

Abstract
Ubiquilin (UBQLN) proteins are a dynamic and versatile family of proteins found in all eukaryotes that function in the regulation of proteostasis. Besides their canonical function as shuttle factors in delivering misfolded proteins to the proteasome and autophagy systems for degradation, there is emerging evidence that UBQLN proteins play broader roles in proteostasis. New information suggests the proteins function as chaperones in protein folding, protecting proteins prior to membrane insertion, and as guardians for mitochondrial protein import. In this review, we describe the evidence for these different roles, highlighting how different domains of the proteins impart these functions. We also describe how changes in the structure and phase separation properties of UBQLNs may regulate their activity and function. Finally, we discuss the pathogenic mechanisms by which mutations in UBQLN2 cause amyotrophic lateral sclerosis and frontotemporal dementia. We describe the animal model systems made for different UBQLN2 mutations and how lessons learnt from these systems provide fundamental insight into the molecular mechanisms by which UBQLN2 mutations drive disease pathogenesis through disturbances in proteostasis.
Keywords: ALS, mitochondria, OXPHOS, UBQLN2
Introduction
Organisms rely on proper cellular function for their health and survival. Cells, particularly long-lived terminally differentiated ones like neurons, must therefore be capable of buffering insults and adapting to developmental, programmed proteome changes. This proper functional adaptation to proteomic imbalance is referred to as proteostasis [1]. Proteins can be damaged or become nonfunctional for a variety of reasons, including but not limited to errors in translation, aberrant folding, and/or assembly of the protein, abnormal post-translational modifications, and aberrant proteolytic processing. Terminally misfolded proteins need to be removed from cells; otherwise, they can wreak havoc. It is therefore not surprising that cells utilize highly sophisticated quality control systems to recognize, ubiquitin tag, and eliminate damaged and unwanted proteins from cells [2,3]. There is now ample evidence that failures to maintain proteostasis can cause disease, most notably neurodegenerative diseases [4–6]. Here, we review the role ubiquilin (UBQLN) proteins play in maintaining proteostasis and how disruption of this function can cause devastating diseases, as exemplified by mutations in UBQLN2 that cause amyotrophic lateral sclerosis (ALS)/frontotemporal dementia (FTD).
Ubiquilins are a small family of proteins that are highly conserved in mammals, with fewer and more distantly related isoforms found in other species [7]. UBQLN proteins function to dispose ubiquitin-tagged proteins from cells through both the ubiquitin-proteasome (UPS) and the autophagy-lysosomal degradation systems [8–10]. Possibly in association with these functions, mutations in different UBQLN genes have now been linked to different neurodegenerative diseases. In this review, we discuss how different structural elements in UBQLN proteins may impart their different function(s). Then, we define the physiological roles that UBQLNs play in protein quality control (PQC) pathways and explore how ALS/FTD-linked UBQLN2 mutations impact these and other functions. Finally, we discuss future avenues of research that will be important to address in UBQLN biology.
UBQLN genes and proteins
Humans and mice both possess 5 UBQLN genes (1, 2, 3, 4, and L), which vary in their tissue expression patterns [7–9,11,12]. UBQLN1 (PLIC1) is the most ubiquitous and highest expressed UBQLN mRNA in tissues [8,9] followed by UBQLN4 (A1UP, UBIN, CIP75), which is also ubiquitously expressed but at lower levels than UBQLN1 [11,13,14]. UBQLN2 has even more restricted expression, with the highest levels seen in the muscle and brain [8]. Expression of UBQLN3 and L is limited to the testis [12,15].
The inferred UBQLN proteins are all about 600 amino acids (aa) long, with the exception of UBQLNL, which is only 475 aa long (Fig 1A). Sequence comparison of the UBQLN proteins reveals UBQLN1 is most closely related to UBQLN2 (83% identity), followed by UBQLN4 (70% identity), then with UBQLN3 (52% identity), and least with UBQLNL (37% identity; Fig 1B). Sequence alignment also reveals that all UBQLN proteins share the greatest identity in their ubiquitin-like (UBL), ubiquitin-associated (UBA), and two Stress inducible 1 (STI) domains that characterize the UBQLN protein family (Fig 1A). The only exception is UBQLNL, which has poor sequence homology in the second STI1domain-II domain and lacks a complete UBA domain. The other notable conserved region in UBQLN isoforms is located between residues 257 to 324 in UBQLN1, but it has no homology to any known protein domain. Interspersed between the conserved modules are a series of insertions that vary in length and position in different UBQLN isoforms. Of particular note is the presence of 12 proline-rich domain (PXX) repeats in UBQLN2 that is absent in other UBQLN proteins. Many UBQLN2 mutations that cause ALS/FTD map in and around the PXX domain, but the function of the domain is unknown [12,16,17]. There are, however, better-studied functions of the UBA, UBL, and STI1 domains.
Fig. 1.

Sequence alignment and structural domain features of UBQLN proteins. (A) Amino acid sequence alignment of UBQLN1, 2, 3, 4, and L reveals structural similarities in their UBL, UBA, and two STI domains, color-coded as blue, green, and orange, respectively. UBQLN2 uniquely contains a PXX, colored as red, in which a large cluster of ALS/FTD-linked mutations reside. The red circle is a phosphorylated amino acid. (B) Table indicates percent sequence homology and size of each UBQLN protein.
Proteins with UBA and UBL domains function in the UPS degradation pathway [18–21]. The UBA domain is a structural module that binds different ubiquitin moieties typically conjugated to proteins marked for degradation, whereas the UBL domain binds Rpn1, Rpn10, and Rpn13 subunits found in the 19S regulatory cap of the proteasome [22–26]. As a result, UBQLN proteins facilitate proteasomal degradation by acting as shuttle factors that capture ubiquitinated proteins through interaction with their UBA domain and then deliver their cargo by docking with subunits in the proteasome cap via their UBL domain [10,25,27–29]. Surprisingly, UBQLN proteins also function in autophagy, the second major degradation pathway in cells where large protein aggregates and organelles are catabolized [30,31]. The exact mechanism by which UBQLN proteins are recruited to the autophagosome machinery is less clear.
Besides their roles in facilitating protein degradation, there is growing evidence that UBQLN proteins enhance biosynthesis of membrane proteins, possibly by shielding hydrophobic segments in transmembrane proteins prior to membrane insertion [9,13,32–35]. This function has been attributed to its STI1 domains that are known to bind heat shock-related proteins, but the exact mechanism by which they do so remains to be clarified [36–38].
Localization of UBQLN protein in normal and disease states
Antibody staining indicates UBQLN proteins are highly dynamic, localizing to different structures in both the nucleus and cytoplasm [9]. Under normal culture conditions, UBQLN staining is typically diffuse and stronger in the cytoplasm. However, within populations, stronger staining in the nucleus and in puncta in the cytoplasm has been observed in a few cells, although the reason for this difference is not known [9]. The cytoplasmic puncta could correspond to UBQLN function in autophagosomes or to the recent evidence that the proteins can phase separate into membraneless organelles [39–43]. Besides the puncta and nuclei, UBQLN staining has been localized to cytoskeleton networks and to the endoplasmic reticulum (ER), in accord with their attributed functions [8,9,28].
Most localization studies of UBQLN proteins have been performed with antibodies that cross-react with multiple UBQLN isoforms, and therefore information on whether the different isoforms have distinct cellular distributions is lacking. UBQLN proteins contain neither a classic nuclear localization signal nor a nuclear export signal (NES). The proteins (~ 52–68 kDa) are also much larger than the ~ 40 kDa cutoff limit for passive diffusion of proteins through nuclear pores [44]. However, UBQLN proteins are imported into the nucleus in cells exposed to certain stressors [37,45–48], but the mechanism of their import is not known. UBQLN4 is also rapidly recruited to sites of nuclear DNA damage [49], which is regulated by phosphorylation of serine318 in the protein by ATM kinase [49,50]. More is known about UBQLN export from the nucleus. Recently, Hirayama et al. [51] found that UBQLN4 is exported from the nucleus by interaction with the 15 kDa protein polyubiquitinated substrate transporter (POST), which contains multiple NESs, along with its piggybacked ubiquitinated cargo.
Abnormalities in UBQLN staining are common in most neurodegenerative diseases. Initial suggestion that UBQLN proteins were involved in neurodegenerative diseases stemmed from the discovery that UBQLN1 interacts with presenilins and from strong UBQLN staining seen in neurofibrillary tangles in Alzheimer’s (AD) and Lewy bodies in Parkinson’s disease [9]. In addition to these aggregate structures, abnormal and strong UBQLN staining has now been found in other pathologic inclusions that characterize most neurodegenerative diseases, including Marinesco and Hirano bodies in AD, cytoplasmic and nuclear inclusions in polyglutamine-expanded proteins in Huntington’s disease (HD), Spinocerebellar ataxia 1, 2, and 3 diseases, as well as different inclusions linked to Dentatorubral-pallidolysian atrophy and C9ORF72 and UBQLN2 ALS/FTD cases [16,52–59]. Additionally, mass spectroscopy analysis and staining with isoform-specific antibodies have shown UBQLN 1, 2, and 4 isoforms all colocalize with huntingtin (Htt) aggregates in cellular and animal models of HD [47,52,60,61]. However, one study did fail to detect UBQLN1 or 2 in some of these inclusions [60]. A common property of all of these neuropathologic inclusions is that they contain misfolded and aggregated proteins that are heavily modified by ubiquitin [62]. It is likely that UBQLN proteins are recruited to the inclusions via binding of their UBA domain with ubiquitin chains tagged onto the misfolded proteins. This binding may lead to depletion of the functional UBQLN pool in cells causing loss of their function. Such depletion could be particularly problematic because of the age-dependent decline in UBQLN isoform expression in the brain [61]. Alternatively, dysfunction of UBQLN protein may lead to accumulation of the inclusions.
Effects of overexpression and knockout of UBQLN genes
Several studies have explored whether increasing UBQLN expression is beneficial. Most of these studies have indicated that overexpression of UBQLN1 protein, in particular, reduces aggregation and toxicity of pathogenic proteins in different cell, animal, and mouse models of neurodegenerative diseases, including HD, AD, ALS/FTD, expanded polyalanine proteins, and ischemic brain injury [53,57,63–67]. Mechanistically, UBQLN1 overexpression was shown to selectively enhance the turnover of mutant polyglutamine-expanded Htt protein without altering the turnover of Htt proteins containing the normal range of polyglutamine repeats [68]. This discrimination suggests efforts to increase UBQLN1 expression may provide an attractive means to selectively eliminate toxic proteins encoded by the mutant Htt allele without interfering with expression from the wild type (WT) allele. This may be therapeutically beneficial in some of these disorders where eliminating expression of both alleles could be detrimental, as shown for the Htt gene [69]. One noticeable consequence of mice overexpressing UBQLN1 is a reduction in body weight, which was attributed to an increase in the metabolic rate of cells [67,70].
Likewise, overexpression of WT human UBQLN2 (hUBQLN2) was shown to be beneficial in alleviating neurodegeneration in a knock-in (KI) mouse model of HD and in reducing poly glycine-alanine dipeptide repeat-induced toxicity implicated in the pathogenicity of the hexanucleotide repeat mutation in C9ORF72 linked to ALS [47,59]. However, there appears to be a limit to which UBQLN2 overexpression may be tolerable, as very high levels of its expression were found to induce neurodegeneration. For example, high overexpression of hUBQLN2 in rat brain neurons using the CaM kinase 2α promoter caused memory impairments and deposition of ubiquitin-positive inclusions [71]. Similarly, high, but not low overexpression of hUBQLN2 using the prion promoter was found to cause age-dependent retinal degeneration [46]. Interestingly, besides the retinal degeneration, no evidence of neurodegeneration or pathology was observed in the brain of the high and low-expressing UBQLN2 mice, suggesting differential vulnerability of neurons to the level of UBQLN2 overexpression. The exact level above which UBQLN2 overexpression becomes toxic is not known. Two studies where the level of hUBQLN2 overexpression was measured found that expression up to 80% of the level of the endogenous mouse protein was tolerable, causing only minor, if any, behavioral or pathologic abnormalities [72–74]. Taken together, these results suggest that efforts to increase UBQLN expression may have therapeutic benefit, but there may be a threshold when overexpression of certain isoforms becomes detrimental.
The effects of single knockout (KO) of the UBQLN 1, 2, and L genes in rodents have also been reported, with UBQLN2 KO animals having the most prominent behavioral alterations [12,66,71,75]. Although mice with inactivated UBQLN1 gene have no developmental or behavioral abnormalities, they nevertheless have higher accumulation of ubiquitinated proteins in the brain and exhibit poorer recovery to ischemic injury than normal mice [66]. Rats inactivated of UBQLN2 were reported to be unremarkable [76]. By contrast, mice inactivated of the UBQLN2 gene were found to have age-dependent deficits in motor performance, but had no neuronal loss or changes in ubiquitinated proteins [75]. KO of UBQLN L did not affect fecundity of mouse litters, suggesting it is dispensable [12]. In contrast to the mouse studies, gene silencing of the sole UBQLN gene in Drosophila melanogaster and Caenorhabditis elegans by RNA interference reduces animal lifespan by 10–15% [28,77].
The health consequences for the complete KO of UBQLN genes in humans are only known for UBQLN4. Two patients with homozygous mutations in UBQLN4 causing a premature truncation and complete loss of the protein were found to have severe health problems, including growth retardation, facial dysmorphism, and intellectual impairments [49]. Further studies of cells derived from the patients identified major defects in DNA repair and increased susceptibility to genotoxic stressors.
Physiologic functions of UBQLN proteins
Recent evidence suggests UBQLN proteins function as key guardians of proteostasis by functioning in multiple quality control pathways, including in UPS, ER-associated degradation (ERAD), autophagy, and mitochondrial protein surveillance. We will discuss their involvement in each of these processes as well as their less understood roles in cellular trafficking, cytoskeleton organization, chaperone function, and liquid–liquid phase separation (LLPS).
UBQLN proteins are shuttle factors in the ubiquitin-proteasome system
A major pathway by which misfolded proteins are canonically disposed of is through the UPS. This process relies on ubiquitination, a post-translational modification in which a single ubiquitin protein (76 aa) or polyubiquitinated chains are attached to the protein targeted for degradation [18,78]. Ubiquitination occurs through a hierarchical ubiquitin E1-E2-E3 cascade, where there are only two identified E1 enzymes that catalyze the first step, and over 40 and 600 known E2 and E3 members, respectively [79]. Furthermore, proteins can be modified by different types of ubiquitin chain linkages, which encode rules for their fate or to alter function. Proteins targeted for degradation through the UPS are generally targeted with K48-linked polyubiquitin chains and those for autophagy with K63-linked chains, although these linkages can also specify other functions [80–82].
In UPS, UBQLN acts as a shuttle protein by binding proteins tagged with polyubiquitin chains and delivering them to the 26S proteasome for degradation (Fig 2). The 26S proteasome is a macromolecular complex with two main components: a 20S core and 19S regulatory cap [83]. The core is a highly symmetrical structure consisting of proteolytically active subunits and is responsible for the main breakdown of the substrates, which enter the core through the 19S regulatory caps. Ubiquitinated proteins dock to the proteasome via Rpn1, Rpn10, or Rpn13 subunits that act as receptors to bind polyubiquitin chains [23,84,85]. K48-linked chains have been shown to be almost exclusively targeted to the proteasome through interaction with Rpn10 [26]. Another subunit, Rpn11, then cleaves the polyubiquitin chain prior to the substrate entering into the proteasomal core and allows for ubiquitin chain proteins to be recycled back into the cellular pool [86,87]. Both this cleaving action on the ubiquitin chains and the translocation of the substrate through the core are ATP-dependent processes.
Fig. 2.
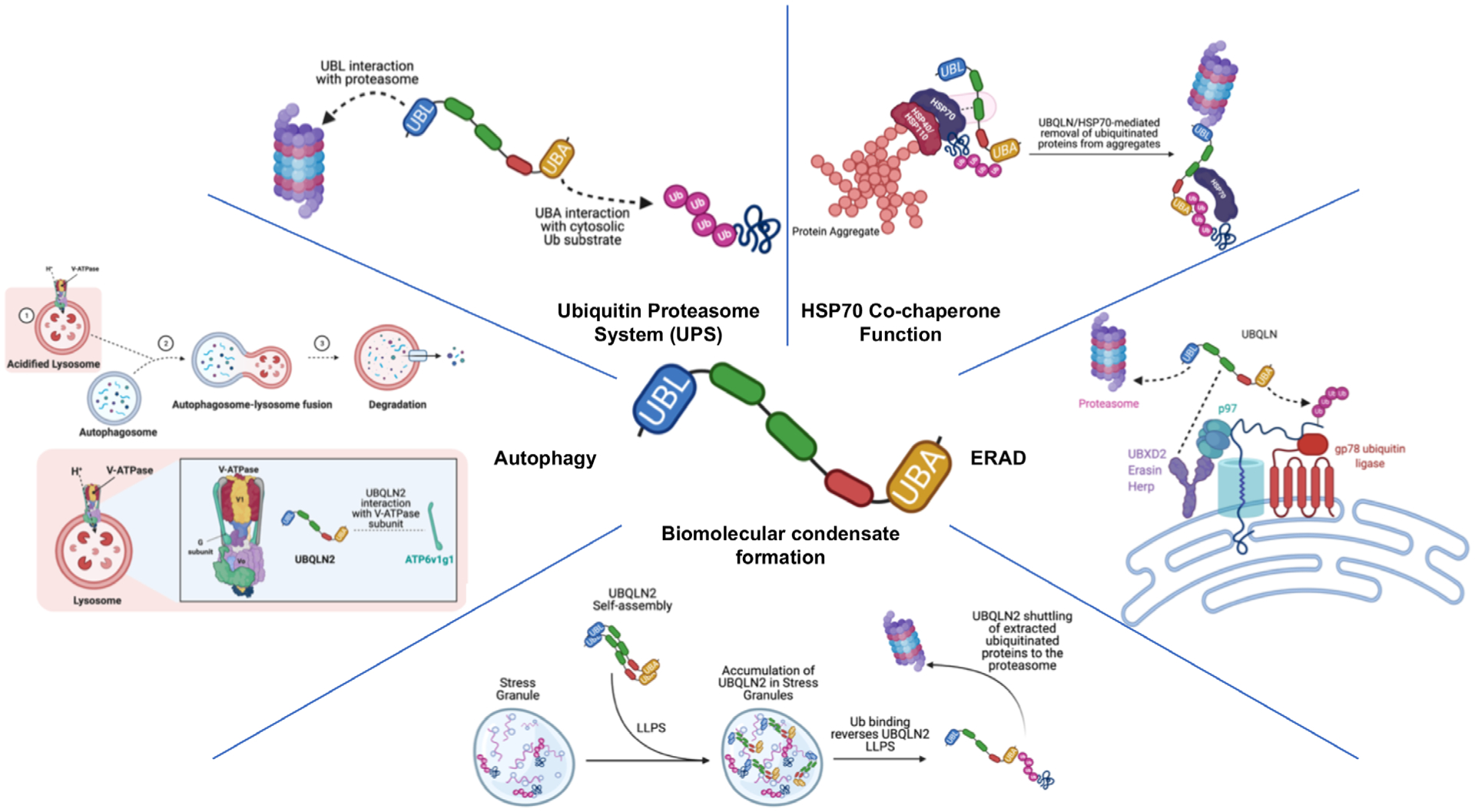
Physiologic functions of UBQLN proteins. UBQLN proteins function as key guardians of proteostasis by functioning in multiple quality control pathways, including the UPS, autophagy, and ERAD and through their physiological roles as HSP70 co-chaperones and in biomolecular condensate formations. While function in the UPS, ERAD, and HSP70 chaperone systems extend to multiple UBQLN proteins, depicted functions in autophagy and biomolecular condensate formation have only been described for UBQLN2. Created with Biorender.com.
UBQLN is one of several shuttle factors involved in delivery of ubiquitinated proteins to the proteasome [19]. The isolated UBA domain of UBQLN1 has particularly high binding affinity for single ubiquitin molecules and different ubiquitin chain moieties compared to most UBA domains [88–90]. However, studies have shown that although the isolated UBA domain displays little specificity in binding different ubiquitin moieties, full-length UBQLN1 exhibits much stronger binding with K63-linked chains than K48 chains, suggesting that sequences besides the UBA domain regulate ubiquitin-binding specificity [91]. For example, deletion of the UBL of UBQLN1 also impairs binding of ubiquitinated chains; however, this may also be due to improper folding of the truncated protein [91]. The functional reason for the preferential binding of UBQLN proteins with certain types of ubiquitin linkages is not known, but it may suggest they are more poised for disposal of autophagy substrates over proteasome substrates. This preference does not imply lack of involvement in proteasomal degradation.
Indeed, UBQLN1 and 2 proteins have been shown to bind the Rpn10 (S5a) and Rpn13 subunits of the proteasome [29,91,92]. Additionally, structural studies have defined the manner by which the UBL domain of UBQLN 1 and 2 dock with the subunits [25,27,93]. Knockdown of the subunits in mice impaired recruitment of UBQLN proteins to the proteasome, demonstrating their necessity in UBQLN-mediated clearance of ubiquitinated substrates through the UPS [94].
UBQLN proteins facilitate ERAD
Approximately one-third of all eukaryotic proteins are targeted to the secretory pathway, which sorts and packages proteins into vesicles for trafficking to other cellular compartments, or for secretion across the plasma membrane [95]. These proteins are first synthesized in the ER, which houses a host of chaperone and modifying enzymes that aid in correct folding and assembly of the proteins [96]. Even so, protein folding is inherently error-prone, with an estimate that one-third of all nascent proteins are misfolded [95,97]. Misfolded proteins in the ER are recognized by the stringent quality control systems that operate there and retrotranslocate the proteins to the cytoplasm for ubiquitination and proteasomal degradation in a regulated process called ERAD [98,99]. UBQLN proteins are components of ERAD complexes (Fig 2) [28,100]. UBQLN proteins bind to ER-anchored proteins such as Herp, erasin (UBXN4), and UBXD8 (UBXN3B), facilitating ERAD by coupling transfer of the dislocated ubiquitinated protein to the proteasome for degradation [28,100,101]. The importance of UBQLN proteins for proper ERAD function was demonstrated by siRNA knockdown of the proteins, which resulted in reduced turnover of ERAD substrates [28,100]. A buildup of ERAD substrates induces the unfolded protein response (UPR), an integrated signaling pathway that functions to restore ER homeostasis [102]. However, if uncorrected, this causes ER stress, chronic induction of which is known to be pathogenic and has been implicated in the etiology of many diseases, including neurodegenerative diseases [102,103]. Consistent with this effect, knockdown of the sole UBQLN gene in C. elegans induces ER stress [28]. On the contrary, agents that induce ER stress increase UBQLN expression, suggesting UBQLNs are stress response genes [28]. Ultimately, UBQLNs appear to function in ERAD as shuttle factors, but it is not known whether they are dedicated to the removal of particular ER clients or whether they function indiscriminately in the disposal of all ERAD substrates. Interestingly, UBQLN1 binds to erasin, whereas the related shuttle factor called hHR23A does not bind to erasin, suggesting different shuttle factors may function in distinct ERAD complexes [28].
UBQLN proteins regulate autophagy
Autophagy is the second major pathway used in cells to catabolize proteins [30,31,104]. Unlike the UPS, which only degrades proteins that can be unfolded and threaded through the catalytic proteasome chamber, autophagy is capable of degrading large macro-protein complexes, organelles, and insoluble protein aggregates. The degradation of such cellular cargo is accomplished by their engulfment in double-membrane structures termed autophagosomes, which subsequently fuse with lysosomes that contain catabolic hydrolases [30,31,104]. Hydrolase activity is activated by a decrease in the pH environment of the autophagosome through the action of the vacuolar-ATPase proton pump (V-ATPase) [105]. Recently, evidence suggests UBQLN proteins play a critical role in autophagosome acidification via regulation of V-ATPase function [34,106].
Prior to elucidation of the physiological functions of UBQLN proteins in autophagy, UBQLN1 was identified as an interactor of mTOR kinase [107], the master regulator in cells that signals either an increase in protein biosynthesis or induction of autophagy in response to environmental cues [108,109]. However, overexpression of UBQLN1 neither affected mTOR kinase activity nor was the protein phosphorylated by mTOR [107]. However, subsequent studies found UBQLN2 to be an activator of mTORC1 [110]. Stronger indication that UBQLN proteins function in autophagy came from immunostaining and EM localization studies showing their presence in autophagosomes and from studies showing overexpression of UBQLN proteins increases autophagosome number and stimulates autophagy, whereas silencing of UBQLN expression had the opposite effect [34,111,112]. UBQLN1 was also shown to be required for xenophagy of mycobacterium tuberculosis [113]. In addition, most UBQLN isoforms have been found to coimmunoprecipitate with LC3, the autophagosome-specific marker [112,114]. Moreover, Wu et al. [34] found UBQLN2 and LC3 also interact with one another in autophagosomes using a split Venus reporter system. However, only UBQLN4 has been shown to bind directly with LC3 [114]. Furthermore, by utilizing a tandem-tagged fluorescent reporter system to track autophagosome maturation to autolysosomes, two independent groups found that deleting either the sole UBQLN gene in Drosophila or UBQLN2 from HeLa cells blocks autophagosome maturation [34,106].
Besides their role in macroautophagy, UBQLN proteins are also substrates for chaperone-mediated autophagy (CMA), a chaperone-assisted process in cells whereby proteins are actively transported into lysosomes for degradation [112,115,116]. Proteins degraded by CMA contain KFERQ-like sequences and their transport typically requires interaction with heat shock protein (HSP) 70-like proteins. Interestingly, the human UBQLN 1, 2, 3, and 4 isoforms all contain two KFREQ-like sequences and the proteins also bind Hsp70-like proteins consistent with the requirements of CMA. However, how CMA regulates UBQLN function, or vice versa is not known.
UBQLNs as guardians of mitochondrial protein quality control
Mitochondria are unique as their proteome is comprised of proteins encoded by both the nuclear and mitochondrial genomes. Interestingly, the mitochondrial genome only encodes 13 proteins, all of which are components of the electron transport chain that is involved in ATP generation by oxidative phosphorylation (OXPHOS). The remaining mitochondrial proteins (> 1000 proteins) must all be imported. Cells utilize a complex system of signaling and factors to coordinate the type, amount, and quality of proteins to import into mitochondria [117]. UBQLN proteins were found to act as guardians in mitochondrial PQC [33].
The role for UBQLN proteins in mitochondrial PQC was discovered through the identification of an interaction between nuclear-encoded Omp25, an outer mitochondrial membrane protein and UBQLN1/4 proteins [33]. The importance of this binding was shown by an excessive buildup of Omp25 precursors in UBQLN 1, 2, and 4 triple KO HEK293 cells [33]. Mechanistic studies further revealed that UBQLN proteins bind the TMDs in Omp25 proteins, facilitating proteasomal degradation of the ubiquitinated and terminally misfolded forms of the protein [33]. The capacity of UBQLN proteins to bind the TMDs was proposed as a novel function of UBQLNs to shield the precursors from aggregation during their transit from the cytosol to the mitochondria [33].
Consistent with this model, UBQLN2 KO mice were found to have major alterations in abundance of mitochondrial proteins [75]. Furthermore, KO of UBQLN1 in B-cell lymphoma-derived cells increased accumulation of several mitochondrial proteins in the cytosol [118]. This misaccumulation of mitochondria proteins did not present with altered mitochondrial respiration in the UBQLN-deficient cells at least when cultured in standard glucose medium [118]. However, Lin et al. [35] recently found UBQLN2 KO HeLa and NSC34 mouse neuron cells have major respiration deficits when cultured in galactose instead of glucose medium, to force energy reliance on the OXPHOS system. The prior lack of detection of the defect may have been obscured due to glucose inhibition of OXPHOS activity by the Crabtree effect [119]. Interestingly, Lin et al. also noted that their UBQLN2 KO HeLa cells had shorter mitochondria and that the dynamics of mitochondrial fusion were slower compared to the parental WT HeLa line. Collectively, these studies implicate UBQLN proteins as novel factors in mitochondrial PQC.
UBQLN and chaperone function
The capacity of UBQLN proteins to bind proteins containing TMDs suggests properties of a chaperone, the classic example of which are the HSP family that bind exposed hydrophobic patches in proteins [120]. Indeed, many of their other biochemical and biological activities are consistent with UBQLN proteins being unusual chaperones, possessing broad, unique, as well as indispensable chaperone-like activities. First, Stieren et al. [121] showed UBQLN1 is a bona fide chaperone as it stimulates refolding of proteins in vitro and can prevent aggregation of the amyloid precursor protein both in vitro and in vivo. Second, both UBQLN1 and UBQLN2 were shown to stimulate biogenesis of presenilin, ATP6v1g1, and TIMM44 in a dose-dependent manner [9,34,35,88]. Third, UBQLN4 was shown to bind the signal sequence of ER proteins [13]. Fourth, UBQLN proteins bind TMD-containing proteins such as mitochondrial and BCLb proteins [32,33]. Fifth, UBQLN proteins are required for proper targeting of mitochondrial proteins [33,35,118]. Sixth, inactivation of UBQLN2 expression leads to reduced protein accumulation of the ATP6v1g1 and TIMM44 subunits of the V-ATPase pump and mitochondria import channel, respectively, leading to disruption of their functions [34,35].
A key question regarding the putative chaperone function of UBQLN proteins is what involvement other proteins like Hsp70 proteins play in this activity, particularly since UBQLN contains STI1 domains that are known to bind HSP70-like proteins [36,37]. Interestingly, the binding region for the TMD segments in UBQLN proteins has been mapped to their STI1 domains [13,32], but whether the TMD segments and HSP70 chaperones are recruited at the same time is not clear. It is entirely possible that both bind simultaneously to UBQLN through an independent interaction with one of the two STI1 domains. Alternatively, UBQLN2 may chaperone TMD-containing proteins indirectly through mediation of Hsp70 rather than through a direct interaction with the TMD protein.
Besides its role in chaperone function, the central domain of UBQLNs also appears to be the binding site of several additional factors, including itself, and thus may be involved in several other regulatory roles. For example, mapping studies have shown that the central domain of UBQLN proteins binds mTOR, erasin, and hnRNP proteins [28,45,107]. In the case of erasin, this binding is essential for UBQLN recruitment to form a functional ERAD complex [28]. Biochemical studies have suggested that UBQLN proteins form dimers in solution and that homo- and hetero-interactions between UBQLN polypeptides are mainly driven by interactions between their central domains, rather than by interactions between their UBA and UBL domains [122]. Consistent with this proposal, proteins with STI1 domains were proposed to be an important module for protein dimerization [123]. Interestingly, studies have shown that the UBQLN1 monomer, but not the dimer, binds presenilins, suggesting the dimer may represent the inert state of UBQLN proteins [122]. It will be important in the future to discover if this is the correct manner by which UBQLN proteins are maintained in an inert state and what the molecular switches are that cause activation of the proteins to bind particular targets.
UBQLN proteins provide a link between proteostasis and RNA metabolism in stress granules
Liquid–liquid phase separation (LLPS) is a biophysical process driven by multivalent weak interactions between protein domains, low-complexity protein regions, or RNA binding proteins (RBPs) and results in the formation of membraneless structures in the cytosol such as stress granules (SGs) [124,125]. SGs form in response to environmental stressors, such as high temperature or exposure to chemicals, and behave as dynamic liquid droplets within the cell [124,126]. A primary function of SGs is to protect mRNA transcripts when stress conditions have limited the cell’s ability to initiate translation. Thus, mRNA and RBPs are major components of SGs, although they have also been shown to contain other PQC components such as Hsp70 and valosin-containing protein (VCP), which help clear misfolded proteins from the SGs [126–128].
It was recently discovered that UBQLN2 is capable of undergoing LLPS at physiologically relevant temperatures and salt concentrations [39]. The PXX domain, along with the central domains of UBQLNs, is considered to be intrinsically disordered and plays a role in modulating the protein’s LLPS behavior [39,129]. Deletion analysis suggests that the STI1 containing region, and in particular the STI1-II domain, is required for LLPS and SG localization [39,40]. Removal of the UBA domain inhibits LLPS, whereas removal of the UBL domain enhances LLPS [39,130]. Additionally, ALS mutations in UBQLN2 have been linked to altered phase separation properties compared to WT UBQLN2 protein [41,43]. Furthermore, UBQLN2 has been shown to interact with stress granule components, including RNPs and undergo LLPS into SGs [39,40,45,131]. Recently, UBQLN2 protein tagged with the mCherry fluorescent protein was shown to undergo LLPS under conditions known to induce SG formation, but the UBQLN2 droplets segregated independently from SG markers, raising caution as to whether tagging of the protein is reliable for accurately monitoring the intracellular dynamics of UBQLN2 in cells [43]. A remarkable finding of the in vitro assembled UBQLN2 droplets is that addition of purified ubiquitin dissolved the droplets, suggesting that binding of ubiquitin-tagged proteins to UBQLN2 may regulate LLPS assembly/disassembly [39]. Thus, changes in ubiquitination of proteins in cells may trigger alterations in UBQLN2 LLPS behavior or vice versa (Fig 2) [39,132]. Other studies have proposed that UBQLN2 is involved in negatively regulating SG formation and the solubility of their components, thus protecting the cell from abnormal SG formation [40]. More recent studies have shown that UBQLN1 and UBQLN4 proteins also undergo LLPS [42]. Comparison of the fluidity of liquid droplets formed by the three UBQLN proteins showed UBQLN1 is most liquid-like, followed by UBQLN2, with UBQLN4 most gel-like as well as most aggregate prone [42]. These differences may reflect distinct roles that different UBQLN isoforms play in cellular processes and their increased propensity to aggregate upon mutation. Further studies are required to understand the dynamics and role that UBQLN LLPS behavior plays in cellular function.
UBQLN interactions with intermediate filaments and membrane trafficking
In addition to its role in PQC pathways and chaperone functions, UBQLNs have been shown to interact with cellular components of the cytoskeleton and endocytic pathway. Early characterization of UBQLN proteins was in the context of their interaction with integrin-associated protein, resulting in their identification as human proteins linking IAP with cytoskeleton 1 and 2 (hPLIC1 and hPLIC2) [8]. Through their interaction with IAP, UBQLN1 and UBQLN2 are proposed to mediate the interaction of vimentin intermediate filaments from the cytoskeleton with the plasma membrane [8]. UBQLN2 has also been shown to play a role in recognizing and shuttling misfolded intermediate filaments in skeletal muscle to the proteasome for degradation via formation of a complex with myotubularin-1 [133].
The endosomal system is necessary for regulating, sorting, and degrading proteins via autophagy and UPS degradation pathways [134]. UBQLN1 has been shown to interact with the ubiquitin-interacting motif (UIM) domain of several key endocytic proteins, including Eps15, Eps15R, and Hrs proteins, via its UBL domain [135]. By contrast, UBQLN2, but not UBQLN1, was found to bind to epsin 1 and 2 through similar domain interactions [135,136]. Epsins and Eps15-related proteins have been implicated in membrane curvature and internalization of different cargos/receptors through clathrin-mediated endocytosis [137,138]. Consistent with its binding to endocytic machinery, UBQLN proteins have been found to regulate expression of many cell surface receptors, including G-protein coupled receptors, γ-aminobutyric acid A receptors, and nicotinic acetylcholine receptors [136,139,140]. Curiously, however, UBQLN proteins function to increase cell surface expression of the receptors, suggesting it is a negative regulator of endocytosis. The interaction of UBQLN proteins with Hrs proteins is intriguing considering Hrs is involved in formation of the late endosome organelle called the multivesicular body (MVB) [141]. UBQLN-related proteins in yeast have been found to regulate anterograde sorting of proteins to the MVB [142]. The contents of MVB are either secreted in exosomes or degraded by fusion of the organelle with lysosomes or autophagosomes [143]. Taken together, these findings suggest that UBQLNs function at the crossroads of endosomal-lysosomal trafficking. Further studies are needed to better understand UBQLN regulation of endocytosis and MVB formation.
UBQLN2 and ALS/FTD
Amyotrophic lateral sclerosis is the most common motor neuron disease [144]. The disease results in the progressive degeneration of upper and lower motor neurons of the brain and spinal cord which subsequently leads to muscle denervation and atrophy [144,145]. Mutations in and around the PXX region of UBQLN2 are linked to ALS and FTD, the second most common form of dementia in individuals below age 65 [16,146–154]. Mutations are inherited in an X-linked dominant fashion and appear to affect males earlier and more aggressively than females [16,17]. Postmortem analysis of brain and spinal cord tissue from UBQLN2-linked cases of ALS/FTD revealed significant accumulation of UBQLN2 inclusions. These inclusions, which are most prominent in the hippocampus of the brain, have been shown to be positive for ubiquitin and p62, suggesting the inclusions may result from failure of protein degradation pathways due to UBQLN2 mutations [16,155]. Notably, this UBQLN2 pathology is also present in ALS and dementia cases without linkage to UBQLN2 mutations, and the accumulation of UBQLN2 inclusions in the hippocampus of the brain strongly correlates with patients that develop ALS/FTD compared to cases of ALS without dementia [16]. UBQLN2 inclusions are also known to co-aggregate with TAR DNA-binding protein 43 (TDP-43), which is found in aggregates in approximately 50% of FTD and 97% of ALS cases [16,156–158]. The discovery of ALS/FTD-linked mutations in UBQLN2, and other proteins involved in PQC, has highlighted impaired protein homeostasis as a critical part of disease pathogenesis. However, the mechanisms by which these mutations lead to disease remain elusive. In particular, identifying whether impaired proteostasis is the primary driver of disease rather than a downstream effect will be critical in producing effective therapies for ALS/FTD. In the following sections, we discuss recent findings of the effects of ALS/FTD-linked mutations in UBQLN2 on key degradation and cellular pathways implicated in disease and highlight the current challenges in linking these findings to human disease and therapeutic developments.
ALS/FTD-linked UBQLN2 mutations impair delivery of ERAD and cytosolic substrates to the proteasome
Amyotrophic lateral sclerosis/FTD-linked UBQLN2 mutations are closely linked to an impairment in proteasomal degradation [16,29]. Though UBQLN2 mutations are not directly present in the UBL domain, ALS-linked UBQLN2 mutations affect binding with the S5a subunit of the proteasome [29]. The same mutations do not however impair binding of polyubiquitinated substrates, and some evidence even suggests that mutant UBQLN2 binds stronger to these ubiquitinated cargos compared to WT UBQLN2 [159]. Collectively, these results suggest that UBQLN2 mutations do not prevent binding of ubiquitinated substrates, but instead impair delivery of the cargo to the proteasome. Subsequently, improper degradation of these cargos can lead to sequestration of these substrates into insoluble aggregates, positive for ubiquitin, and mutant UBQLN2 protein [37]. Canonically, UBQLN2 interacts with Hsp70 as part of the Hsp70-Hsp110 disaggregase complex to extract ubiquitinated components from insoluble protein aggregates and degrade them through the proteasome [160]. However, in a KI mouse of the UBQLN2P506T (P520T in mouse), mutant UBQLN2 is unable to interact with Hsp70 and is, therefore, unable to aid in the degradation of misfolded and aggregated Hsp70 client proteins [37]. This impaired interaction with Hsp70 was also seen in ALS patient cells with other mutations in UBQLN2 (P494L, P497H, and P506A) [153].
Another mechanism by which UBQLN2 mutations have been proposed to interfere with protein degradation is through disruption of proteasome assembly [48]. Previous studies had shown that proteasome subunits misaccumulate with UBQLN2 aggregates in mouse models of ALS/FTD [41,161]. Recently, Zhang et al. [48] also found proteasome subunits are mislocalized in their rat P497H UBQLN2 animal model, which they showed correlated with an age-dependent decline in proteasome activity.
Impaired delivery of ERAD substrates to the UPS for degradation has also been linked to UBQLN2 mutations. Similar to earlier findings that loss of UBQLN proteins results in accumulation of ERAD substrates within the ER, expression of mutant P497H UBQLN2 caused greater accumulation of the ERAD substrate NHK than WT UBQLN2 [101]. Interestingly, ERAD function is disturbed by both UBQLN2 overexpression and knockdown, with the loss of UBQLN2 causing a greater accumulation of NHK than overexpression of either WT or mutant UBQLN2 [101]. These findings indicate not only that the level of UBQLN2 expression plays an important role in the proper disposal of ERAD substrates, but also that defects in ERAD arise chiefly from a loss of proper UBQLN2 function in the pathway. The accumulation of ERAD substrates induces ER stress and triggers stress responses such as the UPR, which signals for cell death when cells are exposed to irreversible or prolonged ER stress [102,162]. Impaired ERAD and ER stress are common disturbances seen in neurodegenerative diseases [163]. Furthermore, neurons identified as more vulnerable to cell death in ALS are selectively prone to ER stress in SOD1 mouse models of familial ALS [164,165]. In these models, vulnerable neurons initiated UPR and triggered microglial activation 25–30 days prior to the first observed denervation in these animals, whereas resistant neurons from the same animals developed ER stress much later in the disease progression, suggesting that ER stress actively contributes to neuronal death [164,165].
Following proper folding in the ER, proteins are packaged into COPII coated vesicles and transported to the Golgi apparatus for further processing [166]. Recently, P497H and P506T mutations in UBQLN2 were shown to inhibit protein transport from the ER to the Golgi in neuronal cells [167]. Another recent study also highlighted ER/Golgi transmembrane proteins and trafficking vesicles as major protein groups that interact with UBQLN2 [40]. Taken together, these studies highlight a critical role for UBQLN2 in maintaining protein homeostasis of ER-targeted proteins both through the degradation of misfolded proteins via ERAD and through its involvement in ER to Golgi trafficking. Further characterization of the role of UBQLN2 in ER to Golgi protein transport is an intriguing route of study as impaired cellular trafficking could be particularly harmful to neuronal functions such as axonal transport and synaptic transmission [167,168].
ALS/FTD-linked UBQLN2 mutations impair autophagy
Early indications that UBQLN2 mutations may disturb autophagy came from pathologic findings in UBQLN2-associated ALS/FTD cases, which show p62 localization to UBQLN2 inclusions that accumulate in the spinal cord and brain [16]. p62 is a key autophagy adaptor that recruits ubiquitinated cargo to autophagosomes by binding to LC3, a membrane-tethered autophagosome-specific protein [169,170]. Abnormal accumulation of p62, particularly in aggregates, is a prominent feature seen in many neurodegenerative diseases linked to defective autophagy [171]. Further support that the mutations perturb autophagy was evident from the large increase in p62 levels and its strong colocalization with UBQLN2 inclusions in mouse models with different UBQLN2 mutations [16,34,37,75,76,161,172]. In addition, an increase in p62 levels was detected in lymphoblast cells cultured from patients carrying UBQLN2 mutations [153].
Recently, strong evidence that loss of function due to UBQLN2 mutations impedes autophagy by blocking autophagosome acidification was reported by two different groups [34,106]. Reduction in the pH environment of autophagosomes is essential for activating the acid hydrolases involved in catabolizing proteins [170]. The pH change is brought about by the action of the V-ATPase proton pump [173]. Both groups found UBQLN proteins are needed for proper regulation of V-ATPase subunit levels, but of different subunits and through opposite mechanisms. The Bellen group found that deletion of the sole ubqn gene in Drosophila leads to loss of autophagosome acidification, proposing that it is due to aberrant accumulation of toxic ATP6v0a1 subunit fragments [106]. The Monteiro group found that deletion of UBQLN2 in HeLa cells causes a block in autophagosome acidification due to decreased accumulation of the ATP6v1g1 subunit [34]. They also found levels of the same subunit were decreased in the P497S mouse model of ALS/FTD, but found no change in the levels of the ATP6v0a1 subunit. Furthermore, they discovered that overexpression of WT UBQLN2, but none of the five ALS/FTD mutant UBQLN2 proteins they analyzed, rescued the acidification defect in UBQLN2 KO cells. This inability to rescue suggests the UBQLN2 mutations cause loss of function in autophagy [34]. Finally, the group showed UBQLN2 binds directly with the ATP6v1g1 subunit, proposing that UBQLN2 is a chaperone for the protein. As a result of these findings, the Monteiro group cautioned that efforts aimed to stimulate autophagy for treatment of UBQLN2 cases may be futile because the autophagy defect lies downstream of the whole pathway. Instead, they advise that efforts should focus on restoration of the acidification defect.
The reason for the difference in the mode of regulation of the V-ATPase function found by the two groups is not clear. One possibility is the use of different model systems, which remains a key challenge in discerning the mechanism by which UBQLN2 mutations induce and drive ALS/FTD pathogenesis. We discuss variations in animal models in detail in a subsequent section of this review.
Other evidence suggests UBQLN2 mutations interfere with TANK-binding kinase 1 (TBK1)/interferon regulatory factor 3 signaling [174]. Previous studies had shown that upregulation of UBQLN2 regulates nuclear factor kappa-B signaling in inflammation and pathogenicity [175]. More recently, Chen et al. [174] found UBQLN2 binds directly with TBK1, promoting phosphorylation and activation of the kinase and interferon signaling.
Mitochondrial deficits associated with ALS/FTD-linked UBQLN2 mutations
Aside from regulating cytosolic protein homeostasis, UBQLNs also participate in maintaining mitochondrial protein homeostasis as loss of UBQLNs leads to accumulation of cytostatic mitochondrial precursor proteins [33,118]. While loss of UBQLN isoforms mildly impacts mitochondrial function [33], little was known about the effect of ALS/FTD-linked UBQLN2 mutations on mitochondria physiology until recently. A large-scale proteomics analysis on the hUBQLN2P497S mouse model revealed a global downregulation of mitochondrial proteins in hUBQLN2P497S compared to hUBQLN2WT and nontransgenic animals [75]. A recent study in our laboratory has revealed that mitochondria in the motor neurons of the lumbar spinal cord of the UBQLN2P497S mouse model have significant cristae deterioration and are of shorter length. Despite the smaller mitochondria, levels of mitochondrial dynamic fusion proteins, such as mitofusins (Mfn1/2), are elevated in the spinal cord of UBQLN2P497S animals [35]. Mitochondrial fusion assays revealed that the decreased mitochondria length may be attributed to an impaired capacity for mitochondrial fusion. Spinal cord and hippocampal mitochondria derived from the mutant mice also indicate an age-dependent deficit in mitochondrial ADP-stimulated phosphorylating State 3 respiration and impaired complex II and IV activity [35]. Finally, Lin et al. [35] found that mitochondria targeting of TIMM44, a nuclear-encoded mitochondrial protein that functions in mitochondrial protein import, misaccumulates in UBQLN2 KO cells, but that the defect can be rescued by re-expression of WT but not ALS/FTD mutant UBQLN2 proteins, suggesting the ALS mutants cause loss of function.
Protein aggregation
Cumulatively, the findings that ALS/FTD-linked UBQLN2 mutations impair the function of multiple PQC pathways provide strong evidence that impaired proteostasis is a key pathological event. Dysfunction in PQC pathways also suggests a mechanism by which the inclusions associated with ALS/FTD, as well as many other neurodegenerative disorders, accumulate. The cell’s inability to dispose of damaged or misfolded proteins could lead to a significant accumulation of toxic protein aggregates. Consistent with this possibility, Sharkey et al. [41] found a direct correlation between mutant UBQLN2 aggregation and neurotoxicity. Recent findings from the Shen group also showed that WT UBQLN2 is involved in the degradation of pathologic GA protein aggregates associated with C9orf72-ALS/FTD via interaction with Hsp70 [59]. They additionally show that ALS mutations in UBQLN2 impair the protein’s ability to bind Hsp70 and knockdown or mutation of Hsp70 impairs this disaggregase function.
Numerous proteins linked to ALS/FTD pathogenesis have been found to co-aggregate with UBQLN2. Importantly, TDP-43, an RBP which mislocalizes from the nucleus to the cytosol in ALS/FTD [176], has been shown to accumulate in some UBQLN2 positive aggregates [16,73], though the functional significance of this colocalization is poorly understood. In addition, our laboratory recently discovered that serine protease inhibitors (serpins) accumulate and co-aggregate with UBQLN2 inclusions in the brain and spinal cord of the P497S mutant UBQLN2 mouse model of ALS/FTD [74]. Serpin proteins also formed insoluble aggregates in UBQLN2 KO cells, suggesting that loss of UBQLN2 function, likely in protein degradation pathways, contributes to this aberrant protein aggregation [74].
The formation of protein-rich droplets, such as SGs, provides another possible mechanism by which protein aggregates form in ALS/FTD and other neurodegenerative diseases. SGs are normally resolved upon mitigation of cellular stress. However, prolonged exposure to stress causes SGs to persist within the cell leading to aberrant phase transition of the liquid droplets into less dynamic gel-like formations and stable protein aggregates [39,41,124,126]. Interestingly, a number of proteins implicated in ALS, including UBQLN2, have been shown to localize to SGs [39–41]. Furthermore, investigation of the effect of pathogenic UBQLN2 mutations on LLPS behavior and SG formation has shown that the mutations alter droplet morphology and significantly reduce the liquid-like dynamics of these droplets [41,43,129].
Animal models of UBQLN2
Using in vivo systems to investigate ALS/FTD is essential for studying the pathological mechanisms that underlie human disease and for testing experimental therapeutic targets prior to clinical trials. Many UBQLN2 ALS animal models have been established, each with their unique advantages, but also shortcomings. The earliest model established in 2014 was a transgenic mouse expressing hUBQLN2P497H under the hUBQLN2 promoter [161]. These animals exhibit dendritic spinopathy, cognitive impairments, and hippocampal UBQLN2 inclusions, but do not present with motor neuron loss. A separate group expressed the same UBQLN2 P497H mutation in rats, and these animals exhibited spatial learning deficiencies and hippocampal inclusions as early as 20 days of age [76]. In contrast to the hUBQLN2P497H mouse model, these transgenic rats were generated through expressing hUBQLN2P497H under a TRE promoter and do exhibit neuronal loss at ~ 4 months of age, accompanied by evidence of impaired autophagy and endocytosis. In addition, another group selectively expressed UBQLN2P497H in motor neurons by crossing the TRE-UBQLN2P497H line with ChATtTA-9 transgenic rats, and these rats exhibited similar motor neuron degeneration and UBQLN2 inclusion formation [172].
Aside from the difference in species, a key differentiating factor that seems to impact phenotypes of the hUBQLN2P497H mouse line vs the latter two described rat lines, is expression level of the hUBQLN2P497H. The former had lower expression of UBQLN2P497H under the hUBQLN2 promoter, which did not result in neuronal death, while the latter two were overexpression models. Overexpression animal models seem to successfully capture key UBQLN2 pathology and behavioral phenotypes, such as neuronal loss, behavioral deficits, and protein aggregation. One model that used recombinant-adeno-associated virus vectors to express ALS-linked UBQLN2 mutants (P497H, P497S, and P506T) in mice developed behavioral deficits and protein pathology, without any evidence of neurodegeneration or neuronal loss [72]. Another mouse model overexpressing UBQLN2P497H to 20% of the endogenous mouse UBQLN2 level using the NF-H neuron promoter was reported as having no noticeable phenotype [177]. However, when crossed with mutant TDP-43 mice, the double transgenic animals were found to have increased motor neuron loss [177]. Transgenic mice expressing UBQLN2P506T under the mouse prion promoter also failed to develop signs of neurodegeneration or behavioral changes. However, the expression was associated with changes in PQC factors and robust UBQLN2 aggregation of both UBQLN2P506T and endogenous mouse UBQLN2, suggesting that mutant UBQLN2 is capable of sequestering soluble UBQLN2, a potentially deleterious effect [46]. A Drosophila model that expresses UBQLN2P497H or UBQLN2P525S also exhibits proteinopathy, eye degeneration, and behavioral deficits [178]. Similarly, transgenic C. elegans models constructed by pan-neuronal expression of human WT and ALS/FTD UBQLN2P506T and P497H mutants were all found to have decreased motor phenotypes with the lines expressing the mutants exhibiting the most profound defects [179]. All of the different organisms used to model UBQLN2 mutations have distinct disadvantages and advantages over one other. For example, Drosophila and C. elegans both possess a single UBQLN gene that is less than 50% identical to hUBQLN2 and are also missing the PXX motif, which may preclude identification of mechanisms by which human mutations in the motif cause disease. On the other hand, these organisms are genetically more tractable for conducting genetic screens compared to rodents whose UBQLN2 gene shares greater homology to hUBQLN2. Clever use of these different models will likely accelerate discovery of the mechanisms by which UBQLN2 mutations trigger pathogenesis.
Knock-in techniques are theoretically favorable for generating models of disease as these exclude effects of overexpressing nonphysiological levels of the gene. One group generated constitutive UBQLN2P506T KI mice, and these mice developed cognitive deficits and hippocampal inclusions, but failed to exhibit any motor deficits through gait and rotarod analysis [37]. The P506T mutation is a highly aggressive mutation causing ALS/FTD early in life [16,74]. There could be two possible reasons for why the mutation does not cause motor neuron disease in mice. First, the mouse protein is different from the human, and therefore, modeling the human mutation in the context of the mouse protein (P520T) may be problematic. Second, the short lifespan of mice is not sufficient to model the human disease. Nevertheless, development of animal models that express UBQLN2 mutations at more physiological levels may be particularly instructive in translating findings to human disease.
Furthermore, KO models of the disease have yielded variable phenotypes. One UBQLN2 KO rat line was generated through TALEN-assisted genetic modification, but no pathology was seen in these animals up to 300 days [76]. In UBQLN2 KO mouse lines, the animals exhibit age-dependent motor deficits, but do not exhibit neurodegeneration [75]. Drosophila only express a single UBQLN gene, and in a Drosophila ubqn KO line, the flies exhibit neuronal and glial degeneration along with impairments to ER and autophagy systems [106]. KO of the sole ubqn gene in Drosophila may also suggest some degree of functional redundancy of UBQLN proteins in other animal models. Taken together, these KO models therefore reinforce the idea that UBQLN2-linked ALS/FTD is driven by a combination of both loss-of-function and gain-of-function toxicity.
Perhaps one of the more translational models of UBQLN2-linked ALS is the UBQLN2P497S mouse model derived from expression of the hUBQLN2P497S transgene via a neuron-specific Thy1 promoter [73]. Mutant mice in this model display motor neuron disease, behavioral deficits, cognitive impairments, and TDP-43 proteinopathy. TDP-43 proteinopathy is characterized by depletion of TDP-43 from the nucleus and, in some cases, accumulation of the protein in the cytoplasm. The pathology is seen in > 95% of all ALS cases, the main exceptions being in cases linked to mutations in SOD1 and FUS [157,176,180,181]. The SOD1G93A mouse model of ALS that is widely used for ALS research does not develop TDP-43 pathology [182]. The UBQLN2P497S mouse model may therefore be a useful tool for studying the molecular mechanisms that trigger TDP-43 proteinopathy. Interestingly, crossing an overexpressing UBQLN1 mouse line with the UBQLN2P497S transgenic line can rescue the motor neuron loss, behavioral deficits, aggregation, and TDP-43 proteinopathy [67]. However, an important limitation exists in this model, as transgene expression is restricted to neuronal cell types and any noncell autonomous disease phenomena will be missed.
Current challenges and future directions
In this review, we have summarized the diverse physiologic functions that UBQLN proteins play in cells. Although they primarily function as guardians of PQC, facilitating disposal of ubiquitin-tagged and misfolded proteins through the proteasome and autophagy pathway, there is increasing evidence that the proteins are involved in many other critical cellular functions, including chaperoning, membrane trafficking, stress responses, and DNA repair. The dire consequence of UBQLN dysfunction is perhaps best illustrated by mutations in UBQLN2, which cause ALS/FTD. However, the multifaceted nature of the proteins makes it challenging to discern the key pathological events by which UBQLN2 mutations in the proteins cause pathogenesis.
Many key questions about the physiological functions of UBQLN proteins still remain unanswered. What role do the different UBQLN isoforms play in cells? Do they have redundant or unique functions? What are the signals that specify and integrate UBQLN function in different PQC pathways? UBQLN2 aids in degradation of ubiquitinated substrates through both the autophagy and proteasomal systems. While the ubiquitination linkage (K48 vs K63) of the protein may provide some clue as to targeting of specific pathways, these linkages are not exclusive, so how do UBQLN proteins decide which pathway to target? One possibility is that changes to UBQLN’s structural properties may alter its protein–protein interactions and thereby dictate their particular activity. However, very little is known about the cellular and environmental signals that trigger activation of UBQLN proteins in different functional pathways. The recent finding that phosphorylation is a trigger for DNA repair suggests post-translational modification could play a central role in this regulation [49]. Another possibility is that changes in self- and co-assembly of the proteins, including biophysical changes in their phase separation properties, may play a role in the regulation of their activity and/or function [183]. A critical gap in our knowledge is the structural conformation UBQLN proteins adopt in their inactive and active states. Further structural information is needed to understand the impact that disease-linked mutations in UBQLN2 have on its own folding, self-assembly, and oligomerization as they relate to its disruption of its physiological functions.
Another fundamental question is how do different domains of UBQLN proteins sense and coordinate its different functions? While the UBL and UBA domains have been well-studied due to the vast number of studies on UBL-UBA family members, there is a significant void in both the structural and functional understanding of their regulation in the context of the whole protein. The central domain of the isoforms contains common as well as unique motifs, but the dynamics of protein–protein interaction with them is still poorly understood. A particular challenge is to uncover the function of the PXX motif in UBQLN2 where most ALS/FTD mutations cluster. Further structural studies are necessary to uncover how single amino acid mutations in this region impair UBL domain function, and possibly the UBA domain. In addition, biophysical studies have revealed that LLPS behavior of UBQLN2 is intrinsically tied to disordered regions of the protein [39,129]. This behavior allows for UBQLN2 to enter SGs, but its precise role in SG function is unclear.
Another outstanding question is whether aggregation of UBQLN2 results in disease pathogenesis through a toxic gain-of-function of the aggregates themselves or a loss-of-function mechanism resulting from sequestration of the protein from its normal cellular functions. Disease is unlikely to arise from a simple loss of function because UBQLN2 KO animal models do not recapitulate human disease [75,76]. Current research in the field suggests that a combination of both occurs. Determining the composition of these aggregates may be the first important step to understanding their contribution to neurodegeneration.
Moreover, does dysfunction in PQC pathways stemming from ALS/FTD-linked UBQLN2 mutations drive disease directly or does collapse of these pathways merely exacerbate an underlying disease mechanism? Recent studies indicated that defects in autophagy in mutant UBQLN2 disease models are attributed to impaired biogenesis and regulation of the ATP6v1g1 subunit of the V-ATPase, which leads to impaired autophagosome acidification. Further studies are required to address whether increasing expression of the subunit can rescue the autophagy defect or whether there is an additional upstream impairment. Moreover, mitochondrial dysfunction has been presented as a possible driver of disease due to its particular importance in supplying energy to fulfill the high bioenergetic demands of neurons [184]. However, how UBQLN2 mutations impair mitochondrial health remains inconclusive though defective mitochondrial protein import has been suggested as a possible mechanism. Additionally, a key pathological finding in ALS is TDP-43 proteinopathy, and this pathology has been observed in the UBQLN2 P497S and P506T mouse models [73]. It is clear that TDP-43 must be downstream of UBQLN2 mutations. An important question then becomes how do UBQLN2 mutations cause TDP-43 pathology?
Cellular and animal models have proven to be essential systems for understanding UBQLN2-linked ALS/FTD. However, a potential confounding variable that must be carefully considered is the expression levels of UBQLN2 as both KO and overexpression of mutant and WT UBQLN2 have been associated with toxicity. The mechanisms by which UBQLN2 expression levels are regulated physiologically remain largely unknown. It is possible that the protein’s self-assembly and phase separation properties play a role in the cell’s ability to sense UBQLN2 levels. Additionally, the micro-RNA MIR-155 has been shown to down-regulate UBQLN1 and UBQLN2 protein levels [185], while exogenous treatment of cells or animals with GM1 ganglioside or nialamide, respectively, increases UBQLN1 expression [186,187]; these methods may be useful for manipulating levels of UBQLN proteins in future studies. Discerning the relationship between UBQLN2 mutations and disease pathogenesis will require study of the mutant proteins at physiological concentrations rather than in overexpression systems. Moreover, a useful tool to study ALS has been induced pluripotent stem cells (iPSCs), which have been generated through donors who carry ALS-linked mutations in genes like C9ORF72 and FUS. However, at present, no patient-derived iPSC UBQLN2 lines have been created. Hopefully in the near future, one will arise, providing a valuable platform to study the effects of UBQLN2 mutants on cell-type specific pathways, such as those of differentiated isogenic motor neurons.
UBQLNs function at the center of the cell’s key systems for maintaining cellular proteostasis. Future studies may uncover additional surprising functions of this unique multifaceted protein family and add to a strong foundation for developing potential therapies against UBQLN-linked diseases.
Acknowledgements
This work was supported by The Robert Packard Center for ALS Research at Johns Hopkins and NIH NINDS grants (grant numbers R01NS098243, R01NS100008) to MJM.
Abbreviations
- aa
amino acids
- AD
Alzheimer’s disease
- ALS
amyotrophic lateral sclerosis
- C elegans
Caenorhabditis elegans
- CMA
chaperone-mediated autophagy
- Drosophila
Drosophila melanogaster
- ER
endoplasmic reticulum
- ERAD
ER-associated degradation
- FTD
frontotemporal dementia
- HD
Huntington’s disease
- HSP
heat shock protein
- Htt
huntingtin
- hUBQLN2
human UBQLN2
- KI
knock-in
- KO
knockout
- LLPS
liquid–liquid phase separation
- MVB
multivesicular body
- NES
nuclear export signal
- OXPHOS
oxidative phosphorylation
- PQC
protein quality control
- RBPs
RNA binding proteins
- SGs
stress granules
- STI1
stress inducible 1 domain
- TBK1
TANK-binding kinase 1
- TDP-43
TAR DNA-binding protein 43
- UBA
ubiquitin-associated domain
- UBL
ubiquitin-like domain
- UBQLN
ubiquilin
- UPR
unfolded protein response
- UPS
ubiquitin-proteasome system
- V-ATPase
vacuolar-ATPase
- WT
wild type
Footnotes
Conflict of interest
The authors declare no conflict of interest.
References
- 1.Balch WE, Morimoto RI, Dillin A & Kelly JW (2008) Adapting proteostasis for disease intervention. Science 319, 916–919. [DOI] [PubMed] [Google Scholar]
- 2.Chen B, Retzlaff M, Roos T & Frydman J (2011) Cellular strategies of protein quality control. Cold Spring Harb Perspect Biol 3, a004374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Amm I, Sommer T & Wolf DH (2014) Protein quality control and elimination of protein waste: the role of the ubiquitin-proteasome system. Biochim Biophys Acta 1843, 182–196. [DOI] [PubMed] [Google Scholar]
- 4.Labbadia J & Morimoto RI (2015) The biology of proteostasis in aging and disease. Annu Rev Biochem 84, 435–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yerbury JJ, Ooi L, Dillin A, Saunders DN, Hatters DM, Beart PM, Cashman NR, Wilson MR & Ecroyd H (2016) Walking the tightrope: proteostasis and neurodegenerative disease. J Neurochem 137, 489–505. [DOI] [PubMed] [Google Scholar]
- 6.Hipp MS, Kasturi P & Hartl FU (2019) The proteostasis network and its decline in ageing. Nat Rev Mol Cell Biol 20, 421–435. [DOI] [PubMed] [Google Scholar]
- 7.Marin I (2014) The ubiquilin gene family: evolutionary patterns and functional insights. BMC Evol Biol 14, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu AL, Wang J, Zheleznyak A & Brown EJ (1999) Ubiquitin-related proteins regulate interaction of vimentin intermediate filaments with the plasma membrane. Mol Cell 4, 619–625. [DOI] [PubMed] [Google Scholar]
- 9.Mah AL, Perry G, Smith MA & Monteiro MJ (2000) Identification of ubiquilin, a novel presenilin interactor that increases presenilin protein accumulation. J Cell Biol 151, 847–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kleijnen MF, Shih AH, Zhou P, Kumar S, Soccio RE, Kedersha NL, Gill G & Howley PM (2000) The hPLIC proteins may provide a link between the ubiquitination machinery and the proteasome. Mol Cell 6, 409–419. [DOI] [PubMed] [Google Scholar]
- 11.Davidson JD, Riley B, Burright EN, Duvick LA, Zoghbi HY & Orr HT (2000) Identification and characterization of an ataxin-1-interacting protein: A1Up, a ubiquitin-like nuclear protein. Hum Mol Genet 9, 2305–2312. [DOI] [PubMed] [Google Scholar]
- 12.Yuan S, Swiggin HM, Zheng H & Yan W (2015) A testis-specific gene, Ubqlnl, is dispensable for mouse embryonic development and spermatogenesis. Mol Reprod Dev 82, 408–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Matsuda M, Koide T, Yorihuzi T, Hosokawa N & Nagata K (2001) Molecular cloning of a novel ubiquitin-like protein, UBIN, that binds to ER targeting signal sequences. Biochem Biophys Res Commun 280, 535–540. [DOI] [PubMed] [Google Scholar]
- 14.Su V, Nakagawa R, Koval M & Lau AF (2010) Ubiquitin-independent proteasomal degradation of endoplasmic reticulum-localized connexin43 mediated by CIP75. J Biol Chem 285, 40979–40990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Conklin D, Holderman S, Whitmore TE, Maurer M & Feldhaus AL (2000) Molecular cloning, chromosome mapping and characterization of UBQLN3 a testis-specific gene that contains an ubiquitin-like domain. Gene 249, 91–98. [DOI] [PubMed] [Google Scholar]
- 16.Deng HX, Chen W, Hong ST, Boycott KM, Gorrie GH, Siddique N, Yang Y, Fecto F, Shi Y, Zhai H et al. (2011) Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 477, 211–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Higgins N, Lin B & Monteiro MJ (2019) Lou Gehrig’s disease (ALS): UBQLN2 mutations strike out of phase. Structure 27, 879–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hershko A & Ciechanover A (1998) The ubiquitin system. Annu Rev Biochem 67, 425–479. [DOI] [PubMed] [Google Scholar]
- 19.Elsasser S & Finley D (2005) Delivery of ubiquitinated substrates to protein-unfolding machines. Nat Cell Biol 7, 742–749. [DOI] [PubMed] [Google Scholar]
- 20.Husnjak K & Dikic I (2012) Ubiquitin-binding proteins: decoders of ubiquitin-mediated cellular functions. Annu Rev Biochem 81, 291–322. [DOI] [PubMed] [Google Scholar]
- 21.Su V & Lau AF (2009) Ubiquitin-like and ubiquitin-associated domain proteins: significance in proteasomal degradation. Cell Mol Life Sci 66, 2819–2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hartmann-Petersen R & Gordon C (2004) Integral UBL domain proteins: a family of proteasome interacting proteins. Semin Cell Dev Biol 15, 247–259. [DOI] [PubMed] [Google Scholar]
- 23.Husnjak K, Elsasser S, Zhang N, Chen X, Randles L, Shi Y, Hofmann K, Walters KJ, Finley D & Dikic I (2008) Proteasome subunit Rpn13 is a novel ubiquitin receptor. Nature 453, 481–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dikic I, Wakatsuki S & Walters KJ (2009) Ubiquitin-binding domains - from structures to functions. Nat Rev Mol Cell Biol 10, 659–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen X, Ebelle DL, Wright BJ, Sridharan V, Hooper E & Walters KJ (2019) Structure of hRpn10 Bound to UBQLN2 UBL illustrates basis for complementarity between shuttle factors and substrates at the proteasome. J Mol Biol 431, 939–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martinez-Fonts K, Davis C, Tomita T, Elsasser S, Nager AR, Shi Y, Finley D & Matouschek A (2020) The proteasome 19S cap and its ubiquitin receptors provide a versatile recognition platform for substrates. Nat Commun 11, 477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Walters KJ, Kleijnen MF, Goh AM, Wagner G & Howley PM (2002) Structural studies of the interaction between ubiquitin family proteins and proteasome subunit S5a. Biochemistry 41, 1767–1777. [DOI] [PubMed] [Google Scholar]
- 28.Lim PJ, Danner R, Liang J, Doong H, Harman C, Srinivasan D, Rothenberg C, Wang H, Ye Y, Fang S et al. (2009) Ubiquilin and p97/VCP bind erasin, forming a complex involved in ERAD. J Cell Biol 187, 201–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chang L & Monteiro MJ (2015) Defective proteasome delivery of polyubiquitinated proteins by ubiquilin-2 proteins containing ALS mutations. PLoS One 10, e0130162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Levine B & Kroemer G (2008) Autophagy in the pathogenesis of disease. Cell 132, 27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dikic I & Elazar Z (2018) Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol 19, 349–364. [DOI] [PubMed] [Google Scholar]
- 32.Kurlawala Z, Shah PP, Shah C & Beverly LJ (2017) The STI and UBA domains of UBQLN1 are critical determinants of substrate interaction and proteostasis. J Cell Biochem 118, 2261–2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Itakura E, Zavodszky E, Shao S, Wohlever ML, Keenan RJ & Hegde RS (2016) Ubiquilins chaperone and triage mitochondrial membrane proteins for degradation. Mol Cell 63, 21–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu JJ, Cai A, Greenslade JE, Higgins NR, Fan C, Le NTT, Tatman M, Whiteley AM, Prado MA, Dieriks BV et al. (2020) ALS/FTD mutations in UBQLN2 impede autophagy by reducing autophagosome acidification through loss of function. Proc Natl Acad Sci USA 117, 15230–15241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lin BC, Phung TH, Higgins NR, Greenslade JE, Prado MA, Finley D, Karbowski M, Polster BM & Monteiro MJ (2021) ALS/FTD mutations in UBQLN2 are linked to mitochondrial dysfunction through loss-of-function in mitochondrial protein import. Hum Mol Genet 30, 1230–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaye FJ, Modi S, Ivanovska I, Koonin EV, Thress K, Kubo A, Kornbluth S & Rose MD (2000) A family of ubiquitin-like proteins binds the ATPase domain of Hsp70-like Stch. FEBS Lett 467, 348–355. [DOI] [PubMed] [Google Scholar]
- 37.Hjerpe R, Bett JS, Keuss MJ, Solovyova A, McWilliams TG, Johnson C, Sahu I, Varghese J, Wood N, Wightman M et al. (2016) UBQLN2 mediates autophagy-independent protein aggregate clearance by the proteasome. Cell 166, 935–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fry MY, Saladi SM & Clemons WM Jr (2021) The STI1-domain is a flexible alpha-helical fold with a hydrophobic groove. Protein Sci 30, 882–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dao TP, Kolaitis RM, Kim HJ, O’Donovan K, Martyniak B, Colicino E, Hehnly H, Taylor JP & Castaneda CA (2018) Ubiquitin modulates liquid-liquid phase separation of UBQLN2 via disruption of multivalent interactions. Mol Cell 69, 965–978.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alexander EJ, Ghanbari Niaki A, Zhang T, Sarkar J, Liu Y, Nirujogi RS, Pandey A, Myong S & Wang J (2018) Ubiquilin 2 modulates ALS/FTD-linked FUS-RNA complex dynamics and stress granule formation. Proc Natl Acad Sci USA 115, E11485–E11494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sharkey LM, Safren N, Pithadia AS, Gerson JE, Dulchavsky M, Fischer S, Patel R, Lantis G, Ashraf N, Kim JH et al. (2018) Mutant UBQLN2 promotes toxicity by modulating intrinsic self-assembly. Proc Natl Acad Sci US A 115, E10495–E10504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gerson JE, Linton H, Xing J, Sutter AB, Kakos FS, Ryou J, Liggans N, Sharkey LM, Safren N, Paulson HL et al. (2021) Shared and divergent phase separation and aggregation properties of brain-expressed ubiquilins. Sci Rep 11, 287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Riley JF, Fioramonti PJ, Rusnock AK, Hehnly H & Castaneda CA (2021) ALS-linked mutations impair UBQLN2 stress-induced biomolecular condensate assembly in cells. J Neurochem in press. 10.1111/jnc.15453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stewart M (2007) Molecular mechanism of the nuclear protein import cycle. Nat Rev Mol Cell Biol 8, 195–208. [DOI] [PubMed] [Google Scholar]
- 45.Gilpin KM, Chang L & Monteiro MJ (2015) ALS-linked mutations in ubiquilin-2 or hnRNPA1 reduce interaction between ubiquilin-2 and hnRNPA1. Hum Mol Genet 24, 2565–2577. [DOI] [PubMed] [Google Scholar]
- 46.Sharkey LM, Sandoval-Pistorius SS, Moore SJ, Gerson JE, Komlo R, Fischer S, Negron-Rios KY, Crowley EV, Padron F, Patel R et al. (2020) Modeling UBQLN2-mediated neurodegenerative disease in mice: Shared and divergent properties of wild type and mutant UBQLN2 in phase separation, subcellular localization, altered proteostasis pathways, and selective cytotoxicity. Neurobiol Dis 143, 105016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gerson JE, Safren N, Fischer S, Patel R, Crowley EV, Welday JP, Windle AK, Barmada S, Paulson HL & Sharkey LM (2020) Ubiquilin-2 differentially regulates polyglutamine disease proteins. Hum Mol Genet 29, 2596–2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang W, Huang B, Gao L & Huang C (2021) Impaired 26S proteasome assembly precedes neuronal loss in mutant UBQLN2 rats. Int J Mol Sci 22. in press. 10.3390/ijms22094319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jachimowicz RD, Beleggia F, Isensee J, Velpula BB, Goergens J, Bustos MA, Doll MA, Shenoy A, Checa-Rodriguez C, Wiederstein JL et al. (2019) UBQLN4 represses homologous recombination and is overexpressed in aggressive tumors. Cell 176, 505–519.e22. [DOI] [PubMed] [Google Scholar]
- 50.Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER 3rd, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y et al. (2007) ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 316, 1160–1166. [DOI] [PubMed] [Google Scholar]
- 51.Hirayama S, Sugihara M, Morito D, Iemura SI, Natsume T, Murata S & Nagata K (2018) Nuclear export of ubiquitinated proteins via the UBIN-POST system. Proc Natl Acad Sci USA 115, E4199–E4208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Doi H, Mitsui K, Kurosawa M, Machida Y, Kuroiwa Y & Nukina N (2004) Identification of ubiquitin-interacting proteins in purified polyglutamine aggregates. FEBS Lett 571, 171–176. [DOI] [PubMed] [Google Scholar]
- 53.Wang H, Lim PJ, Yin C, Rieckher M, Vogel BE & Monteiro MJ (2006) Suppression of polyglutamine-induced toxicity in cell and animal models of Huntington’s disease by ubiquilin. Hum Mol Genet 15, 1025–1041. [DOI] [PubMed] [Google Scholar]
- 54.Mori F, Tanji K, Odagiri S, Toyoshima Y, Yoshida M, Ikeda T, Sasaki H, Kakita A, Takahashi H & Wakabayashi K (2012) Ubiquilin immunoreactivity in cytoplasmic and nuclear inclusions in synucleinopathies, polyglutamine diseases and intranuclear inclusion body disease. Acta Neuropathol 124, 149–151. [DOI] [PubMed] [Google Scholar]
- 55.Satoh J, Tabunoki H, Ishida T, Saito Y & Arima K (2013) Ubiquilin-1 immunoreactivity is concentrated on Hirano bodies and dystrophic neurites in Alzheimer’s disease brains. Neuropathol Appl Neurobiol 39, 817–830. [DOI] [PubMed] [Google Scholar]
- 56.Mizukami K, Abrahamson EE, Mi Z, Ishikawa M, Watanabe K, Kinoshita S, Asada T & Ikonomovic MD (2014) Immunohistochemical analysis of ubiquilin-1 in the human hippocampus: association with neurofibrillary tangle pathology. Neuropathology 34, 11–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Safren N, El Ayadi A, Chang L, Terrillion CE, Gould TD, Boehning DF & Monteiro MJ (2014) Ubiquilin-1 overexpression increases the lifespan and delays accumulation of Huntingtin aggregates in the R6/2 mouse model of Huntington’s disease. PLoS One 9, e87513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brettschneider J, Van Deerlin VM, Robinson JL, Kwong L, Lee EB, Ali YO, Safren N, Monteiro MJ, Toledo JB, Elman L et al. (2012) Pattern of ubiquilin pathology in ALS and FTLD indicates presence of C9ORF72 hexanucleotide expansion. Acta Neuropathol 123, 825–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang K, Wang A, Zhong K, Qi S, Wei C, Shu X, Tu WY, Xu W, Xia C, Xiao Y et al. (2021) UBQLN2-HSP70 axis reduces poly-Gly-Ala aggregates and alleviates behavioral defects in the C9ORF72 animal model. Neuron 109, 1949–1962.e6. [DOI] [PubMed] [Google Scholar]
- 60.Rutherford NJ, Lewis J, Clippinger AK, Thomas MA, Adamson J, Cruz PE, Cannon A, Xu G, Golde TE, Shaw G et al. (2013) Unbiased screen reveals ubiquilin-1 and −2 highly associated with huntingtin inclusions. Brain Res 1524, 62–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Safren N, Chang L, Dziki KM & Monteiro MJ (2015) Signature changes in ubiquilin expression in the R6/2 mouse model of Huntington’s disease. Brain Res 1597, 37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ciechanover A & Brundin P (2003) The ubiquitin proteasome system in neurodegenerative diseases: sometimes the chicken, sometimes the egg. Neuron 40, 427–446. [DOI] [PubMed] [Google Scholar]
- 63.Wang H & Monteiro MJ (2007) Ubiquilin overexpression reduces GFP-polyalanine-induced protein aggregates and toxicity. Exp Cell Res 313, 2810–2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liu Y, Qiao F & Wang H (2017) Enhanced proteostasis in post-ischemic stroke mouse brains by ubiquilin-1 promotes functional recovery. Cell Mol Neurobiol 37, 1325–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Adegoke OO, Qiao F, Liu Y, Longley K, Feng S & Wang H (2017) Overexpression of ubiquilin-1 alleviates Alzheimer’s disease-caused cognitive and motor deficits and reduces amyloid-beta accumulation in mice. J Alzheimers Dis 59, 575–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liu Y, Lu L, Hettinger CL, Dong G, Zhang D, Rezvani K, Wang X & Wang H (2014) Ubiquilin-1 protects cells from oxidative stress and ischemic stroke caused tissue injury in mice. J Neurosci 34, 2813–2821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang S, Tatman M & Monteiro MJ (2020) Overexpression of UBQLN1 reduces neuropathology in the P497S UBQLN2 mouse model of ALS/FTD. Acta Neuropathol Commun 8, 164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang H & Monteiro MJ (2007) Ubiquilin interacts and enhances the degradation of expanded-polyglutamine proteins. Biochem Biophys Res Commun 360, 423–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schulte J & Littleton JT (2011) The biological function of the Huntingtin protein and its relevance to Huntington’s Disease pathology. Curr Trends Neurol 5, 65–78. [PMC free article] [PubMed] [Google Scholar]
- 70.Qiao F, Longley KR, Feng S, Schnack S, Gao H, Li Y, Schlenker EH & Wang H (2017) Reduced body weight gain in ubiquilin-1 transgenic mice is associated with increased expression of energy-sensing proteins. Physiol Rep 5, e13260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Huang B, Wu Q, Zhou H, Huang C & Xia XG (2016) Increased Ubqln2 expression causes neuron death in transgenic rats. J Neurochem 139, 258–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ceballos-Diaz C, Rosario AM, Park HJ, Chakrabarty P, Sacino A, Cruz PE, Siemienski Z, Lara N, Moran C, Ravelo N et al. (2015) Viral expression of ALS-linked ubiquilin-2 mutants causes inclusion pathology and behavioral deficits in mice. Mol Neurodegener 10, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Le NT, Chang L, Kovlyagina I, Georgiou P, Safren N, Braunstein KE, Kvarta MD, Van Dyke AM, LeGates TA, Philips T et al. (2016) Motor neuron disease, TDP-43 pathology, and memory deficits in mice expressing ALS-FTD-linked UBQLN2 mutations. Proc Natl Acad Sci USA 113, E7580–E7589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Higgins NR, Greenslade JE, Wu JJ, Miranda E, Galliciotti G & Monteiro MJ (2021) Serpin neuropathology in the P497S UBQLN2 mouse model of ALS/FTD. Brain Pathol e12948. in press. 10.1111/bpa.12948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Whiteley AM, Prado MA, de Poot SAH, Paulo JA, Ashton M, Dominguez S, Weber M, Ngu H, Szpyt J, Jedrychowski MP et al. (2021) Global proteomics of Ubqln2-based murine models of ALS. J Biol Chem 296, 100153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wu Q, Liu M, Huang C, Liu X, Huang B, Li N, Zhou H & Xia XG (2015) Pathogenic Ubqln2 gains toxic properties to induce neuron death. Acta Neuropathol 129, 417–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kim SH, Shi Y, Hanson KA, Williams LM, Sakasai R, Bowler MJ & Tibbetts RS (2008) Potentiation of ALS-associated TDP-43 aggregation by the proteasome-targeting factor, Ubiquilin 1. J Biol Chem 284, 8083–8092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schwartz AL & Ciechanover A (1999) The ubiquitin-proteasome pathway and pathogenesis of human diseases. Annu Rev Med 50, 57–74. [DOI] [PubMed] [Google Scholar]
- 79.Lipkowitz S & Weissman AM (2011) RINGs of good and evil: RING finger ubiquitin ligases at the crossroads of tumour suppression and oncogenesis. Nat Rev Cancer 11, 629–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Akutsu M, Dikic I & Bremm A (2016) Ubiquitin chain diversity at a glance. J Cell Sci 129, 875–880. [DOI] [PubMed] [Google Scholar]
- 81.Swatek KN & Komander D (2016) Ubiquitin modifications. Cell Res 26, 399–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yau R & Rape M (2016) The increasing complexity of the ubiquitin code. Nat Cell Biol 18, 579–586. [DOI] [PubMed] [Google Scholar]
- 83.Bard JAM, Goodall EA, Greene ER, Jonsson E, Dong KC & Martin A (2018) Structure and function of the 26S proteasome. Annu Rev Biochem 87, 697–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Deveraux Q, Ustrell V, Pickart C & Rechsteiner M (1994) A 26 S protease subunit that binds ubiquitin conjugates. J Biol Chem 269, 7059–7061. [PubMed] [Google Scholar]
- 85.Shi Y, Chen X, Elsasser S, Stocks BB, Tian G, Lee BH, Shi Y, Zhang N, de Poot SA, Tuebing F et al. (2016) Rpn1 provides adjacent receptor sites for substrate binding and deubiquitination by the proteasome. Science 351, 1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yao T & Cohen RE (2002) A cryptic protease couples deubiquitination and degradation by the proteasome. Nature 419, 403–407. [DOI] [PubMed] [Google Scholar]
- 87.Verma R, Aravind L, Oania R, McDonald WH, Yates JR, Koonin EV & Deshaies RJ (2002) Role of Rpn11 metalloprotease in deubiquitination and degradation by the 26S proteasome. Science 298, 611–615. [DOI] [PubMed] [Google Scholar]
- 88.Massey LK, Mah AL, Ford DL, Miller J, Liang J, Doong H & Monteiro MJ (2004) Overexpression of ubiquilin decreases ubiquitination and degradation of presenilin proteins. J Alzheimers Dis 6, 79–92. [DOI] [PubMed] [Google Scholar]
- 89.Raasi S, Varadan R, Fushman D & Pickart CM (2005) Diverse polyubiquitin interaction properties of ubiquitin-associated domains. Nat Struct Mol Biol 12, 708–714. [DOI] [PubMed] [Google Scholar]
- 90.Zhang D, Raasi S & Fushman D (2008) Affinity makes the difference: nonselective interaction of the UBA domain of Ubiquilin-1 with monomeric ubiquitin and polyubiquitin chains. J Mol Biol 377, 162–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Harman CA & Monteiro MJ (2019) The specificity of ubiquitin binding to ubiquilin-1 is regulated by sequences besides its UBA domain. Biochim Biophys Acta Gen Subj 1863, 1568–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ko HS, Uehara T, Tsuruma K & Nomura Y (2004) Ubiquilin interacts with ubiquitylated proteins and proteasome through its ubiquitin-associated and ubiquitin-like domains. FEBS Lett 566, 110–114. [DOI] [PubMed] [Google Scholar]
- 93.Chen X, Randles L, Shi K, Tarasov SG, Aihara H & Walters KJ (2016) Structures of Rpn1 T1:Rad23 and hRpn13:hPLIC2 reveal distinct binding mechanisms between substrate receptors and shuttle factors of the proteasome. Structure 24, 1257–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hamazaki J, Hirayama S & Murata S (2015) Redundant roles of Rpn10 and Rpn13 in recognition of ubiquitinated proteins and cellular homeostasis. PLoS Genet 11, e1005401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sun Z & Brodsky JL (2019) Protein quality control in the secretory pathway. J Cell Biol 218, 3171–3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Araki K & Nagata K (2011) Protein folding and quality control in the ER. Cold Spring Harb Perspect Biol 3, a007526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Schubert U, Anton LC, Gibbs J, Norbury CC, Yewdell JW & Bennink JR (2000) Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature 404, 770–774. [DOI] [PubMed] [Google Scholar]
- 98.Vembar SS & Brodsky JL (2008) One step at a time: endoplasmic reticulum-associated degradation. Nat Rev Mol Cell Biol 9, 944–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hirsch C, Gauss R, Horn SC, Neuber O & Sommer T (2009) The ubiquitylation machinery of the endoplasmic reticulum. Nature 458, 453–460. [DOI] [PubMed] [Google Scholar]
- 100.Kim TY, Kim E, Yoon SK & Yoon JB (2008) Herp enhances ER-associated protein degradation by recruiting ubiquilins. Biochem Biophys Res Commun 369, 741–746. [DOI] [PubMed] [Google Scholar]
- 101.Xia Y, Yan LH, Huang B, Liu M, Liu X & Huang C (2014) Pathogenic mutation of UBQLN2 impairs its interaction with UBXD8 and disrupts endoplasmic reticulum-associated protein degradation. J Neurochem 129, 99–106. [DOI] [PubMed] [Google Scholar]
- 102.Ron D & Walter P (2007) Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 8, 519–529. [DOI] [PubMed] [Google Scholar]
- 103.Hetz C, Zhang K & Kaufman RJ (2020) Mechanisms, regulation and functions of the unfolded protein response. Nat Rev Mol Cell Biol 21, 421–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Levine B & Kroemer G (2019) Biological functions of autophagy genes: a disease perspective. Cell 176, 11–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Collins MP & Forgac M (2018) Regulation of V-ATPase assembly in nutrient sensing and function of V-ATPases in breast cancer metastasis. Front Physiol 9, 902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Senturk M, Lin G, Zuo Z, Mao D, Watson E, Mikos AG & Bellen HJ (2019) Ubiquilins regulate autophagic flux through mTOR signalling and lysosomal acidification. Nat Cell Biol 21, 384–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wu S, Mikhailov A, Kallo-Hosein H, Hara K, Yonezawa K & Avruch J (2002) Characterization of ubiquilin 1, an mTOR-interacting protein. Biochim Biophys Acta 1542, 41–56. [DOI] [PubMed] [Google Scholar]
- 108.Dunlop EA & Tee AR (2014) mTOR and autophagy: a dynamic relationship governed by nutrients and energy. Semin Cell Dev Biol 36, 121–212. [DOI] [PubMed] [Google Scholar]
- 109.Saxton RA & Sabatini DM (2017) mTOR signaling in growth, metabolism, and disease. Cell 169, 361–371. [DOI] [PubMed] [Google Scholar]
- 110.Coffey RT, Shi Y, Long MJ, Marr MT 2nd & Hedstrom L (2016) Ubiquilin-mediated small molecule inhibition of mammalian target of rapamycin complex 1 (mTORC1) signaling. J Biol Chem 291, 5221–5233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.N’Diaye EN, Kajihara KK, Hsieh I, Morisaki H, Debnath J & Brown EJ (2009) PLIC proteins or ubiquilins regulate autophagy-dependent cell survival during nutrient starvation. EMBO Rep 10, 173–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Rothenberg C, Srinivasan D, Mah L, Kaushik S, Peterhoff CM, Ugolino J, Fang S, Cuervo AM, Nixon RA & Monteiro MJ (2010) Ubiquilin functions in autophagy and is degraded by chaperone-mediated autophagy. Hum Mol Genet 19, 3219–3232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sakowski ET, Koster S, Portal Celhay C, Park HS, Shrestha E, Hetzenecker SE, Maurer K, Cadwell K & Philips JA (2015) Ubiquilin 1 promotes IFN-gamma-induced Xenophagy of Mycobacterium tuberculosis. PLoS Pathog 11, e1005076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lee DY, Arnott D & Brown EJ (2013) Ubiquilin4 is an adaptor protein that recruits Ubiquilin1 to the autophagy machinery. EMBO Rep 14, 373–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kaushik S, Bandyopadhyay U, Sridhar S, Kiffin R, Martinez-Vicente M, Kon M, Orenstein SJ, Wong E & Cuervo AM (2011) Chaperone-mediated autophagy at a glance. J Cell Sci 124, 495–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kaushik S & Cuervo AM (2018) The coming of age of chaperone-mediated autophagy. Nat Rev Mol Cell Biol 19, 365–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Pfanner N, Warscheid B & Wiedemann N (2019) Mitochondrial proteins: from biogenesis to functional networks. Nat Rev Mol Cell Biol 20, 267–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Whiteley AM, Prado MA, Peng I, Abbas AR, Haley B, Paulo JA, Reichelt M, Katakam A, Sagolla M, Modrusan Z et al. (2017) Ubiquilin1 promotes antigen-receptor mediated proliferation by eliminating mislocalized mitochondrial proteins. Elife 6, e26435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ibsen KH (1961) The Crabtree effect: a review. Cancer Res 21, 829–841. [PubMed] [Google Scholar]
- 120.Hartl FU, Bracher A & Hayer-Hartl M (2011) Molecular chaperones in protein folding and proteostasis. Nature 475, 324–332. [DOI] [PubMed] [Google Scholar]
- 121.Stieren ES, El Ayadi A, Xiao Y, Siller E, Landsverk ML, Oberhauser AF, Barral JM & Boehning D (2011) Ubiquilin-1 is a molecular chaperone for the amyloid precursor protein. J Biol Chem 286, 35689–35698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ford DL & Monteiro MJ (2006) Dimerization of ubiquilin is dependent upon the central region of the protein: evidence that the monomer, but not the dimer, is involved in binding presenilins. Biochem J 399, 397–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bertolini M, Fenzl K, Kats I, Wruck F, Tippmann F, Schmitt J, Auburger JJ, Tans S, Bukau B & Kramer G (2021) Interactions between nascent proteins translated by adjacent ribosomes drive homomer assembly. Science 371, 57–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Molliex A, Temirov J, Lee J, Coughlin M, Kanagaraj AP, Kim HJ, Mittag T, Taylor JP & Designed JPTAM (2015) Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 163, 123–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Banani SF, Lee HO, Hyman AA & Rosen MK (2017) Biomolecular condensates: organizers of cellular biochemistry. Nat Rev Mol Cell Biol 18, 285–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Protter DSW & Parker R (2016) Principles and properties of stress granules. Trends Cell Biol 26, 668–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Walters RW & Parker R (2015) Coupling of ribostasis and proteostasis: Hsp70 Proteins in mRNA metabolism. Trends Biochem Sci 40, 552–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Jain S, Wheeler JR, Walters RW, Agrawal A, Barsic A & Parker R (2016) ATPase-modulated stress granules contain a diverse proteome and substructure. Cell 164, 487–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Dao TP, Martyniak B, Canning AJ, Lei Y, Colicino EG, Cosgrove MS, Hehnly H & Castaneda CA (2019) ALS-linked mutations affect UBQLN2 oligomerization and phase separation in a position- and amino acid-dependent manner. Structure 27, 937–951.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zheng T, Galagedera SKK & Castaneda CA (2021) Previously uncharacterized interactions between the folded and intrinsically disordered domains impart asymmetric effects on UBQLN2 phase separation. Protein Sci 30, 1467–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Cassel JA & Reitz AB (2013) Ubiquilin-2 (UBQLN2) binds with high affinity to the C-terminal region of TDP-43 and modulates TDP-43 levels in H4 cells: characterization of inhibition by nucleic acids and 4-aminoquinolines. Biochim Biophys Acta 1834, 964–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Subudhi I & Shorter J (2018) Ubiquilin 2: shuttling clients out of phase? Mol Cell 69, 919–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Gavriilidis C, Laredj L, Solinhac R, Messaddeq N, Viaud J, Laporte J, Sumara I & Hnia K (2018) The MTM1-UBQLN2-HSP complex mediates degradation of misfolded intermediate filaments in skeletal muscle. Nat Cell Biol 20, 198–210. [DOI] [PubMed] [Google Scholar]
- 134.Korolchuk VI, Menzies FM & Rubinsztein DC (2010) Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett 584, 1393–1398. [DOI] [PubMed] [Google Scholar]
- 135.Regan-Klapisz E, Sorokina I, Voortman J, de Keizer P, Roovers RC, Verheesen P, Urbe S, Fallon L, Fon EA, Verkleij A et al. (2005) Ubiquilin recruits Eps15 into ubiquitin-rich cytoplasmic aggregates via a UIM-UBL interaction. J Cell Sci 118, 4437–4450. [DOI] [PubMed] [Google Scholar]
- 136.N’Diaye EN, Hanyaloglu AC, Kajihara KK, Puthenveedu MA, Wu P, von Zastrow M & Brown EJ (2008) The ubiquitin-like protein PLIC-2 is a negative regulator of G protein-coupled receptor endocytosis. Mol Biol Cell 19, 1252–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.McMahon HT & Boucrot E (2011) Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nat Rev Mol Cell Biol 12, 517–533. [DOI] [PubMed] [Google Scholar]
- 138.Van Bergen En Henegouwen PMP (2009) Eps15: a multifunctional adaptor protein regulating intracellular trafficking. Cell Commun Signal 7, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Bedford FK, Kittler JT, Muller E, Thomas P, Uren JM, Merlo D, Wisden W, Triller A, Smart TG & Moss SJ (2001) GABA(A) receptor cell surface number and subunit stability are regulated by the ubiquitin-like protein Plic-1. Nat Neurosci 4, 908–916. [DOI] [PubMed] [Google Scholar]
- 140.Ficklin MB, Zhao S & Feng G (2005) Ubiquilin-1 regulates nicotine-induced up-regulation of neuronal nicotinic acetylcholine receptors. J Biol Chem 280, 34088–34095. [DOI] [PubMed] [Google Scholar]
- 141.Bache KG, Brech A, Mehlum A & Stenmark H (2003) Hrs regulates multivesicular body formation via ESCRT recruitment to endosomes. J Cell Biol 162, 435–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kama R, Gabriely G, Kanneganti V & Gerst JE (2018) Cdc48 and ubiquilins confer selective anterograde protein sorting and entry into the multivesicular body in yeast. Mol Biol Cell 29, 948–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Xu J, Camfield R & Gorski SM (2018) The interplay between exosomes and autophagy - partners in crime. J Cell Sci 131, jcs215210. 10.1242/jcs.215210 [DOI] [PubMed] [Google Scholar]
- 144.Brown RH & Al-Chalabi A (2017) Amyotrophic lateral sclerosis. N Engl J Med 377, 162–172. [DOI] [PubMed] [Google Scholar]
- 145.van Es MA, Hardiman O, Chio A, Al-Chalabi A, Pasterkamp RJ, Veldink JH & van den Berg LH (2017) Amyotrophic lateral sclerosis. Lancet 390, 2084–2098. [DOI] [PubMed] [Google Scholar]
- 146.Synofzik M, Maetzler W, Grehl T, Prudlo J, Vom Hagen JM, Haack T, Rebassoo P, Munz M, Schols L & Biskup S (2012) Screening in ALS and FTD patients reveals 3 novel UBQLN2 mutations outside the PXX domain and a pure FTD phenotype. Neurobiol Aging 33, 2949.e13–7. [DOI] [PubMed] [Google Scholar]
- 147.Williams KL, Warraich ST, Yang S, Solski JA, Fernando R, Rouleau GA, Nicholson GA & Blair IP (2012) UBQLN2/ubiquilin 2 mutation and pathology in familial amyotrophic lateral sclerosis. Neurobiol Aging 33, 2527.e3–10. [DOI] [PubMed] [Google Scholar]
- 148.Vengoechea J, David MP, Yaghi SR, Carpenter L & Rudnicki SA (2013) Clinical variability and female penetrance in X-linked familial FTD/ALS caused by a P506S mutation in UBQLN2. Amyotroph Lateral Scler Frontotemporal Degener 14, 615–619. [DOI] [PubMed] [Google Scholar]
- 149.Gellera C, Tiloca C, Del Bo R, Corrado L, Pensato V, Agostini J, Cereda C, Ratti A, Castellotti B, Corti S et al. (2013) Ubiquilin 2 mutations in Italian patients with amyotrophic lateral sclerosis and frontotemporal dementia. J Neurol Neurosurg Psychiatry 84, 183–187. [DOI] [PubMed] [Google Scholar]
- 150.Fahed AC, McDonough B, Gouvion CM, Newell KL, Dure LS, Bebin M, Bick AG, Seidman JG, Harter DH & Seidman CE (2014) UBQLN2 mutation causing heterogeneous X-linked dominant neurodegeneration. Ann Neurol 75, 793–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Kotan D, Iskender C, Ozoguz Erimis A & Basak AN (2016) A Turkish family with a familial ALS-positive UBQLN2-S340I mutation. Noro Psikiyatr Ars 53, 283–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Scotter EL, Smyth L, Bailey J, Wong CH, de Majo M, Vance CA, Synek BJ, Turner C, Pereira J, Charleston A et al. (2017) C9ORF72 and UBQLN2 mutations are causes of amyotrophic lateral sclerosis in New Zealand: a genetic and pathologic study using banked human brain tissue. Neurobiol Aging 49, 214 e1–214 e5. [DOI] [PubMed] [Google Scholar]
- 153.Teyssou E, Chartier L, Amador MD, Lam R, Lautrette G, Nicol M, Machat S, Da Barroca S, Moigneu C, Mairey M et al. (2017) Novel UBQLN2 mutations linked to amyotrophic lateral sclerosis and atypical hereditary spastic paraplegia phenotype through defective HSP70-mediated proteolysis. Neurobiol Aging 58, 239 e11–239 e20. [DOI] [PubMed] [Google Scholar]
- 154.Gkazi SA, Troakes C, Topp S, Miller JW, Vance CA, Sreedharan J, Al-Chalabi A, Kirby J, Shaw PJ, Al-Sarraj S et al. (2019) Striking phenotypic variation in a family with the P506S UBQLN2 mutation including amyotrophic lateral sclerosis, spastic paraplegia, and frontotemporal dementia. Neurobiol Aging 73, 229 e5–229 e9. [DOI] [PubMed] [Google Scholar]
- 155.Fecto F & Siddique T (2012) UBQLN2/P62 cellular recycling pathways in amyotrophic lateral sclerosis and frontotemporal dementia. Muscle Nerve 45, 157–162. [DOI] [PubMed] [Google Scholar]
- 156.Neumann M, Kwong LK, Truax AC, Vanmassenhove B, Kretzschmar HA, Van Deerlin VM, Clark CM, Grossman M, Miller BL, Trojanowski JQ et al. (2007) TDP-43-positive white matter pathology in frontotemporal lobar degeneration with ubiquitin-positive inclusions. J Neuropathol Exp Neurol 66, 177–183. [DOI] [PubMed] [Google Scholar]
- 157.Mackenzie IR, Bigio EH, Ince PG, Geser F, Neumann M, Cairns NJ, Kwong LK, Forman MS, Ravits J, Stewart H et al. (2007) Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann Neurol 61, 427–434. [DOI] [PubMed] [Google Scholar]
- 158.Steinacker P, Barschke P & Otto M (2019) Biomarkers for diseases with TDP-43 pathology. Mol Cell Neurosci 97, 43–59. [DOI] [PubMed] [Google Scholar]
- 159.Osaka M, Ito D & Suzuki N (2016) Disturbance of proteasomal and autophagic protein degradation pathways by amyotrophic lateral sclerosis-linked mutations in ubiquilin 2. Biochem Biophys Res Commun 472, 324–331. [DOI] [PubMed] [Google Scholar]
- 160.Kandasamy G & Andréasson C (2018) Hsp70-Hsp110 chaperones deliver ubiquitin-dependent and - independent substrates to the 26S proteasome for proteolysis in yeast. J Cell Sci 131, jcs210948. 10.1242/jcs.210948 [DOI] [PubMed] [Google Scholar]
- 161.Gorrie GH, Fecto F, Radzicki D, Weiss C, Shi Y, Dong H, Zhai H, Fu R, Liu E, Li S et al. (2014) Dendritic spinopathy in transgenic mice expressing ALS/dementia-linked mutant UBQLN2. Proc Natl Acad Sci USA 111, 14524–14529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Lin JH, Walter P & Yen TS (2008) Endoplasmic reticulum stress in disease pathogenesis. Annu Rev Pathol 3, 399–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Hetz C & Saxena S (2017) ER stress and the unfolded protein response in neurodegeneration. Nat Rev Neurol 13, 477–491. [DOI] [PubMed] [Google Scholar]
- 164.Saxena S, Cabuy E & Caroni P (2009) A role for motoneuron subtype-selective ER stress in disease manifestations of FALS mice. Nat Neurosci 12, 627–636. [DOI] [PubMed] [Google Scholar]
- 165.Sun S, Sun Y, Ling SC, Ferraiuolo L, McAlonis-Downes M, Zou Y, Drenner K, Wang Y, Ditsworth D, Tokunaga S et al. (2015) Translational profiling identifies a cascade of damage initiated in motor neurons and spreading to glia in mutant SOD1-mediated ALS. Proc Natl Acad Sci USA 112, E6993–E7002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Lippincott-Schwartz J, Roberts TH & Hirschberg K (2000) Secretory protein trafficking and organelle dynamics in living cells. Ann Rev Cell Dev Biol 16, 557–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Halloran M, Ragagnin AMG, Vidal M, Parakh S, Yang S, Heng B, Grima N, Shahheydari H, Soo KY, Blair I et al. (2019) Amyotrophic lateral sclerosis-linked UBQLN2 mutants inhibit endoplasmic reticulum to Golgi transport, leading to Golgi fragmentation and ER stress. Cell Mol Life Sci 77, 3859–3873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Atkin JD, Farg MA, Soo KY, Walker AK, Halloran M, Turner BJ, Nagley P & Horne MK (2014) Mutant SOD1 inhibits ER-Golgi transport in amyotrophic lateral sclerosis. J Neurochem 129, 190–204. [DOI] [PubMed] [Google Scholar]
- 169.Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjorkoy G & Johansen T (2007) p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 282, 24131–24145. [DOI] [PubMed] [Google Scholar]
- 170.Yoshii SR & Mizushima N (2017) Monitoring and measuring autophagy. Int J Mol Sci 18, e1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Ma S, Attarwala IY & Xie XQ (2019) SQSTM1/p62: a potential target for neurodegenerative disease. ACS Chem Neurosci 10, 2094–2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Chen T, Huang B, Shi X, Gao L & Huang C (2018) Mutant UBQLN2(P497H) in motor neurons leads to ALS-like phenotypes and defective autophagy in rats. Acta Neuropathol Commun 6, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Collins MP & Forgac M (2020) Regulation and function of V-ATPases in physiology and disease. Biochim Biophys Acta Biomembr 1862, 183341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Chen T, Zhang W, Huang B, Chen X & Huang C (2020) UBQLN2 promotes the production of type I interferon via the TBK1-IRF3 pathway. Cells 9, 1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Picher-Martel V, Dutta K, Phaneuf D, Sobue G & Julien JP (2015) Ubiquilin-2 drives NF-kappaB activity and cytosolic TDP-43 aggregation in neuronal cells. Mol Brain 8, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM et al. (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133. [DOI] [PubMed] [Google Scholar]
- 177.Picher-Martel V, Renaud L, Bareil C & Julien JP (2019) Neuronal expression of UBQLN2(P497H) exacerbates TDP-43 pathology in TDP-43(G348C) mice through interaction with ubiquitin. Mol Neurobiol 56, 4680–4696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Kim SH, Stiles SG, Feichtmeier JM, Ramesh N, Zhan L, Scalf MA, Smith LM, Pandey UB & Tibbetts RS (2018) Mutation-dependent aggregation and toxicity in a Drosophila model for UBQLN2-associated ALS. Hum Mol Genet 27, 322–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Saxton AD & Kraemer BC (2021) Human Ubiquilin 2 and TDP-43 co-pathology drives neurodegeneration in transgenic C. elegans. G3 (Bethesda) jkab158. in press. 10.1093/g3journal/jkab158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Mackenzie IR & Rademakers R (2008) The role of transactive response DNA-binding protein-43 in amyotrophic lateral sclerosis and frontotemporal dementia. Curr Opin Neurol 21, 693–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Scotter EL, Chen HJ & Shaw CE (2015) TDP-43 proteinopathy and ALS: insights into disease mechanisms and therapeutic targets. Neurotherapeutics 12, 352–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX et al. (1994) Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science 264, 1772–1775. [DOI] [PubMed] [Google Scholar]
- 183.Zheng T, Yang Y & Castaneda CA (2020) Structure, dynamics and functions of UBQLNs: at the crossroads of protein quality control machinery. Biochem J 477, 3471–3497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Smith EF, Shaw PJ & De Vos KJ (2019) The role of mitochondria in amyotrophic lateral sclerosis. Neurosci Lett 710, 132933. [DOI] [PubMed] [Google Scholar]
- 185.Yadav S, Singh N, Shah PP, Rowbotham DA, Malik D, Srivastav A, Shankar J, Lam WL, Lockwood WW & Beverly LJ (2017) MIR155 regulation of ubiquilin1 and ubiquilin 2: implications in cellular protection and tumorigenesis. Neoplasia 19, 321–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Liu Z, Ruan Y, Yue W, Zhu Z, Hartmann T, Beyreuther K & Zhang D (2006) GM1 up-regulates Ubiquilin 1 expression in human neuroblastoma cells and rat cortical neurons. Neurosci Lett 407, 59–63. [DOI] [PubMed] [Google Scholar]
- 187.Liu Y, Feng S, Subedi K & Wang H (2020) Attenuation of ischemic stroke-caused brain injury by a monoamine oxidase inhibitor involves improved proteostasis and reduced neuroinflammation. Mol Neurobiol 57, 937–948. [DOI] [PMC free article] [PubMed] [Google Scholar]