

ABSTRACT
Salmonella enterica serovar Typhimurium invades the intestinal epithelium and induces inflammatory diarrhea using the Salmonella pathogenicity island 1 (SPI1) type III secretion system (T3SS). Expression of the SPI1 T3SS is controlled by three AraC-like regulators, HilD, HilC, and RtsA, which form a feed-forward regulatory loop that leads to activation of hilA, encoding the main transcriptional regulator of the T3SS structural genes. This complex system is affected by numerous regulatory proteins and environmental signals, many of which act at the level of hilD mRNA translation or HilD protein function. Here, we show that the sRNA MicC blocks translation of the hilD mRNA by base pairing near the ribosome binding site. MicC does not induce degradation of the hilD message. Our data indicate that micC is transcriptionally activated by SlyA, and SlyA feeds into the SPI1 regulatory network solely through MicC. Transcription of micC is negatively regulated by the OmpR/EnvZ two-component system, but this regulation is dependent on SlyA. OmpR/EnvZ control SPI1 expression partially through MicC but also affect expression through other pathways, including an EnvZ-dependent, OmpR-independent mechanism. MicC-mediated regulation plays a role during infection, as evidenced by an SPI1 T3SS-dependent increase in Salmonella fitness in the intestine in the micC deletion mutant. These results further elucidate the complex regulatory network controlling SPI1 expression and add to the list of sRNAs that control this primary virulence factor.
IMPORTANCE The Salmonella pathogenicity island 1 (SPI1) type III secretion system (T3SS) is the primary virulence factor required for causing intestinal disease and initiating systemic infection. The system is regulated in response to a large variety of environmental and physiological factors such that the T3SS is expressed at only the appropriate time and place in the host during infection. Here, we show how the sRNA MicC affects expression of the system. This work adds to our detailed mechanistic studies aimed at a complete understanding of the regulatory circuit.
KEYWORDS: EnvZ, MicC, OmpR, SPI1, Salmonella, SlyA
INTRODUCTION
Salmonella enterica serovar Typhimurium causes inflammatory diarrhea and potentially life-threatening systemic infection, with an estimated global burden of ∼95 million cases per year worldwide (1). Upon oral ingestion, Salmonella transits through the stomach to reach the distal ileum of the small intestine, the initial site of colonization (2, 3). Salmonella invades the intestinal epithelium of the host and induces inflammatory diarrhea using the Salmonella pathogenicity island 1 (SPI1) type III secretion system (T3SS), a needle-like structure that injects effector proteins into the host cell cytosol (4).
The SPI1 T3SS is controlled by three AraC-like transcriptional regulators, HilD, HilC, and RtsA, which constitute a complex feed-forward regulatory loop, each activating transcription of the hilD, hilC, and rtsA genes, as well as activating hilA, encoding the transcriptional regulator of T3SS structural genes (Fig. 1) (5). This system is controlled in response to numerous regulatory proteins and environmental signals, many of which act at the level of hilD mRNA translation, hilD mRNA stability, or HilD protein activity (6, 7). This includes regulation by a number of small RNAs (8).
FIG 1.

Simplified model of the SPI1 T3SS regulatory circuit. Arrows indicate positive regulation, blunt ends indicate negative regulation, blue lines indicate transcriptional regulation, and green lines indicate posttranscriptional regulation.
Small RNAs (sRNAs), generally 50 to 450 bp long, are increasingly recognized as critical regulators of gene expression (9). Over 300 sRNAs are expressed in Salmonella. They play important roles in the regulation of envelope stress responses, metabolism, and virulence. However, the function of most of these sRNAs is unknown or only partially characterized. Many sRNAs control gene expression by imperfect base pairing with an mRNA near the ribosome binding site (RBS), mediated by the RNA chaperone Hfq (9, 10). This blocks access to the 30S ribosomal subunit, inhibiting translation initiation. In some cases, this also leads to RNase E-mediated degradation of the message (11).
The OmpR/EnvZ two-component regulatory system was originally characterized as the regulator of the OmpF and OmpC porins in response to changes in osmolarity (12). OmpR/EnvZ is now understood to be a global regulator of virulence in Salmonella, activating both the SPI2 and SPI1 type III secretion systems, despite the fact that these systems are primarily required in different niches (13–15). The transmembrane histidine kinase EnvZ autophosphorylates and transfers a phosphoryl group to the response regulator OmpR. At low concentrations of OmpR-P, ompF is activated, while at high concentrations of OmpR-P, ompF is repressed and ompC is activated (16). The sRNA MicF is transcribed upstream and antisense to ompC by OmpR. MicF, one of the first sRNAs identified (17, 18), base pairs with the ompF mRNA to block translation and facilitate the transition from producing OmpF to OmpC porin in high osmolarity. More recently, the MicC sRNA was identified and characterized as a regulator of the outer membrane porin OmpC in Escherichia coli that acts by binding to the ompC mRNA near the RBS to prevent 30S ribosome loading (19). In Salmonella, MicC downregulates both OmpC and OmpD, binding in the ompD coding sequence to initiate RNase E-dependent mRNA degradation (20). Chen et al. (19) reported that micC transcription is negatively regulated by OmpR/EnvZ in E. coli. Transcriptomic data suggested that micC is regulated by OmpR/EnvZ, RpoS, and SlyA in Salmonella (21), but regulation of micC has not been characterized in detail. SlyA is a member of MarR/SlyA family of bacterial transcriptional regulators. In Salmonella, slyA mutants are significantly attenuated in the mouse model of infection (22). SlyA acts both positively and negatively to control expression of some 30 genes (23–26). SlyA controls some genes independently but often functions in concert with other transcriptional regulators, including PhoP and OmpR (23, 27, 28).
In this study, we define a new regulatory role for MicC sRNA, controlling the SPI1 T3SS in Salmonella. We found that MicC base pairs with the leader sequence of hilD mRNA to negatively regulate translation of hilD. MicC-mediated SPI1 regulation is dependent on environmental signals and regulated through both SlyA and the OmpR/EnvZ two-component system, which acts through or in conjunction with SlyA. We also show that MicC-dependent regulation of SPI1 is important during intestinal infection.
RESULTS
The small RNA MicC downregulates the expression of HilD.
Several regulatory proteins and signals affecting expression of the SPI1 T3SS act at the level of hilD mRNA translation (7). In the few cases that have been characterized, this regulation is mediated either by the RNA binding protein CsrA (29) or sRNAs (30). We previously screened a set of highly conserved sRNAs for those that decrease hilD translation when overproduced and subsequently characterized regulation of hilD translation by FnrS and ArcZ (8). This screen also identified the 109-nucleotide (nt) sRNA MicC, first characterized as a regulator of ompC, encoding the OmpC porin protein in E. coli (19). MicC is encoded in the intergenic region downstream of the pyruvate-flavodoxin oxidoreductase gene (nifJ) in both E. coli and Salmonella and is conserved in the Enterobacteriaceae (Fig. 2A). In Salmonella, MicC downregulates translation of the ompC and ompD mRNAs by base pairing using the highly conserved 5′ 20 to 30 nucleotides (20).
FIG 2.

The conserved small RNA MicC negatively regulates the SPI1 T3SS by repressing hilD translation in Salmonella. (A) Alignment of the MicC sequences from various Enterobacteriaceae. The asterisks indicate sequence identity. Sequences corresponding to the regions of MicC that base pair to ompC, ompD, and hilD are underlined. (B) β-Gal (β-galactosidase) activity in Salmonella (SM) strains harboring a hilD′-′lacZ translational fusion or a hilA′-lacZ+ transcriptional fusion, or an E. coli (EC) strain harboring a hilD′-′lacZ translational fusion under the control of an arabinose-inducible promoter. Each background contains the pBR-plac vector or pMicC plasmid. β-Gal activity units are defined as (μmol of ONP formed min−1) × 106/(OD600 × ml of cell suspension). Results are shown as the median with interquartile range, and asterisks indicate significant differences between the data sets (n ≥ 4, P < 0.05, using a Mann-Whitney test). The strains used were JS749, JS892, and JMS6500, with plasmids pBR-plac vector or pMicC.
To understand the regulation of the SPI1 T3SS system of Salmonella by MicC, we overexpressed MicC from the pBR-plac plasmid (31) in Salmonella strains harboring either an in-locus hilD′-′lacZ translational fusion or a hilA′-lacZ+ transcriptional fusion. Note that the hilD fusion strain is a hilD null. Thus, this fusion represents the basal level of transcription and is not autoregulated. The Salmonella cultures were inoculated in no-salt LB (NSLB) overnight and subcultured in high-salt LB (HSLB) for 3 h to induce SPI1. The expression of hilD was downregulated ∼3-fold in the pMicC strain (Fig. 2B). Expression of hilA was decreased 10-fold by MicC (Fig. 2B). These data suggest that MicC negatively regulates HilD expression, leading to a more dramatic effect on hilA transcription, consistent with the feed-forward loop model (Fig. 1). To ensure that this is a direct effect on hilD, we introduced the MicC plasmid into an E. coli strain containing a PBAD-hilD′-′lacZ translational fusion with an arabinose-inducible promoter. The fusion consists of the 35-nt 5′ untranslated region (UTR) and the first 11 codons of hilD fused in-frame to lacZ. Overexpression of MicC in E. coli downregulated the expression of hilD more than 2-fold (Fig. 2B). These results suggest that MicC acts directly on the hilD mRNA to inhibit translation.
MicC targets the 5′ UTR of hilD mRNA by direct base pairing.
Bioinformatic analysis using IntaRNA (http://rna.informatik.uni-freiburg.de/IntaRNA/Input.jsp) suggested that MicC has a binding site in the hilD mRNA immediately upstream of the ribosome binding site (RBS) (Fig. 3A). Based on this prediction, we mutated nucleotides 1 to 5 and 11 to 14 as indicated in the pMicC plasmid. We measured β-galactosidase activity in the Salmonella hilD′-′lacZ translational fusion strain expressing the MicC mutant (pMicC-mt). The pMicC-mt plasmid conferred no significant repression of hilD (Fig. 3B). We then introduced the pMicC-mt plasmid into the E. coli PBAD-hilD′-′lacZ fusion strain. Consistent with the result in Salmonella, overexpression of MicC-mt did not regulate hilD expression (Fig. 3C). Based on the predicted interaction, we introduced compensatory mutations in nucleotides –18 to –26 of hilD in the PBAD-hilD′-′lacZ fusion (Fig. 3A). Overexpression of the wild-type MicC had no significant effect on this fusion, whereas the MicC-mt downregulated the mutant hilD mRNA (Fig. 3C). These data support the proposed base-pairing interaction between MicC and the hilD mRNA. It should be noted that several point mutations and double mutations did not affect the interaction (see Fig. S1 in the supplemental material), suggesting that the base pairing is relatively robust. Similar robust base-pairing interactions were also observed between sRNA MicC and ompC mRNA (19) and ompD mRNA (20) targets.
FIG 3.

MicC negatively regulates hilD translation by base pairing near the RBS of the hilD mRNA. (A) Predicted base-pairing interaction between MicC and hilD mRNA. The RBS is underlined; boxes represent the nucleotides changed in the complementary mutations, with changes indicated in bold. (B) β-Gal activity in the Salmonella (SM) strain harboring the wild-type hilD′-′lacZ translational fusion with vector pBR-plac, wild-type pMicC, or mutated pMicC-mt plasmid. (C) β-Gal activity in E. coli (EC) strains harboring either the wild-type or mutated hilD′-′lacZ translational fusion with empty vector, wild-type pMicC, or mutated pMicC-mt plasmid. Results are shown as the median with interquartile range. Asterisks indicate significant differences between the data sets (n ≥ 4, P < 0.05, using a Mann-Whitney test). The strains used were JS892, JMS6500, and JMS6510, with plasmids pBR-plac vector, pMicC, or pMicC-mt.
The data described above show that MicC affects translation of hilD. We also tested the effect of overexpressed MicC on hilC and rtsA using translational LacZ fusions in Salmonella. In hilD+ strains, expression of MicC led to a decrease in expression of both hilC and rtsA (Fig. 4A). However, there was no effect in the hilD null background, consistent with the fact that MicC inhibits hilD translation (Fig. 4B). Reduced levels of HilD protein decreased transcription of hilC and rtsA, consistent with the feed-forward loop model (Fig. 1). MicC also did not directly affect translation of either hilC or rtsA in E. coli (Fig. 4C). MicC downregulates hilA transcription via HilD (Fig. 2B). To confirm that MicC does not affect hilA translation, we overexpressed MicC in an E. coli strain containing a hilA′-′lacZ translational fusion. MicC had no effect on the expression of this fusion (Fig. 4C). All of these results are consistent with MicC solely regulating hilD translation to affect transcription of hilC, rtsA, and hilA.
FIG 4.

MicC does not regulate HilC, RtsA, or HilA. (A and B) Relative β-gal activity in Salmonella hilC or hilA transcriptional fusion strains that are hilD+ (A) or hilD-null (B). (C) Relative β-gal activity in E. coli hilC′-′lacZ, rtsA′-′lacZ, or hilA′-′lacZ translational fusion strains. All strains include either pBR-plac vector or pMicC plasmid. Results are normalized to each strain containing the vector and are shown as the median with interquartile range. Asterisks indicate significant differences between the data sets (n = 4, P < 0.05, using a Mann-Whitney test). The strains used were JS2187, JS2196, JS2551, JS2552, JMS6503, JMS6504, and JMS6505, with plasmids pBR-plac vector or pMicC.
MicC requires Hfq but not RNase E for hilD mRNA regulation.
The RNA chaperone Hfq is a vital facilitator of sRNA-mRNA imperfect base pairing (32). To test if the MicC-hilD mRNA interaction is dependent on Hfq, we measured hilD expression levels in an hfq mutant Salmonella strain after introducing the MicC plasmid. There was no significant regulation mediated by MicC in the hfq background, suggesting that the hilD mRNA-MicC interaction requires the Hfq chaperone protein (Fig. 5A). Indeed, MicC is known to be unstable in an hfq background (20) and, consistent with our result, Hfq is essential for MicC-dependent regulation of OmpC in E. coli (19) and OmpD in Salmonella (20).
FIG 5.
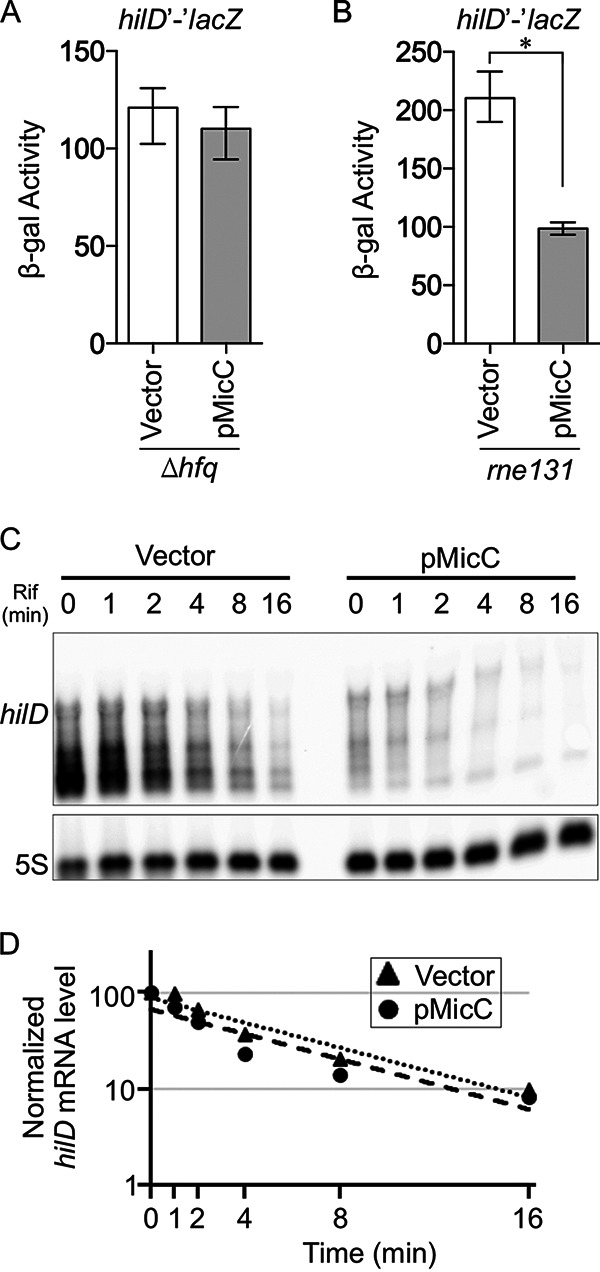
MicC inhibits translation of the hilD mRNA but does not induce degradation. (A and B) β-Gal activity in Salmonella hfq (A) or rne131 (B) strains harboring the wild-type hilD′-′lacZ translational fusion with vector pBR-plac or wild-type pMicC. Results are shown as the median with interquartile range, and asterisks indicate significant differences between the data sets (n = 4, P < 0.05, using a Mann-Whitney test). (C) Northern analysis of hilD mRNA levels. Salmonella strains were grown in SPI1 inducing conditions for 3 h. RNA was isolated at various time points after addition of rifampicin (Rif). The blot was probed for hilD (top panel), and after stripping, probed again for 5S RNA (bottom panel). (D) The intensities of the full-length hilD mRNA bands were quantified and normalized to the 5S bands. The vector or pMicC bands at the zero time point were considered to be 100%, and mRNA decay curves were created based on band intensities. The strains used were 14028, JS2118, and JS2119, with plasmids pBR-plac vector or pMicC.
Negative regulation by sRNAs can be due to simple inhibition of translation initiation or initiation of mRNA degradation by RNase E. We measured the effects of MicC overproduction on the Salmonella hilD′-′lacZ fusion in an rne131 background strain, which has a defect in RNA degradosome assembly (8, 33–35). Although absolute expression of the hilD fusion was increased in the rne131 background, MicC was still able to regulate (Fig. 5B). We then measured the hilD mRNA half-life by Northern analysis (Fig. 5C and D). The levels of hilD mRNA were decreased by overexpression of MicC, as expected, given that HilD is autoregulated. However, the mRNA half-life was unchanged in the presence of the MicC plasmid, consistent with the rne131 result. Therefore, we conclude that MicC base pairing to the hilD mRNA blocks translation initiation but does not induce mRNA degradation.
MicC expression is activated by SlyA and repressed by OmpR/EnvZ.
Studies of E. coli (19) and transcriptomic analysis of Salmonella (21) suggested that MicC expression is repressed by OmpR/EnvZ and activated by SlyA. To characterize this regulation in more detail, we examined expression of a micC′-lacZ+ fusion. Deletion of either envZ or ompR in the fusion strain resulted in increased transcription of MicC in the exponential growth phase (Fig. 6A), confirming that the OmpR/EnvZ two-component system negatively regulates MicC. Deletion of slyA, in contrast, caused a 3-fold decrease in expression, showing that SlyA is a positive regulator of micC expression. Importantly, in the absence of SlyA, deletion of ompR or envZ had no effect, suggesting that OmpR/EnvZ function through, or are at least dependent on, SlyA for their control of micC transcription (Fig. 6A).
FIG 6.
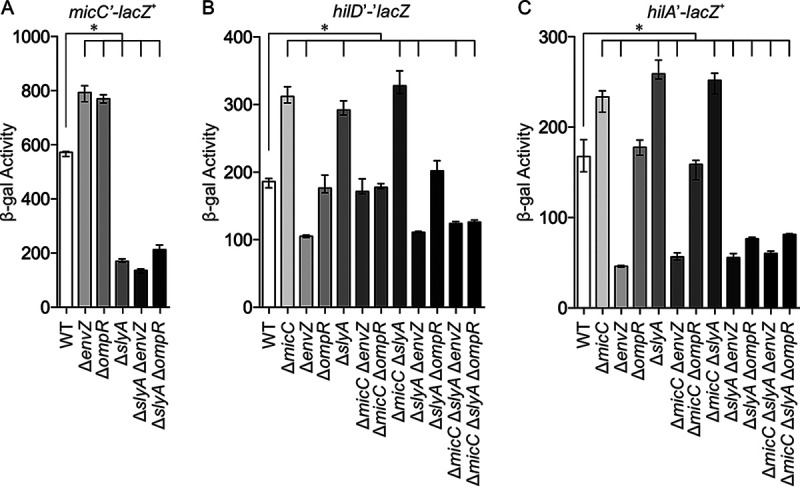
SlyA and EnvZ/OmpR regulate micC and the SPI1 T3SS. β-Gal activity in Salmonella strains with a micC′-lacZ+ transcriptional fusion (A), hilD′-′lacZ translational fusion (B), or hilA′-lacZ+ transcriptional fusion (C) in backgrounds containing the indicated mutations. The results are shown as the median with interquartile range, and asterisks indicate significant differences between the data sets (n ≥ 4, P < 0.05, using a Kruskal-Wallis test followed by post hoc Dunn’s multiple comparisons). The strains used were JS749, JS892, JS2523, JS2524, JS2525, JS2526, JS2527, JS2528, JS2529, JS2530, JS2531, JS2532, JS2533, JS2534, JS2535, JS2536, JS2537, JS2538, JS2539, JS2540, JS2541, JS2542, JS2543, JS2544, JS2545, JS2546, JS2547, JS2548, JS2549, and JS2550.
To determine how this regulation affects SPI1 gene expression, we examined both a hilD′-′lacZ translational (Fig. 6B) and hilA′-lacZ+ transcriptional fusion (Fig. 6C). Deletion of micC caused increased expression of both the hilD and hilA fusions, showing that MicC is affecting hilD translation under these conditions. As expected, deletion of slyA also led to an increase in expression of the hilD and hilA fusions, and deletion of micC in the slyA background had no further effect. These results show that SlyA is affecting SPI1 expression through MicC-mediated control of hilD translation.
Regulation by OmpR/EnvZ is more complicated. Deletion of envZ led to decreased expression of the hilD′-′lacZ fusion, but this decreased expression was also seen in the envZ slyA micC background (Fig. 6B). It is interesting to note that deletion of envZ has a greater effect than mutations in ompR, as noted previously (7, 13). To test this further, we reconstructed the nonpolar in-frame ompR101 allele (36). Expression of the hilA′-lacZ+ transcriptional fusion was significantly decreased in a ΔompR envZ background but was unaffected in the ompR101 background (Fig. S2). Thus, loss of envZ affects SPI1 expression independent of OmpR and MicC.
Deletion of envZ also caused decreased expression of hilA (Fig. 6C) that is independent of SlyA and MicC. Loss of OmpR (and EnvZ) has no effect under these conditions. Our previous data (7) showed that the primary effect of the envZ mutation in the late stationary phase was to decrease HilD protein activity, leading to decreased expression of hilA. In the exponential-phase data shown here, we also see an apparent effect on hilD transcription in the envZ mutant; we do not understand the mechanism. Thus, OmpR/EnvZ, although controlling micC expression, also affect SPI1 independent of MicC, apparently through several mechanisms, which complicates interpretation of the data.
Transcriptomic data also implicated the sigma factor RpoS in the regulation of micC (21). As shown in Fig. S3A, deletion of rpoS caused increased expression of micC in the early stationary phase but has no effect in the exponential phase. Given that RpoS is acting negatively to control micC, this is likely an indirect effect. Deletion of rpoS also led to increased hilD transcription, particularly in the stationary phase. This regulation was unaffected by loss of MicC (Fig. S3B). Thus, RpoS negatively regulates hilD by an unknown, and likely indirect, mechanism (Fig. S3B), but this regulation is independent of MicC.
MicC is negatively regulated by OmpR/EnvZ and negatively regulates OmpC translation. Therefore, it was proposed to play a role in the differential osmoregulation of the porin proteins (19). As such, one would predict that MicC would be preferentially expressed at low osmolarity. We tested this hypothesis by examining expression of the micC′-lacZ+ fusion in low- and high-salt LB. As shown in Fig. S4, expression of micC is increased in high salt. Moreover, this regulation is largely independent of OmpR/EnvZ. Thus, overall regulation of micC is inconsistent with a simple role in regulation of the porins in response to osmolarity.
Deletion of MicC enhances SPI1-dependent fitness in vivo.
MicC regulates expression of the SPI1 T3SS via direct base pairing with the hilD mRNA. In vitro, this regulation is evident at the mid- to late-exponential phase (Fig. 6). To determine if MicC affects SPI1 regulation in vivo in a manner that affects virulence, we performed competition assays using BALB/c mice. In oral infections, the ΔmicC strain outcompeted the wild-type strain in both the upper small intestine (including the duodenum and jejunum) and lower small intestine (including the ileum) (Fig. 7A), the primary site of Salmonella invasion into epithelial cells (2, 3, 37). There was no significant fitness advantage for bacteria recovered from the spleen after either oral or intraperitoneal (IP) infection (Fig. 7B); systemic infection does not require SPI1 (5, 38). To determine whether the observed effects in the intestine were due to changes in SPI1 expression, we also performed competition assays in strains lacking the SPI1 T3SS. In the Δspi1 background, deletion of micC had no significant effect in the competition assay (Fig. 7C). These data are consistent with MicC having a significant regulatory role on hilD translation during intestinal infection.
FIG 7.
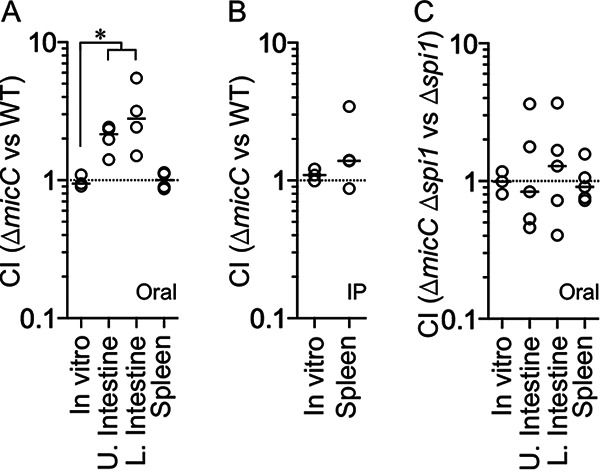
Deletion of MicC provides a fitness advantage in vivo. Competitive index (CI) for in vitro and in vivo infections comparing the following strains: ΔmicC to wild type (WT) after oral infection (A), ΔmicC to WT after intraperitoneal (IP) infection (B), or ΔmicC Δspi1 to Δspi1 after oral infection (C). Upper small intestine (contains duodenum and jejunum), lower small intestine (contains ileum), and spleen were harvested after oral infections, whereas only the spleen was harvested after IP infections. Each circle represents the CI from a single mouse. For in vitro competitions, n = 3; panel A, n = 4; panels B and C, n = 5. The horizontal bars indicate the median of each data set, and the asterisk indicates significant difference (P < 0.05) using a Mann-Whitney test. The strains used were JS135, JS2553, JS2554, and JS2555.
DISCUSSION
The SPI1 T3SS is regulated in response to a plethora of physiological and environmental factors to ensure that this critical virulence factor is expressed at the appropriate time and place in the host and to optimize that expression. In this study, we identified the sRNA MicC as a repressor of hilD translation. MicC was previously identified as a negative regulator of the outer membrane porin proteins OmpC (19) and OmpD (20). MicC acts in the canonical fashion to control hilD translation, base pairing just upstream of the ribosome binding site (Fig. 3A). This binding blocks translation per se rather than initiating mRNA degradation (Fig. 3B). MicC also base pairs just upstream of the ribosome binding site in the ompC mRNA to block translation (19). Interestingly, in the case of ompD, MicC base pairs starting at 67 nucleotides downstream of the AUG and acts by initiating mRNA degradation versus blocking translation (20). MicC does not directly regulate hilC, rtsA, or hilA (Fig. 4), showing that MicC-mediated downregulation of SPI1 T3SS is solely through regulation of hilD mRNA translation. These data reinforce HilD as the primary site of signal integration in the SPI1 regulatory circuit (Fig. 1).
Expression data in E. coli and transcriptome data in Salmonella suggested that micC transcription is controlled by SlyA, OmpR/EnvZ, and RpoS (21). Our data suggest that the primary transcriptional activator of micC is SlyA (Fig. 6A). OmpR/EnvZ negatively regulate micC transcription by affecting SlyA activation. Whether this regulation is all occurring directly at the micC promoter will require further investigation, but SlyA often works in conjunction with other transcriptional regulators in the control of gene expression (23, 24, 27, 28). Comparison of the sequence upstream of micC in various Enterobacteriaceae reveals a strikingly conserved sequence between –31 and –50 from the transcription start site (Fig. 2A). This suggests that this sequence is important for regulation, but it matches neither the SlyA (39–41) nor the OmpR consensus sequence (42). We also show that RpoS negatively regulates micC transcription in the early stationary phase. This is almost certainly an indirect effect, and determining the mechanism will also require further analyses.
The physiological signals that influence SlyA are unclear, although salicylate binds to and inactivates SlyA, and loss of SlyA affects the overall response to reactive oxygen species (25, 43, 44). SlyA is strongly induced when Salmonella is replicating in macrophages, and slyA mutants are not able to survive in macrophages and are, therefore, attenuated for virulence (28). Our data show that SlyA increases the expression of MicC, which helps to repress the SPI1 T3SS. This regulation is apparently evident in the intestine with the micC mutant outcompeting the wild type, consistent with increased expression of the SPI1 T3SS. The SPI1 T3SS is neither expressed nor required during systemic infection and replication in macrophages (5, 45). Our previous data suggest that this strong negative regulation of the system is mediated primarily by PhoPQ (46), but activation of MicC by SlyA may contribute to the downregulation of hilD transcription in the intracellular environment.
MicC, which is negatively regulated by OmpR and blocks ompC translation, was proposed to facilitate regulation of OmpF and OmpC in response to osmolarity (19). OmpR/EnvZ differentially regulate transcription of the porin genes such that ompF is preferentially transcribed in low osmolarity and ompC is preferentially transcribed in high osmolarity (12, 47). The sRNA MicF is cotranscribed with ompC and blocks ompF translation (18). Logic would dictate that MicC should be produced preferentially in low osmolarity to downregulate OmpC expression under these conditions. Interestingly, our results show that micC transcription increases with osmolarity (at least under the conditions we examined; Fig. S4) and that this regulation is independent of OmpR. Thus, the simple model does not hold. Indeed, OmpR is now known to be a global transcriptional regulator, and most genes in the OmpR regulon are not osmoregulated (48) but, rather, are activated at some threshold level of OmpR-P. Only if that threshold level is high, as in the case of ompC, is the gene preferentially activated at high osmolarity. Transcriptional regulation of ompF is more complex and apparently unique, being activated at low levels of OmpR-P but then actively repressed by OmpR-P at higher levels (47–49). Negative regulation of micC by OmpR/EnvZ is via SlyA and the overall osmoregulation of micC is independent of the two-component system.
OmpR/EnvZ regulation of SPI1 is more complicated. Deletion of EnvZ leads to a significant decrease in hilA transcription (Fig. 6), and this effect is surprisingly independent of OmpR (Fig. S2). It has long been known that loss of EnvZ affects SPI1 expression (5, 7, 14). We previously showed that this envZ phenotype is mediated through control of HilD protein activity (7), which is consistent with the data shown here. However, those previous experiments were performed in the late stationary phase. In the exponential-phase experiments shown here, we also see an effect on hilD expression in the hilD null strain. Thus, EnvZ is affecting HilD protein activity (7) and perhaps hilD transcription/translation through unknown mechanisms. To our knowledge, this is the first example of EnvZ acting independently of OmpR.
These results emphasize the role of HilD as the major signal integration point for control of the SPI1 T3SS. Most regulatory input is via posttranscriptional control of HilD, affecting HilD activity via protein-protein interactions, hilD translation, or mRNA stability (6–8, 29, 50), although the mechanisms are understood in only a few cases. We know that hilD translation is affected by binding of the RNA binding protein CsrA in the 39-nt hilD 5′ UTR (29, 51). Translation initiation is also controlled by the sRNAs FnrS, ArcZ (8), and MicC, all of which base pair at the ribosome binding site. All three of these sRNAs affect SPI1 expression during intestinal infection in the animal. More detailed analyses are required to determine the mechanisms and physiological role of additional sRNAs identified as affecting overall control of the T3SS (8).
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The bacterial strains and plasmids used in this study are described in Table S1 in the supplemental material. All Salmonella enterica serovar Typhimurium strains are isogenic derivatives of strain 14028 (American Type Culture Collection [ATCC]) and were constructed using P22 HT105/1 int-201 (P22)-mediated transduction. Deletion of genes or insertion of antibiotic resistance cassettes was performed using λ-red-mediated recombination (52, 53). Insertions and deletions were confirmed by PCR, and mutations were transferred into unmutagenized backgrounds by P22 transduction. In some cases, antibiotic resistance cassettes were removed using the temperature-sensitive plasmid pCP20 carrying the FLP recombinase (54). To create transcriptional lacZ fusions to MicC, the insertion mutation in MicC was converted to a transcriptional lac fusion using FLP-mediated recombination with plasmid pKG136 as previously described (53). The translational lacZ reporter fusions in E. coli were constructed using lambda Red-mediated recombination in strain PM1205, as described previously (8, 31).
Strains were routinely cultured in lysogeny broth (LB; 10% tryptone, 5% yeast extract, 5% NaCl). For SPI1 expression experiments, cells were grown in no-salt LB (NSLB; 10% tryptone, 5% yeast extract, 0% NaCl) or high-salt LB (HSLB; 10% tryptone, 5% yeast extract, 10% NaCl). All strains were grown at 37°C with aeration, except for the strains containing the temperature-sensitive plasmid pCP20 or pKD46, which were grown at 30°C. Antibiotics were used at the following final concentrations: ampicillin (Ap), 100 μg/mL, kanamycin (Km), 50 μg/mL, chloramphenicol (Cm), 20 μg/mL, apramycin (Apr), 50 μg/mL, and tetracycline (Tet), 10 μg/mL.
Construction of plasmids and site-directed mutagenesis.
The MicC small RNA was amplified from the S. Typhimurium 14028 genome using oligonucleotide pairs F-AatII-MicC and R-EcoRI-MicC and cloned into the pBR-plac vector (31) after digestion with AatII and EcoRI restriction enzymes, creating pMicC. The hilD mRNA/MicC sRNA interactions were predicted using the IntaRNA RNA-RNA interaction tool (55). Mutations were introduced into the pMicC plasmid using a QuikChange Lightning site-directed mutagenesis kit (StrataGene). The oligonucleotides used in this study are listed in Table S2.
β-Galactosidase assays.
β-Galactosidase assays were performed using a microtiter plate assay as previously described (49). Briefly, Salmonella strains were inoculated in NSLB medium and grown overnight at 37°C on a roller drum. These cultures were subsequently diluted 1:100 into 2 mL of HSLB medium and grown at 37°C on a roller drum for 3 h or 8 h (where indicated). For E. coli cultures, strains were initially inoculated into LB and grown overnight, then subcultured 1:100 into 2 mL of LB medium with 100 μM isopropyl β-D-1-thiogalactopyranoside (IPTG) and 0.001% arabinose, and grown at 37°C on a roller drum for 3 h. β-Galactosidase activity units are defined as (μmol of orthonitrophenol [ONP] formed min−1) × 106/(optical density at 600 nm [OD600] × mL of cell suspension).
Northern blot analysis.
Overnight NSLB cultures were subcultured 1/100 into 20 mL of HSLB in a flask and incubated for 3 h on a platform shaker at 200 rpm. For the measurement of the half-life of hilD mRNA, 500 μg of rifampicin was added to the bacterial cultures (56). Immediately upon addition of rifampicin (time zero), and at 1, 2, 4, 8, and 16 min after the treatment, 800-μL aliquots of bacterial cells were collected and immediately added to tubes containing 915 μL of phenol (Invitrogen) with 115 μL of lysis buffer (0.3 M NaOAc [pH 5.2], 8% SDS, and 0.02 M EDTA [pH 8.0]). The samples were then incubated at 65°C for 10 min (57). After centrifugation at 13,000 rpm for 10 min, the aqueous fractions were collected and added to 500 μl of a phenol:chloroform:isoamyl alcohol (P:C:I, 25:24:1) solution (pH 6.6; Invitrogen) and mixed. After centrifugation, the aqueous portions were each transferred into 1.3 mL of ice-chilled ethanol and incubated at −80°C for at least 2 h. After centrifugation at 13,000 rpm for 10 min, the supernatants were carefully removed, and the pellets were washed with 1 mL of 70% ethanol. The RNA pellets were allowed to air dry and were suspended in 25 μL of diethyl pyrocarbonate (DEPC)-treated H2O.
A radiolabeled hilD mRNA probe was generated using the MAXIscript T7 transcription kit (Invitrogen) from 1 μg of a PCR product corresponding to a portion of the hilD open reading frame (Table S2). The 5S RNA was detected with an oligonucleotide probe radiolabeled using the KinaseMax 5′ end-labeling kit (Invitrogen). For each sample, 5 μg of total RNA was denatured in 10× MOPS buffer (5%), formamide (50%) and 37% formaldehyde (17.5%) and incubated at 65°C for 15 min before adding 2× loading dye with 1× EtBr. The RNA was separated on a 1% agarose gel with 1× MOPS (morpholinepropanesulfonic acid) buffer for 1.5 h at 90 V. RNA was transferred to a BrightStar-Plus positively charged nylon membrane (Invitrogen) by capillary transfer with NorthernMAX transfer buffer (Invitrogen). The RNA was cross-linked to the membrane with 0.12 J/cm2 UV light. The membrane was hybridized with radiolabeled hilD probe at 60°C in ULTRAhyb hybridization buffer (Invitrogen). After washing, the radiolabel was visualized on a phosphorimager (Fuji FLA-3000) and quantified using the Image Quant image analyzer (ImageGauge V4.22). The membrane was stripped, and the procedure was repeated after hybridization with the 5S RNA probe overnight at 42°C.
In vitro and in vivo competition assays.
All animal work was reviewed and approved by the University of Illinois Institutional Animal Care and Use Committee (IACUC). Procedures were performed in our AAALAC-accredited facility in accordance with university and PHS guidelines under protocol 15214. Competition assays in vivo and in vitro were performed using isogenic wild type and ΔmicC, or ΔSPI1 and ΔmicC ΔSPI1 mutants. Briefly, strains were grown overnight in LB. For oral infections, strains were mixed 1:1, washed, and suspended in 0.1 M phosphate-buffered saline (pH 8) to an adjusted CFU mL−1 of 5 × 108 (for wild type versus ΔmicC) or 109 (for ΔSPI1 versus ΔmicC ΔSPI1). Before infection, food and water were withheld for 4 h, and then mice were inoculated with 200 μL of cell suspension by oral gavage. For intraperitoneal infections, 1:1 cell mixtures were diluted to 103 CFU mL−1 in phosphate-buffered saline (pH 7). Mice were inoculated with 200 μL of cell suspension by intraperitoneal (IP) injection. All inocula were diluted and plated on LB and then replica plated to appropriate antibiotic medium to determine the exact ratios of strains. After 3.5 days of infection, mice were sacrificed by CO2 asphyxiation, and the spleens and small intestines were dissected from orally infected mice, or the spleens were dissected from IP infected mice. Tissues were mechanically homogenized in phosphate-buffered saline (PBS) with 15% glycerol, and appropriate dilutions were plated on LB containing the appropriate antibiotics and subsequently replica plated to determine the ratio of strains recovered. In vitro competitions were performed simultaneously by subculturing 103 CFU of the same inocula used for the in vivo experiments into 2 mL of LB. The cultures were incubated overnight at 37°C with aeration, diluted, and plated on LB. The resulting colonies were replica plated onto LB containing the appropriate antibiotics. The results are presented as a competitive index (CI), calculated as (percentage of strain A recovered/percentage of strain B recovered)/(percentage of strain A inoculated/percentage of strain B inoculated).
ACKNOWLEDGMENTS
This work was funded by NIH grant R01 GM120182 to J.M.S. and C.K.V.
We thank Sabrina Abdulla for help with the Northern analysis.
Footnotes
Supplemental material is available online only.
Contributor Information
James M. Slauch, Email: slauch@illinois.edu.
Laurie E. Comstock, Duchossois Family Institute
REFERENCES
- 1.GBD 2017 Non-Typhoidal Salmonella Invasive Disease Collaborators. 2019. The global burden of non-typhoidal Salmonella invasive disease: a systematic analysis for the Global Burden of Disease Study 2017. Lancet Infect Dis 19:1312–1324. 10.1016/S1473-3099(19)30418-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carter PB, Collins FM. 1974. The route of enteric infection in normal mice. J Exp Med 139:1189–1203. 10.1084/jem.139.5.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Clark MA, Jepson MA, Simmons NL, Hirst BH. 1994. Preferential interaction of Salmonella typhimurium with mouse Peyer’s patch M cells. Res Microbiol 145:543–552. 10.1016/0923-2508(94)90031-0. [DOI] [PubMed] [Google Scholar]
- 4.Galan JE, Lara-Tejero M, Marlovits TC, Wagner S. 2014. Bacterial type III secretion systems: specialized nanomachines for protein delivery into target cells. Annu Rev Microbiol 68:415–438. 10.1146/annurev-micro-092412-155725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ellermeier CD, Ellermeier JR, Slauch JM. 2005. HilD, HilC and RtsA constitute a feed forward loop that controls expression of the SPI1 type three secretion system regulator hilA in Salmonella enterica serovar Typhimurium. Mol Microbiol 57:691–705. 10.1111/j.1365-2958.2005.04737.x. [DOI] [PubMed] [Google Scholar]
- 6.Grenz JR, Cott Chubiz JE, Thaprawat P, Slauch JM. 2018. HilE regulates HilD by blocking DNA binding in Salmonella enterica serovar Typhimurium. J Bacteriol 200:e00750-17. 10.1128/JB.00750-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Golubeva YA, Sadik AY, Ellermeier JR, Slauch JM. 2012. Integrating global regulatory input into the Salmonella pathogenicity island 1 type III secretion system. Genetics 190:79–90. 10.1534/genetics.111.132779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim K, Golubeva YA, Vanderpool CK, Slauch JM. 2019. Oxygen-dependent regulation of SPI1 type three secretion system by small RNAs in Salmonella enterica serovar Typhimurium. Mol Microbiol 111:570–587. 10.1111/mmi.14174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hor J, Matera G, Vogel J, Gottesman S, Storz G. 2020. Trans-acting small RNAs and their effects on gene expression in Escherichia coli and Salmonella enterica. EcoSal Plus 9. 10.1128/ecosalplus.ESP-0030-2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moller T, Franch T, Hojrup P, Keene DR, Bachinger HP, Brennan RG, Valentin-Hansen P. 2002. Hfq: a bacterial Sm-like protein that mediates RNA-RNA interaction. Mol Cell 9:23–30. 10.1016/S1097-2765(01)00436-1. [DOI] [PubMed] [Google Scholar]
- 11.De Lay N, Schu DJ, Gottesman S. 2013. Bacterial small RNA-based negative regulation: Hfq and its accomplices. J Biol Chem 288:7996–8003. 10.1074/jbc.R112.441386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Slauch JM, Silhavy TJ. 1997. The porin regulon: a paradigm for the two-component regulatory systems, p 383–417. In Lin ECC, Lynch AS (ed), Regulation of gene expression in Escherichia coli. R.G. Landes Co., Austin, TX. [Google Scholar]
- 13.Lee AK, Detweiler CS, Falkow S. 2000. OmpR regulates the two-component system SsrA-SsrB in Salmonella pathogenicity island 2. J Bacteriol 182:771–781. 10.1128/JB.182.3.771-781.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lucas RL, Lee CA. 2001. Roles of HilC and HilD in regulation of hilA expression in Salmonella enterica serovar Typhimurium. J Bacteriol 183:2733–2745. 10.1128/JB.183.9.2733-2745.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feng X, Oropeza R, Kenney LJ. 2003. Dual regulation by phospho-OmpR of ssrA/B gene expression in Salmonella pathogenicity island 2. Mol Microbiol 48:1131–1143. 10.1046/j.1365-2958.2003.03502.x. [DOI] [PubMed] [Google Scholar]
- 16.Mattison K, Kenney LJ. 2002. Phosphorylation alters the interaction of the response regulator OmpR with its sensor kinase EnvZ. J Biol Chem 277:11143–11148. 10.1074/jbc.M111128200. [DOI] [PubMed] [Google Scholar]
- 17.Andersen J, Delihas N, Ikenaka K, Green PJ, Pines O, Ilercil O, Inouye M. 1987. The isolation and characterization of RNA coded by the micF gene in Escherichia coli. Nucleic Acids Res 15:2089–2101. 10.1093/nar/15.5.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aiba H, Matsuyama S, Mizuno T, Mizushima S. 1987. Function of MicF as an antisense RNA in osmoregulatory expression of the ompF gene in Escherichia coli. J Bacteriol 169:3007–3012. 10.1128/jb.169.7.3007-3012.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen S, Zhang A, Blyn LB, Storz G. 2004. MicC, a second small-RNA regulator of Omp protein expression in Escherichia coli. J Bacteriol 186:6689–6697. 10.1128/JB.186.20.6689-6697.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pfeiffer V, Papenfort K, Lucchini S, Hinton JC, Vogel J. 2009. Coding sequence targeting by MicC RNA reveals bacterial mRNA silencing downstream of translational initiation. Nat Struct Mol Biol 16:840–846. 10.1038/nsmb.1631. [DOI] [PubMed] [Google Scholar]
- 21.Colgan AM, Kroger C, Diard M, Hardt WD, Puente JL, Sivasankaran SK, Hokamp K, Hinton JC. 2016. The impact of 18 ancestral and horizontally-acquired regulatory proteins upon the transcriptome and sRNA landscape of Salmonella enterica serovar Typhimurium. PLoS Genet 12:e1006258. 10.1371/journal.pgen.1006258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Libby SJ, Goebel W, Ludwig A, Buchmeier N, Bowe F, Fang FC, Guiney DG, Songer JG, Heffron F. 1994. A cytolysin encoded by Salmonella is required for survival within macrophages. Proc Natl Acad Sci USA 91:489–493. 10.1073/pnas.91.2.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Navarre WW, Halsey TA, Walthers D, Frye J, McClelland M, Potter JL, Kenney LJ, Gunn JS, Fang FC, Libby SJ. 2005. Co-regulation of Salmonella enterica genes required for virulence and resistance to antimicrobial peptides by SlyA and PhoP/PhoQ. Mol Microbiol 56:492–508. 10.1111/j.1365-2958.2005.04553.x. [DOI] [PubMed] [Google Scholar]
- 24.Spory A, Bosserhoff A, von Rhein C, Goebel W, Ludwig A. 2002. Differential regulation of multiple proteins of Escherichia coli and Salmonella enterica serovar Typhimurium by the transcriptional regulator SlyA. J Bacteriol 184:3549–3559. 10.1128/JB.184.13.3549-3559.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Will WR, Brzovic P, Le Trong I, Stenkamp RE, Lawrenz MB, Karlinsey JE, Navarre WW, Main-Hester K, Miller VL, Libby SJ, Fang FC. 2019. The evolution of SlyA/RovA transcription factors from repressors to countersilencers in Enterobacteriaceae. mBio 10:e00009-19. 10.1128/mBio.00009-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ellison DW, Miller VL. 2006. Regulation of virulence by members of the MarR/SlyA family. Curr Opin Microbiol 9:153–159. 10.1016/j.mib.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 27.Shi Y, Latifi T, Cromie MJ, Groisman EA. 2004. Transcriptional control of the antimicrobial peptide resistance ugtL gene by the Salmonella PhoP and SlyA regulatory proteins. J Biol Chem 279:38618–38625. 10.1074/jbc.M406149200. [DOI] [PubMed] [Google Scholar]
- 28.Linehan SA, Rytkonen A, Yu XJ, Liu M, Holden DW. 2005. SlyA regulates function of Salmonella pathogenicity island 2 (SPI-2) and expression of SPI-2-associated genes. Infect Immun 73:4354–4362. 10.1128/IAI.73.7.4354-4362.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martinez LC, Yakhnin H, Camacho MI, Georgellis D, Babitzke P, Puente JL, Bustamante VH. 2011. Integration of a complex regulatory cascade involving the SirA/BarA and Csr global regulatory systems that controls expression of the Salmonella SPI-1 and SPI-2 virulence regulons through HilD. Mol Microbiol 80:1637–1656. 10.1111/j.1365-2958.2011.07674.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim K, Palmer AD, Vanderpool CK, Slauch JM. 2019. The small RNA PinT contributes to PhoP-mediated regulation of the Salmonella pathogenicity island 1 type III secretion system in Salmonella enterica serovar Typhimurium. J Bacteriol 201:e00312-19. 10.1128/JB.00312-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mandin P, Gottesman S. 2009. A genetic approach for finding small RNAs regulators of genes of interest identifies RybC as regulating the DpiA/DpiB two-component system. Mol Microbiol 72:551–565. 10.1111/j.1365-2958.2009.06665.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vogel J, Luisi BF. 2011. Hfq and its constellation of RNA. Nat Rev Microbiol 9:578–589. 10.1038/nrmicro2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vanzo NF, Li YS, Py B, Blum E, Higgins CF, Raynal LC, Krisch HM, Carpousis AJ. 1998. Ribonuclease E organizes the protein interactions in the Escherichia coli RNA degradosome. Genes Dev 12:2770–2781. 10.1101/gad.12.17.2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lopez PJ, Marchand I, Joyce SA, Dreyfus M. 1999. The C-terminal half of RNase E, which organizes the Escherichia coli degradosome, participates in mRNA degradation but not rRNA processing in vivo. Mol Microbiol 33:188–199. 10.1046/j.1365-2958.1999.01465.x. [DOI] [PubMed] [Google Scholar]
- 35.Viegas SC, Pfeiffer V, Sittka A, Silva IJ, Vogel J, Arraiano CM. 2007. Characterization of the role of ribonucleases in Salmonella small RNA decay. Nucleic Acids Res 35:7651–7664. 10.1093/nar/gkm916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nara F, Matsuyama S, Mizuno T, Mizushima S. 1986. Molecular analysis of mutant ompR genes exhibiting different phenotypes as to osmoregulation of the ompF and ompC genes of Escherichia coli. Mol Gen Genet 202:194–199. 10.1007/BF00331636. [DOI] [PubMed] [Google Scholar]
- 37.Jones BD, Ghori N, Falkow S. 1994. Salmonella typhimurium initiates murine infection by penetrating and destroying the specialized epithelial M cells of the Peyer’s patches. J Exp Med 180:15–23. 10.1084/jem.180.1.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Murray RA, Lee CA. 2000. Invasion genes are not required for Salmonella enterica serovar Typhimurium to breach the intestinal epithelium: evidence that Salmonella pathogenicity island 1 has alternative functions during infection. Infect Immun 68:5050–5055. 10.1128/IAI.68.9.5050-5055.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ballesteros MF, Torrez Lamberti MF, Farizano JV, Pescaretti MM, Delgado MA. 2019. Regulatory effect of SlyA on rcsB expression in Salmonella enterica serovar Typhimurium. J Bacteriol 201:e00673-18. 10.1128/JB.00673-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Haider F, Lithgow JK, Stapleton MR, Norte VA, Roberts RE, Green J. 2008. DNA recognition by the Salmonella enterica serovar Typhimurium transcription factor SlyA. Int Microbiol 11:245–250. [DOI] [PubMed] [Google Scholar]
- 41.Stapleton MR, Norte VA, Read RC, Green J. 2002. Interaction of the Salmonella typhimurium transcription and virulence factor SlyA with target DNA and identification of members of the SlyA regulon. J Biol Chem 277:17630–17637. 10.1074/jbc.M110178200. [DOI] [PubMed] [Google Scholar]
- 42.Huang KJ, Igo MM. 1996. Identification of the bases in the ompF regulatory region, which interact with the transcription factor OmpR. J Mol Biol 262:615–628. 10.1006/jmbi.1996.0540. [DOI] [PubMed] [Google Scholar]
- 43.Dolan KT, Duguid EM, He C. 2011. Crystal structures of SlyA protein, a master virulence regulator of Salmonella, in free and DNA-bound states. J Biol Chem 286:22178–22185. 10.1074/jbc.M111.245258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cabezas CE, Briones AC, Aguirre C, Pardo-Este C, Castro-Severyn J, Salinas CR, Baquedano MS, Hidalgo AA, Fuentes JA, Morales EH, Meneses CA, Castro-Nallar E, Saavedra CP. 2018. The transcription factor SlyA from Salmonella Typhimurium regulates genes in response to hydrogen peroxide and sodium hypochlorite. Res Microbiol 169:263–278. 10.1016/j.resmic.2018.04.003. [DOI] [PubMed] [Google Scholar]
- 45.Srikumar S, Kroger C, Hebrard M, Colgan A, Owen SV, Sivasankaran SK, Cameron AD, Hokamp K, Hinton JC. 2015. RNA-seq brings new insights to the intra-macrophage transcriptome of Salmonella Typhimurium. PLoS Pathog 11:e1005262. 10.1371/journal.ppat.1005262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Palmer AD, Kim K, Slauch JM. 2019. PhoP-mediated repression of the SPI1 Type 3 secretion system in Salmonella enterica serovar Typhimurium. J Bacteriol 201:e00264-19. 10.1128/JB.00264-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Slauch JM, Silhavy TJ. 1989. Genetic analysis of the switch that controls porin gene expression in Escherichia coli K-12. J Mol Biol 210:281–292. 10.1016/0022-2836(89)90330-6. [DOI] [PubMed] [Google Scholar]
- 48.Kenney LJ, Anand GS. 2020. EnvZ/OmpR two-component signaling: an archetype system that can function noncanonically. EcoSal Plus 9. 10.1128/ecosalplus.ESP-0001-2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Slauch JM, Silhavy TJ. 1991. Cis-acting ompF mutations that result in OmpR-dependent constitutive expression. J Bacteriol 173:4039–4048. 10.1128/jb.173.13.4039-4048.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chubiz JE, Golubeva YA, Lin D, Miller LD, Slauch JM. 2010. FliZ regulates expression of the Salmonella pathogenicity island 1 invasion locus by controlling HilD protein activity in Salmonella enterica serovar typhimurium. J Bacteriol 192:6261–6270. 10.1128/JB.00635-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Altier C, Suyemoto M, Lawhon SD. 2000. Regulation of Salmonella enterica serovar Typhimurium invasion genes by CsrA. Infect Immun 68:6790–6797. 10.1128/IAI.68.12.6790-6797.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA 97:6640–6645. 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ellermeier CD, Janakiraman A, Slauch JM. 2002. Construction of targeted single copy lac fusions using lambda Red and FLP-mediated site-specific recombination in bacteria. Gene 290:153–161. 10.1016/S0378-1119(02)00551-6. [DOI] [PubMed] [Google Scholar]
- 54.Cherepanov PP, Wackernagel W. 1995. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 158:9–14. 10.1016/0378-1119(95)00193-A. [DOI] [PubMed] [Google Scholar]
- 55.Mann M, Wright PR, Backofen R. 2017. IntaRNA 2.0: enhanced and customizable prediction of RNA-RNA interactions. Nucleic Acids Res 45:W435–W439. 10.1093/nar/gkx279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Holmqvist E, Li L, Bischler T, Barquist L, Vogel J. 2018. Global maps of ProQ binding in vivo reveal target recognition via RNA structure and stability control at mRNA 3′ ends. Mol Cell 70:971–982.e6. 10.1016/j.molcel.2018.04.017. [DOI] [PubMed] [Google Scholar]
- 57.Ares M. 2012. Bacterial RNA isolation. Cold Spring Harb Protoc 2012:1024–1027. 10.1101/pdb.prot071068. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 to S4 and Tables S1 and S2. Download JB.00378-21-s0001.pdf, PDF file, 1.0 MB (1MB, pdf)