

Abstract
Pyrrolizidine alkaloids (PAs) are common phytotoxins with both hepatotoxicity and pneumotoxicity. Hepatic cytochrome P450 enzymes are known to bioactivate PAs into reactive metabolites, which can interact with proteins to form pyrrole-protein adducts and cause intrahepatic cytotoxicity. However, the metabolic and initiation biochemical mechanisms underlying PA-induced pneumotoxicity remain unclear. To investigate the in vivo metabolism basis for PA-induced lung injury, this study used mice with conditional deletion of the cytochrome P450 reductase (Cpr) gene and resultant tissue-selective ablation of microsomal P450 enzyme activities. After oral exposure to monocrotaline (MCT), a pneumotoxic PA widely used to establish animal lung injury models, liver-specific Cpr-null (LCN) mice, but not extrahepatic Cpr-low (xh-CL) mice, had significantly lower level of pyrrole-protein adducts in the serum, liver and lungs compared with wild-type (WT) mice. While MCT-exposed LCN mice had significantly higher blood concentration of intact MCT, compared to MCT-exposed WT or xh-CL mice. Consistent with the MCT in vivo bioactivation data, MCT induced lung injury, represented by vasculature damage, in WT and xh-CL mice but not LCN mice. Furthermore, reactive metabolites of MCT were confirmed to exist in the blood efflux from the hepatic veins of MCT-exposed rats. Our results provide the first mode-of-action evidence that hepatic P450s are essential for the bioactivation of MCT, and blood circulating reactive metabolites of MCT to the lung causes pneumotoxicity. Collectively, this study presents the scientific basis for the application of MCT in animal lung injury models, and more importantly, warrants public awareness and further investigations of lung diseases associated with exposure to not only MCT but also different PAs.
Keywords: Pyrrolizidine alkaloids, monocrotaline, lung injury, metabolism-based pneumotoxicity, cytochromes P450, Cpr-null mice
Introduction
Pyrrolizidine alkaloids (PAs) are most common phytotoxins produced by over 6,000 plant species worldwide. To date, more than 660 PAs and their N-oxide have been identified, of which over one half are toxic (Edgar et al. 2015; Stegelmeier et al. 1999). Humans are frequently exposed to PAs through the ingestion of PA-containing herbal medicines, particularly in China (Lin et al. 2011; Ruan et al. 2015; Yang et al. 2017), or the unwitting consumption of PA-contaminated foodstuffs such as grains, tea, and honey (Edgar et al. 2011; Prakash et al. 1999).
PAs are widely reported to exert hepatotoxicity, with the underlying mechanism well studied (Ebmeyer et al. 2020; Lu et al. 2018; Yang et al. 2019; Yang et al. 2020). Outbreaks of PA poisoning in humans, mostly involving acute liver injury, have been continuously reported, with most of the cases due to the ingestion of PA-containing herbal products or PA-contaminated grains (Lin et al. 2011; Mohabbat et al. 1976; Ridker et al. 1985). Apart from hepatotoxicity, PAs are also pneumotoxic. Previous reports as well as our study revealed that PAs with different structures can commonly induce lung injury, characterized by pulmonary endothelial injury and vascular medial hypertrophy (Heath et al. 1975; Kay et al. 1971; Song et al. 2020). Monocrotaline (MCT), a typical pneumotoxic PA, has been used in numerous studies to establish the pulmonary arterial hypertension (PAH) model in rodents (Hill et al. 2017). PAH, initiated by pulmonary vasculature damage, is a life-threatening disease featured with progressive pulmonary arterial remodeling and right heart failure at the late stage (Yuan and Rubin 2005). In fact, PAs are not only PAH modelling compounds but also reported to be possibly associated with human PAH cases (Gyorik and Stricker 2009; Heath et al. 1975). Thus it is important to study the initial biochemical mechanism of PA-induced lung injury.
Native forms of PAs are not toxic, and metabolic activation of PAs mediated by cytochrome P450 enzymes is required to elicit toxicity (Ruan et al. 2014). The reactive metabolites of PAs, namely dehydro-pyrrolizidine alkaloids (dehydro-PAs) and (±)-6,7-dihydro-7-hydroxy-1-hydroxymethyl-5H-pyrrolizine (DHP), which is formed by hydrolysis of dehydro-PAs, are highly electrophilic and capable of binding with proteins to form pyrrole-protein adducts, leading to protein dysfunction and cytotoxicity (He et al. 2019; Ma et al. 2019; Ruan et al. 2014). It is widely reported that PA-induced liver injury is caused primarily by dehydro-PAs generated from in situ hepatic P450s metabolism (Lu et al. 2019). Whereas, for PA-induced lung injury, which was also mediated by the reactive metabolites of PAs (Song et al. 2020), it remains unexplored whether the pneumotoxic PA metabolites are mainly generated in the liver by hepatic P450s or in other tissues by extrahepatic P450s. In addition, it also remains largely unknown how these reactive metabolites reach the lungs to elicit pneumotoxicity. A previous study demonstrated a significant increase of the reactive metabolites of MCT in the lungs after the addition of red blood cells (RBCs) into the in vitro liver-lung tandem preparation, suggesting that RBCs might contribute to the transport of the reactive metabolites of MCT from the liver to the lungs (Lafranconi and Huxtable 1984). However, direct evidence is required to reveal the nature of the reactive metabolites of MCT that migrate from the liver to the lungs.
In the present study, we investigated the respective roles of hepatic and extrahepatic P450s in PA metabolic activation by using Cpr gene knockout mice. A functional microsomal P450 system relies on the electron donor, NADPH-dependent cytochrome P450 reductase (CPR). Unlike the multiplicity of the Cyp genes encoding various P450 enzymes, there is only one Cpr gene in mammals, thus deletion of Cpr abrogates the activities of all microsomal P450 enzymes (Gu et al. 2003). Here we used the liver-specific Cpr-null (LCN) mice with abrogated hepatic P450 activity (Gu et al. 2003) and the extrahepatic Cpr-low (xh-CL) mice with diminished extrahepatic P450 activity (Wei et al. 2010). After exposing the Cpr-null mouse models and wild type (WT) mice to MCT, we compared the formation of pyrrole-protein adducts, which reflects the rate of P450s-mediated PA metabolic activation, kinetic profile of MCT in mouse blood, and toxicological effects in mouse livers and lungs. Moreover, we investigated the in vivo presence of the reactive metabolites of MCT in blood circulating from the liver to the lungs in rats.
Materials and methods
Chemicals and animals
MCT (CAS#315–22-0) was purchased from Sigma Aldrich (St. Louis, MO). LCN and xh-CL mice (both on C57BL/6J background), as well as WT C57BL/6J mice, were obtained from breeding stocks maintained at the University of Arizona. Validation data of LCN mice are available in Supplementary material. All procedures involving mice were approved by the Institutional Animal Care and Use Committee of the University of Arizona. Male Sprague-Dawley rats were obtained from Laboratory Animal Services Centre at the Chinese University of Hong Kong (CUHK). Protocols involving rats were approved by the Animal Experimental Ethics Committee CUHK, under the regulations of Hong Kong SAR government, and all animals were provided with a certified standard diet and tap water ad libitum during the experiments.
Pyrrole-protein adducts analysis
Mice were exposed to a single dose of MCT at 120 mg/kg by oral gavage and euthanized at 48 h after the MCT exposure. Using our previously established derivatization methods (Lin et al. 2011; Ruan et al. 2015), we determined pyrrole-protein adducts, the biomarker for PA intoxication, in serum, liver and lung samples collected from adult (2–3-month old) male and female mice (n=5 per group). Briefly, serum and tissue homogenates were mixed with two volumes of acetonitrile and three volumes of methanol to precipitate proteins, and the mixtures were centrifuged at 15,000 × g for 10 min. The protein pellets were then mixed with 100 μL of acidic silver nitrate ethanol solution, which contained 20 mg/mL silver nitrate and 5% trifuoroacetic acid. The mixture was shaken for 30 min at room temperature, and then centrifuged at 15,000 × g for 10 min. The resultant supernatant (20 μL) was incubated with 4-(dimethylamino)benzaldehyde (20 mg/mL) in ethanol containing 1% perchloric acid (80 μL) at 55°C for 10 min. The reaction mixture was further subjected to liquid chromatography-mass spectrometry (LC-MS) detection for the derivatized pyrrole-protein adducts. Detailed LC-MS parameters are available in Supplementary material.
Toxicokinetic study
Blood samples (20–25 μL) were collected from the tail vein of each mouse (n=3 per group) at the designed time intervals (0, 5 min, 15 min, 30 min, 60 min, 120 min, 240 min, 480 min, and 1440 min) after the oral administration of MCT (120 mg/kg). LC-MS analysis was performed to determine the serum level of MCT with detailed methods available in Supplementary material.
Toxicological study
Liver and lung samples were collected from mice (n=5 per group) at 48 h after a single oral administration of MCT (120 mg/kg). Acute liver injury was detected by measuring alanine aminotransferase (ALT) activities in serum samples using a kit obtained from BioAssay Systems (Hayward, CA). Histological changes were inspected using H&E stained sections of formalin-fixed paraffin-embedded mouse liver and lung samples. The degree of smooth muscle wall thickening in pulmonary arteries was scored using H&E-stained mouse lung sections according to a modified method described previously (Shi et al. 2018). Briefly, five cross sections of pulmonary arteries for each mouse (n=5 per group) were randomly selected for measuring wall thickness, and the results were calculated as follows: Score of relative thickness of pulmonary arterial medial wall = (external diameter − internal diameter)/external diameter. Immunohistochemistry (IHC) assay was performed on formalin-fixed mouse lung sections to investigate the integrity of pulmonary vascular endothelium, which was stained with an anti-CD31 antibody (Cell Signaling Technology, Danvers, MA) and counterstained with hematoxylin.
Detection of the reactive metabolites of MCT
To demonstrate the existence of the reactive metabolites of MCT in the blood efflux from the hepatic vein, cannulation in the inferior vena cava was performed with adult male Sprague Dawley rats (200–220 g) as previously described (Lebrec et al. 1980). Rats (n=3 per group) were then intravenously injected with 65 mg/kg of MCT dissolved in saline. Blood (~0.4 mL), circulating from the liver to lungs, was collected at designated time points (3, 5, 10, 15, 30, 60, 120, and 240 min). The collected blood samples were immediately divided into RBCs and plasma, both of which were further divided in half. Glutathione (GSH, 0.1 M) and paired saline vehicle were added into the corresponding half of the RBC or plasma partition followed by incubation for 2 min at 37 °C to trap the reactive metabolites of MCT and form pyrrole-GSH conjugates, including pyrrole-monoGSH conjugate (7-pyrrole-GSH conjugate) and pyrrole-diGSH conjugate, which were then measured by LC-MS analysis with detailed methods described in Supplementary material.
Statistical and data analysis
Kinetic parameters were calculated with the WinNonlin program version 4.0 (Pharsight Corporation, Mountain View, CA), using a non-compartmental model. Statistical analysis was performed using Prism 7 software (GraphPad, San Diego, CA). Statistical significance of parameter differences among experimental groups was analyzed by Kruskal-Wallis test or Friedman test for comparison of multiple variables. A p value less than 0.05 was considered statistically significant.
Results
Hepatic P450 enzymes are essential for in vivo bioactivation of MCT
The formation of pyrrole-protein adducts is the indicator of P450s-mediated metabolic activation of PAs (Ruan et al. 2014). The respective roles of the hepatic and extrahepatic P450s in the conversion of MCT into dehydro-MCT were determined by comparing levels of pyrrole-protein adducts in WT, LCN, and xh-CL mice. After MCT exposure, comparable levels of pyrrole-protein adducts were found in the respective serum (Fig. 1a), liver (Fig. 1b) and lung (Fig. 1c) samples from MCT-exposed WT mice and xh-CL mice, indicating that the substantial decrease in extrahepatic Cpr expression and P450 activity in xh-CL mice did not alter the in vivo generation of dehydro-MCT. On the other hand, compared with WT mice and xh-CL mice, LCN mice had significantly lower levels (by >60%) of pyrrole-protein adducts in all (serum, liver and lung) samples, a result indicating that liver Cpr deletion inhibited the metabolic activation of MCT in vivo. Moreover, WT and xh-CL mice, with competent hepatic P450s, had significantly higher amounts of pyrrole-protein adducts (by >3-fold) detected in the liver than those in the lungs, indicating the liver as the main site of MCT bioactivation. These data demonstrated that in vivo production of dehydro-MCT was mainly mediated by hepatic P450s in the liver, while with minimal contributions by extrahepatic P450s. Furthermore, regardless of the Cpr expression status, no marked difference in pyrrole-protein adducts level was found between male and female mice, indicating absence of gender variance in the bioactivation of MCT.
Fig. 1.
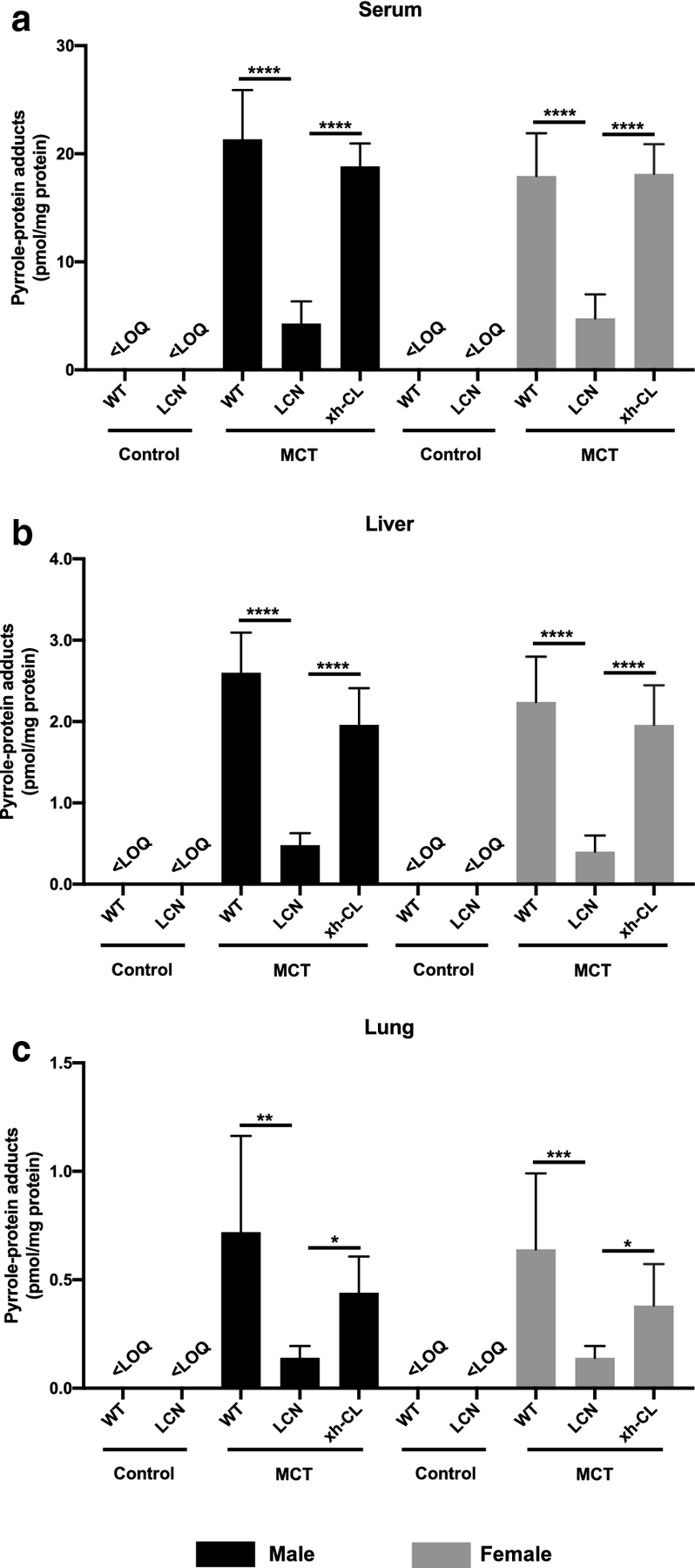
Pyrrole-protein adducts levels in serum, liver and lung samples of WT, LCN, and xh-CL mice. Adult male (a–c) and female (d–f) mice of various genotypes, all on the C57BL/6 genetic background, were treated with a single oral administration of MCT via gavage at 120 mg/kg or saline (vehicle control). Data are presented as means ± SD (n=5). Friedman test was used. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. <LOQ, below limit of quantitation.
Hepatic P450s significantly influence the systemic clearance of MCT
A kinetic study was performed to investigate the effect of hepatic P450 deficiency on circulating levels of MCT in MCT-exposed mice. As shown in Fig. 2, after oral administration, MCT was rapidly absorbed in all mouse groups to reach the maximum serum concentration at approximately 30 min. In comparison with WT mice, LCN mice showed markedly higher serum MCT levels at multiple time points examined. The determined kinetic parameters (Table 1) are also compared among the three genotypic groups. LCN mice had over 2-fold higher Cmax and AUC values compared with WT mice, a result consistent with an anticipated, large decrease in hepatic MCT metabolism in LCN mice. Besides, compared with xh-CL mice, LCN mice also showed significantly higher Cmax and AUC values, whereas xh-CL mice had no significant difference in any PK parameters compared with WT mice. The data indicated that dysfunctional hepatic P450s largely decreased MCT metabolism, while diminished extrahepatic P450 activity did not alter MCT metabolism. Consistently, LCN mice also had lower CL/F values compared with WT mice (by >3-fold in males and >2-fold in females) and xh-CL mice (by >2-fold in males and >1.5-fold in females), further confirming that deficiency of hepatic P450s resulted in lower efficiency of eliminating MCT. Besides, male WT mice demonstrated higher CL/F compared with female WT mice, indicating higher MCT elimination efficiency in male WT mice. Apart from WT mice, no significant difference in the serum level and elimination rate of MCT was found between male and female mice of the same genotype (LCN or xh-CL).
Fig. 2.
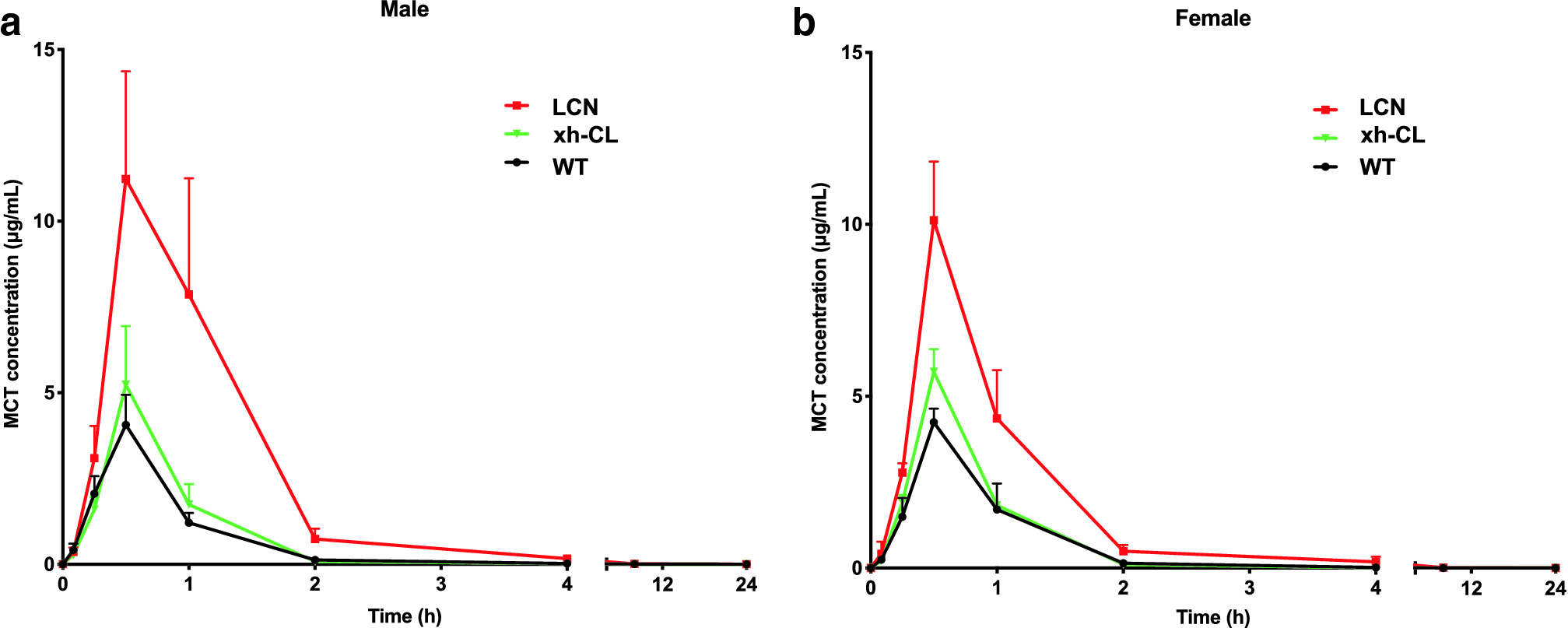
Toxicokinetic profiles of serum MCT in WT, LCN, and xh-CL mice. Adult male (a) and female (b) mice of various genotypes were treated with a single oral administration of MCT via gavage at 120 mg/kg. Blood (20–25 μL) was collected via the tail vein at various times (0–24 h) for the analysis of serum MCT levels. Data are presented as means ± SD (n=3).
Table 1.
Toxicokinetic parameters of serum MCT after a single oral administration of MCT at 120 mg/kg
| Male |
Female |
|||||
|---|---|---|---|---|---|---|
| WT | LCN | xh-CL | WT | LCN | xh-CL | |
| Cmax (μg/mL) | 4.06 ± 0.87 | 11.2 ± 3.1a, b | 5.23 ± 1.71 | 4.24 ± 0.40 | 10.1 ± 1.7 c | 5.71 ± 0.66 |
| AUC0-t (min×μg/mL) | 189 ± 23 | 746 ± 258 d, e | 227 ± 34 | 207 ± 41 | 526 ± 46 f | 246 ± 36 |
| t1/2 (min) | 56.5 ± 38.4 | 60.5 ± 16.0 | 42.6 ± 34.0 | 55.1 ± 36.4 | 48.2 ± 23.1 | 38.0 ± 31.9 |
| CL/F (mL/min) | 24.2 ± 2.1 | 6.62 ± 1.9 g, h | 17.8 ± 1.3 | 17.4 ± 3.2i | 6.92 ± 0.29 j, k | 12.8 ± 1.4 |
Data from Fig. 2 are used to calculate toxicokinetic parameters.
Cmax: maximum peak concentration. AUC0-t: the area under the concentration versus time curve from zero to the last time point tested. t1/2 half-life of serum MCT concentration. CL/F: apparent clearance rate. Data are presented as mean±SD (n=3). Friedman test was used.
p=0.0023
p=0.0005
p<0.0001, vs. WT male mice
p=0.0094
p=0.0010
p=0.0001, vs. xh-CL male mice
p=0.0109
p=0.0381
p=0.0002, vs. WT female mice
p=0.0098, vs. WT male mice
p=0.0245, vs. xh-CL female mice.
Liver-specific Cpr deletion abolishes MCT-induced lung injury
Toxicological assays were performed in parallel to investigate the toxicity outcome of MCT exposure. As shown in Fig. 3a, MCT-exposed WT male mice showed slightly elevated serum ALT activity compared to vehicle-treated WT males, but the value was within the normal physiological range (Okamoto et al. 2001). Apart from WT male mice, serum ALT activity levels were not significantly elevated after MCT exposure in any of the other mouse groups. Consistently, no obvious pathological changes were found in H&E-stained liver sections from any of the MCT-exposed mice (Fig. 3b). It should be noted that some vacuoles (presumably lipid droplets, as was reported previously in LCN mouse (Xue et al. 2011)) were observed in hepatocytes from both control and MCT exposed LCN mice, which should not be considered as MCT-induced liver lesions.
Fig. 3.
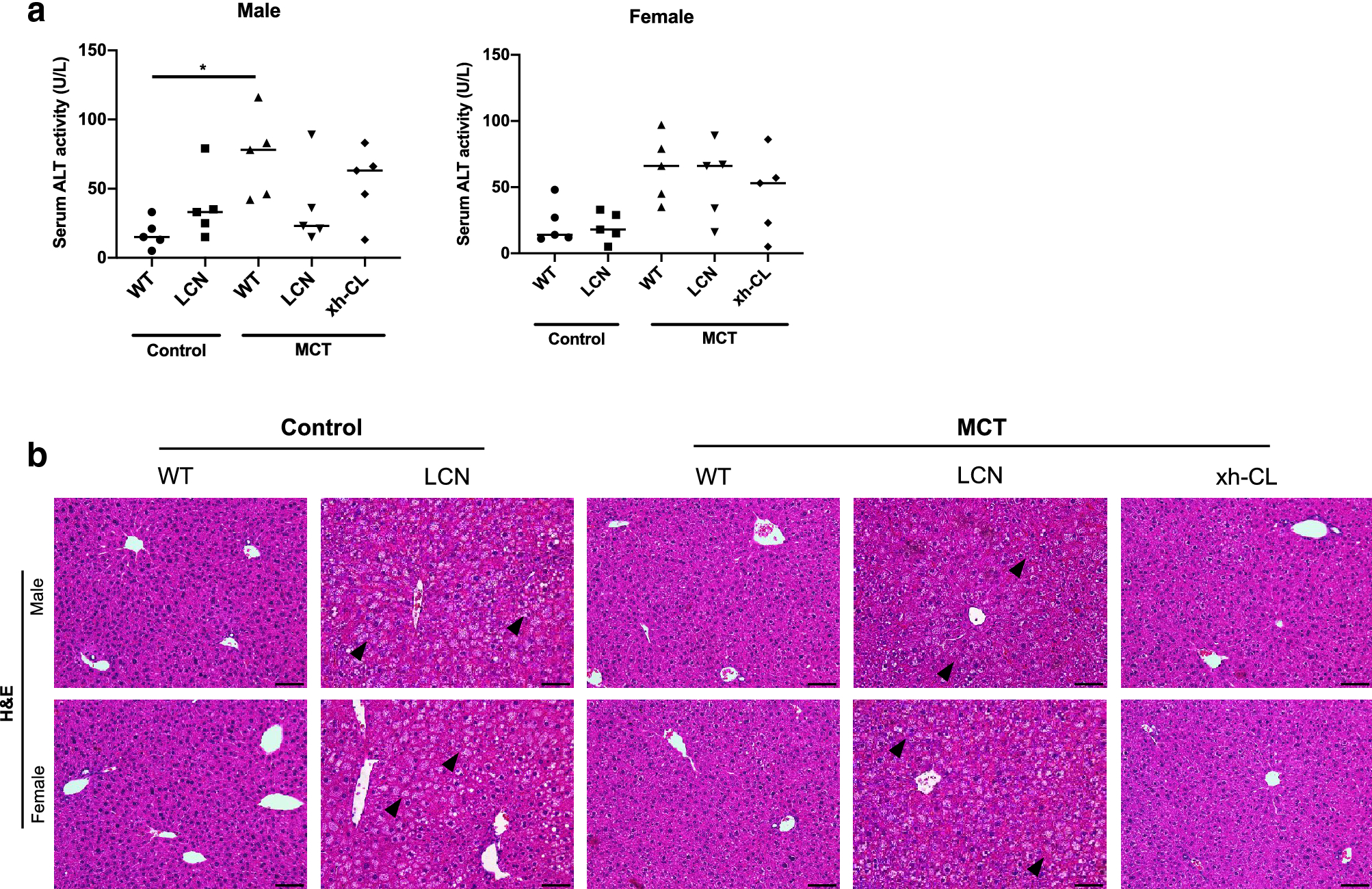
Toxicological assay on control or MCT-exposed mouse liver. Blood and liver were collected from adult (2–3 months old) male and female mice (n=5 per group) at 48 h after a single oral administration of MCT (120 mg/kg) or saline (vehicle control). (a) Serum ALT activity. Kruskal-Wallis test was used. *p<0.05. (b) Representative histopathological changes indicated by H&E staining on control or MCT-exposed mouse liver sections. Scale bars = 200 μm. Arrowheads point to vacuoles in hepatocytes of LCN mice.
Under the same conditions that were non-toxic to the liver, MCT exposure caused pulmonary toxicity. Analyses of H&E-stained lung sections revealed that both WT and xh-CL mice, but not LCN mice, sustained significant lung injury represented by congestion and adventitia disruption in pulmonary arteries (Fig. 4a), arterioles (Fig. 4b), and pulmonary veins (Fig. 4c). The genotypic difference in sensitivity to MCT-induced pulmonary injury was further confirmed by quantitative analysis of the degree of smooth muscle wall thickening in pulmonary arteries, which is the typical lesion of PA-induced lung injury (Heath et al. 1975). As shown in Fig. 4d, compared to saline-treated control mice, MCT exposure significantly increased pulmonary artery wall thickness in WT (by 50% in males and 41% in females) mice, but not in LCN mice. Besides, the degree of arterial wall thickening in xh-CL mice was also higher than that in LCN mice.
Fig. 4.
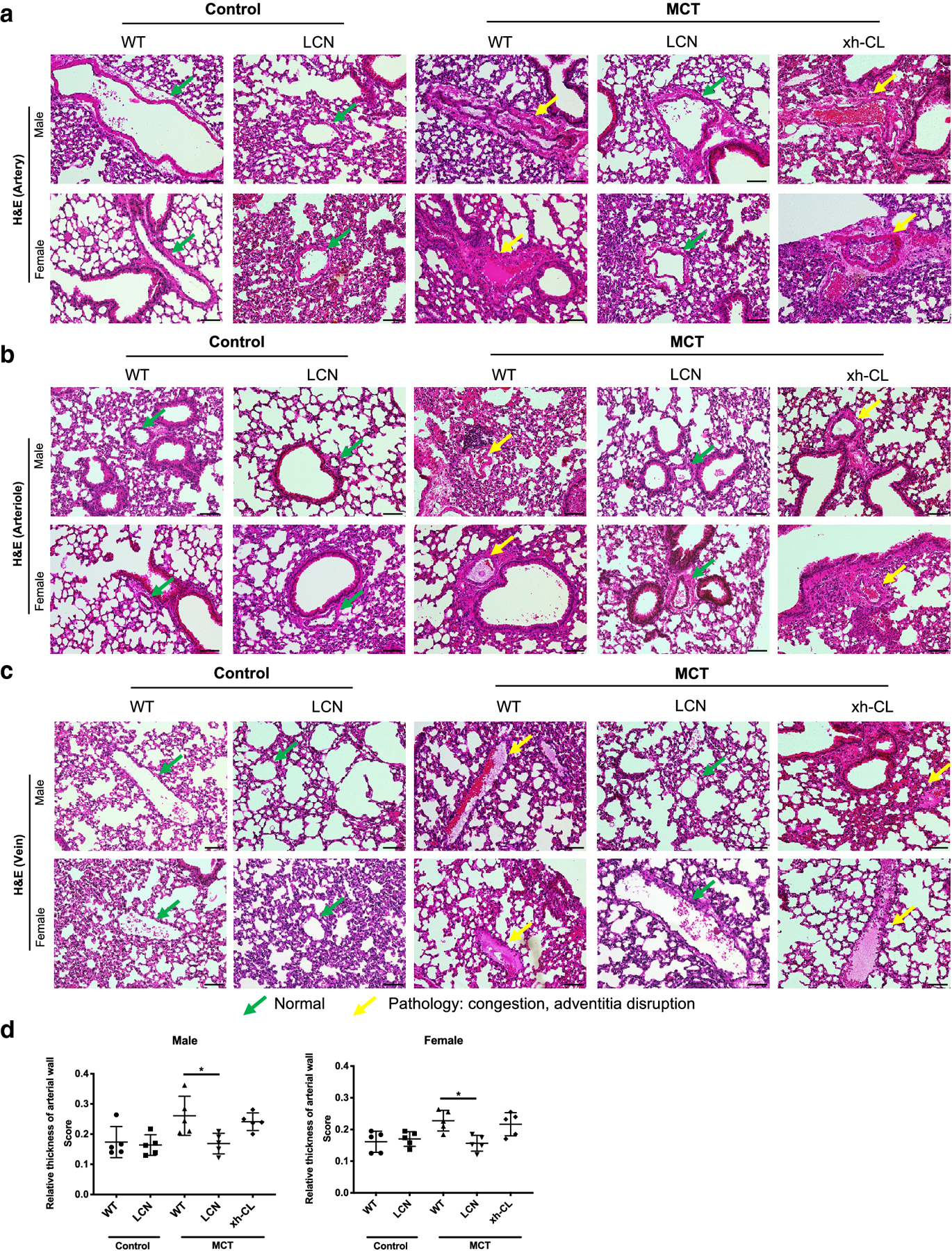
Histopathological analysis of MCT-induced changes in pulmonary vasculature. Lungs were collected from adult (2–3 months old) male and female mice at 48 h after a single oral administration of MCT (120 mg/kg) or saline (vehicle control). Representative data from H&E stained lung sections are shown for pulmonary arteries (a), arterioles (b) and veins (c). Normal pulmonary vasculature (green arrows) and characteristic MCT-induced pathologic changes (yellow arrows) are indicated. Scale bars=200 μm. (d) Scoring on the relative thickness of pulmonary arterial wall of control or MCT-exposed mice. Data are presented as means ± SD (n=5). Kruskal-Wallis test was used. *p<0.05.
Moreover, because PA exposure characteristically induce endothelial cell damage, IHC assays were performed to investigate the integrity of pulmonary vascular endothelium, with use of antibodies to CD31, a specific marker for endothelial cells. The results demonstrated that MCT-exposed WT and xh-CL mice, but not LCN mice, had damaged (CD31-positive) endothelium in pulmonary arteries (Fig. 5a) and veins (Fig. 5b). Taken together, competent hepatic P450s, but not extrahepatic P450s, were indispensable for MCT-induced lung injury.
Figure 5.
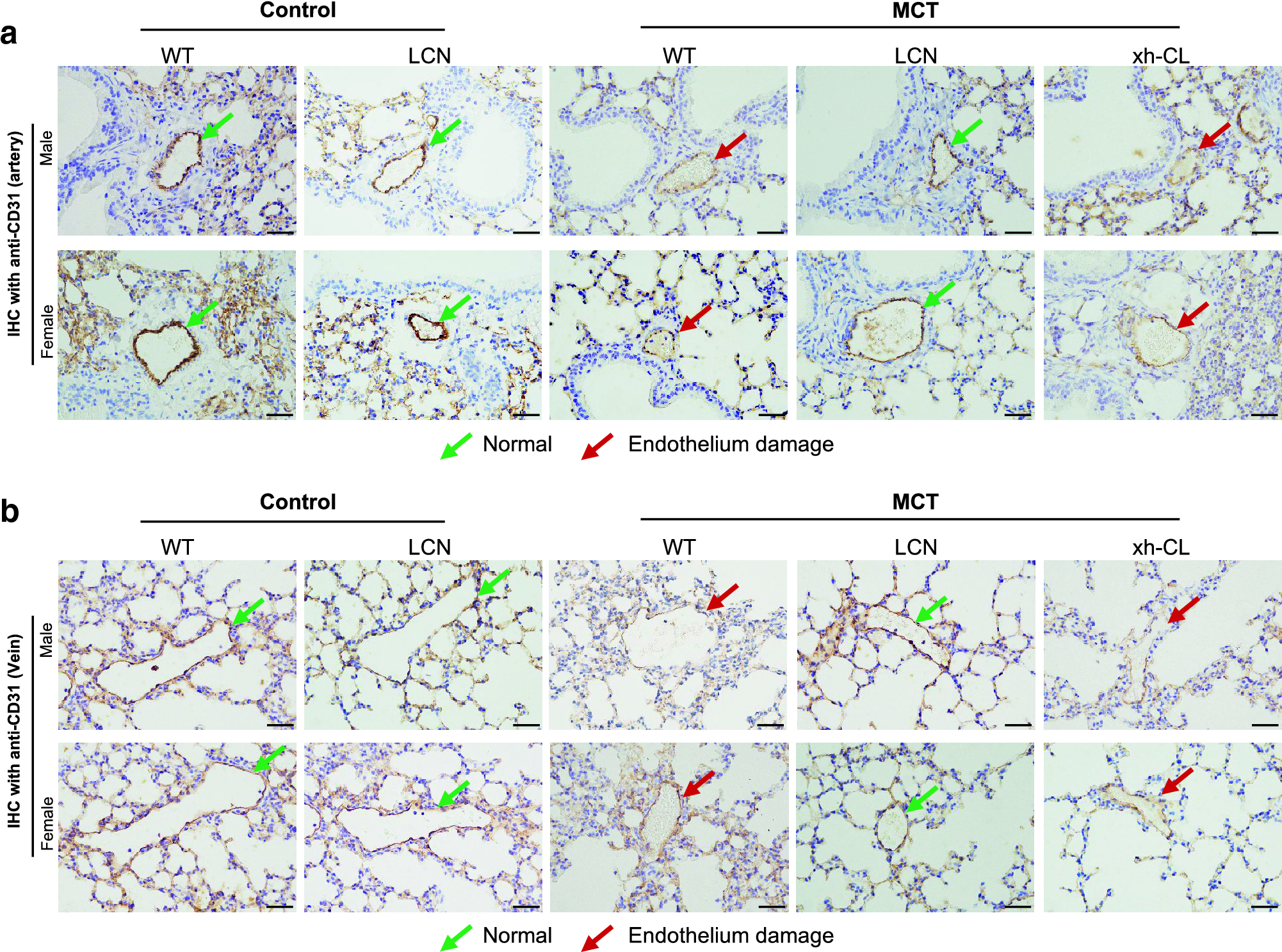
IHC detection of MCT-induced endothelium damage in pulmonary vasculature. Paraffin-embedded lung sections were immunostained with an antibody to CD31, a marker for endothelial cells. The sections were counterstained with hematoxylin (purple). CD31-positive endothelial cells were stained brown. Representative data are shown for pulmonary arteries (a) and veins (b). Green arrows indicate normal intact endothelium of the pulmonary vasculature, whereas red arrows indicate damaged endothelium with loss of CD31-positive endothelial cells. Scale bars=100 μm.
Reactive metabolites of MCT generated in the liver are transported into the lungs via circulating blood
The fact that the amount of hepatic pyrrole-protein adducts was remarkably greater than that of pulmonary pyrrole-protein adducts (Fig. 1), and that the level of pulmonary pyrrole-protein adducts was substantially reduced when hepatic P450 activity was markedly diminished (in LCN mice) supports the hypothesis that liver-derived dehydro-PAs and their hydrolyzed product DHP were transported via circulating blood from the liver into the lungs. To directly prove this hypothesis, we exposed rats to MCT. Rats were used, instead of mice, to facilitate surgical collection of hepatic blood perfusion.
The reactive metabolites of PAs are extremely unstable and nearly impossible to directly detect them by analytical instruments (Huxtable et al. 1996; Pan et al. 1993). We determined the reactive metabolites of MCT, including dehydro-MCT and DHP, by adding GSH as a trapping reagent (Mattocks and Jukes 1990) into freshly-collected blood to form stable pyrrole-GSH conjugates for the analysis (Fig. 6a). LC-MS detection of both pyrrole-monoGSH conjugate and pyrrole-diGSH conjugate confirmed the presence of the reactive metabolites of MCT in the blood. Over the 240-min sampling period, pyrrole-monoGSH and pyrrole-diGSH conjugates were detected in both saline-added RBCs and plasma, indicating the endogenous formation of pyrrole-GSH conjugates induced by MCT exposure. Compared with the saline-added counterparts, the exogenous addition of GSH to RBCs and plasma of MCT-exposed rats led to significant increases in the detected amounts of pyrrole-GSH conjugates (Fig. 6b, 6c, & 6e), although the difference of pyrrole-monoGSH conjugate detected in GSH- and saline-added RBCs was not statistically significant (Friedman test), while apparently higher level of pyrrole-monoGSH conjugate was detected in GSH-added RBCs over the 240-min sampling period (Fig. 6d). The results confirmed the presence of the reactive metabolites of MCT in blood circulation. Interestingly, despite the coexistence of pyrrole-monoGSH conjugate and pyrrole-diGSH conjugate in blood samples of MCT-exposed rats, the level of pyrrole-monoGSH conjugate was approximately 100-fold higher than that of pyrrole-diGSH conjugate. Collectively, these data confirmed that the reactive metabolites of MCT produced in the liver were transported via circulating blood into the lungs.
Fig. 6.
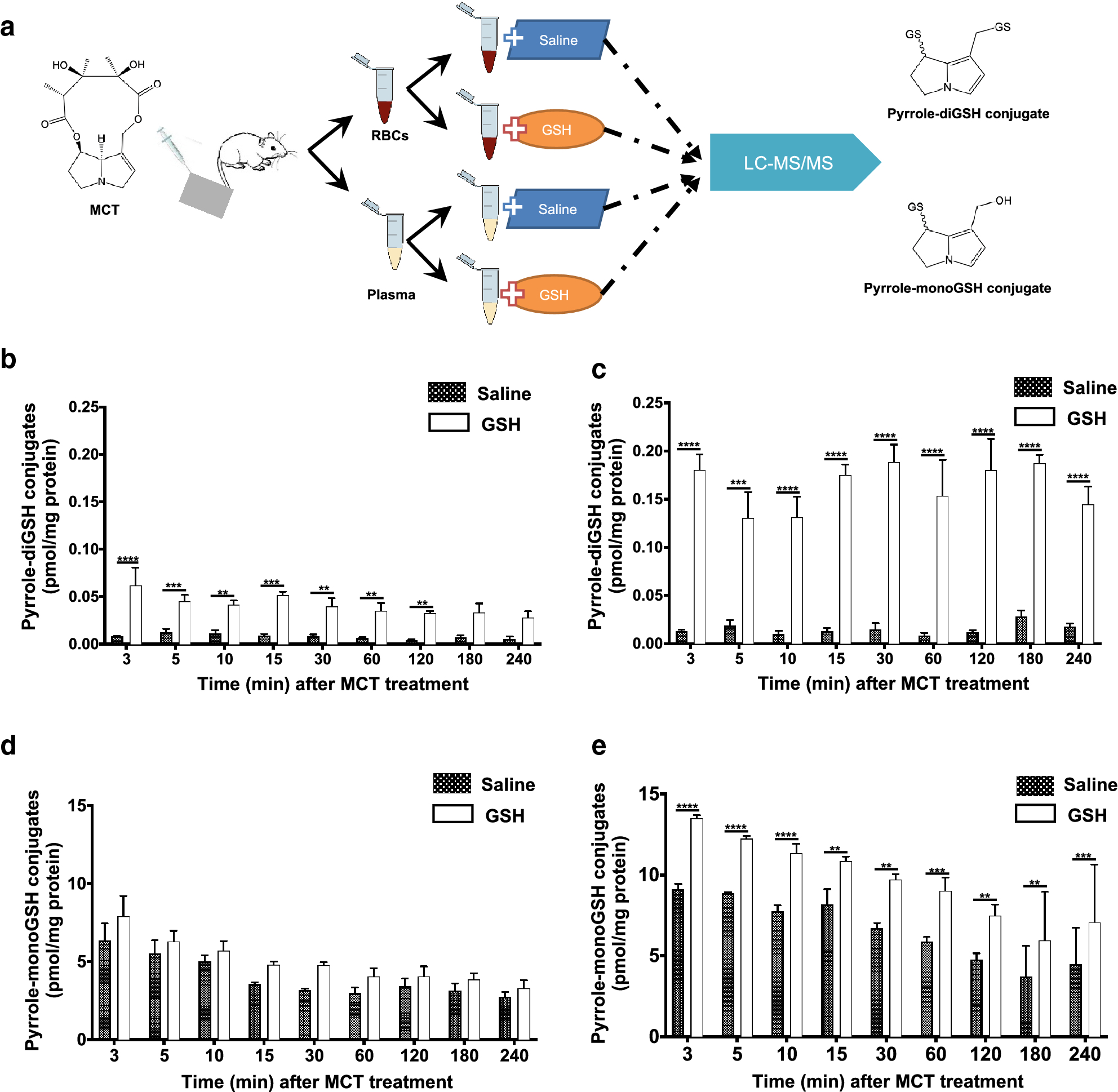
Detection of reactive metabolites of MCT in liver efflux blood of MCT-treated rats. Adult male SD rats (200–220 g) were anesthetized and treated with a single administration of MCT via intravenous injection at 65 mg/kg, and blood efflux from the liver (400 μL) was collected via the cannulated vein at various times (0–240 min) for further processing as illustrated (a). Pyrrole-diGSH conjugates (b, c) and pyrrole-monoGSH conjugates (d, e) were determined in RBCs (b, d) or plasma (c, e). Data are presented as means ± SD (n=3). Friedman test was used to compare GSH-added RBCs or plasma with saline-vehicle RBCs or plasma. **p<0.01, ***p<0.001, ****p<0.0001.
Discussion
PAH is a public health problem with poor survival rate (Gerges et al. 2015). The development of PAH is attributed to multifactorial etiologies such as exposure to pneumotoxins (Yuan and Rubin 2005). MCT, a PA mostly produced by plants from Crotalaria spp., has been extensively used to establish the PAH model in rodents for many years (Hill et al. 2017). In addition, various other PAs can also induce lung injury (Song et al. 2020). In the present study, MCT, a representative pneumotoxic PA, was investigated to delineate the initial biochemical mechanism of PA-induced PAH. Considering that P450s-mediated metabolic activation of PAs is required for their toxic effect (Ruan et al. 2014), it is thus important to investigate the metabolic mechanism of PA-induced lung injury. Although P450s are most abundant in the liver, some extrahepatic tissues, such as the lungs and small intestine, have been reported to be highly efficient in target-tissue bioactivation of certain toxins (Ding and Kaminsky 2003). The Cpr-knockout mouse models enable the delineation of the roles of organ-specific P450s in PA metabolism (Gu et al. 2003; Hessel-Pras et al. 2020; Wei et al. 2010). The present study for the first time directly demonstrated the influence of tissue-specific suppression of P450 activity in the liver versus extrahepatic tissues on PA metabolism, especially metabolic activation, and resultant lung toxicity.
Pyrrole-protein adducts, which are derived from the covalent binding of the reactive metabolites of PAs with various proteins, are the biomarker of PA exposure and are proportionally related to toxicity severity (Lin et al. 2011). The amount of pyrrole-protein adducts is positively correlated with the generation of the reactive metabolites of PAs, thereby reflecting the rate of P450s-mediated PA bioactivation (Ruan et al. 2014). After MCT exposure, we found comparable levels of pyrrole-protein adducts in the blood, liver and lungs of WT and xh-CL mice, a result indicating that diminished extrahepatic P450 activity did not affect the generation of the reactive metabolites of MCT. Compared to WT and xh-CL mice, the significant decrease of levels of pyrrole-protein adducts in LCN mice demonstrated that the loss of hepatic P450 function led to remarkable inhibition of the metabolic activation of PAs. However, the small amount of residual pyrrole-protein adducts found in MCT-exposed LCN mice may reflect minor contributions by extrahepatic P450s or other enzymes to the production of the reactive metabolites of MCT. Nevertheless, our findings provided the first direct evidence that hepatic P450s play a predominant role in the metabolic activation of PAs in vivo.
Results of the toxicokinetic study were consistent with the findings of pyrrole-protein adduct analysis. Compared with WT and xh-CL mice, hepatic P450 deficiency in LCN mice led to dramatically reduced PA metabolism, manifest as lower clearance and higher serum concentration of MCT. On the contrary, the suppression of extrahepatic P450 activity in xh-CL mice did not influence MCT metabolism, as indicated by the essentially comparable kinetic parameters between xh-CL and WT mice. Combined with the marked decrease in metabolic activation of MCT and the resultant decrease in the formation of pyrrole-protein adducts in LCN mice, the kinetic data confirmed that competency of hepatic P450s, not extrahepatic P450s, is essential for systemic metabolism and bioactivation of orally ingested MCT.
Significant lung injury, represented by vasculature damage, was observed in MCT-exposed WT and xh-CL mice but not in LCN mice. The reduction in cytotoxicity and lung injury in MCT-exposed LCN mice was consistent with the largely decreased hepatic MCT metabolism and markedly lowered levels of serum, liver and lung pyrrole-protein adducts in LCN mice, compared to WT and xh-CL mice. These results firmly support that PA-induced lung injury is critically dependent on the metabolic activation of PAs mediated by hepatic P450s.
Our data further support the concept that liver-produced the reactive metabolites of PAs are responsible for the MCT-induced lung injury. The presence of pyrrole-protein adducts in the serum of MCT-treated mice suggested that the reactive metabolites of MCT generated from the liver were released into blood, where they reacted with proteins in serum and/or hepatocytes and subsequently released pyrrole-protein adducts into the blood. Further studies using blood efflux from the hepatic veins of MCT-exposed rats confirmed the presence of reactive metabolites of MCT capable of forming pyrrole-GSH conjugates with endogenously added GSH in both RBCs and plasma (Fig. 6). The fact that the reactive MCT metabolites are detected in both plasma and RBCs suggested that proteins in both compartment (e.g., hemoglobin in RBCs and albumin in plasma) may serve as the carriers for transport of the reactive metabolites of MCT from the liver to the lungs (Fig. 7).
Fig. 7.
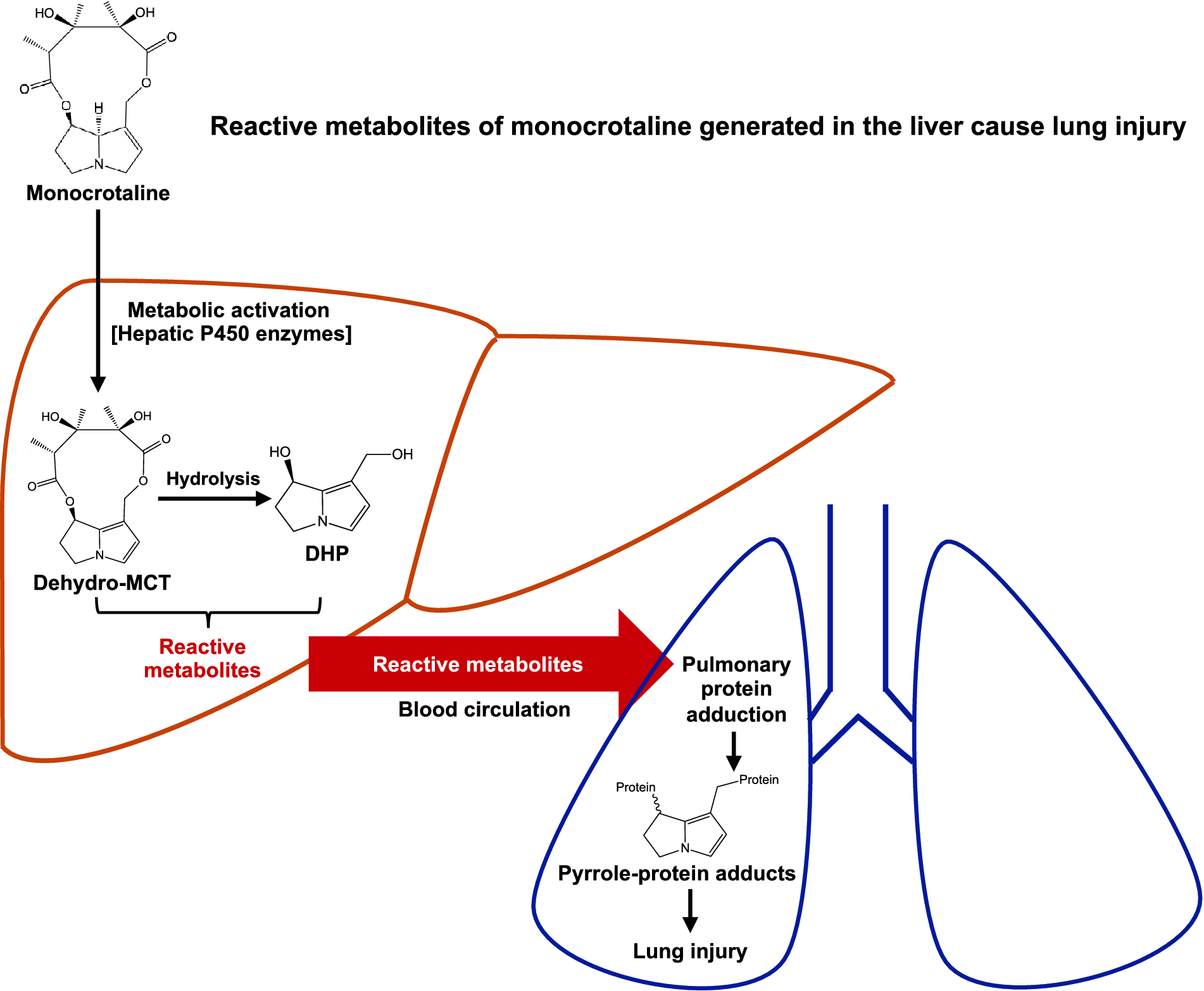
Schematic illustration of the initial biochemical mechanism of MCT-induced lung injury. The metabolic activation of the ingested MCT is mediated by hepatic P450 enzymes. The reactive metabolites of MCT generated in the liver are carried by blood efflux from the liver and transported into the lungs to form pulmonary pyrrole-protein adducts, thereby causing lung injury.
This study also provides an interesting insight regarding stability of the reactive metabolites of MCT in the blood. The experimental procedure, from the blood collection and processing to GSH addition, took at least 5 min to complete, after which, the reactive metabolites of MCT were still intact and able to form conjugates with GSH. This finding, suggesting relatively high stability of these reactive metabolites in blood, is contrary to previous reports, which revealed that the reactive metabolites of PAs, especially dehydro-PAs, were highly unstable in vitro with half-life ranging from 1.8 sec to 2.7 sec (Mattocks and Jukes 1990). The apparent contradiction between previous in vitro results and the present in vivo findings may be explained by the possibility that blood components including RBCs and plasma not only transported, but also stabilized the reactive metabolites of PAs in the body, an idea that has been proposed previously using isolated liver and lung preparations (Lafranconi and Huxtable 1984; Pan et al. 1991; Yan and Huxtable 1995) or blood of rats injected with dehydro-MCT (Lame et al. 1997). Similar “Trojan horse” effects have also been reported previously for nanoparticles in RBCs, macrophages, and lymphocytes (Choi et al. 2007; Pan et al. 1991). Apart from dehydro-PAs, there are also secondary metabolites derived from dehydro-PAs. For instance, dehydro-PAs can be hydrolyzed to DHP, while the reactivity of DHP is much less than that of dehydro-PAs. Moreover, dehydro-PAs and DHP can further interreact with GSH and amino acids (e.g., cysteine) to form pyrrole-GSH or pyrrole-amino acid conjugates, and these secondary reactive metabolites were reported to interact with DNA (He et al. 2019; He et al. 2020a; He et al. 2020b; Xia et al. 2018). It is thus extrapolated that these secondary metabolites of PAs are reactive but to a much less extent compared with dehydro-PAs. Nevertheless, a significantly prolonged half-life of all reactive metabolites of MCT in blood would at least partly explain the susceptibility of the lung vasculature to MCT intoxication. Further investigations are warranted to reveal i) the exact cells or proteins responsible for reversible binding with reactive metabolites of PAs in blood; ii) the mechanisms for stabilizing reactive metabolites of PAs; iii) the process of releasing the reactive metabolites of PAs into other organs (e.g., the lungs) and resultant intoxications.
It is noteworthy that oral administration of MCT at 120 mg/kg induced pulmonary vascular damage but not liver injury in WT and xh-CL mice. This result, that MCT-induced lung injury may occur without symptomatic hepatic damage, is of special interest because most reported human PA-poisoning cases were represented by acute liver injury with explicit clinical symptoms, which were most likely caused by exposures to high dosages of PAs (Lin et al. 2011; Mohabbat et al. 1976; Suparmi et al. 2020). It appears that the lung is a preferred target organ in mice with the relatively low-level of MCT exposure, where acute hepatotoxicity manifestations are not apparent. It remains to be determined whether this apparent lung specificity of MCT toxicity is dose-related or species specific, and whether it is due to a greater capacity of the mouse liver than the lung vasculature to protect against the reactive metabolites of MCT. In that regard, MCT has been found to cause both liver and lung toxicity in rats at doses of 70 mg/kg (Song et al. 2020). Interestingly, Song et al. compared the level of pyrrole-protein adducts, which is positively related to the cytotoxicity, in the liver and lungs of rats exposed to different PAs at the same dose. Monocrotaline generates higher adducts level ratio (lung/liver) than that in other PAs, supporting that monocrotaline, in certain exposure level, is more pneumotoxic than hepatotoxic (Song et al. 2020). Additionally, it should be noted that the MCT exposure-induced pulmonary vasculature lesions in mice, including endothelium damage and arterial smooth muscle thickening, have been reported to be initiators for PAH development (Tuder et al. 2013). Further investigation is warranted indeed.
Our study showed significant lung injury after MCT exposure in mice with competent hepatic P450 activity. It should be noted that the dose (120 mg/kg of MCT) used in animal studies is much higher than the estimated average human PA exposure level: 0.019 μg/kg BW for children and 0.026 μg/kg BW for adults in median (Dusemund et al. 2018; EFSA 2016). Therefore, the present findings should be considered in the context of acute severe PA poisonings but not representative for potential risks posed by low dietary exposure in the general population.
To conclude, the present study provides the first mode-of-action evidence for a major role of hepatic P450 enzymes in PA-induced lung injury. We demonstrate that functional hepatic P450s are indispensable for the metabolic activation of PAs. Further, our data support the notion that the reactive metabolites of PAs generated in the liver are carried by blood efflux from the liver and transported into the lungs to exert pneumotoxicity. Given that hepatic P450 functions may vary due to induction, inhibition, or genetic polymorphism, our results provide the mechanistic basis for assessing the sensitivity of individuals to PA-induced lung injury and potential drug targets for medical intervention. Our findings also warrant further investigation into the possible roles of PA exposure in human pulmonary vascular diseases, especially those with unknown or unexplained causes.
Supplementary Material
Acknowledgment
The study was supported by Research Grants Council of Hong Kong Special Administrative Region (GRF Project No. 14160817, to G. Lin), CUHK Direct Grant (Project No. 4054376, to G. Lin), and a grant from the National Institute of Environmental Health Sciences, National Institutes of Health (R01 ES020867, to X. Ding). We thank Ms. Weizhu Yang for assistance with mouse breeding and genotyping analysis, Dr. Lei Yin and Dr. Xiangmeng Wu for assistance with analytical instruments, and Dr. Weiguo Han and Dr. Nataliia Kovalchuk for assistance with histological assays.
Abbreviations in the order of appearance
- PAs
pyrrolizidine alkaloids
- MCT
monocrotaline
- PAH
pulmonary arterial hypertension
- Dehydro-PAs
dehydro-pyrrolizidine alkaloids
- DHP
(±)-6,7-dihydro-7-hydroxy-1-hydroxymethyl-5H-pyrrolizine
- RBCs
red blood cells
- CPR
NADPH-cytochrome P450 reductase
- LCN
liver-specific Cpr-null
- xh-CL
extrahepatic Cpr-low
- WT
wild type
- LC-MS
liquid chromatography-mass spectrometry
- GSH
glutathione
- ALT
alanine aminotransferase
Footnotes
Compliance with ethical standards
Conflict of interest All authors declare that they do not have anything to disclose regarding funding or conflict of interest with respect to this manuscript.
References
- Choi MR, Stanton-Maxey KJ, Stanley JK et al. (2007) A cellular Trojan horse for delivery of therapeutic nanoparticles into tumors. Nano Lett 7(12):3759–3765. doi: 10.1021/nl072209h [DOI] [PubMed] [Google Scholar]
- Ding X, Kaminsky LS (2003) Human extrahepatic cytochromes P450: function in xenobiotic metabolism and tissue-selective chemical toxicity in the respiratory and gastrointestinal tracts. Annu Rev Pharmacol Toxicol 43:149–173. doi: 10.1146/annurev.pharmtox.43.100901.140251 [DOI] [PubMed] [Google Scholar]
- Dusemund B, Nowak N, Sommerfeld C, Lindtner O, Schafer B, Lampen A (2018) Risk assessment of pyrrolizidine alkaloids in food of plant and animal origin. Food Chem Toxicol 115:63–72. doi: 10.1016/j.fct.2018.03.005 [DOI] [PubMed] [Google Scholar]
- Ebmeyer J, Rasinger JD, Hengstler JG et al. (2020) Hepatotoxic pyrrolizidine alkaloids induce DNA damage response in rat liver in a 28-day feeding study. Arch Toxicol 94(5):1739–1751. doi: 10.1007/s00204-020-02779-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar JA, Colegate SM, Boppre M, Molyneux RJ (2011) Pyrrolizidine alkaloids in food: a spectrum of potential health consequences. Food Addit Contam A 28(3):308–324. doi: 10.1080/19440049.2010.547520 [DOI] [PubMed] [Google Scholar]
- Edgar JA, Molyneux RJ, Colegate SM (2015) Pyrrolizidine alkaloids: Potential role in the etiology of cancers, pulmonary hypertension, congenital anomalies, and liver disease. Chem Res Toxicol 28(1):4–20. doi: 10.1021/tx500403t [DOI] [PubMed] [Google Scholar]
- EFSA (2016) Dietary exposure assessment to pyrrolizidine alkaloids in the European population. EFSA J 14(8):1–50. doi: 10.2903/j.efsa.2016.4572 [DOI] [Google Scholar]
- Gerges M, Gerges C, Pistritto AM et al. (2015) Pulmonary hypertension in heart failure epidemiology, right ventricular function, and survival. Am J Respir Crit Care Med 192(10):1234–1246. doi: 10.1164/rccm.201503-0529OC [DOI] [PubMed] [Google Scholar]
- Gu J, Weng Y, Zhang QY et al. (2003) Liver-specific deletion of the NADPH-cytochrome P450 reductase gene - Impact on plasma cholesterol homeostasis and the function and regulation of microsomal cytochrome P450 and heme oxygenase. J Biol Chem 278(28):25895–25901. doi: 10.1074/jbc.M303125200 [DOI] [PubMed] [Google Scholar]
- Gyorik S, Stricker H (2009) Severe pulmonary hypertension possibly due to pyrrolizidine alkaloids in polyphytotherapy. Swiss Med Wkly 139(13–14):210–211. [DOI] [PubMed] [Google Scholar]
- He X, Xia Q, Gamboa da Costa G, Lin G, Fu PP (2019) 1-Formyl-7-hydroxy-6,7-dihydro-5H-pyrrolizine (1-CHO-DHP): A potential proximate carcinogenic metabolite of pyrrolizidine alkaloids. Chem Res Toxicol 32(6):1193–1203. doi: 10.1021/acs.chemrestox.9b00038 [DOI] [PubMed] [Google Scholar]
- He X, Xia Q, Shi Q, Fu PP (2020a) Effects of glutathione and cysteine on pyrrolizidine alkaloid-induced hepatotoxicity and DNA adduct formation in rat primary hepatocytes. J Environ Sci Health C Toxicol Carcinog 38(2):109–123. doi: 10.1080/26896583.2020.1738161 [DOI] [PubMed] [Google Scholar]
- He X, Xia Q, Zhao Y, Fu PP (2020b) 1-Formyl-7-hydroxy-6,7-dihydro-5H-pyrrolizine (1-CHO-DHP)-cysteine conjugates: Metabolic formation and binding to cellular DNA. Chem Res Toxicol 33(8):2139–2146. doi: 10.1021/acs.chemrestox.0c00143 [DOI] [PubMed] [Google Scholar]
- Heath D, Shaba J, Williams A, Smith P, Kombe A (1975) A pulmonary hypertension-producing plant from Tanzania. Thorax 30(4):399–404. doi: 10.1136/thx.30.4.399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hessel-Pras S, Braeuning A, Guenther G et al. (2020) The pyrrolizidine alkaloid senecionine induces CYP-dependent destruction of sinusoidal endothelial cells and cholestasis in mice. Arch Toxicol 94(1):219–229. doi: 10.1007/s00204-019-02582-8 [DOI] [PubMed] [Google Scholar]
- Hill NS, Gillespie MN, McMurtry IF (2017) Fifty years of monocrotaline-induced pulmonary hypertension: What has it meant to the field? Chest 152(6):1106–1108. doi: 10.1016/j.chest.2017.10.007 [DOI] [PubMed] [Google Scholar]
- Huxtable RJ, Yan CC, Wild S, Maxwell S, Cooper R (1996) Physicochemical and metabolic basis for the differing neurotoxicity of the pyrrolizidine alkaloids, trichodesmine and monocrotaline. Neurochem Res 21(2):141–146. doi: 10.1007/BF02529131 [DOI] [PubMed] [Google Scholar]
- Kay JM, Heath D, Smith P, Bras G, Summerell J (1971) Fulvine and the pulmonary circulation. Thorax 26(3):249–261. doi: 10.1136/thx.26.3.249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafranconi WM, Huxtable RJ (1984) Hepatic-metabolism and pulmonary toxicity of monocrotaline using isolated perfused liver and lung. Biochem Pharmacol 33(15):2479–2484. [DOI] [PubMed] [Google Scholar]
- Lame MW, Jones AD, Morin D, Wilson DW, Segall HJ (1997) Association of dehydromonocrotaline with rat red blood cells. Chem Res Toxicol 10(6):694–701. doi: 10.1021/tx960173v [DOI] [PubMed] [Google Scholar]
- Lebrec D, Lacroix S, Benhamou JP (1980) Hepatic vein catheterization in the rat. Pflug Arch Eur J Phy 387(1):67–68. doi:Doi 10.1007/Bf00580846 [DOI] [PubMed] [Google Scholar]
- Lin G, Wang JY, Li N et al. (2011) Hepatic sinusoidal obstruction syndrome associated with consumption of Gynura segetum. J Hepatol 54(4):666–673. doi:DOI 10.1016/j.jhep.2010.07.031 [DOI] [PubMed] [Google Scholar]
- Lu Y, Ma J, Lin G (2019) Development of a two-layer transwell co-culture model for the in vitro investigation of pyrrolizidine alkaloid-induced hepatic sinusoidal damage. Food Chem Toxicol 129:391–398. doi: 10.1016/j.fct.2019.04.057 [DOI] [PubMed] [Google Scholar]
- Lu Y, Ma J, Song ZJ, Ye Y, Fu PP, Lin G (2018) The role of formation of pyrrole-ATP synthase subunit beta adduct in pyrrolizidine alkaloid-induced hepatotoxicity. Arch Toxicol 92(11):3403–3414. doi: 10.1007/s00204-018-2309-6 [DOI] [PubMed] [Google Scholar]
- Ma J, Ruan J, Chen X et al. (2019) Pyrrole-hemoglobin adducts, a more feasible potential biomarker of pyrrolizidine alkaloid exposure. Chem Res Toxicol 32(6):1027–1039. doi: 10.1021/acs.chemrestox.8b00369 [DOI] [PubMed] [Google Scholar]
- Mattocks AR, Jukes R (1990) Trapping and measurement of short-lived alkylating-agents in a recirculating flow system. Chem Biol Interact 76(1):19–30. [DOI] [PubMed] [Google Scholar]
- Mohabbat O, Younos MS, Merzad AA, Srivastava RN, Sediq GG, Aram GN (1976) An outbreak of hepatic veno-occlusive disease in north-western Afghanistan. Lancet 308(7980):269–271. [DOI] [PubMed] [Google Scholar]
- Okamoto T, Yoshida S, Kobayashi T, Okabe S (2001) Inhibition of concanavalin A-induced mice hepatitis by coumarin derivatives. Jpn J Pharmacol 85(1):95–97. doi: 10.1254/jjp.85.95 [DOI] [PubMed] [Google Scholar]
- Pan LC, Lame MW, Morin D, Wilson DW, Segall HJ (1991) Red-blood-cells augment transport of reactive metabolites of monocrotaline from liver to lung in isolated and tandem liver and lung preparations. Toxicol Appl Pharmacol 110(2):336–346. doi:Doi 10.1016/S0041-008x(05)80016-X [DOI] [PubMed] [Google Scholar]
- Pan LC, Wilson DW, Lame MW, Jones AD, Segall HJ (1993) COR pulmonale is caused by monocrotaline and dehydromonocrotaline, but not by glutathione or cysteine conjugates of dihydropyrrolizine. Toxicol Appl Pharmacol 118(1):87–97. doi: 10.1006/taap.1993.1013 [DOI] [PubMed] [Google Scholar]
- Prakash AS, Pereira TN, Reilly PEB, Seawright AA (1999) Pyrrolizidine alkaloids in human diet. Mutat Res-Gen Tox En 443(1–2):53–67. doi:Doi 10.1016/S1383-5742(99)00010-1 [DOI] [PubMed] [Google Scholar]
- Ridker PM, Ohkuma S, McDermott WV, Trey C, Huxtable RJ (1985) Hepatic venocclusive disease associated with the consumption of pyrrolizidine-containing dietary supplements. Gastroenterology 88(4):1050–1054. doi: 10.1016/s0016-5085(85)80027-5 [DOI] [PubMed] [Google Scholar]
- Ruan J, Gao H, Li N et al. (2015) Blood pyrrole-protein adducts--A biomarker of pyrrolizidine alkaloid-induced liver Injury in humans. J Environ Sci Health C 33(4):404–421. doi: 10.1080/10590501.2015.1096882 [DOI] [PubMed] [Google Scholar]
- Ruan J, Yang M, Fu P, Ye Y, Lin G (2014) Metabolic activation of pyrrolizidine alkaloids: insights into the structural and enzymatic basis. Chem Res Toxicol 27(6):1030–1039. doi: 10.1021/tx500071q [DOI] [PubMed] [Google Scholar]
- Shi RZ, Wei ZH, Zhu DY et al. (2018) Baicalein attenuates monocrotaline-induced pulmonary arterial hypertension by inhibiting vascular remodeling in rats. Pulm Pharmacol Ther 48:124–135. doi: 10.1016/j.pupt.2017.11.03 [DOI] [PubMed] [Google Scholar]
- Song Z, He Y, Ma J, Fu PP, Lin G (2020) Pulmonary toxicity is a common phenomenon of pyrrolizidine alkaloids. J Environ Sci Health C 38(2):124–140. doi: 10.1080/26896583.2020.1743608 [DOI] [PubMed] [Google Scholar]
- Stegelmeier BL, Edgar JA, Colegate SM et al. (1999) Pyrrolizidine alkaloid plants, metabolism and toxicity. J Nat Toxins 8(1):95–116. [PubMed] [Google Scholar]
- Suparmi S, Wesseling S, Rietjens I (2020) Monocrotaline-induced liver toxicity in rat predicted by a combined in vitro physiologically based kinetic modeling approach. Arch Toxicol 94:3281–3295. doi: 10.1007/s00204-020-02798-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuder RM, Archer SL, Dorfmuller P et al. (2013) Relevant issues in the pathology and pathobiology of pulmonary hypertension. J Am Coll Cardiol 62(25):D4–D12. doi: 10.1016/j.jacc.2013.10.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei YA, Zhou X, Fang C et al. (2010) Generation of a mouse model with a reversible hypomorphic cytochrome P450 reductase gene: Utility for tissue-specific rescue of the reductase expression, and insights from a resultant mouse model with gobal suppression of P450 reductase expression in extrahepatic tissues. J Pharmacol Exp Ther 334(1):69–77. doi: 10.1124/jpet.110.167411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Q, He X, Ma L, Chen S, Fu PP (2018) Pyrrolizidine alkaloid secondary pyrrolic metabolites construct multiple activation pathways leading to DNA adduct formation and potential liver tumor initiation. Chem Res Toxicol 31(7):619–628. doi: 10.1021/acs.chemrestox.8b00096 [DOI] [PubMed] [Google Scholar]
- Xue X, Gong L, Qi X et al. (2011) Knockout of hepatic P450 reductase aggravates triptolide-induced toxicity. Toxicol Lett 205(1):47–54. doi: 10.1016/j.toxlet.2011.05.003 [DOI] [PubMed] [Google Scholar]
- Yan CC, Huxtable RJ (1995) Effect of the pyrrolizidine alkaloid, monocrotaline, on bile composition of the isolated, perfused rat liver. Life Sci 57(6):617–26. doi: 10.1016/0024-3205(95)00312-t [DOI] [PubMed] [Google Scholar]
- Yang M, Ma J, Ruan J, Ye Y, Fu PP, Lin G (2019) Intestinal and hepatic biotransformation of pyrrolizidine alkaloid N-oxides to toxic pyrrolizidine alkaloids. Arch Toxicol 93(8):2197–2209. doi: 10.1007/s00204-019-02499-2 [DOI] [PubMed] [Google Scholar]
- Yang M, Ma J, Ruan J et al. (2020) Absorption difference between hepatotoxic pyrrolizidine alkaloids and their N-oxides - Mechanism and its potential toxic impact. J Ethnopharmacol 249:112421. doi: 10.1016/j.jep.2019.112421 [DOI] [PubMed] [Google Scholar]
- Yang M, Ruan J, Gao H et al. (2017) First evidence of pyrrolizidine alkaloid N-oxide-induced hepatic sinusoidal obstruction syndrome in humans. Arch Toxicol 91(12):3913–3925. doi: 10.1007/s00204-017-2013-y [DOI] [PubMed] [Google Scholar]
- Yuan JXJ, Rubin LJ (2005) Pathogenesis of pulmonary arterial hypertension - The need for multiple hits. Circulation 111(5):534–538. doi: 10.1161/01.Cir.0000156326.48823.55 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.