

Abstract
Understanding the exquisitely complex nature of the three-dimensional organization of the genome and how it affects gene regulation remains a central question in biology. Recent advances in sequencing- and imaging-based approaches in decoding the three-dimensional chromatin landscape have enabled a systematic characterization of gene regulatory architecture. In this review, we outline how chromatin architecture provides a reference atlas to predict the functional consequences of non-coding variants associated with human traits and disease. High-throughput perturbation assays such as massively parallel reporter assays (MPRA) and CRISPR-based genome engineering in combination with a reference atlas opened an avenue for going beyond observational studies to experimentally validating the regulatory principles of the genome. We conclude by providing a suggested path forward by calling attention to barriers that can be addressed for a more complete understanding of the regulatory landscape of the human brain.
Keywords: Hi-C, chromatin architecture, gene regulatory landscape, SNP-gene relationship, GWAS, variant effects on gene regulation
1. Introduction
When measured linearly, the approximately 3 billion DNA base pairs comprising the human genome extend 2 meters in length. Despite the relatively large size as compared to other organisms, it contains a similar number of protein coding genes which account for just 2% of its total content. Traditionally, it was thought the other 98% of DNA sequences were “junk DNA”. However, it has become increasingly understood that many of these non-coding regions orchestrate gene regulation via functioning as enhancers, silencers, insulators, and/or non-coding RNAs. It is now estimated that these non-coding regions harbor ~90% of genetic variants associated with human diseases [1], and therefore, detailed characterization of the non-coding genome is imperative for advancing our understanding of disease biology. While large-scale efforts to characterize the non-coding genome such as ENCODE are actively underway [2], deciphering the functional consequences of non-coding variation has been challenging due to its vast size, limited conservation across species, and lack of a generalizable rubric to predict effects from sequence variation. The advent of next generation sequencing technologies and adaptations of genome engineering tools have enabled development of various multi-omic approaches that can be used to decode the regulatory grammar of the genome, or “regulome” [3]. One key approach for better interpreting the non-coding genome is generation of three-dimensional (3-D) contact maps of chromatin conformation, as the coordinated manner in which 2 meter-sized DNA is compacted into a nucleus approximately 200,000 times smaller in size is reflective of gene regulatory principles.
In this review, we highlight recent advances in available techniques for assaying chromosome conformation, which provides a roadmap to interrogate the functional consequences of non-coding genetic variants. We also introduce perturbation assays that complement chromatin architecture by experimentally validating variant effects on gene regulation. We conclude by describing key gaps in our knowledge of the dynamic gene regulatory landscape and how filling these gaps can provide a path forward for gleaning mechanistic understanding of disease pathogenesis.
2. Mapping Three-Dimensional Chromatin Configuration
Decoding the chromatin configuration remains a fundamental question in molecular biology. To date, one of the most popular genomic approaches to identify chromatin configuration is to use chromosome conformation capture (3C)-based techniques [4]. Hi-C is one such technique which profiles chromosome conformation at a genome-wide scale [5], and relies on fixation of DNA to preserve chromosome configuration, followed by restriction enzyme mediated digestion and proximity ligation to link points of contact. The resulting genomic libraries are then subjected to next generation sequencing (NGS).
Development of Hi-C has led to detailed characterization of chromatin organization. Intrachromosomal DNA interactions occur more frequently than inter-chromosomal interactions, which is consistent with chromosome territories [6]. These intra-chromosomal interactions can be further broken into different types of architectural units. A and B compartments, which are megabase-sized cell-type specific chromatin structures, mark open and closed regions of chromatin, respectively [5]. Compartments can be further partitioned into finer domain structure called topologically associating domains (TADs) [7]. TADs are demarcated by specific boundary elements such as CCCTC-binding factor (CTCF) and cohesin, and represent regions in which chromosomal contacts occur more frequently with one another compared to nearby regions in the genome [8]. At higher resolution, TADs often exhibit nested structures and the emergence of subTADs have been observed [9]. SubTADs are similar to TADs; however, their insulation strength is much weaker, thus limiting their ability to hinder long-range contacts between domains [10]. In addition, frequently interacting regions (FIREs) have been recently described. As evidenced by their name, FIREs have significantly more chromatin interactions than would be expected by random chance, and are thought to play an integral role in gene regulation via acting as interaction hubs [11,12]. So far, the finest structures identified by Hi-C are loops, which are formed between two genomic loci coming in contact with one another. One of the most well characterized DNA loops include enhancer-promoter interactions, which play a central function in gene regulation [13]. DNA loops are highly cell-type specific and can form transiently, thus representing a dynamic process whereby segments of DNA are brought into contact in a coordinated manner [14]. Collectively, these findings indicate that chromatin is organized in a way that it defines the gene regulatory principles.
Despite the fact that Hi-C has played a pivotal role in deciphering the chromatin architectural units, key factors such as resolution, sequencing depth, and cost have limited its utility. Additionally, the large number of cells required to generate a high-resolution contact map has prohibited investigation of rare cell types and cell populations. To address these remaining challenges, modifications have been made to traditional Hi-C protocols (Figure 1).
Figure 1: Hi-C and its derivative techniques.
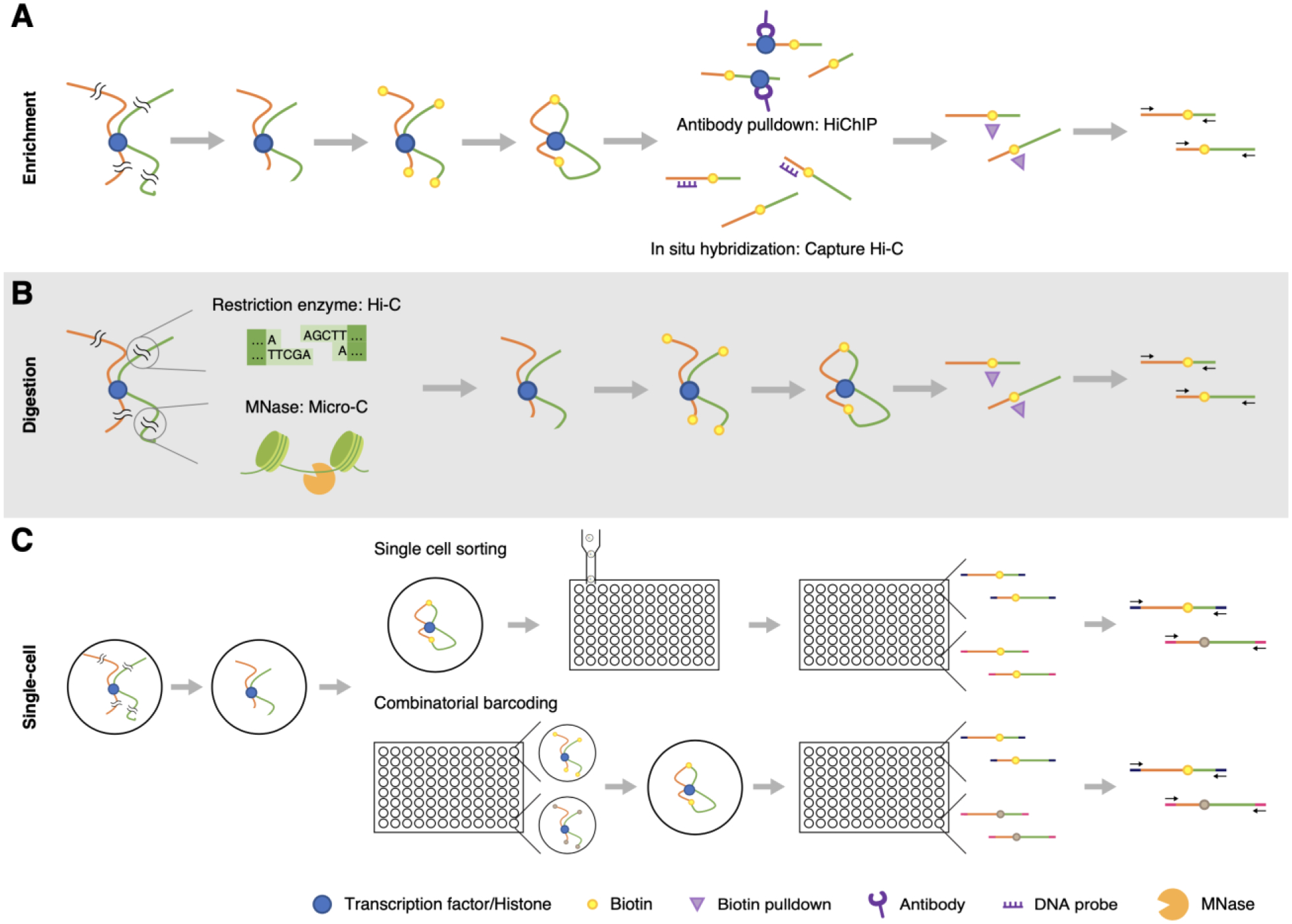
A. Enrichment strategies using either an antibody or in situ hybridization. B. Digestion strategies using either restriction enzyme sites or MNase for unbiased fragmentation at nucleosome resolution. C. Single-cell Hi-C approaches based on flow sorting or combinatorial indexing.
High resolution maps with relatively low sequencing depth can be acquired by enriching for chromatin interactions of interest. HiChIP and PLAC-seq utilize this approach by pulling down chromatin interactions bound to specific antibodies (Figure 1A) [15,16]. Anti-H3K27ac and anti-H3K4me3 are frequently used to capture active gene regulatory elements. Alternatively, capture Hi-C employs a hybridization step to enrich chromatin interactions anchored to specific sequences of interest (e.g. promoter sequences, genetic risk factors, Figure 1A) [17]. In addition to increasing the resolution at a relatively lower cost, these techniques provide an avenue for multi-omic profiling of gene-regulatory relationships. Recently, another multi-omic approach that combined Hi-C, ATAC-seq, and RNA-seq has been proposed [18]. This technique, called HiCAR, can be used to survey transcriptomic signatures simultaneously with gene-regulatory chromatin architecture.
Different strategies for DNA digestion can also be used to increase the resolution. One such technology is Micro-C, which utilizes an MNase digestion to enable unbiased fragmentation at a single-nucleosome resolution (Figure 1B). Employing this technology, researchers have now begun to detect more proximal interactions than what is possible using Hi-C [19,20]. Micro-C also enables the investigation of chromatin interactions that involve regions depleted of restriction enzyme sites, allowing for a more complete genome-wide chromosome conformation map.
In addition to modifications to experimental procedures, computational frameworks to construct high-resolution contact maps from low-resolution data have been developed. Hi-C Interaction Frequency Inference (HIFI) algorithms leverage the dependencies between neighboring restriction fragments to reduce the stochastic nature of interaction frequencies, and can reliably infer interaction frequencies at restriction fragment resolution in contrast to traditional methods that rely on binning raw data into intervals of a fixed size [21]. DeepHiC is another computational framework that uses a generative adversarial network to refine the resolution of low-coverage contact matrices, and is capable of successfully reconstructing high-resolution matrices with as few as 1% downsampled reads [22]. A third computational program to enhance the resolution of coarse Hi-C datasets is HiCPlus. This program utilizes a convolutional neural network that is trained on high resolution (10kb with deep sequencing) Hi-C data [23]. HiCPlus is capable of accurately mapping low resolution datasets using 1/16th of the reads of higher resolution datasets. These computational frameworks collectively allow for reduced sequencing costs since fewer reads are sufficient to generate high-resolution matrices, thus addressing a major barrier in widely adopting Hi-C approaches.
Alternative approaches that are independent of 3C-based techniques have also been developed to interrogate chromatin configuration, such as Genome Architecture Mapping (GAM) [24] and split-pool recognition of interactions by tag extension (SPRITE) [25]. GAM is based on ultra-thin cryosectioning of tissue samples followed by laser capture microdissection of nuclear profiles. These samples are then amplified and subjected to NGS to identify DNA segments in close contact. The development of GAM provided the first genome-wide chromosome conformation maps without relying on proximity ligation. Similarly, SPRITE provides a proximity-ligation free approach to mapping 3-D genome organization. However, it does not require any specialized equipment. Instead, SPRITE incorporates a barcode to each molecule within a cross-linked complex, and the samples are continuously split across 96-well plates such that each complex contains a unique set of molecular tags through sequential barcode additions. In contrast to GAM, SPRITE does not require whole genome amplification, providing the flexibility to incorporate RNA contacts in addition to DNA [25]. As GAM and SPRITE do not rely on pairwise interactions, they can capture higher-order chromatin configuration such as multivalent interactions [24,25]. A more comprehensive comparison of Hi-C, GAM, and SPRITE can be found here [26].
Complementary to genomic approaches, rapid advances in imaging techniques allowed visualization and validation of chromatin architectural units [27]. Super-resolution microscopy enabled the detection of structures as small as 10nm, which corresponds to the size of an individual nucleosome. Super-resolution imaging combined with fluorescence in situ hybridization (FISH) has therefore emerged as a promising avenue to visualize chromatin configuration inside of the nucleus. Oligopaint is one such super-resolution chromatin tracing tool [28]. It employs short single-stranded oligos labeled with a fluorophore to visualize configuration of genomic regions that span from a few kb to tens of kb in size [29]. Importantly, imaging intrinsically characterizes chromatin architecture at a single cell resolution, hence providing a direct measure for cell-to-cell variability in chromosome conformation. For example, super-resolution FISH demonstrated that TADs are not conserved at the level of individual cells but rather an emergent property of a population of cells [30]. Further advances in imaging-based assays have emerged to enable multi-omic investigation within single cells. One such approach, DNA seqFISH+, allows for the simultaneous profiling of chromatin architecture, chromatin marks, and expression of RNA [31].
To better understand the cell-to-cell variability in chromosome conformation and to bridge the gap between high-throughput bulk Hi-C techniques and low-throughput single-cell imaging techniques, different iterations of single-cell Hi-C have been developed. The first successful attempt of single-cell Hi-C relied on traditional steps of Hi-C (e.g., crosslinking, digestion, biotin labeling, and proximity ligation), with these steps being performed in nuclei rather than nuclear lysates. After isolation under a microscope and physical separation into individual tubes, 74 nuclei were coupled to a unique cellular barcode [32]. This proof of principle method set the stage for future modifications that have massively increased the throughput of this technique. For example, utilizing flow cytometry to isolate individual cells and introducing a partially automated library preparation significantly increased throughput (Figure 1C) [33]. Another advance was made by the application of a combinatorial indexing strategy with Hi-C, called single-cell combinatorial indexed Hi-C (sci-HiC) [34]. Sci-HiC obviates the need for physically separating nuclei by implementing successive rounds of split-pool barcoding, by which each nucleus receives a unique molecular index (Figure 1C). This strategy drastically increased the scale and throughput at which single-cell Hi-C could be performed, such that the chromosome conformation could be interrogated in thousands of individual cells simultaneously [34]. Since its original development, modifications have been made to simplify the protocol and lower the cost [35]. This methodology may enable systematic characterization of chromosome conformation at a single-cell resolution, which can be expanded to construct cell-type specific chromosome conformation atlases [36,37]. Single-cell Hi-C can be further coupled with other epigenomic profiling that can complement the Hi-C approach to inform cell types. Methyl-HiC and single-nucleus methyl-3C sequencing (sn-m3C-seq) are such techniques that simultaneously capture methylome and chromosome conformation [38,39]. ChIA-drop is a droplet based multiplexing approach that enables simultaneous enrichment for fragments containing specific histone marks or RNA Polymerase II while also mapping points in physical contact [40]. Methylation or histone signatures can be used to identify the cell type, based on which chromosome conformation can be aggregated to construct cell-type specific chromosome conformation atlases.
Collectively, recent advances in the genomic toolbox allow us to study the full complexity of genome organization. Once decoded, chromatin configuration can become a roadmap to understand the functional impact of non-coding regulatory elements and variants associated with human traits and diseases.
3. Gene regulatory architecture functions as a blueprint for inferring SNP-Gene Relationships
Genome-wide association studies (GWAS) and whole genome sequencing (WGS) studies have identified hundreds to thousands of genetic variants associated with various human traits and diseases [41,42]. The vast majority of these variants are located in the non-coding genome, and high-resolution maps of chromatin architecture may function as a reference atlas to decipher the functional consequences of these risk variants. The basic assumption is that risk variants can regulate distal genes when they are in physical contact via chromatin loops. Therefore, identification of genes that physically interact with risk variants is often the first step to predict their functional consequences.
Here, we outline how chromatin architecture serves as a reference atlas to infer variant-gene relationships. Variants that are associated with a phenotype of interest can be queried by GWAS, a type of study that investigates the association between single nucleotide polymorphisms (SNPs) and a particular trait. To infer variant-gene relationships from GWAS, we first need to identify likely causal variants. This is because GWAS identifies genome-wide significant (GWS) loci (p<5×10−8) rather than individual variants that are associated with a given trait. Each GWS locus contains up to hundreds of genetic risk variants with unknown functional consequences. Statistical finemapping analyses predict a credible set of SNPs that represent likely causal variants [43,44]. Once credible SNPs are determined, chromatin loops overlapping with these SNPs can be used to identify putative target genes by looking at the contacts on the other side of the loop. While this approach is powerful for identifying the cognate genes of non-coding variants, it relies on computational finemapping algorithms to predict credible SNPs. However, different finemapping algorithms can give different sets of credible SNPs [45] and identifying which of the SNPs is functional requires high-throughput experimental validation (see section 4 for more detail). Moreover, only a small proportion of heritability can be explained by GWS variants [1], hence functional annotation of GWS loci is limited in providing the full picture of disease mechanisms. To address this, tools have been developed to investigate the contribution of SNPs to diseases or traits in a genome-wide fashion.
Multimarker analysis of genomic annotation (MAGMA) is an unbiased approach that has been developed to functionally annotate GWAS in a genome-wide manner [46,47]. MAGMA leverages gene-SNP relationships to aggregate SNP-based association statistics to corresponding genes. The resulting gene-level association statistics could (1) partially explain the heritability of traits arising from loci that do not meet the GWS threshold and (2) easily be translated to disease biology. As original MAGMA simply assigned SNPs to their nearest genes without accounting for gene regulatory relationships, it has since been coupled with Hi-C data (H-MAGMA) to take into account the 3-D organization of chromatin architecture in converting SNP-level summary statistics to gene-level associations [48]. H-MAGMA often assigned SNPs as interacting with distal genes, showing the importance of factoring in chromatin architecture in predicting the functional outcome of non-coding risk variants. Further improvements can be made by incorporating additional functional genomic datasets such as cis-regulatory elements (CREs) defined on the basis of chromatin accessibility and/or histone modification profiles. Since genome-wide chromatin interactions do not necessarily distinguish active versus repressive interactions, adding CREs can further refine gene-SNP relationships. CREs can help improve the resolution of Hi-C data because compared to Hi-C (high resolution Hi-C has 5–10kb windows), CREs are defined in narrower windows (100–500bp). Furthermore, ATAC-seq data is now available in more diverse cell types compared to Hi-C, allowing the development of cell-type specific gene-SNP relationships by integrating Hi-C with ATAC-seq data. For example, neuronal Hi-C data can be coupled with glutamatergic and GABAergic H3K27ac data to investigate neuronal subtype specific disease mechanisms [49].
Similarly, the newly developed activity by contact (ABC) model leverages a multi-omic framework to infer enhancer-gene relationships [50]. The model accurately predicts the effect of enhancers on gene expression by taking into account the enhancer activity (measured by the peak size of ATAC-seq or ChIP-seq) and chromatin contact frequency between enhancer and gene pairs (measured by Hi-C). The regulatory impact of a genomic element on a given gene was then measured by CRISPRi-FlowFISH (discussed in section 4) to build a predictive model for enhancer-gene relationship. Notably, the ABC model was shown to perform substantially better than other methodologies in inferring enhancer-gene relationships, such as assigning an enhancer to its nearest target gene based on linear genomic distance and other predictive algorithms such as TargetFinder or JEME [50]. Based on its strong predictive power, the ABC model has been recently proposed to predict SNP-gene relationship [51]. The ABC model is an example of how integrative analyses of multiple genomic datasets can better refine SNP-gene relationships.
Other approaches are available to integrate chromosomal conformation data with expression quantitative trait loci (eQTLs) to further characterize SNP function. One such approach, Contextualising Developmental SNPs in Three Dimensions (CoDeS3D), integrates Hi-C data with SNPs identified through GWAS to first identify SNP-gene interactions. Once completed, these SNP-gene pairs are integrated with eQTL to infer SNPs that alter gene expression [52].
Collectively, integration of multi-layered functional genomic data that encompass CREs and chromatin interactions can aid in the accurate prediction of variant effects on genome function.
4. Experimental Validation to Elucidate Functional Impact of SNPs
Despite tremendous strides in predictive methodologies for inferring SNP-gene relationships, a gap remains in experimentally validating the functional outcome of these relationships. Therefore, the next fundamental step is to functionally characterize the variant effects on gene regulation. However, traditional approaches such as luciferase assays and single gene knock-out experiments are no longer suitable for systematic characterization of thousands of genetic variants associated with human traits and disease. Recent advances in high-throughput technologies have enabled systematic evaluation of the impact of genetic variation on gene regulation.
Massively parallel reporter assays (MPRA) are such technologies that can quantify gene regulatory activity of putative regulatory elements and variants in a high-throughput manner (Figure 2). MPRA introduces a vector containing a putative regulatory element, promoter of interest, reporter gene, and unique molecular barcode to the cells. RNA is extracted from the cells that take up the vector, and molecular barcodes, which provide a direct readout for reporter gene expression, are quantified via NGS. Each molecular barcode can be paired with the regulatory element, so that it allows quantification of regulatory activity of thousands of regulatory elements in a single experiment. Depending on the design of regulatory elements inserted into the vector, MPRA can be used to study enhancer activity of putative regulatory elements [53], repressor activity of putative silencers [54], and variant effects on gene expression [55–57]. Alternatively, different promoter sequences can be inserted to screen for promoter activity [58] and different combinations of enhancers and promoters can be used to study enhancer-promoter interactions. Furthermore, MPRA can be coupled with CRISPR-mediated perturbation of transcription factors (TFs) to investigate trans-regulatory effects of TFs on enhancers [59]. Adeno-associated virus (AAV) and lentivirus based MPRA have been developed [60,61] which have opened the door to performing MPRA within cells that more closely mirror the endogenous context of the cells harboring the variant of interest. However, limitations still exist. One such drawback is the limitation in size of the regulatory element being explored. MPRA typically uses 150–200bp synthetic oligos which may not fully capture the entire regulatory region. This limitation can be partially overcome by adopting tiling strategies to lengthen the region of interest [62]. Another drawback is the inability of these assays to (1) fully recapitulate the 3-D chromatin architecture surrounding the regulatory region and (2) identify target genes influenced by the regulatory elements. Therefore, 3-D genome architecture can be used in parallel with MPRA to delineate the regulatory context and putative target genes of functionally characterized elements and/or variation. Lastly, traditional MPRA experiments performed in vitro may fail to mirror the complex cellular interactions occurring in tissues. Recent advances in the MPRA design that can be administered in vivo can overcome this issue and fully capture cellular dynamics of regulatory elements of interest [60,63,64].
Figure 2. MPRA can characterize variant effects on gene regulation in a high-throughput manner.
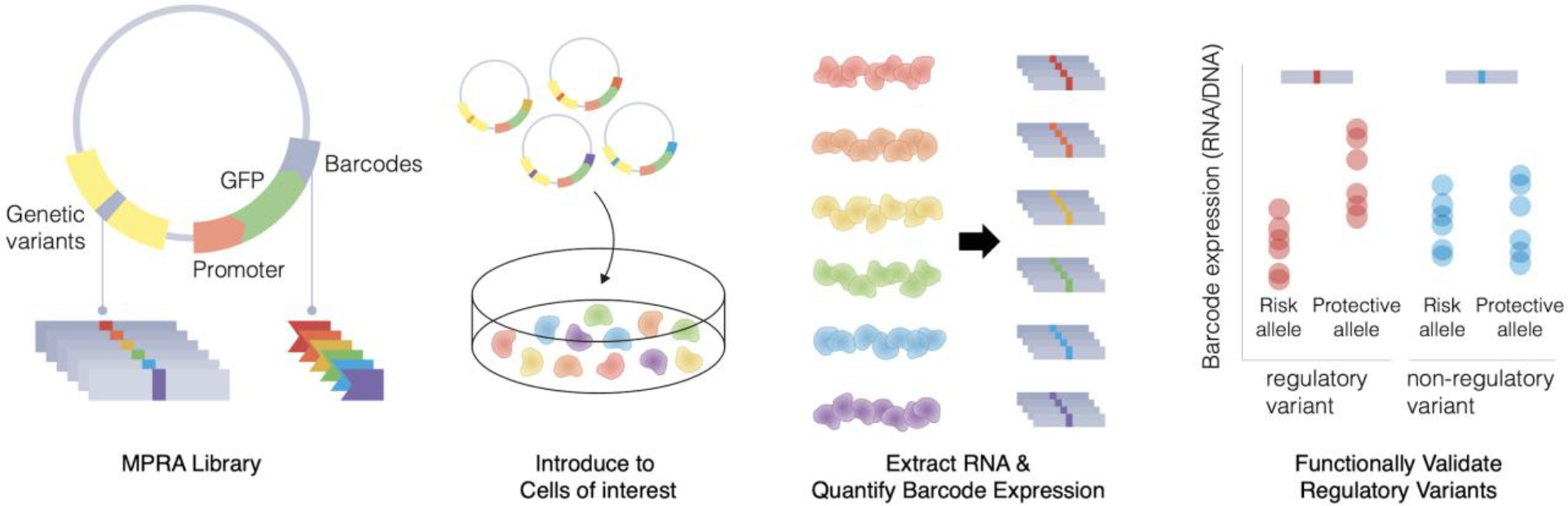
MPRA libraries consist of a regulatory element that contains a genetic variant, promoter, reporter gene (GFP), and 10–20bp barcodes. These libraries are introduced to the cells of interest, from which RNA is extracted. Barcode expression is quantified to measure the variant effects on reporter gene expression.
Advances in CRISPR engineering provide additional complementary approaches for studying gene regulation. CRISPR techniques, such as CRISPRi-FlowFISH, have been developed to provide a functional readout following perturbations. For example, this assay utilizes cells expressing catalytically inactive Cas9 (dCas9) fused to a transcriptionally repressive Krüppel associated box domain (KRAB). Guide RNAs (gRNA) delivered to the cells direct the dCas9-KRAB complex to putative regulatory elements, rendering the elements inactive. Next, RNA FISH is used with probes targeting a particular gene of interest, and cells are flow sorted into bins based on levels of fluorescence. This strategy enables the quantitative measure of the effects of inhibiting putative regulatory elements on a given gene [50]. Until recently, however, the precise introduction of genetic variants has been challenging, obfuscating its application in the study of SNP-gene relationships. A newly developed method, prime editing, utilizes dCas9 fused to a reverse transcriptase engineered to contain a prime editing gRNA [65]. This enables the detection of precise genomic locations while encoding instructions to generate the desired edit. This has shown to be efficacious in human cells, and successfully introduced a large number of targeted genomic variations including insertions, deletions, and point mutations without inducing double-strand breaks. Prime editing opens an avenue to correct nearly 90% of known genetic variants associated with human disease, and can be widely used to study the effect of a SNP on gene function without knocking out the gene in its entirety.
In addition to the use of CRISPR in functional validation of gene regulatory relationships, it can be used to change the 3-D architecture of chromatin folding. Light-activated dynamic looping (LADL) utilizes dCas9 fused to a truncated version of the CIB1 protein (CIBN). Once two gRNAs that target distal sites are introduced, dCas9-CIBN is recruited to those target sites. Upon exposure to blue light, cryptochrome 2 (CRY2), a blue light receptor, gets activated and heterodimerizes with CIBN, then self oligomerizes to bring two genomic regions into physical contact. Therefore, this technique enables precise spatiotemporal manipulations to the 3-D genome, which can be used to interrogate the impact of chromatin architectural changes on gene regulation [66].
Taken together, these approaches can begin to validate SNP-gene relationships that have been inferred based on chromatin structure. For example, the utility of prime editing to precisely insert variation of interest, followed by transcriptomic profiling and/or cellular assays will not only help in assigning variation to genes, but also elucidate the consequence of variation on gene regulation and cellular function. In future studies, large consortia using the combination of these approaches can begin to catalog SNP-gene relationships en masse in an attempt to better understand the contribution of individual variation to genome function and human traits. Importantly, this opens the possibility of building predictive models that can be extended to SNPs not yet characterized.
5. Toward a More Complete Map of Gene Regulatory Architecture in the Human Brain
Chromatin configuration of the human brain has been profiled across key developmental epochs and cell types (Table 1). These rich datasets of 3-D chromatin architecture generated from the human brain provided a cornerstone for predicting the functional impact of non-coding variants and elucidating biological underpinnings of various psychiatric and neurodegenerative disorders [48,67–70]. Despite these resources, we are far from understanding the comprehensive landscape of gene regulation in the human brain.
Table 1.
Available resources of chromosome conformation in the human brain.
| Reference | Technique | Brain Tissue | Cell types | Stage |
|---|---|---|---|---|
| Won et al. [71] | Hi-C | Frontoparietal cortex | Germinal zone, Cortical plate | Fetal: GW 17–18 |
| Song et al. [72] | PLAC-seq | Cortex | Radial glia, IPC, Excitatory and inhibitory neuron | Fetal: GW 17–21 |
| Nott et al. [69] | PLAC-seq | Frontal, Temporal, Parietal cortex | Neuron, Microglia, Oligodendrocyte | Pediatric: 5 Months – 17 Years |
| Wang et al. [67] | Hi-C | DLPFC | Brain homogenate | Adult: 36–64 Years |
| Jung et al. [73] | Promoter- capture Hi-C | DLPFC, Hippocampus | Brain homogenate | Adult |
| Hu et al. [49] | Hi-C | DLPFC | Neuron, Glia | Adult: 36–64 Years |
| Espeso-Gil et al. [74] | Hi-C | Midbrain | Dopaminergic neuron, Glia | Adult: 35–60 Years |
GW, gestation week; DLPFC, dorsolateral prefrontal cortex; IPC, intermediate progenitor cells.
Currently, the majority of Hi-C data has been obtained from the cortex due to the tissue availability and its role in various brain disorders (Table 1). However, a range of subcortical regions may also contribute to human traits and disease. For example, subcortical regions such as the ventral tegmental area (VTA) and nucleus accumbens (NAc) are essential components of the reward circuitry and play an integral role in substance use disorders [75]. Similarly, the basal ganglia circuitry was found to be the central neurocircuitry underlying Parkinson’s disease [76]. Recently reported gene regulatory landscapes of midbrain dopaminergic neurons provides an example of how 3-D genome architecture of previously uncharacterized brain regions and cell types may yield novel insights into the pathogenic mechanisms of brain disorders [74].
Another limitation for currently available Hi-C data is the limited cell-type resolution it can provide. So far, enhancer-promoter interactions have been constructed from four cell types (radial glia, intermediate progenitor cells, excitatory and inhibitory neurons) in the fetal cortex [72], three cell types (neurons, oligodendrocytes, microglia) in the pediatric cortex [69] and two cell types (neurons and glia) in the adult cortex [49]. This reconstruction of cell-type specific 3-D regulome was dependent on a FACS-based cell sorting procedure. However, molecular classification of cell types informed by single-cell genomics has revealed a more complex compendium of cellular subtypes. To date, at least 16 neuronal subtypes with distinct molecular architecture and disease associations have been identified [77]. Similarly, multiple astrocytic populations have been observed through scRNA-seq analyses, from which disease-associated astrocytes form a molecularly distinct cluster [78]. Collectively, over 50 different cell types were detected in the mouse brain [79], suggesting that the cell type diversity of the human brain is far more complex than our current understanding. These more refined cellular subtypes are challenging to sort into distinct populations via FACS, resulting in cell-type specific Hi-C maps still being representative of the average architecture amongst all classes of a given cell type (e.g. neurons). Therefore, the development of single-cell Hi-C methods coupled with other omic approaches is necessary to disentangle the full complexity of cell-type specific chromatin architecture and its relationship with cell-type specific disease vulnerability.
In addition to cellular complexity, another important property of brain cells is that they are responsive to activity, whether it be from neuromodulation, stress, and/or signaling pathways. Despite this, our understanding of the chromatin architecture in the brain is largely based on static pictures. Temporal changes in chromatin architecture upon neuronal activation have been reported, suggesting that neuronal activation-induced gene regulation is coupled with activity-dependent chromatin modification [80]. These findings highlight the importance of investigating brain regulome under the context of environmental stimulation.
Additional important bottlenecks in building comprehensive chromatin architecture of the human brain stem from developmental changes in the gene regulatory landscape. To date, available Hi-C datasets have primarily been limited to mid-gestational and adult brain samples (Table 1). These stages do not encompass the critical window of many neuropsychiatric and neurodevelopmental disorders. For example, schizophrenia is an adolescence-onset disorder, while chromatin architecture around this critical developmental period has not been well characterized. Moreover, early postnatal brain development was shown to have predictive power for later autism diagnosis [81], whereas Hi-C data from this developmental stage is lacking. To address this gap, systematic characterization of 3-D genomic architecture across brain development is imperative.
For the developmental epochs that are challenging to profile, organoids, three-dimensional tissues that self-organize from stem cells in culture, can provide an attractive alternative. Organoids have proven to have great utility in studying brain disease and development [82], but the degree to which organoids accurately model biological processes in humans warrants further investigation. In one study, single-cell RNA-seq was employed to determine the fidelity of cortical cell types in organoids compared with the developing human cortex. While organoids showed molecular signatures of broad cell classes, there was a notable impairment in subtype specification. This finding prompted the authors to urge researchers to consider this lack of specificity when using organoids as a model for studying cell-type specific disease phenotypes [83]. Somewhat contradictory to this finding, a comprehensive epigenomic comparison between primary human forebrain tissue and 3-D forebrain organoids demonstrated that chromatin accessibility during forebrain development is well-modeled in organoids. Chromatin accessibility profiles in organoids were used to annotate genetic variants implicated in neuropsychiatric disorders, as well as to identify key transcription factors potentially driving a robust change in chromatin remodeling during cortical development [84]. Taken together, the degree to which organoids mirror primary tissue is an active area of investigation, and organoids that can faithfully model human brain development can open a promising avenue to study chromatin architecture across a fine-grained developmental trajectory [85].
In summary, addressing these gaps in our knowledge will refine the cellulo-spatio-temporal resolution of 3-D gene regulatory architecture of the human brain. Moving forward, the generation of atlas level resources in different brain regions, at different developmental epochs, across molecularly refined cell types, and under activated cellular states will serve as a foundation for elucidating mechanistic underpinnings of neuropsychiatric disorders, which in turn provide avenues for unexplored therapeutic and translational studies.
Acknowledgements
This review was supported by the National Institute of Mental Health (R00MH113823, DP2MH122403, H.W.), the National Institute on Drug Abuse (R21DA051921, H.W.), the Pharmacological Sciences T32 Training Program (5T32GM135095, B.M.P.), and the NARSAD Young Investigator Award from the Brain and Behavior Research Foundation (H.W.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Competing Interest
Authors declare no conflict of interest.
Reference
- [1].Watanabe K, Stringer S, Frei O, Umićević Mirkov M, de Leeuw C, Polderman TJC, van der Sluis S, Andreassen OA, Neale BM, Posthuma D, A global overview of pleiotropy and genetic architecture in complex traits, Nat. Genet 51 (2019) 1339–1348. [DOI] [PubMed] [Google Scholar]
- [2].ENCODE Project Consortium, Moore JE, Purcaro MJ, Pratt HE, Epstein CB, Shoresh N, Adrian J, Kawli T, Davis CA, Dobin A, Kaul R, Halow J, Van Nostrand EL, Freese P, Gorkin DU, Shen Y, He Y, Mackiewicz M, Pauli-Behn F, Williams BA, Mortazavi A, Keller CA, Zhang X-O, Elhajjajy SI, Huey J, Dickel DE, Snetkova V, Wei X, Wang X, Rivera-Mulia JC, Rozowsky J, Zhang J, Chhetri SB, Zhang J, Victorsen A, White KP, Visel A, Yeo GW, Burge CB, Lécuyer E, Gilbert DM, Dekker J, Rinn J, Mendenhall EM, Ecker JR, Kellis M, Klein RJ, Noble WS, Kundaje A, Guigó R, Farnham PJ, Cherry JM, Myers RM, Ren B, Graveley BR, Gerstein MB, Pennacchio LA, Snyder MP, Bernstein BE, Wold B, Hardison RC, Gingeras TR, Stamatoyannopoulos JA, Weng Z, Expanded encyclopaedias of DNA elements in the human and mouse genomes, Nature. 583 (2020) 699–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Consortium, Roadmap Epigenomics, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, Ziller MJ, Amin V, Whitaker JW, Schultz MD, Ward LD, Sarkar A, Quon G, Sandstrom RS, Eaton ML, Wu YC, Pfenning AR, Wang X, Claussnitzer M, Liu Y, Coarfa C, Harris RA, Shoresh N, Epstein CB, Gjoneska E, Leung D, Xie W, Hawkins RD, Lister R, Hong C, Gascard P, Mungall AJ, Moore R, Chuah E, Tam A, Canfield TK, Hansen RS, Kaul R, Sabo PJ, Bansal MS, Carles A, Dixon JR, Farh KH, Feizi S, Karlic R, Kim AR, Kulkarni A, Li D, Lowdon R, Elliott G, Mercer TR, Neph SJ, Onuchic V, Polak P, Rajagopal N, Ray P, Sallari RC, Siebenthall KT, Sinnott-Armstrong NA, Stevens M, Thurman RE, Wu J, Zhang B, Zhou X, Beaudet AE, Boyer LA, De Jager PL, Farnham PJ, Fisher SJ, Haussler D, Jones SJ, Li W, Marra MA, McManus MT, Sunyaev S, Thomson JA, Tlsty TD, Tsai LH, Wang W, Waterland RA, Zhang MQ, Chadwick LH, Bernstein BE, Costello JF, Ecker JR, Hirst M, Meissner A, Milosavljevic A, Ren B, Stamatoyannopoulos JA, Wang T, Kellis M, Integrative analysis of 111 reference human epigenomes, Nature. 518 (2015) 317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Dekker J, Marti-Renom MA, Mirny LA, Exploring the three-dimensional organization of genomes: interpreting chromatin interaction data, Nat. Rev. Genet 14 (2013) 390–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, Sandstrom R, Bernstein B, Bender MA, Groudine M, Gnirke A, Stamatoyannopoulos J, Mirny LA, Lander ES, Dekker J, Comprehensive mapping of long-range interactions reveals folding principles of the human genome, Science. 326 (2009) 289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Cremer T, Cremer M, Chromosome territories, Cold Spring Harb. Perspect. Biol 2 (2010) a003889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B, Topological domains in mammalian genomes identified by analysis of chromatin interactions, Nature. 485 (2012) 376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Fudenberg G, Imakaev M, Lu C, Goloborodko A, Abdennur N, Mirny LA, Formation of Chromosomal Domains by Loop Extrusion, Cell Rep. 15 (2016) 2038–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Phillips-Cremins JE, Sauria MEG, Sanyal A, Gerasimova TI, Lajoie BR, Bell JSK, Ong C-T, Hookway TA, Guo C, Sun Y, Bland MJ, Wagstaff W, Dalton S, McDevitt TC, Sen R, Dekker J, Taylor J, Corces VG, Architectural protein subclasses shape 3D organization of genomes during lineage commitment, Cell. 153 (2013) 1281–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Beagan JA, Phillips-Cremins JE, On the existence and functionality of topologically associating domains, Nat. Genet 52 (2020) 8–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Schmitt AD, Hu M, Jung I, Xu Z, Qiu Y, Tan CL, Li Y, Lin S, Lin Y, Barr CL, Ren B, A Compendium of Chromatin Contact Maps Reveals Spatially Active Regions in the Human Genome, Cell Rep. 17 (2016) 2042–2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Crowley C, Yang Y, Qiu Y, Hu B, Abnousi A, Lipiński J, Plewczyński D, Wu D, Won H, Ren B, Hu M, Li Y, FIREcaller: Detecting Frequently Interacting Regions from Hi-C Data, Comput. Struct. Biotechnol. J (2020). 10.1016/j.csbj.2020.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Rao SSP, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, Aiden EL, A 3D Map of the Human Genome at Kilobase Resolution Reveals Principles of Chromatin Looping, Cell. 159 (2014) 1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Eagen KP, Principles of Chromosome Architecture Revealed by Hi-C, Trends Biochem. Sci 43 (2018) 469–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Mumbach MR, Satpathy AT, Boyle EA, Dai C, Gowen BG, Cho SW, Nguyen ML, Rubin AJ, Granja JM, Kazane KR, Wei Y, Nguyen T, Greenside PG, Corces MR, Tycko J, Simeonov DR, Suliman N, Li R, Xu J, Flynn RA, Kundaje A, Khavari PA, Marson A, Corn JE, Quertermous T, Greenleaf WJ, Chang HY, Enhancer connectome in primary human cells identifies target genes of disease-associated DNA elements, Nat. Genet 49 (2017) 1602–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Fang R, Yu M, Li G, Chee S, Liu T, Schmitt AD, Ren B, Mapping of long-range chromatin interactions by proximity ligation-assisted ChIP-seq, Cell Res. 26 (2016) 1345–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Mifsud B, Tavares-Cadete F, Young AN, Sugar R, Schoenfelder S, Ferreira L, Wingett SW, Andrews S, Grey W, Ewels PA, Herman B, Happe S, Higgs A, LeProust E, Follows GA, Fraser P, Luscombe NM, Osborne CS, Mapping long-range promoter contacts in human cells with high-resolution capture Hi-C, Nat. Genet 47 (2015) 598–606. [DOI] [PubMed] [Google Scholar]
- [18].Wei X, Xiang Y, Shan R, Peters DT, Sun T, Lin X, Li W, Diao Y, Multi-omics analysis of chromatin accessibility and interactions with transcriptome by HiCAR, Cold Spring Harbor Laboratory. (2020) 2020.11.02.366062. 10.1101/2020.11.02.366062. [DOI] [Google Scholar]
- [19].Hsieh T-HS, Cattoglio C, Slobodyanyuk E, Hansen AS, Rando OJ, Tjian R, Darzacq X, Resolving the 3D Landscape of Transcription-Linked Mammalian Chromatin Folding, Mol. Cell 78 (2020) 539–553.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Hsieh T-HS, Fudenberg G, Goloborodko A, Rando OJ, Micro-C XL: assaying chromosome conformation from the nucleosome to the entire genome, Nat. Methods 13 (2016) 1009–1011. [DOI] [PubMed] [Google Scholar]
- [21].Cameron CJ, Dostie J, Blanchette M, HIFI: estimating DNA-DNA interaction frequency from Hi-C data at restriction-fragment resolution, Genome Biol. 21 (2020) 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Hong H, Jiang S, Li H, Du G, Sun Y, Tao H, Quan C, Zhao C, Li R, Li W, Yin X, Huang Y, Li C, Chen H, Bo X, DeepHiC: A generative adversarial network for enhancing Hi-C data resolution, PLoS Comput. Biol 16 (2020) e1007287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Zhang Y, An L, Xu J, Zhang B, Zheng WJ, Hu M, Tang J, Yue F, Enhancing Hi-C data resolution with deep convolutional neural network HiCPlus, Nat. Commun 9 (2018) 750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Beagrie RA, Scialdone A, Schueler M, Kraemer DCA, Chotalia M, Xie SQ, Barbieri M, de Santiago I, Lavitas L-M, Branco MR, Fraser J, Dostie J, Game L, Dillon N, Edwards PAW, Nicodemi M, Pombo A, Complex multi-enhancer contacts captured by genome architecture mapping, Nature. 543 (2017) 519–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Quinodoz SA, Ollikainen N, Tabak B, Palla A, Schmidt JM, Detmar E, Lai MM, Shishkin AA, Bhat P, Takei Y, Trinh V, Aznauryan E, Russell P, Cheng C, Jovanovic M, Chow A, Cai L, McDonel P, Garber M, Guttman M, Higher-Order Inter-chromosomal Hubs Shape 3D Genome Organization in the Nucleus, Cell. 174 (2018) 744–757.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Fiorillo L, Musella F, Conte M, Kempfer R, Chiariello AM, Bianco S, Kukalev A, Irastorza-Azcarate I, Esposito A, Abraham A, Prisco A, Pombo A, Nicodemi M, Comparison of the Hi-C, GAM and SPRITE methods using polymer models of chromatin, Nat. Methods 18 (2021) 482–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Xie L, Liu Z, Single-cell imaging of genome organization and dynamics, Mol. Syst. Biol 17 (2021) e9653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Beliveau BJ, Joyce EF, Apostolopoulos N, Yilmaz F, Fonseka CY, McCole RB, Chang Y, Li JB, Senaratne TN, Williams BR, Rouillard J-M, Wu C-T, Versatile design and synthesis platform for visualizing genomes with Oligopaint FISH probes, Proc. Natl. Acad. Sci. U. S. A 109 (2012) 21301–21306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Beliveau BJ, Boettiger AN, Nir G, Bintu B, Yin P, Zhuang X, Wu C-T, In Situ Super-Resolution Imaging of Genomic DNA with OligoSTORM and OligoDNA-PAINT, Methods Mol. Biol 1663 (2017) 231–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Bintu B, Mateo LJ, Su J-H, Sinnott-Armstrong NA, Parker M, Kinrot S, Yamaya K, Boettiger AN, Zhuang X, Super-resolution chromatin tracing reveals domains and cooperative interactions in single cells, Science. 362 (2018). 10.1126/science.aau1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Takei Y, Yun J, Zheng S, Ollikainen N, Pierson N, White J, Shah S, Thomassie J, Suo S, Eng C-HL, Guttman M, Yuan G-C, Cai L, Integrated spatial genomics reveals global architecture of single nuclei, Nature. (2021). 10.1038/s41586-020-03126-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Nagano T, Lubling Y, Stevens TJ, Schoenfelder S, Yaffe E, Dean W, Laue ED, Tanay A, Fraser P, Single-cell Hi-C reveals cell-to-cell variability in chromosome structure, Nature. 502 (2013) 59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Nagano T, Lubling Y, Várnai C, Dudley C, Leung W, Baran Y, Mendelson Cohen N, Wingett S, Fraser P, Tanay A, Cell-cycle dynamics of chromosomal organization at single-cell resolution, Nature. 547 (2017) 61–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Ramani V, Deng X, Qiu R, Gunderson KL, Steemers FJ, Disteche CM, Noble WS, Duan Z, Shendure J, Massively multiplex single-cell Hi-C, Nat. Methods 14 (2017) 263–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Ramani V, Deng X, Qiu R, Lee C, Disteche CM, Noble WS, Shendure J, Duan Z, Sci-Hi-C: A single-cell Hi-C method for mapping 3D genome organization in large number of single cells, Methods. 170 (2020) 61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Cao J, O’Day DR, Pliner HA, Kingsley PD, Deng M, Daza RM, Zager MA, Aldinger KA, Blecher-Gonen R, Zhang F, Spielmann M, Palis J, Doherty D, Steemers FJ, Glass IA, Trapnell C, Shendure J, A human cell atlas of fetal gene expression, Science. 370 (2020). 10.1126/science.aba7721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Domcke S, Hill AJ, Daza RM, Cao J, O’Day DR, Pliner HA, Aldinger KA, Pokholok D, Zhang F, Milbank JH, Zager MA, Glass IA, Steemers FJ, Doherty D, Trapnell C, Cusanovich DA, Shendure J, A human cell atlas of fetal chromatin accessibility, Science. 370 (2020). 10.1126/science.aba7612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Li G, Liu Y, Zhang Y, Kubo N, Yu M, Fang R, Kellis M, Ren B, Joint profiling of DNA methylation and chromatin architecture in single cells, Nat. Methods 16 (2019) 991–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Lee D-S, Luo C, Zhou J, Chandran S, Rivkin A, Bartlett A, Nery JR, Fitzpatrick C, O’Connor C, Dixon JR, Ecker JR, Simultaneous profiling of 3D genome structure and DNA methylation in single human cells, Nat. Methods 16 (2019) 999–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Zheng M, Tian SZ, Capurso D, Kim M, Maurya R, Lee B, Piecuch E, Gong L, Zhu JJ, Li Z, Wong CH, Ngan CY, Wang P, Ruan X, Wei C-L, Ruan Y, Multiplex chromatin interactions with single-molecule precision | Nature, Nature. 566 (2019) 558–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].MacArthur J, Bowler E, Cerezo M, Gil L, Hall P, Hastings E, Junkins H, McMahon A, Milano A, Morales J, Pendlington ZM, Welter D, Burdett T, Hindorff L, Flicek P, Cunningham F, Parkinson H, The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog), Nucleic Acids Res. 45 (2017) D896–D901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Claussnitzer M, Cho JH, Collins R, Cox NJ, Dermitzakis ET, Hurles ME, Kathiresan S, Kenny EE, Lindgren CM, MacArthur DG, North KN, Plon SE, Rehm HL, Risch N, Rotimi CN, Shendure J, Soranzo N, McCarthy MI, A brief history of human disease genetics, Nature. 577 (2020) 179–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Benner C, Spencer CC, Havulinna AS, Salomaa V, Ripatti S, Pirinen M, FINEMAP: efficient variable selection using summary data from genome-wide association studies, Bioinformatics. 32 (2016) 1493–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Hormozdiari F, Kostem E, Kang EY, Pasaniuc B, Eskin E, Identifying causal variants at loci with multiple signals of association, Genetics. 198 (2014) 497–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Mah W, Won H, The three-dimensional landscape of the genome in human brain tissue unveils regulatory mechanisms leading to schizophrenia risk, Schizophr. Res (2019). 10.1016/j.schres.2019.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].de Leeuw CA, Mooij JM, Heskes T, Posthuma D, MAGMA: generalized gene-set analysis of GWAS data, PLoS Comput. Biol 11 (2015) e1004219–e1004219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].de Leeuw C, Sey NYA, Posthuma D, Won H, A response to Yurko et al: H-MAGMA, inheriting a shaky statistical foundation, yields excess false positives, (2020) 2020.09.25.310722. 10.1101/2020.09.25.310722. [DOI] [PubMed] [Google Scholar]
- [48].Sey NYA, Hu B, Mah W, Fauni H, McAfee JC, Rajarajan P, Brennand KJ, Akbarian S, Won H, A computational tool (H-MAGMA) for improved prediction of brain-disorder risk genes by incorporating brain chromatin interaction profiles, Nat. Neurosci 23 (2020) 583–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Hu B, Won H, Mah W, Park RB, Kassim B, Spiess K, Kozlenkov A, Crowley CA, Pochareddy S, PsychENCODE Consortium, Li Y, Dracheva S, Sestan N, Akbarian S, Geschwind DH, Neuronal and glial 3D chromatin architecture informs the cellular etiology of brain disorders Nat. Commun, 12 (2021), p. 3968, 10.1038/s41467-021-24243-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Fulco CP, Nasser J, Jones TR, Munson G, Bergman DT, Subramanian V, Grossman SR, Anyoha R, Doughty BR, Patwardhan TA, Nguyen TH, Kane M, Perez EM, Durand NC, Lareau CA, Stamenova EK, Aiden EL, Lander ES, Engreitz JM, Activity-by-contact model of enhancer-promoter regulation from thousands of CRISPR perturbations, Nat. Genet 51 (2019) 1664–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Nasser J, Bergman DT, Fulco CP, Guckelberger P, Doughty BR, Patwardhan TA, Jones TR, Nguyen TH, Ulirsch JC, Lekschas F, Mualim K, Natri HM, Weeks EM, Munson G, Kane M, Kang HY, Cui A, Ray JP, Eisenhaure TM, Collins RL, Dey K, Pfister H, Price AL, Epstein CB, Kundaje A, Xavier RJ, Daly MJ, Huang H, Finucane HK, Hacohen N, Lander ES, Engreitz JM, Genome-wide enhancer maps link risk variants to disease genes, Nature. 593 (2021) 238–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Fadason T, Ekblad C, Ingram JR, Schierding WS, O’Sullivan JM, Physical Interactions and Expression Quantitative Traits Loci Identify Regulatory Connections for Obesity and Type 2 Diabetes Associated SNPs, Front. Genet 8 (2017) 150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Klein JC, Keith A, Agarwal V, Durham T, Shendure J, Functional characterization of enhancer evolution in the primate lineage, Genome Biol. 19 (2018) 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Doni Jayavelu N, Jajodia A, Mishra A, Hawkins RD, Candidate silencer elements for the human and mouse genomes, Nat. Commun 11 (2020) 1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Tewhey R, Kotliar D, Park DS, Liu B, Winnicki S, Reilly SK, Andersen KG, Mikkelsen TS, Lander ES, Schaffner SF, Sabeti PC, Direct Identification of Hundreds of Expression-Modulating Variants using a Multiplexed Reporter Assay, Cell. 165 (2016) 1519–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Matoba N, Liang D, Sun H, Aygün N, McAfee JC, Davis JE, Raffield LM, Qian H, Piven J, Li Y, Kosuri S, Won H, Stein JL, Common genetic risk variants identified in the SPARK cohort support DDHD2 as a candidate risk gene for autism, Transl. Psychiatry 10 (2020) 265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Kircher M, Xiong C, Martin B, Schubach M, Inoue F, Bell RJA, Costello JF, Shendure J, Ahituv N, Saturation mutagenesis of twenty disease-associated regulatory elements at single base-pair resolution, Nat. Commun 10 (2019) 3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].de Boer CG, Vaishnav ED, Sadeh R, Abeyta EL, Friedman N, Regev A, Deciphering eukaryotic gene-regulatory logic with 100 million random promoters, Nat. Biotechnol 38 (2020) 56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Calderon D, Ellis A, Daza RM, Martin B, Tome JM, Chen W, Chardon FM, Leith A, Lee C, Trapnell C, Shendure J, TransMPRA: A framework for assaying the role of many trans-acting factors at many enhancers, Cold Spring Harbor Laboratory. (2020) 2020.09.30.321323. 10.1101/2020.09.30.321323. [DOI] [Google Scholar]
- [60].Shen SQ, Myers CA, Hughes AEO, Byrne LC, Flannery JG, Corbo JC, Massively parallel cis-regulatory analysis in the mammalian central nervous system, Genome Res. 26 (2016) 238–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Gordon MG, Inoue F, Martin B, Schubach M, Agarwal V, Whalen S, Feng S, Zhao J, Ashuach T, Ziffra R, Kreimer A, Georgakopoulos-Soares I, Yosef N, Ye CJ, Pollard KS, Shendure J, Kircher M, Ahituv N, lentiMPRA and MPRAflow for high-throughput functional characterization of gene regulatory elements, Nat. Protoc 15 (2020) 2387–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Ernst J, Melnikov A, Zhang X, Wang L, Rogov P, Mikkelsen T, Kellis M, Genome-scale high-resolution mapping of activating and repressive nucleotides in regulatory regions, Nat. Biotechnol (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Lagunas T, Plassmeyer SP, Friedman RZ, Rieger MA, Fischer AD, Aguilar Lucero AF, An J-Y, Sanders SJ, Cohen BA, Dougherty JD, A Cre-dependent massively parallel reporter assay allows for cell-type specific assessment of the functional effects of genetic variants in vivo, bioRxiv. (2021) 2021.05.17.444514. 10.1101/2021.05.17.444514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Shen SQ, Kim-Han JS, Cheng L, Xu D, Gokcumen O, Hughes AEO, Myers CA, Corbo JC, A candidate causal variant underlying both higher intelligence and increased risk of bipolar disorder, bioRxiv. (2019) 580258. 10.1101/580258. [DOI] [Google Scholar]
- [65].Anzalone AV, Randolph PB, Davis JR, Sousa AA, Koblan LW, Levy JM, Chen PJ, Wilson C, Newby GA, Raguram A, Liu DR, Search-and-replace genome editing without double-strand breaks or donor DNA, Nature. 576 (2019) 149–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Kim JH, Rege M, Valeri J, Dunagin MC, Metzger A, Titus KR, Gilgenast TG, Gong W, Beagan JA, Raj A, Phillips-Cremins JE, LADL: light-activated dynamic looping for endogenous gene expression control, Nat. Methods 16 (2019) 633–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Wang D, Liu S, Warrell J, Won H, Shi X, Navarro FCP, Clarke D, Gu M, Emani P, Yang YT, Xu M, Gandal MJ, Lou S, Zhang J, Park JJ, Yan C, Rhie SK, Manakongtreecheep K, Zhou H, Nathan A, Peters M, Mattei E, Fitzgerald D, Brunetti T, Moore J, Jiang Y, Girdhar K, Hoffman GE, Kalayci S, Gümüş ZH, Crawford GE, PsychENCODE Consortium, Roussos P, Akbarian S, Jaffe AE, White KP, Weng Z, Sestan N, Geschwind DH, Knowles JA, Gerstein MB, Comprehensive functional genomic resource and integrative model for the human brain, Science. 362 (2018). 10.1126/science.aat8464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Matoba N, Quiroga IY, Phanstiel DH, Won H, Mapping Alzheimer’s Disease Variants to Their Target Genes Using Computational Analysis of Chromatin Configuration, J. Vis. Exp (2020). 10.3791/60428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Nott A, Holtman IR, Coufal NG, Schlachetzki JCM, Yu M, Hu R, Han CZ, Pena M, Xiao J, Wu Y, Keulen Z, Pasillas MP, O’Connor C, Nickl CK, Schafer ST, Shen Z, Rissman RA, Brewer JB, Gosselin D, Gonda DD, Levy ML, Rosenfeld MG, McVicker G, Gage FH, Ren B, Glass CK, Brain cell type-specific enhancer-promoter interactome maps and disease-risk association, Science. 366 (2019) 1134–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Watanabe K, Taskesen E, van Bochoven A, Posthuma D, Functional mapping and annotation of genetic associations with FUMA, Nat. Commun 8 (2017) 1826–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Won H, de la Torre-Ubieta L, Stein JL, Parikshak NN, Huang J, Opland CK, Gandal MJ, Sutton GJ, Hormozdiari F, Lu D, Lee C, Eskin E, Voineagu I, Ernst J, Geschwind DH, Chromosome conformation elucidates regulatory relationships in developing human brain, Nature. 538 (2016) 523–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Song M, Pebworth M-P, Yang X, Abnousi A, Fan C, Wen J, Rosen JD, Choudhary MNK, Cui X, Jones IR, Bergenholtz S, Eze UC, Juric I, Li B, Maliskova L, Lee J, Liu W, Pollen AA, Li Y, Wang T, Hu M, Kriegstein AR, Shen Y, Cell-type-specific 3D epigenomes in the developing human cortex, Nature. 587 (2020) 644–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Jung I, Schmitt A, Diao Y, Lee AJ, Liu T, Yang D, Tan C, Eom J, Chan M, Chee S, Chiang Z, Kim C, Masliah E, Barr CL, Li B, Kuan S, Kim D, Ren B, A compendium of promoter-centered long-range chromatin interactions in the human genome, Nat. Genet 51 (2019) 1442–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Espeso-Gil S, Halene T, Bendl J, Kassim B, Ben Hutta G, Iskhakova M, Shokrian N, Auluck P, Javidfar B, Rajarajan P, Chandrasekaran S, Peter CJ, Cote A, Birnbaum R, Liao W, Borrman T, Wiseman J, Bell A, Bannon MJ, Roussos P, Crary JF, Weng Z, Marenco S, Lipska B, Tsankova NM, Huckins L, Jiang Y, Akbarian S, A chromosomal connectome for psychiatric and metabolic risk variants in adult dopaminergic neurons, Genome Med. 12 (2020) 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Koob GF, Volkow ND, Neurobiology of addiction: a neurocircuitry analysis, Lancet Psychiatry. 3 (2016) 760–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Blandini F, Nappi G, Tassorelli C, Martignoni E, Functional changes of the basal ganglia circuitry in Parkinson’s disease, Prog. Neurobiol 62 (2000) 63–88. [DOI] [PubMed] [Google Scholar]
- [77].Lake BB, Ai R, Kaeser GE, Salathia NS, Yung YC, Liu R, Wildberg A, Gao D, Fung HL, Chen S, Vijayaraghavan R, Wong J, Chen A, Sheng X, Kaper F, Shen R, Ronaghi M, Fan JB, Wang W, Chun J, Zhang K, Neuronal subtypes and diversity revealed by single-nucleus RNA sequencing of the human brain, Science. 352 (2016) 1586–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Habib N, McCabe C, Medina S, Varshavsky M, Kitsberg D, Dvir-Szternfeld R, Green G, Dionne D, Nguyen L, Marshall JL, Chen F, Zhang F, Kaplan T, Regev A, Schwartz M, Disease-associated astrocytes in Alzheimer’s disease and aging, Nat. Neurosci 23 (2020) 701–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Tasic B, Yao Z, Graybuck LT, Smith KA, Nguyen TN, Bertagnolli D, Goldy J, Garren E, Economo MN, Viswanathan S, Penn O, Bakken T, Menon V, Miller J, Fong O, Hirokawa KE, Lathia K, Rimorin C, Tieu M, Larsen R, Casper T, Barkan E, Kroll M, Parry S, Shapovalova NV, Hirschstein D, Pendergraft J, Sullivan HA, Kim TK, Szafer A, Dee N, Groblewski P, Wickersham I, Cetin A, Harris JA, Levi BP, Sunkin SM, Madisen L, Daigle TL, Looger L, Bernard A, Phillips J, Lein E, Hawrylycz M, Svoboda K, Jones AR, Koch C, Zeng H, Shared and distinct transcriptomic cell types across neocortical areas, Nature. 563 (2018) 72–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Beagan JA, Pastuzyn ED, Fernandez LR, Guo MH, Feng K, Titus KR, Chandrashekar H, Shepherd JD, Phillips-Cremins JE, Three-dimensional genome restructuring across timescales of activity-induced neuronal gene expression, Nat. Neurosci 23 (2020) 707–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Hazlett HC, Gu H, Munsell BC, Kim SH, Styner M, Wolff JJ, Elison JT, Swanson MR, Zhu H, Botteron KN, Collins DL, Constantino JN, Dager SR, Estes AM, Evans AC, Fonov VS, Gerig G, Kostopoulos P, McKinstry RC, Pandey J, Paterson S, Pruett JR, Schultz RT, Shaw DW, Zwaigenbaum L, Piven J, Network, Ibis, Clinical, Sites, Data Coordinating, Center, C. Image Processing, A. Statistical, Early brain development in infants at high risk for autism spectrum disorder, Nature. 542 (2017) 348–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Chiaradia I, Lancaster MA, Brain organoids for the study of human neurobiology at the interface of in vitro and in vivo, Nat. Neurosci 23 (2020) 1496–1508. [DOI] [PubMed] [Google Scholar]
- [83].Bhaduri A, Andrews MG, Mancia Leon W, Jung D, Shin D, Allen D, Jung D, Schmunk G, Haeussler M, Salma J, Pollen AA, Nowakowski TJ, Kriegstein AR, Cell stress in cortical organoids impairs molecular subtype specification, Nature. 578 (2020) 142–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Trevino AE, Sinnott-Armstrong N, Andersen J, Yoon S-J, Huber N, Pritchard JK, Chang HY, Greenleaf WJ, Pașca SP, Chromatin accessibility dynamics in a model of human forebrain development, Science. 367 (2020). 10.1126/science.aay1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Bock C, Boutros M, Camp JG, Clarke L, Clevers H, Knoblich JA, Liberali P, Regev A, Rios AC, Stegle O, Stunnenberg HG, Teichmann SA, Treutlein B, Vries RGJ, Human Cell Atlas “Biological Network” Organoids, The Organoid Cell Atlas, Nat. Biotechnol (2020). 10.1038/s41587-020-00762-x. [DOI] [PMC free article] [PubMed] [Google Scholar]