

Abstract
Neonatal seizures pose a clinical challenge in their early detection, acute management, and long-term comorbidities. They are often caused by hypoxic-ischemic encephalopathy and are frequently refractory to the first-line anti-seizure medication phenobarbital. One proposed mechanism for phenobarbital inefficacy during neonatal seizures is the reduced abundance and function of the neuron-specific K+/Cl− cotransporter 2 (KCC2), which maintains chloride homeostasis and promotes GABAergic inhibition upon its phosphorylation during postnatal development. Here, we investigated whether this mechanism is causal and whether it can be rescued by KCC2 functional enhancement. In a CD-1 mouse model of refractory ischemic neonatal seizures, treatment with the KCC2 functional enhancer CLP290 rescued phenobarbital efficacy, increased KCC2 abundance, and prevented the development of epileptogenesis, as quantified by video electroencephalogram monitoring. These effects were prevented by knock-in expression of nonphosphorylatable mutants of KCC2 (S940A or T906A and T1007A), indicating that KCC2 phosphorylation regulates both neonatal seizure susceptibility and CLP290-mediated KCC2 functional enhancement. Our findings therefore validate KCC2 as a clinically relevant target for refractory neonatal seizures and provide insights for future drug development.
Introduction
Neonatal seizures occur in an estimated 1 to 3.5 per 1000 live births in the term infant. Hypoxic-ischemic encephalopathy is a major cause of acute neonatal seizures (1). The management of these seizures are a major clinical challenge, because they are often refractory to an initial loading dose of the first-line anti-seizure medication (ASM) phenobarbital (PB) as well as adjunct ASMs (2, 3). Compared to seizures at older ages, neonatal seizures differ in their etiology, semiology, electrographic signature, and response to ASMs (1, 4). There is an urgent need to identify the developmental mechanisms underlying seizure susceptibility and ineffective ASM response in the neonatal brain.
The neonatal brain has low expression of its chief chloride extruder the neuron-specific K+-Cl− cotransporter 2 (KCC2) and a high neuronal intracellular chloride concentration ([Cl−]i) (5–7). GABA is the primary inhibitory neurotransmitter in the mature brain; however, in the neonatal brain, the activation of GABAA receptors (GABARs) results in depolarizing actions on immature neurons (8). KCC2 expression and function increase during development, resulting in a lower [Cl−]i that coincides with a developmental shift from depolarizing to hyperpolarizing GABAergic signaling. The importance of KCC2 function in seizure susceptibility is supported by emerging evidence from human genetics, as pathogenic variants in SLC12A5 are associated with the development of idiopathic generalized epilepsy and early infantile epileptic encephalopathy (OMIM #616685 and #616645, respectively) (9).
Despite the introduction of many new ASMs into clinical practice over the past 20 years, the incidence of refractory seizures has remained unchanged (10). KCC2 hypofunction is increasingly associated with pharmaco-resistant epilepsies and is a proposed cause of disinhibition (11, 12). GABAAR mediated fast synaptic inhibition and the anti-seizure efficacy of PB (a positive allosteric modulator of GABAARs) are both dependent upon active neuronal Cl− extrusion (13, 14). In a preclinical CD-1 mouse model of ischemic neonatal seizures, KCC2 is transiently downregulated and associated with PB-resistant seizures (15). In this model, post-ischemic inhibition of tropomyosin receptor kinase (TrkB) rescued PB-resistant neonatal seizures and KCC2 expression (16, 17). This acute rescue of KCC2 hypofunction by TrkB inhibition improved long-term outcomes after ischemic neonatal seizures (18–20). These results suggest KCC2 is a novel therapeutic target for refractory neonatal seizures.
A published high-throughput screen for compounds that reduce [Cl−]i in a cell line with low KCC2 expression identified the compound CLP257 as a KCC2 functional enhancer (21). Here, we used CLP257 and the carbamate prodrug CLP290 (21) in a well-characterized CD-1 mouse model and S940A+/+ and T1007A+/+ point-mutation mice to test whether functional enhancement of KCC2 can rescue the emergence of PB-resistant ischemic neonatal seizures, if there are differences in their efficacy corresponding to developmental changes in KCC2 expression (15), whether they can prevent the development of epilepsy associated with neonatal seizures (22, 23), and whether these effects are dependent on development-associated KCC2 phosphorylation (24–27).
Results
CLP290 rescues phenobarbital-resistant seizures at P7
The true burden of acute ischemic seizures in mouse pups cannot be identified by behavioral scoring parameters alone and requires continuous vEEG recordings, because their presentation can range from entirely electrographic to generalized convulsive (7). In this CD-1 mouse model of unilateral carotid ligation without transection, pups presented with PB-resistant ischemic neonatal seizures at postnatal day 7 (P7) (Fig. 1A–D), an established characteristic of the model (15–17, 28). This model represents transient ischemia followed by reperfusion that results in cerebral edema at 24 hours with no discernable infarcts when evaluated at P18 (15). All pups underwent 1 hour of baseline vEEG recording after unilateral carotid ligation. After that hour, all pups received a loading dose of PB with a subsequent hour of vEEG recording (Fig. 1, A to D). Pups that only received a PB-loading dose were resistant to PB, given that their second-hour seizure burden remained high and was similar to first-hour levels (Fig. 1D and fig. S1, A to B).
Fig. 1. CLP290 rescued PB-resistant neonatal seizures in P7 CD1 mice.
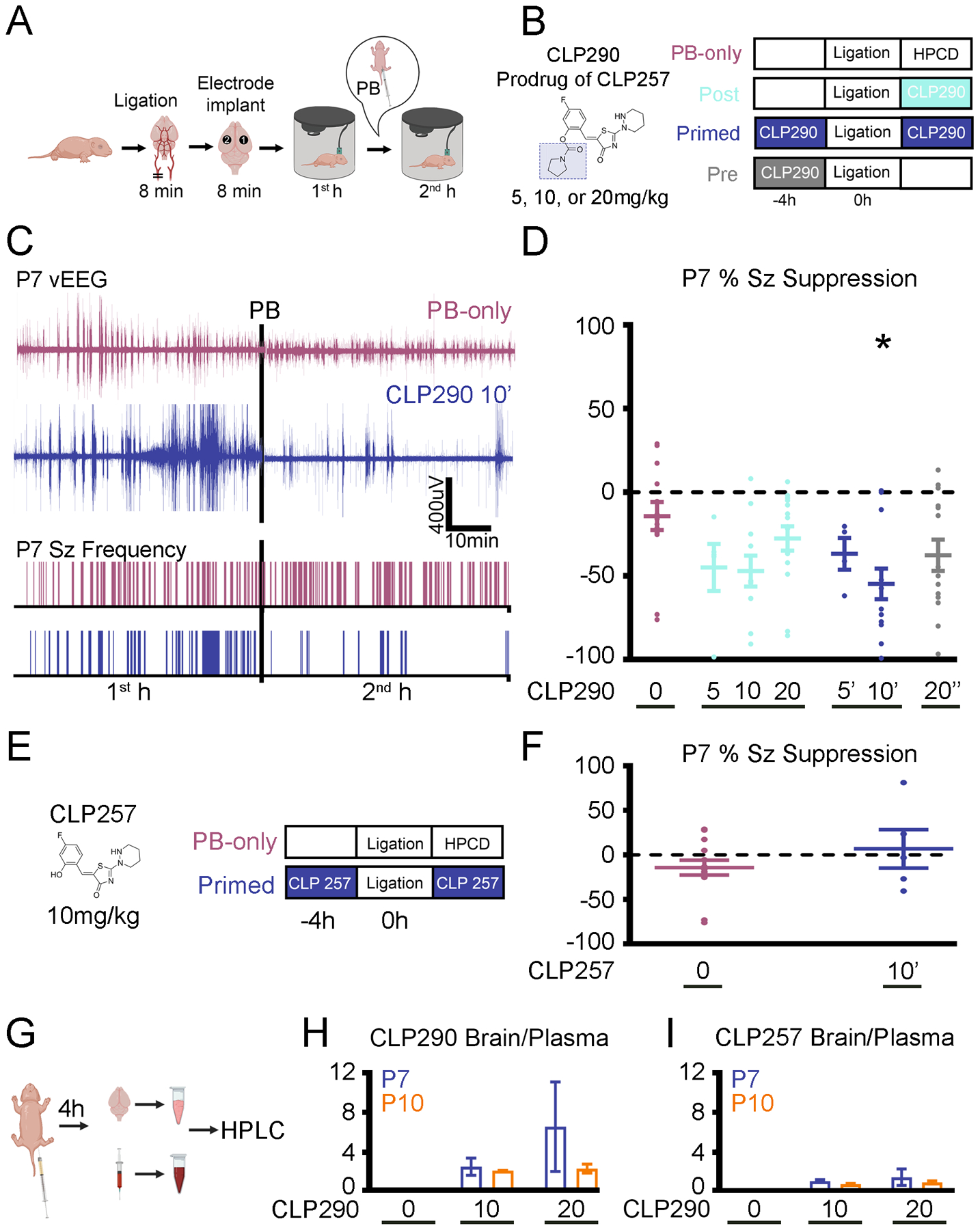
(A) Experimental design of a P7 CD-1 mouse model of ischemic neonatal seizures with continuous vEEG. Unilateral carotid ligation without transection, marking where the recording (“1”) and reference (“2”) electrodes were placed over bilateral parietal cortices with a ground electrode over the rostrum. (B) Doses and treatment protocols for CLP290 experiments. The prodrug’s full structure and that of CLP257 (shaded portion) is shown for reference. Post treatment groups (treated immediately after ligation) are denoted with the dose, such as 10. Pre-treatment groups (treated 4 hours before ligation) are denoted with (“) and primed (treated 4 hours before ligation and immediately after ligation) with (‘) alongside the dose administered, such as 20” and 10’. HPCD, 2-hydroxypropyl-β-cyclodextrin (CLP257/290 dissolving vehicle). (C) Representative EEG traces and seizure (Sz) frequency raster plots of a PB-only and a 10 mg/kg CLP290 pre- and post–treated (“primed”; 10’) pup. Black bars represent the point of PB administration (25mg/kg; i.p.). (D) Analysis of EEGs represented and described in (C), assessing percent suppression of first- (1st) and second (2nd)-hour seizures after unilateral carotid ligation in P7 pups. *P<0.05 by one-way ANOVA vs. PB-only. PB-only group, n=14 pups; CLP290 groups, n=5 to 15 pups. Associated seizure burden and ictal events are shown in figure S1. (E) As in (B), for CLP257 experiments. (F) As described in (D), by CLP257 in a 10 mg/kg primed protocol. Data are from n=5 pups each. Note that the PB-only “0” data are the same as shown for comparison the CLP290-treatment groups in panel (D); groups assessed simultaneously. (G) Experimental paradigm to investigate the pharmacokinetic profile of CLP290 and CLP257. For characteristic peaks of CLP290 and CLP257 on HPLC, see fig. S2. (H and I) Brain to plasma ratio of CLP290 (H) or CLP257 (I) after i.p. administration. n=2 pups per group. Plots show all data points with means ± SEM. *P<0.05; two-way ANOVA.
To investigate if the KCC2 functional enhancer CLP290 could rescue PB-resistant seizures, CLP290 was administered intraperitoneally to P7 pups in three groups: “Pre” (treated with the specified dose—5, 10 or 20 mg/kg— 4 hours before ligation), “Post” (treated immediately after ligation), and “Primed” (both 4 hours before ligation and immediately after ligation) (Fig. 1B). Administration of 10 mg/kg CLP290 i.p. to P7 pups 4 hours before ligation (“primed” treatment; referred to as 10’ in the figures) significantly reduced total burden, duration, and frequency of second-hour seizure events (Fig. 1D and fig. S1, A to B) and significantly increased seizure suppression after PB administration, thereby rescuing P7 PB-resistance (Fig. 1D). In contrast to the 10’ treatment, bolus administration of 20” (20mg/kg CLP290 4 hours before ligation) reduced first-hour baseline seizure burden (fig. S1C).
Bioavailability of the pro-drug CLP290 in the neonatal mouse brain
A major limiting factor for epilepsy drug development is the in vivo brain availability of candidate compounds identified in vitro (29, 30). CLP290 is a carbamate prodrug of CLP257 and has an improved half-life (t1/2) from <15min (CLP257) to 5 hours (CLP290) in blood samples from adult rats (21). At P7, CLP257 was unable to reduce second hour seizure burden (Fig. 1, E and F, and fig. S1, D and E) when administered systemically at the 10’ dose likely due to its poor t1/2. To our knowledge, the brain bioavailability of systemically administered CLP290 in neonatal mice is not yet reported. To determine if CLP290 anti-seizure efficacy was associated with brain bioavailability at 4 hours, we performed HPLC to analyze brain and plasma levels of CLP290 after intraperitoneal (i.p.) delivery. Additionally, the metabolism of CLP290 into its active form CLP257, in the plasma and brain at 4 hours was also quantified in the same samples (Fig. 1, G to I, and fig. S2). At P7 and P10, CLP290 administration of 10 or 20 mg/kg demonstrated adequate brain bioavailability at 4 hours. (Fig. 1H). The concomitant CLP257 brain-plasma ratios were lower compared to the prodrug CLP290 (Fig. 1I).
To further investigate the effect of CLP290, naïve CD-1 pups underwent CLP290 10’ treatment and the cortices harvested for WB analysis 4 hours later (fig. S3, A to D). CLP290 did not significantly increase total KCC2 or Ser940 phosphorylation levels at either P7 or P10 in naïve cortex. These results demonstrate that CLP290 10’ was readily bioavailable in the P7 neonatal brain at 4 hours after systemic delivery, and CLP290 10’ did not significantly increase total KCC2 and Ser940 phosphorylation in naïve brains with the same protocol. In summary, these results suggest that the therapeutic effect of CLP290-mediated functional KCC2 enhancement is beneficial when it is bioavailable at the onset of KCC2 hypofunction following the ischemic insult and does not depend on increasing the total expression KCC2 by pre-treatment.
CLP290 efficacy on phenobarbital-responsive seizures at P10
As previously reported (15–17, 28), ischemic neonatal seizures after unilateral carotid ligation in P10 CD-1 pups were PB responsive (Fig. 2, A to C). The age-dependent emergence of PB-resistant and PB-responsive seizures at P7 versus P10 is a characteristic of the CD-1 neonatal mouse model that is associated with an increase in KCC2 expression between P7 and P10 (15). Administration of CLP290 at P10 did not further improve the efficacy of PB at any of the doses tested (Fig. 2C and fig. S1, F and G). When ischemic neonatal seizures were PB-responsive at P10, the previously reported TrkB-antagonist ANA12 (17, 28) also did not improve the efficacy of PB. This suggests that the therapeutic benefit of KCC2 functional enhancement is dependent upon the degree of KCC2 hypofunction, which is evident in the emergence of refractoriness at P7 but not at P10.
Fig. 2. Ischemic neonatal seizures in P10 CD1 pups were phenobarbital responsive.
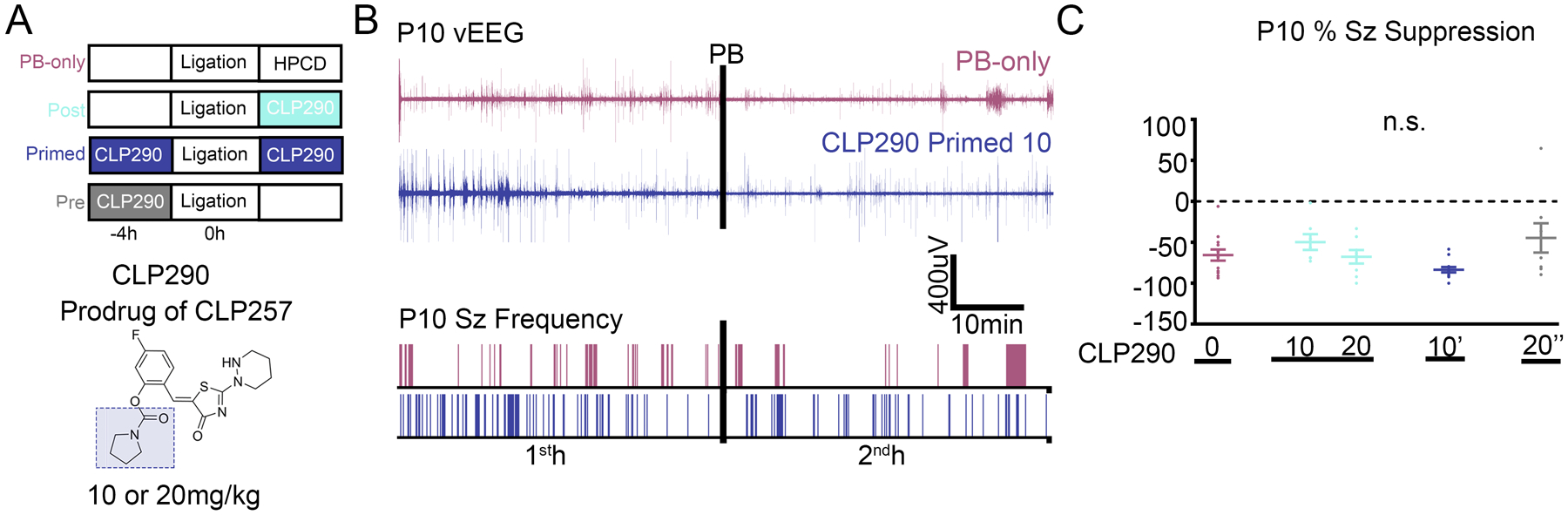
(A) Doses and treatment protocols to evaluate CLP290 in P10 CD-1 pups. (B) Representative EEG traces and seizure frequency raster plots of a P10 phenobarbital-only and CLP290 10’ pup. Black bars represent a loading dose of PB (25 mg/kg i.p.). (C) 1st- and 2nd-hour percent seizure suppression at P10 after unilateral carotid ligation. Percent seizure suppression was analyzed by one-way ANOVA vs. PB-only. PB-only; n=13; CLP290 10 n=7; CLP290 20 n=8; CLP290 10’ n=12; CLP290 20” n=8 pups. For 1st- and 2nd-hour seizure burden and ictal events, see fig. S1.
Acute CLP290 intervention at P7 mitigates epileptogenesis at P12
Hypoxic-ischemic encephalopathy seizures are transient within the first week of life (31, 32) and the long-term consequences of neonatal seizures are difficult to isolate from the consequences of prolonged and/or inefficacious anti-seizure therapy in the clinic (23). In this CD-1 mouse model of refractory neonatal ischemic seizures, we have previously documented the emergence of epilepsy in adulthood (33). To assess if the acute rescue of PB-resistant neonatal seizures at P7 by administration of CLP290 had long-term benefits, the same pups underwent a pentylenetetrazol (PTZ) challenge at P12 (Fig. 3, A and B). Previously, a 80 mg/kg dose of the GABAAR antagonist PTZ was shown to induce high seizure burdens that were PB-responsive and upregulated KCC2 at P7 in CD-1 pups (34). In this study, pups were administered three doses of PTZ (20, 20, and 40 mg/kg) 1 hour apart (Fig. 3, A and B). Using this protocol, we characterized seizure susceptibility in P7 naïve, P7 PB-only, and P7 CLP290 10’ treated pups at P12. The initial dose of PTZ induced seizures in the first hour for all groups (Fig. 3, A to D). Naïve pups demonstrated a general reduction in seizures during the second and third PTZ doses, potentially uncovering a homeostatic compensation to help develop resistance to the chemoconvulsant induced seizures. PB-only pups demonstrated an increase in seizure burden after the repeated doses of PTZ, with mice going into status epilepticus (Fig. 3, D to F). Thus, the significant increase in PB-only seizure burdens at P12 was driven by a significant increase in seizure duration (Fig. 3E). CLP290 10’ treated pups had similar P12 seizure burdens as naïve pups and significantly lower than PB-only pups (Fig. 3, A to D). This novel PTZ challenge protocol identified epileptogenesis in the form of heightened susceptibility to seizures at P12 for neonatal pups that underwent standard but inefficacious PB treatment for their refractory seizures at P7. The efficacious CLP290 10’ intervention at P7 resulted in the regression of epileptogenesis at P12.
Fig. 3. CLP290-mediated regression of epileptogenesis detected using a PTZ challenge.
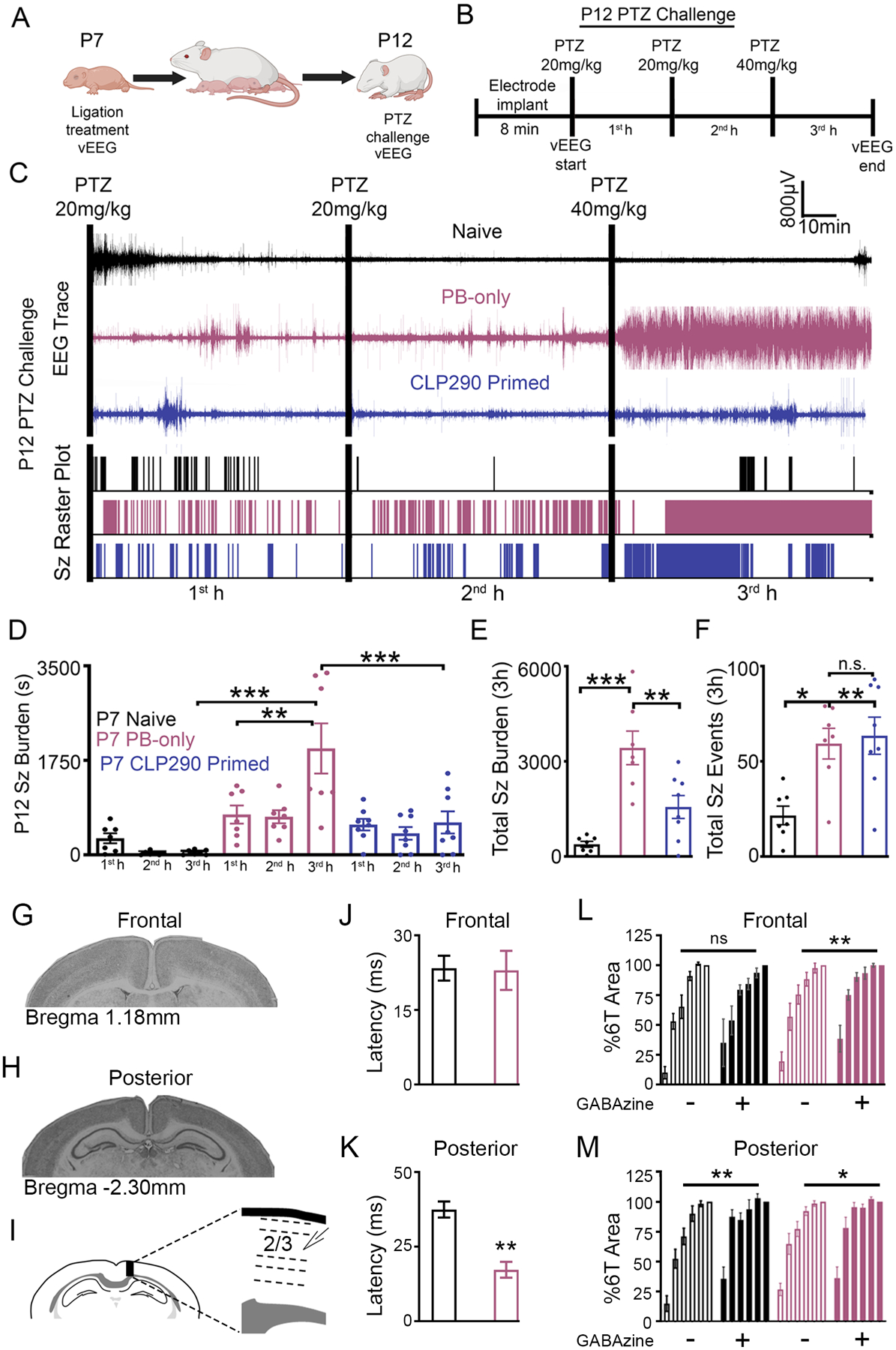
(A) Schematic to investigate the developmental benefits of CLP290 10’ treatment for P7 ischemic seizures. (B) P12 pentylenetetrazol (PTZ) challenge to evaluate epileptogenesis. (C) Representative EEG traces and seizure frequency raster plots for P12 pups that were untreated (naïve) or underwent PB-only or CLP290 10’ treatment at P7. Black bars indicate intraperitoneal PTZ injections. (D) 1st-, 2nd-, and 3rd-hour seizure burdens at P12 in CD-1 mice after PTZ injections. (E) Total electrographic seizure burdens over the three hours of vEEG recording. (F) Total seizure events over three hours of vEEG recording. Data plots show all data points with means ±SEM. *P<0.05; **P<0.01; ***P<0.001 by 2-way ANOVA. Naïve n=7; PB-only n=7; CLP290 10’ n=8 mice. (G to I) Representative coronal sections from frontal (G) and posterior (H) recording areas; line diagram (I) shows the placement of the recording electrode in an intact cortical column from a posterior section. (J and K) Mean 1T latencies from frontal (J) and posterior (K) sections from P12-P15 pups that were untreated (naïve) or PB-treated at P7. (L) Percent 6T area from frontal (L) and posterior (M) sections with and without GABAzine (1 μM). **P<0.01; ***P<0.001 by t-test and 2-way ANOVA. Naïve n=7, and PB-only n=14 mice.
Hyperexcitability after suppression of GABAAR-mediated inhibition has been demonstrated in ex vivo brain slices from experimental models of epilepsy in adulthood (35–38). To determine whether the in vivo PTZ (GABAAR antagonist) challenge represented progressive electrophysiological changes underlying hyperexcitability after P7 ischemic seizures, ex vivo coronal neocortical slices from the ipsilateral cortex of CD-1 mice 5 to 8 days after ischemic seizures (P12 to P15) were investigated in both normal artificial cerebral spinal fluid (ACSF) and after blockade of GABAARs with GABAzine. Evoked field potentials (EFP) were recorded in frontal and posterior sections (Fig. 3, G to I) representing the perfusion territories of the anterior and middle cerebral arteries (ACA and MCA) with and without collateral circulation in this model (35). Brain slices from the posterior neocortex in PB-only pups demonstrated a significantly reduced latency to threshold (Fig. 3, J and K), which was not detected in their anterior brain slice. In normal ACSF, EFPs in both frontal and posterior sections were similar between naïve and PB-only pups, and the responses remained graded from EFP threshold to six times EFP threshold, 1T to 6T (Fig. 3, L and M). In contrast, blockade of GABAARs with GABAzine resulted in larger amplitude EFPs that lacked a graded response in the frontal sections from PB-only mice (Fig. 3, L and M). In contrast, ex vivo sections from naïve mice demonstrated no significant differences between EFPs in normal ACSF and GABAAR blockade (Fig. 3L). The in vivo PTZ challenge and ex vivo EFP results both suggest that GABAergic electrophysiological changes mask underlying hyperexcitability at P12 to P15 following P7 ischemic seizures and become evident when GABAARs were antagonized either by GABAzine in vitro or PTZ in vivo.
Spontaneous epileptiform discharges in S940A+/+ pups
The chloride extrusion capacity of KCC2 is greatly influenced by the phosphorylation status of residues Ser940 and Thr1007 (24–26). KCC2 phospho-mutant mice enabled the investigation of the role of these residues in the emergence of refractory seizures at neonatal ages. It is already known that KCC2 deficient mice die postnatally with generalized seizures and respiratory failure (39, 40). S940A+/+ mice are susceptible to death after kainite-induced status epilepticus in adulthood (41). In patients with idiopathic generalized epilepsy and early childhood onset of febrile seizures, heterozygous missense variants in SLC12A5 have been identified (42, 43) and associated with a reduction in Ser940 phosphorylation (42). However, it is not currently reported whether the prevention of Ser940 phosphorylation alone can induce spontaneous epileptiform activity during the neonatal period. Therefore, S940A+/+ pups underwent vEEG at P7 and P12 (Fig. 4, A to D). S940A+/+ mice had spontaneous epileptiform discharges at both P7 and P12, which failed to respond to CLP290 intervention (Fig. 4, A to D). These results suggest that the inability to phosphorylate the Ser940 residue engenders spontaneous epileptic activity during development.
Fig. 4. Inability to phosphorylate Ser940 or Thr1007 in knock-in pups regulated neonatal seizure susceptibility.
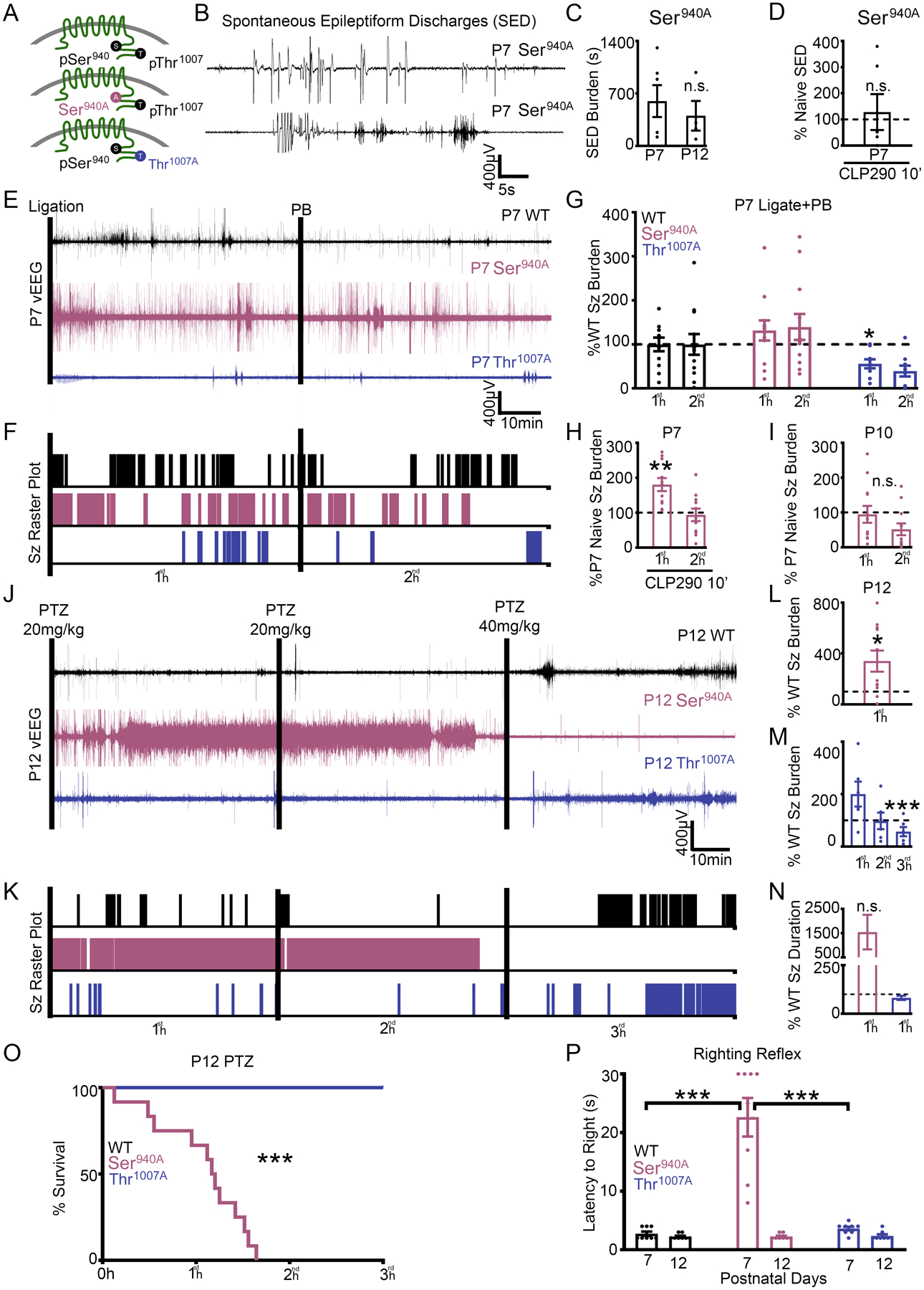
(A) Graphical representation of homozygous S940A knock-in mutant mice (26) and homozygous T1007A knock-in mutant mice (25). (B and C) S940A+/+ mice had spontaneous epileptiform discharges (SEDs) at P7 and P12. (D) Spontaneous epileptiform discharge duration of P7 S940A+/+ pups administered CLP290 10’ treatment. Naïve P7 S940A+/+ n=6, naïve P12 S940A+/+ n=4, and CLP290 10’ P7 S940A+/+ n=6 pups. (E to G) P7 T1007A+/+ pups are resistant to ischemic neonatal seizures. (E) EEG traces and (F) seizure frequency raster plots for P7 WT, S940A+/+, and T1007A+/+pups that underwent unilateral carotid ligation. (G) 1st-hour vs 2nd-hour seizure burdens after unilateral carotid ligation, plotted as percent WT. WT n=12, S940A+/+ n=12, and T1007A+/+ n=9 pups. (H) P7 S940A+/+ ischemic seizures in response to CLP290 (10’). Seizure burden of CLP290 10’ treated P7 S940A+/+ pups after unilateral carotid ligation plotted as P7 percent naïve S940A+/+. CLP290 10’ treated P7 S940A+/+ n=11 pups. (I) Seizure burden of P10 S940A+/+ pups (n=8) after unilateral carotid ligation, plotted as P7 percent naïve S940A+/+. n.s., P=0.57. (J to O) Naïve P12 S940A+/+ mice are susceptible to status epilepticus and mortality. (J) Representative EEG trace and (K) seizure frequency raster plot of naïve P12 WT, S940A+/+, and T1007A+/+ pups. Black bars indicate intraperitoneal pentylenetetrazol (PTZ) injections. (L) Total seizure burden of P12 S940A+/+ pups after PTZ administration plotted as percent WT. (M) 1st-, 2nd-, and 3rd-hour seizure burdens at P12 in T1007A+/+ pups plotted as percent WT. (N) 1st-hour average seizure durations for S940A+/+ and T1007A+/+ P12 pups, plotted as percent WT. (O) Survival plot for P12 WT, T1007A+/+, and S940A+/+ pups during the PTZ challenge. P12 WT n=11; S940A+/+ n=11; and T1007A+/+ n=6 pups. (P) Duration of time to righting reflex at P7 and P12 in naïve WT, T1007A+/+, and S940A+/+ pups. (n=8 pups each group). *P<0.05; **P<0.01; ***P<0.001 by unpaired t-test vs. WT for all seizure data. Survival analysis ***P<0.001 by Mantel-Cox test. Righting Reflex by one-way ANOVA.
T1007A+/+ pups are resistant to ischemic seizures
Thr1007 phosphorylation decreases during development and is associated with an increase in neuronal Cl− extrusion capacity (24, 27, 44). T1007A+/+ knock-in mutant mice have a reduced susceptibility to kainate-induced status epilepticus in adulthood (25). To investigate the role of KCC2 phosphorylation in ischemic neonatal seizures, WT, S940A+/+, and T1007A+/+ P7 pups underwent unilateral carotid ligation with 2 hours of vEEG recordings (Fig. 4, E to G). After P7 unilateral carotid ligation T1007A+/+ pups were significantly resistant to ischemic seizures than WT (Fig. 4, E to G). This result suggests that reducing Thr1007 phosphorylation on KCC2 is a promising therapeutic target. S940A+/+ pups had higher seizure burdens than WT after ischemia (Fig. 4G). CLP290 treatment for 10 min did not improve the efficacy of PB in S940A+/+ mice but significantly increased first-hour seizure burden when compared to untreated S940A+/+ mice (Fig. 4H). During development, mice undergo a reduction in ischemic seizure susceptibility from P7 to P10 that is associated with a robust upregulation of KCC2 and Ser940 phosphorylation (15, 28). S940A+/+ mice did not undergo a developmental decrease in ischemic seizure susceptibility at P10 when compared to P7 S940A+/+ mice (Fig. 4I). These results suggest that both the CLP290-mediated KCC2 functional enhancement and the age-dependent reduction in ischemic seizure susceptibility are dependent upon Ser940 phosphorylation.
S940A+/+ pups are susceptible to status epilepticus and death
WT, S940A+/+, and T1007A+/+ pups at P12 underwent the 3-hour PTZ challenge to determine if KCC2 phosphorylation status influenced seizure susceptibility independent of ischemia. As described above (Fig. 3), pups were administered three doses of PTZ (20, 20, and 40 mg/kg) 1 hour apart. S940A+/+ mice immediately progressed into status epilepticus and died before the second hour (Fig. 4, J to O). This indicated that the prevention of Ser940 phosphorylation increased the risk of sudden unexpected death in epilepsy (SUDEP)-like phenomenon (45). In contrast, T1007A+/+ mice were resistant to the PTZ induced seizures when compared to WT (Fig. 4M). Additionally, the assessment of righting reflex as a neurodevelopmental milestone identified that only S940A+/+ mice had a significantly impaired righting reflex at P7 but not at P12 (Fig. 4P). These data suggest that KCC2 phosphorylation at residues Ser940 and Thr1007 influence seizure susceptibility during development.
In vivo KCC2 inhibition is epileptogenic in the neonatal brain
Micro-infusion of the selective KCC2 inhibitor VU0463271 (VU) has been shown to induce epileptiform discharges in the dorsal hippocampus of the adult mouse in vivo. (46). During spontaneous recurrent ictal-like epileptiform discharges in organotypic hippocampal slices in vitro, VU reduced Cl− extrusion rates and induced status epilepticus (47). These studies highlight the critical role of KCC2 in curbing epileptiform activity in the mature hippocampus. Here, naive P7 CD-1 pups were administered 0.25mg/kg VU (i.p.) at the initiation of vEEG recording with a subsequent dose of 0.5 mg/kg VU (i.p.) at 1hour (Fig. 5A–B). VU was sufficient to induce epileptiform activity (Fig. 5, A to E) at P7. Prolonged and repeated seizures are known to play a role in the reduction of neuronal surface KCC2 expression and function (15, 41, 48, 49). At P7, if post-ischemic KCC2 hypofunction plays a critical role in PB-refractoriness, KCC2 inhibition following repeated ischemic seizures would be expected to further aggravate the seizure burden. To test the effect of KCC2 inhibition following repeated ischemia-induced neonatal seizures at P7, VU 0.25 mg/kg (i.p.) was administered 1 hour after unilateral carotid ligation (Fig. 5F). P7 pups that received VU after 1 hour of ischemic seizures developed a significant aggravation of EEG seizure burden in the second hour (Fig. 5, F to J). Together, the data show that KCC2 inhibition induced epileptiform activity in the naïve neonatal brain and exacerbated ischemic neonatal seizures at P7.
Fig. 5. Selective KCC2 antagonist VU induced spontaneous epileptiform discharges in P7 pups.
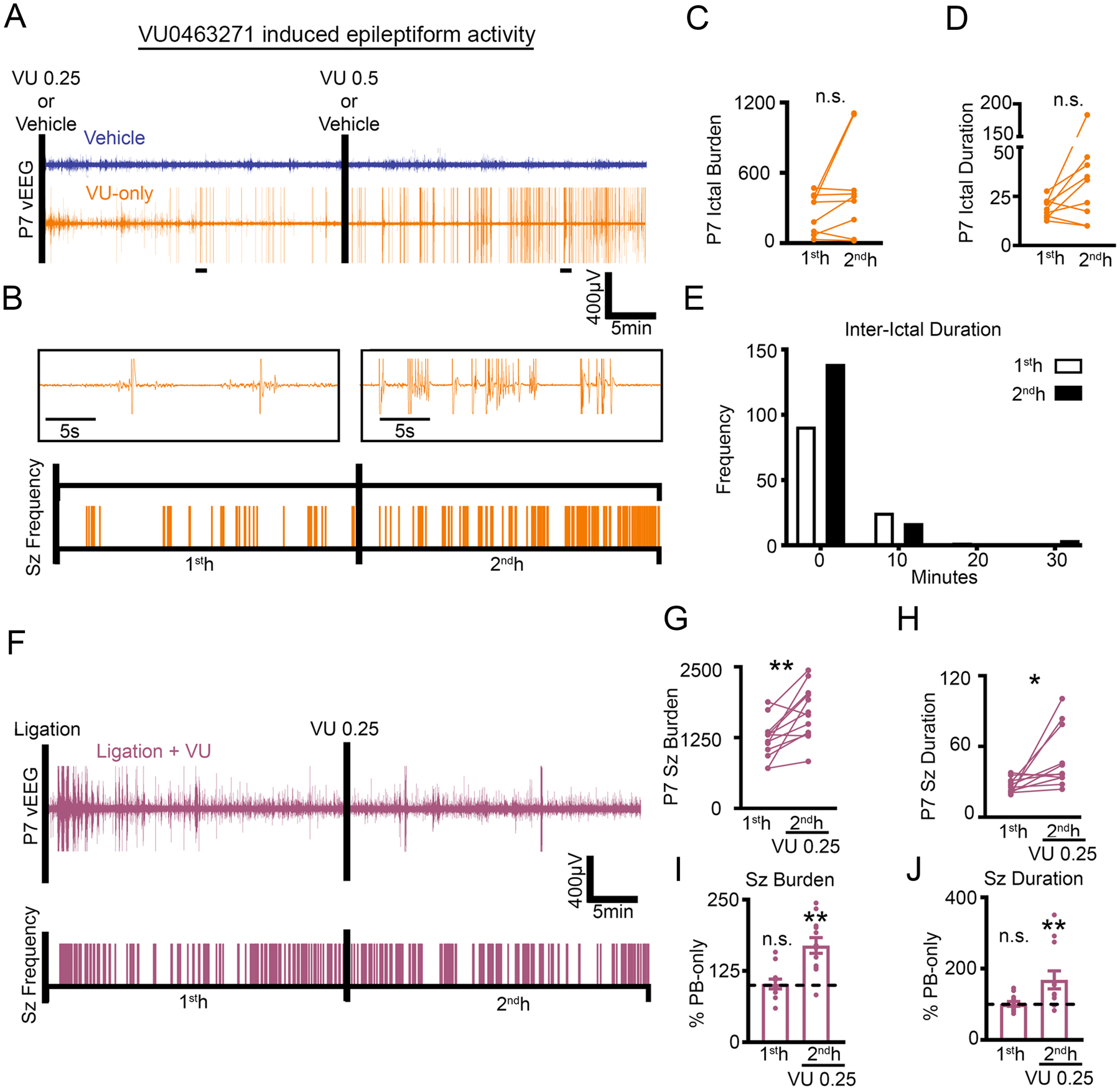
(A to E) Representative EEG traces (A) and ictal event frequency raster plots (B) from P7 CD-1 pups that were administered either vehicle or VU0463271 (VU); black bars note i.p. injections. Expanded timescales show VU-induced epileptiform activity in the 1st- and 2nd-hour. 1st- vs. 2nd-hour ictal burden (C) and duration (D) after VU administration. Total frequency distribution for all interictal durations in P7 CD-1 pups administered VU(E). Vehicle, n=4 pups; VU, n=8 pups. (F to J) Representative EEG trace and seizure frequency raster plot (F) of P7 CD-1 pups that underwent unilateral carotid ligation with administration of VU 0.25 mg/kg at 1 hour (denoted by black bar). 1st- and 2nd-hour seizure burden (G) and duration (H). (I and J) 1st- and 2nd-hour seizure burden and duration plotted as percent PB-only. Data plots show all data points with means ± SEM. **P<0.05 and **P<0.01 by two-tailed paired t-test. Ligation + VU n=12 pups.
CLP290 rescued ipsilateral post-ischemic KCC2 and Ser940 downregulation
Excitotoxic injury results in deficits in KCC2 expression and activity (12, 14, 21, 50–55). PB-resistant ischemic neonatal seizures have been shown to significantly reduce expression of KCC2 and phosphorylation of Ser940 24 hours after ischemia (17). P7 unilateral carotid ligation did not result in an infarct stroke injury, therefore KCC2 degradation at 24 hours is not caused by infarct related cell-death (15). In this model, KCC2 expression undergoes a recovery over 3 to 4 days, a characteristic that is associated with the transient nature of neonatal seizures due to hypoxic-ischemic encephalopathy (31, 32). Twenty-four hours after P7 unilateral carotid ligation, KCC2 abundance and Ser940 phosphorylation was significantly lower in the right hemisphere (ipsilateral to ischemia) than in the left hemisphere (contralateral to ischemia) in the PB-only group (Fig. 6, A to E, and data file S1). Intervention with CLP290 at P7 significantly rescued both KCC2 and Ser940 downregulation in the 10’ group 24 hours after ligation (Fig. 6, B and C). After PB-responsive seizures at P10, KCC2 expression was not significantly decreased at 24 hours (Fig. 6, F to J, and data file S1). However, Ser940 phosphorylation in the PB-only group was significantly decreased (Fig. 6H) and was rescued by CLP290 treatment (Fig. 6H). Therefore, when ischemic neonatal seizures were PB-resistant, CLP290 rescued KCC2 expression and Ser940 phosphorylation, suggesting that the degree of KCC2 hypofunction drives the therapeutic benefit of KCC2 functional enhancement.
Fig. 6. Effect of CLP290-mediated rescue of refractory seizures on post-ischemic KCC2 downregulation.
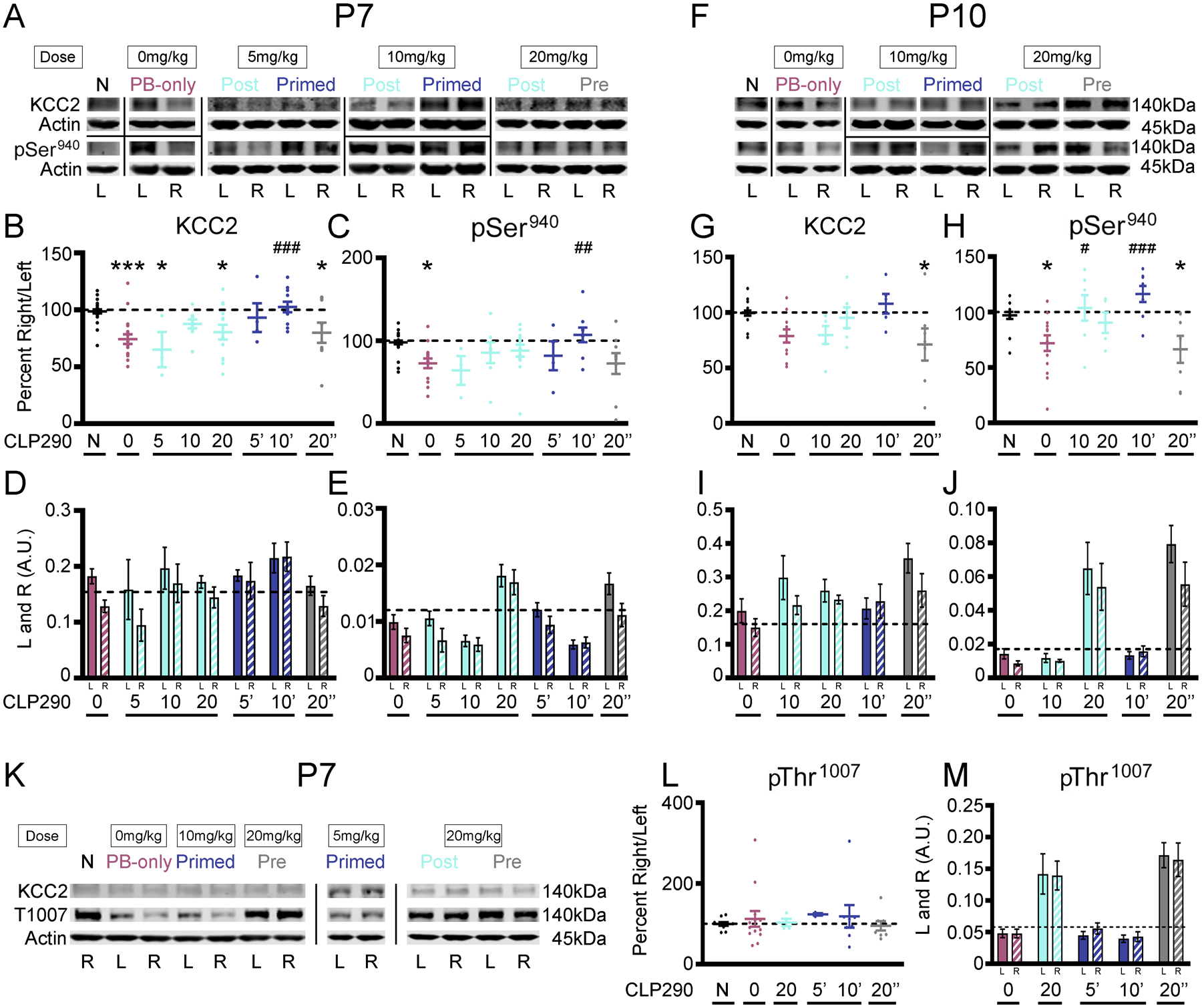
(A) Representative Western blots of KCC2 expression and Ser940 phosphorylation 24 hours after P7 ischemic seizures. N, naïve. (B) KCC2 and (C) Ser940 as percent ipsilateral/contralateral (right/left). (D) KCC2 and (E) Ser940 in left (“L”) and right (“R”) hemispheres. (F) Representative Western blots of KCC2 and Ser940 24 hours after P10 ischemic seizures. (G) KCC2 and (H) Ser940 as percent R/L. (I) KCC2 and (J) Ser940 in left and right hemispheres. (K) Representative western blot of KCC2 and Thr1007 24 hours after ischemic neonatal seizures. (L) Thr1007 phosphorylation as percent R/L. (M) Thr1007 phosphorylation in left and right hemispheres. *P<0.05, **P<0.01, and ***P<0.001 by one-way ANOVA vs. naive. #P<0.05, ##P<0.01 and ###P<0.001 vs. PB-Only. P7 pups: Naïve n=27, PB-only n=18, 5 Post n=3, 10 Post n=9, 20 Post n=13, 5’ n=4, 10’ n=11, 20 Pre n=9 pups. P10 pups: Naïve n=18, PB-only n=11, 10 Post n=6, 20 Post n=6, 10 Primed n=5, 20 Pre n=7 pups.
Ischemic neonatal seizures do not modulate KCC2-Thr1007 phosphorylation
The phosphorylation of KCC2 residue Thr1007 decreases KCC2 function (56, 57). It has been proposed that KCC2 functional enhancers must either increase KCC2 surface stability and/or decrease Thr1007 phosphorylation (58, 59). Therefore, Thr1007 phosphorylation was assessed 24 hours after P7 ischemic neonatal seizures (Fig. 6, K to M, and data file S1). Ischemia did not significantly change Thr1007 phosphorylation at 24 hours in either hemisphere (Fig. 6, K to M, and fig. S4B). The 10-min CLP290 dose also did not significantly modulate Thr1007 phosphorylation; however, bolus administration of 20 mg/kg CLP290 resulted in increased Thr1007 phosphorylation at 24 hours in both 20 mg/kg pre- and post-treatment groups (Fig. 6, K to M, and fig. S4B). Ischemic neonatal seizures at P7 did not significantly modulate the phosphorylation at the Thr1007 residue.
CLP290 rescue of PB-resistant seizures was not mediated through BDNF-TrkB signaling
Ischemic neonatal seizures at P7 induce a significant bilateral increase in TrkB expression and phosphorylation of Tyr816 (17, 28). Similarly here, TrkB expression and Tyr816 phosphorylation were upregulated bilaterally in the PB-only group 24 hours after ischemic neonatal seizures at P7 (figs. S5, A to E, and S6) but not at P10 (figs. S5, F to J, and S6). The bilateral increase in post-ischemic TrkB expression and Tyr816 phosphorylation was detected as previously characterized (17). CLP290 had no effect on the global increase in TrkB expression (fig. S5, B and D). Ipsilateral phosphorylation of the Tyr816 residue was modulated by ischemia at P7 (fig. S5C) and was rescued by CLP290 (fig. S5C). In this model, ischemic neonatal seizures at P10 were PB-responsive and P10 pups did not show activation of TrkB (fig. S5, G to J). These results suggest that the CLP290-mediated seizure suppression in the 10’ group at P7 was independent of TrkB.
In vitro CLP257 incubation increases KCC2 membrane insertion
CLP257 has been shown to increase KCC2 membrane expression, restore chloride extrusion capacity, and reduce the duration of ictal-like discharges in vitro (21, 47). CLP257 and its prodrug CLP290 have been shown to increase KCC2 membrane expression, restore impaired chloride homeostasis, hyperpolarize GABA equilibrium potentials, and improve outcomes in neuropsychiatric mouse models associated with KCC2 hypofunction (21, 54, 55, 60, 61). It has been suggested that the net effect of KCC2 functional enhancement by either CLP257 or CLP290 may emerge from relatively small changes in KCC2 function (11, 21, 62). Further, the neonatal brain tightly regulates KCC2 activity via Ser940 and Thr1007 phosphorylation (24), and it is unknown if age-dependent mechanisms affect KCC2 functional enhancement. Therefore, P7 neonatal brain sections were incubated with graded doses of CLP257 or CLP290 (Fig. 7, A to C). 500μM CLP257 significantly increased membrane KCC2 and Ser940 phosphorylation (Fig. 7B). Thr1007 phosphorylation at the membrane did not show significant modulation when treated with CLP257 (Fig 7B). The phosphorylation of Ser940 increases KCC2 plasma membrane insertion and transport activity (50, 63). To assess if the Ser940 site was necessary for CLP257 mediated KCC2 membrane insertion, brain sections from S940A+/+ knock-in mutant mice (26) were treated with CLP257 (Fig. 7, D to G). Without the ability to phosphorylate Ser940, CLP257 failed to increase KCC2 membrane insertion (Fig. 7F).
Fig. 7. In vitro CLP257 treatment upregulated membrane KCC2 expression and Ser940 phosphorylation.
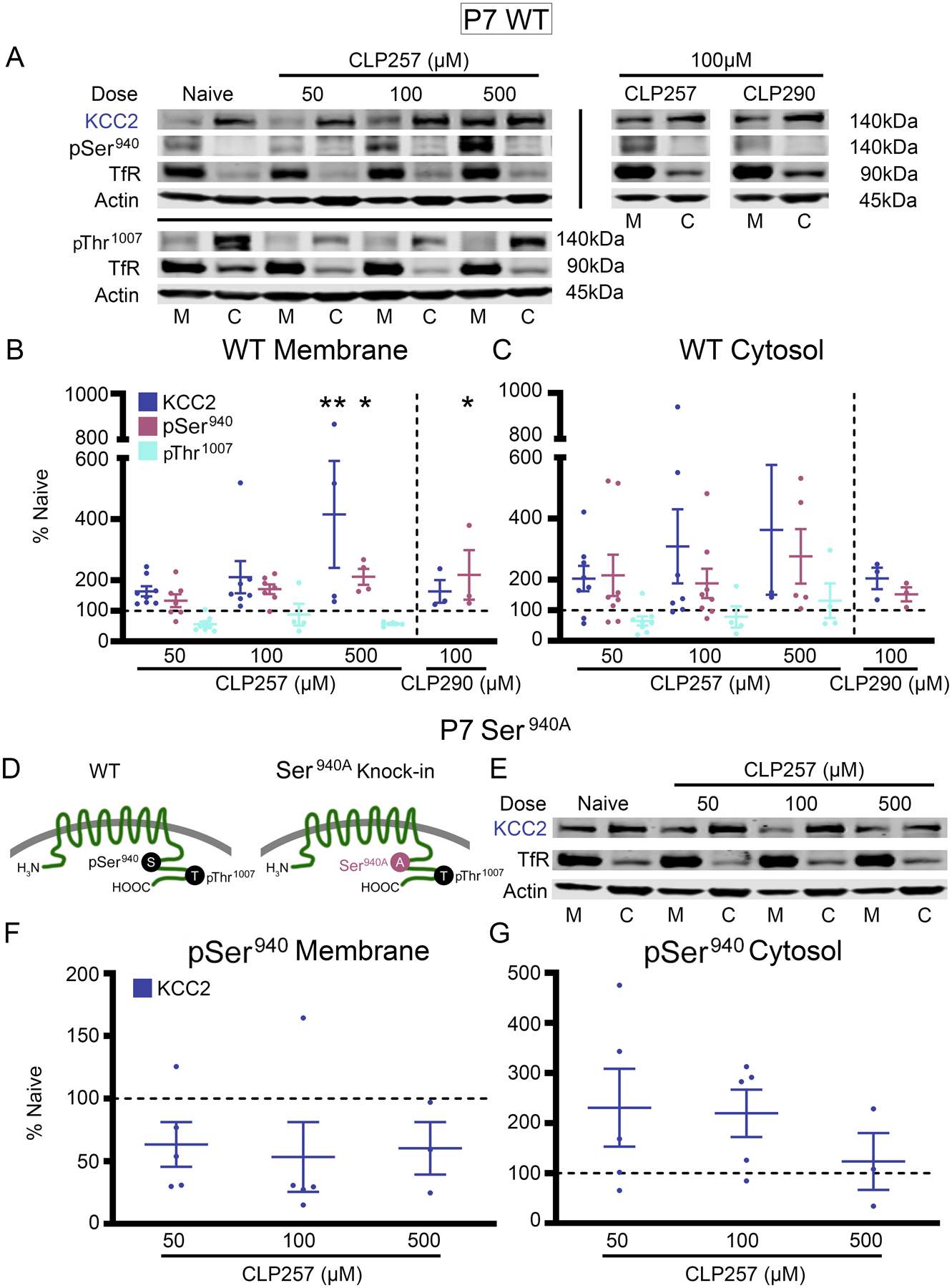
(A) KCC2, Ser940, and Thr1007 in the plasma membrane (M) and cytosol (C) for all treated P7 wild-type (WT) brain slices. (B) KCC2, Ser940, and Thr1007 in the membrane and (C) cytosol for all treatment groups plotted as percent of naïve. Number of WT P7 pups: n=8 (50 μM CLP257), n=7 (100 μM CLP257), n=4 (500 μM CLP257), n=3 (100μM CLP290). (D) Graphical representation of S940A+/+ knock-in mutant mice (41). (E to G) Western blotting for KCC2 expression in the plasma membrane and cytosol for all treated brain slices from S940A+/+ P7 pups (E). Quantified KCC2 expression in the membrane (F) and cytosol (G) for all S940A+/+ treatment groups plotted as percent of naïve. All proteins of interest in the cytosol were normalized to housekeeping protein β-actin; all proteins of interest in the plasma membrane were normalized to transferrin (TfR); and phosphoproteins were normalized to their respective total protein. Data plots show all data points with means ± SEM. * P<0.05 and ** P<0.01 by one-way ANOVA. S940A+/+ P7 pups: n=5 (50μM CLP257), n=5 (100μM CLP257), n=3 brain slices (500μM CLP257).
Discussion
KCC2 is a key regulator of neuronal intracellular chloride (7, 64) and is regulated by diverse mechanisms (11, 12). Acute enhancement of KCC2 membrane stability and function could reestablish synaptic inhibition in seizing neonatal brains when positive GABAR modulators, like PB, fail to curb severe recurrent seizures (15, 65). Documented KCC2 hypofunction, such as in postmortem cerebral samples from preterm infants with white matter injury (66), supports this approach. This acute strategy is distinct from chronic overexpression or enhancement of KCC2 function in neurons with stable endogenous KCC2 expression, which could be detrimental to developing brains (67, 68). Moderate and severe seizures due to hypoxic-ischemic encephalopathy are clinically associated with significant hourly or daily seizure burdens, which tend to cluster into high-seizing and non-seizing periods (4, 69); however, they are transient in nature. An acute protocol of rescuing KCC2 hypofunction during this critical period would help mitigate both the refractory neonatal seizures and their long-term consequences.
Currently, there are very few studies evaluating CLP290 efficacy in models of neurological disease, and there is an urgent need for the discovery of additional novel KCC2 functional enhancers. The De Koninck group originally identified CLP257 as a KCC2 functional enhancer using high-throughput screening (21). The prodrug CLP290 reportedly has improved pharmacokinetics over CLP257 and rescues neuropathic pain in a rat model, and multiple studies have shown that CLP290 improves outcomes in models of neuropathic pain and spinal cord injury associated with KCC2 hypofunction (21, 55, 61). In vitro studies have proposed that the active drug CLP257 does not directly modulate KCC2 activity but potentiates GABAAR activity (70, 71). Here, our results demonstrate that phosphorylation of the Ser940 residue is necessary for the CLP257-mediated increase in KCC2 expression at the membrane, as well as for the CLP290-mediated anti-seizure effect in vivo. These results suggest that the efficacy of CLP290 as a KCC2 functional enhancer is dependent upon the phosphorylation of Ser940. The identification of more efficient KCC2 functional enhancers targeting these phosphorylation sites is needed.
The mechanisms by which CLP290 enhances KCC2 function in neurons are not well understood (21, 70, 71). The ability of CLP290 to rescue both KCC2 expression and Ser940 phosphorylation 24 hours after ischemia in this study are similar to those reported with the TrkB antagonist ANA12 (17); however, our findings are also distinct, given that CLP290 had no effect on subduing the ischemia-induced activation of TrkB. Its ability to increase KCC2 membrane insertion in P7 brain sections and its inability to subdue ischemic seizures in the mutant KCC2 S940A+/+ pups support its role as a KCC2 functional enhancer acting through its Ser940 phosphorylation site. S940A+/+ pups not only showed an increase in seizure susceptibility to the P7 ischemic insult, but importantly we documented the occurrence of spontaneous epileptiform discharges in the naïve mutant pups at P7. S940A+/+ pups did not show the age-dependent (P7 vs. P10) differences in ischemic seizure susceptibility highlighting the developmental role of Ser940 phosphorylation in hyperexcitability of the immature brain. The significance of this finding is highlighted in recent reports of KCC2 mutations in early-onset epileptic syndromes (9), in which mutant KCC2 is associated with a reduction in Ser940 phosphorylation (42) and impaired Cl− extrusion (43, 72). The high seizure burdens and early mortality detected in S940A+/+ pups and low seizure susceptibility of the T1007A+/+ pups to the repeated dose PTZ challenge at P12, may further support the role of the two sites in the evolution of epileptogenesis following P7 neonatal seizures in the CD-1 model.
KCC2 can autoregulate its Cl− extrusion capacity using positive and negative modulators of its multiple phosphoregulation sites (24). Modest KCC2 hypofunction can play a substantial role in the emergence of neurological disorders (62). For example, KCC2-enhancing compounds rescue neurodevelopmental phenotypes in models of Rett syndrome (73), suggesting that genetic strategies to increase KCC2 expression should be compared to functional enhancers like CLP290 in future studies. Overexpression of KCC2 in neurons may not only be detrimental but may also trigger endogenous, negative-feedback homeostatic pathways. Notably, pushing KCC2 function beyond its physiological levels is reportedly difficult in simulation studies (11). The marked reduction in baseline seizure susceptibility and the upregulation of Thr1007 phosphorylation observed here in P7 mice after treatment with the higher dose (20 mg/kg) CLP290—in contrast to the responses at lower doses and in untreated mice—could suggest such a compensatory homeostatic mechanism of KCC2 activation by CLP290. The results here for CLP257-mediated Thr1007 phosphorylation in vitro versus in vivo may indicate a temporal regulation of the homeostatic response but needs further investigation.
Studies have shown that, during induced status epilepticus, reduced Cl− extrusion capacity and exacerbated activity-dependent Cl− loading can result in GABAergic transmission being ictogenic (meaning to generate a seizure) (14). Optogenetic stimulation of GABAergic interneurons in this status, epilepticus-like state enhances the epileptiform activity in a GABAAR dependent manner (74), indicating GABA-mediated depolarization. These findings support data both from our model and clinical reports wherein after an initial loading-dose of PB fails to curb seizures, additional PB doses do not help rescue the refractoriness (2, 75). Additionally, given the toxicity of high-dose PB on neonatal brains, such protocols may be counterproductive in both the short and long term (76–78).
KCC2 hypofunction is emerging as a substantial cause of impaired inhibitory neurotransmission underlying multiple neurological disorders (5, 52, 65). Notably, research into both refractory seizures and refractory spinal nerve pain has identified KCC2 hypofunction as a common underlying cause. Future studies should investigate chloride extrusion dependent and independent functions of KCC2 in neurological disease. In this study, we have shown that enhancing KCC2 function in a well-characterized preclinical model of refractory seizures can rescue not only the acute PB-refractoriness but also help mitigate epileptogenesis with early intervention. Our findings highlight the role of KCC2 hypofunction and its phosphoregulation in hypoxic-ischemic encephalopathy-related refractory seizures and supports the application and development of KCC2 enhancers specifically for the neonatal period.
Materials and Methods
Experimental paradigm and reagents
In CLP290 and CLP257 experiments, all pups (P7 and P10, regardless of treatment group) received a loading dose of PB (25 mg/kg) dissolved in 100% isotonic phosphate-buffered saline (PBS) delivered by intraperitoneal (i.p.) injection at 1 hour. All injections were administered using Hamilton syringes, and all drugs were prepared the day of the experiments. CLP290 and CLP257 were both dissolved in 45/55% 2-Hydroxypropyl-β-cyclodextrin (HPCD)/PBS with a pH between 7.2 and 7.5. Pups were assigned to the PB-only, post, primed, or pre-treatment groups (Fig. 1). PB-only treatment is defined by the single administration of the 25 mg/kg PB at 1 hour after unilateral carotid ligation without further intervention. Post-treatment (post) is defined by administration of 5, 10, or 20 mg/kg i.p. CLP290 (labeled 5, 10, or 20 in the figures) immediately following unilateral carotid ligation, and PB administration at 1 hour. Primed treatment is defined by the administration of 5, 10, or 20 mg/kg CLP290 (labeled 5’, 10’, or 20’ in the figures) 4h preceding unilateral carotid ligation, and another injection of the same dose immediately following unilateral carotid ligation with PB at 1 hour. Pre-treatment is defined by the single administration of 20 mg/kg CLP290 (labeled 20” in the figures) 4 hours before unilateral carotid ligation with PB at 1 hour after ligation. In experiments assessing CLP290 versus CLP257, P7 pups were administered 10 mg/kg CLP257 in a primed-treatment group with PB at 1 hour after unilateral carotid ligation.
For convenience, reagents are tabulated in the supplement (table S1).
Animals
All experimental procedures and protocols were conducted in compliance with guidelines by the Committee on the Ethics of Animal Experiments (Permit Number: A3272–01) and were approved by the Animal Care and Use of Committee of Johns Hopkins University. CD1 litters were purchased from Charles River Laboratories (Wilmington, MA.). Newly born CD-1 litters (n=10) were delivered with a dam at postnatal day three or four and allowed to acclimate. S940A+/+ and T906A/T1007A+/+ mice were a gift from the laboratory of Stephen J. Moss at Tufts University School of Medicine. Equivalent numbers of male and female pups were introduced into the study. All mice were housed on a 12-hour light-dark cycle with food and water provided ad libitum.
Unilateral carotid ligation
A comprehensive protocol for unilateral carotid ligation and neonatal video-EEG recordings has been published (80) At P7 or P10, animals were subjected to permanent unilateral ligation (without transection) of the right common carotid artery using 6–0 surgisilk (Fine Science Tools, BC Canada) under isoflurane anesthesia. The outer skin was closed with 6–0 monofilament nylon (Covidien, MA), and lidocaine was applied as local anesthetic. Animals were implanted with 3 subdermal EEG scalp electrodes: 1 recording and 1 reference overlying the bilateral parietal cortices, and 1 ground electrode overlying the rostrum. Wire electrodes (IVES EEG; Model # SWE-L25 –MA, IVES EEG solutions, USA) were implanted subdermally and fixed in position with cyanoacrylate adhesive (KrazyGlue). Pups recovered from anesthesia over a few minutes. Animals were tethered to a preamplifier within a recording chamber for 2 hours of continuous vEEG recording and were maintained at 36°C with heated isothermal pads. At the end of the recording session, sub-dermal electrodes were removed, and the pups were returned to the dam.
In vivo video-EEG recording and analyses
EEG recordings were acquired using Sirenia Acquisition software with synchronous video capture (Pinnacle Technology Inc.). Data acquisition was done with sampling rates of 400Hz that had a preamplifier gain of 100 and the filters of 0.5Hz high-pass and 50Hz low-pass. The data were scored by binning EEG in 10s epochs. Similar to our previous studies (15), seizures were defined as electrographic ictal events that consisted of rhythmic spikes of high amplitude, diffuse peak frequency of ≥7–8Hz (meaning peak frequency detected by automated spectral power analysis) lasting ≥6s (meaning longer than half of each 10s epoch). Short duration burst activity lasting <6s (brief runs of epileptiform discharges) was not included for seizure burden calculations, as in previous studies with the model. Mean time spent seizing for first hour baseline seizure burdencompared to the second hour post-PB seizure burden was quantified in seconds. Mean seizure suppression was calculated using Equation 1:
Mean ictal events and ictal durations (seconds/event) were calculated for 1sth vs. 2ndh.
CLP290 plasma and brain availability in neonatal mice
Standards for HPLC were created using CLP257 (MilliporeSigma, USA) and CLP290 (Yves De Koninck Lab). P7 and P10 naïve pups of both sexes were administered CLP290 IP as three treatment groups: 10mg/kg, 20mg/kg, or vehicle. After 4h pups were anesthetized with chloral hydrate (90 mg/ml; IP), and transcardiac blood samples (100μL) were collected. The same pups were transcardially perfused with ice cold PBS, and the whole fresh brains harvested. Brain samples were flash frozen in dry ice, homogenized using a sonicator, and stored at −80°C. Blood and brain samples from CLP290 treated and naïve pups were analyzed for CLP290 and CLP257 concentrations via HPLC using a C18 column and 10/90 organic/aqueous mobile phase.
VU 0463271
To assess the role of KCC2 inhibition in neonatal seizure severity at P7 after ligation, the potent and selective KCC2 inhibitor VU 0463271 (VU) was administered at 1 hour (0.5mg/kg; IP) in lieu of PB (Figure 1). VU was dissolved in 20/80% dimethyl sulfoxide (DMSO)/PBS solution. To assess to role of KCC2 antagonism in neonatal seizure occurrence, naïve P7 or P10 pups underwent vEEG with 0.25mg/kg VU administered at the start of recording in lieu of ligation and 0.5mg/kg VU administered at 1 hour in lieu of PB.
P12 PTZ Challenge
To investigate the long-term effects of CLP290 treatment, P12 pups underwent a 3-hour vEEG recording during a challenge with pentylenetetrazole (PTZ; dissolved in 100% PBS). At P7 pups were either naive, PB-only, or P7 CLP290 10’. These P7 pups then underwent a PTZ challenge at P12. All pups were administered PTZ at the beginning of the vEEG recording (20mg/kg; IP), at 1 hour (20mg/kg; IP), and at 2 hours (40mg/kg; IP).
P12-P15 Slice preparation
Coronal brain slices were prepared from CD-1 mice at P12-P15 that underwent either Naïve or PB-only treatment at P7. Pups were deeply anesthetized with isoflurane inhalation and decapitated. The brain was rapidly dissected out and immersed in ice-cold oxygenated cutting solution containing (in mM): 125 NaCl, 3 KCl, 1.25 NaH2PO4, 25 NaHCO3, 0.25 CaCl2, 10 MgSO4 and 11 glucose. After 1–2 min, the brain was glued onto the platform of a specimen syringe and embedded with 1.6% low-melting point agarose (type I-B), quickly chilled and transferred into a buffer tank filled with cutting solution. Coronal slices (350 μm) were cut using a VF-300 microtome (Precisionary Instruments, Inc., Greenville, NC) and separated from the embedding agarose. Slices were kept in oxygenated holding solution containing (in mM): 125 NaCl, 3 KCl, 1.25 NaH2PO4, 25 NaHCO3, 0.5 CaCl2, 5 MgSO4 and 11 glucose, at 34 °C for 45 min and thereafter at room temperature.
Field Potential Recordings
Extracellular field-potential recordings were conducted from layer II/III of the anterior (before hippocampus) and posterior (with hippocampus) cortices, referring to roughly plate 20 and plate 50 in the Paxinos and Watson mouse brain atlas, respectively. Low resistance glass pipettes (1–3 MΩ) filled with aCSF were used to record field potentials. A concentric bipolar stimulating electrode (FHC, Bowdoin, ME) was placed in “off-column” areas of layer IV (i.e., ~100 μm lateral to the hypothetical perpendicular line across the recording site) to deliver synaptic stimulation, through a stimulus generator (STG 4002, Multichannel Systems, MCS GmbH, Reutlingen, Germany). The threshold stimulation (T) was determined by the minimal current intensity to evoke a synaptic response. The input-out responses from 1T-6T were first performed in aCSF, and repeated 10 min after bath application of low concentration (1 μM) of GABAzine. Field potentials were recorded in non-clamp mode (I = 0) at a high gain (α=100), acquired at 10 kHz and low-pass filtered at 2 kHz, analyzed offline using the clampfit software (Molecular Devices, Sunnyvale, CA). All chemicals used in this study were purchased from Sigma-Aldrich (St. Louis, MO).
Western blot analysis at 24 hours post-ligation
All animals for immunochemical characterizations were anesthetized with chloral hydrate (90 mg/ml; IP) before being transcardially perfused with ice cold saline. The whole fresh brains were removed, the cerebellum was discarded, and the left and right hemispheres were separated. . Naïve CLP290 10’ treated mice (fig. S3) were harvested at 4 hours and underwent further microdissection after perfusion to isolate cortex. Brains were stored at −80°C in preparation for further processing. Brain tissue homogenates were made and suspended in TPER cell lysis buffer containing 10% protease/phosphatase inhibitor cocktail. Total protein amounts were measured using the Bradford protein assay (Bio-Rad, Hercules, CA, USA) at 570nm and the samples were diluted in 5x loading buffer for a concentration of 50μg of protein in 20μL. 20μL of protein samples were run on 4–20% gradient tris-glycine gels (Invitrogen, Gand Island, NY, USA) for 120min at 130V and were transferred onto nitrocellulose membranes overnight at 20V. After the transfer, the nitrocellulose membranes underwent a 1h blocking step in Rockland buffer before 6h incubation with primary antibodies (for all antibody RRIDS, see Table S1): mouse α-KCC2 (1:1000, Millipore), rabbit α-phospho-KCC2-S940 (1:1000 Aviva Systems Biology), rabbit α-phospho-KCC2-T1007 (1:1000; Phospho solutions) mouse α-TrkB (1:1000, BD Biosciences), rabbit α-phospho-TrkB-Y816 (1:500, Millipore), and mouse α-actin (1:10000, LI-COR Biosciences). Antibodies were validated as previously described (28). Membranes were then incubated with fluorescent secondary antibodies (1:5000, goat α-rabbit and goat α-mouse, Li-Cor Biosciences, USA). Chemiluminescent protein bands were analyzed using the Odyssey infrared imaging system 2.1 (LI-COR Biosciences). The optical density of each protein sample was normalized to their corresponding actin bands run on each lane for internal control. Mean normalized protein expression levels were then calculated for respective left and right hemispheres. The expression levels of the proteins of interest in ipsilateral hemispheres were normalized to the same in contralateral hemispheres for each pup to examine hemispheric percent change of protein expression.
Surface protein seperation
1mm coronal brain slices were obtained from P7 CD-1 mice and recovered for 45min at 34°C with oxygenation (95%/5% O2/CO2). After recovery, slices were incubated with CLP290 or CLP257 at 34°C with oxygenation for 40min. Slices were placed in TPER cell lysing buffer with HALT protease and phosphatase inhibitors and homogenized via sonication. After 30min incubation on ice, protein lysates were ultracentrifuged at 210,000xg (TLA-120.2 rotor, Beckman Coulter Life Sciences), and supernatants were collected as the cytosolic components. Pellets were resuspended in TPER/HALT buffer and ultracentrifuged; supernatant was discarded as wash fraction. Pellets were resuspended in TPER/HALT buffer and collected as the membrane components. Membrane and cytosolic components underwent Bradford analysis and Western blotting for protein quantification. Plasma membrane proteins were normalized to TfR. Cytosolic proteins were normalized to β-actin.
Statistical analysis
For all experiments, the quantification and analysis of data were performed blinded to the genotype, sex, and treatment conditions. All statistical tests were performed using GraphPad Prism software. Two-way analysis of variance (ANOVA) was performed with Tukey’s post hoc correction. One-way ANOVA was performed with Dunnett’s post hoc correction. Paired and unpaired t-tests were two-tailed. Survival analysis was performed by a Mantel-Cox test. Data are represented as bar graphs representing the mean, with dot plots representing each individual data point. Errors bars are ± 1 standard error of mean. P values ≤0.05 are reported by threshold, marked with asterisks and hashes that are defined in the legends.
Supplementary Material
Fig. 8: Summary.
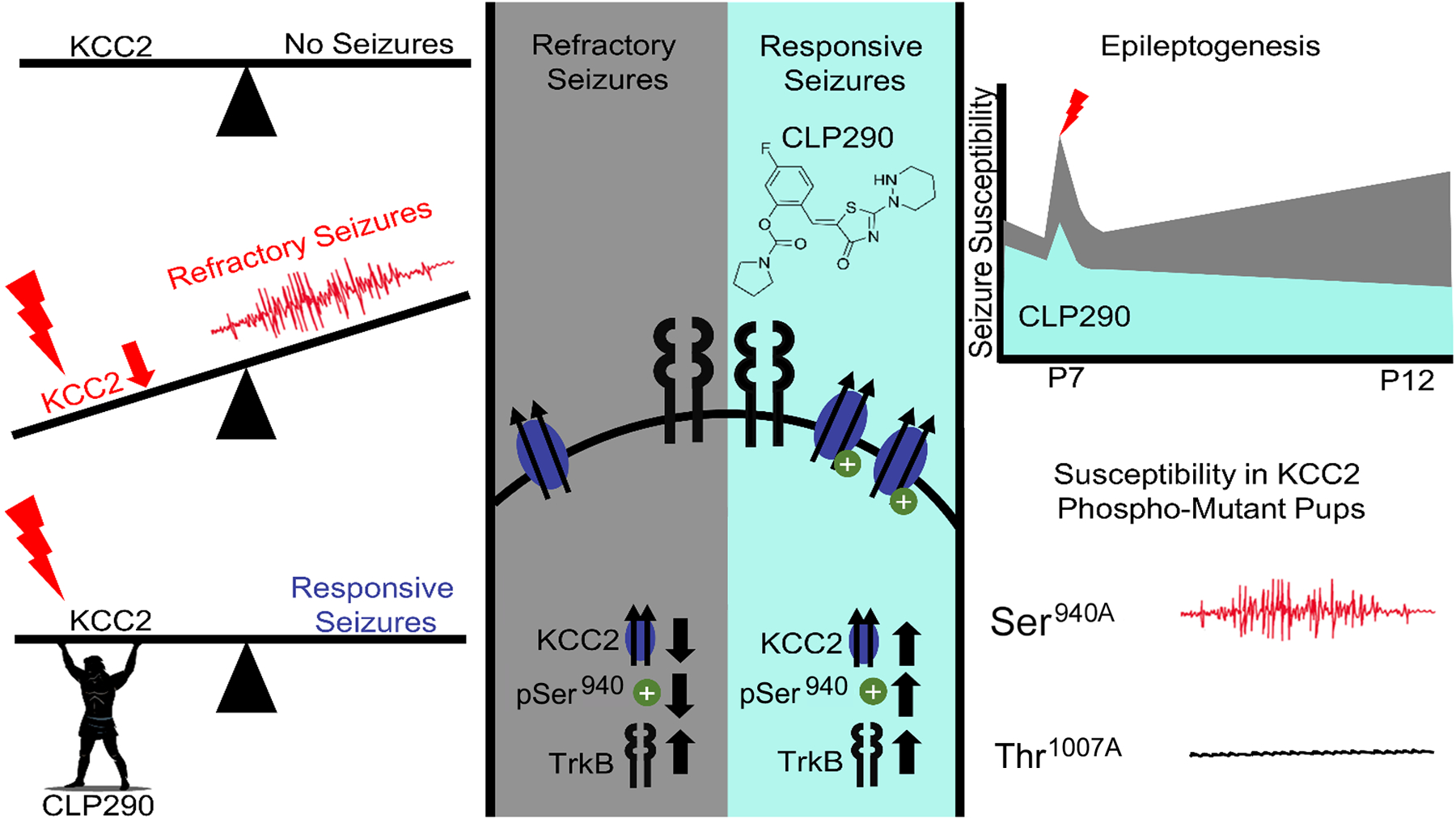
Targeting ischemia-induced KCC2 hypofunction with the KCC2 functional enhancer CLP290 rescued refractory neonatal seizures at P7 and mitigated epileptogenesis in same pups at P12.
Acknowledgements:
We thank Professor Stephen J. Moss (Tufts University School of Medicine) for the generous transfer of the S940A+/+ and T1007A+/+ mice. We thank Dr. Tarek Deeb (Tufts University School of Medicine) and Dr. Yvonne Moore (Tufts University School of Medicine) for useful discussions and technical advice. We thank Professor Yves De Koninck (Université Laval) for generously sharing aliquots of CLP290. We thank Dr. Rana Rais’s laboratory from the Johns Hopkins Drug Discovery Core for running the HPLC experiments. We thank the journal editors and reviewers.
Funding:
This work was supported by the Eunice Kennedy Shriver National Institute of Child Health and Human Development of National Institutes of Health under Grant No. R01HD090884 (SDK).
Footnotes
Competing interests: SDK is listed as an author on US patent 10525024B2, “Methods for rescuing phenobarbital resistance of seizures by ANA-12 or ANA-12 in combination with CLP290.”
Data and materials availability:
All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials. S940A+/+ and T1007A+/+ mice and CLP290 were obtained by materials transfer agreements.
References and Notes
- 1.Volpe J, Inder T, Darras B, de Vries L, du Plessis A, Neill J, Perlman J, Volpe’s Neurology of the Newborn - 6th Edition (Elsevier, ed. 6th, 2017). [Google Scholar]
- 2.Sankar R, Painter MJ, Neonatal seizures: after all these years we still love what doesn’t work. Neurology. 64, 776–777 (2005). [DOI] [PubMed] [Google Scholar]
- 3.Glass HC, Soul JS, Chu CJ, Massey SL, Wusthoff CJ, Chang T, Cilio MR, Bonifacio SL, Abend NS, Thomas C, Lemmon M, McCulloch CE, Shellhaas RA, Response to antiseizure medications in neonates with acute symptomatic seizures. Epilepsia. 60, e20–e24 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boylan GB, Stevenson NJ, Vanhatalo S, Monitoring neonatal seizures. Semin. Neonatal Med 18, 202–208 (2013). [DOI] [PubMed] [Google Scholar]
- 5.Kaila K, Price TJ, Payne JA, Puskarjov M, Voipio J, Cation-chloride cotransporters in neuronal development, plasticity and disease. Nat Rev Neurosci. 15, 637–654 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sedmak G, Jovanov-Milošević N, Puskarjov M, Ulamec M, Krušlin B, Kaila K, Judaš M, Developmental Expression Patterns of KCC2 and Functionally Associated Molecules in the Human Brain. Cereb. Cortex 26, 4574–4589 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Kaila K, Price TJ, Payne JA, Puskarjov M, Voipio J, Cation-chloride cotransporters in neuronal development, plasticity and disease. Nat. Rev. Neurosci 15, 637–654 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ben-Ari Y, Excitatory actions of GABA during development: the nature of the nurture. Nat Rev Neurosci. 3, 728–739 (2002). [DOI] [PubMed] [Google Scholar]
- 9.Duy PQ, David WB, Kahle KT, Identification of KCC2 Mutations in Human Epilepsy Suggests Strategies for Therapeutic Transporter Modulation. Front. Cell. Neurosci 13 (2019), doi: 10.3389/fncel.2019.00515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.French Jacqueline A, Refractory Epilepsy: Clinical Overview. Epilepsia. 48, 3–7 (2007). [DOI] [PubMed] [Google Scholar]
- 11.Doyon N, Vinay L, Prescott SA, De Koninck Y, Chloride Regulation: A Dynamic Equilibrium Crucial for Synaptic Inhibition. Neuron. 89, 1157–1172 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Sullivan BJ, Kadam SD, in Neuronal Chloride Transporters in Health and Disease, Tang X, Ed. (Elsevier, ed. 1, 2020), p. 650. [Google Scholar]
- 13.Nardou R, Yamamoto S, Chazal G, Bhar A, Ferrand N, Dulac O, Ben-Ari Y, Khalilov I, Neuronal chloride accumulation and excitatory GABA underlie aggravation of neonatal epileptiform activities by phenobarbital. Brain J. Neurol 134, 987–1002 (2011). [DOI] [PubMed] [Google Scholar]
- 14.Burman RJ, Selfe JS, Lee JH, van den Berg M, Calin A, Codadu NK, Wright R, Newey SE, Parrish RR, Katz AA, Wilmshurst JM, Akerman CJ, Trevelyan AJ, Raimondo JV, Excitatory GABAergic signalling is associated with benzodiazepine resistance in status epilepticus. Brain J. Neurol 142, 3482–3501 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kang SK, Markowitz GJ, Kim ST, Johnston MV, Kadam SD, Age- and sex-dependent susceptibility to phenobarbital-resistant neonatal seizures: role of chloride co-transporters. Front Cell Neurosci. 9, 173- (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kang SK, Johnston MV, Kadam SD, Acute TrkB inhibition rescues phenobarbital-resistant seizures in a mouse model of neonatal ischemia. Eur. J. Neurosci 42, 2792–2804 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carter BM, Sullivan BJ, Landers JR, Kadam SD, Dose-dependent reversal of KCC2 hypofunction and phenobarbital-resistant neonatal seizures by ANA12. Sci. Rep 8, 11987 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kang SK, Ammanuel S, Thodupunuri S, Adler DA, Johnston MV, Kadam SD, Sleep dysfunction following neonatal ischemic seizures are differential by neonatal age of insult as determined by qEEG in a mouse model. Neurobiol. Dis 116, 1–12 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kang SK, Ammanuel S, Adler DA, Kadam SD, Rescue of PB-resistant neonatal seizures with single-dose of small-molecule TrkB antagonist show long-term benefits. Epilepsy Res. 159, 106249 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sullivan BJ, Kadam SD, Brain-Derived Neurotrophic Factor in Neonatal Seizures. Pediatr. Neurol 118, 35–39 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gagnon M, Bergeron MJ, Lavertu G, Castonguay A, Tripathy S, Bonin RP, Perez-Sanchez J, Boudreau D, Wang B, Dumas L, Valade I, Bachand K, Jacob-Wagner M, Tardif C, Kianicka I, Isenring P, Attardo G, Coull JAM, De Koninck Y, Chloride extrusion enhancers as novel therapeutics for neurological diseases. Nat. Med 19, 1524–1528 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Glass HC, Grinspan ZM, Shellhaas RA, Outcomes after acute symptomatic seizures in neonates. Semin. Fetal. Neonatal Med 23, 218–222 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Kang SK, Kadam SD, Neonatal Seizures: Impact on Neurodevelopmental Outcomes. Front Pediatr. 3, 101- (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moore YE, Conway LC, Brandon NJ, Deeb TZ, Moss SJ, Developmental regulation of KCC2 phosphorylation has long-term impacts on cognitive function. Front. Mol. Neurosci 12 (2019), doi: 10.3389/fnmol.2019.00173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moore YE, Deeb TZ, Chadchankar H, Brandon NJ, Moss SJ, Potentiating KCC2 activity is sufficient to limit the onset and severity of seizures. Proc. Natl. Acad. Sci 115, 10166–10171 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Silayeva L, Deeb TZ, Hines RM, Kelley MR, Munoz MB, Lee HHC, Brandon NJ, Dunlop J, Maguire J, Davies PA, Moss SJ, KCC2 activity is critical in limiting the onset and severity of status epilepticus. Proc. Natl. Acad. Sci. U. S. A 112, 3523–3528 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Watanabe M, Zhang J, Mansuri MS, Duan J, Karimy JK, Delpire E, Alper SL, Lifton RP, Fukuda A, Kahle KT, Developmentally regulated KCC2 phosphorylation is essential for dynamic GABA-mediated inhibition and survival. Sci. Signal 12 (2019), doi: 10.1126/scisignal.aaw9315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kipnis PA, Sullivan BJ, Carter BM, Kadam SD, TrkB agonists prevent postischemic emergence of refractory neonatal seizures in mice. JCI Insight. 5 (2020), doi: 10.1172/jci.insight.136007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kharod SC, Kang SK, Kadam SD, Off-label use of bumetanide for brain disorders: An overview. Front. Neurosci 13 (2019), doi: 10.3389/fnins.2019.00310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Puskarjov M, Kahle KT, Ruusuvuori E, Kaila K, Pharmacotherapeutic targeting of cation-chloride cotransporters in neonatal seizures. Epilepsia. 55, 806–818 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Panayiotopoulos CP, Neonatal Seizures and Neonatal Syndromes (Bladon Medical Publishing, 2005; https://www.ncbi.nlm.nih.gov/books/NBK2599/). [PubMed] [Google Scholar]
- 32.Kossoff E, Neonatal Seizures Due to Hypoxic-Ischemic Encephalopathy: Should We Care? Epilepsy Curr. 11, 147–148 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kang SK, Ammanuel S, Thodupunuri S, Adler DA, Johnston MV, Kadam SD, Sleep dysfunction following neonatal ischemic seizures are differential by neonatal age of insult as determined by qEEG in a mouse model. Neurobiol. Dis 116, 1–12 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kharod SC, Carter BM, Kadam SD, Pharmaco-resistant neonatal seizures: critical mechanistic insights from a chemoconvulsant model. Dev. Neurobiol (2018), doi: 10.1002/dneu.22634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kadam SD, Dudek FE, Temporal progression of evoked field potentials in neocortical slices after unilateral hypoxia-ischemia in perinatal rats: Correlation with cortical epileptogenesis. Neuroscience. 316, 232–248 (2016). [DOI] [PubMed] [Google Scholar]
- 36.Hunt RF, Scheff SW, Smith BN, Posttraumatic epilepsy after controlled cortical impact injury in mice. Exp. Neurol 215, 243–252 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sutula TP, Dudek FE, Unmasking recurrent excitation generated by mossy fiber sprouting in the epileptic dentate gyrus: an emergent property of a complex system. Prog. Brain Res 163, 541–563 (2007). [DOI] [PubMed] [Google Scholar]
- 38.Wuarin J-P, Dudek FE, Electrographic Seizures and New Recurrent Excitatory Circuits in the Dentate Gyrus of Hippocampal Slices from Kainate-Treated Epileptic Rats. J. Neurosci 16, 4438–4448 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Woo N-S, Lu J, England R, McClellan R, Dufour S, Mount DB, Deutch AY, Lovinger DM, Delpire E, Hyperexcitability and epilepsy associated with disruption of the mouse neuronal-specific K–Cl cotransporter gene. Hippocampus. 12, 258–268 (2002). [DOI] [PubMed] [Google Scholar]
- 40.Hubner CA, Disruption of KCC2 reveals an essential role of K-Cl cotransport already in early synaptic inhibition. Neuron. 30, 515–524 (2001). [DOI] [PubMed] [Google Scholar]
- 41.Silayeva L, Deeb TZ, Hines RM, Kelley MR, Munoz MB, Lee HHC, Brandon NJ, Dunlop J, Maguire J, Davies PA, Moss SJ, KCC2 activity is critical in limiting the onset and severity of status epilepticus. Proc. Natl. Acad. Sci. U. S. A 112, 3523–3528 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kahle KT, Merner ND, Friedel P, Silayeva L, Liang B, Khanna A, Shang Y, Lachance-Touchette P, Bourassa C, Levert A, Dion PA, Walcott B, Spiegelman D, Dionne-Laporte A, Hodgkinson A, Awadalla P, Nikbakht H, Majewski J, Cossette P, Deeb TZ, Moss SJ, Medina I, Rouleau GA, Genetically encoded impairment of neuronal KCC2 cotransporter function in human idiopathic generalized epilepsy. EMBO Rep. 15, 766–774 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Puskarjov M, Seja P, Heron SE, Williams TC, Ahmad F, Iona X, Oliver KL, Grinton BE, Vutskits L, Scheffer IE, Petrou S, Blaesse P, Dibbens LM, Berkovic SF, Kaila K, A variant of KCC2 from patients with febrile seizures impairs neuronal Cl− extrusion and dendritic spine formation. EMBO Rep. 15, 723–729 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pisella LI, Gaiarsa J-L, Diabira D, Zhang J, Khalilov I, Duan J, Kahle KT, Medina I, Impaired regulation of KCC2 phosphorylation leads to neuronal network dysfunction and neurodevelopmental pathology. Sci. Signal 12 (2019), doi: 10.1126/scisignal.aay0300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nashef L, So EL, Ryvlin P, Tomson T, Unifying the definitions of sudden unexpected death in epilepsy. Epilepsia. 53, 227–233 (2012). [DOI] [PubMed] [Google Scholar]
- 46.Sivakumaran S, Cardarelli RA, Maguire J, Kelley MR, Silayeva L, Morrow DH, Mukherjee J, Moore YE, Mather RJ, Duggan ME, Brandon NJ, Dunlop J, Zicha S, Moss SJ, Deeb TZ, Selective Inhibition of KCC2 Leads to Hyperexcitability and Epileptiform Discharges in Hippocampal Slices and In Vivo. J. Neurosci 35, 8291–8296 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dzhala VI, Staley KJ, KCC2 chloride transport contributes to the termination of ictal epileptiform activity. eNeuro (2020), doi: 10.1523/ENEURO.0208-20.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rivera C, Voipio J, Thomas-Crusells J, Li H, Emri Z, Sipilä S, Payne JA, Minichiello L, Saarma M, Kaila K, Mechanism of Activity-Dependent Downregulation of the Neuron-Specific K-Cl Cotransporter KCC2. J. Neurosci 24, 4683–4691 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pathak HR, Weissinger F, Terunuma M, Carlson GC, Hsu F-C, Moss SJ, Coulter DA, Disrupted Dentate Granule Cell Chloride Regulation Enhances Synaptic Excitability during Development of Temporal Lobe Epilepsy. J. Neurosci 27, 14012–14022 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee HH, Deeb TZ, Walker JA, Davies PA, Moss SJ, NMDA receptor activity downregulates KCC2 resulting in depolarizing GABA(A) receptor mediated currents. Nat. Neurosci 14, 736–743 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rivera C, Voipio J, Thomas-Crusells J, Li H, Emri Z, Sipilä S, Payne JA, Minichiello L, Saarma M, Kaila K, Mechanism of Activity-Dependent Downregulation of the Neuron-Specific K-Cl Cotransporter KCC2. J. Neurosci 24, 4683–4691 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Barmashenko G, Hefft S, Aertsen A, Kirschstein T, Köhling R, Positive shifts of the GABAA receptor reversal potential due to altered chloride homeostasis is widespread after status epilepticus. Epilepsia. 52, 1570–1578 (2011). [DOI] [PubMed] [Google Scholar]
- 53.Akita T, Fukuda A, Intracellular Cl− dysregulation causing and caused by pathogenic neuronal activity. Pflüg. Arch. - Eur. J. Physiol 472, 977–987 (2020). [DOI] [PubMed] [Google Scholar]
- 54.Lizhnyak PN, Muldoon PP, Pilaka PP, Povlishock J, Ottens AK, Traumatic Brain Injury Temporal Proteome Guides KCC2-targeted Therapy. J. Neurotrauma (2019), doi: 10.1089/neu.2019.6415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen B, Li Y, Yu B, Zhang Z, Brommer B, Williams PR, Liu Y, Hegarty SV, Zhou S, Zhu J, Guo H, Lu Y, Zhang Y, Gu X, He Z, Reactivation of Dormant Relay Pathways in Injured Spinal Cord by KCC2 Manipulations. Cell. 174, 521–535.e13 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rinehart J, Maksimova YD, Tanis JE, Stone KL, Hodson CA, Zhang J, Risinger M, Pan W, Wu D, Colangelo CM, Forbush B, Joiner CH, Gulcicek EE, Gallagher PG, Lifton RP, Sites of regulated phosphorylation that control K-Cl cotransporter activity. Cell. 138, 525–536 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Titz S, Sammler EM, Hormuzdi SG, Could tuning of the inhibitory tone involve graded changes in neuronal chloride transport? Neuropharmacology. 95, 321–331 (2015). [DOI] [PubMed] [Google Scholar]
- 58.Conway LC, Cardarelli RA, Moore YE, Jones K, McWilliams LJ, Baker DJ, Burnham MP, Bürli RW, Wang Q, Brandon NJ, Moss SJ, Deeb TZ, N-Ethylmaleimide increases KCC2 cotransporter activity by modulating transporter phosphorylation. J. Biol. Chem 292, 21253–21263 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Moore YE, Kelley MR, Brandon NJ, Deeb TZ, Moss SJ, Seizing Control of KCC2: A New Therapeutic Target for Epilepsy. Trends Neurosci. 40, 555–571 (2017). [DOI] [PubMed] [Google Scholar]
- 60.Ferrini F, Lorenzo L-E, Godin AG, Quang ML, De Koninck Y, Enhancing KCC2 function counteracts morphine-induced hyperalgesia. Sci. Rep 7, 3870 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mapplebeck JCS, Lorenzo L-E, Lee KY, Gauthier C, Muley MM, De Koninck Y, Prescott SA, Salter MW, Chloride Dysregulation through Downregulation of KCC2 Mediates Neuropathic Pain in Both Sexes. Cell Rep. 28, 590–596.e4 (2019). [DOI] [PubMed] [Google Scholar]
- 62.Doyon N, Prescott SA, De Koninck Y, Mild KCC2 Hypofunction Causes Inconspicuous Chloride Dysregulation that Degrades Neural Coding. Front. Cell. Neurosci 9 (2016), doi: 10.3389/fncel.2015.00516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lee HHC, Walker JA, Williams JR, Goodier RJ, Payne JA, Moss SJ, Direct Protein Kinase C-dependent Phosphorylation Regulates the Cell Surface Stability and Activity of the Potassium Chloride Cotransporter KCC2. J. Biol. Chem 282, 29777–29784 (2007). [DOI] [PubMed] [Google Scholar]
- 64.Gamba G, Molecular Physiology and Pathophysiology of Electroneutral Cation-Chloride Cotransporters. Physiol. Rev 85, 423–493 (2005). [DOI] [PubMed] [Google Scholar]
- 65.Johne M, Römermann K, Hampel P, Gailus B, Theilmann W, Ala-Kurikka T, Kaila K, Löscher W, Phenobarbital and midazolam suppress neonatal seizures in a noninvasive rat model of birth asphyxia, whereas bumetanide is ineffective. Epilepsia (2020), doi: 10.1111/epi.16778. [DOI] [PubMed] [Google Scholar]
- 66.Robinson S, Mikolaenko I, Thompson I, Cohen ML, Goyal M, Loss of Cation-Chloride Cotransporter Expression in Preterm Infants With White Matter Lesions: Implications for the Pathogenesis of Epilepsy. J. Neuropathol. Exp. Neurol 69, 565–572 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kahle KT, Staley KJ, Nahed BV, Gamba G, Hebert SC, Lifton RP, Mount DB, Roles of the cation-chloride cotransporters in neurological disease. Nat Clin Pr. Neuro 4, 490–503 (2008). [DOI] [PubMed] [Google Scholar]
- 68.Hamidi S, Avoli M, KCC2 function modulates in vitro ictogenesis. Neurobiol. Dis 79, 51–58 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Boylan GB, Pressler RM, Neonatal seizures: the journey so far. Semin. Neonatal Med 18, 173–174 (2013). [DOI] [PubMed] [Google Scholar]
- 70.Cardarelli RA, Jones K, Pisella LI, Wobst HJ, McWilliams LJ, Sharpe PM, Burnham MP, Baker DJ, Chudotvorova I, Guyot J, Silayeva L, Morrow DH, Dekker N, Zicha S, Davies PA, Holenz J, Duggan ME, Dunlop J, Mather RJ, Wang Q, Medina I, Brandon NJ, Deeb TZ, Moss SJ, The small molecule CLP257 does not modify activity of the K+-Cl− co-transporter KCC2 but does potentiate GABAA receptor activity. Nat. Med 23, 1394–1396 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gagnon M, Bergeron MJ, Perez-Sanchez J, Plasencia-Fernández I, Lorenzo L-E, Godin AG, Castonguay A, Bonin RP, De Koninck Y, Reply to The small molecule CLP257 does not modify activity of the K + –Cl − co-transporter KCC2 but does potentiate GABA A receptor activity. Nat. Med 23, 1396–1398 (2017). [DOI] [PubMed] [Google Scholar]
- 72.Saitsu H, Watanabe M, Akita T, Ohba C, Sugai K, Ong WP, Shiraishi H, Yuasa S, Matsumoto H, Beng KT, Saitoh S, Miyatake S, Nakashima M, Miyake N, Kato M, Fukuda A, Matsumoto N, Impaired neuronal KCC2 function by biallelic SLC12A5 mutations in migrating focal seizures and severe developmental delay. Sci. Rep 6, 30072 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tang X, Drotar J, Li K, Clairmont CD, Brumm AS, Sullins AJ, Wu H, Liu XS, Wang J, Gray NS, Sur M, Jaenisch R, Pharmacological enhancement of KCC2 gene expression exerts therapeutic effects on human Rett syndrome neurons and Mecp2 mutant mice. Sci. Transl. Med 11, eaau0164 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Magloire V, Cornford J, Lieb A, Kullmann DM, Pavlov I, KCC2 overexpression prevents the paradoxical seizure-promoting action of somatic inhibition. Nat. Commun 10, 1225 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Slaughter LA, Patel AD, Slaughter JL, Pharmacological Treatment of Neonatal Seizures: A Systematic Review. J. Child Neurol 28, 351–364 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Markowitz GJ, Kadam SD, Smith DR, Johnston MV, Comi AM, Different effects of high- and low-dose phenobarbital on post-stroke seizure suppression and recovery in immature CD1 mice. Epilepsy Res. 94, 138–148 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Farwell JR, Lee YJ, Hirtz DG, Sulzbacher SI, Ellenberg JH, Nelson KB, Phenobarbital for Febrile Seizures — Effects on Intelligence and on Seizure Recurrence. N. Engl. J. Med 322, 364–369 (1990). [DOI] [PubMed] [Google Scholar]
- 78.Sulzbacher S, Farwell JR, Temkin N, Lu AS, Hirtz DG, Late cognitive effects of early treatment with phenobarbital. Clin. Pediatr. (Phila.) 38, 387–394 (1999). [DOI] [PubMed] [Google Scholar]
- 79.Tang X, Neuronal Chloride Transporters in Health and Disease (Elsevier, ed. 1, 2020). [Google Scholar]
- 80.Sullivan BJ, Kadam SD, in Experimental and Translational Methods to Screen Drugs Effective Against Seizures and Epilepsy (Humana, New York, NY, 2021), Neuromethods book series. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials. S940A+/+ and T1007A+/+ mice and CLP290 were obtained by materials transfer agreements.