

Summary
Leukodystrophies, genetic neurodevelopmental and/or neurodegenerative disorders of cerebral white matter, result from impaired myelin homeostasis and metabolism. Numerous genes have been implicated in these heterogeneous disorders; however, many individuals remain without a molecular diagnosis. Using whole-exome sequencing, biallelic variants in LSM7 were uncovered in two unrelated individuals, one with a leukodystrophy and the other who died in utero. LSM7 is part of the two principle LSM protein complexes in eukaryotes, namely LSM1-7 and LSM2-8. Here, we investigate the molecular and functional outcomes of these LSM7 biallelic variants in vitro and in vivo. Affinity purification-mass spectrometry of the LSM7 variants showed defects in the assembly of both LSM complexes. Lsm7 knockdown in zebrafish led to central nervous system defects, including impaired oligodendrocyte development and motor behavior. Our findings demonstrate that variants in LSM7 cause misassembly of the LSM complexes, impair neurodevelopment of the zebrafish, and may be implicated in human disease. The identification of more affected individuals is needed before the molecular mechanisms of mRNA decay and splicing regulation are added to the categories of biological dysfunctions implicated in leukodystrophies, neurodevelopmental and/or neurodegenerative diseases.
Keywords: LSM7, LSM1-7 complex, LSM2-8 complex, neurodevelopment, leukodystrophy, leukoencephalopathy
Derksen et al. use molecular and functional studies to demonstrate that variants in LSM7 lead to defects in the assembly of LSM complexes, impair neurodevelopment in zebrafish, and might be implicated in human disease.
Leukodystrophies encompass a group of rare genetic neurodevelopmental and neurodegenerative disorders affecting cerebral white matter. Recent advances in genetics and neuroimaging have led to the discovery of many subtypes that are clinically and genetically heterogeneous.1, 2, 3 These diseases can be classified based on specific brain magnetic resonance imaging (MRI) patterns as either hypomyelinating or non-hypomyelinating. Hypomyelinating leukodystrophies are associated with abnormal myelin formation during development, whereas non-hypomyelinating leukodystrophies are associated with abnormal myelin homeostasis.4,5 These heterogenous diseases collectively affect as many as 1 in 7,600 individuals, with a broad range of onset spanning from prenatal life to late adulthood.6, 7, 8 Although the era of next-generation sequencing (NGS) has facilitated rare disease diagnosis, 20%–30% of individuals with leukodystrophies remain genetically undiagnosed.2,9,10 These individuals and their families often undergo extensive diagnostic odysseys, which adversely impacts their wellbeing and healthcare.11,12
Leukodystrophies were initially identified and classified predominantly as disorders of myelin production or turnover, for example Pelizaeus-Merzbacher disease (MIM: 312080) and metachromatic leukodystrophy (MIM: 250100).8 Recent advances in research have revealed that pathogenic variants in many transcription- and translation-related genes are implicated in white matter pathologies. For example, it is well-established that pathogenic variants in the genes encoding RNA polymerase III subunits (i.e., POLR3A [MIM: 614258], POLR3B [MIM: 614366], POLR1C [MIM: 610060], and POLR3K [MIM: 606007]) as well as tRNA synthetase genes (i.e., EPRS1 [MIM: 138295], RARS1 [MIM: 107820], VARS1 [MIM: 192150], etc.) are associated with white matter disorders.13, 14, 15, 16, 17, 18, 19
LSM proteins (“like Sm”) are a family of proteins that form multi-subunit complexes that interact directly with RNA.20, 21, 22, 23, 24 The 8 main LSM proteins (LSM1-8) form two heteroheptameric ring-shaped complexes composed of different subunits. More specifically, the LSM1 through LSM7 proteins form the LSM1-7 complex, and the LSM2 through LSM8 proteins form the LSM2-8 complex. The LSM1-7 complex interacts with the mRNA processing and degradation factor protein PATL1, localizing to the cytoplasmic processing bodies (P-bodies), and plays a role in mRNA decay.20,21,24 Eukaryotic cytosolic mRNA decay, a well-characterized and highly conserved process, occurs via one of two pathways, both beginning with the shortening of the poly(A) tail and followed by degradation in either the 3′–5′ or 5′–3′ direction.21,23,24 Studies in yeast revealed that the LSM1-7 complex likely acts as an enhancer of decapping in the 5′–3′ pathway, as cells with variants in Lsm1p through Lsm7p block mRNA decay at the decapping step.21,25,26 Conversely, the LSM2-8 complex is localized to the nucleus, where it interacts with U6 small nuclear RNA (snRNA), which is the catalytic component of the primary spliceosome.20,22,23 Splicing is a multi-step reaction involving the interaction and subsequent rearrangements of the 5 primary spliceosomal snRNAs (U1, U2, U4, U5, and U6) and their protein interactors.25, 26, 27, 28, 29 The LSM2-8 complex acts as a chaperone for U6 snRNA, supporting its rearrangements during splicing and its recycling following the spliceosome reaction.30,31 Thus, the LSM1-7 and LSM2-8 complexes play critical roles in the highly regulated cellular mechanisms of mRNA decay and splicing, respectively.20
Here, we report two individuals with biallelic variants in LSM7 (MIM: 607287), one with a clinical diagnosis of leukodystrophy and the other who died in utero. These LSM7 variants were investigated on a molecular and functional level to evaluate their impact on the LSM complexes. A zebrafish model was also created to study the functional impact of wild-type (WT) and mutant LSM7 on the development of the central nervous system (CNS).
This project was approved by the Research Institute of the McGill University Health Centre Research Ethics Board (PED-11-105, 2019-4972), and informed consent was obtained from all participants in this study. The first affected individual was referred at the age of 3 years for neurological assessment at the McGill University Health Center. He presented with spastic quadriparesis, white matter anomalies, developmental delay, progressive sensorineural hearing loss, hypospadias, and feeding difficulties. He is currently 7 years old and wheelchair bound. His MRIs showed cerebellar hypoplasia and progressive white matter signal abnormalities between 17 and 35 months, indicative of a leukodystrophy (Figure 1). To determine the genetic cause, massive parallel (NextGen) sequencing on an Illumina system was performed by GeneDx as previously described,32 using DNA isolated from saliva samples of the affected individual and his parents. The quality threshold of the exome sequencing for this individual was 98.4%, with a mean depth of coverage of 88 reads. Analysis was completed using the general assertion criteria for variant classification and is publicly available on the GeneDx ClinVar submission page. A second analysis of the sequence variants was conducted in-house at the Research Institute of the McGill University Health Centre. Sequence variants were prioritized using the standards and guidelines for interpretation set out by the American College of Medical Genetics (ACMG).33 These analyses revealed a homozygous variant in LSM7 (GenBank: NM_016199.2; GRCh37/hg19) at position c.121G>A (p.Asp41Asn) (chr19: 2324172C>T) as a strong candidate for pathogenicity (Figures 2A and 2B). Targeted Sanger sequencing confirmed the presence of the variant as homozygous in the affected individual, with both parents being carriers (Figure 2D). Subsequently, a second individual was found through GeneMatcher.34,35 This family was referred for genetic investigations following recurrent fetal deaths. The proband died in utero at 32 weeks of gestation and was found to have thick nuchal translucency and encephalocele. Karyotyping was performed in 3 of the pregnancies that ended in fetal death and revealed no chromosomal abnormalities. Duo exome sequencing of the consanguineous parents was conducted as previously described36 and revealed that both parents were carriers of the same variant in LSM7 at position c.206G>C (p.Arg69Pro) (chr19: 2321785C>G). The parents were not found to both be carriers for any known recessive diseases, and no other gene candidate was identified. Their chromosomal analyses were also normal. The multiple fetal deaths are thereby presumed to be caused by the homozygous variant in LSM7. Unfortunately, DNA was not available from the fetuses or the 3 healthy children of this couple for sequencing.
Figure 1.

MRI characteristics of individual 1
Brain MRI of individual 1 at 17 months (A–C) and 35 months of age (D–F).
(A) and (D) Sagittal T1-weighted images at the midline showing cerebellar hypoplasia (thin arrows).
(B, C, E, and F) Axial T2-weighted images at the level of the basal ganglia (B and E) and centrum semiovale (C and F) showing progressive diffuse demyelination (dotted arrow).
Figure 2.

Biallelic variants in LSM7
(A) Genomic organization of LSM7 in humans (UCSC Genome Browser hg19) with the position of the pathogenic variants within the LSM7 gDNA indicated.
(B) Major motifs of the 103 amino acid LSM7 protein with position of variants indicated.
(C) LSM7 variants in affected individuals are at conserved amino acid residues.
(D) LSM7 gene sequencing of affected individual 1 and parents.
(E and F) 3D representations (created with PyMOL) of yeast (E) Lsm1-7 and (F) Lsm2-8 complexes displaying location of variants.
Both LSM7 variants are present in highly conserved amino acid residues (Figure 2C) and are classified as rare. The p.Asp41Asn variant was observed only 9 times as heterozygous in 232,800 alleles in large population cohorts37 (minor allele frequency 3.87e−5), while the p.Arg69Pro variant has never been reported in large population control databases. In silico genetic analysis predict both variants to be deleterious on protein structure and function (p.Asp41Asn: Mutation Taster 0.99, CADD 28.8, Provean −4.61, SIFT 0.001; p.Arg69Pro: Mutation Taster 0.99, CADD 24.9, Provean −7.00, SIFT 0.00) (Table S1).38, 39, 40, 41, 42
To examine these variants at a molecular level, we performed RT-qPCR and immunoblot using RNA and protein extracts (n = 6) from fibroblasts of the first affected individual as well as two age- and sex-matched healthy control individuals. RT-qPCR primers were designed and efficiency tested in accordance with the Minimum Information for Publication of Quantitative Real-Time PCR Experiments (MIQE) guidelines.43,44 All data were normalized to SDHA (MIM: 600857) or RPL30 (MIM: 180467) using the ΔΔCt method, which revealed reduced levels of LSM7 mRNA (p < 0.01) in affected individual 1 compared to control individuals (Figure 3A). Immunoblots were performed on protein extracts using antibodies targeting LSM7 (Abcam 241656, dilution 1:10,000) and beta-tubulin (Abcam 6046, dilution 1:20,000), followed by incubation with a polyclonal goat anti-rabbit IgG (H+L) (Novus Biologicals, dilution 1:5,000) antibody and visualization using Amersham ECL Western Blot Detecting Reagent according to manufacturer’s protocol (GE Life Sciences). Band intensity was quantified using ImageJ software, and statistical significance was determined using a parametric, unpaired t test (two tailed). A decreased amount of LSM7 protein (p < 0.001) was seen in the affected individual as compared to control individuals (Figures 3B and 3C). Samples from the fetuses described in the second family (p.Arg69Pro) were not available for analysis.
Figure 3.
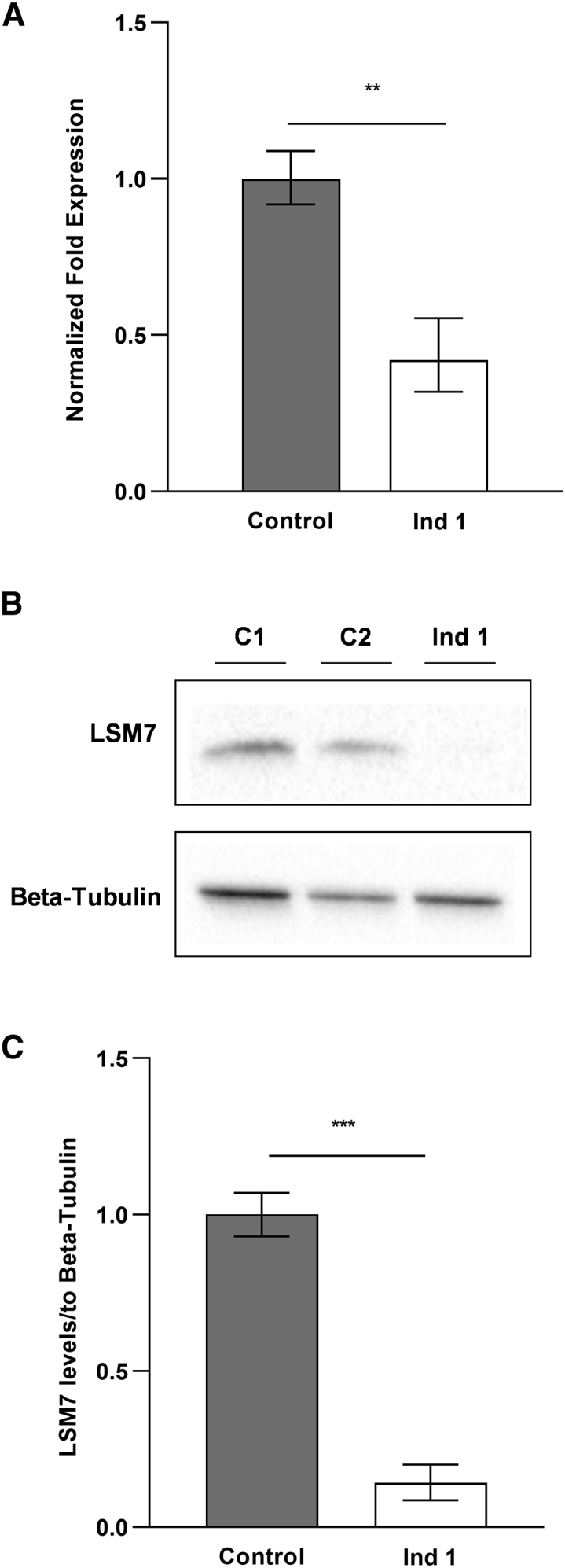
LSM7 mRNA and protein levels are decreased in individual 1 compared to control individuals
(A) RT-qPCR analysis of LSM7 mRNA extracted from individual 1’s fibroblasts and two age- and sex-matched control individuals (n = 6). The results are represented in terms of fold change after normalizing to RPL30 and SDHA mRNAs. Each value represents the mean ± SEM (unpaired t test [two tailed] ∗∗p < 0.01).
(B) Total protein lysates extracted from individual 1 and age- and sex-matched control individual fibroblasts were immunoblotted with anti-LSM7 and anti-beta-tubulin.
(C) The pixel densities of immunoblot bands from at least 3 independent replicates were quantified using ImageJ software, normalized over beta-tubulin immunoblot and represented as mean ± SEM (unpaired t test [two tailed], ∗∗∗p < 0.001).
In order to evaluate the impact of these variants on the formation of the LSM complexes, we generated structural models of wild-type and mutant yeast Lsm1-7 (PDB: 4M75)45 and Lsm2-8 complexes (PDB: 4M77)45 using PyMOL Molecular Graphics System, version 2.0 Schrödinger (Figures 2E and 2F). All the LSM proteins (LSM1 through LSM8) are highly conserved from yeast to humans. Based on their homology, we mapped the human variants at positions Asp41 and Arg69 to their corresponding yeast residues at Asp56 and Arg87, respectively. Using PyMOL, we modeled the effects of the predicted deleterious variants. The first variant, p.Asp56Asn in yeast, is predicted to result in the loss of polar contacts with other Lsm7 residues in both the Lsm1-7 and Lsm2-8 complexes (Figures S1A–S1D). Meanwhile, the second variant, p.Arg87Pro in yeast, is predicted to result in the loss of polar contacts with Lsm7 residues and a residue on the neighboring Lsm5 protein in both the Lsm1-7 and Lsm2-8 complexes (Figures S1E–S1H). Based on this modeling, we predicted that each of these substitutions would lead to a compromised ability of Lsm7 to interact with the other Lsm subunits, and consequently, the assembly of the Lsm1-7 and Lsm2-8 complexes would be significantly impaired.
To determine the potential pathogenic role of these variants, we assessed their impact on the assembly of the LSM1-7 and LSM2-8 complexes in human cells. A FLAG-tagged version of the wild-type LSM7, as well as that containing either the p.Asp41Asn or p.Arg69Pro variant, were produced transiently in HEK293 cells. Anti-FLAG affinity purification was conducted on cell extracts according to standard procedures in 4 independent replicate experiments.46,47 The protein extracts were digested with trypsin, and the resulting tryptic peptides were purified and identified using liquid chromatography-tandem mass spectrometry (LC-MS/MS) with a high-performance liquid chromatography (HPLC) system coupled to Orbitrap fusion mass spectrometer (Thermo Scientific) through a Nanospray Flex Ion Source. Protein database searching was performed using MaxQuant version 1.6.10.4348, 49, 50, 51, 52, 53 against the SwissProt human protein database downloaded on April 4, 2019. Known affinity purification mass spectrometry (AP-MS) protein contaminants including keratins were excluded, and protein intensities were analyzed using Perseus version 1.6.10.43.54, 55, 56, 57 Proteins not present in at least 3 out of 4 replicates were excluded. Wild-type, p.Asp41Asn, and p.Arg69Pro proteins were compared against FLAG empty vector control samples and were labeled as high-confidence interactors when their adjusted p values were under 0.05 and their intensity ratios over 2. The p values were adjusted for multiple hypothesis testing with a permutation-based approach using an s0 correction factor of 0.1 with 10,000 iterations.58 A Student’s t test was performed between p.Asp41Asn and p.Arg69Pro against the wild-type samples. These results revealed that cells producing the wild-type LSM7 protein resulted in the co-precipitation of all the LSM1-7 and LSM2-8 complex subunits, along with multiple other known interactors of these complexes (Figures 4A and 4B). However, pull-downs from lysates of HEK293 cells producing the p.Asp41Asn or p.Arg69Pro variants revealed dramatically reduced association of the mutated LSM7 protein with the other LSM proteins constituting the LSM1-7 and LSM2-8 complexes. Consistent with this defect, most of the known interactors of these complexes were also not pulled down in the mutant strains (Figure 4C). The LSM1-7 and LSM2-8 complexes are known to interact with many mRNA decay and splicing factors, respectively. Relative to the FLAG-tagged wild-type LSM7, the p.Asp41Asn and p.Arg69Pro mutants showed reduced interaction with the mRNA decay factors PATL1 and EDC4 as well as with triple small nuclear ribonucleoprotein (tri-snRNP) factors including PRPF3, PRPF4, SART3, and SNRNP200. These splicing factors are required for the formation of the U4/U6/U5 tri-snRNP complex, an important precursor in the major spliceosome reaction.59, 60, 61 Similarly, PATL1 is a highly conserved interacting partner of the LSM1-7 complex that plays a key role in mRNA decay.62 Thus, these findings suggest that both variants lead to defects in the assembly of the LSM1-7 and LSM2-8 complexes and result in the subsequent loss of key complex interactors.
Figure 4.

Impact of LSM7 variants on LSM1-7 and LSM2-8 complex assembly
(A and B) Volcano plots of the log2-transformed MaxQuant ratios of the p.Asp41Asn or p.Arg69Pro/wild-type (x axis) and the −log10-transformed p values (adjusted with permutation-based false discovery rate) resulting from the two-tailed two-sample Student’s t tests.
(C) Heatmap contains the MaxQuant ratios for the subunits of the LSM complexes and known important complex interactors.
Since the levels of LSM7 protein were found to be decreased in affected individual 1 compared to control individuals (Figure 3C), we predicted that the defect in LSM1-7 and LSM2-8 complex assembly could be further exacerbated by reduced starting levels of the mutant LSM7 protein. In our LC-MS/MS experiments, when the levels of FLAG-tagged mutant LSM7 were normalized to that of the FLAG-tagged wild-type, we found that both mutated subunits pulled down relatively lower amounts of the other LSM complex subunits. Therefore, reduced protein levels of mutant LSM7, as seen in affected individual 1, could potentially intensify the defects we observed in our LC-MS/MS experiments. We also saw reduced levels of LSM7 mRNA in this individual compared to control individuals (Figure 3A). Since the levels of the LSM2-8 complex are expected to be lower in this individual (Figure 4C), reduced LSM7 transcript levels could result from inefficient splicing of LSM7 pre-mRNA. Several other studies have shown that both missense and synonymous variants can impact RNA structure, stability, folding, microRNA (miRNA) binding, splicing regulatory sites, and/or translation efficiency.63, 64, 65 Therefore, we hypothesize that this missense variant could alter the LSM7 pre-mRNA, further targeting it for degradation and thereby reducing transcript levels.
To demonstrate the functional impact of wild-type and mutant LSM7, a zebrafish model was created and used to study the impact of LSM7 hypofunction on CNS development. The zebrafish experiments were performed in strict accordance with relevant institutional and national guidelines and regulations. Adult fish were bred according to standard methods, and embryos were raised at 28.5°C in E3 embryo medium and staged by time and morphology. The zebrafish ortholog lsm7 (ENSDART00000081188.7) and human LSM7 share significant homology. The zebrafish Lsm7 protein has 95% amino acid identity with human LSM7, including conservation of the two amino acid residues of the variants in the affected individuals (p.Asp41Asn and p.Arg69Pro) (Figure 5A). We evaluated lsm7 gene expression using an antisense digoxigenin-uridine triphosphate (UTP) labeled riboprobe, which was synthesized according to the manufacturer’s instructions (Roche). In situ hybridizations were performed as previously described66 from 1 day post-fertilization (dpf) to 5 days post-fertilization, and the color reaction was carried out using NBT/BCIP substrate (Roche). At 1 day post-fertilization and 2 days post-fertilization, the lsm7 transcript was detected in the developing brain, spinal cord, and eye (Figure 5B). By 3 days post-fertilization and persisting through 5 days post-fertilization, expression was present in the olfactory bulb, cerebellum, and hindbrain (Figure 5B).
Figure 5.

The zebrafish Lsm7 ortholog is highly conserved with human LSM7, is expressed in developing nervous system, and is necessary for embryonic morphology, eye size, motor behavior, and survival of oligodendrocytes
(A) Amino acid alignment of human and zebrafish LSM7 sequences. Identical residues are marked by red boxes, and percent similarity is shown by different colors.
(B) In situ hybridization was performed for detecting lsm7 expression during zebrafish embryogenesis. Embryo stages are shown in the bottom left of each panel. All panels display the lateral view, with dorsal to the top and rostral to the left.
(C) Phenotypes of lsm7-Crispants.
(D) Quantification of control and lsm7-Crispants by phenotype severity.
(E) Top left panel shows a cartoon representation of the dissected region of larvae (blue dashed line rectangle). Images of eyes of control, lsm7-Crispant, LSM7full-length cRNA-, LSM7R69P cRNA-, and LSM7D41N cRNA-rescued larvae are shown. The eye diameter was measured as indicated by the magenta dashed line shown on control eye.
(F) Quantification of eye size.
(G) Motor swimming behavior of the control, lsm7-Crispant, LSM7full-length cRNA-, LSM7R69P cRNA-, and LSM7D41N cRNA-rescued larvae at 7 dpf.
(H) Confocal images of dorsal view of the control, lsm7-Crispant, LSM7full-length cRNA-, LSM7R69P cRNA-, and LSM7D41N cRNA-rescued larvae with rostral to the top. Lower panels are enlarged region from upper panels for apoptotic cell quantification.
(I) Quantification of the number of TUNEL and DsRed double-positive cells of (H). Abbreviations: ce, cerebellum; dpf, days post-fertilization; fb, forebrain; hb, hindbrain; mb, midbrain; ob, olfactory bulb; pa, pharyngeal arches; sp, spinal cord. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; n.s., not significant.
To investigate the physiological function of Lsm7 in an animal model of neurodevelopment, we used CRISPR-Cas9 gene editing to develop Lsm7-Crispant zebrafish lacking functional Lsm7 protein. Single guide RNA (sgRNA) target sites for the zebrafish lsm7 gene (Ensembl Zv11: ENSDART00000081188.7) were designed and off-target effects checked. Co-injection of lsm7 exon 3 sgRNA (460 pg) and Cas9 protein (460 pg, PNA BIO) was done on one-cell-stage embryos and efficiently induced insertions and deletions as determined by high-resolution melting analysis (HRMA) PCR (Figure S2).
Embryos injected with lsm7 sgRNA and Cas9 protein showed impaired nervous system development and normal lifespan (1.6% lethality, n = 128). We categorized the surviving embryos into two phenotypes according to the severity: a mild phenotype who displayed normal trunk morphology but brain and eye size reduction, and a moderate phenotype exhibiting a bent trunk, edema in the pericardial sac, and decreased eye and brain size (Figures 5C–5E). At 5 days post-fertilization the fish larvae were fixed, and one eye was dissected out, mounted laterally, and imaged. The eye size was measured along the horizontal axis using ImageJ software (Figure 5E, dashed line) in Lsm7-Crispants (injected with lsm7 sgRNA and Cas9 protein). Eye diameter was found to be significantly reduced in Lsm7-Crispants compared to controls (p < 0.001) (Figures 5E and 5F). We also evaluated motor behavior of Lsm7-Crispants by analyzing the response to light stimuli at 7 days post-fertilization in 96-well square bottom plates (Krackeler Scientific) using video analysis software (Noldus EthoVision). We found that larvae response to light stimuli was significantly reduced in the Lsm7-Crispants (p < 0.001) (Figure 5G).
To explore the effects of the mutant LSM7 proteins, capped RNA (cRNA) encoding the full coding sequence of LSM7full-length, LSM7Asp41Asn, or LSM7Arg69Pro were prepared. The eye phenotype could be rescued by concomitant injection of human LSM7full-length and LSM7Arg69Pro cRNAs, but not by the LSM7Asp41Asn cRNA (Figures 5E and 5F). Since the p.Asp41Asn variant is located on a conserved RNA contacting residue in the Sm motif-I of LSM7, the inability of the LSM7Asp41Asn cRNA to rescue the eye size phenotype of the Lsm7-Crispants implies that RNA processing activity is critical for eye development. Furthermore, the movement deficits observed in the Lsm7-Crispants could only be rescued with the LSM7full-length cRNA but not by either of the mutant cRNAs (LSM7Arg69Pro or LSM7Asp41Asn), indicating that both mutants impact motor behavior (Figure 5G). Overall, these findings demonstrate that lsm7 knockdown zebrafish have phenotypic characteristics similar to those observed in the first affected individual.
On a cellular level using our zebrafish model, we explored the role of LSM7 on the development and survival of oligodendrocytes, the myelin-producing cells of the CNS, which are primarily implicated in leukodystrophies. We used the transgenic line Tg (olig2:dsRed) to label oligodendrocytes, oligodendrocyte precursor cells (OPCs), and motor neurons and compared cell counts in larvae injected with wild-type LSM7full-length cRNA to LSM7Asp41Asn or LSM7Arg69Pro cRNAs. The immunohistochemistry was performed as previously described67 using rabbit anti-dsRed (Clontech, dilution 1:250), Alexa 555 goat anti-rabbit (ThermoFisher Scientific, dilution 1:400), and 4′,6-diamidino-2-phenylindole (DAPI). Additionally, terminal deoxynucleotidyl transferase deoxyuridine triphosphate (dUTP) nick-end labeling (TUNEL) was performed on whole-mount larvae (ApopTag Fluorescein In Situ Apoptosis Detection Kit; Millipore) as previously described.68 Confocal imaging was used and cells counted using Photoshop’s (Adobe) count tool. We found that apoptosis of oligodendrocytes was significantly increased in Lsm7-Crispants (p < 0.01) (Figures 5H and 5I) and could be rescued by concomitant injection of LSM7full-length cRNA but not by the LSM7Asp41Asn or LSM7Arg69Pro cRNAs (Figures 5H and 5I). This suggests a role for LSM7 in oligodendrocyte survival and demonstrates that both variants are associated with an increase in apoptosis of oligodendrocytes. Interestingly, the hypoplastic cerebellum in individual 1 corresponded to the small cerebellar phenotype observed in Lsm7-Crispants. It is possible that deficiency or dysregulation of LSM7 in the cerebellum of the first affected individual at key developmental stages could contribute to the observed cerebellar hypoplasia. Overall, this zebrafish model has allowed us to identify a relationship between LSM7 and CNS development, as well as oligodendrocyte survival, thus providing further evidence of a link to leukodystrophies, genetic neurodevelopmental and/or neurodegenerative disorders.
Our current data do not allow us to understand why one variant (p.Arg69Pro) led to in utero death, while the other variant (p.Asp41Asn) leads to a less severe phenotype. Samples were unavailable from the fetuses described in the second family for mRNA or protein analysis, limiting direct functional studies. However, the p.Arg69Pro variant, but not p.Asp41Asn, is predicted to be located at an exon-intron boundary by MutationTaster,42 which could impair or cause dysfunction in pre-mRNA splicing. For this reason, we hypothesize that perhaps the p.Arg69Pro variant causes splice site changes that are detrimental and cause embryonic lethality. Specific splicing variants can cause cell-specific defects, as in the case of individuals presenting with a unique phenotype of basal ganglia abnormalities and the same pathogenic splicing variant in POLR3A.69, 70, 71, 72, 73 This further supports a possible association between distinct splicing defects and phenotypic heterogeneity within individuals with pathogenic variants in the same gene. While a splicing defect could offer support for the difference in phenotypic severity, we would be unable to confidently conclude that it is indeed the only factor at play. There have been several reports in the literature of individuals with the same variant presenting with large discordances in phenotypic severity. One such report describes a sibling pair with the same compound heterozygous variants in POLR1C, which led to neonatal death in one sibling, while the other remains alive and is much less affected.74 Another example is presented in an inbred family where the same variant in SLC17A5 results in either a mild or severe phenotype.75
In summary, this study identified LSM7, a gene involved in the processes of 5′–3′ mRNA decay and splicing,20 as playing a role in the development of the CNS and in oligodendrocyte survival. Before a definite association between LSM7 variants and leukodystrophy, genetic neurodevelopmental and/or neurodegenerative disease can be made, more affected individuals with biallelic variants in LSM7 need to be identified. Our findings, however, add this gene as a strong gene candidate for these diseases. In recent years, gene discovery has shifted from identifying pathogenic variants in genes for rare diseases to ultra-rare entities with only a handful of affected individuals. Indeed, this is supported by several recently uncovered leukodystrophy genes, in which only a few individuals were identified and published, including EPRS1, POLR3K, RARS1, TMEM106B (MIM: 613413), CNP (MIM:123830), and NKX6-2 (MIM: 605955).13,19,76, 77, 78, 79 LSM7, which encodes a subunit of both the LSM1-7 and LSM2-8 complexes, has not previously been associated with human disease. However, other proteins involved in the processes of mRNA decay and splicing, the primary functions of the LSM1-7 and LSM2-8 complexes, respectively, have been implicated in a variety of diseases.21,23,30,80, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96 Cellular decay pathways play a critical role in regulating mRNA levels, and alterations in these pathways could be the principle factor underlying many diseases,85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95,97,98 including neurological disorders.83,84 Similarly, variants in genes encoding proteins involved in the various steps of the splicing reaction have also been linked to numerous neurodevelopmental and neurodegenerative disorders.59,99, 100, 101, 102, 103, 104 The identification of other individuals with biallelic variants in LSM7 is needed to add with certainty this gene and the pathophysiological mechanisms of mRNA decay and splicing regulation to genes and categories of cellular dysfunction associated with leukodystrophies, genetic neurodevelopmental and/or neurodegenerative disorders.
Declaration of interests
Y.S. is an employee of GeneDx, Inc. J.L.B. reports being on the board of directors for wFluidx; stock ownership of Orchard Therapeutics; consulting for Bluebird, Calico, Denali, Enzyvant, Neurogene, and Passage Bio; and royalties from BioFire (spouse). G.B. reports speaker’s honoraria from Genzyme (2013) and Actelion Pharmaceuticals (2013); being on the scientific advisory boards of Ionis (2019), Shire (2013), Actelion Pharmaceuticals (2011), and Santhera Pharmaceuticals (2011); research grant from Takeda; being on a site PI for studies sponsored by Takeda/Shire (2020-2021), Bluebird Bio (2019), Shire (2016), and Actelion Pharmaceuticals (2017); and consulting for Passage Bio. All other authors declare no competing interests.
Acknowledgments
First and foremost, we would like to thank the individuals and their families for their participation. This study was supported by grants from the Canadian Institutes of Health Research (377869, 426534), Fondation Les Amis d’Elliot, Fondation le Tout pour Loo, and Réseau de Médicine Génétique Appliquée of the Fonds de Recherche du Québec – Santé (FRQS). This research was enabled in part by support provided by Compute Canada. A.D. was supported by the Canadian Institutes of Health Research (CIHR) Canadian Graduates Scholarships – Master’s, the Fondation du Grand Défi Pierre Lavoie Master’s Scholarship, and Heathy Brains for Healthy Lives Masters Fellowship. G.B. has received a Research Scholar Junior 1 award from the Fonds de Recherche du Québec - Santé (2012–2016) and the New Investigator Salary Award from the Canadian Institutes of Health Research (2017–2022). B.C. holds a Bell-Bombardier Research Chair awarded by the IRCM. We would also like to thank the McGill University and Genome Québec Innovation Center, the IRCM Molecular Biology and Functional Genomics Platform, and Denis Faubert, Josée Champagne, and Sylvain Tessier of the IRCM Proteomics Discovery Platform for their services.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xhgg.2021.100034.
Data and code availability
The clinical exomes are not publicly available. The variants generated during this study are available at ClinVar (gene variants; ClinVar: SCV001478394 and SCV001478886).
Web resources
CHOPCHOP, http://chopchop.cbu.uib.no/
Compute Canada, https://www.computecanada.ca/
GeneDx ClinVar submission page, https://www.ncbi.nlm.nih.gov/clinvar/submitters/26957
OMIM, https://omim.org/
PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC, https://www.pymol.org/2/
Supplemental information
References
- 1.Vanderver A., Prust M., Tonduti D., Mochel F., Hussey H.M., Helman G., Garbern J., Eichler F., Labauge P., Aubourg P., et al. GLIA Consortium Case definition and classification of leukodystrophies and leukoencephalopathies. Mol. Genet. Metab. 2015;114:494–500. doi: 10.1016/j.ymgme.2015.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Parikh S., Bernard G., Leventer R.J., van der Knaap M.S., van Hove J., Pizzino A., McNeill N.H., Helman G., Simons C., Schmidt J.L., et al. GLIA Consortium A clinical approach to the diagnosis of patients with leukodystrophies and genetic leukoencephelopathies. Mol. Genet. Metab. 2015;114:501–515. doi: 10.1016/j.ymgme.2014.12.434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ashrafi M.R., Tavasoli A.R. Childhood leukodystrophies: A literature review of updates on new definitions, classification, diagnostic approach and management. Brain Dev. 2017;39:369–385. doi: 10.1016/j.braindev.2017.01.001. [DOI] [PubMed] [Google Scholar]
- 4.Schiffmann R., van der Knaap M.S. Invited article: an MRI-based approach to the diagnosis of white matter disorders. Neurology. 2009;72:750–759. doi: 10.1212/01.wnl.0000343049.00540.c8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Steenweg M.E., Vanderver A., Blaser S., Bizzi A., de Koning T.J., Mancini G.M., van Wieringen W.N., Barkhof F., Wolf N.I., van der Knaap M.S. Magnetic resonance imaging pattern recognition in hypomyelinating disorders. Brain. 2010;133:2971–2982. doi: 10.1093/brain/awq257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bonkowsky J.L., Nelson C., Kingston J.L., Filloux F.M., Mundorff M.B., Srivastava R. The burden of inherited leukodystrophies in children. Neurology. 2010;75:718–725. doi: 10.1212/WNL.0b013e3181eee46b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Adang L.A., Sherbini O., Ball L., Bloom M., Darbari A., Amartino H., DiVito D., Eichler F., Escolar M., Evans S.H., et al. Global Leukodystrophy Initiative (GLIA) Consortium Revised consensus statement on the preventive and symptomatic care of patients with leukodystrophies. Mol. Genet. Metab. 2017;122:18–32. doi: 10.1016/j.ymgme.2017.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van der Knaap M.S., Bugiani M. Leukodystrophies: a proposed classification system based on pathological changes and pathogenetic mechanisms. Acta Neuropathol. 2017;134:351–382. doi: 10.1007/s00401-017-1739-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vanderver A., Simons C., Helman G., Crawford J., Wolf N.I., Bernard G., Pizzino A., Schmidt J.L., Takanohashi A., Miller D., et al. Leukodystrophy Study Group Whole exome sequencing in patients with white matter abnormalities. Ann. Neurol. 2016;79:1031–1037. doi: 10.1002/ana.24650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kevelam S.H., Steenweg M.E., Srivastava S., Helman G., Naidu S., Schiffmann R., Blaser S., Vanderver A., Wolf N.I., van der Knaap M.S. Update on Leukodystrophies: A Historical Perspective and Adapted Definition. Neuropediatrics. 2016;47:349–354. doi: 10.1055/s-0036-1588020. [DOI] [PubMed] [Google Scholar]
- 11.Boycott K.M., Vanstone M.R., Bulman D.E., MacKenzie A.E. Rare-disease genetics in the era of next-generation sequencing: discovery to translation. Nat. Rev. Genet. 2013;14:681–691. doi: 10.1038/nrg3555. [DOI] [PubMed] [Google Scholar]
- 12.Sawyer S.L., Hartley T., Dyment D.A., Beaulieu C.L., Schwartzentruber J., Smith A., Bedford H.M., Bernard G., Bernier F.P., Brais B., et al. FORGE Canada Consortium. Care4Rare Canada Consortium Utility of whole-exome sequencing for those near the end of the diagnostic odyssey: time to address gaps in care. Clin. Genet. 2016;89:275–284. doi: 10.1111/cge.12654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mendes M.I., Gutierrez Salazar M., Guerrero K., Thiffault I., Salomons G.S., Gauquelin L., Tran L.T., Forget D., Gauthier M.S., Waisfisz Q., et al. Bi-allelic Mutations in EPRS, Encoding the Glutamyl-Prolyl-Aminoacyl-tRNA Synthetase, Cause a Hypomyelinating Leukodystrophy. Am. J. Hum. Genet. 2018;102:676–684. doi: 10.1016/j.ajhg.2018.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rezaei Z., Hosseinpour S., Ashrafi M.R., Mahdieh N., Alizadeh H., Mohammadpour M., Khosroshahi N., Amanat M., Tavasoli A.R. Hypomyelinating Leukodystrophy with Spinal Cord Involvement Caused by a Novel Variant in RARS: Report of Two Unrelated Patients. Neuropediatrics. 2019;50:130–134. doi: 10.1055/s-0039-1679911. [DOI] [PubMed] [Google Scholar]
- 15.Friedman J., Smith D.E., Issa M.Y., Stanley V., Wang R., Mendes M.I., Wright M.S., Wigby K., Hildreth A., Crawford J.R., et al. Biallelic mutations in valyl-tRNA synthetase gene VARS are associated with a progressive neurodevelopmental epileptic encephalopathy. Nat. Commun. 2019;10:707. doi: 10.1038/s41467-018-07067-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thiffault I., Wolf N.I., Forget D., Guerrero K., Tran L.T., Choquet K., Lavallée-Adam M., Poitras C., Brais B., Yoon G., et al. Recessive mutations in POLR1C cause a leukodystrophy by impairing biogenesis of RNA polymerase III. Nat. Commun. 2015;6:7623. doi: 10.1038/ncomms8623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bernard G., Chouery E., Putorti M.L., Tétreault M., Takanohashi A., Carosso G., Clément I., Boespflug-Tanguy O., Rodriguez D., Delague V., et al. Mutations of POLR3A encoding a catalytic subunit of RNA polymerase Pol III cause a recessive hypomyelinating leukodystrophy. Am. J. Hum. Genet. 2011;89:415–423. doi: 10.1016/j.ajhg.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tétreault M., Choquet K., Orcesi S., Tonduti D., Balottin U., Teichmann M., Fribourg S., Schiffmann R., Brais B., Vanderver A., Bernard G. Recessive mutations in POLR3B, encoding the second largest subunit of Pol III, cause a rare hypomyelinating leukodystrophy. Am. J. Hum. Genet. 2011;89:652–655. doi: 10.1016/j.ajhg.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dorboz I., Dumay-Odelot H., Boussaid K., Bouyacoub Y., Barreau P., Samaan S., Jmel H., Eymard-Pierre E., Cances C., Bar C., et al. Mutation in POLR3K causes hypomyelinating leukodystrophy and abnormal ribosomal RNA regulation. Neurol. Genet. 2018;4:e289. doi: 10.1212/NXG.0000000000000289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tharun S. Roles of eukaryotic Lsm proteins in the regulation of mRNA function. Int. Rev. Cell Mol. Biol. 2009;272:149–189. doi: 10.1016/S1937-6448(08)01604-3. [DOI] [PubMed] [Google Scholar]
- 21.Tharun S. Lsm1-7-Pat1 complex: a link between 3′ and 5′-ends in mRNA decay? RNA Biol. 2009;6:228–232. doi: 10.4161/rna.6.3.8282. [DOI] [PubMed] [Google Scholar]
- 22.Beggs J.D. Lsm proteins and RNA processing. Biochem. Soc. Trans. 2005;33:433–438. doi: 10.1042/BST0330433. [DOI] [PubMed] [Google Scholar]
- 23.Kufel J., Bousquet-Antonelli C., Beggs J.D., Tollervey D. Nuclear pre-mRNA decapping and 5′ degradation in yeast require the Lsm2-8p complex. Mol. Cell. Biol. 2004;24:9646–9657. doi: 10.1128/MCB.24.21.9646-9657.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sharif H., Conti E. Architecture of the Lsm1-7-Pat1 complex: a conserved assembly in eukaryotic mRNA turnover. Cell Rep. 2013;5:283–291. doi: 10.1016/j.celrep.2013.10.004. [DOI] [PubMed] [Google Scholar]
- 25.Chowdhury A., Mukhopadhyay J., Tharun S. The decapping activator Lsm1p-7p-Pat1p complex has the intrinsic ability to distinguish between oligoadenylated and polyadenylated RNAs. RNA. 2007;13:998–1016. doi: 10.1261/rna.502507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu D., Muhlrad D., Bowler M.W., Jiang S., Liu Z., Parker R., Song H. Lsm2 and Lsm3 bridge the interaction of the Lsm1-7 complex with Pat1 for decapping activation. Cell Res. 2014;24:233–246. doi: 10.1038/cr.2013.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Friesen W.J., Dreyfuss G. Specific sequences of the Sm and Sm-like (Lsm) proteins mediate their interaction with the spinal muscular atrophy disease gene product (SMN) J. Biol. Chem. 2000;275:26370–26375. doi: 10.1074/jbc.M003299200. [DOI] [PubMed] [Google Scholar]
- 28.Will C.L., Lührmann R. Spliceosome structure and function. Cold Spring Harb. Perspect. Biol. 2011;3:a003707. doi: 10.1101/cshperspect.a003707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen W., Moore M.J. Spliceosomes. Curr. Biol. 2015;25:R181–R183. doi: 10.1016/j.cub.2014.11.059. [DOI] [PubMed] [Google Scholar]
- 30.Verdone L., Galardi S., Page D., Beggs J.D. Lsm proteins promote regeneration of pre-mRNA splicing activity. Curr. Biol. 2004;14:1487–1491. doi: 10.1016/j.cub.2004.08.032. [DOI] [PubMed] [Google Scholar]
- 31.Wilusz C.J., Wilusz J. Lsm proteins and Hfq: Life at the 3′ end. RNA Biol. 2013;10:592–601. doi: 10.4161/rna.23695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Retterer K., Juusola J., Cho M.T., Vitazka P., Millan F., Gibellini F., Vertino-Bell A., Smaoui N., Neidich J., Monaghan K.G., et al. Clinical application of whole-exome sequencing across clinical indications. Genet. Med. 2016;18:696–704. doi: 10.1038/gim.2015.148. [DOI] [PubMed] [Google Scholar]
- 33.Richards S., Aziz N., Bale S., Bick D., Das S., Gastier-Foster J., Grody W.W., Hegde M., Lyon E., Spector E., et al. ACMG Laboratory Quality Assurance Committee Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sobreira N., Schiettecatte F., Boehm C., Valle D., Hamosh A. New tools for Mendelian disease gene identification: PhenoDB variant analysis module; and GeneMatcher, a web-based tool for linking investigators with an interest in the same gene. Hum. Mutat. 2015;36:425–431. doi: 10.1002/humu.22769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sobreira N., Schiettecatte F., Valle D., Hamosh A. GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum. Mutat. 2015;36:928–930. doi: 10.1002/humu.22844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Monies D., Abouelhoda M., Assoum M., Moghrabi N., Rafiullah R., Almontashiri N., Alowain M., Alzaidan H., Alsayed M., Subhani S., et al. Lessons Learned from Large-Scale, First-Tier Clinical Exome Sequencing in a Highly Consanguineous Population. Am. J. Hum. Genet. 2019;105:879. doi: 10.1016/j.ajhg.2019.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lek M., Karczewski K.J., Minikel E.V., Samocha K.E., Banks E., Fennell T., O’Donnell-Luria A.H., Ware J.S., Hill A.J., Cummings B.B., et al. Exome Aggregation Consortium Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ng P.C., Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Choi Y., Chan A.P. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics. 2015;31:2745–2747. doi: 10.1093/bioinformatics/btv195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kircher M., Witten D.M., Jain P., O’Roak B.J., Cooper G.M., Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014;46:310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rentzsch P., Witten D., Cooper G.M., Shendure J., Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019;47(D1):D886–D894. doi: 10.1093/nar/gky1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schwarz J.M., Cooper D.N., Schuelke M., Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat. Methods. 2014;11:361–362. doi: 10.1038/nmeth.2890. [DOI] [PubMed] [Google Scholar]
- 43.Taylor S., Wakem M., Dijkman G., Alsarraj M., Nguyen M. A practical approach to RT-qPCR-Publishing data that conform to the MIQE guidelines. Methods. 2010;50:S1–S5. doi: 10.1016/j.ymeth.2010.01.005. [DOI] [PubMed] [Google Scholar]
- 44.Taylor S.C., Mrkusich E.M. The state of RT-quantitative PCR: firsthand observations of implementation of minimum information for the publication of quantitative real-time PCR experiments (MIQE) J. Mol. Microbiol. Biotechnol. 2014;24:46–52. doi: 10.1159/000356189. [DOI] [PubMed] [Google Scholar]
- 45.Zhou L., Hang J., Zhou Y., Wan R., Lu G., Yin P., Yan C., Shi Y. Crystal structures of the Lsm complex bound to the 3′ end sequence of U6 small nuclear RNA. Nature. 2014;506:116–120. doi: 10.1038/nature12803. [DOI] [PubMed] [Google Scholar]
- 46.Chen G.I., Gingras A.C. Affinity-purification mass spectrometry (AP-MS) of serine/threonine phosphatases. Methods. 2007;42:298–305. doi: 10.1016/j.ymeth.2007.02.018. [DOI] [PubMed] [Google Scholar]
- 47.Mellacheruvu D., Wright Z., Couzens A.L., Lambert J.P., St-Denis N.A., Li T., Miteva Y.V., Hauri S., Sardiu M.E., Low T.Y., et al. The CRAPome: a contaminant repository for affinity purification-mass spectrometry data. Nat. Methods. 2013;10:730–736. doi: 10.1038/nmeth.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cox J., Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 49.Cox J., Michalski A., Mann M. Software lock mass by two-dimensional minimization of peptide mass errors. J. Am. Soc. Mass Spectrom. 2011;22:1373–1380. doi: 10.1007/s13361-011-0142-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cox J., Hein M.Y., Luber C.A., Paron I., Nagaraj N., Mann M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteomics. 2014;13:2513–2526. doi: 10.1074/mcp.M113.031591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schaab C., Geiger T., Stoehr G., Cox J., Mann M. Analysis of high accuracy, quantitative proteomics data in the MaxQB database. Mol. Cell Proteomics. 2012;11 doi: 10.1074/mcp.M111.014068. M111.014068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tyanova S., Temu T., Carlson A., Sinitcyn P., Mann M., Cox J. Visualization of LC-MS/MS proteomics data in MaxQuant. Proteomics. 2015;15:1453–1456. doi: 10.1002/pmic.201400449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tyanova S., Temu T., Cox J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016;11:2301–2319. doi: 10.1038/nprot.2016.136. [DOI] [PubMed] [Google Scholar]
- 54.Tyanova S., Temu T., Sinitcyn P., Carlson A., Hein M.Y., Geiger T., Mann M., Cox J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods. 2016;13:731–740. doi: 10.1038/nmeth.3901. [DOI] [PubMed] [Google Scholar]
- 55.Rudolph J.D., Cox J. A Network Module for the Perseus Software for Computational Proteomics Facilitates Proteome Interaction Graph Analysis. J. Proteome Res. 2019;18:2052–2064. doi: 10.1021/acs.jproteome.8b00927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tyanova S., Albrechtsen R., Kronqvist P., Cox J., Mann M., Geiger T. Proteomic maps of breast cancer subtypes. Nat. Commun. 2016;7:10259. doi: 10.1038/ncomms10259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cox J., Mann M. 1D and 2D annotation enrichment: a statistical method integrating quantitative proteomics with complementary high-throughput data. BMC Bioinformatics. 2012;13(Suppl 16):S12. doi: 10.1186/1471-2105-13-S16-S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Choquet K., Pinard M., Yang S., Moir R.D., Poitras C., Dicaire M.J., Sgarioto N., Larivière R., Kleinman C.L., Willis I.M., et al. The leukodystrophy mutation Polr3b R103H causes homozygote mouse embryonic lethality and impairs RNA polymerase III biogenesis. Mol. Brain. 2019;12:59. doi: 10.1186/s13041-019-0479-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vazquez-Arango P., O’Reilly D. Variant snRNPs: New players within the spliceosome system. RNA Biol. 2018;15:17–25. doi: 10.1080/15476286.2017.1373238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Matera A.G., Wang Z. A day in the life of the spliceosome. Nat. Rev. Mol. Cell Biol. 2014;15:108–121. doi: 10.1038/nrm3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Didychuk A.L., Butcher S.E., Brow D.A. The life of U6 small nuclear RNA, from cradle to grave. RNA. 2018;24:437–460. doi: 10.1261/rna.065136.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vindry C., Marnef A., Broomhead H., Twyffels L., Ozgur S., Stoecklin G., Llorian M., Smith C.W., Mata J., Weil D., Standart N. Dual RNA Processing Roles of Pat1b via Cytoplasmic Lsm1-7 and Nuclear Lsm2-8 Complexes. Cell Rep. 2017;20:1187–1200. doi: 10.1016/j.celrep.2017.06.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sabarinathan R., Tafer H., Seemann S.E., Hofacker I.L., Stadler P.F., Gorodkin J. RNAsnp: efficient detection of local RNA secondary structure changes induced by SNPs. Hum. Mutat. 2013;34:546–556. doi: 10.1002/humu.22273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sharma Y., Miladi M., Dukare S., Boulay K., Caudron-Herger M., Groß M., Backofen R., Diederichs S. A pan-cancer analysis of synonymous mutations. Nat. Commun. 2019;10:2569. doi: 10.1038/s41467-019-10489-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Salari R., Kimchi-Sarfaty C., Gottesman M.M., Przytycka T.M. Sensitive measurement of single-nucleotide polymorphism-induced changes of RNA conformation: application to disease studies. Nucleic Acids Res. 2013;41:44–53. doi: 10.1093/nar/gks1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Thisse C., Thisse B. High-resolution in situ hybridization to whole-mount zebrafish embryos. Nat. Protoc. 2008;3:59–69. doi: 10.1038/nprot.2007.514. [DOI] [PubMed] [Google Scholar]
- 67.Bonkowsky J.L., Wang X., Fujimoto E., Lee J.E., Chien C.B., Dorsky R.I. Domain-specific regulation of foxP2 CNS expression by lef1. BMC Dev. Biol. 2008;8:103. doi: 10.1186/1471-213X-8-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lambert A.M., Bonkowsky J.L., Masino M.A. The conserved dopaminergic diencephalospinal tract mediates vertebrate locomotor development in zebrafish larvae. J. Neurosci. 2012;32:13488–13500. doi: 10.1523/JNEUROSCI.1638-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Minnerop M., Kurzwelly D., Wagner H., Soehn A.S., Reichbauer J., Tao F., Rattay T.W., Peitz M., Rehbach K., Giorgetti A., et al. Hypomorphic mutations in POLR3A are a frequent cause of sporadic and recessive spastic ataxia. Brain. 2017;140:1561–1578. doi: 10.1093/brain/awx095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Harting I., Al-Saady M., Krägeloh-Mann I., Bley A., Hempel M., Bierhals T., Karch S., Moog U., Bernard G., Huntsman R., et al. POLR3A variants with striatal involvement and extrapyramidal movement disorder. Neurogenetics. 2020;21:121–133. doi: 10.1007/s10048-019-00602-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hiraide T., Kubota K., Kono Y., Watanabe S., Matsubayashi T., Nakashima M., Kaname T., Fukao T., Shimozawa N., Ogata T., Saitsu H. POLR3A variants in striatal involvement without diffuse hypomyelination. Brain Dev. 2020;42:363–368. doi: 10.1016/j.braindev.2019.12.012. [DOI] [PubMed] [Google Scholar]
- 72.Perrier S., Gauquelin L., Fallet-Bianco C., Dishop M.K., Michell-Robinson M.A., Tran L.T., Guerrero K., Darbelli L., Srour M., Petrecca K., et al. Expanding the phenotypic and molecular spectrum of RNA polymerase III-related leukodystrophy. Neurol. Genet. 2020;6:e425. doi: 10.1212/NXG.0000000000000425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wu S., Bai Z., Dong X., Yang D., Chen H., Hua J., Zhou L., Lv H. Novel mutations of the POLR3A gene caused POLR3-related leukodystrophy in a Chinese family: a case report. BMC Pediatr. 2019;19:289. doi: 10.1186/s12887-019-1656-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gauquelin L., Cayami F.K., Sztriha L., Yoon G., Tran L.T., Guerrero K., Hocke F., van Spaendonk R.M.L., Fung E.L., D’Arrigo S., et al. DDD Study Clinical spectrum of POLR3-related leukodystrophy caused by biallelic POLR1C pathogenic variants. Neurol. Genet. 2019;5:e369. doi: 10.1212/NXG.0000000000000369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Landau D., Cohen D., Shalev H., Pinsk V., Yerushalmi B., Zeigler M., Birk O.S. A novel mutation in the SLC17A5 gene causing both severe and mild phenotypes of free sialic acid storage disease in one inbred Bedouin kindred. Mol. Genet. Metab. 2004;82:167–172. doi: 10.1016/j.ymgme.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 76.Ito Y., Hartley T., Baird S., Venkateswaran S., Simons C., Wolf N.I., Boycott K.M., Dyment D.A., Kernohan K.D. Lysosomal dysfunction in TMEM106B hypomyelinating leukodystrophy. Neurol. Genet. 2018;4:e288. doi: 10.1212/NXG.0000000000000288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wolf N.I., Salomons G.S., Rodenburg R.J., Pouwels P.J., Schieving J.H., Derks T.G., Fock J.M., Rump P., van Beek D.M., van der Knaap M.S., Waisfisz Q. Mutations in RARS cause hypomyelination. Ann. Neurol. 2014;76:134–139. doi: 10.1002/ana.24167. [DOI] [PubMed] [Google Scholar]
- 78.Al-Abdi L., Al Murshedi F., Elmanzalawy A., Al Habsi A., Helaby R., Ganesh A., Ibrahim N., Patel N., Alkuraya F.S. CNP deficiency causes severe hypomyelinating leukodystrophy in humans. Hum. Genet. 2020;139:615–622. doi: 10.1007/s00439-020-02144-4. [DOI] [PubMed] [Google Scholar]
- 79.Chelban V., Patel N., Vandrovcova J., Zanetti M.N., Lynch D.S., Ryten M., Botía J.A., Bello O., Tribollet E., Efthymiou S., et al. SYNAPSE Study Group Mutations in NKX6-2 Cause Progressive Spastic Ataxia and Hypomyelination. Am. J. Hum. Genet. 2017;100:969–977. doi: 10.1016/j.ajhg.2017.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kufel J., Allmang C., Petfalski E., Beggs J., Tollervey D. Lsm Proteins are required for normal processing and stability of ribosomal RNAs. J. Biol. Chem. 2003;278:2147–2156. doi: 10.1074/jbc.M208856200. [DOI] [PubMed] [Google Scholar]
- 81.Ingelfinger D., Arndt-Jovin D.J., Lührmann R., Achsel T. The human LSm1-7 proteins colocalize with the mRNA-degrading enzymes Dcp1/2 and Xrnl in distinct cytoplasmic foci. RNA. 2002;8:1489–1501. [PMC free article] [PubMed] [Google Scholar]
- 82.He W., Parker R. The yeast cytoplasmic LsmI/Pat1p complex protects mRNA 3′ termini from partial degradation. Genetics. 2001;158:1445–1455. doi: 10.1093/genetics/158.4.1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hollams E.M., Giles K.M., Thomson A.M., Leedman P.J. MRNA stability and the control of gene expression: implications for human disease. Neurochem. Res. 2002;27:957–980. doi: 10.1023/a:1020992418511. [DOI] [PubMed] [Google Scholar]
- 84.Linder B., Fischer U., Gehring N.H. mRNA metabolism and neuronal disease. FEBS Lett. 2015;589:1598–1606. doi: 10.1016/j.febslet.2015.04.052. [DOI] [PubMed] [Google Scholar]
- 85.Vlasova-St Louis I., Dickson A.M., Bohjanen P.R., Wilusz C.J. CELFish ways to modulate mRNA decay. Biochim. Biophys. Acta. 2013;1829:695–707. doi: 10.1016/j.bbagrm.2013.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Eberhardt W., Doller A., Akool S., Pfeilschifter J. Modulation of mRNA stability as a novel therapeutic approach. Pharmacol. Ther. 2007;114:56–73. doi: 10.1016/j.pharmthera.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 87.Schoenberg D.R., Maquat L.E. Regulation of cytoplasmic mRNA decay. Nat. Rev. Genet. 2012;13:246–259. doi: 10.1038/nrg3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cheneval D., Kastelic T., Fuerst P., Parker C.N. A review of methods to monitor the modulation of mRNA stability: a novel approach to drug discovery and therapeutic intervention. J. Biomol. Screen. 2010;15:609–622. doi: 10.1177/1087057110365897. [DOI] [PubMed] [Google Scholar]
- 89.Anderson P. Post-transcriptional regulons coordinate the initiation and resolution of inflammation. Nat. Rev. Immunol. 2010;10:24–35. doi: 10.1038/nri2685. [DOI] [PubMed] [Google Scholar]
- 90.Frischmeyer P.A., Dietz H.C. Nonsense-mediated mRNA decay in health and disease. Hum. Mol. Genet. 1999;8:1893–1900. doi: 10.1093/hmg/8.10.1893. [DOI] [PubMed] [Google Scholar]
- 91.Benjamin D., Moroni C. mRNA stability and cancer: an emerging link? Expert Opin. Biol. Ther. 2007;7:1515–1529. doi: 10.1517/14712598.7.10.1515. [DOI] [PubMed] [Google Scholar]
- 92.Khajavi M., Inoue K., Lupski J.R. Nonsense-mediated mRNA decay modulates clinical outcome of genetic disease. Eur. J. Hum. Genet. 2006;14:1074–1081. doi: 10.1038/sj.ejhg.5201649. [DOI] [PubMed] [Google Scholar]
- 93.Miller J.N., Pearce D.A. Nonsense-mediated decay in genetic disease: friend or foe? Mutat. Res. Rev. Mutat. Res. 2014;762:52–64. doi: 10.1016/j.mrrev.2014.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Pashler A.L., Towler B.P., Jones C.I., Newbury S.F. The roles of the exoribonucleases DIS3L2 and XRN1 in human disease. Biochem. Soc. Trans. 2016;44:1377–1384. doi: 10.1042/BST20160107. [DOI] [PubMed] [Google Scholar]
- 95.Morita M., Siddiqui N., Katsumura S., Rouya C., Larsson O., Nagashima T., Hekmatnejad B., Takahashi A., Kiyonari H., Zang M., et al. Hepatic posttranscriptional network comprised of CCR4-NOT deadenylase and FGF21 maintains systemic metabolic homeostasis. Proc. Natl. Acad. Sci. USA. 2019;116:7973–7981. doi: 10.1073/pnas.1816023116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sveen A., Kilpinen S., Ruusulehto A., Lothe R.A., Skotheim R.I. Aberrant RNA splicing in cancer; expression changes and driver mutations of splicing factor genes. Oncogene. 2016;35:2413–2427. doi: 10.1038/onc.2015.318. [DOI] [PubMed] [Google Scholar]
- 97.Weskamp K., Barmada S.J. RNA Degradation in Neurodegenerative Disease. Adv. Neurobiol. 2018;20:103–142. doi: 10.1007/978-3-319-89689-2_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kapur M., Monaghan C.E., Ackerman S.L. Regulation of mRNA Translation in Neurons-A Matter of Life and Death. Neuron. 2017;96:616–637. doi: 10.1016/j.neuron.2017.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Padgett R.A. New connections between splicing and human disease. Trends Genet. 2012;28:147–154. doi: 10.1016/j.tig.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Faustino N.A., Cooper T.A. Pre-mRNA splicing and human disease. Genes Dev. 2003;17:419–437. doi: 10.1101/gad.1048803. [DOI] [PubMed] [Google Scholar]
- 101.Jutzi D., Akinyi M.V., Mechtersheimer J., Frilander M.J., Ruepp M.D. The emerging role of minor intron splicing in neurological disorders. Cell Stress. 2018;2:40–54. doi: 10.15698/cst2018.03.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Pasternack S.M., Refke M., Paknia E., Hennies H.C., Franz T., Schäfer N., Fryer A., van Steensel M., Sweeney E., Just M., et al. Mutations in SNRPE, which encodes a core protein of the spliceosome, cause autosomal-dominant hypotrichosis simplex. Am. J. Hum. Genet. 2013;92:81–87. doi: 10.1016/j.ajhg.2012.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lynch D.C., Revil T., Schwartzentruber J., Bhoj E.J., Innes A.M., Lamont R.E., Lemire E.G., Chodirker B.N., Taylor J.P., Zackai E.H., et al. Care4Rare Canada Disrupted auto-regulation of the spliceosomal gene SNRPB causes cerebro-costo-mandibular syndrome. Nat. Commun. 2014;5:4483. doi: 10.1038/ncomms5483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Bacrot S., Doyard M., Huber C., Alibeu O., Feldhahn N., Lehalle D., Lacombe D., Marlin S., Nitschke P., Petit F., et al. Mutations in SNRPB, encoding components of the core splicing machinery, cause cerebro-costo-mandibular syndrome. Hum. Mutat. 2015;36:187–190. doi: 10.1002/humu.22729. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The clinical exomes are not publicly available. The variants generated during this study are available at ClinVar (gene variants; ClinVar: SCV001478394 and SCV001478886).