

SUMMARY
Repair of genetic damage is coordinated in the context of chromatin, so cells dynamically modulate accessibility at DNA breaks for the recruitment of DNA damage response (DDR) factors. The identification of chromatin factors with roles in DDR has mostly relied on loss-of-function screens while lacking robust high-throughput systems to study DNA repair. In this study, we have developed two high-throughput systems that allow the study of DNA repair kinetics and the recruitment of factors to double-strand breaks in a 384-well plate format. Using a customized gain-of-function open-reading frame library (“ChromORFeome” library), we identify chromatin factors with putative roles in the DDR. Among these, we find the PHF20 factor is excluded from DNA breaks, affecting DNA repair by competing with 53BP1 recruitment. Adaptable for genetic perturbations, small-molecule screens, and large-scale analysis of DNA repair, these resources can aid our understanding and manipulation of DNA repair.
In brief
Martinez-Pastor et al. developed two imaging-based screening platforms for the study of DNA repair kinetics and recruitment of proteins to damaged chromatin. They tested them against an ORF library of chromatin factors and identify PHF20 as a factor excluded from DNA lesions to allow recruitment of 53BP1 and DNA repair.
Graphical abstract
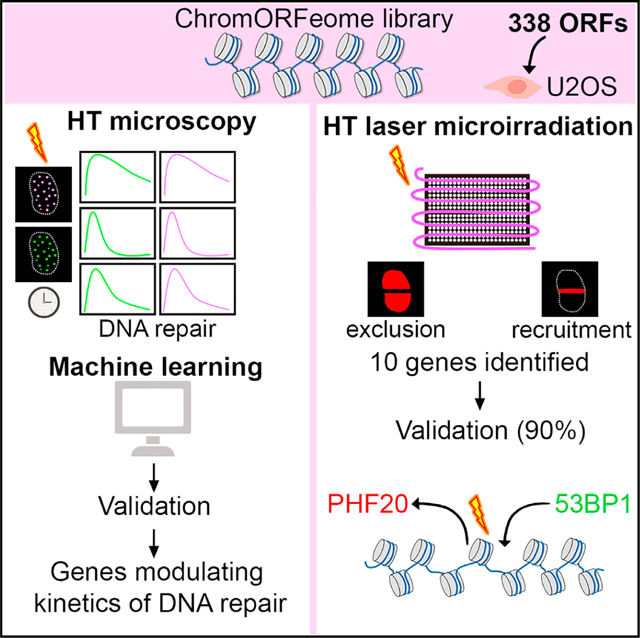
INTRODUCTION
Cells are constantly subjected to DNA damage generated by environmental sources or damage emerging during endogenous cellular metabolic reactions and physiological processes (Ciccia and Elledge, 2010; Tubbs and Nussenzweig, 2017). Accurate repair of such DNA lesions is essential to prevent mutations and chromosomal rearrangements that could both compromise organismal survival and development and that are at the origin of pathologies such as cancer, immunodeficiencies, neurodegeneration, and premature aging (Jackson and Bartek, 2009). To counteract the threats compromising DNA integrity, cells have evolved mechanisms that detect DNA lesions and activate a signaling-transduction pathway to promote DNA repair, termed the DNA damage response (DDR) (Harper and Elledge, 2007; Zhou and Elledge, 2000). One of the earliest events in the DDR is the recruitment of poly(ADP-ribose) polymerase 1 (PARP1) to the site of damage through its binding to DNA, resulting in its activation and the synthesis of poly(ADP-ribose) (PAR) chains on histones, non-histone proteins, and on itself (Jungmichel et al., 2013). PARylation is involved in the early recruitment of DDR proteins, such as the sensor complex MRN (Mre11, Rad50, Nbs1) that in turn recruits ATM to breaks (Haince et al., 2008) leading to its activation. ATM initiates a series of phosphorylation events including the phosphorylation of histone variant H2AX on serine 139 (also termed γH2AX) (Rogakou et al., 1999). Phosphorylation of H2AX by the PI3-K-like kinases ATM, ATR, and DNA-PKcs is required for DNA damage signal amplification and subsequent accumulation of numerous DDR proteins at double-strand break (DSB) sites, including 53BP1, MDC1, and specific components of the main repair pathways homologous recombination (HR) and non-homologous end-joining (NHEJ) (Ciccia and Elledge, 2010).
DNA repair does not occur on naked DNA, but in the context of chromatin. Upon DNA damage, post-translational modifications such as phosphorylation, poly(ADP-ribosyl)ation, acetylation, methylation, ubiquitylation, and sumoylation in histones and in chromatin proteins are key for the ordered recruitment of DNA repair factors (Altmeyer and Lukas, 2013; Clouaire and Legube, 2019; Pinder et al., 2013). For instance, accumulation and retention of p53-binding protein 1 (53BP1) to DNA lesions requires chromatin modifications such as H3 dimethylated at K79 and H4 dimethylated at K20 that are recognized by 53BP1 through its Tudor domains (Botuyan et al., 2006; Huyen et al., 2004) but also de novo histone modifications such as H2A monoubiquitination at K15 (H2AK15Ub) by RNF8 and RNF168 and H2AX phosphorylation (Doil et al., 2009; Fradet-Turcotte et al., 2013; Huen et al., 2007; Kolas et al., 2007; Mailand et al., 2007; Stewart et al., 2009). 53BP1 recruitment to DSBs protects DNA broken ends from resection and promotes NHEJ over HR repair (Bothmer et al., 2010; Bunting et al., 2010). Recent studies identified the Shieldin complex as a major modulator of 53BP1 recruitment (Ghezraoui et al., 2018; Mirman et al., 2018; Noordermeer et al., 2018). On the other hand, acetylation of H4K16 by Tip60 prevents 53BP1 binding to chromatin and promotes HR repair (Tang et al., 2013), a function related to the ability of the MBDT1 subunit of Tip60 to both bind H4K20me2 (and compete with 53BP1) and acetylate H2AK15, which inhibits its ubiquitylation (Jacquet et al., 2016). JMJD2A, by directly competing with 53BP1 for H4K20me2 binding, also prevents the formation of 53BP1 foci after damage when overexpressed (Mallette et al., 2012).
Laser microirradiation and electron microscopy techniques have shown that chromatin undergoes a rapid decondensation at sites of DNA damage (Dellaire et al., 2009; Kruhlak et al., 2006; Strickfaden et al., 2016) accompanied by rapid accumulation of proteins and followed by a re-compaction phase (Burgess et al., 2014) dependent on PARP and chromatin remodelers (Luijsterburg et al., 2016; Sellou et al., 2016; Smith et al., 2019) and involving the displacement of histones at sites of breaks (Strickfaden et al., 2016). In this context, histone acetylation is dynamically regulated at sites of breaks. The histone acetyltransferase Tip60 increases the acetylation of histones and ATM to improve recruitment of repair factors (Kusch et al., 2004; Murr et al., 2006; Sun et al., 2005), while recruitment of the histone deacetylase SIRT6 drives local H3K56 deacetylation and the recruitment of the chromatin remodeler SNF2H for fast decompaction, to also facilitate recruitment of DNA repair factors (Toiber et al., 2013).
Since the seminal identification of DNA repair factors by means of classical mutagenesis in yeast and the discovery of mutations in human cancer, the search for unknown DDR genes has been constant. The use of RNA interference screens led to the discovery of a number of components of the DDR (Adamson et al., 2012; Floyd et al., 2013) despite intrinsic drawbacks such as off-target effects or poor knockdown efficiency. While loss-of-function screens using Crispr/Cas9 KO technology circumvent some limitations of RNAi, the essentiality of a significant fraction of DDR factors may prohibit the further discovery of DDR genes. In this sense, overexpression-based or gain-of-function (GOF) screens, which to date have not been fully exploited for DNA repair studies, offer the possibility to further delineate the order of events and factors in the orchestrated DDR. In addition, the limited throughput of most of the DNA repair assays that are currently in use is a major disadvantage for unbiased screening approaches.
Here, we describe a GOF approach, based on the expression of chromatin factors, to screen for previously unknown modulators of DNA repair using two high-throughput systems. The first one is a specific tailored readout of kinetics of DNA repair based on the sequential accumulation and decrease of γH2AX and 53BP1 at breaks, analyzed in a 384-well format using machine learning, which identified numerous previously unknown factors to influence repair kinetics. The second system is a complementary platform developed specifically to allow the monitoring of the recruitment of tagged-proteins to DNA breaks in an arrayed format. This screen led to the identification of a number of candidates not previously linked to DNA damage that are recruited to or excluded from DNA breaks. We further provide proof-of-principle characterization of the PHD finger protein 20 (PHF20), which we discovered to be excluded from DNA breaks within seconds following damage and independent of PARP activity. Further, we found PHF20 to compete with 53BP1 for binding to H4K20me2, inhibiting recruitment of this factor to DSBs, and altering proper DNA repair. Our strategy defines a system that should allow for the interrogation of novel factors modulating DNA repair in a relatively unbiased way and in a high-throughput manner.
RESULTS
A high content screen for assessing chromatin regulation of DNA repair
To assess the contribution of novel chromatin factors to DNA repair, we first developed a high-throughput microscopy (HTM) assay for the analysis of kinetics of DNA repair in cells. For this purpose, we employed an immunofluorescence (IF) readout for repair based on the quantification of γH2AX and 53BP1 foci kinetics (formation and resolution) in cells that have been previously damaged with 3 Gy of gamma irradiation. The choice of γH2AX and 53BP1 among other markers of DNA breaks was based on their well-established dynamics in the DDR (Ciccia and Elledge, 2010) and the availability of high-quality reagents at low cost with high sensitivity for IF. To evaluate if quantification of nuclear foci over time (dynamics of foci) was a suitable readout for a high content DNA repair screen, we conducted a large-scale pilot experiment in human U2OS cells in a 96-well plate format. Non-irradiated (control) and irradiated cells (fixed at 15 min, 1.5 h, 6 h, and 24 h post-damage) were immunostained and images were acquired in a high content fluorescence microscope followed by analysis with the Cell Profiler Image analysis software (see STAR Methods).
As expected, phosphorylation of H2AX and formation of γH2AX and 53BP1 foci was readily detected 15 min after DNA damage, and foci persisted for at least 90 min, decreased at 6 h, and were only occasionally detected 24 h after damage (Figure 1A). High-throughput (HT) image analysis and quantification of γH2AX intensity, γH2AX foci number, and 53BP1 foci number per nucleus confirmed the kinetics observed (Figure 1B). The number of γH2AX and 53BP1 foci per cell temporally mirrored each other during the time course, since these proteins colocalize at DNA breaks (Bekker-Jensen and Mailand, 2010). As expected, this HT methodology clearly detected the absence of H2AX phosphorylation in irradiated cells pretreated with ATM inhibitor (Figure S1A) and the impairment in 53BP1 foci formation in cells defective for RNF8 (Figure S1B).
Figure 1. A high content screen for assessing chromatin regulation of DNA repair.

(A and B) Proof-of-concept high-throughput microscopy (HTM) assay for kinetics of DNA repair. (A) Accumulation of DNA damage response (DDR) proteins at DNA breaks in nuclei of U2OS cells after exposure to gamma irradiation (3 Gy) and recovery for the indicated times. Representative examples from the HTM images of immunostaining for γH2AX (red) and 53BP1 (green). Nuclei were counterstained with DAPI (blue). Scale bars, 10 μm. (B) HTM-mediated quantification of the kinetics of the DDR. Relative intensity of γH2AX staining per nucleus, number of γH2AX foci per nucleus, and number of 53BP1 foci per nucleus were determined in U2OS cells at the indicated times after DNA damage (3 Gy). Red bars represent the mean.
(C) Schematic of the ChromORFeome HCS for DNA repair.
(D) Diagram showing the kinetics of nuclear γH2AX foci in cells expressing individual ORFs in the ChromORFeome HCS (each well contains a particular ORF-expressing cell line). γH2AX foci number at each well and time point was obtained by getting the average of γH2AX foci per nucleus for all the nuclei in that well, and it is displayed with a color code. In gray, ChromORFeome control wells; in blue, empty wells. These are the results of a single experiment.
(E) Kinetics of nuclear γH2AX intensity in the two ChromORFeome HCS for DNA repair performed (replicate A and replicate B). The collection of all ORF-expressing cells in the ChromORFeome follow the expected kinetics of DNA repair. Nuclear γH2AX intensity for the entire ChromORFeome library-expressing cells was obtained by calculating nuclear γH2AX intensities for cells expressing individual ORFs of the library at each time point (mean ± SD; **p < 0.01; ****p < 0.0001 by one-way ANOVA with multiple comparisons, for each time point vs. non-irradiated control).
We next built an arrayed GOF library of open-reading frames (ORFs) by selecting chromatin-related ORFs from the human ORFeome collection developed at the Broad Institute (Yang et al., 2011), which we referred to as “ChromORFeome” library (Table S1). By conducting a multistep in silico selection, we compiled a list of 338 genes (in 348 clones) based on the following criteria: (1) proteins containing chromatin binding or DNA binding structural domains (such as Bromo-, Chromo-, PWWP-, JmjC, MBD, HMG box, SAND, among others), (2) proteins holding structural domains frequently reported in DDR pathways (such as BRCT, Tudor, MBT, SET, zinc finger, among others), (3) reported nuclear localization, and (4) proteins with a chromatin-related function, such as histone modifiers, histone readers, and chromatin remodelers (Figures S1C and S1D). This preliminary list was curated with (1) ORFeome availability and (2) sequencing validation of the clones.
Upon viral preparation of the library, we first optimized infection conditions in U2OS cells to maximize infection efficiency and minimize toxicity (Figures S1E and S1F). For ChromORFeome high content screen (HCS) for DNA repair (see workflow in Figure 1C), U2OS cells were infected with the ChromORFeome library in arrayed 384-well format (a single ORF per well) in five replicate plates, one per time point. On each plate, several control ORFs known to be irrelevant for kinetics of repair (Luciferase, eGFP, HcRed, and BFP) were included to define normal repair kinetics. After selection of infected cells, all plates except one were irradiated, and cells were fixed at 15 min, 1.5 h, 6 h, and 24 h after DNA damage. An automated immunostaining was performed for detection of γH2AX and 53BP1, and nuclei were counterstained with DAPI. Ten images per well were acquired with the Opera High Content Screening System. Collected images were analyzed with the Columbus platform for the detection and quantification of intranuclear foci up to 24 h after damage in every ORF-expressing cell line (Figure 1C) (see STAR Methods).
Analysis of the HCS for DNA repair revealed an increase in γH2AX intensity, γH2AX foci, and 53BP1 foci 15 min after DNA damage, as expected, for most of the ORF-expressing cell lines in the ChromORFeome and a subsequent decline after 90 min (Figures 1D and S1G). This transient increase and subsequent decrease in DNA damage signaling was also revealed when pooling all ORFs together during the time course of the experiment (Figure 1E) supporting the notion that most wells in the ChromORFeome screen follow the typical kinetics of DNA repair. The experiment was performed twice with high reproducibility (Figure 1E; Tables S2 and S3). Altogether, these results prove the sensitivity, robustness, and reproducibility of our method and support the potential application in determining different DNA repair kinetic patterns using this HT screening platform.
Analysis of the ChromORFeome HCS by machine learning uncovers chromatin factors that influence kinetics of DNA repair
The image analysis of the HCS resulted in a collection of parameters at three different levels: data per well, per nucleus, and per DNA damage focus. Among more than 30 different parameters from over one thousand cells (at each time point), we selected (1) the number of nuclei, (2) the intensity of γH2AX per nucleus, and (3) the number of γH2AX foci and 53BP1 foci per nucleus, for subsequent analysis and interpretation of the HCS.
We performed machine learning (ML) analysis to interpret the data obtained in the HCS (see STAR Methods). Nuclei number per well, mean and median intensities of γH2AX per nucleus, and mean and median number of γH2AX and 53BP1 foci per nucleus of the two replicates were introduced as input data for the ML analysis. Unsupervised ML using k-Means resulted in two distinct clusters. The ML analysis clustered all non-irradiated samples in the same cluster, suggesting the IR versus non-IR status was a main determinant for the clustering of the data even though it was not computed in the algorithm. We hypothesized that cluster 1 contained a non-active DDR and was referred to as DDR OFF. The second cluster contained all wells that significantly differed from cluster 1, as defined by the algorithm, and was named DDR ON. Next, we determined the clustering profiles for each ORF across the non-irradiated and the 15 min, 1.5 h, 6 h, and 24 h timepoints after damage and defined four qualitatively distinct phenotypes of DDR kinetics: A, B, C, and D (Figures 2A and 2B).
Figure 2. Analysis of the ChromORFeome HCS by machine learning uncovers chromatin factors that influence kinetics of repair.

(A) The 384-well plate layout showing the DDR kinetics phenotype for each ORF in the two ChromORFeome HCSs performed (replicate A and replicate B).
(B) Different DDR kinetics phenotypes A (blue), B (red), C (green), D (yellow) were pre-defined by the DDR status ON or OFF across the time points and are shownin a color code in the plate layout. The phenotype E (orange) consists of a different pattern of ON/OFF clusters over the time course that is not represented in the phenotypes A, B, C, or D. The control ORFs (surrounded by a black empty square in the plate layout) show a phenotype B (red) defined by an extinction of the DDR 6 h after DNA damage; we consider phenotype B the standard kinetics of the DDR. Fifteen hits were found under phenotype C (green) in both replicate experiments and are noted as purple circles in the plate layout. Phenotype C is characterized by a later extinction of the DDR (DDR OFF at 24 h after damage). No hits were found with phenotype A (blue) or D (yellow) in either replicate experiment.
(C) Table showing the list of 15 ORFs exhibiting delayed repair kinetics (phenotype C) by ML in the ChromORFeome HCS.
(D) Heatmaps of intensities of γH2AX per nucleus and number of γH2AX and 53BP1 foci per nucleus (mean and median per well) of the control ORFs and the 15 ORF hits found by ML at each time point. CHD2 was excluded by manual curation due to its anomalous γH2AX mean intensity signal.
(E) Representative images of a UV laser microirradiation experiment in U2OS cells stably expressing MAX-V5 and immunostained for γH2AX and V5-tag; nuclei were counterstained with DAPI. Arrows indicate the path of the UV laser inducing DNA breaks. Scale bars, 10 μm.
The most abundant phenotype (phenotype B) was characterized by the activation of DNA damage signaling 15 min after damage (ON), the persistence at 1.5 h (ON), and its extinction at 6 h (OFF), and it represents standard kinetics of DNA repair. Phenotype C showed delayed kinetics of DNA repair and differed from B in the persistence of signaling at the 6-h time point (ON) and its disappearance only 24 h after damage (OFF). Much less abundant among the ORFs in the ChromORFeome was phenotype A, in which the signaling was already extinguished as early as 1.5 h after damage (OFF), suggesting a more efficient repair. Wells under this phenotype were not consistent among replicates, thus precluding further analysis of phenotype A. No candidates showed a permanent activation of DNA damage signaling (phenotype D) across all timepoints. Control ORFs, noted as black squares in the plate representation of the ML results (Figure 2A), followed a typical kinetics of DNA repair (phenotype B), reaffirming the validity of the ML analysis. Fifteen ORFs were clustered under phenotype C in both replicates of the HCS (marked as purple circles in Figure 2A). The 15 ORF hits (ATF7IP, CHD2, FKBP2, H2AFX, H2AFZ, HDAC10, ING2, LBR, MAX, MORF4L1, RBBP8, SETDB1, SETDB2, SMAD1, and TRIM3) followed a kinetics of delayed repair (Figure 2C). Heat maps generated with input data used by the ML analysis revealed that the 15 hits showed an increased number of 53BP1 foci at the 6-h time point after DNA damage, compared with controls (Figures 2D). This was consistent with their classification under a delayed repair phenotype by the ML approach. Notably, the hit list includes factors with known roles in DNA repair, such as H2AX itself (phosphorylated and accumulated at the sites of DNA damage), RBBP8 (also known as CtIP, recruited by MRE11 to DNA breaks to promote resection of DNA ends and HR) (Escribano-Díaz et al., 2013; Paull et al., 2000; Rogakou et al., 1998; Sartori et al., 2007; Yun and Hiom, 2009), and MORF4L1 (partner of the BRCA complex and involved in HR repair) (Figure S2A) (Sy et al., 2009). The fact that both H2AFX and H2AFZ are hits in our screen is supported by their well-established roles in chromatin remodeling after DNA damage (Rogakou et al., 1998; Xu et al., 2012). The presence of such proteins among the identified hits attests to the strength and specificity of this kinetics screen. Our hit list is enriched in proteins that accumulate at DNA breaks (SETDB1, RBBP8, and γH2AX) and further characterization revealed that, for instance, MAX is also recruited to damaged chromatin upon UV laser microirradiation colocalizing with γH2AX (Figure 2E). In addition, in silico analysis of protein interactions among our hits evidenced the association that exists between proteins involved in DNA repair (H2AFX, H2AFZ, RBBP8) and chromatin remodelers and modifiers (MORF4L1, CHD2, ING2, HDAC10) and the connection between SETDB1, SETDB2, and ATF7IP, through the epigenetic mark H3 trimethylated at K9 (Figure S2B).
Downregulation of hits identified in the ChromORFeome HCS impacts DNA repair kinetics
We sought to validate the role of the hits obtained in the ChromORFeome HCS in DNA repair by assessing the impact of their downregulation, an assay that could provide an orthogonal approach to validate the HCS. An esiRNA (endoribonuclease-prepared siRNA) library individually targeting ATF7IP, CHD2, FKBP2, H2AFZ, ING2, LBR, MAX, MORF4L1, RBBP8, SETDB1, SETDB2, SMAD1, and TRIM3 was transduced into U2OS cells (in addition to negative and positive controls), and DNA repair kinetics was evaluated by HTM as described before (see workflow in Figure 3A). Transfection of U2OS cells with esiKIF11 decreased dramatically the number of cells detected by HTM, an expected phenotype upon downregulation of KIF11 (an essential factor of the mitotic spindle), therefore indicating successful transfection of esiRNA in our setup (Figure S3A). Treatment with an ATM inhibitor and transfection with siRNA against RNF8 or esiRNA against RNF168 abolished phosphorylation of H2AX and formation of 53BP1 foci in damaged cells, respectively, as expected (Figure S3B). Downregulation of RBBP8 (a critical player in DNA-end resection, also known as CtIP) with esiRNA enhanced 53BP1 foci formation, indicative of the abolishment of DNA-end resection and the promotion of NHEJ (mediated by 53BP1) over HR (Figure 3B). An increase in phosphorylation of H2AX was observed concomitant to the increase in 53BP1 foci in esiRNA RBBP8-transfected U2OS cells. For the vast majority of hits, the depletion by esiRNA induced a decrease in the number of 53BP1 foci after DNA damage: esiRNA targeting ATF7IP, SETDB1, SETDB2, H2AFZ, ING2, CHD2, LBR, FKBP2, and MAX (Figures 3B and S3C). The decrease in 53BP1 foci was accompanied by increased phosphorylation of H2AX at early time points after damage in cells transfected with esiRNA targeting CHD2, LBR, and FKBP2 or by reduced phosphorylation of H2AX at early time points after damage (esiRNA targeting MAX). Transfection with esiRNAs targeting TRIM3 or SMAD1 had no effect in DNA repair, and U2OS cells transfected with esiRNA against MORF4L1 only showed a transient increase in γH2AX phosphorylation immediately after damage. Of note, of 13 hits analyzed, 9 led to decreased 53BP1 foci, the opposite phenotype observed during the overexpression ChromORFeome screen. These findings validate our HCS strategy and provide support for a role in DNA repair for most of the hits identified in the screen.
Figure 3. Downregulation of hits identified in the ChromORFeome HCS impacts DNA repair kinetics.

(A) Schematic of the esiRNA small screen for DNA repair. An esiRNA library containing esiRNA targeting 13 hits identified in our HCS for DNA repair was transduced in U2OS cells. Untransduced U2OS cells and U2OS cells transduced with an esiRNA targeting Renilla Luciferase (esiRLUC) were used as negative controls. esiRNA against human KIF11 inducing mitotic arrest was used as positive control of transfection. Transfection of U2OS cells with esiRNA targeting RNF168 or siRNA targeting RNF8, as well as treatment with the ATM inhibitor KU55933 (10 μM, 2 h prior to irradiation), were used as positive controls in the assay.
(B) Relative intensity of γH2AX staining per nucleus and number of 53BP1 foci per nucleus were determined at the indicated times after DNA damage (3 Gy) in U2OS cells transfected with the negative control esiRNA (RLUC) or with esiRNA targeting a particular chromatin factor identified by ML in the HCS for DNA repair. The impact in DNA repair kinetics of the downregulation of four hits (RBBP8, ATF7IP, CHD2, MAX) compared to control cells (esiRLUC) is shown: increased γH2AX intensity and increased number of 53BP1 foci after damage (esiRBBP8), decreased 53BP1 foci after damage (esiATF7IP), increased γH2AX intensity at early time points after damage, and decreased 53BP1 foci after damage (esiCHD2) and decreased γH2AX intensity and 53BP1 foci after damage (esiMAX). Red bars represent the mean. (*p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001 by one-way ANOVA with multiple comparisons, for each time point vs. esiRLUC).
Overall, our strategy constitutes a valid platform for the analysis of kinetics of DNA repair in a high-throughput manner. As proof of principle, we used a chromatin ORF library (ChromORFeome) and assessed successfully the involvement of some of these factors in kinetics of repair. This platform could be exploited in a variety of different formats, including genetic and small-molecule screens (see discussion below).
A high-throughput platform to identify factors recruited to damaged chromatin
Recruitment to damaged chromatin is an intrinsic property of DNA repair factors (Aleksandrov et al., 2018). This characteristic is indeed shared by some of our ChromORFeome HCS hits identified by ML (H2AX, RBBP8, SETDB1, MAX). To directly evaluate recruitment of chromatin factors to DNA breaks, we designed an HT screen to identify proteins that localize to DNA lesions. Each clone of the ChromORFeome library carries a tag (V5-tag) that enables the detection of the subcellular localization with a unique specific antibody.
We developed a straightforward HT method based on laser-induced DNA damage to track the recruitment to DNA breaks of all 348 clones in the ChromORFeome library. Although previous studies have performed screens from pooled libraries (for instance, Izhar et al., 2015), our assay successfully attained laser breaks in a 384-well format. U2OS cells were infected in an arrayed 384-well format as described in the kinetics screen of Figure 1, and prior to the microirradiation, cells were pre-sensitized with Hoechst, a DNA intercalating agent, for efficient DNA breakage upon UV light excitement (Figure 4A).
Figure 4. A high-throughput laser microirradiation platform identifies chromatin factors recruited to DNA damage.

(A) Schematic of the high-throughput (HT) laser-induced DNA damage assay employing the ChromORFeome library.
(B) Immunofluorescence image showing γH2AX response (green) to DNA damage induced by 355-nm laser irradiation in a representative well of the 384-well plate. The image shows two consecutive fields within the well acquired with a 103 objective. Nuclei were identified with Hoechst.
(C) Table containing the list of the 10 ORFs that showed a phenotype of recruitment to laser-induced DNA breaks or exclusion from laser-induced DNA breaks in the ChromORFeome 384-well recruitment screen. The position of the 10 hits in the ChromORFeome plate is indicated.
(D) Representative micrographs from the 384-well recruitment screen showing the accumulation of the ChromORFeome protein RNF166 (in well I3 of the ChromORFeome library), immunostained with anti-V5 and shown in red, at laser microirradiation sites colocalizing with γH2AX stripes (green). Scale bars, 10 μm.
(E) Independent UV laser microirradiation experiment confirming the recruitment of RNF166 to DNA breaks in U2OS cells stably expressing the RNF166-V5 ORF construct. γH2AX staining is shown in green and RNF166 staining (anti-V5) is shown in red. Treatment with PARPi (AZD2281, Olaparib) 10 μM for 24 h prior to laser microirradiation abolished the formation of RNF166 stripes at damaged chromatin in U2OS cells. Scale bars, 10 μm.
(F) Representative micrographs from the ChromORFeome 384-well recruitment screen depicting the exclusion pattern showed by the ChromORFeome protein SUV420H1 (in well I23 of the ChromORFeome library), immunostained with anti-V5 and shown in red, at laser microirradiation sites colocalizing with γH2AX stripes (green). Scale bars, 10 μm.
For this screen, we utilized a PALM Microbeam microdissector equipped with a solid-state pulsed laser at 355 nm that allows the generation of large amounts of single-strand and double-strand DNA breaks in restricted subnuclear regions when sensitizing cells with Hoechst. To repurpose the microdissector for its use in an HT configuration, we used the Robosoftware to delineate a defined path of DNA breaks through a 384-well plate. The path of DNA breaks across the wells was detected via immunostaining against γH2AX (see STAR Methods) (Figure 4B). Twenty minutes after microirradiating the ChromORFeome infected cells, these were fixed and processed for immunostaining against V5-tag and γH2AX. The presence of DNA breaks across the 384-well plate was observed with a fluorescence microscope by the accumulation of γH2AX along the laser path. We manually scanned each well to score the colocalization of V5 and γH2AX double-positive stripes.
The performance of the screen was determined by assessing the localization of DNA repair factors known to be recruited to DNA breaks. For instance, the well containing the ORF for DNA ligase III (LIG3), a factor recruited to DNA breaks (Abdou et al., 2015), was clearly identified in the screen (Figure S4A). Image analysis revealed unknown factors recruited to sites of breaks, as detected by V5 and γH2AX colocalization in a striped pattern, and factors that were excluded from γH2AX stripes, showing an anti-stripe pattern (Figure 4C). In the screen, RNF166 (ring finger protein 166), with unknown role in DNA repair, was recruited to DNA breaks by exhibiting a very distinctive pattern of stripes colocalizing with γH2AX accumulation at laser-induced breaks (Figure 4D). Independent experiments showed the recruitment of RNF166 to DNA breaks is dependent on PARP activity (Figure 4E). By contrast, the histone methyltransferase SUV420H1 (KMT5B) showed exclusion from DNA breaks in the screen (Figure 4F). All newly identified hits (RNF166, PHF20, SUV420H1, DBF4B, PIAS4, LIG3, SMARCD1, TCF7L1, ATRIP, and HDAC1) were next validated in independent experiments where U2OS were infected de novo with the ChromORFeome clone and sensitized either with Hoechst or BrdU prior to laser microirradiation (Figure S4B). All the proteins (RNF166, PIAS4, LIG3, SMARCD1, TCF7L1, ATRIP, and HDAC1) reproduced the striped pattern colocalizing with γH2AX confirming the recruitment to DNA breaks, whereas PHF20 and SUV420H1, but not DBF4B, confirmed the exclusion from DNA breaks (Figures 5A andS4B). In summary, the HT platform we developed for localization of proteins to DNA breaks enabled the evaluation of more than 300 individual factors by generating DNA breaks in a single experiment and in a short period of time. Importantly, we detected the recruitment or exclusion of proteins from damaged chromatin in cell populations that presented a vast range of expression levels for the proteins investigated, further proving the high sensitivity of the HT recruitment assay.
Figure 5. PHF20 is excluded from damaged chromatin, regulates the DDR by competing with 53BP1, and is required for DNA repair.

(A) Independent UV laser microirradiation experiment confirming the exclusion from DNA breaks of PHF20 in U2OS cells carrying the PHF20-V5 ORF construct. γH2AX staining is shown in green and PHF20 staining (anti-V5) is shown in red. Scale bars, 10 μm.
(B) Time-lapse video frames from U2OS cells overexpressing PHF20 subjected to live imaging laser-track irradiation (see also Video S1). U2OS cells were transfected with PHF20-Clover; cells were laser microirradiated and imaged. Time-lapse video stills at 15 s and 15 min after irradiation are shown.
(C) Wild-type (WT) and PHF20-KO U2OS cells were exposed to increasing doses of gamma irradiation (IR), and cell viability was determined by colony survival assay (mean ± SD; multiple t test, **p < 0.01; ****p < 0.0001; n = 3).
(D–F) Wild-type (WT) and two PHF20-KO U2OS cell lines were subjected to DNA damage (gamma irradiation 3 Gy), and γH2AX and 53BP1 foci were analyzed by HTM at the indicated times after irradiation. Quantification of γH2AX foci number (D) and quantification of 53BP1 foci number per cell (E) is shown (*p < 0.05; ****p < 0.0001 by one-way ANOVA with multiple comparisons, for each time point compared to WT cells). Red bars represent the mean. (F) Representative images of γH2AX and 53BP1 immunostaining in WT and PHF20-KO cells 24 h after gamma irradiation (3 Gy). Scale bars, 10 μm.
(G) Pull-down competition assay. H4K20me2 peptide was used to pull down the Tudor domain of 53BP1 fused to GFP in the presence of increasing competing amounts of 53BP1 Tudor-GST or PHF20 Tudor-GST (0, 0.05, 0.1, 0.5, 1, 5 μg). Input samples and eluted samples were immunoblotted with anti-GFP antibody.
(H) Quantification of the in vitro competition assay shown in (G); 53BP1 Tudor-GFP levels were measured and normalized to control (0 μg) sample.
(I) Representative images of UV laser microirradiation experiment in U2OS cells stably carrying the PHF20-V5 ORF construct and treated with the indicated siRNA and inhibitors. ATMi (KU-55933) 10 μM for 1 h; ATRi (Toledo et al., 2011) 1 μM for 1 h; DNA PKi (NU-7441) 3 μM for 1 h; PARPi (AZD2281, Olaparib) 10 μM for 24 h. γH2AX staining is shown in green and PHF20 staining (anti-V5) is shown in red. Scale bars, 10 μm.
(J and K) Evaluation of DDR kinetics in WT and PHF20 overexpressing U2OS cells by HTM. U2OS WT cells were transfected with a Crispr-resistant PHF20 fused to GFP (PHF20R-GFP). GFP-positive (transfected) and GFP-negative (non-transfected) cells coexisting in the same well were analyzed for 53BP1 foci (J) and γH2AX foci (K) at the indicated time points after IR (3 Gy). WT cells overexpressing PHF20 (GFP positive cells) showed decreased levels of 53BP1 nuclear foci (at 0.25, 1.5, and 6 h after IR) and increased levels of γH2AX nuclear foci in non-irradiated conditions and at late time points after DNA damage (6 h and 24 h after IR). Red bars represent the mean. (****p < 0.0001 by one-way ANOVA with multiple comparisons, for each time point compared to GFP negative cells).
(L) Left: Schematics of I-SceI assay employing EJ5-GFP cells (adapted from Bennardo et al., 2008). Middle: Representative images of U2OS cells co-transfected with CTRL-mCherry or PHF20R-mCherry (red) and I-SceI enzyme. GFP-positive cells indicate NHEJ, while mCherry represents either CTRL or PHF20 signal. Right: Quantification of NHEJ efficiency (percentage of GFP signal over mCherry signal). *p < 0.05 by Fisher’s exact test. Scale bars, 25 μm.
(M) Quantification of PHF20 levels in U2OS cells transfected with PHF20-Clover at the laser-track site in live imaging experiments, untreated (black line) and treated with the PARPi Olaparib (blue line).
PHF20 is excluded from damaged chromatin
As mentioned earlier, we identified PHF20 as a protein excluded from sites of DNA breaks in the HT recruitment screen (Figures 4C and S5A). PHF20 is a component of the MOF-NSL histone acetyltransferase protein complex with roles in transcriptional activation and acetylation of histone 4 (at lysine 16) and p53 (Cai et al., 2010; Mendjan et al., 2006). PHF20 levels are found altered in different tumor types (Bankovic et al., 2010; Wang et al., 2002), and PHF20 stabilizes and activates p53 (Cui et al., 2012), but no active role of PHF20 in the regulation of DNA repair has been reported. Indeed, Phf20-null mice display a variety of phenotypes and die shortly after birth, presumably as a result of PHF20 functions in transcriptional regulation (Badeaux et al., 2012).
To validate PHF20 exclusion from DNA breaks in our recruitment screen, a freshly generated U2OS cell line overexpressing PHF20-V5 was tested in independent experiments of laser microirradiation confirming the anti-stripe pattern of PHF20 at damaged chromatin (Figure 5A). Although anti-stripes are unusual, previous studies have described an anti-stripe pattern for proteins TAF15, THRAP3, ATAD2, SP140, SAFB1, CIRBP, and RBMX where the exclusion from DNA breaks was, in most cases, preceded by a transient initial recruitment (Adamson et al., 2012; Beli et al., 2012; Chen et al., 2018; Gong et al., 2015; Izhar et al., 2015). Thus, we sought to investigate if PHF20 was transiently recruited before its exclusion observed at 20 min after laser-induced damage. We tracked by live imaging a PHF20 protein fused to the green fluorescent protein variant Clover (PHF20-Clover) upon DNA damage. Notably, we found PHF20 excluded as early as 4 s after damage without evidence of a previous recruitment to DNA lesions (Figure 5B and see Video S1, where exclusion happened already 4 s after irradiation).
We next generated PHF20 knockout cells with CRISPR/Cas9 technology (Figure S5B; see STAR Methods) and assessed the impact of PHF20 deletion on the ability to repair DNA damage. In the absence of PHF20, cells were more sensitive to gamma irradiation (Figures 5C and S5C) and showed increased γH2AX and 53BP1 nuclear foci compared to wild-type cells immediately after damage, a pattern that persisted for at least 24 h after damage (Figures 5D–5F, S5D, and S5E). Ectopic expression of a CRISPR-resistant PHF20-GFP in PHF20-KO cells fully rescued the increase in 53BP1 foci observed in the absence of PHF20 (Figures S5F–S5H). Together, these results argue that PHF20 is important for the regulation of kinetics of the DDR and is required for proper DNA repair.
PHF20 binds H4K20me2 through its Tudor domains (Cui et al., 2012). Interestingly, this histone mark is crucial for 53BP1 binding and stabilization at sites of breaks (Botuyan et al., 2006). The H4K20me2 mark is abundant throughout the genome and not affected by DNA damage, and previous work has suggested H4K20me2 may become exposed locally after damage to allow for 53BP1 binding and stabilization at DNA breaks (Acs et al., 2011; Mallette et al., 2012). Thus, we hypothesized that PHF20 could be masking H4K20me2 to prevent erratic binding of 53BP1. Consistent with this idea, knockout of PHF20 resulted in increased 53BP1 foci even without exogenous damage (Figure 5E). A prediction from such model is that PHF20 should exhibit stronger affinity than 53BP1 for the H4K20me2 histone mark. To test this, we performed an in vitro competition binding assay for H4K20me2 and observed that PHF20 out-competed a 53BP1 peptide (53BP1 Tudor-GFP) better than a full-length 53BP1 (Figures 5G and 5H), indicating that PHF20 binds H4K20me2 with stronger affinity than 53BP1.
Next, we evaluated the dynamics of 53BP1 and PHF20 at damaged chromatin by testing whether 53BP1 binding to DNA breaks displaced PHF20 from damaged chromatin. Downregulation of RNF168, critical for 53BP1 recruitment to DNA breaks, impaired 53BP1 recruitment to laser-induced breaks, as expected, but had no effect on PHF20 (Figures 5I, S5I, and S5J), suggesting that PHF20 displacement from damaged chromatin is upstream of 53BP1 recruitment. We next tested whether enforced expression of PHF20 could inhibit recruitment of 53BP1. Strikingly, acute overexpression of PHF20 prevented the recruitment of 53BP1 to foci after gamma irradiation, while increasing γH2AX foci at late time points, in comparison to wild-type (WT) cells (Figures 5J, 5K, and S5H), indicating unresolved DNA damage. To directly assess whether PHF20 affects DNA repair, we took advantage of an NHEJ-repair assay (Bennardo et al., 2008), a pathway dependent on 53BP1 (Scully et al., 2019). Notably, acute overexpression of PHF20 significantly impaired NHEJ (Figure 5L), providing further support to the importance of a fine balance between PHF20 and 53BP1 for appropriate NHEJ function.
Together these results suggests that (1) the displacement of PHF20 from damaged chromatin may license the correct 53BP1 binding to H4K20me2, which positions PHF20 displacement as a potential checkpoint to avoid spurious activation of the DDR, and (2) in damaged chromatin, PHF20 may buffer transient, physiological exposures of the H4K20me2 mark to raise the threshold for the triggering of the DDR cascade.
To further explore the molecular determinants of PHF20 exclusion from damaged chromatin, we pretreated cells with selective ATM, ATR, or DNA-PK inhibitors before inducing laser breaks. We observed PHF20 exclusion occurred independent of ATM, ATR, DNA-PK signaling and γH2AX accumulation at damaged chromatin (Figures 5I and S5K). These results suggest that PHF20 exclusion might be dictated by a different, non-canonical, upstream signaling in the DDR. In that regard, one of the earliest events in DNA damage signaling and an important mediator of the accumulation of DNA repair factors is the synthesis of PAR chains by PARP at DNA lesions. PAR structures not only serve as docking platforms for accumulation of DNA repair factors but also generate conformational changes in the chromatin that facilitate re-localization of proteins upon damage (Chou et al., 2010; Smith et al., 2019). Remarkably, inhibition of parylation with a PARP inhibitor (Olaparib) (Figures 5I, 5M, and S5L) or downregulation of PARP1 with siRNA (Figures S5M and S5N) did not impair the exclusion of PHF20 from microirradiation tracks in U2OS cells. Taken together, our data show that PHF20 is rapidly excluded from sites of damage independently of other known damage signaling factors, including those of early recruitment to sites of damage, such as ATM, ATR, DNA-PK, PARP1, and 53BP1.
Our model postulates that localized chromatin conformational changes occurring immediately after damage may be responsible for the displacement of PHF20 that enables efficient recruitment of 53BP1. Cells lacking PHF20 are more sensitive to gamma irradiation and retain more damage at later time points, suggesting that PHF20 is an important contributor for the kinetics of the DDR, which provides a strong validation to our HT screen.
DISCUSSION
We have developed two HT screening platforms for the discovery of proteins involved in the repair of DNA lesions. We used a highly sensitive methodology to quantify DNA repair kinetics through γH2AX and 53BP1 nuclear foci, both robust indicators of DSB in individual cells. The imaging platform was coupled to ML tools for computing thousands of data in an unbiased fashion. ML analysis of data across wells, plates, and timepoints allowed the monitoring of foci from a basal control condition, through exogenous DNA damage perturbation and over time of DNA repair, in thousands of cells. A second imaging screen determined the localization of more than 300 proteins to damaged chromatin in a single, rapid experiment by means of HT laser microirradiation of 384-well plates. The scalability of both systems into multiple 384-well plates represents an additional technical advance that will potentiate the utility of both screening platforms. We employed a library of putative DNA and chromatin binding factors tailored for the purpose aimed in this study. The library was in an ORF format, providing several advantages: (1) overexpression systems are under-represented in research and therefore could provide additional discoveries, and (2) it allowed us to track the localization of more than 300 proteins upon DNA damage.
Both platforms demonstrated high sensitivity: (1) in the identification of expected phenotypes under different genetic contexts and inhibitory drugs (gene knockdown, esiRNA library, and treatment with drugs of broad interest in the DNA field) and (2) in the identification of distinct phenotypes across the library, such as delayed and accelerated DNA repair, recruitment, and exclusion from damaged chromatin.
Among the hits identified, we focused on PHF20, a protein we found to be excluded from damaged chromatin. It was previously shown that PHF20 Tudor domains bind the same histone modifications as 53BP1 (H3K9me2 and H4K20me2) (Badeaux et al., 2012). Although only transcriptional roles have been identified so far for PHF20, PHF20 is part of the NSL-MOF complex together with MOF protein, for which functions in DNA repair have been described. We hypothesized that PHF20 could be masking H4K20me2 in non-damaged chromatin to prevent 53BP1 binding, and its exclusion immediately after DNA damage could define a role for PHF20 as a licensing factor for 53BP1. We show that PHF20 competes with 53BP1 (in vitro) for the histone mark H4K20me2, an important regulator for 53BP1 recruitment to damaged chromatin, and that overexpression of PHF20 blocks recruitment of 53BP1 to DSBs, inhibiting NHEJ repair. Moreover, the absence of PHF20 impacts DNA repair by impairing extinction of the DDR and sensitized cells to DNA damage, without affecting cell cycle distribution (Figures S5O and S5P). The fact that other components of the MOF-NSL complex such as ASH2L, MCRS1, and WDR5 do not show exclusion or recruitment to damaged chromatin suggests that PHF20 functions in damaged chromatin may be independent of canonical functions of the MOF-NSL complex. The lack of transcriptional changes in main DDR proteins upon PHF20 depletion (Figure S5Q) supports a function for PHF20 different from its transcriptional role attributed as part of the MOF-NSL complex. In addition to recognizing the same histone modifications, Tudor domains of both PHF20 and 53BP1 bind to the same methylated residues of p53 (Lys370 and Lys382), recognized by MDM2 and leading to degradation of p53 (Cui et al., 2012). We can speculate that 53BP1 and PHF20 binding to H4K20me2 would influence binding to p53 or vice-versa, linking the roles in damaged chromatin to the regulation of p53 activity.
We believe our methodologies could be useful for a large variety of screens to help elucidate the complexity of the cellular response to DNA damage. Both systems can be exploited for the discovery of DNA repair factors by using chemical and genetic libraries in arrayed format. Such libraries could be screened in specific genetic backgrounds and with different genotoxic agents (such as alkylating agents, radiomimetic drugs, topoisomerase II inhibitors), where the efficiency of independent DNA repair pathways could be assessed through dynamics of different DNA repair foci. For instance, RPA foci could be quantified in cyclin B-positive cells upon gamma irradiation for monitoring repair by HR. Chemical and genetic libraries could as well be exploited using the HT platform for recruitment to laser-induced DNA breaks, in order to investigate the molecular requirements for tethering both unidentified and known repair proteins to damaged chromatin, such as 53BP1, ATM, RPA, and BRCA1.
Limitations of the study
During the analysis of the results obtained in the repair screen, we encountered limitations that can be overcome by introducing minor technical modifications in the screen. For instance, discriminating between a defect in DNA repair and a checkpoint adaptation in cells that present an active DDR 24 hours after damage (phenotype D, yellow) is not possible in our experimental conditions. Hence, we recommend an additional immunostaining against phosphorylated H3Ser10 (or cyclin B1) together with γH2AX and 53BP1 staining that will suffice to discern whether those cells have entered mitosis with DNA damage and therefore exhibit checkpoint adaptation.
On the other hand, the HT laser microirradiation generates a clear delay in DNA damage between the individual wells across the 384-well plate. As a consequence, it does not allow the comparability in terms of recruitment time. This minimal caveat does not undermine the potential and significance of the approach to discover proteins recruited to DNA damage since we have identified in a single experiment nine proteins recruited to or excluded from laser breaks, with a validation rate of 90%. The time lapse between DNA damage and fixation was 20 min +/− 28 min for the whole ChromORFeome library. This is consistent with previous works showing that while a small fraction of DDR factors is quickly recruited to DNA lesions and immediately removed, a larger fraction of proteins that is recruited to DNA breaks remain at damaged chromatin for longer times, or is recruited later after damage and remain at DNA lesions up to 120 min after damage (Aleksandrov et al., 2018; Gaudreau-Lapierre et al., 2018; Mistrik et al., 2016). In addition, this methodology is not intended to compare recruitment kinetics of proteins to damaged chromatin nor claim the recruitment status of a protein at a specific time point after damage. Variations of the screen can be performed, by fixing cells at later time points after damage and to identify factors required to reset the chromatin after DNA repair has occurred, something for which we still know very little. As well, if a single time point after damage is required for a given experiment, successive wells could be fixed every 17 s using a microplate washer dispenser as in the DNA repair kinetics screen.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Raul Mostoslavsky (rmostoslavsky@mgh.harvard.edu).
Materials availability
Single DNA clones from the ChromORFeome library can be obtained from the Broad Institute http://www.orfeomecollaboration.org/
Data and code availability
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines were originally obtained from the ATCC bioresource. Cells were maintained in Dulbeccós Modified Eagle Medium with high glucose (Thermo Fisher #11965) supplemented with 10% fetal bovine serum and penicillin-streptomycin and incubated at 37°C and 5% CO2. Cell lines have not been authenticated.
METHOD DETAILS
ChromORFeome viral library preparation and titration
A library of 338 chromatin ORFs and control ORFs (Luciferase, eGFP, HcRed and BFP) was assembled in the lentiviral vector plx304 from the human ORFeome (Yang et al., 2011) and named “ChromORFeome” library. Lentivirus for the ChromORFeome library were prepared by the Broad Institute Genetic Perturbation Platform and provided in 384 well plate format including empty wells as negative controls for the infection.
The accurate volume of virus in order to achieve maximal infection efficiency and minimal toxicity of the ChromORFeome library were determined in optimization experiments using the fluorescent reporter virus plx304-HcRed-V5 (ccsbBroad304_99988), a control ORF from the library. U2OS cells were plated in 384 well plates (Greiner Bio-One # 781090), and infected the following day with different amounts of virus (0, 0.5, 1, 1.5, 2, 2.5 and 3 μl) per well by spin-infection at 2250 rpm for 1 hour at RT. Twenty-four hours after infection, blasticidin (Invivogen # ant-bl-05) was added to the cells in order to reach different concentrations (2, 3, 5, 7.5, 10, 15, 20 μg/ml) in cell culture media and was kept for 5 days. Cells were washed with PBS, fixed with 4% paraformaldehyde (PFA) (Electron Microscopy Sciences # 15710) in PBS and nuclei were counterstained with DAPI. Images were automatically acquired in a BD Pathway 435 high-througput microscope with AttoVision 1.5 software (Becton Dickinson) using a dry 40x magnification lens and analyzed for the detection of HcRed-positive and negative cells using CellProfiler2.0 software (McQuin et al., 2018). Maximal infection efficiency (91% HcRed positive cells) and minimal toxicity was reached with 350 cells per well, 1 μl of virus, 4 μg/ml polybrene and 15 μg/ml blasticidin (Figures S1E and S1F).
ChromORFeome library transduction
U2OS cells were plated in 384-well plates (350 cells per well). The following day, cells were spin-infected with the lentiviral-packaged ChromORFeome library (1 μl of virus per well) in the presence of 4 μg/ml polybrene. Twenty-four hours later, blasticidin was added to cells to a final concentration of 15 μg/ml. Experiments were performed after 5 days of selection with blasticidin.
High Content Screen for DNA repair
A high-throughput microscopy assay for evaluation of DNA repair was developed allowing for the detection and quantification of the number of γH2AX and 53BP1 nuclear foci, as well as γH2AX levels per cell.
For experiments performed in 96-well format (Figures 1A, 1B, S1A, and S1B) 8,000 U2OS cells per well were plated in replicate 96-well plates (Falcon Corning # 353219). The following day, all plates except one (non-irradiated control), were irradiated with 3 Gy of gamma irradiation to induce DNA breaks. All irradiated plates were fixed at different time points after damage 15 min, 1.5 h, 6 h and 24 h, as well as one non-irradiated control plate. Cells were washed with PBS, fixed with 4% paraformaldehyde (PFA) (Electron Microscopy Sciences # 15710) in PBS for 10 min, permeabilized (0.1% NaCitrate, 0.1% Triton X-100) for 5 min at RT, washed with washing solution (PBS with 0.1% Tween-20, 0.25% (w/v) BSA) and incubated with blocking solution (PBS with 0.1% Tween-20, 2.5% (w/v) BSA and 10% goat serum (Sigma-Aldrich #G9023)) for 30 min at RT. Cells were incubated in the following antibodies and dilutions in blocking solution: anti-phospho-H2AX (Ser139) 1:1000 (clone JBW301, Sigma-Aldrich #05–636) and anti-53BP1 (Santa Cruz # sc22760) 1:1000 overnight at 4°C. On the following day, cells were washed with washing solution twice and incubated in the following species-specific fluorophore conjugated secondary antibodies for 1 h at RT in blocking buffer: anti-mouse Alexa Fluor 555 (Thermo Fisher # A-21422) 1:400, anti-rabbit Alexa Fluor 488 (Thermo Fisher # A11008) 1:400. Following secondary antibody incubation, cells were washed with washing solution twice, nuclei were counterstained with DAPI (Sigma-Aldrich # D9542) 300 nM in PBS for 5 min at RT and cells were kept on PBS at 4C until imaging. Pictures were automatically acquired in a BD Pathway 435 high-throughput microscope with AttoVision 1.5 software (Becton Dickinson) using a dry 40x magnification lens. Nine images per well were acquired in the DAPI, γH2AX and 53BP1 channels. Images were analyzed with CellProfiler2.0 sofware. Briefly, nuclei were identified using the “Identify Primary Objects” module in the DAPI channel, 53BP1 foci and γH2AX foci were identified using “Identify Primary Objects” module in the 53BP1 and γH2AX channels. The module “Relate Objects” allowed for the matching of foci with their parental nucleus. Data were exported as .csv files and were analyzed using Excel. Data files contained the parameters measured in each nucleus (foci number and intensity). Data plotting was performed in GraphPad Prism software. A minimum of 600 cells were analyzed for each timepoint.
For High Content Screening, 350 U2OS cells per well were seeded in replicate 384-well plates (Greiner Bio-One # 781090). The following day, cells were spin-infected with the library as described in this section (ChromORFeome library transduction). After a 5-days selection period with blasticidin, all plates except one (non-irradiated control), were irradiated with 3 Gy of gamma irradiation to induce DNA breaks. Irradiated cells were allowed to recover at 37°C, 5% CO2 for the indicated time points after damage (15 min, 1.5 h, 6 h and 24 h). All plates were fixed with 4% PFA in PBS as previously described, washed with PBS and automated immunostaining was performed with a microplate washer-dispenser (Biotek). Permeabilization, blocking and immunostaining for γH2AX and 53BP1 were performed as described before in this section. Nuclei were counterstained with DAPI and cells were kept on PBS at 4°C until imaging. Plates were scanned using the automated confocal microscope Opera High Content Screening System (Perkin Elmer) at 40x magnification at the Rubin’s lab in the Harvard Department of Stem Cell and Regenerative Biology. Ten images per well were acquired in DAPI, γH2AX and 53BP1 channels. Images were analyzed with Columbus software (Columbus Image Data Storage and Analysis System, Perkin Elmer) and several parameters were measured in all nuclei detected across the kinetics experiment and across the different ORFs. Data from γH2AX nuclear intensity, number of 53BP1 foci and number of γH2AX foci per nucleus was utilized in subsequent analysis and data interpretation performed with machine learning. The experiment was performed two times.
Machine learning
For each experiment, an inter-plate normalization was performed before the classification. Machine learning clustering was computed by using as descriptors mean and median intensities for γH2AX and mean and median foci number for γH2AX and 53BP1. This last step was tailored by using k-Means (unsupervised learning algorithm) establishing two clusters. The number of clusters was defined by grouping all non-irradiated samples in one of them. The idea behind it was to find alterations in response to DNA damage and therefore the wells with differences versus non-irradiated wells should appear in the other cluster. For hit nomination, only wells containing more than 100 nuclei and showing up in the same cluster during the two replicate experiments were considered. Heatmaps for the selected variables show the proximity for the controls and hits found. Normalization, machine learning process, heat-maps, and graphs were computed by using Orange data mining software (Demsar et al., 2013).
esiRNA small screen for DNA repair
The esiRNA custom library targeting 13 hits from our ChromORFeome HCS for DNA repair, including transfection control (esiRNA KIF11), positive control (esiRNA RNF168) and negative control (esiRNA RLUC) was purchased from Sigma-Aldrich (MISSION esiFLEX, Eupheria Biotech). U2OS cells were transduced with esiRNA in 384-well plates (Perkin Elmer # 6057302) by reverse transfection using the following amounts per well: 0.125 μl RNAimax; 12.5 ng esiRNA; 15 μl Optimem and 1000 U2OS cells in 35 μl of DMEM 10% FBS without antibiotics. For transfection with RNF8 siRNA: 0.075 μl of RNAimax; 0.2 pmol of a mix of both RNF8 siRNA (CAGAGAAGCUUACAGAUGU and GGAGAUAGCCCAAGGAGAA); 15 μl Optimem and 1000 U2OS cells in 35 μl of DMEM 10% FBS without antibiotics. All transfections were performed in duplicate (2 wells per plate) and in replicate plates (5 plates). Two days after transfection, all replicate plates, except one (non-irradiated sample), were irradiated with 3Gy and let to recover for the indicated times before fixation. As additional positive control, cells were treated with ATM inhibitor ((KU-55933, Selleckem # S1092) 10 μM 2 h prior to irradiation. Analysis of DNA repair kinetics (nuclear γH2AX intensity and 53BP1 nuclear foci over time) was performed as decribed previously in this section, images were acquired in an Opera High Content Screening System (Perkin Elmer) and analyzed in Acapella.2 software.
High throughput UV laser microirradiation
U2OS cells were infected 384-well plates (Greiner Bio-One # 781090) with the ChromORFeome library as described in this section (ChromORFeome library transduction). After a 5 days selection period with blasticidin, cells reached 90% confluence. Cells were pre-sensitized with Hoechst 33258 (Sigma-Aldrich # B2883) (10 μg/ml) for 20 min prior to laser microirradiation. Hoescht incubation and microirradiation experiments were conducted in DMEM without phenol red and supplemented with penicillin/streptomycin and 10% FBS. A Palm Microdissector with Robosoftware was used to induce DNA breaks with a 355 nm solid-state laser (UV-A) in a HT manner across the 384 well plate. First, cells were focused with the 20x objective. Then, under the 2.5x objective, a line was “drawed” accross the middle of all the wells in the first row by using the “Element” tool in the Palm Robosoftware menu. Copy and paste of the line element was performed in all the rows of the plate by using the Palm Robosoftware menu. The 20x objective was used to re-focus the cells and perform the laser irradiation as the following: all the “line elements” created in all rows were selected at a time, or one by one, then the tool “start cutting laser” was selected to initiate the microirradiation. The microdissector generated a UV laser path across all the wells in the first row of the 384 well plate and continued irradiating consecutive rows in an automated manner, while the focus was adjusted manualy during the process. The microdissector took 7 min to irradiate a row. The library was splitted in four 384 well plates that were irradiated consecutively. The conditions used for the laser microbeam were the following: speed 100%, focus 72 and energy 50%. Twenty minutes after microirradiation, cells were washed with PBS, fixed with 4% PFA in PBS for 10 min and immunostaining was performed as previously described in this section using the primary antibodies: anti-phosphoH2AX (Ser139) (Cell Signaling # 9718) 1:1000 and anti-V5 tag (Abcam # 27671) 1:500 overnight at 4°C. Following secondary antibody incubation cells were kept in PBS until imaging. Immunostaining was assessed using a Leica DMI4000 B immunofluorescent microscope. The path of DNA breaks across the nuclei in the plate was detected by the presence of a positive staining of γH2AX stripes in nuclei. In those nuclei with positive staining of γH2AX, the presence of a stripe colocalizing with γH2AX stripe was assessed in the V5 channel. A positive V5 stripe colocalizing with γH2AX in a well would suggest the recruitment of the candidate protein to DNA breaks (or chromatin surrounding a DNA break) while an anti-stripe pattern would suggest the candidate protein moving out from damaged chromatin. Representative images of V5-striped or anti-striped nuclei were acquired using the 40x objective.
Plasmids, lentiviral production and infection, transfection with siRNA
Lentivirus were generated by transfecting 293T cells with the indicated vectors (plx304 or pLentiCRISPRv2) and the packaging plasmids deltaVPR and VSVG following the lipofectamine 2000 (Invitrogen #11668019) transfection protocol. 293T medium was changed 24 h post-transfection. Virus-containing supernatants were harvested 48 h post-transfection and passed throught a 0.45 μm filter. Cells were infected by spin-infection in a centrifuge with equal ratio of supernatant containing viral particles to cell culture medium and 8 mg/ml polybrene in a 6 well plate, at 2250 rpm for 1 hour at RT. Cells were selected with blasticidin (plx304) or puromycin (plko and pLentiCRISPRv2) containing media 48 hours after infection. Lentivirus of plx304 from the human ORFeome (Yang et al., 2011) were produced to generate U2OS cell lines overexpressing candidate hits for validation experiments, lentivirus of plko were used to knockdown human PHF20 and the lentiviral plasmid pLentiCRISPRv2 (Addgene, 52961 ) was used to express Cas9 and sgRNAs for the generation of PHF20-KO U2OS cell lines.
For small interfering RNA (siRNA)-mediated knockdown, U2OS cells were transfected with 30 nM of either RNF168 siRNA (Dharmacon # J-007152–05-0005), PARP1 siRNA (Dharmacon # J-006656–06-0005 and J-006656–07-0005), custom designed RNF8 siRNA (#1 CAGAGAAGCUUACAGAUGU and #2 GGAGAUAGCCCAAGGAGAA) or a control siRNA (Sigma-Aldrich Mission siRNA universal negative control, #SIC001) using the reverse protocol for Lipofectamine RNAiMAX (Invitrogen #13778030) as per manufacturer’s instructions. Assessment of siRNA-knockdown by immunoblotting was performed 48 h after transfection (siPHF20 and siRNF168) or by RT-qPCR (siPARP1).
Generation of PHF20-KO cell lines with CRISPR-Cas9
Two sgRNAs were designed to target the first exon of PHF20 (sg1: AAGCATCCACCTAACAGACG and sg2: CAACCTTCGGGCCCTGG CAA) and cloned into the pLentiCRISPRv2 plasmid. Lentiviral particles containing the pLentiCRISPRv2 construct with the sgRNA targeting PHF20 were produced, and U2OS cells were infected as described above in this Methods section. U2OS PHF20-KO clonal cell lines were obtained by clonal selection of the infected population after selection with puromycin. PHF20-KO clonal cell lines were subjected to western blotting to confirm the absence of PHF20 protein and Sanger sequencing after Topo cloning to analyze their genotype, the allelic frequency and the edited sequence. PHF20-KO clonal cell lines were named after the number of the sg followed by a number unique for that clone (for instance, PHF20 KO 1.1 and PHF20 KO 1.2 were generated with sg1 and KO 2.3 was generated with sg2). When indicated, polyclonal PHF20 KO populations were used in the experiment. Sequencing results confirmed large deletions and frameshift mutations in all alleles of the clones used in this manuscript KO 1.1, KO 1.2 and KO 2.3.
Validation of ORF hits recruited to DNA damage
U2OS cells stably overexpressing an ORF hit were generated by de novo infection with the correspondent plx304-ORF as described before in this section. Validation was conducted by independent UV laser microirradiation experiments either in a Palm Microbeam (Zeiss) or in a MMI CellCut system (Molecular Machines & Industries) and in live imaging laser track irradiation experiments (described in this section).
Immunofluorescence
For quantification of DNA damage foci over time after irradiation, U2OS cells were seeded in 96 well plates (Greiner Bio-One # 655087) (8000 cells per well) exposed to 3 Gy of gamma irradiation and allowed to recover for the indicated time points in a tissue culture incubator at 37°C and 5% CO2. Cells were washed with PBS, fixed with 4 % PFA in PBS for 10 min and immunostained as previously described using the following primary antibodies and dilutions: anti-phospho-H2AX (Ser139) 1:1000 (Cell Signaling # 9718) and anti-53BP1 (BD # 612522) 1:1000 overnight at 4°C. Following secondary antibody incubation with anti-mouse Alexa Fluor 555 (Thermo Fisher # A-21422), and anti-rabbit Alexa Fluor 488 (Thermo Fisher # A11008) or anti-rabbit Alexa Fluor 647 (Thermo Fisher # A21244) 1:400, cells were washed with PBS, nuclei were counterstained with DAPI 300 nM in PBS for 3 min at RT. Plates were scanned in a automated confocal microscope Opera High Content Screening System (Perkin Elmer) at 40x magnification at the CNIO (Madrid, Spain). Thirty images per well were acquired in DAPI, γH2AX and 53BP1 channels. Images were analyzed with Acapella 2.0 software (Perkin Elmer) to determine the number of γH2AX and 53BP1 foci per nucleus and γH2AX nuclear intensity in each cell. A minimum of 500 cells were analyzed per condition.
UV laser microirradiation
U2OS stable cell lines generated by infection with candidate ORFs were seeded in 8-well glass surface chamber slides (Lab-Tek II, Nunc # 154534) at a density of 50 000–80 000 cells per well in order to have cells at 80% confluency at the time of microirradiation. The following day, cells were pre-sensitized with the nucleotide analog BrdU (Sigma-Aldrich # B5002) 10 μM for 24 hours, or alternatively, with the DNA-intercalating dye Hoechst 33258 (Sigma-Aldrich # B2883) 10 μg/ml for 20 min prior to laser microirradiation. During presensitization and microirradiation steps cells were kept in in DMEM without phenol red supplemented with penicillin/streptomycin and 10% FBS. When indicated, the following inhibitors were added to the cells prior to microirradiation and during pre-sensitization: ATMi (KU-55933, Selleckem # S1092) 10 μM for 1 h; DNA PKi (NU7441, Selleckem # S2638) 3 μM for 1 h; PARPi (AZD2281, Selleckem # S1060) 10 μM for 24 h and ATRi (Toledo et al., 2011) 1μM for 1 h. Two alternative microdissection systems were used in UV laser microirradiation experiments: the MMI CellCut system (Molecular Machines & Industries) provided with a 355 nm solid state laser at 4 mW and 56% power using a 40x magnification lens (0.6 aperture) and the Palm Microbeam (Zeiss) provided with a 355 nm, using a 40x magnification lens and 65% focus; 50% laser speed; 30% or 35% laser energy for Hoechst and BrdU pre-treated cells, respectively. After a recovery time of 20 minutes, cells were subjected to immunostaining as described previously in this section. The following primary antibodies and dilutions were used: anti-V5 tag (Abcam # ab27671) 1:1000; anti-phospho-H2AX (Ser139) (Cell Signaling # 9718) 1:1000; anti-phospho-H2AX (Ser139) (Millipore # 05–636) 1:1000; anti-53BP1 (BD # 612522) 1:1000; anti-PAR (Enzo # ALX-210–890A-0100). For the experiment combining the three PI3K inhibitors, the following antibodies were used: mouse IgG2a anti-V5 tag (Abcam # ab27671) 1:1000 followed by anti-mouse IgG2a Alexa488 (Jackson ImmunoResearch #115–547-186) 1:200, rabbit anti-PAR (Enzo # ALX-210–890A-0100) followed by anti-rabbit IgG Alexa 647 (Invitrogen #A-21244) 1:250 and mouse IgG1 phospho-H2AX (Ser139) (Millipore # 05–636) 1:1000 followed by anti-mouse IgG1 Rhodamine Red (Jackson ImmunoResearch #115–297-185) 1:100. Images were captured in a fluorescence microscope under 20x or 40x magnification and manualy analyzed for the detection of V5-positive stripes or anti-stripes at DNA breaks, through colocalization with γH2AX or PAR stripes.
Live imaging laser-track irradiation
For live imaging experiments the plasmid pPHF20-Clover (expressing PHF20 fused to Clover fluorescent protein) was generated by subcloning PHF20 from the plx304-PHF20-V5 (ccsbBroad304_08258) with XhoI and EcoRI into pClover-J1 (gift from Graham Dellaire). One hundred thousand U2OS cells were seeded on a glass bottom 35-mm dish and cultured in DMEM + 10% FBS. Cells were transfected using 2:1 Lipofectamine 2000 (Invitrogen #11668019) to total DNA μg as per manufactureŕs instructions. Plasmid pPHF20-Clover (0.15 μg) was mixed with the non-expressing vector pBluescript (0.6 μg) as a carrier DNA for 0.75 μg of total DNA. On the day of imaging, cells were treated with 2 mM Hoechst 33342 for 20 min and washed 3 times with PBS. Cells were treated with 10 μM of PARP inhibitor (AZD2281, Selleckem # S1060) for at least of 1 hour in live cell media (phenol red-free DMEM supplemented with penicillin/streptomycin, 125 mM HEPES and 10% FBS). Cells were kept at 37°C in a humidified chamber during imaging. Images were captured on a Marianas spinning disk confocal microscope system (Intelligent Imaging Innovations, 3i) based on a Zeiss Axio Cell Observer. UV laser damage was induced by a 100 mW, 405 nM diode laser using a Vector Scan Unit (3i), in which effective light output was measured as ∼8 mW at the objective when using 100% power. A single line scan of the 405 nm laser at 70% power was sufficient to generate DNA DSBs as demonstrated by the rapid recruitment of KU70 (Andrin et al., 2012), which was estimated to be equivalent to ∼40–60 Gy cellular dose by the aforementioned method in (Bekker-Jensen et al., 2006). Cells were observed using a 63x objective (1.4 N.A) lens and images were captured on an Evolve 512 electron-multiplying CCD (EMCCD) (Photometrics) using Slidebook 6 software (3i). Recruitment or exclusion was quantified and normalized against the background within the nucleus. Background was determined as an area far from the damaged site but of the same size and within the same nucleus. Both parameters were first normalized against the image background (outside cell area) to account for variabilities (laser power fluctuations, camera gain adjustments) between live-cell imaging experiments. Twenty two untreated cells and fifteen Olaparib treated cells were used for quantification.
Immunoblotting
Cells were rinsed once with cold PBS and lysed in ice-cold RIPA buffer containing 50 mM Tris-HCL (pH 7.5), 150 mM NaCl, 1% Ige-Pal, 0.1% sodium dodecyl sulfate (SDS), 5 mM EDTA, protease inhibitor cocktail (Complete Mini, Roche Diagnostics), 2 μM sodium orthovanadate (Na3VO4), 1 mM dithiothreitol (DTT), 5 μM trichostatin A (TSA) and 1 mM phenylmethylsulfonyl fluoride (PMSF). Cell lysates were incubated on ice for 20 min and clarified by centrifuging at 16 000 g for 10 min at 4°C. Protein concentration was quantified using Protein Assay Dye Reagent Concentrate (Bio-Rad #5000006).
Alternatively, for the analysis of PHF20-KO clones (Figures S5B and S5E) cells were collected in lysis buffer (50 mM HEPES (pH 7.4), 40 mM NaCl, 2 mM EDTA, 1.5 mM sodium orthovanadate, 50 mM NaF, 10 mM pyrophosphate, 10 mM glycerophosphate and 1% Triton X-100 and one tablet of EDTA-free complete protease inhibitors (Roche) per 25 ml) and protein content was measured with BCA (Bicinchoninic acid) using Pierce BCA protein assay kit (Thermo Scientific # 23227).
Equal amounts of protein were denatured with Laemmli buffer, boiled for 5 min and loaded into Tris-Glycine precast gels (4–20% Mini-PROTEAN® TGX™ precast protein gels from Bio-Rad, or 8% Novex Wedgewell from Invitrogen), resolved by SDS-PAGE and analyzed by immunoblotting. Western blot analysis was performed according to standard procedures.
Transferred proteins were blocked with 5% non-fat milk powder in PBS with 0.1% Tween-20 (Sigma-Aldrich) for 1 h at room temperature and incubated with the corresponding antibodies in TBS with 0.1% Tween-20 with 5% bovine serum albumin (BSA) overnight at 4°C. Primary antibodies were used at the following dilutions: anti-PHF20 (Cell Signaling # 3934) 1:1000; anti-phospho-H2AX (Ser139) (Cell Signaling # 9718) 1:1000; anti-RNF168 (Millipore # ABE367) 1:1000; anti-53BP1(BD Bioscience # 612522) 1:1000; anti-DNAPK (Santa Cruz # sc-5282) 1:1000; anti-CHK1 (Cell Signaling # 2360S) 1:1000; anti-CHK2 (Millipore # 05–649) 1:1000; anti-β-ACTIN (Sigma-Aldrich # A5316) 1:5000, anti-α-TUBULIN (Sigma-Aldrich # T9026) 1:5000 and anti-GAPDH (Sigma-Aldrich # MAB374) 1:5000. Horseradish peroxidase-conjugated secondary antibodies or LI-COR secondary antibodies (IRDYE 800CW, LI-COR # 926–32211 and IRDYE 680RD, LI-COR # 926–68070) were used for immunodetection. Protein quantification was performed using Fiji software (Schindelin et al., 2012) and γH2AX levels were normalized against β-ACTIN for each sample.
Survival assay
WT U2OS, PHF20-KO U2OS clonal cell lines were treated with indicated doses of ionizing radiation and seeded in 6 well plates (150 and 1500 cells per well). Plates were stained with crystal violet solution (0.5% (w/v) crystal violet in 20% methanol) when large colonies consisting of 50 or more cells were visualy detected, around 8–10 days after plating. All experiments were performed using technical triplicates. Colonies were manualy counted and the number adjusted by plating efficiency (number of colonies counted / number of cells plated) and by surviving fraction ((number of colonies counted/number of cells plated) / plating efficiency). Relative survival was normalized against non-damaged controls (set as 100%).
Pull-down competition experiments
GFP-53BP1 Tudor (1144–1709aa) plasmid was transiently transfected into HEK293T cells. Transfected cells were harvested at 95% confluency and lysed in mild immunoprecipitation buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.1% NP-40, 5 mM EDTA, 5 mM EGTA, 15 mM MgCl2) with protease inhibitor cocktail (Roche Diagnostics # 04693124001). GST-53BP1 Tudor and GST-PHF20 Tudor were expressed and purified as previously described (Espejo et al., 2002). For the pull-down experiment, 5 μg of biotinylated H4K20me2 peptide (synthesized by Keck Biotechnology) was pre-conjugated to streptavidin beads (Millipore # 16–126) in mild IP buffer described above, with 2 hours of rocking. Pre-conjugated beads were then washed and incubated with the lysate from the GFP-53BP1 transfected cells, and either GST-53BP1 Tudor or GST-PHF20 Tudor protein for competitive binding. Competition binding included GST-53BP1 Tudor and GST-PHF20 Tudor protein in amounts of 0, 0.05, 0.1, 0.5, 1 and 5 μg. Immunoblotting analysis was performed using an antibody against GFP (Santa Cruz # 9996). Quantification was performed using Fiji software (Schindelin et al., 2012) and 53BP1 GFP-Tudor expression was normalized against 0 μg of either GST-53BP1 Tudor or GST-PHF20 Tudor.
Rescue experiments with a CRISPR-resistant PHF20-GFP (PHF20R-GFP)
To generate a CRISPR-resistant PHF20-GFP (PHF20R-GFP), the eGFPC1-PHF20 plasmid (Biomatik, gift from Mark Bedford) was modified. The PAM sequence targeted by the PHF20 Crispr guide sg#2 (GTTGGAAGCCCGGGACCGTT) was mutated in one nucleotide (C to A) without generating a change in the aminoacid sequence of PHF20 protein (Figure S5F). The point mutation generated in the PAM sequence was confirmed by Sanger sequencing. Additionaly, a 3xFLAG tag was cloned at the N-terminus of the GFP.
For experiments evaluating kinetics of DNA repair, PHF20R-GFP was transfected in WT cells and PHF20-KO cells plated in 96 well plates (Greiner Bio-One # 655087) (4000 cells per well) using 0.3 μl of XtremeGENE9 (Roche # 6365787001) and 0.1 μl of plasmid DNA (per well). All transfections were performed in duplicate (2 wells per plate) and in replicate plates (5 plates). Two days after transfection, all replicate plates, except one (non-irradiated sample), were irradiated with 3Gy and let to recover for the indicated times before fixation. Analysis of DNA repair kinetics was performed as decribed previously in this section, images were acquired in an Opera High Content Screening System (Perkin Elmer) and analyzed in Acapella.2 software as decribed. In a same well, transfected cells (GFP positive) and non transfected cells (GFP negative) were identified during image analysis and evaluated independently for γH2AX and 53BP1 nuclear foci.
DNA repair assay
A U2OS cell line bearing a single copy integration of the reporter EJ5-GFP (cNHEJ) was used to analyze NHEJ repair pathway (Bennardo et al., 2008). For the assay, a PHF20-mCherry plasmid was generated by substituting the GFP sequence of the PHF20RGFP plasmid with the mCherry sequence. To generate CTRL-mCherry, the PHF20 sequence was deleted from the PHF20-mCherry. U2OS cells were plated in 12-well plates (80 000 cells per well) in triplicate, 24 h later cells were co-transfected with 0.4 μg of a CTRL-mCherry or an PHF20-mCherry plasmid and 0.8 μg of I-SceI enzyme by using 3.6 μl of Lipofectamine 2000 (Invitrogen # 11668019) per well, and the medium was changed 4 h later. Two days later cells were imaged using a Nikon Eclipse Ti microscope. An average of 300 cells per replicate were analyzed in the wells transfected with either CTRL-mCherry and I-SceI or PHF20-mCherry and I-SceI. The number of double positive cells (eGFP and mCherry) was determined and normalized against the total of mCherry positive cells. To facilitate the comparison between experiments, the ratio in PHF20-mCherry transfected wells was normalized against the ratio in CTRL-mCherry transfected wells. Unpaired t-test was used for statistical analysis, *p < 0.05.
Cell cycle distribution
U2OS cells were infected with lentiviral particles generated from plko constructs containing shRNA scramble (sh scr) or shRNA targeting human PHF20 (sh PHF20) (GCATGGGATTACTGGAAGAAA). Cell cycle distribution was evaluated by flow cytometry with propidium iodide and data were analyzed using with FlowJO software.
QUANTIFICATION AND STATISTICAL ANALYSIS
Prism 7 and Prism 8 were used. Statistical details including the satistical tests used can be found in figure legends. Values represent the average ± SD or ± SEM. Statistical significance was determined using unpaired Students t-test or one way ANOVA. Differences were considered statistically significant at a value of p < 0.05.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| Mouse monoclonal anti-53BP1 | BD Biosciences | Cat# 612522; RRID: AB_399824 |
| Rabbit anti-53BP1 | Santa Cruz | H-300, Cat# sc-22760; RRID: AB_2256326 |
| Mouse monoclonal anti-Chk1 | Cell Signaling Technology | 2G1D5, Cat# 2360; RRID: AB_2080320 |
| Mouse monoclonal anti-Chk2 | Sigma-Aldrich | Clone 7, Cat# 05-649; RRID: AB_2244941 |
| Mouse monoclonal anti-DNA-PKCS | Santa Cruz | G-4, Cat# sc-5282; RRID: AB_2172848 |
| Mouse monoclonal anti-GFP | Santa Cruz | B-2, Cat# sc-9996; RRID: AB_627695 |
| Mouse monoclonal anti-phospho H2AX (Ser139) | Sigma-Aldrich | Clone JBW301; Cat# 05-636; RRID: AB_309864 |
| Rabbit monoclonal anti-phospho H2AX (Ser139) | Cell Signaling Technology | Clone 20 3 103; Cat# 9718; RRID:_AB_2118009 |
| Rabbit anti-PAR | Enzo Lab Sciences | Cat# ALX-210-890A-0100 |
| Rabbit mouse monoclonal anti-PHF20 | Cell Signaling Technology | Clone D96F6; Cat# 3934; RRID: AB_21655078 |
| Rabbit anti-RNF168 | Sigma-Aldrich | Cat# ABE367; RRID: AB_11212809 |
| Mouse monoclonal IgG2a anti-V5 tag antibody | Abcam | SV5-Pkl, Cat# ab27671; RRID: AB_471093 |
| Mouse monoclonal anti-b-Actin | Sigma-Aldrich | Clone AC-74, Cat# A5316; RRID: AB_476743 |
| Mouse monoclonal anti-GAPDH | Sigma-Aldrich | Clone 6C5, Cat# MAB374; RRID: AB_2107445 |
| Mouse monoclonal anti-a-Tubulin | Sigma-Aldrich | Clone DM1A, Cat# T9026; RRID: AB_477593 |
| Goat anti-rabbit IgG Alexa Fluor 488 | ThermoFisher | Cat# A-11008; RRID: AB_143165 |
| Goat anti-mouse IgG2a Alexa Fluor 488 | Jackson ImmunoResearch | Cat# 115-547-186; RRID: AB_2632535 |
| Goat anti-mouse IgG Alexa Fluor 555 | ThermoFisher | Cat# A-21422; RRID: AB_2535844 |
| Goat anti-mouse IgG1 Rhodamine Red | Jackson ImmunoResearch | Cat# 115-297-185; RRID: AB_2632519 |
| Goat anti-rabbit IgG Alexa Fluor 647 | ThermoFisher | Cat# A-21244; RRID: AB_2535812 |
| IRDye 680RD Goat anti-Mouse IgG | LI-COR | Cat# 926-68070; RRID AB_10956588 |
| IRDye 800CW Goat anti-Rabbit IgG | LI-COR | Cat# 926-32211; RRID AB_621843 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Gibco Opti-MEM I reduced serum medium | Gibco | Cat# 11058021 |
| Lipofectamine RNAiMAX transfection reagent | Invitrogen | Cat# 13778030 |
| Lipofectamine 2000 transfection reagent | Invitrogen | Cat# 11668019 |
| X-tremeGENE 9 transfection reagent | Roche | Cat# 6365787001 |
| Paraformaldehyde 16% aqueous solution | Electron Microscopy Sciences | Cat# 15710 |
| Hoechst 33258 | Sigma-Aldrich | Cat# B2883 |
| BrdU | Sigma-Aldrich | Cat# B5002 |
| DAPI | Sigma-Aldrich | Cat# D9542 |
| KU-55933 (ATM kinase inhibitor) | Selleckem | Cat# S1092 |
| NU7441 (DNA-PK inhibitor) | Selleckem | Cat# S2638 |
| Olaparib (AZD2281; PARP inhibitor) | Selleckem | Cat# S1060 |
| ATR inhibitor | Toledo et al. (2011) | N/A |
| Streptavidin agarose beads | Millipore | Cat# 16-126 |
| Blasticidin | Invivogen | Cat# ant-bl-05 |
| Goat serum | Sigma | Cat# G9023 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Pierce BCA protein assay kit | Thermo Scientific | Cat# 23227 |
| Protein Assay Dye Reagent Concentrate | Bio-Rad | Cat# 5000006 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| Human: U2OS | ATCC | Cat# HTB-96 |
|
| ||
| Oligonucleotides | ||
|
| ||
| RNF8 siRNA_1: CAGAGAAGCUUACAGAUGU | Sigma-Aldrich | N/A |
| RNF8 siRNA_2: GGAGAUAGCCCAAGGAGAA | Sigma-Aldrich | N/A |
| RNF168 siRNA | Dharmacon | Cat# J-007152-05-0005 |
| PARP1 siRNA_1 | Dharmacon | Cat# J-006656-06-0005 |
| PARP1 siRNA_2 | Dharmacon | Cat# J-006656-07-0005 |
| PHF20 siRNA_1 | Dharmacon | Cat# J-015234-11-0005 |
| PHF20 siRNA_2 | Dharmacon | Cat# J-015234-10-0005 |
| PHF20 siRNA_3 | Dharmacon | Cat# J-015234-09-0005 |
| Control siRNA: MISSION siRNA universal negative control | Sigma-Aldrich | Cat# SIC001 |
| ATF7IP esiRNA | Sigma-Aldrich | Cat# EHU071481 |
| CHD2 esiRNA | Sigma-Aldrich | Cat# EHU043581 |
| FKBP2 esiRNA | Sigma-Aldrich | Cat# EHU124351 |
| H2AFZ esiRNA | Sigma-Aldrich | Cat# EHU110691 |
| ING2 esiRNA | Sigma-Aldrich | Cat# EHU048041 |
| LBR esiRNA | Sigma-Aldrich | Cat# EHU072221 |
| MAX esiRNA | Sigma-Aldrich | Cat# EHU096141 |
| MORFL1 esiRNA | Sigma-Aldrich | Cat# EHU127521 |
| RBBP8 esiRNA | Sigma-Aldrich | Cat# EHU070611 |
| SETDB1 esiRNA | Sigma-Aldrich | Cat# EHU046261 |
| SETDB2 esiRNA | Sigma-Aldrich | Cat# EHU140971 |
| SMAD1 esiRNA | Sigma-Aldrich | Cat# EHU051601 |
| TRIM3 esiRNA | Sigma-Aldrich | Cat# EHU088111 |
| RNF168 esiRNA | Sigma-Aldrich | Cat# EHU011891 |
| KIF11 esiRNA | Sigma-Aldrich | Cat# EHU019931 |
| RLUC esiRNA | Sigma-Aldrich | Cat# EHURLUC |
| PARP1-RT-Fw: TGGAAAAGTCCCACACTGGTA | Sigma-Aldrich | N/A |
| PARP1-RT-Rv: AAGCTCAGAGAACCCATCCAC | Sigma-Aldrich | N/A |
| ACTIN-RT-Fw: CATGTACGTTGCTATCCAGGC | Sigma-Aldrich | N/A |
| ACTIN-RT-Rv: CTCCTTAATGTCACGCACGAT | Sigma-Aldrich | N/A |
| PHF20-sg1-Fw: CACCGAAGCATCCACCTAACAGACG | Invitrogen | N/A |
| PHF20-sg1-Rv: AAACCGTCTGTTAGGTGGATGCTTC | Invitrogen | N/A |
| PHF20-sg2-Fw: CACCGAACGGTCCCGGGCTTCCAAC | Invitrogen | N/A |
| PHF20-sg2-Rv: AAACGTTGGAAGCCCGGGACCGTTC | Invitrogen | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| ChromORFome library | This manuscript | Table S1 |
| plasmid lentiCRISPR v2 | Addgene | Plasmid #52961 |
| plasmid plko sh PHF20 | Molecular Profiling Laboratory. MGH | Clone ID #TRCN0000016316 |
| plasmid PHF20-Clover | This manuscript | N/A |
| plasmid PHF20R-GFP | This manuscript | N/A |
| plasmid PHF20-mCherry | This manuscript | N/A |
| plasmid CTRL-mCherry | This manuscript | N/A |
| plasmid EJ5-GFP | Bennardo et al., 2008 | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| CellProfiler v2.0 | McQuin et al. (2018) | https://cellprofiler.org |
| FlowJo | Becton Dickinson | https://www.flowjo.com |
| Prism 7 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Columbus Image Data Storage and Analysis System | Perkin Elmer | https://www.perkinelmer.com/es/product/image-data-storage-and-analysis-system-columbus |
| Orange Data Mining | Demsar et al. (2013) | https://orangedatamining.com/ |
| Palm Robosoftware | Zeiss | https://www.zeiss.com/microscopy/int/products/microscope-software/palm-robosoftware.html |
| Acapella.2 Software | Perkin Elmer | N/A |
| Fiji | ImageJ | https://imagej.net/software/fiji/ |
|
| ||
| Other | ||
|
| ||
| BD Pathway Bioimager 435 with Attovision v1.5 | Becton Dickinson | https://www.bd.com/resource.aspx?IDX=17780 |
| Opera High Content Screening System | Perkin Elmer | https://www.flyrnai.org/supplement/BRO_OperaHighContentScreeningSystem.pdf |
| Palm Microdissector | Zeiss | https://www.zeiss.com/microscopy/int/products/laser-microdissection/microbeam.html |
| MMI CellCut | Molecular Machines and Industries | https://www.molecular-machines.com/products/cellcut |
| Leica DMI4000 B | Leica | https://www.leica-microsystems.com/products/light-microscopes/p/leica-dmi4000-b |
| Marianas spining disk confocal microscope | 3i | https://www.intelligent-imaging.com/marianas |
| Zeiss Axio Observer | Zeiss | https://www.zeiss.com/microscopy/int/products/light-microscopes/axio-observer-for-biology.html |
| Nikon Eclipse Ti microscope | NIkon | https://www.microscope.healthcare.nikon.com/products/inverted-microscopes/eclipse-ti-series |
| NUNC Lab-Tek II 8-well chamber slides | NUNC | Cat# 154534 |
| 96-well plate, cell culture, black, CellStar™ | Greiner Bio-One | Cat# 655087 |
| 96-well black/clear TC-treated imaging microplate | Falcon-Corning | Cat# 353219 |
| 384-well plate, cell culture, black, CellStar™ | Greiner Bio-One | Cat# 781090 |
| 384-well plate, cell culture, black | Perkin Elmer | Cat# 6057302 |
Highlights.
Two high-throughput, imaging-based, DNA repair platforms are developed
Machine learning identifies chromatin genes modulating kinetics of DNA repair
HT laser microirradiation uncovers factors recruited and excluded from DNA lesions
PHF20 is actively removed from DNA breaks to allow 53BP1 recruitment and repair
ACKNOWLEDGMENTS
We would like to thank all the members of the Mostoslavsky lab for helpful discussions and critical reading of the manuscript. We also thank Cari Sagum for technical help, David Root and Federica Piccioni from the Broad Institute Genetics Perturbation Platform for help with developing the ChromORFeome library, and the Bioinformatics Core, Mol. Bio. Department at MGH (NIDDK P30 DK040561). MMI laser cut technology was provided by the Program in Membrane Biology Microscopy Core, which is partially supported by the Boston Area Diabetes Endocrinology Research Center (P30DK057521) and the Massachusetts General Hospital (MGH) Center for the Study of Inflammatory Bowel Disease (P30DK043351). High-throughput laser-induced DNA damage was performed in the Confocal and Specialized Microscopy Shared Resource of the Herbert Irving Comprehensive Cancer Center at Columbia University, supported by NIH grant P30 CAO13696. B.M-P. is supported by an M + Vision Advanced Fellowship Marie Curie COFUND FP7. G.G.S. is supported by an MGH Cancer Center Excellence Award, D.C. was supported by a Nova Scotia Graduate Scholarship, and T.L.C is supported by the Charles King Trust Postdoctoral Fellowship, Simeon J. Fortin Charitable Foundation, Bank of America, N.A., Trustee. A.C. is supported by NIH grant GM117064, and M.T.B. is supported by a CPRIT PAAC grant RP180804. R.M. is the Laurel Schwartz Endowed Chair in Oncology. This work is supported by NIEHS grant R21ES027931 to R.M., a Natural Science and Engineering Research Council of Canada (NSERC) Discovery Grant RGPIN-2020-04034 to G.D, and a EU H-2020 Program Grant (ERC-2014-STG-638891) to A.E.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.110176.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Abdou I, Poirier GG, Hendzel MJ, and Weinfeld M. (2015). DNA ligase III acts as a DNA strand break sensor in the cellular orchestration of DNA strand break repair. Nucleic Acids Res. 43, 875–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acs K, Luijsterburg MS, Ackermann L, Salomons FA, Hoppe T, and Dantuma NP (2011). The AAA-ATPase VCP/p97 promotes 53BP1 recruitment by removing L3MBTL1 from DNA double-strand breaks. Nat. Struct. Mol. Biol. 18, 1345–1350. [DOI] [PubMed] [Google Scholar]
- Adamson B, Smogorzewska A, Sigoillot FD, King RW, and Elledge SJ (2012). A genome-wide homologous recombination screen identifies the RNA-binding protein RBMX as a component of the DNA-damage response. Nat. Cell Biol. 14, 318–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aleksandrov R, Dotchev A, Poser I, Krastev D, Georgiev G, Panova G, Babukov Y, Danovski G, Dyankova T, Hubatsch L, et al. (2018). Protein dynamics in complex DNA lesions. Mol. Cell 69, 1046–1061.e5. [DOI] [PubMed] [Google Scholar]
- Altmeyer M, and Lukas J. (2013). To spread or not to spread-chromatin modifications in response to DNA damage. Curr. Opin. Genet. Dev. 23, 156–165. [DOI] [PubMed] [Google Scholar]
- Andrin C, McDonald D, Attwood KM, Rodrigue A, Ghosh S, Mirzayans R, Masson JY, Dellaire G, and Hendzel MJ (2012). A requirement for polymerized actin in DNA double-strand break repair. Nucl. (United States 3, 14. [DOI] [PubMed] [Google Scholar]
- Badeaux AI, Yang Y, Cardenas K, Vemulapalli V, Chen K, Kusewitt D, Richie E, Li W, and Bedford MT (2012). Loss of the methyl lysine effector protein PHF20 impacts the expression of genes regulated by the lysine acetyltransferase MOF. J. Biol. Chem. 287, 429–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankovic J, Stojsic J, Jovanovic D, Andjelkovic T, Milinkovic V, Ruzdijic S, and Tanic N. (2010). Identification of genes associated with non-small-cell lung cancer promotion and progression. Lung Cancer 67, 151–159. [DOI] [PubMed] [Google Scholar]
- Bekker-Jensen S, and Mailand N. (2010). Assembly and function of DNA double-strand break repair foci in mammalian cells. DNA Repair (Amst) 9, 1219–1228. [DOI] [PubMed] [Google Scholar]
- Bekker-Jensen S, Lukas C, Kitagawa R, Melander F, Kastan MB, Bartek J, and Lukas J. (2006). Spatial organization of the mammalian genome surveillance machinery in response to DNA strand breaks. J. Cell Biol. 173, 195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beli P, Lukashchuk N, Wagner SA, Weinert BT, Olsen JV, Baskcomb L, Mann M, Jackson SP, and Choudhary C. (2012). Proteomic investigations reveal a role for RNA processing factor THRAP3 in the DNA damage response. Mol. Cell 46, 212–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennardo A, Cheng A, Huang N, and Stark JM (2008). Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLos Genet 4(6), e1000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bothmer A, Robbiani DF, Feldhahn N, Gazumyan A, Nussenzweig A, and Nussenzweig MC (2010). 53BP1 regulates DNA resection and the choice between classical and alternative end joining during class switch recombination. J. Exp. Med. 207, 855–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botuyan MV, Lee J, Ward IM, Kim JE, Thompson JR, Chen J, and Mer G. (2006). Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell 127, 1361–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunting SF, Callén E, Wong N, Chen HT, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez-Capetillo O, Cao L, et al. (2010). 53BP1 inhibits homologous recombination in brca1-deficient cells by blocking resection of DNA breaks. Cell 141, 243–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess RC, Burman B, Kruhlak MJ, and Misteli T. (2014). Activation of DNA damage response signaling by condensed chromatin. Cell Rep. 9, 1703–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Y, Jin J, Swanson SK, Cole MD, Choi SH, Florens L, Washburn MP, Conaway JW, and Conaway RC (2010). Subunit composition and substrate specificity of a MOF-containing histone acetyltransferase distinct from the Male-specific Lethal (MSL) complex. J. Biol. Chem. 285, 4268–4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JK, Lin WL, Chen Z, and Liu H. (2018). PARP-1–dependent recruitment of cold-inducible RNA-binding protein promotes double-strand break repair and genome stability. Proc. Natl. Acad. Sci. U S A 115, E1759–E1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou DM, Adamson B, Dephoure NE, Tan X, Nottke AC, Hurov KE, Gygi SP, Colaiácovo MP, and Elledge SJ (2010). A chromatin localization screen reveals poly (ADP ribose)-regulated recruitment of the repressive polycomb and NuRD complexes to sites of DNA damage. Proc. Natl. Acad. Sci. U. S. A. 107, 18475–18480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccia A, and Elledge SJ (2010). The DNA damage response: making it safe to play with knives. Mol. Cell 40, 179–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clouaire T, and Legube G. (2019). A snapshot on the cis chromatin response to DNA double-strand breaks. Trends Genet. 35, 330–345. [DOI] [PubMed] [Google Scholar]
- Cui G, Park S, Badeaux AI, Kim D, Lee J, Thompson JR, Yan F, Kaneko S, Yuan Z, Botuyan MV, et al. (2012). PHF20 is an effector protein of p53 double lysine methylation that stabilizes and activates p53. Nat. Struct. Mol. Biol. 19, 916–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dellaire G, Kepkay R, and Bazett-Jones DP (2009). High resolution imaging of changes in the structure and spatial organization of chromatin, γ-H2A.X and the MRN complex within etoposide-induced DNA repair foci. Cell Cycle 8, 3750–3769. [DOI] [PubMed] [Google Scholar]
- Demsar J, Curk T, Erjavez A, Gorup C, Hocevar T, Milutinovic M, Mozina M, Polajnar M, Toplak M, Staric A, et al. (2013). Orange: data mining toolbox in Python. J. Mach. Learn. Res. 14, 2349–2353. [Google Scholar]
- Doil C,Mailand N, Bekker-Jensen S, Menard P, Larsen DH, Pepperkok R, Ellenberg J, Panier S, Durocher D, Bartek J, et al. (2009). RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell 136, 435–446. [DOI] [PubMed] [Google Scholar]
- Escribano-Díaz C, Orthwein A, Fradet-Turcotte A, Xing M, Young JTF, Tkáč J, Cook MA, Rosebrock AP, Munro M, Canny MD, et al. (2013).A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol. Cell 49, 872–883. [DOI] [PubMed] [Google Scholar]
- Espejo A,Côté J, Bednarek A, Richard S, and Bedford MT (2002). A protein-domain microarray identifies novel protein-protein interactions. Biochem. J. 367, 697–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floyd SR, Pacold ME, Huang Q, Clarke SM, Lam FC, Cannell IG, Bryson BD, Rameseder J, Lee MJ, Blake EJ, et al. (2013). The bromodomain protein Brd4 insulates chromatin from DNA damage signalling. Nature 498, 246–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fradet-Turcotte A, Canny MD, Escribano-Díaz C, Orthwein A, Leung CCY, Huang H, Landry MC, Kitevski-Leblanc J, Noordermeer SM, Sicheri F, et al. (2013). 53BP1 is a reader of the DNA-damage-induced H2A Lys 15ubiquitin mark. Nature 499, 50–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudreau-Lapierre A, Garneau D, Djerir B, Coulombe F, Morin T, and Marechal A. (2018). Investigation of protein recruitment to DNA lesions using 405nm laser micro-irradiation. J. Vis. Exp. 2018, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghezraoui H, Oliveira C, Becker JR, Bilham K, Moralli D, Anzilotti C, Fischer R, Deobagkar-Lele M, Sanchiz-Calvo M, Fueyo-Marcos E, et al. (2018). 53BP1cooperation with the REV7–shieldin complex underpins DNA structure-specific NHEJ. Nature 560, 122–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong F, Chiu LY, Cox B, Aymard F, Clouaire T, Leung JW, Cammarata M, Perez M, Agarwal P, Brodbelt JS, et al. (2015). Screen identifies bromodomain protein ZMYND8 in chromatin recognition of transcription-associated DNA damage that promotes homologous recombination. Genes Dev. 29, 197–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haince JF, McDonald D, Rodrigue A, Déry U, Masson JY, Hendzel MJ, and Poirier GG (2008). PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites. J. Biol. Chem. 283, 1197–1208. [DOI] [PubMed] [Google Scholar]
- Harper JW, and Elledge SJ (2007). The DNA damage response: ten years after. Mol. Cell 28, 739–745. [DOI] [PubMed] [Google Scholar]
- Huen MSY, Grant R, Manke I, Minn K, Yu X, Yaffe MB, and Chen J. (2007). RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell 131, 901–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huyen Y, Zgheib O, DiTullio RA, Gorgoulis VG, Zacharatos P, Petty TJ, Sheston EA, Mellert HS, Stavridi ES, and Halazonetis TD (2004). Methylated lysine 79 of histone H3 targets 53BP1 to DNA double-strand breaks. Nature 432, 406–411. [DOI] [PubMed] [Google Scholar]
- Izhar L, Adamson B, Ciccia A, Lewis J, Pontano-Vaites L, Leng Y, Liang AC, Westbrook TF, Harper JW, and Elledge SJ (2015). A systematic analysis of factors localized to damaged chromatin reveals PARP-dependent recruitment of transcription factors. Cell Rep 11, 1486–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson SP, and Bartek J. (2009). The DNA-damage response in human biology and disease. Nature 461, 1071–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquet K, Fradet-Turcotte A, Avvakumov N, Lambert JP, Roques C, Pandita RK, Paquet E, Herst P, Gingras AC, Pandita TK, et al. (2016). The TIP60 complex regulates bivalent chromatin recognition by 53BP1 through direct H4K20me binding and H2AK15 acetylation. Mol. Cell 62, 409–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungmichel S, Rosenthal F, Altmeyer M, Lukas J, Hottiger MO, and Nielsen ML (2013). Proteome-wide identification of poly(ADP-Ribosyl)ation targets in different genotoxic stress responses. Mol. Cell 52, 272–285. [DOI] [PubMed] [Google Scholar]
- Kolas NK, Chapman JR, Nakada S, Ylanko J, Chahwan R, Sweeney FD, Panier S, Mendez M, Wildenhain J, Thomson TM, et al. (2007). Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science 318, 1637–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruhlak MJ, Celeste A, Dellaire G, Fernandez-Capetillo O, Müller WG, McNally JG, Bazett-Jones DP, and Nussenzweig A. (2006). Changes in chromatin structure and mobility in living cells at sites of DNA double-strand breaks. J. Cell Biol. 172, 823–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusch T, Florens L, MacDonald WH, Swanson SK, Glaser RL, Yates JR, Abmayr SM, Washburn MP, and Workman JL (2004). Acetylation by Tip60 is required for selective histone variant exchange at DNA lesions. Science 306, 2084–2087. [DOI] [PubMed] [Google Scholar]
- Luijsterburg MS, de Krijger I, Wiegant WW, Shah RG, Smeenk G, de Groot AJL, Pines A, Vertegaal ACO, Jacobs JJL, Shah GM, et al. (2016). PARP1 links CHD2-mediated chromatin expansion and H3.3 deposition to DNA repair by non-homologous end-joining. Mol. Cell 61, 547–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mailand N, Bekker-Jensen S, Faustrup H, Melander F, Bartek J, Lukas C, and Lukas J. (2007). RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell 131, 887–900. [DOI] [PubMed] [Google Scholar]
- Mallette FA, Mattiroli F, Cui G, Young LC, Hendzel MJ, Mer G, Sixma TK, and Richard S. (2012). RNF8- and RNF168-dependent degradation of KDM4A/JMJD2A triggers 53BP1 recruitment to DNA damage sites. EMBO J. 31, 1865–1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuin C, Goodman A, Chernyshev V, Kamentsky L, Cimini BA, Karhohs KW, Doan M, Ding L, Rafelski SM, Thirstrup D, et al. (2018). CellProfiler 3.0: next-generation image processing for biology. PLoS Biol. 16, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendjan S, Taipale M, Kind J, Holz H, Gebhardt P, Schelder M, Vermeulen M, Buscaino A, Duncan K, Mueller J, et al. (2006). Nuclear pore components are involved in the transcriptional regulation of dosage compensation in Drosophila. Mol. Cell 21, 811–823. [DOI] [PubMed] [Google Scholar]
- Mirman Z, Lottersberger F, Takai H, Kibe T, Gong Y, Takai K, Bianchi A, Zimmermann M, Durocher D, and de Lange T. (2018). 53BP1–RIF1–shieldin counteracts DSB resection through CST- and Pola-dependent fill-in. Nature 560, 112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mistrik M, Vesela E, Furst T, Hanzlikova H, Frydrych I, Gursky J, Majera D, and Bartek J. (2016). Cells and Stripes: a novel quantitative photo-manipulation technique. Sci. Rep. 6, 19567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murr R, Loizou JI, Yang YG, Cuenin C, Li H, Wang ZQ, and Herceg Z. (2006). Histone acetylation by Trrap-Tip60 modulates loading of repair proteins and repair of DNA double-strand breaks. Nat. Cell Biol. 8, 91–99. [DOI] [PubMed] [Google Scholar]
- Noordermeer SM, Adam S, Setiaputra D, Barazas M, Pettitt SJ, Ling AK, Olivieri M, Álvarez-Quilón A, Moatti N, Zimmermann M, et al. (2018). The shieldin complex mediates 53BP1-dependent DNA repair. Nature 560, 117–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paull TT, Rogakou EP, Yamazaki V, Kirchgessner CU, Gellert M, and Bonner WM (2000). A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr. Biol. 10, 886–895. [DOI] [PubMed] [Google Scholar]
- Pinder JB, Attwood KM, and Dellaire G. (2013). Reading, writing, and repair: the role of ubiquitin and the ubiquitin-like proteins in DNA damage signaling and repair. Front. Genet. 4, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogakou EP, Pilch DR, Orr AH, Ivanova VS, and Bonner WM (1998). DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 273, 5858–5868. [DOI] [PubMed] [Google Scholar]
- Rogakou EP, Boon C, Redon C, and Bonner WM (1999). Megabase chromatin domains involved in DNA double-strand breaks in vivo. J. Cell Biol. 146, 905–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartori AA, Lukas C, Coates J, Mistrik M, Fu S, Bartek J, Baer R, Lukas J, and Jackson SP (2007). Human CtIP promotes DNA end resection. Nature 450, 509–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saafeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scully R, Panday A, Elango R, and Willis NA (2019). DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell. Biol. 20(11), 698–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellou H, Lebeaupin T, Chapuis C, Smith R, Hegele A, Singh HR, Kozlowski M, Bultmann S, Ladurner AG, Timinszky G, et al. (2016). The poly(ADP-ribose)-dependent chromatin remodeler Alc1 induces local chromatin relaxation upon DNA damage. Mol. Biol. Cell 27, 3791–3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith R, Lebeaupin T, Juhász S, Chapuis C, D’Augustin O, Dutertre S, Burkovics P, Biertümpfel C, Timinszky G, and Huet S. (2019). Poly(ADPribose)-dependent chromatin unfolding facilitates the association of DNA-binding proteins with DNA at sites of damage. Nucleic Acids Res. 47, 11250–11267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart GS, Panier S, Townsend K, Al-Hakim AK, Kolas NK, Miller ES, Nakada S, Ylanko J, Olivarius S, Mendez M, et al. (2009). The RIDDLE syndrome protein mediates a ubiquitin-dependent signaling cascade at sites of DNA damage. Cell 136, 420–434. [DOI] [PubMed] [Google Scholar]
- Strickfaden H, McDonald D, Kruhlak MJ, Haince JF, Th’Ng JPH, Rouleau M, Ishibashi T, Corry GN, Ausio J, Underhill DA, et al. (2016). Poly(ADP-ribosyl)ation-dependent transient chromatin decondensation and histone displacement following laser microirradiation. J. Biol. Chem. 291, 1789–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Jiang X, Chen S, Fernandes N, and Price BD (2005). A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc. Natl. Acad. Sci. U. S. A. 102, 13182–13187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sy SMH, Huen MSY, and Chen J. (2009). MRG15 is a novel PALB2-interacting factor involved in homologous recombination. J. Biol. Chem. 284, 21127–21131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J, Cho NW, Cui G, Manion EM, Shanbhag NM, Botuyan MV, Mer G, and Greenberg RA (2013). Acetylation limits 53BP1 association with damaged chromatin to promote homologous recombination. Nat. Struct. Mol. Biol. 20, 317–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toiber D, Erdel F, Bouazoune K, Silberman DM, Zhong L, Mulligan P, Sebastian C, Cosentino C, Martinez-Pastor B, Giacosa S, et al. (2013). SIRT6 recruits SNF2H to DNA break sites, preventing genomic instability through chromatin remodeling. Mol. Cell 51, 454–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo LI, Murga M, Zur R, Soria R, Rodriguez A, Martinez S, Oyarzabal J, Pastor J, Bischoff JR, and Fernandez-Capetillo O. (2011). A cell-based screen identifies ATR inhibitors with synthetic lethal properties for cancer-associated mutations. Nat. Struct. Mol. Biol. 18, 721–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tubbs A, and Nussenzweig A. (2017). Endogenous DNA damage as a source of genomic instability in cancer. Cell 168, 644–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Han K-J, Pang X-W, Vaughan HA, Qu W, Dong X-Y, Peng J-R, Zhao H-T, Rui J-A, Leng X-S, et al. (2002). Large scale identification of human hepatocellular carcinoma-associated antigens by autoantibodies. J. Immunol. 169, 1102–1109. [DOI] [PubMed] [Google Scholar]
- Xu Y, Ayrapetov MK, Xu C, Gursoy-Yuzugullu O, Hu Y, and Price BD (2012). Histone H2A.Z controls a critical chromatin remodeling step required for DNA double-strand break repair. Mol. Cell 48, 723–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Boehm JS, Yang X, Salehi-Ashtiani K, Hao T, Shen Y, Lubonja R, Thomas SR, Alkan O, Bhimdi T, et al. (2011). A public genome-scale lentiviral expression library of human ORFs. Nat. Methods 8, 659–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun MH, and Hiom K. (2009). CtIP-BRCA1 modulates the choice of DNA double-strand-break repair pathway throughout the cell cycle. Nature 459, 460–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou BS, and Elledge SJ (2000). Checkpoints in perspective. Nature 408, 433–439. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.