

Abstract
Iron deficiency impairs skeletal muscle metabolism. The underlying mechanisms are incompletely characterised, but animal and human experiments suggest the involvement of signalling pathways co-dependent upon oxygen and iron availability, including the pathway associated with hypoxia-inducible factor (HIF). We performed a prospective, case–control, clinical physiology study to explore the effects of iron deficiency on human metabolism, using exercise as a stressor. Thirteen iron-deficient (ID) individuals and thirteen iron-replete (IR) control participants each underwent 31P-magnetic resonance spectroscopy of exercising calf muscle to investigate differences in oxidative phosphorylation, followed by whole-body cardiopulmonary exercise testing. Thereafter, individuals were given an intravenous (IV) infusion, randomised to either iron or saline, and the assessments repeated ~ 1 week later. Neither baseline iron status nor IV iron significantly influenced high-energy phosphate metabolism. During submaximal cardiopulmonary exercise, the rate of decline in blood lactate concentration was diminished in the ID group (P = 0.005). Intravenous iron corrected this abnormality. Furthermore, IV iron increased lactate threshold during maximal cardiopulmonary exercise by ~ 10%, regardless of baseline iron status. These findings demonstrate abnormal whole-body energy metabolism in iron-deficient but otherwise healthy humans. Iron deficiency promotes a more glycolytic phenotype without having a detectable effect on mitochondrial bioenergetics.
Subject terms: Energy metabolism, Respiration, Mitochondria, Skeletal muscle
Introduction
Skeletal muscle metabolism is responsible for as much as 25% of resting oxygen consumption (o2) in healthy young adult humans, and o2 may increase 10–15 times above resting values during whole body exercise, even in untrained individuals1. Exercise thus places very considerable demands on those physiological processes concerned with pulmonary uptake of oxygen, its delivery to tissues, and its ultimate fate as the terminal electron acceptor in mitochondrial oxidative phosphorylation. Importantly, iron has indispensable roles in each of these processes.
Many previous studies have addressed the impact of iron deficiency on human exercise capacity. For example, in subjects with profound iron-deficiency anaemia (IDA), maximal treadmill exercise time increased significantly with intravenous (IV) iron supplementation, and post-exercise venous blood lactate concentrations remained similar with successive experiments despite higher workloads2. In iron-deficient (ID) female athletes, whilst oral iron supplementation had no effect on maximal workload on a bicycle ergometer, maximal blood lactate levels decreased following iron administration3.
However, there are major difficulties in interpreting the findings of such studies, which have almost invariably been performed in athletic individuals. Habitual exercise brings about a complex interplay of inflammatory and hypoxic stresses leading to alterations in the master iron-regulatory hormone hepcidin4. The result is that the pathophysiology of iron deficiency in endurance athletes may differ significantly from that of absolute iron deficiency in non-athletes5. Moreover, the use of oral iron over a protracted period inevitably leads to a rise in haemoglobin (Hb) concentration, which is a powerful confounding factor.
The challenge of separating the effects of iron deficiency from the consequences of coexisting anaemia has long been recognised6,7. Although a meta-analysis previously reported iron supplementation to be beneficial for improving maximal oxygen consumption (o2max) in iron-deficient non-anaemic athletes8, iron supplementation predictably increased Hb concentration in the studies included. Given the strong correlation between red cell mass and o2max in health, iron supplementation will inevitably increase o2max over time by promoting a rise in Hb concentration. In a contemporary study where IV iron was administered to elite athletes without precipitating a rise in Hb concentration, no effect on o2max was seen9.
It is possible to limit the confounding effects of Hb concentration using animal models. Isovolaemic transfusion experiments in rodents indicate that iron deficiency: (i) compromises work capacity during whole-body exercise10; (ii) promotes excessive blood lactate accumulation11; and (iii) leads to deranged skeletal muscle oxidative phosphorylation12. Whilst these and other studies7,13,14 indicate that IDA limits oxygen delivery to exercising muscle, they leave unanswered the question of how tissue iron deficiency per se disrupts oxidative metabolism.
A remarkably similar metabolic phenotype is evident in humans with a rare congenital disease of oxygen sensing, Chuvash polycythaemia (CP)15. Adult humans with CP show not only the diminished aerobic exercise capacity and hyperlactataemia of ID rodents, but also similarly disturbed skeletal muscle oxidative phosphorylation. Many of the features of CP result from dysregulation of the hypoxia-inducible factor (HIF) transcription factor pathway16, which regulates integrated physiological responses to changes in oxygen availability, including erythropoietin expression17. The oxygen sensors are HIF-hydroxylase enzymes, which utilise a single ferrous ion coordinated at their active sites. These enzymes hydroxylate the HIF-α subunit at either of two prolyl residues, targeting it for proteasomal degradation, or at an asparaginyl residue, reducing its interaction with a transcriptional coactivator protein18. As would be anticipated, iron availability thus plays a critical role in modulating physiological responses to hypoxia19.
Taking all this together, we hypothesised that clinical iron deficiency would disturb human metabolism in a manner similar to that seen both in rodent models of iron deficiency and in patients with CP. The present study compared healthy iron-replete (IR) participants with their ID – but not frankly anaemic – counterparts. Using an established approach15, we adopted a design involving: (i) a case–control approach for baseline comparisons, employing combined assessment of whole-body cardiopulmonary physiology and small muscle mass high-energy phosphate metabolism; and (ii) repeated assessment a short interval after randomisation to IV iron or saline control, before any change in Hb concentration could occur.
Results
Baseline characteristics of iron-deficient and iron-replete groups
Baseline characteristics of the participants are given in Table 1. Mean serum ferritin in the ID group was 8.3 μg/L, and transferrin saturation (TSat) 10.6%; plasma soluble transferrin receptor (sTfR) values in the ID group were approximately twice those of IR participants, and hepcidin, the major hormone regulating iron uptake and distribution, was very heavily suppressed in the ID group. These values confirm absolute iron deficiency20.
Table 1.
Participant characteristics on enrolment.
| Characteristic | ID Group (n = 13) | IR Group (n = 13) | P-value |
|---|---|---|---|
| Sex, M:F | 1:12 | 1:12 | |
| Age, years | 23 (21.5–38) | 24 (22–25.5) | 0.59 |
| BMI, kg/m2 | 21.8 (20.8–24.1) | 21.1 (20.7–24.1) | 1.0 |
| Resting Spo2, % | 98.8 ± 0.9 | 98.5 ± 1.1 | 0.45 |
| Systolic BP, mmHg | 119 ± 11 | 119 ± 9 | 0.99 |
| Diastolic BP, mmHg | 73 ± 10 | 79 ± 8 | 0.08 |
| FEV1, % predicted | 106 ± 12 | 104 ± 9 | 0.75 |
| Exercise, h/week | 5.0 (2.0–7.5) | 4.0 (1.0–8.5) | 0.70 |
| Plasma CRP, mg/L | 0.4 (0.3–2.6) | 0.5 (0.3–1.6) | 0.96 |
| Serum ferritin, μg/L | 8.3 ± 3.1 | 58.0 ± 38.2 | N/A |
| Serum TSat, % | 10.6 ± 3.9 | 35.2 ± 7.6 | N/A |
| Serum iron, μmol/L | 8.4 ± 3.2 | 22.0 ± 3.6 | < 0.001 |
| Serum transferrin, g/L | 3.6 ± 0.6 | 2.9 ± 0.5 | 0.003 |
| Plasma sTfR, nmol/L | 38.4 (32.7–50.9) | 21.8 (18.4–25.0) | < 0.001 |
| Plasma hepcidin, μg/L | 2.0 (1.3–2.4) | 13.9 (9.9–29.5) | < 0.001 |
| Haemoglobin, g/dl | 12.7 (11.5–13.3) | 13.6 (13.5–14.0) | < 0.001 |
| Haematocrit, % | 39.7 ± 3.2 | 42.0 ± 1.7 | 0.028 |
| Mean cell volume, fl | 86.9 (83.0–91.3) | 92.4 (89.9–94.1) | 0.010 |
For normally distributed data, comparisons are by t-test and values are means ± SD. For non-normally distributed data, comparisons are by Mann Whitney U test and values are medians (IQR). Statistically significant P-values appear in bold. ID, iron deficient; IR, iron replete; BMI, body mass index; Spo2, peripheral oxyhaemoglobin saturation; FEV1, forced expiratory volume in one second; CRP, C-reactive protein; TSat, transferrin saturation; sTfR, soluble transferrin receptor.
Effect of iron infusion on laboratory parameters at the second study visit
Table 2 shows the changes in laboratory parameters between study visits, which were conducted approximately a week apart. There was no significant change in Hb concentration between study visits. Participants randomised to receive iron showed the expected significant increases in ferritin, serum iron, TSat and hepcidin; conversely, transferrin and sTfR fell significantly, also as expected. However, although the direction of effect following IV administration of iron was the same in ID and IR groups for each of these parameters, ID subjects did not attain the same absolute values in in any of the indices compared with IR subjects. Iron administration influenced circulating erythropoietin levels in ID participants, but not IR controls (P = 0.015 for interaction between study visit, baseline iron status and intervention).
Table 2.
Haematological and iron parameters over the course of the study.
| Iron status | Iron deficient (n = 13) | Iron replete (n = 13) | P-value for interactions (RM-ANOVA) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Intervention | Iron (n = 7) | Saline (n = 6) | Iron (n = 7) | Saline (n = 6) | Visit & intervention | Iron status, visit & intervention | ||||
| Visit | 1 | 2 | 1 | 2 | 1 | 2 | 1 | 2 | ||
| Hb, g/dl | 13.1 ± 1.1 | 13.0 ± 1.3 | 12.7 ± 0.6 | 12.5 ± 0.7 | 14.3 ± 0.7 | 13.7 ± 0.9 | 14.1 ± 0.4 | 13.9 ± 0.6 | 0.66 | 0.43 |
| MCV, fl | 84.4 ± 7.6 | 87.1 ± 6.8 | 86.0 ± 3.9 | 86.0 ± 4.3 | 91.9 ± 5.7 | 91.4 ± 2.6 | 90.9 ± 3.2 | 91.3 ± 2.3 | 0.47 | 0.09 |
| Serum ferritin, μg/L | 7.9 ± 3.5 | 561 ± 433 | 6.9 ± 2.4 | 10.6 ± 2.8 | 56.3 ± 35.5 | 840 ± 331 | 29.0 ± 14.3 | 38.7 ± 16.2 | < 0.001 | 0.32 |
| Serum TSat, % | 12.3 ± 4.9 | 37.9 ± 11.2 | 13.7 ± 5.5 | 11.0 ± 2.8 | 29.0 ± 8.8 | 62.1 ± 20.1 | 23.0 ± 5.5 | 24.3 ± 12.5 | < 0.001 | 0.73 |
| Serum iron, μmol/L | 9.3 ± 2.6 | 24.0 ± 9.1 | 11.1 ± 4.8 | 8.7 ± 2.0 | 17.8 ± 4.8 | 31.2 ± 10.4 | 16.2 ± 4.3 | 16.5 ± 8.2 | < 0.001 | 0.54 |
| Serum transferrin, g/L | 3.61 ± 0.77 | 2.90 ± 0.48 | 3.65 ± 0.49 | 3.68 ± 0.44 | 2.83 ± 0.34 | 2.32 ± 0.26 | 3.17 ± 0.40 | 3.13 ± 0.39 | < 0.001 | 0.27 |
| Plasma sTfR, nmol/L | 40.3 ± 12.0 | 33.4 ± 7.6 | 41.8 ± 13.1 | 40.9 ± 14.0 | 24.3 ± 3.5 | 20.7 ± 4.2 | 22.5 ± 2.5 | 21.7 ± 2.0 | 0.007 | 0.30 |
| Serum Epo, mIU/ml | 17.7 ± 9.2 | 13.6 ± 3.9 | 13.8 ± 4.7 | 21.3 ± 7.8 | 8.0 ± 2.8 | 7.6 ± 4.0 | 7.7 ± 2.4 | 7.2 ± 2.1 | 0.016 | 0.015 |
| Plasma hepcidin, μg/L | 2.1 ± 1.2 | 38.5 ± 28.9 | 1.5 ± 0.5 | 2.1 ± 1.0 | 17.2 ± 22.6 | 85.5 ± 54.6 | 7.3 ± 3.3 | 14.6 ± 14.7 | < 0.001 | 0.30 |
Values are means ± SD. Statistically significant P-values appear in bold. RM-ANOVA, repeated measures analysis of variance; MCV, mean cell volume; TSat, transferrin saturation; sTfR, soluble transferrin receptor; Epo, erythropoietin.
Skeletal muscle oxidative phosphorylation
High-energy phosphate metabolism and intracellular pH changes during the first study visit are illustrated in Fig. 1. An analysis of the effect of iron status, workload, and iron infusion based on monoexponential fitting of data from the recovery period after each bout of exercise is given in Table 3. The only significant effect detected was of workload, with end-exercise PCr and pH tending to fall with successive bouts of small-muscle-mass exercise, both as expected. No effect was evident of either baseline iron status or IV iron administration.
Figure 1.

31P-MRS data from first study visit. [PCr] is expressed as a fraction of the mean value during the initial 2-min rest period. Data for ID participants appear as white circles; those for IR participants, black circles. Solid black bars indicate 5-min exercise periods. All participants completed the 3-W exercise bout. Subsequently, several participants ceased exercise prematurely due to fatigue: during the 4-W bout, one IR participant after 1050 s; during the 5-W bout, one ID participant after 1820s, one IR participant after 1720s, and another IR participant after 1775s. For illustrative purposes, recovery data for these participants are shifted to align with cessation of exercise in the other participants; an ‘early recovery’ artefact is thus apparent in the IR group near the end of the 4-W and 5-W bouts. The rest periods include data for all participants. Values are 30-s means; error bars show SE.
Table 3.
Parameters derived from monoexponential fitting of PCr recovery data.
| Parameter | Iron status | Iron deficient (n = 13) | Iron replete (n = 13) | P-value (RM-ANOVA) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Intervention | Iron (n = 7) | Saline (n = 6) | Iron (n = 7) | Saline (n = 6) | ||||||
| Visit | 1 | 2 | 1 | 2 | 1 | 2 | 1 | 2 | ||
| [PCr] | 3 W | 0.72 ± 0.11 | 0.74 ± 0.10 | 0.57 ± 0.15 | 0.62 ± 0.23 | 0.74 ± 0.10 | 0.73 ± 0.09 | 0.60 ± 0.12 | 0.53 ± 0.19 |
Iron status = 0.99 Workload < 0.001 Visit & intervention = 0.60 Iron status, visit & intervention = 0.48 |
| 4 W | 0.59 ± 0.15 | 0.61 ± 0.18 | 0.45 ± 0.17 | 0.47 ± 0.22 | 0.68 ± 0.10 | 0.63 ± 0.12 | 0.44 ± 0.19 | 0.44 ± 0.22 | ||
| 5 W | 0.50 ± 0.20 | 0.52 ± 0.21 | 0.38 ± 0.19 | 0.38 ± 0.24 | 0.50 ± 0.14 | 0.50 ± 0.18 | 0.44 ± 0.20 | 0.32 ± 0.23 | ||
| pH | 3 W | 6.99 ± 0.13 | 7.00 ± 0.06 | 6.95 ± 0.16 | 6.91 ± 0.25 | 7.04 ± 0.03 | 7.04 ± 0.04 | 6.92 ± 0.11 | 6.98 ± 0.10 |
Iron status = 0.75 Workload < 0.001 Visit & intervention = 0.90 Iron status, visit & intervention = 0.37 |
| 4 W | 6.92 ± 0.11 | 6.94 ± 0.11 | 6.90 ± 0.15 | 6.86 ± 0.24 | 6.98 ± 0.10 | 6.97 ± 0.11 | 6.86 ± 0.15 | 6.88 ± 0.07 | ||
| 5 W | 6.90 ± 0.09 | 6.90 ± 0.14 | 6.88 ± 0.18 | 6.87 ± 0.24 | 6.95 ± 0.11 | 6.90 ± 0.14 | 6.87 ± 0.14 | 6.86 ± 0.09 | ||
| τ | 3 W | 35 ± 14 | 32 ± 4 | 40 ± 14 | 51 ± 29 | 32 ± 16 | 29 ± 8 | 43 ± 16 | 49 ± 23 |
Iron status = 0.79 Workload = 0.16 Visit & intervention = 0.14 Iron status, visit & intervention = 0.34 |
| 4 W | 36 ± 13 | 35 ± 6 | 41 ± 15 | 52 ± 31 | 43 ± 16 | 33 ± 11 | 43 ± 16 | 46 ± 15 | ||
| 5 W | 40 ± 15 | 35 ± 9 | 45 ± 19 | 50 ± 19 | 45 ± 15 | 41 ± 14 | 45 ± 15 | 47 ± 19 | ||
| Qmax | 3 W | 0.33 ± 0.08 | 0.37 ± 0.18 | 0.41 ± 0.17 | 0.25 ± 0.07 | 0.41 ± 0.24 | 0.44 ± 0.17 | 0.31 ± 0.03 | 0.49 ± 0.37 |
Iron status = 0.32 Workload < 0.001 Visit & intervention = 0.16 Iron status, visit & intervention = 0.08 |
| 4 W | 0.43 ± 0.16 | 0.44 ± 0.29 | 0.48 ± 0.16 | 0.42 ± 0.32 | 0.47 ± 0.18 | 0.48 ± 0.12 | 0.43 ± 0.11 | 0.54 ± 0.28 | ||
| 5 W | 0.43 ± 0.09 | 0.51 ± 0.24 | 0.46 ± 0.12 | 0.36 ± 0.07 | 0.53 ± 0.11 | 0.52 ± 0.18 | 0.38 ± 0.09 | 0.59 ± 0.30 | ||
Values are given at three different workloads (3 W, 4 W and 5 W) for: (i) [PCr], phosphocreatine concentration at end exercise, expressed as a fraction of the resting level to which it subsequently recovered; (ii) pH at end exercise; (iii) τ, measured in seconds; and (iv) Qmax, measured in mM of ATP per second. For individuals stopping work prematurely owing to fatigue, recovery kinetics were modelled from cessation of exercise. Values are means ± SD. Statistically significant P-values appear in bold.
Exercise to volitional fatigue
Table 4 shows physiological parameters at the point of volitional fatigue during maximal cardiopulmonary exercise testing (CPET), none of which differed according to baseline iron status. A differential effect of IV iron on maximal power output was detected, with the performance of ID individuals receiving iron improving. However, the effect size was only of the order of one percent, which is unlikely to be of clinical significance. Iron administration also increased the oxygen pulse (o2 divided by heart rate) irrespective of iron status. The size of this effect was much larger, of the order of ten percent. There was a trend for IV iron to reduce minute ventilation (E) at maximum power output irrespective of iron status. Data relating to lactate kinetics are presented in Table 5. Individuals randomised to receive IV iron showed an increase in lactate threshold of around ten percent regardless of baseline iron status. There was also a trend for IV iron to reduce peak lactate, again irrespective of iron status. No differential effect of IV iron was detected according to baseline iron status.
Table 4.
Variables measured at the point of volitional fatigue during incremental CPET.
| Iron status | Iron deficient (n = 13) | Iron replete (n = 13) | P-value (RM-ANOVA) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Intervention | Iron (n = 7) | Saline (n = 6) | Iron (n = 7) | Saline (n = 6) | |||||
| Visit | 1 | 2 | 1 | 2 | 1 | 2 | 1 | 2 | |
| Power, W | 180 ± 49 | 181 ± 46 | 182 ± 52 | 178 ± 54 | 216 ± 42 | 212 ± 43 | 184 ± 45 | 186 ± 49 |
Iron status = 0.31 Visit & intervention = 0.62 Iron status, visit & intervention = 0.027 |
| o2, ml/kg | 38.0 ± 11.3 | 38.0 ± 10.9 | 39.0 ± 8.2 | 36.1 ± 8.3 | 41.3 ± 7.7 | 41.4 ± 9.8 | 39.6 ± 4.7 | 39.3 ± 5.6 |
Iron status = 0.45 Visit & intervention = 0.10 Iron status, visit & intervention = 0.22 |
| RER | 1.15 ± 0.08 | 1.16 ± 0.09 | 1.14 ± 0.07 | 1.14 ± 0.06 | 1.15 ± 0.05 | 1.12 ± 0.02 | 1.12 ± 0.04 | 1.12 ± 0.06 |
Iron status = 0.40 Visit & intervention = 0.84 Iron status, visit & intervention = 0.33 |
| E, L/min | 102 ± 42 | 98 ± 40 | 98 ± 21 | 100 ± 16 | 123 ± 33 | 121 ± 32 | 93 ± 26 | 96 ± 28 |
Iron status = 0.50 Visit & intervention = 0.11 Iron status, visit & intervention = 0.86 |
| Oxygen pulse, ml/beat | 12.4 ± 3.1 | 12.8 ± 3.1 | 13.0 ± 3.7 | 12.6 ± 3.5 | 14.9 ± 3.8 | 15.4 ± 3.9 | 13.1 ± 2.9 | 12.6 ± 2.8 |
Iron status = 0.34 Visit & intervention = 0.036 Iron status, visit & intervention = 0.89 |
| RPE | 18.6 ± 1.5 | 18.3 ± 1.1 | 18.0 ± 1.8 | 17.8 ± 2.6 | 19.0 ± 1.0 | 18.9 ± 1.1 | 18.3 ± 1.0 | 18.5 ± 1.0 |
Iron status = 0.37 Visit & intervention = 0.61 Iron status, visit & intervention = 0.82 |
P-values are given for: (i) the effect of baseline iron status; (ii) the interaction between visit and intervention (the effect of IV iron); and (iii) the interaction between baseline iron status, visit, and intervention (whether any effect of IV iron differed according to baseline iron status). Values are means ± SD. Statistically significant P-values appear in bold. RER, respiratory exchange ratio; E, minute ventilation; oxygen pulse is defined as o2 divided by heart rate; RPE, rating of perceived exertion.
Table 5.
Venous lactate kinetics during exhaustive exercise.
| Iron status | Iron deficient (n = 13) | Iron replete (n = 13) | P-value (RM-ANOVA) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Intervention | Iron (n = 7) | Saline (n = 6) | Iron (n = 7) | Saline (n = 6) | Iron status | Visit & intervention | Iron status, visit & intervention | ||||
| Visit | 1 | 2 | 1 | 2 | 1 | 2 | 1 | 2 | |||
| Peak lactate, mmol/L | 6.2 ± 2.4 | 5.7 ± 1.9 | 6.4 ± 1.6 | 7.3 ± 2.3 | 6.7 ± 1.9 | 6.4 ± 2.3 | 6.3 ± 2.0 | 6.5 ± 2.3 | 0.90 | 0.12 | 0.46 |
| o2 lactate threshold, ml/kg | 20.6 ± 6.9 | 23.0 ± 8.0 | 22.5 ± 6.9 | 22.7 ± 7.6 | 24.9 ± 6.3 | 28.0 ± 6.9 | 24.0 ± 4.7 | 24.0 ± 4.9 | 0.24 | 0.039 | 0.72 |
P-values are given for: (i) the effect of baseline iron status; (ii) the interaction between visit and intervention; and (iii) the interaction between baseline iron status, visit, and intervention. Values are means ± SD. Statistically significant P-values appear in bold. The peak lactate is that measured at volitional fatigue.
Submaximal exercise
Both groups exercised at very similar levels of o2 throughout a subsequent 20-min period of submaximal exercise, close to the prescribed target of 65% o2max (mean ± SD: ID group 67 ± 3.0%; IR group 66 ± 2.5%). At the first study visit, the only variable with grossly different behaviour between groups was venous lactate (Fig. 2). In a mixed effects model, lactate concentration fell significantly over the course of the rest and submaximal exercise period (P < 0.001 for time), but the rate of decline in lactate was slower in the ID group (P = 0.005 for interaction between time and iron status).
Figure 2.
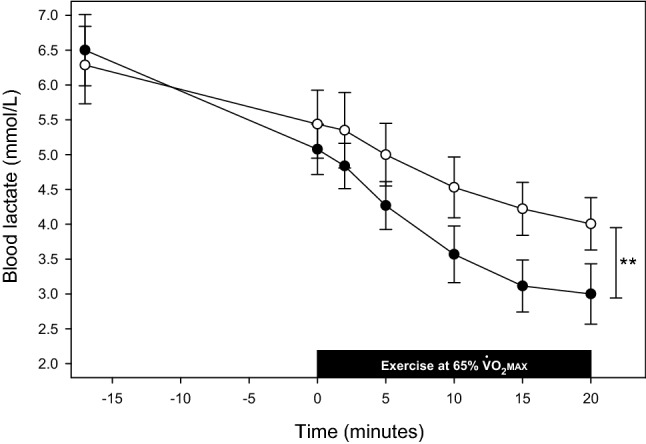
Blood lactate following volitional fatigue and during submaximal exercise at first study visit. Data for ID participants appear as white circles; those for IR participants, black circles. The initial venous lactate value was measured at volitional fatigue during the preceding maximal CPET. Following a 15-min interval, participants returned to the ergometer and measurements were made during two minutes seated at rest. The solid black bar indicates the period of submaximal exercise. A single lactate value was missing for one ID participant at the 2-min timepoint due to a technical issue. Values are means; error bars show SE. **, P = 0.005 for interaction of iron status and time.
For lactate kinetics pre- and post-infusion, there was a significant interaction between baseline iron status, intervention, visit, and time (Fig. 3). Specifically, IV iron had an effect to accelerate the fall in venous blood lactate only in the ID group (P = 0.028). Changes in other cardiorespiratory variables during submaximal exercise are also shown in Fig. 3.
Figure 3.

Cardiorespiratory variables during submaximal exercise at each study visit. Data for ID participants appear as white symbols; those for IR participants, black symbols. Data from the first visit appear as circles; those for the second, squares. Data for the second visit in participants receiving IV iron are shown in grey. E and o2 values for both visits are expressed relative to maximal values at the first visit. Values for parameters other than lactate are means for the previous 30 s; error bars show SE. One ID participant became presyncopal after volitional fatigue at the second visit and did not perform submaximal exercise; data for this individual are excluded from the figure (but not the statistical analysis). Data at the first visit for E, o2 and RER at the 10, 15 and 20-min time points were missing for one IR participant randomised to receive saline due to a technical issue. P = 0.028 for differential effect of IV iron according to baseline iron status; comparisons for all other variables NS. HR, heart rate; E, minute ventilation; RER, respiratory exchange ratio; RPE, rating of perceived exertion.
Skeletal muscle biopsy
Gene expression data from quantitative polymerase chain reaction (qPCR) of skeletal muscle sampled at the first study visit are given in Table 6. No significant differences were detected, although there is some suggestion that LDHA may have been higher in the ID group.
Table 6.
Skeletal muscle mRNA expression levels at first visit.
| Gene | ID group (n = 10) | IR group (n = 6) | P-value (t-test) |
|---|---|---|---|
| LDHA | 1.51 ± 0.18 | 0.98 ± 0.12 | 0.06 |
| PFK | 1.15 ± 0.17 | 1.03 ± 0.15 | 0.63 |
| PDK1 | 1.06 ± 0.13 | 1.07 ± 0.17 | 0.95 |
Expression data are means ± SE at the mRNA level for lactate dehydrogenase A (LDHA), phosphofructokinase (PFK), and pyruvate dehydrogenase kinase isoform 1 (PDK1).
Discussion
Main findings
The main finding of the present study is of abnormal whole-body metabolism in ID individuals, manifest as disturbed blood lactate kinetics during exercise, in the absence of any demonstrable impairment of skeletal muscle oxidative phosphorylation. Iron deficiency appears to promote a shift in favour of anaerobic glycolysis, reflected in sizeable changes in the threshold for anaerobic metabolism, and this effect is not mediated by differences in Hb concentration. Thus, the consequences of clinical iron deficiency in humans are seen to be similar with respect to lactate kinetics to the abnormalities reported in ID rodents7,11 and patients with CP15, without the gross disturbance of high-energy phosphate metabolism seen in these settings. To our knowledge, the present study is the first to describe an iron-mediated action of this sort in healthy humans, distinct from an effect of Hb concentration.
The rate of decline in blood lactate concentration was diminished in ID participants exercising at 65% o2max. This implies that iron deficiency impairs lactate disposal, promotes its production, or causes a combination of these two phenomena. This effect cannot be explained by the marginally lower Hb concentration in the ID group, first, because an individually tailored submaximal work rate was employed for each participant, and second, because the apparent rate of lactate disappearance from the blood increased in ID participants in the absence of any increase in Hb concentration following IV iron. In further support of this view, at the first study visit there was a trend towards lower absolute lactate thresholds in ID participants compared with IR controls. Interestingly, administration of IV iron brought about a striking rise in lactate threshold measured during incremental CPET to volitional fatigue irrespective of baseline iron status, suggesting that the effects of iron status on lactate handling extend into what is considered the physiologically normal range, and are not limited solely to individuals that are iron deficient.
Iron homeostasis
We measured a variety of indices of iron homeostasis over the course of the study. As expected, the ID and IR study entry criteria generated two groups that differed very significantly in iron status, with the mean values observed for serum ferritin and TSat in the ID group at enrolment consistent with depletion of storage iron to the point of absent stainable iron in the bone marrow21,22.
Ferritin is the major mammalian iron storage protein, serum levels of which correlate very strongly with mobilisable iron stores in health, with 1 µg/L serum ferritin corresponding to ~ 8 mg storage iron23. The mean increase in serum ferritin seen in those allocated to iron in the present study exceeded 500 µg/L in ID participants and approached 800 µg/L in the IR group. Given that no more than a 1000 mg dose of IV iron was administered to any participant, it can immediately be seen that these values indicate that soon after IV iron administration the utility of serum ferritin as a marker of storage iron is lost. The likely explanation is the regulation of ferritin mRNA translation by iron24,25, such that soon after IV infusion of a large iron dose, ferritin expression is greatly stimulated; this phenomenon has been observed in previous human physiology studies26–28. That the serum ferritin level achieved in ID participants given iron was lower in absolute terms than that seen in IR participants, suggests that pre-existing iron deficiency constrains ferritin expression by some means. This may be explained by the observation from radioactive isotope studies that iron is directed rapidly towards the bone marrow when infused into individuals with profound iron deficiency29.
Hepcidin is the major hormone regulating iron homeostasis, acting via the cellular iron exporter ferroportin to control intestinal iron absorption, the export of recycled iron from macrophages, and release of iron stored within hepatocytes30. Similar to the trend observed with ferritin, the magnitude of the rise in hepcidin when iron was administered was more substantial in the IR group than in ID participants. The regulation of hepcidin expression by iron is more complicated than ferritin, since it involves signals from developing erythrocytes as well as plasma iron concentration and body iron stores28,30. Nevertheless, our findings imply that existing tissue iron depletion acts as a strong negative regulatory signal for hepcidin expression, even when serum iron levels are acutely grossly elevated.
Finally, IV iron tended to suppress circulating erythropoietin levels in ID, but not IR, participants, implying that iron deficiency acts directly to augment erythropoietin expression, as the suppression occurred prior to any increase in Hb concentration. This finding confirms that the increase in circulating erythropoietin levels seen in heathy humans following iron chelation31 does have a clinical correlate, and is also in keeping with the demonstration in a mouse model that manipulation of iron availability alters the expression of the gene encoding erythropoietin by modulating levels of HIF-2α in renal fibroblasts32.
Strengths and limitations
The main strengths of the present study are four-fold. First, the dual assessment in the same individuals of exercise metabolism using both MRS of a small muscle mass, and whole-body exercise. Second, the randomised use of a substantial dose of IV iron in a double-blind manner. Third, the recruitment of individuals with profound absolute iron deficiency in the absence of marked anaemia. Fourth, the repeat assessment of these individuals after IV iron but before any change in Hb concentration.
The dose of IV iron used was twice that employed in a contemporary study of elite athletes9 and brought about a striking change in circulating markers of iron homeostasis. With the exception of work in patients with CP15, no previous human study has to our knowledge used both CPET and MRS together in the same individuals, and most have been primarily concerned with athletic performance. To our knowledge, only two previous studies have used MRS to investigate the effects of iron status in humans. The first recruited a small, heterogeneous group of anaemic hospital inpatients33; the second studied patients with chronic heart failure and reported that iron deficiency was associated with more marked PCr depletion and profound intracellular acidosis with small muscle mass exercise, leading the authors to speculate that enhanced anaerobic glycolysis was occurring34. Neither whole-body exercise nor measurements of blood lactate were undertaken.
The limited reproducibility of MRS may have restricted our ability to detect a subtle effect of iron deficiency on skeletal muscle oxidative phosphorylation. When assessed using similar apparatus to that employed in the present study, very highly trained individuals were found to have generally reproducible measures of resting high-energy phosphorus metabolites, but the coefficient of variation during exercise for PCr was 27%, and that for PCr half-time, 40%35. Nevertheless, in healthy, non-trained individuals, PCr depletion during exercise appears to be much more highly reproducible, such that biological variability accounts for the vast majority of measurement variability36. Thus, whilst the present study was not powered to exclude a subtle impairment of skeletal muscle oxidative phosphorylation by iron deficiency, and we may have been unable to detect a small effect, gross abnormalities of mitochondrial function of the sort evident in CP patients15 and rodent models of iron deficiency12 are clearly absent.
We sampled venous, rather than arterial blood for real-time analysis of lactate concentration, as this was safer and more acceptable to participants. Measured venous blood lactate levels from antecubital fossa sampling have been shown to correlate closely with arterial values during lower limb exercise37, supporting such an approach. Haemoglobin concentration was slightly lower on enrolment in the ID group. In the case of the MRS data, the small muscle mass exercise used can reasonably be anticipated to be unaffected by any difference in systemic oxygen delivery15. Additionally, because participants were randomised to receive iron or saline, and no change in Hb concentration occurred between study visits, a haemoglobin-mediated effect can confidently be excluded.
It proved difficult to recruit ID males to the present study, such that recruitment fell short of the target sample size. The reasons are not entirely clear, since the same recruitment approach was effective in a previous study38. This should be acknowledged when considering the generalisability of the results. Also, we intentionally avoided recruiting only individuals habituated to aerobic exercise, unlike previous studies that have focused on iron status and athletic performance, so that our participants might better reflect the healthy general adult population. However, one consequence was that the distribution of values for o2max and the anaerobic threshold within each group were rather broad.
Finally, we did not make skeletal muscle biopsy a compulsory part of the protocol because of its invasiveness. Instead, participants were free to decline the biopsy at the second visit having undergone it at the first, or not undergo biopsy at all. Comparisons of qPCR data between groups are therefore based on a subset of the participants at the first study visit; there were insufficient data from the second visit to assess the effect of IV iron.
Underlying mechanisms
One mechanism historically proposed for the metabolic effects of iron deficiency is impaired function of oxidative enzymes requiring iron as a cofactor, particularly cytochrome C11,39–43. However, in rodent models, iron deficiency has typically been induced by severe early life dietary iron restriction, which tends to reduce concentrations of skeletal muscle myoglobin and iron-dependent mitochondrial enzymes12. These latter changes have not been demonstrated in adult humans with iron deficiency44,45, which may explain many of the differences between human iron deficiency and animal models thereof. Our ID participants arguably behaved in a Warburg-like manner; this is the opposite effect to that described in animals with tissue-specific deletion of skeletal muscle HIF-1α46,47, and in endurance athletes, who seem to exhibit downregulation of the HIF pathway48. Significant changes occur in skeletal muscle lactate production and disposal as a consequence of endurance training49, and it is possible that IV iron acted upon the HIF pathway in skeletal muscle mimicking these changes.
The rapid decay of HIF-1α when exposed to euoxic conditions50 precludes reliable direct measurement of levels of the protein itself in human studies in order to elucidate underlying mechanisms. However, the molecular biology of erythropoietin expression and its regulation by HIF are very well understood17, such that erythropoietin expression can be used from cell culture51 to intact healthy humans31 to study the effects of iron availability on the HIF pathway. The observed effects of iron supplementation on circulating erythropoietin levels in the present study are consistent with iron acting on the HIF-hydroxylases within renal fibroblasts32.
The site of action of iron deficiency with respect to lactate kinetics is less clear. The suggestion of increased LDHA expression in skeletal muscle of ID participants is of note, since resynthesis of glycogen within skeletal muscle seems to be a significant sink for lactate accumulated at the conclusion of strenuous exercise52. However, lactate is metabolised in significant quantities during exercise by both the heart and liver53, so alterations in cardiac or hepatic lactate handling may well underlie some of the observed effects in the present study. With respect to the latter possibility, HIF prolyl-hydroxylase 1 deficiency – which is the effective functional state that occurs as a result of iron deficiency – has been shown to increase glycolytic gene expression within the liver in a mouse model54.
The finding of an increased oxygen pulse at o2max following IV iron infusion is of interest since the former is an index of stroke volume (SV) in healthy individuals (oxygen pulse = o2/heart rate = SV × arteriovenous oxygen concentration difference)55. Since iron supplementation attenuates pulmonary vasoconstriction during both hypoxia38,56,57 and aerobic exercise27, another possibility is that an effect on stroke volume arises due to an action on the pulmonary circulation. The findings of a recent study involving IV iron supplementation prior to ascent to very high altitude would support this possibility26. Alternatively, increased tissue oxygen extraction might occur following iron supplementation. It is not possible from an integrated study to determine the relative contribution of these mechanisms.
Clinical implications
Iron deficiency is associated with worse outcomes in chronic health conditions that are extremely prevalent globally, including chronic obstructive pulmonary disease58 and a number of chronic cardiovascular diseases59. Studies have indicated that intravenous iron supplementation can have beneficial effects in such conditions60,61. Of note, one study of patients with idiopathic pulmonary arterial hypertension reported a rise in anaerobic threshold following IV iron along with increased submaximal exercise endurance62, reminiscent of the present study. Importantly, impaired submaximal exercise performance is arguably most functionally relevant for patients with cardiopulmonary disease53, reflecting as it does the impact these conditions can have on everyday life. On the other hand, o2max is an indicator of cardiopulmonary fitness that predicts outcomes, for example, following major surgical procedures63.
The present study furthers our understanding of the mechanisms by which iron deficiency is injurious to human health, and suggests that functional performance might be affected by IV iron in a broader range of conditions than has previously been recognised.
Methods
This was a prospective, case–control, clinical physiology study with subsequent double-blind randomisation. By targeting blood donors, who were offered study information if below the Hb concentration threshold when attending to donate, we enrolled adults with absolute iron deficiency (serum ferritin ≤ 15 ug/L with TSat < 16%)38. Healthy age- and sex-matched IR volunteers (serum ferritin ≥ 20 ug/L with TSat ≥ 20%) served as controls. A screening visit was conducted consisting of medical history, physical examination, spirometry (MicroLab™, CareFusion, UK), and venous bloods. Those with factors likely to disturb cardiopulmonary physiology or iron metabolism were excluded, for example chronic inflammatory disorders, chronic lung disease, haemochromatosis, recent iron supplementation or blood transfusion, and recent long-haul air travel or residence at high altitude. Eligible participants attended two identical study visits approximately a week apart, commencing early in the morning following an overnight fast.
Exercise protocol
Skeletal muscle 31P-MRS was performed using a 3 Tesla MRI scanner (Siemens TIM Trio) with the participant supine and a dual-tuned 31P and 1H 6-cm-diameter surface coil secured under the right calf. The foot was fastened to a custom-built plantarflexion exercise apparatus with the leg straightened, and the calf exercised at 1 Hz in time to a digital metronome, as previously described15. Three 5-min periods of exercise, at 3 W, 4 W and 5 W alternated with 7-min recovery periods. Data were processed offline by a blinded investigator.
Following MRS, a 20-gauge venous cannula (Venflon™, Becton Dickinson) was inserted into a large antecubital fossa vein and blood drawn. Incremental CPET to volitional fatigue was performed on an electronically braked cycle ergometer (ergoselect 100, ergoline GmbH, Germany). Participants wore a close-fitting facemask with respired gases sampled continuously through a catheter and analysed by indirect calorimetry (Metalyzer® 3B CPET System, CORTEX Biophysik GmbH, Germany). Resting data were collected over a 2-min period. Work then began at 50 W, for three minutes, increasing thereafter by 25-W increments at 3-min intervals. Venous blood was sampled via the indwelling cannula during the final 30 s of exercise at each workload and upon cessation of exercise. Samples were analysed using a clinically validated Lactate Pro™ device (Arkray Inc. Japan)64. Participants rated their perception of exertion on a fifteen-point RPE scale, from 6 (rest) to 20 (maximum exertion)65.
After a rest period of 15 min following volitional fatigue, participants returned to the ergometer and 2 min of resting data were recorded. There then followed a further 20 min of exercise at a work rate equivalent to 65% o2max, determined by interpolation of the data just obtained during maximal CPET. During submaximal exercise, blood was sampled at rest, after 2 min, 5 min and every 5 min thereafter. On the second study visit, the same work rate was used during submaximal exercise. Exercise data were processed offline by an investigator blinded to participant iron status and randomisation. The lactate threshold was determined as described by Beaver and colleagues66.
31P magnetic resonance spectroscopy data processing
31P-MRS spectral peaks were fitted using the automated AMARES algorithm within the jMRUI software package, with absolute concentrations of phosphorus metabolites calculated from the spectral data using an established method15,67–69. PCr recovery kinetics were modelled for the period after each exercise bout. A monoexponential relationship was derived70, using a least-squares-fit approach to determine the time-constant (τ), and expressing [PCr] as a function of time (t)71:
The maximum theoretical rate of mitochondrial ATP synthesis (Qmax) was extrapolated from a combination of the end-exercise [ADP] and initial rate of PCr resynthesis, as follows:
where is the initial rate of PCr resynthesis, Km is the [ADP] at which oxidative ATP synthesis is taken to be half maximal (25 µmol/L) and n (2.2) is a Hill coefficient that describes the relationship between and [ADP]35,72,73.
Skeletal muscle biopsy and qPCR
Prior to maximal CPET, participants agreeing to undergo skeletal muscle biopsy reclined comfortably on an examination couch with their right leg fully relaxed. The lateral aspect of the thigh was cleaned with 0.5% chlorhexidine in 70% ethanol, and local anaesthesia procured with ~ 5 ml 1% lidocaine infiltrated using a 22-gauge needle. An 11-blade surgical scalpel was used to penetrate the skin and breach the superficial fascia. A disposable sterile spring-loaded core biopsy instrument (Monopty®, Bard Peripheral Vascular Inc. Tempe, AZ) was inserted through the incision and discharged to take a sample of skeletal muscle, which was immediately flash-frozen in liquid nitrogen. Four passes were made in total. Haemostasis was ensured and a sterile dressing applied.
Total RNA was extracted from 20–30 mg powdered skeletal muscle using the RNeasy® Fibrous Kit (Qiagen, UK). The crushed tissue underwent tissue rupture on ice followed by a DNase treatment step, before complementary DNA was synthesized from 1 μg RNA using the Applied Biosystems High Capacity cDNA Reverse Transcription Kit (Life Technologies, UK). Real-time PCR was performed using an ABI StepOnePlus™ Real-Time PCR System (Applied Biosystems, UK) with TaqMan® Universal PCR Master Mix and TaqMan Gene Expression Assays (choosing manufacturer-recommended assays: Applied Biosystems, UK). Relative mRNA expression levels were determined using the standard curve method and normalised to beta-actin.
Venous blood analyses and infusions
Assays for full blood count, C-reactive protein (CRP), serum ferritin, iron, transferrin and TSat were performed by a University Hospital clinical pathology laboratory. Aliquots of serum and EDTA plasma were obtained by centrifugation and frozen and stored at − 80 °C until the conclusion of the final study visit of the final participant. Serum erythropoietin, plasma sTfR (both Quantikine®, R&D Systems, Abingdon, UK), and plasma hepcidin (Hepcidin 25 HS, DRG, Marburg, Germany) were analysed by enzyme-linked immunosorbent assay. At the end of the first study visit, participants received either ferric carboxymaltose (Ferinject®, Vifor Pharma, Switzerland) 15 mg/kg (maximum 1 g) or 0.9% saline. Block randomisation according to iron status and sex was used. Participants were blindfolded, and the infusion, infusion line and infusion site obscured using an opaque plastic drape.
Study approval
The study received ethical approval from the NHS South Central Oxford B Research Ethics Committee (reference 13/SC/0439). The study was sponsored by the University of Oxford and conducted in accordance with the principles of the declaration of Helsinki. All participants gave written informed consent. The study was registered with ClinicalTrials.gov (NCT02308449).
Statistics
To estimate the sample size required we considered: (i) work in CP patients showing a 1.6 to 1.9 fold greater fall in PCr during calf muscle exercise compared with healthy individuals15; (ii) data from individuals with IDA in whom the ratio of PCr to (PCr + Pi) fell approximately one third more than in healthy individuals during small muscle mass exercise33; and (iii) evidence that the degree of PCr depletion during exercise in non-trained individuals is highly reproducible when measured with with MRS36. We calculated that we would need to study a total of 32 volunteers in order to have 80% power to detect 15% greater PCr depletion in ID compared with IR individuals, with a significance level of P < 0.05.
Data were analysed using SPSS Statistics (version 25, IBM). The two-sided unpaired Student’s t-test and Mann Whitney U test were used for group comparisons. Repeated measures analysis of variance (RM-ANOVA) and mixed-effects modelling were used to determine the effects of exercise and iron infusion within and between groups.
Acknowledgements
The authors would like to thank the volunteers for giving up their time to take part in this study, Dawn Goodwin at NHS Blood and Transplant for her assistance with recruiting iron-deficient blood donors, and Jane Francis at the Oxford Centre for Clinical Magnetic Resonance Research for her expert assistance.
Author contributions
Conceived and designed research – M.C.F., D.A.H., A.W.J., P.J.C., K.C., D.J.T., D.J.R., P.J.R., P.A.R. Performed experiments – M.C.F., D.A.H., Y.C., M.K.C., K.L.D. Analyzed data – M.C.F., D.A.H., A.W.J. Interpreted results of experiments – M.C.F., D.A.H., P.J.C., K.C., D.J.T., D.J.R., P.J.R., K.L.D., P.A.R. Prepared figures – M.C.F. Drafted manuscript – M.C.F., D.A.H. Edited and revised manuscript – M.C.F., D.A.H., A.W.J., Y.C., M.K.C., P.J.C., K.C., D.J.T., D.J.R., P.J.R., K.L.D., P.A.R. Approved final version of manuscript – M.C.F., D.A.H., A.W.J., Y.C., M.K.C., P.J.C., K.C., D.J.T., D.J.R., P.J.R., K.L.D., P.A.R.
Funding
This research was funded by the National Institute for Health Research (NIHR) Oxford Biomedical Research Centre (BRC). The views expressed are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health. MCF was supported by a British Heart Foundation Clinical Research Training Fellowship (FS/14/48/30828), DJT by a BHF Senior Fellowship (FS/14/17/30634), and KLD by the Dunhill Medical Trust (R178/1110).
Data availability
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
Competing interests
PAR has received grant funding from Vifor Pharma for basic science studies of iron biology, including in support of work by MKC outside that presented here. PJR was previously chair of the Research Advisory Board of GlaxoSmithKline. The remaining authors declare no competing interests.
Footnotes
The original online version of this Article was revised: The original version of this Article contained a repeated error where the ‘V̇’ symbol did not display correctly in the Introduction, the Results section under the subheadings ‘Exercise to volitional fatigue’ and ‘Submaximal exercise’, the Discussion section under the subheadings ‘Main findings’, ‘Strengths and limitations’, ‘Underlying mechanisms’ and ‘Clinical implications’, the Methods section, under the subheading ‘Exercise protocol’, Table 4 and 5 and the legends of Figure 3 and Table 4. Full information regarding the corrections made can be found in the correction for this Article.
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Change history
3/1/2022
A Correction to this paper has been published: 10.1038/s41598-022-06694-7
References
- 1.Hawley JA, Hargreaves M, Joyner MJ, Zierath JR. Integrative biology of exercise. Cell. 2014;159:738–749. doi: 10.1016/j.cell.2014.10.029. [DOI] [PubMed] [Google Scholar]
- 2.Ohira Y, et al. Work capacity, heart rate and blood lactate responses to iron treatment. Br. J. Haematol. 1979;41:365–372. doi: 10.1111/j.1365-2141.1979.tb05869.x. [DOI] [PubMed] [Google Scholar]
- 3.Schoene RB, et al. Iron repletion decreases maximal exercise lactate concentrations in female athletes with minimal iron-deficiency anemia. J. Lab. Clin. Med. 1983;102:306–312. [PubMed] [Google Scholar]
- 4.Sim M, et al. Iron considerations for the athlete: A narrative review. Eur. J. Appl. Physiol. 2019;119:1463–1478. doi: 10.1007/s00421-019-04157-y. [DOI] [PubMed] [Google Scholar]
- 5.Zimmermann MB, Hurrell RF. Nutritional iron deficiency. Lancet (London, England) 2007;370:511–520. doi: 10.1016/S0140-6736(07)61235-5. [DOI] [PubMed] [Google Scholar]
- 6.Finch CA, Huebers H. Perspectives in iron metabolism. N. Engl. J. Med. 1982;306:1520–1528. doi: 10.1056/nejm198206243062504. [DOI] [PubMed] [Google Scholar]
- 7.Davies KJ, et al. Distinguishing effects of anemia and muscle iron deficiency on exercise bioenergetics in the rat. Am. J. Physiol. 1984;246:E535–E543. doi: 10.1152/ajpendo.1984.246.6.E535. [DOI] [PubMed] [Google Scholar]
- 8.Burden RJ, Morton K, Richards T, Whyte GP, Pedlar CR. Is iron treatment beneficial in iron-deficient but non-anaemic (IDNA) endurance athletes? A systematic review and meta-analysis. Br. J. Sports Med. 2015;49:1389–1397. doi: 10.1136/bjsports-2014-093624. [DOI] [PubMed] [Google Scholar]
- 9.Burden RJ, et al. Effect of intravenous iron on aerobic capacity and iron metabolism in elite athletes. Med. Sci. Sports Exerc. 2015;47:1399–1407. doi: 10.1249/MSS.0000000000000568. [DOI] [PubMed] [Google Scholar]
- 10.Finch CA, et al. Iron deficiency in the rat. Physiological and biochemical studies of muscle dysfunction. J. Clin. Invest. 1976;58:447–453. doi: 10.1172/jci108489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Finch CA, et al. Lactic acidosis as a result of iron deficiency. J. Clin. Investig. 1979;64:129–137. doi: 10.1172/jci109431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thompson CH, Green YS, Ledingham JG, Radda GK, Rajagopalan B. The effect of iron deficiency on skeletal muscle metabolism of the rat. Acta Physiol. Scand. 1993;147:85–90. doi: 10.1111/j.1748-1716.1993.tb09475.x. [DOI] [PubMed] [Google Scholar]
- 13.Davies KJ, Maguire JJ, Brooks GA, Dallman PR, Packer L. Muscle mitochondrial bioenergetics, oxygen supply, and work capacity during dietary iron deficiency and repletion. Am. J. Physiol. 1982;242:E418–E427. doi: 10.1152/ajpendo.1982.242.6.E418. [DOI] [PubMed] [Google Scholar]
- 14.Perkkio MV, Jansson LT, Brooks GA, Refino CJ, Dallman PR. Work performance in iron deficiency of increasing severity. J. Appl. Physiol. (Bethesda, Md. : 1985) 1985;58:1477–1480. doi: 10.1152/jappl.1985.58.5.1477. [DOI] [PubMed] [Google Scholar]
- 15.Formenti F, et al. Regulation of human metabolism by hypoxia-inducible factor. Proc. Natl. Acad. Sci. USA. 2010;107:12722–12727. doi: 10.1073/pnas.1002339107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ang SO, et al. Disruption of oxygen homeostasis underlies congenital Chuvash polycythemia. Nat. Genet. 2002;32:614–621. doi: 10.1038/ng1019. [DOI] [PubMed] [Google Scholar]
- 17.Bunn HF. Erythropoietin. Cold Spring Harb. Perspect. Med. 2013;3:a011619. doi: 10.1101/cshperspect.a011619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Loenarz C, Schofield CJ. Expanding chemical biology of 2-oxoglutarate oxygenases. Nat. Chem. Biol. 2008;4:152–156. doi: 10.1038/nchembio0308-152. [DOI] [PubMed] [Google Scholar]
- 19.Frise MC, Robbins PA. Iron, oxygen, and the pulmonary circulation. J. Appl. Physiol. (Bethesda, Md. : 1985) 2015;119:1421–1431. doi: 10.1152/japplphysiol.00179.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goodnough LT, Nemeth E, Ganz T. Detection, evaluation, and management of iron-restricted erythropoiesis. Blood. 2010;116:4754–4761. doi: 10.1182/blood-2010-05-286260. [DOI] [PubMed] [Google Scholar]
- 21.Cook JD, Finch CA, Smith NJ. Evaluation of the iron status of a population. Blood. 1976;48:449–455. doi: 10.1182/blood.V48.3.449.449. [DOI] [PubMed] [Google Scholar]
- 22.Cook JD, Skikne BS. Serum ferritin: A possible model for the assessment of nutrient stores. Am. J. Clin. Nutr. 1982;35:1180–1185. doi: 10.1093/ajcn/35.5.1180. [DOI] [PubMed] [Google Scholar]
- 23.Walters GO, Miller FM, Worwood M. Serum ferritin concentration and iron stores in normal subjects. J. Clin. Pathol. 1973;26:770–772. doi: 10.1136/jcp.26.10.770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aziz N, Munro HN. Iron regulates ferritin mRNA translation through a segment of its 5' untranslated region. Proc. Natl. Acad. Sci. USA. 1987;84:8478–8482. doi: 10.1073/pnas.84.23.8478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hentze MW, et al. Identification of the iron-responsive element for the translational regulation of human ferritin mRNA. Science. 1987;238:1570–1573. doi: 10.1126/science.3685996. [DOI] [PubMed] [Google Scholar]
- 26.Holdsworth DA, et al. Iron bioavailability and cardiopulmonary function during ascent to very high altitude. Eur. Respir. J. 2020;56:1902285. doi: 10.1183/13993003.02285-2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cheng HY, et al. Intravenous iron delivers a sustained (8-week) lowering of pulmonary artery pressure during exercise in healthy older humans. Physiol. Rep. 2019;7:e14164. doi: 10.14814/phy2.14164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Talbot NP, et al. Regulation of hepcidin expression at high altitude. Blood. 2012;119:857–860. doi: 10.1182/blood-2011-03-341776. [DOI] [PubMed] [Google Scholar]
- 29.Geisser P, Burckhardt S. The pharmacokinetics and pharmacodynamics of iron preparations. Pharmaceutics. 2011;3:12–33. doi: 10.3390/pharmaceutics3010012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ganz T. Systemic iron homeostasis. Physiol. Rev. 2013;93:1721–1741. doi: 10.1152/physrev.00008.2013. [DOI] [PubMed] [Google Scholar]
- 31.Ren X, Dorrington KL, Maxwell PH, Robbins PA. Effects of desferrioxamine on serum erythropoietin and ventilatory sensitivity to hypoxia in humans. J. Appl. Physiol. (Bethesda, Md. : 1985) 2000;89:680–686. doi: 10.1152/jappl.2000.89.2.680. [DOI] [PubMed] [Google Scholar]
- 32.Suzuki N, et al. Iron attenuates erythropoietin production by decreasing hypoxia-inducible transcription factor 2α concentrations in renal interstitial fibroblasts. Kidney Int. 2018;94:900–911. doi: 10.1016/j.kint.2018.06.028. [DOI] [PubMed] [Google Scholar]
- 33.Thompson CH, Kemp GJ, Taylor DJ, Radda GK, Rajagopalan B. No evidence of mitochondrial abnormality in skeletal muscle of patients with iron-deficient anaemia. J. Intern. Med. 1993;234:149–154. doi: 10.1111/j.1365-2796.1993.tb00724.x. [DOI] [PubMed] [Google Scholar]
- 34.Melenovsky V, et al. Skeletal muscle abnormalities and iron deficiency in chronic heart failurean exercise (31)P magnetic resonance spectroscopy study of calf muscle. Circ. Heart Fail. 2018;11:e004800. doi: 10.1161/CIRCHEARTFAILURE.117.004800. [DOI] [PubMed] [Google Scholar]
- 35.Edwards LM, et al. The reproducibility of 31-phosphorus MRS measures of muscle energetics at 3 Tesla in trained men. PLoS ONE. 2012;7:e37237. doi: 10.1371/journal.pone.0037237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Layec G, et al. Reproducibility assessment of metabolic variables characterizing muscle energetics in vivo: A 31P-MRS study. Magn. Reson. Med. 2009;62:840–854. doi: 10.1002/mrm.22085. [DOI] [PubMed] [Google Scholar]
- 37.Nordrehaug JE, Bjorkhaug A, Danielsen R, Vik-Mo H. Arterial and venous measurement in resting forearm of metabolic indicators during rest and leg exercise. Clin. Physiol. 1991;11:469–476. doi: 10.1111/j.1475-097x.1991.tb00819.x. [DOI] [PubMed] [Google Scholar]
- 38.Frise MC, et al. Clinical iron deficiency disturbs normal human responses to hypoxia. J. Clin. Investig. 2016;126:2139–2150. doi: 10.1172/JCI85715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McLane JA, et al. Physiological and biochemical effects of iron deficiency on rat skeletal muscle. Am. J. Physiol. 1981;241:C47–C54. doi: 10.1152/ajpcell.1981.241.1.C47. [DOI] [PubMed] [Google Scholar]
- 40.McKay RH, Higuchi DA, Winder WW, Fell RD, Brown EB. Tissue effects of iron deficiency in the rat. Biochim. Biophys. Acta. 1983;757:352–358. doi: 10.1016/0304-4165(83)90061-2. [DOI] [PubMed] [Google Scholar]
- 41.Harlan WR, Williams RS. Activity-induced adaptations in skeletal muscles of iron-deficient rabbits. J. Appl. Physiol. (Bethesda, Md. : 1985) 1988;65:782–787. doi: 10.1152/jappl.1988.65.2.782. [DOI] [PubMed] [Google Scholar]
- 42.Willis WT, Brooks GA, Henderson SA, Dallman PR. Effects of iron deficiency and training on mitochondrial enzymes in skeletal muscle. J. Appl. Physiol. (Bethesda, Md. : 1985) 1987;62:2442–2446. doi: 10.1152/jappl.1987.62.6.2442. [DOI] [PubMed] [Google Scholar]
- 43.Willis WT, Dallman PR, Brooks GA. Physiological and biochemical correlates of increased work in trained iron-deficient rats. J. Appl. Physiol. (Bethesda, Md. : 1985) 1988;65:256–263. doi: 10.1152/jappl.1988.65.1.256. [DOI] [PubMed] [Google Scholar]
- 44.Celsing F, Blomstrand E, Werner B, Pihlstedt P, Ekblom B. Effects of iron deficiency on endurance and muscle enzyme activity in man. Med. Sci. Sports Exerc. 1986;18:156–161. doi: 10.1249/00005768-198604000-00002. [DOI] [PubMed] [Google Scholar]
- 45.Celsing F, Ekblom B, Sylven C, Everett J, Astrand PO. Effects of chronic iron deficiency anaemia on myoglobin content, enzyme activity, and capillary density in the human skeletal muscle. Acta Med. Scand. 1988;223:451–457. doi: 10.1111/j.0954-6820.1988.tb15897.x. [DOI] [PubMed] [Google Scholar]
- 46.Mason SD, et al. HIF-1alpha in endurance training: Suppression of oxidative metabolism. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007;293:R2059–R2069. doi: 10.1152/ajpregu.00335.2007. [DOI] [PubMed] [Google Scholar]
- 47.Mason SD, et al. Loss of skeletal muscle HIF-1alpha results in altered exercise endurance. PLoS Biol. 2004;2:e288. doi: 10.1371/journal.pbio.0020288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lindholm ME, et al. Negative regulation of HIF in skeletal muscle of elite endurance athletes: A tentative mechanism promoting oxidative metabolism. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2014;307:R248–R255. doi: 10.1152/ajpregu.00036.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bergman BC, et al. Active muscle and whole body lactate kinetics after endurance training in men. J. Appl. Physiol. (Bethesda, Md. : 1985) 1999;87:1684–1696. doi: 10.1152/jappl.1999.87.5.1684. [DOI] [PubMed] [Google Scholar]
- 50.Yu AY, et al. Temporal, spatial, and oxygen-regulated expression of hypoxia-inducible factor-1 in the lung. Am. J. Physiol. 1998;275:L818–L826. doi: 10.1152/ajplung.1998.275.4.L818. [DOI] [PubMed] [Google Scholar]
- 51.Wang GL, Semenza GL. Desferrioxamine induces erythropoietin gene expression and hypoxia-inducible factor 1 DNA-binding activity: Implications for models of hypoxia signal transduction. Blood. 1993;82:3610–3615. doi: 10.1182/blood.V82.12.3610.3610. [DOI] [PubMed] [Google Scholar]
- 52.Astrand PO, Hultman E, Juhlin-Dannfelt A, Reynolds G. Disposal of lactate during and after strenuous exercise in humans. J. Appl. Physiol. (Bethesda, Md. : 1985) 1986;61:338–343. doi: 10.1152/jappl.1986.61.1.338. [DOI] [PubMed] [Google Scholar]
- 53.Myers J, Ashley E. Dangerous curves. A perspective on exercise, lactate, and the anaerobic threshold. Chest. 1997;111:787–795. doi: 10.1378/chest.111.3.787. [DOI] [PubMed] [Google Scholar]
- 54.Thomas A, et al. Hypoxia-inducible factor prolyl hydroxylase 1 (PHD1) deficiency promotes hepatic steatosis and liver-specific insulin resistance in mice. Sci. Rep. 2016;6:24618. doi: 10.1038/srep24618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stringer WW, Hansen JE, Wasserman K. Cardiac output estimated noninvasively from oxygen uptake during exercise. J. Appl. Physiol. (Bethesda, Md. : 1985) 1997;82:908–912. doi: 10.1152/jappl.1997.82.3.908. [DOI] [PubMed] [Google Scholar]
- 56.Talbot NP, et al. Contrasting effects of ascorbate and iron on the pulmonary vascular response to hypoxia in humans. Physiol. Rep. 2014;2:e12220. doi: 10.14814/phy2.12220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Smith TG, et al. The increase in pulmonary arterial pressure caused by hypoxia depends on iron status. J. Physiol. 2008;586:5999–6005. doi: 10.1113/jphysiol.2008.160960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nickol AH, et al. A cross-sectional study of the prevalence and associations of iron deficiency in a cohort of patients with chronic obstructive pulmonary disease. BMJ Open. 2015;5:e007911. doi: 10.1136/bmjopen-2015-007911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.von Haehling S, Jankowska EA, van Veldhuisen DJ, Ponikowski P, Anker SD. Iron deficiency and cardiovascular disease. Nat. Rev. Cardiol. 2015;12:659–669. doi: 10.1038/nrcardio.2015.109. [DOI] [PubMed] [Google Scholar]
- 60.Ponikowski P, et al. Beneficial effects of long-term intravenous iron therapy with ferric carboxymaltose in patients with symptomatic heart failure and iron deficiency. Eur. Heart J. 2015;36:657–668. doi: 10.1093/eurheartj/ehu385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Santer P, et al. Intravenous iron and chronic obstructive pulmonary disease: A randomised controlled trial. BMJ Open Respir. Res. 2020 doi: 10.1136/bmjresp-2020-000577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ruiter G, et al. Intravenous iron therapy in patients with idiopathic pulmonary arterial hypertension and iron deficiency. Pulm. Circ. 2015;5:466–472. doi: 10.1086/682217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Levett DZ, Grocott MP. Cardiopulmonary exercise testing for risk prediction in major abdominal surgery. Anesthesiol. Clin. 2015;33:1–16. doi: 10.1016/j.anclin.2014.11.001. [DOI] [PubMed] [Google Scholar]
- 64.Pyne DB, Boston T, Martin DT, Logan A. Evaluation of the Lactate Pro blood lactate analyser. Eur. J. Appl. Physiol. 2000;82:112–116. doi: 10.1007/s004210050659. [DOI] [PubMed] [Google Scholar]
- 65.Borg G. Perceived exertion as an indicator of somatic stress. Scand. J. Rehabil. Med. 1970;2:92–98. [PubMed] [Google Scholar]
- 66.Beaver WL, Wasserman K, Whipp BJ. Improved detection of lactate threshold during exercise using a log-log transformation. J. Appl. Physiol. (Bethesda, Md. : 1985) 1985;59:1936–1940. doi: 10.1152/jappl.1985.59.6.1936. [DOI] [PubMed] [Google Scholar]
- 67.Kemp GJ, Meyerspeer M, Moser E. Absolute quantification of phosphorus metabolite concentrations in human muscle in vivo by 31P MRS: A quantitative review. NMR Biomed. 2007;20:555–565. doi: 10.1002/nbm.1192. [DOI] [PubMed] [Google Scholar]
- 68.Golding EM, Teague WE, Jr, Dobson GP. Adjustment of K' to varying pH and pMg for the creatine kinase, adenylate kinase and ATP hydrolysis equilibria permitting quantitative bioenergetic assessment. J. Exp. Biol. 1995;198:1775–1782. doi: 10.1242/jeb.198.8.1775. [DOI] [PubMed] [Google Scholar]
- 69.Kemp GJ, Ahmad RE, Nicolay K, Prompers JJ. Quantification of skeletal muscle mitochondrial function by 31P magnetic resonance spectroscopy techniques: a quantitative review. Acta Physiol. (Oxf.) 2015;213:107–144. doi: 10.1111/apha.12307. [DOI] [PubMed] [Google Scholar]
- 70.Meyer RA. A linear model of muscle respiration explains monoexponential phosphocreatine changes. Am. J. Physiol. 1988;254:C548–C553. doi: 10.1152/ajpcell.1988.254.4.C548. [DOI] [PubMed] [Google Scholar]
- 71.Khushu S, Rana P, Sekhri T, Sripathy G, Tripathi RP. Bio-energetic impairment in human calf muscle in thyroid disorders: A 31P MRS study. Magn. Reson. Imaging. 2010;28:683–689. doi: 10.1016/j.mri.2010.01.006. [DOI] [PubMed] [Google Scholar]
- 72.Trenell MI, Sue CM, Kemp GJ, Sachinwalla T, Thompson CH. Aerobic exercise and muscle metabolism in patients with mitochondrial myopathy. Muscle Nerve. 2006;33:524–531. doi: 10.1002/mus.20484. [DOI] [PubMed] [Google Scholar]
- 73.Jeneson JA, Wiseman RW, Westerhoff HV, Kushmerick MJ. The signal transduction function for oxidative phosphorylation is at least second order in ADP. J. Biol. Chem. 1996;271:27995–27998. doi: 10.1074/jbc.271.45.27995. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.