

Abstract
It is still unclear why pathological amyloid deposition initiates in specific brain regions or why some cells or tissues are more susceptible than others. Amyloid deposition is determined by the self‐assembly of short protein segments called aggregation‐prone regions (APRs) that favour cross‐β structure. Here, we investigated whether Aβ amyloid assembly can be modified by heterotypic interactions between Aβ APRs and short homologous segments in otherwise unrelated human proteins. Mining existing proteomics data of Aβ plaques from AD patients revealed an enrichment in proteins that harbour such homologous sequences to the Aβ APRs, suggesting heterotypic amyloid interactions may occur in patients. We identified homologous APRs from such proteins and show that they can modify Aβ assembly kinetics, fibril morphology and deposition pattern in vitro. Moreover, we found three of these proteins upon transient expression in an Aβ reporter cell line promote Aβ amyloid aggregation. Strikingly, we did not find a bias towards heterotypic interactions in plaques from AD mouse models where Aβ self‐aggregation is observed. Based on these data, we propose that heterotypic APR interactions may play a hitherto unrealized role in amyloid‐deposition diseases.
Keywords: Alzheimer’s disease, amyloid beta, heterotypic aggregation, toxicity
Subject Categories: Neuroscience
Interactions between aggregation‐prone regions in Aβ and short homologous segments in unrelated human proteins modulate Aβ assembly kinetics, fibril morphology and deposition.
Introduction
Neurodegenerative amyloid diseases are a diverse group of pathologies that present very different symptoms and progressions in different areas of the brain (Walsh & Selkoe, 2016; Chiti & Dobson, 2017; Goedert et al, 2017). Simultaneously these diseases share common hallmarks that remain poorly explained. First, their initiation is characterized by the deposition of particular proteins in specific cells or brain regions (Gan et al, 2018). Second, this process is associated with functional and homeostatic dysregulation of affected cells ultimately resulting in neuronal death (Zaman et al, 2019). Third, from the site of initiation the disease progresses in a stereotypical manner by propagation of amyloid deposition to anatomically connected cells and brain regions with symptoms that match their function (Taylor et al, 2002). Together these properties suggest specific neuronal and regional vulnerability of the brain to the aggregation, propagation and toxicity of particular amyloidogenic proteins (Muratore et al, 2017; Fu et al, 2018). Factors enhancing these vulnerabilities not only include physiological ageing but also disease‐specific familial mutations and population risk factors (Hipp et al, 2019; Silva et al, 2019). This illustrates how neuronal susceptibility is favoured by general (protein) homeostatic ageing but that disease initiation and its effects are highly context dependent. It is still unclear which cellular interactions contribute to the modulation of neuronal susceptibility either by sensitizing or protecting particular neurons or brain regions to aggregation. It is also not known whether amyloid interactions in each of these diseases are purely idiosyncratic or whether cross‐β amyloid structure also favours canonical modes of interaction that provide generic mechanisms for amyloid gain‐of‐function.
Amyloid structures from different proteins grown either in vitro or in vivo share a common cross‐β architecture (Landreh et al, 2016; Riek & Eisenberg, 2016; Lutter et al, 2019; Gallardo et al, 2020). Structural analysis of disease‐associated amyloid structures and their polymorphs revealed that they are not uniformly stable but that some regions dominate the thermodynamic stability of the amyloid (preprint: van der Kant et al, 2021). Interestingly stable regions correspond to those previously identified as the amyloid nucleating segments of these proteins (Ventura et al, 2004; Teng & Eisenberg, 2009; Ganesan et al, 2016; Marshall et al, 2016). These aggregation‐prone regions (APRs) consist of short sequence segments, 5–15 residues in length (Fernandez‐Escamilla et al, 2004a; Rousseau et al, 2006b; Goldschmidt et al, 2010) and their thermodynamic stability results from their high propensity to adopt the cross‐β conformation (Rousseau et al, 2006a; Louros et al, 2020; preprint: van der Kant et al, 2021). APRs assemble into stable amyloids both on their own as peptides or in the context of full‐length proteins underlining their essential (Ganesan et al, 2016; Marshall et al, 2016) and dominant role (Ventura et al, 2004; Teng & Eisenberg, 2009). Because of these favourable conformational properties, APRs constitute good protein interaction interfaces favouring amyloid self‐assembly, that is, through their affinity for binding to their own sequence (Krebs et al, 2004; O'Nuallain et al, 2004, 2005; Vanik et al, 2004; Wetzel, 2006). Recent evidence, however, suggests that amyloid self‐specificity is not absolute and that disease amyloids can engage heterotypic interactions resulting in cross‐seeding and co‐aggregation (Giasson et al, 2003; Oskarsson et al, 2015; Lutter et al, 2019; Konstantoulea et al, 2021; Ly et al, 2021) that are relevant to the pathophysiology of these disease (Colom‐Cadena et al, 2013; Vasconcelos et al, 2016; Pham et al, 2019; Gallardo et al, 2020; Sampson et al, 2020). The fact that sequence similarity is apparent in many of these cross‐interactions (Konstantoulea et al, 2021)—and especially with APRs—suggests that APRs constitute favoured protein interaction interfaces for heterotypic protein interactions.
Here, we investigated the potential of amyloid core APRs to engage in heterotypic amyloid interactions with human proteins that share local sequence homology with amyloid APRs. Next, we evaluated the potential of such interactions to modify the structure and kinetics of assembly of amyloids. In order to do this, we used the Alzheimer beta‐peptide Aβ1‐42 as a paradigm as it is a relatively short amyloid peptide sequence the kinetics of which are well‐documented. Aβ harbours two APRs: APR1 encompassing residues (16–21) where several familial AD mutations cluster and APR2 at the C‐terminal region (29‐end), whose variable length is an important factor in the development AD (Fernandez‐Escamilla et al, 2004a; Vandersteen et al, 2012; Marshall et al, 2016). Both regions have a high aggregation propensity due to a high hydrophobicity and beta‐sheet propensity (Fernandez‐Escamilla et al, 2004b) and readily form amyloid‐like aggregates by themselves as peptides (de la Paz & Serrano, 2004). The importance of these APRs for the amyloid formation of Aβ was further demonstrated by a variant form of Aβ1‐42 that was designed to suppress both APRs by introducing a single amino acid substitution in each region (Marshall et al, 2016). This variant, carrying two mutations in total, was shown to no longer aggregate, which also rescued the neurotoxicity of Aβ (Marshall et al, 2016), showing that both APRs are indeed key determinants of the kinetics of amyloid formation of Aβ.
We identified several peptides with homology to Aβ1‐42 APR derived from human proteins including proteins expressed in the brain and demonstrated that they are able to interact with Aβ1‐42 and alter its aggregation kinetics and fibril morphology. Moreover, we showed that in the context of the full‐length protein these same sequences favour Aβ1‐42 aggregation in a reporter cell line. Importantly, reanalysing deep proteomics data of human Aβ plaques (Xiong et al, 2019a) we showed that proteins with homology to Aβ APRs are over‐represented in amyloid plaques from AD patients and that they cluster in gene ontologies related to synaptic organization and regulation of vesicle‐mediated transport. An over‐representation that is not seen in mouse APP overexpression models. Together our analysis demonstrates that at least in the case of Aβ amyloid assembly interfaces provided by APRs also allow for heterotypic interactions with other proteins by a mechanism of local sequence homology and that such interactions have the potential to modify amyloid nucleation, elongation and fibril morphology and co‐opt such proteins into plaques.
Results
Nomenclature
Aβ: The Alzheimer beta‐peptide, which exists as a mixture of different lengths due to carboxy‐ and amino‐terminal heterogeneity resulting from its proteolytic generation.
Aβ1‐42: A single form of the Aβ peptide, starting from canonical position 1 and ending in position 42. This species is particularly enriched in the plaques of patients with sporadic AD.
Mapping Aβ self‐interactions using peptide arrays
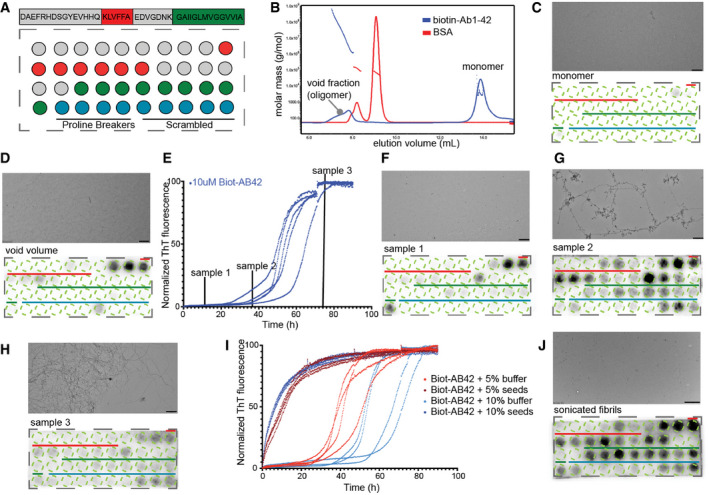
To set up a method to investigate self‐interactions between Aβ molecules, we turned to a method that was previously successfully used for analysing self‐interactions of the yeast prion Sup35 (Tessier & Lindquist, 2007), namely peptide arrays in which peptides correspond to a sliding window over a target protein. By exposing these arrays to an aggregation‐prone protein, the location of self‐interaction sites in the sequence can be observed directly, provided the interacting residues form a contiguous stretch as in the APR model. For this purpose, we synthesized in‐house peptide arrays on a cellulose membrane, by using a sliding window scan over the sequence of Aβ1‐42 of length 12 and step size 1 (Fig 1A, Appendix Table S1). Given that the most widely used antibodies against Aβ have linear epitopes and thus would show cross‐binding on such arrays, we resorted to using a biotinylated derivative of Aβ1‐42 (Biot‐Aβ1‐42, rPeptide) that avoids interference during detection. Upon dissolving Biot‐Aβ1‐42, we performed Size Exclusion Chromatography (SEC, S75, GE Healthcare) using an inline Multiple Angle Light Scattering detector (MALS, Wyatt) to determine the molecular size of the eluents. This revealed a monomeric peak eluting around 14 ml and an oligomeric peak, eluting in the void volume (Fig 1B). Then we exposed a peptide array to the monomeric fraction of Biot‐Aβ1‐42 (100 nM) for 1 h and detected binding of Aβ to the spotted peptides using streptavidin‐HRP, but we observed no significant binding to any of the peptide spots (Fig 1C). However, detection with the 4G8 (Chang et al, 2007), 6E10 and 12F4 antibodies that recognize a central 18–22 epitope in Aβ, the N‐terminus and the C‐terminus, respectively, showed clearly that these peptide sequences are present, confirming also the quality of synthesis (Appendix Fig S1A). When we exposed a fresh membrane to the void volume fraction in the same way as the monomer, we observed clear binding to the N‐terminal fraction of APR1 (Fig 1D), but not APR2. We left monomeric Biot‐Aβ1‐42 samples to aggregate (at 10 μM) while monitoring their aggregation kinetics using Thioflavin‐T (ThT) fluorescence (Fig 1E). At three different time points during the course of the aggregation, we took samples, incubated them in parallel without ThT and put them on a fresh peptide microarray (at 100 nM). Aggregating species collected during the lag phase of aggregation (sample 1 on Fig 1E) showed again a binding pattern in the amino‐terminal peptides of APR1, similar to the void volume species observed during SEC‐MALS (Fig 1F). Aggregating species, taken later in the kinetic from the early elongation phase (sample 2), showed binding throughout both APR1 and APR2 (Fig 1G). Finally, the predominantly fibrillar aggregates present during the plateau phase (sample 3), showed only very weak binding to the Aβ peptides (Fig 1H). Finally, we generated amyloid seeds in the reverse reaction, using 15 cycles of 30 s sonication to generate fragments from mature amyloid fibrils that were obtained after 14 days of incubation. We used the aggregation kinetics to confirm that the sonication protocol led to the formation of functional amyloid seeds, and indeed we found this sample produced a notable reduction in the lag‐phase of aggregation of Biot‐Aβ1‐42 at 5 or 10% molar ratio in monomeric units (Fig 1I), whereas the mature fibrils did not have this effect. These “reverse seeds” (i.e. generated from mature fibrils) indeed also showed an interaction pattern with the membrane that was similar to that of the late oligomers of the elongation phase, with interactions across both APR regions (Fig 1J).
Figure 1. Differential binding of Aβ‐aggregating species in Aβ cellulose peptide microarrays.
-
AAβ1‐42 sliding window membrane setup. Red indicates where the KLVFFA starts presented whole. Green where GAIIGL presented whole. Blue indicates the controls (4 proline breakers, 5 scrambled Aβ peptides, sequences Appendix Table S1).
-
BSEC‐MALS of Biot‐Aβ1‐42 preparation with 7 M GnHCL showing a clear monomeric peak and a smaller oligomeric.
-
C100 nM of Biot‐Aβ1‐42 monomers show no binding on membrane (down panel), TEM image shows no aggregating species in the sample (upper panel). Scale bar: 500 nm.
-
DVoid fraction (oligomers) shows strong binding on first APR of Aβ1‐42. Scale bar: 500 nm.
-
ENormalized ThT kinetics of 10 μM Biot‐Aβ1‐42 with timepoints of samples that incubated with Aβ1‐42 membranes.
-
F–HBinding of different aggregating samples to Aβ membranes and their TEM images. 100 nM of sample1 (early oligomers) binds strongly to middle APR (F), 100 nM of sample 2 (late oligomers) binds in both middle and C‐terminal APR of Aβ1‐42 (down panel) while TEM images show fibrillar structures (upper panel) (G), 100 nM of sample 3 shows no specific binding to Aβ1‐42 membranes (H). Scale bars: 500 nm.
-
IThT kinetics of Biot‐Aβ1‐42 seeding. 10 μM of Biot‐Aβ1‐42 incubated with 0.5 or 1 μM of Biot‐Aβ1‐42 seeds.
-
J100 nM of Biot‐Aβ1‐42 seeds show a strong binding in both APRs. TEM image (upper panel) slow clear fragmentation of fibrils. Scale bar: 1 μm.
The difference in binding between monomeric and oligomeric Biot‐Aβ1–42 species is consistent with the nucleation growth kinetics of amyloid aggregation, in which the rate‐limiting step is the formation of the oligomers, to which monomer addition is then relatively rapid (Dobson, 1999). In structural terms, the rate‐limiting step entails the formation of a stable intermolecular beta‐sheet, to which additional strands can be added rapidly. Hence a reaction mixture containing preformed oligomers bypasses this step and incorporates additional monomers rapidly at the sheet edges. Since mature fibrils still only have two growth sites at opposite ends of the beta‐stack, the number of interaction sites per mass of aggregate declines with fibril length. Interestingly, the peptide array data also show that the early Biot‐Aβ1‐42 oligomeric intermediates (found in the fresh sample or formed from the monomer fraction) engage in different molecular interactions than later oligomeric species, which essentially behave as the fibril fragments generated by sonication. Consistent with the notion that the APRs are the kinetic determinants of Aβ aggregation, we indeed found the oligomers to interact mainly with peptides on the membranes corresponding to these regions. Sequences from APR1 from the central region of Aβ seems to form more interactions with early oligomers, whereas later oligomeric species also interacted with APR2 from the C‐terminus.
To probe the specificity of the observed interactions, we went back to the sup35NM domain originally used to develop this assay (Tessier & Lindquist, 2007), and exposed a fresh membrane to reverse amyloid seeds prepared by sonication of mature sup35NM fibrils (Appendix Fig S1B and C). Moreover, we included in the membranes control peptides consisting of single proline substitutions of peptides 12, 15, 24 and 26, respectively, as well as scrambled versions of peptides 11, 14, 25, 27 and 29. These peptides were chosen to sample the positive regions from the central and the C‐terminal APRs. The sup35NM oligomers showed only very weak binding, including to spots positive for Aβ. Also, all proline substitutions were sufficient to suppress binding, as was scrambling in the central region. However, scrambling in the carboxyterminal region appears to reduce the strength of the interaction, but does not completely suppress it. This highly hydrophobic region is part of the transmembrane region of APP and interacts only with relatively late species in the aggregation pathway, which might indicate an aspecific contribution to binding in this region, perhaps via hydrophobic surfaces on the larger aggregates. On the other hand, the proline substitutions that effectively suppress interaction conserve hydrophobicity, but disrupt secondary structure propensity, suggesting that perhaps the low sequence entropy in this region renders scrambling a blunt tool.
Protein fragments with local sequence similarity that bind to Aβ APRs occurs throughout the proteome
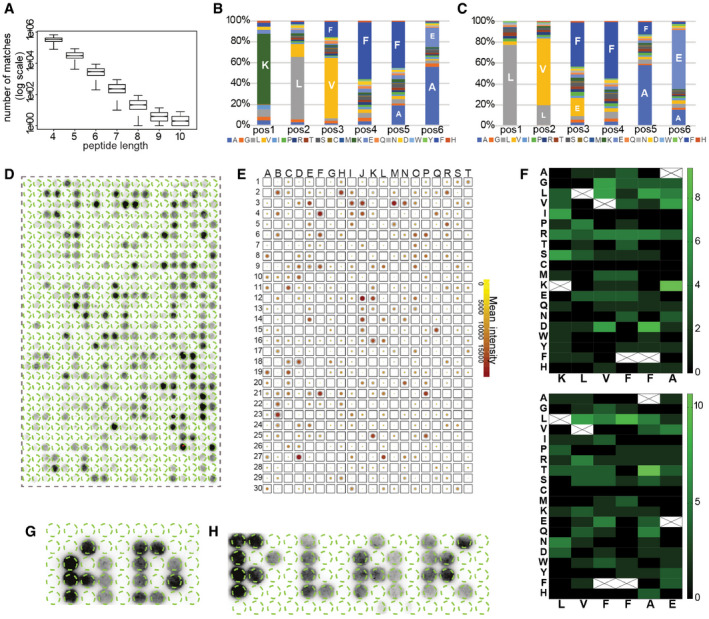
The APR regions of Aβ vary in length from 6 to more than 10 amino acids, a size distribution similar to that of APRs in other amyloids. Although most sequences of length seven amino acids and longer are unique within the human proteome (Ganesan et al, 2015), we wondered how much sequence similarity exist within the proteome when considering mismatches. To this end, we plotted the number of similar sequence matches found in the human proteome (up to 2 mismatches) in function of fragment length, based on 1,000 randomly selected human protein fragments per length, allowing up to 2 mismatches (Fig 2A) (proteome obtained from UniProt Consortium (2008), reviewed entries and filtered for 90% redundancy using CD‐Hit Fu et al (2012)). This plot shows that the number of sequence matches drops exponentially with the length of the fragment and levels off at a length of nine amino acid residues. Incidentally, this is the length that the immune system employs for self‐discrimination, that is, the length of the peptides displayed by the Major Histocompatibility complex (MHC) starts at nine amino acids. Given this strong dependence on fragment length of the number of homologous matches found for any given sequence in the proteome, we chose to set this parameter as fixed in order to compare between APRs. Hence, we decided to use a relatively low fragment length of six to combine some specificity with a large candidate pool. Thus, we performed a search of the human proteome for hexapeptides matching KLVFFA and LVFFAE, allowing up to 2 mismatches, yielding 4,390 matches, not filtering for isoforms. We chose the middle region of the APR peptides, since in our peptide microarrays showed strong binding with both early and late oligomers. Apart from Aβ and the parental amyloid precursor protein (APP), the only other proteins with an identical match are the Potassium voltage‐gated channel subfamily B, members 1 and 2, which have a perfect match to LVFFAE towards the extracellular region of a transmembrane region. In addition, we identified 61 matches with a single mutation and 4,318 matches with 2 mutations. The composition of the mismatches appears to follow a fairly random distribution (Fig 2B and C), with most amino acids appearing at each position.
Figure 2. Aβ binding to APR homologues derived from human proteins.
-
ASequence similarity in combination with peptide length for 1,000 random proteins derived from human proteome. Graph: bottom and top of the boxes are the first and third quartiles, central band represents median, whiskers encompass minimum and maximum.
-
B, CDistribution of amino acids in homologues to Aβ KLVFFA (B) and LVFFAE (C) proteins.
-
DBinding of Biot‐Aβ1‐42 to homologue peptides derived from ˜ 520 randomly selected proteins.
-
ESummary of Biot‐Aβ1‐42 binding throughout eight membranes, colour indicates the mean between membranes and the size of the outline of the standard deviation.
-
FHeatmap of amino acid substitutions in membrane hits.
-
G, HMembrane top binders spell AD and PLAK, white space consists of random sequences. (sequences in Appendix Table S2).
Since for technical reasons the maximum number of sequences we can currently include on our cellulose array format is 600, we randomly selected this number of fragments from the initial list. We then generated a new membrane with these fragments across the proteome, and to take the immediate sequence context into account, we included 2 N‐amino acid and 3 C‐amino acid flanks from the matching protein. We exposed this membrane to oligomeric Biot‐Aβ1‐42 coming from the void fraction of SEC (Fig 1D), and detected the binding pattern to the large membrane using streptavidin‐HRP (Fig 2D). This revealed strong binding with some sequences, whereas others showed no or little binding. The binding pattern was reproducible between independently generated replicates of the same membrane and we also generated replicates with the same sequences but in a randomized order (Appendix Fig S2, Dataset EV1). We calculated the overall binders by identifying manually the lowest positive value and calculating its Z‐score (named Z‐cutoff). Every spot with Z‐score > Z‐cutoff in all eight membranes (at least two repeats for three randomizations) identified as Biot‐Aβ1‐42 interactor. This analysis identified 126 consistent binders from this analysis (21%) that bound consistently to oligomeric Biot‐Aβ1‐42 in all 8 membranes. A summary of the membrane interactions is shown by averaging the binding intensity and standard deviation from eight membranes (Fig 2E). When we analysed the sequence composition of the bound sequences (Fig 2F), we found that some substitutions were better tolerated than others, for example, R in KLVFFA homologues or L in LVFFAE. However, the homologous peptides have single or double mutations to Aβ APRs, which adds a level of complexity in identifying the most favourable mutations.
These experiments show that the presence of specific peptides on the cellulose surface determine where on the membrane oligomeric Biot‐Aβ1‐42 deposits. To explore this point further, we printed a new membrane in which we spelled the pseudo‐words “AD” and “PLAK” using peptide spots from the top 50 binders identified in the previous membranes and surrounded them with peptide spots from random sequences from the human proteome that did not share similarity with Aβ sequences (Fig 2G and H, and Appendix Table S2). This confirmed the observations above, that the binding patterns are consistent. Although a cellulose membrane is a poor two‐dimensional representation of what may be occurring in a complex tissue such as the brain, these consistent binding patterns show that Aβ amyloids can engage in heterotypic interactions with homologous fragments of otherwise unrelated proteins.
Heterotypic APR interactions modify Aβ1–42 amyloid formation in solution
In order to investigate if the interactions that we detected on the cellulose membrane could affect amyloid aggregation of Aβ1‐42 in solution, we generated soluble versions of 32 peptides selected from the membrane (Table 1). We monitored the aggregation kinetics of recombinant rAβ1‐42 by ThT fluorescence in the presence of equal amounts (1:1 molar ratio in monomeric units) of these peptides and compared them to peptide alone (Fig 3A–C and Appendix Figs S3 and S4). We quantified these curves by curve fitting in terms of the lag phase of aggregation (Tlag), the time at which half the aggregation amplitude is reached (T1/2), the total aggregation amplitude (Amp) and the elongation rate (ke) (Dataset EV2). We found that most effects occurred in the lag phase, that is, where mostly oligomers are populated. We found that 7 peptides showed a statistically significant increase in the lag phase, so slowed down the aggregation of rAβ1‐42 (Fig 3D), whereas 12 peptides decreased the lag phase, that is, accelerated rAβ1‐42 kinetics. In addition, seven peptides showed significant differences in fluorescence amplitude (Fig 3E).
Table 1.
Summary of peptides used in the cellulose peptide array.
| ID | Sequence | UniProt ID | Protein Name | Membrane hits | Lag phase | Amplitude | Fibril Length (TEM) | Dye binding | Fibril morphology (AFM) | Biosensor cells | Amyloid plaques (Xiong et al(2019a, b) | Comments |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P1 | HRKSVFFVGQL | Q562E7‐1 | WD repeat‐containing protein 81 | Yes | *** ↑ | ns | ns | ns | nt | nt | Yes | Endolysosomal, involved in aggrephagy |
| P2 | NPKLDFFKNFL | O94822 | E3 ubiquitin‐protein ligase listerin | Yes | **** ↓ | * ↓ | ns | ns | nt | nt | Yes | Part of ribosomal quality control complex |
| P3 | AVLRFFNEVFK | P23786 | Carnitine O‐palmitoyltransferase 2 | Yes | **** ↓ | ns | **** ↑ | ns | x | ****↑ | Yes | Mitochondrial |
| P4 | SQRLVGFALRR | P51790‐2 | H(+)/Cl(‐) exchange transporter 3 | Yes | **** ↑ | ns | ns | ns | nt | *↑ | Yes | Endolysosomal, involved in acidification |
| P5 | FMMTVFFAKKL | O95196‐1 | Chondroitin sulphate proteoglycan 5 | Yes | **** ↑ | **** ↓ | ** ↑ | * (curcumin) | x | ns | Neurogenesis, trans‐synaptic signalling | |
| P6 | KHKPVAFAVRT | P54284‐1 | Voltage‐dependent L‐type calcium channel subunit beta‐3 | Yes | ns | ns | ns | ns | nt | ns | Yes | Presynaptic depolarization and calcium channel opening |
| P7 | KGTVFFDEFTF | O95497 | Pantetheinase | Yes | ns | ns | ** ↑ | ns | nt | ns | Inflammatory response | |
| P8 | LPLVIFHELTK | O15354 | Prosaposin receptor GPR37 | Yes | *** ↓ | **** ↓ | ** ↑ | ns | x | nt | Receptor for neuroprotective factor | |
| P9 | DKLQFFEERRR | Q15772‐5 | Striated muscle preferentially expressed protein kinase | No | ns | ns | ns | ns | nt | nt | Possible growth regulator in muscle | |
| P10 | PLSRVFFASWR | P05164‐1 | Myeloperoxidase | Yes | *** ↑ | ns | ns | ns | nt | nt | Response to oxidative stress | |
| P11 | DFRVFFQELVE | Q92985‐2 | Interferon regulatory factor 7 | Yes | *** ↓ | ns | ns | ns | nt | nt | Transcription factor regulating inflammation | |
| P12 | QRLVGFALRRD | Q6ZNZ2 | cDNA FLJ26854 | Yes | *** ↑ | ns | **** ↓ | ns | x | nt | unknown | |
| P13 | SGLSLFAETIW | O00322 | Uroplakin‐1a | Yes | ns | ns | ns | ns | nt | nt | Component of asymmetric unit membrane | |
| P14 | SNLQFKAERIK | Q8TDM6‐1 | Disks large homolog 5 | Yes | ns | ns | ** ↓ | ns | nt | nt | Hippo regulator, synaptogenesis | |
| P15 | LLAVFFALGLE | Q08462 | Adenylate cyclase type 2 | No | ns | *** ↑ | ns | ns | nt | nt | Yes | Catalyses cAMP in response to G‐protein signalling |
| P16 | LRKLVRGATLD | O95273‐1 | Cyclin‐D1‐binding protein 1 | Yes | ns | ns | **** ↓ | ** (pFTAA) | nt | nt | Negative regulator of cell cycle progression | |
| P17 | PRKLDFFRSEK | O15031 | Plexin‐B2 | Yes | ns | ns | ns | ** (pFTAA) | nt | nt | Yes | Cell surface receptor, synapse assembly |
| P18 | FYLFFFTEKIL | Q15043‐1 | Metal cation symporter ZIP14 | Yes | ** ↓ | ns | ns | ns | nt | nt | Yes | Metal ion membrane transporter |
| P19 | FIFLRFFAPAI | Q14644 | Ras GTPase‐activating protein 3 | Yes | ** ↓ | ns | **** ↓ | ns | nt | nt | Yes | Inhibitory regulator of the Ras‐cyclic AMP pathway |
| P20 | YNNLVSFASPL | Q96Q15‐1 | Serine/threonine‐protein kinase SMG1 | No | ** ↓ | ns | **** ↓ | ns | nt | nt | Genotoxic stress response | |
| P21 | SRRLVPFAQFI | Q71RG8 | FP2025 | Yes | ** ↓ | ns | **** ↓ | ns | nt | nt | Unknown | |
| P22 | ADKLVFFVNGR | P47989 | Xanthine dehydrogenase/oxidase | Yes | ** ↓ | ns | ns | ** (pFTAA) | nt | nt | Contributes to the generation of reactive oxygen species | |
| P23 | APQLVFAARAV | Q9BZ82 | FKSG39 | No | ** ↓ | ns | ns | *** (pFTAA) | nt | nt | Unknown | |
| P24 | VKTLVFFFLES | B1AMT5 | Cohesin subunit SA‐2 | No | * ↓ | ns | ns | ns | nt | nt | DNA replication | |
| P25 | IPKLVNFATLG | P08922 | Proto‐oncogene tyrosine‐protein kinase ROS | Yes | **** ↓ | * ↓ | ns | ns | nt | nt | Orphan receptor tyrosine kinase (RTK) | |
| P26 | SKLLVFFRTEA | A2RRP1‐1;A2RRP1‐2 | Neuroblastoma‐amplified sequence | Yes | ns | **** ↓ | ns | * (curcumin) | nt | nt | Involved in Golgi‐to‐endoplasmic reticulum (ER) retrograde transport | |
| P27 | VGLLVQFAFRE | Q99698‐1 | Lysosomal‐trafficking regulator | Yes | **** ↑ | ns | ns | ns | nt | nt | Regulates and/or fission of intracellular vesicles such as lysosomes | |
| P28 | PVQLVNFAYRD | Q2M3C6‐1;Q2M3C6‐2 | Transmembrane protein 266 | No | ns | ns | ns | ns | nt | ns | Post‐synaptic voltage‐sensor protein | |
| P29 | IFSLVFTAVER | Q99463;B4DRU5 | Putative neuropeptide Y receptor type 6 | No | ns | ns | **** ↑ | ns | nt | nt | Ligand unknown | |
| P30 | GYVLVFDAWTK | P51828 | Adenylate cyclase type 7 | Yes | ns | *** ↓ | ns | ns | nt | nt | Catalyses cAMP in response to G‐protein signalling | |
| P31 | DWRLVFGAKEI | P10323 | Acrosin | Yes | ns | ns | ns | ns | nt | nt | Major protease of mammalian spermatozoa | |
| P32 | LALLVFFGDVG | Q5GH73‐1;Q96KT3 | XK‐related protein 6 | No | **** ↑ | ns | ns | ns | nt | nt | Integral membrane protein, enriched in brain |
The ID is an arbitrary number assigned to each peptide, the full sequence is shown. The UniProt ID (UniProt Consortium, 2008) of the protein from which the segment was taken is provided, as well as its name. Then a summary is provided of its results in the various assays: binding on the peptide array, lag phase and amplitude in the Aβ aggregation kinetics assay using ThT, fibril length determined by TEM, fluorescence emission spectra of amyloid sensor dyes, fibril morphology by AFM and Aβ aggregation in the sensor cell line. Finally, the presence of the protein in patient plaque‐derived data set from Xiong et al (2019a, b) is shown, and finally some comments on function and subcellular localization. * indicates significance (*P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001), ↑ increase compared to Aβ, ↓ decreased compared to Aβ, ns: no significance, nt: not tested, x: observed difference
Figure 3. Homologous peptides can affect rAβ1‐42 kinetics and resulting fibril morphology.
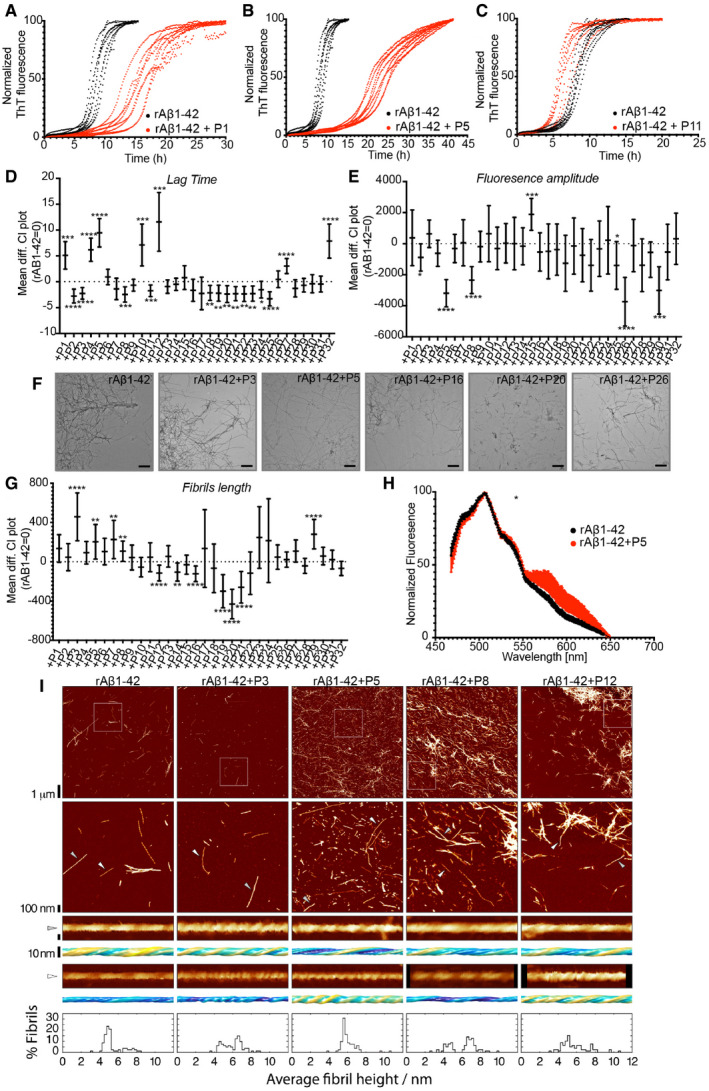
- A–C
-
DLag time difference between rAβ1‐42 alone (rAβ1‐42 = 0) and in the presence of peptides (Statistics: Brown–Forsythe and Welch ANOVA tests with Dunnett’s T3 multiple comparison corrections, n = 2 independent experiments with 4 repeats).Graph: Mean difference and 95% CI. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001.
-
EFluorescence amplitude difference between rAβ1‐42 alone (rAβ1‐42 = 0) and in the presence of the peptides (Statistics: Brown–Forsythe and Welch ANOVA tests with Dunnett’s T3 multiple comparison correction, n = 2 independent experiments with 4 repeats). Graph: Mean difference and 95% CI. *P ≤ 0.05, ***P ≤ 0.001, ****P ≤ 0.0001.
-
FRepresentative TEM images of fibrils made in the presence of 1:1 rAβ1‐42:peptides. Scale bars: 500 nm.
-
GFibril length difference between Aβ alone and in the presence of peptides (Statistics: Brown–Forsythe and Welch ANOVA test with Games‐Howell multiple comparison correction). Graph: Mean difference and 99% CI. **P ≤ 0.01, ****P ≤ 0.0001. At least 9 different positions on grid and at least 100 fibrils were counted for each condition (except Aβ+P23, Aβ+P24). Fibril length distribution in Appendix Fig S7.
-
HCurcumin binding to Aβ fibrils alone or in presence of P5. (n = 2, at least 4 repeats, statistics: Kolmogorov‐Smirnov test). Graph: mean ± SD. P = 0.049. Reused image in Appendix Fig S9.
-
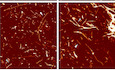
IRepresentative AFM height images of Aβ fibrils alone or in a 1:1 mixture with P3, P5, P8 and P12 peptides are shown in the top row. The boxes indicate the magnified regions shown in the second row. Arrows indicate the locations of representative individual fibrils shown in magnified detail, each shown as a 200 nm digitally straightened segment and a 100 nm segment of the corresponding 3D surface envelope model that was calculated from the image data. The scale bar for each row is shown to the left, with both the 3D model and the straightened image data representing 10 nm. The colour scale of the 3D models from blue to yellow indicates the distance (from low to high) between the fibril surface and fibril centre axis to demonstrate their twist patterns. The average fibril height distribution of around 80 manually selected filaments per sample that showed twist patterns characteristic of single, not fragmented, amyloid fibrils are shown in the bottom row.
To investigate the effect of the peptides on the rAβ1‐42 mature fibrils, we first analysed the morphology of amyloid fibrils formed in the presence of peptides using Transmission Electron Microscopy (TEM, Fig 3F and Appendix Figs S5 and S6) and compared it to Aβ fibrils in the absence of peptides. We analysed 10 positions and measured the length of at least 100 fully traced fibrils for each grid to ensure objective quantification (Dataset EV3). Based on the objective quantification of the fibril length in these images, we found 11 peptides that modified the length of the fibrils (Fig 3G and Appendix Fig S7). Furthermore, we studied the binding to conformationally sensitive amyloid reporter dyes, which alter their emission spectrum depending on the structural detail in the fibril (pFTAA and curcumin, Fig 3H and Appendix Figs S8 and S9). Dye binding showed significant differences between rAβ1‐42 alone and in presence of peptides for four peptides by pFTAA and two by curcumin, suggesting a change in the amyloid structure formed (Appendix Figs S8 and S9). Moreover, to confirm our observations, we performed Atomic Force Microscopy (AFM) of rAβ1‐42 fibrils with a selected number of peptides, which allows the width and morphology of individual filaments to be precisely measured. Interestingly, four of the peptides induced alterations in the mesoscopic arrangement of the rAβ1‐42 aggregates as well as the morphologies of individual fibrils (Fig 3I). The presence of the peptides resulted in a change in the width distribution of the fibrils compared to rAβ1‐42 alone. The average width increased in the presence of these peptides, which could suggest that some fibrils are composed of a greater number of protofilaments or changed protofilament arrangements compared to Aβ alone. Individual fibril surface envelope reconstructions (Aubrey et al, 2020; Lutter et al, 2020) of well‐separated fibrils observed in the AFM images confirm that the morphological details of individual fibril structures formed in the presence of the peptides are indeed different to the rAβ1‐42 fibrils formed in the absence of the peptides (Fig 3I), including some fibrils with a higher twist periodic frequency than in the Aβ only sample.
Our data show that peptide fragments of human proteins with local homology to one of the APRs of rAβ1‐42 can modify Aβ aggregation kinetics as well as the fibril morphology, under conditions where the two molecules have ample opportunity to interact. The number of peptides analysed here is too low to enable meaningful analysis of mismatch patterns that lead to different outcomes, such as acceleration or inhibition of the kinetics or the modification of the fibril morphology. For now, the only trend that appears to emerge from this data set is that the terminal positions are most tolerant, and that positions 3 and 5 are least tolerant of substitution, which is reminiscent of earlier observations on the sequence determinants of amyloid formation of hexapeptides (Lopez de la Paz & Serrano, 2004; Maurer‐Stroh et al, 2010).
Proteins with local sequence homology to Aβ APRs are enriched in human Aβ plaques
In order to determine if proteins containing segments with high sequence similarity to the APR regions of Aβ42 may be enriched in Alzheimer’s disease‐associated amyloid plaques, we analysed a proteomics data set of hippocampal amyloid plaques of AD patients generated from Xiong et al (2019a, b). This study provides high‐quality proteomics profiling at unsurpassed depth of amyloid plaques (AP) and, importantly, nearby control tissue, obtained by tag‐labelling high‐throughput mass spectrometry, which is a highly quantitative method. We searched the proteins identified in amyloid plaques (1125 AP proteins) and adjacent non‐plaque regions for segments with sequence homology to Aβ42 in an unbiased manner: We divided the Aβ sequence into hexapeptides using a sliding window approach and searched the proteins for homologous segments, allowing up to two mutations. To identify if some Aβ segments were over‐represented in amyloid plaques, we studied the occurrence of homologous segments to each Aβ region in amyloid plaques and compared to the control region proteins to calculate enrichment values. Our analysis found six hexapeptide segments of Aβ to be over‐represented in AP proteins, compared to tissue proteins (Fig 4A). Interestingly, those positions are nearly perfectly overlapping with the APR regions of Aβ APR, as would be expected if the enrichment had resulted from heterotypic APR interactions. Two of these hexapeptides reside in the central region, and partially cover the KLVFFA APR, and four additional hexapeptides reside in the C‐terminal APR. To further test if the observed over‐representation is caused by biases in the background proteins, or form set‐size imbalances between the AP and control sets, we employed random subsampling to estimate the distribution of homologous regions in the background (in a so‐called bootstrapping approach). We used the proteins identified in the tissue and created 1,000 random samples with protein numbers equal to the AP proteins (Fig 4B). In a similar way, we also created 1,000 random samples of the same size taken from the whole human proteome and a random plaque sample for each sample (Fig 4C). Both controls showed that the regions identified to be over‐represented in the plaque are residing in the tails or well outside of the random distributions, supporting the notion that the enrichment did not occur by chance.
Figure 4. Presence of proteins with homologous regions to Aβ APRs in human amyloid plaques.
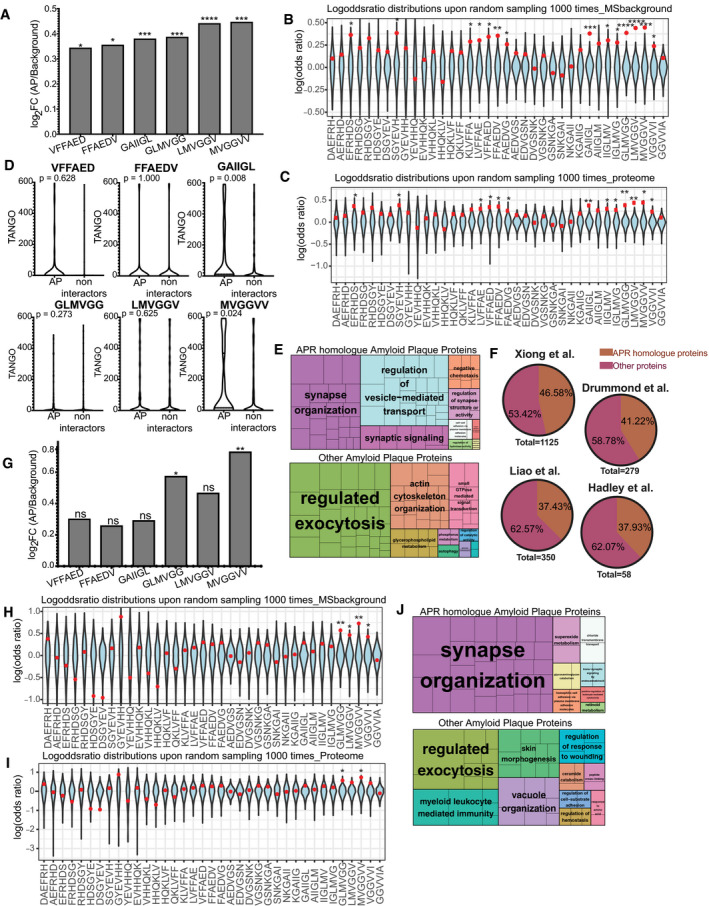
- Log‐odd ratio of random sampling from mass spectrometry background. In blue is the distribution upon random sampling (×1,000) from tissue proteins. Red dot indicates the true values of analysis. In APR regions the actual value (red) resides either in the edges or outside of the random distribution. (statistics: Z‐test). *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001.
- Log‐odd ratio of random sampling from human proteome. In blue is the distribution upon random sampling (x1000) from proteome. Red dot indicates the true values of analysis. (statistics: Z‐test) *P ≤ 0.05, **P ≤ 0.01.
- TANGO scores of homologue APRs in amyloid plaques and non‐amyloid plaques proteins. (statistics: Kolmogorov–Smirnov test).
- Biological pathways enrichment of Aβ APR homologue‐related and non‐related proteins derived from AD amyloid plaques.
- APR homologue proteins identified in other MS studies.
- Log‐odd ratio of random sampling from mass spectrometry background. In blue is the distribution upon random sampling (×1,000) from tissue proteins. Red dot indicates the true values of analysis. (statistics: Z‐test) *P ≤ 0.05, **P ≤ 0.01.
- Log‐odd ratio of random sampling from human proteome. In blue is the distribution upon random sampling (×1,000) from proteome. Red dot indicates the true values of analysis. (statistics: Z‐test) *P ≤ 0.05, **P ≤ 0.01.
- Biological pathways enrichment in of Aβ APR homologue‐related and non‐related proteins derived from non‐AD amyloid plaques.
After we identify the regions that are over‐represented in amyloid plaques, we wondered if the AP proteins containing homologous segments to the Aβ APRs had a higher aggregation propensity than proteins found in the control tissue that also contain homologous segments, but that are not found in the plaques. To do so we used the TANGO algorithm to analyse the protein segments with homology to Aβ regions identified before as over‐represented. Our analysis showed that homologous regions from AP proteins showed a higher aggregation propensity than the ones not found in the plaques, with two regions (GAIIGL and MVGGVV) showing statistically significant differences (Fig 4D). These results suggest again that heterotypic APR interactions may be involved in the enrichment of these proteins in the plaques.
Because proteins associated with amyloid plaques may be involved in high‐risk pathways for AD, we sought to identify the pathways that proteins with homology to Aβ APRs are involved. The two previous groups of amyloid plaques were searched against Gene Ontology Biological process pathways and the significantly enriched pathways were isolated (Fig 4E). Interestingly, AP proteins with homology to Aβ APRs were found to play a role in “synaptic organization, structure and activity”, pathways highly relevant to AD, since synaptic dysfunction is known to play an important role in AD progression. Finally, we wanted to test if a similar occurrence of homologue to Aβ APR proteins exists in other proteomic studies of AP. Indeed, a similar trend is observed in other AP proteomic profiles, with a range of 35–45% of proteins found in APs to have a sequence homology to Aβ APRs (Fig 4F).
To investigate if a similar over‐representation is seen in the amyloid plaques from the brains of cognitively healthy elderly people (non‐AD) and the APP/PS1 mouse model, we analysed them in a similar way (Figs 4G and 5A). In the case of amyloid plaques from non‐AD brains obtained in the same way as before (Xiong et al, 2019a, b), two of the six previously identified regions were found to be over‐represented (Fig 4G–I). Since two of the previously identified six positions are found to be over‐represented, we hypothesized that proteins may still be interacting through the other four positions but without reaching the high levels observed in AD brains yet. So, we used the AP proteins found in non‐AD plaques that have homology to all six regions and the other AP proteins to do the enrichment analysis of Gene Ontology biological processes, like previously. Interestingly, AP homologue proteins of non‐AD brains were also found to be involved in synaptic pathways (Fig 4J).
Figure 5. No over‐representation is observed in amyloid plaques from APP/PS1 mouse or in Glial cytoplasmic inclusions from.
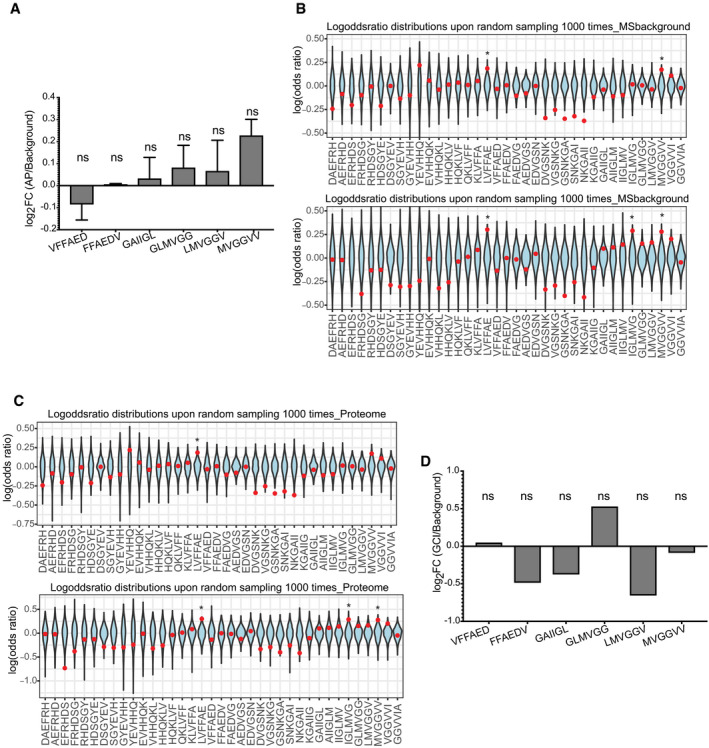
- Over‐representation of Aβ APRs in amyloid plaques of APP/PS1 mouse brains (mean ± SD) from two biological replicates. (statistics: hypergeometric test with Bonferroni correction).
- Log‐odd ratio of random sampling from mass spectrometry background for both replicates. Red dot indicates the true values of analysis. (statistics: Z‐test) *P ≤ 0.05.
- Log‐odd ratio of random sampling from mouse proteome for both replicates. Red dot indicates the true values of analysis. (statistics: Z‐test) *P ≤ 0.05.
- No over‐representation of Aβ APRs was observed in proteins from Glial cytoplasmic inclusions (α‐synuclein aggregates). (statistics: hypergeometric test with Bonferroni correction).
Furthermore, we analysed proteomic data of amyloid plaques from APP/PS1 mouse AD model obtained using a similar method as the human amyloid plaques (Xiong et al, 2019a, b). To do so we analysed in a similar way as previously two biological replicates. Remarkably, the over‐representation of Aβ homologue regions was completely abolished in both replicates (Fig 5A). However, the random distribution identified two to three positions slightly significantly over‐represented but not in the extend observed in human AD brains (Fig 5B and C). Mouse models usually are overexpressing Aβ which leads to rapid aggregation and deposition compared to the slow process found in humans. So, the lack of over‐representation in the mouse model suggests that self‐aggregation is promoted in the mouse model, reducing the opportunity for heterotypic interactions, thereby potentially explaining the differential toxicity that is observed between humans and mouse.
To test if the observed over‐representation is exclusively seen in amyloid plaques and not aggregates from other proteins, we sought to analyse proteomic data from other pathological aggregates. We chose to analyse a study of Glial cytoplasmic inclusions (GCIs), which are mainly composed from α‐synuclein, from Multiple system atrophy (MSA) brains (McCormack et al, 2019a, b). These GCIs were isolated from Basal Ganglia of five MSA brains and the proteins identified in at least four cases were used as the aggregation‐related proteins. Since, this analysis comes from the purification of aggregates, no normal tissue was analysed. To overcome this problem, we used as tissue control, proteins identified in a proteomic study of Basal ganglia (Fernandez‐Irigoyen et al, 2014a, b). From our analysis, no over‐representation of any Aβ region was observed in those α‐synuclein‐enriched aggregates (Fig 5D).
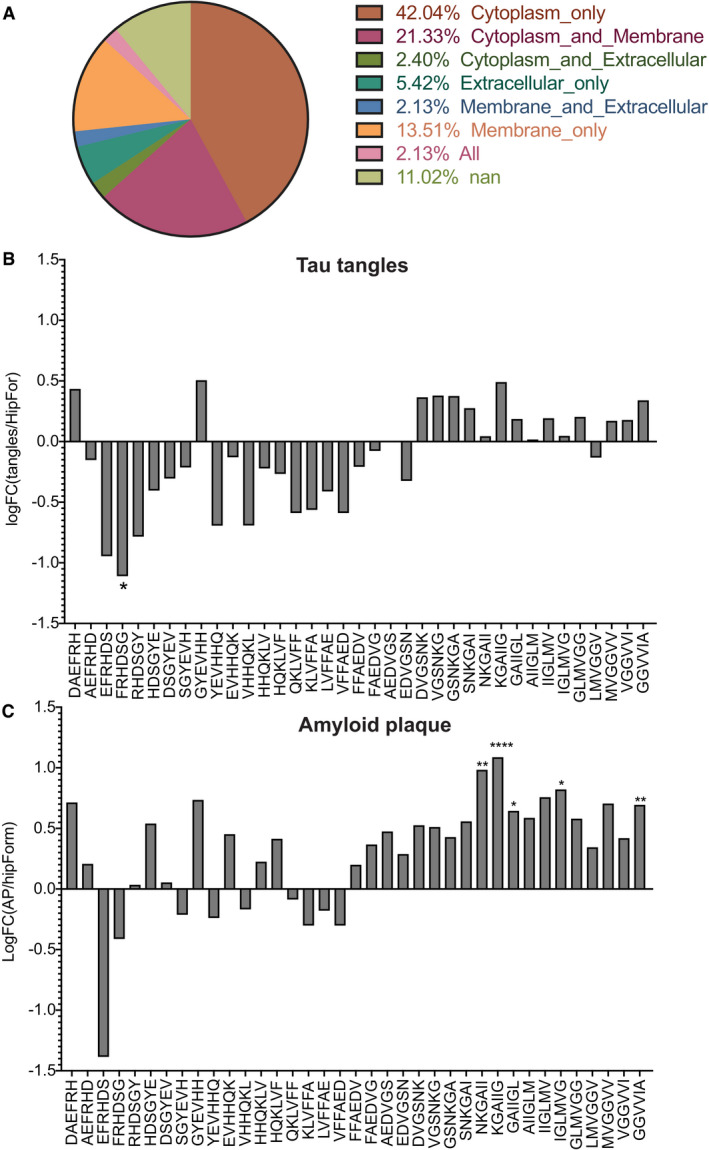
Finally, we analysed a proteomic data set derived from tau tangles (Drummond et al, 2017a, b). Neurofibrillary tangles were isolated from hippocampus and entorhinal cortex of seven sporadic Alzheimer’s disease patients. We analysed those tau aggregates in a similar way as before with taking as background tissue proteins expressed in Hippocampal Formation according to Human Protein Atlas (Uhlen et al, 2015). From our analysis, we did not find any over‐representation of Aβ regions in those aggregates (Fig EV1B). Finally, we analysed another amyloid plaque data set produced from the same group as the tau tangles data set and in similar fashion (Drummond et al, 2017a, b). In this study, amyloid plaques were isolated from the hippocampus of 22 sporadic and 22 rapidly progressive Alzheimer’s disease brains. We analysed the proteins that were consistently found in all of these cases taking as background the proteins of Hippocampal formation. Our analysis showed that regions in the C‐terminus APR of Aβ were over‐represented in amyloid plaques (Fig EV1C). These results indicate that the proteins with Aβ homology regions are primarily found in amyloid plaques and are not significantly over‐represented in aggregates driven by other proteins.
Figure EV1. Over‐representation of Aβ APRs in tau tangles and amyloid plaques.
Proteins containing local homology to Aβ APRs that favour the initiation of Aβ1‐42 aggregation in a biosensor cell line
To test the potential effect by full‐length proteins on Aβ1‐42 aggregation of these heterotypic interactions in a more complex biological environment, we implemented a simplified model system that allows to investigate the potential of full‐length proteins to modulate Aβ aggregation. To that purpose, we created a biosensor cell line in HEK293T, in a similar fashion to a previous line by the Prusiner (Aoyagi et al, 2019) and Diamond laboratories (Kaufman et al, 2016), that stably expresses a fusion construct between Aβ1‐42 and mCherry tag at the N‐terminal (Fig 6A). In untreated cells of this line, diffused mCherry fluorescence is observed throughout the cytoplasm of >95% of the cells. However, when we prepared seeds of rAβ1‐42 by sonicating mature amyloid fibrils and adding them to the cells by transfection, we observed the appearance of a punctate pattern of the RFP fluorescence (Fig 6B). Automated high‐content image analysis revealed that the diffuse to punctate transition occurred in a dose‐responsive manner (Fig 6C). We also confirmed that the puncta were protein aggregates using Fluorescence Recovery After Photobleaching (FRAP) in a region of increased fluorescence: bleaching of this region resulted in limited recovery supporting that the observed spots were indeed Aβ1‐42 aggregates (Fig 6C and Appendix Fig S10B and C).
Figure 6. Proteins with homologues to Aβ regions can induce the aggregation of Aβ1‐42 in HEK293T cells Aβ biosensor.
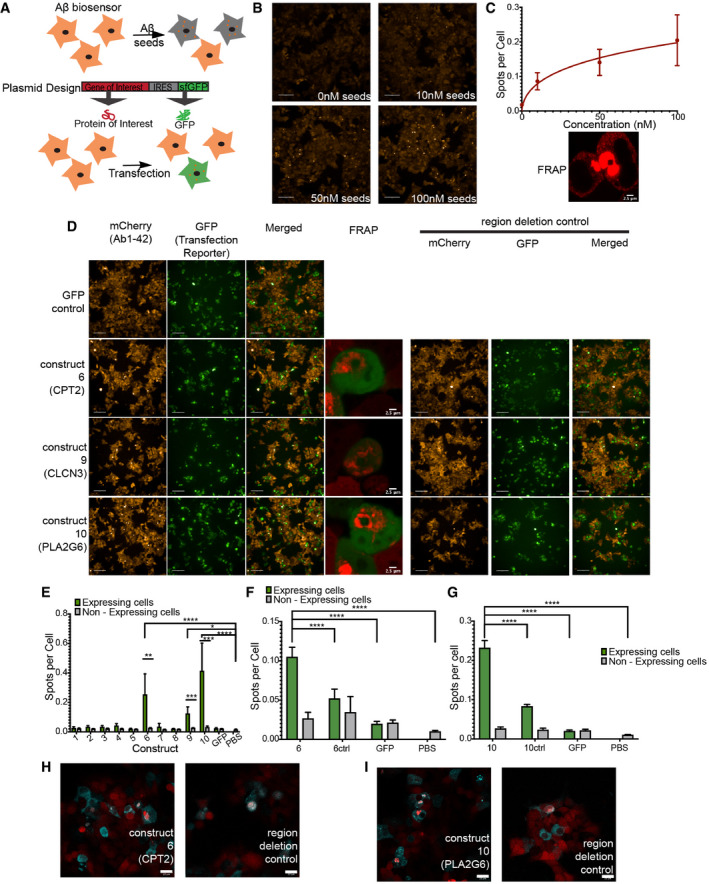
-
AExperimental setup of inducing aggregation in Aβ biosensor cell line.
-
BRepresentative images of Aβ biosensor seeding with three different seed concentration (10, 50, 100 nM) Scale bars: 100 μm.
-
CTreating Aβ biosensor with different concentrations of rAβ1‐42 seeds induces the aggregation of mCherry‐Aβ1‐42 in a dose‐dependent matter (n = 3 independent experiments, graph: mean and 95% CI). FRAP of Aβ spots shows limited recovery confirming that are aggregates (in detail at Appendix Fig S10B and C). Scale bar: 2.5 μm.
-
DRepresentative images of three proteins that can induce aggregation of Aβ1‐42 in biosensor cell. Increased aggregation is observed in cells expressing the construct but not GFP alone (left panels). Scale bar: 100 μm. FRAP of the resulting aggregates shows no recovery (in detail at Appendix Fig S11). Scale bar: 2.5 μm Removal of the homologue regions resulting in reduced aggregation (right panels). (n = 3 independent experiments).
-
EQuantification of a number of spots per cell in cells expressing/not expressing the construct (identified by GFP, transfection reporter) (n = 3 independent experiments, statistics: ordinary one‐way ANOVA with Dunnett’s T3 multiple comparison correction, unpaired t‐test for transfected/non‐transfected cells). Bar plot: mean with 95% CI. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001.
-
F, GQuantification of Spots per cell for construct6 and construct6 control (6Ctrl, removal of homologue region) (F) and construct10 and construct10 control (10ctrl, removal of homologue region) (G). (n = 4 independent experiments, statistics: ordinary one‐way ANOVA). Bar plot: mean with 95% CI. ****P ≤ 0.0001.
-
H, IConfocal images of colocalization of protein of interest with Aβ aggregates. (Contrast of images were enhanced to 0.1% saturated pixels using Fiji). Scale bars: 22 μm.
To test if proteins with homologous regions to the Aβ APR segments are capable of inducing Aβ1‐42 aggregation in a similar way than seeds did, 10 expression constructs were generated containing each a gene of interest as well as a fluorescent reporter (GFP) separated by an Internal Ribosome Entry Site (IRES) (Fig 6A). This setup allows us to quantify Aβ1‐42 aggregates in cells that are expressing our protein of interest and compare it directly with non‐transfected cells from the same well. The 10 proteins were chosen based on their binding signal in our peptide microarrays, their synthesis potential and/or their connection to brain or neurodegenerative diseases (Table 2). For 9 out of the 10 proteins, the corresponding peptide showed a positive signal on the peptide array (Fig 2D and E), whereas the remaining (construct 8) was taken along as a control. Five of these proteins were found back in plaques of AD patients in the Xiong data set. From those 10 constructs, 8 correspond to the full‐length protein and 2 had to be cut slightly due to size limitations of the synthesis method (TWIST bioscience). When we transfected the biosensor cells with these constructs and compared the number of spots per cell in transfected and non‐transfected cells as a measure of Aβ1‐42 aggregation, we identified three proteins (correspondent to constructs 6, 9 and 10) that by mere overexpression induced a significant increase in Aβ1‐42 aggregation (Fig 6D and E and Appendix Fig S12D). As further controls, we ensured that we did not observe induction of puncta in mock transfected cells (PBS), nor in cells transfected with a control plasmid expressing only GFP. Moreover, FRAP showed that also these spots were Aβ1‐42 aggregates since no recovery was observed after bleaching (Fig 6D and Appendix Fig S11A and B). Interestingly, these constructs correspond to peptides that were both positive on the microarray and proteins that accumulated in the patient plaques analysed by Xiong et al (2019a, b). For two of the constructs (6 and 9) the corresponding peptide fragments were included in the detailed biophysical study (peptides P3 and P4 in Fig 3 respectively), and showed strong effects on fibril morphology and assembly kinetics of Aβ1‐42.
Table 2.
Summary of constructs transiently expressed in the Aβ aggregation biosensor line.
| No C(onstruct | Uniprot ID | Gene | Construct | Protein | Peptide ID | Amyloid plaques Xiong et al (2019a, b) | Comments |
|---|---|---|---|---|---|---|---|
| 1 | Q02641‐1 | CACNB1 | HA‐CACNB1‐3xFLAG‐IRES‐sfGFP | Voltage‐dependent L‐type calcium channel subunit beta‐1 | – | Subunit of calcium type L‐ type. GO cellular response to amyloid‐beta | |
| 2 | Q9BY11‐1 | PASCIN1 | HA‐PACSIN1‐3xFLAG‐IRES‐sfGFP | Protein kinase C and casein kinase substrate in neurons protein 1 | – | Role in organization of microtubules. Role in synaptic vesicle endocytosis | |
| 3 | O95196‐1 | CSPG5 | HA‐CSPG5‐3xFLAG‐IRES‐sfGFP | Chondroitin sulphate proteoglycan 5 | 5 | Neurogenesis, trans‐synaptic signalling | |
| 4 | P54284‐1 | CACNB3 | HA‐CACNB3‐3xFLAG‐IRES‐sfGFP | Voltage‐dependent L‐type calcium channel subunit beta‐3 | 6 | Yes | Presynaptic depolarization and calcium channel opening |
| 5 | P04083‐1 | ANXA1 | HA‐ANXA1‐3xFLAG‐IRES‐sfGFP | Annexin A1 | – | Yes | Role in immune response |
| 6 | P23786‐1 | CPT2 | HA‐CPT2(2‐600aa)‐3xFLAG‐IRES‐sfGFP | Carnitine O‐palmitoyltransferase 2, mitochondrial | 3 | Yes | Mitochondrial |
| 7 | O95497‐1 | VNN1 | HA‐VNN1‐3xFLAG‐IRES‐sfGFP | Pantetheinase | 7 | Inflammatory response | |
| 8 | Q2M3C6‐1 | TMEM266 | HA‐TMEM266‐3xFLAG‐IRES‐sfGFP | Transmembrane protein 266 | 28 | Post‐synaptic voltage‐sensor protein | |
| 9 | P51790‐1 | CLCN3 | HA‐CLCN3(2‐720aa)‐3xFLAG‐IRES‐sfGFP | H(+)/Cl(−) exchange transporter 3 | 4 | Yes | Endolysosomal, involved in acidification |
| 10 | O60733‐1 | PLPL9 | HA‐PLPL9‐3xFLAG‐IRES‐sfGFP | 85/88 kDa calcium‐independent phospholipase A2 | ‐ | Yes | Phospholipase involved in mitochondria integrity, cellular membrane homeostasis and signal transduction. |
| GFP | P42212‐1 | GFP | HA‐GFP‐3xFLAG‐IRES‐sfGFP | Green fluorescence protein |
The constructs are arbitrarily numbered from 1 to 10, the UniProt ID of the corresponding protein is provided, as well as the gene and protein names. Finally, the corresponding peptide ID from Table 1 is provided, as well as some comments on function and subcellular localization. Half of the proteins listed here occur at the patient plaque data set from Xiong et al (2019a, b). aa: amino acids.
To test our hypothesis that the effects we observed on Aβ1‐42 aggregation were due to the presence of the sequence segments that are homologous to Aβ APRs, we synthesized additional constructs in which we deleted 10–60 aa (amino acids) containing the Aβ APR homologues. We tried very short deletions, corresponding to the homologous segment, but we also made larger deletions. The latter is because APRs are typically part of the hydrophobic core of a globular folded domain, and hence there are typically one to three other elements of the structure that have been evolutionarily optimized to interact with the APR. Hence, deletion of just the APR promotes 3D domain swapping type of interactions, where the APR in Aβ would interact with the remaining compatible regions in the rest of the domain, which we have previously shown promotes aggregation through that mechanism (Rousseau et al, 2001). Construct 6 expresses 1‐600 aa of CPT2 protein, containing the peptide P3 from Table 1 that significantly affected the kinetics and morphology of Aβ1‐42 aggregation (Fig 3) and as a full‐length protein increased the aggregation of Aβ1‐42 in our biosensor cell line. We design a control construct, 6Ctrl by removing the homologous region (aa 379‐388) and expressed in Aβ1‐42 biosensor. The absence of homology significantly decreased Aβ aggregation when compared to initial construct (Fig 6D and F). Moreover, immunofluorescence confirmed that the majority of aggregates exist in cells expressing CPT2 and partially colocalized with Aβ aggregates (Fig 6H). However, that was not the case for the control (Fig 6H). PLA2G6 (Construct 10) showed the most acute increase in Aβ1‐42 aggregation in our biosensor. PLA2G6 control, 10Ctrl, was made by removing the ANK7 domain (aa 349–378). Expression of this control in an Aβ biosensor reduced the aggregation of Aβ1‐42 significantly (Fig 6D and G). Indeed, the presence of PLA2G6 induced the aggregation of Aβ1‐42 as seen by immunofluorescence. Moreover, PLA2G6’s increased signal is observed in Aβ1‐42 aggregates (Fig 6I). This is not observed in the control (Fig 6I). Finally, the third protein that induced an aggregation of Aβ1‐42 is CLCN3 (Construct 9) (Fig 6D and E). Two controls, Ctrl1 and Ctrl2, were made by removing a 63‐aa and 20‐aa domain respectively. Removal of the homologous region only showed a minor decrease in the aggregation of Aβ1‐42 (Appendix Fig S12A and B). Moreover, we could not detect CLCN3 in the Aβ1‐42 aggregates by immunofluorescence (Appendix Fig S12C), and we found another homologous region elsewhere in the CLCN3 sequence, further complicating the analysis. This suggests that the effect of CLCN3 expression on Aβ1‐42 aggregation that we observed is indirect or only partially resulted from heterotypic amyloid interactions.
The weakness of this cellular reporter is that it does not capture the correct subcellular localization of Aβ as plaque are an extracellular phenomenon and Aβ is cytoplasmic in our model. This means that interactions that are observed in this model will need to be followed up further in more sophisticated models. It was explicitly not our goal to identify proteins that co‐aggregate with Aβ in a disease context, but to provide evidence for the potential impact of the mechanism of heterotypic aggregation. However, it is also important to note that the majority of the proteins found in patient plaques discussed above reside in various intracellular localizations in healthy cells (Fig EV1A), suggesting that mis‐colocalization, perhaps as a result of cell death, is part of the formation process. Although our main hits in the cellular reporter are annotated as endolysomal, mitochondrial and cytoplasmic membrane, they were found back in the plaque data set by Xiong et al (2019a, b).
Given that the subcellular localization issue raises the possibility of artefacts resulting from the overexpression, we decided to further probe the specificity of our findings via two additional rounds of control experiments:
First, to address the possibility that overexpression of aggregation‐prone constructs might induce aggregation of the Aβ sensor in an aspecific fashion, we overexpressed well‐known aggregating proteins in this line: wild‐type alpha‐synuclein, the repeat domain of AD‐relevant P301S mutant of Tau and the most frequently occurring Super Oxide Dismutase 1 (SOD1) mutation occurring in familial cases of Amyotrophic Lateral Sclerosis, namely A4V. We only observed a slight increase in the aggregation of the Aβ reporter with these constructs (Fig EV2A and B), and at a similar level as the untransfected cells used as background in all wells, showing that the effect likely stems from the transfection stress, not the overexpression itself.
Figure EV2. Effect of expression of amyloidogenic proteins in aggregation of Aβ.
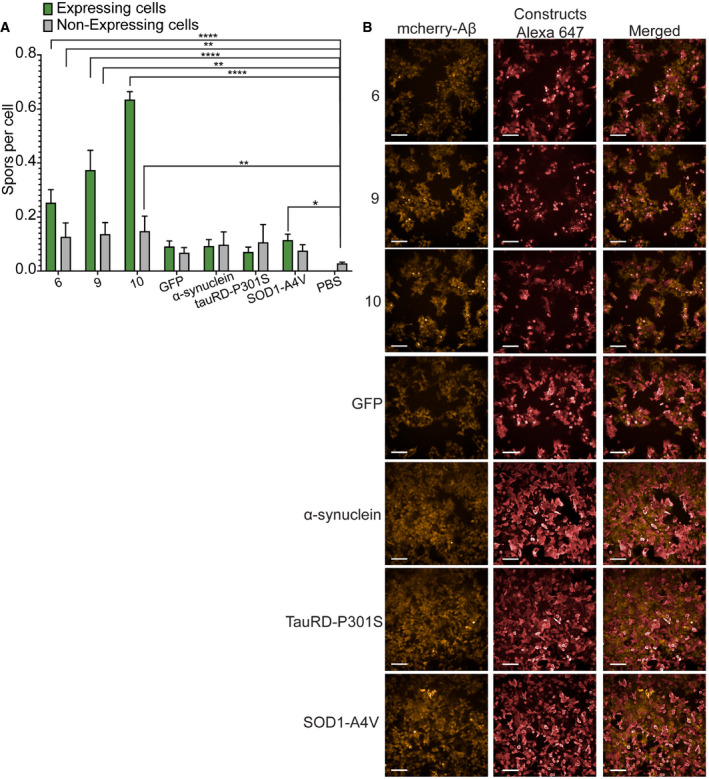
- Quantification of spots per cell for aggregation inducer constructs (6, 9, 10) and known amyloidogenic proteins (α‐synuclein, tauRD‐P301S, SOD1‐A4V). Graph: mean with 95% CI. (n = 4 independent experiments, statistics: ordinary one‐way ANOVA). *P ≤ 0.05, **P ≤ 0.01, ****P ≤ 0.0001.
- Representative images of cells expressing the different constructs. mCherry indicates Aβ, Alexa647 the constructs. Scale bars: 100 μm.
Second, to address the possibility that our constructs could be a particularly potent burden on the proteostasis network, thereby inducing the aggregation of other proteins by depletion of essential cellular chaperones and proteases, we overexpressed our key constructs (6, 9 and 10) in two additional cell lines, namely the tau biosensor Hek293 line developed by Marc Diamond (Sanders et al, 2014) and the sup35NM N2A line developed by Ina Vorberg (Hofmann et al, 2013). Although we could successfully induce the aggregation of each reporter by treating the cells with preformed tau and sup35NM seeds, respectively, overexpression of constructs 6, 9 and 10 did not have a potent effect on sup35 aggregation (Appendix Fig S13A and B) and only a minor effect on tau aggregation (Appendix Fig S13C and D).
Together these results suggest that aspecific effects resulting from overloading the proteostasis network in our reporter line may certainly occur, but their contribution is very limited compared to the specific interactions we observed between our constructs and Aβ.
Discussion
Understanding selective neuronal and regional vulnerability requires knowledge of both loss‐ and gain‐of‐function effects associated with amyloid deposition. Much of our understanding on the role of amyloids in neurodegenerative diseases derives from improvements in our knowledge of the physiological function of these proteins. At the same time, it remains hard to contextualize the role of amyloid deposition and in particular the specific interactions they engage and how these contribute to disease. Historically, a lot of the mechanistic thinking on the role of amyloids in disease was inspired by their common structural properties and the presumption that amyloids, therefore, also possess generic modes of interaction with their environment (Bucciantini et al, 2002, 2004; Campioni et al, 2010; Flagmeier et al, 2020). Yet the overall view that emerged is that of rather promiscuous amyloids that easily interact with various lipids and membranes or nucleic acids and that co‐precipitate in an unspecific manner with many other proteins (Olzscha et al, 2011). This view has been complemented by the realization that protein expression of many proteins—and particularly in the brain—leads to their supersaturation (Tartaglia et al, 2007; Ciryam et al, 2015) while proteostatic regulation erodes with ageing (Labbadia & Morimoto, 2015) together setting the scene of a metastable proteome that becomes increasingly prone to collapse. While these findings have vastly increased our understanding of the existence of misfolding and aggregation diseases and the conditions favouring their development, they do not explain the specific neuronal vulnerabilities characterizing each of these diseases.
We here show that Aβ oligomers can interact with various short APR homologous segments of otherwise unrelated human proteins and that such interactions modify Aβ aggregation kinetics and fibril morphology. Such heterotypic interactions de facto modify the cellular vulnerability of a reporter cell line for spontaneous Aβ aggregation. While this cellular model does not aim to mimic the pathological context of amyloid initiation in Alzheimer’s disease it does in a simplified manner illustrate how cellular vulnerability for amyloid initiation can be shaped by specific interactions of a disease amyloid with its proteomic background. While the cross‐β propensity of APRs favours self‐assembly it also allows some degree of “off target” interaction which can further facilitate or inhibit amyloid assembly thereby sensitizing or protecting cells from amyloid nucleation. Our findings are supported by the increasing observation of heterotypic amyloid assembly both in disease and for the functional regulation of biological processes (Zhou et al, 2012; Louros et al, 2016). These experiments have further revealed the importance of local sequence homology in heterotypic amyloid assembly, indicating that the selectivity of these interactions is related to the sequence similarity level shared between constituent elements (O'Nuallain et al, 2004; Yan et al, 2007; Xu et al, 2011; Wang & Fersht, 2015; Kehrloesser et al, 2016; preprint: van der Kant et al, 2021). Co‐assembly often results in accelerated and more severe pathological outcomes, as reported for Aβ and α‐synuclein in AD and PD patients (Mandal et al, 2006). Both proteins have been shown to hold a nucleation effect towards Tau (Colom‐Cadena et al, 2013), whereas huntingtin may as well be involved in cross‐fibrillation mechanisms, leading to polyglutamine disorders (Furukawa et al, 2009). Heterotypic assembly has also been associated with amyloid transmissibility of neurodegenerative disorders and as a causative agent for the progression of certain forms of systemic amyloidosis (Westermark & Westermark, 2010).
The observation that heterotypic interactions affect the mesoscopic structure of Aβ fibrils in vitro suggests that such interactions can potentially also contribute to polymorphic bias in disease. Thus, next to specific post‐translational modifications and interaction with non‐proteinaceous prosthetic ligands observed in amyloid cryoEM structures, heterotypic amyloid interactions could represent yet another way in which amyloid polymorphism can be affected by and possibly also affect the specific environment in which they are formed (Arakhamia et al, 2020; Scheres et al, 2020; Wesseling et al, 2020).
To evaluate whether heterotypic interactions occur in a neurodegenerative context we re‐evaluated deep proteomics data of human Aβ plaques (Xiong et al, 2019a). We found that sequences homologous to Aβ APRs are enriched in plaques while this is not the case for non‐APR segments of Aβ. While Aβ plaques are of course mostly composed of Aβ itself, the enrichment of these “contaminants” suggests that heterotypic interactions occur in the process leading to plaque formation. Interestingly we do not find such enrichment in the Aβ overexpression APP/PS1 mouse model. Possibly this could mean that in this case overexpression is the dominant driver of plaque formation thereby outcompeting any possibility for heterotypic interaction. When analysing the function of heterotypic plaque components, we find that they mainly cluster in gene ontologies related to synaptic regulation and the regulation of vesicle‐mediated transport suggesting that heterotypic interactions observed in plaques are associated with the synaptopathology of Alzheimer’s disease (Forner et al, 2017). This raises the question of the nature of such an association. Does heterotypic plaque composition reflect interactions that follow synaptic damage or do such interactions directly participate in the pathological chain of events resulting in synaptic breakdown? In the latter case the gain‐of‐function effects of heterotypic interactions could be bidirectional whereby protein interaction with Aβ facilitates Aβ initiation while Aβ also perturbs the normal function of these proteins. Previous work with synthetic amyloids (Betti et al, 2016; Gallardo et al, 2016; Michiels et al, 2020b), the inhibition of homologues by p53 tumour suppressor (Xu et al, 2011) or the inhibition of mammalian necroptosis by viral proteins supports the possibility that heterotypic amyloid interactions (Pham et al, 2019) can result in the functional knockdown of target proteins. It is also probable that heterotypic interactions will be favoured in situations where proteins are (partially) unfolded, for example, during translation, translocation or due to physiological ageing implying additional spatial and temporal context.
It remains to be seen how the net result of the simultaneous expression of many homologous sequences adds up and to what degree these contribute to shaping neurodegenerative diseases. It also remains to be explored whether and how heterotypic amyloid interactions relate to genetic risk factors and to the complex pathophysiologic alterations observed in amyloid‐associated neurodegenerative diseases. For now, we here present evidence for a generic molecular mechanism predisposing Aβ amyloid structures to local sequence‐specific gain‐of‐function binding interfaces allowing them to interact with specific proteins in a complex proteome. Thereby, these proteins affect the aggregation of Aβ which in turn may functionally affect these proteins by promoting co‐assembly in amyloid deposits.
Materials and Methods
In silico screening of Aβ homologous APR
For the analysis in Fig 2A, a proteome assembly of human proteins was obtained from UniProt Consortium (2008). The set was redundancy filtered at 90% sequence identity using the CD‐Hit algorithm (Fu et al, 2012). Per length analysed (4–10) 1,000 random fragments were selected as follows: first random protein was selected, then a random position within it. The number of matches were calculated using a basic string search algorithm. The human proteome (UniProt Consortium, 2008) was a computational screen for protein sequence fragments that are highly similar to the APR of the Aβ peptide (KLVFFA and LVFFAE), allowing a maximum of two mismatches within a hexapeptide. An inhouse generated algorithm was used to identify these homologue sequences. Approximately 600 homologous peptides were randomly picked from the human genome and further in‐house synthetized/printed in membrane peptide arrays.
Homologous peptide membrane arrays and Aβ1‐42 binding assay
The peptide arrays were developed through SPOT synthesis on acid‐stable cellulose membranes using the Intavis Multipep RSi synthesis robot. The peptides were synthetized, from the C‐terminus to the N‐terminus, starting with a GGS linker and containing a PEG spacer (Aims‐Scientific). The obtained peptide array membranes were first incubated in 50% methanol for 10 min, followed by three short washes in PBS‐T (PBS, 0.05% Tween‐20). The membranes were blocked overnight in 1% BSA in PBS‐T, then washed for three times for 5 min first in PBS‐T and next in the incubation buffer (10 mM MES, 150 mM NaCl, 0.05% Tween‐20, pH 5.5). Then the membranes were incubated with Biot‐Aβ1‐42 in incubation buffer supplemented with 100 mM threhalose, for 1 h at room temperature.
The used Biot‐Aβ1‐42 sample was obtained by solubilizing 0.1 mg Biot‐Aβ1‐42 (rPeptide) in HFIP for 1 h, followed by a 10‐min water bath sonication, and finally drying under a N2 stream. The film was re‐dissolved in 8 M urea or 7 M GnHCl in 50 mM Tris, pH 7.4 and the sample was run over a Superdex 75 Increase 10/300 GL gel filtration column (GE Healthcare) equilibrated on the same buffer supplemented with or without 150 mM NaCl, 8 M Urea respectively. The void peak of Biot‐Aβ1‐42 was diluted 1:32 in the incubation buffer, with a final concentration of 0.25 M urea. 10 μM of monomeric Biotin‐Aβ42 was left to aggregate while measuring the ThT kinetics and 100 nM of monomeric or different aggregating species were incubated in the membrane.
After the incubation with Biot‐Aβ1‐42, membranes were washed four times 5 min in Aβ incubation buffer, then in PBS‐T, and then incubated with Streptavidin‐poly HRP (Pierce 22140), diluted 1:100,000 in PBS‐T, for 1 h. Finally, membranes were washed in PBS‐T for three times for 5 min and developed through chemiluminescence using a ChemiDoc XRS (Bio‐Rad).
In a similar way Aβ sliding window membranes were incubated with Sup35‐NM seeds. Detection was done with His‐HRP antibody (Biolegend 652504). Sup35‐NM seeds preparation was described before (Michiels et al, 2020a).
SEC‐MALS analysis
The MW of the Biot‐Aβ1‐42 sample used in peptide membrane assays was studied using multi‐angle light scattering (MALS) on a DAWN HELEOS MALS instrument from Wyatt Technology (Santa Barbara, CA, U.S.A.) with an incident laser wavelength of 658 nm. The proteins were separated using a Superdex 75 Increase 10/300 GL gel filtration column (GE Healthcare) connected to an LC‐10 Prominence HPLC system (Shimadzu), equilibrated with 50 mM Tris, pH 7.4 containing 150 mM NaCl, at a flow of 0.3 ml/min at RT. First, 25 μl of a 2.0 mg/ml bovine serum albumin standard (Pierce) was injected. The scattering intensities at different angles were collected, corrected for the refractive indices of glass and solvent and normalized using the standard. Then, a 0.1 mg Biot‐Aβ1‐42 HFIP film was re‐dissolved in 250 μl 8 M urea in 50 mM Tris, pH 7.4, passed through a 0.2 μm Spartan filter (Whatman) and 100 μl was injected on the column. The value of dn/dc (wherein n is the refractive index of the solution and c the solute concentration) was set to 0.185 ml/g and the scattering data (collected at an interval of 0.5 s) were then fitted according to Zimm formulation.
Membranes analysis
The signal for each spot in membrane was quantified using ImageLab (Biorad). One hundred spots in the borders of the membrane were also quantified and the mean and SD of the background was calculated. We identified manually the lowest positive value for each membrane and calculated the . This Z score was rounded up in an attempt to exclude the faint spots and labeled as Z‐cutoff. Each Z value of spots was calculated and the ones higher than the Z‐cutoff identified as hits. Hits consistent in all eight membranes were identified as interactors of Aβ.
Purification of Met‐Aβ1‐42 (rAβ1‐42)
The purification of the recombinant Met‐Aβ1‐42 peptide was performed in‐house based on the previously reported protocol (Walsh et al, 2009), using the human Met‐Aβ1‐42 expression plasmid, a kind gift from C. Gomes (FCT, Lisbon). Briefly, the Met‐Aβ1‐42 plasmid was expressed overnight in E. coli, and used to inoculate 1 l of M9 culture medium, freshly supplemented with 50 mg/ml ampicillin and chloramphenicol, 2 mM MgSo4, 0.1 mM Ca2Cl and 20% glucose. After reaching an OD600: 0.6–0.8, the expression of the plasmid was induced with 0.5 M IPTG and left to grow for 4 h. The collected pellet was suspended in 10 mM Tris, 1 mM EDTA pH8, sonicated and centrifuged at 30,966 g and the final pellet was dissolved in the same buffer supplemented with 8 M. After being diluted to 2 M urea, a first ion‐exchange chromatography was performed in a DEAE Sepharose resin (GEHealthcare), using the suspension buffer supplemented with 25 mM NaCl, as a binding buffer, and with 125 mM NaCl as an elution buffer. The purified solution was filtered in 30KD spin columns (GE Healthcare), further concentrated in a 3KD ones and the final sample lyophilized in vials with 1 mg or 0.65 mg.
Prior to each experiment, the lyophilized sample was suspended for 1 h at room temperature in 800 μl of 7 M GuHCl in 50 mM Tris pH8, centrifuged 5 min at 15,000 rpm at 4°C and the supernatant injected (using 1 ml injection loop) in a Superdex 75 10/300 GL gel filtration column (GE Healthcare), previously equilibrated with 50 mM Tris pH8 buffer. The fraction containing monomeric Met‐Aβ1‐42 was collected and kept on ice, the concentration was determined in a NanoDrop 2000 (Thermo Fisher Scientific), using a molecular weight of 4645 Da and an extinction coefficient of 1.49. The sample was immediately used in several assays.
Seeds
Biot‐Aβ1‐42 or rAβ1‐42 seeds were obtained by sonicating mature fibrils for 15 min (30 s on, 30 s off) at 10°C using Bioruptor Pico.
ThT kinetic assay
A total of 10 μM of monomeric Biot‐Aβ1‐42 in 50 mM Tris pH7.4 was pipetted to µclear medium binding half area plates (Greiner, #675096) and ThT was added to a final concentration of 25 µM. ThT binding was measured over time (through excitation at 440 nm and emission at 480), using a Fluostar fluorescence plate reader (BMG Labtech) at 30°C. ThT kinetics for biotin‐Aβ42 was done in a similar way by adding 5 or 10% of biotin seeds.
The monomeric samples of Met‐Aβ1‐42 obtained after the purification protocol, described above, with a concentration of 10 µM was incubated at 30°C with a constant shaking for 4 days.
All used peptides were purchased from Genscript, and in their design scheme have a GGS on the C‐terminus, a PEG2 on both termini and are acetylated and amidated, respectively, on the N‐ and C‐terminus. Peptides were solubilized in HFIP, aliquoted in 0.25 mg vials, dried under N2 stream and stored at −20°C. Peptide HFIP films were dissolved in 50 mM Tris pH 8 buffer, filtered through a 0.22 um Millex‐GV spin filter and diluted to 20 µM. In‐house purified Met‐Aβ1‐42, described above, was diluted to 20 µM. Peptide and Met‐Aβ1‐42 were mixed 1:1 with a final concentration of 10 µM each, in 50 mM Tris pH8 buffer. Mixtures containing only peptide or Met‐Aβ1‐42 were used as controls.
The mixtures of Aβ1‐42 and the peptides were pipetted to a µclear medium binding half area plates (Greiner, #675096) and ThT was added to a final concentration of 25 µM. ThT binding was measured over time (through excitation at 440 nm and emission at 480), using a Fluostar fluorescence plate reader (BMG Labtech) at 30°C, with a readout every 10 min and 10 s of shaking before each readout. Similar peptide:Met‐Aβ1‐42 samples, but without ThT, were included for further TEM imaging and pFTAA end‐point measurements.
Data were normalized and fitted in ThT kinetics Fitting formula.
T1/2 and k was calculated from the formula above. Fluorescence amplitude was identified as the highest value of kinetics and lag time from, lagtime = t1/2‐(2‐k). Statistical analysis was performed using Brown–Forsythe and Welch ANOVA test with Dunnett’s T3 multiple comparison correction and 95% confidence interval. Mean difference and 95% CI of difference was plotted. GraphPad was used for statistics and graphs.
Transmission electron microscopy
Once the ThT signal reach a plateau, the resulting fibrils from the peptide:rAβ1‐42 samples were analysed for their structural characteristics. Therefore, 10 μl of each sample was spotted in a copper grid (Formvar/Carbon on 400 Mesh Copper ‐ AGAR SCI, AGS162‐4), previously glow discharged. The sample was adsorbed for 3 min. Afterwards the grids were washed by contact with three drops of MQ water, negative stained with one drop of uranyl acetate (2% w/v) for 1 min and finally washed in a drop of MQ water. The grids were examined using a JEM‐1400 120 kV transmission electron microscope (Jeol, Japan), at accelerating voltage of 80 keV. At least nine positions on the grid was used for quantification. Fibrils were quantified if they were able to be traced from start to end. More than 100 fibrils were quantified in most cases (except rAβ1‐42+P23, rAβ1‐42+P24) (Dataset EV3). Length was measured by using the freehand line of Fiji and tracing the fibrils from start to end. Statistical analysis was performed using Brown–Forsythe and Welch ANOVA test with Games‐Howell multiple comparison correction and 99% confidence intervals. GraphPad was used for statistics and graphs.
Atomic force microscopy imaging
Fibril samples were deposited on freshly cleaved mica for AFM imaging. Each sample was adjusted using a solution of HCl at a predetermined concentration to result in the sample reaching pH 2. Immediately afterwards, 20 μl samples were deposited onto freshly cleaved mica surfaces (Agar scientific, F7013) and incubated for 5 min. Following incubation, the sample was washed with 1 ml of filter sterilized milli‐Q water and then dried using a stream of nitrogen gas. Fibrils were imaged using a Multimode AFM with a Nanoscope V (Bruker) controller operating under peak‐force tapping mode using ScanAsyst probes (silicon nitride triangular tip with tip height = 2.5–2.8 μm, nominal tip radius = 2 nm, nominal spring constant 0.4 N/m, Bruker). Each collected image was scanned at either 4 × 4 μm and 2,048 × 2,048 pixels or 8 × 8 μm and 4,096 × 4,096 pixels. Therefore, the same pixel density was maintained for all images within the data set. A scan rate of 0.203 Hz was used. A noise threshold of 0.5 nm was used, and the Z limit was reduced to 1.5 μm. Nanoscope analysis software (Version 1.5, Bruker) were used to process the image data by flattening the height topology data to remove tilt and scanner bow. Fibrils were traced (Xue et al, 2009; Aubrey et al, 2020), digitally straightened (Egelman, 1986) and surface envelope reconstructed as previously described (Aubrey et al, 2020; Lutter et al, 2020) using an in‐house application. The height profile for each fibril was extracted from the centre contour line of the straightened fibrils from which the average height of each fibril was calculated.
pFTAA and curcumin measurements
pFTAA and curcumin end‐point measurements were performed on 3‐day‐old peptide:Met‐Aβ1‐42 samples, prepared as described above, using a final pFTAA concentration of 0.5 µM and curcumin 5 μM. Measurements were done in a low‐volume black 384‐well plate (Corning) using a ClarioStar fluorescence plate reader (BMG Labtech). After excitation at 440 nM, the emission spectra of pFTAA or curcumin were measured between 468 nm and 650 nm. Curcumin spectra were normalized and tested for significance with Kolmogorov–Smirnov test using GraphPad. The ratio of the two pFTAA picks was calculated average(505,506,507)/average(530,539,540) and the statistical significance was calculated using Brown–Forsythe and Welch ANOVA test with Dunnett’s T3 multiple comparison correction and 95% confidence interval using GraphPad.
HEK Aβ1‐42 biosensor cell line
The Aβ1‐42 biosensor cell line was developed in house. Briefly, a Aβ1‐42 gene block was cloned in the multiple cloning sites of a lentivirus plasmid, containing mCherry tag at the N‐terminus and CMV as promoter. The plasmid was transfected in HEK293T cells, together with the packaging plasmids (pCMV‐deltaR 8.9 and pCMV‐VSV‐G), for the production of viral particles. HEK cells were transduced with these viral particles and sorted for mCherry. Cells were diluted in 96‐well plates and grown as single cell colonies. A colony showing diffused expression of mCherry‐Aβ1‐42 was expanded and stored, to further be used as a biosensor cell line in Aβ seeding assays.
Seeding and transfection assay of biosensor Aβ1‐42 cell line
The biosensor mCherry‐Aβ1‐42 HEK293T cell line was cultured in DMEM medium, supplemented with 10% FBS at 37°C, and a 5% CO2 atmosphere. The seeding assay was performed by transfecting these cells with freshly prepared rAβ1‐42 seeds, described above, and the quantification of the formed Aβ1‐42 inclusions, resulted from the Aβ1‐42 aggregation.
Briefly, the assay was performed in a 96‐well plate (PerkinElmer), previously coated for 30’ with poly‐L‐lysine at 37°C and washed three times with PBS. Adhered cells were passed twice through a 22G needle and plated at 15.000 cells/well and 5 h later were transfected with 0, 10, 50, 100 nM seeds or 100 ng of DNA per well, using Lipofectamine 3000 (Invitrogen) according to the manufacturer.
After 17 h of seed transfection and 41 h of DNA transfection, the cells were fixed with 4% formaldehyde in PBS for 10 min. Cells were washed with PBS, blocked and permeabilized with 1% BSA, 0.2% TritonX‐100 in PBS for 1 h. Cells were nuclei stained with 3 μM Draq7 in 1% BSA in PBS for 1 h. Cells were washed and plates were imaged using Operetta CLS. For seeded cells: For each well, 17 fields were imaged by using the channels Digital Phase Contrast, mCherry (Ex:530‐560, Em:570‐650), DRAQ7 (Ex:615‐645, Em: 655‐705). The images were analysed by Operetta CLS. Nuclei were detected with DRAQ7, cytoplasm with Digital Phase Contrast. Spots measured on ROIs: Nuclei and Cell. For transfected cells: For each well, 17 fields were imaged by using the channels Digital Phase Contrast, mCherry (Ex:530‐560, Em:570‐650), DRAQ7 (Ex:615‐645, Em: 655‐705), EGFP (Ex:460‐490, Em: 500‐550). The images were analysed by Operetta CLS. Nuclei were detected with DRAQ7, cytoplasm with Digital Phase Contrast. Spots measured on ROIs: Nuclei and Cell. EGFP intensity was measured for each cell identified. The baseline EGFP was calculated in PBS‐treated cells. Every cell with higher fluorescence was identified as transfected with our plasmids. The number of spots were identified in cells with and without EGFP fluorescence. Number of spots per cell was calculated from number of spots/number of cells for EGFP‐positive and ‐negative cells. Statistical significance was calculated using Ordinary one‐way ANOVA with Dunett’s multiple comparison correction and unpaired t‐test for comparison between transfected/non‐transfected cells. GraphPad was used for statistics and graphs.
FRAP
Images were acquired on Nikon A1R Eclipse Ti confocal with Plan APO VC 60× oil lens. For mCherry excitation we used 561.6 nm laser line and emission was collected at 570–620 nm. A pre‐bleached image was acquired and ROI was bleached for 0.06, 0.62, 1.25 s at 100% power with 30 image acquisition for 1.91 s after each bleaching and 50 image acquisition after last bleaching. For seeds a similar protocol was used with bleaching for 0.06, 0.62, 1.25 s and 30 image acquisition of 0.97 s after each bleaching and 50 image acquisition after last bleaching. Live cell imaging was done at 37°C and CO2.
Immunofluorescence
Cells were fixed with 4% formaldehyde in PBS for 10 min. Cells were washed with PBS, block and permeabilize with 1% BSA, 0.2% TritonX‐100 in PBS for 1 h. Cells were stained with Alexa647 (ThermoFisher A‐21245) 1:1,000 in 1% BSA in PBS for 1 h or 6E10 Aβ antibody (Biolegend 803001). Images were acquired on Nikon A1R Eclipse Ti confocal with Plan APO VC 60× oil lens. For mCherry excitation, we used a 561.6 nm laser line and emission was collected at 570–620 nm, and for Alexa647 we used a 640.8 laser and the emission was measured at 663–738 nm.
Proteomic analysis
Aβ sequence was divided into hexapeptides with a sliding window. Proteins with absent values, #Unique peptides < 1 and Score Sequest HT < 10 were filtered out from the proteomic data set (Xiong et al, 2019a, b). The remaining proteins were searched for homology to Aβ sequence with up to two mutations using an inhouse algorithm. Proteins with a fold change > 1.186 was previously characterized as upregulated in APs, and defined the AP proteins. Enrichment ratios were calculated by:
Significance was calculated with hypergeometric test with Bonferroni correction for multiple comparisons. A similar approach was used for analysing non‐AD proteomic data, mouse data. For Glial Cytoplasmic Inclusions (McCormack et al, 2019a, b) we used as background proteins the proteins identified in Basal ganglia (Fernandez‐Irigoyen et al, 2014a, b) For tau tangles (Drummond et al, 2020a, b) and Drummond amyloid plaques (Drummond et al, 2017a, b), we used as background the Hippocampal formation proteins (http://www.proteinatlas.org) (Uhlen et al, 2015).
In order to verify the significance of the observed enrichments of homologue peptides in AD plaque proteins, we asked how (un)likely it would be to observe these enrichments if the assignment of “AD‐plaque‐enriched” proteins was entirely random. To do so, we generated 1,000 random samples of the same size of the set of proteins found to be enriched in AD plaques by Xiong et al (2019a, b). For each of these random samples, we calculated logFCs for the homologue peptides versus background (either MS data set background or the entire proteome, as indicated). We then calculated P‐values for the observed logFCs versus the distribution obtained through random sampling, assuming normality. Same approach was used for non‐AD and mouse plaques.
Aggregation propensity of hexapeptides with homology to over‐represented regions of Aβ from AP and non‐AP proteins was calculated using TANGO. Statistical analysis was performed using Kolmogorov–Smirnov test. GraphPad Prism was used for statistical analysis.
Proteins identified in other MS studies were searched for homology to over‐represented Aβ APRs, and pie charts of the proteins with homology versus proteins with no homology were plotted using GraphPad Prism.
Gene ontologies of AP proteins with and without homology to Aβ APRs were identified by using ClueGO(Bindea et al, 2009) plug‐in of Cytoscape. Ontology used GO_BiologicalProcess‐EBI‐UniProt‐GOA_18.09.2019. Statistical analysis was performed with right‐sided hypergeometric test with Bonferroni step down multiple correction. Identified pathways and their P‐values were imported in REVIGO (Supek et al, 2011). Pathways were summarized using Homo Sapiens database and SimRel as semantic similarity measure. TreeMap R script was downloaded and the most significant pathway in the group was used as representation. A similar approach was used for GO identification of non‐AD brain AP proteins.
Glial cytoplasmic inclusions analysis was performed as previously stated, using as AP proteins the ones identified in purified Lewy Bodies from at least four MSA cases (McCormack et al, 2019a). As background proteins we used proteins identified previously in human basal ganglia (Fernandez‐Irigoyen et al, 2014a).
Author contributions
JS and FR conceived the study. KK, PG, MR, NL, JdW, WFX, JS and FR designed experiments. KK, PG, MR, NL, LA, BH, EM, MD, YL and LR performed experiments. KK, FR, JS, NL and WFX performed analyses. KK, PG, FR and JS wrote the manuscript. All authors proofread and corrected the manuscript. JdW, WFX, NL, JS and FR provided supervision and funding acquisition.
Conflict of interest
Joost Schymkowitz and Frederic Rousseau are the scientific founders of, and scientific consultants to, Aelin Therapeutics NV. The Switch Laboratory is engaged in a collaboration research agreement with Aelin Therapeutics.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Acknowledgements
The authors gratefully acknowledge the Electron Microscopy Platform and the VIB Screening Core/C‐BIOS facility, the Department of Neurosciences KU Leuven, and the VIB – KU Leuven Center for Brain & Disease Research for their support and assistance in this work. The Switch Laboratory was supported by the Flanders Institute for Biotechnology (VIB); KU Leuven; the European Research Council under the European Union's Horizon 2020 Framework Programme ERC Grant agreement 647458 (MANGO) to JS; the Fund for Scientific Research Flanders (FWO, project grant G0C2818N, FWO/Hercules Foundation equipment grant NextGenQBio ‐ AH2016.133 and Postdoctoral Fellowships 12P0919N and 12P0922N to NL); the Stichting Alzheimer Onderzoek (SAO‐FRA 2019/0015, SAO‐FRA 2020/0009 and SAO‐FRA 2020/0013); and the Flanders Innovation & Entrepreneurship Agency (VLAIO, Innovation Mandate HBC.2020.2854 to EM). LDA and WFX were supported by Biotechnology and Biological Sciences Research Council (BBSRC), UK grant BB/S003312/1. LFR was supported by FWO 12N0319N and JdW by Stichting Alzheimer Onderzoek grant SAO‐FRA 2019/0013 and Methusalem Grant of KU Leuven/Flemish Government. The Nikon A1R Eclipse Ti confocal was acquired through an FWO Hercules type 1 AKUL/097/037 grant to Wim Annaert.
The EMBO Journal (2022) 41: e108591
Contributor Information
Joost Schymkowitz, Email: joost.schymkowitz@kuleuven.be.
Frederic Rousseau, Email: frederic.rousseau@kuleuven.be.
Data availability
This study includes no data deposited in external repositories.
References
- Aoyagi A, Condello C, Stohr J, Yue W, Rivera BM, Lee JC, Woerman AL, Halliday G, van Duinen S, Ingelsson M et al (2019) Abeta and tau prion‐like activities decline with longevity in the Alzheimer's disease human brain. Sci Transl Med 11: eaat8462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arakhamia T, Lee CE, Carlomagno Y, Duong DM, Kundinger SR, Wang K, Williams D, DeTure M, Dickson DW, Cook CN et al (2020) Posttranslational modifications mediate the structural diversity of tauopathy strains. Cell 180: 633–644.e612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubrey LD, Blakeman BJF, Lutter L, Serpell CJ, Tuite MF, Serpell LC, Xue WF (2020) Quantification of amyloid fibril polymorphism by nano‐morphometry reveals the individuality of filament assembly. Commun Chem 3: 125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betti C, Vanhoutte I, Coutuer S, De Rycke RM, Mishev K, Vuylsteke M, Aesaert S, Rombaut D, Gallardo R, De Smet F et al (2016) Sequence‐specific protein aggregation generates defined protein knockdowns in plants. Plant Physiol 171: 773–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bindea G, Mlecnik B, Hackl H, Charoentong P, Tosolini M, Kirilovsky A, Fridman WH, Pages F, Trajanoski Z, Galon J (2009) ClueGO: a Cytoscape plug‐in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 25: 1091–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucciantini M, Calloni G, Chiti F, Formigli L, Nosi D, Dobson CM, Stefani M (2004) Pre‐fibrillar amyloid protein aggregates share common features of cytotoxicity. J Biol Chem 279: 31374–31382 [DOI] [PubMed] [Google Scholar]
- Bucciantini M, Giannoni E, Chiti F, Baroni F, Formigli L, Zurdo J, Taddei N, Ramponi G, Dobson CM, Stefani M (2002) Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 416: 507–511 [DOI] [PubMed] [Google Scholar]
- Campioni S, Mannini B, Zampagni M, Pensalfini A, Parrini C, Evangelisti E, Relini A, Stefani M, Dobson CM, Cecchi C et al (2010) A causative link between the structure of aberrant protein oligomers and their toxicity. Nat Chem Biol 6: 140–147 [DOI] [PubMed] [Google Scholar]
- Chang WP, Downs D, Huang XP, Da H, Fung KM, Tang J (2007) Amyloid‐beta reduction by memapsin 2 (beta‐secretase) immunization. FASEB J 21: 3184–3196 [DOI] [PubMed] [Google Scholar]
- Chiti F, Dobson CM (2017) Protein misfolding, amyloid formation, and human disease: a summary of progress over the last decade. Annu Rev Biochem 86: 27–68 [DOI] [PubMed] [Google Scholar]
- Ciryam P, Kundra R, Morimoto RI, Dobson CM, Vendruscolo M (2015) Supersaturation is a major driving force for protein aggregation in neurodegenerative diseases. Trends Pharmacol Sci 36: 72–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colom‐Cadena M, Gelpi E, Charif S, Belbin O, Blesa R, Marti MJ, Clarimon J, Lleo A (2013) Confluence of alpha‐synuclein, tau, and beta‐amyloid pathologies in dementia with Lewy bodies. J Neuropathol Exp Neurol 72: 1203–1212 [DOI] [PubMed] [Google Scholar]
- Dobson CM (1999) Protein misfolding, evolution and disease. Trends Biochem Sci 24: 329–332 [DOI] [PubMed] [Google Scholar]
- Drummond E, Nayak S, Faustin A, Pires G, Hickman RA, Askenazi M, Cohen M, Haldiman T, Kim C, Han X et al (2017a) Proteomic differences in amyloid plaques in rapidly progressive and sporadic Alzheimer's disease. Acta Neuropathol 133: 933–954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond E, Nayak S, Faustin A, Pires G, Hickman RA, Askenazi M, Cohen M, Haldiman T, Kim C, Han X et al (2017b) Proteomic differences in amyloid plaques in rapidly progressive and sporadic Alzheimer's disease. Acta Neuropathol 133: 933–954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond E, Pires G, MacMurray C, Askenazi M, Nayak S, Bourdon M, Safar J, Ueberheide B, Wisniewski T (2020a) Phosphorylated tau interactome in the human Alzheimer's disease brain. Brain 143: 2803–2817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond E, Pires G, MacMurray C, Askenazi M, Nayak S, Bourdon M, Safar J, Ueberheide B, Wisniewski T (2020b) Phosphorylated tau interactome in the human Alzheimer's disease brain. Brain 143: 2803–2817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egelman EH (1986) An algorithm for straightening images of curved filamentous structures. Ultramicroscopy 19: 367–373 [DOI] [PubMed] [Google Scholar]
- Fernandez‐Escamilla AM, Rousseau F, Schymkowitz J, Serrano L (2004a) Prediction of sequence‐dependent and mutational effects on the aggregation of peptides and proteins. Nat Biotechnol 22: 1302–1306 [DOI] [PubMed] [Google Scholar]
- Fernandez‐Escamilla AM, Rousseau F, Schymkowitz J, Serrano L (2004b) Prediction of sequence‐dependent and mutational effects on the aggregation of peptides and proteins. Nat Biotechnol 22: 1302–1306 [DOI] [PubMed] [Google Scholar]
- Fernandez‐Irigoyen J, Zelaya MV, Tunon T, Santamaria E (2014a) Anatomo‐proteomic characterization of human basal ganglia: focus on striatum and globus pallidus. Mol Brain 7: 83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez‐Irigoyen J, Zelaya MV, Tunon T, Santamaria E (2014b) Anatomo‐proteomic characterization of human basal ganglia: focus on striatum and globus pallidus. Mol Brain 7: 83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flagmeier P, De S, Michaels TCT, Yang X, Dear AJ, Emanuelsson C, Vendruscolo M, Linse S, Klenerman D, Knowles TPJ et al (2020) Direct measurement of lipid membrane disruption connects kinetics and toxicity of Abeta42 aggregation. Nat Struct Mol Biol 27: 886–891 [DOI] [PubMed] [Google Scholar]
- Forner S, Baglietto‐Vargas D, Martini AC, Trujillo‐Estrada L, LaFerla FM (2017) Synaptic impairment in Alzheimer's disease: a dysregulated symphony. Trends Neurosci 40: 347–357 [DOI] [PubMed] [Google Scholar]
- Fu H, Hardy J, Duff KE (2018) Selective vulnerability in neurodegenerative diseases. Nat Neurosci 21: 1350–1358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu L, Niu B, Zhu Z, Wu S, Li W (2012) CD‐HIT: accelerated for clustering the next‐generation sequencing data. Bioinformatics 28: 3150–3152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa Y, Kaneko K, Matsumoto G, Kurosawa M, Nukina N (2009) Cross‐seeding fibrillation of Q/N‐rich proteins offers new pathomechanism of polyglutamine diseases. J Neurosci 29: 5153–5162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallardo R, Ramakers M, De Smet F, Claes F, Khodaparast L, Khodaparast L, Couceiro JR, Langenberg T, Siemons M, Nyström S et al (2016) De novo design of a biologically active amyloid. Science 354: aah4949 [DOI] [PubMed] [Google Scholar]
- Gallardo R, Ranson NA, Radford SE (2020) Amyloid structures: much more than just a cross‐beta fold. Curr Opin Struct Biol 60: 7–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan L, Cookson MR, Petrucelli L, La Spada AR (2018) Converging pathways in neurodegeneration, from genetics to mechanisms. Nat Neurosci 21: 1300–1309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganesan A, Debulpaep M, Wilkinson H, Van Durme J, De Baets G, Jonckheere W, Ramakers M, Ivarsson Y, Zimmermann P, Van Eldere J et al (2015) Selectivity of aggregation‐determining interactions. J Mol Biol 427: 236–247 [DOI] [PubMed] [Google Scholar]
- Ganesan A, Siekierska A, Beerten J, Brams M, Van Durme J, De Baets G, Van der Kant R, Gallardo R, Ramakers M, Langenberg T et al (2016) Structural hot spots for the solubility of globular proteins. Nat Commun 7: 10816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giasson BI, Forman MS, Higuchi M, Golbe LI, Graves CL, Kotzbauer PT, Trojanowski JQ, Lee VM‐Y (2003) Initiation and synergistic fibrillization of tau and alpha‐synuclein. Science 300: 636–640 [DOI] [PubMed] [Google Scholar]
- Goedert M, Eisenberg DS, Crowther RA (2017) Propagation of tau aggregates and neurodegeneration. Annu Rev Neurosci 40: 189–210 [DOI] [PubMed] [Google Scholar]
- Goldschmidt L, Teng PK, Riek R, Eisenberg D (2010) Identifying the amylome, proteins capable of forming amyloid‐like fibrils. Proc Natl Acad Sci USA 107: 3487–3492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hipp MS, Kasturi P, Hartl FU (2019) The proteostasis network and its decline in ageing. Nat Rev Mol Cell Biol 20: 421–435 [DOI] [PubMed] [Google Scholar]
- Hofmann JP, Denner P, Nussbaum‐Krammer C, Kuhn PH, Suhre MH, Scheibel T, Lichtenthaler SF, Schatzl HM, Bano D, Vorberg IM (2013) Cell‐to‐cell propagation of infectious cytosolic protein aggregates. Proc Natl Acad Sci U S A 110: 5951–5956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Kant R, Louros N, Schymkowitz J, Rousseau F (2021) A structural analysis of amyloid polymorphism in disease: clues for selective vulnerability? bioRxiv 10.1101/2021.03.01.433317 [PREPRINT] [DOI] [Google Scholar]
- Kaufman SK, Sanders DW, Thomas TL, Ruchinskas AJ, Vaquer‐Alicea J, Sharma AM, Miller TM, Diamond MI (2016) Tau prion strains dictate patterns of cell pathology, progression rate, and regional vulnerability in vivo . Neuron 92: 796–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehrloesser S, Osterburg C, Tuppi M, Schafer B, Vousden KH, Dotsch V (2016) Intrinsic aggregation propensity of the p63 and p73 TI domains correlates with p53R175H interaction and suggests further significance of aggregation events in the p53 family. Cell Death Differ 23: 1952–1960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konstantoulea K, Louros N, Rousseau F, Schymkowitz J (2021) Heterotypic interactions in amyloid function and disease. FEBS J 10.1111/febs.15719 [DOI] [PubMed] [Google Scholar]
- Krebs MR, Morozova‐Roche LA, Daniel K, Robinson CV, Dobson CM (2004) Observation of sequence specificity in the seeding of protein amyloid fibrils. Protein Sci 13: 1933–1938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labbadia J, Morimoto RI (2015) The biology of proteostasis in aging and disease. Annu Rev Biochem 84: 435–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landreh M, Sawaya MR, Hipp MS, Eisenberg DS, Wuthrich K, Hartl FU (2016) The formation, function and regulation of amyloids: insights from structural biology. J Intern Med 280: 164–176 [DOI] [PubMed] [Google Scholar]
- Lopez de la Paz M, Serrano L (2004) Sequence determinants of amyloid fibril formation. Proc Natl Acad Sci U S A 101: 87–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louros NN, Chrysina ED, Baltatzis GE, Patsouris ES, Hamodrakas SJ, Iconomidou VA (2016) A common 'aggregation‐prone' interface possibly participates in the self‐assembly of human zona pellucida proteins. FEBS Lett 590: 619–630 [DOI] [PubMed] [Google Scholar]
- Louros N, Konstantoulea K, De Vleeschouwer M, Ramakers M, Schymkowitz J, Rousseau F (2020) WALTZ‐DB 2.0: an updated database containing structural information of experimentally determined amyloid‐forming peptides. Nucleic Acids Res 48: D389–D393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutter L, Serpell CJ, Tuite MF, Xue WF (2019) The molecular lifecycle of amyloid ‐ Mechanism of assembly, mesoscopic organisation, polymorphism, suprastructures, and biological consequences. Biochim Biophys Acta Proteins Proteom 1867: 140257 [DOI] [PubMed] [Google Scholar]
- Lutter L, Serpell CJ, Tuite MF, Serpell LC, Xue WF (2020) Three‐dimensional reconstruction of individual helical nano‐filament structures from atomic force microscopy topographs. Biomol Concepts 11: 102–115 [DOI] [PubMed] [Google Scholar]
- Ly H, Verma N, Sharma S, Kotiya D, Despa S, Abner EL, Nelson PT, Jicha GA, Wilcock DM, Goldstein LB et al (2021) The association of circulating amylin with β‐amyloid in familial Alzheimer's disease. Alzheimers Dement 7: e12130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal PK, Pettegrew JW, Masliah E, Hamilton RL, Mandal R (2006) Interaction between Abeta peptide and alpha synuclein: molecular mechanisms in overlapping pathology of Alzheimer's and Parkinson's in dementia with Lewy body disease. Neurochem Res 31: 1153–1162 [DOI] [PubMed] [Google Scholar]
- Marshall KE, Vadukul DM, Dahal L, Theisen A, Fowler MW, Al‐Hilaly Y, Ford L, Kemenes G, Day IJ, Staras K et al (2016) A critical role for the self‐assembly of Amyloid‐beta1‐42 in neurodegeneration. Sci Rep 6: 30182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurer‐Stroh S, Debulpaep M, Kuemmerer N, de la Paz ML, Martins IC, Reumers J, Morris KL, Copland A, Serpell L, Serrano L et al (2010) Exploring the sequence determinants of amyloid structure using position‐specific scoring matrices. Nat Methods 7: 237–242 [DOI] [PubMed] [Google Scholar]
- McCormack A, Keating DJ, Chegeni N, Colella A, Wang JJ, Chataway T (2019a) Abundance of synaptic vesicle‐related proteins in alpha‐synuclein‐containing protein inclusions suggests a targeted formation mechanism. Neurotox Res 35: 883–897 [DOI] [PubMed] [Google Scholar]
- McCormack A, Keating DJ, Chegeni N, Colella A, Wang JJ, Chataway T (2019b) Abundance of synaptic vesicle‐related proteins in alpha‐synuclein‐containing protein inclusions suggests a targeted formation mechanism. Neurotox Res 35: 883–897 [DOI] [PubMed] [Google Scholar]
- Michiels E, Liu S, Gallardo R, Louros N, Mathelie‐Guinlet M, Dufrene Y, Schymkowitz J, Vorberg I, Rousseau F (2020a) Entropic bristles tune the seeding efficiency of prion‐nucleating fragments. Cell Rep 30: 2834–2845.e2833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michiels E, Roose K, Gallardo R, Khodaparast L, Khodaparast L, van der Kant R, Siemons M, Houben B, Ramakers M, Wilkinson H et al (2020b) Reverse engineering synthetic antiviral amyloids. Nat Commun 11: 2832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muratore CR, Zhou C, Liao M, Fernandez MA, Taylor WM, Lagomarsino VN, Pearse RV 2nd, Rice HC, Negri JM, He A et al (2017) Cell‐type dependent Alzheimer's disease phenotypes: probing the biology of selective neuronal vulnerability. Stem Cell Reports 9: 1868–1884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olzscha H, Schermann SM, Woerner AC, Pinkert S, Hecht MH, Tartaglia GG, Vendruscolo M, Hayer‐Hartl M, Hartl FU, Vabulas RM (2011) Amyloid‐like aggregates sequester numerous metastable proteins with essential cellular functions. Cell 144: 67–78 [DOI] [PubMed] [Google Scholar]
- O'Nuallain B, Shivaprasad S, Kheterpal I, Wetzel R (2005) Thermodynamics of A beta(1–40) amyloid fibril elongation. Biochemistry 44: 12709–12718 [DOI] [PubMed] [Google Scholar]
- O'Nuallain B, Williams AD, Westermark P, Wetzel R (2004) Seeding specificity in amyloid growth induced by heterologous fibrils. J Biol Chem 279: 17490–17499 [DOI] [PubMed] [Google Scholar]
- Oskarsson ME, Paulsson JF, Schultz SW, Ingelsson M, Westermark P, Westermark GT (2015) In vivo seeding and cross‐seeding of localized amyloidosis: a molecular link between type 2 diabetes and Alzheimer disease. Am J Pathol 185: 834–846 [DOI] [PubMed] [Google Scholar]
- de la Paz ML, Serrano L (2004) Sequence determinants of amyloid fibril formation. Proc Natl Acad Sci U S A 101: 87–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham CL, Shanmugam N, Strange M, O'Carroll A, Brown JW, Sierecki E, Gambin Y, Steain M, Sunde M (2019) Viral M45 and necroptosis‐associated proteins form heteromeric amyloid assemblies. EMBO Rep 20: e46518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riek R, Eisenberg DS (2016) The activities of amyloids from a structural perspective. Nature 539: 227–235 [DOI] [PubMed] [Google Scholar]
- Rousseau F, Schymkowitz JW, Wilkinson HR, Itzhaki LS (2001) Three‐dimensional domain swapping in p13suc1 occurs in the unfolded state and is controlled by conserved proline residues. Proc Natl Acad Sci U S A 98: 5596–5601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseau F, Schymkowitz J, Serrano L (2006a) Protein aggregation and amyloidosis: confusion of the kinds? Curr Opin Struct Biol 16: 118–126 [DOI] [PubMed] [Google Scholar]
- Rousseau F, Serrano L, Schymkowitz JW (2006b) How evolutionary pressure against protein aggregation shaped chaperone specificity. J Mol Biol 355: 1037–1047 [DOI] [PubMed] [Google Scholar]
- Sampson TR, Challis C, Jain N, Moiseyenko A, Ladinsky MS, Shastri GG, Thron T, Needham BD, Horvath I, Debelius JW et al (2020) A gut bacterial amyloid promotes alpha‐synuclein aggregation and motor impairment in mice. eLife 9: e53111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders D, Kaufman S, DeVos S, Sharma A, Mirbaha H, Li A, Barker S, Foley A, Thorpe J, Serpell L et al (2014) Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron 82: 1271–1288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres SH, Zhang W, Falcon B, Goedert M (2020) Cryo‐EM structures of tau filaments. Curr Opin Struct Biol 64: 17–25 [DOI] [PubMed] [Google Scholar]
- Silva MVF, Loures CMG, Alves LCV, de Souza LC, Borges KBG, Carvalho MDG (2019) Alzheimer's disease: risk factors and potentially protective measures. J Biomed Sci 26: 33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supek F, Bosnjak M, Skunca N, Smuc T (2011) REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One 6: e21800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tartaglia GG, Pechmann S, Dobson CM, Vendruscolo M (2007) Life on the edge: a link between gene expression levels and aggregation rates of human proteins. Trends Biochem Sci 32: 204–206 [DOI] [PubMed] [Google Scholar]
- Taylor JP, Hardy J, Fischbeck KH (2002) Toxic proteins in neurodegenerative disease. Science 296: 1991–1995 [DOI] [PubMed] [Google Scholar]
- Teng PK, Eisenberg D (2009) Short protein segments can drive a non‐fibrillizing protein into the amyloid state. Protein Eng Des Sel 22: 531–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tessier PM, Lindquist S (2007) Prion recognition elements govern nucleation, strain specificity and species barriers. Nature 447: 556–561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson Å, Kampf C, Sjöstedt E, Asplund A et al (2015) Proteomics. Tissue‐based map of the human proteome. Science 347: 1260419 [DOI] [PubMed] [Google Scholar]
- UniProt Consortium (2008) The universal protein resource (UniProt). Nucleic Acids Res 36: D190–D195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandersteen A, Masman MF, De Baets G, Jonckheere W, van der Werf K, Marrink SJ, Rozenski J, Benilova I, De Strooper B, Subramaniam V et al (2012) Molecular plasticity regulates oligomerization and cytotoxicity of the multipeptide‐length amyloid‐beta peptide pool. J Biol Chem 287: 36732–36743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanik DL, Surewicz KA, Surewicz WK (2004) Molecular basis of barriers for interspecies transmissibility of mammalian prions. Mol Cell 14: 139–145 [DOI] [PubMed] [Google Scholar]
- Vasconcelos B, Stancu I‐C, Buist A, Bird M, Wang P, Vanoosthuyse A, Van Kolen K, Verheyen AN, Kienlen‐Campard P, Octave J‐N et al (2016) Heterotypic seeding of Tau fibrillization by pre‐aggregated Abeta provides potent seeds for prion‐like seeding and propagation of Tau‐pathology in vivo . Acta Neuropathol 131: 549–569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura S, Zurdo J, Narayanan S, Parreno M, Mangues R, Reif B, Chiti F, Giannoni E, Dobson CM, Aviles FX et al (2004) Short amino acid stretches can mediate amyloid formation in globular proteins: the Src homology 3 (SH3) case. Proc Natl Acad Sci U S A 101: 7258–7263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DM, Selkoe DJ (2016) A critical appraisal of the pathogenic protein spread hypothesis of neurodegeneration. Nat Rev Neurosci 17: 251–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DM, Thulin E, Minogue AM, Gustavsson N, Pang E, Teplow DB, Linse S (2009) A facile method for expression and purification of the Alzheimer's disease‐associated amyloid beta‐peptide. FEBS J 276: 1266–1281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Fersht AR (2015) Propagation of aggregated p53: Cross‐reaction and coaggregation vs. seeding. Proc Natl Acad Sci U S A 112: 2443–2448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesseling H, Mair W, Kumar M, Schlaffner CN, Tang S, Beerepoot P, Fatou B, Guise AJ, Cheng L, Takeda S et al (2020) Tau PTM profiles identify patient heterogeneity and stages of Alzheimer's disease. Cell 183: 1699–1713.e1613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermark GT, Westermark P (2010) Prion‐like aggregates: infectious agents in human disease. Trends Mol Med 16: 501–507 [DOI] [PubMed] [Google Scholar]
- Wetzel R (2006) Kinetics and thermodynamics of amyloid fibril assembly. Acc Chem Res 39: 671–679 [DOI] [PubMed] [Google Scholar]
- Xiong F, Ge W, Ma C (2019a) Quantitative proteomics reveals distinct composition of amyloid plaques in Alzheimer's disease. Alzheimers Dement 15: 429–440 [DOI] [PubMed] [Google Scholar]
- Xiong F, Ge W, Ma C (2019b) Quantitative proteomics reveals distinct composition of amyloid plaques in Alzheimer's disease. Alzheimers Dement 15: 429–440 [DOI] [PubMed] [Google Scholar]
- Xu J, Reumers J, Couceiro JR, De Smet F, Gallardo R, Rudyak S, Cornelis A, Rozenski J, Zwolinska A, Marine J‐C et al (2011) Gain of function of mutant p53 by coaggregation with multiple tumor suppressors. Nat Chem Biol 7: 285–295 [DOI] [PubMed] [Google Scholar]
- Xue WF, Homans SW, Radford SE (2009) Amyloid fibril length distribution quantified by atomic force microscopy single‐particle image analysis. Protein Eng Des Sel 22: 489–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan L‐M, Velkova A, Tatarek‐Nossol M, Andreetto E, Kapurniotu A (2007) IAPP mimic blocks Abeta cytotoxic self‐assembly: cross‐suppression of amyloid toxicity of Abeta and IAPP suggests a molecular link between Alzheimer's disease and type II diabetes. Angew Chem Int Ed Engl 46: 1246–1252 [DOI] [PubMed] [Google Scholar]
- Zaman M, Khan AN, Wahiduzzaman , Zakariya SM, Khan RH (2019) Protein misfolding, aggregation and mechanism of amyloid cytotoxicity: An overview and therapeutic strategies to inhibit aggregation. Int J Biol Macromol 134: 1022–1037 [DOI] [PubMed] [Google Scholar]
- Zhou Y, Smith D, Leong BJ, Brannstrom K, Almqvist F, Chapman MR (2012) Promiscuous cross‐seeding between bacterial amyloids promotes interspecies biofilms. J Biol Chem 287: 35092–35103 [DOI] [PMC free article] [PubMed] [Google Scholar]