

ABSTRACT
Background
DNA methylation–based epigenetic age measures have been used as biological aging markers and are associated with a healthy lifespan. Few population-based studies have examined the relation between diet and epigenetic age acceleration.
Objectives
We aimed to investigate the relation between diet quality and epigenetic age acceleration.
Methods
We analyzed data from 1995 participants (mean age, 67 years; 55% women) of the Framingham Heart Study Offspring Cohort. Cross-sectional associations between the Dietary Approaches to Stop Hypertension (DASH) score and 3 whole-blood DNA methylation–derived epigenetic age acceleration measures—Dunedin Pace of Aging Methylation (DunedinPoAm), GrimAge acceleration (GrimAA), and PhenoAge acceleration (PhenoAA)—were examined. A mediation analysis was conducted to assess the mediating role of epigenetic age acceleration in relation to DASH and all-cause mortality.
Results
A higher DASH score was associated with lower levels of DunedinPoAm (β = −0.05; SE = 0.02; P = 0.007), GrimAA (β = −0.09; SE = 0.02; P < 0.001), and PhenoAA (β = −0.07; SE = 0.02; P = 0.001). All 3 epigenetic measures mediated the association between the DASH score and all-cause mortality, with mean proportions of 22.1% for DunedinPoAm (Pmediation = 0.04), 45.1% for GrimAA (Pmediation = 0.001), and 22.9% for PhenoAA (Pmediation = 0.03). An interaction was observed between the DASH score and smoking status in relation to the epigenetic aging markers. The association between the DASH score and epigenetic aging markers tended to be stronger in “ever-smokers” (former and current smokers) compared to “never-smokers.” The proportions of mediation were 31.3% for DunedinPoAm, 46.8% for GrimAA, and 10.3% for PhenoAA in ever-smokers, whereas no significant mediation was observed in never-smokers.
Conclusions
Higher diet quality is associated with slower epigenetic age acceleration, which partially explains the beneficial effect of diet quality on the lifespan. Our findings emphasize that adopting a healthy diet is crucial for maintaining healthy aging.
Keywords: diet quality, DNA methylation, epigenetic age acceleration, smoking, all-cause mortality
See corresponding editorial on page 6 and article on page 171.
Introduction
Diet quality is an important lifestyle factor affecting human health (1). Several diet quality scores have been developed—for example, the Alternate Healthy Eating Index (AHEI), the Mediterranean-style diet score (MDS), and the Dietary Approaches to Stop Hypertension (DASH) score—which have been associated with a variety of chronic diseases and mortality (2–4).
DNA methylation is the most studied epigenetic mechanism. A number of algorithms have been developed to predict lifespan based on DNA methylation status, including the Hannum (5) and the Horvath (6) epigenetic clocks, DNA methylation GrimAge (7), DNA methylation PhenoAge (8), and Dunedin Pace of Aging Methylation (DunedinPoAm) (9). Epigenetic aging markers derived from these algorithms have been linked to the time to disease progression and death occurrence independent of chronological age; therefore, these markers can be used as proxies to reflect the biological aging process (7–9). Food and nutrients may modify DNA methylation patterns by modulating enzyme activity and altering substrates and cofactors (10). A few epidemiological studies observed associations of individual nutrients, including ω-3 PUFA supplementation (7), and foods, including vegetables, fish, and poultry (7, 11), with epigenetic aging markers. Diet quality scores capture the synergistic effects of individual food and nutrients; however, the relationship between overall diet quality and epigenetic aging markers has not been fully explored.
Exploring the association of overall diet quality with epigenetic aging markers in a large population‐based study may provide molecular insights into the beneficial effects of a healthy diet on the lifespan in humans. Further, examining whether the epigenetic aging measures mediate the relationship between diet quality and all-cause mortality may facilitate a better understanding of the potential clinical impacts of the epigenetic aging measures. In the present study, we utilized data from the Framingham Heart Study (FHS) to examine the associations between diet quality and epigenetic aging measures and whether the epigenetic measures mediate the associations between diet quality and all-cause mortality.
Methods
Study participants
The present study was focused on participants who attended the eighth (2005‒2008) examination of the FHS Offspring Cohort (12) and whose DNA methylation data were generated by the Framingham Offspring Exam 8 DNA Methylation Study [n = 2590; data available at the database of Genotypes and Phenotypes (dbGaP); phs000724.v7.p11] (13). All participants were white. The final analysis was comprised of 1995 participants after excluding those who had missing information on diet and covariates. Inclusion and exclusion of study participants are depicted in Figure 1. The FHS protocols and procedures were approved by the Institutional Review Board for Human Research at Boston University Medical Center, and all participants provided written informed consent. The current study was approved by the Institutional Review Board at Tufts University.
FIGURE 1.
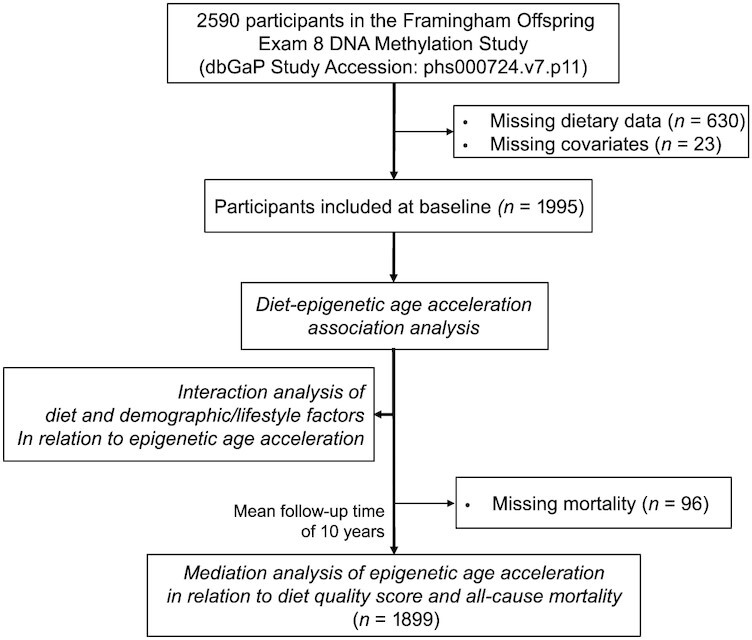
The flow diagram of participant selection and study overview.
Diet quality score
A validated 126-item semiquantitative FFQ (14) was used to assess habitual dietary intake. Participants completed the FFQ at the eighth examination (baseline) to report their habitual consumption of each food item during the past year. Dietary data were excluded if the reported energy intake was <2.5 MJ/day (600 kcal/day) or ≥16.7 MJ/day (4000 kcal/day) for women and <2.5 MJ/day (600 kcal/day) or ≥17.5 MJ/day (4200 kcal/day) for men, or when participants left ≥13 food items blank on the FFQ (15). Qualified dietary data were used to compute the DASH score (16, 17). We calculated the DASH score based on 8 dietary components: higher intake of vegetables, fruits, nuts and legumes, whole grains, and low-fat dairy and lower intake of red and processed meat, sugar-sweetened beverages, and sodium. Energy intake–adjusted residuals for each component were first computed and classified into quintiles. A score of 1 to 5 was then given for each component, according to its quintile rank, for foods where high intake was favorable, while the quintile ranking was reversed for the components where low intake was desired. The sum of the component scores resulted in a DASH score ranging from 8 to 40, with higher scores indicating better diet quality. To reduce the burden of multiple testing and for easier interpretation, the primary analysis focused on the DASH score. Nonetheless, in a sensitivity analysis, the AHEI (18) and a modified MDS (19) were used to investigate the impacts of different diet quality measures. Calculations of the AHEI and MDS scores are summarized in the Supplemental Methods.
Epigenetic aging markers
Blood samples used for measurement of DNA methylation status were collected at the eighth examination. A detailed description of DNA methylation profiling has been described elsewhere (20). Briefly, DNA were extracted from whole-blood buffy coat samples. DNA methylation was assayed using the Illumina Infinium HumanMethylation450 BeadChip platform, followed by quality control and normalization procedures. The methylation status at each cytosine-phosphate-guanine (CpG) site was quantified as a β value, calculated as the proportion of methylated signal intensity.
We estimated 3 epigenetic aging measures: DunedinPoAm (9), GrimAA (7), and PhenoAA (8). DunedinPoAm is a whole-blood DNA methylation–based marker for the pace of aging, with a higher value representing more deteriorated biological aging: that is, an accelerated aging process (9). DunedinPoAm was developed based on changes in 18 blood-chemistry and organ-system function biomarkers over 12 years (9). DunedinPoAm was calculated using the R code accessible at https://github.com/danbelsky/DunedinPoAm38. GrimAge and PhenoAge are whole-blood DNA methylation–derived predictors of a healthy lifespan using an online calculator (https://dnamage.genetics.ucla.edu/new) (5). GrimAge was developed based on 8 plasma proteins and smoking pack-years (7), and PhenoAge was created based on 9 clinical biomarkers and chronological age (8). By adjusting GrimAge and PhenoAge for the chronological age, GrimAge acceleration (GrimAA) and PhenoAge acceleration (PhenoAA) were calculated and used as markers for the state of biological aging (7, 8). Similar to DunedinPoAm, higher values of GrimAA and PhenoAA represent accelerated aging. DunedinPoAm is a unit-free measure, while GrimAA and PhenoAA have units of years. All 3 markers have been associated with age-related morbidity and mortality (7–9). The clinical and biological factors used to develop the 3 epigenetic aging measures are summarized in Supplemental Table 1.
Clinical outcome ascertainment
The primary clinical outcome was all-cause mortality, with data obtained from continuous surveillance after the eighth examination cycle. A panel of 3 physicians reviewed and adjudicated by examining all pertinent information, including medical and hospital records, death certificates, communication with personal physicians, and next-of-kin interviews (21).
Covariates
Demographic, lifestyle, and anthropometric data were obtained in accordance with standard protocols (22). BMI was calculated as weight divided by height squared (kg/m2). Self-reported smoking status was used to characterize participants as never, former, or current smokers. We defined current smokers as individuals who had smoked at least 1 cigarette per day in the year prior to the examination and former smokers as those who had stopped smoking at least 1 year prior to the examination. A physical activity score was generated using the intensity and time spent for 5 types of activities, assessed via a physical activity questionnaire (23). Alcohol consumption was calculated based on the self-reported total quantity of alcohol consumed (grams) per day (24).
Statistical analysis
We examined cross-sectional associations between the DASH score and the 3 measures of epigenetic age acceleration. The primary outcome variables were DunedinPoAm, GrimAA, and PhenoAA. To account for family relatedness in our study sample, a linear mixed-effect model was applied. Epigenetic age acceleration measures were outcome variables and the DASH score was the exposure variable. Family structure was considered as a random effect. Models were adjusted for age, sex, smoking status, physical activity score, alcohol consumption, and BMI. Geometric means and 95% CIs were estimated for epigenetic age acceleration measures according to quartile categories of the DASH score. A test for a linear trend was performed using the median value for each quartile category of the DASH score as a continuous variable in the model. To compare the magnitude of the association between the DASH score and epigenetic age acceleration, the DASH score and the measures of epigenetic age acceleration were standardized to a mean of 0 and SD of 1 to facilitate better comparison of the results. We applied a similar statistical approach for each of the 8 individual components of the DASH score. For sensitivity analyses, we further adjusted for the modified DASH score by excluding the tested individual components from the total DASH score.
We also evaluated effect modification of the DASH score by age, sex, BMI, physical activity, and smoking status (25) on epigenetic age acceleration with the addition of interaction terms in the models. In a previous study of smoking-associated DNA methylation profiles (26), many common differentially methylated CpGs (i.e., DNA methylation sites) were observed in former and current smokers. We therefore combined former and current smokers as “ever-smokers” in the primary interaction analysis for smoking status. Interaction analyses using the 3 smoking categories (never, former, and current smokers) were also conducted in a sensitivity analysis. A log likelihood ratio test was used to estimate the significance level (P) for interaction terms. A Bonferroni-corrected P-value threshold (0.01; i.e., 0.05/5 potential effect modifiers) was used to account for multiple comparisons. When an interaction term was significant, we conducted a stratified analysis by the effect modifier.
A mediation analysis was conducted to investigate to what extent the association between the DASH score and all-cause mortality may be mediated by epigenetic age acceleration. The primary outcome variable was all-cause mortality. A modified mediation analytical approach was applied (27). A linear mixed-effect model was used to estimate the association between the DASH score and epigenetic age acceleration, and a mixed-effect Cox proportional hazard model was subsequently adopted to estimate the association of epigenetic age acceleration and all-cause mortality, with adjustments for the DASH score and covariates (age, sex, energy intake, smoking status, physical activity score, alcohol consumption, BMI, systolic blood pressure, use of hypertension medications, HDL cholesterol, total cholesterol, type 2 diabetes, and history of cardiovascular disease and cancer). We calculated 95% CIs and the significance levels (P values) for natural indirect (i.e., mediation) effects using a resampling method that takes random draws from a multivariate normal distribution of estimates. The proportion of mediation was calculated as the ratio of indirect effects to the sum of both direct and indirect effects. All statistical analyses were conducted using R statistical analysis software (version 4.0), and the results were considered statistically significant at a P value < 0.05 unless otherwise stated.
Two additional sensitivity analyses were carried out to verify the robustness of the analyses described above. First, we tested the impacts of different diet quality measures: the AHEI and MDS. Second, we tested the impacts of blood cell counts; that is, models were further adjusted for blood cell counts estimated using the Houseman's method (28), including the proportions of CD8 + T cells, CD4 + T cells, natural killer cells, B cells, and granulocytes.
Results
Characteristics among the 1995 participants according to quartiles of the DASH score are shown in Table 1. Participants with a higher DASH score were more likely to be women, were less likely to be ever-smokers (i.e., current and former smokers), had less alcohol consumption, had lower BMIs, had higher HDL cholesterol, and were less likely to take hypertension medication. The DASH score was moderately correlated with MDS and AHEI, with Pearson r values of 0.69 and 0.75, respectively (both P values < 0.0001; Supplemental Figure 1). GrimAA and DunedinPoAm were moderately correlated, with a Pearson r of 0.68, whereas the correlations of PhenoAA with GrimAA and DunedinPoAm were relatively low, with Pearson r values of 0.43 and 0.37, respectively (all P values < 0.0001; Supplemental Figure 1).
TABLE 1.
Baseline characteristics of participants according to quartiles of DASH score (n = 1995)1
| Q1, n = 522 | Q2, n = 562 | Q3, n = 501 | Q4, n = 410 | P-trend2 | |
|---|---|---|---|---|---|
| Median (range) | 18 (9‒20) | 23 (21‒24) | 26 (25‒28) | 31 (29‒40) | <0.0001 |
| Age, y | 67 ± 9 | 67 ± 9 | 68 ± 9 | 66 ± 9 | 0.85 |
| Women, % (n) | 219 (42) | 277 (49) | 307 (61) | 295 (72) | <0.0001 |
| Ever smokers,3 % (n) | 261 (50) | 255 (45) | 184 (37) | 146 (36) | <0.0001 |
| Current smokers, % (n) | 14 (72) | 8 (43) | 6 (29) | 3 (14) | 0.004 |
| Former smokers, % (n) | 36 (188) | 38 (212) | 31 (155) | 32 (132) | <0.0001 |
| Alcohol consumption, g/d | 6 ± 10 | 5 ± 9 | 5 ± 6 | 4 ± 6 | <0.0001 |
| Physical activity score4 | 35 ± 6 | 35 ± 5 | 36 ± 5 | 36 ± 5 | 0.001 |
| BMI, kg/m2 | 29 ± 6 | 29 ± 5 | 28 ± 5 | 27 ± 5 | <0.0001 |
| Systolic blood pressure, mmHg | 129 ± 17 | 128 ± 17 | 128 ± 17 | 128 ± 18 | 0.16 |
| Total cholesterol, mg/dl | 182 ± 35 | 185 ± 39 | 186 ± 38 | 189 ± 36 | 0.01 |
| HDL cholesterol, mg/dl | 55 ± 18 | 56 ± 17 | 59 ± 19 | 62 ± 18 | <0.0001 |
| Energy intake, kcal/d | 1920 ± 679 | 1782 ± 632 | 1830 ± 621 | 2009 ± 531 | 0.04 |
| Diet quality score | |||||
| AHEI score | 46 ± 8 | 54 ± 8 | 61 ± 9 | 70 ± 9 | <0.0001 |
| MDS score | 9 ± 3 | 11 ± 3 | 14 ± 3 | 17 ± 3 | <0.0001 |
| HTN meds, % (n) | 319 (61) | 327 (58) | 246 (49) | 186 (45) | <0.0001 |
| T2D, % (n) | 63 (12) | 83 (15) | 70 (14) | 46 (11) | 0.70 |
Abbreviations: AHEI, Alternate Healthy Eating Index score; DASH, Dietary Approaches to Stop Hypertension; HTN meds, hypertension medications; MDS, Mediterranean-style diet score; T2D, type 2 diabetes.
Data were expressed as means (SDs) or percentages (numbers).
The P value for the linear trend was tested by treating the median value of each quartile as a continuous variable. Corresponding P values for trends were calculated using linear mixed-effects models for continuous variables and Cochran-Armitage trend tests for categorical variables.
An ever-smoker was defined as a current or former smoker.
A physical activity score was generated using the intensity and time spent performing each type of activity, assessed by a physical activity questionnaire.
Association of diet quality and epigenetic age acceleration
A higher DASH score was associated with less epigenetic age acceleration in the model adjusting for age, sex, smoking status, BMI, physical activity score, alcohol consumption, and energy intake (Figure 2; Supplemental Table 2). For a 1 SD increase of the DASH score (5.34 units), the standardized epigenetic aging markers (regression coefficient) were reduced, by 0.05 (95% CI: −0.09 to −0.01; P = 0.007) for DunedinPoAm, 0.09 (95% CI: −0.12 to −0.05; P < 0.001) for GrimAA, and 0.07 (95% CI: −0.12 to −0.03; P = 0.001) for PhenoAA. The DunedinPoAm is not associated with a unit; however, as shown in Figure 2, the differences in GrimAA and PhenoAA between the highest and the lowest DASH quartile scores were 1.04 years and 1.18 years, respectively. Sensitivity analyses showed that the AHEI and MDS were also associated with the 3 epigenetic aging measures, with the same direction and similar magnitudes (all P values ≤ 0.002; Supplemental Table 2). Additional adjustments for blood cell counts did not substantially modify the observed associations between diet quality scores and epigenetic aging measures (Supplemental Table 3).
FIGURE 2.

Association between DASH score and epigenetic age acceleration measures (n = 1995). The DASH score was categorized into quartiles, with the first quartile representing a tendency toward an unhealthy diet and the fourth quartile representing a tendency toward a healthier diet. GrimAA and PhenoAA are scaled in units of years, while DunedinPoAm is a unit-free measure. The filled circles and lines indicate least-square means and 95% CIs, respectively. A linear mixed-effects model was adjusted for age, sex, BMI, smoking status, physical activity score, alcohol consumption, and energy intake. Abbreviations: DASH, Dietary Approaches to Stop Hypertension; DunedinPoAm, Dunedin Pace of Aging Methylation; GrimAA, DNA methylation GrimAge Acceleration; PhenoAA, DNA methylation PhenoAge Acceleration.
Using a Bonferroni-corrected P-value threshold of 0.01, significant interactions were observed between the DASH score and smoking status in relation to DunedinPoAm and GrimAA (both Pinteraction values < 0.0001; Supplemental Table 4). However, there was no significant interaction of diet quality with age, sex, BMI, or physical activity (Supplemental Table 4). When GrimAA was recomputed by regressing the GrimAge on the chronological age and smoking pack-years, the significant diet-smoking interaction remained. Figure 3 and Supplemental Table 5 showed that the association of the DASH score with epigenetic age acceleration was stronger in ever-smokers. For example, in ever-smokers, geometric means for GrimAA were 4.09 years (95% CI: 3.63–4.55 years) and 0.81 years (95% CI: 0.19–1.43 years) in the lowest and highest DASH score quartiles, respectively, whereas in never-smokers, the geometric means were −1.90 years (95% CI: −2.36 to −1.44 years) and −2.18 years (95% CI: −2.65 to −1.72 years) in the lowest and highest DASH score quartiles, respectively. A sensitivity analysis according to the 3 categories of smoking status (never, former, and current) showed that the interaction patterns with the DASH score in relation to the 3 measures of epigenetic age acceleration were not substantially changed (Supplemental Table 4). The magnitude of the inverse association between the DASH score and epigenetic age acceleration seems stronger in current smokers compared to that in former smokers (Supplemental Table 5).
FIGURE 3.

Association between the DASH score and epigenetic age acceleration according to smoking status (never, n = 1149; ever, n = 846). The DASH score was categorized into quartiles, with the first quartile representing a tendency toward an unhealthy diet and the fourth quartile representing a tendency toward a healthier diet. GrimAA and PhenoAA are scaled in units of years, while DunedinPoAm is a unit-free measure. An ever-smoker was defined as a current or former smoker. Symbols and bars are least-square means and 95% CIs, respectively. Linear mixed-effects models were adjusted for age, sex, BMI, physical activity score, alcohol consumption, and energy intake. Abbreviations: DASH, Dietary Approaches to Stop Hypertension; DunedinPoAm, Dunedin Pace of Aging Methylation; GrimAA, DNA methylation GrimAge Acceleration; PhenoAA, DNA methylation PhenoAge Acceleration.
Mediation analysis of epigenetic age acceleration
During a mean follow-up of 10 years, a total of 297 deaths were documented. The DASH score and the 3 epigenetic age acceleration measures were significantly associated with all-cause mortality (P < 0.05; Supplemental Table 6). All 3 measures of epigenetic age acceleration significantly mediated the association of the DASH score with all-cause mortality, with Pmediation values of 0.04 for DunedinPoAm, 0.001 for GrimAA, and 0.03 for PhenoAA (Table 2). Proportions of mediation were 22.1%, 45.1%, and 22.9% by DunedinPoAm, GrimAA, and PhenoAA, respectively. In a stratified analysis by smoking status, mediation was more pronounced in ever-smokers: the proportions of mediation in ever-smokers were 31.3% by DunedinPoAm (Pmediation = 0.004), 46.8% by GrimAA (Pmediation = 0.0001), and 10.3% by PhenoAA (Pmediation = 0.049). No significant mediation effect was observed in never-smokers. Similar proportions of mediation were observed in sensitivity analyses using the AHEI and MDS (Supplemental Table 7). In a sensitivity analysis, we further adjusted for metformin use (n = 111) and found the association remained the same (Supplemental Table 8).
TABLE 2.
Mediation analysis of epigenetic age acceleration on the associations of DASH score with all-cause mortality1
| Indirect association | |||
|---|---|---|---|
| HR (95% CI) | P | Proportion mediated, % | |
| All, n = 1995 | |||
| DunedinPoAm | 0.981 (0.963–0.996) | 0.038 | 22.1 |
| GrimAA | 0.964 (0.945–0.981) | 0.001 | 45.1 |
| PhenoAA | 0.982 (0.966–0.995) | 0.025 | 22.9 |
| Never smoker, n = 1149 | |||
| DunedinPoAm | 0.995 (0.975–1.012) | 0.505 | 14.9 |
| GrimAA | 0.989 (0.971–1.004) | 0.204 | 0.0 |
| PhenoAA | 0.986 (0.966–1.000) | 0.133 | 96.5 |
| Ever smoker, n = 846 | |||
| DunedinPoAm | 0.925 (0.880–0.963) | 0.004 | 31.3 |
| GrimAA | 0.890 (0.843–0.932) | <0.001 | 46.8 |
| PhenoAA | 0.971 (0.943–0.994) | 0.049 | 10.3 |
Abbreviations: DASH, Dietary Approaches to Stop Hypertension; DunedinPoAm, Dunedin Pace of Aging Methylation; GrimAA, DNA methylation GrimAge Acceleration; HR, hazard ratio; PhenoAA, DNA methylation PhenoAge Acceleration.
An ever-smoker was defined as a current or former smoker. HRs per 1-SD increase of the DASH score in standardized z-score and P values were derived from mixed-effect Cox proportional hazard models. A linear mixed-effect model was estimated for epigenetic age acceleration (mediator), conditional on the DASH score (exposure) and covariates. A mixed-effect Cox proportional hazard model was estimated for all-cause mortality (outcome), conditional on the DASH score, epigenetic age acceleration, and covariates to estimate the indirect (mediation) effect. The proportion of mediation was calculated as the ratio of an indirect effect to the sum of both direct and indirect effects. Models were adjusted for age, sex, smoking status, physical activity score, alcohol consumption, energy intake, BMI, systolic blood pressure, hypertension medications, total and HDL cholesterol, type 2 diabetes, and history of cardiovascular disease and cancer. For a stratified analysis on smoking status, the same covariates were used as in the models, except for smoking status.
Analysis for individual DASH components
Among the 8 components of the DASH score, higher intakes of vegetables (P = 0.020), fruits (P = 0.002), nuts and legumes (P = 0.021), and whole grains (P = 0.044) were associated with lower GrimAA, while higher intakes of red and processed meat (P = 0.008) and sodium (P = 0.001) were associated with higher GrimAA (Supplemental Table 9). Higher intake of nuts and legumes was also associated with lower DunedinPoAm (P = 0.001) and PhenoAA (P = 0.011; Supplemental Table 9). The observed associations remained similar after further adjustment for the modified DASH score (Supplemental Table 9). In addition, we observed more instances of significant associations between the DASH score components and epigenetic age acceleration in ever-smokers than in never-smokers (Supplemental Table 10). In ever-smokers, 8 components of the DASH score were associated with GrimAA (P < 0.05) and 5 components were associated with DunedinPoAm (P < 0.05), whereas 1 component was associated with GrimAA in never-smokers.
Discussion
The present study showed a strong association between higher diet quality and decelerated DNA methylation–based epigenetic aging markers. A mediation analysis further demonstrated that DNA methylation may partly explain the beneficial relation between a healthy diet and all-cause mortality, particularly in individuals who have a history of smoking (i.e., former and current smokers). Taken together, our study emphasizes that improving diet quality is important to delay the aging process.
Utilizing biomarkers may facilitate a better understanding of the association between diet and disease risks (29). In the present study, we exploited the so-called second-generation epigenetic aging markers. Different from the first-generation DNA methylation–based markers, such as intrinsic epigenetic age acceleration (IEAA) (5) and extrinsic epigenetic age acceleration (EEAA) (6), the second-generation markers were developed using a 2-stage approach with the integration of additional clinical and functional biomarkers (7–9). Previous studies have shown that the epigenetic aging markers used by the present study have stronger predictive abilities for the time to chronic diseases and death compared to the first-generation epigenetic aging markers (7). The present study demonstrated that the novel whole blood–derived DNA methylation–based epigenetic aging markers are useful biomarkers for assessing the effects of diet on health in observational studies, and potentially can be applied to intervention studies (30). Further research is needed to explore whether these biomarkers can evaluate individual-level diet quality. Such information may contribute to tailored nutritional recommendations.
It is reported that GrimAA and PhenoAA were negatively correlated with plasma carotenoid levels—an indicator of fruit and vegetable consumption—in the Women's Health Initiative cohort of multiethnic US postmenopausal women (7, 8). In the meta-analysis of the Women's Health Initiative and Invecchiare nel Chianti (InCHIANTI) studies, IEAA and EEAA exhibited significant negative correlations with poultry and fish intake (11). A combined supplementation of folic acid and vitamin B12 led to a significant reduction in IEAA in a 2-year, randomized controlled study among 44 Dutch elderly subjects aged 65–75 years (31). Relative to individual food and nutrients, diet quality scores may represent the synergistic effects of all dietary components. Consistent with our findings, a significant decrease in IEAA was observed after a 1-year Mediterranean-style dietary intervention in 120 healthy older adults aged 65–79 years (32). Nonetheless, the number of studies examining the relation of overall diet quality and epigenetic aging markers is limited. Our study therefore contributes novel evidence to support a strong relation between overall diet quality and epigenetic aging markers in the general population.
A possibility was raised that epigenetic age acceleration may be altered by potentially interactive effects between lifestyle factors (33). We found that the association of diet quality with GrimAA and DunedinPoAm appears to be greater for ever-smokers than never-smokers. Although no significant interaction was detected, the diet-PhenoAA relation was also stronger in ever-smokers than in never-smokers. Smoking was an important lifestyle factor associated with DNA methylation changes (34), and has been associated with cardiovascular and all-cause mortality (35). Our findings on the stronger association between diet quality and all-cause mortality amongst ever-smokers was also in line with a recent publication of 2 Swedish cohort studies (total n = 68,273) (36). In Swedish adults aged 45–83 years, a significant interaction was reported between the anti‐inflammatory diet index and smoking status on all-cause mortality, with the strongest inverse association between this index and mortality amongst current smokers. Therefore, our findings emphasize that adopting a healthy diet is crucial for maintaining healthy aging.
This favorable relation between higher diet quality scores and epigenetic age deceleration may be related to reductions in oxidative and inflammatory stress (37). It was demonstrated that epigenetic age acceleration relates to inflammatory biomarkers and postprandial lipid levels in white individuals (38). A meta-analysis of 5 US and European population-based cohorts also showed that diet-associated differential DNA methylation can be linked to metabolic and inflammatory pathways, which indicates the importance of diet-induced epigenetic changes on health outcomes (39). We also found that all 3 measures of epigenetic aging acceleration were associated with the consumption of nuts and legumes, which can be linked to reductions in oxidative and inflammatory processes prohibited by bioactive compounds such as MUFA, PUFA, phenolic compounds, tocopherols, and carotenoids (40).
The strengths of the present study included the use of a relatively large sample size of a community-based cohort with detailed information on a broad range of covariates, as well as the use of multiple measures of accelerated aging and sufficient follow-up data for clinical outcomes. Nonetheless, there are some limitations to be considered in the interpretation of our study. In the present study, we examined dietary data and epigenetic age measures collected at 1 time point; therefore, we were unable to capture causal associations of dietary changes on epigenetic age acceleration. The study participants were middle-aged and older white adults: therefore, our findings may not be generalizable to other populations. Misclassification and measurement errors might occur because of the use of self-reported data on dietary intake and smoking status. Although multiple potential confounders were adjusted for in the present analysis, residual confounding could not be completely ruled out.
Our findings demonstrate that better diet quality was associated with decelerated biological aging, providing a promising avenue to explore the beneficial effects of diet on prolonged lifespans. This effect seems to be more prominent for those who have a history of smoking. Further studies are warranted to validate our findings, including studies with larger sample sizes, studies with racially and ethnically diverse populations, and studies investigating the causal role of dietary factors on epigenetic regulations.
Supplementary Material
Acknowledgments
The authors’ responsibilities were as follows—JM: designed research and had primary responsibility for the final content; DL: directed and supervised the project; TH and RJ: conducted the analyses; NMM: critically reviewed the manuscript; SH: contributed to the methodology used in the paper; YK: performed the statistical analysis; YK and JM: interpreted the results and wrote the manuscript; and all authors: read and approved the final manuscript.
Author disclosures: The authors report no conflicts of interest.
Notes
This research was supported by grants from the National Heart, Lung and Blood Institute's Framingham Heart Study (contract number N01-HC-25195) and by the National Heart, Lung, and Blood Institute Career Transition Award (1K22HL135075-01).
The funding sources had no role in the study design, data collection and analysis, preparation of the manuscript, or publication of the study.
Supplemental Figure 1, Supplemental Tables 1–10, and Supplemental Methods are available from the “Supplementary data” link in the online posting of the article and from the same link in the online table of contents at https://academic.oup.com/ajcn/.
Abbreviations used: AHEI, Alternate Healthy Eating Index; DASH, Dietary Approaches to Stop Hypertension; DunedinPoAm, Dunedin Pace of Aging Methylation; EEAA, extrinsic epigenetic age acceleration; FHS, Framingham Heart Study; GrimAA, DNA methylation GrimAge Acceleration; IEAA, intrinsic epigenetic age acceleration; MDS, Mediterranean-style Diet Score; PhenoAA, DNA methylation PhenoAge Acceleration.
Contributor Information
Youjin Kim, Nutrition Epidemiology and Data Science, Friedman School of Nutrition Science and Policy, Tufts University, Boston, MA, USA.
Tianxiao Huan, Department of Ophthalmology and Visual Sciences, University of Massachusetts Medical School, Worcester, MA, USA.
Roby Joehanes, Population Sciences Branch, National Heart, Lung, and Blood Institute, National Institutes of Health, Bethesda, MD & Framingham Heart Study, Framingham, MA, USA.
Nicola M McKeown, Nutrition Epidemiology and Data Science, Friedman School of Nutrition Science and Policy, Tufts University, Boston, MA, USA; Nutritional Epidemiology Laboratory, Jean Mayer USDA Human Nutrition Research Center on Aging, Tufts University, Boston, MA, USA.
Steve Horvath, Department of Human Genetics, David Geffen School of Medicine, University of California Los Angeles, Los Angeles, CA, USA; Department of Biostatistics, Fielding School of Public Health, University of California Los Angeles, Los Angeles, CA, USA.
Daniel Levy, Population Sciences Branch, National Heart, Lung, and Blood Institute, National Institutes of Health, Bethesda, MD & Framingham Heart Study, Framingham, MA, USA.
Jiantao Ma, Nutrition Epidemiology and Data Science, Friedman School of Nutrition Science and Policy, Tufts University, Boston, MA, USA.
Data Availability
The data sets analyzed in this study are available in the database of Genotypes and Phenotypes (dbGaP) repository with accession number phs000724.v7.p11.
References
- 1. Schwingshackl L, Bogensberger B, Hoffmann G. Diet quality as assessed by the Healthy Eating Index, Alternate Healthy Eating Index, Dietary Approaches to Stop Hypertension Score, and health outcomes: an updated systematic review and meta-analysis of cohort studies. J Acad Nutr Diet. 2018;118(1):74–100.e11. [DOI] [PubMed] [Google Scholar]
- 2. Sotos-Prieto M, Bhupathiraju SN, Mattei J, Fung TT, Li Y, Pan A, Willett WC, Rimm EB, Hu FB. Association of changes in diet quality with total and cause-specific mortality. N Engl J Med. 2017;377(2):143–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Harmon BE, Boushey CJ, Shvetsov YB, Ettienne R, Reedy J, Wilkens LR, Le Marchand L, Henderson BE, Kolonel LN. Associations of key diet-quality indexes with mortality in the multiethnic cohort: the Dietary Patterns Methods Project. Am J Clin Nutr. 2015;101(3):587–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Reedy J, Krebs-Smith SM, Miller PE, Liese AD, Kahle LL, Park Y, Subar AF. Higher diet quality is associated with decreased risk of all-cause, cardiovascular disease, and cancer mortality among older adults. J Nutr. 2014;144(6):881–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hannum G, Guinney J, Zhao L, Zhang L, Hughes G, Sadda S, Klotzle B, Bibikova M, Fan JB, Gao Y et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol Cell. 2013;49(2):359–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013;14(10):R115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lu AT, Quach A, Wilson JG, Reiner AP, Aviv A, Raj K, Hou L, Baccarelli AA, Li Y, Stewart JD et al. DNA methylation GrimAge strongly predicts lifespan and healthspan. Aging. 2019;11(2):303–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Levine ME, Lu AT, Quach A, Chen BH, Assimes TL, Bandinelli S, Hou L, Baccarelli AA, Stewart JD, Li Y et al. An epigenetic biomarker of aging for lifespan and healthspan. Aging. 2018;10(4):573–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Belsky DW, Caspi A, Arseneault L, Baccarelli A, Corcoran DL, Gao X, Hannon E, Harrington HL, Rasmussen LJ, Houts R et al. Quantification of the pace of biological aging in humans through a blood test, the DunedinPoAm DNA methylation algorithm. Elife. 2020;9:e54870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kadayifci FZ, Zheng S, Pan YX. Molecular mechanisms underlying the link between diet and DNA methylation. Int J Mol Sci. 2018;19(12):4055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Quach A, Levine ME, Tanaka T, Lu AT, Chen BH, Ferrucci L, Ritz B, Bandinelli S, Neuhouser ML, Beasley JM et al. Epigenetic clock analysis of diet, exercise, education, and lifestyle factors. Aging. 2017;9(2):419–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kannel WB, Feinleib M, McNamara PM, Garrison RJ, Castelli WP. An investigation of coronary heart disease in families. The Framingham Offspring study. Am J Epidemiol. 1979;110(3):281–90. [DOI] [PubMed] [Google Scholar]
- 13. Yin X, Subramanian S, Hwang SJ, O'Donnell CJ, Fox CS, Courchesne P, Muntendam P, Gordon N, Adourian A, Juhasz P et al. Protein biomarkers of new-onset cardiovascular disease: prospective study from the systems approach to biomarker research in cardiovascular disease initiative. Arterioscler Thromb Vasc Biol. 2014;34(4):939–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rimm EB, Giovannucci EL, Stampfer MJ, Colditz GA, Litin LB, Willett WC. Reproducibility and validity of an expanded self-administered semiquantitative food frequency questionnaire among male health professionals. Am J Epidemiol. 1992;135(10):1114–26.; discussion 1127–36. [DOI] [PubMed] [Google Scholar]
- 15. Willett W. Implications of total energy intake for epidemiologic analyses. 3rd. ed. In: Nutritional epidemiology. New York (NY): Oxford University Press; 2012.260–86. [Google Scholar]
- 16. Djoussé L, Ho YL, Nguyen XT, Gagnon DR, Wilson PWF, Cho K, Gaziano JM; VA Million Veteran Program . DASH score and subsequent risk of coronary artery disease: the findings from Million Veteran Program. J Am Heart Assoc. 2018;7(9):e008089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fung TT, Chiuve SE, McCullough ML, Rexrode KM, Logroscino G, Hu FB. Adherence to a DASH-style diet and risk of coronary heart disease and stroke in women. Arch Intern Med. 2008;168(7):713–20. [DOI] [PubMed] [Google Scholar]
- 18. Chiuve SE, Fung TT, Rimm EB, Hu FB, McCullough ML, Wang M, Stampfer MJ, Willett WC. Alternative dietary indices both strongly predict risk of chronic disease. J Nutr. 2012;142(6):1009–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fung TT, Rexrode KM, Mantzoros CS, Manson JE, Willett WC, Hu FB. Mediterranean diet and incidence of and mortality from coronary heart disease and stroke in women. Circulation. 2009;119(8):1093–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Huan T, Joehanes R, Song C, Peng F, Guo Y, Mendelson M, Yao C, Liu C, Ma J, Richard M et al. Genome-wide identification of DNA methylation QTLs in whole blood highlights pathways for cardiovascular disease. Nat Commun. 2019;10(1):4267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wilson PW, D'Agostino RB, Levy D, Belanger AM, Silbershatz H, Kannel WB. Prediction of coronary heart disease using risk factor categories. Circulation. 1998;97(18):1837–47. [DOI] [PubMed] [Google Scholar]
- 22. Splansky GL, Corey D, Yang Q, Atwood LD, Cupples LA, Benjamin EJ, D'Agostino RB Sr., Fox CS, Larson MG, Murabito JM et al. The third generation cohort of the National Heart, Lung, and Blood Institute's Framingham Heart Study: design, recruitment, and initial examination. Am J Epidemiol. 2007;165(11):1328–35. [DOI] [PubMed] [Google Scholar]
- 23. Kannel WB, Belanger A, D'Agostino R, Israel I. Physical activity and physical demand on the job and risk of cardiovascular disease and death: the Framingham Study. Am Heart J. 1986;112(4):820–5. [DOI] [PubMed] [Google Scholar]
- 24. Walsh CR, Larson MG, Evans JC, Djousse L, Ellison RC, Vasan RS, Levy D. Alcohol consumption and risk for congestive heart failure in the Framingham Heart Study. Ann Intern Med. 2002;136(3):181–91. [DOI] [PubMed] [Google Scholar]
- 25. Dhingra R, Nwanaji-Enwerem JC, Samet M, Ward-Caviness CK. DNA methylation age-environmental influences, health impacts, and its role in environmental epidemiology. Curr Environ Health Rep. 2018;5(3):317–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Joehanes R, Just AC, Marioni RE, Pilling LC, Reynolds LM, Mandaviya PR, Guan W, Xu T, Elks CE, Aslibekyan S et al. Epigenetic signatures of cigarette smoking. Circ Cardiovasc Genet. 2016;9(5):436–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Huang YT, Yang HI. Causal mediation analysis of survival outcome with multiple mediators. Epidemiology. 2017;28(3):370–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, Wiencke JK, Kelsey KT. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics. 2012;13(1):86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jenab M, Slimani N, Bictash M, Ferrari P, Bingham SA. Biomarkers in nutritional epidemiology: applications, needs and new horizons. Hum Genet. 2009;125(5–6):507–25. [DOI] [PubMed] [Google Scholar]
- 30. Dragsted LO, Gao Q, Pratico G, Manach C, Wishart DS, Scalbert A, Feskens EJM. Dietary and health biomarkers–time for an update. Genes Nutr. 2017;12(1):24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sae-Lee C, Corsi S, Barrow TM, Kuhnle GGC, Bollati V, Mathers JC, Byun HM. Dietary intervention modifies DNA methylation age assessed by the epigenetic clock. Mol Nutr Food Res. 2018;62(23):e1800092. [DOI] [PubMed] [Google Scholar]
- 32. Gensous N, Garagnani P, Santoro A, Giuliani C, Ostan R, Fabbri C, Milazzo M, Gentilini D, di Blasio AM, Pietruszka B et al. One-year Mediterranean diet promotes epigenetic rejuvenation with country- and sex-specific effects: a pilot study from the NU-AGE project. Geroscience. 2020;42(2):687–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Benayoun BA, Pollina EA, Brunet A. Epigenetic regulation of ageing: linking environmental inputs to genomic stability. Nat Rev Mol Cell Biol. 2015;16(10):593–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lee KW, Pausova Z. Cigarette smoking and DNA methylation. Front Genet. 2013;4:132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhang Y, Schottker B, Florath I, Stock C, Butterbach K, Holleczek B, Mons U, Brenner H. Smoking-associated DNA methylation biomarkers and their predictive value for all-cause and cardiovascular mortality. Environ Health Perspect. 2016;124(1):67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kaluza J, Hakansson N, Harris HR, Orsini N, Michaelsson K, Wolk A. Influence of anti-inflammatory diet and smoking on mortality and survival in men and women: two prospective cohort studies. J Intern Med. 2019;285(1):75–91. [DOI] [PubMed] [Google Scholar]
- 37. Schwingshackl L, Hoffmann G. Mediterranean dietary pattern, inflammation and endothelial function: a systematic review and meta-analysis of intervention trials. Nutr Metab Cardiovasc Dis. 2014;24(9):929–39. [DOI] [PubMed] [Google Scholar]
- 38. Irvin MR, Aslibekyan S, Do A, Zhi D, Hidalgo B, Claas SA, Srinivasasainagendra V, Horvath S, Tiwari HK, Absher DM et al. Metabolic and inflammatory biomarkers are associated with epigenetic aging acceleration estimates in the GOLDN study. Clin Epigenetics. 2018;10(1):56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ma J, Rebholz CM, Braun KVE, Reynolds LM, Aslibekyan S, Xia R, Biligowda NG, Huan T, Liu C, Mendelson MM et al. Whole blood DNA methylation signatures of diet are associated with cardiovascular disease risk factors and all-cause mortality. Circ Genom Precis Med. 2020;13(4):e002766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Souza RG, Gomes AC, Naves MM, Mota JF. Nuts and legume seeds for cardiovascular risk reduction: scientific evidence and mechanisms of action. Nutr Rev. 2015;73(6):335–47. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data sets analyzed in this study are available in the database of Genotypes and Phenotypes (dbGaP) repository with accession number phs000724.v7.p11.