

Abstract
Obstructive sleep apnea (OSA) is characterized by chronic intermittent hypoxia (CIH) and sleep fragmentation and deprivation. Exposure to CIH results in oxidative stress in the cortex, hippocampus and basal forebrain of rats and mice. We show that sustained and intermittent hypoxia induces antioxidant responses, an indicator of oxidative stress, in the rat cerebellum and pons. Increased glutathione reductase (GR) activity and thiobarbituric acid reactive substance (TBARS) levels were observed in the pons and cerebellum of rats exposed to CIH or chronic sustained hypoxia (CSH) compared with room air (RA) controls. Exposure to CIH or CSH increased GR activity in the pons, while exposure to CSH increased the level of TBARS in the cerebellum. The level of TBARS was increased to a greater extent after exposure to CSH than to CIH in the cerebellum and pons. Increased superoxide dismutase activity (SOD) and decreased total glutathione (GSHt) levels were observed after exposure to CIH compared with CSH only in the pons. We have previously shown that prolonged sleep deprivation decreased SOD activity in the rat hippocampus and brainstem, without affecting the cerebellum, cortex or hypothalamus. We therefore conclude that sleep deprivation and hypoxia differentially affect antioxidant responses in different brain regions.
Keywords: antioxidant responses, cerebellum, chronic intermittent hypoxia, chronic sustained hypoxia, pons, sleep deprivation
Obstructive sleep apnea (OSA) is a condition characterized by repeated upper airway obstruction during sleep. It affects ~ 4% of adult men and 2% of women (Young et al. 1993; Loredo et al. 2001). OSA causes neurocognitive and behavioral deficits including excessive daytime sleepiness, impaired short-term memory, and problems with language comprehension and expression, all of which are compatible with global executive dysfunction (Bedard et al. 1993; Naegele et al. 1998; Gozal et al. 2001; Beebe and Gozal 2002). OSA also leads to increased cardiovascular morbidity, including hypertension, cardiovascular and cerebrovascular disease and increased risk for sudden death (Zaninelli et al. 1991; Seppala et al. 1991; Fletcher et al. 1992; Lavie et al. 1993; Fletcher 1995; Silverberg and Oksenberg 1997; Greenberg et al. 1999). Furthermore, magnetic resonance imaging studies in patients with OSA revealed significant gray matter loss in several brain regions including the cortex, hippocampus and cerebellum, suggesting that sleep apnea might result in neuronal damage, impairing cognitive function (Macey et al. 2002). Of note, localized gray matter loss was also seen unilaterally in normally well-perfused regions involved in motor regulation of the upper airway, suggesting that underlying brain damage may also precipitate the occurrence of OSA in some of these patients.
OSA produces both chronic intermittent hypoxia (CIH) and sleep fragmentation and deprivation. Rats subjected to CIH, of similar magnitude to that experienced by sleep apnea patients, showed time-dependent increases in neuronal apoptosis within the CA1 hippocampal region and neocortex (Gozal et al. 2001). Based on these findings, it was proposed that hypoxia-induced damage might result from oxidative stress. Oxidative stress occurs when free radicals are produced at a greater rate than they are removed, resulting not only in changes in the activities of antioxidative enzymes and the levels of endogenous antioxidants, but ultimately in oxidative damage to lipids, proteins and/or nucleic acids. As further evidence of the viability of the oxidative stress hypothesis, Row et al. (2003) reported that the antioxidant PNU-101033E significantly attenuated the behavioral and oxidative changes associated with CIH (Row et al. 2003). Transgenic mice over-expressing Cu/Zn-superoxide dismutase (SOD1; EC 1.15.1.1) showed lower cortical free radical production and reduced apoptosis compared with normal mice exposed to CIH (Xu et al. 2004). Veasey et al. (2004) recently showed that 8 weeks of CIH increased lipid oxidation, protein oxidation and mRNA levels of several antioxidant enzymes in the rostral basal forebrain. CIH also increased iNOS mRNA and protein expression, as well as NOS activity, nitrotyrosine residue formation and nitrate/nitrite production (Li et al. 2004). We previously reported that prolonged sleep deprivation (5–11 days) decreased Cu/Zn-SOD activity in the rat hippocampus and brainstem (Ramanathan et al. 2002). This finding is consistent with the hypothesis that enzymatic damage results from oxidative stress, whereby the overproduction of free radicals decreases SOD activity (Davies et al. 1987; Yunoki et al. 1998).
In this study, we investigated the effects of hypoxia on antioxidant responses, as an indicator of oxidative stress, in the rat cerebellum and pons. The cerebellum is of particular interest, because of its role in respiratory and autonomic control (Xu and Frazier 2002), while the pons is involved in sleep–wake regulation and also in hypoxia-induced hypothermia and hyperventilation (Fabris et al. 1999).
Superoxide (O2·−) and hydrogen peroxide (H2O2), which are always present in cells due to normal metabolism, are removed by endogenous antioxidative enzymes and antioxidants. There are many enzymes that contribute significantly to the antioxidant responses. In this study we measured the activities of Cu/Zn-SOD, glutathione reductase (GR; EC 1.8.1.7) and total glutathione (GSHt) levels. We also analyzed the levels of the lipid oxidation product, thiobarbituric acid reactive substances (TBARS). Changes in these markers reflect changes in the levels of free radicals, and are therefore indicative of oxidative stress.
Materials and methods
Young adult male Sprague–Dawley rats (45–50 days of age) were used for all experiments. The experimental protocols were approved by our Institutional Animal Use and Care Committee and conform to the National Institutes of Health guide for the care and use of laboratory animals.
Hypoxic exposures
Rats were randomly assigned to three experimental groups consisting of: (i) CIH (n = 6/time point); (ii) CSH (n = 6/time point) for 6 h, 1, 3, 7, 14 or 30 days; or (iii) room air (RA, n = 6). Animals were placed in four identical commercially designed chambers (30 × 20 × 20 in.; Oxycycler model A44XO; Biospheryx, Redfield, NY, USA). The chambers were operated under a 12:12 h light/dark cycle (light 06.00 to 18.00). Gas was circulated around each of the chambers at 60 L/min (i.e. one complete change every 10 s). The O2 concentration was continuously measured by an O2 analyzer and regulated throughout the 12 h of light time by a computerized system controlling the gas valve outlets. Deviation from the desired concentration was corrected by addition of N2 or O2 through solenoid valves. For the remaining 12 h of night, oxygen concentrations were kept at the RA concentration of 21%. Ambient CO2 in the chamber was periodically monitored and maintained at < 0.01% by adjusting overall chamber basal ventilation. Hypocapnic hypoxia models most aspects of the OSA syndrome (Gozal et al. 2001). The gas was circulated through a molecular sieve (type 3A; Scientific Instrument Services, Ringoes, NJ, USA) to remove ammonia. Humidity was measured and maintained at 40–50% by circulating the gas through a freezer and silica gel. Ambient temperature was kept at 22–24°C. The CIH profile consisted of exposing rats to alternating RA and 10% O2 every 90 s during the light phase (06.00 to 18.00). The CSH profile consisted of exposing rats to 10% oxygen throughout the light phase. The rats were exposed to RA for the remaining 12 h (18.00 to 06.00). Hence, the CIH group was re-oxygenated every 90 s during the light phase, while the CSH group was re-oxygenated at the onset of the light phase and then again at the onset of the dark phase. Rats exposed to RA throughout the experimental time period served as controls.
Biochemical analysis
Animals were killed at the end of the designated duration of hypoxia exposure and the cerebellum and pons were dissected on ice and stored at −80°C until analyzed. The cerebellum and pons were divided into two portions, one portion was homogenized in a handheld homogenizer with 12 strokes in cold homogenizing buffer (50 mm Tris–HCl, pH 7.5, 50 mm MgCl2 and 5 mm EDTA) containing protease inhibitors (Roche Diagnostics, Mannheim, Germany) to make a 10% homogenate (w/v). The homogenate was centrifuged in an Eppendorf microcentrifuge (5415C) at 320 g for 10 min at 4°C. The pellet was discarded and the supernatant recentrifuged at 14 000 g for 30 min at 4°C. This supernatant was used for determining the activities of Cu/Zn-SOD, GR and the levels of GSHt. The remaining portion of the cerebellum and pons was homogenized with 12 strokes in cold homogenizing buffer containing 20 mm phosphate buffer (pH 7.4) to which 1% of 0.5 M butylated hydroxytoluene had been added to prevent oxidation during sample preparation. The homogenate (10%, w/v) was centrifuged in an Eppendorf microcentrifuge at 2900 g for 10 min at 4°C. The pellet was discarded and the supernatant was used for determining the level of the lipid oxidation product, TBARS.
Prior to determining the enzyme activities and the level of GSHt and TBARS, the protein content of the samples was determined with a protein assay kit (Bio-Rad Laboratories, Richmond, CA, USA) using bovine plasma gamma globulin as the standard. The amount of protein in the standard and in the samples was determined on a microtitre plate reader at a wavelength of 750 nm.
Superoxide dismutase activity
SOD activity was measured according to the method of Misra and Fridovich (1972). Tissue extract was added to carbonate buffer (50 mm, pH 10.2 containing 0.1 mm EDTA) and the reaction initiated with epinephrine (30 mm in 0.05% acetic acid). The rate of auto-oxidation of epinephrine was measured at 480 nm for 180 s on a Hitachi U2000 spectrophotometer. SOD activity was expressed as units (U) of SOD/mg of protein, where 1 U of SOD is defined as the amount of enzyme present that inhibits the auto-oxidation of epinephrine by 50%.
Glutathione reductase activity
GR activity was determined according to the method of Somani and Husain (1997). Tissue extract was added to the reaction mixture containing phosphate buffer (50 mm), 50 μL of GSSG (20 mm) and 100 μL of NADPH (2 mm in 10 mm Tris–HCl pH 7.0). The rate of NADPH oxidation was measured at 340 nm for 180 s. The molar extinction coefficient of 6.22 × 103 (mcm)−1 was used to determine GR activity. GR activity was expressed as mm NADPH oxidized/min/mg protein.
Total glutathione levels
GSHt was measured by the enzymatic recycling procedure in which GSH is sequentially oxidized by 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB) to GSSG, which is then reduced by NADPH in the presence of GR back to GSH (Griffith 1980). To 800 μL of NADPH (0.3 mm) and 100 μL of DTNB (6 mm), 100 μL of either tissue extract or known amounts of GSH standard were added. The reaction was initiated with 10 μL of GR (50 units/mL). All solutions were made up in stock buffer (pH 7.5) containing sodium phosphate (125 mm) and sodium-EDTA (6.3 mm). The rate of DTNB reduction was measured at 412 nm continuously until it exceeds a value of 2.0 (~ 120 s). Total glutathione levels were expressed as nmoles GSH/g tissue.
Lipid oxidation assay
Lipid oxidation was determined as TBARS according to the method of Ohkawa et al. (1979). Tissue extract (100 μL) was added to a mixture containing 50 μL of sodium dodecyl sulfate (8.1%), 375 μL of acetic acid (20%, pH 3.5) and 375 μL of aqueous thiobarbituric acid (0.8%). The samples were heated in a boiling water bath for 60 min. The samples were allowed to cool at room temperature and then 1.25 mL of n-butanol/pyridine (15 : 1, v/v) was added. The mixture was shaken vigorously and centrifuged at 1300 g for 10 min. The colored upper organic layer was removed and read at 532 nm. Tetraethoxypropane was used as an external standard, and the level of lipid peroxides was expressed as nmoles malondialdehyde/mg protein.
Statistical analysis
Each type of assay was carried out on a different day, and so in order to minimize the differences due to sample and/or reagent preparation, we normalized the data to the mean of each set. For each assay (SOD, GR, GSHt and TBARS), values from duplicate samples were averaged to obtain 1-value point. The specific SOD activity for each sample was first calculated as USOD/mg protein. Each assay set was run at one time and included 13 samples: RA (1 sample), CIH (6 samples) and CSH (6 samples). Each sample from a particular assay set was normalized by dividing the specific SOD activity of that sample by the mean specific SOD activity of the assay set. The average of six assay sets was then used to determine the final normalized specific SOD activity for each sample. The results are expressed as fold increase in SOD activity ± SEM. The activity of GR as well as the levels of GSHt and TBARS, were analyzed and expressed in the same way. Two-way analyses of variance (treatment and time) with post-hoc Bonferroni analysis were used to determine statistical significance, which was set at a level of p < 0.05.
Results
There was a significant effect of time (F6,77 = 6.17, p < 0.001) and treatment (F1,77 = 8.13, p < 0.05) on the level of TBARS in the cerebellum of rats exposed to hypoxia (Fig. 1a). TBARS was significantly higher after 1 day (42%, t = 2.90, d.f. = 5, p < 0.05), 7 days (39%, t = 2.72, d.f. = 5, p < 0.05), 14 days (65%, t = 4.51, d.f. = 5, p < 0.001) and 30 days (69%, t = 4.78, d.f. = 5, p < 0.001) of exposure to CSH compared with RA controls. There was no significant time difference in the amount of TBARS in rats exposed to CIH compared with RA controls. Also a greater amount of TBARS was produced in the cerebellum of rats exposed to 1 day (26%, t = 2.03, d.f. = 5, p < 0.05), 14 days (26%, t = 2.37, d.f. = 5, p < 0.05) and 30 days (30%, t = 2.72, d.f. = 5, p < 0.01) of CSH compared with CIH (Fig. 1a). There was a significant interaction between time and treatment on the level of TBARS in the pons (F6,77 = 2.35, p < 0.05). In other words, the effect of different levels of time depended on what level of treatment was present. TBARS levels were significantly higher in CSH animals than CIH animals after 30 days of exposure (24%, t = 3.15, d.f. = 5, p < 0.01, Fig. 1b).
Fig. 1.
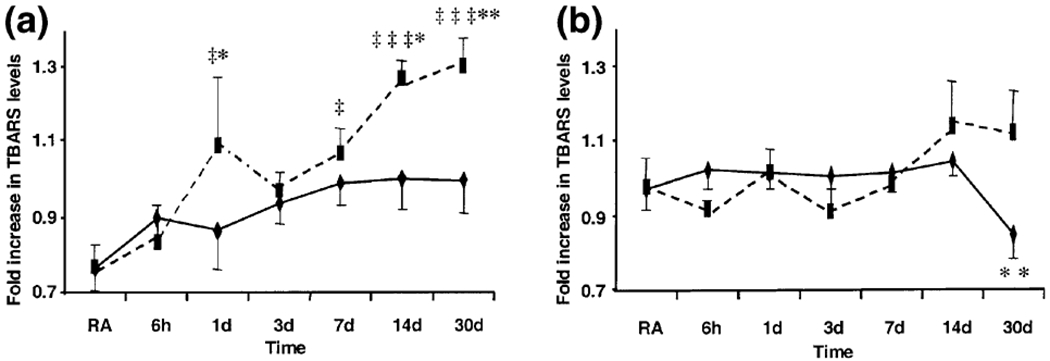
Thiobarbituric acid reactive substance (TBARS) levels in the (a) cerebellum and (b) pons of rats exposed to room air (RA, n = 6), chronic intermittent hypoxia (CIH, diamonds, n = 6/time point) or chronic sustained hypoxia (CSH, squares, n = 6/time point). Bars represent standard error. (‡p < 0.05, ‡‡‡p < 0.001, CSH vs. RA; *p < 0.05, **p < 0.01 CSH vs. CIH, post-hoc Bonferroni.) Normalized RA values for cerebellum = 0.77 ± 0.06 and pons = 0.97 ± 0.06.
There was a significant effect of time (F6,77 = 3.39, p < 0.005) but not of treatment (F1,77 = 0.8, p > 0.05) on GR activity in the pons (Fig. 2a). GR activity was significantly higher after 14 days (37%, t = 3.09, d.f. = 5, p < 0.05) of exposure to CIH, and after 6 h (34%, t = 2.85, d.f. = 5, p < 0.05) and 1 day (35%, t = 2.85, d.f. = 5, p < 0.05) of exposure to CSH compared with RA controls (Fig. 2a). There was no significant effect of either time (compared with RA) or treatment (CIH vs. CSH) on GR activity in the cerebellum.
Fig. 2.
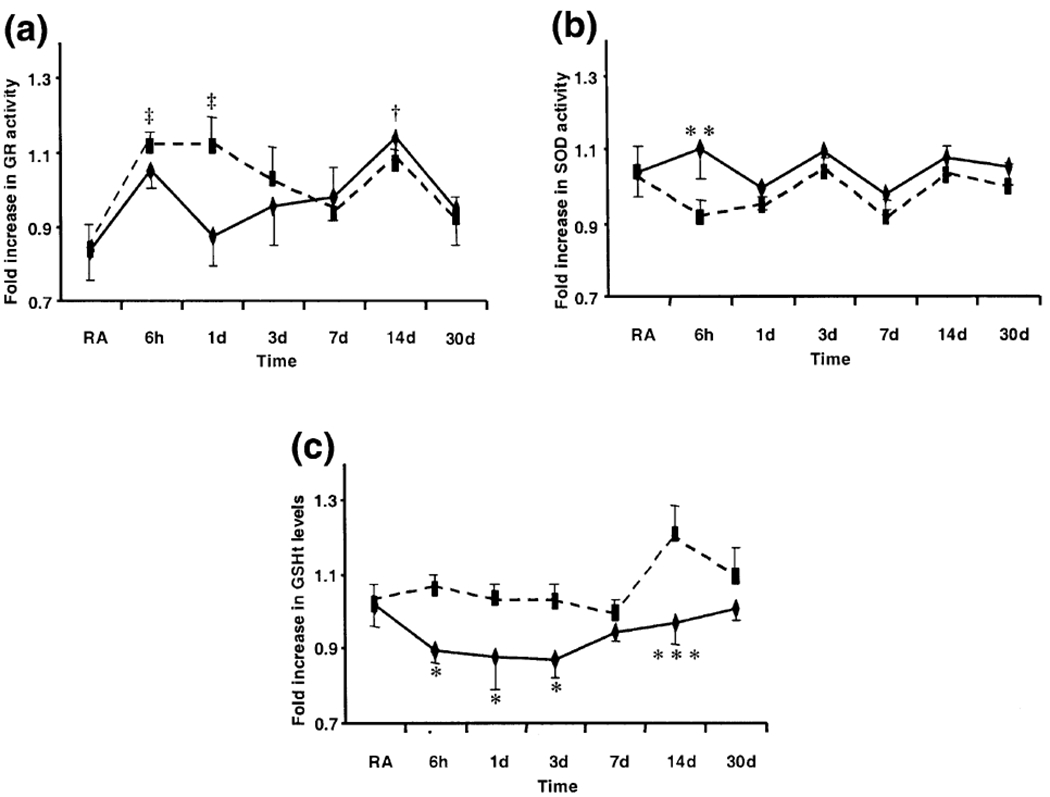
(a) Glutathione reductase (GR) activity (b) superoxide dismutase (SOD) activity and (c) total glutathione (GSHt) levels in the pons of rats exposed to room air (RA, n = 6), chronic intermittent hypoxia (CIH, diamonds, n = 6/time point) or chronic sustained hypoxia (CSH, squares, n = 6/time point). Bars represent standard error. (‡p < 0.05, CSH vs. RA; †p < 0.05, CIH vs. RA, post-hoc Bonferroni.) (*p < 0.05, **p < 0.01, ***p < 0.001, CSH vs. CIH, post-hoc Bonferroni). Normalized RA values for GR activity = 0.83 ± 0.07, SOD activity = 1.04 ± 0.07 and GSHt levels = 1.02 ± 0.06.
There was a significant effect of treatment (F1,77 = 5.81, p < 0.05) but not time (F6,77 = 1.36, p > 0.05) on SOD activity in the pons (Fig. 2b). SOD activity was significantly higher after 6 h of exposure to CIH compared with CSH (21%, t = 2.60, d.f. = 5, p < 0.01). There was no significant effect of either time or treatment on SOD activity in the cerebellum.
Similarly, there was a significant effect of treatment (F1,77 = 20.29, p < 0.001) but not time (F6,77 = 2.13, p > 0.05) on GSHt levels in the pons (Fig. 2c). GSHt levels were significantly lower after 6 h (15%, t = 2.29, d.f. = 5, p < 0.05), 1 day (15%, t = 2.12, d.f. = 5, p < 0.05), 3 days (15%, t = 2.22, d.f. = 5, p < 0.05) and 14 days (20%, t = 3.42, d.f. = 5, p < 0.001) of exposure to CIH compared with CSH (Fig. 2c). There was no significant effect of either time or treatment on GSHt levels in the cerebellum.
Discussion
In this study we show that hypoxia, at levels commonly occurring in human patients with sleep apnea, induces antioxidant responses in the rat cerebellum and pons. This is indicated by increased activities of the antioxidant enzymes, SOD and GR, decreased levels of the endogenous antioxidant, GSHt, and elevated levels of the lipid oxidation product, TBARS.
Hypoxia significantly increased GR activity in the pons and TBARS levels in the cerebellum compared with RA controls. The increased GR activity indicates an increase in the levels of hydrogen peroxide (H2O2), which may arise from: (i) the reaction of superoxide (O2·−) with superoxide dismutase; (ii) as a normal byproduct of the activity of several enzymes, including monoamine oxidase, tyrosine hydroxylase and l-amino oxidase; or (iii) via the autoxidation of endogenous substances, such as ascorbic acid and catecholamines. The increased levels of H2O2, if not removed by GR, will ultimately lead to increased levels of the lipid oxidation product, TBARS, which is observed in the cerebellum. Increased H2O2 levels could also elevate levels of oxidized proteins and/or nucleic acids, which may have been altered in the pons, but were not investigated in this study.
Cerebellar structures play an essential role in the onset of inspiration after apnea (Lutherer and Williams 1986; Gozal et al. 1995), in recovery from extremes of blood pressure changes (Lutherer et al. 1983), in carbon dioxide regulation (Xu and Frazier 1997) and in gasping (Smejkal et al. 1999; Peiffer et al. 2001). Macey et al. (2002) speculated that the cerebellar damage, linked to disordered breathing during sleep, results from the inappropriate coordination of upper airway muscles. Bernett et al. (2003) showed that perinatal asphyxia, resembling the clinical situation of intrauterine hypoxia-ischemia, caused neurodegeneration, neuronal loss and gliosis in the cerebellum of the adult guinea pig. Hypoxia also caused a significant increase in Fos-immunoreactivity in several pontine nuclei of fetal sheep (Breen et al. 1997). In addition, Fabris et al. (1999) showed that nitric oxide in the locus coeruleus was responsible for the inhibitory modulation of the ventilatory response to hypoxia in adult rats.
Hypoxia differentially affects antioxidant responses in the cerebellum and pons across the period observed. Regional differences in antioxidant responses have similarly been observed after sleep deprivation (Ramanathan et al. 2002). OSA produces both CIH as well as sleep fragmentation and deprivation. We compared antioxidant responses due to sleep deprivation with that produced by CIH and CSH (Table 1). We show that hypoxia and sleep deprivation differentially affect antioxidant responses in different brain regions and may interact in the progression of OSA.
Table 1.
Antioxidant responses in different brain regions as a result of prolonged (5–11 days) sleep deprivation (SD), chronic intermittent hypoxia (CIH) or chronic sustained hypoxia (CSH)
| SD | CIH | CSH | |
|---|---|---|---|
| Brainstem | decreased SOD activity | increased GR activity | increased GR activity |
| Hippocampus | decreased SOD activity | increased RNA damage | not done |
| Cerebellum | no change | no change | increased TBARS |
| Cortex | no change | increased MDA and isoprostane | not done |
| Hypothalamus | no change | not done | not done |
Furthermore, CSH significantly increased the levels of TBARS compared with CIH in the cerebellum. However, CIH significantly decreased GSHt levels compared with CSH in the pons. Several studies have shown decreased GSHt content under various oxidative stress conditions, such as after repeated inhalation exposure to manganese sulfate (Dobson et al. 2003), hypoxiareoxygenation induced damage to astroglia-rich cells (Makarov et al. 2002) and after 96 h of REM sleep deprivation (D’Almeida et al. 1998).
The differences in antioxidant responses as a function of region, time and treatment may be due to: (i) how well the brain areas are perfused, (ii) different brain areas being differentially resistant to hypoxia, (iii) different brain areas being more resistant to CIH than CSH, and/or (iv) different antioxidant capacity of different brain areas. These antioxidant responses could initiate a positive feedback loop whereby hypoxia causes cellular damage which then produces further respiratory and sleep/wake impairment. In particular, the CIH profile producing blood oxygenation changes comparable with those seen in sleep apnea, may explain the brain damage and cognitive effects seen in the sleep apnea syndrome.
Acknowledgements
This research was supported by HL41370, 1P50 HL060296 and the VA Medical Research Service to JMS and HL63912 and HL69932 to DG.
Abbreviations used:
- CIH
chronic intermittent hypoxia
- CSH
chronic sustained hypoxia
- DTNB
5,5′-dithiobis-(2-nitrobenzoic acid)
- GR
glutathione reductase
- GSHt
total glutathione
- GSSG
oxidized glutathione
- GSH
reduced glutathione
- OSA
obstructive sleep apnea
- RA
room air
- SOD
superoxide dismutase
- TBARS
thiobarbituric acid reactive substances
References
- Bedard MA, Montplaisir J, Malo J, Richer F and Rouleau I (1993) Persistent neuropsychological deficits and vigilance impairment in sleep apnea syndrome after treatment with continuous positive airways pressure (CPAP). J. Clin. Exp. Neuropsychol 15, 330–341. [DOI] [PubMed] [Google Scholar]
- Beebe DW and Gozal D (2002) Obstructive sleep apnea and the prefrontal cortex: towards a comprehensive model linking nocturnal airway obstruction to daytime cognitive and behavioral deficits. J. Sleep Res 11, 1–16. [DOI] [PubMed] [Google Scholar]
- Bernett G, Hoeger H, Mosgoeller W, Stolzlechner D and Lubec B (2003) Neurodegeneration, neuronal loss, and neurotransmitter changes in the adult guinea pig with perinatal asphyxia. Pediatr. Res 54, 523–528. [DOI] [PubMed] [Google Scholar]
- Breen S, Rees S and Walker D (1997) Identification of brainstem neurons responding to hypoxia in fetal and newborn sheep. Brain Res 748, 107–121. [DOI] [PubMed] [Google Scholar]
- D’Almeida V, Lobo LL, Hipolide DC, de Oliveira AC, Nobrega JN and Tufik S (1998) Sleep deprivation induces brain region-specific decreases in glutathione levels. Neuroreport 9, 2853–2856. [DOI] [PubMed] [Google Scholar]
- Davies KJ, Lin SW and Pacifici RE (1987) Protein damage and degradation by oxygen radicals. IV. Degradation and denatured proteins. J. Biol. Chem 262, 9914–9920. [PubMed] [Google Scholar]
- Dobson AW, Weber S, Dorman DC, Lash LK, Erikson KM and Aschner M (2003) Oxidative stress is induced in the rat brain following repeated inhalation exposure to manganese sulfate. Biol. Trace Elem. Res 93, 113–126. [DOI] [PubMed] [Google Scholar]
- Fabris G, Anselmo-Franci J and Branco LG (1999) Role ofnitric oxide in hypoxia-induced hyperventilation and hypothermia: participation of the locus coeruleus. Braz. J. Med. Biol. Res 32, 1389–1398. [DOI] [PubMed] [Google Scholar]
- Fletcher EC (1995) An animal model of the relationship between systemic hypertension and repetitive hypoxia as seen in sleep apnea. J. Sleep Res 4, 71–77. [DOI] [PubMed] [Google Scholar]
- Fletcher EC, Lesske J, Qian W, Miller CC and Unger T (1992) Repetitive episodic hypoxia causes diurnal elevations of systematic blood pressure in rats. Hypertension 19, 555–561. [DOI] [PubMed] [Google Scholar]
- Gozal D, Omidvar O, Kirlew KAT, Hathout GM, Hamilton R, Lufkin RB and Harper RM (1995) Identification of human brain regions underlying responses to inspiratory loading with functional magnetic resonance imaging. Proc. Natl Acad. Sci. USA 92, 6607–6611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gozal D, Daniel JM and Dohanich GP (2001) Behavioral and anatomical correlates of chronic episodic hypoxia during sleep in the rat. J. Neurosci 21, 2442–2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg HE, Sica AL, Scharf SM and Ruggiero DA (1999) Expression of c-fos in the rat brainstem after chronic intermittent hypoxia. Brain Res 816, 638–645. [DOI] [PubMed] [Google Scholar]
- Griffith OW (1980) Determination of glutathione and glutathione disulfide using glutathione reductase and 2-vinylpyridine. Anal. Biochem 106, 207–212. [DOI] [PubMed] [Google Scholar]
- Lavie P, Yoffe N, Berger I and Peled R (1993) The relationship between the severity of sleep apnea syndrome and 24-h blood pressure values in patients with obstructive sleep apnea. Chest 103, 717–721. [DOI] [PubMed] [Google Scholar]
- Li RC, Row BW, Kheirandish L, Brittian KR, Gozal E, Guo SZ, Sachleben LR Jr and Gozal D (2004) Nitric oxide synthase and intermittent hypoxia-induced spatial learning deficits in the rat. Neurobiol. Dis 17, 44–53. [DOI] [PubMed] [Google Scholar]
- Loredo JS, Clausen JL, Nelesen RA, Ancoli-Israel S, Ziegler MG and Dimsdale JE (2001) Obstructive sleep apnea and hypertension: are peripheral chemoreceptors involved? Med. Hypotheses 56, 17–19. [DOI] [PubMed] [Google Scholar]
- Lutherer LO,Lutherer BC,Dormer KJ, Janssen HF andBarnes CD (1983) Bilateral lesions of the fastigial nucleus prevent the recovery of blood pressure following hypotension induced by hemorrhage or administration of endotoxin. Brain Res 269, 251–257. [DOI] [PubMed] [Google Scholar]
- Lutherer LO and Williams JL (1986) Stimulating fastigial nucleus pressor region elicits patterned respiratory responses. Am. J. Physiol 250, R418–R426. [DOI] [PubMed] [Google Scholar]
- Macey PM, Henderson LA, Macey KE, Alger JR, Frysinger RC, Woo MA, Harper RK, Yan-Go FL and Harper RM (2002) Brain morphology associated with obstructive sleep apnea. Am. J. Respir. Crit. Care Med 166, 1382–1387. [DOI] [PubMed] [Google Scholar]
- Makarov PR, Wiswedel I, Augustin W and Schild L (2002) Hypoxia/reoxygenation-induced damage to mitochondrial activity is determined by glutathione threshold in astroglia-rich cell cultures. Brain Res 933, 91–97. [DOI] [PubMed] [Google Scholar]
- Misra HP and Fridovich I (1972) The role of superoxide anion in the autoxidation of epinephrine and a simple assay for superoxide dismutase. J. Biol. Chem 247, 3170–3175. [PubMed] [Google Scholar]
- Naegele B, Pepin JL, Levy P, Bonnet C, Pellat J and Feuerstein C (1998) Cognitive executive dysfunction in patients with obstructive sleep apnea syndrome (OSAS) after CPAP treatment. Sleep 21,392–397. [DOI] [PubMed] [Google Scholar]
- Ohkawa H, Ohishi N and Yagi K (1979) Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal. Biochem 95, 351–358. [DOI] [PubMed] [Google Scholar]
- Peiffer C, Poline JB, Thivard L, Aubier M and Samson Y (2001) Neural substrates for the perception of acutely induced dyspnea. Am. J. Respir. Crit. Care Med 163, 951–957. [DOI] [PubMed] [Google Scholar]
- Ramanathan L, Gulyani S, Nienhuis R and Siegel JM (2002) Sleep deprivation decreases superoxide dismutase activity in rat hippocampus and brainstem. Neuroreport 13, 1387–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Row BW, Liu R, Xu W, Kheirandish L and Gozal D (2003) Intermittent hypoxia is associated with oxidative stress and spatial learning deficits in the rat. Am. J. Respir. Crit. Care Med 167, 1548–1553. [DOI] [PubMed] [Google Scholar]
- Seppala T, Partinen M, Penttila A, Aspholm R, Tiainen E and Kaukianen A (1991) Sudden death and sleeping history among Finnish men. J. Intern. Med 229, 23–28. [DOI] [PubMed] [Google Scholar]
- Silverberg DS and Oksenberg A (1997) Essential hypertension and abnormal upper airway resistance during sleep. Sleep 20, 794–806. [PubMed] [Google Scholar]
- Smejkal V, Druga R. and Tintera J. (1999) Control of breathing and brain activation in human subjects seen by functional magnetic resonance imaging. Physiol. Res 48, 21–25. [PubMed] [Google Scholar]
- Somani SM and Husain K (1997) Interaction of exercise training and chronic ethanol ingestion on antioxidant system of rat brain regions. J. Appl Toxicol 17, 329–336. [DOI] [PubMed] [Google Scholar]
- Veasey SC, Davis CW, Fenik P, Zhan G, Hsu YJ, Pratico D. and Gow A (2004) Long-term intermittent hypoxia in mice: protracted hypersomnolence with oxidative injury to sleep-wake brain regions. Sleep 27, 194–201. [DOI] [PubMed] [Google Scholar]
- Xu F and Frazier DT (1997) Respiratory-related neurons of the fastigial nucleus in response to chemical and mechanical challenges. J. Appl. Physiol 82, 1177–1184. [DOI] [PubMed] [Google Scholar]
- Xu F and Frazier DT (2002) Role of the cerebellar deep nuclei in respiratory modulation. Cerebellum 1, 35–40. [DOI] [PubMed] [Google Scholar]
- Xu W, Chi L, Row BW, Xu R, Ke Y, Xu B, Luo C, Kheirandish L, Gozal D and Liu R (2004) Increased oxidative stress is associated with chronic intermittent hypoxia-mediated brain cortical neuronal cell apoptosis in a mouse model of sleep apnea. Neuroscience 126, 313–323. [DOI] [PubMed] [Google Scholar]
- Young T, Palta M, Dempsey J, Skatrud J, Weber S and Badr S (1993) Occurrence of sleep-disordered breathing among middle-aged adults. New Engl. J. Med 328, 1230–1235. [DOI] [PubMed] [Google Scholar]
- Yunoki M, Kawauchi M, Ukita N, Noguchi Y, Nishio S, Ono Y, Asari S, Ohmoto T, Asanuma M and Ogawa N (1998) Effects of lecithinized SOD and sequential change in SOD activity after cerebral contusion. Acta Neurochir 71, 142–145. [DOI] [PubMed] [Google Scholar]
- Zaninelli A, Fariello R, Boni E, Corda L, Alicandri C and Grassi V (1991) Snoring and risk of cardiovascular disease. Int. J. Cardiol 32, 347–351. [DOI] [PubMed] [Google Scholar]