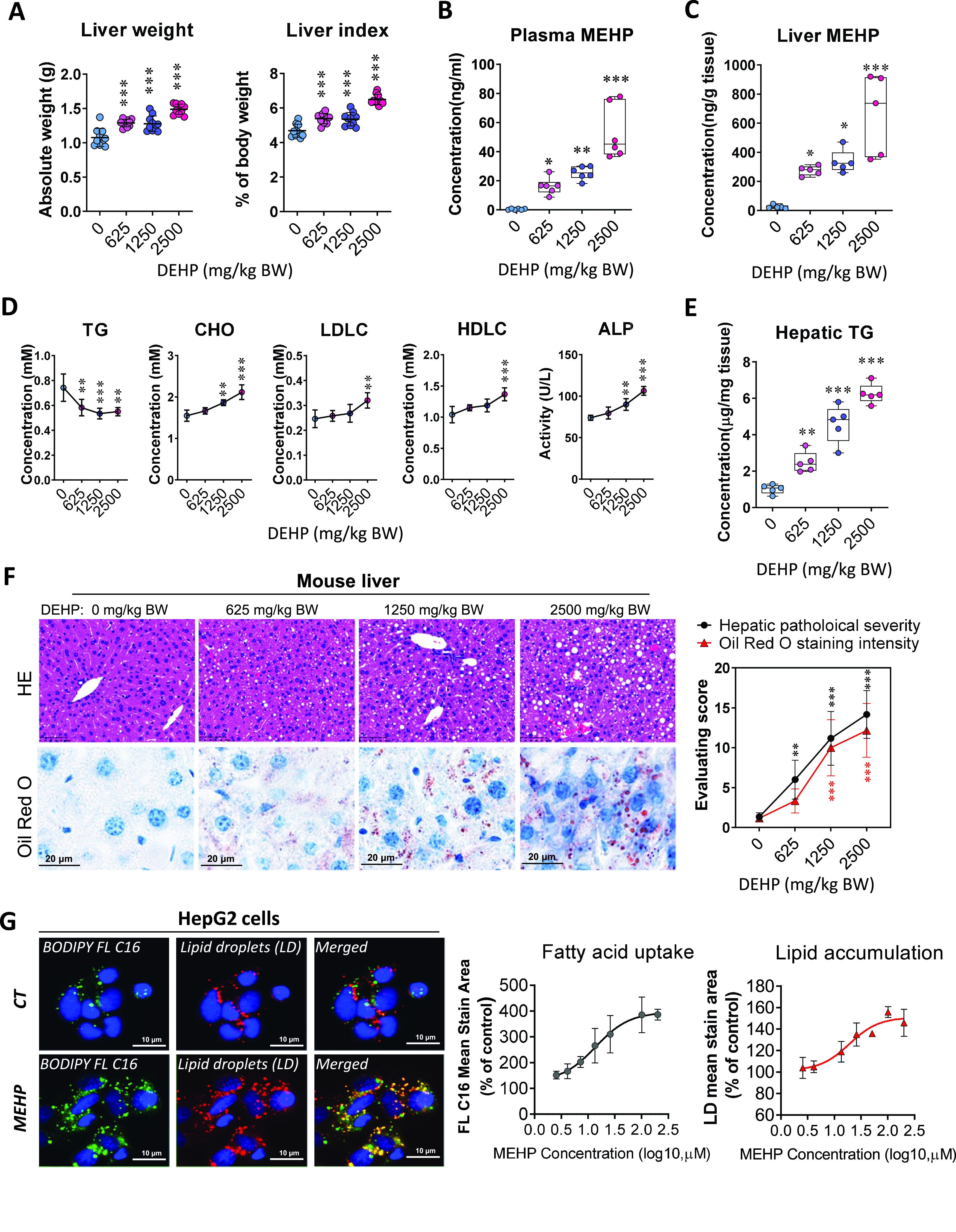
Effect of DEHP on hepatic lipid metabolism in mouse liver and human hepatocyte HepG2 cells. C57BL/6J male mice were treated with 0.5% (wt/vol) sodium carboxymethylcellulose (CMC; vehicle control) or different doses of DEHP (625, 1,250, and BW) by daily gavage for 28 d. (A) Liver weight and liver index at the end of the treatment period (). The data are provided in Table S2. (B) Plasma level of MEHP at the end of treatment (). (C) Tissue level of MEHP in the mouse liver at the end of treatment (). The data for (B,C) are provided in Table S3. (D) Plasma levels of triglycerides (TGs), cholesterol (CHO), high-density lipoprotein cholesterol (HDLC), low-density lipoprotein cholesterol (LDLC), and alkaline phosphatase (ALP) at the end of treatment (). The data are provided in Table S4. (E) TG concentration in mouse liver at the end of treatment (). The data are provided in Table S5. (F) Representative images of histology and Oil Red O staining of mouse liver at the end of treatment (left). Hepatic steatosis and lipid accumulation were quantified by calculating the score of pathological severity and Oil Red O staining intensity, respectively (right, ). The data are provided in Table S6. (G) Representative images of lipid uptake and lipid droplets (LDs) in HepG2 cells (left). HepG2 cells were treated in replicate wells () with various concentrations of MEHP () for 24 h and then incubated with the fluorescent dyes BODIPY FL (green) and LipidTOX Neutral Lipid Stain (red). Dose–response curves (right) were determined by comparing the mean fluorescent staining area to that of the control cells (DMSO treatment). The data are provided in Table S7. All data except for (B,C,E) are expressed as . Box plots (B,C,E) show the distribution of individual detections for each sample. The upper and lower limits in box plots represent the maximum to minimum values, with the horizontal lines in the boxes representing the median. The data were analyzed using one-way ANOVA followed by Dunnett’s multiple comparisons test. *, **, *** compared with the vehicle control. Note: ANOVA, analysis of variance; BW, body weight; CT, control; DEHP, diethylhexyl phthalate; DMSO, dimethyl sulfoxide; HE, hematoxylin and eosin; MEHP, mono(2-ethylhexyl) phthalate; SD, standard deviation.
Interaction of DEHP and MEHP with Different PPARs Isotypes
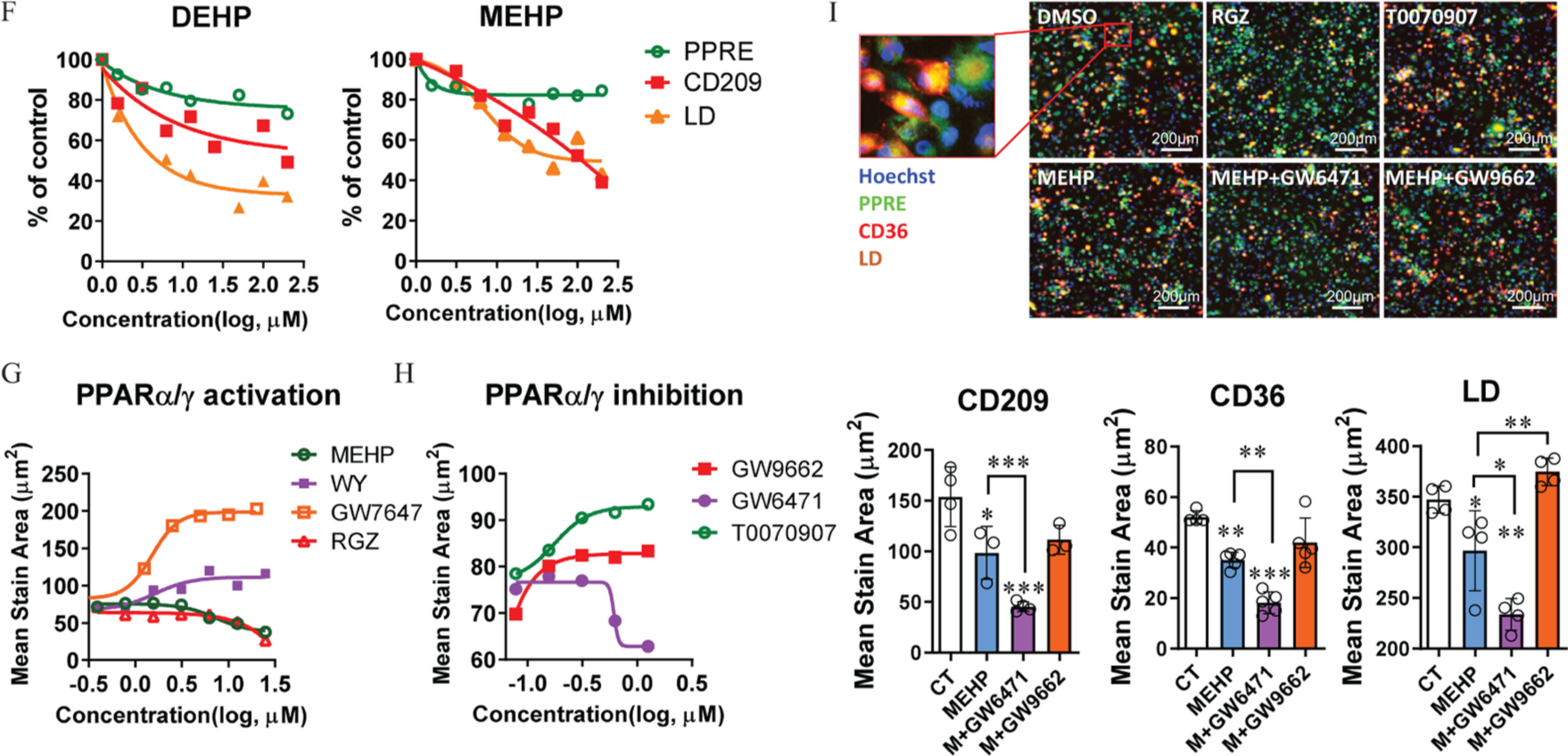
To examine the interaction of DEHP and its main metabolite, MEHP, with different human PPARs isotypes at the molecular level, we determined their binding activities using surface plasmon resonance (SPR) analysis. The result demonstrated that the RUs of both and were significantly responsive to MEHP at various concentrations (100, 200, 400, and ), but no significant RU changes were observed in response to DEHP (Figure 2A; Tables S8 and S9). No binding activity was detected between DEHP and MEHP with human (Figure S3A). The calculated binding affinity of MEHP with [dissociation constant ] was higher compared with () (Figure 2A; Tables S8 and S9). In addition, the protein–ligand docking and dynamic simulation analysis structurally predicted the binding interaction of MEHP with and (Figure 2B; Figure S3B). Although and have similar structures of binding pockets, there are still differences in key residues (e.g., H440 in and H449 in ) that may affect the binding with MEHP (Figure 2B). The calculated binding energy of MEHP with () was lower in comparison with () (Figure 2B; Figure S3C), which was consistent with the SPR results.
Figure 2.
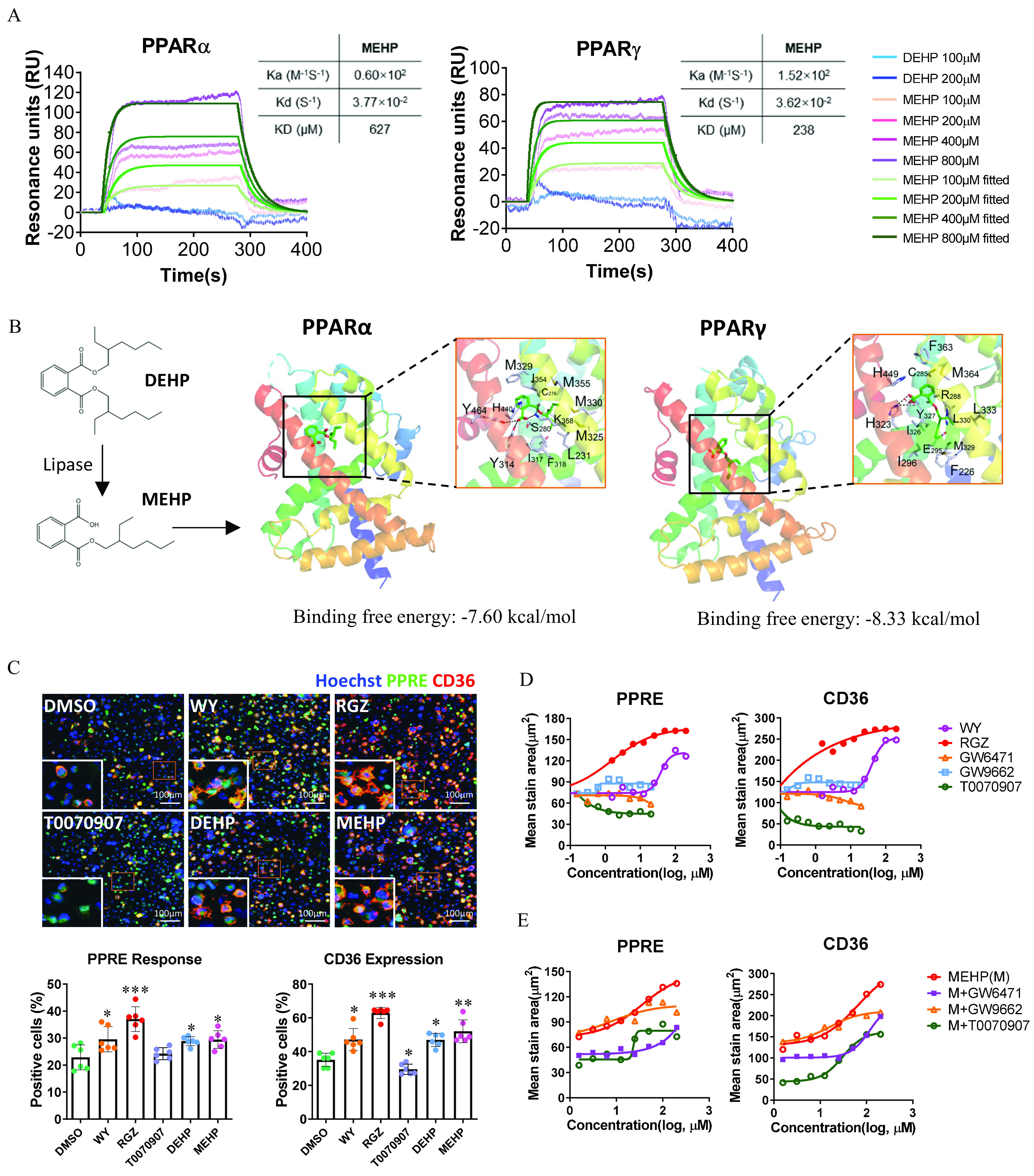
Determination of human and activation by DEHP and MEHP at the molecular and cellular levels. (A) Sensorgrams are shown of the binding responses of DEHP and MEHP with human or proteins. The binding affinity was determined using surface plasmon resonance (SPR). The data are provided in Tables S8 and S9. (B) Molecular docking simulation for the ligand–protein binding of MEHP with human and . The proteins are displayed as ribbons, and small molecules are displayed as sticks. (C) High-content imaging (up) and quantification (down) of PPRE response and CD36 expression in the PPRE-eGEP-THP-1–derived macrophages. The cells were treated with Wy14643 (WY; ), rosiglitazone (RGZ; ), T0070907 (), DEHP (), and MEHP () for 24 h. The number of cells with positive staining of PPRE activation (green) and CD36 expression (red) were normalized to the total cells (blue, stained with Hoechst 33342) in each of triplicate wells (the plots contain all technical replicates from two imaging sites of each well). Data are expressed as . The data are provided in Table S10. The data were analyzed using one-way ANOVA followed by Dunnett’s multiple comparisons test. *, **, *** compared with the vehicle control (DMSO). (D) Dose–response curves of PPRE activation and CD36 expression for selective PPARs ligands. The PPRE-eGEP-THP-1–derived macrophages were treated with selective PPARs ligands at various concentrations of WY (), RGZ (), GW6471 (), GW9662 (), or T0070907 () in replicate wells () for 24 h. The summary data are provided in Tables S11 and S12. (E) Dose–response curves of PPRE activation and CD36 expression for MEHP. PPRE-eGEP-THP-1–derived macrophages were treated with MEHP (M; ) alone or in combination with GW6471 (), GW9662 (), or T0070907 () in replicate wells () for 24 h. The summary data are provided in Table S13. The fluorescent stain of PPRE-eGFP and CD36 were analyzed using high-content cellular imaging, as described in the “Methods” section. values of concentrations were used. Note: ANOVA, analysis of variance; DEHP, diethylhexyl phthalate; DMSO, dimethyl sulfoxide; MEHP, mono(2-ethylhexyl) phthalate; PPAR, peroxisome proliferator-activated receptor; PPRE, peroxisome proliferator-activated receptor response element; RU, resonance unit; SD, standard deviation.
Next, the interaction between DEHP/MEHP and was evaluated at the cellular level in the human monocytic THP-1 cells stably transfected with PPRE promoter-H2B-eGFP. Using a high-content cellular imaging system, we observed that both PPRE transactivation and CD36 expression in the cells treated with DEHP and MEHP were significantly higher compared with the control cells (Figure 2C; Table S10). This was consistent with the responses in the cells treated with the specific agonist of (WY) and (RGZ) (Figure 2C; Table S10). In addition, the dose–response of the PPARs agonists (WY and RGZ) or antagonists (GW6471, GW9662, and T0070907) on PPRE transactivation paralleled with their effects on the protein expression of PPARs target gene CD36 (Figure 2D; Tables S11 and S12). Furthermore, when compared with the dose–response curves of MEHP, the co-treatment with the specific antagonist of (GW6471) or (T0070907) resulted in notably lower levels of PPRE transactivation and CD36 expression (Figure 2E; Table S13).
Effect of Hepatocyte-Specific Knockout in DEHP-Induced Fatty Liver
To evaluate the role of hepatocyte-specific in the dysregulation of hepatic lipid metabolism induced by DEHP, we conducted the 28-d repeated dose study in mice with conditioned knockout of in the hepatocytes (, Hep-KO). At the end of the treatment, both liver weight and liver index in the Hep-KO mice treated with DEHP were significantly higher compared with the control mice treated with CMC, but no significant difference was observed between the WT and Hep-KO mice (Figure 3A; Table S14). The differences between the DEHP and control group on plasma levels of TG, CHO, CHE, ALP, HDLC, and LDLC in the WT mice were comparable to those in the Hep-KO mice (Figure 3B; Table S15). In addition, the hepatic TG concentrations in the WT and Hep-KO mice treated with DEHP were both significantly higher than for the control mice (Figure 3C; Table S16). Similarly, the significant differences between DEHP and control group on hepatic steatosis in the WT mice was also observed in the Hep-KO mice (Figure 3D; Table S17). Therefore, there was no significant difference between WT and Hep-KO mice in response to the treatment with DEHP. The IHC with mouse liver showed clear nuclear translocation of in the hepatocyte after DEHP treatment in the WT mice (Figure 3E). It is worth noting that there was also strong staining of in the non-hepatocyte cells (Figure 3E). Next, we performed the immunofluorescence staining of liver resident macrophages and Kupffer cells with F4/80 and CLEC4F, respectively. The quantitative analysis indicated that the number of Kupffer cells were remarkably lower in both WT and Hep-KO mice treated with DEHP compared with the control mice (Figure 3F; Table S18). However, there was no significant difference in the total hepatic macrophages between the DEHP and CT group in either WT or Hep-KO mice (Figure 3F; Table S18).
Figure 3.
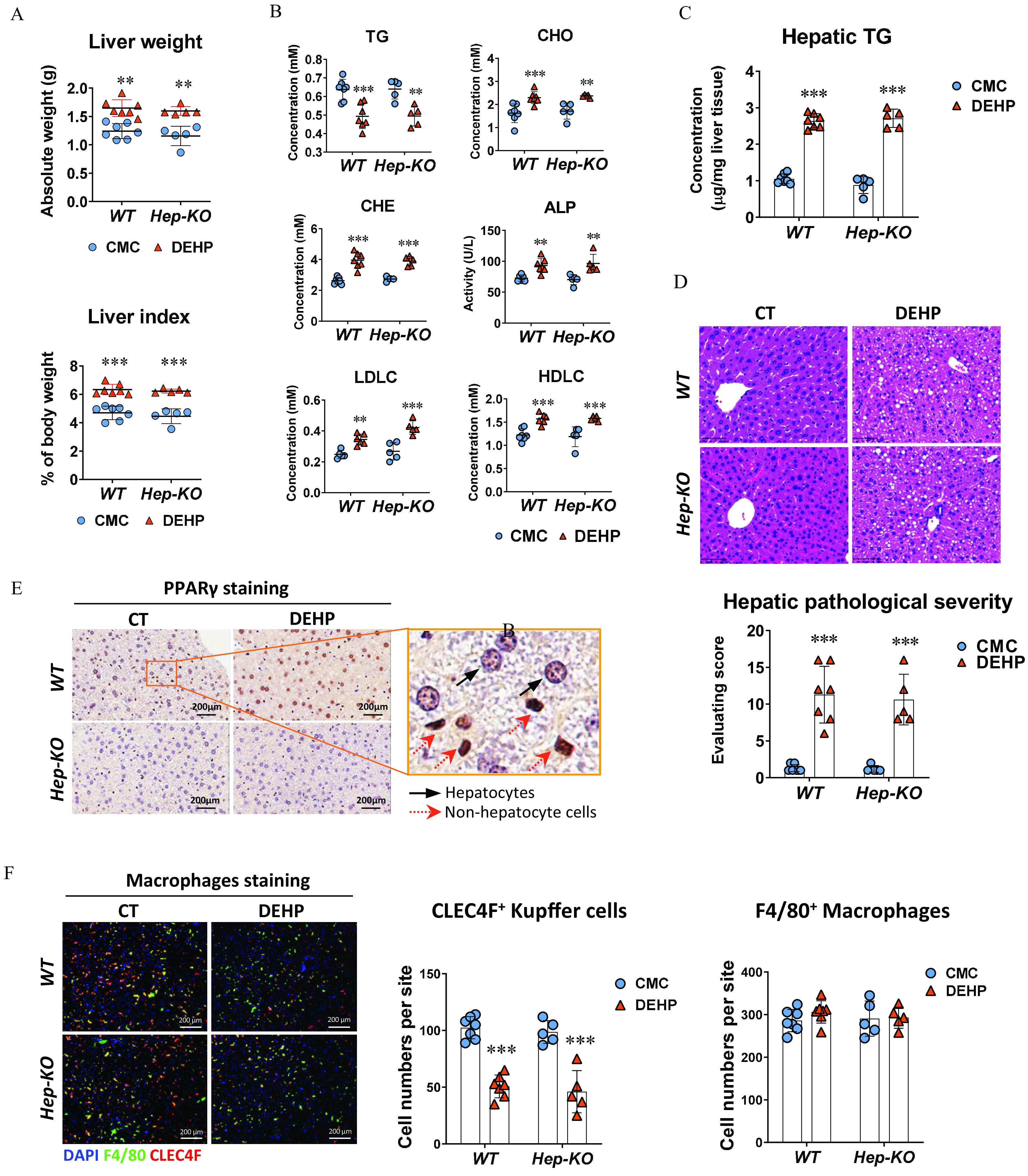
Effect of hepatocyte-specific knockout in DEHP-induced fatty liver. Wild-type (WT; ) and hepatocyte-specific knockout (Hep-KO; ) C57BL/6J male mice were treated with 0.5% (wt/vol) sodium carboxymethylcellulose (CMC; vehicle control) or DEHP ( BW) by daily gavage for 28 d. (A) Liver weight and liver index of mice at the end of the treatment. The data are provided in Table S14. (B) Plasma level of triglycerides (TGs), cholesterol (CHO), high-density lipoprotein cholesterol (HDLC), low-density lipoprotein cholesterol (LDLC), alkaline phosphatase (ALP), and cholinesterase (CHE) of WT () and Hep-KO () mice at the end of the treatment. The data are provided in Table S15. (C) Hepatic TG concentration in the liver of WT () and Hep-KO () mice at the end of the treatment. The data are provided in Table S16. (D) Representative histology images (up) and quantification of hepatic steatosis (down) in the WT () and Hep-KO () mice at the end of the treatment. The data are provided in Table S17. (E) Representative immunohistochemistry (IHC) images of expression in the liver of WT and Hep-KO mice at the end of the treatment. (F) Immunofluorescence staining of CLEC4F (red) and F4/80 (green) in the mouse liver (left). Cell nuclei were stained with DAPI (blue). Cells with positive stain in the liver sections of WT () and Hep-KO () mice were quantified using the ImageXpress system under . The data are provided in Table S18. All data are expressed as . The data were analyzed using one-way ANOVA followed by Bonferroni’s multiple comparisons test. *, **, *** compared with the vehicle control (CMC). Note: ANOVA, analysis of variance; BW, body weight; CT, control; DAPI, 4′,6-diamidino-2-phenylindole; DEHP, diethylhexyl phthalate; Hep-KO, hepatic PPAR knockout mice; Mac-KO, macrophage-specific PPAR knockout mice; PPAR, peroxisome proliferator-activated receptor; SD, standard deviation.
Effect of Macrophage-Specific Knockout in DEHP-Induced Fatty Liver
Next, we aimed to evaluate whether macrophage-specific contributes to the fatty liver induction by DEHP. To this end, mice were crossed with mice to construct the specific knockout of in mature macrophages (, Mac-KO). After treatment with DEHP ( BW) by daily gavage for 28 d, both liver weight and liver index of mice were significantly higher than for the WT control mice. In contrast, no significant difference between the DEHP and control group was observed in the Mac-KO mice (Figure 4A; Table S19). The differences between DEHP and control group on the plasma levels of TG, CHO, CHE, ALP, LDLC, and HDLC in the WT mice were not observed in the Mac-KO mice (Figure S4A; Table S49). Although hepatic TG concentration (Figure 4B; Table S20), pathological steatosis severity, and Oil Red O staining intensity (Figure 4C; Table S21) were higher in the Mac-KO mice treated with DEHP compared with the control mice, these levels were significantly lower when compared with the WT mice treated with DEHP.
Figure 4.
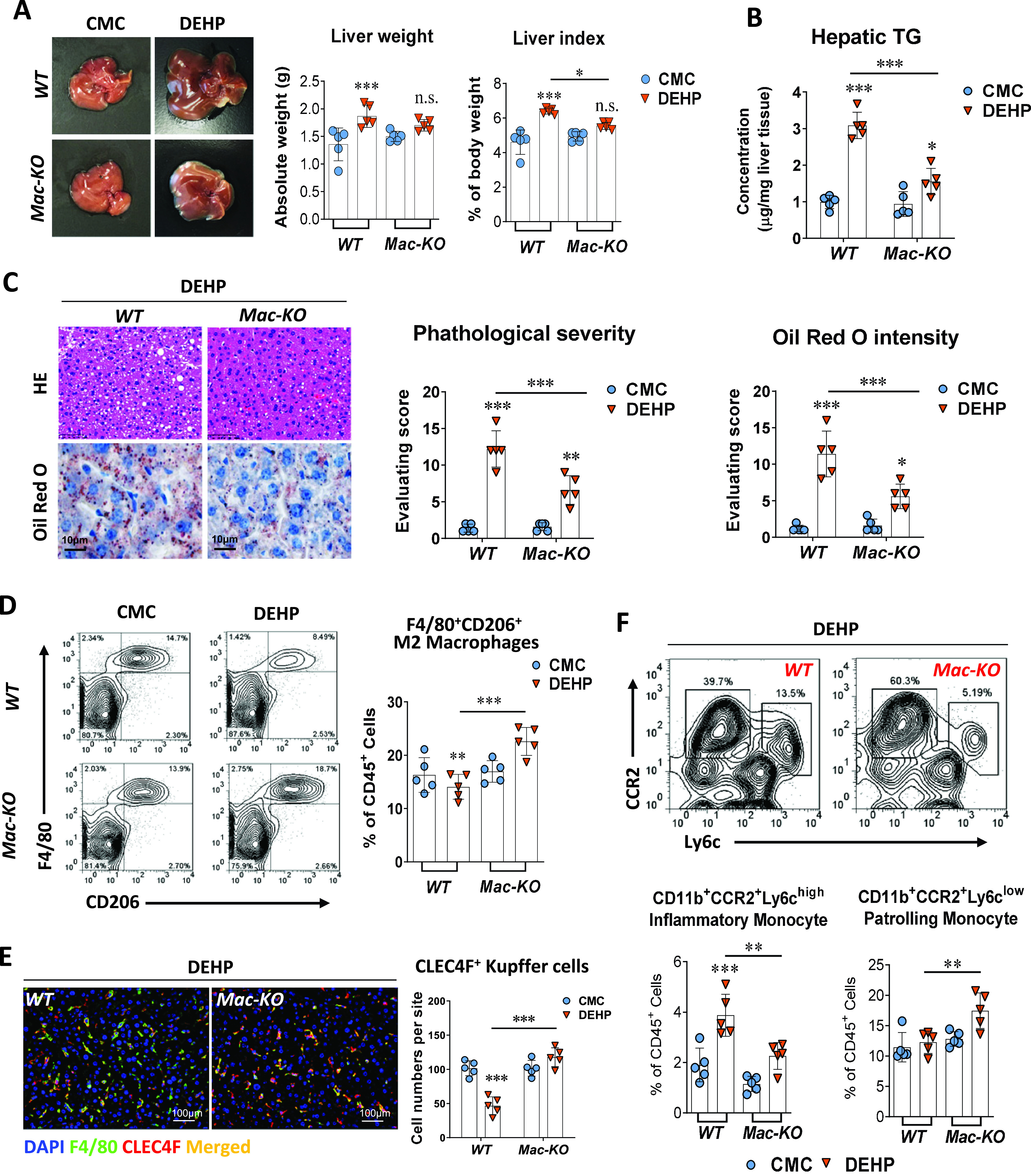
Effect of macrophage-specific knockout in DEHP-induced fatty liver. Wild-type (WT, ) and macrophage-specific knockout (Mac-KO, ) C57BL/6J male mice were treated with 0.5% (wt/vol) sodium carboxymethylcellulose (CMC; vehicle control) or DEHP ( BW) by daily gavage for 28 d. (A) Representative image of mice livers and quantification of liver weight and liver index at the end of the treatment (). The data are provided in Table S19. (B) Hepatic TG concentration in the liver tissue of WT () and Mac-KO () mice at the end of the treatment. The data are provided in Table S20. (C) Representative images of histology and Oil Red O staining of mouse liver at the end of the treatment (left). Hepatic steatosis and lipid accumulation were quantified by calculating the score of pathological severity and Oil Red O staining intensity, respectively (right, ). The data are provided in Table S21. (D) Representative flow cytometry gating graph and quantification of hepatic M2 macrophages () in the WT or Mac-KO () mice at the end of the treatment. The data are provided in Table S22. (E) Immunofluorescence staining of CLEC4F (red) and F4/80 (green) in the mouse liver of WT or Mac-KO mice () at the end of the treatment. Cells with positive stain of CLEC4F in the liver sections of WT and Mac-KO mice () were quantified using the ImageXpress system under . The data are provided in Table S23. (F) Representative flow cytometry gating graph and quantification of inflammatory monocyte () and patrolling monocyte () in the liver of WT or Mac-KO mice () at the end of the treatment. The data are provided in Table S24. All data are expressed as . The data were analyzed using two-way ANOVA followed by Tukey’s multiple comparisons test. *, **, *** compared with the vehicle control (i.e., CMC); significance of comparison between WT and Mac-KO groups of DEHP treatment was labeled above lines. Note: ANOVA, analysis of variance; BW, body weight; DAPI, 4′,6-diamidino-2-phenylindole; DEHP, diethylhexyl phthalate; HE, hematoxylin and eosin; SD, standard deviation; TG, triglyceride.
To explore the mechanism by which macrophage-specific regulates hepatic lipid metabolism, we determined the macrophage subpopulations in the liver of WT and Mac-KO mice using flow cytometry (Figure S4B; Table S50). First, the proportion of M2-type macrophages were notably lower in the liver of WT mice treated with DEHP compared with the control mice. However, the proportion of hepatic M2 macrophages in the Mac-KO mice was significantly higher in the DEHP group than in the control group (Figure 4D; Table S22). In contrast to the results from experiments with the WT and Hep-KO mice models, the number of Kupffer cells in the liver of Mac-KO mice treated with DEHP was markedly higher compared with the control mice (Figure 4E; Table S23).
We also analyzed monocytes in the liver, which can give rise to the self-renewal of hepatic resident macrophages. After treatment with DEHP, the proportion of inflammatory monocytes in the liver of Mac-KO mice was significantly lower compared with the WT mice (Figure 4F; Table S24). In contrast, the proportion of patrolling monocytes was higher in the Mac-KO mice than in the WT mice (Figure 4F; Table S24). This was in parallel with the result on the determination of hepatic M2 macrophages (Figure 4D; Table S22). In agreement, similar results were also observed in the determination of cytokines associated with the M2-type macrophages (IL-10 and IL-23) and the M1-type proinflammatory macrophages (IL-12p70 and IL-18) (Figure S4C and Table S51).
The Role of and in Regulating Macrophages’ M2 Polarization
To further validate the effect of DEHP on the regulation of macrophages’ M2 polarization in the presence and absence of , we isolated bone marrow-derived monocytes from the (WT) and (Mac-KO) mice, differentiated into BMDMs and induced polarization into the M1-type () or M2-type () macrophages in vitro (Figure S5A). In agreement with the results from in vivo study with mice, DEHP and MEHP treatments resulted in significantly lower levels of BMDMs polarizing into M2-type macrophages in the WT cells, whereas the proportions of M2 macrophages in the Mac-KO cells treated with DEHP or MEHP were significantly higher compared with the WT cells of the same treatment (Figure 5A; Table S25). In contrast, the proportion of M1-polarized macrophages were not different between the WT and Mac-KO cells treated with either DEHP or MEHP (Figure S5B and Table S52).
Figure 5.
Regulation of macrophages’ M2 polarization by and . (A) Percentage of cells in the bone marrow-derived macrophages (BMDMs). Bone marrow monocytes were isolated from the wild-type (WT; ) and macrophage-specific knockout (Mac-KO; ) C57BL/6J male mice and then differentiated into BMDMs. BMDMs were treated with 0.1% DMSO control (CT), DEHP (), or MEHP () in combination with murine IL-4 () and IL-13 () for 24 h. Cells with positive stain of CD206 were detected using the ImageXpress system and normalized to the number of total cells (%) in each of the triplicate wells (the plots contain all technical replicates from five imaging sites of each well).The data are provided in Table S25. (B) Heatmap of differently expressed genes (DEGs) associated with M2 macrophages. THP-1–derived M0 macrophages were treated with MEHP () during the induction of M1 and M2 polarization. Cells from triplicate wells () were analyzed using RNA sequencing at the end of the treatment. The data are provided in Table S26. (C) Heatmap of DEGs associated with PPAR signaling pathway. The data are provided in Table S27. (D) Representative images for the fluorescent staining of CD209, CD36, and PPRE in human M0, M1, and M2 macrophages derived from PPRE-eGEP-THP-1 cells. (E) Quantification of protein expression of CD209, CD36, PPRE response and lipid droplets (LDs) in the M0, M1, and M2 macrophages from duplicate wells (). The data are provided in Table S28. (F) Dose–response curves for the effect of DEHP and MEHP on PPRE activation, CD209 expression, and LD level during macrophage M2 polarization. THP-1–derived M0 macrophages were treated with various concentrations of DEHP () or MEHP () in combination with human IL-4 () and IL-13 () for 24 h. The fluorescent stain area in the cells treated in triplicate wells () were quantified using the ImageXpress system and normalized by comparing with the control well treated with DMSO. The data are provided in Table S29. (G) Dose–response curves for the effect of activation on CD209 expression. THP-1–derived M0 macrophages were treated in triplicate wells () with various concentrations () of MEHP, Wy14643 (WY), GW7647, and rosiglitazone (RGZ) in combination with human IL-4 () and IL-13 () for 24 h. The data are provided in Table S30. (H) Dose–response curves for the effect of inhibition on CD209 expression. THP-1–derived M0 macrophages were treated with various concentrations () of GW9662, GW6471, and T0070907 in combination with human IL-4 () and IL-13 () for 24 h. The fluorescent stain area of CD209 in the cells were quantified using the ImageXpress system and normalized to the number of total cells (%) in each of the replicate wells (). The data are provided in Table S31. (I) Representative fluorescence staining images and quantification of CD36, CD209, and LDs in the macrophages treated with MEHP (M; ) alone or in combination with GW9662 () and GW6471 () during the M2 polarization (). The data are provided in Table S32. All data are expressed as . The data were analyzed using one-way ANOVA followed by Bonferroni’s multiple comparisons test. *, **, *** compared with the vehicle CT or as indicated by lines. Note: ANOVA, analysis of variance; DEHP, diethylhexyl phthalate; DMSO, dimethyl sulfoxide; IL, interleukin; MEHP, mono(2-ethylhexyl) phthalate; PPAR, peroxisome proliferator-activated receptor; PPRE, peroxisome proliferator-activated receptor response element; SD, standard deviation.
Furthermore, the effect of MEHP on PPARs activation and macrophages’ polarization was evaluated in a human cellular model derived from monocytic THP-1 cells. First, RNA-sequencing data showed that the expression levels of a broad spectrum of M2 macrophage-associated genes, including Kupffer cell core genes (CLEC1B, IL18BP, PCOLCE, SLC40A1, and TIMD4), were notably lower in the polarized M2 macrophages treated with MEHP compared with the control cells (Figure 5B; Table S26). In contrast, the expression of genes associated with M1-type macrophages were not evidently different between the cells treated with MEHP and the control cells (Figure S5C and Table S53). Notably, the expression pattern of genes associated with the PPAR signaling pathway was divergent between the M1-type and M2-type macrophages under the treatment with MEHP (Figure 5C; Table S27). In this cellular model, the levels of PPRE transactivation, CD36 expression, and the intracellular LDs were remarkably higher in the M2 macrophages compared with M0 and M1 macrophages, paralleling the protein level of the human M2 macrophages’ marker gene CD209 (Figure 5D,E; Table S28). The difference was consistent with the higher levels of CD36 and LDs in the M2 macrophages of the mouse model (Figure S5D and Table S54). More importantly, in line with our results from animal studies, the proportion of M2 macrophages was lower in the polarizing cells treated with DEHP and MEHP when compared with the control cells, and the responses appeared to be dose dependent (Figure 5F; Table S29). Similar results were also found in the LD levels, whereas the PPRE response was not evident (Figure 5F; Table S29).
Next, we compared the effect of MEHP on the polarization of M2 macrophages with selective PPARs ligands. First, treatment with the agonists WY and GW7647 resulted in significantly higher levels of CD209 expression in the macrophages, but the expression levels of CD209 in the cells treated with agonist, RGZ, were lower than for the control cells, resembling the dose-dependent manner in the cells treated with MEHP (Figure 5G; Table S30). On the contrary, treatments with the antagonist GW9662 or T0070907 resulted in higher CD209 expression levels in a dose-dependent manner when compared with the treatment with antagonist GW6471 (Figure 5H; Table S31). Next, we validated the respective role of and in the action of MEHP on macrophages’ M2 polarization. Compared with the treatment with MEHP alone, the co-treatment with GW6471 resulted in significantly lower levels of CD209 and CD36 in M2 macrophages, whereas no evident difference was observed in the treatment with GW9662 (Figure 5I; Table S32). The intracellular LD level was significantly lower in the cells treated with MEHP and GW6471 compared with the cells treated with MEHP alone, whereas co-treatment with GW9662 resulted in higher LD levels compared with the treatment with MEHP alone (Figure 5I; Table S32).
Integrative Analysis of Transcriptomic and Lipidomic Data from WT, Hep-KO, and Mac-KO Mice Models
The transcriptomic profiles with RNA sequencing showed that the DEGs in the mice treated with DEHP vs. the control mice in the WT model were divergently overlapped by the Hep-KO model (19.1%) and the Mac-KO model (11%) (Figure 6A; Table S33). The major portion (95/141, 67.4%) of DEGs in the Hep-KO model was shared by the WT model, whereas only a small part (55/230, 23.9%) of DEGs in the Mac-KO model was overlapped by the WT model. Importantly, a cluster of genes associated with lipid metabolism showed higher expression in the mice of the DEHP treatment compared with the control mice in both WT and Hep-KO model, but they were not evidently different in the Mac-KO model (Figure 6B; Table S34). Moreover, pathway enrichment analysis with the DEGs showed that the enrichment scores of PPAR signaling pathway, fatty acid degradation, and retinol metabolism in the three models were higher than for the others among the top 10 significantly enriched KEGG pathways (Figure 6C; Table S35). Notably, the enrichment score of oxidative phosphorylation (OXPHOS) was markedly lower in the Mac-KO model than for the WT and Hep-KO (Figure 6C; Table S35). In addition, Gene Ontology analysis showed that the biological processes—including the long-chain fatty acid metabolic process, TG biosynthetic process, and acyl-CoA metabolic process—were enriched in the WT and Hep-KO model, but not in the Mac-KO model. In contrast, the biological processes related to negative regulation of lipid biosynthesis and storage were particularly enriched ( and TOP 20) in the Mac-KO model (Figure 6D; Table S36).
Figure 6.
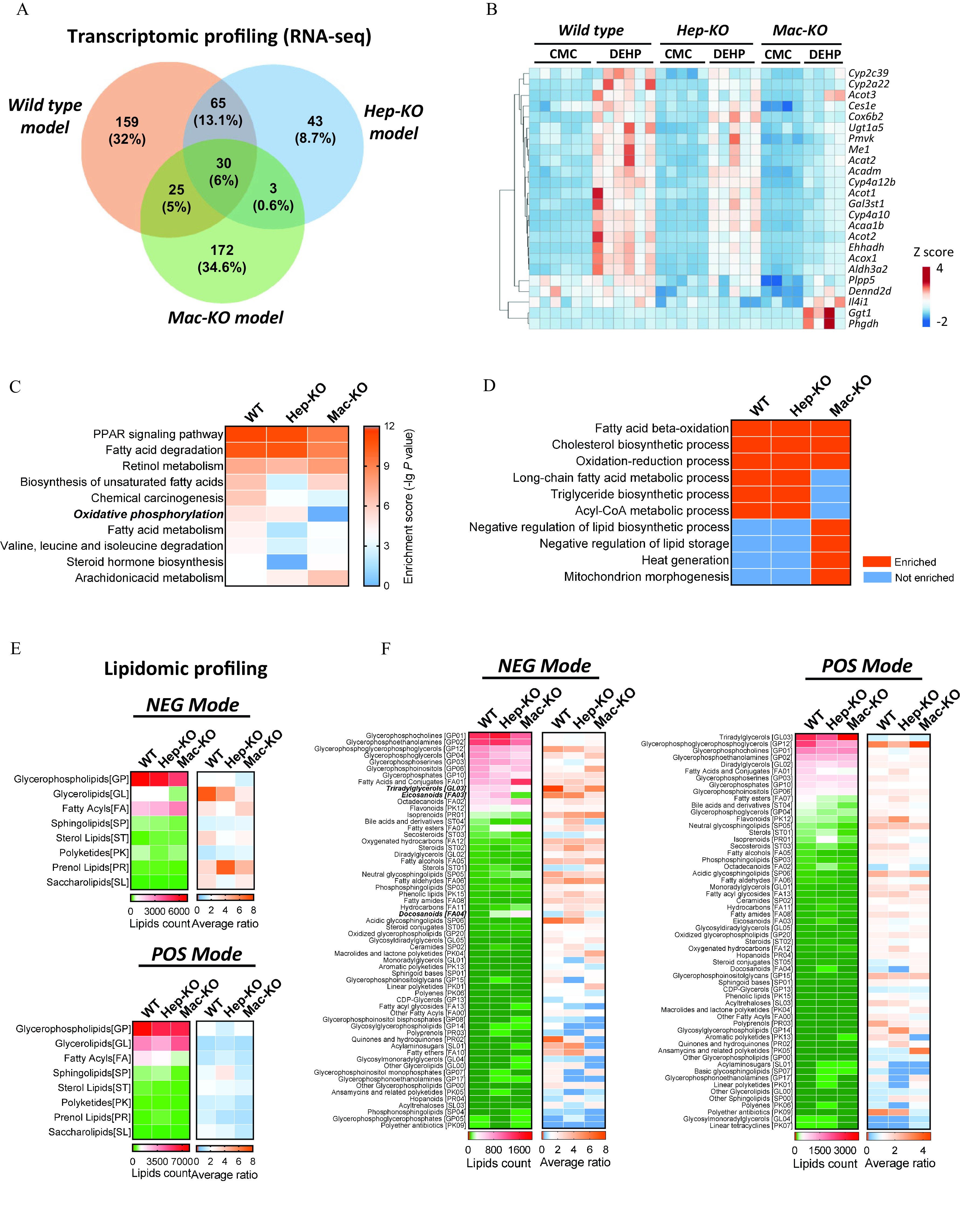
Transcriptomic and lipidomic analysis of DEHP-induced fatty liver. RNA sequencing was performed with the liver of wild-type (WT; ), hepatocyte-specific knockout (Hep-KO; ), and macrophage-specific knockout (Mac-KO; ) mice treated with 0.5% (wt/vol) sodium carboxymethylcellulose (CMC; vehicle control) or DEHP ( BW) by daily gavage for 28 d. (A) Venn diagram of differently expressed genes (DEGs) between DEHP and control (CT) group in the mice models. The data are provided in Table S33. (B) Heatmap of DEGs induced by DEHP in the WT, Hep-KO, and Mac-KO mice models. The data are provided in Table S34. (C) Integrated comparison of pathway enrichment (top 10) across the WT, Hep-KO, and Mac-KO mice models. The data are provided in Table S35. (D) Integrated comparison of Gene Ontology (GO) Biological Process enrichment across the WT, Hep-KO, and Mac-KO mice models. The criteria for “enriched” item is and TOP 20. The data are provided in Table S36. (E) Integrated comparison of DEHP-induced lipidomic changes in the WT, Hep-KO, and Mac-KO mice models. Lipidomic profiling was conducted with the liver of WT (), Hep-KO (), and Mac-KO () mice in the negative ion (NEG) and positive ion (POS) mode. The heatmap shows lipid counts and average fold change (DEHP/CT ratio) of lipid metabolites at the “superclass” level. The data are provided in Table S37. (F) Integrated comparison of DEHP-induced lipidomic changes in the WT, Hep-KO, and Mac-KO mice models at the “class” level. The heatmap shows lipid counts and average fold change in DEHP group compared with the CT group. The data are provided in Table S38. Note: BW, body weight; DEHP, diethylhexyl phthalate; PPAR, peroxisome proliferator-activated receptor.
Lipidomic analysis showed that, in comparison with the control group in the WT mice model, DEHP treatment resulted in remarkably higher levels (fold ) of a wide spectrum of lipid metabolites, which were primarily constituted (in lipid counts) by glycerophospholipids (GPs), glycerolipids (GLs), and fatty acyls (FAs) (Figure 6E; Table S37). Importantly, the average fold changes (DEHP/CT ratio) of GP and GL under the NEG mode were notably lower in the Mac-KO model than for the WT and Hep-KO model (Figure 6E; Table S37). The difference was in parallel with the reduced lipid count and lower ratio of triacylglycerol (GL03) in the Mac-KO model when compared with the WT and Hep-KO model (Figure 6F; Table S38). In addition, the level of fatty acids such as eicosanoids (FA03) and docosanoids (FA04) in the Mac-KO model was also significantly different from those in the WT and Hep-KO model (Figure 6F; Table S38).
The Impact of M2-Type Macrophages on Mitochondrial OXPHOS in Hepatocytes
After answering the question of how macrophage polarization is regulated by MEHP through the activation of and , we next aimed to validate the impact of M2 macrophages on the OXPHOS and lipid metabolic dysregulation. First, the MMP as determined using TMRE was significantly higher in the HepG2 cells treated with MEHP compared with the control cells, whereas the co-treatment with mitochondrial OXPHOS inhibitors (IACS, VLX, and 3-NP) resulted in evidently lower levels of TMRE compared with the treatment with MEHP alone (Figure 7A; Table S39). Similar results were observed in the co-treatment with M2 macrophage-specific cytokine IL-1RA. Importantly, the levels of fatty acid uptake [as determined using BODIPY FL (Figure 7A; Table S39)] and LDs (Figure 7B; Table S39) were closely correlated with the TMRE levels in the cells treated with MEHP and OXPHOS inhibitors. In addition, the coculture with M2 macrophages also resulted in significantly lower levels of TMRE, mitochondrial reactive oxygen species (i.e., MitoSOX), and intracellular LDs compared with the treatment with MEHP alone (Figure 7C; Table S40).
Figure 7.
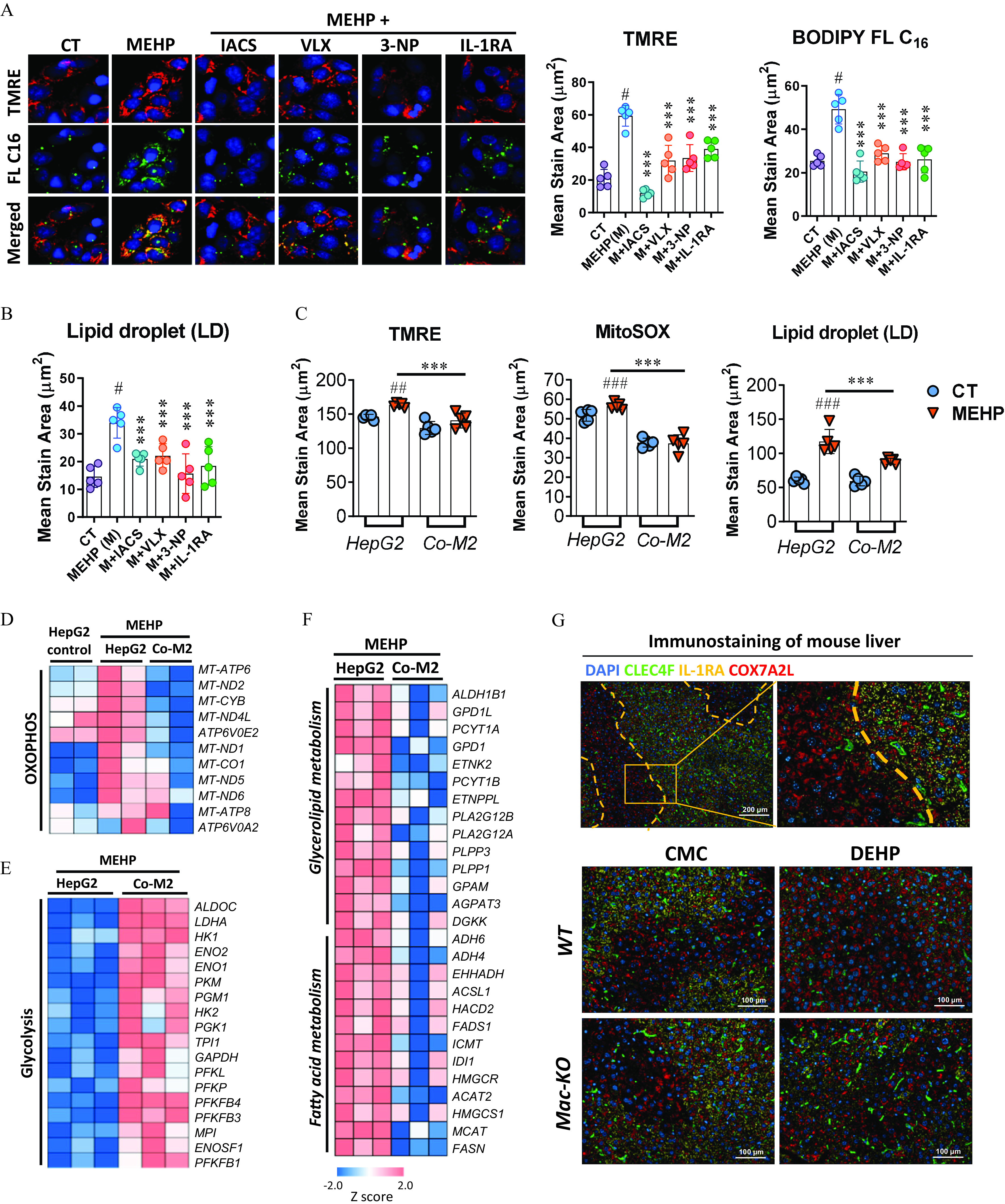
Effect of M2 macrophages on OXPHOS and lipid metabolism in the hepatocytes. (A,B) HepG2 cells were treated with MEHP (M; ) alone or in combination with IACS-10759 (IACS; ), VLX600 (VLX; ), 3-nitropropanoic acid (3-NP; ), and IL-1RA () in replicate wells () for 24 h. Cells were incubated with the fluorescent dyes BODIPY FL and tetramethylrhodamine ethyl ester (TMRE), or LipidTOX Neutral Lipid Stain at the end of the treatment. (A) Representative images (left) and quantification (right) of lipid uptake (FL ) and mitochondrial membrane potential (TMRE) in HepG2 cells at the end of the treatment. (B) Quantification of lipid droplets (LDs) at the end of the treatment. The data for (A,B) are provided in Table S39. (C) Quantification of TMRE, Mitochondrial Superoxide (MitoSOX) and LD in the HepG2 cells treated with MEHP () alone or cocultured with M2 macrophages for 24h (). The data are provided in Table S40. HepG2 Cells in an independent experiment with the same treatment as (C) were used for RNA sequencing (). Heatmaps show differently expressed genes associated with (D) OXPHOS, (E) glycolysis, and (F) lipid metabolism between different treatments. The data for (D–F) are provided in Tables S41–S43. (G) Immunofluorescence staining of CLEC4F (green), IL-1RA (yellow), and COX7A2L (red) in the liver tissues of wild-type (WT) and macrophage-specific knockout (Mac-KO) mice treated with 0.5% (wt/vol) sodium carboxymethylcellulose (CMC; vehicle control) or DEHP ( BW) by daily gavage for 28 d. Data for (A–C) are expressed as . The data were analyzed using one-way (A,B) or two-way (C) ANOVA followed by Tukey’s multiple comparisons test. #, ##, ### compared with the vehicle control (CT); ***, compared with the MEHP group. Note: ANOVA, analysis of variance; BW, body weight; DAPI, 4′,6-diamidino-2-phenylindole; DEHP, diethylhexyl phthalate; IL, interleukin; MEHP, mono(2-ethylhexyl) phthalate; OXPHOS, oxidative phosphorylation; SD, standard deviation.
RNA-sequencing data from the cellular model showed that the expression of OXPHOS-associated genes was notably lower in the HepG2 cells cocultured with M2 macrophages compared with the cells treated with MEHP alone (Figure 7D; Table S41). In contrast, the expression of genes associated with glycolysis was significantly higher in the HepG2 cells cocultured with M2 macrophages when compared with MEHP group (Figure 7E; Table S42). Furthermore, the coculture with M2 macrophages also resulted in lower levels of gene expression associated with lipid metabolism compared with the treatment with MEHP alone (Figure 7F; Table S43). In addition, the immunofluorescence staining with mouse liver demonstrated that the expression of OXPHOS marker protein COX7A2L in the hepatocytes was inversely correlated with the tissue distribution of M2 macrophages’ marker cytokine IL-1RA and the Kupffer cell marker CLEC4F in the Mac-KO mice model (Figure 7G).
Discussion
In the present study, we compared the respective roles of hepatocyte- and macrophage-specific in the development of fatty liver induced by the typical phthalate, DEHP, in mice. Our data showed that DEHP-induced lipid accumulation in the liver was tightly associated with the depletion of Kupffer cells. In addition, the orchestrated activation of and by MEHP may be an important driver for hepatic macrophage polarization. Interestingly, blockade of macrophage-specific appeared to facilitate the M2-type polarization of macrophages and to lead to the amelioration of fatty liver in the mice treated with DEHP. Mechanistic study demonstrated that DEHP-induced lipid dysregulation in the hepatocytes was strongly attenuated by M2 macrophages, possibly through the constraint of mitochondrial OXPHOS (Figure 8). These findings may have important implications for understanding the biology of hepatic macrophages and the role of cell-specific PPAR signaling pathway in metabolic disorders induced by DEHP.
Figure 8.

Schematic illustration for the impact of macrophage-specific and activation on the fatty liver induction by DEHP. Note: DEHP, diethylhexyl phthalate; FAO, fatty acid oxidation; IL, interleukin; MEHP, mono(2-ethylhexyl) phthalate; OXPHOS, oxidative phosphorylation; PPAR, peroxisome proliferator-activated receptor; TCA, tricarboxylic acid cycle; TNF, tumor necrosis factor.
Although the discussion on the health concerns of phthalates started half a century ago (Ganning et al. 1984), the chronic toxicity for humans still remains unclear. DEHP-induced hepatotoxicity was previously recognized to be dependent on in a feeding study with mice (Ward et al. 1998), but later studies reported that DEHP increased the incidence of liver tumors in mice (Ito et al. 2007; Takashima et al. 2008), implying the involvement of other factors. Increasing evidence has suggested that macrophages are critically involved in metabolic homeostasis (Hotamisligil 2017; Krenkel and Tacke 2017). Therefore, the central role of the PPAR signaling pathway in regulating macrophage functions may render hepatic macrophages an important target cell type in the hepatotoxicity induced by DEHP. However, the potential impact of PPAR activation in macrophages on the adverse effect of DEHP has not been evaluated. In the present study, we showed the proficient activation of both human and by MEHP at the molecular and cellular levels (Figure 2). But the binding affinity of MEHP with appeared to be stronger than (Figure 2A,B), therefore we focused on the role of in hepatotoxicity induced by DEHP. Interestingly, blockade of hepatocyte- or macrophage-specific resulted in the divergent progression of lipid metabolic disorder in mice (Figures 3 and 4). These results suggest that DEHP-induced responses were evidently different across cell types. Our findings highlight the role of hepatic macrophages in fatty liver induction by DEHP and may aid in explaining the controversial results from studies. Recently, phthalate exposure has been shown to be associated with metabolic disorders in human populations (Radke et al. 2019). Thus, our data might provide a novel mechanism by which phthalates interact with cell-specific PPARs and disrupt metabolic homeostasis.
Our previous in vitro study reported the important role of in the M2 polarization of human macrophages derived from THP-1 cells (Xu et al. 2021). In the present study, our data provide evidence supporting the concept that and may act in concert to achieve macrophages subtypes in responding to exogenous stimuli, such as DEHP exposure. The relation between and macrophages’ M2 polarization has been extensively investigated, but the results remain controversial. Based on the mRNA expression of M2 marker genes such as Arg1, Mrc1, and Mgl1, earlier studies have proposed that primed macrophages alternative activation in BMDMs (Odegaard et al. 2007), peripheral monocytes (Bouhlel et al. 2007), and adipose tissue macrophages (Bouhlel et al. 2007). On the contrary, activation was also suggested to be dispensable (Szanto et al. 2010) or adversely correlated (Ji et al. 2018) with macrophages alternative (M2) activation. Multiple factors can contribute to the discrepancy among these results. On one hand, PPREs that are contained in the promoter region of certain M2 marker genes are transcriptionally controlled by all the types of PPARs, hence the observed changes at the mRNA level of these M2 markers might be attributed to the orchestrated regulation by different isoforms other than one single type PPAR. Our in vitro data suggest that the suppression of M2 polarization by MEHP is tightly associated with the activation of but that the process is strongly reversed when is abrogated. This was possibly due to the unrestricted activation of by MEHP, given that MEHP acts as a dual agonist for and (Figure 2). On the other hand, MEHP-induced activation of and may govern different subsets of genes associated with macrophages’ polarization (Figure 5C), for example, , which has been proposed as a critical regulator for the M2 activation of Kupffer cells (Odegaard et al. 2008).
The regulatory functions of hepatic macrophages in NAFLD are not completely understood. It is suggested that the pro- or anti-inflammatory mediators such as , IL-6, and IL-10 secreted by macrophages may play a crucial role (Tacke 2017). Insulin resistance is known to be closely related to and IL-6 levels (Tilg and Moschen 2008). However, the involvement of other cytokines remains less well characterized. In the present study, we showed that the M2 macrophages featured cytokine IL-1RA efficiently constricted mitochondrial OXPHOS and fatty acid oxidation in the neighboring hepatocytes in the cellular model. This is in line with the previous report showing that the deficiency of IL-1RA deteriorated fatty liver development under an atherogenic diet (Isoda et al. 2005). The protective role of IL-1RA might be related to the anti-oxidative function through the nuclear factor erythroid 2-related factor 2/hemeoxygenase-1 pathway (Jin et al. 2019). However, the intracellular process linking IL-1RA with OXPHOS warrants more precise studies.
Although the toxicological effects of DEHP have been extensively investigated in rats and mice at various dosage levels, the evaluation of internal exposure (i.e., MEHP) in these animal studies was rare, hindering the extrapolation from human biomonitoring levels (urine or plasma) to the realistic experimental doses. In the present work, the oral doses of DEHP were relatively high but the plasma level of MEHP in the mice treated with DEHP was comparable to the blood levels of MEHP in human studies () (Araki et al. 2014; Silva et al. 2003). However, the tissue level of MEHP in mice liver appeared to be much higher than the plasma level (Figure 1B,C). It has been suggested that the liver tumor response induced by DEHP in rodents is not relevant to humans (Corton et al. 2018; Rusyn et al. 2006). Hence, the human exposure data of MEHP at target tissues (e.g., liver) would be of vital importance to evaluating species difference on DEHP metabolism and the health risk in human population. Moreover, our results showed that the antagonism of by GW9662 and T0070907 in human macrophages showed a dose-related effect (Figure 5H), whereas deficiency in the mouse BMDMs did not reveal a profound effect (Figure 5A), suggesting an interspecies difference in the role of macrophage-specific . The extrapolation of these findings to the human liver warrants more investigation with optimal models, for example, using macrophage-specific PPARs-humanized mice models.
In conclusion, the present study evaluated the respective role of hepatocyte- and macrophage-specific in the hepatotoxicity induced by DEHP in mice. Our data suggest that the orchestrated activation of and by MEHP may reprogram macrophage polarization, thereby affecting lipid metabolism in the liver. Although this conclusion is based on studies conducted in mice and in vitro, these findings may aid in elucidating the health effect of environmental phthalates.
Supplementary Material
Acknowledgments
We thank C.S. Yang from Rutgers, The State University of New Jersey, for the critical reading of the manuscript. This study was funded by the National Key Research and Development Program of China (2018YFC1603102), the National Natural Science Foundation of China (81773437), and the CAMS Innovation Fund for Medical Science (CIFMS 2019-I2M-5-024). The determination of DEHP and MEHP in mouse plasma and liver was supported by S. Fan and N. Zhang from the Beijing Center for Disease Prevention and Control (China).
RNA-sequencing data can be found in the Gene Expression Omnibus repository under accession numbers GSE159120, GSE160004, GSE160373, and GSE160826. Further information and requests for resources should be directed to and will be fulfilled by the lead contact, H.Y. (yanghui@cfsa.net.cn).
References
- Araki A, Mitsui T, Miyashita C, Nakajima T, Naito H, Ito S, et al. 2014. Association between maternal exposure to di(2-ethylhexyl) phthalate and reproductive hormone levels in fetal blood: the Hokkaido study on environment and children’s health. PLoS One 9(10):e109039, PMID: , 10.1371/journal.pone.0109039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouhlel MA, Derudas B, Rigamonti E, Dièvart R, Brozek J, Haulon S, et al. 2007. PPARγ activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab 6(2):137–143, PMID: , 10.1016/j.cmet.2007.06.010. [DOI] [PubMed] [Google Scholar]
- Chawla A. 2010. Control of macrophage activation and function by PPARs. Circ Res 106(10):1559–1569, PMID: , 10.1161/CIRCRESAHA.110.216523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corton JC, Cunningham ML, Hummer BT, Lau C, Meek B, Peters JM, et al. 2014. Mode of action framework analysis for receptor-mediated toxicity: the peroxisome proliferator-activated receptor alpha (PPARα) as a case study. Crit Rev Toxicol 44(1):1–49, PMID: , 10.3109/10408444.2013.835784. [DOI] [PubMed] [Google Scholar]
- Corton JC, Peters JM, Klaunig JE. 2018. The PPARα-dependent rodent liver tumor response is not relevant to humans: addressing misconceptions. Arch Toxicol 92(1):83–119, PMID: , 10.1007/s00204-017-2094-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degrelle SA, Shoaito H, Fournier T. 2017. New transcriptional reporters to quantify and monitor PPARγ activity. PPAR Res 2017:6139107, PMID: , 10.1155/2017/6139107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Y, Yan Z, Shen R, Wang M, Huang Y, Ren H, et al. 2020. Microplastics release phthalate esters and cause aggravated adverse effects in the mouse gut. Environ Int 143:105916, PMID: , 10.1016/j.envint.2020.105916. [DOI] [PubMed] [Google Scholar]
- EFSA CEP Panel (European Food Safety Authority Panel on Food Contact Materials, Enzymes and Processing Aids), Silano V, Barat Baviera JM, Bolognesi C, Chesson A, Cocconcelli PS, et al. 2019. Update of the risk assessment of di-butylphthalate (DBP), butyl-benzyl-phthalate (BBP), bis(2-ethylhexyl)phthalate (DEHP), di-isononylphthalate (DINP) and di-isodecylphthalate (DIDP) for use in food contact materials. EFSA J 17:e05838, PMID: , 10.2903/j.efsa.2019.5838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feige JN, Gerber A, Casals-Casas C, Yang Q, Winkler C, Bedu E, et al. 2010. The pollutant diethylhexyl phthalate regulates hepatic energy metabolism via species-specific PPARα-dependent mechanisms. Environ Health Perspect 118(2):234–241, PMID: , 10.1289/ehp.0901217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francque S, Szabo G, Abdelmalek MF, Byrne CD, Cusi K, Dufour JF, et al. 2021. Nonalcoholic steatohepatitis: the role of peroxisome proliferator-activated receptors. Nat Rev Gastroenterol Hepatol 18(1):24–39, PMID: , 10.1038/s41575-020-00366-5. [DOI] [PubMed] [Google Scholar]
- Ganning AE, Brunk U, Dallner G. 1984. Phthalate esters and their effect on the liver. Hepatology 4(3):541–547, PMID: , 10.1002/hep.1840040331. [DOI] [PubMed] [Google Scholar]
- Giannakis N, Sansbury BE, Patsalos A, Hays TT, Riley CO, Han X, et al. 2019. Dynamic changes to lipid mediators support transitions among macrophage subtypes during muscle regeneration. Nat Immunol 20(5):626–636, PMID: , 10.1038/s41590-019-0356-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil GS. 2017. Inflammation, metaflammation and immunometabolic disorders. Nature 542(7640):177–185, PMID: , 10.1038/nature21363. [DOI] [PubMed] [Google Scholar]
- IARC (International Agency for Research on Cancer Working Group on the Evaluation of Carcinogenic Risks to Humans). 2013. Some chemicals present in industrial and consumer products, food and drinking-water. IARC Monogr Eval Carcinog Risks Hum 101:9–549, PMID: . [PMC free article] [PubMed] [Google Scholar]
- Isoda K, Sawada S, Ayaori M, Matsuki T, Horai R, Kagata Y, et al. 2005. Deficiency of interleukin-1 receptor antagonist deteriorates fatty liver and cholesterol metabolism in hypercholesterolemic mice. J Biol Chem 280(8):7002–7009, PMID: , 10.1074/jbc.M412220200. [DOI] [PubMed] [Google Scholar]
- Issemann I, Green S. 1990. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature 347(6294):645–650, PMID: , 10.1038/347645a0. [DOI] [PubMed] [Google Scholar]
- Ito Y, Yamanoshita O, Asaeda N, Tagawa Y, Lee CH, Aoyama T, et al. 2007. Di(2-ethylhexyl)phthalate induces hepatic tumorigenesis through a peroxisome proliferator-activated receptor α-independent pathway. J Occup Health 49(3):172–182, PMID: , 10.1539/joh.49.172. [DOI] [PubMed] [Google Scholar]
- Ji J, Xue TF, Guo XD, Yang J, Guo RB, Wang J, et al. 2018. Antagonizing peroxisome proliferator-activated receptor γ facilitates M1-to-M2 shift of microglia by enhancing autophagy via the LKB1-AMPK signaling pathway. Aging Cell 17(4):e12774, PMID: , 10.1111/acel.12774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin C, Fu WL, Zhang DD, Xing WW, Xia WR, Wei Z, et al. 2019. The protective role of IL-1Ra on intestinal ischemia reperfusion injury by anti-oxidative stress via Nrf2/HO-1 pathway in rat. Biomed J 42(1):36–45, PMID: , 10.1016/j.bj.2018.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Langmead B, Salzberg SL. 2015. HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12(4):357–360, PMID: , 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krenkel O, Tacke F. 2017. Liver macrophages in tissue homeostasis and disease. Nat Rev Immunol 17(5):306–321, PMID: , 10.1038/nri.2017.11. [DOI] [PubMed] [Google Scholar]
- Li Y, Zhang Q, Fang J, Ma N, Geng X, Xu M, et al. 2020. Hepatotoxicity study of combined exposure of DEHP and ethanol: a comprehensive analysis of transcriptomics and metabolomics. Food Chem Toxicol 141:111370, PMID: , 10.1016/j.fct.2020.111370. [DOI] [PubMed] [Google Scholar]
- Liang X, Feswick A, Simmons D, Martyniuk CJ. 2018. Environmental toxicology and omics: a question of sex. J Proteomics 172:152–164, PMID: , 10.1016/j.jprot.2017.09.010. [DOI] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W. 2014. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30(7):923–930, PMID: , 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- Liu Z, Dai X, Zhang H, Shi R, Hui Y, Jin X, et al. 2020. Gut microbiota mediates intermittent-fasting alleviation of diabetes-induced cognitive impairment. Nat Commun 11(1):855, PMID: , 10.1038/s41467-020-14676-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manteiga S, Lee K. 2017. Monoethylhexyl phthalate elicits an inflammatory response in adipocytes characterized by alterations in lipid and cytokine pathways. Environ Health Perspect 125(4):615–622, PMID: , 10.1289/EHP464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maradonna F, Carnevali O. 2018. Lipid metabolism alteration by endocrine disruptors in animal models: an overview. Front Endocrinol (Lausanne) 9:654, PMID: , 10.3389/fendo.2018.00654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariana M, Feiteiro J, Verde I, Cairrao E. 2016. The effects of phthalates in the cardiovascular and reproductive systems: a review. Environ Int 94:758–776, PMID: , 10.1016/j.envint.2016.07.004. [DOI] [PubMed] [Google Scholar]
- Miao H, Huang Y, Ma C, Li J, Zhao Y, Wu Y. 2018. Ultra-high-performance liquid chromatography-isotope dilution tandem mass spectrometry for the determination of phthalate secondary metabolites in human serum based on solid-phase extraction. J AOAC Int 102(1):271–277, PMID: , 10.5740/jaoacint.18-0025. [DOI] [PubMed] [Google Scholar]
- NTP (National Toxicology Program). 1982. Carcinogenesis bioassay of di(2-ethylhexyl)phthalate (CAS No. 117-81-7) in F344 rats and B6C3F1 mice (feed studies). Natl Toxicol Program Tech Rep Ser 217:1–127, PMID: . [PubMed] [Google Scholar]
- Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L, et al. 2007. Macrophage-specific PPARγ controls alternative activation and improves insulin resistance. Nature 447(7148):1116–1120, PMID: , 10.1038/nature05894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odegaard JI, Ricardo-Gonzalez RR, Red Eagle A, Vats D, Morel CR, Goforth MH, et al. 2008. Alternative M2 activation of Kupffer cells by PPARδ ameliorates obesity-induced insulin resistance. Cell Metab 7(6):496–507, PMID: , 10.1016/j.cmet.2008.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radke EG, Galizia A, Thayer KA, Cooper GS. 2019. Phthalate exposure and metabolic effects: a systematic review of the human epidemiological evidence. Environ Int 132:104768, PMID: , 10.1016/j.envint.2019.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusyn I, Peters JM, Cunningham ML. 2006. Modes of action and species-specific effects of di-(2-ethylhexyl)phthalate in the liver. Crit Rev Toxicol 36(5):459–479, PMID: , 10.1080/10408440600779065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva MJ, Barr DB, Reidy JA, Kato K, Malek NA, Hodge CC, et al. 2003. Glucuronidation patterns of common urinary and serum monoester phthalate metabolites. Arch Toxicol 77(10):561–567, PMID: , 10.1007/s00204-003-0486-3. [DOI] [PubMed] [Google Scholar]
- Szanto A, Balint BL, Nagy ZS, Barta E, Dezso B, Pap A, et al. 2010. STAT6 transcription factor is a facilitator of the nuclear receptor PPARγ-regulated gene expression in macrophages and dendritic cells. Immunity 33(5):699–712, PMID: , 10.1016/j.immuni.2010.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tacke F. 2017. Targeting hepatic macrophages to treat liver diseases. J Hepatol 66(6):1300–1312, PMID: , 10.1016/j.jhep.2017.02.026. [DOI] [PubMed] [Google Scholar]
- Takashima K, Ito Y, Gonzalez FJ, Nakajima T. 2008. Different mechanisms of DEHP-induced hepatocellular adenoma tumorigenesis in wild-type and Pparα-null mice. J Occup Health 50(2):169–180, PMID: , 10.1539/joh.l7105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tassinari R, Tait S, Busani L, Martinelli A, Narciso L, Valeri M, et al. 2021. Metabolic, reproductive and thyroid effects of bis(2-ethylhexyl) phthalate (DEHP) orally administered to male and female juvenile rats at dose levels derived from children biomonitoring study. Toxicology 449:152653, PMID: , 10.1016/j.tox.2020.152653. [DOI] [PubMed] [Google Scholar]
- Tilg H, Moschen AR. 2008. Insulin resistance, inflammation, and non-alcoholic fatty liver disease. Trends Endocrinol Metab 19(10):371–379, PMID: , 10.1016/j.tem.2008.08.005. [DOI] [PubMed] [Google Scholar]
- Wahli W, Michalik L. 2012. PPARs at the crossroads of lipid signaling and inflammation. Trends Endocrinol Metab 23(7):351–363, PMID: , 10.1016/j.tem.2012.05.001. [DOI] [PubMed] [Google Scholar]
- Ward JM, Peters JM, Perella CM, Gonzalez FJ. 1998. Receptor and nonreceptor-mediated organ-specific toxicity of di(2-ethylhexyl)phthalate (DEHP) in peroxisome proliferator-activated receptorα-null mice. Toxicol Pathol 26(2):240–246, PMID: , 10.1177/019262339802600208. [DOI] [PubMed] [Google Scholar]
- Xu M, Wang X, Li Y, Geng X, Jia X, Zhang L, et al. 2021. Arachidonic acid metabolism controls macrophage alternative activation through regulating oxidative phosphorylation in PPARγ dependent manner. Front Immunol 12:618501, PMID: , 10.3389/fimmu.2021.618501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu T, Lim YT, Chen L, Zhao H, Low JH, Xia Y, et al. 2020. A novel mechanism of monoethylhexyl phthalate in lipid accumulation via inhibiting fatty acid beta-oxidation on hepatic cells. Environ Sci Technol 54(24):15925–15934, PMID: , 10.1021/acs.est.0c01073. [DOI] [PubMed] [Google Scholar]
- Yang H, Zhang Q, Xu M, Wang L, Chen X, Feng Y, et al. 2020. CCL2-CCR2 axis recruits tumor associated macrophages to induce immune evasion through PD-1 signaling in esophageal carcinogenesis. Mol Cancer 19(1):41, PMID: , 10.1186/s12943-020-01165-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zota AR, Calafat AM, Woodruff TJ. 2014. Temporal trends in phthalate exposures: findings from the National Health and Nutrition Examination Survey, 2001–2010. Environ Health Perspect 122(3):235–241, PMID: , 10.1289/ehp.1306681. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.