

Abstract
One of the most powerful and commonly used approaches for detecting local adaptation in the genome is the identification of extreme allele frequency differences between populations. In this article, we present a new maximum likelihood method for finding regions under positive selection. It is based on a Gaussian approximation to allele frequency changes and it incorporates admixture between populations. The method can analyze multiple populations simultaneously and retains power to detect selection signatures specific to ancestry components that are not representative of any extant populations. Using simulated data, we compare our method to related approaches, and show that it is orders of magnitude faster than the state-of-the-art, while retaining similar or higher power for most simulation scenarios. We also apply it to human genomic data and identify loci with extreme genetic differentiation between major geographic groups. Many of the genes identified are previously known selected loci relating to hair pigmentation and morphology, skin, and eye pigmentation. We also identify new candidate regions, including various selected loci in the Native American component of admixed Mexican-Americans. These involve diverse biological functions, such as immunity, fat distribution, food intake, vision, and hair development.
Keywords: positive selection, admixture, population structure, human evolution, selective sweeps
Introduction
The emergence of population genomic data has facilitated fine-scale detection of regions under recent positive selection in humans and other species. There are multiple different methods for carrying out such selection scans. Some of these rely on patterns of long-range linkage disequilibrium (Voight et al. 2006; Sabeti et al. 2007), one of the characteristic genomic footprints left by a selective sweep (Kim and Stephan 2002; Kim and Nielsen 2004; McVean 2007). However, this pattern fades rapidly over time, and these methods are, consequently, best suited for detecting very recent selective sweeps from de novo mutations. Other techniques, based on distortions in the allele frequency spectrum caused by positive selection, can allow for the detection of more ancient events, but are generally only applicable to one population at a time (Tajima 1989; Fu and Li 1993; Fay and Wu 2000; Nielsen 2005; DeGiorgio et al. 2016; Huber et al. 2016).
A different class of methods for detecting selection involves analyzing patterns of allele frequency differentiation between populations. The basic idea is that regions that have experienced episodes of positive selection will display frequency differences between populations that are stronger than what would be expected under pure genetic drift. For example, one can compute Wright’s fixation index (FST) locally across different regions of a genome and look for extreme outliers (Beaumont and Nichols 1996; Akey et al. 2002; Beaumont and Balding 2004). Population differentiation methods can detect more ancient selective events than linkage disequilibrium-based methods (Sabeti et al. 2006), and are sensitive to different types of positive selection events, including sweeps from a de novo mutation, sweeps from standing variation, incomplete sweeps, and adaptive introgression (Bonhomme et al. 2010; Yi et al. 2010; Fumagalli et al. 2015; Racimo et al. 2017). Recent methods have allowed researchers to detect excess local differentiation on particular branches of a three-population tree (Yi et al. 2010; Racimo 2016), a four-population tree (Cheng, Xu, et al. 2017), or an arbitrarily large tree (Librado and Orlando 2018), albeit without modeling postsplit admixture events.
A generalization of these approaches was developed by Coop et al. (2010), Günther and Coop (2013), and Gautier (2015). It involves the detection of genomically local distortions from a genome-wide covariance matrix, which is used as a neutral baseline. An advantage of this approach is that one can apply it to an arbitrary number of populations. Other researchers have used hierarchical Bayesian models (Foll and Gaggiotti 2008; Foll et al. 2014) or principal component analysis (Duforet-Frebourg et al. 2016; Luu et al. 2017) to model patterns of population differentiation to identify local distortions across the genome. Another method extended single-locus differentiation-based methods to the analysis of haplotype differentiation (Fariello et al. 2013). More recently, Mathieson et al. (2015) developed an admixture-aware selection test based on a linear model and applied it to human data. The analysis took advantage of the fact that present-day European populations could be modeled as a mixture of three highly differentiated ancestral components. Regions of the genome that exhibited strong deviations from the genome-wide mixture proportions were therefore strong candidates for positive selection. Finally, Refoyo-Martínez et al. (2019) developed a method to test for selection on an admixture graph, which represents the history of divergence and admixture events among populations. Although useful for detecting selection in the presence of admixture, it still requires the user to specify which individuals belong to which populations, and to infer the graph in advance.
Here, we introduce a new selection detection framework that can explicitly model admixture and detect selection from populations of admixed ancestries. It can simultaneously compare arbitrarily many populations and ancestry components and is encoded in a flexible framework for testing selection on a specific lineage or set of lineages. The method allows the user to identify signals of positive selection via population differentiation, without relying on self-reported ancestry or admixture correction to group individuals into populations. The method can also determine if a selective event is specific to a particular population or shared among different populations.
Unlike previous methods, we fully take advantage of admixed populations, and we do not require the user to a priori categorize samples into populations, or to correct allele frequencies to account for recent admixture. Thus, the selection scan does not rely on user-supplied sample labels or ancestry compositions. These methods identify positive selection by searching for loci showing distortions in the population covariance matrix, relative to the genome-wide baseline. It provides a flexible framework to specifically test for selection on individual components or sets of components. This functionality allows researchers to accommodate specific evolutionary scenarios into the range of testable hypotheses, including local adaptation, adaptive introgression, and convergent selection. The method first coestimates the population structure of the input panel and the allele frequencies of the ancestral admixture components through an unsupervised learning process (Cheng, Mailund, et al. 2017), before testing for selection on the ancestral components themselves. Researchers can also use the method to examine estimated population structure and visualize trees connecting the ancestral components using plotting functionalities provided by our software package, Ohana, as part of the analysis pipeline.
Results
Simulations
Power to Detect Selection
We first evaluated the performance of our method against comparable methods in detecting whether a locus has evolved under positive selection (fig. 1a–c). For all tests, we use the empirical null distribution to find the threshold associated with 5% false positive rate (FPR). We compute power as the proportion of simulations with test statistic exceeding this threshold. For all three methods, the test statistic was the site statistic with the maximum value across the whole 2 Mbp locus; with Ohana, the test statistic used was the log-likelihood ratio (testing for selection in the specified ancestry group); with BayPass, the test statistic was the “XtX” statistic; and with pcadapt, the test statistic was the selection test P value, using K = 3 PCs (because there are four ancestral populations in each case). For all three methods, power increased uniformly with the value of the selection coefficient (fig. 1a–c). However, different demographic scenarios result in different power levels; for example, all methods were better-powered under the simple demographic scenario (model 1, figs. 1a and 2a) compared with selection preadmixture (model 1, figs. 1b and 2b) or in the human demographic model (model 3, figs. 1c and 2c).
Fig. 1.

Simulation tests of Ohana performance and efficiency in detecting and mapping selected sites. (a–c) Power to detect selection relative to two comparable methods, BayPass and pcadapt. Error bars are 95% CIs; (d–f) efficacy of Ohana to fine-map the causal site; (g) computational efficiency compared with that of BayPass. Error bars are 5th to 95th percentiles.
Fig. 2.

Illustration of simulation models. (a) Model 1, a basic model of four-population split with no admixture. (b) Model 2, a four-population split with subsequent admixture. (c) Model 3, a four-population model mimicking human demographic models. Population size changes in model 3 are omitted from the visualization for simplicity. Selection is simulated to operate on the branch that has a larger width.
pcadapt was significantly less well-powered than Ohana in most scenarios, for example, 72% versus 88% and 90% versus 98% power under moderate and strong selection, respectively (see fig. 1a). In most scenarios, we found Ohana to have power equivalent to or greater than that of BayPass; for example, in models 1 and 2, we fail to reject that Ohana and BayPass have different power curves with 95% confidence (fig. 1a and b). However, in one simulation scenario, we found Ohana had significantly higher power than Baypass, under a model of human demography (fig. 1c). However, we note that although BayPass and Ohana have similar power, Ohana’s test is by design more specific, as it is testing for selection in a specified ancestry group; hence power calculations are inherently more lenient for BayPass than Ohana. In figure 1a–c, we show results assuming , and present full results illustrating the entire ROC curve (i.e., not conditioned on FPR = 0.05) and for other values of f, in figure 3 and supplementary figures S1 and S2, Supplementary Material online. Also note the overall low power in figure 3. The main reason for this low power is that selection is acting in a relatively short period in the past, and that the population has experienced 50% admixture after selection. The strong admixture after selection tends to obscure much of the selection signal.
Fig. 3.

ROC curves for Ohana versus state-of-the-art methods, assessed using simulations with various values of the initial allele frequency at the beginning of selection (f) and different selection coefficients (s). Here the demographic model used was our human model (with selection in the Native American lineage), versus other demographic models considered (i.e., basic tree without and with admixture, supplementary figs. S1 and S2, Supplementary Material online, respectively).
Efficacy for Fine-Mapping the Causal Site
We also considered the performance of Ohana for fine mapping the position of the causal site (fig. 1d–f). We considered the distribution of the distance between the site of the test statistics described above (i.e., the locus-wide max statistic) and the causal site (in the center of the 2 Mbp locus). We plot the empirical cumulative distribution of these distances for different values of the selection coefficient under each demographic model. We found that in cases where Ohana is well-powered to detect selection, there is considerable power to narrow the position of the causal site down to ±10 kb of the max test statistic (e.g., >40% power for under model 1 and power for s = 0.05 under models 2 and 3; see fig. 1d–f). Interestingly, under models 1 and 2, there is similar power to fine-map sites with moderate and strong selection (s = 0.02 vs. 0.05, see fig. 1d and e); in contrast, under model 3, there is significantly higher power to fine-map sites with strong selection (fig. 1f); this dramatic difference may be due to the effects of demography on the pattern of hitchhiking surrounding the causal site. In figure 1d–f, we show results assuming and present full results illustrating other values of f in supplementary figures S3–S5, Supplementary Material online.
Computational Efficiency
In addition to comparing power to detect selection, we compared computational efficiency of Ohana and BayPass, which we showed in previous sections was the most competitive method in terms of statistical power (fig. 1g). We found that Ohana was >250× faster than BayPass (mean selection scan runtimes: Ohana, 0.626 s [±0.008 s]; BayPass, 168 s [±2 s]; N = 1,000 replicates). We reiterate that our power comparison revealed Ohana to generally have comparable power to that of BayPass, despite multiple orders of magnitude improvement in computational efficiency.
Analysis of Real Data
We identified regions in the genome that are likely to have been under the influence of positive selection using a merged data set containing several population panels from phase 3 of the 1000 Genomes Project (1000 Genomes Project Consortium et al. 2015). We randomly selected 64 genomes from each of four populations from the 1000 Genomes project: the British from Great Britain (GBR), the Han Chinese from Beijing (CHB), the Yoruba Africans (YRI), and the admixed Mexican-Americans from Los Angeles (MXL) (the number 64 was chosen because it was the size of the smallest panel). We only included variable sites with no missing data and a minimum allele frequency of 0.05 across the entire merged panel. In total, we analyzed 5,601,710 variable sites across the autosomal genome. We inferred genome-wide allele frequencies and covariances for the latent ancestry components as described in Materials and Methods section, using K = 4. To scan for covariance outliers, we performed four hypothesis-driven scans, in which we specifically searched for selection separately in each of the four inferred ancestry components in our data set (fig. 4 and table 1).
Fig. 4.
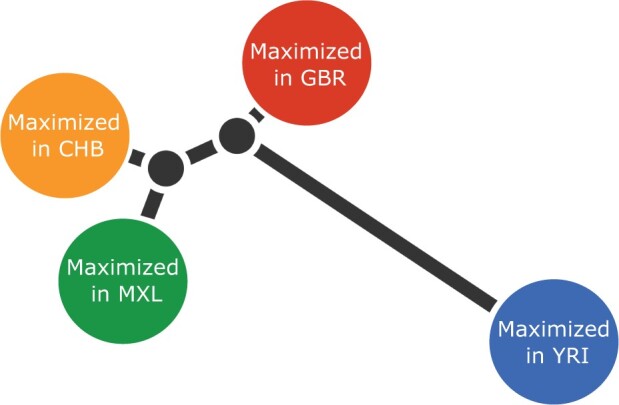
Inferred unrooted tree of latent ancestry components for the analysis including the CHB, YRI, MXL, and GBR genomic panels. We label each component by the population in which it is maximized, but emphasize that the components and the populations are not equivalent entities.
Table 1.
Top Ten Most Differentiated SNPs from Each of the Ancestry-Specific Scans.
| Chr | Pos | Rsid | LLRS | Target Ancestry | Nearest Gene |
|---|---|---|---|---|---|
| 5 | 33951693 | rs16891982 | 22.085902 | European | SLC45A2 |
| 15 | 48426484 | rs1426654 | 19.707464 | European | SLC24A5 |
| 15 | 28356859 | rs1129038 | 19.290553 | European | HERC2 |
| 15 | 28495956 | rs12912427 | 18.270213 | European | HERC2 |
| 9 | 16792200 | rs10962596 | 15.819739 | European | BNC2 |
| 1 | 1385211 | rs1312568 | 15.066101 | European | ATAD3C |
| 2 | 136407479 | rs1446585 | 14.957582 | European | R3HDM1 |
| 2 | 136616754 | rs182549 | 14.629386 | European | MCM6 |
| 1 | 204784969 | rs3940119 | 14.393216 | European | NFASC |
| 4 | 38798648 | rs5743618 | 14.38681 | European | TLR1 |
| 16 | 48258198 | rs17822931 | 23.271759 | CHB | ABCC11 |
| 16 | 48375777 | rs6500380 | 22.474103 | CHB | LONP2 |
| 1 | 234635790 | rs2175591 | 20.95541 | CHB | TARBP1 |
| 4 | 100142780 | rs75721934 | 20.453247 | CHB | LOC100507053 |
| 11 | 61579427 | rs72643557 | 20.114033 | CHB | FADS1 |
| 11 | 120154631 | rs12224052 | 19.696284 | CHB | POU2F3 |
| 21 | 43974948 | rs228088 | 19.518001 | CHB | SLC37A1 |
| 11 | 133043841 | rs79802711 | 19.157192 | CHB | OPCML |
| 5 | 128016573 | rs79478220 | 18.476104 | CHB | FBN2 |
| 19 | 51441759 | rs11084040 | 18.158963 | CHB | KLK5 |
| 14 | 46745012 | rs140736443 | 32.730697 | Native American | LINC00871 |
| 9 | 82968379 | rs6559543 | 27.584847 | Native American | LINC01507 |
| 16 | 80619307 | rs2316155 | 27.399123 | Native American | LINC01227 |
| 14 | 21647765 | rs77549780 | 27.355769 | Native American | LINC00641 |
| 12 | 14189549 | rs12425115 | 25.867367 | Native American | GRIN2B |
| 10 | 8150713 | rs10508343 | 25.609772 | Native American | GATA3 |
| 15 | 34936250 | rs16959274 | 25.424824 | Native American | GOLGA8B |
| 8 | 4490837 | rs71523639 | 24.59957 | Native American | CSMD1 |
| 1 | 14301862 | rs72640512 | 24.455822 | Native American | PRDM2 |
| 12 | 29817716 | rs12580697 | 23.967094 | Native American | TMTC1 |
| 8 | 145639681 | rs1871534 | 11.906794 | Yoruba/Ancestral Non-African | SLC39A4 |
| 5 | 178626609 | rs6869589 | 11.541667 | Yoruba/Ancestral Non-African | ADAMTS2 |
| 15 | 29427400 | rs10152250 | 11.48232 | Yoruba/Ancestral Non-African | FAM189A1 |
| 1 | 1106112 | rs6670693 | 11.447873 | Yoruba/Ancestral Non-African | TTLL10 |
| 4 | 3666494 | rs58827274 | 11.341367 | Yoruba/Ancestral Non-African | LOC100133461 |
| 17 | 2631985 | rs4790359 | 11.118134 | Yoruba/Ancestral Non-African | PAFAH1B1 |
| 9 | 136769888 | rs2789823 | 11.031687 | Yoruba/Ancestral Non-African | VAV2 |
| 6 | 169656029 | rs6930377 | 10.824098 | Yoruba/Ancestral Non-African | THBS2 |
| 17 | 29350769 | rs8073072 | 10.794224 | Yoruba/Ancestral Non-African | RNF135 |
| 5 | 173642871 | rs10067518 | 10.787147 | Yoruba/Ancestral Non-African | HMP19 |
LLRS = log-likelihood ratio score for positive selection.
After running these scans, we queried the CADD server (Rentzsch et al. 2019) to obtain functional, conservation, and regulatory annotations for the top candidate SNPs, including SIFT (Sim et al. 2012), PolyPhen (Adzhubei et al. 2013), GERP (Davydov et al. 2010), PhastCons (Siepel et al. 2005), PhyloP (Pollard et al. 2010), and Segway (Hoffman et al. 2012) annotations, so as to find the changes most likely to be disruptive. We discuss some of these below. We also queried the GTEx cis-eQTL database (Lonsdale et al. 2013), the UK Biobank GeneATLAS (Canela-Xandri et al. 2018), and the GWAS catalog (MacArthur et al. 2017), to look for trait-associated SNPs. We particularly focus on SNPs that have both high log-likelihood ratios in favor of positive selection (LLRS > 15) and high CADD scores in favor of functional disruption (>10).
Below, we describe some of the top SNPs with high LLRS and their surrounding regions, for those cases in which available genic, expression, or regulatory information can provide us some clue as to the possible organismal function that may have been affected by the selective event. We particularly focus on the Native American ancestry scan (fig. 5), as few selection scans have been performed in this population, but also briefly summarize the results from the other scans.
Fig. 5.

Top 5 annotated peaks in each of the ancestry-specific selection studies. MXL-specific = scan for selection in Native American ancestry of MXL. GBR-specific = scan for selection in European ancestry of GBR. CHB-specific = scan for selection in CHB ancestry of CHB. YRI-specific = scan for selection in Yoruba African ancestry or ancestral non-African ancestry. We analyzed 5,601,710 variable sites across the autosomal genomes. We inferred genome-wide allele frequencies and covariances as described in Materials and Methods section. We applied a likelihood model for each SNP by rescaling all variances and covariances by a scalar multiplier α. Descriptions of each candidate region are in table 1. LLR, log-likelihood ratio score.
European Ancestry Scan
Results for the top 30 loci in the European ancestry scan are presented in supplementary table S1, Supplementary Material online. Most loci have been previously shown to be under selection in Europeans populations, including SLC45A2, SLC24A5, BNC2, the OCA2/HERC2 region, the LCT/MCM6 region, and the TLR region (Bersaglieri et al. 2004; Voight et al. 2006; Barreiro et al. 2009; Vernot and Akey 2014; Mathieson et al. 2015). We notice that, in several cases, the presumed causal SNP previously identified in the literature coincides with the SNP with the strongest selection signal. This is the case, for example, for rs1426654 (SLC24A5) (Lamason et al. 2005; Kimura et al. 2009) and for rs16891982 (SLC45A2) (Branicki et al. 2008). This suggest that the top SNPs for other loci, for which the causal SNPs are not yet known, may be good candidates for further tests of functional effects.
East Asian Ancestry Scan
We also performed a scan where we sought to recover SNPs that were candidates for selection in the ancestry component that is prevalent among our CHB samples. Results for the top 30 loci in this scan are in supplementary table S2, Supplementary Material online. Here, we also recover several candidate regions that have been previously reported in East Asian selection scans, including ABCC11, POU2F3, ADH1B, FADS1, and TARBP1 (Peng et al. 2010; Ohashi et al. 2011; Vernot and Akey 2014; Liu et al. 2018; Refoyo-Martínez et al. 2019). Here, as in the previous scan, the top-scoring SNPs also tend to have the strongest phenotypic associations. For example, the highest scoring SNP (rs17822931) is the well-known missense variant in ABCC11, which is involved in sweat and earwax production (Yoshiura et al. 2006).
Yoruba/Ancestral Non-African Ancestry Scan
Because our algorithm relies on an unrooted tree of the ancestry components (fig. 4), we cannot distinguish between SNPs under positive selection in the terminal branch leading to the Yoruba/Sub-Saharan Africans and the ancestral non-African branch (supplementary table S3, Supplementary Material online). Nevertheless, more careful study of the allele frequencies of these SNPs in other populations may serve to distinguish among these scenarios in the future. As in the other ancestry scans, we also retrieve several genes that have been previously reported in positive selection studies. For example, the highest-scoring SNP is a missense variant in SLC39A4 (rs1871534) that has been reported to be under selection in Sub-Saharan Africa and to be causal for zinc deficiency (Engelken et al. 2014).
Native American Ancestry Scan
The Native American ancestry scan yielded several novel candidates for positive selection (supplementary table S4, Supplementary Material online). As this ancestry has been less studied than the other aforementioned populations in the selection scan literature, we highlight some of the more interesting regions here.
The top SNP (rs140736443) is located in an intron of LINC00871. This SNP does not have a high CADD score (=1.125), but is very close to an SNP (rs10133371) with a very high LLRS (=16.54) and CADD score (=15.99). This SNP is also intronic but is highly conserved in primates (PhastCons = 0.972) and is located in a GERP conserved element (P = 1.92e−21). LINC00871 is a long noncoding RNA gene that has been associated with number of children born (Barban et al. 2016), although the specific trait-associated SNP in that study does not have a high LLRS. This gene also contains a suggestive association to longevity in females (Zeng et al. 2018), although this study was under-powered to retrieve genome-wide significant associations.
The third top SNP (rs2316155) has a low CADD score (=0.633) but is located near two SNPs with high LLRS (rs1466182, rs1466183) that overlap a regulatory region (ENSR00000088366) and have high CADD scores (=16.8 and 19.5, respectively). Both of these SNPs have high PhastCons conservation scores across primates, mammals and vertebrates, and both overlap a GERP conserved element.
The sixth top SNP (rs10508343) has a low CADD score but lies very close to another SNP (rs17143255) with a high LLRS and a very high CADD score (=14.16). The latter is an intergenic SNP overlapping a GERP conserved element between LINC00708 and GATA3, which has been shown to lead to abnormal hair shape and growth in mice when mutated (Kaufman et al. 2003). Interestingly, SNPs overlapping LINC00708 have been recently associated with hair shape in a GWAS of admixed Latin Americans (Adhikari et al. 2016). There is also a high-LLRS SNP in this region that is significantly associated with the response to treatment for acute lymphoblastic leukemia (rs10508343) (Yang et al. 2009).
The seventh top SNP (rs16959274) is a GTEx eQTL for GOLGA8A for tibial artery and skeletal muscle, and for GOLGA8B in pancreas. These two genes are members of the same gene family, and code for an autoantigen localized in the surface of the Golgi complex (Eystathioy et al. 2000).
The tenth top SNP (rs12580697) is a GTEx eQTL for TMTC1 in whole blood and has a moderately high CADD score (=8.676). TMTC1 codes for an endoplasmic reticulum transmembrane protein that is involved in calcium homeostasis (Sunryd et al. 2014).
The 11th top SNP (rs75607199) has a low CADD score but lies near three other SNPs (rs41325445, rs4901738, and rs59250732) with almost equally high LLRS and high CADD scores (=13.49, 19.7, and 12.67, respectively). All of these SNPs are intronic and overlap OTX2-AS1, a long noncoding RNA gene. The SNP with the highest CADD score (rs4901738) is located in a GERP conserved element and has high PhastCons conservation scores across primates and mammals (>0.98). They all lie upstream of OTX2, coding for a developmental transcription factor implicated in microphthalmia (Ragge et al. 2005), retinal dystrophy (Vincent et al. 2014), and pituitary hormone deficiency (Diaczok et al. 2008). In mice, this gene has been found to be involved in the embryonic development of the brain (Boncinelli et al. 1993), photoreceptor development (Nishida et al. 2003), and susceptibility to stress (Peña et al. 2017).
The 14th top SNP (rs78441257) has a fairly high CADD score (=12.72) and lies in a GERP conserved element of the 3′ UTR of LRAT. This gene is implicated in retinal dystrophy (Thompson et al. 2001) and retinitis pigmentosa (Sénéchal et al. 2006).
The 15th top SNP (rs1919550) is a GTEx eQTL for FBXO40 in whole blood, but does not have a high CADD score. However, it lies near an SNP (rs9813391) with a high LLRS that leads to a nonsynonymous change (R145Q) in ARGFX—a homeobox gene—and another SNP (rs4676737) with both a high LLRS and high CADD score (=14.07) overlapping a repressor region in an intron of FBXO40. The latter SNP is a GTEx eQTL for IQCB1 in fibroblasts, muscular esophagus, and thyroid. IQCB1 is associated with Senior-Loken syndrome (Otto et al. 2005), a ciliopathic eye disorder.
The 22nd top SNP (rs4946567) is an eQTL of TBC1D32 in cerebellar brain. This SNP has a high CADD score (=11.02) and is conserved across vertebrates (vertebrate PhyloP = 0.916 and vertebrate PhastCons = 0.747). Interestingly, the region in which it is located also harbors signature of selection in Yucatan miniature pigs (Kim et al. 2015; Kwon et al. 2019). TBC1D32 plays a role in cilia assembly (Ko et al. 2010) and may be involved in ciliopathic congenital abnormalities, including midline cleft, microcephaly, and microphthalmia (Adly et al. 2014).
The 23rd and 24th top SNPs (rs5758430 and rs4822061) are close to each other and lie in a large region with several high-LLRS SNPs. They are both linked GTEx eQTLs to several genes in a variety of different tissues. They are also both significantly associated with several traits related to body fat, food intake and white blood cells in the UK Biobank GeneATLAS (P). Although these SNPs do not have particularly high CADD scores, there are several neighboring linked high-LLRS, high-CADD SNPs with significant associations to the same traits, including splice site and missense mutations. We also find two significantly associated SNPs in the GWAS catalog in this region (P): rs4822024 is associated with Vitiligo (Jin et al. 2012) and rs13054099 is associated with neuroticism (Nagel et al. 2018).
We also repurposed our aforementioned neutral simulations under human demography to estimate the false discovery rate (FDR) of these selected variants in aggregate. We estimate the expected number of SNPs to exceed a threshold log LR T, assuming a genome length of bp, a simple LD structure of 2 Mbp blocks, and ascertaining the SNP with the top log LR within each block. Under this approach, we find that at the cutoffs of top 1, 5, 10, 20, and 30 SNPs, the FDR is approximately 0.0% (i.e., up to simulation precision), 15.1%, 22.6%, 30.1%, and 42.6%, respectively. We encourage users of the program to do similar simulations for estimating FDRs for inferences made on their specific data sets.
Signals of Selection in Mexican Ancestry
We wanted to verify that our method was picking up signals of selection that were supported by alternative methods not explicitly relying on single-SNP patterns of population differentiation. For this, we used the program CLUES (Stern et al. 2019), which relies on a likelihood approach based on reconstructed approximation to the ancestral recombination graph along the genome (supplementary table S5, Supplementary Material online). We applied CLUES using parameters corresponding to the demographic history of Mexican-ancestry (MXL) individuals in the 1000 Genomes Project (i.e., effective population size inferred by the method Relate [Speidel et al. 2019]) to the set of hits identified using Ohana with selection acting on the Native American branch. We found that nine out of the ten tested SNPs showed significant (P < 0.05) signals of positive selection in MXL, under the asymptotic interpretation of the log-likelihood ratio statistics, supporting the evidence that these top hits in Native American ancestry have been targets of selection.
To learn more about the mode and time-frame of selection in these loci, we also used CLUES (Stern et al. 2019) to estimate the trajectory of allele frequency changes for the ten loci in the Native American component mentioned in table 1 (supplementary fig. S6, Supplementary Material online). In all cases, the estimated allele frequency trajectory was compatible with relative old selection leading to alleles with current day intermediate frequencies, typically between 0.4 and 0.6, that is, incomplete sweeps. The fact that we only detect incomplete sweep might be related to the filtering procedure we have used to eliminate SNPs with small MAF. The fastest change in allele frequency is found for the SNP in CSMD1 (s71523639), which currently is at frequency close to 0.5 but was at a frequency of approximately zero 700 generation ago, suggesting relative strong selection on a de novo mutation.
Discussion
We describe a new modeling framework that can detect signals of positive selection on ancestry components, using allele frequency patterns across admixed populations. It models admixture explicitly and works with an arbitrary number of populations with or without admixed ancestries. It also does not rely on labeling of samples into particular populations, and allows for testing of different positive selection models reflecting different historical adaptive hypotheses. It is in many ways similar to the Bayesian methods by Coop et al. (2010) and Günther and Coop (2013) in the structure of the likelihood function. The major differences being the use of optimization of the likelihood function in Ohana instead of Markov Chain Monte Carlo (MCMC) used by Coop et al. (2010) and Günther and Coop (2013), which provides some computational advantages. The methods also differ in other ways, including the enforcement of a tree-structure in Ohana, the use of ancestry components to model selection in hypothesized ancestral populations in Ohana, and the functionality to perform branch-specific detection of selection, or detection of selection in multiple branches if one has an a priori selection hypothesis one wants to test.
The run-time complexity of our method is linear in the number of markers, but we still recommend a high-performance cluster to be used in a typical genomic analysis. With parallelization, a selection scan takes <10 min to analyze a 6 Mbp genome for <10 ancestry components using 100 cores. An example of how to perform this parallelization can be found on the project’s wiki page on GitHub: https://github.com/jade-cheng/ohana.
Our method works by testing for selection in specific components of the ancestry covariance matrix. We also explored what would occur if we used a likelihood model in which the ancestry covariance matrix was multiplied by a scalar, so as to find “global” candidates for selection rather than testing for selection in particular ancestries. We found, however, that this was not an optimal way to detect candidates for selection, as it is biased toward finding many variants in highly drifted populations, likely because the excess variance in the Wright–Fisher process is not well modeled by the multivariate Gaussian assumption, especially at the boundaries of fixation and extinction.
We note, however, that the latent ancestry components inferred by Ohana and other similar programs cannot be strictly interpreted as corresponding to existing populations (now or in the past) and that the labels we assign to them (“European,” “Asian,” “African,” etc.) are largely for convenience. This is especially true when the studied individuals are not descended from recent admixture events among highly differentiated populations, so care should be taken in the interpretation of the identity of these components. We refer the reader to Lawson et al. (2018); Mathieson and Scally (2020) for more in-depth studies and discussions on the assumptions and limitations of latent ancestry inference methods.
We note that there is currently some debate in the field on the possibility that FST outliers could be caused by negative selection in various forms (see, e.g., Matthey-Doret and Whitlock 2019; Johri et al. 2020; Schrider 2020). Although it has been argued that such an effect is unlikely to explain FST outliers in real data (Matthey-Doret and Whitlock 2019; Schrider 2020), our method will be similarly challenged by this effect, as the information used is very similar to that of FST outlier scans.
When specifically testing for candidates for selection in the “European,” “East Asian,” and “Sub-Saharan African” components, we identified several well-known candidates under positive selection, including OCA2, SLC24A5, SLC45A2, ABCC1, and SLC39A4. Many of our top scoring SNPs were also previously known to be causal for particular traits, as in the case of rs17822931 in ABCC11 in East Asians, rs16891982 in SLC45A2 in Europeans, rs1426654 in SLC24A5 in Europeans, and rs1871534 in SLC39A4 in Sub-Saharan Africans.
Our scan for positive selection in the Native American ancestry component of Latin Americans yielded several novel candidates for adaptation in the human past. We found signatures of selection near genes involved in fertility (LINC00871), hair shape and growth (LINC00708), immunity (GOLGA8A/GOLGA8B and IRAK4), vision (OTX2 and LRAT), the nervous system (MDGA2), and various ciliopathies (IQCB1 and TBC1D32). Several of the highest-scoring SNPs in the candidate regions are known to be cis-eQTLs to their nearby genes, as is the case for rs12580697/TMTC1 (involved in calcium homeostasis) and rs4676737/IQCB1 (involved in ciliopathies). We also found individual SNPs with high likelihood ratio scores in favor of selection that are associated with a variety of phenotypes, including rs12426688 (fat percentage), rs10508343 (response to leukemia treatment), rs34670506 (insomnia), and the cluster of high-scoring SNPs that include rs5758430 and rs4822061, among other SNPs. This particular cluster is especially interesting, as the SNPs in the region are associated with a variety of traits related to body fat distribution, food intake and white blood cells, suggesting a possible underlying phenotype related to these traits that may have driven an adaptive event. Estimates of the FDR suggest that the lion’s share of these SNPs are selected, especially toward the higher end (e.g., the top eight SNPs have an FDR of <10%).
We provide a list of functional annotations for all the SNPs with high LLRS (>15) within a 2 Mb region surrounding each of the top genome-wide SNPs, including CADD, conservation, regulatory, and protein deleteriousness scores, which we hope will guide future functional validation studies in these regions of the genome (supplementary table S6, Supplementary Material online).
In conclusion, Ohana provides a fast and flexible selection–detection and hypothesis-testing framework. It is easy to use and has in-built visualization functionalities to explore patterns on a genome-wide and locus-specific scale. We believe that it will be a useful tool for biologists aiming to study positive selection and understanding the genomic basis of adaptation, particularly in cases where demographic histories are complex or not well characterized.
Materials and Methods
Basic Model
The new method is based on the Ohana inference framework (Cheng, Mailund, et al. 2017), which works with both genotype calls and genotype likelihoods. In brief, the classical structure model (Pritchard et al. 2000) is used to infer allele frequencies, ancestry components, and admixture proportions using maximum likelihood (ML). Then a covariance matrix among components is inferred using a multivariate Gaussian distribution while enforcing constraints imposed by the assumption of a tree structure. The covariance between leaf nodes is proportional to the amount of shared phylogenetic history between the nodes. Consider, for example, the example of the matrix and corresponding tree in the left side of figure 6. In this tree, all branches have length 0.1 and the tree is rooted in node A. The covariance between node E and node B is then 0.1, because B and E share one edge in the path from A. However, the covariance between node C and E is 0.2 because they share two edges in common in the path from A. The covariance matrix, , can be converted into a distance matrix, , using the rule . Treeness can then be tested using the four point condition applied to d.
Fig. 6.

Selection hypotheses and their encodings as covariance matrices. In this example, the ancestry component E is assumed to be the potential target of selection. The entry E: E in the covariance matrix is therefore allowed to deviate from the globally estimated value.
This system is underdetermined because the tree can be rooted in any node (see, e.g., Felsenstein 1985), and the same joint probability distribution is obtained no matter which rooting is chosen. We root the tree in one of the ancestry components and condition on the allele frequencies in this component when calculating the joint distribution of allele frequencies in the other components. This idea is similar to Felsenstein’s restricted ML approach (Felsenstein 1985). We emphasize that the rooting is arbitrary but that it does not imply any assumptions about this component actually being ancestral.
We estimate the covariance matrix Ω via ML. This matrix has size , where K is the number of populations assuming a joint density of allele frequencies given by
| (1) |
where faj is the allele frequency in the ancestry component arbitrarily assigned as ancestral and fj is a vector of the allele frequencies in the other K− 1 components, at SNP j. μj is the mean allele frequency for SNP j (averaged over all components). Note that this model of joint allele frequencies is similar to the model implemented in TreeMix (Pickrell and Pritchard 2012) which also uses a Gaussian approximation to allele frequency change. The full-likelihood function is obtained by taking the product of equation (1) over all SNPs in the genome. The method for optimizing this function is described in a subsequent section.
Selection Model
Following the genome-wide estimation of Ω, a natural extension of this framework is to detect SNPs that deviate strongly from the globally estimated covariance structure. The idea of testing for deviations from a Gaussian distribution follows Günther and Coop (2013), but differs in the use of an enforced tree-structure, an ML inference framework and fast optimization algorithms, thereby avoiding some of the computational challenges associated with MCMC. We also note that admixture is incorporated into the inference framework, thereby enabling the possibility to test for positive selection that acted on the ancestral components of a panel, before interbreeding occurred between the ancestors of the sampled individuals.
Ohana uses a likelihood ratio test that identifies SNPs with allele frequency patterns that are poorly described by the genome-wide pattern. After estimating Ω jointly for all SNPs, each SNP is then independently tested for deviations from this model, using a scalar factor introduced to certain elements of the covariance matrix. This scalar factor can be introduced in different ways depending on which selection hypotheses are tested. In our analyses, we chose to scale the covariance matrix such that one of its diagonal values is multiplied by a scalar, α, corresponding to differences in allele frequency in one of the ancestry components relative to the rest, for example:
| (2) |
The value of α is then estimated via ML using equation (1) (assuming all other values in is fixed at the genomic ML estimates) and a likelihood ratio is formed by testing the hypothesis of α = 1 against the alternative of . A significantly high likelihood ratio indicates a larger deviation in allele frequency in a focal component than expected under the globally estimated null-model. Figure 6 shows an example. This test can also be implemented to test selection on ancestral nonterminal lineages by multiplying the corresponding values in the covariance matrix by a scaling factor.
Under the null-hypothesis, the likelihood ratio test statistic is expected to approximately follow a 50:50 mixture between a -distribution and a point mass at zero (Self and Liang 1987) because α is bounded at 1, and we use this asymptotic distribution to calculate P values.
In summary, we estimate a scaling factor for one or more components of the covariance matrix in a multivariate normal model of allele frequency distribution among populations. For each candidate SNP, we then compare the estimated covariance matrix to that obtained genome-wide, using a likelihood ratio test.
Optimization
To estimate allele frequencies, we assume a classical structure/admixture model (Pritchard et al. 2000) and first estimate Q, a matrix of admixture proportions for each individual, and F, the matrix of allele frequencies for all loci, using a quadratic programming algorithm described in full detail in Cheng, Mailund, et al. (2017) and we refer the reader to the description in this paper. This method can also incorporate genotype likelihoods.
Conditional on these estimates of values of fj and faj for all j, we then maximize the likelihood in equation (1) for Ω. This optimization is done using the Nelder–Mead simplex method (Nelder and Mead 1965). It uses Cholesky decomposition (Cholesky 1910) to determine the positive semidefiniteness of a matrix and to compute matrix inverses and determinants. For the initial starting point, we use sample covariances:
| (3) |
To enforce treeness, instead of using a costly constrained optimization, we convert the covariance matrix into a distance matrix, , which is converted into a tree using the Neighbor-Joining algorithm (Saitou and Nei 1987). We then use the covariance matrix induced by this procedure. For estimating α during a selection scan for a single SNP, conditionally on the globally estimated value of Ω, we use a simple Golden-section search algorithm (Kiefer 1953).
Simulations
We conducted population genetic simulations using the forward simulator SLiM 3 (Haller and Messer 2019). We consider three distinct demographic models (fig. 2):
A basic four-population tree with no admixture (fig. 2a): An ancestral population splits into four subpopulations at times 4,000, 2,000, and 800 generations before present, following the topology in figure 2a. Selection is simulated on the yellow branch in figure 2a. Tests for selection are conducted for yellow ancestry (i.e., the main ancestry component in the third branch).
A four-population tree with admixture (fig. 2b): The same model as in (1), but split times are shifted backwards in time by 100 generations; at 100 generations before present, selection is turned off, and each population is supplanted by a (1/3,1/3,1/3) mixture of the other three populations. Tests for selection are conducted for yellow ancestry (i.e., the most depleted ancestry component in the third branch).
A model based on human demography of Mexican (MXL), Northwestern European (CEU), CHB (EAS), and African Yoruba (YRI) populations (fig. 2c): The model is based on parameter estimates from Gravel et al. (2011) and Gutenkunst et al. (2009). MXL is modeled as a (1/2,1/2) mixture of CEU and Native American (NA) ancestry. We simulate selection only in the ancestral NA population (i.e., no ongoing selection in MXL). We use Ohana to test for selection in this NA ancestry component, which is only observed in the admixed MXL individuals.
In simulations (1) and (2), we assume all populations are constant in size with . For all simulations, we simulate a locus of 2 Mbp with mutation and recombination rates per bp per generation. In all cases, we sample 20 diploid individuals from each extant population (i.e., 160 chromosomes sampled). We simulate a single selected site occurring within a ±10 kb window of the center of the simulated locus. In order to simulate selection during particular time periods, we simulate sweeps from standing variation (an initial frequency f), although we consider such low frequencies (down to f = 0.0001) that these should produce indistinguishable patterns from those produced by hard sweeps (Przeworski et al. 2005). For each demographic scenario, we consider four different selection coefficients (s = 0, 0.01, 0.02, and 0.05) and three different ranges of starting frequencies for the selected allele (f in , and ). (Simulations under model (3) exclude sweeps with f < 0.001 because the ancestral NA population size is too small for any such variation at that low frequency.) We use a neutral burn-in phase of 100,000 generations. For all simulations, as is typical in forward simulations, we scale times down by a factor of 10, and scale up the selection coefficients and mutation and recombination rates by a factor of 10, in order to ease computational burden. In all simulation scenarios we use 1,000 independent replicates. Open-source implementations of each model are provided at https://github.com/35ajstern/ohana_simulation_models.
We compared Ohana’s performance to that of two other state-of-the-art methods: pcadapt and BayPass (Gautier 2015; Duforet-Frebourg et al. 2016). Like Ohana, both methods depend on some sort of empirical null model. To this end, we simulated three 20 Mb-long neutral regions under otherwise the same settings as previously described, with s = 0, in order to generate a null data set for calibrating each method. In the case of Ohana and BayPass, this null data set is used to estimate the covariance matrix for each population; in pcadapt, we append this null data set to each region we test for selection (we do this because the pcadapt package does not have an equivalent two-step process for calculating PCs in one region and testing for deviation from these PCs in a separate region). In all cases, we filter out SNPs with MAF <0.05 prior to any analysis. In Ohana, we test for selection in specific ancestry groups; in contrast, BayPass and pcadapt test for any significant deviation from the empirical covariance matrix (BayPass models population-level covariance, whereas pcadapt models individual-level covariance). In this sense, it is important to keep in mind that Ohana is performing a more specific test for selection, and can be used to methodologically attribute selection to a particular ancestral component/branch.
Supplementary Material
Supplementary data are available at Molecular Biology and Evolution online.
Supplementary Material
Acknowledgments
The authors are grateful to Thomas Mailund, Mikkel Schierup, Christian Storm Pedersen, and the GenomeDK staff for their support during the course of this research. We also thank Leo Speidel for providing coalescence time and effective population size estimates for the 1000 Genomes Project. FR was funded by a Villum Fonden Young Investigator award (project no. 00025300). This research was supported by NIH grant number R01GM138634.
Data Availability
All data analyzed in this manuscript are previously published publicly available data. The software Ohana described in the article is open source and available at https://github.com/jade-cheng/ohana.
References
- Adzhubei I, Jordan DM, Sunyaev SR.. 2013. Predicting functional effect of human missense mutations using polyphen-2. Curr Protocols Hum Genet. 76(1):7–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akey JM, Zhang G, Zhang K, Jin L, Shriver MD.. 2002. Interrogating a high-density SNP map for signatures of natural selection. Genome Res. 12(12):1805–1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barban N, Jansen R, De Vlaming R, Vaez A, Mandemakers JJ, Tropf FC, Shen X, Wilson JF, Chasman DI, Nolte IM, et al. ; LifeLines Cohort Study. 2016. Genome-wide analysis identifies 12 loci influencing human reproductive behavior. Nat Genet. 48(12):1462–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barreiro LB, Ben-Ali M, Quach H, Laval G, Patin E, Pickrell JK, Bouchier C, Tichit M, Neyrolles O, Gicquel B, et al. 2009. Evolutionary dynamics of human toll-like receptors and their different contributions to host defense. PLoS Genet. 5(7):e1000562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaumont MA, Balding DJ.. 2004. Identifying adaptive genetic divergence among populations from genome scans. Mol Ecol. 13(4):969–980. [DOI] [PubMed] [Google Scholar]
- Beaumont MA, Nichols RA.. 1996. Evaluating loci for use in the genetic analysis of population structure. Proc R Soc Lond Ser B: Biol Sci. 263(1377):1619–1626. [Google Scholar]
- Bersaglieri T, Sabeti PC, Patterson N, Vanderploeg T, Schaffner SF, Drake JA, Rhodes M, Reich DE, Hirschhorn JN.. 2004. Genetic signatures of strong recent positive selection at the lactase gene. Am J Hum Genet. 74(6):1111–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boncinelli E, Gulisano M, Broccoli V.. 1993. Emx and Otx homeobox genes in the developing mouse brain. J Neurobiol. 24(10):1356–1366. [DOI] [PubMed] [Google Scholar]
- Bonhomme M, Chevalet C, Servin B, Boitard S, Abdallah J, Blott S, SanCristobal M.. 2010. Detecting selection in population trees: the Lewontin and Krakauer test extended. Genetics 186(1):241–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branicki W, Brudnik U, Draus-Barini J, Kupiec T, Wojas-Pelc A.. 2008. Association of the SLC45A2 gene with physiological human hair colour variation. J Hum Genet. 53(11–12):966–971. [DOI] [PubMed] [Google Scholar]
- Canela-Xandri O, Rawlik K, Tenesa A.. 2018. An atlas of genetic associations in UK biobank. Nat Genet. 50(11):1593–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng JY, Mailund T, Nielsen R.. 2017. Fast admixture analysis and population tree estimation for SNP and NGS data. Bioinformatics 33(14):2148–2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X, Xu C, DeGiorgio M.. 2017b. Fast and robust detection of ancestral selective sweeps. Mol Ecol. 26(24):6871–6891. [DOI] [PubMed] [Google Scholar]
- Cholesky A-L. 1910. Sur la résolution numérique des systèmes d’équations linéaires. Bulletin de la Sabix. Société Des Amis de la Bibliothèque et de L’Histoire de L’École Polytechnique (39):81–95. [Google Scholar]
- 1000 Genomes Project Consortium, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, et al. 2015. A global reference for human genetic variation. Nature 526(7571):68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coop G, Witonsky D, Di Rienzo A, Pritchard JK.. 2010. Using environmental correlations to identify loci underlying local adaptation. Genetics 185(4):1411–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davydov EV, Goode DL, Sirota M, Cooper GM, Sidow A, Batzoglou S.. 2010. Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput Biol. 6(12):e1001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeGiorgio M, Huber CD, Hubisz MJ, Hellmann I, Nielsen R.. 2016. SweepFinder2: increased sensitivity, robustness and flexibility. Bioinformatics 32(12):1895–1897. [DOI] [PubMed] [Google Scholar]
- Diaczok D, Romero C, Zunich J, Marshall I, Radovick S.. 2008. A novel dominant negative mutation of OTX2 associated with combined pituitary hormone deficiency. J Clin Endocrinol Metab. 93(11):4351–4359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duforet-Frebourg N, Luu K, Laval G, Bazin E, Blum MG.. 2016. Detecting genomic signatures of natural selection with principal component analysis: application to the 1000 genomes data. Mol Biol Evol. 33(4):1082–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelken J, Carnero-Montoro E, Pybus M, Andrews GK, Lalueza-Fox C, Comas D, Sekler I, de la Rasilla M, Rosas A, Stoneking M, et al. 2014. Extreme population differences in the human zinc transporter zip4 (SLC39A4) are explained by positive selection in sub-Saharan Africa. PLoS Genet. 10(2):e1004128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eystathioy T, Jakymiw A, Fujita DJ, Fritzler MJ, Chan EK.. 2000. Human autoantibodies to a novel Golgi protein golgin-67: high similarity with golgin-95/gm 130 autoantigen. J Autoimmun. 14(2):179–187. [DOI] [PubMed] [Google Scholar]
- Fariello MI, Boitard S, Naya H, SanCristobal M, Servin B.. 2013. Detecting signatures of selection through haplotype differentiation among hierarchically structured populations. Genetics 193(3):929–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay JC, Wu C-I.. 2000. Hitchhiking under positive Darwinian selection. Genetics 155(3):1405–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J. 1985. Phylogenies and the comparative method. Am Nat. 125(1):1–15. [Google Scholar]
- Foll M, Gaggiotti O.. 2008. A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: a Bayesian perspective. Genetics 180(2):977–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foll M, Gaggiotti OE, Daub JT, Vatsiou A, Excoffier L.. 2014. Widespread signals of convergent adaptation to high altitude in Asia and America. Am J Hum Genet. 95(4):394–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y-X, Li W-H.. 1993. Statistical tests of neutrality of mutations. Genetics 133(3):693–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumagalli M, Moltke I, Grarup N, Racimo F, Bjerregaard P, Jørgensen ME, Korneliussen TS, Gerbault P, Skotte L, Linneberg A, et al. 2015. Greenlandic Inuit show genetic signatures of diet and climate adaptation. Science 349(6254):1343–1347. [DOI] [PubMed] [Google Scholar]
- Gautier M. 2015. Genome-wide scan for adaptive divergence and association with population-specific covariates. Genetics 201(4):1555–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gravel S, Henn BM, Gutenkunst RN, Indap AR, Marth GT, Clark AG, Yu F, Gibbs RA, Bustamante CD, Project G, et al. ; 1000 Genomes Project. 2011. Demographic history and rare allele sharing among human populations. Proc Natl Acad Sci U S A. 108(29):11983–11988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Günther T, Coop G.. 2013. Robust identification of local adaptation from allele frequencies. Genetics 195(1):205–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutenkunst RN, Hernandez RD, Williamson SH, Bustamante CD.. 2009. Inferring the joint demographic history of multiple populations from multidimensional SNP frequency data. PLoS Genet. 5(10):e1000695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haller BC, Messer PW.. 2019. Slim 3: forward genetic simulations beyond the Wright–Fisher model. Mol Biol Evol. 36(3):632–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman MM, Buske OJ, Wang J, Weng Z, Bilmes JA, Noble WS.. 2012. Unsupervised pattern discovery in human chromatin structure through genomic segmentation. Nat Methods 9(5):473–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber CD, DeGiorgio M, Hellmann I, Nielsen R.. 2016. Detecting recent selective sweeps while controlling for mutation rate and background selection. Mol Ecol. 25(1):142–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y, Birlea SA, Fain PR, Ferrara TM, Ben S, Riccardi SL, Cole JB, Gowan K, Holland PJ, Bennett DC, et al. 2012. Genome-wide association analyses identify 13 new susceptibility loci for generalized vitiligo. Nat Genet. 44(6):676–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johri P, Charlesworth B, Howell EK, Lynch M, Jensen JD.. 2020. Revisiting the notion of deleterious sweeps. Genetics. iyab094, doi: 10.1093/genetics/iyab094. [DOI] [PMC free article] [PubMed]
- Kaufman CK, Zhou P, Pasolli HA, Rendl M, Bolotin D, Lim K-C, Dai X, Alegre M-L, Fuchs E.. 2003. Gata-3: an unexpected regulator of cell lineage determination in skin. Genes Dev. 17(17):2108–2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiefer J. 1953. Sequential minimax search for a maximum. Proc Am Math Soc. 4(3):502–506. [Google Scholar]
- Kim H, Song KD, Kim HJ, Park W, Kim J, Lee T, Shin D-H, Kwak W, Kwon Y-J, Sung S, et al. 2015. Exploring the genetic signature of body size in Yucatan miniature pig. PLoS One 10(4):e0121732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Nielsen R.. 2004. Linkage disequilibrium as a signature of selective sweeps. Genetics 167(3):1513–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Stephan W.. 2002. Detecting a local signature of genetic hitchhiking along a recombining chromosome. Genetics 160(2):765–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura R, Yamaguchi T, Takeda M, Kondo O, Toma T, Haneji K, Hanihara T, Matsukusa H, Kawamura S, Maki K, et al. 2009. A common variation in Edar is a genetic determinant of shovel-shaped incisors. Am J Hum Genet. 85(4):528–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko HW, Norman RX, Tran J, Fuller KP, Fukuda M, Eggenschwiler JT.. 2010. Broad-minded links cell cycle-related kinase to cilia assembly and hedgehog signal transduction. Dev Cell 18(2):237–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon D-J, Lee Y-S, Shin D, Won K-H, Song K-D.. 2019. Genome analysis of Yucatan miniature pigs to assess their potential as biomedical model animals. Asian-Aust J Anim Sci. 32(2):290–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamason RL, Mohideen M-AP, Mest JR, Wong AC, Norton HL, Aros MC, Jurynec MJ, Mao X, Humphreville VR, Humbert JE, et al. 2005. SLC24A5, a putative cation exchanger, affects pigmentation in zebrafish and humans. Science 310(5755):1782–1786. [DOI] [PubMed] [Google Scholar]
- Lawson DJ, Van Dorp L, Falush D.. 2018. A tutorial on how not to over-interpret structure and admixture bar plots. Nat Commun. 9(1):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Librado P, Orlando L.. 2018. Detecting signatures of positive selection along defined branches of a population tree using LSD. Mol Biol Evol. 35(6):1520–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Huang S, Chen F, Zhao L, Yuan Y, Francis SS, Fang L, Li Z, Lin L, Liu R, et al. 2018. Genomic analyses from non-invasive prenatal testing reveal genetic associations, patterns of viral infections, and Chinese population history. Cell 175(2):347–359. [DOI] [PubMed] [Google Scholar]
- Lonsdale J, Thomas J, Salvatore M, Phillips R, Lo E, Shad S, Hasz R, Walters G, Garcia F, Young N, et al. 2013. The genotype-tissue expression (GTEx) project. Nat Genet. 45(6):580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luu K, Bazin E, Blum MG.. 2017. pcadapt: an r package to perform genome scans for selection based on principal component analysis. Mol Ecol Resour. 17(1):67–77. [DOI] [PubMed] [Google Scholar]
- MacArthur J, Bowler E, Cerezo M, Gil L, Hall P, Hastings E, Junkins H, McMahon A, Milano A, Morales J, et al. 2017. The new NHGRI-EBI catalog of published genome-wide association studies (GWAS catalog). Nucleic Acids Res. 45(D1):D896–D901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathieson I, Lazaridis I, Rohland N, Mallick S, Patterson N, Roodenberg SA, Harney E, Stewardson K, Fernandes D, Novak M, et al. 2015. Genome-wide patterns of selection in 230 ancient Eurasians. Nature 528(7583):499–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathieson I, Scally A.. 2020. What is ancestry? PLoS Genet. 16(3):e1008624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthey-Doret R, Whitlock MC.. 2019. Background selection and Fst: consequences for detecting local adaptation. Mol Ecol. 28(17):3902–3914. [DOI] [PubMed] [Google Scholar]
- McVean G. 2007. The structure of linkage disequilibrium around a selective sweep. Genetics 175(3):1395–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagel M, Jansen PR, Stringer S, Watanabe K, de Leeuw CA, Bryois J, Savage JE, Hammerschlag AR, Skene NG, Muñoz-Manchado AB, et al. ; 23andMe Research Team. 2018. Meta-analysis of genome-wide association studies for neuroticism in 449,484 individuals identifies novel genetic loci and pathways. Nat Genet. 50(7):920–927. [DOI] [PubMed] [Google Scholar]
- Nelder JA, Mead R.. 1965. A simplex method for function minimization. Comput J. 7(4):308–313. [Google Scholar]
- Nielsen R. 2005. Molecular signatures of natural selection. Annu Rev Genet. 39:197–218. [DOI] [PubMed] [Google Scholar]
- Nishida A, Furukawa A, Koike C, Tano Y, Aizawa S, Matsuo I, Furukawa T.. 2003. Otx2 homeobox gene controls retinal photoreceptor cell fate and pineal gland development. Nat Neurosci. 6(12):1255–1263. [DOI] [PubMed] [Google Scholar]
- Ohashi J, Naka I, Tsuchiya N.. 2011. The impact of natural selection on an abcc11 SNP determining earwax type. Mol Biol Evol. 28(1):849–857. [DOI] [PubMed] [Google Scholar]
- Otto EA, Loeys B, Khanna H, Hellemans J, Sudbrak R, Fan S, Muerb U, O'Toole JF, Helou J, Attanasio M, et al. 2005. Nephrocystin-5, a ciliary IQ domain protein, is mutated in Senior-Loken syndrome and interacts with RPGR and calmodulin. Nat Genet. 37(3):282–288. [DOI] [PubMed] [Google Scholar]
- Peña CJ, Kronman HG, Walker DM, Cates HM, Bagot RC, Purushothaman I, Issler O, Loh Y-HE, Leong T, Kiraly DD, et al. 2017. Early life stress confers lifelong stress susceptibility in mice via ventral tegmental area otx2. Science 356(6343):1185–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y, Shi H, Qi X-B, Xiao C-J, Zhong H, Run-lin ZM, Su B.. 2010. The ADH1B Arg47His polymorphism in East Asian populations and expansion of rice domestication in history. BMC Evol Biol. 10(1):15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell J, Pritchard J.. 2012. Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genet. 8(11):e1002967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard KS, Hubisz MJ, Rosenbloom KR, Siepel A.. 2010. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 20(1):110–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard JK, Stephens M, Donnelly P.. 2000. Inference of population structure using multilocus genotype data. Genetics 155(2):945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przeworski M, Coop G, Wall JD.. 2005. The signature of positive selection on standing genetic variation. Evolution 59(11):2312–2323. [PubMed] [Google Scholar]
- Racimo F. 2016. Testing for ancient selection using cross-population allele frequency differentiation. Genetics 202(2):733–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racimo F, Marnetto D, Huerta-Sánchez E.. 2017. Signatures of archaic adaptive introgression in present-day human populations. Mol Biol Evol. 34(2):296–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragge NK, Brown AG, Poloschek CM, Lorenz B, Henderson RA, Clarke MP, Russell-Eggitt I, Fielder A, Gerrelli D, Martinez-Barbera JP, et al. 2005. Heterozygous mutations of OTX2 cause severe ocular malformations. Am J Hum Genet. 76(6):1008–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Refoyo-Martínez A, da Fonseca RR, Halldórsdóttir K, Árnason E, Mailund T, Racimo F.. 2019. Identifying loci under positive selection in complex population histories. Genome Res. 29(9):1506–1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M.. 2019. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 47(D1):D886–D894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabeti PC, Schaffner SF, Fry B, Lohmueller J, Varilly P, Shamovsky O, Palma A, Mikkelsen T, Altshuler D, Lander E.. 2006. Positive natural selection in the human lineage. Science 312(5780):1614–1620. [DOI] [PubMed] [Google Scholar]
- Sabeti PC, Varilly P, Fry B, Lohmueller J, Hostetter E, Cotsapas C, Xie X, Byrne EH, McCarroll SA, Gaudet R, et al. ; International HapMap Consortium. 2007. Genome-wide detection and characterization of positive selection in human populations. Nature 449(7164):913–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitou N, Nei M.. 1987. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 4(4):406–425. [DOI] [PubMed] [Google Scholar]
- Schrider DR. 2020. Background selection does not mimic the patterns of genetic diversity produced by selective sweeps. Genetics 216(2):499–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Self SG, Liang K-Y.. 1987. Asymptotic properties of maximum likelihood estimators and likelihood ratio tests under nonstandard conditions. J Am Stat Assoc. 82(398):605–610. [Google Scholar]
- Sénéchal A, Humbert G, Surget M-O, Bazalgette C, Bazalgette C, Arnaud B, Arndt C, Laurent E, Brabet P, Hamel CP.. 2006. Screening genes of the retinoid metabolism: novel LRAT mutation in leber congenital amaurosis. Am J Ophthalmol. 142(4):702–704. [DOI] [PubMed] [Google Scholar]
- Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S, et al. 2005. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 15(8):1034–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sim N-L, Kumar P, Hu J, Henikoff S, Schneider G, Ng PC.. 2012. Sift web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 40(Web Server issue):W452–W457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speidel L, Forest M, Shi S, Myers SR.. 2019. A method for genome-wide genealogy estimation for thousands of samples. Nat Genet. 51(9):1321–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern AJ, Wilton PR, Nielsen R.. 2019. An approximate full-likelihood method for inferring selection and allele frequency trajectories from DNA sequence data. PLoS Genet. 15(9):e1008384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunryd JC, Cheon B, Graham JB, Giorda KM, Fissore RA, Hebert DN.. 2014. Tmtc1 and tmtc2 are novel endoplasmic reticulum tetratricopeptide repeat-containing adapter proteins involved in calcium homeostasis. J Biol Chem. 289(23):16085–16099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima F. 1989. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123(3):585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson DA, Li Y, McHenry CL, Carlson TJ, Ding X, Sieving PA, Apfelstedt-Sylla E, Gal A.. 2001. Mutations in the gene encoding lecithin retinol acyltransferase are associated with early-onset severe retinal dystrophy. Nat Genet. 28(2):123–124. [DOI] [PubMed] [Google Scholar]
- Vernot B, Akey JM.. 2014. Resurrecting surviving neandertal lineages from modern human genomes. Science 343(6174):1017–1021. [DOI] [PubMed] [Google Scholar]
- Vincent A, Forster N, Maynes JT, Paton TA, Billingsley G, Roslin NM, Ali A, Sutherland J, Wright T, Westall CA, et al. ; FORGE Canada Consortium. 2014. OTX2 mutations cause autosomal dominant pattern dystrophy of the retinal pigment epithelium. J Med Genet. 51(12):797–805. [DOI] [PubMed] [Google Scholar]
- Voight BF, Kudaravalli S, Wen X, Pritchard JK.. 2006. A map of recent positive selection in the human genome. PLoS Biol. 4(3):e72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JJ, Cheng C, Yang W, Pei D, Cao X, Fan Y, Pounds SB, Neale G, Treviño LR, French D, et al. 2009. Genome-wide interrogation of germline genetic variation associated with treatment response in childhood acute lymphoblastic leukemia. JAMA 301(4):393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi X, Liang Y, Huerta-Sanchez E, Jin X, Cuo ZXP, Pool JE, Xu X, Jiang H, Vinckenbosch N, Korneliussen TS, et al. 2010. Sequencing of 50 human exomes reveals adaptation to high altitude. Science 329(5987):75–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshiura K-I, Kinoshita A, Ishida T, Ninokata A, Ishikawa T, Kaname T, Bannai M, Tokunaga K, Sonoda S, Komaki R, et al. 2006. A SNP in the abcc11 gene is the determinant of human earwax type. Nat Genet. 38(3):324–330. [DOI] [PubMed] [Google Scholar]
- Zeng Y, Nie C, Min J, Chen H, Liu X, Ye R, Chen Z, Bai C, Xie E, Yin Z, et al. 2018. Sex differences in genetic associations with longevity. JAMA Netw Open. 1(4):e181670. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data analyzed in this manuscript are previously published publicly available data. The software Ohana described in the article is open source and available at https://github.com/jade-cheng/ohana.