

Abstract
Background:
Initial evidence that OPRM1 genotype moderates the clinical response to naltrexone has not been replicated in prospective clinical trials. However, the use of traditional statistical analyses and clinical endpoints might limit sensitivity for studying pharmacogenetic associations, whereas the use of intensive daily assessments and person-centered analytic methods might increase sensitivity. This study leveraged person-centered analyses and daily measures of alcohol use, craving, and medication adherence to investigate OPRM1 as a moderator of changes in clinical outcomes during naltrexone treatment.
Methods:
Treatment-seeking participants with alcohol use disorder (n=58; Mage=38 years; 71% male) provided daily cellphone reports of craving and consumption while taking naltrexone as part of a mobile health trial. Daily medication adherence was measured remotely using electronic pillcap recordings. Multilevel modeling and multilevel structural equation modeling analyses evaluated the hypotheses that OPRM1 genotype would moderate prospective reductions in daily alcohol use and craving, and would also moderate within-person associations of daily adherence with same-day craving and consumption.
Results:
OPRM1 genotype moderated the association of daily adherence with reduced same-day consumption (p = .007) and craving (p = .06), with these associations being stronger for participants with the 118G variant. OPRM1 genotype did not moderate changes in craving and consumption over time.
Conclusions:
These findings suggest that high-density assessments and person-centered analytic approaches, including modeling within-person variation in medication adherence, could be advantageous for pharmacogenetic studies.
Keywords: pharmacotherapy, precision medicine, medication adherence, mu opioid receptor, treatment
Introduction
Pharmacotherapy for alcohol use disorder (AUD) remains relatively underutilized in clinical practice (Heilig et al., 2011, Jonas et al., 2014). Perceptions of limited efficacy have been noted as one barrier to pharmacotherapy uptake (Garbutt et al., 2014, Kranzler and Soyka, 2018, Mark et al., 2009), perhaps reflecting the modest overall effect sizes observed in clinical trials of AUD medications (Jonas et al., 2014, Kranzler and Soyka, 2018). Given significant heterogeneity in AUD treatment response (Litten et al., 2015), identifying individual difference factors that predict improved treatment efficacy (i.e., a precision medicine approach) is one approach for maximizing therapeutic response in particular subgroups. For example, while naltrexone (a μ-opioid receptor antagonist and front-line AUD treatment) has been in clinical use for over two decades, recent work has continued to identify biological and behavioral factors accounting for individual differences in treatment response (Heilig et al., 2011, Kranzler et al., 2013, Maisel et al., 2013, Mann et al., 2018, Schacht et al., 2017, Witkiewitz et al., 2019).
Efforts to advance precision medicine approaches for AUD have focused largely on between-person factors predictive of treatment response (Heilig et al., 2011, Mann et al., 2018; Kranzler and McKay, 2012), with a particular focus on genetic variants. Of numerous candidate genes examined to date (Hartwell and Kranzler, 2019, Kranzler and Edenberg, 2010), the mu opioid receptor gene (OPRM1) has received the most attention. The OPRM1 A118G variant (Asn40Asp; rs1799971), located in exon 1 of the OPRM1 gene, is a single nucleotide polymorphism (SNP) resulting in substitution of an asparagine (Asn) residue with an aspartic acid (Asp) residue. While the precise functional consequences of OPRM1 A118G have yet to be confirmed, substantial preclinical and clinical evidence points to a functional role of this variant (or its non-human orthologs) in moderating responses to psychoactive drugs, including alcohol (Heilig et al., 2011; Mague and Blendy, 2010). For instance, rigorous preclinical studies support functional associations of OPRM1 with indices of alcohol reward and motivation (Barr et al., 2007, Bilbao et al., 2015, Ramchandani et al., 2011), with some findings also translating to human studies (Ramchandani et al., 2011).
Importantly, non-human primate studies showed that a functional ortholog of the human A118G variant moderated naltrexone-induced reductions in ethanol self-administration (Barr et al., 2010, Vallender et al., 2010). This finding was consistent with early retrospective analyses from naltrexone clinical trials, which reported that patients with the 118G variant (i.e., GG or GA genotypes) showed reduced consumption or relapse likelihood during treatment with naltrexone vs. placebo, relative to patients homozygous for the 118A variant (i.e., AA genotype; Anton et al., 2008, Oslin et al., 2003). Given additional evidence for OPRM1 genotype differences in alcohol-related craving and subjective responses to alcohol (for a review, see Hartwell and Kranzler, 2019, Ray et al., 2012), which are putative treatment mechanisms of naltrexone, greater medication-related reductions in alcohol craving or reinforcement among 118G carriers (relative to 118A homozygotes) is one explanation for potential genotype differences in treatment response. Consistent with this possibility, one randomized trial found that participants with the 118G variant showed a relatively stronger daily association between evening desire to drink and subsequent consumption, as well as relatively greater attenuation of this association by naltrexone, relative to 118A homozygotes (Kranzler et al., 2013).
Although these findings were considered promising, subsequent naltrexone trials using prospective genotyping failed to confirm that OPRM1 genotype moderates treatment response (Oslin et al., 2015, Schacht et al., 2017), and there is presently insufficient evidence for precision medicine applications involving OPRM1. Nevertheless, the failure of initial findings to replicate in prospective trials does not preclude the biological plausibility of this association, or negate its study in clinical contexts. In particular, cross-species evidence that OPRM1 moderates reductions in consumption during naltrexone treatment (Barr et al., 2010, Bilbao et al., 2015, Ramchandani et al., 2011, Ray and Heilig, 2013) supports the basic validity of this association, and these studies have proven useful for heuristic models of AUD pharmacogenetics (Bilbao et al., 2015, Heilig et al., 2011, Ray and Heilig, 2013). Even absent evidence for clinical utility, such studies can contribute to mechanistic understanding of pharmacogenetic associations, and can inform future research.
An important methodological consideration is that most pharmacogenetic studies have focused on between-person differences in traditional clinical endpoints (e.g., mean levels of drinking or proportion of patients who return to heavy drinking), typically using aggregate outcomes measured over weeks or months. In contrast, intensive micro-longitudinal assessment methods, coupled with person-centered analytic approaches that probe within-person effects, might increase sensitivity for detecting genetic moderation of treatment processes. For example, in a naltrexone trial examining OPRM1 in relation to daily changes in drinking and craving (Kranzler et al., 2013), conventional between-person analyses found no main effects of genotype or treatment-by genotype interactions. However, within-person analyses detected a moderating role of OPRM1 A118G, such that the association between evening craving and subsequent consumption was stronger for those with the 118G variant (Kranzler et al., 2013). Moreover, naltrexone (vs. placebo) selectively attenuated the within-person association of craving and consumption in this group, consistent with prior evidence for improved treatment response in those with the 118G variant (Anton et al., 2008, Oslin et al., 2003). These findings indicate that a “within-person approach to phenotyping” might detect pharmacogenetic associations that go unobserved when applying traditional analytic methods and clinical endpoints (Kranzler et al., 2013, p. 199).
Other studies have similarly capitalized on intensive daily assessments to study genetic moderators of consumption and/or treatment response. In a re-analysis of a naltrexone trial involving ecological momentary assessment (EMA), researchers examined daily drinking and craving as a function of treatment condition and DRD4 genotype (Miranda et al., 2018). Results indicated that craving during non-drinking moments mediated naltrexone effects on reduced same-day heavy drinking, with this effect being specific to participants with the DRD4-L allele (Miranda et al., 2018). In another study using EMA, participants with the OPRM1 118G variant consumed more drinks per episode relative to 118A homozygotes, and these groups showed differential associations between urge and subsequent consumption (Ray et al., 2010). As these studies illustrate, combining intensive daily assessments with person-centered analyses has advantages for AUD clinical trials and pharmacogenetic studies. These advantages include the generation of high-density longitudinal data, resulting in the ability to model within-person processes relevant to treatment outcome with high resolution.
Because detecting pharmacogenetic associations requires sufficient medication exposure, another important consideration is medication adherence. Adherence to AUD medications in clinical settings is typically poor (Mark et al., 2003, 2009) and declines over the course of treatment (Dermody et al., 2018), presenting challenges for pharmacogenetic studies. Importantly, naltrexone’s neuropharmacological profile suggests that daily changes in adherence could have implications for fluctuations in craving and consumption. A single dose leads to rapid peak plasma levels and receptor occupancy (Lee et al., 1988), and is sufficient to reduce short-term craving and consumption in laboratory settings (Hendershot et al., 2017), implying that daily changes in drinking or craving could be sensitive to fluctuations in adherence. Whereas most clinical trials estimate adherence with aggregate, retrospective measures (i.e., proportion of pills taken over the course of treatment), modeling daily variability in adherence could be advantageous for studying pharmacogenetic associations.
Using data from a completed naltrexone trial, the present study capitalized on intensive daily measurements and within-person analytic approaches to investigate changes in daily craving and consumption as a function of OPRM1 genotype and medication adherence. In addition to collecting daily measures of drinking and craving via smartphone, daily adherence was measured via remote pill cap recordings. Multilevel models (MLM) and multilevel structural equation models (MSEM) evaluated the predictions that: 1) daily alcohol use and craving would decrease over time, with larger decreases for participants with the 118G variant, 2) daily naltrexone adherence would predict reduced same-day craving and consumption, and 3) OPRM1 would moderate within-person associations of adherence with reductions in craving and consumption, with these associations being strongest for those with the 118G variant. Given some evidence for lower naltrexone adherence among participants with the 118G variant (Oslin et al., 2015), we also examined daily changes in adherence as a function of genotype.
Materials and Methods
Participants and Setting
Participants were recruited for a randomized trial to investigate a mobile health (mHealth) intervention as a means of improving AUD medication adherence (ClinicalTrials.gov Identifier: NCT01349985) (Stoner et al., 2015, Stoner and Hendershot, 2012). The parent study included 76 treatment-seeking participants with AUD who sought to reduce their drinking while enrolled in an open-label naltrexone study. Briefly, the primary results of the trial showed that an automated text message system did not improve overall adherence during treatment, but that intervention participants maintained adequate adherence (80%) for relatively longer than control participants (Stoner et al., 2015). The study was carried out at the Mind Research Network (Albuquerque, NM). Ethics approval was obtained from the University of New Mexico Human Research Review Committee and Quorum Review (Seattle, WA).
The primary inclusion criteria included being 21–55 years of age, seeking assistance to reduce or stop drinking alcohol, and being willing to take naltrexone (52.6% of randomized participants reported an abstinence goal). Participants were excluded if they reported illicit drug use (other than cannabis) in the prior 30 days; recent or current prescription opioid use; history of opioid dependence or psychotic disorder; current use of psychiatric medication other than antidepressants; concurrent treatment for alcohol use other than Alcoholics Anonymous; potential pregnancy; or a legal requirement to attend treatment. Due to a high degree of missing adherence data during weeks 5–8 (reflecting considerable attrition and a high proportion of non-compliance in returning MEMS caps among those participants), the present investigation focuses on the subsample of participants (n = 58) for whom daily adherence data were available through the first month of treatment, which required participants to remain enrolled in the trial and return their MEMS electronic pill caps at a 4-week follow-up visit.
Procedures
Participants were recruited using local print and radio advertisements targeting heavy drinkers with interest in cutting down or quitting drinking. After a preliminary telephone screening, participants attended a baseline assessment visit to determine eligibility, including an assessment by the study physician. Eligible participants provided informed consent and proceeded to complete self-report measures before being randomized to condition. All randomized participants received an initial 4-week supply of naltrexone (50mg/day) and a smartphone (Android operating system) with unlimited service. Participants were instructed on using the phone to respond to daily survey prompts (described below). Participants were instructed to take medication daily in the morning, and were scheduled for a medication refill and follow-up assessments at week 4, prior to a final follow-up appointment at week 8.
As described previously (Stoner et al., 2015, Stoner and Hendershot, 2012), the mobile health intervention involved automated text message reminders to facilitate adherence. Participants were randomly assigned to the text message reminder intervention (n=37) or a control condition (n=39), in which participants completed daily assessments but did not receive medication reminders. All participants received a daily SMS prompt with a hyperlink to a brief questionnaire assessing alcohol use and craving for the prior day. These assessments, which were standard across intervention conditions (to avoid the possibility that assessment reactivity would differentially affect drinking across intervention arms), provide the basis for the present analyses.
Baseline Measures and Daily Assessments
Self-report measures completed at baseline included the Timeline Follow-back (TLFB) calendar method (Sobell and Sobell, 1992), which was used to quantify alcohol use during the 90 days preceding baseline, including average standard drinks per drinking day. This method has been shown to have adequate reliability and validity in a number of studies (Sobell and Sobell, 1992, Sobell and Sobell, 1995). Daily drinking and craving levels were queried once per day via smartphone using text message reminders. Upon responding to the daily text message prompt, participants were asked how many standard drinks they consumed on the prior day (response options ranged from 0 to 13+). Prior-day craving was assessed with a single item from the Penn Alcohol Craving Scale: “At its most severe point, how strong was your craving to drink alcohol?” The 7 response options ranged from “none at all” to “strong urge and would have drunk alcohol if it were available.”
Naltrexone adherence was assessed using the Medication Event Monitoring System (MEMS, MWV Healthcare). Participants received their medication in a pill bottle equipped with a microchip-embedded cap that logged all openings. Recorded pill bottle openings served as the index of daily medication adherence (with non-adherence inferred based on absence of a recorded opening). Participants were asked to bring pill bottles to the 4-week laboratory visit, at which point the research assistant uploaded MEMS data. Although efforts were made to retrieve caps from all randomized participants, some caps could not be retrieved (e.g., reflecting attrition and/or missed follow-up appointments). Given the once per day dosing schedule for naltrexone, MEMS data were cleaned to ensure that no more than one opening per day was counted as a taken dose. In total, week 4 MEMS adherence data were available from 60 participants who returned their pill bottles. Two participants who disclosed information invalidating their MEMS data (discarding pills, taking half- or double-doses) were omitted from further analyses, which resulted in a final sample of 58 participants for the present analyses.
Genotyping
Saliva samples were collected for purposes of DNA extraction. Following extraction, the OPRM1 A118G variant (rs1799971) was genotyped using the C___8950074_1_ TaqMan® pre-designed assay (Life Technologies, Burlington, ON, Canada) according to manufacturer’s directions. Post-amplification products were analyzed on the ViiA™ 7 Real-Time PCR System. Genotypes were assigned automatically and manually verified by two lab personnel. For quality control purposes, a subset of samples were genotyped an additional time by a second lab personnel. Consistent with prior work, participants were grouped according to presence or absence of the 118G allele (GA/GG genotypes, n=16; AA genotype, n=42).
Statistical Analyses
All models were conducted in a multilevel framework using Mplus version 8.3 (Muthén and Muthén, 1998–2012). Maximum likelihood estimation with robust standard errors (MLR) was used to estimate all models. MLR adjusts standard error estimates to be robust to violations of the normality assumption (Asparouhov and Muthén, 2007) and handles missing data using full information maximum likelihood (FIML). FIML has been shown to produce the least biased naltrexone effect estimates and standard errors when compared to other missing data approaches such as complete case analysis, last observation carried forward, and making worst case assumptions about missing data (Hallgren and Witkiewitz, 2013).
To evaluate the hypothesis that daily alcohol use and craving would decrease over time, with larger decreases for individuals with the 118G variant, multilevel models were utilized. Specifically, these models tested associations between OPRM1 A118G with changes in daily alcohol use and daily craving (in separate models) over time. We first specified trends in changes over time by entering successive polynomial effects of time (linear then quadratic) as level 1 predictors of daily alcohol use and craving, along with corresponding random intercepts and slopes for these time effects. To facilitate model estimation, time was scaled such that each day was coded as a proportion of the four-week interval between randomization and the week 4 follow-up visit (i.e., Day 1=0.00, Day 2=0.04, Day 3 = 0.07, …, Day 28=1.00). Nested model testing was used to determine which fixed and random effects for time should be included in the model by retaining those that significantly improved model fit. Nested models were compared using log-likelihood tests of the deviance statistics (reported in the Results section as Delta −2LL) and scaling correction factors obtained with MLR estimation (Satorra and Bentler, 2010). Once the trajectory was fitted, genotype was included as a level-2 (between-person) predictor of the intercept and slope for the alcohol use and craving trajectories. Genotype was coded as a dichotomous variable (GA/GG = 1, AA = 0). These models controlled for intervention condition and race (each grand-mean centered to facilitate interpretation of level 2 intercept and slope means as a weighted-average across the covariates) at level 2.
To evaluate the hypothesis that daily naltrexone adherence would predict reduced same-day craving and consumption, the second set of analyses focused on relations between daily-level (within-person) naltrexone adherence and same-day alcohol use and craving using Multilevel Structural Equation Modeling (MSEM). MSEM uses latent variable modeling to disaggregate within-person and between-person associations between the independent and dependent variables of interest, thereby allowing for an examination of individual differences in day-to-day fluctuations in treatment outcomes. An advantage of MSEM is that it results in more reliable estimates of between-person associations relative to the traditional MLM approach of manually centering variables (Lüdtke et al., 2008). In these analyses (Figure 1), daily naltrexone adherence (coded as 0=MEMS cap opening not recorded, 1=MEMS cap opening recorded) was examined as a level 1 predictor of number of drinks per day and daily craving reports in separate models. Analyses controlled for between-person associations of adherence with alcohol use and craving, which helps to isolate the daily-level associations between adherence and use/craving from the influence of between-person factors (e.g., individual difference characteristics) that might relate both to overall likelihood of adherence and overall alcohol use and craving. Random intercepts and slopes for the within-person associations of adherence and same-day alcohol use and craving were evaluated.
Figure 1:
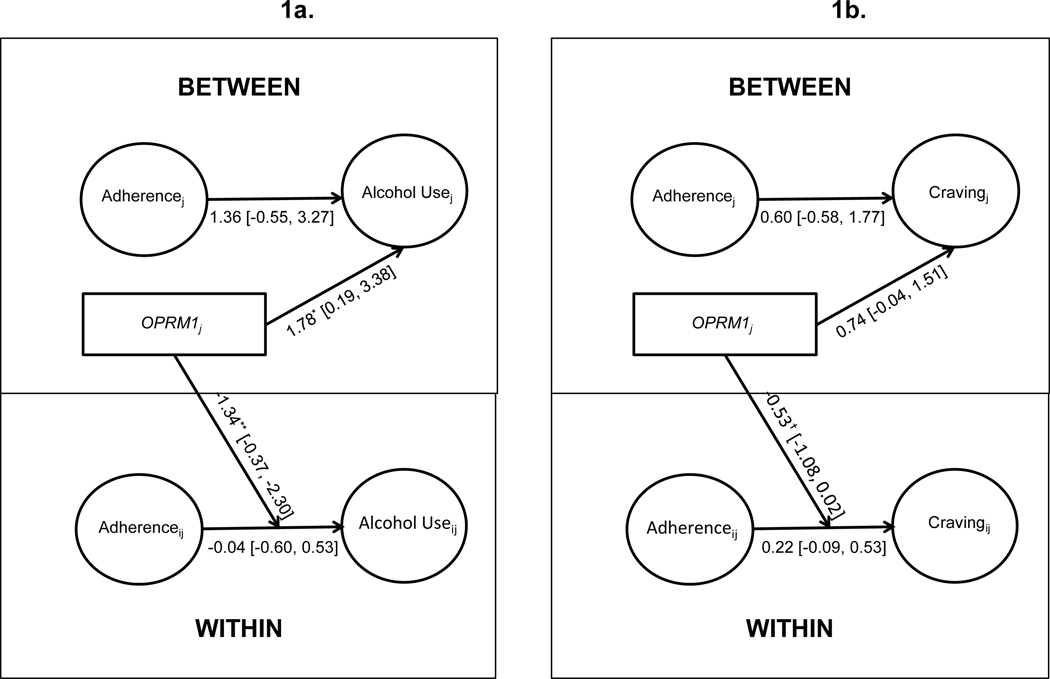
Multilevel structural equation models (MSEM) evaluating between- and within-person associations of daily naltrexone adherence with same-day alcohol use (1a) and craving (1b) and moderation by OPRM1 genotype. The moderation effect represents the regression of variance in the random slopes for within-person associations (i.e., adherence to drinking/craving) on OPRM1. Unstandardized parameter estimates shown. The associations of OPRM1 with the intercept represent OPRM1 differences in mean drinking levels, adjusted for intervention condition, race, and overall adherence. The OPRM1 moderation of within-person daily-level association between taking naltrexone and same-day values of the outcome adjusted for intervention condition and race. This figure depicts a simplified version of the model in which the random slopes are omitted. OPRM1 was coded GA/GG = 1, AA = 0.
†p<.10; *p<.05; **p<.01; ***p<.001.
Subsequently, in order to test the hypothesis that OPRM1 would moderate within-person associations of adherence and craving/consumption (with this association being strongest for 118G carriers) we examined whether the same-day associations differed based on genotype. Specifically, genotype was added as a predictor of the within-person associations (i.e., the random slope factors), along with covariates. In addition to the level 2 covariates described above, weekend day (Friday, Saturday, Sunday) versus weekday at level 1 (group-mean centered to facilitate parameter interpretation as a weighted-average across weekend vs weekday) was controlled for to account for day-of-the-week fluctuations in alcohol use, craving, and adherence previously identified in this sample (Dermody et al., 2018).
Results
Sample characteristics
Table 1 includes descriptive characteristics of the randomized participants included in the present analyses (n = 58); this subset did not differ from the primary sample (n = 76) on baseline measures (ps > .05) or genotype (ps > .05). The majority of individuals endorsed Caucasian race (n = 46), with the remaining identifying as Native American or Alaskan Native (n = 11) or Asian (n = 1). On average, during the first 28 treatment days, MEMS caps openings were recorded on 74% of days (SD = 28%); pill bottle openings did not differ based on genotype (p > .05). MEMS cap open-times suggest that the medication was typically taken in the mid-morning (mean = 11:30 AM, SD = 4:55; median = 10:05 AM), consistent with the physician’s instructions. On average, 75.80% (SD = 24.67) of the daily mobile phone assessments were completed for alcohol use and 76.54% (SD = 24.75) for craving, which did not differ based on genotype (ps > .05). Based on daily mobile phone assessments, participants reported drinking alcohol on 42% of these days (SD = 35%), averaging 4.96 (SD = 1.76) drinks per drinking day, and reported an average craving rating of 2.16 (SD = 1.22) on a scale of 0 to 6. Aggregate (between-person) drinks per drinking day and craving also did not differ based on OPMR1 genotype (ps > .05). Genotype status (GA/GG vs. AA) did not differ by racial group (Caucasian vs. non-Caucasian), χ2(57) = .90, p > .05.
Table 1.
Baseline characteristics of participants
Means (M) and standard deviations (SD) are reported for continuous variables along with corresponding t-test comparing values between the genotype groups. Similarly, sample size (N) and percentages are provided for dichotomous variables alongside chi-square (χ2) test statistics for differences between the genotype groups.
| AA (n = 42) | GA/GG (n = 16) | ||
|---|---|---|---|
|
| |||
| M (SD) | t-test | ||
| Age (years) | 37.36 (8.81) | 41.19 (9.50) | −1.45 |
| Drinks per drinking day | 9.91 (5.87) | 10.08 (4.71) | −.10 |
|
| |||
| N (%) | χ2 | ||
|
| |||
| Sex (female) | 15 (35.7) | 2 (12.5) | 3.01† |
| Income (above $20,000) | 25 (59.5) | 8 (53.3) | .17 |
| Race (Caucasian) | 32 (76.2) | 10 (87.5) | .90 |
| Education (Bachelor’s or higher) | 10 (23.8) | 2 (12.5) | .90 |
| DSM-IV-TR Alcohol Dependent | 27 (65.9) | 13 (86.7) | 2.31 |
p<.10
p<.05
p<.01
p<.001.
Changes in alcohol use and craving over time
We first fit linear and quadratic time trends to model changes in daily alcohol use. Modeling a random linear slope significantly improved model fit relative to a random intercept model only (Delta −2LL = 12.08, df = 2, p < .05); however, including the quadratic effect of time (along with a random slope factor) did not improve model fit relative to a linear time effect only (Delta −2LL = 1.92, df = 3, p > .05). Thus, change over time was modeled with a random linear slope only. The final model for the effects of time indicated that, on average, there was a non-significant reduction in drinks per day through week 4 (b = −.13, SE = .36, p = .73), but there was variability across individuals in the extent of reduction based on the random slope differed by genotype (random slope variance = 3.44, SE = 1.77, p = .05).
We similarly fit linear and quadratic time trends to model changes in daily craving. The model with the random linear time effect was retained, as the model with the random quadratic effect did not significantly improve fit (Delta −2LL = 1.27, df = 3, p > .05). The final model for the effect of time suggested that, on average, daily craving declined non-significantly over time (b = −.33, SE = .23, p = .15), and there was significant variability across participants in the linear slope (variance = 2.24, SE = .66, p = .001).
OPRM1 as a predictor of changes in alcohol use and craving over time
Controlling for intervention group, race, and weekend/weekday effects, OPRM1 was tested as a predictor of the linear drinking trajectory. OPRM1 was not a significant predictor of the intercept (i.e., drinks per day on day 1) or slope for drinks per day through week 4 (Table 2). Similarly, OPRM1 was not a significant predictor of the intercept (i.e., craving on day 1) or slope for craving through week 4. These results indicated no main effects of genotype on changes in craving or consumption during naltrexone treatment.
Table 2.
Multilevel model of associations of OPRM1 genotype with standard drinks consumed and craving over time
Unstandardized parameter estimates shown. Time was modeled as a random linear slope. The random intercept was centered at Day 1. Coefficients represent relationships between OPRM1 and the change in alcohol use or craving over time (random linear slope) as well as individual differences in adherence on Day 1 (random intercept). Time was coded with each day represented as proportion of the total trial length (i.e., by dividing each day by 28). Analyses controlled for effects of the intervention condition and race on the random linear intercept and slope (level 2) and weekend versus weekday on the daily outcome variable (level 1).
| Dependent Variable | Independent Variable | B | SE | p | 95% CI |
|---|---|---|---|---|---|
| Alcohol Use | |||||
|
| |||||
| Random Linear Slope | OPRM1 (G allele = 1) | 1.31 | .84 | .12 | [−.33, 2.96] |
| Random Intercept | OPRM1 (G allele = 1) | .21 | .72 | .77 | [−1.19, 1.61] |
|
| |||||
| Craving | |||||
|
| |||||
| Random Linear Slope | OPRM1 (G allele = 1) | .17 | .64 | .79 | [−1.09, 1.42] |
| Random Intercept | OPRM1 (G allele = 1) | .30 | .41 | .46 | [−.49, 1.10] |
Daily-level associations between adherence and alcohol use and craving
Contrary to hypotheses, there was no significant within-person association between daily adherence (based on MEMS openings vs. non-openings) and same-day alcohol use (slope = −0.44, SE = 0.30, p = .15; variance = 0.82, SE = .76, p = .28). There was also no between-person association between the proportion of days naltrexone was taken and proportion of drinking days (b = 1.42, SE = 1.08, p = .19). Similarly, analyses of daily craving indicated there was no within-person association between taking naltrexone and same-day craving (slope = 0.12, SE = 0.14, p = .40), and no between-person association between naltrexone taking and craving (b = 0.23, SE = 0.57, p = .68).
OPRM1 as a moderator of daily-level associations between adherence and alcohol use/craving
Intervention condition (p = .82), race (p = .50), and overall adherence (p = .22) were not significantly associated with average drinking levels (i.e., the random intercept for drinks per day) during the study. However, controlling for covariates, OPRM1 was associated with average drinking levels (p = .03; Figure 1a). Specifically, 118G participants (i.e., GA/GG genotypes) reported higher average drinks per day during the study than participants with the AA genotype. Results of moderation tests also showed a significant moderating effect of OPRM1 on the daily association between naltrexone adherence and same-day alcohol use (i.e., genotype significantly predicted variance in the random slope for the within-person association (p = .007; Fig. 1a). Controlling for covariates, there was a significant within-person association between daily adherence and reduced same-day consumption for GA/GG individuals (simple slope = −1.37, p = .002). This within-person association was not significant for AA participants (simple slope = −.04, p = .90). Simple slopes are depicted in Figure 2.
Figure 2:
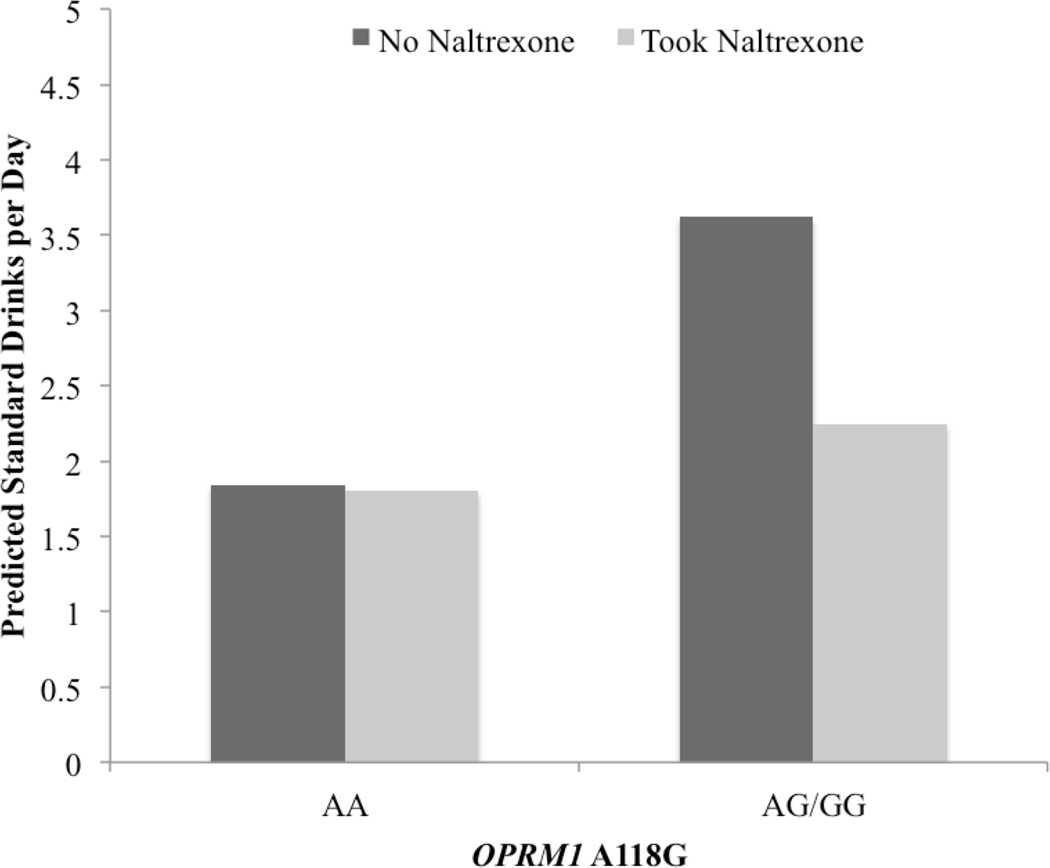
Simple slopes are depicted of within-person association of naltrexone adherence (as measured by medication bottle openings) with drinks per day that was moderated by OPRM1 (p = .007). The predicted means for each subgroup can be interpreted as the average standard drinks reported each day when individuals took naltrexone versus did not take naltrexone for individuals with GA/GG genotypes or AA genotype.
Intervention condition (p = .15), race (p = .56), and overall adherence (p = .41) were not significantly associated with average craving levels (i.e., the random intercept for craving each day) during the study. Controlling for covariates, there was a trend for GA/GG participants to report higher average craving levels (i.e., the random intercept for alcohol craving) during the trial (p = .06; Figure 1b). Examination of OPRM1 as a moderator of the daily association between adherence and same-day craving (based on the association between genotype and the random slope factor) also revealed a trend-level effect; p = .06), such that the association of daily adherence with decreases in craving was relatively stronger for GA/GG participants (Figure 1b).
Discussion
This study leveraged high-density within-subjects data, including remote daily assessments of alcohol, craving, and medication adherence, to examine the moderating role of OPRM1 A118G on changes in alcohol craving and consumption during naltrexone treatment. Consistent with most AUD clinical trials, results indicated significant variability in drinks per drinking day and craving during treatment, suggesting the importance of characterizing factors contributing to such variability. Results from person-centered analyses indicated that, after controlling for covariates, OPRM1 moderated associations of daily naltrexone adherence with same-day alcohol use. Specifically, naltrexone adherence was significantly associated with decreased same-day consumption among individuals with the 118G variant, but not among 118A homozygotes. A similar pattern, though not statistically significant, was evident for the daily adherence-craving association. Results from daily reports also indicated higher mean drinks per day and marginally higher craving levels among 118G carriers during treatment, although no genotype differences in prospective changes in drinking were observed.
The finding that naltrexone adherence was associated with reduced same-day alcohol use only among 118G carriers is partly consistent with initial (Oslin et al., 2003) and subsequent (Anton et al., 2008, Chamorro et al., 2012, Kranzler et al., 2013) evidence for improved naltrexone treatment outcomes in this group. In contrast, two more recent trials involving prospective genotyping failed to confirm differential treatment response as a function of OPRM1 (Oslin et al., 2015, Schacht et al., 2017). Aspects of the current design and/or statistical approach may have facilitated its ability to detect a moderating role of genotype. First, daily reports of alcohol consumption may be more sensitive, and less susceptible to recall bias, relative to retrospective reports aggregated over longer intervals. Consistent with this idea are findings from a prior naltrexone trial involving daily assessments and person-centered analyses, which detected a moderating role of OPRM1 genotype. Using nightly interactive voice response assessments during a 12-week randomized trial, Kranzler and colleagues showed that OPRM1 moderated naltrexone effects: when reporting high evening desire to drink, participants with the 118G variant (but not AA homozygotes) reported significant increases in consumption, an effect that was attenuated in the naltrexone condition (Kranzler et al., 2013). Notably, no moderating effect of OPRM1 was detected when using traditional (between-person) analyses (Kranzler et al., 2013).
A second consideration is that daily assessments and person-centered analyses are better able to capture complex, dynamic changes in consumption and craving, including variability that may occur secondary to fluctuations in medication adherence. Examining genotype as moderating these daily processes could potentially reveal effects that would go undetected when using aggregate measures of craving, consumption or adherence collected over weeks or months. The present findings are consistent with this possibility with respect to the moderating effect of genotype on the daily association between adherence and consumption. However, contrary to hypotheses, we did not observe differential reductions in drinking over time as a function of genotype. This finding should be interpreted in light of the fact that these analyses found no significant overall reductions in drinking and craving across groups during the initial weeks of treatment. This result differs from our prior observation of significant reductions in drinking and craving during the study (Stoner et al., 2015, Stoner and Hendershot, 2012), perhaps reflecting that the largest reductions in drinking appeared to occur between baseline (pre-medication) and week 4, whereas the present analyses modeled drinking based on daily reports collected once medication began.
The OPRM1 A118G variant has received considerable attention in pharmacogenetic studies, in part reflecting preclinical and human evidence for functional significance of this variant with respect to alcohol responses (including differential reductions in consumption during naltrexone exposure). While these findings generated substantial interest in precision medicine applications related to OPRM1, the present evidence base—which includes negative results from two prospective trials (Oslin et al., 2015, Schacht et al., 2017) – is inconsistent overall. Given the expectation of small effect sizes and replication difficulties in candidate gene studies, these inconsistencies are perhaps not surprising. Mixed findings and issues with replicability are commonplace in candidate gene studies (Dick et al., 2015). Moreover, given the high heterogeneity of AUD presentation, the modest effect sizes of AUD medications, and the complex nature of behavior change during treatment, developing precision medicine applications in a candidate gene context is a challenging endeavor. Polygenic investigations are a necessary next step, and to date there have been relatively few such attempts in pharmacogenetic studies (for an exception, see Gelernter et al., 2007). Research into genetic composite scores comprising variants in the δ- and/or κ-opioid receptors – also potentially relevant for naltrexone, albeit to a lesser extent than the μ-opioid receptor – could be promising. Further, to capture genetic contributions within and beyond the opioidergic system, future work should look to high-powered scans across the genome provided by genome-wide association studies (GWAS).
Several limitations should be considered when interpreting the present findings. First, because the aim of the parent study was to test an adherence intervention (rather than the efficacy of naltrexone), this was an open-label study, making it difficult to rule out alternative explanations for the association of adherence with consumption in 118G carriers. For instance, we cannot rule out the possibility that expectancies about medication effects could contribute to this association. Relatedly, although medication adherence events (inferred from pill cap readings) were time-stamped, and typically occurred in the mid-morning, pill cap openings remain an indirect measure of medication adherence (and timing of ingestion), and participants’ reports of peak daily craving were not linked to a specific time point. This leaves open the possibility that the marginal association of adherence and same-day craving in the 118G variant group could have reflected adherence in response to elevated craving. The use of multiple daily craving measurements (e.g., via ecological momentary assessment) is one means of addressing this question in future studies. Another limitation, also reflecting the aims of the parent study, is that this was a relatively short-term analysis focused on the initial weeks of treatment. This trial also did not include any adjunct behavioral treatment. While this may minimize comparability to other clinical contexts and AUD medication trials, prior findings suggest that moderating effects of OPRM1 may be more evident in the context of a minimal behavioral intervention (Anton et al., 2008).
Another important limitation is the modest sample size of this study. Although the small sample size is an important limitation, it should be noted that the use of intensive daily assessments and within-person analyses would nonetheless be expected to increase power for detecting the hypothesized associations. For example, guidelines for power estimates in multilevel analyses (Arend and Schafer, 2019) suggest that in the context of this daily-sampling design, this sample size would likely have yielded good power (80%) for detecting small effect sizes for within-person associations (e.g., the daily association of adherence with craving), although power may only be sufficient to detect medium to large effect sizes for cross-level interactions (i.e., the moderating effect of OPRM1). In contrast, more conventional approaches (e.g., between-person analyses involving two or three time points) would have rendered this study substantially underpowered. It should be noted that these estimates are retrospective, and that estimating power for multilevel models is complex (see Arend and Schafer, 2019).
It should also be noted that, to date, there remain no pharmacogenetic applications with clinical utility in AUD treatment settings, including applications related to OPRM1 (Hartwell and Kranzler, 2019, Oslin et al., 2015). Despite limited clinical or public health implications of pharmacogenetic research for AUD treatment thus far, work in this area has nonetheless proven instructive for characterizing biological moderators of treatment response from a mechanistic standpoint. In this respect, findings from the present study and others (e.g., Kranzler et al., 2013, Miranda et al., 2018) support the notion that daily assessments and person-centered analytic approaches might improve sensitivity for detecting pharmacogenetics effects. This “within-person phenotyping”approach appears a promising method for studying the moderating role of genotype in pharmacogenetic studies (Kranzler et al., 2013). This strategy would also apply to other individual difference factors studied as treatment moderators within a precision medicine framework (e.g., Garbutt et al., 2014, Kranzler and McKay, 2012, Mann et al., 2018). The current findings also suggest the potential importance of modeling within-person variability in medication adherence. Given efforts to augment naltrexone with other pharmacotherapies (Naglich et al., 2018), incorporating these approaches in studies of combined pharmacotherapies is another potential direction. Overall, precision medicine studies – and pharmacotherapy trials more broadly – might benefit from more frequent incorporation of high-density assessments in order to model behavior change and clinical outcomes with greater temporal precision, allowing for process-based investigations of treatment mechanisms and response.
Acknowledgements
This research was supported by a Small Business Innovation Research contract no. HHSN275201000011C from the National Institute on Alcohol Abuse and Alcoholism (NIAAA) to Talaria, Inc. (Principal Investigator, S.A.S.). This research was also supported by the Canada Research Chairs Program (C.S.H.) and by postdoctoral fellowships awarded to S.S.D. and J.D.W. from the Canadian Institutes of Health Research. The authors express appreciation to Dr. Pamela Arenella for clinical coverage, and to Natalie Freeman for assistance with genotyping. NIAAA had no role in the design, conduct, or reporting of this study. Talaria, Inc., has closed and donated its technology to the Alcohol and Drug Abuse Institute at the University of Washington. Neither the funders nor the owners of Talaria, Inc., had any role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The authors declare no conflicts of interest with respect to the funding or publication of this research.
References
- Anton RF, Oroszi G, O’Malley S, Couper D, Swift R, Pettinati H, Goldman D (2008) An evaluation of μ-opioid receptor (OPRM1) as a predictor of naltrexone response in the treatment of alcohol dependence: results from the combined pharmacotherapies and behavioral interventions for alcohol dependence (COMBINE) study. Arch Gen Psychiatry 65:135–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arend MG, Schafer T (2019) Statistical power in two-level models: a tutorial based on Monte Carlo simulation. Psychol Methods 24:1–19. [DOI] [PubMed] [Google Scholar]
- Asparouhov T, Muthén B (2007) Constructing covariates in multilevel regression. Version 2. [Google Scholar]
- Barr CS, Chen SA, Schwandt ML, Lindell SG, Sun H, Suomi SJ, Heilig M (2010) Suppression of alcohol preference by naltrexone in the rhesus macaque: a critical role of genetic variation at the μ-opioid receptor gene locus. Biol Psychiatry 67:78–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr CS, Schwandt M, Lindell SG, Chen SA, Goldman D, Suomi SJ, Higley JD, Heilig M (2007) Association of a functional polymorphism in the μ-opioid receptor gene with alcohol response and consumption in male rhesus macaques. Arch Gen Psychiatry 64:369–376. [DOI] [PubMed] [Google Scholar]
- Bilbao A, Robinson JE, Heilig M, Malanga CJ, Spanagel R, Sommer WH, Thorsell A (2015) A pharmacogenetic determinant of mu-opioid receptor antagonist effects on alcohol reward and consumption: evidence from humanized mice. Biol Psychiatry 77:850–858. [DOI] [PubMed] [Google Scholar]
- Chamorro A-J, Marcos M, Mirón-Canelo J-A, Pastor I, González-Sarmiento R, Laso F-J (2012) Association of μ-opioid receptor (OPRM1) gene polymorphism with response to naltrexone in alcohol dependence: a systematic review and meta-analysis. Addict Biol 17:505–512. [DOI] [PubMed] [Google Scholar]
- Dermody SS, Wardell JD, Stoner SA, Hendershot CS (2018) Predictors of daily adherence to naltrexone for alcohol use disorder treatment during a mobile health intervention. Ann Behav Med 52:787–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick DM, Agrawal A, Keller MC, Adkins A, Aliev F, Monroe S, Hewitt JK, Kendler KS, Sher KJ (2015) Candidate gene–environment interaction research: reflections and recommendations. Perspect Psychol Sci 10:37–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbutt JC, Greenblatt AM, West SL, Morgan LC, Kampov-Polevoy A, Jordan HS, Bobashev GV (2014) Clinical and biological moderators of response to naltrexone in alcohol dependence: a systematic review of the evidence. Addiction 109:1274–1284. [DOI] [PubMed] [Google Scholar]
- Gelernter J, Gueorguieva R, Kranzler HR, Zhang H, Cramer J, Rosenheck R, Krystal JH (2007) Opioid receptor gene (OPRM1, OPRK1, and OPRD1) variants and response to naltrexone treatment for alcohol dependence: results from the VA Cooperative Study. Alcohol Clin Exp Res 31:555–563. [DOI] [PubMed] [Google Scholar]
- Hallgren KA, Witkiewitz K (2013) Missing data in alcohol clinical trials: a comparison of methods. Alcohol Clin Exp Res 37:2152–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwell EE, Kranzler HR (2019) Pharmacogenetics of alcohol use disorder treatments: an update. Expert Opin Drug Metab Toxicol 15:553–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heilig M, Goldman D, Berrettini W, O’Brien CP (2011) Pharmacogenetic approaches to the treatment of alcohol addiction. Nat Rev Neurosci 12:670–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendershot CS, Wardell JD, Samokhvalov AV, Rehm J (2017) Effects of naltrexone on alcohol self-administration and craving: meta-analysis of human laboratory studies. Addict Biol 22:1515–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas DE, Amick HR, Feltner C, Bobashev G, Thomas K, Wines R, Kim MM, Shanahan E, Gass CE, Rowe CJ, Garbutt JC (2014) Pharmacotherapy for adults with alcohol use disorders in outpatient settings: a systematic review and meta-analysis. JAMA 311:1889–1900. [DOI] [PubMed] [Google Scholar]
- Kranzler HR, Armeli S, Covault J, Tennen H (2013) Variation in OPRM1 moderates the effect of desire to drink on subsequent drinking and its attenuation by naltrexone treatment. Addict Biol 18:193–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranzler HR, Edenberg HJ (2010) Pharmacogenetics of alcohol and alcohol dependence treatment. Curr Pharm Des 16:2141–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranzler HR, McKay JR (2012) Personalized treatment of alcohol dependence. Curr Psychiatry Rep 14:486–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranzler HR, Soyka M (2018) Diagnosis and pharmacotherapy of alcohol use disorder: A review. JAMA 320:815–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MC, Wagner HN Jr., Tanada S, Frost JJ, Bice AN, Dannals RF (1988) Duration of occupancy of opiate receptors by naltrexone. J Nucl Med 29:1207–11. [PubMed] [Google Scholar]
- Litten RZ, Ryan ML, Falk DE, Reilly M, Fertig JB, Koob GF (2015) Heterogeneity of alcohol use disorder: understanding mechanisms to advance personalized treatment. Alcohol Clin Exp Res 39:579–84. [DOI] [PubMed] [Google Scholar]
- Lüdtke O, Marsh HW, Robitzsch A, Trautwein U, Asparouhov T, Muthén B (2008) The multilevel latent covariate model: a new, more reliable approach to group-level effects in contextual studies. Psychol Methods 13:203–229. [DOI] [PubMed] [Google Scholar]
- Mague SD, Blendy JA (2010) OPRM1 SNP (A118G): involvement in disease development, treatment response, and animal models. Drug Alcohol Depend 108:172–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maisel NC, Blodgett JC, Wilbourne PL, Humphreys K, Finney JW (2013) Meta-analysis of naltrexone and acamprosate for treating alcohol use disorders: When are these medications most helpful? Addiction 108:275–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann K, Roos CR, Hoffmann S, Nakovics H, Lemenager T, Heinz A, Witkiewitz K (2018) Precision medicine in alcohol dependence: a controlled trial testing pharmacotherapy response among reward and relief drinking phenotypes. Neuropsychopharmacology 43:891–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark TL, Kassed CA, Vandivort-Warren R, Levit KR, Kranzler HR (2009) Alcohol and opioid dependence medications: prescription trends, overall and by physician specialty. Drug Alcohol Depend 99:345–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark TL, Kranzler HR, Song X (2003) Understanding US addiction physicians’ low rate of naltrexone prescription. Drug Alcohol Depend 71:219–228. [DOI] [PubMed] [Google Scholar]
- Miranda R, Treloar Padovano H, Gray JC, Wemm SE, Blanchard A (2018) Real-time assessment of alcohol craving and naltrexone treatment responsiveness in a randomized clinical trial. Addict Behav 83:72–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muthén LK, Muthén BO (1998-2012) Mplus User’s Guide. 7th ed. Muthén & Muthén, Los Angeles, CA. [Google Scholar]
- Naglich AC, Lin A, Wakhlu S, Adinoff BH (2018) Systematic review of combined pharmacotherapy for the treatment of alcohol use disorder in patients without comorbid conditions. CNS Drugs 32:13–31. [DOI] [PubMed] [Google Scholar]
- Oslin DW, Berrettini W, Kranzler HR, Pettinati H, Gelernter J, Volpicelli JR, O’Brien CP (2003) A functional polymorphism of the μ-opioid receptor gene is associated with naltrexone response in alcohol-dependent patients. Neuropsychopharmacology 28:1546–1552. [DOI] [PubMed] [Google Scholar]
- Oslin DW, Leong SH, Lynch KG, Berrettini W, O’Brien CP, Gordon AJ, Rukstalis M (2015) Naltrexone vs placebo for the treatment of alcohol dependence: a randomized clinical trial. JAMA Psychiatry 72:430–437. [DOI] [PubMed] [Google Scholar]
- Ramchandani VA, Umhau J, Pavon FJ, Ruiz-Velasco V, Margas W, Sun H, Damadzic R, Eskay R, Schoor M, Thorsell A, Schwandt ML, Sommer WH, George DT, Parsons LH, Herscovitch P, Hommer D, Heilig M (2011) A genetic determinant of the striatal dopamine response to alcohol in men. Mol Psychiatry 16:809–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray LA, Barr CS, Blendy JA, Oslin D, Goldman D, Anton RF (2012) The role of the Asn40Asp polymorphism of the mu opioid receptor gene (OPRM1) on alcoholism etiology and treatment: a critical review. Alcohol Clin Exp Res 36:385–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray LA, Heilig M (2013) Subjective responses to alcohol as an endophenotype: implications for alcoholism etiology and treatment development, in Genetic Influences on Addiction: An Intermediate Phenotype Approach (MacKillop J, Munafò MR eds), pp 97–120. MIT Press, Cambridge, MA. [Google Scholar]
- Ray LA, Miranda R Jr, Tidey JW, McGeary JE, MacKillop J, Gwaltney CJ, Rohsenow DJ, Swift RM, Monti PM (2010) Polymorphisms of the μ-opioid receptor and dopamine D₄ receptor genes and subjective responses to alcohol in the natural environment. J Abnorm Psychol 119:115–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satorra A, Bentler PM (2010) Ensuring positiveness of the scaled difference chi-square test statistic. Psychometrika 75:243–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schacht JP, Randall PK, Latham PK, Voronin KE, Book SW, Myrick H, Anton RF (2017) Predictors of naltrexone response in a randomized trial: reward-related brain activation, OPRM1 genotype, and smoking status. Neuropsychopharmacology 42:2640–2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobell LC, Sobell MB (1992) Timeline follow-back: a technique for assessing self-reported alcohol consumption, in Measuring Alcohol Consumption: Psychosocial and Biochemical Methods (Litten RZ, Allen JP eds), pp 41–72. Humana Press, Totowa, NJ. [Google Scholar]
- Sobell LC, Sobell MB (1995) Alcohol consumption measures, in Assessing Alcohol Problems: A Guide for Clinicians and Researchers (Allen JP, Columbus M eds), pp 75–99. National Institute on Alcohol Abuse and Alcoholism, Bethesda, MD. [Google Scholar]
- Stoner SA, Arenella PB, Hendershot CS (2015) Randomized controlled trial of a mobile phone intervention for improving adherence to naltrexone for alcohol use disorders. PLoS One 10:e0124613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoner SA, Hendershot CS (2012) A randomized trial evaluating an mHealth system to monitor and enhance adherence to pharmacotherapy for alcohol use disorders. Addict Sci Clin Pract 7:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallender EJ, Rüedi-Bettschen D, Miller GM, Platt DM (2010) A pharmacogenetic model of naltrexone-induced attenuation of alcohol consumption in rhesus monkeys. Drug Alcohol Depend 109:252–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witkiewitz K, Roos CR, Mann K, Kranzler HR (2019) Advancing precision medicine for alcohol use disorder: replication and extension of reward drinking as a predictor of naltrexone response. Alcohol Clin Exp Res 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]