

Abstract
Two major subclasses of mononuclear non-heme ferrous enzymes use two electron donating organic cofactors (alpha-ketoglutarate or pterin) to activate O2 to form FeIV=O intermediates that further react with their substrates through hydrogen atom abstraction or electrophilic aromatic substitution. New spectroscopic methodologies have been developed enabling the study of the active sites in these enzymes and their oxygen intermediates. Coupled to electronic structure calculations the results of these spectroscopies provide fundamental insight into mechanism; this Prospective summarizes the results of these studies in elucidating the mechanism of dioxygen activation to form the FeIV=O intermediate and the geometric and electronic structure of this intermediate that enables its high reactivity and selectivity in product formation.
Graphical Abstract
Introduction
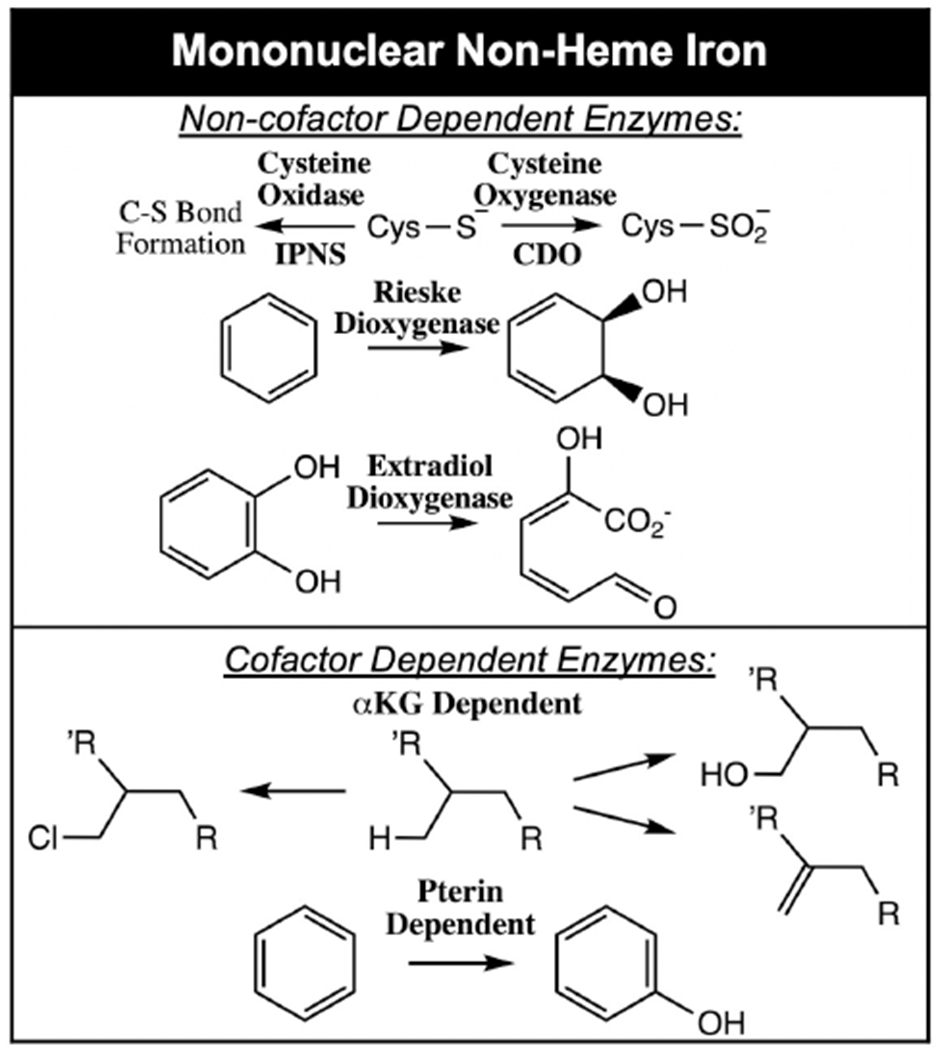
Iron enzymes play a major role in O2 utilization by nature.1–3 Their diverse range of functions include antibiotic, 4,5 neurotransmitter6,7 and natural product biosynthesis, 8 hypoxia regulation, 9 DNA repair, 10 bioremediation 11,12 and drug metabolism 13,14 and are related to a number of genetic diseases.15,16 On a molecular level, they perform H-atom abstraction (HAA) leading to hydroxylation,17,18 halogenation,8 desaturation,19 ring closure20 and expansion21 and electrophilic attack on aromatics (electrophilic aromatic substitution, EAS)22 and on heteroatoms.23,24 O2 activating Fe enzymes have either heme or non-heme active sites, where the latter can be mononuclear or binuclear. Here we focus on the mononuclear non-heme iron (NHFe) enzymes. These are divided into two classes based on whether or not they utilize a 2 e− organic cofactor (either alpha ketoglutarate (αkg) or tetrahydrobiopterin (pterin)).25 In both classes, the majority of the enzymes utilize a facial triad (2 histidines, 1 carboxylate) ligand set,26 however in several enzymes, the carboxylate is replaced by a third histidine27 and in the halogenases, it is replaced by a halide.28 NHFe enzymes have been challenging to study as they lack the intense spectroscopic features of the porphyrin ring characteristic of heme enzymes. While both non-heme and heme Fe enzymes are accessible by Mössbauer spectroscopy, this technique mainly probes redox and spin state. It does not provide detailed geometric and electronic structure insight to complement crystallography of the ferrous (FeII) sites and fully define intermediates, and thus the catalytic mechanism. Our focus has been to develop variable temperature variable field (VTVH) magnetic circular dichroism (MCD) spectroscopy to study the ferrous sites and their interactions with cosubstrates,29 nuclear resonance vibrational spectroscopy (NRVS) to define the geometric structures of intermediates30–32 and MCD on these intermediates to define their electronic structure and frontier molecular orbitals (FMOs) for reactivity.33 These spectroscopic methods are presented in detail in references 29–33 and here we focus on their application to provide chemical insights into non-heme iron enzyme reaction mechanisms.
From Table 1, there are three subclasses of FeII facial triad enzymes that do not use organic cofactors: those having non-redox active, cysteine containing substrates (isopenicillin N synthase (IPNS),34 cysteine dioxygenase (CDO),27 persulfide dioxygenase (ETHE1)),35 those having 1 e− donating Fe2S2 Rieske centers (Rieske dioxygenases (RDO))11 and those having redox active catecholate substrates (the extradiol dioxygenases (EDO)).36 Recent studies have utilized the above-mentioned combination of spectroscopic methods coupled to density functional theory (DFT) calculations to evaluate O2 activation by all three subclasses.37–39 Investigation into the oxygen-activated intermediates in these subclasses yielded two possible intermediates, an FeIII-hydroperoxo and an FeIII-superoxide. While FeIII-hydroperoxo species can be trapped and spectroscopically and structurally defined in the RDO40,41 and EDO, 42 the barriers for their attack on their corresponding substrates are too high to be catalytically relevant.37,38 Alternatively, FeIII-superoxide intermediates in all three subclasses have significantly lower reaction barriers that are consistent with observed catalytic rates.37–39 In the RDO, O2-activation to form an FeIII-O2− is endergonic but the reaction is driven by proton coupled electron transfer from the Rieske center following superoxo attack on the aromatic ring of the substrate (which is bound in the protein pocket oriented above the O2 binding site).38,43 In the two other subclasses, the cysteine containing39 and catecholate37 substrates coordinate the Fe(II) as strong donors, lowering its reduction potential such that FeIII-superoxide formation is exergonic and has been observed both in IPNS44 and in an EDO with a less reactive substrate.42 From spectroscopy supported calculations, the lower energy barriers for substrate attack in all three subclasses derive from the overlap of the highest occupied molecular orbital (HOMO) of the substrate with the frontier molecular orbital (FMO) of the FeIII-superoxo, which is the unoccupied π* orbital of the superoxide (Figure 1). This FMO is highly anisotropic with respect to the FeO2 plane and must be oriented by the ligation on the Fe for effective electrophilic attack on the substrate.37–39
Table 1:
O2 activating mononuclear non-heme Fe enzyme classes.
|
Figure 1:
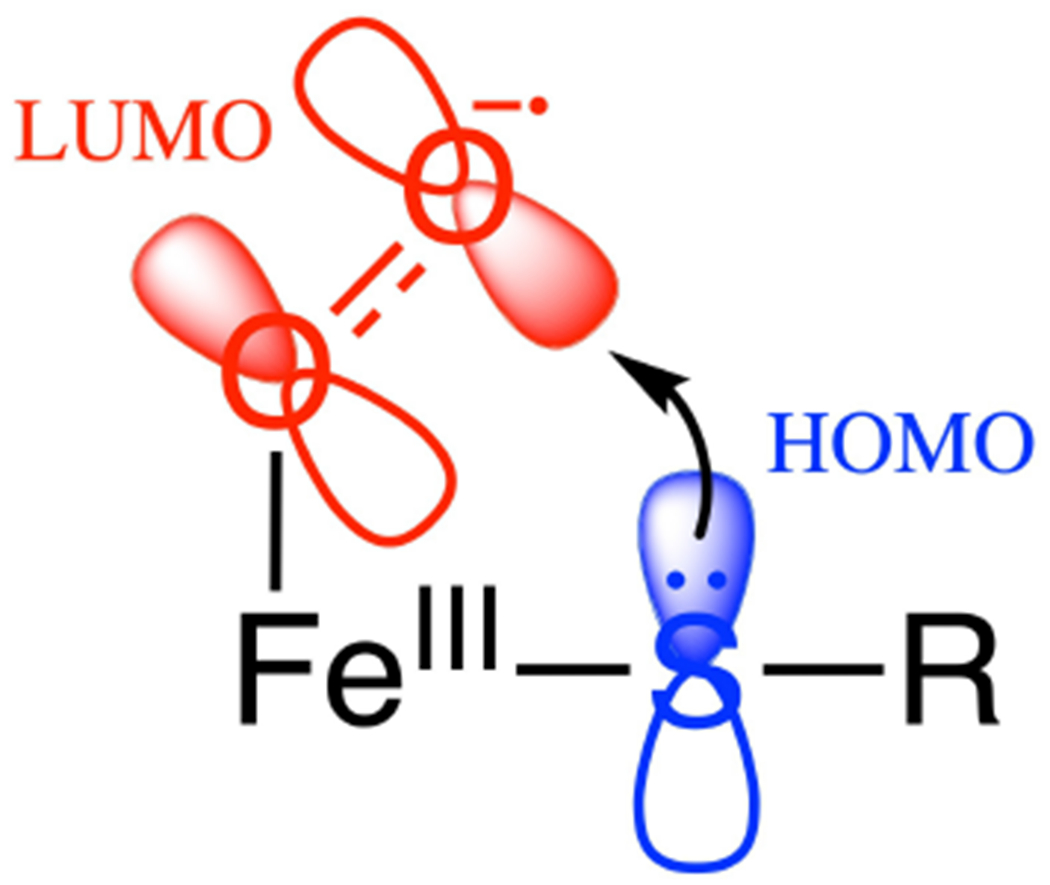
FeIII-superoxo π* FMO (i.e. the LUMO) for electrophilic attack on substrate.
Again, from Table 1, there are two subclasses of NHFe enzymes that use organic cofactors: the αkg and the pterin dependent enzymes. (Note from Scheme 1 that the αkg and pterin are, in fact, cosubstrates as one atom of the O2 is initially incorporated into the oxidized product). These enzymes use an FeII facial triad site plus the 2e− donating organic cofactor/cosubstrate to reduce O2 by four electrons to generate a high spin S = 2 FeIV=O intermediate25,45,46 that goes on to perform a wide range of reactions (Scheme 1).
Scheme 1:
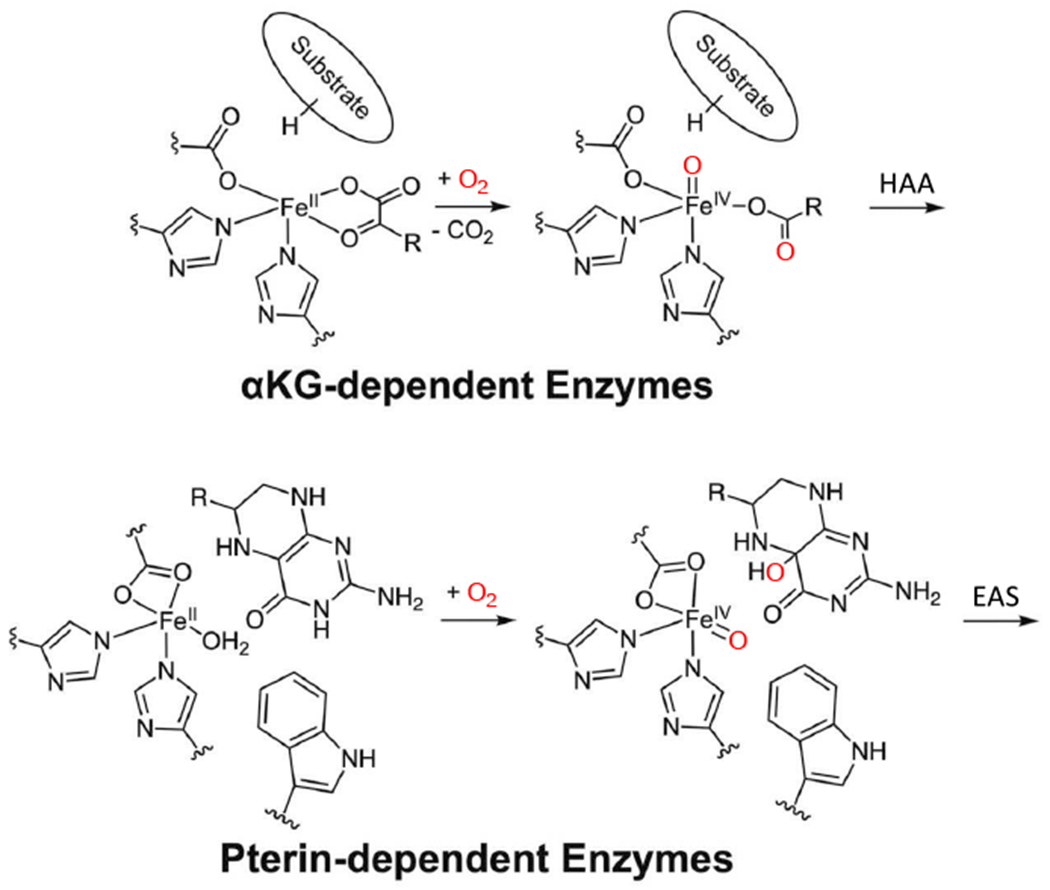
Reaction mechanisms for cofactor (αkg and pterin) dependent non-heme iron enzymes. Adapted from [Iyer, S. R. et al. (2021) Direct coordination of pterin to FeII enables neurotransmitter biosynthesis in the pterin-dependent hydroxylases. Proceedings of the National Academy of Sciences 118.] Copyright [2021].
The αkg dependent FeII enzyme reactions47 involve HAA followed by hydroxylation, halogenation, desaturation or the related ring closure or expansion, important in a variety of functions, including antibiotic biosynthesis,4,5 hypoxia regulation9 and DNA repair.10 In the pterin dependent enzymes, the FeIV=O reaction involves EAS leading to aromatic amino acid hydroxylation such as in neurotransmitter biosynthesis.48 In this Prospective, we focus on the recent advances made in understanding: 1) the mechanism of O2 activation by the cofactor dependent NHFe enzymes to form the FeIV=O intermediate, and 2) the factors that enable the FeIV=O reactivity and selectivity, derived from the application of spectroscopies, kinetics and electronic structure calculations.
1. O2 Activation Mechanism
1.1. Sequential vs. Concerted
MCD spectroscopy has proven to be a powerful method to study ferrous active sites and their interactions with cosubstrates. In particular, MCD enables the study of d-d transitions, i.e., ligand field (LF6) transitions that are too weak to observe in the absorption spectrum of metalloproteins, but directly probe the geometric structure of the high spin ferrous active sites.49 The application of MCD spectroscopy to a range of different NHFeII enzymes has led to the general mechanistic strategy50 illustrated in Scheme 2 and demonstrated by the representative MCD data on the enzyme FIH51 (factor inhibiting HIF (hypoxia inducible factor)) in Figure 2A. The resting enzyme has two LF transitions in the 10,000 cm−1 region split by ~2000 cm−1 indicative of a six coordinate (6C) FeII site (see references 48 and 49 by for spectroscopic details).
Scheme 2:
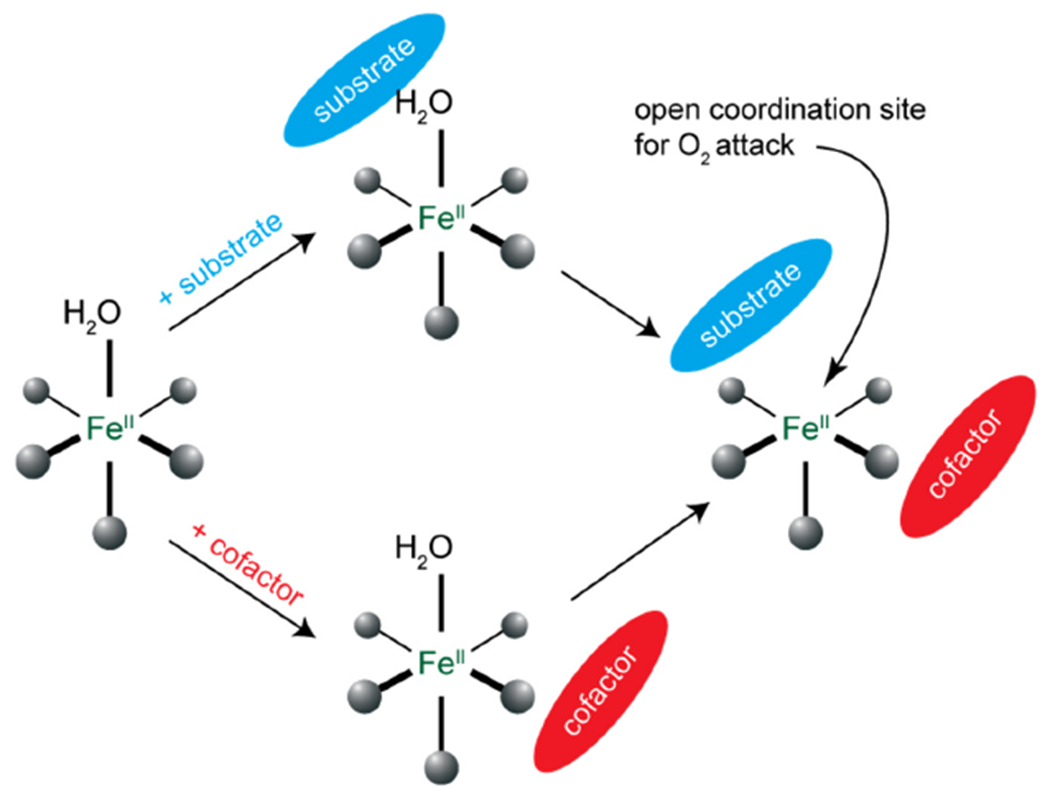
General mechanistic strategy for cofactor dependent NHFe enzymes.
Figure 2:
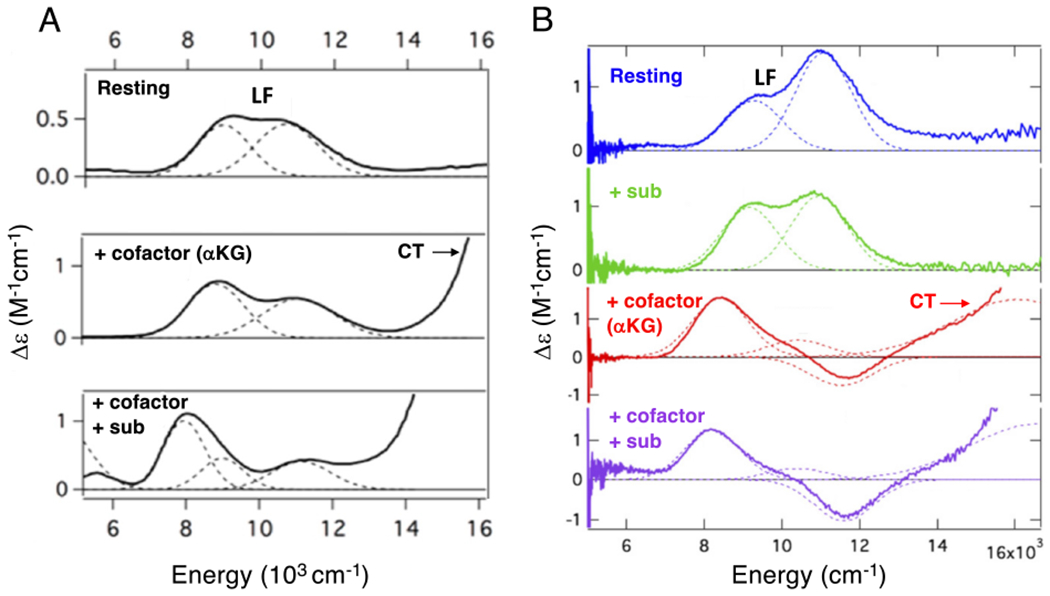
Near IR MCD spectra of FeII sites in FIH (A) and DAOCS (B) for resting enzyme (top), substrate bound (right second down), akg bound (left middle and right third down) and both akg and substrate bound (bottom). Ferrous ligand field (LF) transitions and charge transfer (CT) transitions. Figure adapted from [Iyer, S. R. et al. (2018) O2 activation by non-heme FeII α-ketoglutarate-dependent enzyme variants: Elucidating the role of the facial triad carboxylate in FIH. Journal of the American Chemical Society 140, 11777–11783.] and [Goudarzi, S. et al. (2020) Evaluation of a concerted vs. sequential oxygen activation mechanism in α-ketoglutarate—dependent nonheme ferrous enzymes. Proceedings of the National Academy of Sciences 117, 5152–5159]. Copyright [2018] and [2020] respectively.
Binding substrate to the protein pocket minimally perturbs these transitions (not included in Fig 2A) showing that the substrate does not directly coordinate to the FeII. Alternatively, when αkg is bound new transitions appear at higher energy (Fig 2A middle).52 These are charge transfer (CT) transitions and demonstrate that the αkg is bound to the FeII (bidentate from their analysis52 and from crystal structures53). Importantly, from Fig 2A, the FeII site still has two LF transitions in the 10,000 cm−1 region split by 2,000 cm−1 indicating that the FeII site remains 6C when αkg is bound. Thus, while all four electrons required for O2 reduction to form the FeIV=O intermediate are available (two from the FeII and two from the αkg), the FeII is coordinatively saturated and unreactive with O2. However, when the substrate binds to the protein pocket near the αkg bound FeII center, the LF transitions show large changes (Fig 2A bottom), indicating that the FeII site is now five coordinate (5C) and can bind and react with O2 in the presence of substrate.
However, an alternative sequential mechanism54 has been proposed based on crystallography and calculations, initially on the enzyme Deacetoxycephalosporin-C synthase (DAOCS) and then applied to human DNA oxidative demethylase ABH255 and (S)-2-hydroxypropyl-phosphonic acid epoxidase.56 This proposal involves αkg binding to the FeII active site, reaction with O2 to generate a ferrous-peroxysuccinate intermediate and upon subsequent substrate binding, its conversion to an FeIV=O that reacts to form product. MCD studies on FeII-DAOCS have defined a behavior of the FeII active site that enabled the experimental evaluation of a possible sequential mechanism.57 As shown in Fig 2B, the resting FeII site is 6C and only minimally perturbed by substrate (penicillin-G) binding in the vicinity of the FeII. Importantly, when αkg alone is bound (demonstrated by the CT transitions in the higher energy region in Fig 2B the third spectrum down), in contrast to the behavior of other αkg dependent NHFe enzymes, as in Fig 2A, the LF transitions of DAOCS show a major change (including the negative feature at 11,600 cm−1), indicating that a component (~45%) of the FeII sites has become 5C. Thus this site has become coordinatively unsaturated and since αkg is bound, it can now react with O2 in the absence of substrate. Note from Fig 2B bottom, that substrate addition to the αkg bound DAOCS FeII site further changes the MCD spectrum indicating that more of the FeII site (~60%) become 5C in this ternary complex.
In order to evaluate the concerted versus sequential reaction mechanism in DAOCS, stopped flow absorption and rapid freeze quench (RFQ) Mössbauer and electron paramagnetic resonance (EPR) studies were pursued on the O2 reaction of αkg bound FeII-DAOCS both in absence and presence of substrate. The results of these studies presented in work by Goudarzi et al.57 are summarized in Scheme 3. In the absence of substrate (Scheme 3A), a succinate bound FeIII-OH species rapidly forms. Its subsequent reaction with penicillin G resulted in only the unproductive one electron oxidation of the substrate. Thus, in the absence of prebound substrate, the reactive O2 intermediate has rapidly dissipated, and a sequential mechanism is not operative. Alternatively, when the O2 reaction is performed with both the αkg and substrate simultaneously bound (Scheme 3B), FeIV=O formation is observed that rapidly reacts with the bound substrate to form an FeIII-OH/succinate and substrate radical that concertedly decay with the productive formation of the ring expanded cephalosporin product. Note that the ring expansion rearranges the radical away from the FeIII-OH precluding rebound hydroxylation and resulting in desaturation.
Scheme 3:
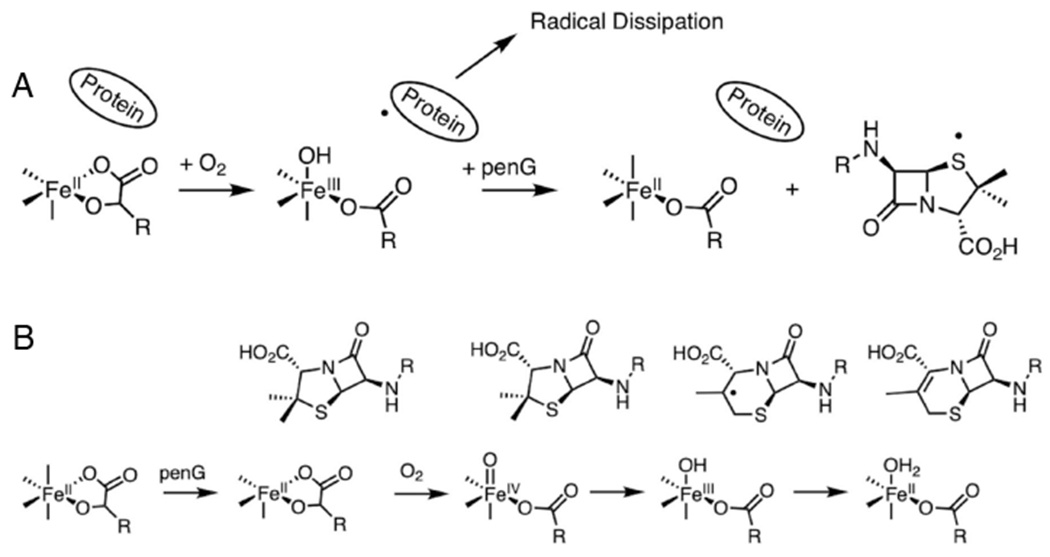
FeII-DAOCS/αkg reaction with O2 in the absence (A) and presence (B) of bound substrate (penicillin G). Adapted from [Goudarzi, S. et al. (2020) Evaluation of a concerted vs. sequential oxygen activation mechanism in α-ketoglutarate—dependent nonheme ferrous enzymes. Proceedings of the National Academy of Sciences 117, 5152–5159.] Copyright [2020].
Thus, the general mechanistic strategy where both cofactor and substrate are bound to generate a 5C FeII site for O2 activation is an effective gating mechanism utilized by NHFe enzymes for coupled catalysis (Scheme 2).
1.2. Cofactor Coordination and Mechanism of FeIV=O Formation
As presented in the Introduction, for both the αkg and pterin dependent NHFe enzymes, the reactions of the substrate and cofactor bound FeII sites have been shown by Mössbauer spectroscopy to form FeIV=O intermediates.25,45,46 For the pterin dependent enzyme tryptophan hydroxylase (TPH), we have recently trapped a precursor intermediate that experimentally defines a common mechanism for both subclasses of cofactor dependent NHFe enzymes in FeIV=O formation.58 From the inset in Figure 3A bottom, addition of substrate to FeII-TPH preloaded with the pterin cofactor results in changes in the LF region of the MCD spectrum, indicating a 6C to 5C conversion, paralleling the results summarized above for most αkg dependent enzymes (Fig 2A).
Figure 3:
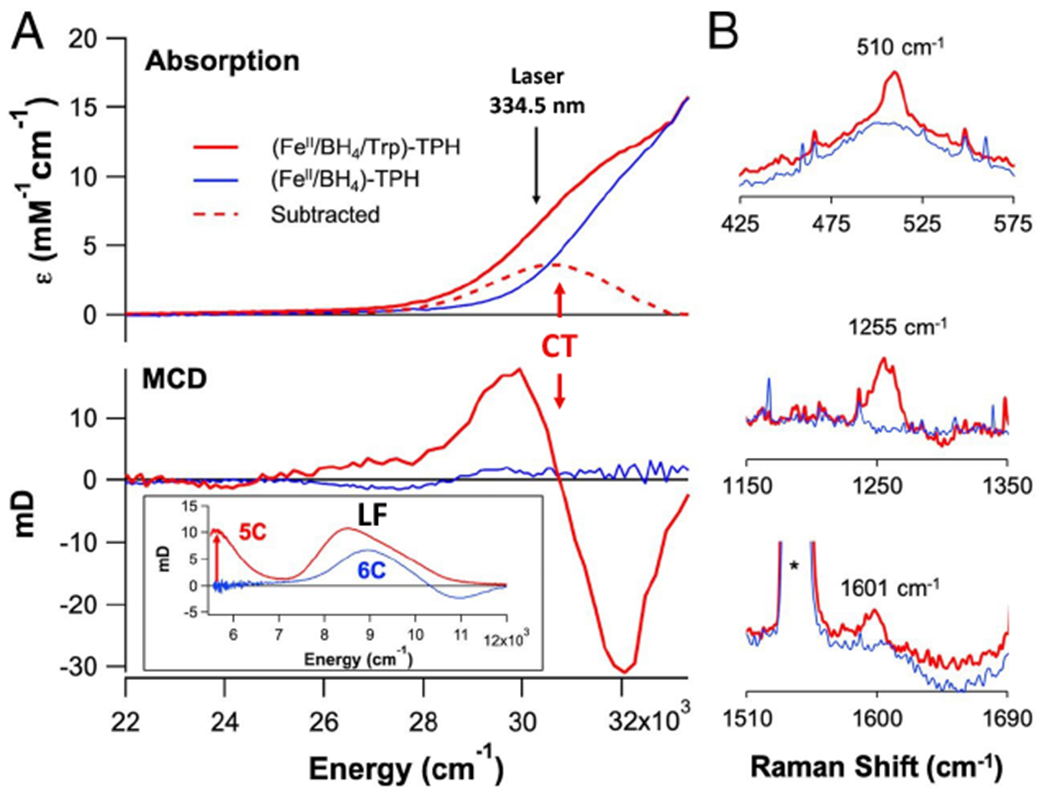
Substrate binding (red) to pterin-bound (blue) FeII tryptophan hydroxylase (TPH). A. Abs (top) and MCD (bottom, near-IR MCD inset). B. Resonance Raman spectra (excitation at 334.5 nm). Adapted from [Iyer, S. R. et al. (2021) Direct coordination of pterin to FeII enables neurotransmitter biosynthesis in the pterin-dependent hydroxylases. Proceedings of the National Academy of Sciences 118.] Copyright [2021].
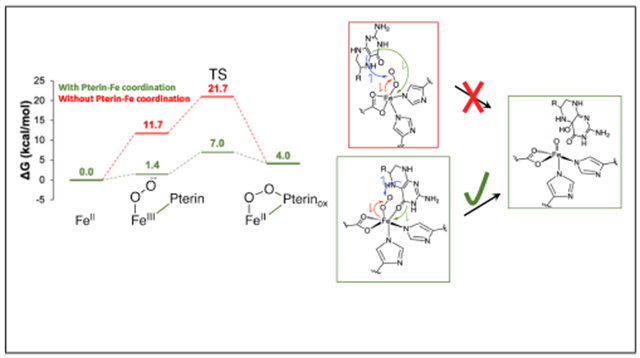
Importantly, when the substrate is also bound (Fig 3A red), a new CT feature appears at 330 nm (30,000 cm−1) in the absorption spectrum that is intense and derivative shaped in MCD, indicating that it is associated with a paramagnetic center. Thus, this is a CT to the FeII, and from the resonance Raman (rR) data in Fig 3B, it is from the pterin with its carbonyl bound to the FeII (the C=O stretch is at 1601 cm−1, decreased from 1695 cm−1 by coordination). This 330 nm CT requires that the pterin is bound to the FeII in this ternary complex. Previously, based on crystallography,59 it had been generally thought that the pterin does not bind to the FeII which, as presented below, would have resulted in too high of a barrier for the O2 activation reaction.
From Figure 4A, reaction of the substrate plus pterin bound 5C FeII site with O2 eliminates the 330 nm CT and initiates the growth of a new CT at 442 nm. This 442 nm intermediate has a Mössbauer spectrum (Fig 4C) indicating that it has a high spin FeII center, while the rR (Fig 4D) and other spectroscopic data indicate that this intermediate is a peroxy-oxidized pterin with both the peroxide and the carbonyl of the pterin bound to the FeII. Specifically, the rR shows the 1570 cm−1 ν(C=O) is again shifted down in energy due to coordination, and the 18O2 isotope perturbation is distributed over the stretches and bonds of the ring formed by this bidetate peroxy-pterin coordination. The 442 nm peroxy intermediate decays with a solvent kinetic isotope effect of about four (Fig 2B), indicating a proton-induced O-O cleavage to produce hydroxybiopterin and the subsequent FeIV=O intermediate. This is the first time a precursor to the FeIV=O intermediate has been spectroscopically defined in either cofactor dependent subclass and elucidates the mechanism of FeIV=O formation.
Figure 4:
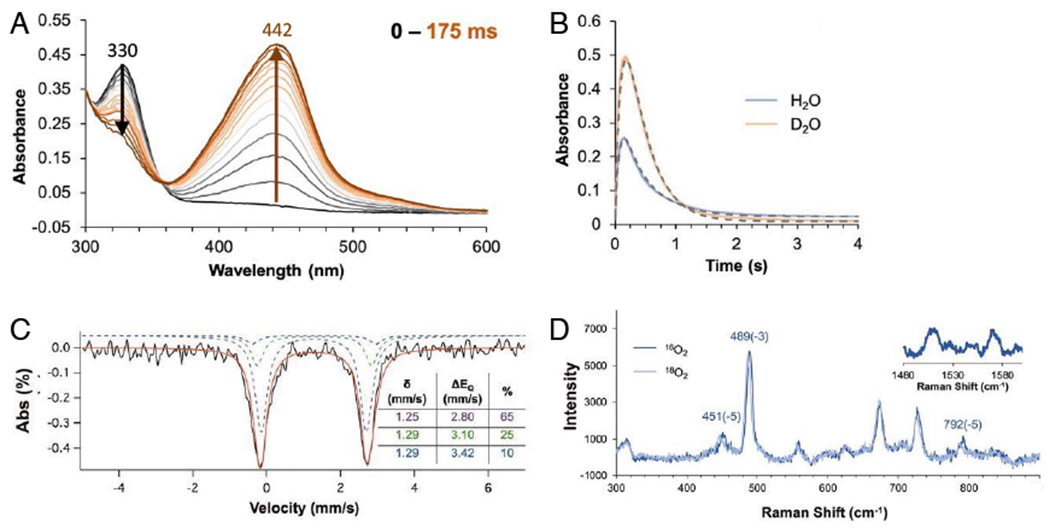
Reaction of substrate and pterin bound FeII-TPH with O2. A. Kinetics of first 175 ms of the reaction. B. Formation and decay kinetics at 442 nm in H2O and D2O. C. Mössbauer spectrum of 442 nm intermediate. D. Resonance Raman spectrum of 442 nm intermediate (excitation at 457.9 nm). Adapted from [Iyer, S. R. et al. (2021) Direct coordination of pterin to FeII enables neurotransmitter biosynthesis in the pterin-dependent hydroxylases. Proceedings of the National Academy of Sciences 118.] Copyright [2021].
As shown in Figure 5A, O2 binding to the open coordination position of the FeII results in some FeIII-superoxo character. Attack of the distal O of the nascent superoxo on the C4α position of the pterin results in the doubly oxidized peroxy pterin bound to an FeII (the 442 nm intermediate). Thus, the pterin needs to donate one electron to the superoxo and a second electron to the Fe to form this peroxy-ferrous intermediate.
Figure 5:
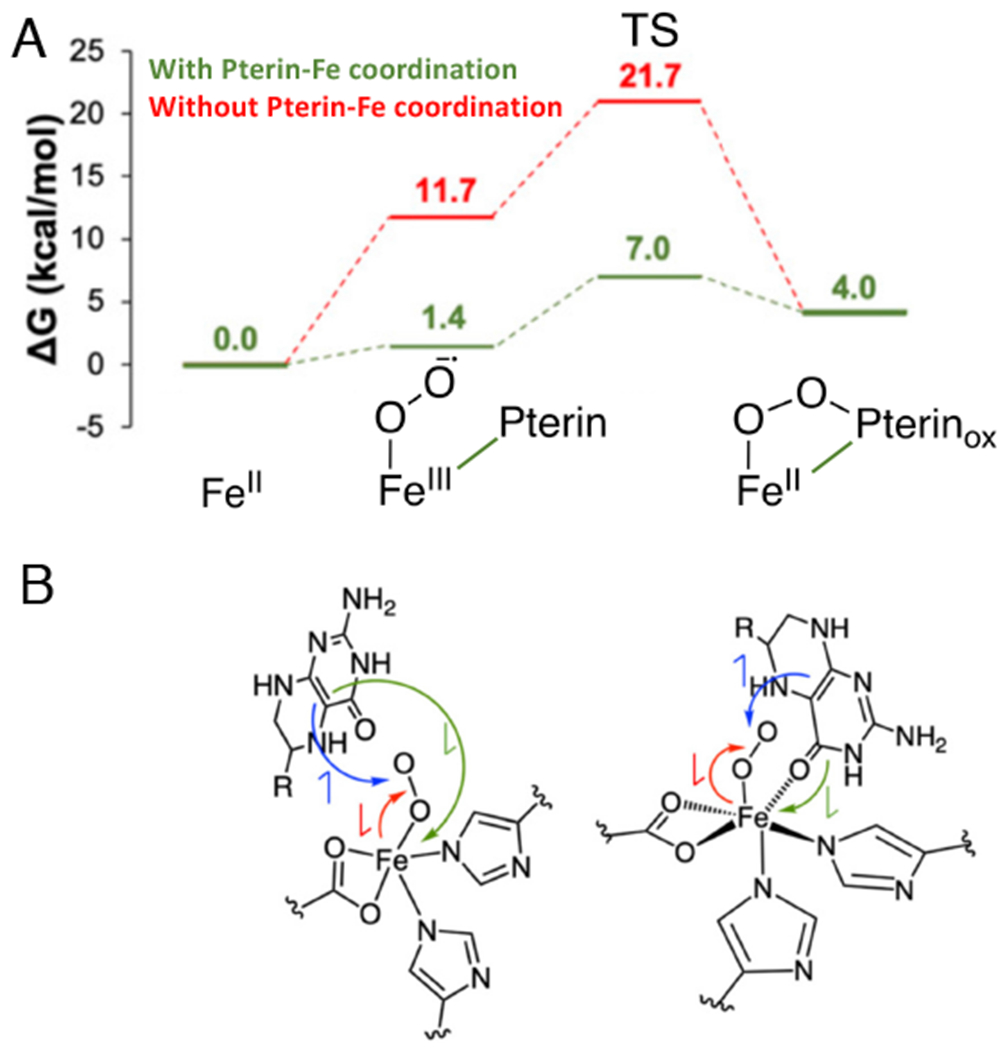
Reaction Coordinate for FeII-pterin TPH. A. Effect of pterin binding to FeII on reaction energetics (red to green). B. Schematics of electronic structure contributions of pterin carbonyl coordination on the energetics in A. Red arrow indicates e-donation from FeII to O2, blue arrow indicates e- donation from pterin to O2; the green arrow indicates donation of a second e- from pterin to Fe (to compensate for the Fe donation to the O2). The green electron donation is facilitated by pterin coordination to the Fe (depicted on the right). Adapted from [Iyer, S. R. et al. (2021) Direct coordination of pterin to FeII enables neurotransmitter biosynthesis in the pterin-dependent hydroxylases. Proceedings of the National Academy of Sciences 118.] Copyright [2021].
As shown in Fig 5B right, pterin coordination to the Fe enables this second electron to transfer directly to the Fe (green arrow) and lowers the barrier for reactivity by 14.7 kcal/mol (Fig 5A green vs. red). Since the rate determining step for the reaction is product release with a barrier of 16 kcal/mol,60,61 if the pterin were not bound to the Fe, the 21 kcal/mol calculated barrier would result in 104 slower turnover, thus enzymatic dysregulation that would impact the metabolic pathways necessary for proper brain function.62 Thus, there is a common mechanism utilized by both the αkg and pterin dependent NHFe enzymes to form the FeIV=O intermediate. Concerted substrate plus cofactor binding to the active site pocket opens a coordination position on the FeII for O2-activation. For both subclasses, the cofactor binds directly to the Fe enabling the electron transfer to the FeO2 required for FeII-peroxy-oxidized cofactor formation.
2. FeIV=O Structure and Reactivity
Proceeding from the FeII-peroxy-oxidized cofactor now observed by spectroscopy in the pterin dependent enzymes58 and predicted by crystallography63 for the αkg dependent enzymes, protonation of the distal O or direct O-O cleavage leads to the hydroxybiopterin or bound succinate, respectively, and the FeIV=O S = 2 intermediate. In the αkg enzymes, the FeIV=O performs HAA followed by rebound hydroxylation,17,18 halogenation8 (in halogenases) or desaturation19 and related reactions. In the pterin dependent enzymes, the FeIV=O performs EAS on aromatic amino acid substrates. M. Bollinger, C. Krebs and colleagues provided high concentration, high quality samples of the FeIV=O S = 2 intermediates in the hydroxylating TauD (degradation of sulfonic acids) and the halogenating SyrB2. These samples enabled geometric structural determination by NRVS 64,65 and, combined with parallel studies on the structurally defined S = 2 FeIV=O model of L. Que and colleagues,66 electronic structure determination by VTVH MCD.33,67
NRVS uses a synchrotron to scan the 57Fe nuclear Mössbauer transition with a focus on the vibrational sidebands at higher energy,68 where the NRVS intensity reflects Fe motion in a normal mode at the observed energy. The NRVS spectrum of the FeIV=O intermediate in TauD is given in Figure 6; that of SyrB2 is very similar.
Figure 6:
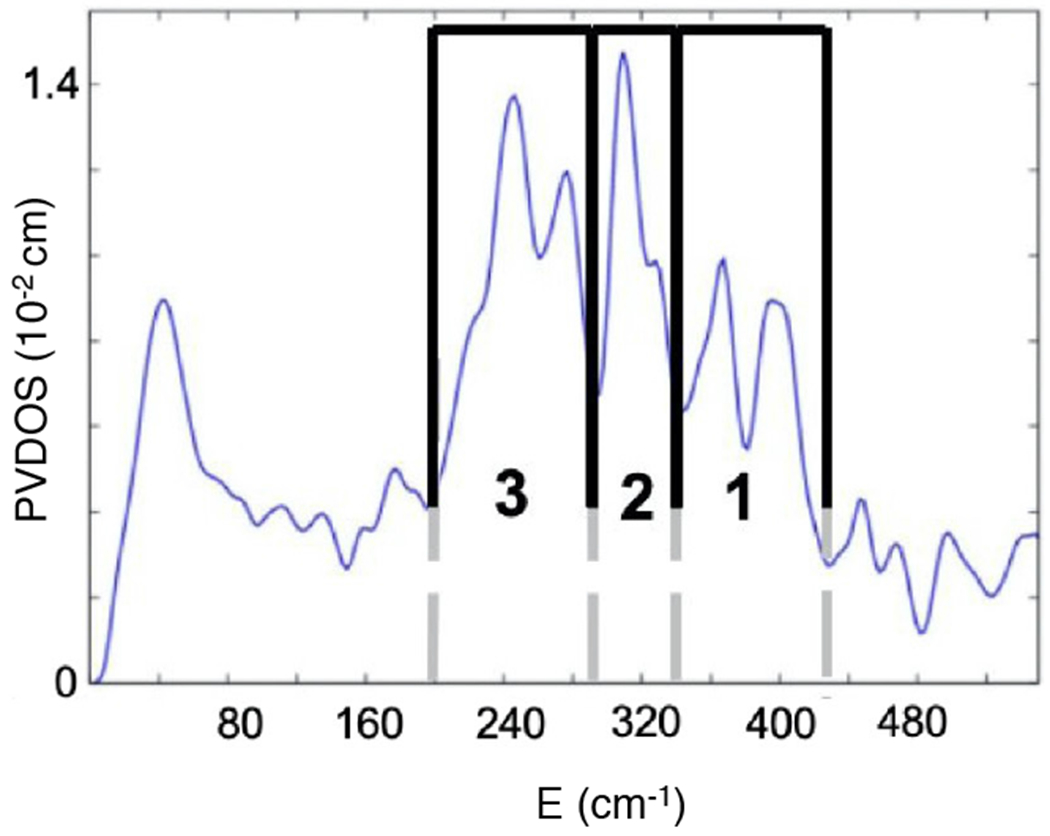
NRVS spectrum of the FeIV=O intermediate of TauD. Adapted from [Srnec, M. et al. (2020) Nuclear Resonance Vibrational Spectroscopic Definition of the Facial Triad FeIV=O Intermediate in Taurine Dioxygenase: Evaluation of Structural Contributions to Hydrogen Atom Abstraction. Journal of the American Chemical Society 142, 18886–18896.] Copyright [2020].
The features in region 3 are the His N-Fe stretches, the intense feature in region 2 is the trans axial bend of the FeIV=O, and the two peaks in region 1 are carboxylate O-Fe stretches. To contribute to this high energy region 1, the two carboxylates (one from the facial triad and one from the succinate formed by decarboxylation) must both be coordinated monodentate, as bidentate carboxylates have longer Fe-O bonds resulting in lower energy stretches shifted into region 2. Thus, both the carboxylate of the facial traid and that of the succinate are monodentate and the FeIV=O is 5C. Simulations of the NRVS data with a series of possible 6C and 5C structures showed that the Fe=O intermediate in TauD (and in SyrB2) is trigonal bipyramidal (TBP) with the oxo along the C3 axis.64 A schematic of this structure is shown in Figure 7A.
Figure 7:
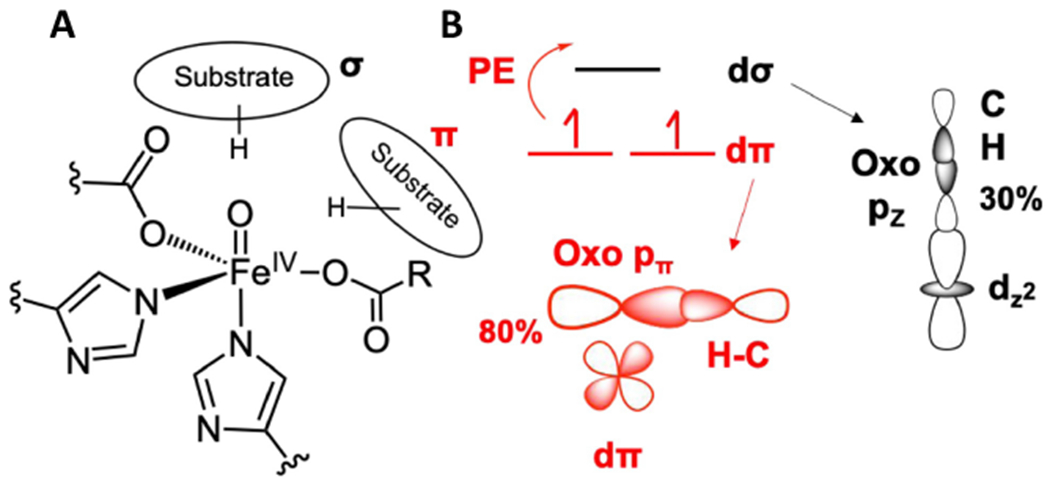
FeIV=O structure and FMOs. A. Schematic of FeIV=O intermediate with possible substrate orientations along (σ) and perpendicular (π) to the Fe-oxo bond. B. σ (black) and π (red) FMOs enabling HAA along (σ) and perpendicular to (π) the Fe-oxo bond. PE indicates promotion energy required to reach the dπ FMO. Adapted from [Srnec, M. et al. (2020) Nuclear Resonance Vibrational Spectroscopic Definition of the Facial Triad FeIV=O Intermediate in Taurine Dioxygenase: Evaluation of Structural Contributions to Hydrogen Atom Abstraction. Journal of the American Chemical Society 142, 18886–18896.] Copyright [2020].
Due to the short Fe-O bond (1.62 Å) of this TBP FeIV=O S = 2 site, the LF results in the dz2/dσ orbital at highest energy and unoccupied (FeIV is d4), thus this is the FMO for the HAA reaction and, based on orbital overlap, this would occur along the Fe-O bond (Fig 7B, black). However, VTVH MCD spectra of the FeIV=O intermediate in SyrB267 and of the TBP FeIV=O Que model33 , Figure 8A (note these spectra are very similar further supporting the TBP enzyme intermediate structure from NRVS), show a low-lying electronic transition near 12,000 cm−1 (red double arrow). This transition is very weak in absorption but intense in MCD and from this intensity ratio, its derivative shape, its temperature dependence, and its vibronic structure in the Fe=O stretch vibration, this is the dπ* to dσ* LF transition. As shown in Fig 7B red, this dπ* to dσ* transition creates a hole in the dπ* orbital, an FMO activated for attack on the substrate perpendicular to the Fe-O bond. From calculations64 correlated to the MCD data, this dπ* FMO has 80% oxo (i.e., oxyl) character and from Fig 8B, rapidly comes down in energy as the Fe-O bond is elongated to reach the TS for HAA (1.82 Å).
Figure 8:
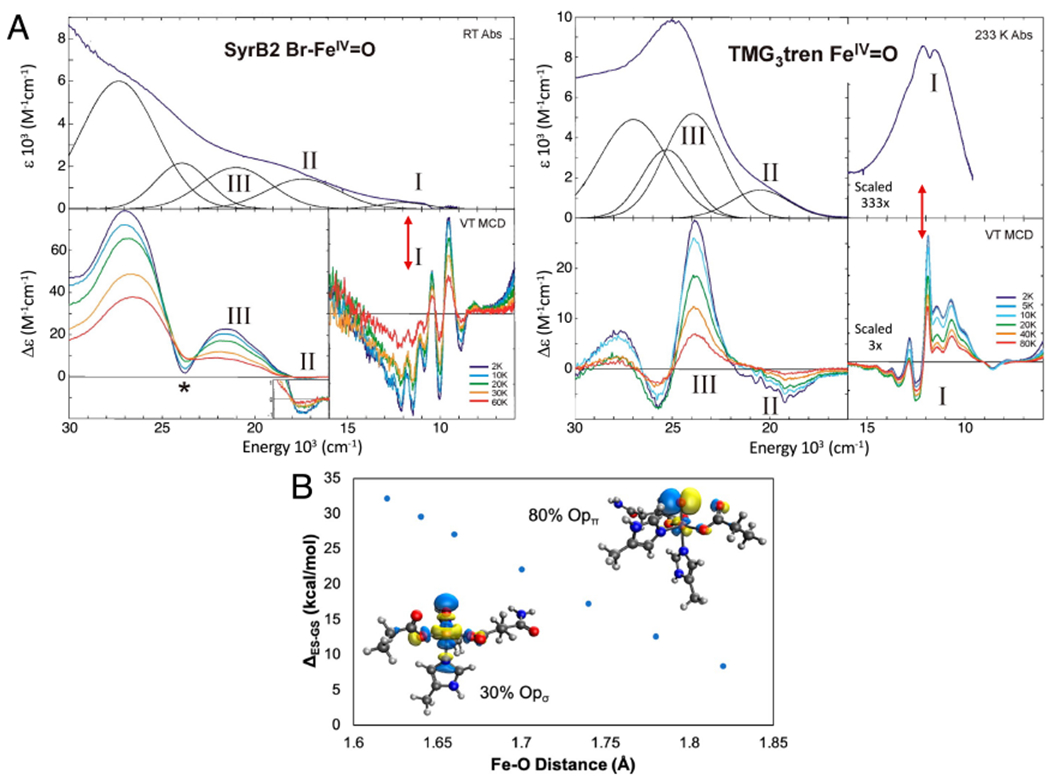
Experimental insight into dπ* FMO. A. Abs (top) and MCD (bottom) definition of FeIV=O FMOs in SyrB2 (left) and its TMG3tren model complex (right). Red double arrows indicate dπ* to dσ* LF excitation at an Fe=O bond length of 1.62 Å. B. Dependence of the dπ* to dσ* excitation energy (the promotion energy (PE) in Fig. 7B) on Fe=O bond length (1.82 Å at the TS). Insets show the σ and π FMOS at an Fe=O distance of 1.62 Å. Adapted from [Srnec, M. et al. (2020) Nuclear Resonance Vibrational Spectroscopic Definition of the Facial Triad FeIV=O Intermediate in Taurine Dioxygenase: Evaluation of Structural Contributions to Hydrogen Atom Abstraction. Journal of the American Chemical Society 142, 18886–18896.] and [Srnec, M. et al. (2016) Electronic Structure of the Ferryl Intermediate in the alpha-Ketoglutarate Dependent Non-Heme Iron Halogenase SyrB2: Contributions to H Atom Abstraction Reactivity. Journal of the American Chemical Society 138, 5110–5122.] Copyright [2020] and [2016] respectively.
Thus, the promotion energy (PE) to reach this dπ* FMO in Fig 7B is only 7 kcal/mol, and, due to its high oxyl character (inset in Fig 8B, right), the TS is earlier in C-H elongation (i.e., better overlap) resulting in less distortion energy (relative to σ attack along the FeIV=O bond). The lower distortion energy compensates for the promotion energy and both the π and σ FMOs in Fig 7B are found to have comparable HAA reactivity.64 However, due to its orientation towards the substrate the dσ* FMO can only enable rebound hydroxylation (assuming the substrate radical does not rearrange as in DAOCS (Scheme 3B) or decay69). Alternatively, the π FMO enables selectivity in rebound, thus far elucidated for the halogenases, as summarized below.
For SyrB2, reaction coordinate calculations65 correlated to the NRVS data lead to an FeIV=O oriented perpendicular to the C-H bond of the substrate, L-threonine (Fig 7A, π substrate orientation). From this orientation, HAA results in an HS FeIII-OH with the substrate radical oriented above the plane containing both the FeIII-OH and the FeIII-Cl bonds (Fig 9A left). From this orientation, while rebound hydroxylation is 12 kcal/mol more favorable (Figure 9A, red), the halogenation and hydroxylation are found to have very similar barriers (Fig 9A, halogenation (green) favored).70 While the thermodynamics of hydroxylation is found to lower the barrier by 4 kcal/mol, the halogenation rebound has a 4.5 kcal/mol lower (thermodynamically corrected) intrinsic barrier.
Figure 9:
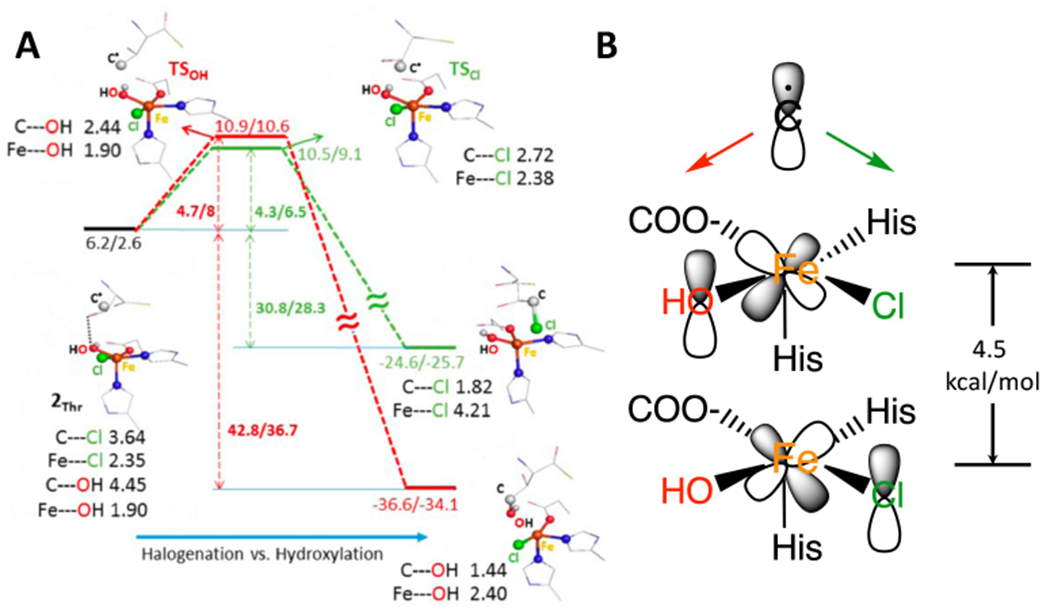
Rebound halogenation vs hydroxylation. A. After HAA by SyrB2 the resultant FeIII-OH has the substrate radical oriented above the HO-FeIII-Cl plane. Energetics of rebound hydroxylation in red and rebound halogenation in green. B. FMOs for rebound, dπ* of HO-FeIII is 4.5 kcal/mol higher than dπ* of Cl-FeIII in energy. Adapted from [Srnec, M. et al. (2017) Frontier Molecular Orbital Contributions to Chlorination versus Hydroxylation Selectivity in the Non-Heme Iron Halogenase SyrB2. Journal of the American Chemical Society 139, 2396–2407.] Copyright [2017].
This reflects the lower energy of the halide dπ* orbital relative to the hydroxo dπ* orbital of the FeIII (Fig 9B right, from their difference in LF), where the rebound mechanism involves two asynchronous transfers: electron transfer from the substrate radical to the appropriate dπ FMO on the Fe followed by late ligand (Cl− or OH−) transfer to the substrate. Thus, the thermodynamic and intrinsic barrier contributions to the rebound barrier cancel and the product of rebound is determined by the relative overlap of the substrate radical with the two dπ* FMOs (Fig 9B), and thus varies with the positioning of the substrate as observed experimentally.71 This emphasizes the important role of the second coordination sphere of the protein in positioning the substrate to control the selectivity.
3. Concluding Comments
These spectroscopic and experimentally calibrated DFT insights into the reactivity of cofactor-dependent non-heme iron enzymes sets the stage for fundamental insight into the reaction mechanisms of these and related enzymes. For the αkg dependent enzymes, we need to evaluate whether the π and σ FMO concepts extend to differences in selectivity in desaturation vs hydroxylation. For the pterin dependent enzymes, an outstanding issue is whether the π FMO enables reaction with the substrate that must be oriented perpendicular to the Fe=O bond formed by O-O cleavage of the FeII-peroxy-pterin intermediate. Further, in comparing the two enzyme subclasses, it is important to determine whether the FeIV=O intermediate in the pterin enzymes is selectively activated for EAS based on its ligation differences relative to the FeIV=O of the αkg dependent enzymes (no bound succinate and bidentate coordination of the facial triad carboxylate). Finally, studies are underway to determine the nature of FeIV-oxo activation in the binuclear NHFe intermediate Q in soluble methane monooxygenase and in Compound I and II in heme enzymes and how these correlate to the mononuclear NHFe enzyme catalysis summarized above.
Acknowledgement
We thank our collaborators listed in the publications cited for their significant contributions, in particular Dr. Shyam Iyer and Prof. Martin Srnec. We would also like to acknowledge the NIH, Grant GM 40392 for support of this research.
References
- (1).Solomon EI, Light KM, Liu LV, Srnec M, and Wong SD (2013) Geometric and Electronic Structure Contributions to Function in Non-heme Iron Enzymes. Accounts of Chemical Research 46, 2725–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Jasniewski AJ, and Que L Jr. (2018) Dioxygen Activation by Nonheme Diiron Enzymes: Diverse Dioxygen Adducts, High-Valent Intermediates, and Related Model Complexes. Chemical Reviews 118, 2554–2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Poulos TL (2014) Heme Enzyme Structure and Function. Chemical Reviews 114, 3919–3962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Baldwin JE, and Abraham E (1988) The Biosynthesis of Penicillins and Cephalosporins. Natural Product Reports 5, 129–145. [DOI] [PubMed] [Google Scholar]
- (5).Busby RW, and Townsend CA (1996) A single monomeric iron center in clavaminate synthase catalyzes three nonsuccessive oxidative transformations. Bioorganic Medicinal Chemistry 4, 1059–1064. [DOI] [PubMed] [Google Scholar]
- (6).Fitzpatrick PF (2000) The aromatic amino acid hydroxylases. Advances in Enzymology 74, 235–294. [DOI] [PubMed] [Google Scholar]
- (7).Spector S, Sjoerdsm A, and Udenfried S (1965) Blockade of endogenous norepinephrine synthesis by alpha-methyl-tyrosine an inhibitor of tyrosine hydroxylase. Journal of Pharmacology and Experimental Therapeutics 147, 86–95. [PubMed] [Google Scholar]
- (8).Vaillancourt FH, Yin J, and Walsh CT (2005) SyrB2 in syringomycin E biosynthesis is a nonheme Fe-II alpha-ketoglutarate- and O-2-dependent halogenase. Proceedings of the National Academy of Sciences of the United States of America 102, 10111–10116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Mahon PC, Hirota K, and Semenza GL (2001) FIH-1: a novel protein that interacts with HIF-1 alpha and VHL to mediate repression of HIF-1 transcriptional activity. Genes Development 15, 2675–2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Falnes PO, Klungland A, and Alseth I (2007) Repair of methyl lesions in DNA and RNA by oxidative demethylation. Neuroscience 145, 1222–1232. [DOI] [PubMed] [Google Scholar]
- (11).Barry SM, and Challis GL (2013) Mechanism and catalytic diversity of Rieske non-heme iron-dependent oxygenases. ACS Catalysis 3, 2362–2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Fetzner S (2012) Ring-Cleaving Dioxygenases with a Cupin Fold. Applied and Environmental Microbiology 78, 2505–2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Ortiz de Montellano PR Cytochrome P450: structure, mechanism, and biochemistry; Springer Science & Business Media, 2015; Chapter 9. Human Cytochrome P450 Enzymes. [Google Scholar]
- (14).Guengerich FP, Waterman MR, and Egli M (2016) Recent Structural Insights into Cytochrome P450 Function. Trends in Pharmacological Sciences 37, 625–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Jaffe EK, Stith L, Lawrence SH, Andrake M, and Dunbrack RL (2013) A new model for allosteric regulation of phenylalanine hydroxylase: Implications for disease and therapeutics. Archives of Biochemistry and Biophysics 530, 73–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Tiranti V, D’Adamo P, Briem E, Ferrari G, Mineri R, Lamantea E, Mandel H, Balestri P, Garcia-Silva MT, Vollmer B, Rinaldo P, Hahn SH, Leonard J, Rahman S, Dionisi-Vici C, Garavaglia B, Gasparini P, and Zeviani M (2004) Ethylmalonic encephalopathy is caused by mutations in ETHE1, a gene encoding a mitochondrial matrix protein. American Journal of Human Genetics 74, 239–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Schofield C, and Hausinger R 2-Oxoglutarate-dependent oxygenases; Royal Society of Chemistry, 2015; Chapter 1. Biochemical Diversity of 2-Oxoglutarate-Dependent Oxygenases, pp 1–58. [Google Scholar]
- (18).Martinez S, and Hausinger RP (2015) Catalytic Mechanisms of Fe(II)-and 2-Oxoglutarate-dependent Oxygenases. Journal of Biological Chemistry 290, 20702–20711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Krol WJ, Basak A, Salowe SP, and Townsend CA (1989) Oxidative Cyclization Chemistry Catalyzed By Clavaminate Synthase. Journal of the American Chemical Society 111, 7625–7627. [Google Scholar]
- (20).Baldwin JE, Adlington RM, Moroney SE, Field LD, and Ting HH (1984) Stepwise Ring-Closure In Penicillin Biosynthesis - Initial Beta-Lactam Formation. Journal of the Chemical Society-Chemical Communications 984–986. [Google Scholar]
- (21).Tarhonskaya H, Szollossi A, Leung IKH, Bush JT, Henry L, Chowdhury R, Iqbal A, Claridge TDW, Schofield CJ, and Flashman E (2014) Studies on deacetoxycephalosporin C synthase support a consensus mechanism for 2-oxoglutarate dependent oxygenases. Biochemistry 53, 2483–93. [DOI] [PubMed] [Google Scholar]
- (22).Hillas PJ, and Fitzpatrick PF (1996) A mechanism for hydroxylation by tyrosine hydroxylase based on partitioning of substituted phenylalanines. Biochemistry 35, 6969–6975. [DOI] [PubMed] [Google Scholar]
- (23).Komor AJ, Rivard BS, Fan RX, Guo YS, Que L Jr., and Lipscomb JD (2016) Mechanism for Six-Electron Aryl-N-Oxygenation by the Non-Heme Diiron Enzyme CmII. Journal of the American Chemical Society 138, 7411–7421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Simurdiak M, Lee J, and Zhao HM (2006) A new class of arylamine oxygenases: Evidence that p-aminobenzoate N-oxygenase (AurF) is a di-iron enzyme and further mechanistic studies. Chembiochem 7, 1169–1172. [DOI] [PubMed] [Google Scholar]
- (25).Krebs C, Fujimori DG, Walsh CT, and Bollinger JM Jr. (2007) Non-heme Fe(IV)-oxo intermediates. Accounts of Chemical Research 40, 484–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Hegg EL, and Que L Jr. (1997) The 2-His-1-carboxylate facial triad - An emerging structural motif in mononuclear non-heme iron(II) enzymes. European Journal of Biochemistry 250, 625–629. [DOI] [PubMed] [Google Scholar]
- (27).Joseph CA, and Maroney MJ (2007) Cysteine dioxygenase: structure and mechanism. Chemical Communications 3338–3349. [DOI] [PubMed] [Google Scholar]
- (28).Blasiak LC, Vaillancourt FH, Walsh CT, and Drennan CL (2006) Crystal structure of the non-haem iron halogenase SyrB2 in syringomycin biosynthesis. Nature 440, 368–371. [DOI] [PubMed] [Google Scholar]
- (29).Solomon EI, Brunold TC, Davis MI, Kemsley JN, Lee SK, Lehnert N, Neese F, Skulan AJ, Yang YS, and Zhou J (2000) Geometric and electronic structure/function correlations in non-heme iron enzymes. Chemical Reviews 100, 235–349. [DOI] [PubMed] [Google Scholar]
- (30).Liu LV, Bell I, Caleb B, Wong SD, Wilson SA, Kwak Y, Chow MS, Zhao J, Hodgson KO, Hedman B, and Solomon EI (2010) Definition of the intermediates and mechanism of the anticancer drug bleomycin using nuclear resonance vibrational spectroscopy and related methods. Proceedings of the National Academy of Sciences of the United States of America 107, 22419–22424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Wong SD, Bell I, Caleb B, Liu LV, Kwak Y, England J, Alp EE, Zhao J, Que L Jr., and Solomon EI (2011) Nuclear Resonance Vibrational Spectroscopy on the Fe-IV=O S=2 Non-Heme Site in TMG(3)tren: Experimentally Calibrated Insights into Reactivity. Angewandte Chemie-International Edition 50, 3215–3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Bell CB III, Wong SD, Xiao Y, Klinker EJ, Tenderholt AL, Smith MC, Rohde JU, Que L Jr., Cramer SP, and Solomon EI (2008) A Combined NRVS and DFT Study of Fe-IV=O Model Complexes: A Diagnostic Method for the Elucidation of Non-Heme Iron Enzyme Intermediates. Angewandte Chemie-International Edition 47, 9071–9074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Srnec M, Wong SD, England J, Que L Jr., and Solomon EI (2012) pi-Frontier molecular orbitals in S=2 ferryl species and elucidation of their contributions to reactivity. Proceedings of the National Academy of Sciences of the United States of America 109, 14326–14331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Roach PL, Clifton IJ, Fulop V, Harlos K, Barton GJ, Hajdu J, Andersson I, Schofield CJ, and Baldwin JE (1995) Crystal Structure of Isopenicillin N-Synthase is the First from a New Structural Family of Enzymes. Nature 375, 700–704. [DOI] [PubMed] [Google Scholar]
- (35).Kabil O, and Banerjee R (2012) Characterization of Patient Mutations in Human Persulfide Dioxygenase (ETHE1) Involved in H2S Catabolism. Journal of Biological Chemistry 287, 44561–44567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Lipscomb JD (2008) Mechanism of extradiol aromatic ring-cleaving dioxygenases. Current Opinion in Structural Biology 18, 644–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Sutherlin KD, Wasada-Tsutsui Y, Mbughuni MM, Rogers MS, Park K, Liu LV, Kwak Y, Srnec M, Bottger LH, Frenette M, Yoda Y, Kobayashi Y, Kurokuzu M, Saito M, Seto M, Hu M, Zhao J, Alpan EE, Lipscomb JD, and Solomon EI (2018) Nuclear resonance vibrational spectroscopy definition of O2 intermediates in an extradiol dioxygenase: correlation to crystallography and reactivity. Journal of the American Chemical Society 140, 16495–16513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Sutherlin KD, Rivard BS, Böttger LH, Liu LV, Rogers MS, Srnec M, Park K, Yoda Y, Kitao S, Kobayashi Y, Saito M, Seto M, Hu M, Zhao J, Lipscomb JD, and Solomon EI (2018) NRVS studies of the peroxide shunt intermediate in a Rieske dioxygenase and its relation to the native FeII O2 reaction. Journal of the American Chemical Society 140, 5544–5559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Goudarzi S, Babicz JT Jr, Kabil O, Banerjee R, and Solomon EI (2018) Spectroscopic and electronic structure study of ETHE1: elucidating the factors influencing sulfur oxidation and oxygenation in mononuclear nonheme iron enzymes. Journal of the American Chemical Society 140, 14887–14902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Neibergall MB, Stubna A, Mekmouche Y, Munck E, and Lipscomb JD (2007) Hydrogen peroxide dependent cis-dihydroxylation of benzoate by fully oxidized benzoate 1,2-dioxygenase. Biochemistry 46, 8004–8016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Karlsson A, Parales JV, Parales RE, Gibson DT, Eklund H, and Ramaswamy S (2003) Crystal structure of naphthalene dioxygenase: Side-on binding of dioxygen to iron. Science 299, 1039–1042. [DOI] [PubMed] [Google Scholar]
- (42).Kovaleva EG, and Lipscomb JD (2007) Crystal structures of Fe2+ dioxygenase superoxo, alkylperoxo, and bound product intermediates. Science 316, 453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Rivard BS, Rogers MS, Marell DJ, Neibergall MB, Chakrabarty S, Cramer CJ, and Lipscomb JD (2015) Rate-Determining Attack on Substrate Precedes Rieske Cluster Oxidation during Cis-Dihydroxylation by Benzoate Dioxygenase. Biochemistry 54 , 4652–4664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Tamanaha E, Zhang B, Guo YS, Chang WC, Barr EW, Xing G, St Clair J, Ye SF, Neese F, Bollinger JM, and Krebs C (2016) Spectroscopic Evidence for the Two C-H-Cleaving Intermediates of Aspergillus nidulans Isopenicillin N Synthase. Journal of the American Chemical Society 138, 8862–8874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Eser BE, Barr EW, Frantorn PA, Saleh L, Bollinger JM Jr., Krebs C, and Fitzpatrick PF (2007) Direct spectroscopic evidence for a high-spin Fe(IV) intermediate in tyrosine hydroxylase. Journal of the American Chemical Society 129, 11334–11335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Panay AJ, Lee M, Krebs C, Bollinger JM, and Fitzpatrick PF (2011) Evidence for a High-Spin Fe(IV) Species in the Catalytic Cycle of a Bacterial Phenylalanine Hydroxylase. Biochemistry 50, 1928–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Schofield CJ, and Zhang Z (1999) Structural and mechanistic studies on 2-oxoglutarate-dependent oxygenases and related enzymes. Current opinion in structural biology 9, 722–731. [DOI] [PubMed] [Google Scholar]
- (48).Kappock TJ, and Caradonna JP (1996) Pterin-dependent amino acid hydroxylases. Chemical Reviews 96, 2659–2756. [DOI] [PubMed] [Google Scholar]
- (49).Solomon EI, Pavel EG, Loeb KE, and Campochiaro C (1995) Magnetic circular dichroism spectroscopy as a probe of the geometric and electronic structure of non-heme ferrous enzymes. Coordination Chemistry Reviews 144, 369–460. [Google Scholar]
- (50).Solomon EI, Goudarzi S, and Sutherlin KD (2016) O2 Activation by Non-Heme Iron Enzymes. Biochemistry 55, 6363–6374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Iyer SR, Chaplin VD, Knapp MJ, and Solomon EI (2018) O2 activation by nonheme FeII α-ketoglutarate-dependent enzyme variants: Elucidating the role of the facial triad carboxylate in FIH. Journal of the American Chemical Society 140, 11777–11783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Pavel EG, Zhou J, Busby RW, Gunsior M, Townsend CA, and Solomon EI (1998) Circular dichroism and magnetic circular dichroism spectroscopic studies of the non-heme ferrous active site in clavaminate synthase and its interaction with alpha-ketoglutarate cosubstrate. Journal of the American Chemical Society 120, 743–753. [Google Scholar]
- (53).Elkins JM, Ryle MJ, Clifton IJ, Dunning Hotopp JC, Lloyd JS, Burzlaff NI, Baldwin JE, Hausinger RP, and Roach PL (2002) X-ray crystal structure of Escherichia coli taurine/α-ketoglutarate dioxygenase complexed to ferrous iron and substrates. Biochemistry 41, 5185–5192. [DOI] [PubMed] [Google Scholar]
- (54).Valegard K, van Scheltinga ACT, Dubus A, Ranghino G, Oster LM, Hajdu J, and Andersson I (2004) The structural basis of cephalosporin formation in a mononuclear ferrous enzyme. Nature Structural Molecular Biology 11, 95–101. [DOI] [PubMed] [Google Scholar]
- (55).Girl NC, Sun H, Chen H, Costa M, and Maroney MJ (2011) X-ray absorption spectroscopy structural investigation of early intermediates in the mechanism of DNA repair by human ABH2. Biochemistry 50, 5067–5076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Higgins LJ, Yan F, Liu P, Liu H, and Drennan CL (2005) Structural insight into antibiotic fosfomycin biosynthesis by a mononuclear iron enzyme. Nature 437, 838–844. [DOI] [PubMed] [Google Scholar]
- (57).Goudarzi S, Iyer SR, Babicz JT, Yan JJ, Peters GH, Christensen HE, Hedman B, Hodgson KO, and Solomon EI (2020) Evaluation of a concerted vs. sequential oxygen activation mechanism in α-ketoglutarate—dependent nonheme ferrous enzymes. Proceedings of the National Academy of Sciences 117, 5152–5159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Iyer SR, Tidemand KD, Babicz JT, Jacobs AB, Gee LB, Haahr LT, Yoda Y, Kurokuzu M, Kitao S, Saito M, Seto M, Christensen HEM, Peters GHJ, and Solomon EI (2021) Direct coordination of pterin to FeII enables neurotransmitter biosynthesis in the pterin-dependent hydroxylases. Proceedings of the National Academy of Sciences 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Andersen OA, Stokka AJ, Flatmark T, and Hough E (2003) 2.0 Å Resolution crystal structures of the ternary complexes of human phenylalanine hydroxylase catalytic domain with tetrahydrobiopterin and 3-(2-thienyl)-L-alanine or L-norleucine: substrate specificity and molecular motions related to substrate binding. Journal of molecular biology 333, 747–757. [DOI] [PubMed] [Google Scholar]
- (60).Pavon JA, Eser B, Huynh MT, and Fitzpatrick PF (2010) Single Turnover Kinetics of Tryptophan Hydroxylase: Evidence for a New Intermediate in the Reaction of the Aromatic Amino Acid Hydroxylases. Biochemistry 49, 7563–7571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Roberts KM, Pavon JA, and Fitzpatrick PF (2013) Kinetic mechanism of phenylalanine hydroxylase: Intrinsic binding and rate constants from single-turnover experiments. Biochemistry 52, 1062–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Fitzpatrick PF (1999) Tetrahydropterin-dependent amino acid hydroxylases. Annual Review of Biochemistry 68, 355–381. [DOI] [PubMed] [Google Scholar]
- (63).Mitchell AJ, Dunham NP, Martinie RJ, Martinie RJ, Bergman JA, Pollock CJ, Hu K, Allen BD, Chang W, Silakov A, Bollinger JM Jr., Krebs C, and Boal AK (2017) Visualizing the reaction cycle in an iron (II)-and 2-(oxo)-glutarate-dependent hydroxylase. Journal of the American Chemical Society 139, 13830–13836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Srnec M, Iyer SR, Dassama LM, Park K, Wong SD, Sutherlin KD, Yoda Y, Kobayashi Y, Kurokuzu M, Saito M, Seto M, Krebs C, Bollinger JM Jr., and Solomon EI (2020) Nuclear Resonance Vibrational Spectroscopic Definition of the Facial Triad FeIV=O Intermediate in Taurine Dioxygenase: Evaluation of Structural Contributions to Hydrogen Atom Abstraction. Journal of the American Chemical Society 142, 18886–18896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Wong SD, Srnec M, Matthews ML, Liu LV, Kwak Y, Park K, Bell I, Caleb B, Alp EE, Zhao J, Yoda Y, Kitao S, Seto M, Krebs C, Bollinger JM Jr., and Solomon EI (2013) Elucidation of the Fe(IV)=O intermediate in the catalytic cycle of the halogenase SyrB2. Nature 499, 320–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).England J, Guo Y, Farquhar ER, Young J, Victor G, Münck E, and Que L Jr. (2010) The Crystal Structure of a High-Spin Oxoiron(IV) Complex and Characterization of Its Self-Decay Pathway. Journal of the American Chemical Society 132, 8635–8644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Srnec M, Wong SD, Matthews ML, Krebs C, Bollinger JM, and Solomon EI (2016) Electronic Structure of the Ferryl Intermediate in the alpha-Ketoglutarate Dependent Non-Heme Iron Halogenase SyrB2: Contributions to H Atom Abstraction Reactivity. Journal of the American Chemical Society 138, 5110–5122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Sturhahn W, Toellner TS, Alp EE, Zhang X, Ando M, Yoda Y, Kikuta S, Seto M, Kimball CW, and Dabrowski B (1995) Phonon Density-of-States Measured by Inelastic Nuclear Resonant Scattering. Physical Review Letters 74, 3832–3835. [DOI] [PubMed] [Google Scholar]
- (69).Dunham NP, Chang W, Mitchell AJ, Martinie RJ, Zhang B, Bergman JA, Rajakovich LJ, Wang B, Silakov A, Krebs C, Boal AK, and Bollinger JM Jr. (2018) Two distinct mechanisms for C—C desaturation by Iron (II)-and 2-(Oxo) glutarate-Dependent oxygenases: importance of α-heteroatom Assistance. Journal of the American Chemical Society 140, 7116–7126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Srnec M, and Solomon EI (2017) Frontier Molecular Orbital Contributions to Chlorination versus Hydroxylation Selectivity in the Non-Heme Iron Halogenase SyrB2. Journal of the American Chemical Society 139, 2396–2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Matthews ML, Neumann CS, Miles LA, Grove TL, Booker SJ, Krebs C, Walsh CT, and Bollinger JM Jr. (2009) Substrate positioning controls the partition between halogenation and hydroxylation in the aliphatic halogenase, SyrB2. Proceedings of the National Academy of Sciences of the United States of America 106, 17723–17728. [DOI] [PMC free article] [PubMed] [Google Scholar]