

Abstract
Iron has been emerging as a key contributor to aging-associated, chronic disorders due to the propensity for generating reactive oxygen species. To date, there are a limited number of publications exploring the role of iron in the pathogenesis of primary/age-related osteoarthritis (OA). The objective of this study was to determine whether reduced iron via pharmacologic iron chelation with deferoxamine (DFO) affected the development and/or severity of cartilage lesions in a primary OA model. At 12-weeks-of-age, 15 male Dunkin-Hartley guinea pigs received either 46 mg/kg DFO (n=8) or vehicle control (n=7) injected subcutaneously twice daily for five days each week. Movement changes, captured via overhead enclosure monitoring, were also determined. Termination occurred at 30-weeks-of-age. Iron was quantified in serum, urine, liver, and femoral head articular cartilage. Left knees were evaluated for structural changes using histopathology guidelines and immunohistochemistry (IHC). Gene expression analysis was conducted on right knee articular cartilage. DFO reduced iron levels in femoral head articular cartilage (p=0.0006) and liver (p=0.02), and increased iron within urine (p=0.04) and serum (p=0.0009). Mobility of control animals declined, while the DFO group maintained activity levels similar to the first month of treatment (p=0.05). OA-associated cartilage lesions were reduced in knees of DFO animals (p=0.0001), with chondrocyte hypocellularity a key histologic difference between groups (p<0.0001). DFO-receiving animals had increased immunostaining for phosphorylated adenosine monophosphate activated protein kinase alpha within knee articular cartilage; lower transcript counts of several proapoptotic genes (p=0.04–0.0004) and matrix-degrading enzymes (p=0.02-<0.0001), and increased expression of the anti-apoptotic gene Bcl-2 (p<0.0001) and a tissue inhibitor of matrix-metalloproteinases (p=0.03) were also observed. These results suggest that iron chelation delayed the progression of primary OA in an animal model and could hold potential as a translational intervention. These findings provide expanded insight into factors that may contribute to the pathogenesis of primary OA.
Keywords: Osteoarthritis, Iron, Aging, Knee, Articular Cartilage
Graphical Abstract
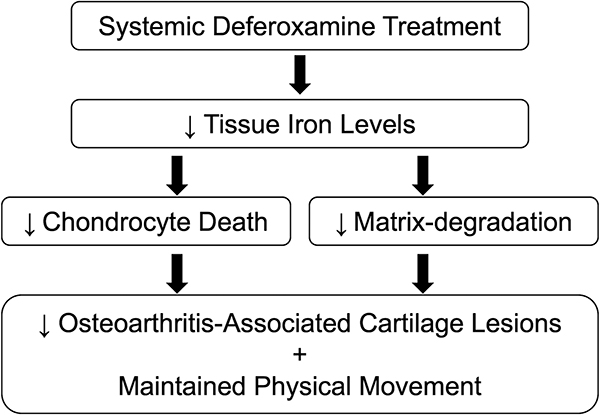
Introduction.
Iron is essential for numerous physiologic processes including oxygen transport, DNA synthesis, and ATP production. Mammals have developed complex systems to appropriately absorb, store, transport, and utilize this mineral, as unbound or partially-liganded iron can participate in redox chemistry [1–2]. These reactions generate harmful reactive oxygen species (ROS) that can elicit or perpetuate inflammatory responses, damage DNA and cellular components, and promote cell death, which collectively encourage tissue damage [3–5]. Thus, the risk for iron-mediated toxicity is not limited to overt excess and can also arise from conditions where intracellular iron becomes delocalized and/or inappropriately stored, causing the labile iron pool containing catalytically-active iron to expand [6–8]. Despite this, mammals do not have a physiologically regulated mechanism for excreting iron and the majority of the iron absorbed is continuously recycled throughout the body [9]. Indeed, iron progressively accumulates throughout the aging process [10–11] and has been linked to numerous aging-associated chronic diseases, including atherosclerosis [12], neurodegenerative disorders [13], cancer [14], and type II diabetes [15]. Excess and/or improperly managed iron has also been implicated in several disorders with associated arthropathies, including hemophilic arthropathy [16], traumatic arthropathy, hereditary hemochromatosis [17], and rheumatoid arthritis (RA) [18]. In these afflictions, ironloaded human synoviocytes have been demonstrated to release proinflammatory mediators that stimulate the catabolic activity of chondrocytes and contribute to joint degradation [19].
Although the role of iron has been explored in the arthropathies above, it has yet to be widely investigated in the context of primary/idiopathic osteoarthritis (OA). OA is a progressive joint disorder with the degradation and subsequent loss of articular cartilage as a central feature [20]; this is accompanied by synovial hyperplasia, osteophyte formation and tissue remodeling, and narrowing of the joint space. These alterations cause pain and reduced mobility, resulting in increased susceptibility to developing comorbidities such as obesity, cardiovascular diseases, and depression [21]. Chondrocytes, the only cell type present in articular cartilage, secrete macromolecules that comprise the extracellular matrix (ECM) structure and collectively maintain the integrity of the tissue through a delicate balance of catabolic and anabolic events [22]. In OA, this homeostasis becomes disrupted and inappropriately shifts in favor of catabolic events that degrade the ECM and promote cell death, further contributing to disease progression. Additionally, as adult cartilage is unable to regenerate, the depletion of chondrocytes themselves due to ROS and other stressors has also been implicated in OA pathogenesis [20,23]. While the above findings are accepted, the underlying molecular mechanisms driving the development of OA remain loosely described.
There have been a handful of publications exploring the role of iron in primary OA. One human study reported that synovial fluid iron concentration was significantly higher in OA-affected patients relative to both healthy controls and individuals with RA [24]. Another study found that serum ferritin levels were positively correlated with the severity of cartilage damage in people affected by OA; this finding was independent of age, sex, and BMI [25]. Finally, enhanced iron deposition in the synovium has been reported in patients with OA [26]. These studies suggest that iron may be a contributing factor in knee OA and may provide mechanistic insight to the development of the disorder.
As advancing age is the leading risk factor for developing primary OA [27], we theorize that age-related iron accumulation in joint tissues may contribute to the development of the disorder. Recently, we determined that administering excess iron systemically exacerbated the development of OA-like lesions in the knee joints of disease-resistant Strain 13 guinea pigs [28]. To build on this finding, we designed a study to assess the effects of systemic iron reduction on the development of primary knee OA in an animal model of the disorder: the Dunkin-Hartley guinea pig. This well-described model develops bilateral, age-related knee OA with a predictable progression that closely mimics the pathology observed in humans [29–30]. As in humans, these spontaneous lesions tend to develop first within the medial compartment of the knee, with the tibia being more severely affected before the femur [30]. Histopathologic evidence of knee OA can be detected after 2 to 3 months-of-age, with severe, late-stage OA occurring between 15–18 months-of-age [30].
For this work, systemic iron reduction was achieved by administration of the pharmacologic iron chelator deferoxamine (DFO). DFO has a high affinity for binding iron relative to other transition metals and has been used to treat acute iron toxicity and iron overload conditions since the 1960s. Based on our previous findings [28], we hypothesized that systemic administration of DFO would decrease the development of OA-like lesions in the knees of Dunkin-Hartley guinea pigs.
Materials and Methods.
Animals.
All procedures were approved by the Institutional Animal Care and Use Committee and performed in accordance with the NIH Guide for the Care and Use of Laboratory Animals. Group size was determined from a pilot study with the primary outcome being the histologic assessment of OA. Using a within group error of 0.5 with a detectable difference between means of 1.0, power associated with an alpha of 0.5 (two sided) was calculated as 0.9 with a sample size of 6 animals per group. To ensure adequate power for all study outcomes, a total of 16, 8-week-old male Dunkin-Hartley guinea pigs were purchased from Charles River Laboratories (Wilmington, MA). Guinea pigs were individually housed in solid bottom cages with appropriate bedding and were monitored daily by a veterinarian. Iron chelation therapy was supplied to reduce, but not deplete, systemic iron levels and encourage iron mobilization out of storage in tissues. As such, animals were allowed unlimited access to standard guinea pig chow that was replete in iron; hay cubes and water were also provided ad libitum.
Deferoxamine (DFO) Injections.
Injections were initiated at 12-weeks-of-age. Eight guinea pigs were randomly assigned by cage card number to the DFO group, with the remainder in the control group. Using the lowest dose and dosing frequency utilized in humans with chronic iron overload [31], allometric scaling was conducted to determine the equivalent dose for use in the guinea pig [32]. Animals within the DFO group received 46 mg/kg of DFO (Fresenius Kabi, Lake Zurich, IL) injected subcutaneously twice daily for 5 consecutive days, followed by 2 days without receiving any treatment. Control animals received an equivalent dose of sodium lactate solution (Pfizer, Lake Forest, IL) subcutaneously at the same frequency. Injections were given for 18 weeks. Body weights were recorded prior to starting treatments, as well as weekly throughout the study. One animal in the control group was lost prior to study termination due to the presence of underlying pathologies unrelated to the study at hand. This animal was excluded from all analyses; n=7 animals were evaluated for the control group.
Specimen Collection.
The study was terminated when animals were 30-weeks-of-age, with the final treatment occurring the night prior. Animals were placed under isoflurane anesthesia to collect whole blood via direct cardiac puncture. Urine was saved following involuntary voiding during the initial anesthetic transition. After fluids were collected, animals were transferred to a CO2 chamber for euthanasia. Serum was separated and, along with urine, submitted for iron quantification using the Roche Cobas 6000 (Basel, Switzerland). Complete blood counts (CBCs) were performed using the Advia 120 hematology analyzer (Siemans, Munich, Germany) with instrument settings and software specifically designed for guinea pig samples; serum biochemistry profiles were also determined (Roche Cobas 6000; Basel, Switzerland). The liver from each animal was collected into 10% (v/v) neutral buffered formalin (NBF) for 48 hours for iron quantification.
Hind limbs were removed at the coxofemoral joints. The left limb was placed into 10% NBF for 48 hours and subsequently transferred to a solution of 12.5% (w/v) ethylenediaminetetracetic acid (pH 7.00) for decalcification and histologic evaluation. The right hind limb was dissected to expose the knee joint and cartilage was collected from the articular surface of the patella and the weight-bearing regions of the femoral condyles and tibial plateaus. Cartilage was stored in RNAlater (Qiagen, Hilden, Germany) for gene expression analysis. The length of the right tibia was measured with calipers. Articular cartilage was isolated from the right femoral head and stored in 10% NBF for 48 hours for iron quantification.
Iron quantification by atomic absorption spectroscopy (AAS).
Iron quantification was performed on samples of NBF-fixed liver tissue and femoral head articular cartilage, as previously described [28]. The coxofemoral joint was utilized as knee articular cartilage was reserved for gene expression analysis. Briefly, dried tissue was weighed, ashed, sonicated in nitric acid, and diluted 30-fold with deionized water [33]. Diluted samples were analyzed using a Model 240 AA flame atomic absorption spectrometer and SpectrAA software (Agilent Technologies, Santa Clara, CA) [34]. Iron levels were reported as parts per million (ppm) dry weight [35].
Histologic evaluation of knee joints.
Following decalcification, knee joints were divided into sagittal sections of the medial and lateral joint compartments and embedded in paraffin wax. Five-micron segments were stained with toluidine blue, and medial and lateral femoral condyles and tibias were scored in a blinded fashion by two assessors (LHB and KSS) using the Osteoarthritis Research Society International (OARSI) guidelines [30]. One slide from the medial and lateral compartment of each knee joint was evaluated, for a total of 2 slides per animal. Values from the 4 anatomic locations were summed to obtain a whole joint knee OA score.
Gene expression of articular cartilage using NanoString technology.
As previously described [28], total RNA was isolated from knee articular cartilage using the RNeasy Lipid Tissue Mini Kit (Qiagen) and was sent to the University of Arizona Genetics Core (University of Arizona, Tucson, AZ) for analysis. A custom set of guinea pig-specific probes were designed and manufactured by NanoString Technologies (Seattle, WA) for the following genes: B-cell lymphoma 2 (BCL-2), BCL-2-associated death promoter (BAD), BCL-2-associated x protein (BAX), BCL-2 homologous antagonist killer (BAK), caspase-3, caspase-8, caspase-9, aggrecan (ACAN), type II collagen (COL2A1), matrixmetalloproteinase-2 (MMP-2), MMP-9, MMP-13, tissue inhibitor of matrixmetalloproteinases-2 (TIMP-2), transferrin receptor 1 (TFR1), divalent metal transporter 1 (SLC11A2/DMT1), ZRT/IRT-like protein 14 (SLC39A14/ZIP14), ferroportin (SLC40A1/FPN), ferritin heavy chain (FTH), and hepcidin (HAMP). Target sequences are presented in Supplemental Table S1. Based on initial RNA quantification (Invitrogen Qubit 2.0 Fluorometer and RNA High Sensitivity Assay Kit, Thermo Fisher Scientific, Waltham, MA) and fragment analysis quality control subsets (Fragment Analyzer Automated CE System and High Sensitivity RNA Assay Kit, Agilent Technologies), the optimal amount of total RNA (150–400 ng) was hybridized with the custom codeset in an overnight incubation at 65°C, followed by processing on the NanoString nCounter® FLEX Analysis System (NanoString Technologies). Results are reported as absolute transcript counts normalized to 2 housekeeping genes, β-actin (ACTB) and eukaryotic elongation factor 1α1 (EEF1A1). Any potential sample input variance was normalized by use of housekeeping genes and application of a sample-specific correction factor to all target probes. Data analysis was conducted using nSolver™ software (NanoString Technologies).
Immunohistochemistry (IHC) of articular cartilage for 4-hydroxynonenal and phosphorylated adenosine monophosphate activated protein kinase alpha.
Medial knee joint compartments embedded in paraffin wax were sectioned in 5-micron segments and mounted onto slides for immunostaining against 4-hydroxynonenal (4-HNE) adducts and phosphorylated adenosine monophosphate activated protein kinase alpha (p-AMPKα). Briefly, slides were deparaffinized with xylene, rehydrated with ethanol, and antigen retrieval was performed with sodium citrate buffer (pH = 6.00) heated to 55°C in a desert chamber. Slides were blocked with 5% goat serum (SeraCare Life Sciences Inc., Milford, MA) and then incubated with either 2.7 μg/mL of rabbit anti-4-hydroxynonenal (4-HNE; Abcam, Cambridge, MA) or 2.0 μg/mL of rabbit anti-AMPKα1 (phospho T183)+AMPKα2 (phospho T172) antibody (p-AMPKα; Abcam, Cambridge, MA) overnight at 4°C; isotype negative control slides received the equivalent concentration of rabbit IgG antibody (Invitrogen). Slides were then stained with Alexa Fluor 647 (Invitrogen) for 60 minutes at room temperature. Coverslips were added using mounting medium containing 4′,6-diamidino-2-phenylindole (DAPI; Vectashield, Vector Laboratories, Burlingame, CA) to visualize nuclei. Stained tissue was immediately imaged using a confocal microscope (model DMI4000B, Leica Microsystems, Wetzlar, Germany) with the same microscope settings applied across all samples to ensure comparable relative fluorescence intensities. Images were acquired using Leica Application Suite X 3.7.1.21655 (Leica Microsystems).
Analysis of confocal microscope images was conducted using Fiji [36], an image processing package of ImageJ [37]. Regions of interest (ROI) were determined from phase contrast images to exclude calcified cartilage, and 3 consistent areas of articular cartilage on the medial tibial plateau (MTP) were evaluated for each animal. Chondrocytes located within the articular cartilage ROI were identified by DAPI staining above a set threshold minimum across all samples. Chondrocytes were counted with a new overlay created for each cell detected. Semiquantitative analysis of 4-HNE and p-AMPKα expression was determined by the presence of Cy5 staining above threshold limit within the previously created cellular overlays. The percent positive cell count was determined as a ratio of the number of cells with positive immunostaining to the total number of cells present in the articular cartilage ROI.
Overhead enclosure monitoring.
Animal movement was monitored using ANY-maze behavioral tracking software (Stoelting Co., Wood Dale, IL). To conduct overhead enclosure monitoring, guinea pigs were placed into an open top apparatus containing a habitat hut, with a camera positioned above the enclosure. The activity of animals was recorded during 10-minute sessions occurring once per month throughout the study. Baseline parameters were collected at the one-month time point to allow guinea pigs to acclimate to the system. Results are presented as the difference in activity levels from the first month to the final month of treatment.
Statistical Analyses.
Statistical analyses were performed with GraphPad Prism 8.4.2 (La Jolla, CA). Rationale for the exclusion of an entire animal from the study were determined a priori and included the presence of any pathologies and/or the inability to complete the study for any reason. Prior to conducting analyses, the authors determined that individual points would be excluded from data sets if: a sample did not pass quality control parameters set for an experimental method, the integrity of a sample was compromised, or an appropriate sample was unable to be obtained for analysis. Urine and cartilage iron quantification are incomplete data sets due to the inability to acquire adequate urine at euthanasia and absence of available tissue, respectively. For chondrocyte quantification and 4-HNE IHC, one control animal was unable to have one region of the MTP evaluated due to loss of sample integrity on the slides within the area of interest. As such, one control animal had 2 MTP regions evaluated for chondrocyte quantification with DAPI and 4-HNE staining. Similarly, appropriate tissue sections were unavailable for one control animal in conducting subsequent p-AMPKα IHC, resulting in n=6 control animals evaluated for this protein. Authors were not blinded to group allocation during data analysis. The distribution and variance of data sets were determined with the Shapiro-Wilk and F-Test, respectively. Normally distributed data with similar variance were compared using parametric t-tests◆. Normally distributed data with significant differences in variance were compared using parametric t-tests with Welch’s correction◇. Data with non-Gaussian distribution was compared using non-parametric Mann-Whitney U-test×. Statistical tests are noted in figure legends using designated superscripts. For data analyzed by parametric t-tests, black lines on graphs represent mean values. Black lines on graphs represent median values for data analyzed by Mann-Whitney U-tests. Statistical significance was set at P ≤ 0.05.
Results.
General Description of Animals.
Animals receiving iron chelation therapy appeared clinically healthy; no changes in cage behavior were observed at any point during the study. At the time of termination, mean total body weight was 1116.00 g in control animals and 1084.00 g in DFO-treated animals (95% confidence interval [CI]: −155.10–90.04 g; p=0.58; data not shown). Body weights of the animals throughout the study are provided in Supplemental Figure S2. Mean tibia length was similar between animals in the control group (49.06 millimeters, mm) and in animals receiving DFO (49.59 mm), indicating that iron chelation therapy did not alter the skeletal growth of these animals (95% CI: −1.28–2.33 mm; p=0.53; data not shown). CBC profiles showed minimal evidence of clinically-relevant iron deficiency or anemia and suggested that that DFO-treated animals had sufficient iron to maintain physiologic processes (Supplemental Figure S3).
Iron Quantification.
Administration of DFO allowed for iron to be chelated and eliminated via the urine. Relative to the control group, animals receiving iron chelation therapy had increased urinary iron levels (p=0.04; Figure 1A). Consistent with increased iron mobilization, DFO-treated animals had significantly higher serum iron concentration than control animals (p=0.0009; Figure 1B). Overall, iron elimination was also reflected in tissue iron quantification. Iron AAS revealed that animals receiving DFO had significantly lower iron concentration in both the liver (p=0.02; Figure 1C) and femoral head articular cartilage (p=0.0006; Figure 1D).
Figure 1. Tissue iron quantification.
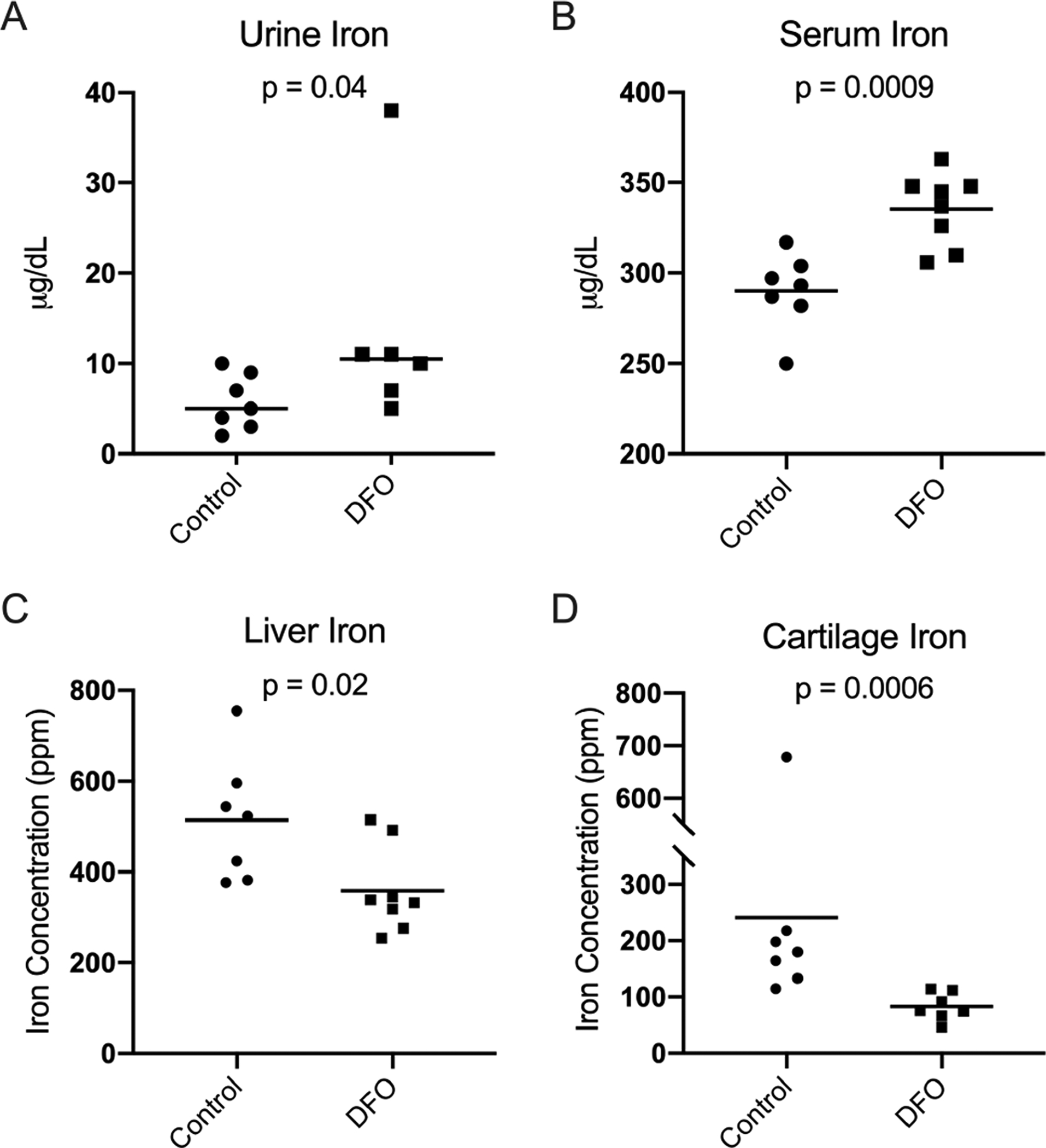
[A] Median urine iron× concentration was 5.00 micrograms/deciliter (μg/dL) in the control group and 10.50 μg/dL in the DFO group. [B] Mean serum iron concentration◆ was 290.00 μg/dL in the control group and 335.40 μg/dL in the DFO group. [C] Mean liver iron concentration◆ was 514.40 ppm in the control group and 358.90 in the DFO group. [D] Median femoral head cartilage iron concentration× was 180.00 ppm in the control group and 75.80 ppm in the DFO group.
Semiquantitative Histologic Scoring of Knee Joints Using OARSI Guidelines.
Histologic evaluation of knee joints [30] demonstrated significantly lower total joint OA scores, and therefore decreased OA development, in DFO-treated animals (p=0.0001; Figure 2A). Reduced OA in DFO animals was maintained when medial and lateral compartments were evaluated separately (p=0.0002 and p=0.0012, respectively; Figure 3A–3B). Representative photomicrographs depicting articular cartilage in the medial compartment are provided in Figures 2B–C. The representative image from a control animal showed a disrupted tibial surface with fissures and proteoglycan loss in the superficial zone and chondrocyte hypocellularity distributed throughout ECM (Figure 2B). Conversely, the DFO-treated animal had cartilage surface with mild irregularities, uniform proteoglycan content, and slight chondrocyte hypercellularity (Figure 2C). Figure 3 provides the four main parameters that contributed to the whole joint OA score. Relative to the control group, animals receiving DFO had significantly lower values for articular cartilage structure (p=0.0007), proteoglycan content (p=0.003), and cellularity (p<0.0001) (Figure 3C–E). There was no notable difference in the tidemark integrity between groups (>0.9999; Figure 3F). Histologic evidence of osteophytes was not documented in any animals evaluated.
Figure 2. Histologic evaluation of knee joints.
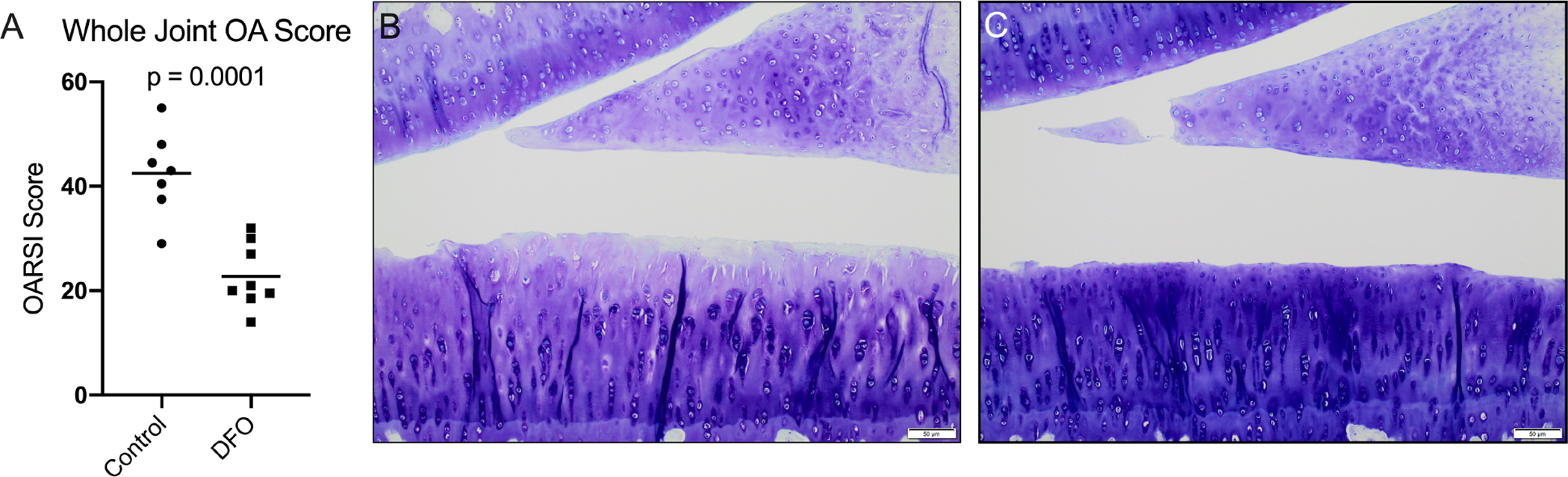
[A] Mean whole joint OA score◆ was 42.50 in the control group and 22.75 in the DFO group. [B] 10X representative histologic image of a control animal knee joint demonstrating a disrupted tibial surface with fissures, proteoglycan loss in the superficial zone, and chondrocyte hypocellularity throughout the ECM. [C] 10X representative histologic image of a knee joint from an animal treated with DFO demonstrating mild surface irregularities, uniform proteoglycan content, and slight chondrocyte hypercellularity.
Figure 3. Contributions to histologic whole joint OA score.
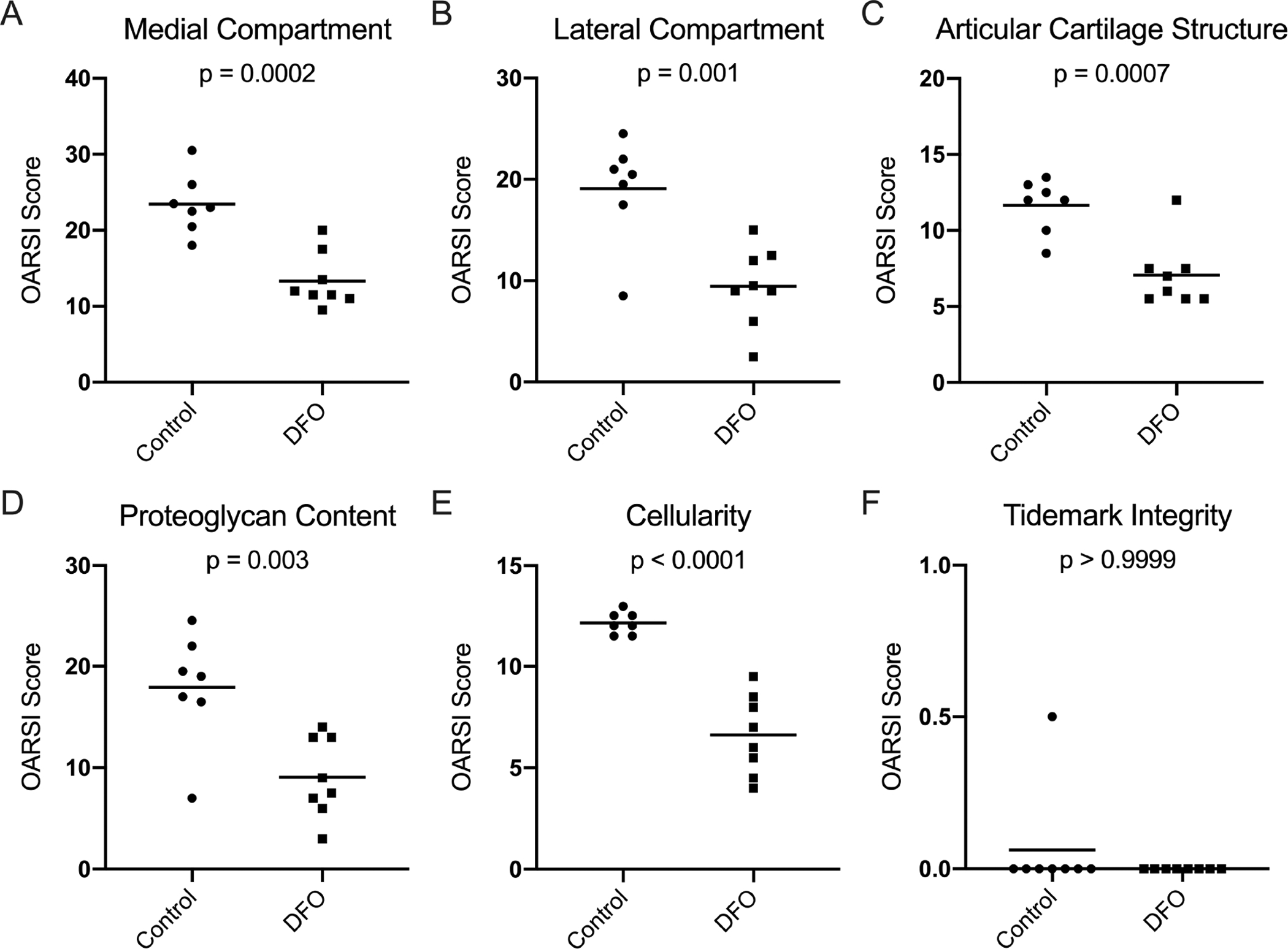
[A] Mean OARSI score in the medial compartment◆ was 23.43 in the control group and 13.31 in the DFO group. [B] Mean OARSI score in the lateral compartment◆ was 19.07 in the control group and 9.44 in the DFO group. [C] Mean whole joint score for articular cartilage structure◆ was 11.64 in the control group and 7.06 in the DFO group. [D] Mean whole joint score for proteoglycan content◆ was 17.93 in the control group and 9.06 in the DFO group. [E] Mean whole joint score for chondrocyte cellularity◇ was 12.14 in the control group and 6.63 in the DFO group. [F] Median whole joint score for tidemark integrity× was 0.00 in both the control and DFO group.
Immunohistochemistry of 4-hydroxynonenal.
4-HNE is a major lipid peroxidation product generated by the interaction of hydroxyl radicals and other ROS with polyunsaturated fatty acids abundant in cell membranes [38]. Somewhat unexpectedly, the percentage of cells positive for 4-HNE was higher in the DFO group than the control group (p=0.001; Figure 4G). Representative images are provided in Figures 4A–F. Consistent with what was observed in the histologic evaluation of knee joints, animals within the control group exhibited lower chondrocyte density in the MTP than animals treated with DFO (p=0.02; Figure 4G). Overall, control articular cartilage displayed minimal to no isogenic cell groups, and the remaining chondrocytes were often individualized with shrunken and/or irregular boarders (Figures 2B and 4A–C). Conversely, DFO-treated animals had chondrocytes distributed throughout the extracellular matrix with chondrones present in the mid-to-deep zones of articular cartilage (Figures 2C and 4D–F).
Figure 4. 4-HNE immunohistochemistry of MTP articular cartilage.
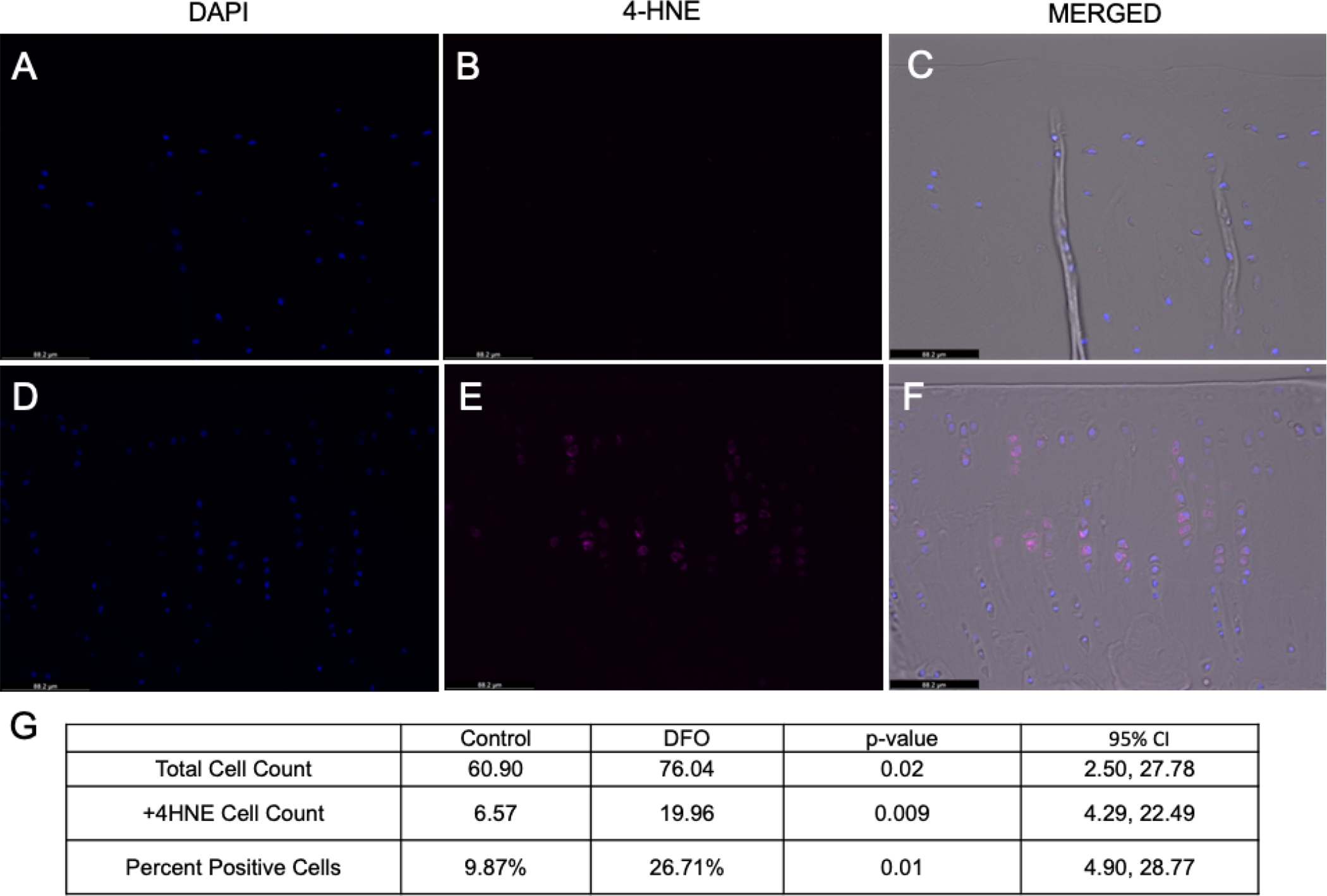
[A-C] 20X Representative images of MTP articular cartilage from a control animal. [A] Chondrocyte nuclei stained with DAPI. [B] Positive 4-HNE staining (Cy5 channel). [C] Merged image with phase contrast, DAPI, and 4-HNE (Cy5) channels. [D-F] 20X Representative images of MTP articular cartilage from a DFO-treated animal. [D] Chondrocyte nuclei stained with DAPI. [E] Positive 4-HNE staining (Cy5 channel). [F] Merged image with phase contrast, DAPI, and 4-HNE (Cy5) channels. [G] Mean values for total cell count, 4-HNE positive cell count, and the percentage of total cells positive for 4-HNE adducts.
Immunohistochemistry of phosphorylated AMPKα.
AMPK expression has been identified as playing a pivotal role in maintaining chondrocyte homeostasis and viability [39]. Comprised of 3 subunits (α, β, and γ), phosphorylation of the alpha subunit is critical for AMPK activity [40]. As such, the activity of AMPK can be assessed by measuring the phosphorylation of AMPKα (p-AMPKα) [41]. The percentage of chondrocytes with p-AMPKα immunostaining was higher in the articular cartilage of DFO-treated animals than controls, indicating greater p-AMPKα activation within the iron chelation group (p=0.001; Figure 5A–G). Notably, Pearson’s correlation revealed that the percentage of p-AMPKα positive cells was inversely correlated with the histologic whole joint OA score (r=−0.79, p=0.0008; Figure 5H), suggesting that AMPK may influence articular cartilage lesions. The percentage of cells positive for p-AMPKα also significantly correlated with the individual histologic scores for articular cartilage structure (r=−0.63, p=0.02), proteoglycan content (r=−0.74, p=0.0025), and chondrocyte cellularity (r=−0.83, p=0.0002), which contribute to the whole joint OA score (Supplemental Figure 5).
Figure 5. p-AMPKα immunohistochemistry of MTP articular cartilage.
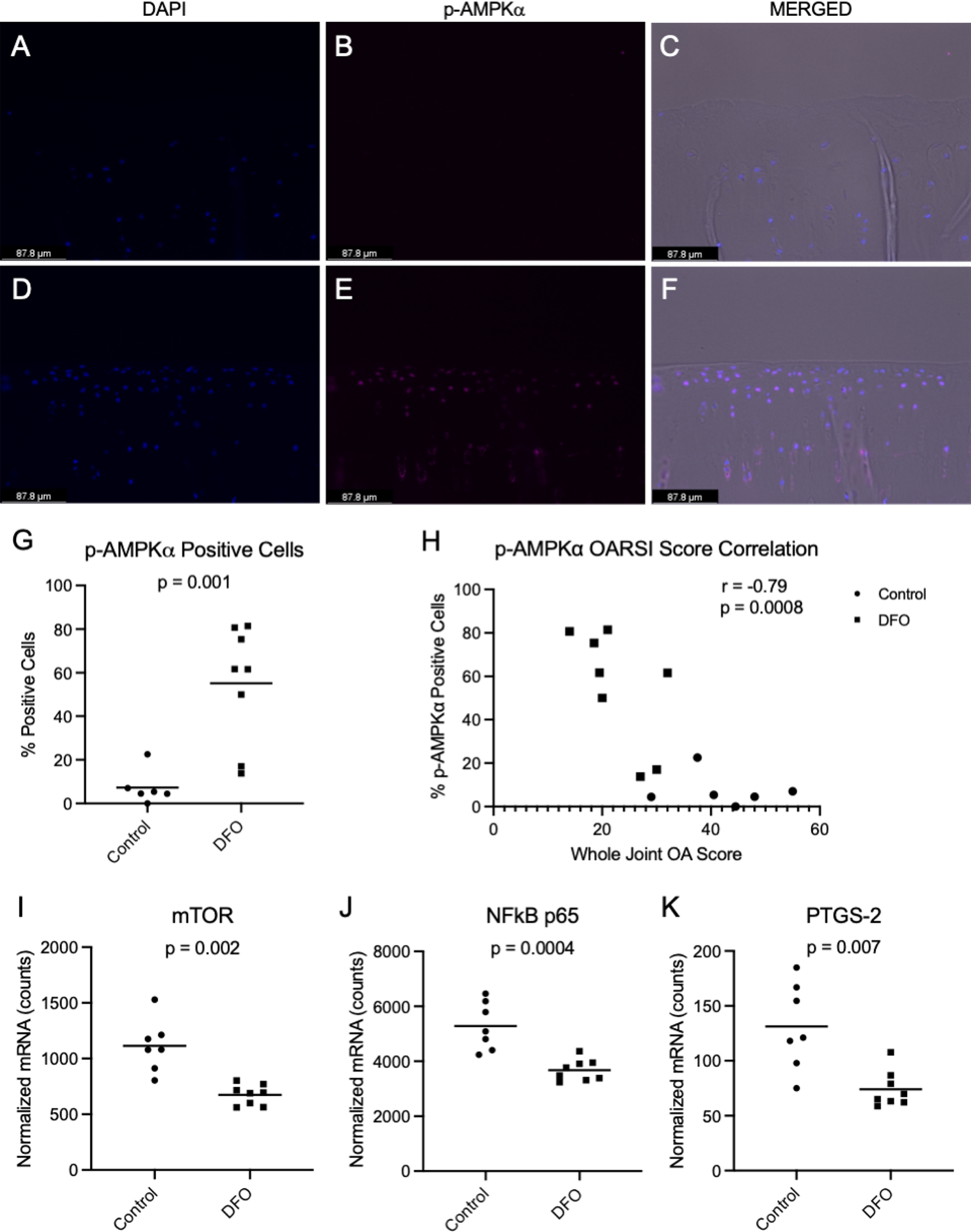
[A-C] 20X Representative images of MTP articular cartilage from a control animal. [A] Chondrocyte nuclei stained with DAPI. [B] Positive p-AMPKα staining (Cy5 channel). [C] Merged image with phase contrast, DAPI, and p-AMPKα (Cy5) channels. [D-F] 20X Representative images of MTP articular cartilage from a DFO-treated animal. [D] Chondrocyte nuclei stained with DAPI. [E] Positive p-AMPKα staining (Cy5 channel). [F] Merged image with phase contrast, DAPI, and p-AMPKα (Cy5) channels. [G] Mean percentage of p-AMPKα positive cells◇ was 7.33% in the control group and 55.22% in the DFO group. [H] Correlation of the percentage of p-AMPKα positive cells with whole joint OA score. [I] Mean transcript counts for mTOR◇ were 1113.00 in the control group and 674.80 in the DFO group. [J] Mean transcript counts for NF-κB p65◆ were 5282.00 in the control group and 3676.00 in the DFO group. [K] Mean transcript counts for PTGS-2◇ were 131.20 in the control group and 74.08 in the DFO group.
Consistent with increased p-AMPKα activity, the gene transcript counts for select downstream catabolic mediators inhibited by AMPK expression were decreased. Specifically, treatment with DFO decreased the transcript counts of mammalian target of rapamycin (mTOR; p=0.002, Figure 5I), nuclear factor kappa B p65 (NF-κB p65; p=0.0004, Figure 5J), and the gene coding for cyclooxygenase-2: prostaglandin-endoperoxide synthase 2 (PTGS-2; p=0.007, Figure 5K). Additional transcript differences that may also be explained by p-AMPKα activity are presented in the sections below.
Gene Expression Analysis of Knee Articular Cartilage.
Cell Death-Related Genes.
Several genes associated with cell death were differentially expressed in animals treated with DFO. Relative to control animals, gene transcript counts for the proapoptotic genes BAD (p=0.0004), BAX (p=0.008), and BAK (p=0.04) were significantly reduced in DFO-treated animals (Figure 6A–C). Additionally, expression of the antiapoptotic gene BCL-2 was significantly higher in the DFO group (p<0.0001; Figure 6D). Animals treated with DFO displayed significantly decreased mRNA expression of the apoptosis initiator caspase-9 (p=0.002) and effector caspase-3 (p=0.007) (Figure 6E–F). Of note, there was not a substantial difference in caspase-8 transcripts between groups (95% CI: −8.13–16.88 normalized mRNA counts; p=0.4; data not shown).
Figure 6. Normalized mRNA counts for cell death-related genes in knee articular cartilage.
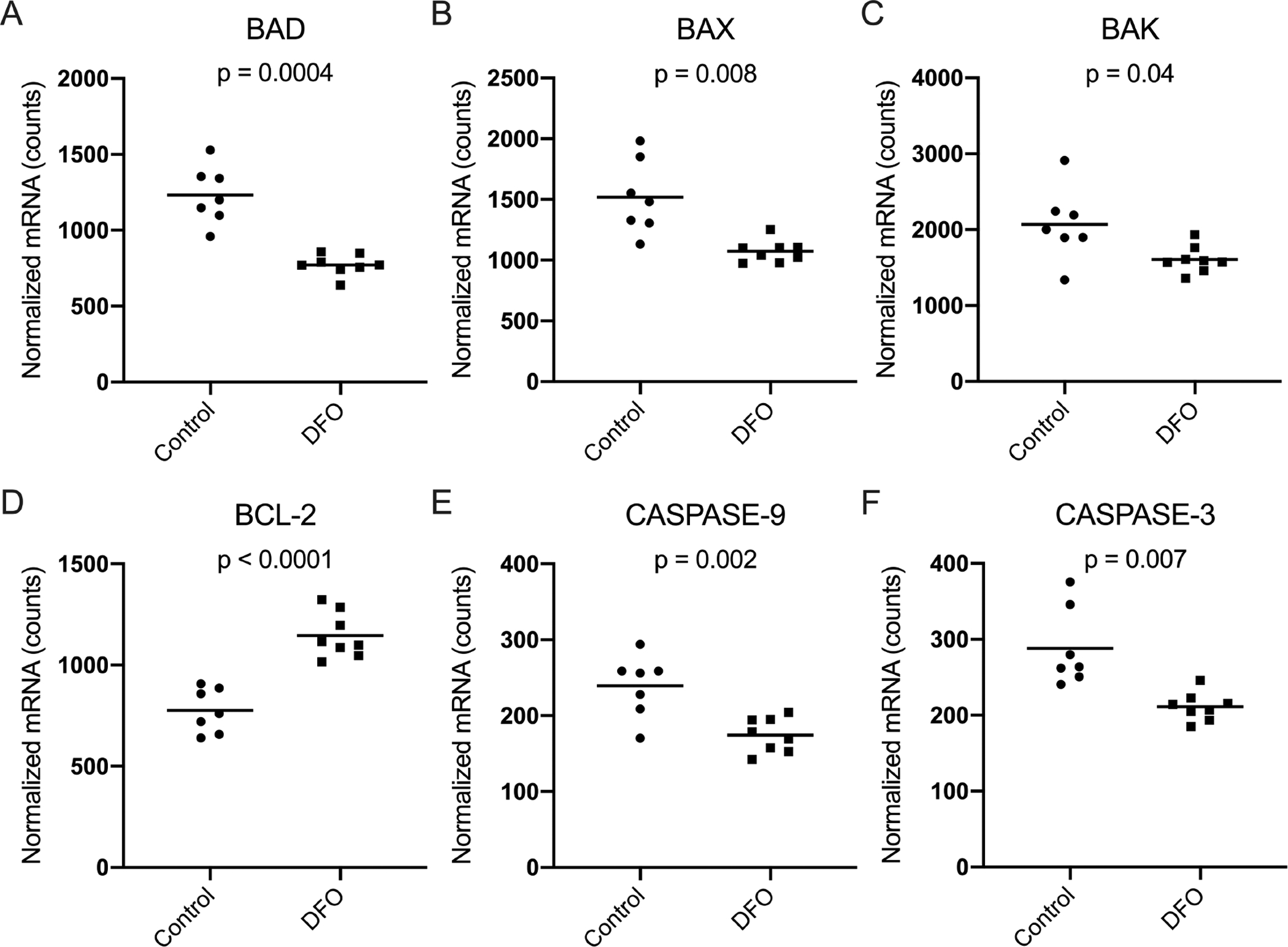
[A] Mean transcript counts of BAD◇ were 1233.00 in the control group and 771.80 in the DFO group. [B] Mean transcript counts of BAX◇ were 1519.00 in the control group and 1073.00 in the DFO group. [C] Mean transcript counts of BAK◇ were 2068.00 in the control group and 1608.00 in the DFO group. [D] Mean transcript counts of BCL-2◆ were 775.90 in the control group and 1146.00 in the DFO group. [E] Mean transcript counts of caspase-9◆ were 239.20 in the control group and 174.20 in the DFO group. [F] Mean transcript counts of caspase-3◇ were 288.40 in the control group and 211.10 in the DFO group.
Genes Related to Articular Cartilage Structure.
Type II collagen and aggrecan are two of the most abundant molecules within the cartilage ECM [22] and contribute to cartilage structure and proteoglycan content, respectively. Compared to the control group, animals treated with DFO had significantly lower transcript counts of COL2A1 (p=0.0003) and ACAN (p=0.002) (Figure 7A–B). Likewise, administration of the iron chelator resulted in decreased expression of several genes for MMPs that degrade the ECM of articular cartilage, including MMP-2 (p<0.0001), MMP-9 (p=0.02), and MMP-13 (p=0.02) (Figure 7C–E). Finally, treatment with DFO resulted in a significant increase in gene expression for the MMP inhibitor, TIMP-2 (p=0.03; Figure 7F).
Figure 7. Normalized mRNA counts for genes related to the structure of knee articular cartilage.
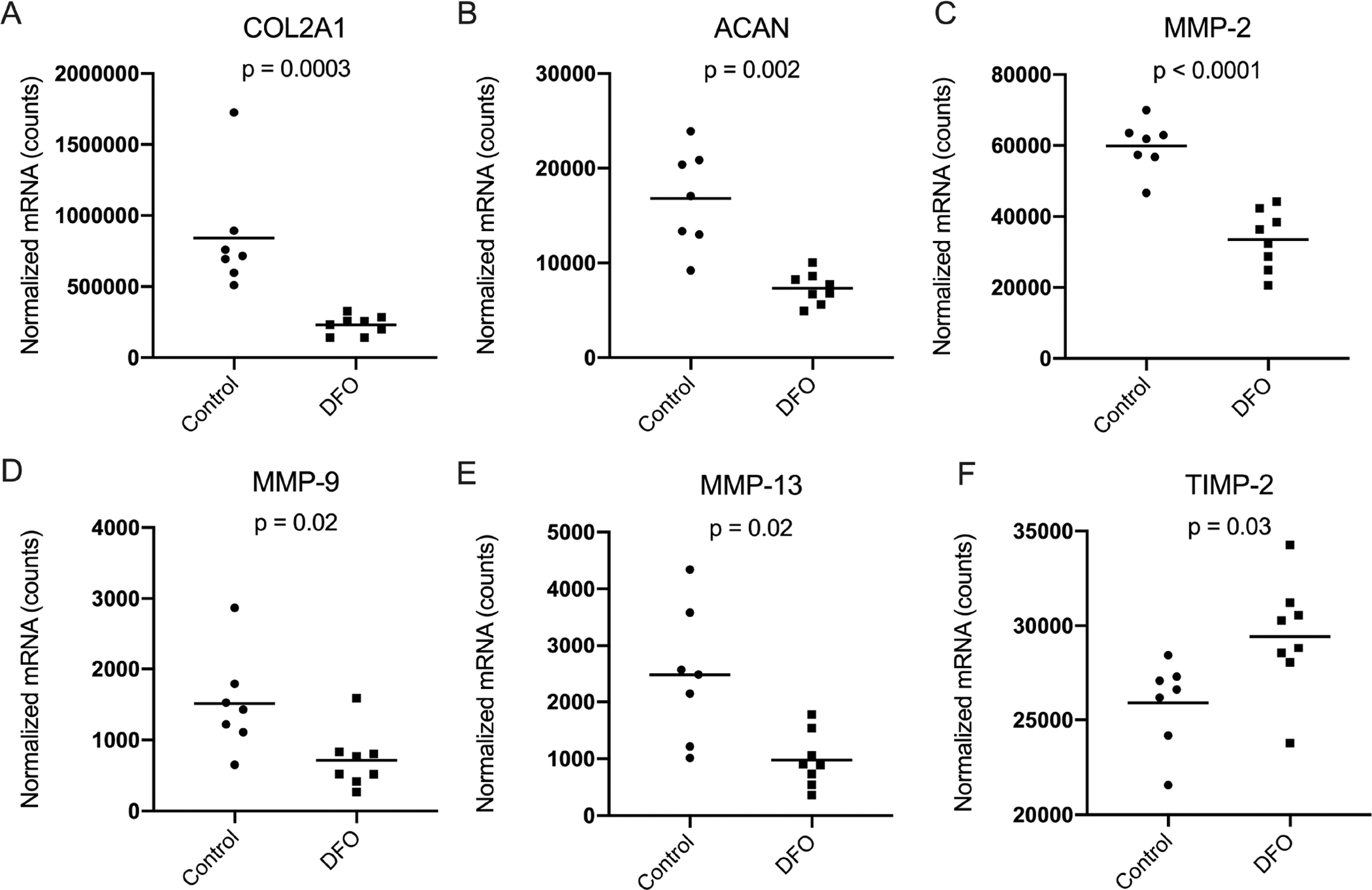
[A] Median transcript counts for type II collagen (COL2A1) × were 715415.00 in the control group and 156550.00 in the DFO group. [B] Mean transcript counts for aggrecan (ACAN) ◇ were 16828.00 in the control group and 7331 in the DFO group. [C] Mean transcript counts for MMP-2◆ were 59889.00 in the control group and 33501.00 in the DFO group. [D] Mean transcript counts for MMP-9◆ were 1514.00 in the control group and 715.20 in the DFO group. [E] Mean transcript counts for MMP-13◇ were 2478.00 in the control group and 976.50 in the DFO group. [F] Mean transcript counts for TIMP-2◆ were 25897.00 in the control group and 29433.00 in the DFO group.
Overhead Enclosure Monitoring.
Overhead enclosure monitoring revealed that, relative to the first month of the study, control animals moved a mean distance of 5.71 meters (m) less during the final monitored activity session, while the DFO group maintained their mean distance traveled (p=0.05; Figure 8A). Likewise, control animals moved an average of 9.7 millimeters/second (mm/s) slower by study termination, while DFO-treated animals sustained the mean speed of travel initially recorded (p=0.05; Figure 8B).
Figure 8. Movement parameters from overhead enclosure monitoring.
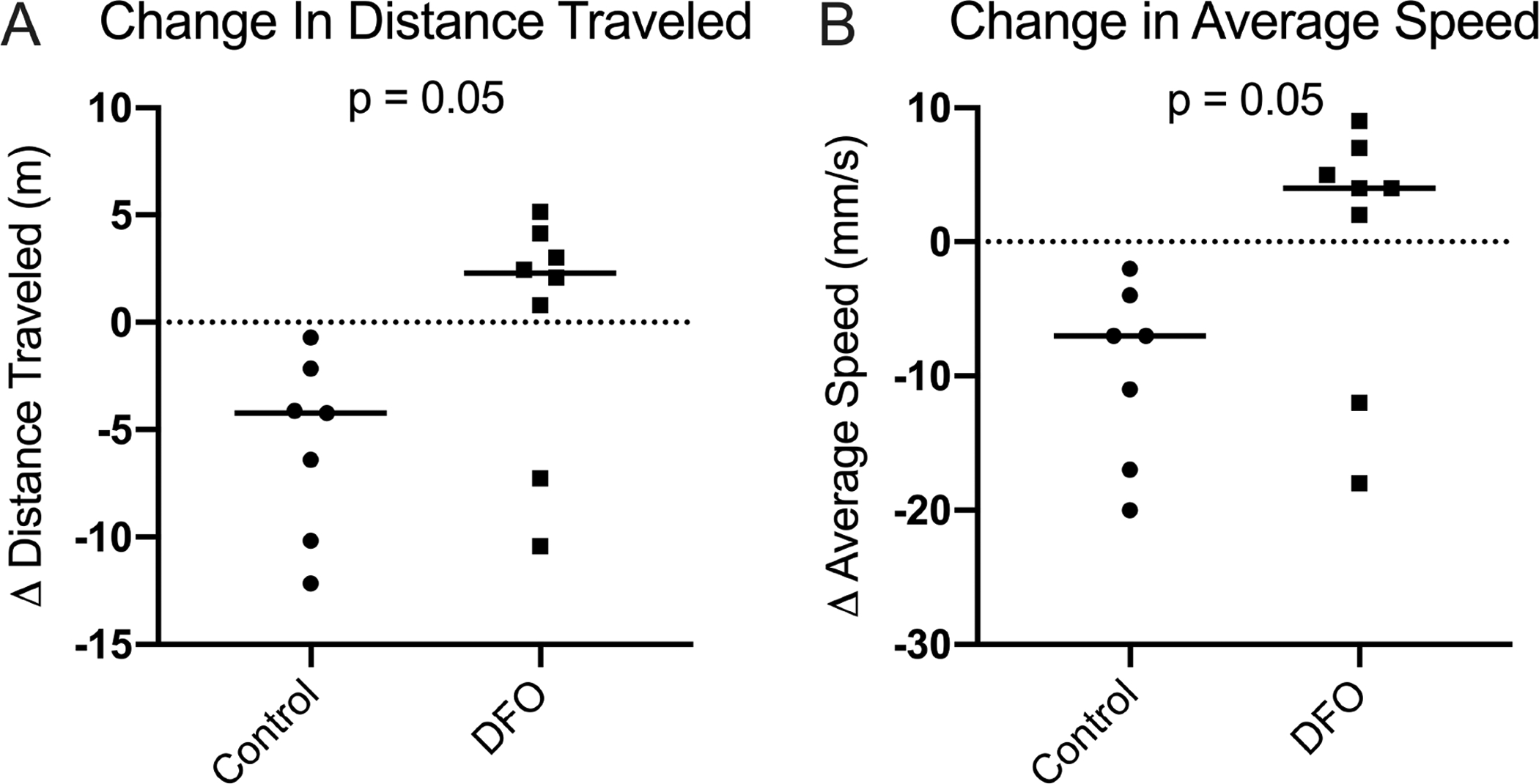
[A] Change in distance traveled×. During the final monitoring session, control animals were traveling a median distance of 4.22 m less than what was recorded in the first monitoring session. Conversely, DFO-treated animals traveled a median distance of 2.28 m more in the final monitoring session relative to the first activity session. [B] Change in the average speed of travel×. During the final monitoring session, control animals were moving a median speed× of 7.00 mm/s slower than values recorded in the first session. DFO treated animals moved at a median speed of 4.00 mm/s faster than what was recorded in the first activity session.
Discussion
In the present work, we demonstrated that systemic iron reduction decreased the development of OA-associated cartilage lesions in the knee joints of male Hartley guinea pigs. To our knowledge, this is the first published study reporting the effects of extraarticular pharmacologic iron chelation on primary knee OA in a spontaneous/idiopathic animal model. The results indicate that lowering iron levels by administration of DFO is beneficial to knee joint articular cartilage and may suggest that systemic and/or local iron concentrations be considered as a factor in primary OA development.
Treatment with DFO was successful at decreasing tissue iron content both systemically (measured within the liver) and in a diarthrodial joint environment (femoral head articular cartilage). The enhanced urine iron concentration with DFO treatment reflects the major mechanism of iron elimination by this compound [42] and thus provides an explanation for reduced tissue iron within these animals. Although iron levels were reduced in the DFO group, animals were still regularly receiving iron through the standard, iron-replete diet, and CBC data indicated these animals contained sufficient iron for normal physiologic processes. An interesting finding from this study was the increased serum iron concentration in chelated animals, despite an overall decrease in iron within the organs investigated. A potential explanation for increased serum iron with treatment is that DFO encouraged iron to be mobilized out of tissues into systemic rotation; this hypothesis is supported by the altered expression of several iron transport genes within knee articular cartilage. Correspondingly, transcript counts for the iron import genes TFR1, DMT1, and ZIP14 were decreased, suggesting reduced iron uptake by chondrocytes (Supplemental Figure S4). Additionally, higher expression of the iron export gene FPN was observed in this same tissue, further supporting increased iron removal from these cells (Supplemental Figure S4). Additional iron-related genes are presented in Supplemental Figure S4.
Relative to control animals, iron chelation therapy reduced the development and/or severity of OA-associated cartilage lesions within the knee joint. Analysis of the individual OARSI score components revealed that variations in chondrocyte cellularity was the largest contributor to the histologic differences present between groups. Within the medial compartment, all control animals exhibited tibial hypocellularity; this was only observed in 2 of the 8 DFO animals evaluated. Quantitative analysis of chondrocytes stained with DAPI confirmed that animals treated with DFO had significantly more chondrocytes in the MTP than controls. The beneficial retention of chondrocyte density within the DFO group was supported by alterations in gene expression of knee articular cartilage. In particular, iron chelated animals displayed decreased expression of several genes associated with promoting the intrinsic pathway of apoptosis, while simultaneously showing a significant increase in the antiapoptotic gene BCL-2. Indeed, iron has been shown to induce chondrocyte apoptosis [44] resulting in accelerated degradation of the ECM [43]. It is interesting that caspase-8, which is required to progress apoptosis triggered by external stimuli, did not have significant changes in gene expression with iron chelation treatment. When considered with the reduced transcript expression of BAX, BAK, and caspase-3, the relative consistency in caspase-8 supports that the intrinsic pathway of apoptosis was modulated in chondrocytes from treated animals more than the extrinsic pathway. However, additional work is required to determine the mechanisms by which iron promotes chondrocyte apoptosis and the type(s) of cell death observed in OA.
Because iron can catalyze reactions leading to free radical formation, the authors were interested to see if there were differences in oxidant damage between groups. Interestingly, animals treated with DFO had increased staining for 4-HNE protein adducts relative to control animals. This finding was unexpected, as 4-HNE is a major product of iron-mediated oxidant damage [38]. As 4-HNE has been cited to induce apoptosis at higher concentrations [38,45], chondrocytes within control animals may have already proceeded through cell death (driven by 4-HNE and/or other contributing factors) and therefore may not be detectable at this particular, later time point. Investigations at a preceding stage of disease pathology may clarify the impact of iron chelation on 4-HNE formation in chondrocytes and should be pursued in the future.
To provide an explanation for the structural results observed in this study, immunostaining for p-AMPKα was conducted. In addition to being documented in OA, AMPK activity has been implicated in the general aging process and many of the aging-related disorders also affected by iron accumulation, including cardiovascular disease, type II diabetes, atherosclerosis, cancers, and neurodegenerative disorders [46–48]. Notably, immunostaining for p-AMPKα was increased in the articular cartilage of animals treated with DFO. As a master energy sensor, AMPK has a widespread role in maintaining cartilage homeostasis by regulating energy expenditure, influencing chondrocyte differentiation and hypertrophy, promoting autophagy, and mediating the production of inflammatory molecules that can contribute to ECM destruction and decreased cell viability [40,46]. While AMPK is constitutively expressed in normal cartilage, a reduction in AMPK activation has been documented in OA cartilage in humans and animal models of the disorder [40, 50–51]; this shift has been associated with the increased production of catabolic mediators (such as MMPs and proinflammatory mediators) [39,52], cellular senescence [47], and chondrocyte apoptosis [53]. In the present work, DFO may have enhanced AMPK activation by chelating the iron required for degrading the upstream hypoxia inducible factor alpha 1 (HIF-1α) protein [54]. As HIF-1α is responsible for phosphorylating AMPKα [55], the sustained expression of HIF-1α by DFO may be responsible for increasing p-AMPKα in OA-prone animals. Indeed, increased AMPK activity has been documented in skeletal muscle of iron deficient rats [56] as well as in chondrocytes treated with DFO in vitro [51]. Collectively, AMPK activity decreases the catabolic activity of chondrocytes, including those occurring in response to injury or proinflammatory mediators [46, 50], thereby helping to maintain chondrocyte viability and preserve the ECM. In support of this, transcript counts for type II collagen and aggrecan were significantly reduced in DFO-treated animals, suggesting that less matrix synthesis was required to replace degraded molecules. The increase in AMPK activation was substantiated by decreased transcript expression of select downstream targets inhibited by AMPK activity: the autophagy inhibitor mTOR, as well as the inflammation-related genes NF-κB p65 and PTGS-2.
Additionally, the preservation of knee articular cartilage may be attributed to the decreased gene expression of several matrix degrading proteinases, which are also influenced by AMPK expression. Augmented enzymatic digestion of proteoglycans and collagens is a major mechanism of ECM degradation in OA [57] and represents one of several catabolic events inappropriately upregulated in the disorder. Within the DFO group, there were significantly lower transcript counts of MMP-2, −9, and −13; this was accompanied by an increased expression of the MMP inhibitor TIMP-2. Indeed, enhanced expression of these MMPs has been documented in OA-affected human chondrocytes [57–58] and MMP production decreases with AMPK activation [46]. The altered expression of MMPs and TIMP-2 hint that DFO treatment may have helped restore the imbalanced expression of these enzymes occurring in OA pathogenesis.
While the activity of control animals declined by study termination, 8-month-old animals with reduced systemic iron exhibited mobility similar to that measured at 3-months-of-age. This finding is of interest as a main consequence of knee OA is reduced movement [59], leading to an increased risk of developing comorbidities [21] and ultimately impacting an individual’s quality of life. Conversely, physical movement has been demonstrated to reduce pain and increase bodily function by up to 40% in individuals already affected by arthritis [60], and therefore may help combat both the deleterious effects of OA itself and the development of other chronic disorders. Regardless of the rationale, maintaining physical activity throughout aging is undeniably beneficial, as it allows for the maintenance of strength that helps prevent functional decline and lowers susceptibility to morbidity and mortality [61]. At present, it is not clear whether systemic DFO had ramifications beyond preserving the knee joint cartilage that contributed to conserved animal motion. As such, there is the distinct possibility that the activity level observed with treatment may have also been influenced by the effects of DFO outside of the joint, cumulatively resulting in these animals performing better overall.
There are confines to this work worthy of discussion. First, although DFO has been primarily used for its iron chelating properties, there has been evidence that the parent compound can also act as a radical scavenger through interaction with hydroxyl radicals, superoxide radicals, and peroxy-radicals [62–63]. Given this, it is unclear whether the results observed in this study were due to iron removal by DFO alone and/or through another direct mechanism of the pharmacologic agent itself. Future work should focus on whether the effects observed in this study were from a reduction in ROS at earlier stages of disease development, through other processes influenced by reduced iron levels, or an effect of DFO itself. Next, the authors recognize that changes in gene expression do not necessarily correlate to protein abundance and/or activity; indeed, the limited availability of guinea pig antibody-based reagents remains an unfortunate hurdle. While we were unable to provide corresponding protein data for all transcripts discussed in the present study, it is noteworthy that the expression of the genes provided was consistent with what was observed during histologic analysis of knee joints. The authors aim to establish immunostaining protocols for select genes presented in this work (BAD, BAX, BCL-2, caspase-3, and iron-related genes), as well as related proteins for elucidating mechanisms of chondrocyte cell death (such as phosphorylated p53), as reagents become available. While the results from this study are promising, the pursuit of more localized and/or clinically feasible iron chelators would enhance the translatability of this research for the treatment of OA. Finally, to elucidate the role that sex may play in this work, the authors have recently finished a study to examine the relationship between systemic iron levels and OA development in females.
Supplementary Material
The mass of animals was similar between groups throughout the duration of the study.
[A] Mean cell volume (MCV) ◆ was an average of 84.43 femtoliters/cell in the control group and 81.88 femtoliters/cell in the DFO group. Reference interval range is 70.00–90.00 femtoliters/cell [64]. [B] Mean hematocrit (HCT) ◆ volume was 48.29% red blood cells (RBCs) in the control group and 46.75% RBCs in the DFO group. Reference interval range is 30.00–50.00% RBCs [64]. [C] Median values for mean corpuscular hemoglobin concentration (MCHC) × were 32.00 g/dL in the control group and 32.00 g/dL in the DFO group. Reference interval range is 25.00–40.00 [64]. [D] Mean platelet count◆ was 427.60 × 103/microliter in the control group and 593.00 × 103/microliter in the DFO group. Reference interval range 250.00 − 850.00 × 103/microliter [64].
[A] Mean transcript counts for TFR1◆ were 932.10 in the control group and 757.70 in the DFO group. [B] Mean transcript counts for DMT1◆ were 1089.00 in the control group and 858.90 in the DFO group. [C] Mean transcript counts for ZIP14◇ were 1703.00 in the control group and 901.60 in the DFO Group. [D] Mean transcript counts for FPN◆ were 1127.00 in the control group and 1313.00 in the DFO group. [E] Mean transcript counts for FTH◆ were 89350.00 in the control group and 82526.00 in the DFO group. FTH is a subunit of the major iron storage protein ferritin. In addition to storing iron, FTH exhibits ferroxidase activity and is responsible for the exchange of iron between stable storage and the redox-active iron pool within cells. [F] Mean transcript counts for HAMP◆ were 21.65 in the control group and 16.23 in the DFO group. HAMP is a peptide hormone that regulates iron metabolism by inhibiting FPN expression, thereby preventing the export of iron out of cells.
[A] The percentage of p-AMPKα positive cells were inversely correlated to the histologic score for articular cartilage surface. [B] The percentage of p-AMPKα positive cells were inversely correlated to the histologic score for the proteoglycan content of articular cartilage. [C] The percentage of p-AMPKα positive cells were inversely correlated to the histologic score for chondrocyte cellularity within articular cartilage.
The length of all target sequences is 99 base pairs.
Manuscript Highlights:
Deferoxamine reduced the concentration of iron in several tissues.
Osteoarthritis-associated cartilage lesions were reduced with deferoxamine.
Pharmacologic iron chelation preserved chondrocyte cellularity in knee cartilage.
Gene expression changes supported reduced cell death with deferoxamine.
Deferoxamine treatment helped prevent mobility decline observed in control animals.
Acknowledgements.
We would like to thank Crystal Richt and the staff at the University of Arizona Genetics Core, as well as Kevin Daniels and Zaria Torres-Poche at Colorado State University Veterinary Diagnostic Laboratories, for their assistance in generating experimental data. Additionally, we would like to thank the following members from the Santangelo Research Team at Colorado State University for their assistance in administering treatments to animals: Richard Martinez, Ariel Timkovich, Kendra Andrie, Joseph Sanford, Sydney Bork, Sara Wist, Zori Oberle, and Michelle Cornwall. Dr. Andrie also independently corroborated the cellular morphology/appearance of chondrocytes in articular cartilage. Finally, we would like to thank the Laboratory Animal Resources staff at Colorado State University for their excellent commitment and care provided to the animals used in this study.
Funding Acknowledgement:
This research was supported by funding from the National Institute on Aging, National Institute of Health by the funding mechanism 5R21AG056807. This work was not funded or supported by any commercial source.
Abbreviations:
- OA
Osteoarthritis
- DFO
Deferoxamine
- ECM
Extracellular Matrix
- CBC
Complete Blood Count
- NBF
Neutral Buffered Formalin
- AAS
Atomic Absorption Spectroscopy
- OARSI
Osteoarthritis Research Society International
- BCL-2
B-cell lymphoma 2
- BAD
BCL-2-associated death promoter
- BAX
BCL-2-associated x protein
- BAK
BCL-2 homologous antagonist killer
- ACAN
Aggrecan
- COL2A1
Type II collagen
- MMP
Matrixmetalloproteinase
- TIMP-2
Tissue inhibitor of matrixmetalloproteinases-2
- TFR1
Transferrin receptor 1
- SLC11A2/DMT1
Divalent metal transporter 1
- SLC39A14/ZIP14
ZRT/IRT-like protein 14
- SLC40A1/FPN
Ferroportin
- FTH
Ferritin heavy chain
- HAMP
hepcidin
- ACTB
β-actin
- EEF1A1
Eukaryotic elongation factor 1α1
- 4-HNE
4-hydroxynonenal
- MTP
Medial tibial plateau
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest.
The authors have no conflicts of interest to report.
References.
- [1].Winterbourn CC. Toxicity of iron and hydrogen peroxide: the Fenton reaction. Toxicol Lett. 1995; 82–83:969–74. doi: 10.1016/0378-4274(95)03532-x. [DOI] [PubMed] [Google Scholar]
- [2].Koppenol WH, Hider RH. Iron and redox cycling. Do’s and don’ts. Free Radic Biol Med. 2019; 133: 3–10. doi: 10.1016/j.freeradbiomed.2018.09.022. [DOI] [PubMed] [Google Scholar]
- [3].Voest EE, Vreugdenhil G, Marx JJ. Iron-chelating agents in non-iron overload conditions. Ann Intern Med. 1994; 120(6): 490–9. doi: 10.7326/0003-4819-120-6-199403150-00008. [DOI] [PubMed] [Google Scholar]
- [4].Dixon SJ, Stockwell BR. The role of iron and reactive oxygen species in cell death. Nat Chem Biol. 2014; 10(1): 9–17. doi: 10.1038/nchembio.1416. [DOI] [PubMed] [Google Scholar]
- [5].Raven EP, Lu PH, Tishler TA, Heydari P, Bartzokis G. Increased iron levels and decreased tissue integrity in hippocampus of Alzheimer’s disease detected in vivo with magnetic resonance imaging. J Alzheimers Dis. 2013; 37(1): 127–36. doi: 10.3233/JAD-130209. [DOI] [PubMed] [Google Scholar]
- [6].Xu J, Marzetti E, Seo AY, Kim JS, Prolla TA, Leeuwenburgh C. The emerging role of iron dyshomeostasis in the mitochondrial decay of aging. Mech Ageing Dev. 2010; 131(7–8): 487–93. doi: 10.1016/j.mad.2010.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Paesano R, Natalizi T, Berlutti F, Valenti P. Body iron delocalization: the serious drawback in iron disorders in both developing and developed countries. Pathog Glob Health. 2012; 106(4): 200–16. doi: 10.1179/2047773212Y.0000000043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Wessling-Resnick M Iron homeostasis and the inflammatory response. Annu Rev Nutr. 2010; 30: 105–22. doi: 10.1146/annurev.nutr.012809.104804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Silva B, Faustino P. An overview of molecular basis of iron metabolism regulation and the associated pathologies. Biochim Biophys Acta. 2015; 1852(7): 1347–59. doi: 10.1016/j.bbadis.2015.03.011. [DOI] [PubMed] [Google Scholar]
- [10].Cook CI, Yu BP. Iron accumulation in aging: modulation by dietary restriction. Mech Ageing Dev. 1998; 102(1): 1–13. doi: 10.1016/s0047-6374(98)00005-0. [DOI] [PubMed] [Google Scholar]
- [11].Zacharski LR, Ornstein DL, Woloshin S, Schwartz LM. Association of age, sex, and race with body iron stores in adults: analysis of NHANES III data. Am Heart J. 2000; 140(1): 98–104. doi: 10.1067/mhj.2000.106646. [DOI] [PubMed] [Google Scholar]
- [12].Stadler N, Lindner RA, Davies MJ. Direct detection and quantification of transition metal ions in human atherosclerotic plaques: evidence for the presence of elevated levels of iron and copper. Arterioscler Thromb Vasc Biol. 2004; 24(5): 949–54. doi: 10.1161/01.ATV.0000124892.90999.cb. [DOI] [PubMed] [Google Scholar]
- [13].Smith MA, Harris PL, Sayre LM, Perry G. Iron accumulation in Alzheimer disease is a source of redox-generated free radicals. Proc Natl Acad Sci. 1997; 94(18): 9866–8. doi: 10.1073/pnas.94.18.9866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Stevens RG, Graubard BI, Micozzi MS, Neriishi K, Blumberg BS. Moderate elevation of body iron level and increased risk of cancer occurrence and death. Int J Cancer. 1994; 56(3): 364–9. doi: 10.1002/ijc.2910560312. [DOI] [PubMed] [Google Scholar]
- [15].Ford ES, Cogswell ME. Diabetes and serum ferritin concentration among U.S. adults. Diabetes Care. 1999; 22(12): 1978–83. doi: 10.2337/diacare.22.12.1978. [DOI] [PubMed] [Google Scholar]
- [16].Roosendaal G, Vianen ME, Venting MJ, van Rinsum AC, van den Berg HM, Lafeber FP. Iron deposits and catabolic properties of synovial tissue from patients with haemophilia. J Bone Joint Surg Br. 1998; 80(3): 540–5. doi: 10.1302/0301-620x.80b3.7807. [DOI] [PubMed] [Google Scholar]
- [17].Richette P, Ottaviani S, Vicaut E, Bardin T. Musculoskeletal complications of hereditary hemochromatosis: a case-control study. J Rheumatol. 2010; 37(10): 2145–50. doi: 10.3899/jrheum.100234. [DOI] [PubMed] [Google Scholar]
- [18].Biemond P, Swaak AJ, van Eijk HG, Koster JF. Intraarticular ferritin-bound iron in rheumatoid arthritis. A factor that increases oxygen free radical-induced tissue destruction. Arthritis Rheum. 1986; 29(10): 1187–93. doi: 10.1002/art.1780291002. [DOI] [PubMed] [Google Scholar]
- [19].Nieuwenhuizen L, Schutgens REG, van Asbeck BS, Wenting MJ, van Veghel K, Roosendaal G, et al. Identification and expression of iron regulators in human synovium: evidence for upregulation in haemophilic arthropathy compared to rheumatoid arthritis, osteoarthritis, and healthy controls. Haemophilia. 2013; 19(4): e218–27. doi: 10.1111/hae.12208. [DOI] [PubMed] [Google Scholar]
- [20].Henrotin Y, Kurz B, Aigner T. Oxygen and reactive oxygen species in cartilage degradation: friends or foes? Osteoarthritis Cartilage. 2005; 13(8): 643–54. doi: 10.1016/j.joca.2005.04.002. [DOI] [PubMed] [Google Scholar]
- [21].Hunter DJ, Neogi T, Hochberg MC. Quality of osteoarthritis management and the need for reform in the US. Arthritis Care Res. 2011; 63(1): 31–8. doi: 10.1002/acr.20278. [DOI] [PubMed] [Google Scholar]
- [22].Fox AJS, Bedi A, Rodeo SA. The basic science of articular cartilage: structure, composition, and function. Sports Health. 2009; 1(6): 461–8. doi: 10.1177/1941738109350438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Carlo MD Jr, Loeser RF. Increased oxidative stress with aging reduces chondrocyte survival: correlation with intracellular glutathione levels. Arthritis Rheum. 2003; 48(12): 3419–30. doi: 10.1002/art.11338. [DOI] [PubMed] [Google Scholar]
- [24].Yazer M, Sarban S, Kocyigit A, Isikan UE. Synovial fluid and plasma selenium, copper, zinc, and iron concentrations in patients with rheumatoid arthritis and osteoarthritis. Biol Trace Elem Res. 2005;106(2): 123–32. doi: 10.1385/BTER:106:2:123. [DOI] [PubMed] [Google Scholar]
- [25].Nugzar O, Zandman-Goddard G, Oz H, Lakstein D, Feldbrin Z, Shargorodsky M. The role of ferritin and adiponectin as predictors of cartilage damage assessed by arthroscopy in patients with symptomatic knee osteoarthritis. Best Pract Rest Clin Rheumatol. 2018; 32(5): 662–8. doi: 10.1016/j.berh.2019.04.004. [DOI] [PubMed] [Google Scholar]
- [26].Ogilvie-Harris DJ, Fornasier VL. Synovial iron deposition in osteoarthritis and rheumatoid arthritis. J Rheumatol. 1980; 7(1): 30–6. [PubMed] [Google Scholar]
- [27].Martin JA, Buckwalter JA. Aging, articular cartilage chondrocyte senescence and osteoarthritis. Biogerontology. 2002; 3(5): 257–64. doi: 10.1023/a:1020185404126. [DOI] [PubMed] [Google Scholar]
- [28].Burton LH, Radakovich LB, Marolf AJ, Santangelo KS. Systemic iron overload exacerbates osteoarthritis in the strain 13 guinea pig. Osteoarthritis Cartilage. 2020; 28(9): 1265–75. doi: 10.1016/j.joca.2020.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Tessier JJ, Bowyer J, Brownrigg NJ, Peers IS, Westwood FR, Waterton JC, et al. Characterisation of the guinea pig model of osteoarthritis by in vivo three-dimensional magnetic resonance imaging. Osteoarthritis Cartilage. 2003; 11(12): 845–53. doi: 10.1016/s1063-4584(03)00162-6. [DOI] [PubMed] [Google Scholar]
- [30].Kraus VB, Huebner JL, DeGroot J, Bendele A. The OARSI histopathology initiative—recommendations for histological assessments of osteoarthritis in the guinea pig. Osteoarthritis Cartilage. 2010; 18 Suppl 3(Suppl 3): S35–52. doi: 10.1016/j.joca.2010.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Ehlers KH, Giardina PJ, Lesser ML, Engle MA, Hilgartner MW. Prolonged survival in patients with beta-thalassemia major treated with deferoxamine. J Pediatr. 1991; 118(4 Pt 1): 540–5. doi: 10.1016/s0022-3476(05)83374-8. [DOI] [PubMed] [Google Scholar]
- [32].Nair AB, Jacob S. A simple practice guide for dose conversion between animals and human. J Basic Clin Pharm. 2016; 7(2): 27–31. doi: 10.4103/0976-0105.177703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Helrich K, eds; Association of Official Analytical Chemists. Official Methods of Analysis of AOAC International. 15th ed. Gaithersburg, MD, USA; AOAC International; 1990. Official method 968.08D. [Google Scholar]
- [34].Helrich K, eds; Association of Official Analytical Chemists. Official Methods of Analysis of AOAC International. 15th ed. Gaithersburg, MD, USA; AOAC International; 1990. Official method 974.27A, B, E and F. [Google Scholar]
- [35].Helrich K, eds; Association of Official Analytical Chemists. Official Methods of Analysis of AOAC International. 15th ed. Gaithersburg, MD, USA; AOAC International; 1990. Official method 985.40D. [Google Scholar]
- [36].Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012; 9(7): 676–82. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012; 9(7):671–5. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Dalleau S, Baradat M, Gueraud F, Huc L. Cell death and diseases related to oxidative stress: 4-hydroxynonenal (HNE) in the balance. Cell Death Differ. 2013; 20(12): 1615–30. doi: 10.1038/cdd.2013.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Yi D, Yu H, Lu K, Ruan C, Ding C, Tong L et al. AMPK signaling in energy control, cartilage biology, and osteoarthritis. Front Cell Dev Biol. 2021;9:696602. doi: 10.3389/fcell.2021.696602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Wu J, Qian Y, Chen C, Feng F, Pan L, Yang L et al. Hesperetin exhibits anti-inflammatory effects on chondrocytes via the AMPK pathway to attenuate anterior cruciate ligament transection-induced osteoarthritis. Front Pharmacol. 2021;12:735087. doi: 10.3389/fphar.2021.735087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Stein SC, Woods A, Jones NA, Davison MD, Carling D. The regulation of AMP-activated protein kinase by phosphorylation. Biochem J. 2000;345 Pt3(Pt3):437–43. [PMC free article] [PubMed] [Google Scholar]
- [42].Gerhardsson L, Kazantzis G. Diagnosis and treatment of metal poisoning: general aspects. In: Nordberg GF, Fowler BA, Nordberg M, editors. Handbook on the toxicology of metals. Amsterdam: Academic Press; 2015. p. 487–505. doi: 10.1016/B978-0-444-59453-2.00023-8. [DOI] [Google Scholar]
- [43].Madhock R, Bennet D, Sturrock RD, Forbes CD. Mechanisms of joint damage in an experimental model of hemophilic arthritis. Arthritis Rheum. 1988; 31(9): 1148–55. doi: 10.1002/art.1780310910 [DOI] [PubMed] [Google Scholar]
- [44].Ferreira AV, Duarte TL, Marques S, Costa P, Neves SC, dos Santos T et al. Iron triggers the early stages of cartilage degeneration in vitro: the role of articular chondrocytes. Osteoarthritis Cartilage Open. 2021:3(2):100145. 10.1016/j.ocarto.2021.100145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Vaillancourt F, Fahmi H, Shi Q, Lavigne P, Ranger P, Fernandes JC, et al. 4-hydroxynonenal induces apoptosis in human osteoarthritic chondrocytes: the protective role of glutathione-S-transferase. Arthritis Res Ther. 2008; 10(5): R107. doi: 10.1186/ar2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Wang J, Li J, Song D, Ni J, Ding M, Huang J et al. AMPK: implications in osteoarthritis and therapeutic targets. Am J Transl Res. 2020;12(12):7670–81. [PMC free article] [PubMed] [Google Scholar]
- [47].Zhou S, Lu W, Chen L, Ge Q, Chen D, Xu Z et al. AMPK deficiency in chondrocytes accelerated the progression of instability-induced and ageing-associated osteoarthritis in adult mice. Sci Rep. 2017;7:43245. doi: 10.1038/srep43245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Li J, Zhang B, Liu WX, Lu K, Pan H, Wang T et al. Metformin limits osteoarthritis development and progression through activation of AMPK signalling. Ann Rheum Dis. 2020;79(5):635–45. doi: 10.1136/annrheumdis-2019-216713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Terkeltaub R, Yang B, Lotz M, Liu-Bryan R. Chondrocyte AMP-activated protein kinase activity suppresses matrix degradation responses to proinflammatory cytokines interleukin-1B and tumer necrosis factor a. Arthritis Rheum. 2011;63(7):1928–37. doi: 10.1002/art.30333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Petursson F, Husa M, June R, Lotz M, Terkeltaub R, Liu-Bryan R. Linked decreases in liver kinase B1 and AMP-activated protein kinase activity modulate matrix catabolic responses to biomechanical injury in chondrocytes. Arthritis Res Ther. 2013;15(4):R77. doi: 10.1186/ar4254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Tchetina EV, Markova GA, Poole AR, Zukor DJ, Antoniou J, Makarov SA et al. Deferoxamine suppresses collagen cleavage and protease, cytokine, and COL10A1 expression and upregulates AMPK and Krebs cycle genes in human osteoarthritic cartilage. Int J Rheumatol. 2016;2016:6432867. doi: 10.1155/2016/6432867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Ma CH, Chiua YC, Wu CH, Jou IM, Tu YK, Hung CH et al. Homocysteine causes dysfunction of chondrocytes and oxidative stress through repression of SIRT1/AMPK pathway: a possible link between hyperhomocysteinemia and osteoarthritis. Redox Biol. 2018;15:504–12. doi: 10.1016/j.redox.2018.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Wang C, Yao Z, Zhang Y, Yang Y, Liu J, Shi Y et al. Metformin mitigates cartilage degradation by activating AMPK/SIRT1-mediated autophagy in a mouse osteoarthritis model. Front Pharmacol. 2020;11:1114. doi: 10.3389/fphar.2020.01114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Wang CL, Semenza GL. Desferrioxamine induces erythropoietin gene expression and hypoxia-inducible factor 1 DNA-binding activity: implications for models of hypoxia signal transduction. Blood. 1993;82(12):3610–5. [PubMed] [Google Scholar]
- [55].Bohensky J, Leshinsky S, Srinivas V, Shapiro IM. Chondrocyte autophagy is stimulated by HIF-1 dependent AMPK activation and mTOR suppression. Pediatr Nephrol. 2010;25(4):633–42. doi: 10.1007/s00467-009-1310-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Han DH, Hancock CR, Jung SR, Higashida K, Kim SH, Holloszy JO. Deficiency of the mitochondrial electron transport chain in muscle does not cause insulin resistance. PLoS One. 2011;6(5):e19739. doi: 10.1371/journal.pone.0019739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Lipari L, Gerbino A. Expression of gelatinases (MMP-2, MMP-9) in human articular cartilage. Int J Immunopathol Pharmacol. 2013;26(3):817–23. doi: 10.1177/039463201302600331. [DOI] [PubMed] [Google Scholar]
- [58].Bau B, Gebhard PM, Haag J, Knorr T, Bartnik E, Aigner T. Relative messenger RNA expression profiling of collagenases and aggrecanases in human articular chondrocytes in vivo and in vitro. Arthritis Rheum. 2002;46(10):2648–57. doi: 10.1002/art.10531. [DOI] [PubMed] [Google Scholar]
- [59].Litwic A, Edwards MH, Dennison EM, Cooper C. Epidemiology and burden of osteoarthritis. Br Med Bull. 2013;105:185–99. doi: 10.1093/bmb/lds038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Barbour KE, Helmick CG, Boring M, Brady TJ. Vital signs: prevalence of doctor-diagnosed arthritis and arthritis-attributable activity limitation—United States, 2013–2015. MMWR Morb Mortal Wkly Rep. 2017;66(9):246–253. doi: 10.15585/mmwr.mm6609e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].World Health Organization. Health in older age. In: World report on ageing and health. Geneva: World Health Organization; 2015. p. 43–74. [Google Scholar]
- [62].Halliwell B Protection against tissue damage in vivo by desferrioxamine: what is its mechanism of action? Free Radic Biol Med. 1989;7(6):645–51. doi: 10.1016/0891-5849(89)90145-7. [DOI] [PubMed] [Google Scholar]
- [63].Zhu BZ, Har-El R, Kitrossky N, Chevion M. New modes of action of desferrioxamine: scavenging of semiquinone radical and stimulation of hydrolysis of tetrachlorohydroquinone. Free Radic Biol Med. 1998;24(2):390–9. doi: 10.1016/s0891-5849(97)00220-7. [DOI] [PubMed] [Google Scholar]
- [64].Washington IM, Van Hoosier G. Clinical biochemistry and hematology. In: Suckow MA, Stevens KA, Wilson RP, editors. The laboratory rabbit, guinea pig, hamster, and other rodents. Amsterdam: Academic Press; 2012. p. 57–116. doi: 10.1016/B978-0-12-380920-9.00003-1 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The mass of animals was similar between groups throughout the duration of the study.
[A] Mean cell volume (MCV) ◆ was an average of 84.43 femtoliters/cell in the control group and 81.88 femtoliters/cell in the DFO group. Reference interval range is 70.00–90.00 femtoliters/cell [64]. [B] Mean hematocrit (HCT) ◆ volume was 48.29% red blood cells (RBCs) in the control group and 46.75% RBCs in the DFO group. Reference interval range is 30.00–50.00% RBCs [64]. [C] Median values for mean corpuscular hemoglobin concentration (MCHC) × were 32.00 g/dL in the control group and 32.00 g/dL in the DFO group. Reference interval range is 25.00–40.00 [64]. [D] Mean platelet count◆ was 427.60 × 103/microliter in the control group and 593.00 × 103/microliter in the DFO group. Reference interval range 250.00 − 850.00 × 103/microliter [64].
[A] Mean transcript counts for TFR1◆ were 932.10 in the control group and 757.70 in the DFO group. [B] Mean transcript counts for DMT1◆ were 1089.00 in the control group and 858.90 in the DFO group. [C] Mean transcript counts for ZIP14◇ were 1703.00 in the control group and 901.60 in the DFO Group. [D] Mean transcript counts for FPN◆ were 1127.00 in the control group and 1313.00 in the DFO group. [E] Mean transcript counts for FTH◆ were 89350.00 in the control group and 82526.00 in the DFO group. FTH is a subunit of the major iron storage protein ferritin. In addition to storing iron, FTH exhibits ferroxidase activity and is responsible for the exchange of iron between stable storage and the redox-active iron pool within cells. [F] Mean transcript counts for HAMP◆ were 21.65 in the control group and 16.23 in the DFO group. HAMP is a peptide hormone that regulates iron metabolism by inhibiting FPN expression, thereby preventing the export of iron out of cells.
[A] The percentage of p-AMPKα positive cells were inversely correlated to the histologic score for articular cartilage surface. [B] The percentage of p-AMPKα positive cells were inversely correlated to the histologic score for the proteoglycan content of articular cartilage. [C] The percentage of p-AMPKα positive cells were inversely correlated to the histologic score for chondrocyte cellularity within articular cartilage.
The length of all target sequences is 99 base pairs.