

Abstract
Pancreatic ductal adenocarcinoma (PDAC) is an intractable malignancy with a dismal survival rate. Recent combination therapies have had a major impact on the improvement of PDAC prognosis. Nevertheless, clinically used combination regimens such as FOLFIRINOX, and Gemcitabine/nab-Paclitaxel still face major challenges due to lack of the safe and ratiometric delivery of multiple drugs. Here, we report on a rationally designed mesoporous silica nanoparticle (MSN)-based platform for the target-specific, spatiotemporal, ratiometric, and safe co-delivery of gemcitabine (Gem) and cisplatin (cisPt). We show that systemic administration of the nanoparticles results in synergistic therapeutic outcome in a syngeneic and clinically relevant genetically engineered PDAC mouse model that has rarely been used for the therapeutic evaluation of nanomedicine. This synergism is associated with a strategic engineering approach, in which nanoparticles provide redox-responsive controlled delivery and in situ differential release of Gem/cisPt drugs with the goal of overcoming resistance to Pt-based drugs. Our platform is also rendered with additional tumor-specificity via a novel tumor-associated mucin1 (tMUC1)-specific antibody, TAB004. Overall, our platform suppresses tumor growth and eliminates the of-target toxicities of a highly toxic chemotherapy combination.
Keywords: Pancreatic cancer, Spatiotemporal release, targeted drug delivery, combinatorial therapy
Graphical Abstract
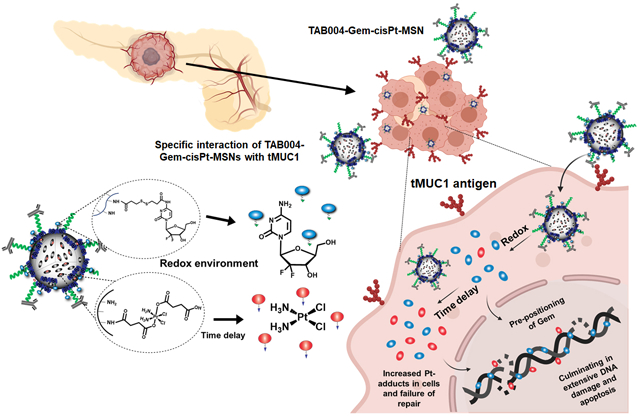
A multifunctional mesoporous silica nanoparticle (MSN)-based platform for the target-specific, spatiotemporal, ratiometric, and safe co-delivery of gemcitabine (Gem) and cisplatin (cisPt) has been developed. This platform efficiently suppresses tumor growth and eliminates the of-target toxicities of this highly toxic chemotherapy combination in a syngeneic and clinically relevant genetically engineered PDAC mouse model.
1. Introduction
Pancreatic ductal adenocarcinoma (PDAC) disproportionately ranks high on the list of cancer-related mortality, relative to its incidence. Despite enormous scientific efforts, PDAC has the worst prognosis, with a 5-year survival rate of 9% for all the stages combined, a trend that has not improved significantly over decades.[1–3] Multiple reasons contribute to the ineffectiveness of current PDAC treatments, underscoring the need for developing novel and effective therapeutic options aimed at improving PDAC prognosis.[4, 5] Basic as well as clinical research, have recently turned towards combination therapies with the FDA’s approval of FOLFIRINOX (5-fluorouracil, leucovorin, irinotecan, and oxaliplatin), gemcitabine (Gem) plus nab-paclitaxel, and ONIVYDE (liposomal formulation of irinotecan) used in combination with 5-fluorouracil and leucovorin.[6–8] A number of other combinations, including multidrug chemotherapy and molecular agents are also currently under investigation.[9, 10] Recent insights gained from the characterization of recurrent genetic alterations has revealed that a subset of PDACs, linked to germline-based mutations, can benefit from Pt-based agents, including cisplatin (cisPt).[11, 12] This new genetic understanding of PDAC has motivated the addition of Pt-based drugs to chemotherapy combinations in some ongoing clinical trials.[13, 14] Inspired by these scientific advancements; herein, we report on the application of Gem/cisPt combination with the added benefit of advanced nanoengineering approach, that can potentially be an excellent therapeutic option to the subset of PDAC associated with germline-mutations.
Traditional multidrug regimens suffer from limitations of spatiotemporal and ratiometric control due to the distinct physicochemical and biological properties of the constituent drugs, yet each exhibits high toxicity profiles.[15–17] Developing versatile delivery systems that can simultaneously transport and release multiple drugs in a controlled fashion is vital in order to achieve the full potential of combination therapies.[17–19] Nanoparticulate delivery systems offer several advantages for combinatorial delivery: they can unify the biodistribution and pharmacokinetics of chemically dissimilar drugs and improve the stability of the drugs. Nanoplatforms that have been investigated for this purpose include liposomes, polymeric nanoparticles, mesoporous silica nanoparticles (MSNs), and self-assembled systems.[20–22] However, earlier platforms suffer from limitations in combining drugs with distinct physicochemical properties, controlling spatio-temporal release under tumor-specific stimuli, controlling action-based drug sequence, and preventing systemic leakage of the drugs. Here we have used MSNs for Gem/cisPt multidrug combination, due to the unique properties of these nanoparticles, including their high surface area, tunable chemical and morphological properties, and selective modification of internal pores and external surfaces.[23–25] Though multidrug regimens are usually designed to achieve an optimal therapeutic outcome through a ratiometric combination, there is also a huge benefit from a timely sequence-based delivery according to the mechanism of action for each drug.[19, 26] In particular, Gem preceding cisPt regimen is advantageous, as Gem inhibits the enzymes involved in the mechanisms of DNA damage repair (DDR); moreover, pre-positioning of the Gem molecules in the DNA physically inhibits the repair of Pt-adduct patch.[27, 28] Thus, the delivery of Gem/cisPt with use of a precisely engineered approach, in which the release of Gem is followed by cisPt can potentiate the cells, enhances the cytotoxic effect. In this work, we investigated a strategic MSN-based nanoengineering approach to implement such a in situ differential drug release pattern.
TAB004 is a novel antibody with a high specificity towards tMUC1, as has been demonstrated in preclinical models of breast cancer and PDAC, as well as in tumor samples from PDAC patients.[29–32] We have also previously demonstrated the efficiency of TAB004-functionalized MSNs for tMUC1-specific tumor imaging in the genetically engineered mouse (GEM) model of breast cancer.[33] Target-specific drug delivery is highly beneficial for facilitating the specific and safe homing of nanoparticles, and increasing their retention at the tumor site and uptake by tumor cells. This is especially vital in PDAC, which possesses a high degree of stochasticity to intratumoral drug delivery, and has led to a compromised efficacy of nanomedicine-based approaches.[34] For this reason, we have additionally functionalized our MSN-based platform with the TAB004 antibody, to specifically target our nanoplatform to PDAC cells.
We present here a novel, target-specific nanoplatform for the co-delivery of Gem and cisPt, that has the additional benefits of chemical conjugation of drugs for controlled and redox-responsive release (Figure 1a). The resulting material was stable in blood and showed increased target-specific accumulation in the tumor (Figure 1b); this not only increased tumor inhibition, but also reduced off-target toxicity (Figure 1c). Following receptor-mediated endocytosis in PDAC cells, our platform dictates an “in situ differential release” of Gem/cisPt that further improves the therapeutic performance of this synergistic drug combination. We show that this nanoengineering approach successfully afforded the conjugation of drugs at a specific ratio along with high drug content and reproducibility. The material displayed enhanced DNA damage and apoptosis as a result of the ratiometric as well as the in situ differential release of Gem/cisPt (Figure 1d). These advantages of our platform yield improved tumor inhibition and disease control in two clinically relevant models: KCM syngeneic mice and GEM PDA.MUC1 mice.
Figure 1.
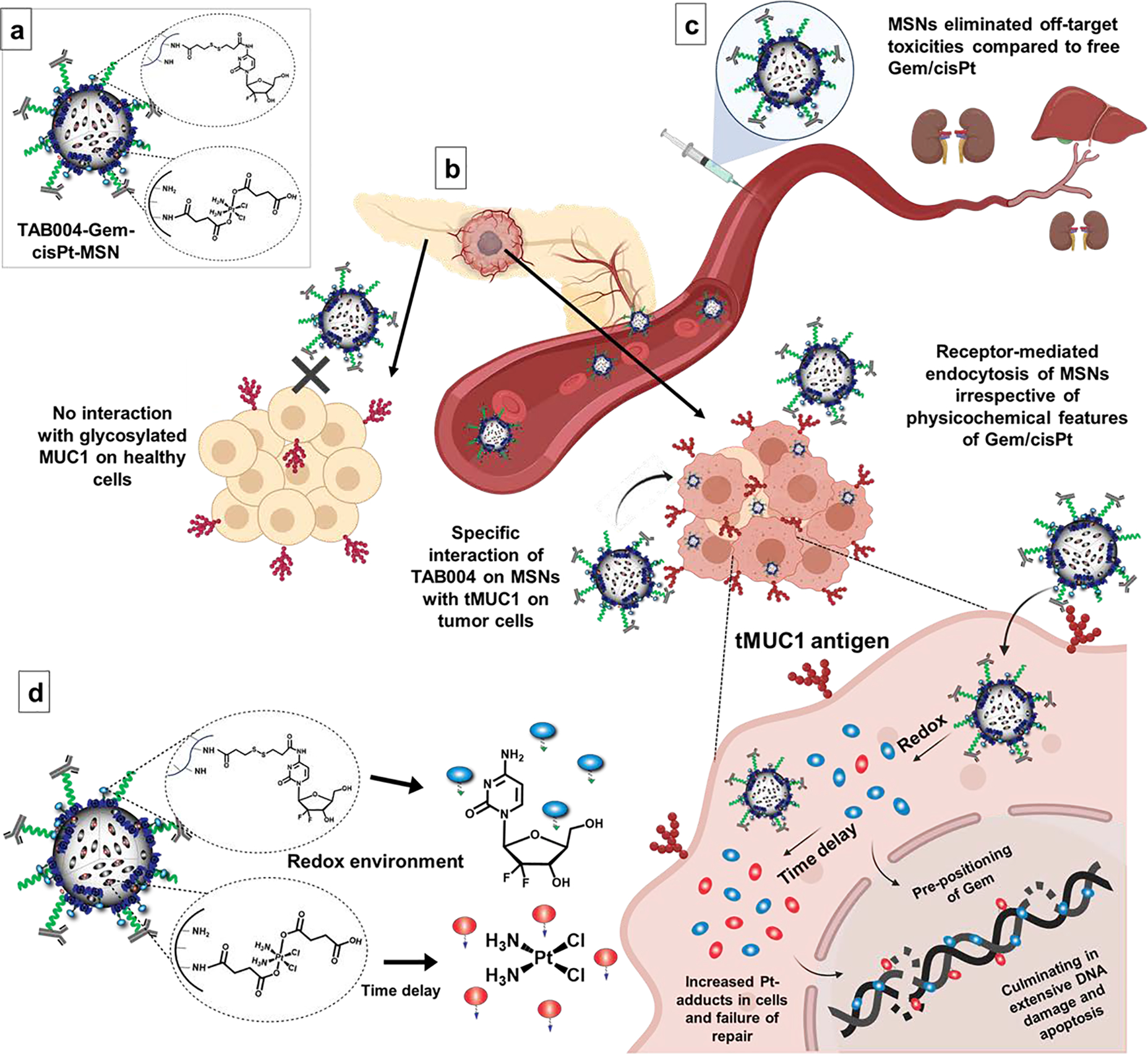
Schematic representation of the relevant features of TAB004-Gem-cisPt-MSNs. (a) The rational design of the nanoplatform yielded a redox-responsive MSN-based combinatorial system with chemically conjugated cisPt and Gem into the interior and exterior surface, respectively. (b and c) TAB004-Gem-cisPt-MSNs are stable in blood circulation, target tMUC1-positive PDAC tumors, and have decreased off-target toxicities. (d) The unique design of our nanoplatform exhibits ratiometric and in-situ differential release, enhancing DNA damage and apoptosis in PDAC cells.
2. Experimental procedures.
2.1. Synthesis and characterization of Gem/cisPt prodrugs and TAB004-Gem-cisPt-MSNs.
Synthesis and characterization of Gem and cisPt prodrugs is reported in the ESI (Section S2.1). Fabrication, characterization and optimization of TAB004-Gem-cisPt-MSNs are also reported in the ESI (Sections S2.2–S2.7 and S6).
2.2. Cytotoxicity of MSN materials in PDAC cells.
The following PDAC cells were used for the cytotoxicity analysis: KCM, HPAF II, AsPC1, and Capan-1 (Table S1). Normal pancreatic ductal cells (HPDE) were also used as control. For this experiment, all cell types were seeded in 96-well plates. KCM cells were seeded at a cell density of 500 cells per well and Capan-1 cells at 1000 cells per well. HPAF II, AsPC1 and HPDE cells were seeded a density of 2000 cells per well. After incubated for 24 h, the cells were then treated with different MSNs at concentrations ranging from 0.1 to 100 μg mL−1. The MSN materials used were: PEI-MSNs, cisPt-MSNs, Gem-MSNs, Gem-cisPt-MSNs and a physical mixture of Gem-MSNs plus cisPt-MSNs at equivalent drug concentrations (Table S2 and S3). The cisPt-MSNs used for in vitro studies were coated with PEI, to avoid differences in cellular uptake due to the surface properties of Gem-MSNs or Gem-cisPt-MSNs (ζ-potential, Table S4). The MSNs were dispersed in complete cell culture media, and cells were treated for 48 h. The cells were then washed once with PBS, replaced with fresh media and allowed to recover for another 24 h. Post-treatment, cell viability was evaluated using the MTS assay. The results are reported as average IC50 values for the three independent experiments.
2.3. Analysis of apoptotic cells after treatment with Gem-cisPt-MSNs.
KCM cells were seeded at a density of 20,000 cells per well in a 24-well plate and incubated for 48 h. The cells were inoculated for 24 h with 20 μg mL−1 of Gem-cisPt-MSNs and other MSN materials at the equivalent drug concentrations (Table S3). Cells were washed with PBS, detached using trypsin, and collected by centrifugation. Cells were then washed once with PBS, and with 1 mL of 1X binding buffer (BD Pharmingen 556547). Cells were resuspended in 200 μL of binding buffer followed by the addition of 5 μL of Annexin V-FITC solution. Finally, the cell suspensions were incubated for 15 min at room temperature in the dark and then washed once with binding buffer. Cells were resuspended in 200 μL of binding buffer and transferred to flow cytometry tubes. Propidium iodide (PI) solution (5 μL) was then added to each tube and incubated for 5–10 min at room temperature in the dark. Cells were immediately analyzed using flow cytometry. Healthy cells were negative for both Annexin V-FITC and PI (FITC-/PI-); early apoptotic cells were Annexin V-FITC positive and PI negative (FITC+/PI-); late apoptotic cells were positive for both Annexin V-FITC and PI (FITC+/PI+); and necrotic cells were Annexin V-FITC negative and PI positive (FITC-/PI+). Data are presented as percentage apoptotic cells (early plus late apoptotic cells) and results are reported as the average ± SD of three independent experiments.
2.4. Analysis of γH2AX after treatment with Gem-cisPt-MSNs.
KCM cells were seeded at a density of 20,000 cells per well in a 24-well plate, incubated for 48 h and inoculated for 24 h with 20 μg mL−1 of Gem-cisPt-MSNs and other MSN materials at the equivalent drug concentrations (Table S3). Cells were then washed with PBS, detached using trypsin, and collected by centrifugation. Cells were washed once with PBS, resuspended in the fixative solution (Sigma Aldrich 17–344) and incubated on ice for 20 min. Cells were then washed twice with PBS and mixed with 100 μL of 1X permeabilization solution (Sigma Aldrich 17–344) and incubated for 5 min. FITC-conjugated anti-γH2AX antibody (3.5 μL) was added and cells were incubated on ice for 20 min. After a final wash with 1X wash buffer, cells were resuspended in PBS and analyzed using flow cytometry. Results are reported as the percentage of γH2AX positive cells (average ± SD) obtained from three independent experiments.
2.5. Analysis of Pt adducts after treatment with Gem-cisPt-MSNs.
KCM cells were seeded at a density of 20,000 cells per well in a 24-well plate. After 48 h of incubation, cells were inoculated for 24 h with 20 μg mL−1 of Gem-cisPt-MSNs and other MSN materials at the equivalent drug concentrations (Table S3). Cells were washed with PBS, detached using trypsin, and collected using centrifugation. Cells were then washed once with PBS, resuspended in the fixative solution (Sigma Aldrich 17–344) and incubated on ice for 20 min. Cells were then washed twice with PBS, mixed with the 100 μL of 1X permeabilization solution (Sigma Aldrich 17–344) and incubated for 5 min. Cells were incubated with the primary antibody (5 μg/mL, Abcam ab103261) for 18 h at 4 °C. Cells were then washed with PBS once, dispersed in PBS and incubated with 1:200 secondary antibody (Abcam ab6730) for 2 h. After a final wash with PBS, cells were immediately analyzed using flow cytometry. Results are reported as the percentage of Pt-adduct positive cells (average ± SD) obtained from three independent experiments.
2.6. Targeting ability of TAB004-MSNs in PDAC cells.
Procedures for the synthesis of TAB004-MSNs and PEG-MSNs is described in detail in ESI (Section S2.4). Cellular uptake was quantified with the use of using flow cytometry. Briefly, KCM, KCKO, and HPAF II cells were seeded at a density of 20,000 cells per well in a 24-well plate and incubated for 24 h. TAB004-MSNs or PEG-MSNs (20, 40, and 80 μg mL−1) were dispersed in complete cell culture media, added to the cells and incubated for 24 h. Cells were washed with PBS three times, detached using trypsin, and collected using centrifugation. Cells were dispersed in PBS and immediately analyzed using flow cytometry. Results are reported as the percentage of TRITC positive cells (average ± SD) obtained from three independent experiments.
Qualitative cellular uptake analysis was done using confocal microscopy. KCM cells were seeded at a density of 50,000 cells per well on micro cover glass placed in a 6-well plate and incubated for 24 h. TAB004-MSNs or PEG-MSNs (40 μg mL−1) were dispersed in complete cell culture media and added to the cells for incubation for 24 h. Cells were washed with PBS three times, and cell nuclei were stained with DAPI for 30 min. Cells were washed with PBS and the micro cover glass was mounted on a glass slide separated by a spacer, and imaged on an Olympus Fluoview FV 1000 confocal microscope.
2.7. Targeting ability of TAB004-Gem-cisPt-MSNs in syngeneic KCM mice.
All animal experiments were reviewed and approved by the University of North Carolina Charlotte (Charlotte, NC) Institutional Animal Care and Use Committee under protocols 14–015 and 17–015. The targeting ability of the TAB004-Gem-cisPt-MSNs was evaluated in the syngeneic KCM mice 8 days post cell implantation. KCM tumor-bearing mice were injected intravenously with TAB004-Gem-cisPt-MSNs or PEG-Gem-cisPt-MSNs (40 mg kg−1). Mice were imaged prior to administering MSN material and at 30 min, 1, 4, 24, 48 and 96 h post injection using the IVIS® Spectrum imaging system (PerkinElmer). Images were analyzed using the Living Image® Software (version 4.5.5, PerkinElmer). Mice were euthanized after 96 h, and major organs including liver, lungs, kidneys, spleen and tumor were collected. The ex vivo fluorescence associated with the organs was measured using the IVIS imaging system.
2.8. Therapeutic efficacy of TAB004-Gem-cisPt-MSNs in KCM syngeneic mice.
The therapeutic efficacy of TAB004-Gem-cisPt-MSNs was determined in mice bearing KCM tumors that were randomly divided into 4 groups (n=5) and treated with PBS, free Gem/cisPt, PEG-Gem-cisPt-MSNs or TAB004-Gem-cisPt-MSNs. Tumors reached approximately 100 mm3 at 8 days post KCM cell implantation at which point mice were intravenously injected with PBS, free Gem/cisPt (9.52/2.05 mg kg−1), PEG-Gem-cisPt-MSNs (40 mg/kg, 9.52/2.05 mg kg−1) or TAB004-Gem-cisPt-MSNs (40 mg kg−1, 9.52/2.05 mg kg−1). Each treatment consisted of 6 injections with a 4 days-interval between each injection. Tumor growth was monitored with a caliper every other day. On day 34 post-KCM cell implantation, mice were euthanized, and major organs including liver, lungs, kidneys, spleen, heart, and tumor were collected. The fluorescence associated with each organ was evaluated using the IVIS imaging system. A portion of each organ and of the tumors was fixed in buffered formalin for histological analysis.
2.9. Targeting ability of TAB004-MSNs in PDA.MUC1 mice.
PDA.MUC1 mice with 8, 12, and 27 weeks post-tamoxifen induction were injected intravenously with 40 mg kg−1 of TAB004-MSNs. Mice were imaged prior to the administration of the MSN material and at 30 min, 1, 4, 24, 48 and 96 h post injection using the IVIS® Spectrum imaging system (PerkinElmer). Images were analyzed using the Living Image® Software (version 4.5.5, PerkinElmer). Mice were euthanized after 96 h and the major organs including liver, lungs, kidneys, spleen and pancreas were collected. The ex vivo fluorescence associated with the pancreas was measured using the IVIS imaging system and was fixed in formalin. The pancreas was sectioned at a thickness of 4 μm, at various tissue depths. Four of these sections were stained with H&E and imaged to determine the PanIN lesion and PDAC stage.
2.10. Therapeutic efficacy of TAB004-Gem-cisPt-MSNs in PDA.MUC1 mice.
To determine the therapeutic efficacy of TAB004-Gem-cisPt-MSNs, PDA.MUC1 mice with approximately 30 weeks post-tamoxifen induction were used. Mice were injected with TAB004-Gem-cisPt-MSNs at a dose of 40 mg kg−1. The therapeutic regimen consisted of a total of 6 injections (one every 15 day). After the treatment, mice were euthanized, liver, lungs, kidneys, spleen and pancreas were collected. The fluorescence associated with each organ was measured using the IVIS imaging system. Each Pancreas was weighed, fixed in formalin and sectioned at a thickness of 4 μm. Four sections with a distribution spanning the entire organ were stained with H&E and imaged, to evaluate the presence of PanIN lesions and to determine the PDAC stage. Slides were also imaged using confocal microscopy to localize the MSNs.
2.11. Statistical analysis.
All data are represented as mean ± SD unless noted otherwise. The hydrodynamic size and ζ-potential analysis using DLS were performed in triplicates. The amount of drug conjugated to MSNs is reported as the average of 5 independent batches (n=5). The drug release studies were performed in triplicates (n=3). For cell viability studies, GraphPad prism was used to calculate the IC50 values (n=6). DNA damage, DNA damage response markers, apoptosis, and Pt-adduct studies were performed in triplicates (n=3). Statistical analysis for DNA damage, apoptosis, and Pt-adducts was performed with One-way ANOVA using Tukey’s multiple comparison test. Dose-escalation and safety studies were evaluated using n=3 mice per group. The targeting ability studies in KCM syngeneic mice were evaluated using n=3 mice per group. The therapeutic experiments in KCM syngeneic mice were evaluated using n=5 mice per group. Tumor volumes were reported as mean ± SEM and Two-way ANOVA with Tukey’s multiple comparison test was used for statistical analysis. The statistical differences between groups for tumor weights were assessed via the unpaired t-test. The NIR fluorescence, and the content of Si and Pt in organs were evaluated using n=5 per group. The therapeutic experiments in PDA.MUC1 mice were evaluated using n=3 mice per group. All statistical analyses were performed using GraphPad Prism (v8.2.0 for Windows, La Jolla California, CA, USA) with α=0.05 and reported as stars assigned to the p values; ****p≤0.0001, ***p≤0.001, ** p≤0.01, * p≤0.05 and ns p>0.05.
3. Results and discussions.
3.1. Strategic nanoengineering approach for the synthesis of TAB004-Gem-cisPt-MSNs.
Our therapeutic nanoplatform was designed to implement four key considerations: (i) co-delivering the Gem/cisPt combinations using a single nanocarrier; (ii) maintaining an optimal therapeutic ratio between Gem and cisPt along with high drug loading; (iii) using chemical conjugation approach to impart stimuli-responsivity, reduce off-target toxicities, and guarantee reproducibility; and (iv) in situ differential releasing of Gem followed by cisPt to enhance the performance of the Gem/cisPt combination based on each one’s mechanism of action. These features address some of the critical limitations in traditional multidrug regimens and drug delivery systems.[17, 18] To facilitate chemical conjugation of the drugs, we synthesized the bio-reversible Gem and cisPt prodrugs, which contain stimulus-responsive moieties that will allow the release of drugs in reducing environment (details in ESI section S2.1, Schemes S1 and S2). The nanoengineering approach involved the localization of cisPt and Gem prodrugs to the inner and outer surface of the MSNs, respectively, based on the unique structural properties of MSNs.[35] The selective functionalization of MSNs was achieved via the optimized multi-step procedure depicted in Figure 2a (details for synthesis and optimization in ESI sections S2.2 and S6, and in Figure S2). The cisPt prodrug was conjugated to the interior surface of MSNs to afford cisPt-MSNs with 5.13 ± 0.9 wt% (n=3) loading of cisPt (Figure S4a). To obtain a synergistic molar ratio of 4–5.7 (Gem/cisPt), as determined from the combination index (CI) studies in PDAC cells (see ESI section S5 and Figure S1), we required a Gem loading of 18–25 wt% on the surface of MSNs. We thus carried out an optimization to maximize the number of reactive sites on the MSNs’ surface, in order to effectively conjugate Gem prodrug (see ESI section S6 and Figure S6). Our results revealed that the best approach was to coat PEI polymer on the surface of cisPt-MSNs via electrostatic interaction between previously grafted phosphonate groups on the surface of MSNs and PEI polymer. Chemical conjugation of the Gem prodrug was performed using the heterobifunctional linker, N-succinimidyl-3-(2-pyridyldithio) propionate (SPDP), via a redox-responsive disulfide bond (Figure S4b). In this way, both drugs are chemically conjugated to the MSNs via redox-responsive linkages. These bonds are broken once the NPs reach a highly reducing environment, which is commonly observed in cancer cells (Figure S4c). This strategy afforded Gem-cisPt-MSNs with 23.9 ± 2.6 wt% Gem loading (n=5). The nanomaterial was thoroughly characterized for hydrodynamic size (Dh), surface charge, TEM, N2 isotherms, and drug content (Figures 2c–e and S3, and Table S4).
Figure 2.
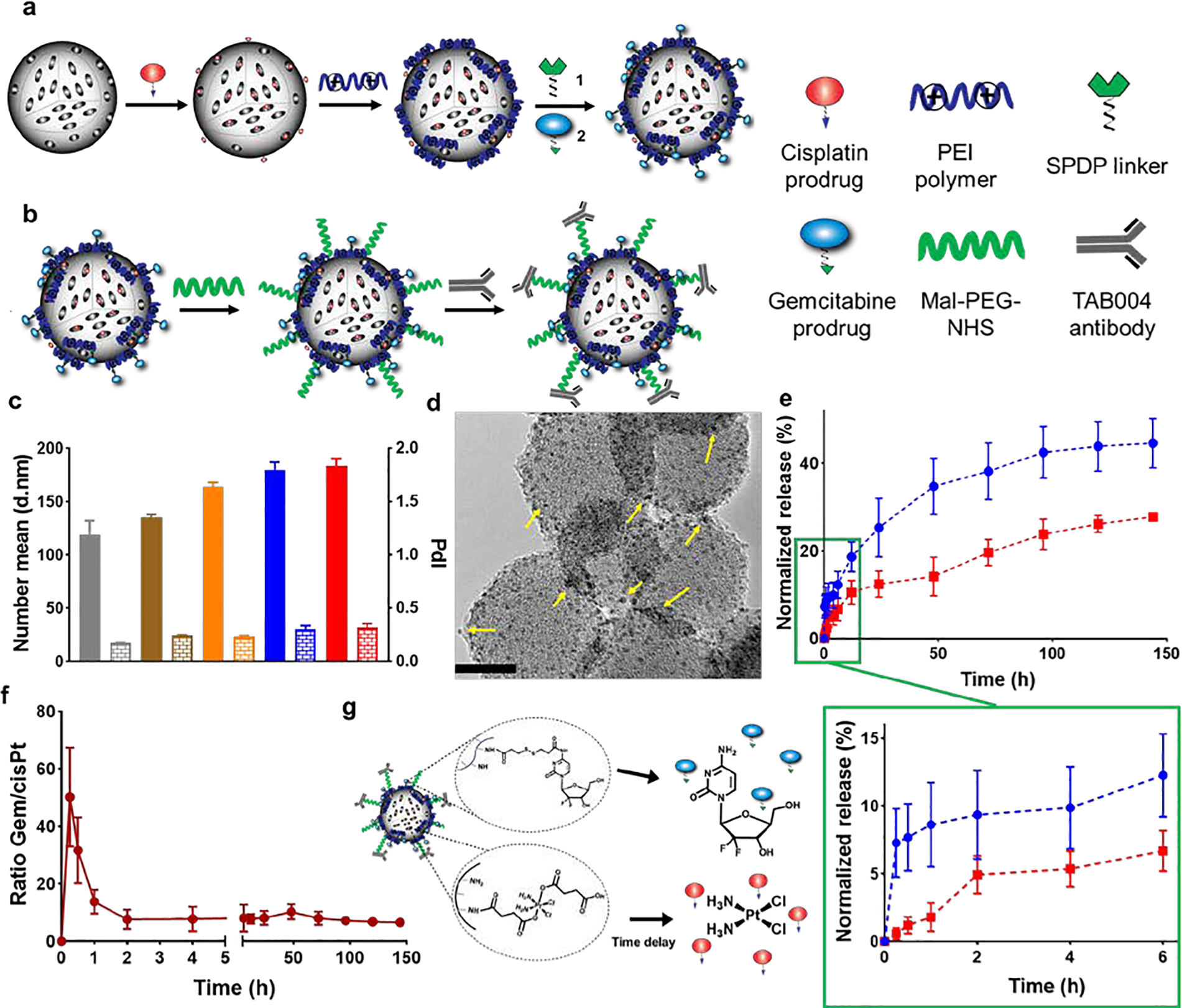
Synthesis and physicochemical characterization of TAB004-Gem-cisPt-MSNs. (a, b) Schematic representation of the multistep procedure used for the synthesis of TAB004-Gem-cisPt-MSNs. (c) Hydrodynamic sizes (Dh) and polydispersity index (PdI) of AP-MSNs (gray), cisPt-MSNs (brown), Gem-cisPt-MSNs (orange), PEG-Gem-cisPt-MSNs (blue), and TAB004-Gem-cisPt-MSNs (red) in cell culture media supplemented with serum. Data represents the mean ± SD of three independent experiments (n=3). (d) TEM image of negatively stained TAB004-Gem-cisPt-MSNs, showing the functionalized antibody (yellow arrows). Scale bar = 20 nm. (e) Release profile of Gem (blue) and cisPt (red), in the presence of reducing agent. (below) Zoom-in of the first 6 hours showing fast release of Gem and the delay in cisPt release, which is characteristic of in situ differential release. Data represents the mean ± SD of three independent experiments (n=3). (f) Molar ratio of Gem/cisPt calculated at different times, showed a rapid release of Gem in the first 60 min afforded a high ratio of Gem/cisPt. However, the molar ratio reached a plateau after 2 h, and was then maintained value at which synergism was observed from CI studies. (g) Schematic representation of the drug release from Gem-cisPt-MSNs depicting the fast release of Gem followed by the delayed release of cisPt as dictated by the design features of the material.
As envisioned, the rational design of our nanoplatform led to Gem-cisPt-MSNs carrying two physiochemically distinct drugs at a high drug content, as well as at a consistent synergistic molar ratio of 5.3 ± 0.8 (Gem/cisPt). The fabrication procedure was highly reproducible, and the drug loading and Gem/cisPt ratio was consistent, over several independent batches (Table S5). The single drug content of our nanoplatform was comparable to the drug loading reported in other platforms; nevertheless, it is one of the highest for combinatorial nanocarriers.[20–22, 36, 37] To the best of our knowledge, our nanoplatform is the first reported system that employs chemical conjugation of dual drugs.
Conjugation of Gem/cisPt prodrugs via stimulus-responsive chemical linkages provides control over the drug release in cancer tissue and cells (Figure S4c).[38–41] In the last years, the difference in redox potential in tumor and normal tissue has been successfully explored as a potential target for tumor treatment leading to the development of redox-responsive drug delivery systems.[42–44] We also documented that selective localization of Gem and cisPt into the exterior and interior surfaces of MSNs renders in situ differential release properties to this platform as demonstrated by evaluating the release profile of Gem and cisPt in solution, mimicking the reductive environment of the cancer cells (Figures 2e–g). After the addition of reducing agent (DTT), the profile at early time points showed a burst release of Gem in the first 30 min followed by a constant rate of release during the subsequent 6 h, while a small amount of cisPt release was observed in the early time points that built up to a constant rate after 2 h (Figure 2e zoom-in). As expected, this differential release led to a time-dependent change in the ratio of Gem/cisPt with a molar ratio of >15 for the first 60 min (Figure 2f). The ratio values rapidly decreased after that, and were maintained at fairly constant values close to the predetermined synergistic ratio of Gem/cisPt. These release data certainly confirmed the in situ differential release property of the Gem-cisPt-MSN platform where Gem (localized at the exterior surface of the material) showed a fast release followed by a delay in cisPt release, to reach the optimal ratio as cisPt was incorporated into the interior surface of MSNs (Figure 2g). Total drug release after six days under reducing condition reached 44 ± 9 % and 27 ± 3 % for Gem and cisPt, respectively. In contrast, in the absence of a reducing agent, the control release experiment exhibited a total of 5 ± 3 % and 1.9 ± 0.5 % Gem and cisPt release, respectively, confirming the redox-responsive properties of the delivery platform (Figure S8).
Several reports indicate that the combination of Gem/cisPt exhibits both ratio- and sequence-dependent synergy.[45, 46] Our own experiments in vitro confirm that the sequential treatment of Gem followed by cisPt potentiated the cells improving the effect of Gem/cisPt combination in PDAC cells (Table S6). Our nanoplatform thus has an inherent advantage whereby a single nanoplatform carries and releases the Gem/cisPt combination in ratiometric amounts, and also possesses the in situ differential release property such that the release of Gem precedes that of cisPt at early times. This release behavior offers a therapeutic advantage in that it overcomes the DNA damage repair (DDR) associated with cisPt resistance.[27, 28]
To render Gem-cisPt-MSNs that are stable in blood circulation and have tumor-targeting capability, PEG polymer and TAB004 antibody were chemically functionalized to the MSN surface. Heterobifunctional Mal-PEG-NHS polymer was conjugated to the amine groups on the surface of Gem-cisPt-MSNs to afford Mal-PEG-Gem-cisPt-MSNs (Figures 2b and S10). These MSNs were further functionalized with the TAB004 antibody via maleimide-thiol chemistry, whereby the antibody was previously modified to incorporate free sulfhydryl groups. Control MSN material, PEG-Gem-cisPt-MSNs was also synthesized using MeO-PEG-NHS polymer. The amount of TAB004 chemically attached to the surface of MSNs was 7.4 ± 2.5 μg of antibody per mg of MSNs (n=3), equivalent to 3–5 molecules of TAB004 antibody per nanoparticle (Figure S10). The MSN materials modified with PEG and TAB004 were thoroughly characterized using TEM, DLS, and ζ-potential (Figures 2c–d and Table S4). The material also exhibited good colloidal stability in cell culture media supplemented with serum for more than 24 h (Figure S11).
Overall, the rational nanoengineering design of our MSN-based platform yielded a single nanocarrier with a high drug content, reproducible ratiometric loading and release of Gem/cisPt. Our material is stimuli-responsive, exhibits in situ differential release of Gem/cisPt, and offers unique benefits for the Gem/cisPt combination based on its therapeutic mechanism of action. The combinatorial MSN platform is functionalized with TAB004 (a tMUC1-specific antibody) for targeted drug delivery to PDAC.
3.2. Gem-cisPt-MSNs exhibit increased DNA damage and Pt-adducts accumulation in PDAC cells ending in cell apoptosis.
To evaluate the in vitro performance of the nanoplatform, the MTS assay was employed to test the cytotoxicity of Gem-cisPt-MSNs, of monotherapies (cisPt-MSNs or Gem-MSNs), or of the physical mixture (Gem-MSNs plus cisPt-MSNs) in a panel of PDAC cells. The panel included KCM, AsPC1, HPAF II, and Capan1 cells, to reflect the high degree of heterogeneity observed in PDAC (Table S1). KCM and HPAF II cell lines overexpress human tMUC1 (see section S3 in the ESI).[29, 32, 47] The cytotoxicity data (Figures 3a, S12 and S13) show that Gem-cisPt-MSNs significantly enhanced cytotoxicity in all cell lines, compared to their MSN monotherapeutic counterparts, or their physical mixture. IC50 values were determined by curve fitting for the dose-response plot for each material and cell line (Figure S13). These values highlighted that at least 1.5–2.0 times lower drug concentration is required when the PDAC cells were treated with Gem-cisPt-MSNs, relative to their monotherapeutic counterparts. Also, the requirement for total drug concentration was 2.5–3.5 times lower when cells were treated with Gem-cisPt-MSNs relative to the physical mixture (Figure S13). To demonstrate that the chemical conjugation of drugs is a beneficial strategy compared to physical loading, we fabricated MSNs with physically encapsulated Gem/cisPt (PL-Gem-cisPt-MSNs; details in ESI section S2.3 and Figure S14a). The PL-Gem-cisPt-MSNs material showed no major cytotoxic effect in KCM or HPAF II cells (IC50 >> 100 μg mL−1) (Figure S14b). Together, these results corroborate that, chemically conjugating Gem and cisPt drugs at a synergistic ratio through redox responsive linkers in Gem-cisPt-MSNs provides therapeutic benefit compared to their monotherapeutic counterparts, a physical mixture, and by physically loaded drugs.
Figure 3.
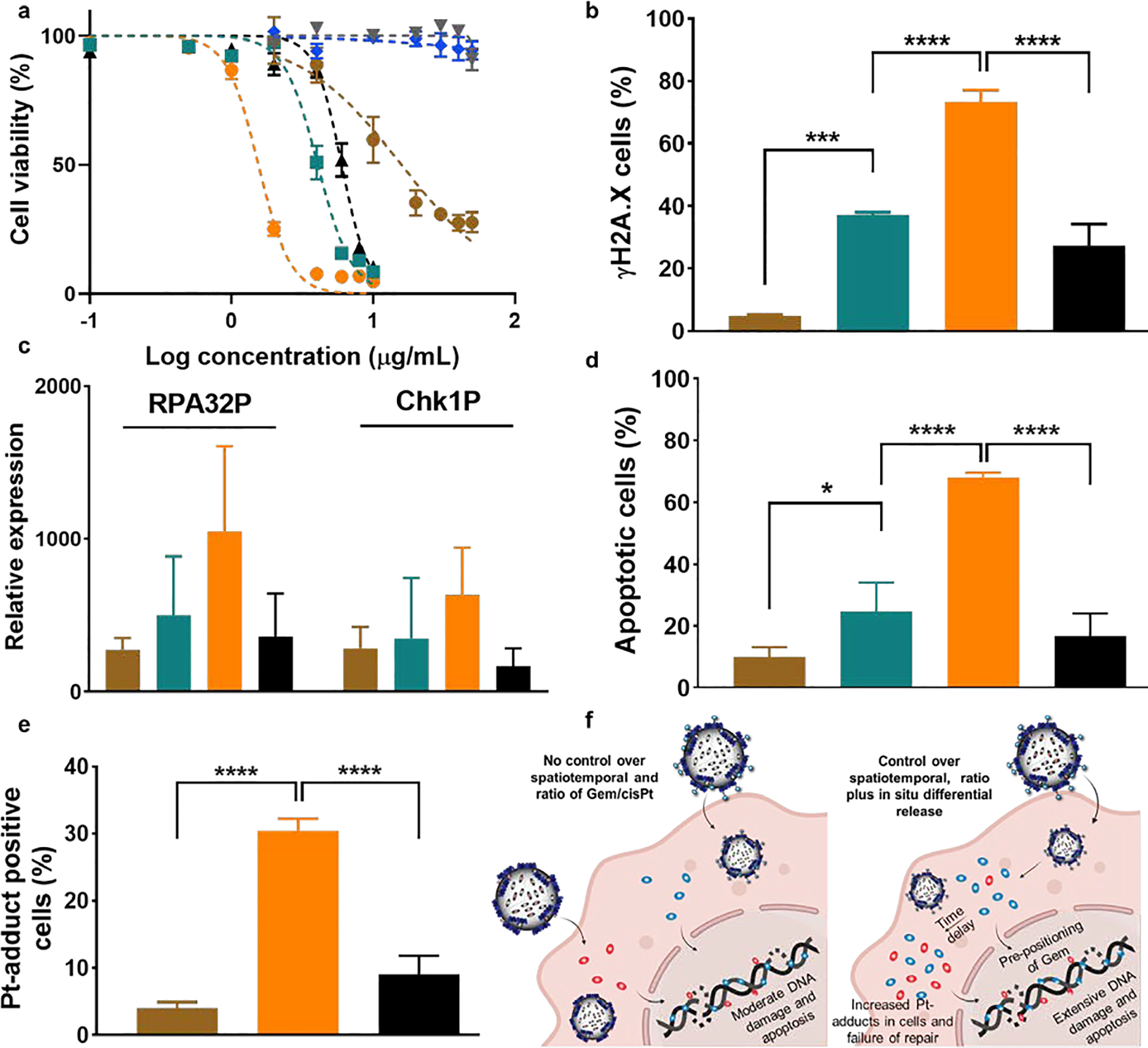
In vitro evaluation of Gem-cisPt-MSNs in a panel of PDAC cells. (a) Dose-response curve of KCM cells treated with AP-MSNs (gray), PEI-MSNs (blue), cisPt-MSNs (brown), Gem-MSNs (green), Gem-cisPt-MSNs (orange), and a physical mixture of Gem-MSNs plus cisPt-MSNs (black). Data represents the mean ± SD of three independent experiments (n=3). (b) Percentage of γH2AX positive cells; (c) western blot analysis of the phosphorylation status of RPA32 and Chk1 proteins (RPA32P indicates RPA32 phosphorylation at the Ser33 and Chk1P indicates Chk1 phosphorylation at the Ser345); (d) percentage of apoptotic cells; and (e) percentage of cells positive for Pt-adduct after treating KCM cells with cisPt-MSNs (brown), Gem-MSNs (green), Gem-cisPt-MSNs (orange) or a physical mixture of Gem-MSNs plus cisPt-MSNs (black). (f) Schematic representation of the mechanism that underlies the enhanced cytotoxic benefit of Gem-cisPt-MSN on PDAC cells. Data represents the mean ± SD of three independent experiments (n=3). One-way ANOVA using Tukey’s multiple comparison test was performed between different groups to determine statistical differences. Statistics: ****p≤0.0001, ***p≤0.001, ** p≤0.01, * p≤0.05 and not significant (ns) p>0.05.
The improved cytotoxicity exhibited by Gem-cisPt-MSNs led us to investigate key steps in the cellular mechanism underlying for the synergistic therapeutic effect of Gem/cisPt combination on PDAC cells. Gem and cisPt drugs have different, but complementary mechanisms for DNA damage: Gem is incorporated into the DNA as a deoxycytidine analog, while cisPt forms Pt-DNA adducts.[46] The following steps are critical events for the transition from DNA damage to cell apoptosis when the drugs enter the nucleus (Figure S15): rapid phosphorylation of the histone protein γH2AX at serine 139 at the damaged site marks the early response to DNA damage;[48, 49] this indices various proteins, including replication protein A (RPA) and checkpoint kinase 1/2 (ChK1/Chk2), to participate as cell cycle regulators, and to act as critical sensors at different stages, in order to induce cell cycle arrest, DNA repair, or cell apoptosis, depending on the extent of DNA damage.[50, 51] Subsequent mechanistic studies were designed to evaluate these events in KCM cells, following Gem-cisPt-MSN treatment.
Flow cytometry results based on use of the FITC-labeled anti-γH2AX antibody indicates that treatment with Gem-cisPt-MSNs produced 73.3 ± 3.8 % γH2AX positive cells, this number is higher than that following treatment with only Gem-MSNs (37.3 ± 0.7 %) or only cisPt-MSNs (7.3 ± 0.7 %) (p≤0.001; Figure 3b). Notably, the physical mixture (cisPt-MSNs plus Gem-MSNs) showed only 27.3 ± 7.0 % γH2AX positive cells, further underscoring the advantage of co-delivering Gem/cisPt in a single nanocarrier. Western blot analysis further confirmed the significant increase in γH2AX protein, when KCM cells are treated with Gem-cisPt-MSNs versus other control groups (Figure S16a). We also evaluated the expression of DNA damage effectors like RPA and ATM/ATR-triggered Chk1, using protein analysis. Western blotting results showed a significant increase of phosphorylated RPA and Chk1 protein in KCM cells treated with Gem-cisPt-MSNs, compared to those treated with control MSN materials (Figures 3c, S16b and S16c). These findings indicate that the Gem-cisPt-MSNs lead to greater DNA damage, causing an efficient relay in the downstream DNA damage effectors compared to monotherapy MSNs or the physical mixture.
To determine if the increased DNA damage associated with Gem-cisPt-MSN treatment leads to cell apoptosis, we used the Annexin V-FITC/PI double staining assay to evaluate the percentage of apoptotic cells.[52] After treating KCM cells with MSN materials, the highest percent of apoptotic cells was observed following treatment with Gem-cisPt-MSNs (70 ± 1.6%; p≤0.001; Figures 3d and S17) followed by Gem-MSNs (24.6 ± 9.3%), physical mixture cisPt-MSNs plus Gem-MSNs (16.7 ± 7.2 %) and cisPt-MSNs (9.8 ± 3.2 %). These data indicate that the enhanced DNA damage produced by Gem-cisPt-MSNs culminates in effective cell death via apoptosis.
The synergism between Gem and cisPt results in the enhancement of Pt-adducts in the DNA. Gem inhibits the DNA repair mechanism enzymatically and also, physically by getting incorporated in the vicinity of the Pt-adduct DNA region.[27, 28] We thus evaluated the Pt-adduct accumulation in cells following Gem-cisPt-MSN treatment, and observed a remarkable increase in Pt-adduct positive cells in the group treated with Gem-cisPt-MSNs (30.4 ± 1.8 %). The increase was at least three times higher compared to 4.0 ± 0.8 and 9.0 ± 2.7 % of Pt-adduct positive cells treated with cisPt-MSNs or the physical mixture of cisPt-MSNs and Gem-MSNs, respectively (Figure 3e). The enhanced Pt-adduct accumulation confirms the therapeutic benefit, due to the unique features of Gem-cisPt-MSNs. In relation to the release properties, the increased ratio of Gem/cisPt at early time points led to pre-positioning of Gem in DNA and increased the inhibitory functions of GEM; this, along with the delayed release of cisPt led to a higher Pt-adduct accumulation, causing the cells to be more sensitive to cisPt and ultimately increasing the cytotoxic effect of the drug (Figure 3f). Cells treated with only cisPt-MSNs showed low Pt-adduct accumulation, likely due to the efficient repair of Pt-adducts that is characteristic of PDAC cells.[53] The physical mixture of cisPt-MSNs plus Gem-MSNs improve Pt-adduct accumulation; but not as much as with Gem-cisPt-MSN material. While the physical mixture has the same concentration and ratio of Gem/cisPt drugs as does Gem-cisPt-MSNs; however, the mixture cannot deliver the optimal ratio inside the cells. Also, the time-dependent release of Gem and cisPt are not the same as in the Gem-cisPt-MSN material (Figure 3f). As evidenced in the treatment of other cancers, the Gem release preceding the release of cisPt potentiates the cancer cells toward cisPt damage, which otherwise would effectively repair the Pt-adducts.[27, 28] The controlled spatio-temporal and in-situ differential delivery of Gem/cisPt associated with our nanoplatform thus confers a unique advantage to Gem/cisPt combination therapy.
Our comprehensive in vitro studies successfully demonstrate that the Gem-cisPt-MSNs increase DNA damage, trigger more efficient activation of the ATM/ATR-mediated DDR pathway, and result in extensive apoptosis, all of which indicate that the cellular machinery failed to repair the DNA damage. This correlates with the synergistic therapeutic advantage of Gem-cisPt-MSNs, owing to the rational design of the material. The extensive and detailed in vitro findings highlighted the superior performance of Gem-cisPt-MSN relative to its monotherapy counterparts and their physical mixture. Based on this finding, we selected only the Gem-cisPt-MSN material for functionalization with TAB004 and for subsequent in vivo studies.
3.3. TAB004-Gem-cisPt-MSNs specifically recognize the tMUC1 receptor on PDAC cells, thereby leading to increased cellular uptake and cytotoxic benefit.
Given the synergistic properties of Gem-cisPt-MSNs for the treatment of PDAC cells, we investigated whether functionalizing this nanoplatform with TAB004 antibody confers a further benefit as a targeting moiety. As a proof of concept, MSNs without drugs were modified with TAB004 antibody (TAB004-MSNs, for details of the synthesis in ESI section S2.4), and we used flow cytometry and confocal microscopy to study their target-specific intracellular uptake. Two PDAC cell lines, KCM and HPAF II, which express human tMUC1 were used for these experiments.[29, 47] Cells were incubated with varying concentrations of TRITC labeled TAB004-MSNs or with PEG-MSNs. Cellular internalization of TAB004-MSNs in KCM and HPAF II cells was 5- and 2.5-fold higher, respectively, compared to that for the control PEG-MSNs (Figures 4a and S18a). These results confirm that the functionalization of the MSNs with the TAB004 antibody enhances the tMUC1-dependent uptake of the nanoparticles (Figure 4b).
Figure 4.
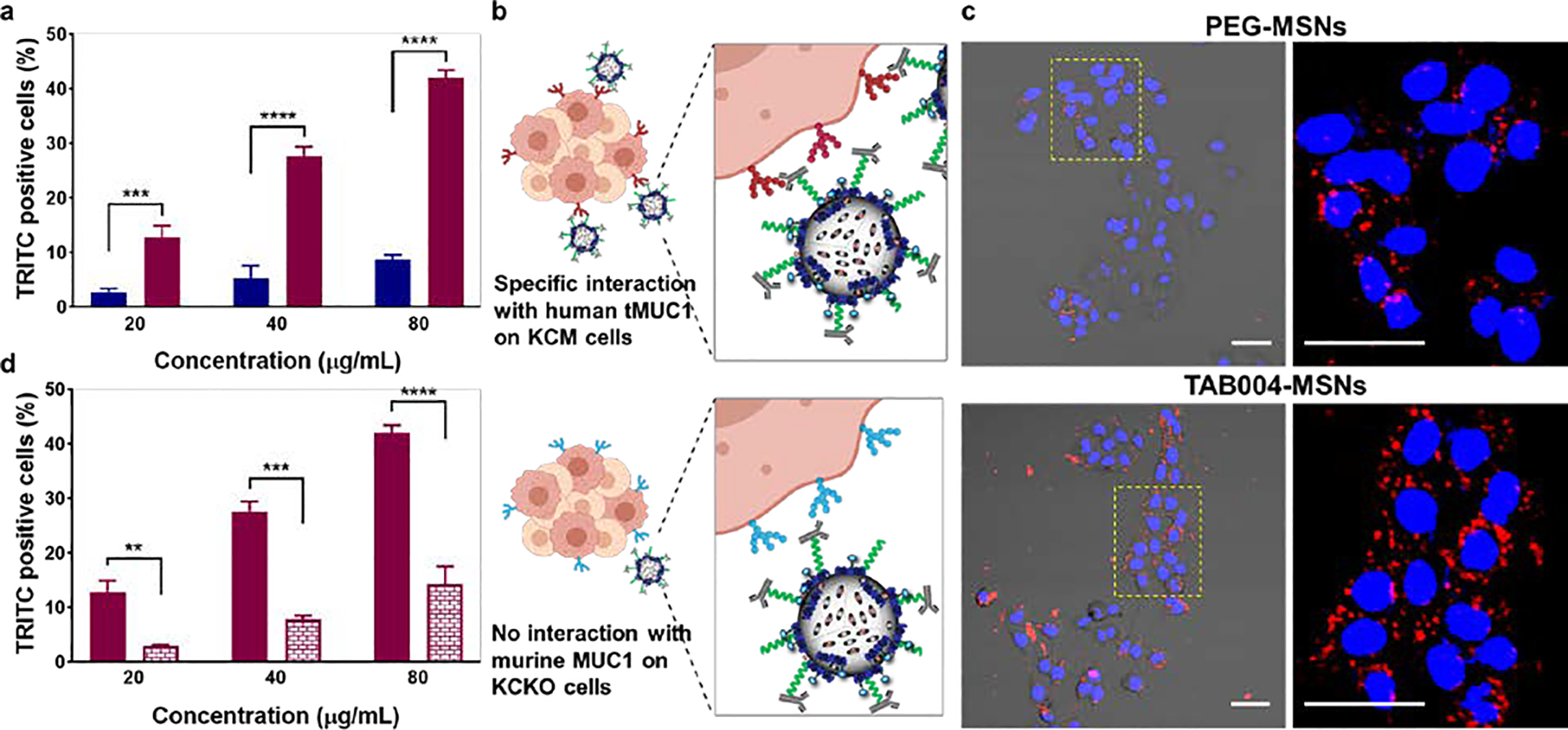
Targeting of TAB004-MSNs to PDAC cells. (a) Percentage TRITC positive KCM cells after treatment with PEG-MSNs (blue) and TAB004-MSNs (red) at various concentrations. Data represents the mean ± SD of three independent experiments (n=3). (b) Specific interaction between human tMUC1 on KCM cells and TAB004 functionalized on the MSN surface leads to increased cellular uptake. (c) Confocal images of KCM cells treated with PEG-MSNs or with TAB004-MSNs (40 μg mL−1). Overlay of the blue channel, which shows the nuclei stained with Hoechst dye; the red channel that depicts the TRITC fluorescence associated to the MSNs; and the DIC channel depicts the cell morphology. Scale bar = 20 μm. (d) Percentage of TRITC positive cells after treating KCM cells (solid red) and KCKO cells (brick-patterned red) with various concentrations of TAB004-MSNs. Data represents the mean ± SD of three independent experiments (n=3). One-way ANOVA using Tukey’s multiple comparison test was performed between different groups to determine statistical difference. Statistics: ****p≤0.0001, ***p≤0.001, ** p≤0.01, * p≤0.05 and not significant (ns) p>0.05.
The cellular uptake of TAB004-MSNs was further corroborated using confocal microscopy: KCM cells treated with TAB004-MSNs exhibited an increased amount of the material internalized by the cells relative to cells inoculated with PEG-MSNs (Figure 4c). To fully demonstrate that the specificity of TAB004-MSNs was due to the interaction of TAB004 antibody with the tMUC1 glycoprotein, cells that lacked tMUC1 expression (KCKO cells) were used for comparison (Figure 4b).[32, 47] KCM and KCKO cells were each treated with different concentrations of TAB004-MSNs or PEG-MSNs. Based on flow cytometry data, uptake of TAB004-MSNs by KCM cells was 3.5-fold higher as compared to that in KCKO cells (Figure 4d); no statistical difference was detected between the cellular uptake of TAB004-MSNs and PEG-MSNs in KCKO cells (Figure S18b). Overall, these data confirm that the targeting ability of TAB004-MSNs is driven by the specific interaction of the TAB004 antibody with tMUC1 protein on the PDAC cell surface, most likely via a receptor-mediated endocytic pathway.[54]
To corroborate the cytotoxic benefit of the targeting ability rendered by the TAB004 antibody, we evaluated the cytotoxicity of TAB004-Gem-cisPt-MSNs in KCM and HPAF II cells. A normal pancreatic ductal cell line (HPDE) was included as control. Treatment withTAB004-Gem-cisPt-MSNs had a higher therapeutic effect in both KCM and HPAF II cells compared to PEG-Gem-cisPt-MSNs with the IC50 values being 2-fold higher (Figures S19a, S19b and S19d). Notably, cytotoxicity was minimal in HPDE cells in the presence of TAB004-Gem-cisPt-MSNs or PEG-Gem-cisPt-MSNs (Figures S19c and 19d). This lack of cytotoxicity in the HPDE cells is likely due to the differences in metabolism and cell division between cancerous and normal cells that decreases the internalization of nanoparticles.[55] Taken together these data show that TAB004-Gem-cisPt-MSNs not only show an increased cytotoxicity in PDAC cells compared to PEG-Gem-cisPt-MSNs, but also are safe toward normal pancreatic ductal cells. The cellular uptake and cytotoxicity of the tMUC1-specific MSN version, provides a strong evidence that nanoparticle functionalization with TAB004 imparts a better therapeutic outcome. TAB004-Gem-cisPt-MSNs can also be associated with safe homing to PDAC tumors that overexpress tMUC1, and a decrease in off-target toxicities. These features are relevant for the treatment of PDAC since multiple physiological barriers associated with this disease present major obstacles for the extravasation of nanoparticles.
3.4. TAB004-Gem-cisPt-MSNs exhibit a good safety profile, enhanced targeting ability and therapeutic efficacy in KCM syngeneic mice.
Mitigation of potential off-target toxicities is vital for translating nanomedicine from the bench to the bedside. MSN-based nanocarriers have been widely investigated, and show convincing preclinical evidence of their safety and biocompatibility.[56–58] In particular, our group has demonstrated the safety and functionality of TAB004-MSNs in preclinical models of breast cancer.[33] Here, we evaluate our new MSN-based platform, which carries a drug combination that has not been evaluated before in terms of its safety and biocompatibility. Dose-escalation studies with PEG-Gem-cisPt-MSNs were undertaken in immune-competent non-tumor bearing C57BL/6 mice. A single dose of NIR-labeled PEG-Gem-cisPt-MSNs was administered to the mice at 20, 40 and 60 mg kg−1 (n=3 per group). The animals were euthanized 10 days post nanoparticle administration and major organs including lungs, spleen, liver, heart and kidneys were excised. NIR fluorescence associated with the organs demonstrated that the PEG-Gem-cisPt-MSNs were distributed primarily in liver and spleen, as expected from previous reports (Figure S20a).[56] Body weights were decreased by approximately 15%, 10% and 5% during the first 3 days for the groups administered with 60, 40 and 20 mg kg−1, respectively; but were fully recovered after six days (Figure S20b). Histological analyses showed no toxic effects of the combination drug on major organs in any of the groups (Figure S20c). Based on these dose-escalation studies, we selected 40 mg kg−1 as the optimal dose for studies on targeting and efficacy.
3.4.1. TAB004-Gem-cisPt-MSNs exhibit enhanced targeting at the tumor site.
Targeting ability and therapeutic efficacy were investigated in the KCM syngeneic mouse model, which was developed by implanting KCM cells derived from spontaneous, triple transgenic PDA.MUC1 mice into immune-competent C57BL/6 mice.[47] This stringent mouse model mimics the genetics of human PDAC, as well as the aggressive growth characteristics, and expression of human tMUC1 that is associated with PDAC (Figure S24). The model also retains the complete immune system, which offers an opportunity for comprehensive evaluation of the performance of TAB004-Gem-cisPt-MSNs.[59, 60] The KCM syngeneic mouse model is thus an advantageous cell-based PDAC model for testing the targeting ability and efficacy of nanoparticles.
The biodistribution of our platform was tested in the KCM syngeneic mice after a one-time i.v administration of 40 mg/kg NIR-labeled TAB004-Gem-cisPt-MSNs or PEG-Gem-cisPt-MSNs (Figure 5a). Real-time IVIS imaging of mice at various time-points post MSN administration uncovered a prominent increase in NIR signaling at the tumor site in the TAB004-targeted nanoparticles relative to the untargeted version (Figures 5b and S21a). Ex vivo fluorescence associated with the nanoparticles accumulated in tumor tissue was quantified using the Living Image® software and showed stronger signaling intensity in tumors of mice injected with TAB004-Gem-cisPt-MSNs compared that in mice administered with PEG-Gem-cisPt-MSNs (p≤0.01) (Figures 5c, 5d and S21b). Ex vivo NIR fluorescence associated with the tumors after 96 h of nanoparticle administration aligned with the real-time IVIS imaging. To corroborate that the NIR signaling intensity in the tumors in fact does reflect MSNs accumulation, we quantified Si with the use of ICP-OES. Si content was higher in the tumor tissue in the TAB004-targeted group compared to that in the untargeted group (p≤0.05) (Figure S21c). Functionalization of MSNs with TAB004 showed no appreciable changes in the accumulation of MSNs in other major organs such as liver and spleen (Figures S21d and S21e). These results highlight that functionalizing MSNs with the TAB004 antibody enhances their accumulation in the tumor tissue (Figure 5e).
Figure 5.
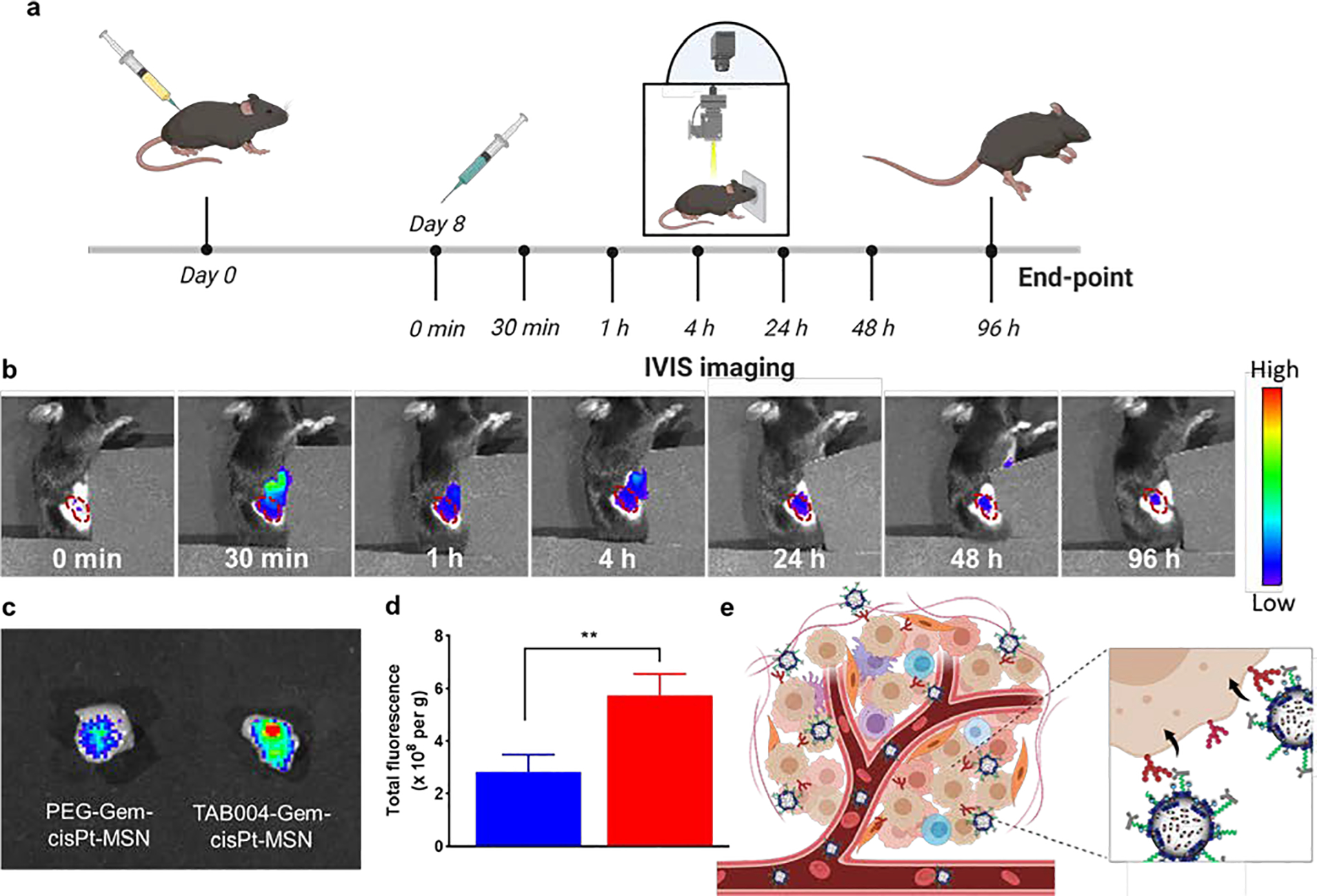
In vivo targeting of TAB004-Gem-cisPt-MSNs in syngeneic KCM mice. (a) Schematic illustration showing the regimen for administration of MSNs and the subsequent imaging schedule. (b) Representative IVIS images at 0, and 30 min, and at 1, 4, 24, 48, and 96 h after intravenous injection of mouse with TAB004-Gem-cisPt-MSNs (region outlined in red denotes subQ tumor location). (c) Tumors excised after 96 h exhibited the difference in accumulation of TAB004-Gem-cisPt-MSNs versus PEG-Gem-cisPt-MSNs. (d) Quantification of fluorescence in tumors excised from mice injected with PEG-Gem-cisPt-MSNs (blue) or TAB004-Gem-cisPt-MSNs (red) (n=3). Data represents the mean ± SD of three independent experiments (n=3) and t-test was performed between the two groups to determine the statistical difference. (e) Schematic representation of the specific interaction between TAB004 functionalized on the surface of MSNs with the tMUC1 expressed on KCM cells. Statistics: ****p≤0.0001, ***p≤0.001, ** p≤0.01, * p≤0.05 and not significant (ns) p>0.05.
3.4.2. TAB004-Gem-cisPt-MSNs exhibit enhanced tumor inhibition.
To evaluate the therapeutic efficacy of TAB004-Gem-cisPt-MSNs, four experimental treatment groups were assessed: those administered PBS, free Gem/cisPt, PEG-Gem-cisPt-MSNs, or TAB004-Gem-cisPt-MSNs. Syngeneic KCM mice were intravenously administered with MSN materials (40 mg kg−1) or free Gem/cisPt (9.52 mg kg−1 Gem and 2.05 mg kg−1 cisPt) every 4 days for a total of 6 injections (Figure 6a). Tumor growth was measured throughout the treatment, to determine the therapeutic efficacy of the platform. TAB004-Gem-cisPt-MSNs showed a greater inhibition tumor growth compared to PBS only (p≤0.0001), free Gem/cisPt (p≤0.0001), or PEG-Gem-cisPt-MSNs (p≤0.05) (Figure 6b). At the end-point, weights of the excised tumor confirmed that TAB004-Gem-cisPt-MSNs have a significantly enhanced therapeutic efficacy compared to that of the other treatments (Figure 6c). TAB004-Gem-cisPt-MSNs showed 79% tumor inhibition compared to 58% and 15% inhibition observed in PEG-Gem-cisPt-MSNs and free drug groups, respectively. Of note, one animal in the TAB004-Gem-cisPt-MSN group showed complete remission. The increased antitumor efficacy of TAB004-Gem-cisPt-MSNs was also confirmed by analyzing the number of apoptotic cells in the tumor tissue. The results indicate that treatment with TAB004-Gem-cisPt-MSNs increases the percentage of apoptotic cells relative to the other treatments (Figure 6d). We further investigated the γH2AX and phosphorylated Chk1 (Chk1P) proteins in the tumor tissues post treatment. Consistent with our in vitro findings, these proteins involved in DNA damage were increased in the TAB004-Gem-cisPt-MSNs, followed by PEG-Gem-cisPt-MSNs treatment group evident by the brown stained nuclei compared to the purple stained nuclei (yellow arrows, Figures 6e and 6f). The tumors from mice treated with free drugs showed negligible IHC staining for both γH2AX and Chk1P (Figures 6e and 6f). These data illustrate the effectiveness of the MSN carrier in efficiently transporting and co-delivering the Gem/cisPt combination, compared with the free drug regimen, and show that functionalizing Gem-cisPt-MSNs with TAB004 antibody confers a clear advantage. The specific interaction of TAB004 with the tMUC1 antigen on tumor cells enhances the therapeutic outcome.
Figure 6.
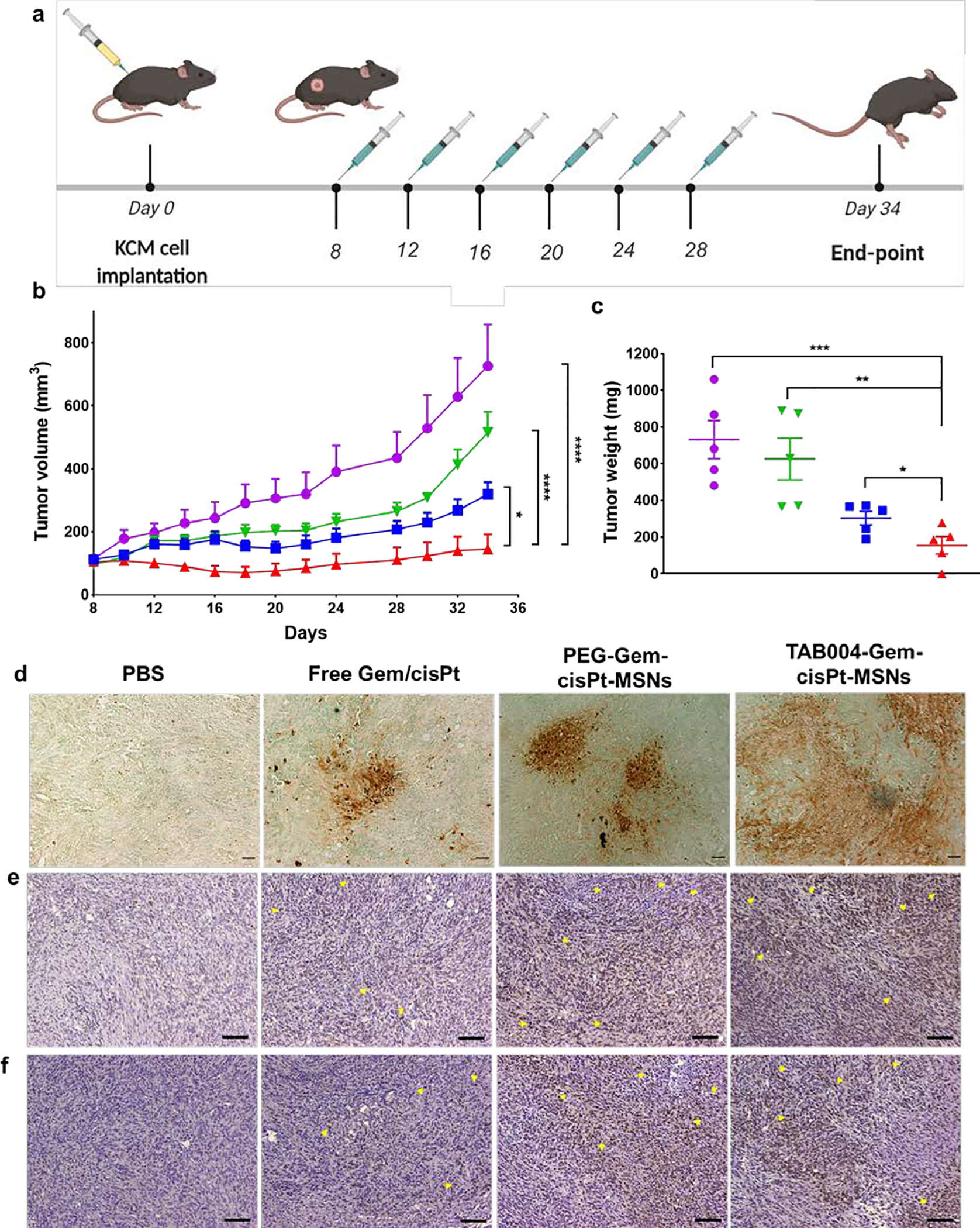
In vivo therapeutic efficacy of TAB004-Gem-cisPt-MSNs in syngeneic KCM mice. (a) Schematic representation of the treatment regimen: mice were injected with MSN materials or free drugs 6 times with a 4-day interval between each injection. (b) Tumor volume was measured throughout the study in the following treatment groups: PBS (purple), Free Gem/cisPt (green), PEG-Gem-cisPt-MSNs (blue), and TAB004-Gem-cisPt-MSNs (red) (n=5 mice per group). Two-way ANOVA was performed between the groups and time points, to determine whether outcomes were statistically different. (c) Tumor weights measured at the endpoint of the efficacy studies. T-test was performed to determine whether the weights between groups were statistically different. Statistics: ****p≤0.0001, ***p≤0.001, ** p≤0.01, * p≤0.05 and not significant (ns) p>0.05. Ex vivo analysis of tumor sections assessed to show the number of apoptotic cells (d), γH2AX positive cells (e), phosphorylated Chk1 (Chk1P) positive cells (f) in tumors post treatment. Scale bar = 100 μm (d) and 200 μm (e and f).
Notably, treatment that uses free drugs shows negligible tumor inhibition compared to PBS (p > 0.05) and induces less apoptotic cells compared to that which uses MSN materials. This can be attributed to the short half-life in blood and the rapid metabolization associated with free Gem and cisPt, due to which they are rapidly cleared from the body. The different pharmacokinetics of Gem and cisPt also present as a challenge for the drugs in terms of reaching the tumor site in a time- and ratio-dependent manner.[18] Overall, the rational approach we have used to fabricate the target-specific MSN-platform increases their stability in the blood, enhances their accumulation in tumors, and finally yields an improved therapeutic effect against PDAC tumors.
3.4.3. TAB004-Gem-cisPt-MSNs exhibit a biocompatible profile.
Another important consideration for the success of combination therapies is the elimination or reduction of off-target toxicities associated to the drugs. Nanomaterials are known to reduce the side effects of the drugs by controlling the pharmacokinetics and biodistribution of the drugs.[61] We assessed the systemic biodistribution and safety of our nanoplatform, by performing a thorough end-point investigation using ex vivo fluorescence imaging, Si content, organ histology, and blood serum analysis. Ex vivo NIR fluorescence associated with liver, lungs, kidneys, spleen, heart, and tumor indicated that both TAB004-Gem-cisPt-MSNs and PEG-Gem-cisPt-MSNs are primarily accumulated in the spleen followed by the liver and lungs (Figures S22a and S22b). This trend was confirmed by analyzing the total Si content in different organs, using ICP-OES (Figure S22c). Previous reports have shown that MSNs are mainly cleared through the hepatobiliary excretion pathway:[58] our findings also indicate that accumulation and clearance of the TAB004-Gem-cisPt-MSN platform follows this same pathway. Out of note, the organ retention of PEG-Gem-cisPt-MSNs is higher than that of TAB004-Gem-cisPt-MSNs (Figure S22a–c). We believe that this difference in retention between the materials is due to the presence of the TAB004 antibody, which potentially triggers the immune system to increase the clearance of nanoparticles.
We also evaluated the post treatment toxicity of TAB004- and PEG-Gem-cisPt-MSNs by analyzing changes in animal behavior, body weight, blood chemistry, and organ histopathology. We found no noticeable behavioral changes, nor any significant changes in body weights of mice throughout the treatment for any of the groups (Figure S23a). We also did not encounter any histopathological changes related to any treatment in H&E-stained sections of liver, kidneys, lungs, heart, and spleen of mice treated with either PEG- or TAB004-Gem-cisPt-MSNs (Figure S23b), corroborating the biocompatibility of the MSN platform, as reported.[56] However, in the free drugs group, we observed characteristic histological changes related to edema of Bowman’s capsule in the kidney tissue (Figures 7a and S23b). Blood serum analysis confirmed the toxicity related to administering free drugs. The biomarker ratio of BUN/creatine, a routinely used measure for kidney function, was observed to higher in the free drugs group relative to the PBS group, indicating compromised kidney functions (Figure 7b). The biomarker ratio AST/ALT, which reflect liver function, was also higher for the free drugs compared to that for the PBS group (Figure 7c), indicating possible toxicity to liver.[20] The BUN/creatine and AST/ALT values obtained for both the MSN materials were comparable to those for the PBS group, further corroborating the biocompatibility of the nanoparticles. Given that the free drugs are mainly eliminated via renal excretion, the kidney is the major accumulation organ, and thus nephrotoxicity is a common side effect.[62] To assess whether accumulation of cisPt is higher in the livers and kidneys of the free drugs group, we quantified the actual Pt content in these organs, using ICP-MS (Figure 7d). We found that the amount of Pt per gram of tissue was indeed higher in both organs from mice treated with free drugs as compared with those form mice treated with the MSN materials, thus pointing to a clear role of cisPt drug as a significant contributor to the off-target toxicities.[63] Notably, TAB004-Gem-cisPt-MSNs showed lower amount of Pt than the PEGylated version, in accordance with the biodistribution results described above. Likely, the Pt detected in the organs associated with the MSN platform is still chemically attached to the nanoparticles, as the cisPt prodrug. These results support our strategy of using drugs chemically conjugated to the nanocarrier in order to reduce off-target toxicity in combination therapy.
Figure 7.
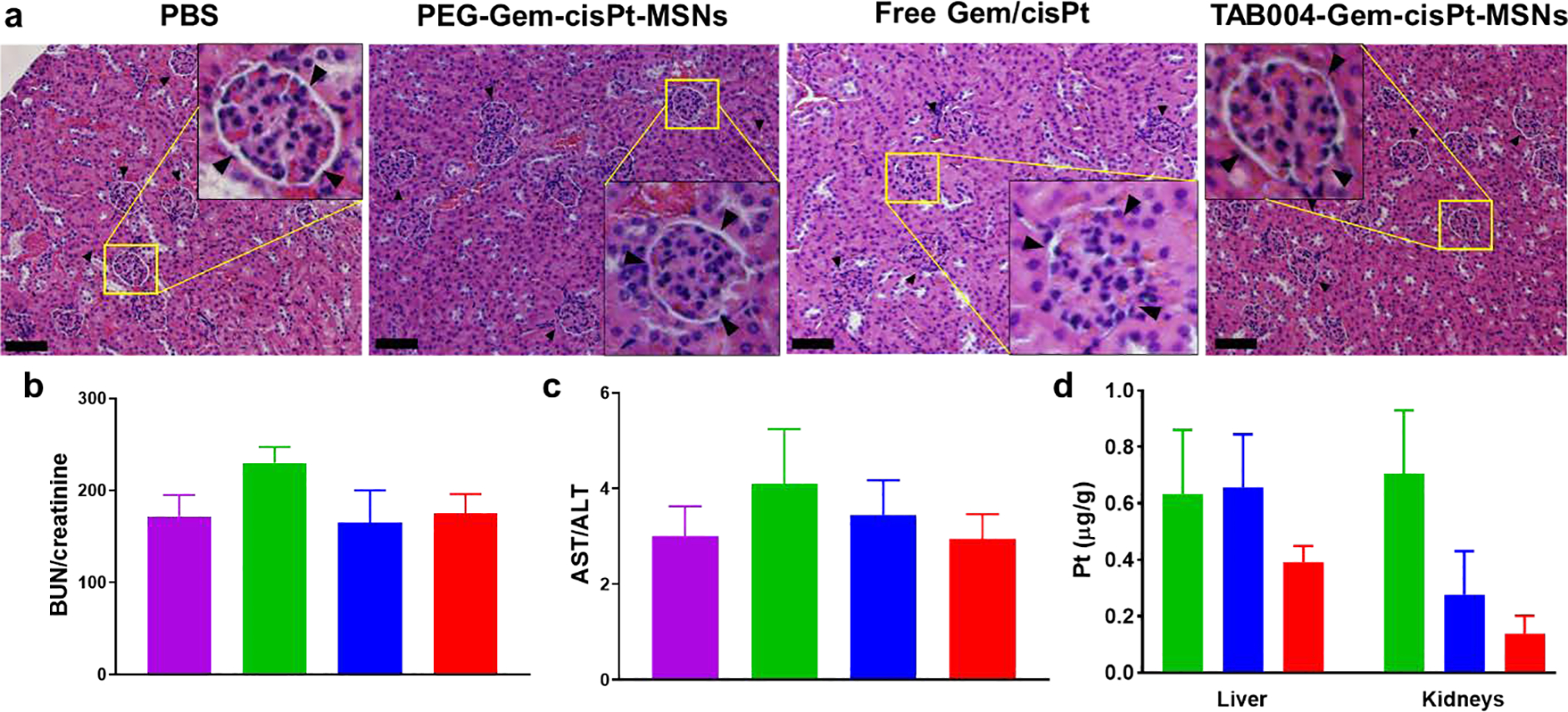
End-point analysis demonstrating the safe and biocompatible profile of TAB004-Gem-cisPt-MSNs. (a) Histological analysis of kidney tissue sections showing toxicities related to the different treatment groups. Bowman’s capsule and edema are shown in the inset images. Scale bar = 200 μm. Blood serum biomarker ratios (b) BUN/creatinine, (c) AST/ALT associated with kidney and liver function, and (d) Pt content analysis in liver and kidneys for different treatment groups PBS (purple); free Gem/cisPt (green), PEG-Gem-cisPt-MSNs (blue) and TAB004-Gem-cisPt-MSNs (red). Data represents the mean ± SD (n=5).
The remarkable in vivo targeting and therapeutic efficacy of TAB004-Gem-cisPt-MSNs in the KCM syngeneic mouse model supports the conclusion that the outstanding performance is a direct translation of the rational design features of our platform. The chemical conjugation of drugs to the MSNs contributes to the stable circulation and reduces drug leakage from the nanoparticles - this, in turn, enables their safe homing at the tumor site, further facilitated by tMUC1-specific TAB004 antibody. The increased accumulation in the tumor, along with the synergistic cytotoxic effect due to ratiometric and in situ differential release of the Gem/cisPt combination in the tumor cells, contributes to the superior performance of TAB004-Gem-cisPt-MSNs against PDAC.
3.5. TAB004-Gem-cisPt-MSNs exhibit enhanced targeting and therapeutic efficacy in the clinically relevant PDA.MUC1 mouse model.
Though cell-line based pre-clinical models have some predictive value, they lack the genetic and phenotypic heterogeneity of PDAC, and thus their ability to precisely predict the therapeutic responses in clinical settings is limited.[60, 64] GEM models recapitulate the progression of PDAC disease in humans, and thus are more accurate predictors of drug response, and better models for biomarker and therapeutic discovery.[65, 66] We have generated triple transgenic PDA.MUC1 mice by crossing LSL-KRASG12D with tamoxifen-inducible P48Cre to a human MUC1.Tg mice (Figure S24). PDA.MUC1 mice develop the full spectrum of PDAC symptoms, including preinvasive lesions (pancreatic intraepithelial neoplasia - PanIN), invasive PDA, and metastasis; they also express tMUC1 in a pattern similar to that observed in human PDAC.[32, 47, 67, 68] Here, we use this unique GEM PDA.MUC1 model to obtain preliminary data on the targeting or therapeutic capability of our MSN-based platform. We achieved this by first assessing the potential of TAB004-MSNs to accumulate in the pancreas at different stages of the disease, by using PDA.MUC1 mice at various times (8-,12- and 27-weeks) following tamoxifen administration (Figure S25a). These intervals fairly represent different grades of PanIN lesions and stage 1 PDAC that is observed in the clinical setting.[69] The position of the pancreas makes it difficult to derive reliable information about accumulation of TAB004-MSNs in the pancreatic region from whole-body fluorescence images (Figures S25b–d). Hence, we used end-point ex vivo NIR fluorescence from the pancreas to determine the accumulation of TAB004-MSNs, which indicated that TAB004-MSNs are accumulated in the pancreas of PDA.MUC1 mice as early as 8-weeks post-tamoxifen injection. At 12- and 27-weeks post-tamoxifen injection the mice also showed enhanced accumulation of the nanoparticles (Figure S26a). The pancreas from PDA.MUC1 mice that were not administered nanoparticle, or from healthy C57BL/6 mice injected with TAB004-MSNs, did not exhibit NIR fluorescence associated with the nanoparticles, indicating clearly that localization of nanoparticles in the pancreas of PDA.MUC1 mice that were injected with TAB004-MSNs is a function of the stage of the disease as well as the targeting ability of the nanoparticles. Histopathological analysis of the pancreas confirmed the presence of PanIN 1 and PanIN 2 grade lesions in mice at 8- and 12-weeks post tamoxifen, respectively (Figure S26b) whereas at 27-weeks post tamoxifen, we detected high-grade PanIN 3 lesions as well as invasive PDA. Thus, our preliminary data show strong evidence that the TAB004-MSNs selectively accumulate in the regions of the pancreas associated with stages as early as PanIN 1 and PanIN 2 lesions.
Having confirmed that TAB004-MSNs can selectively accumulate in the pancreas of PDA.MUC1 mice, we next investigated the therapeutic efficacy of TAB004-Gem-cisPt-MSNs. Currently, PDAC patients usually present with advanced stages of the disease at the time of diagnosis. Thus, we designed therapeutic studies in PDA.MUC1 mice at 32-weeks post-tamoxifen that are good models for the study of high-grade disease. We evaluated three PDA.MUC1 mice per group for two experimental groups; control and TAB004-Gem-cisPt-MSNs. PDA.MUC1 mice were injected i.v. with TAB004-Gem-cisPt-MSNs (40 mg kg−1) every 15 days, over three months, for a total of six injections (Figure 8a). The idea was to observe therapeutic efficacy based on the progression of invasive PDAC in the PDA.MUC1 model. At the endpoint, mice were euthanized and the pancreas was excised, weighed, fixed and sectioned to determine the stage of the disease. The pancreas weight gives a direct measurement of the tumor growth since in this animal model the tumor tissue cannot be accurately separated from the whole pancreas. The weights showed that TAB004-Gem-cisPt-MSNs treatment results in a greater decrease of the PDAC burden (142.0 ± 8.0 mg), relative to the untreated group (193.0 ± 7.0 mg) (Figure 8b and Table S7). Histopathological analysis of pancreatic tissue depicted extensive, highly differentiated, invasive PDA regions (depicted by characteristic glandular cells[70]) in the untreated group, while the treated group revealed a reduction in the invasive regions (Figure 8c and S27). To determine the therapeutic effect on PDAC progression, the area of PanIN and invasive PDA regions were analyzed, and quantified with use of SketchandCalc (Table S8). The results showed that TAB004-Gem-cisPt-MSNs treatment significantly reduced the percentage of the area associated with PanIN and invasive PDA regions to 4.0 and 11.3 %, respectively. The pancreas in the untreated group had 9.5 and 36.7 % regions of PanIN and invasive PDA, respectively. The area of invasive PDA after treatment with TAB004-Gem-cisPt-MSNs was almost four times smaller than that observed in the untreated group. To demonstrate that the MSN material was localized in the invasive PDA regions, H&E-stained sections through the pancreas were imaged using confocal microscopy, and confirmed that the TAB004-Gem-cisPt-MSNs are indeed present in the pancreas, are especially co-localized in the invasive PDA regions (Figure 8d).
Figure 8.
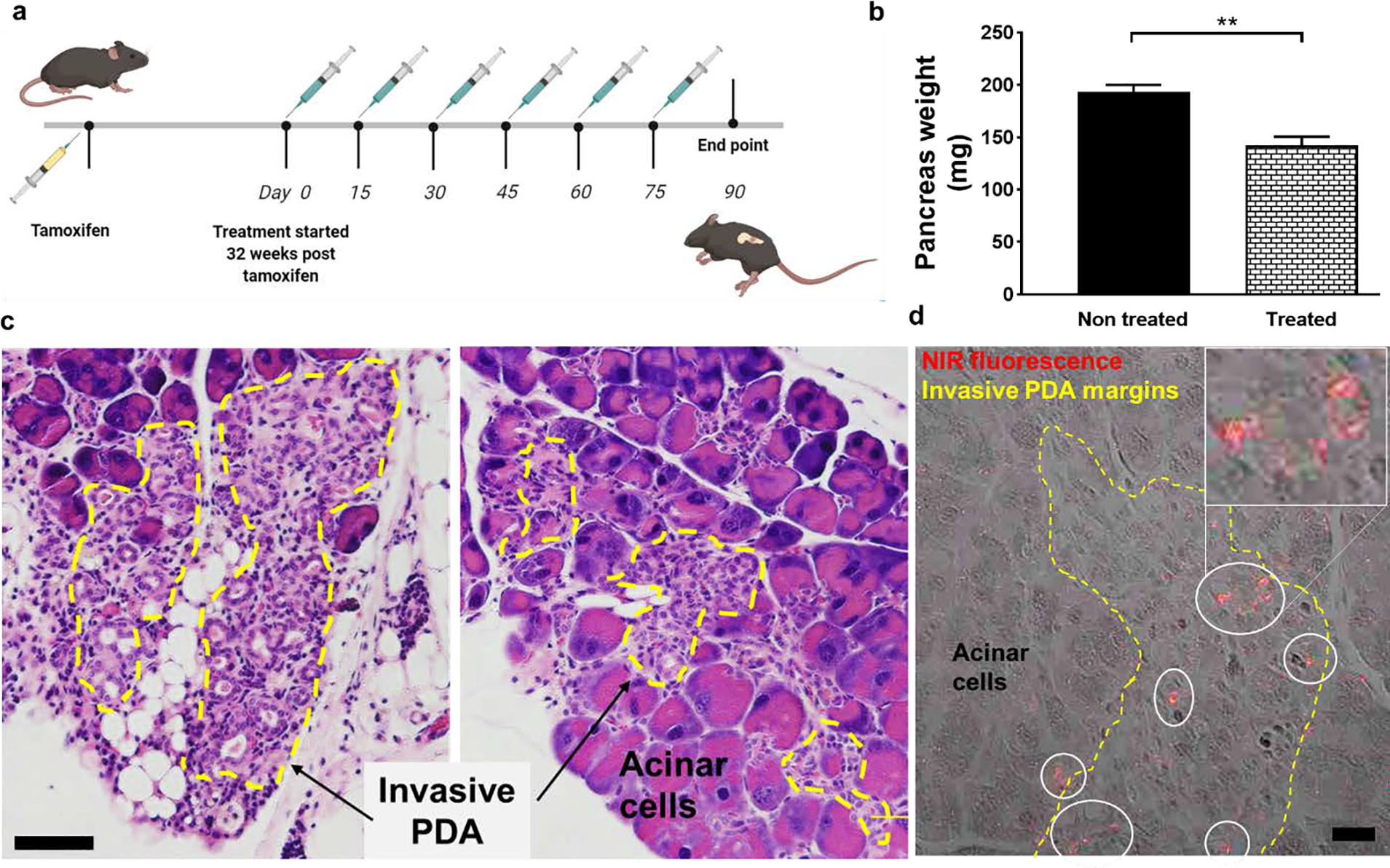
Therapeutic efficacy of TAB004-Gem-cisPt-MSNs in PDA.MUC1 mice. (a) Schematic representation of the treatment regimen. Thirty-two weeks after the tamoxifen injection, PDA.MUC1 mice were injected with 6 doses of 40 mg kg−1 of TAB004-Gem-cisPt-MSNs each, with an interval of 15-days between consecutive doses. (b) Weight of pancreas harvested from PDA.MUC1 mice from control and treatment groups (n=3). (c) Representative micrographs of pancreas tissue from mice in the untreated control group (left) and in TAB004-Gem-cisPt-MSNs treated PDA.MUC1 mice (right). PDA regions are significantly less invasive in the tissue sections from the treated group compared to the controls. Yellow margins represent the invasive PDA regions. Scale bar = 200 μm (d) Confocal micrograph of the H&E-stained pancreatic tissue from the treated group, showing extensive co-localization of TAB004-Gem-cisPt-MSNs (red) in the invasive PDA regions (yellow margins). Scale bar = 20 μm.
Overall, the preliminary therapeutic efficacy of TAB004-Gem-cisPt-MSNs against PDAC demonstrated in this clinically relevant model supports the use of this platform for translation to clinical applications and supports the potential application of this system in the early diagnosis of PDAC, which is one of the major contributors of the poor prognosis in PDAC patients.
Conclusions and outlook.
We have developed and successfully documented the performance of a novel MSN-based nanoplatform for the Gem/cisPt combination therapy in PDAC. Our nanoplatform is designed with spatially localized, chemical conjugation of Gem and cisPt to MSNs via redox-responsive linkers; this conjugation confers highly desirable features on the material. The synergistic performance of our platform was further studied in a panel of PDAC cell lines. The in situ differential release of Gem inside the cells inhibits DNA repair mechanisms, and enhances Pt-adducts and cell apoptosis. Moreover, the functionalization of the MSN material with TAB004 antibody, which targets tMUC1 antigen, significantly enhances its therapeutic performance in vivo, increases tumor growth suppression and reduces the off-target toxicities. The unique spatial localization of the drugs in the MSNs and the advantage of having a stimulus-responsive controlled release makes this platform attractive for other combination therapies, not only for the treatment of PDAC but also for other malignancies that require drug combinations such as paclitaxel/carboplatin, Gem/paclitaxel, and others.
PDAC classification and patient stratification based on the biomarkers is an important criterion for personalized therapies. Germline alterations in the DNA damage repair resulting in genomic instability have recently shown to be present in a subset of 5–6% of PDAC patients. This population can benefit from DNA damaging agents and their combinations; in particular, Pt-based agents. We envision that our Gem/cisPt nanoplatform can be an excellent candidate to improve the prognosis of this subset of PDAC. Certainly, another important factor limiting the efficacy of current treatments for PDAC is its unique desmoplastic stroma. Our group is also investigating the combination of stroma modulation agents and TAB004-Gem-cisPt-MSNs to further improve the therapeutic efficacy of this platform.
Supplementary Material
ACKNOWLEDGMENT
M. T. received financial support from the Thomas Reynolds Graduate Research Award. Work reported here was supported by the National Institute of Cancer of the National Institutes of Health under Award Number R15CA192160 (to J.V.-E. and P.M.). The Yan lab was supported, in part, by a grant from the NIH/NCI (R01CA225637). The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. Authors appreciate Ms. Hemapriyadarshini Vadarevu help with in vitro experiments. Authors would also like to thank the staff of the Dr. Chandra Williams Vivarium at UNC Charlotte, and Dr. Didier Dréau for assistance with animal experiments and data analysis.
Funding Sources.
CBES award (UNC Charlotte); National Institute of Cancer of the National Institutes of Health under Award Number R15CA192160 and R01CA225637.
Footnotes
The authors declare no competing financial interest. Dr. Mukherjee is Founder of OncoTAb Inc.
Supporting Information. Materials and extended experimental section including synthesis and characterization of Gem and cisPt prodrugs, stepwise synthesis of TAB004-Gem-cisPt-MSNs and control MSN materials, characterization of MSN materials, and drug release. In vitro approaches including the cell culture, combination index studies, DNA damage and protein analysis in PDAC cells. In vivo approaches including KCM syngeneic mice and post-treatment analysis. Supporting Figures S1–S25 and Table S1–S5 are available free of charge.
Contributor Information
Mubin Tarannum, Department of Chemistry, University of North Carolina Charlotte, Charlotte NC 28223, U.S.A.; Nanoscale Science Program, University of North Carolina Charlotte, Charlotte NC 28223, U.S.A.
Md Akram Hossain, Department of Biological Sciences, University of North Carolina Charlotte, Charlotte NC 28223, U.S.A..
Bryce Holmes, Analytical Research Laboratory, North Carolina A&T State University, Greensboro NC, U.S.A. 27411, U.S.A..
Shan Yan, Department of Biological Sciences, University of North Carolina Charlotte, Charlotte NC 28223, U.S.A..
Pinku Mukherjee, Department of Biological Sciences, University of North Carolina Charlotte, Charlotte NC 28223, U.S.A..
Juan L. Vivero-Escoto, Department of Chemistry, University of North Carolina Charlotte, Charlotte NC 28223, U.S.A. Center for Biomedical Engineering and Science, University of North Carolina Charlotte, Charlotte NC 28223, U.S.A.
References.
- 1.Bray F; Ferlay J; Soerjomataram I; Siegel RL; Torre LA; Jemal A, CA Cancer J Clin 2018, 68 (6), 394–424. DOI 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Rawla P; Sunkara T; Gaduputi V, World J Oncol 2019, 10 (1), 10–27. DOI 10.14740/wjon1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Siegel RL; Miller KD; Jemal A, CA Cancer J Clin 2020, 70 (1), 7–30. DOI 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- 4.Adamska A; Domenichini A; Falasca M, Int J Mol Sci 2017, 18 (7). DOI 10.3390/ijms18071338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Oberstein PE; Olive KP, Therap Adv Gastroenterol 2013, 6 (4), 321–37. DOI 10.1177/1756283X13478680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Von Hoff DD; Ramanathan RK; Borad MJ; Laheru DA; Smith LS; Wood TE; Korn RL; Desai N; Trieu V; Iglesias JL; Zhang H; Soon-Shiong P; Shi T; Rajeshkumar NV; Maitra A; Hidalgo M, J Clin Oncol 2011, 29 (34), 4548–54. DOI 10.1200/JCO.2011.36.5742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Von Hoff DD; Ervin T; Arena FP; Chiorean EG; Infante J; Moore M; Seay T; Tjulandin SA; Ma WW; Saleh MN; Harris M; Reni M; Dowden S; Laheru D; Bahary N; Ramanathan RK; Tabernero J; Hidalgo M; Goldstein D; Van Cutsem E; Wei X; Iglesias J; Renschler MF, N Engl J Med 2013, 369 (18), 1691–703. DOI 10.1056/NEJMoa1304369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Conroy T; Desseigne F; Ychou M; Bouché O; Guimbaud R; Bécouarn Y; Adenis A; Raoul JL; Gourgou-Bourgade S; de la Fouchardière C; Bennouna J; Bachet JB; Khemissa-Akouz F; Péré-Vergé D; Delbaldo C; Assenat E; Chauffert B; Michel P; Montoto-Grillot C; Ducreux M; Unicancer, G. T. D. o.; Intergroup, P., N Engl J Med 2011, 364 (19), 1817–25. DOI 10.1056/NEJMoa1011923. [DOI] [PubMed] [Google Scholar]
- 9.Zhang Y; Yang C; Cheng H; Fan Z; Huang Q; Lu Y; Fan K; Luo G; Jin K; Wang Z; Liu C; Yu X, J Hematol Oncol 2018, 11 (1), 14. DOI 10.1186/s13045-017-0551-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miller AL; Garcia PL; Yoon KJ, Pharmacol Res 2020, 155, 104740. DOI 10.1016/j.phrs.2020.104740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cancer Genome Atlas Research Network. Electronic address, a. a. d. h. e.; Cancer Genome Atlas Research, N., Cancer Cell 2017, 32 (2), 185–203.e13. DOI 10.1016/j.ccell.2017.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roberts NJ; Norris AL; Petersen GM; Bondy ML; Brand R; Gallinger S; Kurtz RC; Olson SH; Rustgi AK; Schwartz AG; Stoffel E; Syngal S; Zogopoulos G; Ali SZ; Axilbund J; Chaffee KG; Chen YC; Cote ML; Childs EJ; Douville C; Goes FS; Herman JM; Iacobuzio-Donahue C; Kramer M; Makohon-Moore A; McCombie RW; McMahon KW; Niknafs N; Parla J; Pirooznia M; Potash JB; Rhim AD; Smith AL; Wang Y; Wolfgang CL; Wood LD; Zandi PP; Goggins M; Karchin R; Eshleman JR; Papadopoulos N; Kinzler KW; Vogelstein B; Hruban RH; Klein AP, Cancer Discov 2016, 6 (2), 166–75. DOI 10.1158/2159-8290.CD-15-0402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jameson GS; Borazanci E; Babiker HM; Poplin E; Niewiarowska AA; Gordon MS; Barrett MT; Rosenthal A; Stoll-D’Astice A; Crowley J; Shemanski L; Korn RL; Ansaldo K; Lebron L; Ramanathan RK; Von Hoff DD, JAMA Oncol 2019. DOI 10.1001/jamaoncol.2019.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reni M; Zanon S; Balzano G; Passoni P; Pircher C; Chiaravalli M; Fugazza C; Ceraulo D; Nicoletti R; Arcidiacono PG; Macchini M; Peretti U; Castoldi R; Doglioni C; Falconi M; Partelli S; Gianni L, Eur J Cancer 2018, 102, 95–102. DOI 10.1016/j.ejca.2018.07.007. [DOI] [PubMed] [Google Scholar]
- 15.Mayer LD; Harasym TO; Tardi PG; Harasym NL; Shew CR; Johnstone SA; Ramsay EC; Bally MB; Janoff AS, Molecular cancer therapeutics 2006, 5 (7), 1854–63. DOI 10.1158/1535-7163.mct-06-0118. [DOI] [PubMed] [Google Scholar]
- 16.Meng F; Han N; Yeo Y, Expert Opin Drug Deliv 2017, 14 (3), 427–446. DOI 10.1080/17425247.2016.1218464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liboiron BD; Mayer LD, Ther Deliv 2014, 5 (2), 149–71. DOI 10.4155/tde.13.149. [DOI] [PubMed] [Google Scholar]
- 18.Hu Q; Sun W; Wang C; Gu Z, Adv Drug Deliv Rev 2016, 98, 19–34. DOI 10.1016/j.addr.2015.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miao L; Guo S; Lin CM; Liu Q; Huang L, Adv Drug Deliv Rev 2017, 115, 3–22. DOI 10.1016/j.addr.2017.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miao L; Guo S; Zhang J; Kim WY; Huang L, Adv Funct Mater 2014, 24 (42), 6601–6611. DOI 10.1002/adfm.201401076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meng H; Wang M; Liu H; Liu X; Situ A; Wu B; Ji Z; Chang CH; Nel AE, ACS Nano 2015, 9 (4), 3540–57. DOI 10.1021/acsnano.5b00510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Poon C; He C; Liu D; Lu K; Lin W, J Control Release 2015, 201, 90–9. DOI 10.1016/j.jconrel.2015.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kankala RK; Han YH; Na J; Lee CH; Sun Z; Wang SB; Kimura T; Ok YS; Yamauchi Y; Chen AZ; Wu KC, Adv Mater 2020, 32 (23), e1907035. DOI 10.1002/adma.201907035. [DOI] [PubMed] [Google Scholar]
- 24.Sábio RM; Meneguin AB; Ribeiro TC; Silva RR; Chorilli M, Int J Pharm 2019, 564, 379–409. DOI 10.1016/j.ijpharm.2019.04.067. [DOI] [PubMed] [Google Scholar]
- 25.Vallet-Regí M; Colilla M; Izquierdo-Barba I; Manzano M, Molecules 2017, 23 (1). DOI 10.3390/molecules23010047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang RX; Li J; Zhang T; Amini MA; He C; Lu B; Ahmed T; Lip H; Rauth AM; Wu XY, Acta Pharmacol Sin 2018, 39 (5), 825–844. DOI 10.1038/aps.2018.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ledermann JA; Gabra H; Jayson GC; Spanswick VJ; Rustin GJ; Jitlal M; James LE; Hartley JA, Clin Cancer Res 2010, 16 (19), 4899–905. DOI 10.1158/1078-0432.CCR-10-0832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moufarij MA; Phillips DR; Cullinane C, Mol Pharmacol 2003, 63 (4), 862–9. DOI 10.1124/mol.63.4.862. [DOI] [PubMed] [Google Scholar]
- 29.Curry JM; Thompson KJ; Rao SG; Besmer DM; Murphy AM; Grdzelishvili VZ; Ahrens WA; McKillop IH; Sindram D; Iannitti DA; Martinie JB; Mukherjee P, J Surg Oncol 2013, 107 (7), 713–22. DOI 10.1002/jso.23316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kelly VJ; Wu ST; Gottumukkala V; Coelho R; Palmer K; Nair S; Erick T; Puri R; Ilovich O; Mukherjee P, Theranostics 2020, 10 (15), 6946–6958. DOI 10.7150/thno.38236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roy LD; Dillon LM; Zhou R; Moore LJ; Livasy C; El-Khoury JM; Puri R; Mukherjee P, Genes Cancer 2017, 8 (3–4), 536–549. DOI 10.18632/genesandcancer.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu ST; Williams CD; Grover PA; Moore LJ; Mukherjee P, PLoS One 2018, 13 (2), e0193260. DOI 10.1371/journal.pone.0193260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dréau D; Moore LJ; Alvarez-Berrios MP; Tarannum M; Mukherjee P; Vivero-Escoto JL, J Biomed Nanotechnol 2016, 12 (12), 2172–2184. DOI 10.1166/jbn.2016.2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Medina OP; Tower RJ; Medina TP; Ashkenani F; Appold L; Bötcher M; Huber L; Will O; Ling Q; Hauser C; Rohwedder A; Heneweer C; Peschke E; Hövener JB; Lüdtke-Buzug K; Boretius S; Mentlein R; Kairemo K; Glüer CC; Sebens S; Kalthoff H, Curr Pharm Des 2020. DOI 10.2174/1381612826666200717084846. [DOI] [PubMed] [Google Scholar]
- 35.Vivero-Escoto JL; Slowing II; Trewyn BG; Lin VS, Small 2010, 6 (18), 1952–67. DOI 10.1002/smll.200901789. [DOI] [PubMed] [Google Scholar]
- 36.Liu X; Situ A; Kang Y; Villabroza KR; Liao Y; Chang CH; Donahue T; Nel AE; Meng H, ACS Nano 2016, 10 (2), 2702–15. DOI 10.1021/acsnano.5b07781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Das M; Li J; Bao M; Huang L, AAPS J 2020, 22 (4), 88. DOI 10.1208/s12248-020-00467-8. [DOI] [PubMed] [Google Scholar]
- 38.Mi P, Theranostics 2020, 10 (10), 4557–4588. DOI 10.7150/thno.38069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tang Q; Yu B; Gao L; Cong H; Song N; Lu C, Curr Med Chem 2018, 25 (16), 1837–1866. DOI 10.2174/0929867325666180111095913. [DOI] [PubMed] [Google Scholar]
- 40.Ding C; Tong L; Feng J; Fu J, Molecules 2016, 21 (12). DOI 10.3390/molecules21121715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alvarez-Berríos MP; Vivero-Escoto JL, Int J Nanomedicine 2016, 11, 6251–6265. DOI 10.2147/IJN.S118196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mirhadi E; Mashreghi M; Faal Maleki M; Alavizadeh SH; Arabi L; Badiee A; Jaafari MR, Int J Pharm 2020, 589, 119882. DOI 10.1016/j.ijpharm.2020.119882. [DOI] [PubMed] [Google Scholar]
- 43.Li R; Peng F; Cai J; Yang D; Zhang P, Asian J Pharm Sci 2020, 15 (3), 311–325. DOI 10.1016/j.ajps.2019.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Raza A; Hayat U; Rasheed T; Bilal M; Iqbal HMN, Eur J Med Chem 2018, 157, 705–715. DOI 10.1016/j.ejmech.2018.08.034. [DOI] [PubMed] [Google Scholar]
- 45.Tang Y; Wang Y; Teng X, Mol Med Rep 2013, 8 (1), 221–6. DOI 10.3892/mmr.2013.1495. [DOI] [PubMed] [Google Scholar]
- 46.Besançon OG; Tytgat GA; Meinsma R; Leen R; Hoebink J; Kalayda GV; Jaehde U; Caron HN; van Kuilenburg AB, Cancer Lett 2012, 319 (1), 23–30. DOI 10.1016/j.canlet.2011.12.016. [DOI] [PubMed] [Google Scholar]
- 47.Besmer DM; Curry JM; Roy LD; Tinder TL; Sahraei M; Schettini J; Hwang SI; Lee YY; Gendler SJ; Mukherjee P, Cancer Res 2011, 71 (13), 4432–42. DOI 10.1158/0008-5472.CAN-10-4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sharma A; Singh K; Almasan A, Methods Mol Biol 2012, 920, 613–26. DOI 10.1007/978-1-61779-998-3_40. [DOI] [PubMed] [Google Scholar]
- 49.Mah LJ; El-Osta A; Karagiannis TC, Leukemia 2010, 24 (4), 679–86. DOI 10.1038/leu.2010.6. [DOI] [PubMed] [Google Scholar]
- 50.Zhou BB; Elledge SJ, Nature 2000, 408 (6811), 433–9. DOI 10.1038/35044005. [DOI] [PubMed] [Google Scholar]
- 51.Giglia-Mari G; Zotter A; Vermeulen W, Cold Spring Harb Perspect Biol 2011, 3 (1), a000745. DOI 10.1101/cshperspect.a000745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.van Engeland M; Nieland LJ; Ramaekers FC; Schutte B; Reutelingsperger CP, Cytometry 1998, 31 (1), 1–9. DOI . [DOI] [PubMed] [Google Scholar]
- 53.Modi S; Kir D; Giri B; Majumder K; Arora N; Dudeja V; Banerjee S; Saluja AK, J Gastrointest Surg 2016, 20 (1), 13–23; discussion 23–4. DOI 10.1007/s11605-015-3000-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xu S; Olenyuk BZ; Okamoto CT; Hamm-Alvarez SF, Adv Drug Deliv Rev 2013, 65 (1), 121–38. DOI 10.1016/j.addr.2012.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Behzadi S; Serpooshan V; Tao W; Hamaly MA; Alkawareek MY; Dreaden EC; Brown D; Alkilany AM; Farokhzad OC; Mahmoudi M, Chem Soc Rev 2017, 46 (14), 4218–4244. DOI 10.1039/c6cs00636a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li Z; Zhang Y; Feng N, Expert Opin Drug Deliv 2019, 16 (3), 219–237. DOI 10.1080/17425247.2019.1575806. [DOI] [PubMed] [Google Scholar]
- 57.Lu J; Liong M; Li Z; Zink JI; Tamanoi F, Small 2010, 6 (16), 1794–805. DOI 10.1002/smll.201000538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dogra P; Adolphi NL; Wang Z; Lin YS; Butler KS; Durfee PN; Croissant JG; Noureddine A; Coker EN; Bearer EL; Cristini V; Brinker CJ, Nat Commun 2018, 9 (1), 4551. DOI 10.1038/s41467-018-06730-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Labrijn AF; Meesters JI; Bunce M; Armstrong AA; Somani S; Nesspor TC; Chiu ML; Altintaş I; Verploegen S; Schuurman J; Parren PWHI, Sci Rep 2017, 7 (1), 2476. DOI 10.1038/s41598-017-02823-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kong K; Guo M; Liu Y; Zheng J, J Cancer 2020, 11 (6), 1555–1567. DOI 10.7150/jca.37529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gurunathan S; Kang MH; Qasim M; Kim JH, Int J Mol Sci 2018, 19 (10). DOI 10.3390/ijms19103264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kobayashi S; Ueno M; Ohkawa S; Irie K; Goda Y; Morimoto M, Oncology 2014, 87 (1), 30–9. DOI 10.1159/000362604. [DOI] [PubMed] [Google Scholar]
- 63.Dugbartey GJ; Peppone LJ; de Graaf IA, Toxicology 2016, 371, 58–66. DOI 10.1016/j.tox.2016.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sun D; Zhou S; Gao W, ACS Nano 2020, 14 (10), 12281–12290. DOI 10.1021/acsnano.9b09713. [DOI] [PubMed] [Google Scholar]
- 65.Mohammed A; Janakiram NB; Lightfoot S; Gali H; Vibhudutta A; Rao CV, Curr Med Chem 2012, 19 (22), 3701–13. DOI 10.2174/092986712801661095. [DOI] [PubMed] [Google Scholar]
- 66.Krempley BD; Yu KH, Chin Clin Oncol 2017, 6 (3), 25. DOI 10.21037/cco.2017.06.15. [DOI] [PubMed] [Google Scholar]
- 67.Wang P; Yoo B; Sherman S; Mukherjee P; Ross A; Pantazopoulos P; Petkova V; Farrar C; Medarova Z; Moore A, Int J Cancer 2016, 139 (3), 712–8. DOI 10.1002/ijc.30098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tinder TL; Subramani DB; Basu GD; Bradley JM; Schettini J; Million A; Skaar T; Mukherjee P, J Immunol 2008, 181 (5), 3116–25. DOI 10.4049/jimmunol.181.5.3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Goggins M, Clin Cancer Res 2011, 17 (4), 635–7. DOI 10.1158/1078-0432.CCR-10-3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hingorani SR; Petricoin EF; Maitra A; Rajapakse V; King C; Jacobetz MA; Ross S; Conrads TP; Veenstra TD; Hitt BA; Kawaguchi Y; Johann D; Liotta LA; Crawford HC; Putt ME; Jacks T; Wright CV; Hruban RH; Lowy AM; Tuveson DA, Cancer Cell 2003, 4 (6), 437–50. DOI 10.1016/s1535-6108(03)00309-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.