

Abstract
SAMHD1 is a potent HIV-1 restriction factor that blocks reverse transcription in monocytes, dendritic cells and resting CD4+ T cells by decreasing intracellular dNTP pools. However, SAMHD1 may diminish innate immune sensing and antigen presentation, resulting in a weaker adaptive immune response. To date, the role of SAMHD1 on antiretroviral immunity remains unclear, as mouse SAMHD1 had no impact on murine retrovirus replication in prior in vivo studies. Here, we show that SAMHD1 significantly inhibits acute Friend retrovirus infection in mice. Pre-treatment with lipopolysaccharide (LPS), a significant driver of inflammation during HIV-1 infection, further unmasked a role for SAMHD1 in influencing immune responses. LPS treatment in vivo doubled the intracellular dNTP levels in immune compartments of SAMHD1 KO, but not WT mice. SAMHD1 KO mice exhibited higher plasma infectious viremia and proviral DNA loads than WT mice at 7 days post-infection (dpi), and proviral loads inversely correlated with a stronger CD8+ T cell response. SAMHD1 deficiency was also associated with weaker NK, CD4+ T and CD8+ T cell responses by 14 dpi and weaker neutralizing antibody responses by 28 dpi. Intriguingly, SAMHD1 influenced these cell-mediated immune (14 dpi) and neutralizing antibody (28 dpi) responses in male, but not female mice. Our findings formally demonstrate SAMHD1 as an antiretroviral factor in vivo that could promote adaptive immune responses in a sex-dependent manner. The requirement for LPS to unravel the SAMHD1 immunological phenotype suggests that co-morbidities associated with a ‘leaky’ gut barrier may influence the antiviral function of SAMHD1 in vivo.
Introduction
The existence of restriction factors and counterpart retroviral antagonists illustrate the long-standing evolutionary relationship between retroviruses and mammalian hosts (1, 2). Many simian immunodeficiency viruses (SIVs), including those in the HIV-2/SIVsm lineage, encode the Vpx/Vpr proteins to counteract a restriction factor known as SAMHD1 (3, 4). However, Vpx has not evolved in the HIV-1/SIVcpz lineage, and the Vpr proteins in this lineage do not antagonize SAMHD1 (5). SAMHD1 inhibits HIV-1 reverse transcription in monocytes, dendritic cells (DCs) and resting CD4 T cells by depleting the intracellular deoxynucleotide (dNTP) pool through its dNTP triphosphohydrolase activity (6–10). Under this scenario, SAMHD1 protects against HIV-1 infection by reducing overall virus replication. However, a counter-theory posits that enhanced virus replication in SAMHD1-deficient myeloid cells such as DCs may promote antigen presentation (11) and innate sensing via the cGAS/STING pathway (12) to direct a stronger adaptive immune response. Thus, a lack of ‘enforced viral replication’ in DCs with functional SAMHD1 may weaken adaptive immune responses. Such a mechanism could contribute to weaker adaptive immune responses in HIV-1 (where SAMHD1 is functionally restrictive) compared to HIV-2 infection (where SAMHD1 is antagonized by Vpx). In other words, SAMHD1 could be protective or pathogenic during retrovirus infection, depending on how it influences subsequent adaptive immune responses.
Investigating a potential role of SAMHD1 in shaping adaptive immunity would require a suitable in vivo model. Individuals with homozygous mutations in SAMHD1 were found in the human population. These individuals are at high risk for an autoimmune encephalopathy known as Aicardi-Goutierres Syndrome (AGS), which is associated with a persistent type I interferon (IFN-I) response (13, 14). However, individuals with AGS due to SAMHD1 mutations are extremely rare and would be difficult to study with respect to HIV-1 infection. Studies on rhesus macaques infected with SIV lacking Vpx may be a way to evaluate the role of SAMHD1 in vivo, but Vpx may have additional activities such as degrading the HUSH Complex that inhibits reverse transcription (15–18). Thus, to directly assess the effects of SAMHD1 in vivo, efforts were made to develop mice lacking SAMHD1 to evaluate their susceptibility to murine retroviruses (19, 20).
Previously, knockouts (KO) of the murine orthologues of HIV-1 restriction factors APOBEC3G and/or Tetherin/BST2 supported higher infection levels of Friend retrovirus (FV) complex, Moloney murine leukemia virus (MLV), murine AIDS (LP-BM5) and/or mouse mammary tumor virus (21–27). Studies on FV were particularly informative in documenting how these restriction factors influence adaptive immune responses (28–32). FV is a complex of a replication-competent helper virus, Friend MLV (F-MLV) and a replication-defective spleen focus-forming virus (SFFV). SFFV encodes an altered envelope protein (gp55R) that promotes the rapid proliferation of erythroblasts in mice with the Fv2-susceptibility allele, leading to severe splenomegaly (33). Murine Apobec3 and Tetherin/BST2 were shown to promote neutralizing antibody and cell-mediated immune responses against FV, respectively, even in genetic backgrounds such as B6 that were Fv2-resistant (22, 31). To date, the impact of SAMHD1 on adaptive immunity following infection with replication-competent retroviruses remain unknown.
C57BL/6 (B6) SAMHD1 KO mice showed elevated dNTP levels and modestly increased expression of IFN-I-stimulated genes (ISGs) compared to B6 wild-type (WT) mice, but Moloney MLV and FV infection were similar between WT and SAMHD1 KO mice (19, 20). By contrast, HIV-1 pseudotyped with the pan-tropic vesicular stomatitis virus G protein showed higher infection levels in SAMHD1 KO mice (19, 20). One explanation was that unlike lentiviruses that can replicate in nondividing cells that normally harbor low dNTP levels, most murine oncoretroviruses require active cell division to propagate (34); dNTP levels are relatively higher in actively dividing cells (35). While these results are interesting, HIV-1 is not an endemic retrovirus in mice, as it encounters blocks at multiple stages in murine cells both in vitro and in vivo (36). By contrast, MLVs have co-evolved with mice about 1 million years ago (37). Notably, murine SAMHD1 exhibited a modest inhibitory effect against Moloney MLV in vitro (38). In fact, an MLV-GFP vector exhibited 10-fold higher reverse transcription levels in bone marrow-derived DCs from SAMHD1 KO mice versus WT mice (39). An earlier study demonstrated that Vpx could alleviate an intrinsic restriction for MLV in nondividing cells (40), though there may be a further block in nuclear import of viral cDNA (41). Interestingly, FV was reported to infect DCs both in vitro and in vivo (42). These findings raised the possibility that murine SAMHD1 may restrict replication-competent retroviruses in vivo under certain conditions.
We reported that SAMHD1 limits NFκB and IFN-I signaling in human macrophages following treatment with the gram-negative bacterial cell wall component, LPS (43, 44). Ex vivo, primary macrophages from SAMHD1 KO mice expressed higher levels of the inflammatory cytokine TNFα than macrophages from SAMHD1 heterozygous mice following treatment with monophosphoryl lipid A (MPL-A), a vaccine adjuvant derived from LPS (43). Moreover, mouse SAMHD1 was reported to alter immunoglobulin somatic hypermutation (SHM) (45) and class switch recombination (CSR) (46) following LPS activation of B cells ex vivo. Notably, LPS is elevated in plasma from persons with chronic HIV-1 infection (47). This was likely due to the loss of Th17 cells that protect the integrity of the gut epithelial barrier, resulting in the translocation of microbes and/or microbial products from the gut lumen to the systemic circulation (48, 49). Importantly, LPS levels correlated with inflammation and immune activation during HIV-1 infection (50). Thus, a ‘leaky’ gut is a physiologically-relevant feature of chronic HIV-1 infection (51, 52). We therefore tested if pre-treatment of mice with LPS could unravel a role for SAMHD1 during FV infection.
Materials and Methods
Mice.
C57BL/6J (B6) and BALB/cJ mice were purchased from The Jackson Laboratory (Bar Harbor, ME). B6 mice are susceptible to FV infection and succumb to acute viremia, but recover from viremia by 14 dpi due multiple resistance alleles that govern erythroblast proliferation (Fv2r/r), cell-mediated immunity (H-2b/b) and neutralizing antibody responses (Rfv3r/r) (33, 53). SAMHD1 KO mice were generated in the B6 background from embryonic stem cells from the International Knockout Mouse Consortium as previously described (19). It encodes a gene trap cassette in intron 1 of the SAMHD1, resulting in exon 2 deletion and subsequent frameshift and premature stops in the SAMHD1 gene. Upon arrival at the University of Colorado, SAMHD1 KO mice were crossed to B6 mice and heterozygous mice were mated to each other, and progeny mice were genotyped to generate WT and SAMHD1 KO cohorts. This mating strategy was adopted to reduce potential genetic variation between the mouse cohorts. The mice used in the study ranged from 11 to 25 weeks of age. This study was approved by the University of Colorado Institutional Care and Use Committee Permit Number 00054.
FV infection.
Mice were infected intravenously with 10,000 spleen-focus forming units of FV complex, which is composed of a replication-competent F-MLV and replication-defective spleen-focus forming virus SFFV. The stock used in this study, a kind gift from Dr. Kim Hasenkrug (Rocky Mountain Laboratories, Hamilton, MT, USA), lacked a contaminant in early studies, lactate-dehydrogenase-elevating virus (54). Assays for virus replication described below specifically focused on F-MLV, which is critical for FV replication. By contrast, the inoculum used for in vivo infections were based on spleen-focus forming counts that were assayed after infection of BALB/c mice (55).
Measurement of intracellular dNTP levels.
dNTP measurement was conducted as previously described (56). Briefly, intracellular dNTPs were extracted by methanol-based extraction from frozen 2×106 cell pellets sent to Emory University and applied to the RT-based dNTP assay. For quantification, the products were run on a 14% urea-PAGE gel then analyzed using a PharosFX molecular imager and ImageJ software. The dNTP amount (pmole) in each sample was normalized by 1×106 cells for comparisons.
Plasma viral load.
Total RNA was extracted from 50 µl plasma using the EZNA Total RNA Kit I (Omega Bio-Tek), eluting in 100 µl water. 10 µl of RNA was added into a 1× 1-step TaqMan Reverse Transcriptase PCR reaction mix (Applied Biosystems) that included 10 pmol of F-MLV-specific primers: F-MLV sense, 5’-GGACAGAAACTACCGCCCTG-3’; F-MLV antisense, 5’-ACAA- CCTCAGACAACGAAGTAAGA-3’; and F-MLV probe, FAM-TCGCCACCCAGCAGTTTCAGCAGC-TAMRA. Quantitative PCR (qPCR) conditions in a Bio-Rad CFX96 cycler were as follows: 48°C for 15 min, 95°C for 10 min, 40 cycles of 95°C for 15 s, 60°C for 1 min. In vitro transcribed F-MLV-RNA of known copy number was used as a standard.
Proviral load.
Spleen and BM DNA were extracted using the EZNA Tissue DNA Kit (Omega Bio-Tek). 100 ng of DNA was added into a qPCR reaction containing the primers noted above and thermocycling conditions to measure F-MLV DNA copy numbers. Plasmid DNA containing F-MLV target sequences were used as a standard.
Infectious viremia.
Infectious viremia was measured as previously described (57). Briefly, plasma (5µl) was added into Mus dunni cells in a 48-well plate (~4000–8000 cells/well) in the presence of 4 µg/ml polybrene. After 48 h, DNA was extracted from the cells using the EZNA Tissue DNA Kit (Omega Bio-Tek), and proviral DNA copy numbers were measured by qPCR as described above.
Quantification of IgM and IgG subclasses in plasma.
Immulon-4 HBX plates (Thermo-Scientific) were coated with 200 ng/well with goat, anti-mouse kappa light chain (Bethyl) and incubated overnight at 4°C. The following day, the plates were blocked with Superblock (Pierce) for 2 h at room temperature, and serial dilutions of IgM, IgG1, IgG2b, IgG2c and IgG3 standards (Bethyl) were added using PBS as a diluent and incubated for 1.5 h. The plate was washed six times with PBS containing 0.25% Tween-20, then HRP-labeled conjugates were added and incubated for 1.5 h at room temperature. The conjugate concentrations were: IgM, 1:2000 (Bethyl); IgG1, 1:1000 (Bethyl); IgG2b, 1:2000 (Bethyl), IgG2c, 1:2000 (Southern Biotechnology); IgG3, 1:10,000 (Bethyl). After incubating for 1.5 h at room temperature, the plates were washed six times with PBS containing 0.25% Tween-20, and developed by adding TMB substrate (BioFX) and stopped with 0.3M sulfuric acid. Plates were read at 450 nm using a Victor X5 plate reader. Ig concentrations were interpolated from linear regression curves generated with the standards using GraphPad Prism 8.0.
Virus-specific antibody titers.
F-MLV virions generated from Mus dunni cells (57) were coated into Immobilon-4 HBX plates (Thermo-Scientific) at 200 ng/well. Plates were blocked with Superblock (Pierce) for 2 h then serial 2 to 10-fold dilutions of plasma were incubated for 1 to 1.5 h at room temperature. Plasma dilutions started at 1:100 for total IgG and IgG1 and 1:1000 for IgM, IgG2b and IgG2c. After 4 to 6 washes with PBS with 0.25% Tween-20, goat anti-mouse antibodies conjugated to HRP were added and incubate for 30 to 50 min at room temperature. The conjugate concentrations were as follows: total IgG, 1:4000 (Southern Biotechnology); IgM, 1:5000 (Bethyl); IgG1, 1:1000 (Bethyl); IgG2b 1:400 (Bethyl); IgG2c 1:400 (Southern Biotechnology). After 6 washes with PBS-T, 1:4000 streptavidin-conjugated HRP was added. The plates were developed by adding TMB substrate (BioFX) and stopped with 0.3M sulfuric acid. Plates were read at 450 nm using a Victor X5 plate reader. Binding curves were generated in Prism 8.0 using a one-site total equation and reciprocal titers at an OD450nm=0.25 or 0.5 were calculated.
Neutralizing antibody.
Neutralizing antibody titers were evaluated as previously described (31, 57). Briefly, 4-fold dilutions of plasma were incubated with ~50 infectious foci of F-MLV for 1 h at 37°C. The virus/plasma mixture was then added into Mus dunni cells in a 48-well plate. After 48 h, the cells were fixed briefly with ethanol, then incubated overnight with Mab 720, a monoclonal antibody that specifically binds to F-MLV envelope protein (58). The number of foci were counted using ImageJ software, and the percentage of foci relative to mock or the highest plasma dilution (1:5120) was calculated. Inhibitory concentrations (80%) were calculated using GraphPad Prism 8.0 using a one-site total equation.
Flow cytometry.
Spleen and bone marrow cells (4 x 106) were disaggregated into single-cell suspensions, treated with RBC lysis buffer (eBioscience) then stimulated with PMA (25 ng/ml) and ionomycin (1 µg/ml) (Sigma). After stimulation, the cells were stained with NK-1.1-allophycocyanin (PK136), CD3-AlexaFluor700 (17A2), CD8α-FITC (53–6.7) and CD4-PE-CF594 (RM4–5) (all from BD Biosciences) for 30 min. The cells were permeabilized in Perm/Fix buffer (BD Biosciences) then stained with IFNγ-PE (XMG1.2) (BD Biosciences). All cells were fixed in 1% paraformaldehyde before analysis with an LSR-II flow cytometer (BD Biosciences). Up to 500,000 events per sample was captured for data analysis using the FlowJo software (Treestar). Isotype controls (BD Biosciences) were used for gating.
Statistical analyses.
Differences between WT and SAMHD1 cohorts were evaluated using GraphPad Prism 8.0. Datasets were first evaluated for skewness using the Kolmogorov-Smirnov test. If the distribution of data values were not normal, a 2-tailed nonparametric Mann-Whitney U test was used. If distribution was normal, a 2-tailed Student’s t-test was used. Comparisons with p<0.05 were considered statistically significant, though comparisons with p-values between 0.05 and 0.10 were described as trending towards significance and noted in the graphs.
Results
SAMHD1 restricts a replication-competent retrovirus in mice.
Previous studies have not shown a role for SAMHD1 in murine retrovirus replication in vivo, as WT and SAMHD1 KO mice had similar levels of Friend retrovirus (FV) (19) and Moloney MLV replication (20). These data seemed inconsistent with in vitro studies demonstrating mouse SAMHD1 antiviral activity against murine retroviruses (38–40). However, the prior FV infection study (19) measured virus at 10 days post-infection (dpi), which was past the initial peak of virus replication (7 dpi). We therefore evaluated if SAMHD1 can inhibit FV infection at 7 dpi. WT and SAMHD1 KO mice were infected with 104 spleen-focus forming units (SFFU) of FV complex, and plasma and splenocytes were harvested to measure FV infection levels. To quantify FV replication, assays based on the helper virus (F-MLV) were used (57). Notably, SAMHD1 WT mice had significantly lower splenic proviral loads compared to SAMHD1 KO mice (Fig. 1A), and this was observed for both male and female mice (Fig. S1A). Plasma infectious titers, measured by quantifying proviral DNA levels in Mus dunni target cells 48 h after incubation with plasma, were also higher in SAMHD1 KO mice (Fig. 1B), although the difference did not reach statistical significance (p=0.066).
Figure 1.
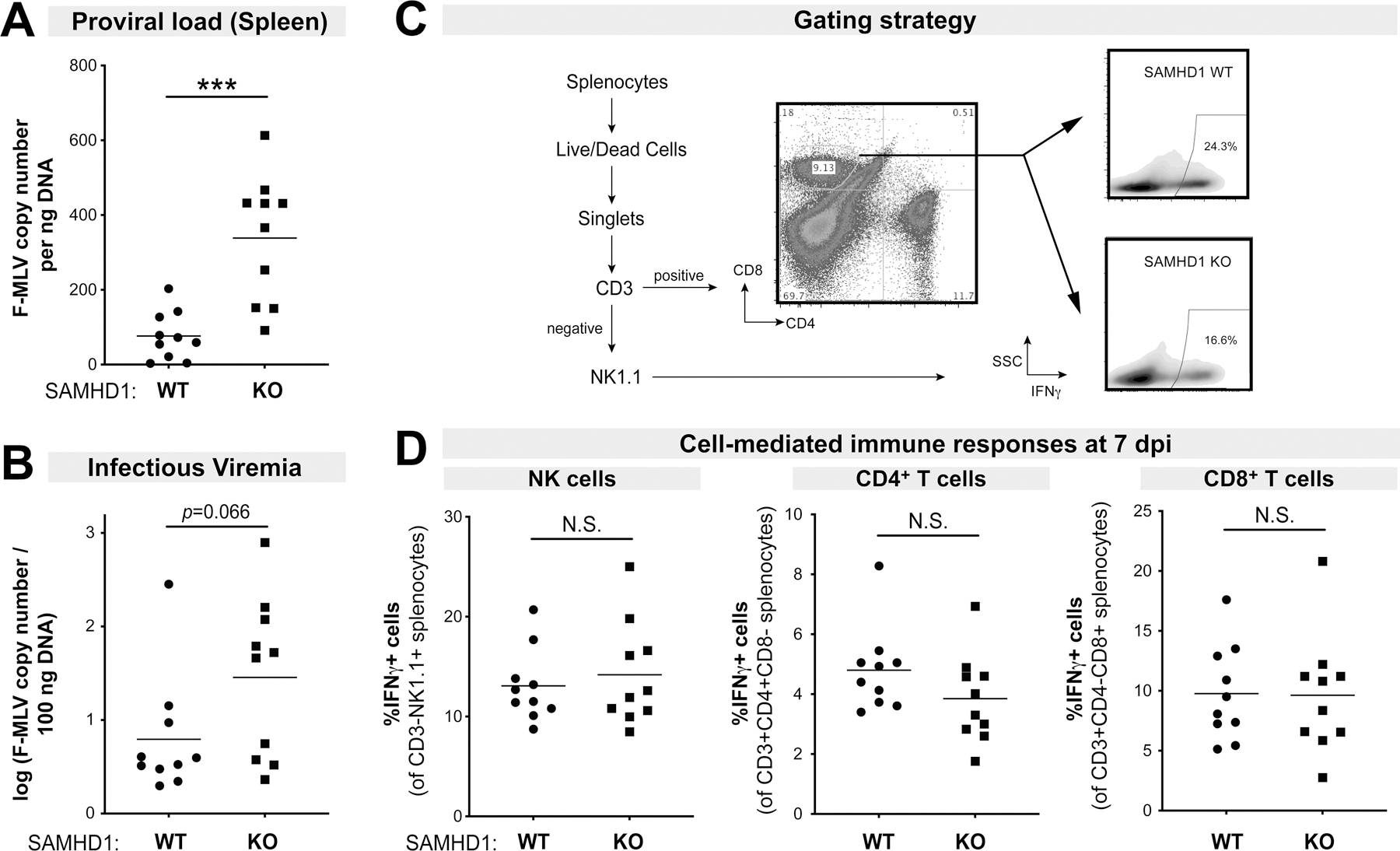
Effects of SAMHD1 on FV infection and cell-mediated immune responses in the absence of LPS. B6 SAMHD1 WT (n=10) and KO mice (n=10) were infected with 104 spleen focus forming units of FV without LPS pre-treatment, and at 7 dpi, spleen and plasma were harvested. FV infection were evaluated for (A) Proviral loads by qPCR of spleen DNA and (B) infectious viremia by measuring viral DNA levels in target Mus dunni cells after 48 h following incubation with 5µl plasma. Splenocytes were subjected to flow cytometry as shown in the (C) gating strategy. The WT and KO panels were derived from samples in Figure 4C. (D) Splenocytes were evaluated for IFNγ expression in (left) NK cells; (middle) CD4+ T cells and (right) CD8+ T cells by flow cytometry. For all panels, each dot corresponds to a mouse. Data were combined from 2 independent mouse cohorts that included both male and female mice (Fig. S1A). Two-way comparisons were evaluated using a 2-tailed Student’s t-test. ***, p<0.001; NS, not significant.
The mechanism for SAMHD1 restriction in vivo could be due to lower intracellular dNTP levels, as previously reported in WT versus SAMHD1 KO mice (19, 20). However, an improved adaptive immune response may also contribute to the antiviral effect. Specifically, cell-mediated immune responses could be readily detected in FV-infected B6 mice by 7 dpi (21, 22, 59). We therefore evaluated cell-mediated immune responses in the spleen by quantifying the percentage of natural killer (NK), CD4+ T and CD8+ T cells that express IFNγ by flow cytometry, using a similar gating strategy as we previously reported (22) (Fig. 1C). NK cell, CD4+ T cell and CD8+ T cell responses were not significantly different between WT and KO mice at 7 dpi (Fig. 1D). SAMHD1 also did not influence cell-mediated immune responses at 14 dpi (Fig. S1B). Thus, SAMHD1 functions as a retrovirus restriction factor in mice, but other factors may be required to unravel an effect of SAMHD1 on cell-mediated immunity.
Pre-treatment with LPS doubled intracellular dNTP levels in SAMHD1 KO mice.
LPS induced higher levels of TNFα production in SAMHD1-deficient myeloid cells, suggesting that SAMHD1 could inhibit inflammatory responses (43). We therefore hypothesized that pre-treatment with LPS, a bacterial component present in the systemic circulation of persons with HIV- 1 infection, may unveil a role for SAMHD1 in antiretroviral immunity. As SAMHD1 is a known dNTPase, we first tested how LPS exposure in vivo would impact SAMHD1-dependent intracellular dNTP levels. WT and SAMHD1 KO mice were treated intraperitoneally with 10 µg of LPS and splenocytes and bone marrow (BM) cells were harvested after 24 h. BM and spleen were chosen as these are the primary tissue compartments for FV replication (60). A 24 h timepoint was chosen, as inflammatory cytokines TNFα and IL6 triggered by LPS treatment of B6 mice peak at 2 h, wane by 6 h (61, 62), and normalize by 24 h (62).
As previously reported (19, 20), in the absence of LPS, SAMHD1 KO mice exhibited higher dNTP levels in both spleen and the BM compared to WT mice (Fig. 2). These differences were most pronounced for dATP, dGTP and dTTP. LPS treatment had no effect on dNTP levels in WT mice (p>0.05 for all pairwise comparisons). By contrast, in the presence of LPS, the fold-difference in dNTP levels between WT and SAMHD1 KO mice were significantly higher. Specifically, in vivo LPS treatment of BM and spleen cells doubled the intracellular dNTP levels (mean=2.1×; range: 1.5× spleen dCTP to 3.0× spleen dTTP) in both immune compartments of SAMHD1 KO mice (Fig. 2A–B). These findings reveal that LPS pre-treatment augmented the impact of SAMHD1 deficiency on the intracellular dNTP pool.
Figure 2.
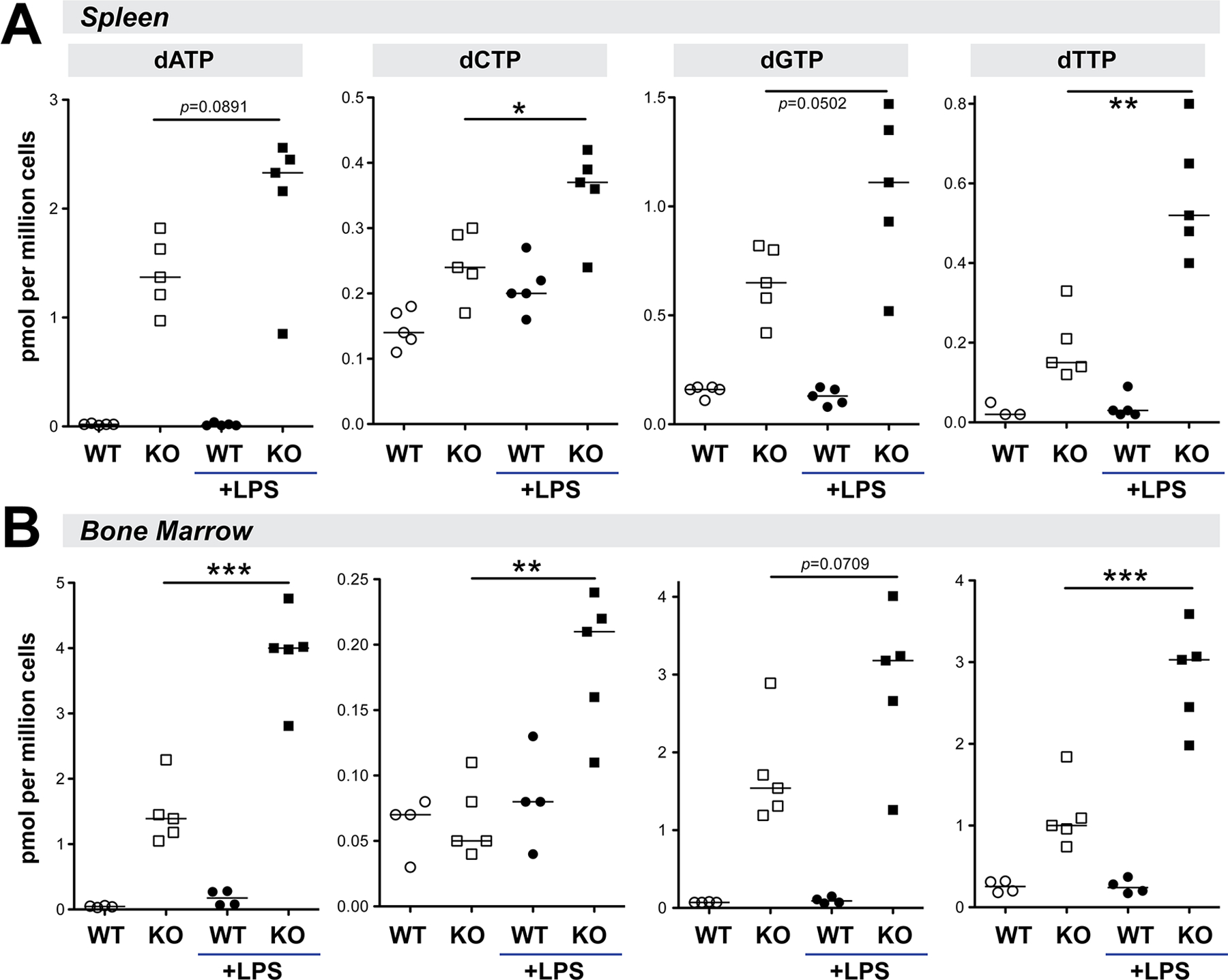
The effect of SAMHD1 deficiency on intracellular dNTP levels is more pronounced in the context of LPS exposure. WT and SAMHD1 KO mice were treated with 10µg LPS i.p. and (A) splenocytes and (B) BM cells were harvested after 24 h. Intracellular dNTP levels were quantified using an RT-based assay. Each dot corresponds to mice from one cohort. Both male and female mice were included in the cohorts. Differences between dNTP levels in SAMHD1 KO mice treated or not treated with LPS was evaluated using a 2-tailed Student’s t-test; *, p<0.05; **, p<0.01; ***, p<0.001.
SAMHD1 inhibits proviral DNA loads and virion infectivity in LPS-treated mice.
Administration of LPS in SIV-infected monkeys resulted in higher viral loads (63, 64), possibly due to heightened inflammation that increased the numbers of activated cellular targets. We thus tested how LPS pre-treatment affects acute FV replication in mice with or without SAMHD1. WT and SAMHD1 KO mice were infected with 104 SFFU of FV complex as in Fig. 1, then immediately treated with 10 µg LPS intraperitoneally. At 7 dpi, plasma, splenocytes and BM cells were harvested to measure FV infection levels. As expected, pre-treatment with LPS resulted in significantly higher splenic proviral DNA loads and plasma infectious viremia in WT mice (Fig. S1C).
In the context of LPS exposure, splenic proviral DNA loads were significantly higher in SAMHD1 KO mice (Fig. 3A); a similar pattern was observed in the BM (Fig. 3B). Plasma infectious viremia was significantly higher in SAMHD1 KO mice compared to WT mice (Fig. 3C). However, plasma viral loads (F-MLV RNA copy numbers) were not significantly different between WT and SAMHD1 KO mice at 7 dpi (Fig. 3D). This suggested that the infectivity of virions, taken as the ratio of infectious titers over plasma viral RNA copy numbers, were significantly lower in WT mice (Fig. 3E). Of note, splenic proviral DNA loads were higher in both male and female SAMHD1 KO mice compared to WT counterparts (Fig. 2F), similar to our findings in mice that were not pre-treated with LPS (Fig. S1A). Interestingly, the effect of SAMHD1 on infectious viremia, virion infectivity and BM proviral load were more pronounced in males (Fig. 2G–I). These findings confirm that SAMHD1 acts as a potent retrovirus restriction factor even in the setting of elevated inflammation due to LPS exposure.
Figure 3.
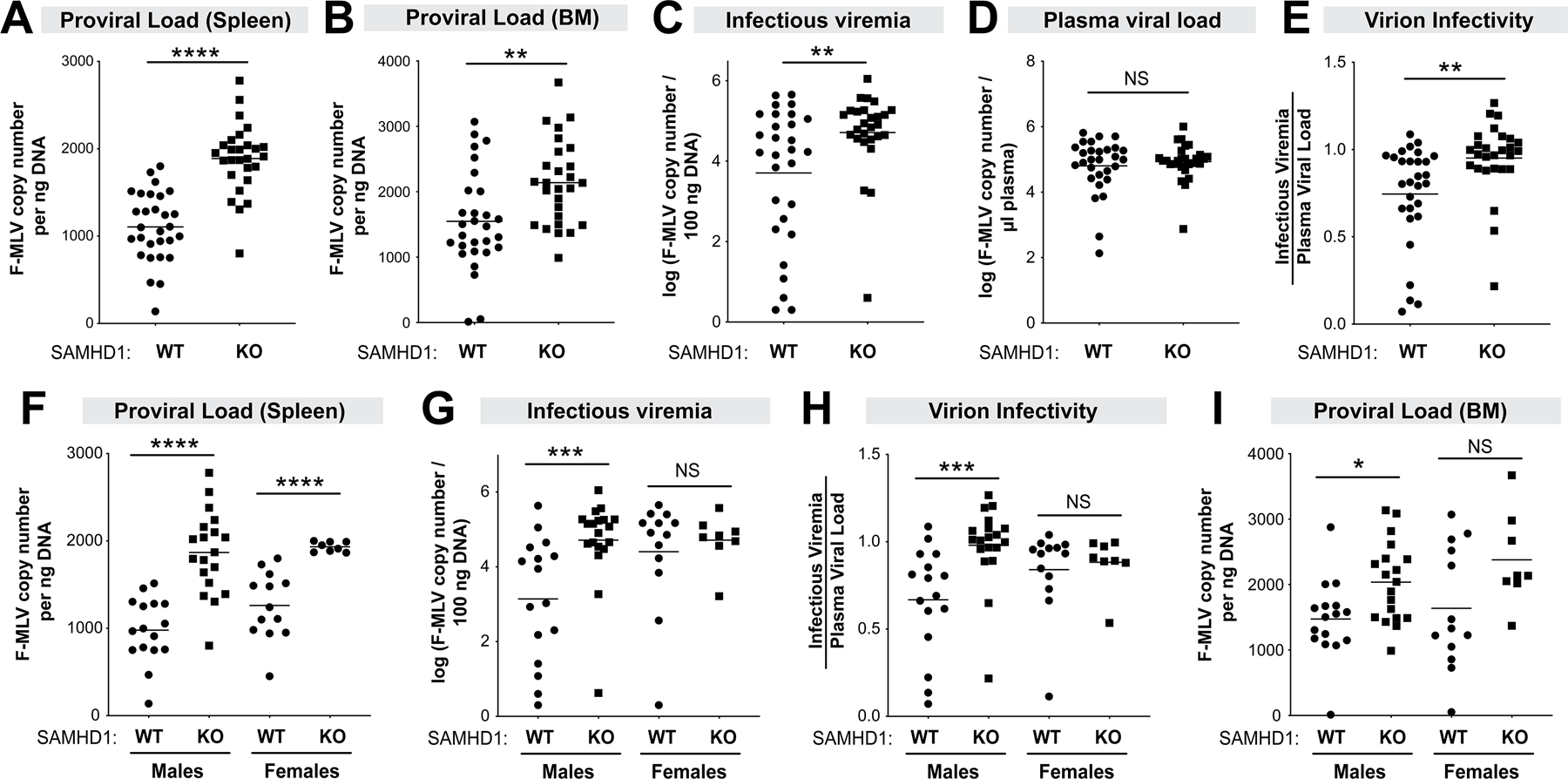
SAMHD1 protects against FV infection in mice pre-treated with LPS. WT (n=29; 16 males, 13 females) and SAMHD1 KO (n=27; 19 males, 8 females) mice were infected with 104 SFFU of FV complex. Plasma, spleen and BM samples were harvested at 7 dpi to evaluate FV infection levels using different methods. Proviral DNA load in the (A) spleen and (B) bone marrow were measured by qPCR and normalized to ng of input DNA. (C) Infectious viremia were determined by co-incubating 5µl plasma with Mus dunni cells for 48 h and measuring F-MLV DNA copy numbers; (D) Plasma viral load was measured using F-MLV qPCR. (E) Virion infectivity was calculated by taking the ratio of log-transformed plasma infectious titer (C) and viral load (D). (Lower panels) Data from panels A, C, E and B were analyzed separately for males and females for (F) proviral load in the spleen, (G) infectious viremia, (H) virion infectivity) and (I) proviral load in the bone marrow, respectively. For all panels, each data point corresponds to a mouse. The data in the upper panels were pooled from 5 independent cohorts of WT and SAMHD1 KO mice. Each experiment consisted of WT (n=5–8) and SAMHD1 KO (n=3–8) mice. Log-transformed data were analyzed using a 2-tailed unpaired Student’s t-test. *, p<0.05; **, p<0.01; ***, p<0.001; ****, p<0.0001; NS, not significant (p>0.05).
SAMHD1 promotes CD8+ T cell responses in LPS-treated mice at 7 dpi that inversely correlated with proviral DNA loads.
We next evaluated if SAMHD1 could influence cell-mediated immune responses in the context of LPS exposure. The percentage of natural killer (NK), CD4+ T and CD8+ T cells that express IFNγ were evaluated in splenocytes of WT and SAMHD1 KO mice by flow cytometry at 7 dpi (Fig. 1C). We found no significant difference in NK cell responses (Fig. 4A) and a trend (p=0.09) for higher CD4+ T cell responses (Fig. 4B) between WT and SAMHD1 KO mice at 7 dpi. Interestingly, SAMHD1 WT mice had significantly stronger CD8+ T cell responses compared to KO mice (Fig. 4C), and this CD8+ T cell response inversely correlated with proviral DNA loads from the same compartment and time point (Fig. 4D). The differences in CD8+ T cell responses between WT and SAMHD1 KO mice, and the inverse correlation with spleen proviral loads, were observed in both male and female mice (Fig. S2). These data suggest that SAMHD1 can promote early antiretroviral CD8+ T cell responses in the context of LPS exposure.
Figure 4.
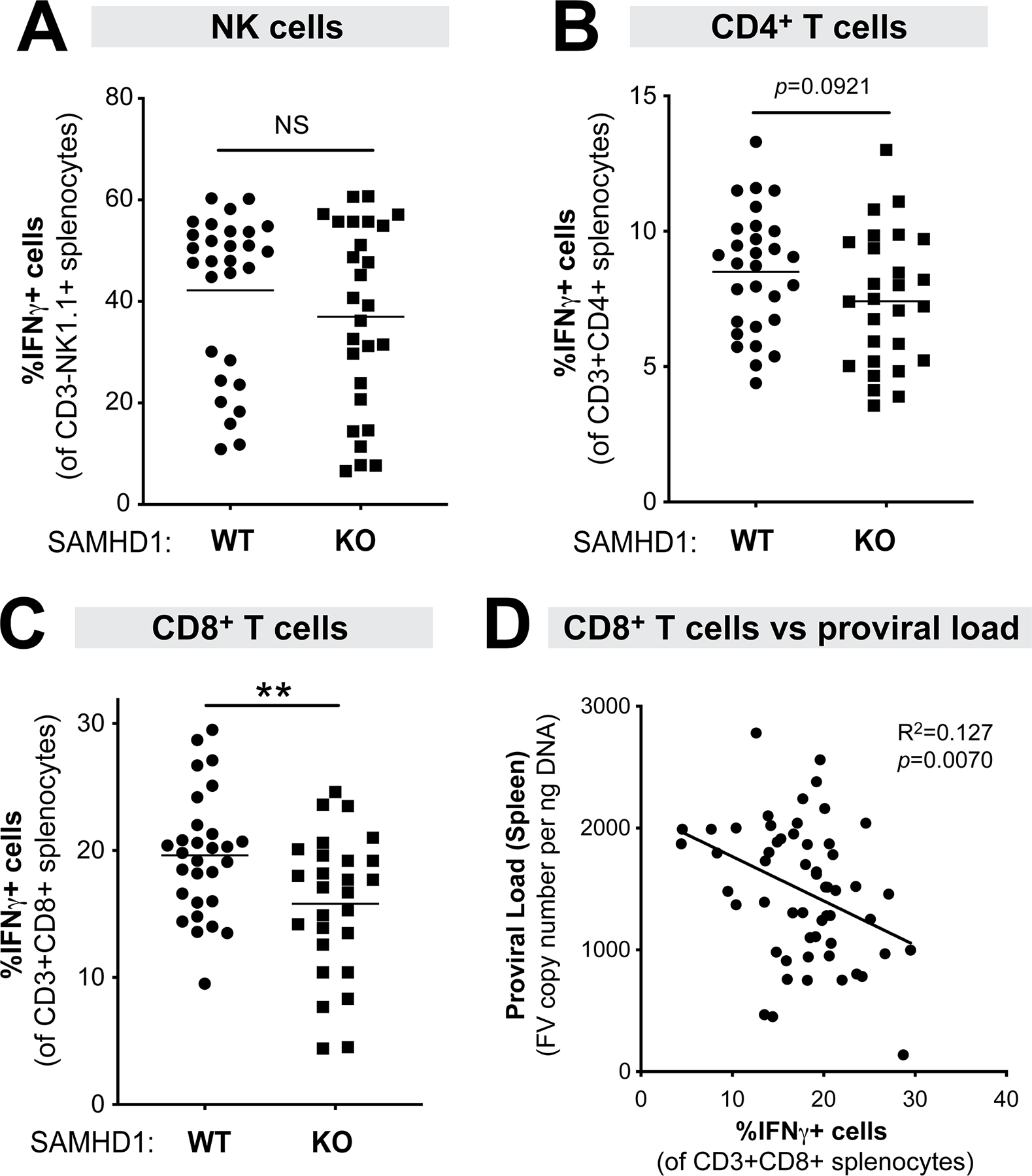
Cell-mediated immune responses in LPS-treated WT versus SAMHD1 KO mice at 7 dpi. Splenocytes from WT (n=29; 16 males, 13 females) and SAMHD1 KO (n=27; 19 males, 8 females) mice were subjected to flow cytometry using the gating strategy shown in Fig. 1C, evaluating the percentage of IFNγ+ cells in (A) NK cells (CD3-NK1.1+); (B) CD4 T cells (CD3+CD4+) and (C) CD8+ T cells (CD3+CD8+). (D) An inverse correlation was observed between CD8+ T cell responses and splenic proviral DNA load (from Fig. 3A) using linear regression. For all panels, each data point corresponds to a mouse. The data were pooled from 5 independent cohorts of WT and SAMHD1 KO mice. Each experiment consisted of WT (n=5–8) and SAMHD1 KO (n=3–8) mice. For panels A-C, data were analyzed using a 2-tailed unpaired Student’s t-test. **, p<0.01; NS, not significant (p>0.05).
SAMHD1 promoted cell-mediated immune responses in a sex-dependent manner at 14 dpi.
We next evaluated if these cell-mediated immunity profiles were maintained at a later time point. SAMHD1 WT and KO mice were treated with LPS, infected with 104 SFFU of FV complex and spleens were harvested at 14 dpi. At 14 dpi, multiple resistance genes in the B6 background (Fv2r/r, H-2b/b and Rfv3r/r) result in significant recovery from viremia (33, 53). The mean proviral DNA loads dropped 4.5-fold in WT and 6.4-fold in SAMHD1 KO mice from 7 dpi (Fig. 3A) to 14 dpi (Fig. S3A). Although the mean proviral DNA loads in SAMHD1 KO mice were higher than WT mice, these differences were not significantly different, even if partitioned between males and females (Fig. S3B). Nevertheless, differences in adaptive immune responses may still be observed at this later time point as a consequence of SAMHD1 effects during the acute stages (7 dpi). Flow cytometry was performed as in the 7 dpi time point (Figs. 1C–D and 4) to determine the percentage of IFNγ-expressing NK cell, CD4+ T cell and CD8+ T cells. SAMHD1 KO mice exhibited significantly weaker NK cell, CD4+ T cell and CD8+ T cell responses (Figs. 5A–C, left panels). Interestingly, the difference between SAMHD1 WT and KO mice were driven primary by male mice (Figs. 5A–C, middle panels) and not female mice (Figs. 5A–C, right panels). As expected, CD4+ and CD8+ T cell responses did not inversely correlate with proviral DNA loads (p>0.05 in both cases). Interestingly, we detected an inverse correlation between NK cell responses and proviral DNA loads at 14 dpi (Fig. S3C), and this was reflected in males, though the correlation did not achieve statistical significance (p=0.06). These data demonstrate a sex-dependent effect of SAMHD1 on cell-mediated immune responses in the context of LPS exposure.
Figure 5.
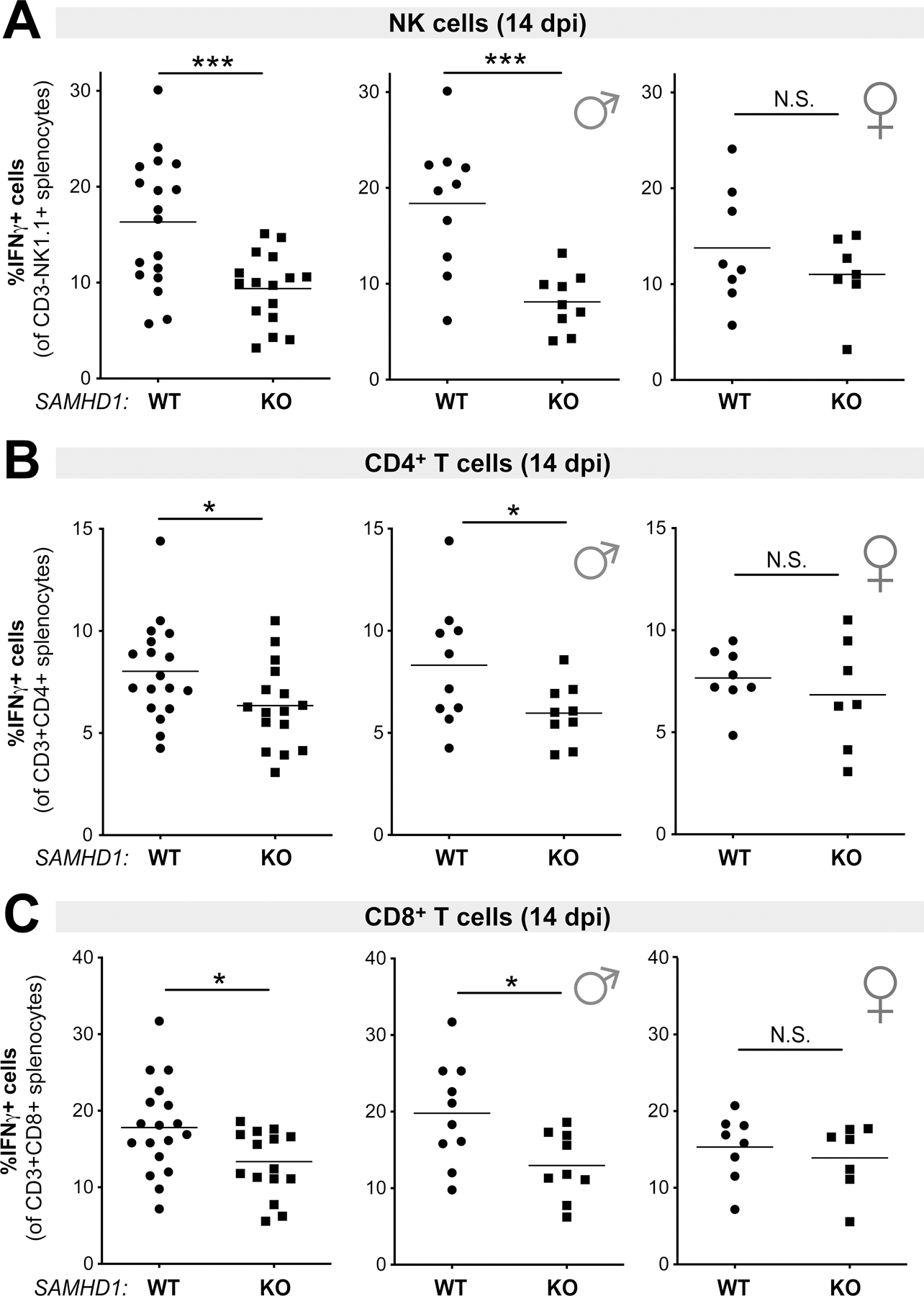
Cell-mediated immune responses in LPS-treated WT versus SAMHD1 KO mice at 14 dpi. Splenocytes from WT (n=18) and SAMHD1 KO (n=16) mice were subjected to flow cytometry, evaluating the percentage of IFNγ+ cells in (A) NK cells (CD3-NK1.1+); (B) CD4 T cells (CD3+CD4+) and (C) CD8+ T cells (CD3+CD8+). Left panels correspond to the full cohort, whereas middle and right panels partitioned the data into males and females, respectively. For all panels, each data point corresponds to a mouse. The data were pooled from 3 independent cohorts of WT and SAMHD1 KO mice. Each experiment consisted of WT (n=5–7) and SAMHD1 KO (n=5–6) mice. Data were analyzed using a 2-tailed unpaired Student’s t-test. *, p<0.05, ***, p<0.001; NS, not significant (p>0.05).
SAMHD1 promotes neutralizing antibody responses but not virus-specific IgG binding titers by 28 dpi.
SAMHD1 was recently reported to influence Ig SHM in B cells pre-treated with LPS (45). We thus tested if SAMHD1 had an effect on the humoral immune response at 28 dpi in the context of LPS pre-treatment. We first evaluated IgG binding titers against native F-MLV virions in an ELISA (Fig. 6A). IgG binding titers were similar between WT and SAMHD1 KO mice (Fig. 6B), in both male and female mice (Fig. 6C). By contrast, neutralizing antibody (NAb) responses were significantly weaker in SAMHD1 KO mice (Fig. 6E). Interestingly, the SAMHD1-dependent NAb response was biased towards male and not female mice (Fig. 6F). Thus, SAMHD1 did not influence the development of IgG antibodies that bind to the virus, but the neutralization capacity of those antibodies was impaired in the absence of SAMHD1 in male mice.
Figure 6.
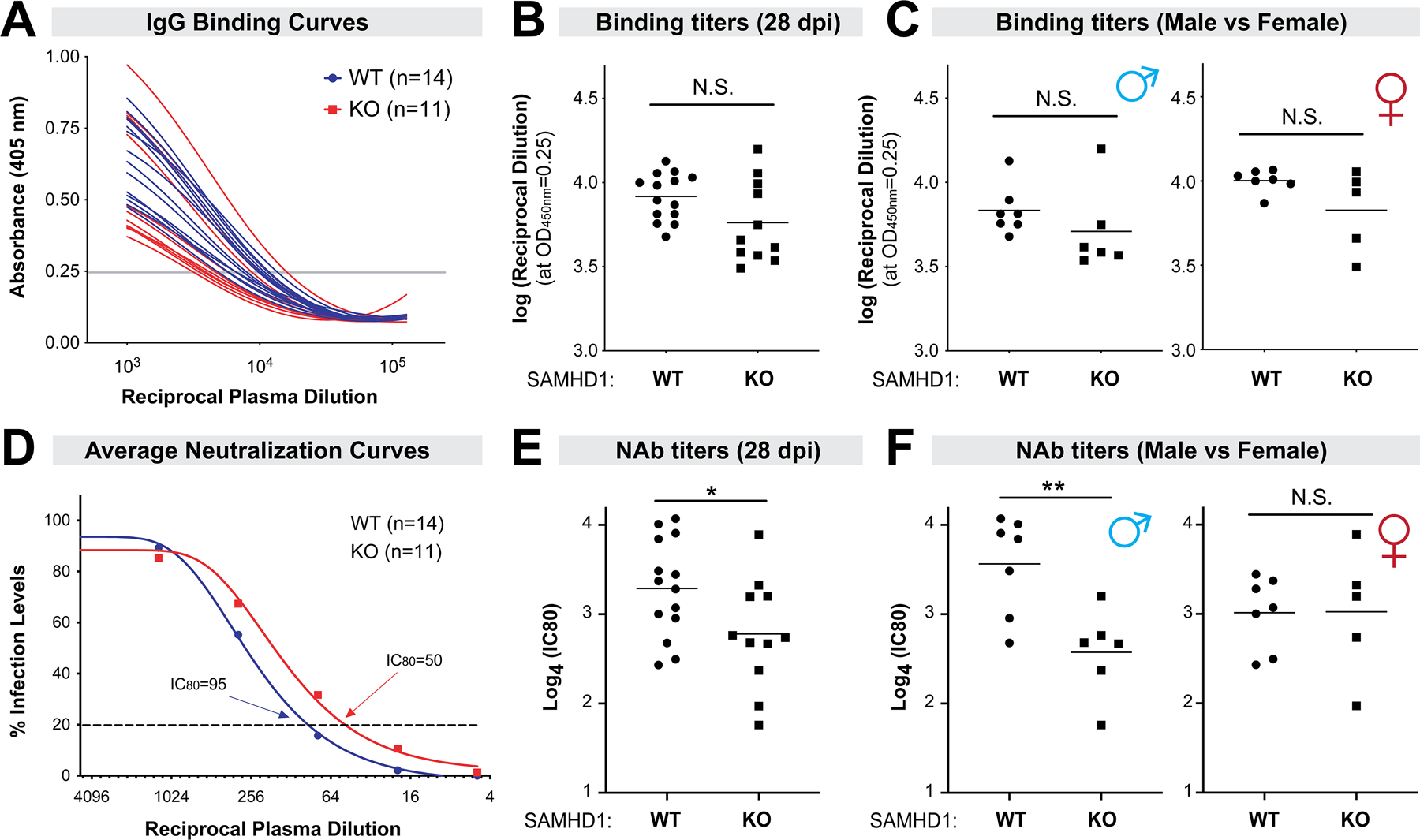
SAMHD1 promoted NAb responses at 28 dpi in a sex-dependent manner. Plasma samples were collected in FV-infected mice pre-treated with LPS. (A) FV-specific IgG binding titers were determined by ELISA using plates coated with native F-MLV virions. Binding titers were determined based on an arbitrary absorbance cut-off (0.25) from a best-fit curve and plotted in (B). (C) The data were partitioned between male and female mice. (D) Neutralizing antibody responses were evaluated by serially diluting plasma 4-fold with a known amount of F-MLV and plating on Mus dunni cells for 2 days. Foci of infection were counted using ImageJ software and neutralization curves were constructed. Shown here were the average %infection values for WT versus SAMHD1 KO mice. (E) 80% inhibitory concentrations (IC80) were calculated and compared between the cohorts of mice. (F) IC80 were subdivided between male and female mice. For all panels, each data point corresponds to a mouse. The data were pooled from 2 independent cohorts of WT and SAMHD1 KO mice. Experiment 1 had 7 WT and 6 SAMHD1 KO mice, and experiment 2 had 7 WT and 5 SAMHD1 KO mice. Log-transformed data were analyzed using a 2-tailed unpaired Student’s t-test. *, p<0.05; **, p<0.01; NS, not significant (p>0.05).
Since SAMHD1 was recently shown to influence Ig CSR (46), we next measured the levels of IgM, IgG1, IgG2b, IgG2c and IgG3 in the 28 dpi plasma from LPS pre-treated mice (Fig. 7A–E, Fig. S4A–E). We observed lower levels of IgG1 (Fig. 7B) and IgG2c (Fig. 7D) in SAMHD1 KO mice, and these trends were reflected in both male and female mice (Fig. S4B, S4D). As lower levels of IgG1 and IgG2c may explain why SAMHD1 KO mice had weaker neutralizing antibodies, we next determined the titers of IgM and IgG-subclass antibodies that reacted to FV virions in an ELISA (Fig. 7F–I and Fig. S4F–I). FV-specific IgG3 antibodies in most samples were near background levels, precluding robust comparisons (not shown). We did not observe a significant decrease in virus-reactive IgM, IgG1 and IgG2b binding titers in SAMHD1 KO mice (Fig. 7F–H). There was a trend for lower virus-reactive IgG2c in SAMHD1 KO mice (Fig. 7I), but these data were driven by female mice (Fig. S4I), where SAMHD1 had no significant effect on NAb responses (Fig. 6F). Thus, the decrease in neutralizing antibody responses in SAMHD1 KO mice (Fig. 6D–E) did not correlate with the effects of SAMHD1 on Ig CSR.
Figure 7.
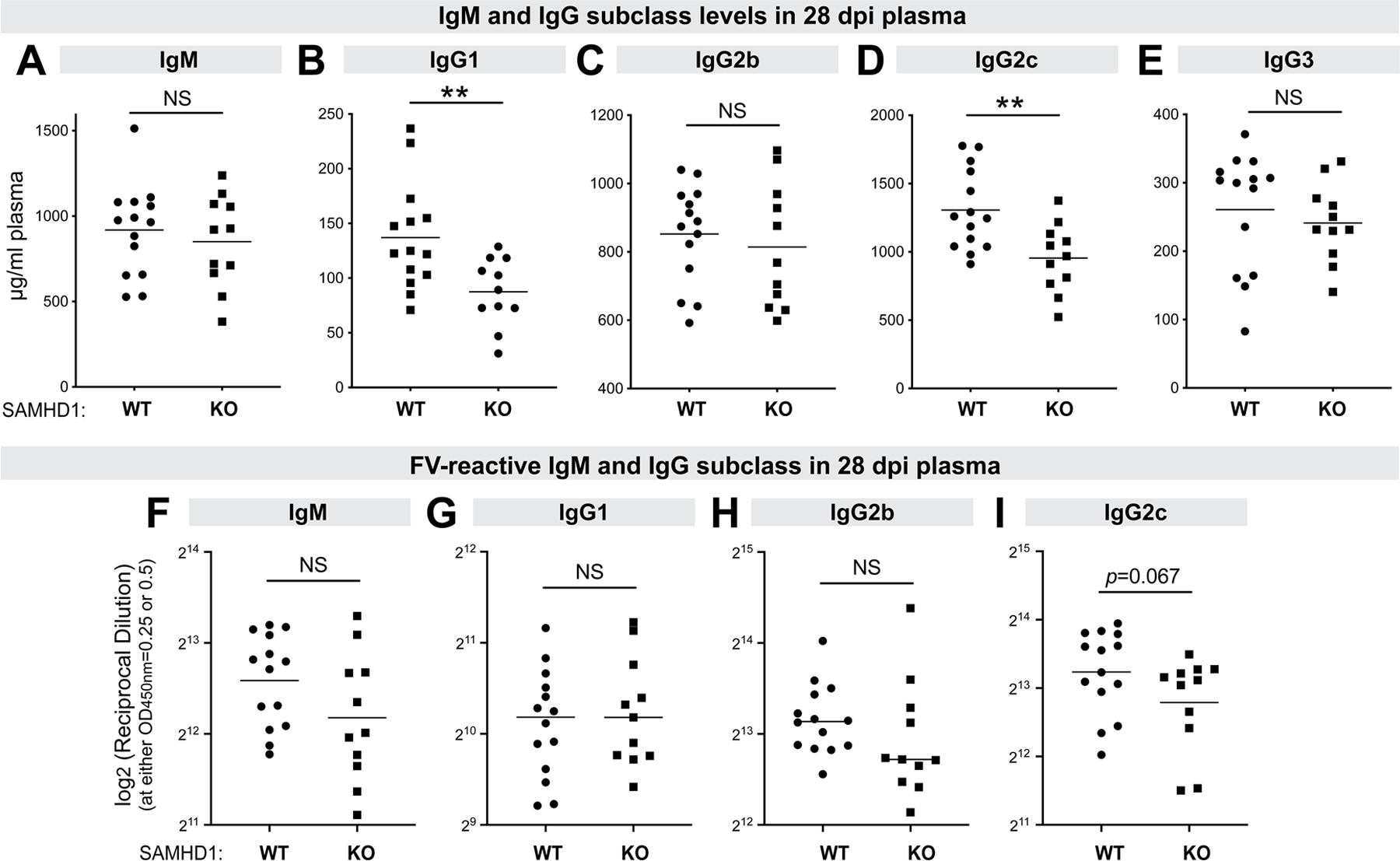
IgM and IgG subclass responses to FV. Plasma at 28 days post-infection in WT and SAMHD1 KO mice pre-treated with LPS were evaluated for (A-E) IgM and IgG subclass levels; and (F-I) IgM and IgG subclass binding titers to FV virions. (A-E) The concentrations of IgM, IgG1, IgG2b, IgG2c and IgG3 were measured in an ELISA. The data were interpolated from the respective antibody standard curves. (F-I) FV-reactive IgM, IgG1, IgG2b and IgG2c were measured in ELISA plates coated with FV virions using serial dilutions of plasma as noted in Fig. 5A–B. Reciprocal binding titers corresponding to an arbitrary absorbance cut-off (0.25 for IgM and IgG2c, and 0.5 for IgG1 and IgG2b) from a best-fit nonlinear regression curve were plotted in log2 scale. Each dot corresponds to a mouse and the data were pooled from 2 independent cohorts of WT and SAMHD1 KO mice. Experiment 1 had 7 WT and 6 SAMHD1 KO mice, and experiment 2 had 7 WT and 5 SAMHD1 KO mice. Differences between the cohorts were analyzed using a 2-tailed unpaired Student’s t-test or Mann-Whitney U test depending on whether the data distribution was normal. **, p<0.01; *, p<0.05; NS, not significant (p>0.05).
Discussion
Restriction factors initially identified to potently restrict HIV-1 – APOBEC3G and tetherin/BST2 – have orthologues in mice that could inhibit replication-competent murine retroviruses in vivo (21–27). However, to date, this did not appear to extend towards mouse SAMHD1 (19, 20), despite emerging evidence that SAMHD1 could modulate both innate and adaptive immune responses (65). Here, we demonstrate that murine SAMHD1 protects against Friend retrovirus infection in vivo. Proviral DNA loads at 7 dpi were significantly higher in SAMHD1 KO versus WT mice, independent of LPS treatment. These findings strengthen the case for SAMHD1 as a retrovirus restriction factor that is evolutionarily conserved from mice to humans in vivo. However, in the absence of LPS treatment, we did not detect an effect of SAMHD1 on cell-mediated immune responses. We showed in this report that LPS treatment doubled intracellular dNTP levels in SAMHD1 KO mice, but not WT mice. Previously, we showed that human SAMHD1 inhibited LPS-mediated NFκB activation by directly interacting with NFκB1/2 and inhibiting IκBα phosphorylation (43, 44). Following MPL-A treatment, inflammatory cytokine production (TNFα) was also augmented in murine SAMHD1 KO macrophages compared to heterozygous counterparts (43). LPS treatment of murine B cells ex vivo also unraveled an effect on Ig CSR and SHM (45, 46). Thus, LPS treatment may have activated additional pathways to potentiate an effect of SAMHD1 on antiretroviral immune responses (see next paragraph). Of note, heightened levels of plasma LPS is a common feature of chronic HIV-1 infection as well as in individuals with inflammatory bowel diseases, COVID-19 and cancer (52, 66, 67). It would be of interest to determine how SAMHD1 affects virus infections in established mouse models of mucosal barrier dysfunction.
The antiretroviral effect of SAMHD1 in vivo by 7 dpi could be due to its dNTPase activity that would maintain lower intracellular dNTP concentrations in critical target cells. However, in the context of LPS exposure, we also observed a negative correlation between proviral loads and CD8+ T cell responses at this time point. CD8+ T cell responses were stronger in WT compared to SAMHD1 KO mice at 7 dpi, and may have contributed to the overall antiretroviral effect of SAMHD1 in vivo. Based on studies on HIV-1 and human monocyte-derived DCs (11) and nonreplicating HIV-1 encoding a SIINFEKL epitope in SAMHD1 KO mice (12), we speculate that the effect of SAMHD1 on CD8+ T cell responses in vivo maybe due to an effect on antigen-presenting cells (APCs) such as DCs. FV infection was shown to inhibit DC maturation (42), although FV infection is usually not cytopathic. Higher FV replication in APCs due to SAMHD1 deficiency may have augmented the immunosuppressive effects of this virus, resulting in a weaker CD8+ T cell response, as well as weaker NK cell and CD4+ T cell responses in SAMHD1 KO mice at 14 dpi. A similar scenario may operate in HIV-1 infection. HIV-1 can suppress the maturation of primary DCs in vitro, and removal of SAMHD1 (through Vpx) increased HIV-1 replication and further suppressed DC maturation (68). We note that at this time, the specificity of the SAMHD1-dependent CD4+ and CD8+ T cell responses remains to be determined. Additional T cell epitopes of FV have been identified (69), and responses to individual epitopes could be altered by SAMHD1 deficiency if DCs are involved. Further studies that probe the role of SAMHD1 in antigen presentation and virus-specific T cell responses should provide more mechanistic insight. Importantly, the effects of SAMHD1 on cell-mediated immunity may also be NK- and T-cell intrinsic. Altered dNTP levels due to SAMHD1 deficiency in these cells could affect cellular metabolism, activation and proliferation. Overall, our work reinforces the model that SAMHD1 could significantly influence cell-mediated immunity during retrovirus infection in the context of LPS exposure. Further studies on the underlying mechanism(s) should provide interesting immunological insights on SAMHD1 biology in vivo.
SAMHD1 was previously implicated as a potential modulator of humoral immunity (45, 46). A study done using transgenic B cells specific to hen-egg lysozyme revealed that imbalances in the dNTP pool in SAMHD1-deficient B cells may affect transversion rates and reduce immunoglobulin somatic hypermutation (Ig SHM). In that study, CRISPR-engineered B cells were stimulated with LPS ex vivo then injected into congenic mice (45). SAMHD1 is involved in DNA repair and in resolving entanglements in DNA replication forks (70, 71). Thus, SAMHD1 deficiency may perturb the resolution of double-stranded DNA breaks induced by Activation Induced Cytidine Deaminase in B cells. In fact, a recent study showed that SAMHD1 localized to the Ig switch region and SAMHD1 deficiency resulted in lower class-switching of IgM to IgA, IgG1 and IgG3 in LPS-activated B cells ex vivo (46). To date, it remains unknown whether these effects of SAMHD1 on Ig SHM and CSR would extend to virus infections. Here, we demonstrate that SAMHD1 promoted FV-specific neutralizing antibody responses in vivo. The levels of IgG1 and IgG2c antibodies in SAMHD1 KO mice were decreased, consistent with the reported effect of SAMHD1 on Ig CSR. These findings were intriguing, as IgG2c antibodies primarily mediate antibody neutralization against FV, and passive protection from 28 dpi FV antisera was diminished in mice lacking FcγRIV (29, 72). However, FV-reactive IgG1 and IgG2c were similar between WT and SAMHD1 KO mice. Given that there was no impact on the titers of virus-reactive binding IgG antibodies, we speculate that the effects of SAMHD1 on neutralizing antibody responses may be at the level of Ig SHM. Aside from potential effects of SAMHD1 on dNTP levels and DNA repair, SAMHD1 also promoted the release of noninfectious virions at 7 dpi. We reported a similar phenomenon with Apobec3/Rfv3, and proposed that the release of Apobec3+ noninfectious virions during acute infection in mice may help prime a stronger antibody response (57). Of note, SAMHD1 could restrict HIV-1 not only at the reverse transcription step, but also during endogenous reverse transcription in the virion. HIV-1 virions with lower levels of endogenous reverse transcription due to SAMHD1 activity were found to be less infectious in vitro (14, 73). Thus, multiple mechanisms could explain how SAMHD1 influenced neutralizing antibody responses that could be tested in subsequent work. Follow-up studies should also help confirm if LPS is required for SAMHD1 to influence neutralizing antibody responses against retroviruses, as well as virus families for which SAMHD1 had no in vitro restriction activity.
The current study unraveled a sex-dependent effect of SAMHD1 on humoral and cell-mediated immune responses. In HIV-1 infection, sex-dependent differences in viral load and innate immunity have been reported (74). In fact, macrophages derived from peripheral blood of male donors were more susceptible to HIV-1 infection than macrophages from female donors (75). SAMHD1 restriction activity is inhibited by phosphorylation at T592 by cyclin-dependent kinases CDK1 and CDK2 (76–78). The authors reported that SAMHD1 in male macrophages expressed higher levels of CDK1 and were hyperphosphorylated at position T592 compared to that of females (75). This would suggest that removal of SAMHD1 in males should have no phenotypic effect on immune responses, as SAMHD1 was already inactivated due to hyperphosphorylation. However, we observed the opposite effect in mice, as removal of SAMHD1 had a significant effect on adaptive immunity in males, but not females. Mouse SAMHD1 is phosphorylated at multiple sites, including an analogous site (T634) to human SAMHD1 T592 (38, 39). It would be of interest to document the phosphorylation status of murine SAMHD1 in APCs from male versus female mice pre-treated with LPS, how it potentially affects antigen presentation during FV infection in the context of LPS exposure, and subsequent cell-mediated and neutralizing antibody responses. Of note, sexual dimorphism in viral infections and autoimmunity were previously documented in mice, and have been linked to the IFN-I response (79, 80). SAMHD1 KO mice express modestly elevated levels of IFN-stimulated genes (19, 20). One hypothesis to pursue in future work will be to test if X-linked immune genes such as TLR7, a critical innate sensor for murine retroviruses in vivo (81, 82), could direct divergent IFN-I responses in male versus female mice. In fact, TLR7 signaling has been linked to the development of T-Bet+ B cells (83) that could also explain the sex-dependent difference in neutralizing antibody responses that we observed.
To conclude, we provide evidence that SAMHD1 acts as a retrovirus restriction factor that could promote stronger adaptive immune responses in vivo, confirming its role as a protective antiretroviral factor. An immunological phenotype was uncovered after pre-exposure of mice to LPS. The mechanism for how LPS unraveled an immunological phenotype of SAMHD1 in vivo was linked to the disproportionate increase in dNTP levels (in this study) and inflammatory cytokines such as TNFα (43) in SAMHD1-deficient versus SAMHD1-sufficient mice. Interestingly, adaptive immune responses that were influenced by SAMHD1 at later time points were sex-dependent. Further investigation on the underlying mechanisms governing the diverse SAMHD1 phenotypes in the FV infection model may unravel useful insights on how to harness SAMHD1 biology for better HIV-1 control.
Supplementary Material
Key Points.
LPS augmented the impact of SAMHD1 deficiency on intracellular dNTP levels
SAMHD1 protected mice from acute Friend retrovirus replication in vivo
SAMHD1 promoted immune responses in a sex-dependent manner in vivo
Acknowledgments
We thank the University of Colorado Anschutz Medical Campus (CU-AMC) vivarium for assistance with mouse husbandry.
Funding support
The work was supported by NIH R01 grants AI116603 (MLS), AI136581 (BK), AI162633 (BK) and AI141495 (LW), Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) 369799452-TRR237 and BE 5877/2-1 (RB) and Institutional funds from the CU-AMC Department of Medicine (MLS). STJ was supported by an Immunology T32 grant AI007405.
References
- 1.Duggal NK, and Emerman M 2012. Evolutionary conflicts between viruses and restriction factors shape immunity. Nat Rev Immunol 12: 687–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Malim MH, and Bieniasz PD 2012. HIV Restriction Factors and Mechanisms of Evasion. Cold Spring Harb Perspect Med 2: a006940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Laguette N, Sobhian B, Casartelli N, Ringeard M, Chable-Bessia C, Segeral E, Yatim A, Emiliani S, Schwartz O, and Benkirane M 2011. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature 474: 654–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hrecka K, Hao C, Gierszewska M, Swanson SK, Kesik-Brodacka M, Srivastava S, Florens L, Washburn MP, and Skowronski J 2011. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature 474: 658–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lim ES, Fregoso OI, McCoy CO, Matsen FA, Malik HS, and Emerman M 2012. The ability of primate lentiviruses to degrade the monocyte restriction factor SAMHD1 preceded the birth of the viral accessory protein Vpx. Cell host & microbe 11: 194–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lahouassa H, Daddacha W, Hofmann H, Ayinde D, Logue EC, Dragin L, Bloch N, Maudet C, Bertrand M, Gramberg T, Pancino G, Priet S, Canard B, Laguette N, Benkirane M, Transy C, Landau NR, Kim B, and Margottin-Goguet F 2012. SAMHD1 restricts the replication of human immunodeficiency virus type 1 by depleting the intracellular pool of deoxynucleoside triphosphates. Nat Immunol 13: 223–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Descours B, Cribier A, Chable-Bessia C, Ayinde D, Rice G, Crow Y, Yatim A, Schwartz O, Laguette N, and Benkirane M 2012. SAMHD1 restricts HIV-1 reverse transcription in quiescent CD4(+) T-cells. Retrovirology 9: 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baldauf HM, Pan X, Erikson E, Schmidt S, Daddacha W, Burggraf M, Schenkova K, Ambiel I, Wabnitz G, Gramberg T, Panitz S, Flory E, Landau NR, Sertel S, Rutsch F, Lasitschka F, Kim B, Konig R, Fackler OT, and Keppler OT 2012. SAMHD1 restricts HIV-1 infection in resting CD4(+) T cells. Nature Med 18: 1682–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gelais C St, de Silva S, Amie SM, Coleman CM, Hoy H, Hollenbaugh JA, Kim B, and Wu L 2012. SAMHD1 restricts HIV-1 infection in dendritic cells (DCs) by dNTP depletion, but its expression in DCs and primary CD4+ T-lymphocytes cannot be upregulated by interferons. Retrovirology 9: 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goldstone DC, Ennis-Adeniran V, Hedden JJ, Groom HC, Rice GI, Christodoulou E, Walker PA, Kelly G, Haire LF, Yap MW, de Carvalho LP, Stoye JP, Crow YJ, Taylor IA, and Webb M 2011. HIV-1 restriction factor SAMHD1 is a deoxynucleoside triphosphate triphosphohydrolase. Nature 480: 379–382. [DOI] [PubMed] [Google Scholar]
- 11.Ayinde D, Bruel T, Cardinaud S, Porrot F, Prado JG, Moris A, and Schwartz O 2015. SAMHD1 limits HIV-1 antigen presentation by monocyte-derived dendritic cells. J Virol 89: 6994–7006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maelfait J, Bridgeman A, Benlahrech A, Cursi C, and Rehwinkel J 2016. Restriction by SAMHD1 Limits cGAS/STING-Dependent Innate and Adaptive Immune Responses to HIV-1. Cell Rep 16: 1492–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rice GI, Bond J, Asipu A, Brunette RL, Manfield IW, Carr IM, Fuller JC, Jackson RM, Lamb T, Briggs TA, Ali M, Gornall H, Couthard LR, Aeby A, Attard-Montalto SP, Bertini E, Bodemer C, Brockmann K, Brueton LA, Corry PC, Desguerre I, Fazzi E, Cazorla AG, Gener B, Hamel BC, Heiberg A, Hunter M, van der Knaap MS, Kumar R, Lagae L, Landrieu PG, Lourenco CM, Marom D, McDermott MF, van der Merwe W, Orcesi S, Prendiville JS, Rasmussen M, Shalev SA, Soler DM, Shinawi M, Spiegel R, Tan TY, Vanderver A, Wakeling EL, Wassmer E, Whittaker E, Lebon P, Stetson DB, Bonthron DT, and Crow YJ 2009. Mutations involved in Aicardi-Goutieres syndrome implicate SAMHD1 as regulator of the innate immune response. Nat Genet 41: 829–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Coggins SA, Mahboubi B, Schinazi RF, and Kim B 2020. SAMHD1 Functions and Human Diseases. Viruses 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fujita M, Nomaguchi M, Adachi A, and Otsuka M 2012. SAMHD1-Dependent and -Independent Functions of HIV-2/SIV Vpx Protein. Front Microbiol 3: 297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chougui G, Munir-Matloob S, Matkovic R, Martin MM, Morel M, Lahouassa H, Leduc M, Ramirez BC, Etienne L, and Margottin-Goguet F 2018. HIV-2/SIV viral protein X counteracts HUSH repressor complex. Nat Microbiol 3: 891–897. [DOI] [PubMed] [Google Scholar]
- 17.Baldauf HM, Stegmann L, Schwarz SM, Ambiel I, Trotard M, Martin M, Burggraf M, Lenzi GM, Lejk H, Pan X, Fregoso OI, Lim ES, Abraham L, Nguyen LA, Rutsch F, Konig R, Kim B, Emerman M, Fackler OT, and Keppler OT 2017. Vpx overcomes a SAMHD1-independent block to HIV reverse transcription that is specific to resting CD4 T cells. Proc Natl Acad Sci USA 114: 2729–2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yurkovetskiy L, Guney MH, Kim K, Goh SL, McCauley S, Dauphin A, Diehl WE, and Luban J 2018. Primate immunodeficiency virus proteins Vpx and Vpr counteract transcriptional repression of proviruses by the HUSH complex. Nat Microbiol 3: 1354–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Behrendt R, Schumann T, Gerbaulet A, Nguyen LA, Schubert N, Alexopoulou D, Berka U, Lienenklaus S, Peschke K, Gibbert K, Wittmann S, Lindemann D, Weiss S, Dahl A, Naumann R, Dittmer U, Kim B, Mueller W, Gramberg T, and Roers A 2013. Mouse SAMHD1 has antiretroviral activity and suppresses a spontaneous cell-intrinsic antiviral response. Cell Rep 4: 689–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rehwinkel J, Maelfait J, Bridgeman A, Rigby R, Hayward B, Liberatore RA, Bieniasz PD, Towers GJ, Moita LF, Crow YJ, Bonthron DT, and Reis e Sousa C 2013. SAMHD1-dependent retroviral control and escape in mice. EMBO J 32: 2454–2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barrett BS, Smith DS, Li SX, Guo K, Hasenkrug KJ, and Santiago ML 2012. A single nucleotide polymorphism in tetherin promotes retrovirus restriction in vivo. PLoS Pathog 8: e1002596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li SX, Barrett BS, Heilman KJ, Messer RJ, Liberatore RA, Bieniasz PD, Kassiotis G, Hasenkrug KJ, and Santiago ML 2014. Tetherin Promotes the Innate and Adaptive Cell-Mediated Immune Response against Retrovirus Infection In Vivo. J Immunol 193: 306–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Santiago ML, Smith DS, Barrett BS, Montano M, Benitez RL, Pelanda R, Hasenkrug KJ, and Greene WC 2011. Persistent Friend virus replication and disease in Apobec3-deficient mice expressing functional B-cell-activating factor receptor. J Virol 85: 189–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Okeoma CM, Lovsin N, Peterlin BM, and Ross SR 2007. APOBEC3 inhibits mouse mammary tumour virus replication in vivo. Nature 445: 927–930. [DOI] [PubMed] [Google Scholar]
- 25.Low A, Okeoma CM, Lovsin N, de las Heras M, Taylor TH, Peterlin BM, Ross SR, and Fan H 2009. Enhanced replication and pathogenesis of Moloney murine leukemia virus in mice defective in the murine APOBEC3 gene. Virology 385: 455–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Takeda E, Tsuji-Kawahara S, Sakamoto M, Langlois MA, Neuberger MS, Rada C, and Miyazawa M 2008. Mouse APOBEC3 restricts friend leukemia virus infection and pathogenesis in vivo. J Virol 82: 10998–11008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liberatore RA, and Bieniasz PD 2011. Tetherin is a key effector of the antiretroviral activity of type I interferon in vitro and in vivo. Proc Natl Acad Sci USA 108: 18097–18101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li SX, Barrett BS, Guo K, Kassiotis G, Hasenkrug KJ, Dittmer U, Gibbert K, and Santiago ML 2016. Tetherin/BST-2 promotes dendritic cell activation and function during acute retrovirus infection. Sci Rep 6: 20425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Halemano K, Barrett BS, Heilman KJ, Morrison TE, and Santiago ML 2015. Requirement for Fc effector mechanisms in the APOBEC3/Rfv3-dependent neutralizing antibody response. J Virol 89: 4011–4014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Halemano K, Guo K, Heilman KJ, Barrett BS, Smith DS, Hasenkrug KJ, and Santiago ML 2014. Immunoglobulin somatic hypermutation by APOBEC3/Rfv3 during retroviral infection. Proc Natl Acad Sci USA 111: 7759–7764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Santiago ML, Montano M, Benitez R, Messer RJ, Yonemoto W, Chesebro B, Hasenkrug KJ, and Greene WC 2008. Apobec3 encodes Rfv3, a gene influencing neutralizing antibody control of retrovirus infection. Science 321: 1343–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Santiago ML, Benitez RL, Montano M, Hasenkrug KJ, and Greene WC 2010. Innate retroviral restriction by Apobec3 promotes antibody affinity maturation in vivo. J Immunol 185: 1114–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dittmer U, Sutter K, Kassiotis G, Zelinskyy G, Banki Z, Stoiber H, Santiago ML, and Hasenkrug KJ 2019. Friend retrovirus studies reveal complex interactions between intrinsic, innate and adaptive immunity. FEMS Microbiol Rev 43: 435–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yamashita M, and Emerman M 2006. Retroviral infection of non-dividing cells: old and new perspectives. Virology 344: 88–93. [DOI] [PubMed] [Google Scholar]
- 35.Amie SM, Noble E, and Kim B 2013. Intracellular nucleotide levels and the control of retroviral infections. Virology 436: 247–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bieniasz PD, and Cullen BR 2000. Multiple blocks to human immunodeficiency virus type 1 replication in rodent cells. J Virol 74: 9868–9877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bamunusinghe D, Naghashfar Z, Buckler-White A, Plishka R, Baliji S, Liu Q, Kassner J, Oler AJ, Hartley J, and Kozak CA 2016. Sequence Diversity, Intersubgroup Relationships, and Origins of the Mouse Leukemia Gammaretroviruses of Laboratory and Wild Mice. J Virol 90: 4186–4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang F, Gelais C St, de Silva S, Zhang H, Geng Y, Shepard C, Kim B, Yount JS, and Wu L 2016. Phosphorylation of mouse SAMHD1 regulates its restriction of human immunodeficiency virus type 1 infection, but not murine leukemia virus infection. Virology 487: 273–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wittmann S, Behrendt R, Eissmann K, Volkmann B, Thomas D, Ebert T, Cribier A, Benkirane M, Hornung V, Bouzas NF, and Gramberg T 2015. Phosphorylation of murine SAMHD1 regulates its antiretroviral activity. Retrovirology 12: 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kaushik R, Zhu X, Stranska R, Wu Y, and Stevenson M 2009. A cellular restriction dictates the permissivity of nondividing monocytes/macrophages to lentivirus and gammaretrovirus infection. Cell host & microbe 6: 68–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gramberg T, Kahle T, Bloch N, Wittmann S, Mullers E, Daddacha W, Hofmann H, Kim B, Lindemann D, and Landau NR 2013. Restriction of diverse retroviruses by SAMHD1. Retrovirology 10: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Balkow S, Krux F, Loser K, Becker JU, Grabbe S, and Dittmer U 2007. Friend retrovirus infection of myeloid dendritic cells impairs maturation, prolongs contact to naive T cells, and favors expansion of regulatory T cells. Blood 110: 3949–3958. [DOI] [PubMed] [Google Scholar]
- 43.Chen S, Bonifati S, Qin Z, Gelais C St, Kodigepalli KM, Barrett BS, Kim SH, Antonucci JM, Ladner KJ, Buzovetsky O, Knecht KM, Xiong Y, Yount JS, Guttridge DC, Santiago ML, and Wu L 2018. SAMHD1 suppresses innate immune responses to viral infections and inflammatory stimuli by inhibiting the NF-kappaB and interferon pathways. Proc Natl Acad Sci USA 115: E3798–E3807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Espada CE, Gelais C St, Bonifati S, Maksimova VV, Cahill MP, Kim SH, and Wu L 2021. TRAF6 and TAK1 Contribute to SAMHD1-Mediated Negative Regulation of NF-kappaB Signaling. J Virol 895: e01970–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thientosapol ES, Bosnjak D, Durack T, Stevanovski I, van Geldermalsen M, Holst J, Jahan Z, Shepard C, Weninger W, Kim B, Brink R, and Jolly CJ 2018. SAMHD1 enhances immunoglobulin hypermutation by promoting transversion mutation. Proc Natl Acad Sci USA 115: 4921–4926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Husain A, Xu J, Fujii H, Nakata M, Kobayashi M, Wang JY, Rehwinkel J, Honjo T, and Begum NA 2020. SAMHD1-mediated dNTP degradation is required for efficient DNA repair during antibody class switch recombination. EMBO J 39: e102931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brenchley JM, Price DA, Schacker TW, Asher TE, Silvestri G, Rao S, Kazzaz Z, Bornstein E, Lambotte O, Altmann D, Blazar BR, Rodriguez B, Teixeira-Johnson L, Landay A, Martin JN, Hecht FM, Picker LJ, Lederman MM, Deeks SG, and Douek DC 2006. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nature Med 12: 1365–1371. [DOI] [PubMed] [Google Scholar]
- 48.Cecchinato V, Trindade CJ, Laurence A, Heraud JM, Brenchley JM, Ferrari MG, Zaffiri L, Tryniszewska E, Tsai WP, Vaccari M, Parks RW, Venzon D, Douek DC, O’Shea JJ, and Franchini G 2008. Altered balance between Th17 and Th1 cells at mucosal sites predicts AIDS progression in simian immunodeficiency virus-infected macaques. Mucosal Immunol 1: 279–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brenchley JM, Paiardini M, Knox KS, Asher AI, Cervasi B, Asher TE, Scheinberg P, Price DA, Hage CA, Kholi LM, Khoruts A, Frank I, Else J, Schacker T, Silvestri G, and Douek DC 2008. Differential Th17 CD4 T-cell depletion in pathogenic and nonpathogenic lentiviral infections. Blood 112: 2826–2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vassallo M, Mercie P, Cottalorda J, Ticchioni M, and Dellamonica P 2012. The role of lipopolysaccharide as a marker of immune activation in HIV-1 infected patients: a systematic literature review. Virol J 9: 174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dillon SM, Santiago ML, and Wilson CC 2016. HIV-1 pathogenesis in the gut. In Encyclopedia of AIDS Hope TJ, Stevenson M, and Richman D, eds. Springer, New York. 1–8. [Google Scholar]
- 52.Marchetti G, Tincati C, and Silvestri G 2013. Microbial translocation in the pathogenesis of HIV infection and AIDS. Clin Microbiol Rev 26: 2–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Halemano K, Harper MS, Guo K, Li SX, Heilman KJ, Barrett BS, and Santiago ML 2013. Humoral immunity in the Friend retrovirus infection model. Immunol Res 55: 249–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Robertson SJ, Ammann CG, Messer RJ, Carmody AB, Myers L, Dittmer U, Nair S, Gerlach N, Evans LH, Cafruny WA, and Hasenkrug KJ 2008. Suppression of acute anti-friend virus CD8+ T-cell responses by coinfection with lactate dehydrogenase-elevating virus. J Virol 8 82: 408–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lilly F, and Steeves RA 1973. B-tropic Friend virus: a host-range pseudotype of spleen focus-forming virus (SFFV). Virology 55: 363–370. [DOI] [PubMed] [Google Scholar]
- 56.Diamond TL, Roshal M, Jamburuthugoda VK, Reynolds HM, Merriam AR, Lee KY, Balakrishnan M, Bambara RA, Planelles V, Dewhurst S, and Kim B 2004. Macrophage tropism of HIV-1 depends on efficient cellular dNTP utilization by reverse transcriptase. J Biol Chem 279: 51545–51553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Smith DS, Guo K, Barrett BS, Heilman KJ, Evans LH, Hasenkrug KJ, Greene WC, and Santiago ML 2011. Noninfectious retrovirus particles drive the APOBEC3/Rfv3 dependent neutralizing antibody response. PLoS Pathog 7: e1002284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Robertson MN, Miyazawa M, Mori S, Caughey B, Evans LH, Hayes SF, and Chesebro B 1991. Production of monoclonal antibodies reactive with a denatured form of the Friend murine leukemia virus gp70 envelope protein: use in a focal infectivity assay, immunohistochemical studies, electron microscopy and western blotting. J Virol Methods 34: 255–271. [DOI] [PubMed] [Google Scholar]
- 59.Zelinskyy G, Kraft AR, Schimmer S, Arndt T, and Dittmer U 2006. Kinetics of CD8+ effector T cell responses and induced CD4+ regulatory T cell responses during Friend retrovirus infection. Eur J Immunol 36: 2658–2670. [DOI] [PubMed] [Google Scholar]
- 60.Zelinskyy G, Dietze KK, Husecken YP, Schimmer S, Nair S, Werner T, Gibbert K, Kershaw O, Gruber AD, Sparwasser T, and Dittmer U 2009. The regulatory T-cell response during acute retroviral infection is locally defined and controls the magnitude and duration of the virus-specific cytotoxic T-cell response. Blood 114: 3199–3207. [DOI] [PubMed] [Google Scholar]
- 61.Michaud JP, Halle M, Lampron A, Theriault P, Prefontaine P, Filali M, Tribout-Jover P, Lanteigne AM, Jodoin R, Cluff C, Brichard V, Palmantier R, Pilorget A, Larocque D, and Rivest S 2013. Toll-like receptor 4 stimulation with the detoxified ligand monophosphoryl lipid A improves Alzheimer’s disease-related pathology. Proc Natl Acad Sci USA 110: 1941–1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Copeland S, Warren HS, Lowry SF, Calvano SE, Remick D, Inflammation, and I. the Host Response to Injury. 2005. Acute inflammatory response to endotoxin in mice and humans. Clin Diagn Lab Immunol 12: 60–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pandrea I, Gaufin T, Brenchley JM, Gautam R, Monjure C, Gautam A, Coleman C, Lackner AA, Ribeiro RM, Douek DC, and Apetrei C 2008. Cutting edge: Experimentally induced immune activation in natural hosts of simian immunodeficiency virus induces significant increases in viral replication and CD4+ T cell depletion. J Immunol 181: 6687–6691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bao R, Zhuang K, Liu J, Wu J, Li J, Wang X, and Ho WZ 2014. Lipopolysaccharide induces immune activation and SIV replication in rhesus macaques of Chinese origin. PloS one 9: e98636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen S, Bonifati S, Qin Z, Gelais C St, and Wu L 2019. SAMHD1 Suppression of Antiviral Immune Responses. Trends Microbiol 27: 254–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Arunachalam PS, Wimmers F, Mok CKP, Perera R, Scott M, Hagan T, Sigal N, Feng Y, Bristow L, Tak-Yin Tsang O, Wagh D, Coller J, Pellegrini KL, Kazmin D, Alaaeddine G, Leung WS, Chan JMC, Chik TSH, Choi CYC, Huerta C, Paine McCullough M, Lv H, Anderson E, Edupuganti S, Upadhyay AA, Bosinger SE, Maecker HT, Khatri P, Rouphael N, Peiris M, and Pulendran B 2020. Systems biological assessment of immunity to mild versus severe COVID-19 infection in humans. Science 369: 1210–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Landy J, Ronde E, English N, Clark SK, Hart AL, Knight SC, Ciclitira PJ, and Al-Hassi HO 2016. Tight junctions in inflammatory bowel diseases and inflammatory bowel disease associated colorectal cancer. World J Gastroenterol 22: 3117–3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hertoghs N, van der Aar AM, Setiawan LC, Kootstra NA, Gringhuis SI, and Geijtenbeek TB 2015. SAMHD1 degradation enhances active suppression of dendritic cell maturation by HIV-1. J Immunol 194: 4431–4437. [DOI] [PubMed] [Google Scholar]
- 69.Messer RJ, Lavender KJ, and Hasenkrug KJ 2014. Mice of the resistant H-2(b) haplotype mount broad CD4(+) T cell responses against 9 distinct Friend virus epitopes. Virology 456–457: 139–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Coquel F, Silva MJ, Techer H, Zadorozhny K, Sharma S, Nieminuszczy J, Mettling C, Dardillac E, Barthe A, Schmitz AL, Promonet A, Cribier A, Sarrazin A, Niedzwiedz W, Lopez B, Costanzo V, Krejci L, Chabes A, Benkirane M, Lin YL, and Pasero P 2018. SAMHD1 acts at stalled replication forks to prevent interferon induction. Nature 557: 57–61. [DOI] [PubMed] [Google Scholar]
- 71.Daddacha W, Koyen AE, Bastien AJ, Head PE, Dhere VR, Nabeta GN, Connolly EC, Werner E, Madden MZ, Daly MB, Minten EV, Whelan DR, Schlafstein AJ, Zhang H, Anand R, Doronio C, Withers AE, Shepard C, Sundaram RK, Deng X, Dynan WS, Wang Y, Bindra RS, Cejka P, Rothenberg E, Doetsch PW, Kim B, and Yu DS 2017. SAMHD1 Promotes DNA End Resection to Facilitate DNA Repair by Homologous Recombination. Cell Rep 20: 1921–1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nimmerjahn F, Lux A, Albert H, Woigk M, Lehmann C, Dudziak D, Smith P, and Ravetch JV 2010. FcgammaRIV deletion reveals its central role for IgG2a and IgG2b activity in vivo. Proc Natl Acad Sci USA 107: 19396–19401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mahboubi B, Gavegnano C, Kim DH, Schinazi RF, and Kim B 2018. Host SAMHD1 protein restricts endogenous reverse transcription of HIV-1 in nondividing macrophages. Retrovirology 15: 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Addo MM, and Altfeld M 2014. Sex-based differences in HIV type 1 pathogenesis. J Inf Dis 209 Suppl 3: S86–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Szaniawski MA, Spivak AM, Bosque A, and Planelles V 2018. Sex influences SAMHD1 activity and susceptibility to HIV-1 in primary human macrophages. J Inf Dis 219: 777–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.White TE, Brandariz-Nunez A, Valle-Casuso JC, Amie S, Nguyen LA, Kim B, Tuzova M, and Diaz-Griffero F 2013. The retroviral restriction ability of SAMHD1, but not its deoxynucleotide triphosphohydrolase activity, is regulated by phosphorylation. Cell host & microbe 13: 441–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cribier A, Descours B, Valadao AL, Laguette N, and Benkirane M 2013. Phosphorylation of SAMHD1 by cyclin A2/CDK1 regulates its restriction activity toward HIV-1. Cell reports 3: 1036–1043. [DOI] [PubMed] [Google Scholar]
- 78.Gelais C St, de Silva S, Hach JC, White TE, Diaz-Griffero F, Yount JS, and Wu L 2014. Identification of cellular proteins interacting with the retroviral restriction factor SAMHD1. J Virol 88: 5834–5844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Geurs TL, Hill EB, Lippold DM, and French AR 2012. Sex differences in murine susceptibility to systemic viral infections. J Autoimmun 38: J245–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rubtsova K, Marrack P, and Rubtsov AV 2015. Sexual dimorphism in autoimmunity. J Clin Invest 125: 2187–2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Browne EP 2011. Toll-like receptor 7 controls the anti-retroviral germinal center response. PLoS Pathog 7: e1002293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kane M, Case LK, Wang C, Yurkovetskiy L, Dikiy S, and Golovkina TV 2011. Innate immune sensing of retroviral infection via Toll-like receptor 7 occurs upon viral entry. Immunity 35: 135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rubtsova K, Rubtsov AV, van Dyk LF, Kappler JW, and Marrack P 2013. T-box transcription factor T-bet, a key player in a unique type of B-cell activation essential for effective viral clearance. Proc Natl Acad Sci USA 110: E3216–3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.