


Abstract
Histone variants have been implicated in regulating chromatin dynamics and genome functions. Previously, we have shown that histone variant H3.3 actively marks enhancers and cooperates with H2A.Z at promoters to prime the genes into a poised state in mouse embryonic stem cells (mESCs). However, how these two important histone variants collaboratively function in this process still remains elusive. In this study, we found that depletion of different components of HIRA complex, a specific chaperone of H3.3, results in significant decreases of H2A.Z enrichment at genome scale. In addition, CUT&Tag data revealed a genomic colocalization between HIRA complex and SRCAP complex. In vivo and in vitro biochemical assays verified that HIRA complex could interact with SRCAP complex through the Hira subunit. Furthermore, our chromatin accessibility and transcription analyses demonstrated that HIRA complex contributed to preset a defined chromatin feature around TSS region for poising gene transcription. In summary, our results unveiled that while regulating the H3.3 incorporation in the regulatory regions, HIRA complex also collaborates with SRCAP to deposit H2A.Z onto the promoters, which cooperatively determines the transcriptional potential of the poised genes in mESCs.
INTRODUCTION
In eukaryotic cells, genomic DNA is hierarchically organized into chromatin with multi-level structures from the nucleosome to higher order structures (1). The basic structural and functional unit of chromatin is the nucleosome (2). Nucleosome comprises 147 bp of DNA fragment wrapping around a histone octamer, consisting of a H3/H4 heterotetramer and two histone H2A/H2B heterodimers, about 1.7 superhelical turns in a left-handed manner (3). In addition to canonical histones, a number of histone variants have been identified to provide chromatin with diverse characteristics in different species (4). Different histone chaperones have been found to specifically deposit and replace canonical histones with their variants into distinct genomic regions for fulfilling various biological functions (5–7). These dynamic histone variant substitutions could modulate the structure and dynamics of the nucleosome and chromatin fibers, and subsequently set up distinct chromatin states to cater for various of cellular processes throughout the cell cycle (8).
Over the past two decades, many studies have emphasized the biological functions of different histone variants in chromatin dynamics and transcriptional regulation. Unlike canonical histones, histone variants, such as H3.3 and H2A.Z, are expressed continuously throughout the cell cycle and assembled into chromatin in a DNA replication-independent manner (9). This difference empowers histone variants with particularly biological functions in DNA-related cellular processes. For instance, both H3.3 and H2A.Z have been shown to play a crucial role in the regulation of chromatin dynamics and gene transcription in mammalian cells (10–14).
H3.3 differs from H3.1 with only four or five amino acid residues but exhibits unique functions in many cellular processes (15). While H3.3 has been identified mainly as an active marker in gene transcription, it was also shown to regulate heterochromatin formation in some vague manners (15–17). Histone regulator A (HIRA) complex and death domain-associated protein/α-thalassemia/mental retardation syndrome X-linked (DAXX/ATRX) complex are two major chaperones that specifically recognize and deposit H3.3 into distinct genomic regions (15,18,19). HIRA complex mainly loads H3.3 to regulatory elements in euchromatin (15), whereas DAXX/ATRX complex deposits H3.3 to retrotransposons in heterochromatin (17,20,21). HIRA complex, which is highly conserved among species, is composed of three core subunits: Hira, ubinuclein 1 (Ubn1), and calcineurin-binding protein 1 (Cabin1), collaborating with anti-silencing function 1A/B (Asf1a/b) (22–25). Our recent study confirmed that Ubn2, a paralog and alternative of Ubn1 in HIRA complex, could also deposit H3.3 on regulatory elements in mESCs (26). However, the difference of biological functions between Ubn1 and Ubn2 is not yet clear (26,27). It's also verified that H3.3 became disoriented in Hira-, Ubn1- or Ubn2-deleted mESCs (26). Accompanied with HIRA complex, H3.3 plays important roles in many embryonic development stages including gametogenesis, fertilization, early embryonic development, and tissue formation (19).
H2A.Z is another one of the most widespread histone variants and closely related to genome integrity, transcriptional regulation and DNA replication origin selection and activation (28–32). It has about 60% sequence similarity with canonical histone H2A and displays extensive functionality. It has been widely recognized that the occupancy and dynamic change of H2A.Z at transcribed regions and DNA damage sites are mainly mediated by two adenosine triphosphate (ATP)-dependent chromatin remodelers, Snf2-related CREBBP activator protein (SRCAP) complex and INO80 complex in mammalian cells (33–35). SRCAP complex is a member of INO80 remodeler subfamily and involved in a variety of epigenetic events including chromatin remodeling and transcription regulation (36,37). SRCAP complex binds the nucleosome via YL1 subunit and unwraps the outer ring of DNA to replace H2A with H2A.Z (38). INO80 complex does not only evict H2A.Z from the nucleosome (34,39,40), but also slides the nucleosome on DNA to involve in the DNA repair, DNA replication and transcription (41–44).
H2A.Z exhibits both repressive and active functions in gene activation (10,11,28,29,45,46). Many biophysical and biochemical assays have indicated that H2A.Z can enhance the stability of the nucleosome and facilitate compaction of high-order chromatin structure (16,30). In addition, our previous in vitro transcription assay demonstrated that the incorporation of H2A.Z could inhibit transcriptional activity at the chromatin level (16). These data agree well with the repressive role of H2A.Z in gene transcription. Apparently, contradictory and complicated roles of H2A.Z might be generated by the collaboration with other histone variants or regulators. Previous studies have found that H2A.Z co-localized with H3.3 at the regulatory regions of highly transcribed genes, especially active promoters (13,16). The nucleosome arrays containing both these two variants impaired the compaction of higher-ordered chromatin structure and regulated the transcriptional activity at chromatin template in vitro (16). Surprisingly, using an all-trans retinoic acid (tRA)-induced transcription model system, we demonstrated that H2A.Z performed comprehensive functions in transcriptional regulation in mESCs (31). H3.3 actively marks enhancers and collaborates with H2A.Z at promoters to prime genes into a poised state for the quick induction of gene expression. Therefore, we hypothesize that an unknown crosstalk between HIRA-H3.3 and SRCAP-H2A.Z pathways might exist in vivo for maintaining gene transcriptional potentials in mESCs. To test this hypothesis, we initially investigated the dynamic oscillation of H2A.Z deposition after the deletion/knocking-down of components of HIRA complex at genome scale using chromatin immunoprecipitation sequencing (ChIP-seq) with exogenous chromatin from drosophila as spike-in control. Interestingly, our results showed that depletion of HIRA complex significantly impaired H2A.Z deposition on genome wide in H3.3-dependent or H3.3-independent manner. We found a genomic colocalization of HIRA complex and SRCAP complex through CUT&Tag assay with also spike-in control as described (47–49). Our biochemical interaction assays also demonstrated that HIRA complex interacts with SRCAP complex through the Hira subunit. In contrast, knockdown of H2A.Z or SRCAP did not obviously affect the H3.3 incorporation, supporting the upstream function of HIRA complex in this regulatory process. In addition, through partial chromatin digestion using our GST (glutathione S-transferase) tagged micrococcal nuclease followed by deep sequencing (GST-MNase-seq) and ATAC-seq assays (50,51), we showed that H2A.Z incorporation mediated by HIRA complex coupling with SRACP pathway contributed to the maintenance of high-order chromatin compaction at gene promoters. Furthermore, functional enrichment analysis found that the genes that could not be activated under tRA induction in HIRA complex deleted cell lines were significantly enriched in transcriptional regulation during neuronal development, which was largely supported by the phenotypic and molecular verifications. As expected, activation of these genes and the induction of neural progenitor cells (NPC) from mESC both failed due to the depletion of HIRA. Together, in this study, we provided a molecular mechanism of the crosstalk between the HIRA-H3.3 and SRCAP-H2A.Z pathways, which is important to maintain the poised states of developmental genes in mESCs.
MATERIALS AND METHODS
This study was approved by the Animal Ethics Committee of Institute of Biophysics, Chinese Academy of Sciences, Beijing, China. The study did not involve experiments on live participants, embryos and tissues. No ethic permits were therefore needed for this report, which complied with all the relevant regulations.
Cell culture and maintenance
Mouse embryonic stem cell, R1 cell line (ATCC® SCRC-1011™) was obtained from American Type Culture Collection (ATCC). This cell line was only used for biochemical, cellular and genomic analyses. R1 cells were cultured in the medium with 80% DMEM, 15% FBS, 1% l-glutamine, 1% nucleosides, 1% nonessential amino acids, 1% 2-mercaptoethanol, 1% Pen/Strep, and 1000 U/ml leukemia inhibitory factor (LIF) in standard incubator with 5% CO2 at 37°C.For immunofluorescence staining of neural progenitor cells, the mESCs were plated onto 0.1% gelatin-coated plates at a density of 2–4 × 104/cm2 in N2-B27 medium. N2-B27 medium is a 1:1 mixture of DMEM/F12 (Gibco, 11320–033) supplemented with N2 (Gibco, 17502-048) and Neurobasal medium (Gibco, 21103–049) supplemented with B27 (Gibco, 17504-044), 25 μg/ml insulin, and 50 μg/ml BSA. The medium was renewed every day during 7 days of culture.
Genome editing of mESCs
The knockout and mutant cell lines were generated by CRIPSR-Cas9 design tool as described previously (26). In R1 WT and these defective cell lines, the sequence of 6 × HA was added into the N terminus of Srcap gene by CRIPSR-Cas9 design tool. The donor plasmid containing the homologous arms for recombination was constructed as described (52). The homologous arm containing the PAM sequence of SpCas9 target site was mutated to disrupt the PAM sequence. The donor plasmid and the pX260-Cas9 plasmid were co-transfected into mESCs using Lipofectamine 3000 (Invitrogen, L3000015) according to the manufacturer's instructions. Next, cells were seeded into 10-cm dish at low density, and 12 h later 200 μg/ml hygromycin (InvivoGen) was added to select clones for 1–2 weeks. Clones were then picked out and screened by PCR and agarose gel electrophoresis, then validated by western blot and Sanger sequencing. DNA sequences of the targets used for genome editing were provided in Supplementary Table S1.
siRNA/shRNA knockdown
For knocking down specific genes, cells were grown in 6 cm dish and were then transfected with 20 pmol targeting siRNAs (siUbn1, siH2A.Z and siNT) three times per 24 h. These siRNAs were ordered from Gene Pharma (CHN). After 72 h transfection, total RNA or whole cell extract was prepared for RT-qPCR, RNA-seq and western blot analyses. The pSUPER-shSrcap was ordered from Gene Pharma (CHN). The retroviruses were generated in HEK293T cells. Per 10 mL viral supernatant was enriched into 1 ml concentrate. In ChIP-seq and immunofluorescence assays, mESCs were infected with the enriched retrovirus (1 ml/10 cm culture plate) for knocking down Srcap. The sequences of siRNA and shRNAs were provided in Supplementary Table S1.
ChIP-seq and ChIP-qPCR
For ChIP analyses, as decribed previously (26), R1 WT, Hira-KO, Ubn2-KO/Ubn1-KD, Ubn1/2-DM and H3.3-KO mESCs were crosslinked with 1% formaldehyde in DMEM medium for 10 min at room temperature (RT), then terminated with 125mM glycine for 5min at RT. Crosslinked cells were lysed in Buffer I (50 mM HEPES pH 7.5, 140 mM NaCl, 1 mM EDTA, 10% glycerol, 0.5% Nonidet P-40, 0.25% Triton X-100, 1 × Protease inhibitor cocktail (Roche, 4693132001) for 10 min at 4°C, then incubated in Buffer II (10 mM Tris–HCl pH 7.5, 200 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 1 × Protease inhibitor cocktail) for 10 min at RT. Then the nuclei were re-suspended in Buffer III (10 mM Tris–HCl pH 7.5, 1 mM EDTA, 0.5 mM EGTA, 0.5% N-lauroyl-sarcosine, 1 × Protease inhibitor cocktail) and were sonicated using a Bioruptor (Diagenode, UCD-200). Following crosslink reversal, the main fragment size should be about 200–500 bp checked by agarose electrophoresis. The same procedures were performed for preparing of spike-in control sample, drosophila S2 chromatin. In immunoprecipitation procedure, 10 μl Dyna protein A beads (Invitrogen, 10002D) slurry and 10 μl Dyna protein G beads (Invitrogen, 10004D) slurry were mixed and then washed three times by RIPA-150 buffer (50 mM Tris–HCl pH 7.5, 150 mM NaCl, 1 mM EDTA, 0.5% Triton X-100, 1 × Protease inhibitor cocktail). Sonicated chromatin containing 5% S2 chromatin as spike-in control, 3 μg antibody of interest and 0.5 μg spike-in antibody were incubated with beads in RIPA-150 buffer at 4°C for overnight. The mixture was respectively washed twice with RIPA-150, RIPA-500 and RIPA-LiCl at 4°C for 10 min. ChIP DNA fragments were eluted in Elution buffer (10 mM Tris–HCl, 5 mM EDTA, 300 mM NaCl, 0.5% SDS) and extracted using a standard phenol-chloroform extraction procedure. For ChIP sequencing, libraries were prepared according to NEBNext Ultra II DNA Library Prep Kit for Illumina (E7645L) and were sequenced using a HiSeq2000 system at Berry Genomics. The 200–750 bp length of DNA library was collected by running a 1.2% agarose gel. For high-throughput sequencing, purified DNA libraries were performed quality test before sequencing. All ChIP-seq data were provided from two independent replicates. ChIP-qPCR was performed with SYBR dye (Roche) on an Applied Biosystems StepOnePlus system. Antibodies used for ChIP were as follows: H2A.Z antibody (Abcam, ab4174), SRCAP antibody (Kerafast ESL103) and spike-in antibody (Active Motif, 61686). The primers used for ChIP-qPCR analysis were provided in Supplementary Table S3.
CUT&Tag
As described previously (47,48), R1 cells and were harvested and centrifuged for 3 min at 300 × g at RT. Cells were washed twice with 1 mL Wash-150 buffer (20 mM HEPES pH 7.5, 150 mM NaCl, 0.5 mM Spermidine, 1 × Protease inhibitor cocktail, 0.1% BSA) by gentle pipetting and counted. Drosophila S2 cells were used as spike-in control and were treated with the same process of R1 cells. Concanavalin A coated magnetic beads (BioMagPlus, BP531) were washed once with Wash-150 buffer and Binding buffer (20 mM HEPES pH 7.9, 10 mM KCl, 1 mM MnCl2, 1 mM CaCl2) and were then resuspended in original volume of Binding Buffer. 50 000 R1 cells, 5000 S2 cells and 10 μl prepared beads were added per sample and incubated at RT for 10 min and collected by the magnet stand. The cells-beads mixture, 2 μl antibody of interest and 0.5 μl spike-in antibody were resuspended in 100 μl Antibody Buffer (mix 8 μl 0.5 M EDTA with 2 ml Dig-wash-150 buffer (0.05% digitonin in Wash-150 buffer) and were rotated at RT for 2 h. The mixture was collected by magnet stand and gently washed three times with Dig-wash-150 buffer at RT for 5 min and was then incubated in Dig-wash-300 buffer (0.01% digitonin; 20 mM HEPES pH 7.5; 300 mM NaCl; 0.5 mM Spermidine; 1 × Protease inhibitor cocktail; 0.1% BSA) containing 0.4 μM Protein-A coupled Tn5 (PAT). The recombinant PAT was prepared as described (47). The mixture was incubated at RT for 1 h and washed with 500 μl Dig-300-wash buffer three times for 5 min. The PAT was activated in 200 μl Reaction Buffer (5 mM MgCl2 in Dig-wash-300 buffer). The reaction was gently mixed and incubated at 25°C for 1 hr. To stop the reaction, 40 μl 6 × Stopping buffer (120 mM EDTA, 0.3% SDS, 0.2 mg/ml proteinase K) was added followed by incubation at RT for 15 min and then at 55 °C for 1 h. The fragmented DNA in mixture was extracted using a standard phenol-chloroform extraction procedure. To amplify libraries, 2 μl DNA was mixed with 21 μl ddH2O, 25 μl of NEBNext HiFi 2 × PCR Master mix (NEB, E7649), 1 μl each of uniquely barcoded i5 and i7 primer as described in NEBNext Multiplex Oligos for Illumina (Dual Index Primers Set 1). The amplified reaction was performed with the following cycling conditions: 72°C for 5 min; 98°C for 30 s; 15 cycles of 98°C for 10 s and 65°C for 30 s; final extension at 72°C for 1 min and hold at 12°C. The 200–750 bp length of DNA library was collected by running a 1.2% agarose gel. For high-throughput sequencing, purified DNA libraries were performed quality test before sequencing. All CUT&Tag data were provided from two independent replicates. Antibodies used for CUT&Tag were as follows: SRCAP antibody (Kerafast, ESL103), HIRA antibody (Millipore, WC119) and spike-in antibody (Active Motif, 61686).
RT-qPCR and RNA-seq
RNA was extracted using a standard chloroform extraction procedure. For RT-qPCR, mRNA was reverse-transcribed to cDNA using a Perfect Real-Time kit (TaKaRa, RR047A). QPCR was performed by SYBR (Roche, 04707516001) using Applied Biosystems StepOne Plus system. For RNA-seq, libraries were prepared according to the Illumina TruSeq protocol and sequenced using a HiSeq2000 system at Berry Genomics. The primers used for RT-qPCR analysis were provided in Supplementary Table S2.
Co-immunoprecipitation
As described previously (53), R1 cells were cultured to 80%–90% coverage per 15 cm plate. Five plates (about 8–9 × 108 cells) were washed with PBS and collected into 10 mL Buffer 1 (10 mM Tris–HCl pH 7.5, 150 mM NaCl, 0.1% Nonidet P-40, 2.5 mM EDTA, 1 × protease inhibitors cocktails) with 10 min incubation on ice. Then cells were centrifuged (300 g, 4°C, 4 min) to collect nucleus pellet. The nuclei were resuspended in 3mL of Buffer 2 (50 mM Tris–HCl pH 7.5, 300 mM NaCl, 0.5% Nonidet P-40, 2.5 mM MgCl2, 1 × protease inhibitors cocktails) containing 3 μl Benzonase (Sigma, E8263) following by incubation on ice for 30 min. Supernatant was collected by centrifugation (10 min, 13 000 g, 4°C) and diluted with 3 mL of Buffer 3 (50 mM Tris pH 7.5, 100 mM NaCl, 0.5% NP40, 15 mM EDTA, 1 × protease inhibitors cocktails). This lysate was used for immunoprecipitation, severally for input and target proteins. 1 ml input was incubated with 10 μg specific antibody and 100 μl precleared Protein A or Protein G agarose beads (Thermo Scientific, 20333 and 20398) at 4°C overnight. After incubation, the beads were washed three times with Buffer 3 and boiled 10 min in loading buffer. The samples were analyzed by western blot.
Flag-Hira was overexpressed in Ubn2-KO/Ubn1-KD cells. Flag-Ubn1 and Flag-Ubn2 were separately overexpressed in Hira-KO cells. Each plasmid was transfected into cells and maintained 48–72 h. Then the cells were collected and performed with procedure of Co-IP and analysis of western blot as described above. Antibodies and beads used for immunoprecipitation and western blot were as follows: H2A.Z antibody (1:5,000, Active Motif, 39113); Ubn1 antibody (1:3,000, Abcam, ab84953); H2B antibody (1:10,000, Abcam, ab1790); Hira antibody (1:500, Millipore, WC119); HA antibody (1:2,000, CWBIO, CW01245); Ubn1 antibody (1:3,000, Santacruz, SC-515340); Ubn1 antibody (in-house antibody); Ubn2 antibody (1:3,000, in-house antibody); Ino80 antibody (1:2,000, Proteintech, 18810-1-AP); Daxx antibody (1:3,000, Santa cruz, sc-7152); mouse IgG antibody (Santa cruz, sc-2025); Srcap antibody (1:750, Kerafast, ESL103); Flag-M2 (1:3,000, Sigma, F3165); anti-HA agarose beads (Sigma, 104M4753V); Flag antibody (1:5,000, rabbit, huaxingbio, HX1819); GAPDH antibody (1:10,000, Abclonal, AC033). The in-house Ubn1 and Ubn2 antibodies were used for immunoprecipitation.
Protein purification and GST pull-down assay
Human GST fusion proteins were cloned in pGEX-6p-1 vector. Human Asf1a 1–157 truncation was cloned in pRSF-Duet vector. These proteins were expressed in BL21-DE3 cells at 16°C for 16 h. After standard GST- or His-tagged affinity purifications, proteins were dialyzed into Binding Buffer (20 mM HEPES, pH 8.0, 300 mM NaCl, 2 mM DTT). About the purification of SRCAP complex, the ORFs of human SRCAP, DMAP1, YL1, RUVBL1, RUVBL2, ACTL6A, ARP6, ACTIN, GAS41 and ZNHIT1 were subcloned into six modified pMlink vectors. The six plasmids were co-transfected into Expi293 cells using polyethylenimine. Cells were harvested after culturing at 37°C for 3 days, and lysed in lysis buffer (50 mM HEPES pH 8.0, 300 mM NaCl, 0.2% CHAPS, 2 mM MgCl2, 0.5 mM EDTA,1 μg/ml aprotinin, 1 μg/ml pepstatin, 1 μg/ml leupeptin, 1 mM PMSF, and 2 mM DTT) at 4°C for 30 min. The lysate was clarified by centrifugation at 16,000 rpm for 30 min and the supernatant was incubated with IgG resin for 4 h. Then the resin was washed with the buffer containing 20 mM HEPES pH 8.0, 300 mM NaCl, 2 mM MgCl2, 0.5 mM EDTA and 2 mM DTT. The proteins were digested at 4°C overnight and eluted using wash buffer and further loaded onto a Mono Q 5/50 GL column (GE Healthcare) to achieve highly pure SRCAP complex. The peak fractions were collected and concentrated to ∼3 mg/ml for in vitro assays.
For GST pull-down assays, GST fusion proteins were immobilized on Glutathione Sepharose 4 Fast Flow (GE Healthcare) resin, then an equal molar ratio of SRCAP complex was mixed and incubated with the HIRA-bound resins in Binding Buffer with 0.1% NP-40 at 4°C for 8 hr. An equal molar ratio of His-Asf1a 1–157 would be added in HIRA-bound resins for 2 h before mixing of SRCAP complex. The resins were washed five times with 1 ml Binding Buffer. The resins with bound proteins were boiled in 2×SDS Loading Buffer and separated on 6–15% SDS-PAGE gel for western blotting analyses.
Rescue assays for ChIP-seq and ChIP-qPCR of H2A.Z and SRCAP
Full-length Hira and its truncation mutants were exogenously expressed in Hira-KO cell line. After 48 h transfection with pCAG-Flag of target proteins or empty vector, cells underwent the resistance selection for a week. We check the protein level of exogenously overexpressed proteins by western blot before performing ChIP-seq and ChIP-qPCR assays. We performed anti-H2A.Z ChIP-seq with spike-in control in two parallel samples. ChIP-qPCR of H2A.Z, SRCAP and Flag antibodies were respectively performed three times. Antibodies used for ChIP were as follows: H2A.Z antibody (Abcam, ab4174), SRCAP antibody (Kerafast ESL103), anti-Flag M2 agarose beads (Sigma A2220) and spike-in antibody (Active Motif, 61686).
GST-MNase digestion sequencing and ATAC sequencing
The detail steps of this procedure were performed as described previously (51). In briefly, cells were counted and collected under the native conditions. We used GST-tagged MNase which has been shown to digest mononucleosomes with a better hierarchy of spatial discrimination and a more measurable enzymatic reaction rate than the commercial MNase. The native chromatin was digested by a tested time course (2, 4, 8, 16 and 32 min). The mononucleosomes from shorter time digestions, including 2- and 4-min time point, were mixed and prepared for high-throughput sequencing to evaluate the chromatin accessibilities aroundtranscriptional start sites. GST-tagged MNase enzyme was constructed, expressed and purified according to the method in previous study (50). The fully digested products could be collected and performed with procedure of mononucleosome immunoprecipitation and mass spectrometry analysis. ATAC-seq was performed on 1×105 mESCs per reaction, according to the standard procedures (54). All MNase-seq and ATAC-seq data were provided from two independent replicates.
Immunofluorescence
For immunofluorescence analysis of neural progenitor cell markers, mESCs were grown on glass coverslips coated by gelatin in N2-B27 medium. To prepare samples, cells were washed with PBS and then fixed with 4% paraformaldehyde for 15 min, washed three times with PBS, and permeabilized with 0.5% Triton X-100 in PBS for 15 min at RT. Cells were incubated with 3% BSA in PBS containing 0.1% Triton X-100 for 1 h at RT and then incubated with primary antibodies overnight at 4°C. After three washes with PBS containing 0.1% Triton X-100, the cells were incubated with fluorophore conjugated secondary antibodies for 1 hr at RT and stained with DAPI for 15 min. Fluorescent images were collected on an Olympus FV1200 microscope. Antibodies used for Immunofluorescence were as follows: Nanog (1:200, BETHYL, A300-397A); Tuj1 (1:200, Biolegend, 801201).
Sequencing data analysis
Quality of all ChIP-seq/ATAC/Cut&Tag with spike-in and GST-MNase-seq paired-end reads sequenced using Illumina platforms were summarized using FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Adaptor oligos reads with nucleotide quality of ‘Q30’ were trimmed using Cutadapt (v1.13) (parameters: -a AGATCGGAAGAGC -A AGATCGGAAGAGC -m 50 -n 2) (55). While clean tags of ChIP-seq/Cut&Tag with spike-in were mapped to both fly (dm6) and mouse (GRCm38) genomic assemblies using bowtie2 (version 2.2.5) with ‘–no-mixed –no-discordant’ (56), paired mates mapping to both organisms were removed. Clean tags of GST-MNase-seq were mapped to mouse (GRCm38) genomic assemblies using bowtie2 (version 2.2.5) with ‘–no-mixed –no-discordant’ (56). Only one alignment of each multi-mapped read pair was randomly retained.
To detect H2A.Z and H3.3 peaks in R1, biological replicates were merged and peaks were called using MACS (version 2.1.1, –keep-dup 1 -g mm P-value ≤ 1e–5 for H2A.Z and -p 1e-3 –llocal 100 000 –slocal 5000 for H3.3) against genomic input data (57). After that, peaks in blacklist, downloaded from the ENCODE project (58), were removed. The rest H2A.Z peaks were used for all later analysis and annotated against gene structures retrieved from GENCODE (vM10) using anotatePeaks.pl from Homer suite (v4.8) with default parameters (59). H3.3 ChIP-seq data of HIRA subunit deleted cells were downloaded and analyzed as described before (60).
For GST-MNase-seq data, PCR duplicates were marked and removed using MarkDuplicates from Picard (http://broadinstitute.github.io/picard/). all alignments retained were pileupped alongside with genome assembly for different samples in bedgraph format (61) and then normalized using TPM algorithm.
For CUT&Tag data, all mapped read pairs were retained, and peaks were called using MACS (version 2.1.1, –keep-dup all -g mm, P-value ≤ 5e–5 for SRCAP and P-value ≤ 5e–4 HIRA) and filtered as described for H2A.Z and H3.3. Meanwhile, raw counts alongside with genome assembly for different samples were pileupped in bedgraph format (61).
To perform ChIP, CUT&Tag and ATAC signal enrichment comparisons between different cell lines, the spike-in based normalization method was applied. Library sizes of all retained fragments from spike-in data in each sample were processed and used as size factors.
 |
A, the vector of library sizes of spike-in data of all samples. ai, the spike-in library size of sample i. ei, normalization factor for sample i.
Raw fragment counts in peak regions were computed using BEDTools (version 2.17.0) (61) and normalized using the above size factors, as the same with spike-in ChIP-seq, ATAC and CUT&Tag genomic profiles.
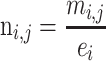 |
mi,j, raw counts for the peak/genomic bin j in sample i. ni,j, normalized density for the peak/genomic bin j in sample i.
RNA-seq data was first cleaned as described as described above. Then, clean data was mapped to mouse (GRCm38) genomic assemblies using HISAT2 (v2.1.0) with default parameters (62). FeatureCount of Subread package (v1.6.3) (63) were applied to yield the count matrix of genes using default parameters, against which normalized expression values and statistical significances were computed using DESeq2 (v1.24.0) with default parameters (64). For transcriptome analysis of tRA induction, the following algorithm was adopted to monitor the time-course transcriptional activation: log2 ((normi,j,k+ 1)/(norm0h,j,k+ 1)), normi,j,k, the normalized expression value of gene j at the time point i in the sample k. Genes with log2(fold change) ≥ 1 were defined as tRA inducible genes. Gene ontology (GO) enrichment analysis of genes were performed at The Gene Ontology project (http://geneontology.org/) using Fisher's exact test (FDR ≤ 0.05).
Data statistics and visualization
Wilcoxon signed-rank test was applied for H2A.Z enrichment and chromatin accessibility differential statistical analysis. All statistical and visualization analyses were achieved using R (https://www.r-project.org/). Heatmap of fragment densities around centers of various enriched regions were summarized using DeepTools (65) and visualized in R. Genome-wide tracks of all high-throughput data were visualized in the IGV (integrative genomics viewer) browser (66).
RESULTS
HIRA complex determines the distribution of H2A.Z on genome-wide
To investigate the functional link between histone variants H3.3 and H2A.Z in genomic events, we respectively constructed Hira knockout (Hira-KO), Ubn2 knockout/Ubn1 knockdown (Ubn2-KO/Ubn1-KD), Ubn1&2 FID/AAA double mutations (Ubn1/2-DM) and H3.3a&b knockout (H3,3-KO) cell lines from mESC R1 cells using CRISPR-Cas9 editor and RNA interference (Figure 1A and B). As shown in our previous study, Hira depletion resulted in decreased protein level of Ubn1/2 and vice versa (26). For this reason, we constructed Ubn1/2 double mutations (Ubn1/2 DM) in which the H3.3-binding sites Phe/Ile/Asp in Hpc2 related domain of both Ubn1 and Ubn2 were blocked (Figure 1A) (26,67). These mutations only impaired H3.3 deposition but not alter the integrity of HIRA complex. We performed anti-H2A.Z ChIP-seq using drosophila S2 chromatin and H2A.V antibody as spike-in control to standardize relative quantification. The spike-in antibody specifically recognized unique C-terminal of H2A.V and exhibited a negligible background in mammalian cells (Supplementary Figure S1A) (68,69). 46,830 H2A.Z peaks were identified in R1 wild type cells. Interestingly, our ChIP-seq analyses showed that both Hira-KO and Ubn2-KO/Ubn1-KD resulted in a significant reduction of H2A.Z enrichment at genome scale, while Ubn1/2-DM and H3.3-KO exhibited a modest effect (Figure 1C). To verify these results, we performed anti-H2A.Z ChIP coupling with quantitative PCR (ChIP-qPCR) at promoter of Nrg2 gene. In line with ChIP-seq analysis, a significant reduction of H2A.Z was triggered by HIRA complex deletion (Supplementary Figure S1B and C). Principal component analysis (PCA) was performed against all H2A.Z peaks to assess the overall impacts of HIRA complex and H3.3 variant on H2A.Z deposition. The PC1 factor explains 80% of the observed variation triggered by HIRA complex (Supplementary Figure S1D). Since the spike-in normalization method used, we selected reduced peaks with a decrease of 1.5-fold change in above defective cell lines and then respectively found 17 754, 15 487, 7501 and 9729 reduced peaks in Hira-KO, Ubn2-KO/Ubn1-KD, Ubn1/2-DM and H3.3-KO cell lines (Figure 1D). Apparently, 9,711 H2A.Z peaks were co-regulated by both Hira and Ubn1/2, which was consistent with the previous findings that Hira and Ubn1 showed strong co-localization at genome-wide (53). Relatively less H2A.Z reduced peaks were caused by Ubn1/2-DM and H3.3-KO, and the majority of H2A.Z reduced peaks in H3.3-KO cell (8144 of 9729) and Ubn1/2-DM cell (5426 of 7501) were highly overlapped with those in HIRA complex functionally defective cells (Figure 1D).
Figure 1.

Defect of HIRA complex results in H2A.Z decrease on genome wide. (A) Schematic diagram of genome editing in defective cell lines. Hira-, Ubn2- and H3.3-knockout cell lines and Ubn1/2 double mutants were generated using CRISPR-Cas9 system. For subsequent experiments, 6 × HA tag was added to the N-terminus of Srcap in R1 wild type and above defective cell lines. (B) Western blot respectively shows the proteins levels of Hira (a), Ubn1&Ubn2 (b), and H3.3 (c) in the defective cell lines. (C) Meta-analysis (left) and heatmap (right) show H2A.Z reads density around summit (upper) and TSS (below) in R1 WT, Hira-KO, Ubn2-KO/Ubn1-KD, Ubn1/2-DM and H3.3-KO cells within a ±3 kb window. (D) Venn diagram shows the overlap of H2A.Z down-regulated peaks between the above defective cell lines. (E) The reduced H2A.Z peaks in either Hira-KO or Ubn2-KO/Ubn1-KD cell lines are defined as HIRA complex regulated (HR) peaks (23 530 peaks). The 5426 HR peaks overlapping with Ubn1/2-DM cell line is defined as HIRA-regulated and H3.3-dependent (HIRA-H3.3D). The other 18 104 HR peaks are HIRA-regulated and H3.3-independent (HIRA-H3.3ID).
To further investigate the difference of regulatory roles of HIRA complex and H3.3 variant in H2A.Z deposition, we merged the reduced H2A.Z peaks in either Hira-KO or Ubn2-KO/Ubn1-KD cell lines and defined them as HIRA complex regulated (HR) peaks (23 530 peaks) (Figure 1D and E). The reduced peaks (7501 peaks) in Ubn1/2-DM cell line was due to the functional loss of HIRA complex in regulating H3.3 deposition, 5426 of them were overlapped with HR peaks and therefore defined as HIRA-regulated and H3.3-dependent (HIRA-H3.3D) peaks. These peaks were under the regulation of both HIRA complex and H3.3 variant. The rest 18,104 HR peaks regulated by HIRA complex in a H3.3-independent manner was defined as HIRA-regulated and H3.3-independent (HIRA-H3.3ID) peaks (Figure 1D and E). These analyses indicated that HIRA complex facilitated the deposition of H2A.Z in either H3.3-dependent or H3.3-independent manner. Furthermore, Cis-element annotation analysis indicated the functional distinction of those H2A.Z mediated by these two distinct pathways. The HIRA-H3.3ID peaks (18,104 peaks) displayed a significantly high percentage (56.4%) with their locations in intergenic region (Supplementary Figure S1E and F). Notably, the HIRA-H3.3D peaks (5,426 peaks) were mainly distributed in core promoters and coding regions (Supplementary Figure S1E), indicating that HIRA complex may regulate gene transcription through coordinating the depositions of histone variants H3.3 and H2A.Z at promoters. Beyond that, we also investigated whether H2A.Z has any similar effect on H3.3 deposition. Because of the lethal effect after H2A.Z knockout (70), we performed instantaneous RNA-interference of H2A.Z and stable knockdown of SRCAP with retrovirus system (Supplementary Figure S1G and H). Interestingly, siH2A.Z or shSrcap exhibited little influence on H3.3 deposition at the genome scale, including active and poised enhancers (Supplementary Figure S1I). Taken together, above results illustrated the dual functions of HIRA complex in histone variant deposition. As a chaperone of H3.3, HIRA complex does not only specifically deposited H3.3 at the regulatory regions (such as enhancers), but also facilitated the recruitment of H2A.Z at the regulatory regions (such as promoters).
HIRA complex facilitates the recruitment of H2A.Z through collaborating with SRCAP complex
As described above, depletion of HIRA complex could result in a marked reduction of H2A.Z enrichment at genome scale, which hints that there might exist some functionally biochemical link between these chaperones and histone variants themselves. It has been known that the exchange of H2A.Z into nucleosomes is mainly mediated by SRCAP complex (33,35). To verify that, we performed anti-H2A.Z ChIP-seq in shRNA-retrovirus mediated Srcap knockdown cells (shSrcap). As expected, depletion of Srcap almost caused a genome wide H2A.Z collapse (Figure 1C). Then, we wanted to detect whether HIRA colocalized with SRCAP at H2A.Z binding sites. We performed anti-Hira and anti-Srcap ChIP-seq but failed to obtain high quality sequencing data even using knock-in tagging instead. To solve this puzzle, we respectively performed anti-Srcap and anti-Hira CUT&Tag with spike-in control under native condition. Under this condition, the spike-in antibody produced negligible background in R1 cells (Supplementary Figure S2A). Compared with H3.3-KO, depletion of HIRA complex resulted in a significant reduction of Srcap on genome, especially at TSS regions (Figure 2A and Supplementary Figure S2B), These patterns are interestingly in line with H2A.Z ChIP-seq analysis. Anti-H2A.Z and anti-Srcap ChIP-qPCR at the representative loci also verified these observations (Figure 2B and C). Then, we compared the genomic coordinates of the 22,841 Hira peaks with 16,301 Srcap peaks, finding that nearly 50% of Srcap peaks were colocalized with Hira peaks (Figure 2D). Notably, at least 66% of those colocalized binding sites were also overlapped H2A.Z peaks (Figure 2D). Interestingly, 86% of these overlapped H2A.Z peaks were located in transcription related regions, especially promoters (Supplementary Figure S2C). The protein level of H2A.Z, Srcap and Ino80 in nuclear extract had no obvious alteration in above defective cells compared with R1 wild type cells (Supplementary Figure S2D). These results further suggested that HIRA complex and SRCAP complex could somehow cooperatively deposit H2A.Z at genome scale.
Figure 2.

HIRA collaborates with SRCAP to deposit H2A.Z on genome. (A) Meta-analysis (left) and heatmap (right) show Srcap reads density around summit (upper) and TSS (below) within a ±3 kb window. (B) Representative loci on Slc25a42 gene show the enrichment of H2A.Z and Srcap. (C) ChIP-qPCR validation of H2A.Z and Srcap enrichment at promoter of Slc35a42 gene. Error bars represent data from three independent experiments. (D) Venn diagram shows the overlap between H2A.Z, Hira and Srcap peaks. (E) Western blot shows the interaction among endogenous HA-Srcap, Hira and Ubn1 from nuclear lysates. Ino80, Daxx, H2A.Z and H3.3 are also detected. (F) Top panel showed schematic presentation of full length and truncation mutants of Hira subunit. Bottom panel showed the biochemical interactions between Hira truncations and SRCAP complex are analyzed by GST pull-down coupled with western blot analyses. (G) Heatmaps (upper) show H2A.Z reads density around H2A.Z peak summits (upper) in R1 WT, Hira-KO and Hira-truncation mutant expressing cells within a ±3 kb window. Representative loci (bottom) show the enrichment of H2A.Z. (H) ChIP-qPCR of H2A.Z, Srcap and Flag at representative genes Nrg2 and Spata4 in the rescued overexpressed cell lines. Error bars represent data from three independent experiments.
To investigate the relationship between HIRA/H3.3 and SRCAP/H2A.Z pathways, we tested the designated interactions among the chaperones of two histone variants by endogenous co-immunoprecipitation in R1 HA-tagged Srcap (HA-Srcap) cells. The results showed that subunits of HIRA complex, including Hira, Ubn1 and Ubn2, could co-precipitate with HA-Srcap, but not with Ino80 (Figure 2E and Supplementary Figure S2E). In addition, there was no obvious interaction between Srcap and Daxx in these co-immunoprecipitation assays (Figure 2E and Supplementary Figure S2E). Moreover, this interaction was not affected by the defect of H3.3 in Ubn1/2-DM cells and H3.3-KO cells (Supplementary Figure S2F and G). Since deleting one subunit of HIRA complex would reduce protein levels of others (26). To find out which subunit of HIRA complex was essential for this interaction, one subunit was exogenously overexpressed in other subunit defective cells and then co-immunoprecipitated with HA-Srcap. As shown in Supplementary Figure S2H, Hira subunit could co-immunoprecipitate with Srcap in Ubn2-KO/Ubn1-KD cells, but Ubn1 or Ubn2 could not co-immunoprecipitate with Srcap in Hira-KO cells. As shown in Figure 2F, Hira subunit was composed of several functional domains, including WD repeats domain, B domain and C-terminal domain (24). N-terminal half including WD repeats domain and B domain was known as the mainly functional domain of HIRA complex. We therefore investigated whether N-terminal half of Hira subunit play a role in the collaboration with SRCAP complex. For this, we purified GST-fusion full-length and truncation mutants of human Hira subunit from E. coli (Supplementary Figure S2I) and performed in vitro GST pull-down assays incubating with the purified human SRCAP complex. As shown in the Figure 2F, the full-length Hira obviously interacted with the SRCAP complex (as shown by the Srcap subunit and YL-1 subunit). In contrast, the truncation mutants of 1–420 amino acids (aa) and 1–481 aa could not interact with the SRCAP complex (Figure 2F). This result indicated that N-terminal half of Hira containing WD repeats domain and B domain was not sufficient for this interaction. Indeed, the defect of WD binding proteins Ubn1 and Ubn2, had no impact on the interaction (Supplementary Figure S2H). Here, we also wondered whether Asf1a, a B domain specific binding protein (24,25), was involved in regulating the interaction between the Hira subunit and SRCAP complex. To this end, we purified His-tag human Asf1a 1–157 aa (Supplementary Figure S2I), a functional truncation mutant could efficiently bind to the Hira B domain (25). We performed the GST pull-down assay by mixing GST tagged Hira truncation mutants, SRCAP complex and His-Asf1a 1–157 aa. Asf1a could bind with full-length Hira or 1–481 truncation mutant, whereas no obvious effect was observed on their interactions with SRCAP complex (Figure 2F). This result indicated that the binding of Asf1a to B domain is dispensable for the interaction of Hira subunit with SRCAP complex.
To further identify the functional domain of Hira subunit, we performed rescue experiments by exogenously overexpressing the Flag-tagged full-length Hira and its different truncation mutants (Flag-tagged 1–420 truncation, 1–476 truncation, and ΔB truncation mutants) in the R1 Hira-KO cell line. Then, we carried out anti-H2A.Z ChIP-seq with spike-in control in R1 WT, Hira-KO and the above cells (Figure 2G). Comparing with R1 WT, both the rescue of full-length Hira and the ΔB truncation mutant could efficiently restore the depositions of H2A.Z and SRCAP on genome wide, but the 1–420 truncation and 1–476 truncation mutants could not (Figure 2G). In line with our ChIP-seq analysis results, anti-H2A.Z and anti-SRCAP ChIP-qPCR analyses on the representative genic regions also displayed the similar rescue effects (Figure 2H). Moreover, anti-Flag ChIP-qPCR assays showed that the full-length Hira and B deletion (ΔB) truncation mutant could be efficiently recruited to the targeted genes, however, the 1–420 truncation and 1–476 truncation mutants could not (Figure 2H). Taken together, our results suggested that the N-terminal half is dispensable for the direct interaction between the Hira subunit and the SRCAP complex, and the C-terminal region of Hira subunit might be important for collaborating with SRCAP to accurately deposit H2A.Z to target sites. However, the precise SRCAP binding sites or regions of the Hira subunit still need to be further investigated in the future.
HIRA complex presets transcriptional potential through establishing poised chromatin state
Our previous in vitro studies have demonstrated that histone variants H3.3 and H2A.Z play critical roles in regulating high-order chromatin structure, that is, H2A.Z can facilitate chromatin compaction, whereas H3.3 impairs chromatin compaction (16). On the other hand, H2A.Z has been known as a functional partner of H3.3 and co-localize with H3.3 at transcribed regulatory regions (13,71). These results suggest that H3.3 and H2A.Z may function together with their chaperones to epigenetically regulate chromatin dynamics during transcriptional activation. To further investigate the crosstalk between HIRA-H3.3 pathway and H2A.Z deposition in chromatin structure, we performed time course digestion of reduced MNase (TC-rMNase) assay to analyze local chromatin accessibility (Supplementary Figure S3A and S3B) (51). As described in our previous study, TC-rMNase was an effective means to measure the local chromatin compaction using time-course digestion with mild enzyme activity of recombinant GST-MNase (51). Here we simplified that method to collect mononucleosomes only digested with 2 and 4 minutes, of which a theoretically and preferentially high ratio derives from active and bivalent regions. Then, DNA fragments were purified for high-throughput sequencing (Supplementary Figure S3C). With these analyses, we monitored the local chromatin structures around TSSs. It showed a significant increase of mononucleosome signal flanking TSS in siH2A.Z cells, which supports the role of H2A.Z in nucleosome stabilization and chromatin compaction (Figure 3A). Similar patterns were observed in both Hira-KO cells and Ubn2-KO/Ubn1-KD cells. Ubn1/2-DM and H3.3-KO exhibited a comparatively weaker effect but a visible enhanced signal at upstream of TSSs (Figure 3A). Interestingly, a more remarkable variation of mononucleosome signal was found at HIRA-H3.3D than HIRA-H3.3ID associated TSSs (Figure 3A). Moreover, the parallel analysis of ATAC-seq also displayed a similar pattern of chromatin accessibilities to that of GST-MNase-seq, particularly at HIRA-H3.3D and HIRA-H3.3ID associated TSSs (Supplementary Figure S3E). These observations suggested that HIRA complex functions together with SRCAP complex to coordinate the depositions of H3.3 and H2A.Z at regulatory regions (enhancers and promoters), which presets the poised chromatin states around TSS and facilitate gene transcriptional activation.
Figure 3.

HIRA-mediated H2A.Z recruitment is crucial for the local chromatin state of TSS and tRA inducible gene transcription. (A) Heatmap (below) and statistical graph (upper) of normalized mononucleosome density on TSSs within a ±0.5 kb window. Each unsaturated MNase signaling contains 2 min and 4 min digested production. The distributions of mononucleosomes are classified into three groups based on H2A.Z content, that is, HIRA-regulated and H3.3-independent (HIRA-H3.3ID) H2A.Z, HIRA-regulated and H3.3-dependent (HIRA-H3.3D) H2A.Z, and no H2A.Z binding (NO H2A.Z) in each cell line. (B) Boxplot shows the normalized H2A.Z density around TSS of tRA inducible gene (iTSS) and other TSS (TSS) in R1 WT cells. These two subsets are further classified into HIRA-H3.3ID H2A.Z, HIRA-H3.3D H2A.Z and NO H2A.Z. P, P-value by two-tailed Wilcoxon rank test. (C) Statistical graph of normalized gene expression in R1 WT cells. The inducible genes are classified into HIRA-H3.3ID iTSS, HIRA-H3.3D iTSS and NO H2A.Z iTSS. P, P-value by two-tailed Wilcoxon rank test. (D) The K-means clustering analysis of time course expression profiles across samples of tRA inducible genes with HIRA-H3.3ID and HIRA-H3.3D H2A.Z in their promoters.
Due to the importance of pluripotency of ESC, we preliminarily followed 70 genes from the pathway of ‘stem cell population maintenance’ (GO: 0019827) with interest, investigating how depletions of HIRA and H3.3 variant impacted H2A.Z depositions and transcription status of them. As shown in Supplementary Figure S4A and S4B, H2A.Z deposition was indeed corrupted after Hira/Ubn knock out, while gene transcriptions still remain rarely changed. These results indicated that the depletion of HIRA complex does not affect the pluripotency of mESC at the transcriptional level. To gain insight into the biological function of HIRA mediated H2A.Z deposition and chromatin state at TSSs in transcriptional regulation, we performed messenger RNA sequencing (RNA-seq) analysis to monitor transcriptome dynamics during the tRA time course treatment, which could induce gene transcriptional activation (16). Initially, we defined those TSSs with their gene expressions up-regulated with at least 2-fold change after tRA induction as inducible TSSs (iTSSs). Totally, we found 8,708 iTSS-related genes in R1 wild type cells. Then, we assessed the dynamics of both histone variants around those iTSSs. Notably, both HIRA-H3.3D and HIRA-H3.3ID associated iTSSs contain more H2A.Z and H3.3 than average, especially HIRA-H3.3D associated iTSSs (Figure 3B and Supplementary Figure S3D). These results suggested that the role of HIRA complex in regulating H2A.Z deposition might be functionally associated, but not necessary, with its activity in H3.3 incorporation. Previously, we found that H3.3 collaborates with H2A.Z at promoters to prime genes into a poised state for the quick induction of gene expression (16,31). Indeed, HIRA-H3.3D H2A.Z associated genes were activated more efficiently than others after tRA treatment (Figure 3C).
Next, we explored functions of the above-mentioned HIRA-facilitated H2A.Z deposition in gene transcription. We initially extracted 1,417 genes with HIRA-H3.3ID or HIRA-H3.3D H2A.Z peaks at their promoters from all tRA inducible genes in R1 wild type cells. Interestingly, a K-means clustering analysis shows that transcriptional activation of 63% (893 genes, cluster C1-C4) of the selected genes were impeded to different extent since the functional corruption of HIRA complex (Figure 3D). To verify the transcriptional activation related to above mentioned mechanism, we performed anti-H2A.Z and anti-Srcap ChIP-qPCR at promoters of HIRA-H3.3D representative inducible genes. The transcriptional activity was positively correlated with the enrichment of H3.3 and H2A.Z (Supplementary Figure S4C). To sum up, HIRA complex collaborates with SRCAP complex to deposit H2A.Z at the promoter, modulating chromatin stability around TSS to poise genes for quick response to tRA induction.
The poised chromatin state contributes to mESCs differentiation
To investigate the biological functions of those genes transcriptionally poised by HIRA-facilitated H3.3 and H2A.Z deposition, we performed gene ontology analysis of the genes in clusters C1-C4 (Figure 3D). Interestingly, these genes are mainly enriched in transcriptional regulation and tissue development especially nervous system development process (Fisher's exact test, FDR ≤ 0.05) (Figure 4A).
Figure 4.

The poised chromatin state is essential for NPC differentiation. (A) The Gene Ontology analysis of genes in cluster C1-C4. Real-time quantitative PCR (B) and immunofluorescent staining (C) of Nanog, Pou5f1, Tubb3 and Nes in 0, 4, 7-day NPCs which derived from R1 WT, Hira-KO, H3.3b-KO/H3.3a-KD and shSrcap cells respectively. *P-value < 0.033, P-value < 0.001, unpaired t test. Nanog and Tuj1 represent proteins encoded by Nanog and Tubb3. Standard deviation (SD) was calculated from three replicates.
We further performed in vitro neuronal differentiation and detected the expression levels of pluripotent genes (Nanog and Pou5f1) and differentiation genes (Tubb3 and Nes). As shown by real-time quantitative PCR and immunofluorescent staining during 7 days of differentiation, depletion of Hira or H3.3 resulted in the constantly high expression level of Nanog and Pou5f1, and the inefficient expression of Tubb3 and Nes (Figure 4B and C), which was consistent with our previous findings (26). As RNA interference was inefficient in long process of NPC differentiation, we used shSrcap instead of siH2A.Z to consistently reduce the expression of H2A.Z in next experiments. Srcap depletion had an evident impact on the expression of Nes, but a limited impact on Tubb3 gene. The occupancy of H3.3 and H2A.Z on Tubb3 suggested that the gene transcription was likely controlled by both H3.3 and H2A.Z (Supplementary Figure S4D). Both pluripotent genes Nanog and Pou5f1 were insensitive to Srcap depletion (Figure 4B and C). This phenotype suggested that HIRA and H3.3 plays critical roles in neural differentiation as previously described (26), which might not only depend on H2A.Z. Based on the above findings, we demonstrated that HIRA complex facilitates H3.3 and H2A.Z depositions for stabilizing the poised chromatin state at promoters, which presets the gene transcriptional potential for quick response to tRA induction during mESC neuronal differentiation.
DISCUSSION AND CONCLUSION
Transcriptional process is finely regulated by precise mechanism under a highly dynamic chromatin environment (72). Histone variants H3.3 and H2A.Z are enriched at the regulatory regions of transcribed genes and show a bimodal distribution around TSSs reflecting H3.3/H2A.Z-containing nucleosomes either side of the TSS (31,73). These hybrid nucleosomes containing double-variants mark promoters and characterize local chromatin state. In our previous study, we have illustrated the functional interaction between histone variants H3.3 and H2A.Z to prime gene transcription via regulating chromatin dynamics at regulatory regions such as enhancers and promoters. Our previous genomic analyses show that the enrichment of H3.3 on enhancers is relatively higher than those at promoters (31). A previous study has also shown that H3.3 co-localizes with HIRA complex at the poised enhancers, the regions enriched of H3K4me1 but lack of H3K27ac and p300 (53). In addition, another recent study has indicated that H2A.Z can co-localize with H3.3 and dynamically incorporate into enhancers during transcriptional activation (74). Most recently, Zhu and colleagues found that mutation of lysine 27 in histone variant H3.3 results in a dramatic decrease of H3K27ac at enhancers, indicating that enhancers are mainly occupied by H3.3 instead of canonical H3.1/H3.2 (75). In this study, among the reduced H2A.Z peaks in Hira-KO cells, a large number were closely associated with intergenic regions. This analysis implied that HIRA complex might affect gene transcription via recruiting double variants (H3.3 and H2A.Z) to adjacent or same nucleosomes flanking TSSs. Together, these studies suggest that the incorporation of H3.3 by HIRA might prime the chromatin state of enhancers into a ‘poised’ state via preventing chromatin compaction as proposed previously (9).
In addition, we and others have shown that H2A.Z are preferentially deposited at the promoter regions to poise genes via facilitating chromatin compaction and stabilizing the nucleosome structure (16). In this study, we further provide a molecular mechanism by which how HIRA complex, a H3.3 specific chaperone, not only is responsible for H3.3 deposition at enhancers, but also facilitates deposition of H2A.Z at promoters through collaborating with SRCAP. In this way, HIRA complex coordinates these two important variants H3.3 and H2A.Z at enhancers and promoters to preset a featured chromatin state for priming and maintaining gene transcriptional potential for quick response to stimuli (Figure 5).
Figure 5.

A model for the HIRA-faciliated the poised chromatin state during gene activation. When gene prepares to be activated, histone variants H3.3 and H2A.Z occupy the transcribed regions such as promoter and enhancer to set up a poised chromatin state (31). HIRA complex, as a chaperone of H3.3, also facilitates H2A.Z deposition through collaborating with SRCAP complex in this process. This molecular mechanism plays an important role in stabilizing the poised chromatin state at promoters to preset gene transcriptional potential during mESC differentiation.
Next, a key question is focused that how the histone variant H3.3 at enhancers communicates with the variant H2A.Z at the promoter regions at least several kilobases away to prime the genes as a poised state. In line with this question, H3.3 and H2A.Z have been found to co-localize with CTCF (CCCTC-binding factor)-binding sites along the genome and might be important for the bindings of CTCF and cohesin to mediate higher-ordered chromatin organization (76,77). Particularly, we and others have shown that H2A.Z could regulate the deposition of unwrapping nucleosomes and CTCF binding at promoters (60). In addition, HIRA complex was found to interact with CTCF and BRG1 (53). These studies suggest that HIRA-mediated histone variants H3.3 and H2A.Z deposition may mediate chromatin looping between enhancers and promoters as proposed previously (31). Therefore, it is great of interest to study in the future, how HIRA/H3.3 and SRCAP/H2A.Z pathway work together to preset a featured 3D chromatin organization between enhancers and promoters for priming genes into a poised state by 3D genomics techniques, such as Hi-ChIP and HiC or micro-C.
DATA AVAILABILITY
The raw sequence data reported in this paper have been deposited in the Genome Sequence Archive in National Genomics Data Center (78), Beijing Institute of Genomics (BIG), Chinese Academy of Sciences, under accession number PRJCA004722 that are publicly accessible at https://bigd.big.ac.cn/gsa.
✦ GSA: Genome Sequence Archive. Genomics, Proteomics & Bioinformatics 2017. [PMID = 28387199]
✦ Database Resources of the National Genomics Data Center in 2020. Nucleic Acids Res 2020, 48(D1): D24–D33. [ PMID = 31702008]
Supplementary Material
ACKNOWLEDGEMENTS
All fluorescence imaging data were collected at the Center for Bioimaging, Core Facility for Protein Sciences, Institute of Biophysics, Chinese Academy of Sciences. We thank Dr. Bing Zhu for providing PAT expressed plasmid. We thank Ms. Ting Yao (National Laboratory of Biomacromolecules, Institute of Biophysics, Chinese Academy of Sciences) for preparing materials and administrating work.
Contributor Information
Yang Yang, School of Life Sciences, Jilin University, 2699 Qianjin Street, Changchun 130012, China; National Laboratory of Biomacromolecules, CAS Center for Excellence in Biomacromolecules, Institute of Biophysics, Chinese Academy of Sciences, Beijing 100101, China.
Liwei Zhang, Key Laboratory of Infection and Immunity, Institute of Biophysics, Chinese Academy of Sciences, Beijing 100101, China.
Chaoyang Xiong, National Laboratory of Biomacromolecules, CAS Center for Excellence in Biomacromolecules, Institute of Biophysics, Chinese Academy of Sciences, Beijing 100101, China.
Jun Chen, National Laboratory of Biomacromolecules, CAS Center for Excellence in Biomacromolecules, Institute of Biophysics, Chinese Academy of Sciences, Beijing 100101, China; Department of Immunology, School of Basic Medical Sciences, Advanced Innovation Center for Human Brain Protection, Capital Medical University, Beijing 100069, China; University of Chinese Academy of Sciences, Beijing, China.
Li Wang, Fudan University Shanghai Cancer Center, Institutes of Biomedical Sciences, Shanghai Medical College of Fudan University, Shanghai 200032, China.
Zengqi Wen, National Laboratory of Biomacromolecules, CAS Center for Excellence in Biomacromolecules, Institute of Biophysics, Chinese Academy of Sciences, Beijing 100101, China; University of Chinese Academy of Sciences, Beijing, China.
Juan Yu, National Laboratory of Biomacromolecules, CAS Center for Excellence in Biomacromolecules, Institute of Biophysics, Chinese Academy of Sciences, Beijing 100101, China.
Ping Chen, Department of Immunology, School of Basic Medical Sciences, Advanced Innovation Center for Human Brain Protection, Capital Medical University, Beijing 100069, China.
Yanhui Xu, Fudan University Shanghai Cancer Center, Institutes of Biomedical Sciences, Shanghai Medical College of Fudan University, Shanghai 200032, China.
Jingji Jin, School of Life Sciences, Jilin University, 2699 Qianjin Street, Changchun 130012, China; National Engineering Laboratory for AIDS Vaccine, Jilin University, 2699 Qianjin Street, Changchun 130012, China; Key Laboratory for Molecular Enzymology and Engineering, The Ministry of Education, Jilin University, 2699 Qianjin Street, Changchun 130012, China.
Yong Cai, School of Life Sciences, Jilin University, 2699 Qianjin Street, Changchun 130012, China; National Engineering Laboratory for AIDS Vaccine, Jilin University, 2699 Qianjin Street, Changchun 130012, China; Key Laboratory for Molecular Enzymology and Engineering, The Ministry of Education, Jilin University, 2699 Qianjin Street, Changchun 130012, China.
Guohong Li, National Laboratory of Biomacromolecules, CAS Center for Excellence in Biomacromolecules, Institute of Biophysics, Chinese Academy of Sciences, Beijing 100101, China; University of Chinese Academy of Sciences, Beijing, China.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Natural Science Foundation of China [31521002, 31991161, 31630041 to G.L.]; Ministry of Science and Technology of China [2017YFA0504202]; CAS Key Research Program on Frontier Science [QYZDY-SSW-SMC020]; Beijing Municipal Science and Technology Commission [Z201100005320013]; Howard Hughes Medical Institute (HHMI) international research scholar grant [55008737 to G.L.]; National Natural Science Foundation of China [31801062 to L.Z.]; Ministry of Science and Technology of China [2018YFE0203302]; National Natural Science Foundation of China [31871290 to P.C.]. Funding for open access charge: bank transfer.
Conflict of interest statement. None declared.
REFERENCES
- 1. Song F., Chen P., Sun D., Wang M., Dong L., Liang D., Xu R.M., Zhu P., Li G.. Cryo-EM study of the chromatin fiber reveals a double helix twisted by tetranucleosomal units. Science. 2014; 344:376–380. [DOI] [PubMed] [Google Scholar]
- 2. Kornberg R.D. Chromatin structure: a repeating unit of histones and DNA. Science. 1974; 184:868–871. [DOI] [PubMed] [Google Scholar]
- 3. Luger K., Mader A.W., Richmond R.K., Sargent D.F., Richmond T.J.. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997; 389:251–260. [DOI] [PubMed] [Google Scholar]
- 4. Talbert P.B., Henikoff S.. Histone variants–ancient wrap artists of the epigenome. Nat. Rev. Mol. Cell Biol. 2010; 11:264–275. [DOI] [PubMed] [Google Scholar]
- 5. Chen P., Zhao J., Li G.. Histone variants in development and diseases. J Genet Genomics. 2013; 40:355–365. [DOI] [PubMed] [Google Scholar]
- 6. Xiong C., Wen Z., Li G.. Histone Variant H3.3: A versatile H3 variant in health and in disease. Sci. China Life Sci. 2016; 59:245–256. [DOI] [PubMed] [Google Scholar]
- 7. Li B., Carey M., Workman J.L.. The role of chromatin during transcription. Cell. 2007; 128:707–719. [DOI] [PubMed] [Google Scholar]
- 8. Venkatesh S., Workman J.L.. Histone exchange, chromatin structure and the regulation of transcription. Nat. Rev. Mol. Cell Biol. 2015; 16:178–189. [DOI] [PubMed] [Google Scholar]
- 9. Henikoff S., Smith M.M.. Histone variants and epigenetics. Cold Spring Harb. Perspect. Biol. 2015; 7:a019364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Santisteban M.S., Kalashnikova T., Smith M.M.. Histone H2A.Z regulates transcription and is partially redundant with nucleosome remodeling complexes. Cell. 2000; 103:411–422. [DOI] [PubMed] [Google Scholar]
- 11. Raisner R.M., Hartley P.D., Meneghini M.D., Bao M.Z., Liu C.L., Schreiber S.L., Rando O.J., Madhani H.D.. Histone variant H2A.Z marks the 5′ ends of both active and inactive genes in euchromatin. Cell. 2005; 123:233–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Creyghton M.P., Markoulaki S., Levine S.S., Hanna J., Lodato M.A., Sha K., Young R.A., Jaenisch R., Boyer L.A.. H2AZ is enriched at polycomb complex target genes in ES cells and is necessary for lineage commitment. Cell. 2008; 135:649–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jin C., Zang C., Wei G., Cui K., Peng W., Zhao K., Felsenfeld G.. H3.3/H2A.Z double variant-containing nucleosomes mark ‘nucleosome-free regions’ of active promoters and other regulatory regions. Nat. Genet. 2009; 41:941–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Weber C.M., Henikoff S.. Histone variants: dynamic punctuation in transcription. Genes Dev. 2014; 28:672–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Goldberg A.D., Banaszynski L.A., Noh K.M., Lewis P.W., Elsaesser S.J., Stadler S., Dewell S., Law M., Guo X., Li X.et al.. Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell. 2010; 140:678–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chen P., Zhao J., Wang Y., Wang M., Long H., Liang D., Huang L., Wen Z., Li W., Li X.et al.. H3.3 actively marks enhancers and primes gene transcription via opening higher-ordered chromatin. Genes Dev. 2013; 27:2109–2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Elsasser S.J., Noh K.M., Diaz N., Allis C.D., Banaszynski L.A.. Histone H3.3 is required for endogenous retroviral element silencing in embryonic stem cells. Nature. 2015; 522:240–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tagami H., Ray-Gallet D., Almouzni G., Nakatani Y.. Histone H3.1 and H3.3 complexes mediate nucleosome assembly pathways dependent or independent of DNA synthesis. Cell. 2004; 116:51–61. [DOI] [PubMed] [Google Scholar]
- 19. Filipescu D., Szenker E., Almouzni G.. Developmental roles of histone H3 variants and their chaperones. Trends in Genetics. 2013; 29:630–640. [DOI] [PubMed] [Google Scholar]
- 20. Drane P., Ouararhni K., Depaux A., Shuaib M., Hamiche A.. The death-associated protein DAXX is a novel histone chaperone involved in the replication-independent deposition of H3.3. Genes Dev. 2010; 24:1253–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lewis P.W., Elsaesser S.J., Noh K.M., Stadler S.C., Allis C.D.. Daxx is an H3.3-specific histone chaperone and cooperates with ATRX in replication-independent chromatin assembly at telomeres. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:14075–14080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Banumathy G., Somaiah N., Zhang R., Tang Y., Hoffmann J., Andrake M., Ceulemans H., Schultz D., Marmorstein R., Adams P.D.. Human UBN1 is an ortholog of yeast Hpc2p and has an essential role in the HIRA/ASF1a chromatin-remodeling pathway in senescent cells. Mol. Cell. Biol. 2009; 29:758–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tang Y., Puri A., Ricketts M.D., Rai T.S., Hoffmann J., Hoi E., Adams P.D., Schultz D.C., Marmorstein R.. Identification of an ubinuclein 1 region required for stability and function of the human HIRA/UBN1/CABIN1/ASF1a histone H3.3 chaperone complex. Biochemistry. 2012; 51:2366–2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ricketts M.D., Marmorstein R.. A molecular prospective for HIRA complex assembly and H3.3-specific histone chaperone function. J. Mol. Biol. 2017; 429:1924–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tang Y., Poustovoitov M.V., Zhao K., Garfinkel M., Canutescu A., Dunbrack R., Adams P.D., Marmorstein R.. Structure of a human ASF1a-HIRA complex and insights into specificity of histone chaperone complex assembly. Nat. Struct. Mol. Biol. 2006; 13:921–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Xiong C., Wen Z., Yu J., Chen J., Liu C.P., Zhang X., Chen P., Xu R.M., Li G.. UBN1/2 of HIRA complex is responsible for recognition and deposition of H3.3 at cis-regulatory elements of genes in mouse ES cells. BMC Biol. 2018; 16:110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Balaji S., Iyer L.M., Aravind L.. HPC2 and ubinuclein define a novel family of histone chaperones conserved throughout eukaryotes. Mol. Biosyst. 2009; 5:269–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Meneghini M.D., Wu M., Madhani H.D.. Conserved histone variant H2A.Z protects euchromatin from the ectopic spread of silent heterochromatin. Cell. 2003; 112:725–736. [DOI] [PubMed] [Google Scholar]
- 29. Gevry N., Chan H.M., Laflamme L., Livingston D.M., Gaudreau L.. p21 transcription is regulated by differential localization of histone H2A.Z. Genes Dev. 2007; 21:1869–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bonisch C., Hake S.B.. Histone H2A variants in nucleosomes and chromatin: more or less stable?. Nucleic. Acids. Res. 2012; 40:10719–10741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chen P., Wang Y., Li G.. Dynamics of histone variant H3.3 and its coregulation with H2A.Z at enhancers and promoters. Nucleus. 2014; 5:21–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Krogan N.J., Baetz K., Keogh M.C., Datta N., Sawa C., Kwok T.C., Thompson N.J., Davey M.G., Pootoolal J., Hughes T.R.et al.. Regulation of chromosome stability by the histone H2A variant Htz1, the Swr1 chromatin remodeling complex, and the histone acetyltransferase NuA4. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:13513–13518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mizuguchi G., Shen X., Landry J., Wu W.H., Sen S., Wu C.. ATP-driven exchange of histone H2AZ variant catalyzed by SWR1 chromatin remodeling complex. Science. 2004; 303:343–348. [DOI] [PubMed] [Google Scholar]
- 34. Papamichos-Chronakis M., Watanabe S., Rando O.J., Peterson C.L.. Global regulation of H2A.Z localization by the INO80 chromatin-remodeling enzyme is essential for genome integrity. Cell. 2011; 144:200–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wong M.M., Cox L.K., Chrivia J.C.. The chromatin remodeling protein, SRCAP, is critical for deposition of the histone variant H2A.Z at promoters. J. Biol. Chem. 2007; 282:26132–26139. [DOI] [PubMed] [Google Scholar]
- 36. Morrison A.J., Shen X.. Chromatin remodelling beyond transcription: the INO80 and SWR1 complexes. Nat. Rev. Mol. Cell Biol. 2009; 10:373–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cai Y., Jin J., Gottschalk A.J., Yao T., Conaway J.W., Conaway R.C.. Purification and assay of the human INO80 and SRCAP chromatin remodeling complexes. Methods. 2006; 40:312–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Liang X., Shan S., Pan L., Zhao J., Ranjan A., Wang F., Zhang Z., Huang Y., Feng H., Wei D.et al.. Structural basis of H2A.Z recognition by SRCAP chromatin-remodeling subunit YL1. Nat. Struct. Mol. Biol. 2016; 23:317–323. [DOI] [PubMed] [Google Scholar]
- 39. Brahma S., Udugama M.I., Kim J., Hada A., Bhardwaj S.K., Hailu S.G., Lee T.H., Bartholomew B.. INO80 exchanges H2A.Z for H2A by translocating on DNA proximal to histone dimers. Nat. Commun. 2017; 8:15616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ayala R., Willhoft O., Aramayo R.J., Wilkinson M., McCormack E.A., Ocloo L., Wigley D.B., Zhang X.. Structure and regulation of the human INO80-nucleosome complex. Nature. 2018; 556:391–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Su J., Sui Y., Ding J., Li F., Shen S., Yang Y., Lu Z., Wang F., Cao L., Liu X.et al.. Human INO80/YY1 chromatin remodeling complex transcriptionally regulates the BRCA2- and CDKN1A-interacting protein (BCCIP) in cells. Protein Cell. 2016; 7:749–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cao L., Ding J., Dong L., Zhao J., Su J., Wang L., Sui Y., Zhao T., Wang F., Jin J.et al.. Negative regulation of p21Waf1/Cip1 by human INO80 chromatin remodeling complex is implicated in cell cycle phase G2/M arrest and abnormal chromosome stability. PLoS One. 2015; 10:e0137411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cai Y., Jin J., Yao T., Gottschalk A.J., Swanson S.K., Wu S., Shi Y., Washburn M.P., Florens L., Conaway R.C.et al.. YY1 functions with INO80 to activate transcription. Nat. Struct. Mol. Biol. 2007; 14:872–874. [DOI] [PubMed] [Google Scholar]
- 44. Ding J., Yu C., Sui Y., Wang L., Yang Y., Wang F., Yao H., Xing F., Liu H., Li Y.et al.. The chromatin remodeling protein INO80 contributes to the removal of H2A.Z at the p53-binding site of the p21 gene in response to doxorubicin. FEBS J. 2018; 285:3270–3285. [DOI] [PubMed] [Google Scholar]
- 45. Hu G., Cui K., Northrup D., Liu C., Wang C., Tang Q., Ge K., Levens D., Crane-Robinson C., Zhao K.. H2A.Z facilitates access of active and repressive complexes to chromatin in embryonic stem cell self-renewal and differentiation. Cell Stem Cell. 2013; 12:180–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Coleman-Derr D., Zilberman D. Deposition of histone variant H2A.Z within gene bodies regulates responsive genes. PLoS Genet. 2012; 8:e1002988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Picelli S., Bjorklund A.K., Reinius B., Sagasser S., Winberg G., Sandberg R.. Tn5 transposase and tagmentation procedures for massively scaled sequencing projects. Genome Res. 2014; 24:2033–2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kaya-Okur H.S., Wu S.J., Codomo C.A., Pledger E.S., Bryson T.D., Henikoff J.G., Ahmad K., Henikoff S.. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat. Commun. 2019; 10:1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang Q., Xiong H., Ai S., Yu X., Liu Y., Zhang J., He A.. CoBATCH for high-throughput single-cell epigenomic profiling. Mol. Cell. 2019; 76:206–216. [DOI] [PubMed] [Google Scholar]
- 50. Zhuo B., Yu J., Chang L., Lei J., Wen Z., Liu C., Mao G., Wang K., Shen J., Xu X.. Quantitative analysis of chromatin accessibility in mouse embryonic fibroblasts. Biochem. Biophys. Res. Commun. 2017; 493:814–820. [DOI] [PubMed] [Google Scholar]
- 51. Yu J., Xiong C., Zhuo B., Wen Z., Shen J., Liu C., Chang L., Wang K., Wang M., Wu C.et al.. Analysis of local chromatin states reveals gene transcription potential during mouse neural progenitor cell differentiation. Cell Rep. 2020; 32:107953. [DOI] [PubMed] [Google Scholar]
- 52. Zhang X., Guo C., Chen Y., Shulha H.P., Schnetz M.P., LaFramboise T., Bartels C.F., Markowitz S., Weng Z., Scacheri P.C.et al.. Epitope tagging of endogenous proteins for genome-wide ChIP-chip studies. Nat. Methods. 2008; 5:163–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Pchelintsev N.A., McBryan T., Rai T.S., van Tuyn J., Ray-Gallet D., Almouzni G., Adams P.D.. Placing the HIRA histone chaperone complex in the chromatin landscape. Cell Rep. 2013; 3:1012–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Shashikant T., Ettensohn C.A.. Genome-wide analysis of chromatin accessibility using ATAC-seq. Methods Cell Biol. 2019; 151:219–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal. 2011; 17:10–12. [Google Scholar]
- 56. Langmead B., Salzberg S.L.. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012; 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W.et al.. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008; 9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kazachenka A., Bertozzi T.M., Sjoberg-Herrera M.K., Walker N., Gardner J., Gunning R., Pahita E., Adams S., Adams D., Ferguson-Smith A.C.. Identification, characterization, and heritability of murine metastable epialleles: implications for non-genetic inheritance. Cell. 2018; 175:1259–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K.. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010; 38:576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wen Z., Zhang L., Ruan H., Li G.. Histone variant H2A.Z regulates nucleosome unwrapping and CTCF binding in mouse ES cells. Nucleic Acids Res. 2020; 48:5939–5952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Quinlan A.R., Hall I.M.. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kim D., Langmead B., Salzberg S.L.. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods. 2015; 12:357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Liao Y., Smyth G.K., Shi W.. The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 2013; 41:e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Love M.I., Huber W., Anders S.. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014; 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ramirez F., Dundar F., Diehl S., Gruning B.A., Manke T.. deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 2014; 42:W187–W191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Thorvaldsdottir H., Robinson J.T., Mesirov J.P.. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform. 2013; 14:178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ricketts M.D., Frederick B., Hoff H., Tang Y., Schultz D.C., Singh Rai T., Grazia Vizioli M., Adams P.D., Marmorstein R. Ubinuclein-1 confers histone H3.3-specific-binding by the HIRA histone chaperone complex. Nat. Commun. 2015; 6:7711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Egan B., Yuan C.C., Craske M.L., Labhart P., Guler G.D., Arnott D., Maile T.M., Busby J., Henry C., Kelly T.K.et al.. An alternative approach to ChIP-Seq normalization enables detection of genome-wide changes in histone H3 lysine 27 trimethylation upon EZH2 inhibition. PLoS One. 2016; 11:e0166438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kloet S.L., Karemaker I.D., van Voorthuijsen L., Lindeboom R.G.H., Baltissen M.P., Edupuganti R.R., Poramba-Liyanage D.W., Jansen P., Vermeulen M.. NuRD-interacting protein ZFP296 regulates genome-wide NuRD localization and differentiation of mouse embryonic stem cells. Nat. Commun. 2018; 9:4588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Long H., Zhang L., Lv M., Wen Z., Zhang W., Chen X., Zhang P., Li T., Chang L., Jin C.et al.. H2A.Z facilitates licensing and activation of early replication origins. Nature. 2020; 577:576–581. [DOI] [PubMed] [Google Scholar]
- 71. Soboleva T.A., Nekrasov M., Ryan D.P., Tremethick D.J.. Histone variants at the transcription start-site. Trends Genet. 2014; 30:199–209. [DOI] [PubMed] [Google Scholar]
- 72. Henikoff S., Greally J.M.. Epigenetics, cellular memory and gene regulation. Curr. Biol. 2016; 26:R644–R648. [DOI] [PubMed] [Google Scholar]
- 73. Henikoff S. Labile H3.3+H2A.Z nucleosomes mark ‘nucleosome-free regions. Nat. Genet. 2009; 41:865–866. [DOI] [PubMed] [Google Scholar]
- 74. Kang J., Kim Y.W., Kim A.. Histone variants H3.3 and H2A.Z are incorporated into the beta-globin locus during transcription activation via different mechanisms. Biochim. Biophys. Acta Gene Regul. Mech. 2018; 1861:637–646. [DOI] [PubMed] [Google Scholar]
- 75. Zhang T., Zhang Z., Dong Q., Xiong J., Zhu B.. Histone H3K27 acetylation is dispensable for enhancer activity in mouse embryonic stem cells. Genome Biol. 2020; 21:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Park Y.J., Dyer P.N., Tremethick D.J., Luger K.. A new fluorescence resonance energy transfer approach demonstrates that the histone variant H2AZ stabilizes the histone octamer within the nucleosome. J. Biol. Chem. 2004; 279:24274–24282. [DOI] [PubMed] [Google Scholar]
- 77. Nekrasov M., Amrichova J., Parker B.J., Soboleva T.A., Jack C., Williams R., Huttley G.A., Tremethick D.J.. Histone H2A.Z inheritance during the cell cycle and its impact on promoter organization and dynamics. Nat. Struct. Mol. Biol. 2012; 19:1076–1083. [DOI] [PubMed] [Google Scholar]
- 78. CNCB-NGDC Members and Partners Database resources of the National Genomics Data Center, China National Center for Bioinformation in 2021. Nucleic Acids Res. 2021; 49:D18–D28. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw sequence data reported in this paper have been deposited in the Genome Sequence Archive in National Genomics Data Center (78), Beijing Institute of Genomics (BIG), Chinese Academy of Sciences, under accession number PRJCA004722 that are publicly accessible at https://bigd.big.ac.cn/gsa.
✦ GSA: Genome Sequence Archive. Genomics, Proteomics & Bioinformatics 2017. [PMID = 28387199]
✦ Database Resources of the National Genomics Data Center in 2020. Nucleic Acids Res 2020, 48(D1): D24–D33. [ PMID = 31702008]