


Abstract
Transcription and genome architecture are interdependent, but it is still unclear how nucleosomes in the chromatin fiber interact with nascent RNA, and which is the relative nuclear distribution of these RNAs and elongating RNA polymerase II (RNAP II). Using super-resolution (SR) microscopy, we visualized the nascent transcriptome, in both nucleoplasm and nucleolus, with nanoscale resolution. We found that nascent RNAs organize in structures we termed RNA nanodomains, whose characteristics are independent of the number of transcripts produced over time. Dual-color SR imaging of nascent RNAs, together with elongating RNAP II and H2B, shows the physical relation between nucleosome clutches, RNAP II, and RNA nanodomains. The distance between nucleosome clutches and RNA nanodomains is larger than the distance measured between elongating RNAP II and RNA nanodomains. Elongating RNAP II stands between nascent RNAs and the small, transcriptionally active, nucleosome clutches. Moreover, RNA factories are small and largely formed by few RNAP II. Finally, we describe a novel approach to quantify the transcriptional activity at an individual gene locus. By measuring local nascent RNA accumulation upon transcriptional activation at single alleles, we confirm the measurements made at the global nuclear level.
Graphical Abstract
Graphical Abstract.
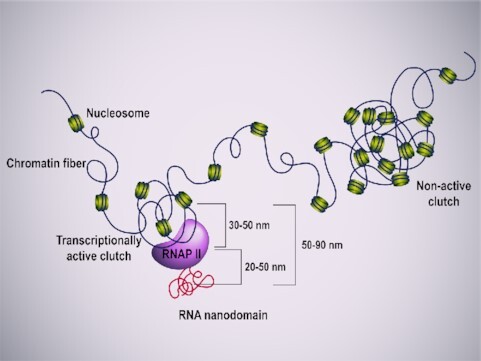
Schematic representation depicting the association between nucleosome clutches, RNAP II clusters and RNA nanodomains. Chromatin (in green) is organized in clutches. Small active nucleosome clutches are associated with elongating RNAP II clusters (in purple) which generates small RNA nanodomains (in red).
INTRODUCTION
Chromatin is highly organized inside the nucleus. At the fine scale level, chromatin is largely composed of 10 nm fibers with different levels of compaction and organization, playing a vital role in cell state (1,2). This organization results in large chromatin compartments that physically segregate into transcriptionally repressed heterochromatin and transcriptionally active euchromatin (3,4).
Our understanding of nuclear organization shows how chromatin structure and gene activity are intimately interdependent. Transcription has been linked to euchromatin organization, with actively transcribed loci being less compacted and with transcribed genes being relocated to active nuclear compartments (5–12). Conversely, gene transcription can shape global genomic organization (13).
Both super-resolution (SR) microscopy and genomic approaches have shown that highly active genomic regions can associate into 3D structures enriched in RNA polymerase II (RNAP II) that connects active genes between them (7,8,14,15). Transcribed genes associate with macromolecular assemblies of transcription factors, RNAP II and RNA transcripts. These assemblies constitute the so-called transcription factories or transcriptionally active pockets (12,16–18). The number and size reported for these structures varied among studies, from 40 to 170 nm of diameter in HeLa cells, depending on the method used (12,18,19). In cultured embryonic fibroblasts ∼1500 transcription factories were reported, while in other cell types the number of transcription factories varied from ∼100 to ∼300 (14). In turn, 300–500 factories were shown by widefield fluorescence microscopy (20), while up to 2100 factories were identified by confocal microscopy (12) and more than 8000 by Electron Microscopy (21). This variation in number and size can be attributed to the dense nuclear environment and to the fact that transcription factories are sub-diffraction limited structures. The use of SR microscopy could provide more accurate measurements on the number, size, and distribution of transcription factories, and RNAP II itself, in the cell.
Recent developments in SR microscopy have introduced exciting opportunities for the fields of nuclear architecture and transcriptomics, bridging the two fields and shedding light on how transcription is directly responsible for the organization of chromatin at multiple levels (1,2,13,22–26). Our previous works demonstrated that nucleosomes form heterogeneous groups, termed nucleosome clutches (1). Furthermore, using SR, we directly linked the epigenetic state of nucleosome clutches to their DNA packing density, defining a distance in which the organization of the DNA is influenced by the clutch (2). Although the epigenetic state of the cell can alter the clutch state, the specific transcriptional activity of individual clutches has not yet been ascertained.
In this work, Stochastic Optical Reconstruction Microscopy (STORM), DNA Points Accumulation for Imaging in Nanoscale Topography (DNA-PAINT), RNA FISH and SunTAg modified CRISPR (STAC) are used to investigate the nuclear organization and the nascent transcriptome with nanometric precision (27–29). We combined Single Molecule Localization Microscopy (SMLM) approaches to obtain super-resolved, single-cell quantitative data of the nucleoplasmic and nucleolar transcriptome and its physical distribution in the nucleus. We found that nascent RNA is organized in discrete, highly dense nanodomains. We linked the features of these nanodomains to the nuclear distribution of elongating RNAP II and to the local compaction of the chromatin fiber. Taking advantage of the quantitative nature of SMLM data, we have developed new analysis methods to precisely measure the inter-relation between nucleosome clutches, RNAP II and nascent RNA. Moreover, we analyzed and measured the transcription factories at nanoscale level excluding the presence of large factories. Finally, we developed a novel approach to quantify the local transcriptional activity at individual loci, measuring local nascent RNA accumulation upon transcriptional activation.
MATERIALS AND METHODS
Cell culture
Human BJ Fibroblasts (hFibs) (Skin Fibroblast, American Type Culture Collection, ATCC CRL-2522) and GP220 cells (RRID:CVCL_S968, kind gift from Bolo's lab) were cultured in Dulbecco's modified Eagle's medium (DMEM, Thermo Fisher Scientific, #41965039) supplemented with 10% fetal bovine serum (FBS, Thermo Fisher Scientific, #10270106), 1% Penicillin–Streptomycin (Thermo Fisher Scientific, #15140122), 1% GlutaMAX (Thermo Fisher Scientific, #35050038), 1% non-essential amino acids (Gibco, #11140050). Cells were grown in a humidified hypoxic incubator at 37°C, 5% CO2 and 5% O2.
Sample preparation and RNA labelling
For imaging purposes cells were plated in borosilicate glass bottom 8-well chambers (Nunc Lab-Tek, #155411 or μSlide Ibidi, #80827) at a confluency of 20 000–30 000 cells/cm2). For RNA labelling experiments, cells were cultured with media supplemented with 1 mM ethynil-uridine (EU) (Thermo Fisher, #E10345) for 5, 10, 20, 30 and 60 min prior to fixation. Due to the short time of the EU pulses, 2 mM EU on pre-heated media was added 1:1 to the cells without media change. When using fiduciary markers for drift correction and nanometric image overlap, growth media was supplemented with 1:800 dilution of 160 nM amino yellow beads (Spherotech, #AFP-0252–2) for 1 h to allow internalization of the beads into the cells prior to fixation. To examine the effect of RNA synthesis inhibition (Supplementary Figure S1A), hFibs were grown for 6 h in complete media with 1 mM EU, without actinomycin D or with 100 nM or 2 μM actinomycin D (Sigma-Aldrich, #A9415). After EU pulse, cells were fixed with PFA 4% (Alfa Aesar, #43368) for 15 min at room temperature (RT) and then rinsed with PBS three times for 5 min each. Cells were permeabilized with 0.4% Triton X-100 (Acros Organics, #327371000) in PBS for 15 min and rinsed with PBS three times for 5 min each. Click chemistry reaction was performed by incubating cells for 30 min at RT in click chemistry buffer [150 mM HEPES pH 8.2, 50 mM Amino Guanidine (Sigma-Aldrich, #396494), 100 mM ascorbic acid (Sigma-Aldrich, #A92902), 1 mM CuSO4 (Sigma-Aldrich, #C1297), 2% glucose (Sigma-Aldrich, #G8270), 0.1% Glox solution (described in STORM imaging) and 10 μM AlexaFluor647 azide (Thermo Fisher Scientific, #A10277)] protected from light. After three washes with PBS, we directly proceeded with STORM imaging for single color RNA imaging experiments (see section STORM imaging) or with immunolabelling for combined RNA and protein imaging experiments (see section Immunolabelling for STORM and STORM-PAINT imaging).
Immunolabelling for STORM and STORM-PAINT imaging
After permeabilization for Single Color STORM imaging, and click chemistry for STORM-PAINT imaging, cells were blocked in blocking buffer (10% BSA (Fisher Scientific, #9048468)–0.01% Triton X-100 in PBS) for 1 h at RT. Cells were incubated with primary antibodies, mouse anti-RNAP II phSer2 (MBL, #mabI0602), rabbit anti-H2B (Abcam, #ab1790) or Rabbit anti-Fibrillarin (Abcam, #ab166630) in incubation buffer (10% BSA–0.01% Triton X-100 in PBS) at 1:50 dilution at 4°C overnight. Cells were washed three times for 5 min each with washing buffer (2% BSA–0.01% Triton X-100 in PBS) and incubated in secondary antibody. For DNA-PAINT imaging, docking strand labelled secondary antibodies (see Table 1) were added at a 1:100 dilution in antibody dilution buffer (Ultivue-2 or Massive Photonics) and were incubated for 1.5 h at RT. For Single Color STORM, home-labelled secondary antibodies (30) (see Table 1) were added at a 1:50 dilution in blocking buffer and were incubated for 1 h at RT. For conventional imaging, commercial antibodies (see Table 1) were added at a 1:300 dilution in blocking buffer and were incubated for 1 h at RT. Cells were washed three times for 5 min each with washing buffer before proceeding to imaging.
Table 1.
Secondary antibodies used
| Antibody | Fluorophore | Brand | Code |
|---|---|---|---|
| Goat anti-Mouse D1 | Unlabelled Docking strand | Ultivue-2 | Ultivue-2 |
| Goat anti-Rabbit D2 | Unlabelled Docking strand | Ultivue-2 | Ultivue-2 |
| Anti-Mouse | Unlabelled Docking strand | Massive Photonics | Massive-AB 2-Plex |
| Goat anti-Mouse Home labelled from IgG | Alexa Fluor 405/Alexa Fluor 647 | Jackson ImmunoResearch | Home made from 115-005-205 |
| Alexa Fluor 488 Donkey-anti-Rabbit | Alexa Fluor 488 | Invitrogen | A21206 |
STELLARIS probe design and RNA FISH
Custom Stellaris® FISH Probes (#SMF-1081-5) were designed against MUC4 by utilizing the Stellaris® RNA FISH Probe Designer (Biosearch Technologies, Inc., Petaluma, CA) available online at www.biosearchtech.com/stellarisdesigner (Version 4.2). The MUC4 probes were coupled to CAL Fluor® Orange 560 Dye. Stellaris RNA FISH Probe set labelled with (Biosearch Technologies, Inc.), following the manufacturer's instructions available online at www.biosearchtech.com/stellarisprotocols.
Stellaris® FISH Probes recognizing Human GAPDH and labelled with Quasar® 570 dye (SMF-2026-1, Biosearch Technologies, Inc.) were hybridized to hFibs, following the manufacturer's instructions (www.biosearchtech.com/stellarisprotocols).
RNA FISH (Figure 4E, Supplementary Figures S3A, S6D) was performed based on the manufacturer's recommended protocol for Sequential IF + FISH in adherent cells. Briefly, cells were fixed with 3.7% formaldehyde (Sigma-Aldrich, #104003) in 1× PBS at RT for 10 min and washed 2 times in 1× PBS. Then, samples were permeabilized with 0.3% Triton X-100 in 1xPBS for 10 min at RT and washed 2 times in 1xPBS. Cells were then incubated with Wash Buffer A (Biosearch Technologies, #SMF-WA1-60) at RT for 5 min. After that, Hybridization buffer (89% Stellaris RNA FISH Hybridization Buffer (Biosearch Technologies, #SMF-HB1-10), 10% Formamide (Sigma, #F9037), 1% 125 nM probe) was added to the samples and incubated for 5 h at 37°C. Then, hybridization buffer was removed, and cells were incubated with Wash Buffer A (Biosearch Technologies, #SMF-WA1-60) for 30 min at 37°C. Subsequently, cells were incubated for other 30 min in Wash Buffer A with 5 ng/ml of DAPI (Meilunbio, #MA0128) at 37°C. Finally, a 5 min rinse with Wash Buffer B (Biosearch Technologies, #SMF-WB1-20) at RT was performed before proceeding to imaging.
Figure 4.

Labelling single gene locus in different states of gene transcription. (A) Schematic representation depicting the strategy for labelling MUC4 and nascent RNA. Cells are transfected with the STAC-MUC4 system (27,29) treated with Interleukin-6 (IL6) for 20 h and with EU for the last 10 or 20 min before fixation and posterior click labelling. (B) Activation of RNA transcription can be visualized using SMLM. SunTag modified CRISPR was used to tag the MUC4 gene locus with GFP in GP220 cells. Cells were labelled with a pulse of 10 min (top) or 20 min (bottom) of EU, and treated with IL6 to stimulate MUC4 expression. For each condition, representative super resolution images of the cropped nucleus are displayed, with a zoom of the yellow box on the right. In the zoom the RNA is color coded according to density and the MUC4 locus is represented as a green point. (C) Quantification of RNA density in a transcriptionally active area of the genome. Measurement of the RNA density as a function of the distance to the MUC4 locus centroid, for both cells treated with a 10 min pulse of EU (dashed line) and a 20 min pulse of EU (solid line). (10 min N = 9 for each condition, 20 min N = 14 for –IL6, N = 17 for +IL6). (D) Measurement of distances between the MUC4 locus and the nearest RNA nanodomains neighbors for 10 (left) and 20 (right) min EU pulses. The columns represent the mean of the distances. The error bars show the SD between each MUC4 locus for that particular neighbor rank. (10 min N = 9 for each condition, 20 min N = 14 for –IL6, N = 17 for +IL6). Asterisks indicate statistical significance of the effect of IL6 treatment according to a two-way ANOVA test. ns P > 0.05; * P < 0.05; ** P < 0.01; *** P < 0.001; **** P < 0.0001. (E) Simultaneous imaging of MUC4 locus, MUC4 RNA and nascent, super-resolved RNA. Cells were transfected with the STAC-MUC4 system (in green), labelled with STELLARIS probes designed against MUC4 mRNA (in red) and treated with a 20 min pulse of EU to visualize the nascent RNA with STORM. For each condition, representative conventional fluorescence images are shown. The nucleus outline is represented by the white-dash line. An active locus of transcription appears after transcriptional activation of the MUC4 gene by IL6 treatment (yellow box). Zoom of the MUC4 locus can be seen, and the region that approximately occupies is represented by a red-dashed circle. Super Resolved nascent RNA overlapping with MUC4 locus is shown (in white).
For RNA FISH combined with STORM (Supplementary Figure S3A, Figure 4E), we performed click chemistry as previously described (see Sample Preparation for RNA labelling) after the permeabilization step. After the click reaction washing, we proceeded with RNA FISH as stated above. Before imaging, 1:800 of fiduciary beads in PBS were added to the sample for further drift correction and overlapping.
STAC-MUC4 labelling
For MUC4 and MUC1 locus labelling (Figure 4B, E, Supplementary Figures S5C, S6A), we followed the strategy described in (27,29). Briefly, the following plasmids were used for transfection:
- pHRdSV40-NLS-dCas9–24xGCN4_v4-NLS-P2A-BFPdWPRE (Addgene, #60910) and pHR-scFv-GCN4-sfGFP-GB1-NLS-dWPRE (Addgene, #60906) were a gift from Ron Vale (29).
- pSLQ1661-sgMUC4-E3(F + E) (Addgene, #51025) was a gift from Bo Huang and Stanley Qi (31).
- pSLQ1661-sgMUC1-E1(F + E) was previously generated in our lab (27).
Transfections were performed in suspension with Fugene HD (Promega, #E2311) for GP220 cells under manufacturer's conditions and with equimolar amounts of plasmids. Transfected cells were directly plated in borosilicate glass bottom 8-well chambers (μSlide Ibidi, #80827) at a density of 3.5 × 105 cells/cm2. Twenty-four hours after transfection, cells were incubated in growth medium with 20 ng/ml Interleukin 6 (Preprotech, #200-06-20) for 20 h. EU was added to the medium as previously described in order to label nascent RNA 10 or 20 min prior to fixation.
Gene expression analysis
RNA from GP220 cells treated with or without IL6 (four biological replicates for each condition) was extracted with RNeasy Mini Kit (QIAGEN), including the on-column DNAse digest. Reverse transcription was carried out with PrimeScript RT Master Mix (Takara, #RR036A) per manufacturer's instructions. 500 ng of RNA was used per RT reaction. qPCR was performed with Tb Green Premix Ex Taq II master (Takara, #RR820A). qPCR reactions were prepared as follows: 2 μl of a 1:10 dilution of cDNA, 0.8 μl primer mix (10 μM each), 10 μl of TB Green Premix Ex Taq II (Tli RNaseH Plus) and 6.4 μl of water. The primers used are listed in Table 2. Samples were run in technical replicates on a QuantStudio 6 (Applied Biosystems) qPCR instrument for 30 s at 95°C and 40 cycles of 5 s at 95°C and 30 s at 60°C followed by melting curve analysis. Relative expression levels were determined using the Comparative Ct method and normalized against 18S levels. 18S gene was normalized against GAPDH.
Table 2.
List of primers used for qPCR
| Gene | Forward primer | Reverse primer |
|---|---|---|
| HPRT | GGCCAGACTTTGTTGGATTTG | TGCGCTCATCTTAGGCTTTGT |
| ACTB | ATAGCAACGTACATGGCTGG | CACCTTCTACAATGAGCTGC |
| 18S | CCATCCAATCGGTAGTAGCG | GTAACCCGTTGAACCCCATT |
| MUC1 | TAAAACGGAAGCAGCCTCTC | CTGGGCAGAGAAAGGAAATG |
| MUC4 | CCTCTTCCTGTCACCGACAC | CCTGTGGATG CTGAGGAAGT |
| GAPDH | TCAAGAAGGTGGTGAAGCAG | ACCAGGAAATGAGCTTGACAA |
| GP130 | AAGACCAAAGATGCCTCAAC | GAATGAAGATCGGGTGGATG |
STORM imaging
SMLM of RNA (Figure 1A, Supplementary Figure S1A–C) was performed on a custom-built inverted microscope based on Nikon Eclipse Ti frame (Nikon Instruments). The excitation module was equipped with four excitation laser lines: 405 nm (100 mW, OBIS Coherent, CA), 488 nm (200 mW, Coherent Sapphire, CA), 561 nm (500 mW MPB Communications, Canada), and 647 nm (500 mW MPB Communications, Canada). Each laser beam power was regulated through AOMs (AAOpto Electonics MT80 A1,5 Vis) and different wavelengths were coupled into an oil immersion 1.49 NA objective (Nikon). An inclined illumination mode (32) was used to obtain the images. The focus was locked through the Perfect Focus System (Nikon). The fluorescence signal was collected by the same objective and imaged onto an EMCCD camera (Andor iXon X3 DU-897, Andor Technologies). Simultaneous imaging acquisition was performed (for every frame, 647 nm reporter laser was used concurrently with 405 nm laser in order to reactivate the reported dye) with 10 ms exposure time for 45 000 frames. 647 nm laser was used at constant ∼2 kW/cm2 power density and 405 nm laser power was gradually increased over the imaging. Single color imaging was performed using a previously described imaging buffer (30), 100 mM Cysteamine MEA (Sigma-Aldrich, #30070), 5% glucose (Sigma-Aldrich, #G8270), 1% Glox (0.5 mg/ml glucose oxidase, 40 mg/ml catalase (Sigma-Aldrich, #G2133 and #C100) in PBS.
Figure 1.

EU labelling enables super-resolution imaging and subsequent analysis of nascent RNA structure in the nucleus. (A) Representative STORM density renderings of nuclear nascent RNA distribution in BJ Fibroblasts (hFibs) after 5, 10, 20, 30, and 60 min EU pulse labelling. Images show the differences in RNA density following the color scale bar (from 0.009 nm−2 in dark red to 0.00001 nm−2 in dark blue). Left: Full nucleus. Right: Zoom up of the yellow box in each condition. (B) Cumulative distribution of the Voronoi Polygon densities for 5 (N = 5), 10 (N = 11), 20 (N = 22), 30 (N = 10) and 60 min (N = 11 cells) pulses of nascent nucleoplasmic RNA labelling. Nucleoli were excluded from the analysis and analyzed separately (See Supplementary Figure S2D). 20 and 30 min are mostly overlapping. The thick lines show the median values and light colors the interquartile range (25–75 percentiles); asterisks indicate statistical significance of the separation between the mean of the medians according to Kruskal-Wallis test followed by Dunn's Multiple comparison test. Values were compared against 20 min. ns P > 0.05; * P < 0.05; ** P < 0.01; *** P < 0.001; **** P < 0.0001. (C–E) Dot plots showing the median number of localizations per nanodomain (C), median area per RNA nanodomain (D) and the Nearest Neighbor Distance (NND) between RNA nanodomains in logarithmic scale (E) for 5 (N = 5), 10 (N = 11), 20 (N = 22), 30 (N = 10) and 60 min (N = 11) pulses of nascent nuclear RNA labelled in hFibs. Nucleolar RNA was excluded and analyzed separately (See Supplementary Figure S2A-C). Asterisks indicate statistical significance of the separation between the mean of the medians according to One-way ANOVA followed by Tukey's multiple comparison test against 20 min. ns P > 0.05; * P < 0.05; ** P < 0.01; *** P < 0.001; **** P < 0.0001.
Combined dual color of RNA together with protein imaging, MUC4/MUC1-STORM imaging and single color RNAP II phSer2 imaging was performed on a NSTORM 4.0 microscope (Nikon) equipped with a CFI HP Apochromat TIRF 100 × 32 1.49 oil objective and an iXon Ultra 897 camera (Andor), using Highly Inclined and Laminated Optical sheet illumination (HILO).
To image RNA together with proteins in two-colors, combined STORM and DNA-PAINT approaches were used (Figure 2). A Dual View system (Optosplit-II Cairn Research housing with a T647lpxr dichroic beamsplitter from Chroma) was used in combination with the imaging strategy described in Otterstrom et al. (2). The dual view allowed to split the image on the full chip of the camera based on emission wavelength. 647 nm laser was used to excite the RNA labelled with AF647-azide using a power density of ∼2 kW/cm2. Simultaneously, to perform DNA-PAINT, the 560 nm laser was used with ∼1 kW/cm2 power density to excite Atto-568 (Massive Photonics) or Cy3- equivalent dye (Ultivue) attached to the imager strands. The 488 nm laser at ∼0.05 kW/cm2 power density was used to illuminate the fiduciary beads, which were used for drift correction and chromatic alignment. Images were acquired at 20 ms per frame in continuous mode. The imaging cycle was composed by 100 frames of simultaneous 560 nm and 647 nm activation interspersed with one frame of 488 nm illumination to a total of 120 000 frames. The yellow fiduciary beads imaged with the 488 nm laser were visible in both the red and orange channel, albeit dimly in the red channel. STORM-PAINT imaging buffer was 100 mM cysteamine MEA, 5% glucose, 1% Glox solution, 0.75 nM Imager strand (I1-560 and I2-560 for mouse and rabbit secondary antibodies respectively, Ultivue) in Ultivue Imaging Buffer or 100 mM cysteamine MEA, 5% glucose, 0.1% Glox solution, 1 nM Imager strand (Atto-568-ImagerStrand for mouse secondary, Massive Photonics) in Massive Photonics Imaging buffer.
Figure 2.

RNAP II phSer2 clusters and nascent RNA associate at small distance. RNA is associated to nucleosome clutches and falls inside the ‘Clutch-DNA’ radius. (A) (Top) Cropped nuclear super-resolution image in hFibs of 20 min EU labelled RNA (magenta) and PAINT image of immunolabelled RNAP II phSer 2 (green) and their merge. (Bottom) Zoom of the region inside the yellow box is shown. Yellow arrows indicate representative examples of local enrichment (clusters) of RNAP II phSer2 signal. Dotted yellow circles indicate representative examples of areas depleted of RNAP II phSer2 signal. (B) (Left) Zoom of the region inside the dotted yellow box in (A, bottom). (Right) Scheme of the analysis of RNAP II phSer2-associated RNA. The centers of RNAP II phSer2 clusters (stars) are the seeds for the Voronoi polygons (black lines) inside which the RNA localizations (black dots) are distributed. Overlaid on top are concentric circles (red) whose radii increase by 10 nm steps. (C) RNA density as a function of the distance from the center of the RNAP II phSer2 cluster (N = 12) The density is measured inside rings of increasing search radii. The dots correspond to the mean, the bars correspond to the standard deviations. (D) (Top) Cropped nuclear super-resolution image in hFibs of 20 min EU labelled RNA (magenta) and DNA-PAINT image of immunolabelled H2B (green) and their overlay. (Bottom) Zoom of the region inside the yellow box is shown. (E) (Left) Zoom of the region inside the dotted yellow box in (D, bottom). (Right) Scheme of the analysis of H2B-associated RNA. The centers of the nucleosome clutches (stars) are the seeds for the Voronoi polygons (black lines) inside which the RNA localizations (black dots) are distributed. Overlaid on top are concentric circles (red) whose radii increase by 10 nm steps. (F) RNA density as a function of the distance from the center of the clutch (N = 6). The density is measured inside rings of increasing search radii. The dots correspond to the mean, the bars correspond to the standard deviations.
MUC4-STORM and MUC1-STORM imaging cycle was composed by 100 frames of 647 nm excitation interspersed with one frame of 488 nm illumination at low power to identify the STAC-MUC4/MUC1 locus during 60 000 frames. Single color RNAP II phSer2 imaging cycle was composed by three frames of 647 nm excitation interspersed with one frame of 405 nm activation for 60 000 frames. Imaging buffer was 100 mM cysteamine MEA (Sigma-Aldrich, #30070), 5% glucose (Sigma-Aldrich, #G8270, 1% Glox (0.5 mg/ml glucose oxidase, 40 mg/ml catalase (Sigma-Aldrich, #G2133 and #C100) in PBS.
RNA-FISH imaging and combined RNA FISH-STORM imaging were performed on the Nanoimager S Mark II from ONI (Oxford Nanoimaging) with the lasers 405 (150 mW), 488 (1000 mW), 560 (500 mW) and 640 (1000 mW), an Olympus 1.4NA 100× oil immersion super apochromatic objective and a Hamamatsu sCMOS Orcaflash 4 V3. The microscope has a built-in beam splitter with a T647lpxr dichroic filter. Images were obtained using HILO. For RNA-FISH STORM imaging, a conventional image of either the MUC4 locus and the MUC4 mRNA or the GAPDH mRNA were obtained using respectively 488 and 560 nm laser. Ten frames were obtained and then averaged using FIJI (ImageJ) in order to diminish the noise and identify clearly the MUC4 transcription loci. Later, we imaged the RNA using STORM. The imaging cycle was composed by 20 frames of 647 nm activation interspersed with: (a) one frame of 488 nm illumination at low power to identify the STAC-MUC4 locus and the fiduciary beads, (b) one frame of 560 nm illumination at low power to identify the RNA FISH signal. Beads were used for drift correction and overlapping with the conventional images. Localizations were extracted from raw images of bead calibration, STORM and STORM-PAINT data using Insight3 standalone software (kind gift of Bo Huang, UCSF). For ONI images, localization lists were obtained using the integrated simultaneous localization software from ONI (ONI NimOS v.10.5) and subsequently converted into Insight3 compatible files for post-processing using a custom-built software in MATLAB 2015a.
Localizations were identified based on a set threshold and fit to a simple Gaussian to determine the X and Y positions for STORM imaging. For combined STORM and DNA-PAINT imaging, we followed the analysis workflow previously described (2).
Voronoi tessellation analysis and cluster analysis
Voronoi tessellation analysis was performed in MATLAB 2015a as previously described (33). X, Y localizations were used to compute a Voronoi diagram using the ‘delaunayTriangulation’ and ‘Voronoidiagram’ functions. The area of the Voronoi cells were obtained with the function ‘polyarea’. The local density in each data point was defined as the inverse value of the area of the corresponding Voronoi polygon. Cumulative Distribution Functions were plotted from the density values (Figure 1B, Supplementary Figure S2D).
To analyze RNA nanodomains, RNAP II phSer2 clusters and nucleosome clutches (Figure 1C–E, 2, 3B–D, 4D, Supplementary Figures S1C, D, S2A–C, S6C), cluster analysis was performed as previously described (1). For RNA, parameters capable of identifying nanodomains across a large range of RNA density values were used (Supplementary Figure S1C).
Figure 3.

Large RNA nanodomains closely associate with RNAP II phSer2 clusters while a wide range of RNA nanodomain size associates with increased distance with small nucleosome clutches. (A) Cumulative RNA distribution as a function of the distance to the centroids of the RNAP II phSer2 clusters (magenta, N = 12 cells) and H2B-containing nucleosome clutches (green, N = 6 cells). Each column represents the cumulative ratio over the total RNA which is localized in a bound circle of the distance to the cluster centers. (B) Global NND distribution between RNAP II phSer2 clusters and RNA nanodomains. Probability distribution of nearest neighbors between RNAP II phSer2 clusters and RNA nanodomains, taking RNAP II as the neighbor of origin. The line represents the kernel density distribution (KDD) of the histogram. (C) Global NND distribution between H2B-containing clutches and RNA nanodomains. Probability distribution of nearest neighbors between H2B-containing clutches and RNA nanodomains, taking H2B as the neighbor of origin. The line represents the kernel density distribution (KDD) of the histogram. (D) Nearest neighboring cluster size comparison. (Up) Matrix of comparison between RNAP II phSer2 clusters and RNA nanodomains pairs. (Bottom) Matrix of comparison between H2B-containing clutches and RNA nanodomain pairs. Clusters are binned according to their size. Color represents mean probability of two clusters pairs being nearest neighbors given their respective cluster size.
For the cluster density analysis (Supplementary Figure S1D), the number of localizations for each cluster was divided by its area in nm2. The global distribution of the cluster density in the population was then plotted with a bin size of 0.005 localizations/nm2.
For Supplementary Figure S4A, B, the cluster area for RNAP II phSer2 single color STORM images was calculated (N = 6), and its distribution was fitted to a kernel function using MATLAB 2015a. The local maxima of the function were calculated and from them the number of RNAP II molecules per cluster was obtained.
Quantification of RNA-free areas
RNA-free area was quantified from RNA STORM images by applying a binary threshold on a Gaussian filtered density map (imbinarize.m MATLAB function, with adaptive threshold, sensitivity of 0.001, pixel size 20 nm, sigma 0.5) (34,35). The analysis script is publicly available at (https://github.com/CosmaLab/Black-Space-Analysis). Percentage of RNA-free areas over the imaged nuclear area were estimated for each nucleus.
Radial density analysis
Radial density analysis was performed using a custom-written script in Python, version 3.7. A previous version has been described in (2). Briefly, raw RNA localizations were used as input, along with the centroids of H2B-containing nucleosome clutches or RNAP II phSer2 clusters, previously obtained by cluster analysis. Coordinates of protein clusters were used as seeds for Voronoi tessellation. We clipped the polygons to a hand-drawn mask to exclude polygons outside of the nucleus. RNA localizations falling inside the Voronoi polygon of a cluster were used for analysis. Disks of increasing radii, in steps of 10 nm, were drawn until fully bound with each Voronoi polygon. The RNA density was then calculated as the ratio between number of RNA localizations and the area of the clipped ring (Figure 2C, F). Cumulative ratio of RNA (Figure 3A) was calculated as RNA localizations falling inside bound circles of increasing radius divided by total amount of RNA localizations. For Supplementary Figure S4C, the cumulative number of localizations as a measure of the radius to the cluster center was calculated, and the percentage of clusters with more than one localization at the given radius was obtained from it. For Supplementary Figure S4D, the RNAP II phSer2 clusters were binned by cluster area, and the median RNA density for the cluster of each size bin was plotted.
For MUC4 and MUC1 radial density analysis (Figure 4C, Supplementary Figure S6B), we used a modified version of our custom-written script. Using the 488 nm frames of our image acquisition, we treated the MUC4/MUC1 locus as a Point Spread Function, and obtained its centroid (Supplementary Figure S5B). The position of the MUC4/MUC1 locus was measured as the geometric center of the total MUC4/MUC1 localizations obtained. Next, we drew disks of increasing radii, in steps of 10 nm, around the centroid of the STAC-MUC4/MUC1 signal.
Dual color NND analysis
For Figure 3B–D, a customized Python script was used to perform the NND analysis (Python version 3.7). To generate the NNDs for RNAP II phSer2 and H2B versus RNA, the script used the centroids obtained from the cluster analysis.
A Ball Tree algorithm from Scikit-learn library was used, with a leaf size of 2. The RNAP II phSer2 cluster centers were fed to the Ball Tree, which paired them to the Nearest Neighbor from the RNA nanodomains centers list, obtaining the distance between them.
A threshold of 200 nm for the NND was set. Kernel Density Estimation (KDE) plots were plotted from the NND distribution (Figure 3B, C). Next, the RNA nanodomains and RNAP II phSer2 clusters pairs were binned by cluster size, and the frequency of each combination of cluster pairs was represented in a heat map. The values were normalized to sum to unity (Figure 3D). Correspondingly, the same process was followed to analyze the NND between H2B and RNA (Figure 3D).
For Figure 4D and Supplementary Figure S6C, the centroid of the MUC4/MUC1 localizations was used as reference. A custom-made script in Python, version 3.7 calculated the distances between the MUC4/MUC1 locus and its 10 nearest neighbor RNA nanodomains. Once the NNDs were collected for all the cells, the mean and the standard deviation were calculated between the distances in the same closeness rank.
For Supplementary Figure S3A and S3B, the centroid of the cytoplasmic GAPDH mRNA localizations from smRNA FISH was similarly used as a reference to pair the cytoplasmic GAPDH mRNA localizations and its closest RNA nanodomain. A cutoff of a maximum of 200 nm was established for the pairs. Once the pairs were collected, we obtained the median number of localizations for the paired RNA nanodomains. We used those as a reference for calculating the size of the nucleoplasmic nanodomains.
For Supplementary Figure S4E and S4F, the cluster areas of the RNA nanodomains, of the H2B clutches and of RNAP II phSer2 clusters were extracted. Then, the probability distribution for every cluster type was calculated. Next, we created a simulated matrix of size-independent probability distribution by multiplying the pair of cluster populations probability distributions. Finally, we subtracted the observed probability distribution (Figure 3D) to the simulated, size-independent probability distribution.
Statistical analysis
Graphad Prism (v.8.1) and MATLAB 2015a were used for statistical analysis. DNA Voronoi density data in Figure 1B and Supplementary Figure S2D were obtained by binning the Voronoi density distributions into 300 logarithmically spaced bins ranging from 10−5 to 1 nm−2. This encompassed the maximal range of the datasets. The median for each bin was calculated and used for Kruskal–Wallis test followed by Dunn's multiple comparison test. For Figure 1C-E and Supplementary Figure S2A-C the median values were calculated and used for one-way ANOVA followed by Tukey's multiple comparison test against 20 min. For Supplementary Figure S2E, the mean density values were calculated and used for one-way ANOVA followed by Dunnet's multiple comparison test against 20 min in the nucleoplasm, and 20 min in the nucleolus, separately. For Supplementary Figure S2F, the mean RNA free area percentage values were calculated and used for One-way ANOVA followed by Dunnet's multiple comparison test. For Supplementary Figure S1D, the median values of the cluster density of the cells were calculated and unpaired two-tailed t-test comparisons was performed. For Supplementary Figure S5A, the mean fold change was calculated and used for a One-Way ANOVA followed by Fisher LSD test. For Supplementary Figure S5D the total number of localizations was divided by the area of the nucleus for each cell to calculate global density and unpaired two-tailed t-test comparisons were performed between -IL6 and + IL6 conditions. For Figure 4D, the P-value of the statistical significance of the IL6 treatment was obtained using a two-way ANOVA analysis. Statistical significance is represented in the following manner: ns P > 0.05, * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001.
Software
Insight3 Software used for STORM image processing has been generated (36) and kindly provided by Dr Bo Huang (UCSF).
ImageJ 2.0.0 software used for STORM and RNA FISH analysis and visualization can be found at: https://imagej.nih.gov/ij/download.html
Graphpad Prism software used for statistical analysis can be found at: https://www.graphpad.com/scientific-software/prism/
MATLAB software used for imaging data analysis can be found at: https://www.mathworks.com/products/matlab.html
NimOS v.10.5 Software used for STORM image processing can be requested to ONI at: https://oni.bio/nanoimager/software/
Python 3.7.0 used for the radial density and the dual color NND analysis can be found at: https://www.python.org/downloads/release/python-370/
RESULTS
EU labelling enables time-dependent, super-resolution imaging of nascent RNA
To visualize RNA using STORM, human BJ Fibroblasts (hFibs) were labelled with the nucleotide analog 5-ethynyl-uridine (EU) (see Materials and Methods) (37). Cells were incubated with 1 mM EU for increasing amounts of time (5, 10, 20, 30 and 60 min) to ensure dense RNA labelling and enable visualization of global RNA distribution. Subsequent fixation and click chemistry with AlexaFluor647 (AF647) allowed STORM imaging of the RNA (Figure 1A). To verify the labelling specificity for nascent RNAs, we treated the cells with increasing concentrations of Actinomycin D (ActD) (Supplementary Figure S1A). ActD inhibits transcription by binding DNA at the transcription initiation complex and preventing the elongation of the RNA chain, blocking RNAP I at low concentration (Supplementary Figure S1A, middle), and both RNAP I and II at higher concentrations (Supplementary Figure S1A, bottom) (38). The number of localizations detected after ActD treatment was negligible, confirming that we specifically labelled newly transcribed RNA. Thus, EU incorporation followed by AF647 labelling provides a novel way to investigate the nascent transcriptome with nanoscale resolution.
Nascent RNAs were heterogeneously distributed across the nucleus, forming local accumulations thereafter called RNA nanodomains (Figure 1A, right). Actively transcribed nucleoplasmic and nucleolar regions displayed different and clearly distinguishable RNA distributions. Nucleolar RNA was densely and homogeneously labelled, forming high density accumulations that colocalize with the nucleolar marker Fibrillarin (Supplementary Figure S1B). In turn, the distribution of nucleoplasmic RNA was more uneven, with areas of exclusion and RNA nanodomains of highly variable size (Figure 1A, Supplementary Figure S1B, C). This might reflect the different transcription rates found in the nucleoplasm and the nucleolus (39,40). Thus, we analyzed the RNA localizations from the nucleolus and the nucleoplasm separately, to extract the specific RNA features associated to each of these nuclear sub-compartments.
We quantified the RNA distribution by applying Voronoi tessellation, which determines the heterogeneity of the nascent RNA distribution, based on the local spatial context, without any a priori imposition of user-defined parameters (33,41). We combined this analysis with our previously described clustering method (1) that allows the identification and quantification of the RNA nanodomains and its characteristics (Supplementary Figure S1C).
The RNA density distribution in the nucleoplasm significantly increased in an EU-pulse time-dependent manner, from 5 min to 20 min. From 20 min onwards, the changes in RNA density were not statistically significant, indicating a saturation effect (Figure 1B).
More RNA nanodomains progressively appeared upon longer EU pulses (Figure 1A). Quantitative cluster analysis of the nucleoplasm (Supplementary Figure S1C) excluding the nucleolus revealed a progressive increase in the median number of localizations (loc) per nanodomain, indicating an accumulation of RNA copies in the nanodomains (Figure 1C). Conversely, the area of nanodomains increased until 20 min of EU pulse (Figure 1D). The increased number of localizations per nanodomain and increased areas of the nanodomains up to 20 min suggest they belong to regions of nascent RNA transcription. From 20 min onwards, the median area of the nanodomains did not increase significantly. At 20 min of EU pulse, we found that RNA nanodomains are of 557 ± 122 nm2 on average (Figure 1D).
Although the area of the nanodomains reaches a saturation, they become slightly denser as more RNA accumulates, ranging from a median density of 1.89 × 10–2 loc/nm2 at 20 min to 2.51 × 10–2 loc/nm2 at 60 min (Supplementary Figure S1D). Additionally, nascent RNA nanodomains showed a wide scale of density of localizations, ranging from 5 × 10–3 to 0.1 loc/nm2 at 20 min of EU pulse, suggesting different rates of transcription for different nanodomains. The increased area and densities of RNA nanodomains, thus the increased number of labelled transcripts, resulted in decreased nearest neighbor distances (NND) between nanodomains (Figure 1E). No significant changes in NND between nanodomains were detected after 10 min, suggesting that the majority of active transcription sites were already identified with short EU pulses.
Although the nucleolus is a denser environment with different structure and distribution of RNAs, the features described for the nucleoplasm were comparable to those in the nucleolus. Median number of localizations per nanodomain (Supplementary Figure S2A), median nanodomain area (Supplementary Figure S2B) and median NND (Supplementary Figure S2C) reached saturation after 20 min of EU pulse. Voronoi analysis of the localizations in the nucleolus showed a highly similar trend with respect to the nucleoplasmic RNA density, although with higher absolute values (Supplementary Figure S2D). Accordingly, transcription of rRNA is very efficient, with approximately 100 polymerases transcribing each rRNA gene at a rate of ∼5700 nucleotides/min in mammalian cells, several times faster than the rate calculated for the genes transcribed by RNAP II (39). In spite of this, the global trend of the newly transcribed RNA accumulation in identifiable nanodomains was comparable inside and outside the nucleolus.
When considering the global density of RNAs we clearly observed how it progressively increased in both nucleoplasm and nucleolus from 5 to 60 min pulses (Supplementary Figure S2E). Furthermore, the percentage of RNA-free area decreased from 5 to 60 min time points (Supplementary Figure S2F), suggesting that the increase in global RNA density correlated with a progressive increase of the RNA signal across the nucleus. These data indicate that the newly transcribed RNA diffuses and moves out from the area of transcription, increasing the global RNA density and the presence of RNA across the whole nucleus, while the areas of nascent transcription reach a plateau at 20 min.
Next, we asked if we could estimate the number of RNAs present within RNA nanodomains. We co-imaged the nascent RNA transcriptome together with a single molecule RNA (smRNA) FISH against GAPDH (Supplementary Figure S3A). We identified single GAPDH RNAs overlapping with sparse RNA signal in the cytoplasm (Supplementary Figure S3A). Assuming that this cytoplasmic RNA signal corresponded to a single GAPDH mRNA, we normalized the nucleoplasmic RNA nanodomains by this value to obtain an approximated number of RNA molecules per nucleoplasmic nanodomain. We obtained a median value of 2.32 ± 1.34 RNAs per RNA nanodomain in the nucleoplasm. Noteworthy, in line with the fact that RNA transcripts vary considerably in length, we detected a broad distribution of RNA nanodomain sizes with up to 15 RNAs per nanodomain, with most nanodomains comprised of only 1–2 RNAs (Supplementary Figure S3B).
SMLM microscopy reveals a tight association between elongating RNAP II and nascent RNA nanodomains
Having quantitatively analyzed the nascent RNAs in the nucleoplasm and nucleolus, we aimed to investigate its distribution with respect to the elongating form of RNAP II, RNAP II phSer2. For this, we used our previously described method of STORM-PAINT imaging (2). Given the results already introduced, we focused on 20 min EU pulses and we simultaneously labelled RNA with click chemistry for STORM imaging, and RNAP II phSer2 using antibodies compatible with DNA-PAINT.
Dual-color SR images of RNAP II phSer2 and RNA revealed a heterogeneous pattern, with regions enriched in and depleted of RNAP II phSer2 within the nucleus, as expected (Figure 2A). RNAP II phSer2 accumulated in clusters (Figure 2A, middle column), distributed across the whole nucleus except in the nucleolus. We did not observe specific nuclear areas with accumulation of drastically bigger clusters or with large areas of cluster exclusion.
To estimate the number of RNAP II phSer2 molecules forming the nuclear clusters, we performed cluster analysis and classified clusters by increasing cluster area. Interestingly, we observed two peaks in the distribution of RNAP II phSer2 cluster areas (Supplementary Figure S4A). By assuming that the first peak of elongating RNAP II is composed of individual RNAP II molecules, we normalized the area of RNAP II phSer2 clusters to the area of the smallest clusters identified (first peak). We observed that clusters are formed by 3.35 ± 0.68 RNAP II molecules on average. We estimated that around 20% (18.91%) of the cluster population corresponds to individual elongating RNAP II molecules, and the remaining clusters are mostly formed by few RNAP II phSer2 molecules (Supplementary Figure S4B).
RNA nanodomains were closely associated to RNAP II phSer2 clusters (Figure 2A, B). The ratio between the number of RNA nanodomains versus RNAP II phSer2 clusters was of 0.91 ± 0.65, showing a clear correlation between the clustering of RNAP II phSer2 and nascent RNA nanodomains due to active transcription. To quantify the spatial relationship between RNAP II phSer2 clusters and RNA nanodomains, we segmented the clusters using our previously described radial clustering algorithm (2). We used the centroid positions of each RNAP II phSer2 cluster as a seed for tessellation analysis. With this, we divided the nuclear space into polygons, with each tessel belonging to a specific RNAP II cluster (Figure 2B). We then segmented the space of each tessel in concentric 10 nm rings to analyze the density of nascent RNAs as a function of their physical relation to RNAP II phSer2 clusters. We previously used a similar analysis to determine the density of DNA associated to individual nucleosome clutches (2). Here, this analysis allowed us to study the density of RNA surrounding RNAP II phSer2 clusters.
Using the radial analysis, we showed RNA nanodomains were closely associated to the elongating RNAP II clusters, with a maximum RNA density at 30–40 nm from RNAP II phSer2 clusters (Figure 2C). RNA density progressively declined until there was barely any RNA-RNAP II phSer2 association from 100 nm onwards.
To define RNAP II phSer2 clusters as transcriptionally active, we investigated how many of the RNAP II clusters had at least one localization of RNA within a distance of 40 nm, which corresponds to the peak of maximum density. We found that 77.4 ± 16.5% of the RNAP II phSer2 clusters were transcriptionally active (Supplementary Figure S4C). Of note, this classification excludes RNA interacting with RNAP II phSer2 clusters at a distance greater than 40 nm, thus the percentage of active RNAP II clusters is potentially underestimated. Conversely, at 70 nm, 88.4 ± 11.5% of the clusters have at least one RNA localization inside its radius (Supplementary Figure S4C).
Lastly, we asked whether RNA localizations could be more tightly associated with large RNAP II phSer2 clusters. To this end, we quantified RNA density as a function of RNAP II phSer2 cluster size. We detected only a slight increase in the association between RNA and larger RNAP II phSer2 clusters (Supplementary Figure S4D).
RNA is associated to small nucleosome clutches and falls inside the ‘Clutch-DNA’ radius
In previous works we have described how the chromatin is organized in discrete clutches and how the median nucleosomal clutch size correlates with cell state (1). We also showed that small clutches are more closely associated with RNAP II (1) and that clutches affect the folding of DNA named ‘Clutch-DNA’ up to 70 nm from the clutch center in hFibs (2). However, the relation between the clutch characteristics and its local transcriptional state has not been characterized yet.
In order to investigate this, we used dual-color SMLM to obtain super-resolved images of nascent RNA together with H2B (Figure 2D). From a qualitative inspection of the images, we first observed that RNA is found close to nucleosome clutches but rarely overlapping with them (Figure 2D, bottom, 2E). This was quantitatively demonstrated by examining the radial analysis results (Figure 2E, F). RNAs were associated with the nucleosome clutches, but the point of maximum RNA density was found at 70–80 nm from the center of nucleosome clutches (Figure 2F), at a larger distance when compared to RNA-RNAP II association (Figure 2C). Interestingly, RNAs were found inside the area in which the clutches affect the DNA folding (2). Of note, close to the center of the clutch (up to 30 nm) the RNA density was lower, possibly indicating the presence of the transcriptional machinery, creating a spatial constraint and thus a distance shift between the center of clutches and nascent RNA.
Radial density values between RNA versus H2B-containing nucleosome clutches and RNAP II phSer2 clusters are not directly comparable, as the density of clusters, global distribution and sizes are different. In order to address this relation, we measured instead the percentage of total RNA around nucleosome clutches and elongating RNAP II clusters in iteratively bound circles (Figure 3A). At a radius of 40 nm from the center of the RNAP II phSer2 clusters, we found ∼40% of the RNA accumulated. At 60 nm, ∼50% of the labelled RNA was found. By contrast, only 26% of RNA was found within 40 nm from nucleosome clutches, and ∼50% of RNA was found only when considering a distance higher than 80 nm from the nucleosome clutch.
RNA nanodomains preferentially interact with small nucleosome clutches and with a broad range of RNAP II phSer2 clusters
As described before, we found that RNA is organized in discrete nanodomains. As such, we next aimed to characterize the inter-relations between RNAP II phSer2 clusters or nucleosome clutches and RNA nanodomains. For this, we analyzed the cells using the cluster analysis previously described (1). Each RNAP II phSer2 cluster/H2B-containing clutch was paired with its closest nearest neighbor RNA nanodomain.
Once paired, we looked at the NND between clusters (Figure 3B, C). We found RNAP II phSer2-RNA pairs associated at a median distance of 55.97 nm, with a maximum peak in the range of 25–50 nm of distance. H2B-RNA pairs were found further away, at a median distance of 79.10 nm, and with a broader distribution, in the range of 50–90 nm of distance (Figure 3C).
Next, to investigate which is the effect of cluster size on the NND pairing, we plotted the pairs of clusters according to their area (Figure 3D). When looking at the normalized frequency of cluster pairs, most RNAP II phSer2 clusters were found in a size range of 200–800 nm2, while RNA nanodomains span a range of 200 to 900 nm2 (Figure 3D, top, Supplementary Figure S4E). By contrast, the H2B-containing nucleosome clutches predominantly interacting with RNA measured only 200–500 nm2, with a narrower size distribution (Figure 3D, bottom, Supplementary Figure S4F). This supports the notion that small histone clutches, generally related with open chromatin states, are preferentially associated with RNA and thus with transcriptionally active chromatin regions.
As a control, we performed the reverse NND analysis, pairing each RNA nanodomain with its closest RNAP II phSer2 cluster or H2B clutch. We found 83.01 ± 14% of RNA nanodomains associated with elongating RNAP II clusters, and 93.4 ± 5% of RNA nanodomains associated to H2B clutches. This further confirms our reported association between nascent RNA nanodomains, elongating RNAP II clusters and H2B clutches.
Imaging transcriptional activation at MUC4 gene
Having observed the reciprocal distribution of RNA nanodomains and RNAP II at the global nuclear level, we next asked if we could detect transcriptional changes at individual genes with SR microscopy. For this, we used a cell line in which we can specifically induce the expression of an endogenous gene efficiently. In GP220 gastric cancer cells, Interleukin 6 (IL6) treatment triggers the specific upregulation of the MUCIN 4 (MUC4) gene (42) as confirmed by qPCR analysis (Supplementary Figure S5A). Thus, we investigated the changes of nascent RNA density at the MUC4 locus upon IL6-mediated activation in order to correlate RNA abundance to changes in transcriptional activity of MUC4.
To label the MUC4 locus, we used STAC (SunTAg modified CRISPR), a protein-tagging system for signal amplification based on a SunTag scaffold fused to CRISPR-dCas9, recognized by multiple scFv-GFPs (27,29). STAC allowed to endogenously label the MUC4 locus in GP220 cells treated with IL6 while concomitant EU pulses enabled the detection of nascent RNA (Figure 4A, B, Supplementary Figure S5B, C).
Using STORM, we specifically measured the transcriptional environment around MUC4 single alleles. Upon IL6 treatment, we observed a net increase in RNA density in close proximity to the MUC4 locus (Figure 4B). While we observed a local accumulation of RNA at the MUC4 locus, we ruled out a possible global increase of transcription upon IL6 treatment (Supplementary Figure S5A, D), further reinforcing the specificity of our observation.
Since the MUC4 locus is visualized as a diffraction-limited spot, we calculated its center position with high precision (Supplementary Figure S5B). This defines the centroid of the targeted region of the MUC4 gene. Although it does not represent the total volume of the gene, it allows for a precise approximation of its localization. From this centroid, we measured the density of RNAs as a function of the distance from MUC4 (Figure 4C, Supplementary Figure S5B). We observed a clear difference in RNA density between IL6+ and IL6– cells, both at 10- and 20-min pulses, although the difference was drastically higher at 20 min. Thus, our observation at the single allele level agreed with our data at the global level, where RNA nanodomains reached saturation of signal after 20 min of EU pulse.
For each cell, we could identify several RNA nanodomains in close proximity to the MUC4 locus. We measured the distance between MUC4 and its closest RNA nanodomains. IL6+ cells showed areas of active transcription significantly closer to the MUC4 locus at 20 min of EU pulse, further reinforcing our previous observations (Figure 4D).
As control we also labelled MUC1 using the STAC system (Supplementary Figure S6A). MUC1 expression is not significatively increased by IL6 treatment (Supplementary Figure S5A). We did not observe any significative difference in the density of RNAs as a function of the distance from MUC1 (Supplementary Figure S6B) and in the distances between MUC1 and its closest RNA nanodomains (Supplementary Figure S6C) when comparing IL6- and IL6 + cells. This result indicates the specificity of the STAC system to measure IL6-dependent MUC4 expression.
To ensure that the areas of active transcription close to MUC4 included MUC4 RNAs upon IL6 induction, we performed RNA FISH experiments. We designed specific Stellaris RNA-FISH probes for MUC4 (see Materials and Methods). In IL6+ cells, one or two loci of expression per cell were clearly observed, while nascent RNA-FISH signal was not detected in IL6– cells (Supplementary Figure S6D). This indicates that MUC4 loci are indeed activated by cytokine treatment and that there is a drastic change in the local mRNA concentration associated to gene activation.
Finally, we combined the SR/STAC approach with RNA FISH imaging and successfully imaged the gene locus, the specific RNA and the nascent transcriptome simultaneously (Figure 4E). We therefore report a novel approach allowing visualization and quantitative measurements of the transcriptional state of a single gene at a single locus with nanometric precision.
DISCUSSION
Visualization of RNA molecules at nanoscale resolution within intact nuclei can reveal how transcription is physically organized and compartmentalized inside the nucleus. Previous studies have identified pockets of transcription preferentially localized in the active compartments of the genome (9,10,14). These accumulations of RNA transcriptional machinery and other proteins were termed transcription factories. Two main approaches were used to image regions of active transcription, namely labelling of RNA or labelling of active RNAP II (12,19,40,43,44). Different methodologies showed highly variable factories in terms of number and size (43–45). Using diffraction-limited microscopy, aggregates of several active transcription areas could be misidentified as single transcription factories, potentially explaining the variability found. As such, SR techniques are ideally suited to study the localization of RNA and RNAP II molecules in the nucleus, properly resolving their structure and size. One such study, using STORM coupled with light sheet microscopy, imaged two populations of RNAP II differentially labelled and showed no overlap between the two populations, thus challenging the existence of transcription factories as aggregates of several RNAPs (46). Notably, recent studies have identified these active transcription pockets as accumulations of RNA-RNA Binding Proteins that form a pattern of microphase domains which are prevented from coalescing into fully phase-separated domains by the tethering of RNA transcripts to chromatin via RNAP II (15).
Intron seqFISH technology is instrumental to investigate the distribution of the nascent transcriptome in the nucleus (47). Using this approach, more than 10000 nascent RNAs have been visualized in single cells, and its relative position has been mapped preferentially to the surface of chromosome territories. Although this approach constitutes a leap in the understanding of single cell transcriptomics, the microscopy method used does not provide quantitative nanometric data about the structure of the active transcription sites and its relation with chromatin and elongating RNAP II structure.
In this work, we have built upon these studies, labelling not only genes but also the global nascent RNA, and looking at the characteristics and changes of active transcription areas in the nucleus upon different labelling times. We also applied our developed methods for dual-color SMLM imaging and analysis to obtain SR images, which allowed us to study the interplay between elongating RNAP II, nucleosome clutches and RNA.
Our analyses revealed that the nascent transcriptome is organized in the nucleus in sub-diffraction limited structures that we termed RNA nanodomains. We observed that RNA nanodomains have a median size of 557 ± 122 nm2, smaller than previously reported sizes for transcription factories (12,18,19), likely because we segmented fine sub-structures, earlier identified as single entities. Importantly, the number of RNAs corresponds to a median of 2.32 ± 1.34 RNAs per nanodomain leading us to conclude that the majority of nanodomains belong to a single actively transcribed gene. Using SR microscopy, we could obtain a previously unachieved resolution on the structure of the nascent transcriptome, with high precision, capturing its physical characteristics.
We studied the relation between RNA nanodomains and active transcription achieving simultaneous imaging of RNA and elongating RNAP II. We found that most nucleoplasmic RNA is closely associated with RNAP II phSer2 clusters. RNA nanodomains distribution has a maximum peak at 20–50 nm from the elongating RNAP II clusters. Interestingly, an important fraction of the RNA nanodomains was associated to small RNAP II clusters, on average corresponding to only one RNAP II. However, the few remaining elongating RNAP II clusters contained two or more RNAP II and only a subset of RNA associated to these large RNAP II clusters. This, coupled with the predominantly small number of RNAs per nanodomain, might suggest that large RNA factories are rare in the nuclei.
Previous single molecule tracking measurements showed residence times of RNAP II of 10 min (40) to 20 min (48). This residence time is in line with our measurements, where we established that we reach extensive labelling of RNA nanodomains within 20 min. Additionally, our measurements give an indication of the residence time of nascent RNA at sites of transcription. Indeed, we observe that RNA nanodomain reach a plateau in size at 20 min, indicating that readily transcribed molecules might translocate from transcription sites to the cytosol within this time window.
We found that RNA nanodomains are physically associated to the periphery of nucleosome clutches, interacting preferentially with small nucleosome clutches. RNA density is slightly reduced at the clutch center, peaking at around 70–80 nm from it. This matches the previously measured “Clutch-DNA” distance in hFibs, showing how the transcribed DNA corresponds to the DNA whose folding is affected by the clutch itself (2). In particular, when pairing H2B-containing nucleosome clutches to its nearest neighboring RNA nanodomains, we find that the clutches associated with RNA nanodomains are preferentially the small clusters, with the size of the RNA nanodomains being quite variable, and the median NND being again 79.10 nm. This indicates that smaller nucleosome clutches are preferentially transcriptionally active. Interestingly, the reduced density of RNA at short distance from the center of the clutches and the higher NND could be due to the high compaction and low accessibility to RNAP II at the clutch center, or due to the RNAP II machinery imposing a physical constraint in the distance between clutches and RNA.
From the combined measurements of distances between H2B, RNAP II and RNA, and considering the distances previously measured between RNAP II and nucleosome clutches of 40 nm on average (1), we propose a clutch transcriptional model. RNAP II is recruited to the periphery of small nucleosome clutches, with the transcribed RNA organized in highly dense domains closely associated to RNAP II and further away from the nucleosome clutch (see graphical abstract). In our model, elongating RNAP II is recruited to existing clutches. Of note, it is unlikely that the nucleosome clutches were formed a posteriori, with the transcriptional activity being responsible for clutch formation, because in such case, only actively transcribed areas would form clutches.
Finally, we also showed a novel method to measure the transcription of a single, endogenous gene locus, and to quantify the distribution of the transcripts around the locus with nanometric precision. With this approach, we directly measured changes of the transcriptional state at nanometric distance from individual loci, confirming what we observed for the global nascent RNA transcriptome. Indeed, upon transcription activation, increased RNA density at the locus (at 20 nm radius) and closer RNA nanodomains were observed within 20 minutes of labelling.
Recently, new methods, such as Oligopaint-derived approaches, have been developed to visualize specific genomic regions with ultra-high spatial resolution (23,49,50). Highly multiplexed visualization of specific nascent mRNAs at single cell level has also been achieved (47,51). A combination of these techniques with the one presented here could help us dissect the precise transcriptional state of specific genes in different genomic regions in single cells with nucleosomal precision.
DATA AVAILABILITY
Custom-made scripts and example datasets are publicly available at Github (https://github.com/CosmaLab/NND_Analysis).
Supplementary Material
ACKNOWLEDGEMENTS
The authors acknowledge Bo Huang (UCSF, USA) for kindly sharing Insight3 software. The authors acknowledge Carme de Bolos for kindly sharing the GP220 cell line. The authors thank Pablo A Gomez-Garcia for his help in conceptualizing analysis for the paper. The authors thank Martina Pesaresi for her suggestion with using the GP220 cell line. We acknowledge the advanced light microscopy (ALMU) from the Centre for Genomic Regulation (CRG) for their technical assitance.
Author contributions: Conceptualization, A.C.G., M.V.N., M.P.C., M.L.; Experimentation, A.C.G. and E.G.A.; Software, I.E., A.C.G., C.V. and J.J.O.; Formal analysis, I.E., A.C.G., C.V, J.O. and E.G.A.; Writing – Original Draft, A.C.G., M.P.C., M.V.N; Writing – Review & Editing E.G.A., I.E., M.L.; Funding Acquisition M.P.C., M.L.; Supervision, M.P.C., M.V.N.
Notes
Present address: Jason Ottestrom, IDEA Bio-Medical, 08860 Castelldefels, Spain.
Contributor Information
Alvaro Castells-Garcia, Bioland Laboratory, Guangzhou Regenerative Medicine and Health Guangdong Laboratory, Guangzhou 510005, China.
Ilyas Ed-daoui, CAS Key Laboratory of Regenerative Biology, Guangdong Provincial Key Laboratory of Stem Cell and Regenerative Medicine, Guangzhou Institutes of Biomedicine and Health, Chinese Academy of Sciences, Guangzhou 510530, China.
Esther González-Almela, Bioland Laboratory, Guangzhou Regenerative Medicine and Health Guangdong Laboratory, Guangzhou 510005, China.
Chiara Vicario, Centre for Genomic Regulation (CRG), The Barcelona Institute of Science and Technology, 08003 Barcelona, Spain.
Jason Ottestrom, ICFO-Institute of Photonic Sciences, The Barcelona Institute of Science and Technology, Barcelona, 08860, Spain.
Melike Lakadamyali, Perelman School of Medicine, Department of Physiology, University of Pennsylvania, Philadelphia, PA 19104, USA; Perelman School of Medicine, Department of Cell and Developmental Biology, University of Pennsylvania, Philadelphia, PA 19104, USA.
Maria Victoria Neguembor, Centre for Genomic Regulation (CRG), The Barcelona Institute of Science and Technology, 08003 Barcelona, Spain.
Maria Pia Cosma, Bioland Laboratory, Guangzhou Regenerative Medicine and Health Guangdong Laboratory, Guangzhou 510005, China; CAS Key Laboratory of Regenerative Biology, Guangdong Provincial Key Laboratory of Stem Cell and Regenerative Medicine, Guangzhou Institutes of Biomedicine and Health, Chinese Academy of Sciences, Guangzhou 510530, China; Centre for Genomic Regulation (CRG), The Barcelona Institute of Science and Technology, 08003 Barcelona, Spain; ICREA, Pg. Lluís Companys 23, 08010 Barcelona, Spain; Universitat Pompeu Fabra (UPF), Dr Aiguader 88, 08003 Barcelona, Spain.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Innovative Team Program of Guangzhou Regenerative Medicine and Health Guangdong Laboratory [2018GZR110103001 to M.P.C.]; Guangzhou Key Projects of Brain Science and Brain-Like Intelligence Technology [20200730009 to M.P.C.]; National Natural Science Foundation of China [31971177 to M.P.C.]; Science and Technology Program of Guangzhou, China [202002030146 to M.P.C.]; European Union's Horizon 2020 Research and Innovation Programme [CellViewer No. 686637 to M.P.C. and M.L.]; Ministerio de Ciencia e Innovación [BFU2017-86760-P (AEI/FEDER, UE) to M.P.C.]; AGAUR grant from Secretaria d’Universitats i Recerca del Departament d’Empresa I Coneixement de la Generalitat de Catalunya [2017 SGR 689 to M.P.C.]; Centro de Excelencia Severo Ochoa [2013–2017 to M.P.C.]; CERCA Programme/Generalitat de Catalunya [to M.P.C.]; People Program (Marie Curie Actions) FP7/2007–2013 under REA grant [608959 to M.V.N.]; Spanish Ministry of Science and Innovation to the European Molecular Biology Laboratory (EMBL) partnership [to M.P.C.]; Juan de la Cierva-Incorporación 2017 [to M.V.N.]. Funding for open access charge: National Natural Science Foundation of China (NSFC) grant [31971177 to M.P.C].
Conflict of interest statement. None declared.
REFERENCES
- 1. Ricci M.A., Manzo C., Garcia-Parajo M.F., Lakadamyali M., Cosma M.P.. Chromatin fibers are formed by heterogeneous groups of nucleosomes in vivo. Cell. 2015; 160:1145–1158. [DOI] [PubMed] [Google Scholar]
- 2. Otterstrom J., Castells-Garcia A., Vicario C., Gomez-Garcia P.A., Cosma M.P., Lakadamyali M.. Super-resolution microscopy reveals how histone tail acetylation affects DNA compaction within nucleosomes in vivo. Nucleic Acids Res. 2019; 47:8470–8484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kempfer R., Pombo A.. Methods for mapping 3D chromosome architecture. Nat. Rev. Genet. 2020; 21:207–226. [DOI] [PubMed] [Google Scholar]
- 4. Solovei I., Thanisch K., Feodorova Y.. How to rule the nucleus: divide et impera. Curr. Opin. Cell Biol. 2016; 40:47–59. [DOI] [PubMed] [Google Scholar]
- 5. Wang S., Su J.H., Beliveau B.J., Bintu B., Moffitt J.R., Wu C.T., Zhuang X.. Spatial organization of chromatin domains and compartments in single chromosomes. Science. 2016; 353:598–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tsukamoto T., Hashiguchi N., Janicki S.M., Tumbar T., Belmont A.S., Spector D.L.. Visualization of gene activity in living cells. Nat. Cell Biol. 2000; 2:871–878. [DOI] [PubMed] [Google Scholar]
- 7. Beagrie R.A., Scialdone A., Schueler M., Kraemer D.C., Chotalia M., Xie S.Q., Barbieri M., de Santiago I., Lavitas L.M., Branco M.R.et al.. Complex multi-enhancer contacts captured by genome architecture mapping. Nature. 2017; 543:519–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Quinodoz S.A., Ollikainen N., Tabak B., Palla A., Schmidt J.M., Detmar E., Lai M.M., Shishkin A.A., Bhat P., Takei Y.et al.. Higher-order inter-chromosomal hubs shape 3D genome organization in the nucleus. Cell. 2018; 174:744–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chambeyron S., Bickmore W.A.. Chromatin decondensation and nuclear reorganization of the HoxB locus upon induction of transcription. Genes Dev. 2004; 18:1119–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cremer T., Cremer M., Hübner B., Strickfaden H., Smeets D., Popken J., Sterr M., Markaki Y., Rippe K., Cremer C.. The 4D nucleome: evidence for a dynamic nuclear landscape based on co-aligned active and inactive nuclear compartments. FEBS Lett. 2015; 589:2931–2943. [DOI] [PubMed] [Google Scholar]
- 11. Boettiger A.N., Bintu B., Moffitt J.R., Wang S., Beliveau B.J., Fudenberg G., Imakaev M., Mirny L.A., Wu C.T., Zhuang X.. Super-resolution imaging reveals distinct chromatin folding for different epigenetic states. Nature. 2016; 529:418–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Iborra F.J., Pombo A., Jackson D.A., Cook P.R.. Active RNA polymerases are localized within discrete transcription ‘factories’ in human nuclei. J. Cell Sci. 1996; 109:1427–1436. [DOI] [PubMed] [Google Scholar]
- 13. Hilbert L., Sato Y., Kuznetsova K., Bianucci T., Kimura H., Jülicher F., Honigmann A., Zaburdaev V., Vastenhouw N.L.. Transcription organizes euchromatin via microphase separation. Nat. Commun. 2021; 12:1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Osborne C.S., Chakalova L., Brown K.E., Carter D., Horton A., Debrand E., Goyenechea B., Mitchell J.A., Lopes S., Reik W.et al.. Active genes dynamically colocalize to shared sites of ongoing transcription. Nat. Genet. 2004; 36:1065–1071. [DOI] [PubMed] [Google Scholar]
- 15. Wei M., Fan X., Ding M., Li R., Shao S., Hou Y., Meng S., Tang F., Li C., Sun Y.. Nuclear actin regulates inducible transcription by enhancing RNA polymerase II clustering. Sci. Adv. 2020; 6:eaay6515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tsai A., Muthusamy A.K., Alves M.R., Lavis L.D., Singer R.H., Stern D.L., Crocker J.. Nuclear microenvironments modulate transcription from low-affinity enhancers. eLife. 2017; 6:e28975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Papantonis A., Kohro T., Baboo S., Larkin J.D., Deng B., Short P., Tsutsumi S., Taylor S., Kanki Y., Kobayashi M.et al.. TNFα signals through specialized factories where responsive coding and miRNA genes are transcribed. EMBO J. 2012; 31:4404–4414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cook P.R. The organization of replication and transcription. Science. 1999; 284:1790–1795. [DOI] [PubMed] [Google Scholar]
- 19. Eskiw C.H., Rapp A., Carter D.R.F., Cook P.R.. RNA polymerase II activity is located on the surface of protein-rich transcription factories. J. Cell Sci. 2008; 121:1999–2007. [DOI] [PubMed] [Google Scholar]
- 20. Jackson D.A., Hassan A.B., Errington R.J., Cook P.R.. Visualization of focal sites of transcription within human nuclei. EMBO J. 1993; 12:1059–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pombo A., Jackson D.A., Hollinshead M., Wang Z., Roeder R.G., Cook P.R.. Regional specialization in human nuclei: visualization of discrete sites of transcription by RNA polymerase III. EMBO J. 1999; 18:2241–2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cardozo Gizzi A.M., Cattoni D.I., Fiche J.-B., Espinola S.M., Gurgo J., Messina O., Houbron C., Ogiyama Y., Papadopoulos G.L., Cavalli G.et al.. Microscopy-Based chromosome conformation capture enables simultaneous visualization of genome organization and transcription in intact organisms. Mol. Cell. 2019; 74:212–222. [DOI] [PubMed] [Google Scholar]
- 23. Beliveau B.J., Boettiger A.N., Avendano M.S., Jungmann R., McCole R.B., Joyce E.F., Kim-Kiselak C., Bantignies F., Fonseka C.Y., Erceg J.et al.. Single-molecule super-resolution imaging of chromosomes and in situ haplotype visualization using Oligopaint FISH probes. Nat. Commun. 2015; 6:7147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Szabo Q., Jost D., Chang J.-M., Cattoni D.I., Papadopoulos G.L., Bonev B., Sexton T., Gurgo J., Jacquier C., Nollmann M.et al.. TADs are 3D structural units of higher-order chromosome organization in Drosophila. Sci. Adv. 2018; 4:eaar8082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cho W.-K., Jayanth N., English B.P., Inoue T., Andrews J.O., Conway W., Grimm J.B., Spille J.-H., Lavis L.D., Lionnet T.et al.. RNA polymerase II cluster dynamics predict mRNA output in living cells. eLife. 2016; 5:e13617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cho W.-K., Spille J.-H., Hecht M., Lee C., Li C., Grube V., Cisse I.I.. Mediator and RNA polymerase II clusters associate in transcription-dependent condensates. Science. 2018; 361:412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Neguembor M.V., Sebastian-Perez R., Aulicino F., Gomez-Garcia P.A., Cosma M.P., Lakadamyali M.. (Po)STAC (Polycistronic SunTAg modified CRISPR) enables live-cell and fixed-cell super-resolution imaging of multiple genes. Nucleic Acids Res. 2018; 46:e30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schnitzbauer J., Strauss M.T., Schlichthaerle T., Schueder F., Jungmann R.. Super-resolution microscopy with DNA-PAINT. Nat. Protoc. 2017; 12:1198. [DOI] [PubMed] [Google Scholar]
- 29. Tanenbaum M.E., Gilbert L.A., Qi L.S., Weissman J.S., Vale R.D.. A protein-tagging system for signal amplification in gene expression and fluorescence imaging. Cell. 2014; 159:635–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bates M., Huang B., Dempsey G.T., Zhuang X.. Multicolor super-resolution imaging with photo-switchable fluorescent probes. Science. 2007; 317:1749–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chen B., Gilbert L.A., Cimini B.A., Schnitzbauer J., Zhang W., Li G.W., Park J., Blackburn E.H., Weissman J.S., Qi L.S.et al.. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell. 2013; 155:1479–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tokunaga M., Imamoto N., Sakata-Sogawa K.. Highly inclined thin illumination enables clear single-molecule imaging in cells. Nat. Methods. 2008; 5:159. [DOI] [PubMed] [Google Scholar]
- 33. Andronov L., Orlov I., Lutz Y., Vonesch J.L., Klaholz B.P.. ClusterViSu, a method for clustering of protein complexes by Voronoi tessellation in super-resolution microscopy. Sci. Rep. 2016; 6:24084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Martin L., Vicario C., Castells-García Á., Lakadamyali M., Neguembor M.V., Cosma M.P.. A protocol to quantify chromatin compaction with confocal and super-resolution microscopy in cultured cells. STAR Protoc. 2021; 2:100865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Neguembor M.V., Martin L., Castells-García Á., Gómez-García P.A., Vicario C., Carnevali D., AlHaj Abed J., Granados A., Sebastian-Perez R., Sottile F.et al.. Transcription-mediated supercoiling regulates genome folding and loop formation. Mol. Cell. 2021; 81:3065–3081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Huang B., Wang W., Bates M., Zhuang X.. Three-dimensional super-resolution imaging by stochastic optical reconstruction microscopy. Science. 2008; 319:810–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jao C.Y., Salic A.. Exploring RNA transcription and turnover in vivo by using click chemistry. PNAS. 2008; 105:15779–15784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Perry R.P., Kelley D.E.. Inhibition of RNA synthesis by actinomycin D: characteristic dose-response of different RNA species. J. Cell. Physiol. 1970; 76:127–139. [DOI] [PubMed] [Google Scholar]
- 39. Dundr M., Hoffmann-Rohrer U., Hu Q., Grummt I., Rothblum L.I., Phair R.D., Misteli T.. A kinetic framework for a mammalian RNA polymerase in vivo. Science. 2002; 298:1623–1626. [DOI] [PubMed] [Google Scholar]
- 40. Darzacq X., Shav-Tal Y., de Turris V., Brody Y., Shenoy S.M., Phair R.D., Singer R.H.. In vivo dynamics of RNA polymerase II transcription. Nat. Struct. Mol. Biol. 2007; 14:796–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Levet F., Hosy E., Kechkar A., Butler C., Beghin A., Choquet D., Sibarita J.B.. SR-Tesseler: a method to segment and quantify localization-based super-resolution microscopy data. Nat. Methods. 2015; 12:1065–1071. [DOI] [PubMed] [Google Scholar]
- 42. Mejias-Luque R., Peiro S., Vincent A., Van Seuningen I., de Bolos C.. IL-6 induces MUC4 expression through gp130/STAT3 pathway in gastric cancer cell lines. Biochim. Biophys. Acta. 2008; 1783:1728–1736. [DOI] [PubMed] [Google Scholar]
- 43. Eskiw C.H., Fraser P.. Ultrastructural study of transcription factories in mouse erythroblasts. J. Cell Sci. 2011; 124:3676–3683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cisse I.I., Izeddin I., Causse S.Z., Boudarene L., Senecal A., Muresan L., Dugast-Darzacq C., Hajj B., Dahan M., Darzacq X.. Real-time dynamics of RNA polymerase II clustering in live human cells. Science. 2013; 341:664–667. [DOI] [PubMed] [Google Scholar]
- 45. Martin S., Pombo A.. Transcription factories: quantitative studies of nanostructures in the mammalian nucleus. Chromosome Res. 2003; 11:461–470. [DOI] [PubMed] [Google Scholar]
- 46. Zhao Z.W., Roy R., Gebhardt J.C.M., Suter D.M., Chapman A.R., Xie X.S.. Spatial organization of RNA polymerase II inside a mammalian cell nucleus revealed by reflected light-sheet superresolution microscopy. PNAS. 2014; 111:681–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Shah S., Takei Y., Zhou W., Lubeck E., Yun J., Eng C.-H.L., Koulena N., Cronin C., Karp C., Liaw E.J.et al.. Dynamics and spatial genomics of the nascent transcriptome by intron seqFISH. Cell. 2018; 174:363–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kimura H., Sugaya K., Cook P.R.. The transcription cycle of RNA polymerase II in living cells. J. Cell Biol. 2002; 159:777–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Beliveau B.J., Boettiger A.N., Nir G., Bintu B., Yin P., Zhuang X., Wu C.T.. In situ super-resolution imaging of genomic DNA with OligoSTORM and OligoDNA-PAINT. Methods Mol. Biol. 2017; 1663:231–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Nir G., Farabella I., Pérez Estrada C., Ebeling C.G., Beliveau B.J., Sasaki H.M., Lee S.D., Nguyen S.C., McCole R.B., Chattoraj S.et al.. Walking along chromosomes with super-resolution imaging, contact maps, and integrative modeling. PLos Genet. 2018; 14:e1007872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Eng C.-H.L., Lawson M., Zhu Q., Dries R., Koulena N., Takei Y., Yun J., Cronin C., Karp C., Yuan G.-C.et al.. Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH+. Nature. 2019; 568:235–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Custom-made scripts and example datasets are publicly available at Github (https://github.com/CosmaLab/NND_Analysis).