

Abstract
Lysine crotonylation (Kcr) is a newly discovered type of protein post-translational modification and has been reported to be involved in various pathophysiological processes. High-resolution mass spectrometry is the primary approach for identification of Kcr sites. However, experimental approaches for identifying Kcr sites are often time-consuming and expensive when compared with computational approaches. To date, several predictors for Kcr site prediction have been developed, most of which are capable of predicting crotonylation sites on either histones alone or mixed histone and nonhistone proteins together. These methods exhibit high diversity in their algorithms, encoding schemes, feature selection techniques and performance assessment strategies. However, none of them were designed for predicting Kcr sites on nonhistone proteins. Therefore, it is desirable to develop an effective predictor for identifying Kcr sites from the large amount of nonhistone sequence data. For this purpose, we first provide a comprehensive review on six methods for predicting crotonylation sites. Second, we develop a novel deep learning-based computational framework termed as CNNrgb for Kcr site prediction on nonhistone proteins by integrating different types of features. We benchmark its performance against multiple commonly used machine learning classifiers (including random forest, logitboost, naïve Bayes and logistic regression) by performing both 10-fold cross-validation and independent test. The results show that the proposed CNNrgb framework achieves the best performance with high computational efficiency on large datasets. Moreover, to facilitate users’ efforts to investigate Kcr sites on human nonhistone proteins, we implement an online server called nhKcr and compare it with other existing tools to illustrate the utility and robustness of our method. The nhKcr web server and all the datasets utilized in this study are freely accessible at http://nhKcr.erc.monash.edu/.
Keywords: deep learning, crotonylation, protein post-translational modification, bioinformatics, nonhistone proteins, sequence analysis
Introduction
As an important type of post-translation modification, lysine crotonylation (Kcr) is evolutionarily conserved, and it was originally identified on histone proteins [1]. Histone crotonylation is enriched on sex chromosomes and can act as a critical indicator for male germ cell differentiation [1]. It also plays an important role in other biological processes such as driving male haploid cell gene expression [2] and stimulating transcription [3]. Recently, several studies have started to explore Kcr in nonhistone proteins as well as its functional implications [4–8]. It has been reported that crotonylated nonhistone proteins are involved in diverse cellular functions and signaling pathways [7].
To understand the functional roles of crotonylated proteins and the regulation mechanism of different enzymes on diverse cellular process, large-scale analysis of Kcr in the proteome has been carried out recently. Wei et al. [5] studied Kcr in the nonhistone proteins of HeLa cells and identified 1185 Kcr sites in 453 proteins after treatment with sodium crotonateb. Xu et al. [7] generated a much larger dataset with 2696 Kcr sites in 1024 proteins. Wu et al. [6] comprehensively studied lysine crotonylome in both histone proteins and nonhistone proteins and identified 10 163 Kcr sites in A549 cells by Suberoylanilide hydroxamic acid (SAHA) treatment. In addition, Huang et al. [4] identified 816 unique Kcr sites in 392 proteins in mammalian cells. Yu et al. [8] identified 14 311 Kcr sites across 3734 proteins in HeLa cells and provided by far the largest Kcr dataset. Although these studies have expanded our understanding of Kcr at a proteomics scale, future investigations are warranted to characterize the functional role of Kcr in diverse cellular pathways.
According to previous researches, there are some specific amino acid preferences adjacent to the crotonylation sites. For instance, it has been discovered that negatively charged glutamate (E) residues were overrepresented at the −1 and +1 positions of Kcr sites [7]. In another work, Li et al. [9] reported that AF9 YEATS domain was a selective histone crotonylation reader. They further demonstrated that the histone acetylation-binding double PHD finger (DPF) domains of human MOZ and DPF2 accommodate a wide range of histone lysine acylations with the strongest preference for Kcr [10]. Meanwhile, Andrews et al. [11] reported the Taf14 YEATS domain engages crotonyllysine and acts as an effective reader of Kcr. Furthermore, Yu et al. [8] identified ‘EKxxxxxK’, ‘KExxxK’ and ‘KxxxEK’ as significantly overrepresented sequence motifs for Kcr sites, and structural analysis revealed that 30% of the Kcr sites were found in helices, 6% were located in strands, while the remaining 64% were seen in disordered coils. These characteristic biases implied that the computational methods for identification of Kcr sites are complementary with the time-consuming and labor-intensive experimental methods.
Compared to the alternative computational methods, experimental methods for identifying Kcr sites are often time-consuming, labor-intensive and expensive. To date, there exist several tools that have been developed to predict crotonylation sites on human proteins. Huang and Zeng [12] proposed the first predictor, named CrotPred, to identify the Kcr sites in proteins. Recently, Qiu et al. [13] proposed a new sequence encoding scheme called the position weight amino acid composition (PWAA) and further developed a support vector machine (SVM)-based approach for predicting Kcr sites in histone proteins. In another recent work, Malebary et al. [14] incorporated various position and composition relative features along with statistical moments (SMs) into the pseudo amino acid composition (PseAAC) to develop a Kcr site prediction tool called iCrotoK-PseAAC. However, all of the above three methods do not provide online servers, which are not convenient for experimental biologists to study crotonylation. Subsequently, Ju et al. [15] proposed CKSAAP_CrotSite to identify Kcr sites based on the composition of k-spaced amino acid pair encoding scheme and SVM. By using PWAA, Qiu et al. [16] presented iKcr-PseEns based on ensemble random forest (RF) algorithm to predict Kcr sites. Most recently, Lv et al. [17] developed a deep learning-based method termed Deep-Kcr for the detection of Kcr sites in both histone and nonhistone proteins and achieved an area under the receiver-operating curve (ROC) curve (AUC) value of 0.859 on the independent test.
Although the performance of previously developed methods was generally good based on the datasets they used, there is a strong need for the development of new methods based on the most recent experimental datasets with improved performance. Such methods can be better applied for the identification of novel Kcr sites on the proteomic scale. Another issue in using the existing methods is that they are designed for predicting Kcr sites on either histone proteins, or those mixed with nonhistone proteins. None of the existing predictors was specifically designed for predicting Kcr sites on human nonhistone proteins. In addition to the crotonylated histone proteins, a massive number of crotonylated nonhistone proteins need to be explored. Hence, a predictor that could identify Kcr sites in nonhistone proteins would be more desirable for experimental biologists. We, herein, propose a predictor named nhKcr which aims to identify Kcr sites in human nonhistone proteins precisely. By designing and using a new deep learning-based framework called CNNrgb, we show that nhKcr achieves an improved performance than previously reported methods. In addition, we also implement an online web server which is publicly available at http://nhKcr.erc.monash.edu/ to enable online high-throughput prediction of Kcr sites in nonhistone proteins. We anticipate that nhKcr will serve as a useful bioinformatics tool for accurate identification of Kcr sites and help to narrow down highly reliable candidates for experimental validation.
Materials and methods
Benchmark dataset construction
In the current study, non-redundant experimentally verified Kcr sites on human nonhistone proteins were collected to construct the benchmark datasets. In total, five different datasets which were originally produced in previous literatures were extracted, including 19 287 Kcr sites identified in 4230 nonhistone proteins across HeLa cell, lung cell, A549 and HCT116 cell [4–8], respectively. All the above protein sequences were then downloaded from the UniProt database [18], using their UniProt IDs. The CD-HIT program [19] was used to remove the sequence redundancy and to avoid the overestimation caused by sample similarity by setting the cutoff threshold of sequence identity to 30%. Then, the processed sequences were truncated into 29-residue-long sequence segments with the residue K located at the center. The segments were defined as positive samples if the central K was crotonylation, and the remaining lysine sites were defined as negative samples. Finally, 15 603 positive samples and 164 709 negative samples were obtained. The negative samples were randomly selected with five times of the positives to construct the training dataset and the independent testing dataset. As a result, 12 262 positive and 60 101 negative segments were subjected to 5-fold cross-validation; 3343 positive and 15 010 negative segments were utilized as the independent test set (the curated datasets can be downloaded at http://nhKcr.erc.monash.edu/). In addition, in order to illustrate the preferences of neighboring residues flanking the crotonylation sites on the histone and nonhistone proteins, the same procedures were applied to the above five datasets and Qiu’s dataset [13] to extract the Kcr sites on human histone proteins.
Feature encoding schemes employed
In order to develop a well-performing machine learning method for Kcr prediction, in this study, a number of different feature encoding schemes have been employed to encode the 21 types of amino acids, including the gap (O) [20]. In the current study, we have applied 10 encoding schemes which can be grouped into three major types. The first type is derived from protein primary sequence, such as binary encoding (BE) scheme, composition of k-space amino acid pairs (CKSAAP) [21], amino acid composition (AAC) [22], enhanced amino acid composition (EAAC) [23], di-peptide composition (DPC) [24] and enhanced grouped amino acids content (EGAAC) [25]. The second type is extracted from physicochemical properties such as amino acid index (AAindex) [26] and Z-scale [27]. The third type is BLOSUM62 [28] which is derived from the protein position-specific scoring matrices.
Protein primary sequence
Binary encoding
BE is the most popular and the easiest encoding method to transform protein sequences into numeric vectors. It simply converts each amino acid by a 21-dimensional binary vector, for example, A (100000000000000000000), C (010000000000000000000), …, O (000000000000000000001), etc. This encoding has been used to encode amino acid sequence as the input feature for training the classifiers in a number of our previous studies [29–32]. Hence, each Kcr site is represented by a fragment of 2n + 1, and the total dimension of the proposed binary feature vector is 21 × 2n (here, ‘n’ represents the maximum length of each side of the Kcr site).
Composition of k-space amino acid pairs
The CKSAAP encoding has been widely used in numerous post-translational modification (PTM) prediction studies [21, 32–34] as it could effectively describe the short-range interaction between the amino acids surrounding the predicted site. The value of k represents the space between two amino acids. When k = 0, there will be 441 0-spaced amino acid pairs (i.e. AA, AC, AD, …, OO). Similarly, ‘AxA, AxC, AxD, …, OxO’ will be for k = 1 and so on. Then the feature vector can be calculated by the following equation:
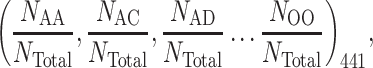 |
where NTotal represents the total number of residue pairs in a fragment [for instance, when a sequence window length L is 51 and space k = 0, 1, 2, 3, 4, 5, then NTotal = (L − k − 1) will be 50, 49, 48, 47, 46 and 45, respectively]. In the present study, the total dimension of the CKSAAP-based feature vector is 441 × (kmax + 1), which is 441 × 6 = 2646.
Amino acid composition and EAAC
AAC is a commonly used encoding scheme for examining substrate site motifs [22]. It calculates the probability of amino acids occurring in the sequence fragment surrounding PTM sites. By calculating the number of each specific amino acid occurring in the fragment, the composition of the 21 amino acids can be transformed to a 21-dimensional numeric vector. EAAC encoding is developed based on AAC. The main difference is that EAAC is calculated in a fixed-length sequence window continuously sliding from the N-terminus to the C-terminus of each peptide [23]. For instance, if the sliding window size was fixed as 6, and the length of the fragment was 51, then there will be 46 (51 − 6 + 1) sliding windows. The dimension of the EAAC encoding was 46 × 21 (amino acids) = 966.
Di-peptide composition
DPC is another widely used encoding scheme for PTM site prediction [24, 35, 36], which reflects the global information about each protein sequence as well as the local order information of amino acids within the protein by calculating the percentages of the 400 (20 × 20) dipeptide combinations. In other words, DPC is identical with the 0-spaced CKSAAP.
Protein physicochemical properties
Enhanced grouped amino acids content
The EGAAC encoding [25] is based on the grouped amino acids content (GAAC) features in which the 20 amino acid types are categorized into five major groups according to their physicochemical properties, including GAVLMI, FYW, KRH, DE and STCPNQ. The frequency of each group is calculated for each position in the flanking region of PTM sites. For the EGAAC features, the GAAC values are calculated in a fixed-length sequence window (the default value is 5) continuously sliding from the N-terminus to the C-terminus of each peptide.
Amino acid index
AAindex [26] is a public database of AAindexes representing various physicochemical and biochemical properties of amino acids (https://www.genome.jp/aaindex/). After the removal of properties with ‘NA’ in the AAindexes, 531 physicochemical properties from the AAindex database remained and were used for further encoding analysis.
Six_letter encoding
Six_letter encoding is another form of BE based on a reduced alphabet [37]. In this encoding scheme, the 20 amino acids are categorized into five groups according to their physical characteristics, which include aliphatic (AVLI), charged (RKDE), polar (STNQ), cyclic (FHYW) and other (GPMC). With the addition of the letter ‘O’ to represent an empty position, we obtained a total of six groups.
Z-scale
In this encoding, each amino acid is characterized by five physiochemical descriptor variables, which were proposed by Sandberg et al. in 1998 [27]. Specifically, each Z-scale represents an amino-acid property as follows: Z1 (lipophilicity), Z2 (steric bulk/polarizability), Z3 (electronic properties) and Z4 and Z5 relate to electronegativity, heat of formation, electrophilicity and hardness.
Protein position-specific scoring matrices
BLOcks SUbstitution Matrix (BLOSUM62)
BLOSUM62 matrix is generally used by the Basic Local Alignment Search Tool (BLAST) program. Here, we adopted it to transform the primary protein sequence to represent the similarity of two sequence fragments. It is a substitution matrix for studying protein sequence conservation in large databases of related proteins. It is generally used to score alignments between evolutionarily divergent protein sequences, and it has been widely used in many predictors [28, 38].
Machine learning algorithms employed
As listed in Table 1, all the computational approaches for crotonylation site prediction were built using well-established machine learning algorithms such as SVM [21], RF [23], LightGBM [39] and artificial neural networks (ANNs) [14]. In the current study, we employed stochastic gradient descent (SGD) [40], multilayer perceptrons (MLPs) [41], RF [23], logistic regression (LR) [42], convolutional neural network (CNN) [43], LightGBM [39, 44, 45] and XGBoost [46] to predict Kcr sites on human nonhistone proteins. These algorithms are briefly described below.
Table 1.
A comprehensive summary of the reviewed predictors for Kcr site prediction
| Tool | Algorithm | Species | Encoding scheme | Evaluation strategy | URL/stand-alone package | Histone or nonhistone | Benchmark dataset | Option of batch prediction | Window size | Published year | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Qiu et al. | SVM | Human Mouse |
PWAA | Jackknife test | No | Histone | 101 Histone proteins, 169 positive, 847 negative | No | 31 | 2017 | [13] |
| CKSAAP CrotSite |
SVM | Human Mouse |
CKSAAP PseAAC |
Jackknife test | 123.206.31.171/ CKSAAP CrotSite/(not available) | Histone | 101 Histone proteins, 169 positive, 847 negative | No | 31 | 2017 | [15] |
| iKcr-PseEns | Ensemble RF | Human Mouse |
PseAAC | Jackknife test | http://www.jci-bioinfo.cn/iKcr-PseEns (available) | Histone | 55 histones, 169 positive 46 Histones, 866 negative |
Yes | 35 | 2018 | [16] |
| LightGBM-CroSite | LightGBM | Human Mouse |
BE PWAA EBGW kNN PsePSSM |
Jackknife test | https://github.com/QUST-AIBBDRC/LightGBM-CroSite/ | Histone | 101 Histone proteins, 159 positive, 847 negative | No | 31 | 2020 | [39] |
| iCrotoK-PseAAC | ANN | Mixed | PseAAC RPRIM PRIM SM SVV FV AAPIV RAAPIV |
10-fold cross-validation | No | Mixed | 378 Positive, 500 negative | No | 41 | 2019 | [14] |
| Deep-Kcr | CNN RF LB NB LR |
Human | CKSAAP PWAA AAindex CTD EBGW |
10-fold cross-validation | http://lin-group.cn/server/Deep-Kcr (available) | Mixed | 3734 Proteins, 9964 positive, 9964 negative | No | 31 | 2020 | [17] |
Abbreviations: EBGW, encoding based on grouped weight; kNN, k nearest neighbors, PsePSSM, pseudo-position specific scoring matrix; CTD, composition, transition and distribution; RPRIM, position relative incidence matrix; SVV, site vicinity vector; FV, frequency vector; AAPIV, accumulative absolute position incidence vector; RAAPIV, reverse accumulative absolute position incidence vector; NB, naïve Bayes; LB, logitboost.
Stochastic gradient descent
SGD [40] is an iterative method that has been applied to many large-scale problems in machine learning and data analysis due to its scalability and efficiency. It is a variation on gradient descent, which is a popular optimization technique. Gradient descent seeks to minimize the cost function by iteratively updating each parameter by a small amount based on the negative gradient of a given dataset. In contrast, SGD modifies the batch gradient descent algorithm by calculating the gradient for only one training example at each iteration to solve the local minimum problem and decreases the computational time.
Random forest
RF [23] is one of the most popular algorithms for addressing many prediction problems because of its flexibility and simplicity. It is essentially an ensemble of decision trees and can be applied to deal with both classification and regression problems. In the RF algorithm, a large number of decision trees are constructed and then the prediction from each of them will be obtained and averaged to overcome the over-fitting. To achieve a better performance or to make the model faster, understanding the hyperparameters of scikit-learn’s built-in RF function is very important. For instance, the n_estimators hyperparameter denotes the number of trees the algorithm builds before taking the maximum voting or taking the averages of predictions. Another important hyperparameter is max_features, which is the maximum number of features RF considers to split a node. The last important hyperparameter is min_sample_leaf, which determines the minimum number of leaves required to split an internal node.
Convolutional neural networks
As one of the most successful deep learning-based methods, CNNs have been applied in many different prediction studies [43]. CNNs are composed of multiple layers, including convolutional layers, non-linear layers, pooling layers and output layers. In addition, there is an activation layer, which applies different layer activation functions, such as ReLu, to increase the non-linearity of the network without affecting the receptive fields of convolutional layers.
Multilayer perceptrons
Multilayer perceptrons (MLP) is flexible to be applied to different types of data and is suitable for both classification problems and regression problems [41, 47, 48]. MLP is composed of more than one perceptron. It is a linear classifier to classify the input samples by separating two categories with a straight line and generating a single output based on several real-valued inputs by forming a linear combination using its input weights. By increasing the number of perceptrons and hidden layers, MLP becomes more powerful in solving difficult classification tasks.
Logistic regression
LR is a popular algorithm for binary classification problems based on a set of independent variables [42]. There are two phases in LR. The first phase is training, which gets the weights and bias term by using SGD and the cross-entropy loss. The second phase is testing, which computes and returns the probability for the predicted label. LR could explain the relationship between one dependent binary variable and one or more independent variables and as such it is very popular.
Extreme gradient boosting algorithm
Extreme gradient boosting algorithm (XGBoost) is a decision-tree based machine learning algorithm [46], which has been widely used for many classification problems and provides state-of-the-art results [49–51]. It is very flexible to use in many different programming languages including R, Python, C, JVM, etc. Before running XGBoost, three types of parameters need to be set, including the general parameters, booster parameters and task parameters. The general parameters determine which boost to use, commonly tree or linear model. Then, different booster parameters need to be selected according to different boosters. The task parameters are used to specify the learning task and the corresponding learning objective. Taken together, XGBoost is efficient in reducing the computing time and the memory cost and thus is very fast compared to other implementations of gradient boosting.
Light Gradient Boosting Machine (LightGBM)
LightGBM [39, 44, 45] is also a boosting algorithm that is very similar to XGBoost. However, they are different from each other in a few specific ways, especially in how the trees are growing: XGBoost applies level-wise tree growth which is horizontal, whereas LightGBM applies leaf-wise tree growth which is vertical. Usually, the leaf-wise approach is mostly faster than the level-wise approach, which is why LightGBM is always faster than XGBoost in practical applications. However, XGBoost has recently introduced a new tree growing method similar to the one LightGBM uses. They both have advantages and disadvantages; choosing the right framework for the right job is crucial [44, 45].
An improved convolutional neural network (CNNrgb)
In this work, we introduce a novel method nhKcr for predicting Kcr sites, which is based on an improved CNN termed CNNrgb. It takes the AAindex, Binary and Blosum62 encoding schemes as three arrays of the matrix of red, green and blue (RGB) color channels. An overview of the architecture of the proposed deep learning framework of nhKcr is provided in Figure 1.
Figure 1 .
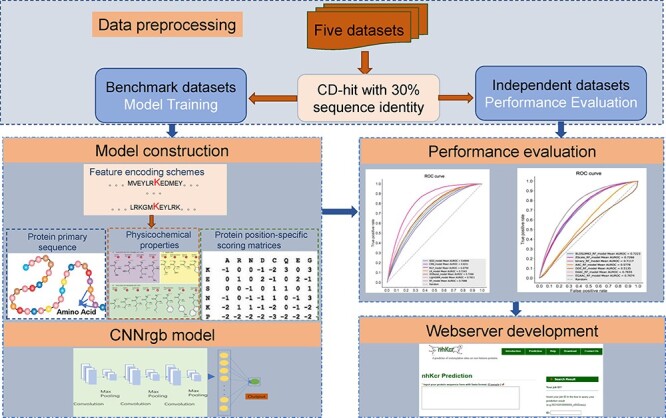
Overview of the nhKcr methodology.
More specifically, for the AAindex encoding scheme, we first normalized it with the z-score method and then utilized 1000 trees in RF to calculate the AUC value for each physicochemical property. Then, the top 29 physicochemical properties were selected to encode the sequence fragment. If duplicated physicochemical properties exist, we will choose the one with the best performance. In our study, the length of sequence fragment was equal to 29, the amino acids surrounding the central K could be represented as a 29 × 29-dimensional matrix. For the BE scheme, to construct the 29 × 29 dimensional matrix, we filled the remaining gaps with the average of the binary vector, which is 0.05. As for BLOSUM62, we first normalized the element value of the matrix in the range of 0–1. Next, we filled the remaining gaps to build the 29 × 29-dimensional matrix with the average of BLOSUM62, which is 0.267. The three types of encoding schemes were deemed as the RGB channels of a color image and were processed by a two-dimensional convolution including three following layers (Figure 2):
Figure 2 .
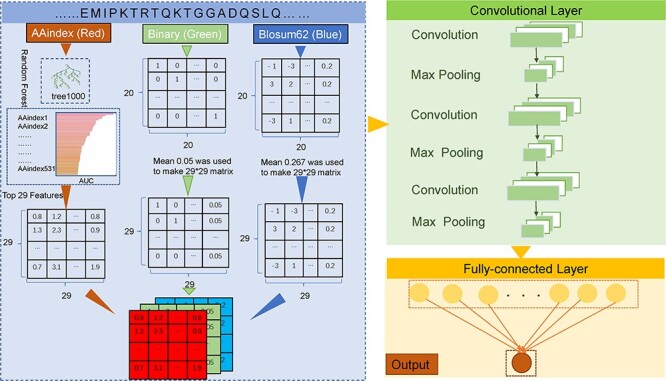
Flowchart of the new proposed CNN-based framework CNNrgb.
(i) Input layer: Three 29 x 29 matrices corresponding to the RGB channels of a color image were utilized as the inputs in this layer.
(ii) Convolutional layers: This layer included three sequentially connected blocks. Each block included a convolution layer and a max pooling layer. The rectified linear unit (ReLU) [52] was considered as its activation function, the number of convolution kernels was set as 128 and the convolution kernel size was set as 5. The size of max pooling size was set as 2.
(iii) Fully connected layer: This layer took the output from the above layers, flattened them and turned them into a single vector, which comprised of 64 neurons and was activated by the ReLU function.
(iv) Output layer: This layer contained only one neuron, which output the final probability score indicating the likelihood of the lysine residue in the center to be crotonylated. The ‘sigmoid’ function was utilized as the activation function in this layer, which was expressed as follows:
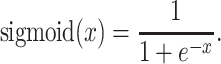 |
The CNN model was developed using PyTorch [53].
Performance evaluation strategies
Two performance evaluation methods, namely 5-fold cross-validation and independent test, were used to derive comparative metrics (values) of our predictors. A detailed interpretation of the evaluation strategies can be found in [54].
Six performance measurements, including sensitivity (Sn), specificity (Sp), Matthew’s correlation coefficient (MCC), accuracy (ACC), AUC and area under precision-recall curve (AUPRC), which are commonly used in other studies, are applied to evaluate the prediction performance. The definitions of Sn, Sp, ACC and MCC are given as follows:
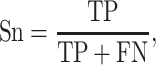 |
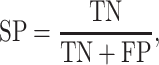 |
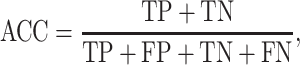 |
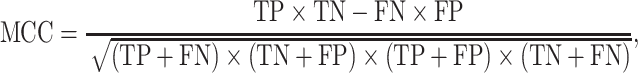 |
where TP, FP, FN and TN denote the numbers of true positives, false positives, false negatives and true negatives, respectively. The MCC value ranges from −1 to 1, and a higher value indicates a better prediction performance, where a coefficient of +1 means a perfect prediction, while −1 indicates a total disagreement between the prediction and the observation. The prediction performance was also measured using the ROC analysis, which plots the true positive rate (i.e. Sn) as a function of the false positive rate (i.e. 1 − Sp) for all possible thresholds. We also calculate the AUC to quantify the prediction performance of the proposed method. Generally, the closer the AUC value to 1, the better the prediction performance of the proposed method.
Results
Motif conservation analysis of Kcr sites in nonhistone proteins
To illustrate the different distribution and preference of the flanking residues of crotonylation sites on nonhistone proteins, we used the Probability Logo Generator (pLogo) [55] algorithm to compare the amino acid sequences around the observed Kcr sites against non-Kcr sites sequences, which is presented in Figure 3A. The default values ±4.08 were used as the thresholds for significantly overrepresented and underrepresented amino acids, respectively. As can be seen from Figure 3, charged residues K, D, F, R and E are predominantly different between Kcr sites and non-Kcr sites. For instance, it was observed that the residue K was more overrepresented at positions +8, +11 and −5 with the frequency equal to 13.6%, 12.5% and 10.4%, respectively. In addition, both residues K and R were most underrepresented on the position −1. We also found that residue E was overrepresented at the −1 and +1 positions of the Kcr sites. Meanwhile, the hydrophobic amino acid P rarely occurred on positions +1, +3 and + 6. It has been reported that the motifs ‘EKxxxxxK’, ‘KExxxK’ and ‘KxxxEK’ were identified as significantly overrepresented hotspots for Kcr sites. A recent crotonylation study [4] also determined the similar enriched motifs within the identified Kcr substrates. These results are in agreement with our observations on the benchmark datasets curated in this study.
Figure 3 .
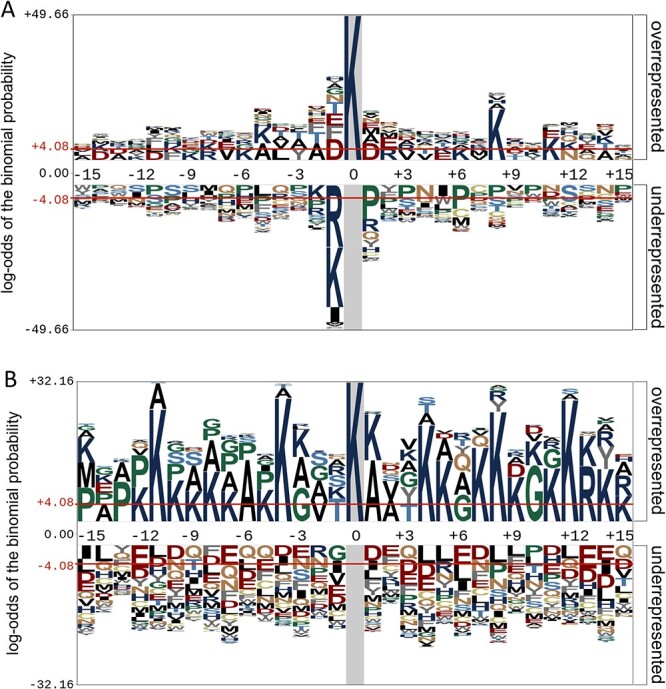
Motif conservation analysis of Kcr sites on (A) human nonhistone proteins and (B) histone proteins. The sequence logos were generated with pLogo with scaled better data visualization. The red horizontal lines on the sequence logos denote the P < 0.05 threshold.
In addition, we used the same strategy to analyze the different frequencies of each type of residue surrounding Kcr sites on histone proteins. The results clearly showed that the Kcr sites in nonhistone proteins (Figure 3A) exhibited different patterns from those in histone proteins (Figure 3B). As it can be seen, the residues K, A, G and P were largely enriched in histone proteins across the majority of the positions (Figure 3B). Such differential patterns in Kcr sites between the nonhistone and histone proteins highlight the need and further motivate us to develop a predictor specifically for the Kcr sites on nonhistone proteins independently.
Performance evaluation on 5-fold cross-validation and independent tests
In this section, we evaluated the prediction performance of 10 different encoding schemes using RF by conducting 5-fold cross-validation. Figure 4A showed the mean ROC curves for each encoding scheme. As can be seen, the area under the EAAC_RF curve (AUC = 0.8228) is remarkably largest among all the encoding schemes. Moreover, we utilized the same evaluation strategy to assess the performance on the BE using seven different machine learning algorithms. We can see that the traditional CNN model achieved the best performance with a mean AUC of 0.8231 (Figure 4B). We also performed 5-fold cross-validation and the independent test to evaluate the prediction performance of nhKcr. The results are provided in Table 2. We can see that nhKcr achieved a remarkable performance, with AUC equal to 0.882 and 0.878, respectively, which is superior to those of the above encoding schemes and machine learning methods.
Figure 4 .
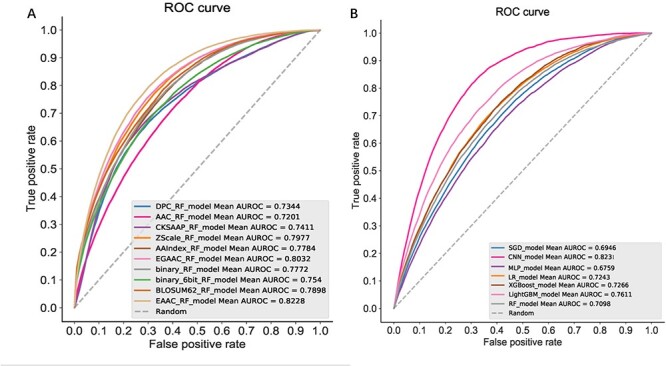
The performance of (A) prediction models trained using different sequence encoding schemes and the performance (B) of the BE-based model trained using different machine learning algorithms. On each panel, colored lines represent the different methods. Diagonal gray dashed lines represent the performance of a random classifier. Each legend box contains the AUC for each method and the average of them.
Table 2.
Prediction performance of nhKcr in terms of five major performance metrics, that is, Acc, Sn, Sp, MCC and AUC
| Acc | Sn | Sp | MCC | AUC | |
|---|---|---|---|---|---|
| 5-fold cross-validation | 85.40 | 62.86 | 90.00 | 0.506 | 0.882 |
| Independent test | 84.33 | 58.90 | 90.00 | 0.482 | 0.878 |
Performance comparison between nhKcr and the state-of-the-art predictors
To illustrate the predictive capability and robustness of nhKcr, we further compared the performance of nhKcr with other state-of-the-art predictors. According to Table 1, there exist six Kcr predictors developed to date. However, only three of them are available now. Deep-Kcr is the latest developed tool for predicting Kcr sites in mixed histone and nonhistone proteins. In this predictor, the authors randomly divided 9964 positive samples and 9964 negative samples into the training dataset and independent test dataset according to the positive-to-negative ratio of 7:3. After removing those Kcr sites from the test dataset that existed in their training dataset, we obtained an independent test set for a fair comparison. Figure 5 shows that our method nhKcr clearly outperformed Deep-Kcr in terms of predicting the Kcr sites on nonhistone proteins. The results again highlight the necessity of developing a precise predictor for the Kcr sites on nonhistone proteins alone. In addition, to better assess and understand the performance of nhKcr, we also used the three encodings as the conventional direct input to train the deep learning model (termed DirectInput-CNN) and compared with nhKcr and Deep-Kcr. The statistical significance between the prediction results of the three models Deep-Kcr, nhKcr and DirectInput-CNN in terms of P-value was calculated to evaluate whether the pair-wise performance comparison between the two methods was statistically significant or not. As can be seen from Figure 5, the AUC of DirectInput-CNN was 0.8392, which was 3% lower than that of nhKcr (P-value = 2.42e-08), which indicates that our proposed algorithm nhKcr performed significantly better than DirectInput-CNN.
Figure 5 .
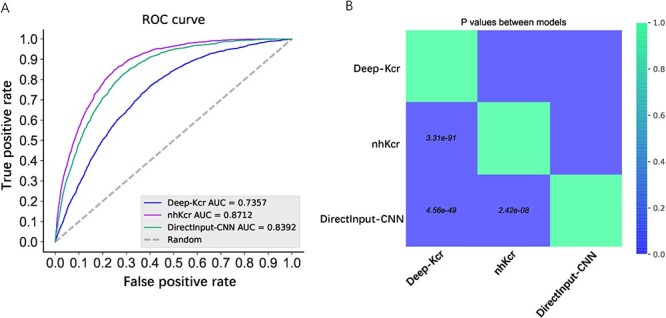
Performance comparison of the proposed nhKcr, the state-of-the-art predictor Deep-Kcr and the DirectInput-CNN. (A) ROC curves. Colored lines represent the different predictors. Diagonal gray dashed lines represent the performance of a random classifier. Legend box contains the AUC for each predictor. (B) pair-wise performance comparison between three methods in terms of P-value.
Taken together, we conclude that nhKcr achieved a remarkable performance due to the following three primary reasons: (1) the use of large-scale training dataset could help us to improve the generalization and robustness of the deep learning model of nhKcr; (2) the idea of converting three encoding schemes into three channels of RGB picture proved to be an effective strategy and (3) our predictor was designed particularly for predicting Kcr sites on nonhistone proteins, and accordingly, it achieved a better performance compared to the other predictor that was designed for predicting the Kcr sites on mixed histone and nonhistone proteins.
Implementation of the nhKcr web server
As an implementation of the proposed methodology, a user-friendly web server has been developed and made publicly accessible at http://nhKcr.erc.monash.edu/. The screen copy of the server user interface together with an example prediction output is displayed in Figure 6. The web server is maintained by the cloud computing facility supported by the eResearch Centre at Monash University and is equipped with 16 cores, 64 GB memory and a 2 TB hard disk. It was developed on the open-source web platform Linux–Apache–MySQL–PHP (LAMP) and has been tested using several commonly used web browsers, including Internet Explorer (≥v.7.0), Microsoft Edge (Microsoft Corp.), Mozilla Firefox, Google Chrome and Safari. The server uses the optimal model to identify Kcr sites from the protein sequences for the submitted tasks. At the index webpage, users can input one or more protein sequences (a maximum number of 100 sequences is allowed for each submission) in the FASTA format in the textbox. To control FP predictions, two different cutoff values are provided (i.e. ‘HIGH’ =90% Sp and ‘MEDIUM’ = 80% Sp). The prediction results can be directly visualized within the web server containing detailed information regarding the positions of predicted modification sites, scores and the prediction results. The generated prediction results can also be downloaded in plain text format for users’ follow-up analysis. Moreover, the curated benchmark datasets and the independent test dataset in this study can be downloaded from the nhKcr web server as well.
Figure 6 .
Screenshots of the developed nhKcr web server for prediction Kcr sites on human nonhistone proteins. (A) The interface of the nhKcr web server, (B) the example prediction output of the web server.
Discussion
An increasing number of deep learning-based methods have been recently developed for predicting PTM sites. For instance, Baisya et al. [56] used a deep learning architecture called DeepPTM for predicting histone protein PTMs from the transcription factor binding data and the primary DNA sequence. DeepPPSite [57] is another deep learning-based model using a stacked long short-term memory recurrent network for predicting phosphorylation sites. Thapa et al. [58] developed DeepSuccinylSite that combined deep learning and embedding to identify the succinylation sites in proteins based on their primary structure. RBPsuite [59], employed two deep learning-based methods iDeepS and CRIP for RNA-protein binding sites prediction. Hong et al. [60] utilized a protein encoding strategy together with a deep learning algorithm to control the false discovery rate for the functional annotation of protein sequence. These studies show that deep learning methods are suitable for PTM prediction problems and can lead to favorable performance compared with traditional machine learning methods.
Up until now, a variety of predictors have been developed for Kcr sites’ prediction. However, none of them were developed specifically for predicting nonhistone Kcr sites. Recently, Kcr substrates have been expanded to nonhistone proteins. However, due to the lack of high-quality pan-antibodies for Kcr in nonhistone proteins, large-scale characterization of Kcr sites at the proteomic level remains a challenge. In this work, we have developed a novel bioinformatics tool called nhKcr which is based on an improved CNN method, termed CNNrgb, for the effective prediction of Kcr sites on human nonhistone proteins. To the best of our knowledge, nhKcr is the first predictor that has been developed specifically for the prediction of crotonylation sites on nonhistone proteins in mammals. We have compared its predictive performance with different encoding schemes and traditional machine learning methods. Benchmarking results on the independent test dataset show that nhKcr achieved the best performance compared to the state-of-the-art predictor, Deep-Kcr, which could predict Kcr sites in both histone proteins and nonhistone proteins based on a typical CNN model. The results also demonstrate that our method exhibited a great superiority in identifying the Kcr sites in nonhistone proteins.
Due to the limited availability of Kcr site data on histone proteins, the predictors such as CKSAAP CrotSite, iKcr-PseEns and LightGBM-CroSite could only use small datasets, including 169 positive and 847 negative samples, to train and test their models, resulting in unsatisfactory prediction performance when tested on the independent test dataset. Although iCrotoK-PseAAC expanded the dataset by adding the Kcr sites on nonhistone proteins, there were only 378 positive samples and 500 negative samples for training and testing, which is still insufficient to develop a robust and accurate predictor. The state-of-the-art predictor, Deep-Kcr, was developed based on a large dataset including 9964 positive and 9964 negative samples and achieved a more competitive performance than the existing predictors. However, it was not designed particularly for predicting the Kcr sites on nonhistone proteins and hence did not achieve a satisfactory performance.
Although our method has achieved a satisfactory performance in Kcr site prediction on nonhistone proteins, there are several ways for further improving the prediction performance. For instance, data redundancy is an important issue to consider prior to model construction. Meanwhile, the window size and the ratio of positive to negative samples are other important aspects that need to be considered when training a robust predictor. Moreover, ensemble learning methods might be useful for improving the prediction performance. For example, ZincExplorer [61] is a zinc-binding site predictor, which integrates the outputs from three individual predictors (i.e. an SVM predictor, a cluster-based predictor and a template-based predictor). In addition, some predictors employed different methods for different purposes. For instance, DeepSVM-fold [62] is a powerful tool for protein fold recognition. It utilized deep learning networks to effectively extract features and generated a new feature vector, which was then fed into a SVM algorithm to construct the predictor. These strategies suggest that the prediction performance may be significantly improved by introducing the ensemble learning strategy via the integration of the outputs of multiple predictors in future work. In addition, a biological sequence is analogous to a sentence composed of language words; as such, emerging natural language processing (NLP)-based models have a great promise to be transformed to sequence-based models using bidirectional encoder representations from transformers (BERT) [63] and transformers [64] or word embedding-based methods, such as Word2Vec [65, 66], ELMo [67] and FastText [68, 69]. Furthermore, CNNs are mostly used in image classification and pattern recognition. Therefore, it would be also of particular interest to combine CNNs and NLP-based models in future studies and to examine the possibility of further improving the prediction performance of crotonylation sites.
Key Points
As an important type of post-translation modification, Kcr can occur on both histone and nonhistone proteins; however, there is currently no predictor specifically developed for predicting Kcr sites in human nonhistone proteins.
We present nhKcr, a new bioinformatics tool based on a novel deep learning method termed CNNrgb to improve the prediction performance of Kcr site prediction in nonhistone proteins.
Comparative analysis of nhKcr and different encoding schemes as well as different machine learning algorithms shows its superior prediction performance.
Benchmarking analysis on the independent test shows that nhKcr outperformed the state-of-the-art predictor Deep-Kcr for Kcr site prediction on nonhistone proteins.
A user-friendly web server of nhKcr is publicly available at http://nhKcr.erc.monash.edu.au/.
Supplementary Material
Funding
Y.-Z.C’s work was supported by the National Natural Science Foundation of China (No. 81772843). J.S.’s work was supported by grants from the National Health and Medical Research Council of Australia (NHMRC) (1092262), the Australian Research Council (ARC) (LP110200333 and DP120104460), the National Institute of Allergy and Infectious Diseases of the National Institutes of Health (R01 AI111965), a major inter-disciplinary research (IDR) project awarded by Monash University.
Conflict of interest
The authors declare that they have no competing interests.
Yong-Zi Chen is currently an assistant researcher in the Laboratory of Tumor Cell Biology, Tianjin Medical University Cancer Institute and Hospital. She conducted her postdoctoral research at the Moffitt Cancer Center, USA. Her research interests are protein bioinformatics, drug model development and biomarker discovery.
Zhuo-Zhi Wang received his Bachelor of Veterinary Medicine from the Inner Mongolia University for Nationalities, China. He is currently a master student in the School of Biomedical Engineering, Tianjin Medical University, China. His research interests are bioinformatics, cancer diagnosis and biomarker discovery.
Yanan Wang received his Master degree in Control Science and Technology from the Shanghai Jiao Tong University, China. He is currently a PhD candidate in the Biomedicine Discovery Institute and the Department of Biochemistry and Molecular Biology at the Monash University, Australia. His research interests are bioinformatics, machine learning and data mining.
Guoguang Ying is currently a professor and director of the Laboratory of Tumor Cell Biology in Tianjin Medical University Cancer Institute and Hospital. His research interests are cancer bioinformatics, biomarker discovery and cancer treatment research.
Zhen Chen is a professor at the Collaborative Innovation Center of Henan Grain Crops, Henan Agricultural University, China. His research interests include protein bioinformatics, machine learning, and analysis of next-generation sequencing data.
Jiangning Song is an associate professor and group leader in the Monash Biomedicine Discovery Institute, Monash University, Australia. He is a member of the Monash Centre for Data Science, Faculty of Information Technology, Monash University. His research interests include bioinformatics, computational biomedicine, machine learning and pattern recognition.
Availability and implementation
To facilitate the cost-effective identification of Kcr sites from the protein sequences and widespread use by the research community, we have provided a user-friendly web server that is freely available at http://nhkcr.erc.monash.edu/. All the data utilized for training and testing nhKcr can be downloaded at this website.
References
- 1. Tan M, Luo H, Lee S, et al. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 2011;146:1016–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Montellier E, Rousseaux S, Zhao Y, et al. Histone crotonylation specifically marks the haploid male germ cell gene expression program: post-meiotic male-specific gene expression. Bioessays 2012;34:187–93. [DOI] [PubMed] [Google Scholar]
- 3. Sabari BR, Tang Z, Huang H, et al. Intracellular crotonyl-CoA stimulates transcription through p300-catalyzed histone crotonylation. Mol Cell 2015;58:203–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Huang H, Wang DL, Zhao Y. Quantitative crotonylome analysis expands the roles of p300 in the regulation of lysine crotonylation pathway. Proteomics 2018;18:e1700230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wei W, Mao A, Tang B, et al. Large-scale identification of protein crotonylation reveals its role in multiple cellular functions. J Proteome Res 2017;16:1743–52. [DOI] [PubMed] [Google Scholar]
- 6. Wu Q, Li W, Wang C, et al. Ultradeep lysine crotonylome reveals the crotonylation enhancement on both histones and nonhistone proteins by SAHA treatment. J Proteome Res 2017;16:3664–71. [DOI] [PubMed] [Google Scholar]
- 7. Xu W, Wan J, Zhan J, et al. Global profiling of crotonylation on non-histone proteins. Cell Res 2017;27:946–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yu H, Bu C, Liu Y, et al. Global crotonylome reveals CDYL-regulated RPA1 crotonylation in homologous recombination-mediated DNA repair. Sci Adv 2020;6:eaay4697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li Y, Sabari BR, Panchenko T, et al. Molecular coupling of histone crotonylation and active transcription by AF9 YEATS domain. Mol Cell 2016;62:181–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Xiong X, Panchenko T, Yang S, et al. Selective recognition of histone crotonylation by double PHD fingers of MOZ and DPF2. Nat Chem Biol 2016;12:1111–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Andrews FH, Shinsky SA, Shanle EK, et al. The Taf14 YEATS domain is a reader of histone crotonylation. Nat Chem Biol 2016;12:396–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Huang GH, Zeng WFA. Discrete hidden Markov model for detecting histone crotonyllysine sites, match-communications in mathematical and in computer. Chemistry 2016;75:717–30. [Google Scholar]
- 13. Qiu WR, Sun BQ, Tang H, et al. Identify and analysis crotonylation sites in histone by using support vector machines. Artif Intell Med 2017;83:75–81. [DOI] [PubMed] [Google Scholar]
- 14. Malebary SJ, Rehman MSU, Khan YD. iCrotoK-PseAAC: identify lysine crotonylation sites by blending position relative statistical features according to the Chou’s 5-step rule. PLoS One 2019;14:e0223993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ju Z, He JJ. Prediction of lysine crotonylation sites by incorporating the composition of k-spaced amino acid pairs into Chou’s general PseAAC. J Mol Graph Model 2017;77:200–4. [DOI] [PubMed] [Google Scholar]
- 16. Qiu WR, Sun BQ, Xiao X, et al. iKcr-PseEns: identify lysine crotonylation sites in histone proteins with pseudo components and ensemble classifier. Genomics 2018;110:239–46. [DOI] [PubMed] [Google Scholar]
- 17. Lv H, Dao FY, Guan ZX, et al. Deep-Kcr: accurate detection of lysine crotonylation sites using deep learning method. Brief Bioinform 2020. doi: 10.1093/bib/bbaa255. [DOI] [PubMed] [Google Scholar]
- 18. Dimmer EC, Huntley RP, Alam-Faruque Y, et al. The UniProt-GO annotation database in 2011. Nucleic Acids Res 2012;40:D565–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Huang Y, Niu B, Gao Y, et al. CD-HIT suite: a web server for clustering and comparing biological sequences. Bioinformatics 2010;26:680–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chen Z, Zhao P, Li C, et al. iLearnPlus: a comprehensive and automated machine-learning platform for nucleic acid and protein sequence analysis, prediction and visualization. Nucleic Acids Res 2021. doi: 10.1093/nar/gkab122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chen YZ, Tang YR, Sheng ZY, et al. Prediction of mucin-type O-glycosylation sites in mammalian proteins using the composition of k-spaced amino acid pairs. BMC Bioinformatics 2008;9:101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kao HJ, Nguyen VN, Huang KY, et al. SuccSite: incorporating amino acid composition and informative k-spaced amino acid pairs to identify protein succinylation sites. Genomics Proteomics Bioinformatics 2020;18(2):208–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chen Z, He N, Huang Y, et al. Integration of a deep learning classifier with a random forest approach for predicting malonylation sites. Genomics Proteomics Bioinformatics 2018;16:451–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Meher PK, Sahu TK, Banchariya A, et al. DIRProt: a computational approach for discriminating insecticide resistant proteins from non-resistant proteins. BMC Bioinformatics 2017;18:190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen Z, Zhao P, Li F, et al. PROSPECT: a web server for predicting protein histidine phosphorylation sites. J Bioinform Comput Biol 2020;18:2050018. [DOI] [PubMed] [Google Scholar]
- 26. Kawashima S, Kanehisa M. AAindex: amino acid index database. Nucleic Acids Res 2000;28:374–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sandberg M, Eriksson L, Jonsson J, et al. New chemical descriptors relevant for the design of biologically active peptides. A multivariate characterization of 87 amino acids. J Med Chem 1998;41:2481–91. [DOI] [PubMed] [Google Scholar]
- 28. Li A, Wang L, Shi Y, et al. Phosphorylation site prediction with a modified k-nearest neighbor algorithm and BLOSUM62 matrix. Conf Proc IEEE Eng Med Biol Soc 2005;2005:6075–8. [DOI] [PubMed] [Google Scholar]
- 29. Song J, Burrage K, Yuan Z, et al. Prediction of cis/trans isomerization in proteins using PSI-BLAST profiles and secondary structure information. BMC Bioinformatics 2006;7:124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Song J, Tan H, Shen H, et al. Cascleave: towards more accurate prediction of caspase substrate cleavage sites. Bioinformatics 2010;26:752–60. [DOI] [PubMed] [Google Scholar]
- 31. Song J, Tan H, Perry AJ, et al. PROSPER: an integrated feature-based tool for predicting protease substrate cleavage sites. PLoS One 2012;7:e50300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen Z, Zhou Y, Song J, et al. hCKSAAP_UbSite: improved prediction of human ubiquitination sites by exploiting amino acid pattern and properties. Biochim Biophys Acta 2013;1834:1461–7. [DOI] [PubMed] [Google Scholar]
- 33. Chen Z, Chen YZ, Wang XF, et al. Prediction of ubiquitination sites by using the composition of k-spaced amino acid pairs. PLoS One 2011;6:e22930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mosharaf MP, Hassan MM, Ahmed FF, et al. Computational prediction of protein ubiquitination sites mapping on Arabidopsis thaliana. Comput Biol Chem 2020;85:107238. [DOI] [PubMed] [Google Scholar]
- 35. Ding Y, Cai Y, Zhang G, et al. The influence of dipeptide composition on protein thermostability. FEBS Lett 2004;569:284–8. [DOI] [PubMed] [Google Scholar]
- 36. Meher PK, Sahu TK, Gahoi S, et al. Ir-HSP: improved recognition of heat shock proteins, their families and sub-types based on g-spaced di-peptide features and support vector machine. Front Genet 2017;8:235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chen YZ, Chen Z, Gong YA, et al. SUMOhydro: a novel method for the prediction of sumoylation sites based on hydrophobic properties. PLoS One 2012;7:e39195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wen YT, Lei HJ, You ZH, et al. Prediction of protein-protein interactions by label propagation with protein evolutionary and chemical information derived from heterogeneous network. J Theor Biol 2017;430:9–20. [DOI] [PubMed] [Google Scholar]
- 39. Liu Y, Yu Z, Chen C, et al. Prediction of protein crotonylation sites through LightGBM classifier based on SMOTE and elastic net. Anal Biochem 2020;609:113903. [DOI] [PubMed] [Google Scholar]
- 40. Allam A, Krauthammer M. PySeqLab: an open source python package for sequence labeling and segmentation. Bioinformatics 2017;33:3497–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yang S, Fu C, Lian X, et al. Understanding human-virus protein-protein interactions using a human protein complex-based analysis framework. mSystems 2019;4:e00303–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Diaz AA, Tomba E, Lennarson R, et al. Prediction of protein solubility in Escherichia coli using logistic regression. Biotechnol Bioeng 2010;105:374–83. [DOI] [PubMed] [Google Scholar]
- 43. Zou Q, Xing P, Wei L, et al. Gene2vec: gene subsequence embedding for prediction of mammalian N (6)-methyladenosine sites from mRNA. RNA 2019;25:205–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sharma A, Singh B. AE-LGBM: sequence-based novel approach to detect interacting protein pairs via ensemble of autoencoder and LightGBM. Comput Biol Med 2020;125:103964. [DOI] [PubMed] [Google Scholar]
- 45. Deng L, Pan J, Xu X, et al. PDRLGB: precise DNA-binding residue prediction using a light gradient boosting machine. BMC Bioinformatics 2018;19:522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chen T, Guestrin C. XGBoost: a scalable tree boosting system. In: Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining. San Francisco, California, USA: ACM, 785–94. [Google Scholar]
- 47. Kiranyaz S, Ince T, Pulkkinen J, et al. Classification and retrieval on macroinvertebrate image databases. Comput Biol Med 2011;41:463–72. [DOI] [PubMed] [Google Scholar]
- 48. Shi Q, Chen W, Huang S, et al. Deep learning for mining protein data. Brief Bioinform 2021;22:194–218. [DOI] [PubMed] [Google Scholar]
- 49. Yu J, Shi S, Zhang F, et al. PredGly: predicting lysine glycation sites for Homo sapiens based on XGboost feature optimization. Bioinformatics 2019;35:2749–56. [DOI] [PubMed] [Google Scholar]
- 50. Chen C, Zhang Q, Yu B, et al. Improving protein-protein interactions prediction accuracy using XGBoost feature selection and stacked ensemble classifier. Comput Biol Med 2020;123:103899. [DOI] [PubMed] [Google Scholar]
- 51. Pang L, Wang J, Zhao L, et al. A novel protein subcellular localization method with CNN-XGBoost model for Alzheimer’s disease. Front Genet 2018;9:751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Nair V, Hinton GE. Rectified linear units improve restricted boltzmann machines. In: ICML'10, University of Toronto, Canada. 2010, 807–14. [Google Scholar]
- 53. Paszke A, Gross S, Francisco F, et al. PyTorch: AnImperative Style, High-Performance Deep Learning Library, 33rd Conference on Neural Information Processing System, Canada, 2019, 8024–35.
- 54. Chen Z, Liu X, Li F, et al. Large-scale comparative assessment of computational predictors for lysine post-translational modification sites. Brief Bioinform 2019;20:2267–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. O'Shea JP, Chou MF, Quader SA, et al. pLogo: a probabilistic approach to visualizing sequence motifs. Nat Methods 2013;10:1211–2. [DOI] [PubMed] [Google Scholar]
- 56. Baisya DR, Lonardi S. Prediction of histone post-translational modifications using deep learning. Bioinformatics 2020;36:5610–7. [DOI] [PubMed] [Google Scholar]
- 57. Ahmed S, Kabir M, Arif M, et al. DeepPPSite: a deep learning-based model for analysis and prediction of phosphorylation sites using efficient sequence information. Anal Biochem 2021;612:113955. [DOI] [PubMed] [Google Scholar]
- 58. Thapa N, Chaudhari M, McManus S, et al. DeepSuccinylSite: a deep learning based approach for protein succinylation site prediction. BMC Bioinformatics 2020;21:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Pan X, Fang Y, Li X, et al. RBPsuite: RNA-protein binding sites prediction suite based on deep learning. BMC Genomics 2020;21:884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hong J, Luo Y, Zhang Y, et al. Protein functional annotation of simultaneously improved stability, accuracy and false discovery rate achieved by a sequence-based deep learning. Brief Bioinform 2020;21:1437–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Chen Z, Wang Y, Zhai YF, et al. ZincExplorer: an accurate hybrid method to improve the prediction of zinc-binding sites from protein sequences. Mol Biosyst 2013;9:2213–22. [DOI] [PubMed] [Google Scholar]
- 62. Liu B, Li CC, Yan K. DeepSVM-fold: protein fold recognition by combining support vector machines and pairwise sequence similarity scores generated by deep learning networks. Brief Bioinform 2020;21:1733–41. [DOI] [PubMed] [Google Scholar]
- 63. Charoenkwan P, Nantasenamat C, Hasan MM, et al. BERT4Bitter: a bidirectional encoder representations from transformers (BERT)-based model for improving the prediction of bitter peptides. Bioinformatics 2021. doi: 10.1093/bioinformatics/btab133. [DOI] [PubMed] [Google Scholar]
- 64. Chen L, Tan X, Wang D, et al. TransformerCPI: improving compound-protein interaction prediction by sequence-based deep learning with self-attention mechanism and label reversal experiments. Bioinformatics 2020;36:4406–14. [DOI] [PubMed] [Google Scholar]
- 65. Smaili FZ, Gao X, Hoehndorf R. OPA2Vec: combining formal and informal content of biomedical ontologies to improve similarity-based prediction. Bioinformatics 2019;35:2133–40. [DOI] [PubMed] [Google Scholar]
- 66. Yi HC, You ZH, Cheng L, et al. Learning distributed representations of RNA and protein sequences and its application for predicting lncRNA-protein interactions. Comput Struct Biotechnol J 2020;18:20–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Heinzinger M, Elnaggar A, Wang Y, et al. Modeling aspects of the language of life through transfer-learning protein sequences. BMC Bioinformatics 2019;20:723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Le NQK, Huynh TT. Identifying SNAREs by incorporating deep learning architecture and amino acid embedding representation. Front Physiol 2019;10:1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Asgari E, McHardy AC, Mofrad MRK. Probabilistic variable-length segmentation of protein sequences for discriminative motif discovery (DiMotif) and sequence embedding (ProtVecX). Sci Rep 2019;9:3577. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.