

Abstract
Desmoplastic solid tumors are characterized by the rapid build-up of extracellular matrix macromolecules, such as Hyaluronic acid (HA). The resulting physiological barrier prevents the infiltration of immune cells and also impedes the delivery of anticancer agents. We report the development of a hypervesiculating E.coli Nissle (ΔECHy) based tumor targeting bacterial system capable of distributing a fusion peptide, Cytolysin A (ClyA)-Hyaluronidase (Hy) via outer membrane vesicles (OMVs). The capability of targeting hypoxic tumors, manufacturing recombinant proteins in-situ and the added advantage of an on-site OMV based distribution system makes the engineered bacterial vector a unique candidate for peptide delivery. The HA degrading potential of Hy for stromal modulation is combined with the cytolytic activity of ClyA followed by testing it within syngeneic cancer models. ΔECHy is combined with immune checkpoint antibodies and tyrosine kinase inhibitors to demonstrate that remodeling the tumor stroma results in the improvement of immunotherapy outcomes and enhancing the efficacy of biological signaling inhibitors. The biocompatibility of ΔECHy is also investigated to show that the engineered bacteria is effectively cleared, elicits minimal inflammatory and immune responses, and therefore could be a reliable candidate as a live biotherapeutic.
Keywords: e.coli nissle, outer membrane vesicle, desmoplastic cancer, stromal remodeling, hyaluronic acid, immunotherapy, targeted therapy
Graphical Abstract
Engineered hypervesiculating E.coli Nissle capable of localizing in tumors is utilized to deliver the Cytolysin A (ClyA)-Hyaluronidase (Hy) fusion protein by outer membrane vesicles to modulate the tumor matrix. Recombinant Hy decreases tumor tissue hyaluronic acid thereby improving the penetrability of anticancer agents and potentiation their effect in mouse models of breast and colon cancer.
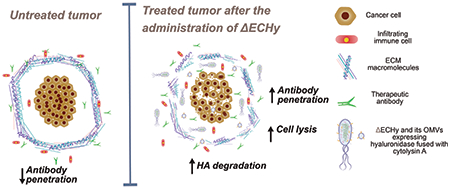
1. Introduction
Solid tumors display a vast heterogeneous milieu that consists of epithelial cells, mutated highly proliferative epithelial cells, abnormal blood vessels, fibroblasts, cancer associated fibroblast (CAFs), proteins, proteoglycans and glycosaminoglycans, all densely swamped in a sea of interstitial fluid[1]. Another significant hallmark of solid tumors is the development of an unorganized network of compressed and highly permeable blood vessels exhibiting abnormal morphology, ultimately resulting in heterogeneous perfusion and deficient drainage[2]. The overgrowth of fibrous connective tissue in a rapidly proliferating tumor matrix works in tandem with poor blood perfusion further impeding the influx and distribution of nutrient supply thereby promoting hypoxic conditions in the tumor microenvironment (TME) [3]. The resulting hypoxia has unfavorable therapeutic implications aiding tumor progression by modulating the apoptotic pathways and mitochondrial activity, inducing autophagy and inactivating the tumor suppressive p53 pathway[4]. Abundantly dispersed within the tumor extracellular matrix (ECM) are CAFs that secrete fibrotic macromolecules that result in a highly dense and difficult to navigate extracellular space[5]. This severe desmoplastic reaction in combination with poor perfusion forms a physical barrier hindering tumor access, which is a major cancer cell resistance mechanism inhibiting the infiltration of cancer-cell eliminating immune cells and therapeutic agents, especially large macromolecules, such as therapeutic antibodies and nanoparticles[6]. Adhesion of cancer cells to ECM elements is essential for their survival and ECM macromolecules have been demonstrated to promote drug resistance via multiple biological mechanisms[7]. For example, Hyaluronic acid (HA) which is a glycosaminoglycan secreted by stromal CAFs has been implicated in mediating resistance to tyrosine kinase inhibitors (TKIs), such as lapatinib, by reducing its accumulation and also improving the apoptotic threshold of cancer cells[8]. Hence modulating and engineering the tumor stroma to penetrate this complex desmoplastic barrier is critical for efficient drug delivery and successful immuno- and targeted therapies.
Targeting ECM macromolecules via CAF inhibition or enhancing their degradation is one way to improve the penetration of immuno- and targeted therapeutic agents. Presently, no therapeutic regimen has received the US Food and Drug Administration’s approval for specifically targeting this desmoplastic reaction. Clinical trials with small molecules such as calcipotriol, nintedanib and metformin that target CAFs are ongoing[7a, 9]. These molecules act on the CAFs thereby inhibiting the synthesis of extracellular macromolecules. An alternative approach is to enhance the degradation of these macromolecules using biologics such as the oncolytic adenovirus VCN-01 carrying the hyaluronidase (Hy) gene, which is also being currently tested in phase I clinical trials[7a, 9–10]. Live biotherapeutics in the form of programmable bacteria offer unique advantages that can be leveraged for delivering recombinant proteins, thereby modulating molecular mechanisms in-vivo and altering cancer cell proliferation and disease progression[11]. A significant amount of intravenously administrated conventional nanoparticulate carriers have been shown to be rapidly eliminated by the hepatobiliary and the renal route of systemic elimination[12]. Facultative anaerobic bacteria on the other hand have been found to localize and multiply in the TME by taking advantage of the leaky tumor vasculature, immunodeficient TME and the hypoxia associated tumor-tropism[13]. Even a minute fraction of bacterial dose reaching the tumor is sufficient to repopulate and allow therapeutic concentrations of peptide drug to be maintained for a long period of time. Moreover, selecting a commensal or a probiotic bacterial strain will help in ensuring biocompatibility. Escherichia coli Nissle (EcN) is an excellent template for bioengineering applications and has been historically used as an oral probiotic and has also entered clinical trials for delivering recombinant proteins in-vivo[11, 14]. The facultative anaerobic nature and amenability for genetic modification allows EcN to be used as a carrier for targeting hypoxic tumors and delivering peptide based drugs in-vivo[14e, 15].
Sophisticated quorum sensing based lytic population control has been previously reported to release therapeutic payload in-situ[16]. We report an alternate strategy where engineered bacteria is used to generate outer membrane vesicles (OMVs) carrying a therapeutic payload for controlling in-situ distribution of the payload in a spatial and temporal fashion, acting analogous to nanoparticulate carriers navigating and distributing the payload within the tumor. OMVs are comparable to mammalian cell derived exosomes, since these are nanosized vesicles that play a functional role in cellular communication[17]. We reasoned that hypervesiculating EcN that have localized at the tumor site would release OMVs packed with therapeutic peptides in-situ, which would enhance tumor penetrability due their nanoscale size, thus delivering the payload deep within the tumor thereby considerably improving the distribution of recombinant peptides, particularly in dense desmoplastic tumors. This concept is explored by designing a hypervesiculating EcN, generating OMVs packaged with Hy (ΔECHy), for degrading extracellular HA in tumors, and fused to a membrane anchoring protein Cytolysin A (ClyA), which is also a hemolytic protein known to be cytolytic towards mammalian cells (the fusion protein abbreviated as CHy) (Figure 1 A)[18]. This system is tested in syngeneic mouse tumor models to induce stromal changes that would aid anticancer agent penetration and promote tumor resolution (Figure 1B). The co-expression of two different classes of recombinant proteins, Hy and ClyA, acting synergistically on ECM and cancer cells, respectively, is expected to take advantage of a programmable microbiome in serving not only as a monotherapy platform but also complementing existing therapies such as immuno- and targeted therapies.
Figure 1:
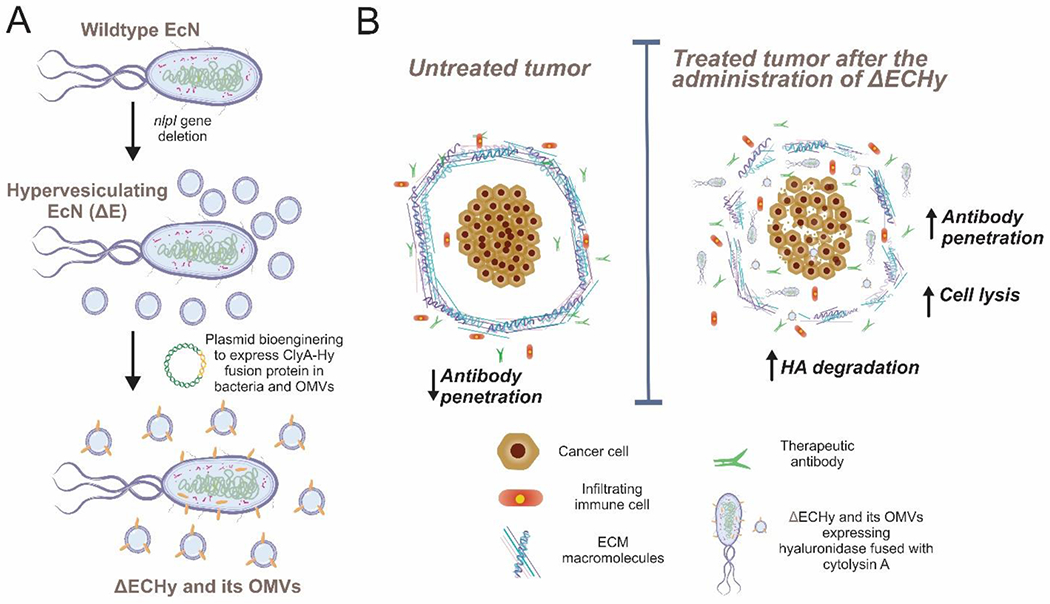
(A) Schematic representing engineering of EcN into ΔECHy. (B) Graphical abstract showing the tumor targeting ability of anaerobic bacteria, ΔECHy after systemic administration. Bacteria produce membrane and OMV confined CHy which is distributed throughout the tumor matrix by OMVs. Recombinant Hy considerably decreases tumor tissue HA thereby improving the penetration of antibodies and immune cells, and decreasing resistance to targeted therapies.
2. Results and Discussion
2.1. Genomic deletion on nlpI from EcN to obtain hypervesiculating ΔE
To develop a hypervesiculating strain of EcN, its genome was modified by deleting the nlpI gene as reported previously for similar strains of Escherichia coli[19]. Deletion of the nlpI gene has been reported to be safe for bacterial membrane integrity and stability[19c, 20]. EcN expressing the λ-red recombination system proteins, gamma, exo and beta, was electroporated with a double stranded DNA encoding the chloramphenicol resistance cassette gene with overhangs homologous to the gene sequences upstream and downstream of the nlpI gene. The λ-red recombineering system excises the nlpl gene from the genomic DNA and replaces it with the electroporated dsDNA containing the chloramphenicol cassette (Figure S1A, Supporting Information)[21]. Bacterial colonies obtained after chloramphenicol based antibiotic plate selection were analyzed via colony polymerase chain reaction (PCR) (Figure S1, Supporting Information) to confirm the integration of the chloramphenicol cassette. The PCR amplified product was further analyzed by Sanger sequencing which reconfirmed the previous results. Thus, the nlpI gene was successfully replaced to obtain a hypervesiculating strain of EcN, ΔE.
2.2. Phenotyping ΔE for enhanced OMV production
ΔE was evaluated phenotypically for assessing the increase in OMV production via protein measurements with the bicinchoninic acid assay (BCA). ΔE produced a higher OMV yield, 1.75±0.2 mg/L which is approximately twice in comparison to the non-hypervesiculating EcN (0.85±0.31 mg/L). ΔE and its OMVs analyzed via Transmission Electron Microscopy (TEM) revealed nanosized vesicles that had diameters ranging from 80 to 400 nm, in agreement with the size range reported elsewhere (Figure 2A and 2B)[22]. The nanosized nature and size distribution of the isolated OMVs was further confirmed via dynamic light scattering (DLS) (Figure 2C). The mean vesicle diameter was calculated as 101.47±25.85 nm while the polydispersity index (PDI), which is a measure of particle size distribution was between narrow to moderately polydisperse (0.2-0.3). The production of OMVs is a natural biological process inherently controlled by its mechanism of formation and release (blebbing of the outer membrane and explosive cell lysis)[22]. Thus, a variation in size distribution and Z-average is reasonably expected with each batch isolated.
Figure 2:
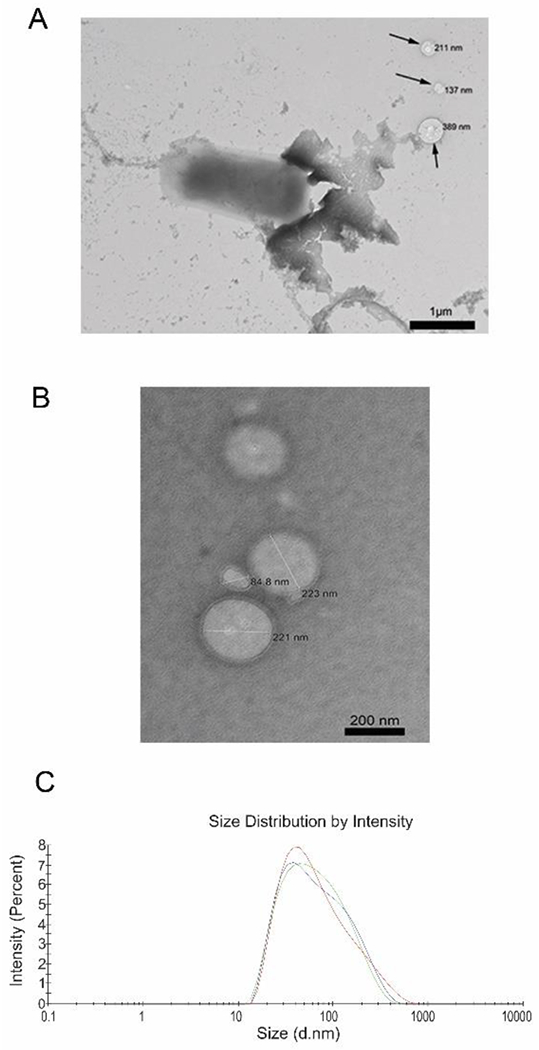
(A, B) TEM images of ΔE and isolated OMVs showing nanosized vesicles. (C) Size analysis of ΔE derived OMVs via DLS. A nanoparticle population with moderate polydispersity was obtained.
2.3. Designing Cytolysin A (ClyA)-Hyaluronidase (Hy) (CHy) fusion protein
Previously, human and bovine forms of Hy, such as the sperm surface protein PH-20 (PH20/SPAM 1, 509 amino acids) have been cloned for therapeutic objectives[23]. For complete catalytic activity, the enzyme needs to undergo post-translational modifications or else the enzyme would perform sub-optimally, especially when being cloned in bacteria[23b]. Hence, an alternate form of Hy of bacterial origin was selected that does not require post-translational modifications. A 217 amino acid sequence of Hy from Streptomyces koganeiensis (UniProtKB#A0A0U2E2J7) has been reported to be enzymatically active against high molecular weight HA which was subsequently selected for cloning and expression[24]. Moreover, its small size (≈24.5 kDa) makes it a favorable candidate for synthetic cloning. Further, the enzyme needs to be translocated onto the bacterial outer membrane which would then be subsequently packaged inside the OMVs as they are being formed. Fusion of proteins at the C terminal of ClyA has been extensively studied for bacterial surface and OMV localization[19a, 19c]. ClyA is 34 kDa transmembrane pore-forming hemolytic protein (UniProtKB#P77335) known to be cytolytic towards mammalian cells and macrophages[15a]. Fusion of fluorescent proteins and enzymes with the ClyA gene has been utilized to transport and localize these protein onto the bacterial membrane and further onto the OMVs without any loss in functional activity (Figure 3A)[19a, 19c, 19d, 25]. ClyA retains its pore forming cytolytic activity even after the addition of peptide fragments on either ends as shown previously[26]. Thus, ClyA serves two roles– primarily as an anchoring protein for Hy and secondarily to exert a cytotoxic effect against cancer cells. Hence, for bacterial membrane localization a fusion protein was designed, CHy encoding ClyA on the 5’end and Hy on the 3’ end with a TEV cleavage site in the middle, having glycine (G4) spacers/linkers on either side (≈56.5 kDa). EcN codon optimized gene blocks were obtained for the fusion protein and incorporated it into a high copy number plasmid to obtain pecol19A-ClyA-Hy (Figure S2, Supporting Information). The designed plasmid has a strong E.coli constitutive promoter, J23119 that would drive the production of the fusion protein without an inducer[27]. The assembled plasmid was sequence verified and subcloned into ΔE to obtain ΔECHy. Further, the metabolic burden due to the production of recombinant proteins can also affect the growth of bacteria. The J23119 promoter and ClyA mediated membrane localization have been reported to have negligible effects on bacterial hosts cellular capacity and growth rate[19c, 28]. The effect of the plasmid on the growth rate of ΔECHy was analyzed and found to be comparable with EcN (Figure S3, Supporting Information).
Figure 3:
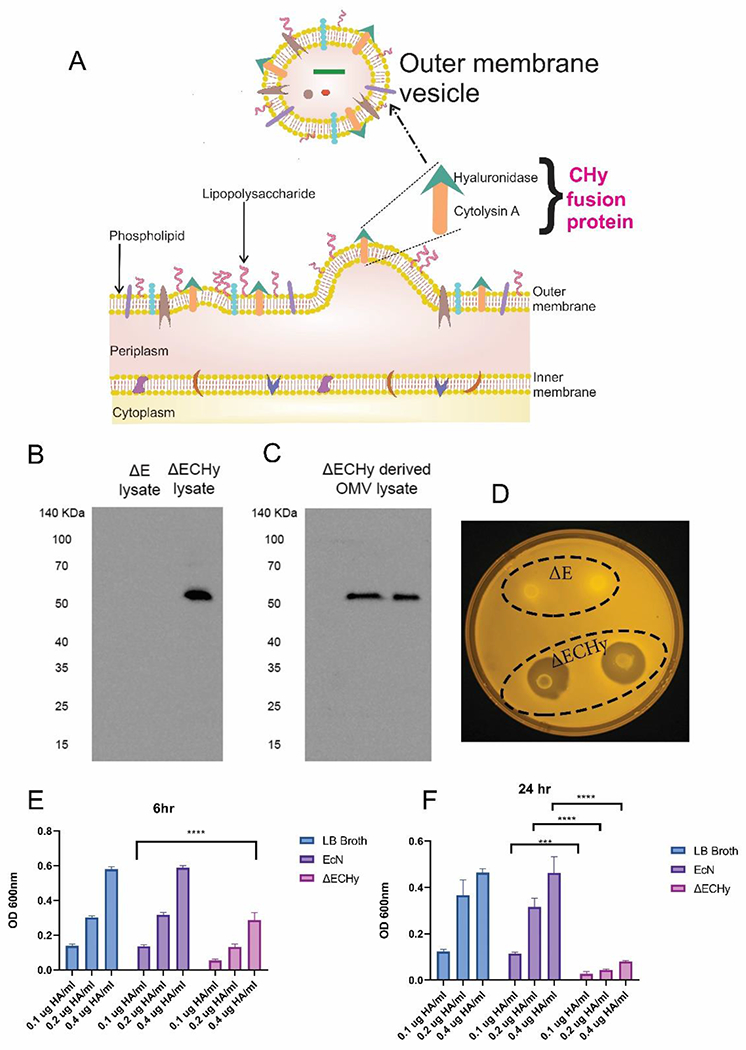
(A) Schematic showing translocation of CHy fusion protein to the bacterial outer membrane and vesiculation to form OMVs. (B, C) Immunoblotting for detecting the presence of CHy protein (56 kDa). ΔE lysate was used as the negative control and ΔECHy lysate and ΔECHy OMVs were analyzed for the presence of CHy. (D) Qualitative analysis for Hy activity using the HA agarose plate assay. Zones of degradation were observed around the ΔECHy bacteria while ΔE showed no activity. (E, F) Quantitative estimation of Hy activity using the HA degradation spectrophotometric assay for three different concentration of HA (0.1, 0.2, 0.4 μg/ml). Significant reduction in HA optical density was observed for the ΔECHy group in comparison to ΔE after 6 hours (p<0.0001) and 24 hours (p<0.0002 for 0.1μg /ml and p<0.0001 for 0.2 and 0.4μg /ml) (two-way anova, data represented as mean±SD, compared using two-way anova (tukey’s multiple comparisons test).
2.4. Localization of functional CHy in ΔECHy OMV’s
Next, ΔECHy and its derived OMVs were analyzed for the presence of recombinant CHy. ΔECHy lysates and its OMVs were tested by SDS-PAGE immunoblotting with antibodies specific for the TEV cleavage site. Bands at the molar mass corresponding to 55.6 kDa were observed in both samples, which were absent in the negative control (i.e., ΔE lysate) (Figure 3B and 3C). This confirmed that ClyA is an efficient carrier protein for translocation of fused Hy on OMV’s. CHy was evaluated for its functionality via a qualitative agarose plate-based HA degradation assay. The plate-based method is a test to analyze HA degradation by observing zones of degradation/clearance around regions of bacterial growth in response to precipitation of HA. Clear regions around the circular bacterial disk of ΔECHy were observed after 24 hours which were transparent in comparison to the region inoculated with ΔE, which was opaque (Figure 3D). A quantitative assay was also performed for analyzing Hy mediated HA degradation using turbidimetry. This spectrophotometric assay is based on the precipitation of solubilized HA using a surfactant, cetyltrimethylammonium bromide (CTAB) followed by measuring the optical density at 600 nm[29]. Three different concentrations of HA 0.1, 0.2 and 0.4 μg/ml were analyzed for three different groups, namely LB broth, ΔE and ΔECHy. After a period of 6 hours, ΔECHy showed an approximate 50% reduction in optical density for all the concentrations tested, which increased to to 80% after a period of 24 hours. Insignificant changes in optical density were observed for the LB Broth negative control and the ΔE groups (Figure 3E and 3F). The assays confirmed the successful synthesis of a hypervesiculating strain of EcN (ΔECHy).
2.5. Tumor screening for the presence of fibrotic elements
Multiple tumor models, both xenograft and syngeneic, in immunocompromised and immunocompetent mice were screened for evaluating the composition of extracellular matrix proteins such as HA, periostin, fibronectin and collagen IV. Tumor specimens of human origin breast (MDA-MB-231) and prostate (PC3) cancers; and murine origin breast (4T1), colon (MC38) and pancreatic (RInk1) cancers were analyzed for the fibrotic markers by immunohistochemistry (IHC), where MDA-MB-231, 4T1, MC38 and RInk1 tumors were shown to contain significantly higher amounts of these fibrotic elements (Figure S4, Supporting Information) over PC3 tumors. We reasoned that targeting one or a combination of these elements in vivo for stromal modulation is expected to improve the perfusion dynamics in tumors and thereby have significant improvement in drug delivery of small molecules and macromolecules.
2.6. Localization and biodistribution of ΔE and OMVs in tumor models
4T1 (breast) and MC38 (colon) were selected as tumor models for further evaluation of ΔECHy due to the syngeneic nature of these models, and the reliable development of a densely fibrotic and hypoxic tumor that is essential to study the localization of engineered facultative anaerobes[30]. ΔElux, a bioluminescent strain containing the constitutively expressed luciferase (lux) reporter gene, was administered intravenously in 4T1/BALB/cJ and MC38/C57BL/6 tumor bearing mice once a tumor size of 200–300 mm3 was attained. After allowing 24 h for tumor seeding and populating, consistent bioluminescence (BLI) signals were observed to originate from the tumors in both the models thereby indicating that ΔElux retains the tumor homing properties of EcN (Figure 4A). The BLI signals were exclusively originating from tumors suggesting successful seeding and proliferation of the bacteria in the immunocompromised TME and the clearance of bacteria from all major organs. While BLI is a highly specific modality to study growth kinetics of living therapeutics non-invasively in-vivo, it is not the most sensitive technique to accurately estimate the biodistribution profile and trace the molecular signatures of these agents in-vivo. Hence, the study was repeated with an intravenous administration of ΔECHy in 4T1 and MC38 tumor models. The mice were euthanized, and various organs were collected 72 hours post administration, processed and the total DNA was analyzed for the presence of pecol19A-ClyA-Hy plasmid via PCR, a highly specific and sensitive modality, albeit invasively. The plasmid sequence was detected exclusively from tumors in both models with no signal from other organs (Figure 4B and C). These results indicate the proficiency of bacteria in selectively populating the tumor matrix in a spatial and temporal fashion with continued propagation and maintenance of their population in the hypoxic and immunocompromised tumor microenvironment.
Figure 4:
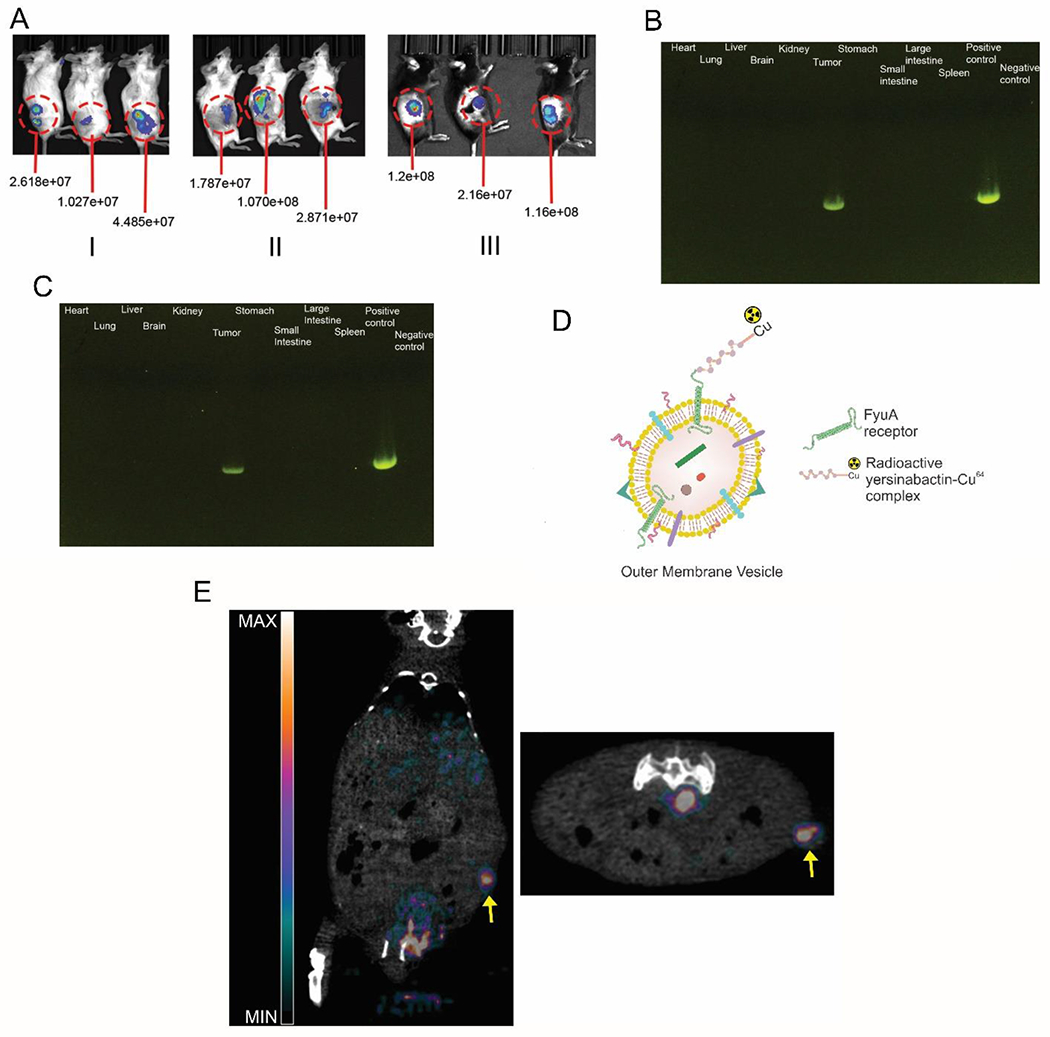
(A) Bioluminescence images showing (I) EcN-luxCDABE accumulation in 4T1 tumors in BALB/cJ mice after intravenous injection at the end of 24 hours (n=5). (II) ΔElux accumulation in 4T1 tumors in BALB/cJ mice after intravenous injection at the end of 24 hours (n=5). (III) ΔElux accumulation in MC38 tumors in C57BL/6 mice after intravenous injection at the end of 24 hours (quantified intensity units in radiance) (n=5). PCR analysis of tissue sample for detecting pecol19A-ClyA-Hy plasmid fragments in 4T1 (B) and MC38 (C) tumors (731bp) (n=4). (D) Schematic depicting the labelling of OMVs with the 64Cu-YbT complex. (E) PET/CT images along the longitudinal (left) and transverse (right) axis showing in-vivo biodistribution of 64Cu-YbT labelled OMVs and preferential uptake in tumor. Arrow points towards the tumor location (n=3).
Considering OMVs are generated from ΔECHy in situ in our strategy, we sought to evaluate the biodistribution profile of OMVs to determine whether they exhibit higher tropism to other organs, over tumor, in the event there is infiltration of OMVs into the systemic circulation from the tumor. OMVs are expected to exhibit tumor-tropism, accumulation and penetration characteristics via the enhanced permeability and retention (EPR) effect, akin to nanoparticle-based carriers. To test this hypothesis and simulate biodistribution of OMVs in the systemic vasculature, OMVs were isolated from the hypervesiculating bacteria and purified before radiolabeling them with copper-64 labeled Yersiniabactin (64Cu-YbT), a radioactive siderophore complex, to track them in vivo using Positron Emission Tomography (PET) imaging [31]. 64Cu-YbT complex binds selectively to the outer membrane FyuA receptor, a siderophore transporter, expressed in multiple species of Escherichia coli and their derived OMVs, including Nissle and has been shown to play a role in bacterial copper acquisition and import[31a, 32]. Hence, the radioactive complex was used to tag OMVs for tracking them in-vivo (Figure 4D). Radiolabeling OMVs with 64Cu-YbT and processing to remove the unbound complex yielded approximately 7.4 MBq of radioactivity in 3 mg of OMVs. A 3-dimensional PET/CT scan was performed 4 hours post intravenous administration of 300 μg of radiolabeled OMVs (0.74 MBq). Strong signals were observed originating from the tumor location, compared to other major organs (Figure 4E). This result illustrates the high tumor-tropism of OMVs and their likely low dissemination potential beyond the tumor site. Thus, OMVs released by hypervesiculating EcN localizing within tumors are likely to be retained within the tumor matrix due to the EPR effect.
2.7. ΔECHy-mediated tumor stromal remodeling and potentiation of lapatinib response
After confirming the tumor homing property of ΔE and demonstrating the expression of functional CHy on the bacterial surface and in the OMVs, the next goal was to evaluate the efficiency of HA degradation and its effect on improving targeted therapeutic outcomes in-vivo. It is advantageous to evaluate ΔECHy in syngeneic models since it would allow testing the live biotherapeutic in immunocompetent mice and take advantage of the native immune responses triggered against the tumor in combination with synergistic therapeutic agents such as TKIs. There are several studies that implicate HA in TKI treatment resistance across several tumor types. For example, HA has been shown to be associated with modifying responses to TKIs by activating the CD44 and the cell-surface RHAMM receptors that also act as co-receptors for activating transmembrane tyrosine kinases (e.g., EGFR, c-MET, PDGFR and ERK) and their multiple pathways downstream (e.g., Rho GTPases and Ras GTPases in the Rho and MAPK signaling pathways)[33]. Stromal HA secreted by CAFs is shown to be essential in developing lapatinib resistance by protecting against the accumulation of drug and the resulting pro-survival CAF signaling pathways such as the JAK2/STAT3 pathway and improving the apoptotic threshold of cancer cells. Intratumoral injection with Hy has been found to significantly retard tumor progression and enhance the sensitivity of cancer cells towards lapatinib[8a]. Thus, for a small molecule like lapatinib, significant improvements can be expected by disrupting the HA-CD44 axis by modulating and reducing stromal HA.
Lapatinib has been shown to have a therapeutic effect in 4T1 tumors at multiple doses ranging from 75-100 mg/kg orally, however with HA linked CD44 signaling, development of resistance has also been reported for the drug[8a, 34]. Thus, ΔECHy-mediated HA degradation should significantly enhance the activity of lapatinib and a therapeutic response could be observed at subcytotoxic doses of the drug. To test the hypothesis, mice were treated orally with lapatinib at a dose of 5 mg/kg (Figure 5A). The following 5 treatment groups were assessed in parallel: control mice with saline injections, non-hypervesciculating ECHy+lapatinib, ΔECHy, lapatinib and ΔECHy+lapatinib. Figure 5B represents the mean tumor volume progression with time (tumor volume-time plot for each mouse for all the groups can be found in Figure S5, Supporting Information). 4T1 tumors grow at a fast rate and as anticipated, mice in the control group (saline) reached their experimental endpoint (tumor size of 1000 mm3) quickly with a median survival of 13 days. The tumor growth rate was found to be comparatively slower in the lapatinib treated group, with a median survival of 20 days, which indicate that even subcytotoxic doses administered were able to attenuate tumor progression. The non-hypervesiculating ECHy and lapatinib combination showed a profile similar to the lapatinib only treatment group with a median survival of 22 days, indicating that a non-hypervesiculating strain with CHy is not able to substantially amplify and complement the therapeutic action of lapatinib. ΔECHy alone also demonstrated a progression profile similar to the two treated control groups, with a median survival of 22 days, indicating that hypervesiculation and OMV mediated ClyA distribution is causing cytolytic activity on tumor cells and concurrently improving tumor penetration of immune cells and potentiating the immune response from Hy activity. The combination of lapatinib with ΔECHy demonstrated the slowest progression in tumor growth in comparison to the other four groups tested, with a significant increase in median survival to up to 29 days (ΔECHy+lapatinib vs control: **p=0.0021, ΔECHy+lapatinib vs lapatinib: *p=0.0421, ΔECHy+lapatinib vs ECHy+lapatinib: *p=0.0173 and ΔECHy+lapatinib vs ΔECHy; **p=0.0019) (Figure 5C). This suggests that hypervesiculation leads to increased production of OMVs, allowing enhanced distribution and penetration of CHy inside the tumor matrix, even if the bacteria are themselves physically immobile and are possibly stuck in the dense stroma. Hence, OMVs generated in-situ distribute Hy more effectively and uniformly inside the tumors, permitting the penetration of drugs and potentiating their effect.
Figure 5:
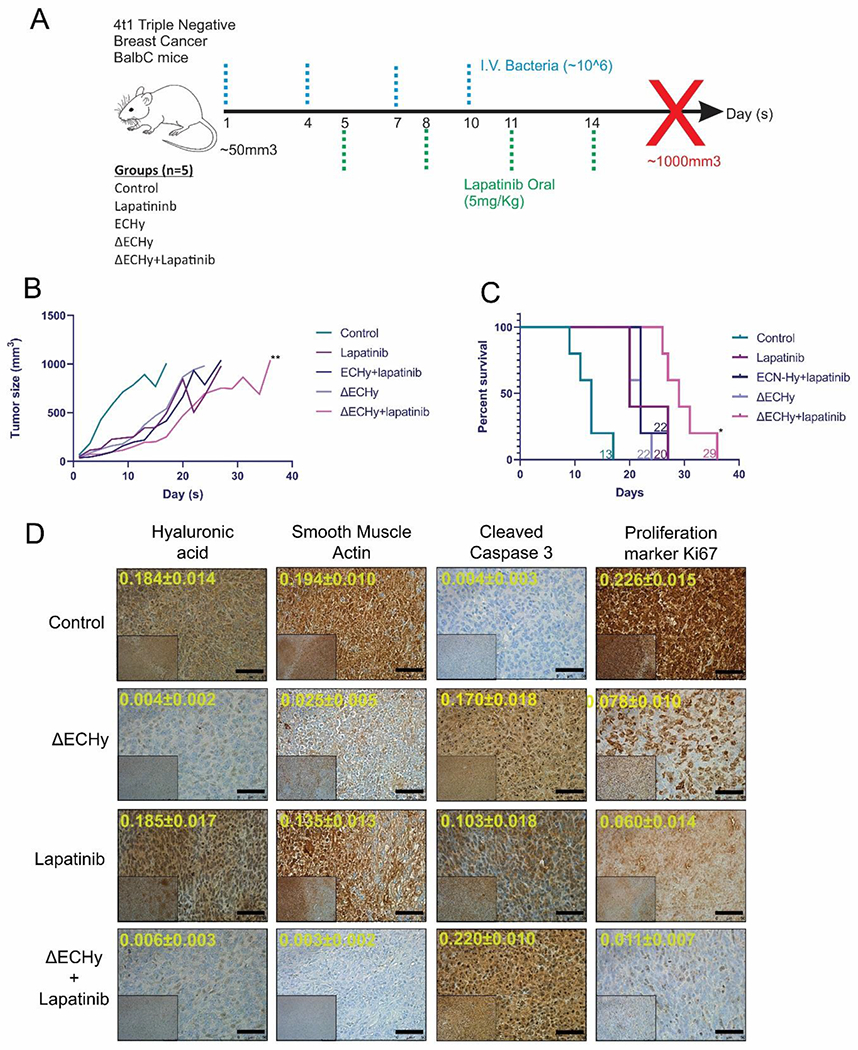
(A) Treatment protocol for the in-vivo analysis of HA degradation in 4T1-BALB/cJ mice. (B) Line plot for different groups showing mean tumor volume over the course of study (n=5). ΔECHy+lapatinib vs lapatinib comparison was performed using wilcoxon matched-pairs signed rank paired t-test, p=0.0078, p-value summary=**, data represented as mean. (C) Kaplan meier survival analysis for different groups tested. The four groups were compared, and the median survival time was calculated using the log-rank (Mantel–Cox) test, which is indicated for each group (corresponding color-coded numerals) (*p=0.0421). (D) Representative images for IHC analysis of tumor tissues from different treatment groups: saline control, ΔECHy, lapatinib and ΔECHy+lapatinib (inset x100 and outset x400). Quantification of markers (arbitrary units) is indicated in yellow digits for each image (data represented as mean±SD, compared using two-way anova (tukey’s multiple comparisons test). Black bar for scale corresponds to 50 μm.
To further elucidate the stromal changes in the different treatment groups, tumor samples were analyzed using IHC (Figure 5D and Figure S6, Supporting Information). ΔECHy treated tumors showed a significant reduction in extracellular HA compared to control tumors. These findings at the tissue level correlate well with the results from the tumor progression study. Previous studies have shown that the downregulation of CAFs in tumors is associated with lowering of smooth muscle actin as a consequence of tumor architecture remodeling[35]. Thus, the tumors were analyzed for smooth muscle actin as a CAFs marker. ΔECHy and lapatinib treated groups showed a higher reduction of smooth muscle actin, which suggests there is a proportional decrease in CAF numbers. Analyzing cellular proliferation is another aspect to qualitatively measure treatment response. As observed in Figure 5D, apart from the control group, all the other treatment groups showed cells with high degree of cleaved caspase 3 (CC3) staining, which is an indicator of cells undergoing apoptosis. Further, the highest reduction in the number of proliferating cells (Ki67+) was also observed in the ΔECHy+lapatinib treatment group. These observations further support the impact of ΔECHy in enhancing tumor tissue remodeling and improving the effect of the targeted therapeutic agent, lapatinib. Quantitative estimation of the stromal markers also indicates an ΔECHy mediated enhancement in the therapeutic effect offered by lapatinib (Figure S6, Supporting Information). These observations clearly support the multipronged impact of ΔECHy as an effective agent for stromal remodeling, cytolytic therapy, and improving the activity of anticancer targeted therapeutics.
2.8. ΔECHy mediated tumor stromal modulation and potentiation of anti-PDL1 immune checkpoint antibody response
The buildup of rigid stroma in the tumor matrix is a physical barrier to macromolecules that reduces diffusion of therapeutic antibodies and immune cells to cancer cells. Hence, stromal modulation is key to enhancing the tumor penetration of macromolecules as demonstrated by previous studies where systemic administration of recombinant hyaluronidase (PEGPH20) improved tumor accumulation of antibodies such as rituximab, trastuzumab and PDL1 antibody[36]. Hy administration has also been shown to induce an immune response against the tumor tissue which could be a direct effect of enhanced infiltration of immune cells[36c, 37]. Thus, to test this hypothesis, 4T1 tumors and MC38 tumors were treated with ΔECHy and immune checkpoint blockade anti-PDL1 antibodies. A subtherapeutic dose of anti-PDL1 antibody was used to resolve the therapeutic potentiation afforded by ΔECHy. Anti-PD-L1 antibody, similar to lapatinib, is tested at a subtherapeutic dose of 5 mg/kg instead of the reported dose of 10 mg/kg[38].In the 4T1 model, the tumor progression for the anti-PDL1 group was similar to the control group, both showing a median survival of 13 days. PD-L1 combination therapy with ΔECHy did show a significant attenuation in tumor growth rate in comparison to anti-PDL1 antibody treatment (**p=0.0078). However this did not translate to an improved survival outcome over what was observed with ΔECHy alone, both with a median survival of 22 days (ΔECHy+anti-PDL1 antibody vs control: *p=0.0112, ΔECHy+anti-PDL1 antibody vs anti-PDL1 antibody: *p=0.0110 and ΔECHy+anti-PDL1 antibody vs ΔECHy; not significant p=0.5164) (Figure 6A and 6B and Figure S7, Supporting Information). 4T1 tumors are typically refractory to immune checkpoint blockade, a probable reason for which could be the vast number of immunosuppressive cells such as the regulatory T-cells and the myeloid derived suppressor cells populating the tumor matrix and lesser number of CD8+ T cells and natural killer (NK) cells infiltrating the tumor[38]. Thus, a possible reason for such an outcome could be the immunosuppressive nature of tumor or the low dose being insufficient to elicit a response[38].
Figure 6:
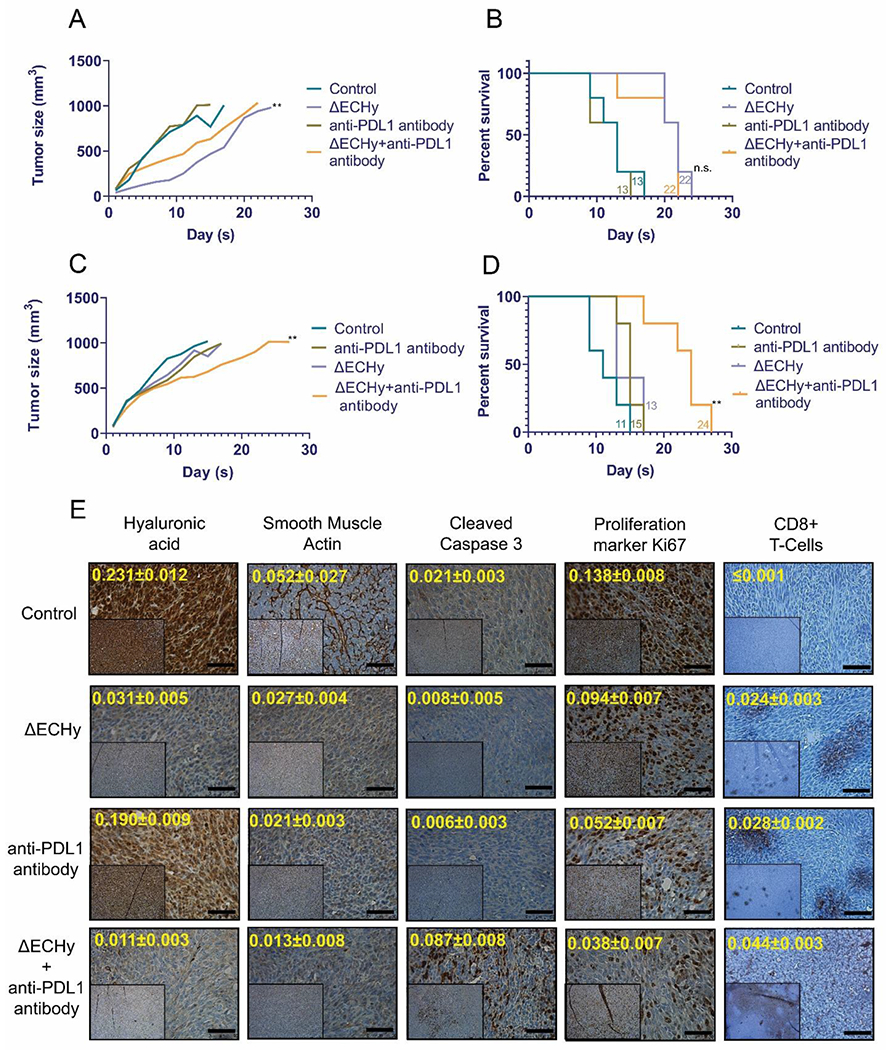
Line plot and kaplan meier survival analysis for different groups combining ΔECHy with PDL1 antibody showing mean tumor volume over the course of study in 4T1 tumors (A and B) and MC38 tumors (C and D) (n=5) (**p=0.0078, wilcoxon matched-pairs signed rank paired t-test). The four groups were compared, and the median survival time was calculated using the Log-rank (Mantel–Cox) test, which is indicated for each group (corresponding color-coded numerals) (**p=0.080). (E) IHC analysis of tumor tissues from different treatment groups in MC38 tumor models: saline control, ΔECHy, anti-PDL1 and ΔECHy+anti-PDL1(inset x100 and outset x400). Quantification of markers (arbitrary units) is indicated in yellow digits for each image (data represented as mean, compared using two-way anova (tukey’s multiple comparisons test). Black bar for scale corresponds to 50 μm.
In MC38 tumors, ΔECHy and anti-PDL1 groups showed a slight reduction in the tumor growth rate in comparison to control (saline) with a modest increase in survival from 11 days (control) to 15 (anti-PDL1) and 13 (ΔECHy) days. However, the ΔECHy and anti-PDL1 combination groups displayed a considerable improvement in tumor survival with the median survival to up to 24 days (ΔECHy+anti-PDL1 antibody vs control: **p=0.0017, ΔECHy+anti-PDL1 antibody vs anti-PDL1 antibody: **p=0.0052 and ΔECHy+anti-PDL1 antibody vs ΔECHy; **p=0.080) (Figure 6C and 6D and Figure S7, Supporting Information). The different treatment groups were further analyzed by IHC (Figure 6E and Figure S8, Supporting Information). Similar to the lapatinib study, HA synthesis was again found to be qualitatively reduced in the ΔECHy treated groups. Smooth muscle actin was also found to be reduced in all the treatments groups in comparison to the untreated control thereby indicating positive stromal modulation. The combination therapy of ΔECHy with the immune checkpoint inhibitor antibody showed a qualitative and quantitative reduction in cellular proliferation. Analysis of the tumor tissue for the cytotoxic T-cell marker, CD8+, and the apoptosis marker, CC3, showed a widespread tissue distribution of cytotoxic T-cells and increased cellular apoptosis in the combination group in comparison to the other groups. MC38 tumors exhibit a vast expansion of T-cell populations and macrophage populations with significant numbers of CD8+ T-cells, CD4+ T-cells and NK cells throughout tumor development[38]. For this reason, in comparison to 4T1 tumors, MC38 showed a definitive improvement in survival with the combination of ΔECHy+PD-L1 antibody, suggesting that stromal engineering clearly potentiated the antibody therapy by enhanced permeation of the therapeutic antibody and subsequently immune cells, even at a subtherapeutic dose.
2.9. Biocompatibility and systemic elimination of ΔECHy
From a regulatory standpoint, a critical aspect of using a live biotherapeutic for therapy is the selection of an appropriate biocompatible and non-toxic cellular carrier. In an ideal scenario, when used for therapeutic applications, the engineered bacteria should exclusively populate tumors and be cleared from the body as the therapy progresses towards completion. Most importantly, it is desirable that neither the bacteria nor the degradation products elicit a detrimental immune response towards the heathy tissues. To better understand the in-vivo biodistribution and fate of ΔE, naive BALB/cJ mice were administered ΔElux intravenously and imaged after a period of 24 hours. No BLI signals were observed from the mice indicating clearance of ΔElux from the body (Figure 7A). BALB/cJ mice are immunocompetent and their immune system is capable of clearing the ΔE strains from the circulation quite easily. Any persistent ΔE in circulation or in the organs could be in very low numbers for repopulation. Nevertheless, at the end of 24 hours, ΔE population is certainly below the detection limits of the bioluminescent imaging system.
Figure 7:
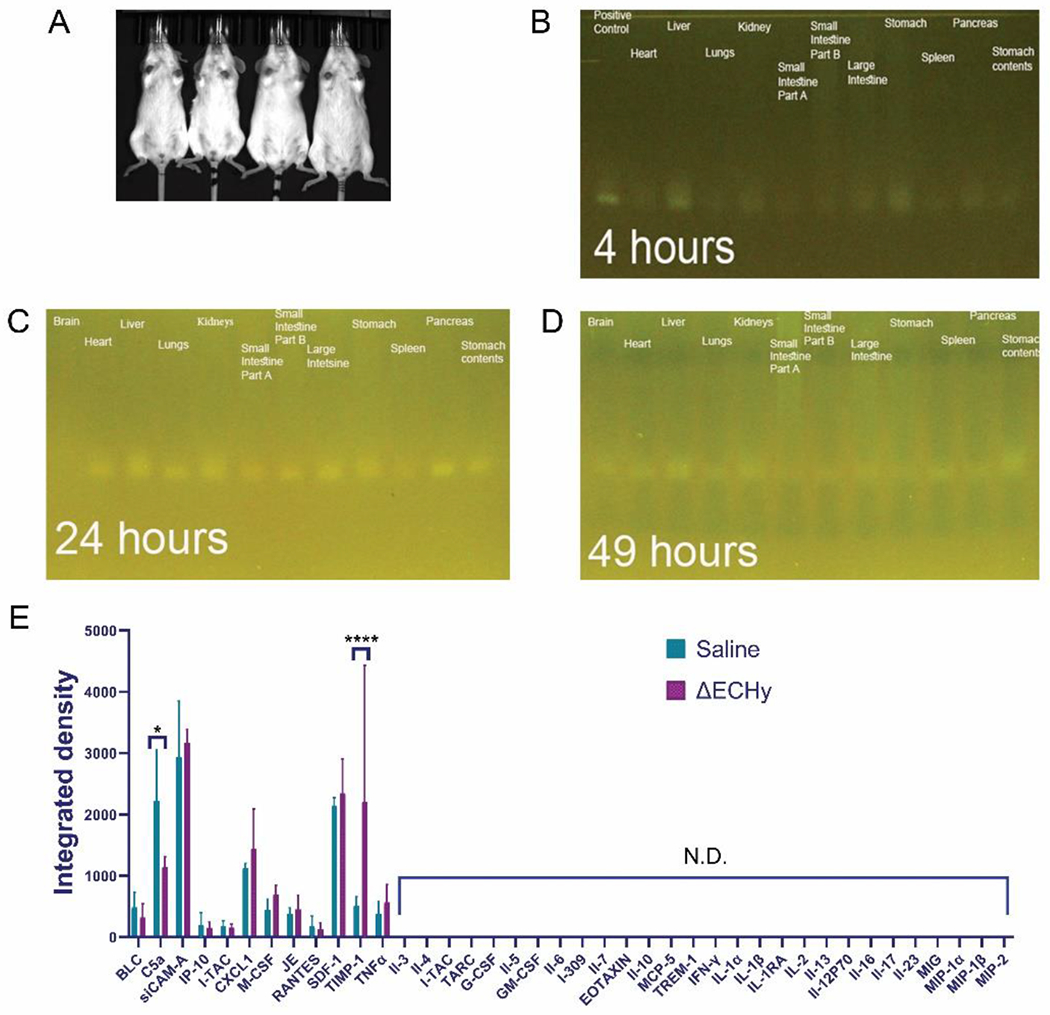
(A) Mice biodistribution study for ΔElux showing no detectable signals in-vivo (n=4). (B, C, D) PCR amplification for detecting the presence of ΔElux containing pakfplux1 plasmid from the microbiome genetic material isolated from each organ (121bp). (E) Graphical plot of cytokine profiling results from the biocompatibility study in mice. ΔECHy was compared with saline and only two cytokines out of the 40 tested were observed to be significantly regulated, complement component 5a (C5a, p=0.016) was downregulated and the tissue inhibitor of metalloproteinases (TIMP-1, p<0.0001) was upregulated. No other inflammatory markers were detected (n=3, data represented as mean±SD and compared using two-way anova (tukey’s multiple comparisons test).
A more sensitive method for detecting the presence of ΔE is to analyze different tissue samples for the presence of genomic components via PCR based amplification. The study was repeated in naive BALB/cJ mice and different organs collected at the end of 4, 24 and 49 hours respectively. The organs were processed for the isolation of total genomic DNA. The genomic contents were analyzed at each time point via PCR based amplification with primers against the pakgfplux luciferin-luciferase plasmid (pakgfplux1). The amplified reaction products were analyzed by nucleic acid gel electrophoresis. At the end of 4 hours a faint band was observed in the liver and stomach (Figure 7B). However, at the end of 24 hours faint bands were also observed in various organs of the digestive tract, heart, liver, lungs and the kidneys (Figure 7C). This could either be due to presence of ΔElux or due to the residual plasmid/genomic contents or their fragments in circulation. 49 hours post intravenous injection the bands started to become weaker (Figure 7D) and disappear eventually. Thus, the in-vivo study in healthy mice shows initial rapid elimination which then gradually declines over time. The remaining small number of bacteria are cleared slowly, and as shown through PCR analysis there was no trace of bacteria in the body.
Analyzing the different markers of immune response and inflammatory mediators is expected to provide a better picture of ΔE biocompatibility. ΔECHy was administered to naive BALB/cJ mice systemically via intravenous injection and 40 major cytokines and chemokines were measured in plasma after a period of 24 hours with a membrane-based antibody sandwich immunoassay. The 24-hour period is a satisfactory time point to measure any acute immune reaction since the biodistribution study showed that the majority of the administered dose is cleared within that time frame. An array plot data from the immunoassay was quantitated to generate a protein profile histogram for the detected proteins (Figure 7E and Figures S9A and S9B, Supporting Information). Among all cytokines and chemokines, only TIMP-1 (p<0.0001) was found to be significantly upregulated and C5a (p=0.0160) downregulated. Complement 5a or C5a is a part of the complement/clotting cascade. Tissue inhibitor of metalloproteinases or TIMP-1 is a metalloproteinase inhibitor and also a signaling cytokine known to attenuate chronic pain. The increased presence of TIMP-1 might be related to the method of asphyxiation induced euthanasia or because of the stress induced during submandibular vein blood collection. The cytokine profiling study, therefore, did not demonstrate any meaningful difference between the control and ΔECHy administered mice, which suggests there is no acute inflammatory and immune responses against ΔECHy. These results indicate that ΔECHy is cleared rapidly, does not induce any off-target toxicity nor has any detrimental effect towards animal health in mouse models. However, further studies are needed before ΔECHy can be deemed completely safe for in-vivo administration and therapy.
3. Conclusion (s)
In this report EcN was engineered to develop a hypervesiculating strain capable of over-generating nanoscale OMVs in-situ and also programmed to express Hy enzyme for degrading HA and ClyA for cytolytic activity. The enzyme and the strain were phenotyped to produce large quantities of OMVs with functional Hy enzyme. Designing a fusion protein with ClyA serves a dual purpose of anchoring Hy and also potentially killing cancer cells. The engineered strain was evaluated in combination with tyrosine kinase inhibitors and immune checkpoint antibodies to demonstrate that ΔECHy can remodel the tumor stroma and induce cancer cell killing that ultimately results in the improvement of immunotherapy outcomes and enhancing the activity of tyrosine kinase inhibitors. The biocompatibility of ΔECHy was investigated in-vivo by examining the cytokine and chemokine profile to demonstrate that the bacteria elicits minimal inflammatory and immune responses, and could be a reliable candidate as a live biotherapeutic.
4. Experimental Section/Methods
Chemicals, Antibodies and Kits
All chemical and reagents used were either of analytical grade or molecular biology grade and were purchased from commercial sources. The chemicals and reagents were stored following the manufacturers recommendations and used without further processing. Lapatinib for the animal studies was obtained from Apex Bio (#A8218). Anticoagulant Heparin for blood collection was obtained from Alfa Aesar (#A16198). For DNA fragment amplification and assembly, Q5 High Fidelity polymerase (NEB#M0491S) and the HiFi DNA Assembly Master Mix (NEB#E2621S) was used. Colony PCR was performed using the DreamTaq Green PCR Master Mix (ThermoFisher#K1081). Commercial kits for plasmid and DNA fragment isolation from PCR amplification reactions, nucleic acid gel electrophoresis and bacteria were used following the manufacturers prescribed protocol (Monarch Plasmid DNA Miniprep Kit, NEB #T1010 and the Monarch PCR & DNA Cleanup Kit, NEB#T1030). For mice plasma analysis and cytokine profiling the Proteome Profiler Mouse Cytokine Array Kit, Panel A (R&D Systems#ARY006) was used as per the manufacturers protocol. For total DNA extraction (including microbial) the tissues were processed using the Qiagen DNEasy Blood and Tissue Kit (#69504) as per the manufacturer’s recommendation.
Antibodies used in this paper are summarized below:
| Antibody | Dilution | Application |
|---|---|---|
| TEV Cleavage Site Monoclonal Antibody (ThermoFisher# MA1-124) | 1:2000 | Western Blots |
| Goat anti-Mouse IgG (H+L) Cross-Adsorbed Secondary Antibody, HRP (ThermoFisher# A16072) | 1:10000 | |
| IHC-plus Polyclonal Rabbit anti-Human ACTA2 / Smooth Muscle Actin Antibody (IHC) (LSBio#LS-B4742) | 1:200 | Immunohistochemistry |
| Anti-Cleaved Caspase-3 antibody (Abcam#ab2302) | 1:100 | |
| anti-Ki-67 (30-9) Rabbit Monoclonal Primary Antibody (Ventana Medical System/Roche#790-4286) | Ready to use dilutions | |
| Fibronectin Antibody (A-11) (Santacruz Biotech#sc-271098) | 1:20 | |
| Anti-Periostin antibody (Abcam# Ab83739) | 1:400 | |
| Collagen IV Monoclonal Antibody (PHM-12) (ThermoFisher# MA5-13255) | 1:50 | |
| Hyaluronic Acid (HA) Antibody (abbexa#abx101090) | 1:200 | |
| Bioxcell In-Vivo MAb anti-mouse PD-L1 (B7-H1) | - | In-vivo antitumor study |
Bacterial strains and cell culture conditions
Bacterial strains and plasmids used in this paper are summarized in the table given below. For general cloning TOP10 cells were used (One Shot TOP10 Chemically Competent E. coli, ThermoFisher# C404010) Strains were grown in LB agar (BD Difco#244520) and LB medium (BD Difco#244620) as appropriate with supplements such as antibiotics for transformant selection or IPTG for induction. Growth rates were measured via optical density measurements at 600nm.
| Strain abbreviation | Genomic modification/plasmid | Comment |
|---|---|---|
| EcN | - | Escherichia coli Nissle 1917 commercially available as Mutaflor (GenBank: CP007799.1) |
| EcN | pKM208 | EcN with λ-red recombination system (gamma, exo and beta) (Addgene Plasmid #13077) |
| TOP10 | pBbS18c-RFP | TOPP10 strain with plasmid for preparing chloramphenicol cassette (Addgene Plasmid #126201) |
| ΔE | Δnlpl | Hypervesiculating EcN |
| DH5α | pOGG005 | High copy number plasmid backbone (Addgene Plasmid #113980) |
| TOP10 | pecol19A-ClyA-Hy | Plasmid cloning |
| ΔElux | pAKgfplux1 | Hypervesiculating EcN with luciferase for BLI in-vivo (Addgene Plasmid #14083) |
| ECHy | pecol19A-ClyA-Hy | EcN expressing membrane confined Cytolysin A-Hyaluronidase (CHy) |
| ΔECHy | pecol19A-ClyA-Hy | Hypervesiculating EcN Expressing membrane confined Chy |
Primer used in the study can be found below (5’-3’):
| Forward primers | Reverse primers | Comments |
|---|---|---|
| TAGCAGCTGCAAAGAGG AGAAACACCATATGACA GAGATTGTTGCGGACAA |
CGGTACCCACGTGGGGCCC GTCGACTCTAGAGGATCCT CACGCAGGTGCAATTGTCGTTA |
Amplify CHy gene blocks |
| AGAGTCGACGGGCCCCA CGTGGGTACCGTTTAAA CCCCGGGTGTACAAGTA CTAAGCTTC |
ATGGTGTTTCTCCTCTTTGCAGC | Amplify vector backbone |
| CTATTGCTGGTCCGATTC TGCCAGGTCATCTTGGTC CTGGCCCAGTTACGCCCC GCCCTG |
ATGAAGCCTTTTTTGCGCT GGTGTTTCGTTGCGACAG CATGATCGGCACGTAAGAGGTTC |
Ds CmpR gene |
| CATGTAGTACGTGTGCC TCAA |
AACCTGGCCTATTTCCCTAAAG | Colony PCR for CmpR gene |
| AGCGAAAGAGATTTGCCGGA | ACGCCACTGCCCTTTATTGA | PCR for luxA fragment |
| CTTGGTGGGCGACATTAAGA | CGACCTCTGGGACTTCAAATAA | PCR for pecol19A-ClyA-Hy fragment |
| ATAATACCGCGCCACATAGC | - | Additional Sequencing primer for CHy |
| TTGACAGCTAGCTCAGTCC | - | |
| ATACCAAAAGCGCCACGGTA | - | |
| - | CCATCTAATTCAACAA GAATTGGGACAAC |
The murine breast cancer 4T1 cells (ATCC CRL-2539, passage number 5 to 15) and human MDA-MB-231 cells (ATCC HTB-26, passage number 5 to 15) were cultured in RPMI (Gibco 21875034) containing 10% fetal bovine serum (Gibco#26140079) and 5% Penicillin-Streptomycin (Gibco#15070063). Pancreatic cancer cells- Human Panc1 (ATCC CRL-1469) and mice derived RInk1 (generated as previously reported by Seeley et al.,)[39] and MC38 colon cancer cells (Kerafast#ENH204-FP) were cultured in DMEM (Gibco# 12430112) containing 10% fetal bovine serum (Gibco#26140079), 1X HEPES (Thermofisher# 15630080) and 5% Penicillin-Streptomycin (Gibco#15070063). All the cells were maintained at 37°C with 5% CO2 in air and sub-cultured twice weekly.
Generating bacterial competent cells
Chemically competent cells were made using the TSS buffer or using the 100mM CaCl2 solution and electrocompetent cells were made using deionized water as reported earlier[40].
Constructing the ΔE strain
For the nlpI gene deletion a chloramphenicol resistance cassette with ends overlapping with upstream and downstream regions of the nlpi gene was amplified (30bp overlapping ends). The dsDNA was electroporated into EcN cells with the λ-red recombinase system and the transformants were selected as per the procedure reported earlier[41].
Constructing pecol19A-ClyA-Hy and ΔECHy
The plasmid pOGG005 was initially modified by introducing an MCS site using restriction digestion (PsiL and PacI). A fusion protein was then designed having a J23119 promoter, followed by an RBS, ClyA (Escherichia coli (strain K12) 303 amino acids, UniProtKB #P77335), TEV site and Hy (Streptomyces koganeiensis, 217 amino acids, UniProtKB#A0A0U2E2J7) and the geneblocks were obtained from IDT. Codon optimization was performed using available online tools[42]. The geneblock and pOGG005-MCS plasmid blackbone were amplified with overlapping ends which were subsequently joined using the HiFI assembly master mix as per the manufacturer’s instruction. The plasmid was cloned into TOP10 cells followed by subcloning in ΔE cells to obtain ΔECHy.
All DNA assembly fragments gene modifications were sequence verified by colony/plasmid PCR, nucleic acid gel electrophoresis (1.2% and 1.5% agarose gel) and Sanger sequencing (DNA Sequencing and Genotyping Core, Cincinnati Children’s Hospital Medical Center). Plasmid designing and sequence alignments were performed using Snapgene and NCBI blast.
OMV Isolation and characterization
A sequential differential centrifugation protocol was developed for isolating the OMVs. In brief, bacteria were grown overnight till they reached the stationary phase and 1ml of this culture was used to inoculate 1L of LB Broth. The bacteria were grown till they reached an OD of approximately 1.5 units. The culture was cleared of bacteria by centrifugation (8000g, 4°C, 15 minutes) followed by concentrating the supernatant using Pierce protein concentrators (30KdA, ThermoFisher#88531). 70 ml of the concentrated supernatant was then ultracentrifuged at 91000g for 4 hours. The pellet obtained was resuspended in PBS pH 7.4, followed by washing with Amicon Ultra-0.5 Centrifugal Filter Unit (Millipore#UFC500308) filters to remove the broth completely. The resulting dispersion was sterile filtered through 0.22um PVDF syringe filters (Cole-Parmer#UX-06060–62). OMVs were then characterized for size and size distribution using Dynamic Light Scattering (Zetasizer Nano ZS, Malvern Instruments) and electron microscopy (Hitachi H-7650 Transmission Electron Microscope). For long term storage, the OMVs were stored at −80°C for further processing downstream.
SDS-PAGE Western Blot
Bacterial lysates were prepared using the Bacterial Protein Extraction Reagent (ThermoFisher#89821) following the manufacturer instruction. Protease and phosphate inhibitors were added as per the general requirements of preparing lysates for western blots. Bacterial and OMV lysates were separated using 8% Bis-Tris precast gels (Thermofisher#NW00080BOX) followed by semi dry transfer onto a 0.45um Nitrocellulose membrane. The membranes were blocked with 5% BSA in PBST followed by incubation with primary and secondary antibodies. Chemiluminescent substrate was added (Thermofisher#34579) and detected using the Biorad ChemiDoc imaging system.
Hyaluronidase assay
HA agarose plate method: The assay method was performed as reported earlier[43]. In brief, a 1% agarose solution (20ml) containing 0.4μg/ml HA and 1% BSA fraction IV was poured onto sterile 30 mm petridishes. 100μl droplet of bacterial strain containing 106 bacteria was dropped onto the plate followed by airdrying and overnight incubation at 37°C. The plates were flooded with 2N glacial acetic acid followed by imaging for transparent and opaque regions for determining the zones of HA degradation.
HA degradation turbidimetric assay: The assay method was performed as reported earlier[29]. The CTAB reagent was prepared by dissolving cetyltrimethylammonium bromide at 2.5% w/v in 100 ml of 2% w/v NaOH solution. Overnight cultures of bacteria containing different concentration of HA (0.1, 0.2 and 0.4 μg/ml) were cleared of the bacteria and 50μl of each sample was mixed with 0.1 M phosphate buffer pH 7 in a 96 well microplate. The plates were incubated at 37°C for 15 minutes followed by the addition of 100 μl of CTAB reagent. The plates were incubated for 10 min at 37°C followed by measuring optical density at 600nm in triplicates.
Animals and tumor models
6-8 weeks old female BALB/cJ and male C57BL/6 mice (Jackson Labs) were used in all the experiments. All animal experiments were conducted by following a protocol approved by the University of Cincinnati Biosafety, Radiation Safety, and Animal Care and Use Committees. For tumor induction 106 of tumor cells/0.1ml saline (4T1, MDA-MB-231, RInk1, PANC1, MC38) were injected subcutaneously in the shaved right flank of anesthetized (2% isoflurane) mice. Tumor measurements were taken every alternate day and tumor volume was calculated using the following formula: 0.5*length*width*width. For therapeutic assessment or bioluminescent imaging, each bacterial dose contained 106 bacteria suspended in 0.1ml saline was administered intravenously. Lapatinib was suspended in a solution containing 0.5% w/v HPMC and 0.1% w/v Tween 80 and each dose delivered 5mg/Kg orally. At the study end point the mice were euthanized via carbon dioxide inhalation and cervical dislocation followed by collection of organs. For cytokine profiling blood withdrawal was performed after a period of 24 hours via the submandibular vein and collected in tubes containing Heparin as an anticoagulant (40U/ml of blood). Collected blood was immediately centrifuged at 2000g, 4C for 15 minutes to obtain the plasma as supernatant. Cytokine profiling was conducted by using a membrane membrane-based sandwich immunoassay kit as per the manufacturer’s instructions. Animals were maintained in an Association for the Assessment and Accreditation of Laboratory Animal Care approved facility (Assurance # D16-00190) in accordance with current regulations of the U.S. Department of Agriculture and Department of Health and Human Services. Experimental methods were approved by and in accordance with institutional guidelines established by the Institutional Animal Care and Use Committee (approved protocol numbers: 20-05-16-01).
Bioluminescence imaging
Bioluminescence images were acquired for 1-2 mins using the Perkin Elmer IVIS Spectrum In-Vivo Imaging System for quantification of radiance of the bioluminescent signals from the regions of interest.
PET imaging of OMVs in-vivo
For PET imaging of OMVs, a 64Cu-YbT complex was prepared as reported in our earlier publication[31a]. OMVs were incubated with 1 mCi of the radioactive complex for 1 hour at 37°C. Size exclusion chromatography was used to remove the free complexes and purify OMVs (PD-10 columns, Cytiva Life Sciences#17085101). Vesicles were concentrated using Pierce Protein Concentrators (30 kDa, Thermo Scientific#88531) before administration in mice (BalbC with 4T1 tumors ≈200 mm3, n=3). 4 hours post intravenous administration small animal PET scan was performed on a μPET scanner (Siemens Inveon). Mice in the supine position under anesthesia were placed on the imaging gantry with continued warming. For anatomical reference overlay, a CT scan was performed (80 kVp, 500μA, at 120 projections) followed by the acquisition of PET images for 15 minutes with real-time reconstruction. Spatial resolution was determined by ordered subset expectation maximization in 2D. Histogramming and reconstruction were applied using the system onboard Inveon software. Post-processing was carried out using the Inveon Research Workplace software.
Immunohistochemistry
Tumor tissue was isolated and fixed with 10% v/v formalin followed by replacement of storage solution with 70% v/v ethanol. Immunohistochemistry slides were prepared via paraffin processing and were developed by the Pathology Research Core at the Cincinnati Children’s Hospital Medical Center. The slides were imaged under x100 and x400 magnification using the Leica DMi8 Widefield Fluorescence/Brighfield Microscope. The images were quantified with ImageJ as reported previously[44].
Statistical analysis
Data has been represented as mean (in the tumor regression graphs) and as mean±SD everywhere else. Sample size for each experiment has been indicated in the figure legends and in the text. The survival cure was analyzed using the nonparametric Log-rank (Mantel-Cox) test. Ordinary two-way ANOVA and/or paired t-test (Wilcoxon) was used to compare means between different groups. Values of p < 0.05 were considered to be statistically significant. All statistical analyses were performed using the GraphPad Prism 8 software.
Supplementary Material
Acknowledgements
The authors would like to thank the Laboratory Animal Medical Services staff at the University of Cincinnati and the core facilities at the Cincinnati Children’s Hospital Medical Center for the instrumental and infrastructural support provided. The authors also acknowledge the help provided by Betsy DiPasquale and Jessica Webster in preparing samples for IHC and electron microscopy and Xiangning Wang for assistance with IVIS and PET animal imaging.
Funding information
This work was supported by funding from the University of Cincinnati Cancer Institute, Give HOPE and BSI foundations, and National Institute of General Medical Sciences of the National Institutes of Health (R21GM137321).
Footnotes
Conflict of Interest
The authors declare no competing financial interest related to this work.
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Shindu C. Thomas, Division of Pharmaceutical Sciences, James L. Winkle College of Pharmacy, University of Cincinnati, 231 Albert Sabin Way, Cincinnati, OH 45267, USA
Tushar Madaan, Division of Pharmaceutical Sciences, James L. Winkle College of Pharmacy, University of Cincinnati, 231 Albert Sabin Way, Cincinnati, OH 45267, USA.
Nitin S. Kamble, Division of Pharmaceutical Sciences, James L. Winkle College of Pharmacy, University of Cincinnati, 231 Albert Sabin Way, Cincinnati, OH 45267, USA
Nabil A. Siddiqui, Division of Pharmaceutical Sciences, James L. Winkle College of Pharmacy, University of Cincinnati, 231 Albert Sabin Way, Cincinnati, OH 45267, USA
Giovanni M. Pauletti, Department of Pharmaceutical and Administrative Sciences, University of Health Sciences and Pharmacy in St. Louis, 1 Pharmacy Place, St. Louis, MO 63110, USA
Nalinikanth Kotagiri, Division of Pharmaceutical Sciences, James L. Winkle College of Pharmacy, University of Cincinnati, 231 Albert Sabin Way, Cincinnati, OH 45267, USA.
References
- [1].a) Valkenburg KC, de Groot AE, Pienta KJ, Nat Rev Clin Oncol 2018, 15, 366; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hynes RO, Science 2009, 326, 1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Baish JW, Stylianopoulos T, Lanning RM, Kamoun WS, Fukumura D, Munn LL, Jain RK, Proc Natl Acad Sci U S A 2011, 108, 1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Martin JD, Seano G, Jain RK, Annu Rev Physiol 2019, 81, 505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Jing X, Yang F, Shao C, Wei K, Xie M, Shen H, Shu Y, Mol Cancer 2019, 18, 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].a) Sahai E, Astsaturov, Cukierman E, DeNardo DG, Egeblad M, Evans RM, Fearon D, Greten FR, Hingorani SR, Hunter T, Hynes RO, Jain RK, Janowitz T, Jorgensen C, Kimmelman AC, Kolonin MG, Maki RG, Powers RS, Pure, Ramirez DC, Scherz-Shouval R, Sherman MH, Stewart S, Tlsty TD, Tuveson DA, Watt FM, Weaver V, Weeraratna AT, Werb Z, Nat Rev Cancer 2020, 20, 174; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Nissen NI, Karsdal M, Willumsen N, J Exp Clin Cancer Res 2019, 38, 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Chan TS, Shaked Y, Tsai KK, Front Oncol 2019, 9, 688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].a) Henke E, Nandigama R, Ergun S, Front Mol Biosci 2019, 6, 160; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Deville SS, Cordes N, Front Oncol 2019, 9, 1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].a) Marusyk A, Tabassum DP, Janiszewska M, Place AE, Trinh, Rozhok AI, Pyne S, Guerriero JL, Shu S, Ekram M, Ishkin A, Cahill DP, Nikolsky Y, Chan TA, Rimawi MF, Hilsenbeck S, Schiff R, Osborne KC, Letai A, Polyak K, Cancer Res 2016, 76, 6495; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zervantonakis IK, Poskus MD, Scott AL, Selfors LM, Lin JR, Dillon DA, Pathania S, Sorger PK, Mills GB, Brugge JS, Proc Natl Acad Sci U S A 2020, 117, 16500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Abyaneh HS, Regenold M, McKee TD, Allen C, Gauthier MA, Theranostics 2020, 10, 1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Guedan S, Rojas JJ, Gros A, Mercade E, Cascallo M, Alemany R, Mol Ther 2010, 18, 1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Charbonneau MR, Isabella VM, Li N, Kurtz CB, Nat Commun 2020, 11, 1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].a) Zhang YN, Poon W, Tavares AJ, McGilvray ID, Chan WCW, J Control Release 2016, 240, 332; [DOI] [PubMed] [Google Scholar]; b) Longmire M, Choyke PL, Kobayashi H, Nanomedicine (Lond) 2008, 3, 703; [DOI] [PubMed] [Google Scholar]; c) Poon W, Zhang YN, Ouyang B, Kingston BR, Wu JLY, Wilhelm S, Chan WCW, ACS Nano 2019, 13, 5785. [DOI] [PubMed] [Google Scholar]
- [13].a) Zhou S, Gravekamp C, Bermudes D, Liu K, Nat Rev Cancer 2018, 18, 727; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ozdemir T, Fedorec AJH, Danino T, Barnes CP, Cell Syst 2018, 7, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].a) Isabella VM, Ha BN, Castillo MJ, Lubkowicz DJ, Rowe SE, Millet YA, Anderson CL, Li N, Fisher AB, West KA, Reeder PJ, Momin MM, Bergeron CG, Guilmain SE, Miller PF, Kurtz CB, Falb D, Nat Biotechnol 2018, 36, 857; [DOI] [PubMed] [Google Scholar]; b) Kurtz CB, Millet YA, Puurunen MK, Perreault M, Charbonneau MR, Isabella VM, Kotula JW, Antipov E, Dagon Y, Denney WS, Wagner DA, West KA, Degar AJ, Brennan AM, Miller PF, Sci Transl Med 2019, 11; [DOI] [PubMed] [Google Scholar]; c) Danino T, Prindle A, Kwong GA, Skalak M, Li H, Allen K, Hasty J, Bhatia SN, Sci Transl Med 2015, 7, 289ra84; [DOI] [PubMed] [Google Scholar]; d) Gurbatri CR, Lia I, Vincent R, Coker C, Castro S, Treuting PM, Hinchliffe TE, Arpaia N, Danino T, Sci Transl Med 2020, 12; [DOI] [PubMed] [Google Scholar]; e) Yu X, Lin C, Yu J, Qi Q, Wang Q, Microb Biotechnol 2020, 13, 629. [DOI] [PubMed] [Google Scholar]
- [15].a) Chiang CJ, Huang PH, Sci Rep 2021, 11, 5853; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Li R, Helbig L, Fu J, Bian X, Herrmann J, Baumann M, Stewart AF, Müller R, Li A, D. J. R. i. m. Zips, 2019, 170, 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Din MO, Danino T, Prindle A, Skalak M, Selimkhanov J, Allen K, Julio E, Atolia E, Tsimring LS, Bhatia SN, Hasty J, Nature 2016, 536, 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Schwechheimer C, Kuehn MJ, Nat Rev Microbiol 2015, 13, 605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Jiang SN, Phan TX, Nam TK, Nguyen VH, Kim HS, Bom HS, Choy HE, Hong Y, Min JJ, Mol Ther 2010, 18, 635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].a) Chen DJ, Osterrieder N, Metzger SM, Buckles E, Doody AM, DeLisa MP, Putnam D, Proc Natl Acad Sci U S A 2010, 107, 3099; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Huang W, Wang S, Yao Y, Xia Y, Yang X, Li K, Sun P, Liu C, Sun W, Bai H, Chu X, Li Y, Ma Y, Sci Rep 2016, 6, 37242; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Kim JY, Doody AM, Chen DJ, Cremona GH, Shuler ML, Putnam D, DeLisa MP, J Mol Biol 2008, 380, 51; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Rappazzo CG, Watkins HC, Guarino CM, Chau A, Lopez JL, DeLisa MP, Leifer CA, Whittaker GR, Putnam D, Vaccine 2016, 34, 1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Schwechheimer C, Rodriguez DL, Kuehn MJ, Microbiologyopen 2015, 4, 375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Caldwell BJ, Bell CE, Prog Biophys Mol Biol 2019, 147, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Toyofuku M, Nomura N, Eberl L, Nat Rev Microbiol 2019, 17, 13. [DOI] [PubMed] [Google Scholar]
- [23].a) Chen KJ, Sabrina S, El-Safory NS, Lee GC, Lee CK, J Biosci Bioeng 2016, 122, 673; [DOI] [PubMed] [Google Scholar]; b) Kaessler A, Olgen S, Jose J, Eur J Pharm Sci 2011, 42, 138. [DOI] [PubMed] [Google Scholar]
- [24].a) Messina L, Gavira JA, Pernagallo S, Unciti-Broceta JD, Sanchez Martin RM, Diaz-Mochon JJ, Vaccaro S, Conejero-Muriel M, Pineda-Molina E, Caruso S, Musumeci L, Di Pasquale R, Pontillo A, Sincinelli F, Pavan M, Secchieri C, FEBS Lett 2016, 590, 2180; [DOI] [PubMed] [Google Scholar]; b) Ebelt ND, Zuniga E, Passi KB, Sobocinski LJ, Manuel ER, Mol Cancer Ther 2020, 19, 706. [DOI] [PubMed] [Google Scholar]
- [25].Wai SN, Lindmark B, Soderblom T, Takade A, Westermark M, Oscarsson J, Jass J, Richter-Dahlfors A, Mizunoe Y, Uhlin BE, Cell 2003, 115, 25. [DOI] [PubMed] [Google Scholar]
- [26].a) Galen JE, Zhao L, Chinchilla M, Wang JY, Pasetti MF, Green J, Levine MM, Infect Immun 2004, 72, 7096; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) del Castillo FJ, Moreno F, del Castillo I, FEMS Microbiol Lett 2001, 204, 281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Yang S, Liu Q, Zhang Y, Du G, Chen J, Kang Z, ACS Synth Biol 2018, 7, 287. [DOI] [PubMed] [Google Scholar]
- [28].Part:BBa_J23119, http://parts.igem.org/Part:BBa_J23119, accessed: 2021.
- [29].Oueslati N, Leblanc P, Harscoat-Schiavo C, Rondags E, Meunier S, Kapel R, Marc I, Carbohydr Polym 2014, 112, 102. [DOI] [PubMed] [Google Scholar]
- [30].a) Lou Y, McDonald PC, Oloumi A, Chia S, Ostlund C, Ahmadi A, Kyle A, Auf dem Keller U, Leung S, Huntsman D, Clarke B, Sutherland BW, Waterhouse D, Bally M, Roskelley C, Overall CM, Minchinton A, Pacchiano F, Carta F, Scozzafava A, Touisni N, Winum JY, Supuran CT, Dedhar S, Cancer Res 2011, 71, 3364; [DOI] [PubMed] [Google Scholar]; b) Takai K, Le A, Weaver VM, Werb Z, Oncotarget 2016, 7, 82889; [DOI] [PubMed] [Google Scholar]; c) Steenbrugge J, Vander Elst N, Demeyere K, De Wever O, Sanders NN, Van Den Broeck W, Dirix L, S. Van Laere, Meyer E, Front Immunol 2019, 10, 2928; [DOI] [PubMed] [Google Scholar]; d) Nocito A, Dahm F, Jochum W, Jang JH, Georgiev P, Bader M, Graf R, Clavien PA, Cancer Res 2008, 68, 5152; [DOI] [PubMed] [Google Scholar]; e) Muller-Edenborn K, Leger K, Glaus Garzon JF, Oertli C, Mirsaidi A, Richards PJ, Rehrauer H, Spielmann P, Hoogewijs D, Borsig L, Hottiger MO, Wenger RH, Oncotarget 2015, 6, 20288. [DOI] [PubMed] [Google Scholar]
- [31].a) Siddiqui NA, Houson HA, Kamble NS, Blanco JR, O’Donnell RE, Hassett DJ, Lapi SE, Kotagiri N, JCI Insight 2021, 6; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wurpel DJ, Moriel DG, Totsika M, Easton DM, Schembri MA, Journal of proteomics 2015, 115, 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Koh EI, Robinson AE, Bandara N, Rogers BE, Henderson JP, Nat Chem Biol 2017, 13, 1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].a) Schwertfeger KL, Cowman MK, Telmer PG, Turley EA, McCarthy JB, Front Immunol 2015, 6, 236; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Price ZK, Lokman NA, Ricciardelli C, Cancers (Basel) 2018, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].a) Wan X, Zheng X, Pang X, Pang Z, Zhao, Zhang Z, Jiang T, Xu W, Zhang Q, Jiang X, Oncotarget 2016, 7, 34038; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Liu CY, Hu MH, Hsu CJ, Huang CT, Wang DS, Tsai WC, Chen YT, Lee CH, Chu PY, Hsu CC, Chen MH, Shiau C, Tseng LM, Chen KF, Oncotarget 2016, 7, 9135; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Morse MA, Wei, Hartman Z, Xia W, Ren XR, Lei G, Barry WT, Osada T, Hobeika AC, Peplinski S, Jiang H, Devi GR, Chen W, Spector N, Amalfitano A, Lyerly HK, Clay TM, Int J Cancer 2010, 126, 2893; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Atiya HI, Dvorkin-Gheva A, Hassell J, Patel S, Parker RL, Hartstone-Rose A, Hodge J, Fan D, Ramsdell AF, Anticancer Res 2019, 39, 2277; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Yang C, Sheng Y, Shi X, Liu Y, He Y, Du Y, Zhang G, Gao F, Cell Death Dis 2020, 11, 831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Bonnans C, Chou J, Werb Z, Nat Rev Mol Cell Biol 2014, 15, 786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].a) Shpilberg O, Jackisch C, Br J Cancer 2013, 109, 1556; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Clift R, Souratha J, Garrovillo SA, Zimmerman S, Blouw B, Cancer Res 2019, 79, 4149; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR, Cancer Cell 2012, 21, 418; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Rahbari NN, Kedrin D, Incio J, Liu H, Ho WW, Nia HT, Edrich CM, Jung K, Daubriac J, Chen I, Heishi T, Martin JD, Huang Y, Maimon N, Reissfelder C, Weitz J, Boucher Y, Clark JW, Grodzinsky AJ, Duda DG, Jain RK, Fukumura D, Sci Transl Med 2016, 8, 360ra135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Thompson CB, Shepard HM, O’Connor PM, Kadhim S, Jiang P, Osgood RJ, Bookbinder LH, Li X, Sugarman BJ, Connor RJ, Nadjsombati S, Frost GI, Mol Cancer Ther 2010, 9, 3052. [DOI] [PubMed] [Google Scholar]
- [38].a) Taylor MA, Hughes AM, Walton J, Coenen-Stass AML, Magiera L, Mooney L, Bell S, Staniszewska AD, Sandin LC, Barry ST, Watkins A, Carnevalli LS, Hardaker EL, J Immunother Cancer 2019, 7, 328; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Grasselly C, Denis M, Bourguignon A, Talhi N, Mathe D, Tourette A, Serre L, Jordheim LP, Matera EL, Dumontet C, Front Immunol 2018, 9, 2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Seeley ES, Carriere C, Goetze T, Longnecker DS, Korc M, Cancer Res 2009, 69, 422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].a) Seidman CE, Struhl K, Sheen J, Jessen T, Curr Protoc Mol Biol 2001, Chapter 1, Unit1 8; [DOI] [PubMed] [Google Scholar]; b) Chung CT, Niemela SL, Miller RH, Proc Natl Acad Sci U S A 1989, 86, 2172. [DOI] [PubMed] [Google Scholar]
- [41].Murphy KC, Campellone KG, BMC Mol Biol 2003, 4, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].http://biocatalysis.uni-graz.at/, Codon Harmonizer, http://biocatalysis.uni-graz.at/sites/codonharmonizer.html, accessed.
- [43].Grenier D, Michaud J, J Clin Microbiol 1993, 31, 1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Crowe A, Zheng W, Miller J, Pahwa S, Alam K, Fung KM, Rubin E, Yin F, Ding K, Yue W, Pharm Res 2019, 36, 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.