

Abstract
Ca2+ homeostasis is essential for cell function and survival. As such, the cytosolic Ca2+ concentration is tightly controlled by a wide number of specialized Ca2+ handling proteins. One among them is the Na+-Ca2+ exchanger (NCX), a ubiquitous plasma membrane transporter that exploits the electrochemical gradient of Na+ to drive Ca2+ out of the cell, against its concentration gradient. In this critical role, this secondary transporter guides vital physiological processes such as Ca2+ homeostasis, muscle contraction, bone formation, and memory to name a few.
Herein, we review the progress made in recent years about the structure of the mammalian NCX and how it relates to function. Particular emphasis will be given to the mammalian cardiac isoform, NCX1.1, due to the extensive studies conducted on this protein. Given the degree of conservation among the eukaryotic exchangers, the information highlighted herein will provide a foundation for our understanding of this transporter family. We will discuss gene structure, alternative splicing, topology, regulatory mechanisms, and NCX’s functional role on cardiac physiology. Throughout this article, we will attempt to highlight important milestones in the field and controversial topics where future studies are required.
Introduction
Cardiac function relies on the spatio-temporal control of intracellular Ca2+ levels. Initiation of the contraction cycle begins with the depolarization of the plasma membrane, which opens voltage-dependent Ca2+ channels permitting an influx of Ca2+ into the cell (15). The elevation of Ca2+ within the dyadic cleft, the narrow space between the plasma membrane and the sarcoplasmic reticulum (SR), triggers the release of Ca2+ from the SR via the opening of the SR resident Ryanodine receptor (RyR). The resulting Ca2+ increase within the cytoplasm activates the contractile machinery of cardiac cells. To relax, intracellular Ca2+ levels need to decrease to its resting concentration. This task is accomplished by the sarcoplasmic reticulum Ca2+-ATPase (SERCA) and the plasma membrane Na+-Ca2+ exchanger (NCX). Both transporters are capable of moving Ca2+ ions against their concentration gradients. To accomplish this process, SERCA hydrolyzes ATP to refill the SR with Ca2+, while NCX is powered by the electrochemical gradient of Na+ to expel Ca2+ from the cell.
A third transporter, the plasma membrane Ca2+-ATPase (PMCA), also extrudes Ca2+ from the cell, although its contribution to Ca2+ removal during excitation-contraction coupling seems limited when compared to NCX (13, 15, 55). More investigations are needed to decipher PMCA role in Ca2+ homeostasis.
This article will summarize our current understanding of the exchanger with special emphasis on the molecular properties and physiological roles of the cardiac isoform NCX1.1, as this is the most widely investigated isoform. We will contextualize its molecular and biophysical properties relative to the other two mammalian isoforms, NCX2 (147) and NCX3 (188), and the crystalized archaebacterial homolog, NCX_Mj (150). Key features of the cardiac exchanger in its native environment will be reviewed, including its distribution in cardiac cells and functional coupling with other plasma membrane proteins essential for contractility. Finally, the ability to modify the genomic properties of mice has opened the possibility to generate animals with either altered expression profiles or transport properties of NCX. We will dedicate the last section of this article to discuss mouse models since they have been instrumental in defining the important physiological impact that NCX plays in cardiac function. The information detailed herein may serve as a platform for those interested in investigating the properties and physiological role of this essential transporter.
General Aspects
The first evidence of Ca2+ movements across the plasma membrane coupled to Na+ concentration gradients is from 1968 with studies from two different groups showing that Ca2+ efflux in cardiac tissue and crab nerve ceased when Na+ was removed from the extracellular media (7, 8, 232). These seminal studies suggested the presence of a protein carrier able to move both Na+ and Ca2+ ions across the membrane. The molecular identity of this hypothesized transporter was revealed 20 years later when a protein of 120 kDa was purified from canine sarcolemma (203). A key milestone followed in 1990 when the first exchanger (NCX1) from canine heart was cloned (185), which enabled manipulation of NCX properties via mutagenesis. In conjunction with heterologous expression systems and the development of the giant patch technique (96, 100, 102), this created the opportunity for elegant and informative studies at the molecular level. The history of NCX investigations is marked by numerous discoveries that have impacted the field, one of the most recent milestones is the resolved crystal structure of the archaebacterial homolog NCX-Mj, as it has provided the first look into the atomic organization of the exchanger family (150).
The NCX family is a subset of the superfamily of Ca2+/cation membrane transporters called solute carriers (SLC). This includes well over 400 membrane transporter proteins organized into 65 families with NCX comprising the SLC8 group (158). Four different SLC8 genes have been identified in eukaryotic cells but three have been reported in mammals (SLC8A1, SLC8A2, and SLC8A3).
All NCX transporters work in a similar fashion: they couple the downhill movement of Na+ ions to the counter transport of Ca2+ ions, classifying them as secondary transporters. During each cycle, three Na+ ions are exchanged for one Ca2+ ion (Figure 1), becoming the dominant Ca2+ extrusion mechanism in cells. These proteins are high-capacity transporters, with Na+ and Ca2+ exchange rates reaching approximately 5000 s−1 (97, 190), but with a relatively low affinity for cytoplasmic Ca2+ (101, 167, 196). This implies that substantial elevations in intracellular Ca2+ are required to fully activate NCX, making it particularly suitable to quickly remove the high Ca2+ levels. This task makes this family of proteins essential for physiological function. Particularly, excitable cells such as neurons and cardiac myocytes, rely on these transporters to rebalance Ca2+ to its resting or preexcitation levels to terminate signaling and contractile events. Malfunction of these proteins has been implicated in pathologies in both tissues (121–123, 157, 174, 175, 208–211, 258). We will focus our review upon NCX’s role in the heart and the reader is directed to other excellent work if interested in learning about the NCX family in the brain (121–123, 157, 174, 175, 258).
Figure 1. Modes of operation of the Na+-Ca2+ exchanger.
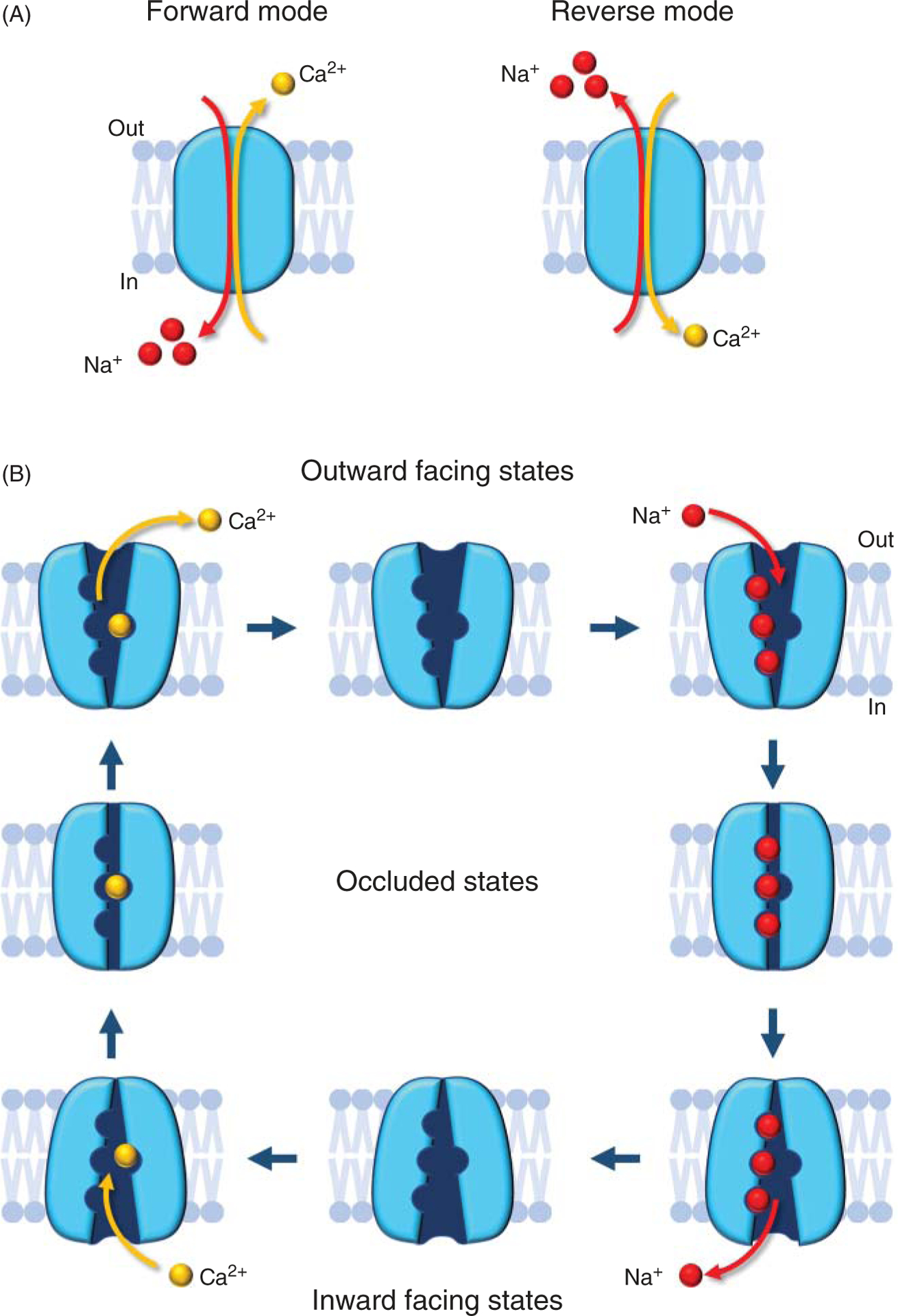
(A) In the forward mode (top) NCX transports one Ca2+ ion out of the cell in exchange for three Na+ ions entering. One net charge is moved across the membrane resulting in a measurable ionic current. This is the most physiologically relevant cycle. In the reverse mode (bottom), one Ca2+ ions enter the cell and three Na+ ions exit. The direction in which Ca2+ is moved is governed by both the ionic gradients of Na+ and Ca2+ and membrane potential (164). (B) The scheme shows NCX transport cycle. This electrogenic transporter transitions between the outward and inward-facing states via intermediate occluding states, leading to the coupled but opposite movements of Na+ and Ca2+ ions across the plasma membrane (164).
Na+-Ca2+ Exchanger Encoding Gene and Alternative Splicing
Mammals have been shown to carry three different genes SLC8A1, SLC8A2, and SLC8A3 encoding exchangers NCX1, NCX2, and NCX3, respectively. These three proteins show high degrees of both sequence and functional homology. A fourth gene, SLC8A4 is found only in the genomes of amphibian, reptilian, and teleosts, including zebra fish, which is routinely used as an animal model (163). A detailed summary of the NCX4 gene and those found in invertebrate species is beyond the scope of this article and the reader is directed to more comprehensive references (191). Herein we will discuss the key features of the mammalian exchanger genes and their regulation with special emphasis on SLC8A1, which encodes for NCX1 including the cardiac isoform (NCX1.1).
SLC8A1: NCX1 protein-encoding gene
NCX1 was originally cloned from dog heart cDNA using expression libraries probed with polyclonal antibodies (185). The single-copy human gene was localized by southern hybridization to a panel of mouse-human somatic cell hybrids and assigned to chromosome 2 (251). It consists of 12 exons, with exon 1 being noncoding (Figure 2A). Similarly, in mice, NCX1 is a single copy gene (133) but has 11 exons, all of which are coding (163). Both the mouse and human SLC8A1 gene have a large exon (exons 1 and 2, respectively) that extends for approximately 500 amino acids and ends at an alternative splice site (Figure 2A). Here there are six exons A, B, C, D, E, and F (136) that can be used in different combinations generating various splice variants. Each exon expression profile has been given a unique identifier, that is, NCX1.1 encodes ACDEF exons (Figure 3). These exons encode a variable region in the C-terminal half of the large cytoplasmic loop (220) (Figure 2A) and this alternative splicing translates into different versions of a highly specific region identified as the Calcium-Binding Domain 2 (CBD2). Exons A and B are mutually exclusive with exon A being expressed in exchangers found in excitable cells, such as the cardiac (NCX1.1) and brain (NCX1.4) isoforms (Figure 3). In contrast exon B (Figure 3) expressing exchangers are found in nonexcitable cells, such as NCX1.3 in the kidney (220). Exon D is expressed in all transcripts while the remaining exons C, E, and F shuffle to create unique cell-specific protein profiles (220) (Figure 3). The functional implications associated with the shuffling of these exons will be detailed in sections titled “Regulation by Cytosolic Ca2+” and “Influence of Cytoplasmic Ca2+ on the Na+ Dependent Inactivation”.
Figure 2. Na+-Ca2+ exchanger gene organization.
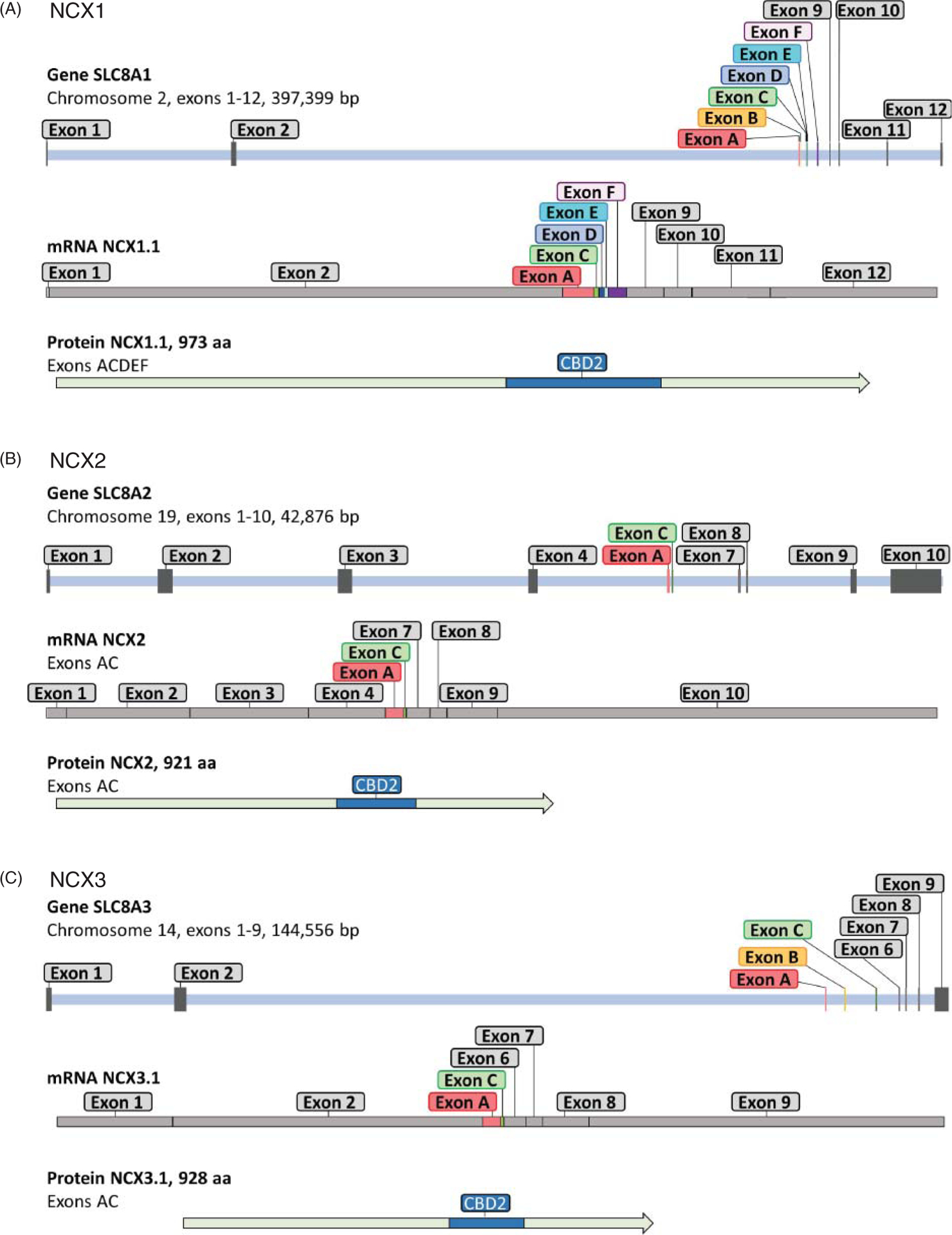
Each panel depicts the chromosomal location, number of exons, size, mRNA example, and corresponding protein of the three human isoforms. Gene exon structures are derived from Ensembl. Exons are indicated as grey bars and include both coding and noncoding exons. Exons undergoing alternative splicing are shown in color and are named in accordance with Quednau and colleagues (220). 5′ and 3′ regions of the gene are not included so that the gene size given extends from the beginning of the first exon and ends at the 3′ end of the last exon. Of special note, NCX2 does not undergo alternative splicing and all exons encode for protein. NCX1 and NCX3 have one noncoding exon, both undergo alternative splicing. An example of mRNA profile of the indicated exchangers is shown under each corresponding gene. Graphs are to scale. The alternative spliced exons (in colors), which at the protein level result in changes in the calcium-binding domain 2 (CBD2) (220), are depicted in blue.
Figure 3. Tissue distribution of the exchanger isoforms.
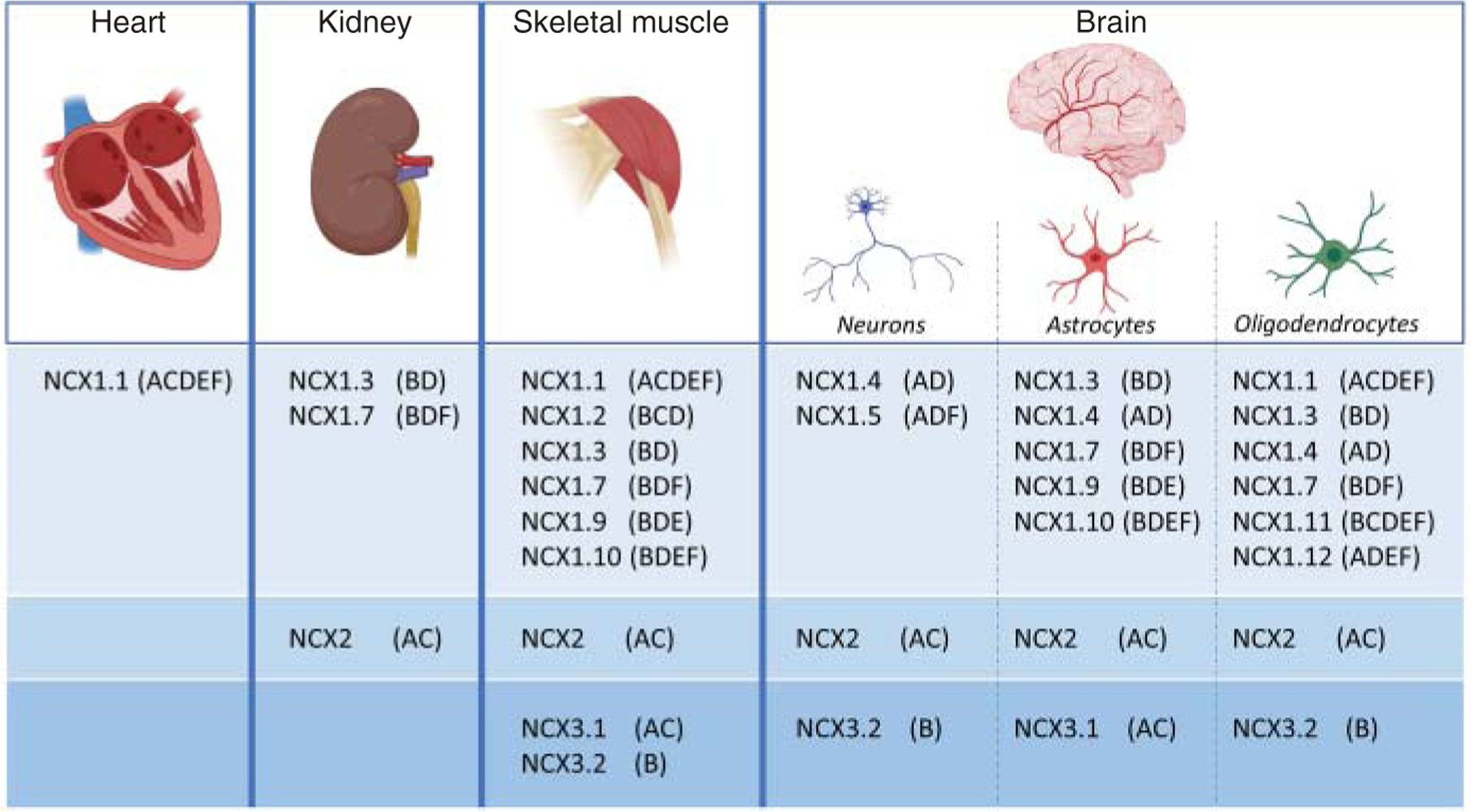
The three exchanger isoforms and their corresponding splice variants are differentially distributed within tissues (133, 136, 143, 220, 221). Examples are given for heart, brain, kidney, and skeletal muscle. The tissue distribution has been assessed using reverse transcriptase-polymerase chain reaction as described in Ref. 220. Numerous evidence indicates that the isoform NCX1.1 (136, 143, 220) is the only exchanger expressed in cardiomyocytes, although a study has identified isoforms NCX1.3 and NCX1.4 (133). The splice variants NCX1.6 (ACD) and NCX3.3 (BC) are absent from the table. These exchangers have been localized in the brain (136, 220), but their cell specific expression is undetermined.
To date, at least 12 different SLC8A1 transcripts, expressed in a tissue-specific fashion have been reported (136, 143, 179, 220) (Figure 3). It is generally accepted that cardiac myocytes exclusively express isoform NCX1.1 (143, 221), although low amounts of isoforms NCX1.3 and NCX1.4 have been reported (133). The different expression profile is due to the presence of three tissue-specific alternative NCX1 promoters (10, 143, 182). These promoters have been identified and designated as H1 (heart), K1 (kidney), and Br (brain), with most evidence for the role of H1 in cardiac expression (178, 182). Sequence analysis shows that the cardiac NCX1 promoter lacks a TATA box but has an initiator site with the consensus sequence YYANWYY and includes sites for tissue-specific factors such as activator protein 1, Specific protein 1 (SP-1), nuclear factor 1 (NF-1), and GATA-4 (181, 182). It has also been shown that NCX expression in neonatal rat ventricular myocytes requires both CArG and GATA boxes (30, 181). Moreover, the cardiac promoter has an E-box consensus site which confers adrenergic sensitivity to NCX1 expression and it may be responsible for NCX1 upregulation during cardiac hypertrophy (30).
In contrast to the cardiac promoter, the kidney transcript has a TATA box as well as AP-1, NF-Y transcription binding sites (182) but shares the presence of two GATA boxes. Finally, the brain NCX1 promoter has multiple SP-1 binding sites, it lacks a TATA box but has a notable CpG island which suggests that it remains active during early embryogenesis (182).
The elucidation of NCX gene structure has opened the door to the determination of the expression profiles of the various isoforms, and in some cases their functional characterization. It has also enabled genetic studies to be carried out in mice including, overexpression, knockout, and CRISPR-based mutagenesis, as detailed in section titled “Mouse Models with Genetically Modified Na+-Ca2+ Exchangers”.
SLC8A2: NCX2 protein-encoding gene
SLC8A2 is the shortest of all the mammalian genes just 30 kb in size. It encodes for a 921 a.a. protein (NCX2) predominately expressed in the brain and skeletal muscle but also found in low amounts in liver, testes, vascular smooth muscle cells, and endothelial cells (220). NCX2 shares 65% similarity with NCX1, and it is expected to follow the predicted topology of NCX1 (147) (Figure 4). In human, SLC8A2 is located on chromosome 19 and consists of 10 exons (Figure 2B), which all contribute to coding regions. Within the central segment, NCX2 gene differs from SLC8A1 gene by lacking exons B, D, E, and F and possessing only exons A and C (163) (Figure 2B). Of the three mammalian isoforms, NCX2 is unique in appearing to lack any alternative splicing (220).
Figure 4. Progression of Na+-Ca2+ exchanger topology.
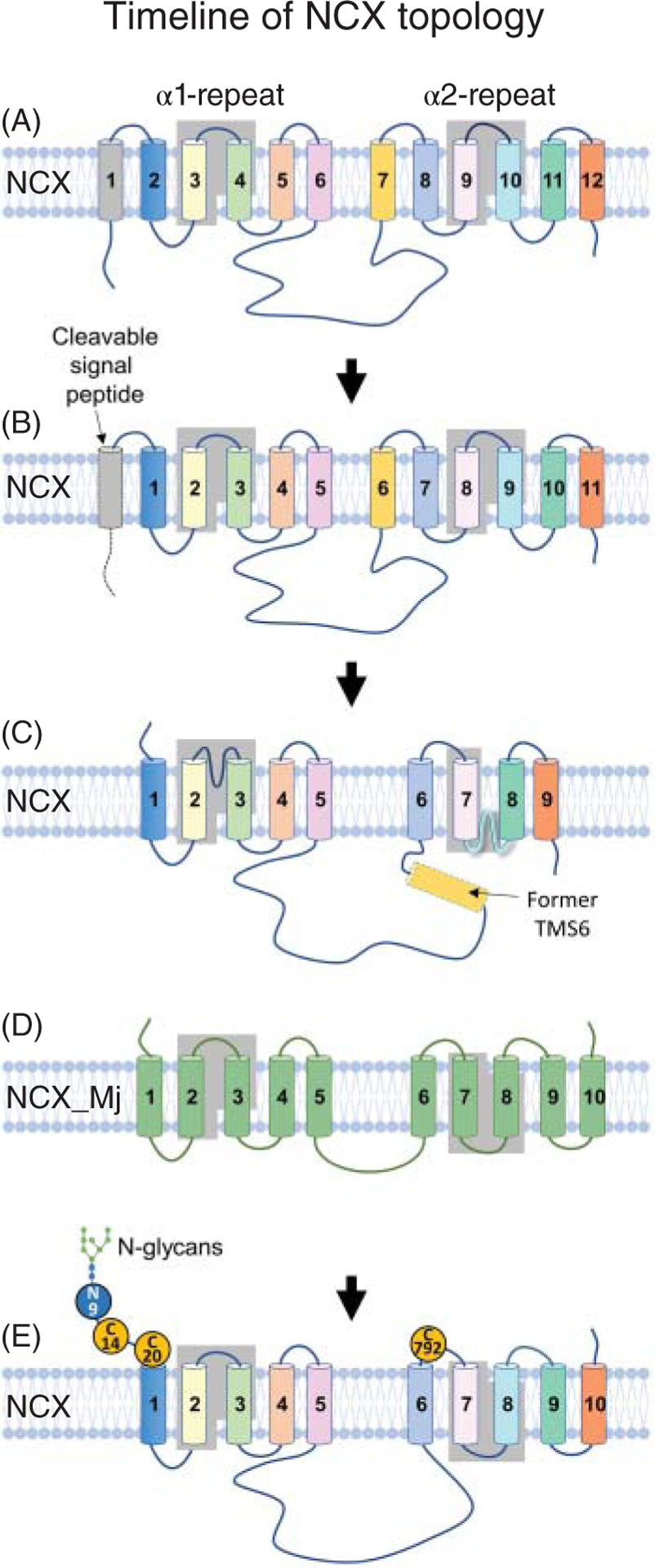
The cartoons show how the topology of the mammalian exchanger has evolved through the years since its cloning in 1990 (185). (A) Initially, NCX1.1 was predicted to be organized into 12 transmembrane segments (TMS). The two highly conserved regions among the exchangers which are known as α-repeats (245) are highlighted. These regions are essential for ion transport (183, 184, 195). (B) Subsequent studies (50, 60, 106, 239) revealed that the first helix was a signal peptide not necessary for activity, restricting the mature protein to 11 TMSs with an extracellular N terminus. (C) Cysteine scanning mutagenesis and epitope tagging were then applied to investigate NCX1.1 architecture. Based on these studies the topology of NCX was reorganized into nine transmembrane segments with an intracellular C-terminus and two reentrant loops between TMSs 2–3 and 7–8 (35, 112, 114, 186, 187, 212). (D) The crystal structure of the archaebacterial exchanger (NCX_Mj) (150) has revealed a protein of 10 TMSs, contrasting with the predicted 9 TMSs for the mammalian exchanger as shown in (C). (E) Topology of the mammalian NCX based on NCX_Mj organization. The new architecture does not include the previously modeled reentrant loops, which are now predicted to be a short extracellular loop between TMSs 2–3, within the α1-repeat, and a new transmembrane segment (TMS 8) in the α2-repeat. Future studies should be conducted to confirm the absence of either or both of these two reentrant structures and to validate that the mammalian exchanger and the archaebacterial are had a similar organization. The position of the cysteine residues involved in the intramolecular disulfide bond (241) is also shown. Amino acids are numbered based on the protein lacking the signal sequence.
SLC8A3: NCX3 protein-encoding gene
The human SLC8A3 gene is located on chromosome 14 and it is composed of 9 exons (Figure 2C), with alternative exons A, B, and C present, but lacking exons D, E, and F, which are found in SLC8A1 (62–64, 163). Similar to SLC8A1, however, SLC8A3 does not utilize exon 1 for coding. A short 250 bp, region upstream of the transcription start site, was identified as the minimal promoter region (63, 64). This region, which contains a TATA box and SP-1 elements, appears to respond to cAMP, Ca2+, the morphogen retinoic acid, and brain-derived neurotrophic factor when transfected into SH-SY5Y cells, a cell line used for neuronal functional studies (64). Whether these regulations occur in vivo has not been determined.
Similarly to SLC8A1, SLC8A3 undergoes alternative splicing to encode for three different transcripts (NCX3.1, NCX3.2, and NCX3.3) (188, 220), which share 73% identity with NCX1 and 75% identity with NCX2 (188). These splice variants, found in brain and skeletal muscle, show developmental patterns of expression, with NCX3.3 mainly found in neonatal tissue, and NCX3.1 and 3.2 in adult (220). In human skeletal muscle, a fourth transcript has been detected (NCX3.4). This splice variant is unique as it lacks much of the cytoplasmic regulatory loop and the last 5 transmembrane segments (TMSs), leading to a protein of 620 amino acid (63). A similar splicing pattern for NCX1 has been reported (146, 274). Two other NCX3 transcripts have been detected in human fetal brain (154). These transcripts, designated as NCX3.tN-1 and tN-2, also translate into dramatically truncated proteins, lacking the first five TMSs and the regulatory large cytoplasmic loop. Whether these truncated exchangers can transport ions remains controversial (192, 274) and more investigations are needed to elucidate the differential physiological impact of the truncated and full-length splice variants.
Organization of the Mammalian Na+-Ca2+ Exchanger
Progression of NCX topology
Since the cloning of the cardiac exchanger (NCX1.1) in 1990 (185), the topological organization of NCX1.1 has been significantly modified and in spite of recent progress and the resolved structure of the homologous archaebacterial exchanger NCX-Mj (150, 151), numerous questions about its architecture remain unanswered.
Based on hydrophobicity analysis, NCX1 was initially modeled to have 12 TMSs, with a large cytoplasmic loop between TMSs 5 and 6 (Figure 4A) (185). This organization was later revised with the demonstration that the first TMS is actually a cleavable signal peptide (50), neither essential for trafficking nor function (60, 106, 239). This suggested that the mature exchanger was composed of 11 TMSs with an extracellular N-terminus (Figure 4B). Immunofluorescence and epitope tagging investigations buttressed this architectural configuration by confirming the orientation of the N-terminus and establishing the intracellular localization of both the large cytoplasmic loop and the C-terminus (Figure 4B) (35, 212).
A major turning point in elucidating NCX topology occurred when cysteine scanning mutagenesis analysis was applied to assess its organization (112, 114, 186, 187). This technique involves replacement of residues thought to be at strategic locations within the protein with cysteine residues. Reactivity of the cysteine sulfhydryl group to membrane-impermeable sulfhydryl reagents then defines the accessibility of these amino acids to either the intracellular or extracellular aqueous environment. Figure 5 summarizes NCX1.1 residues investigated with this approach and found accessible to either the external or cytoplasmic side of the plasma membrane (112, 114, 186, 187). Please note that the amino acid (a.a.) numbering presented in this figure and throughout the review refers to the canine sequence, lacking the cleaved (32 a.a.) N-terminal signal sequence.
Figure 5. Inconsistencies between the topology of the Na+-Ca2+ exchanger and the archaebacterial exchanger.
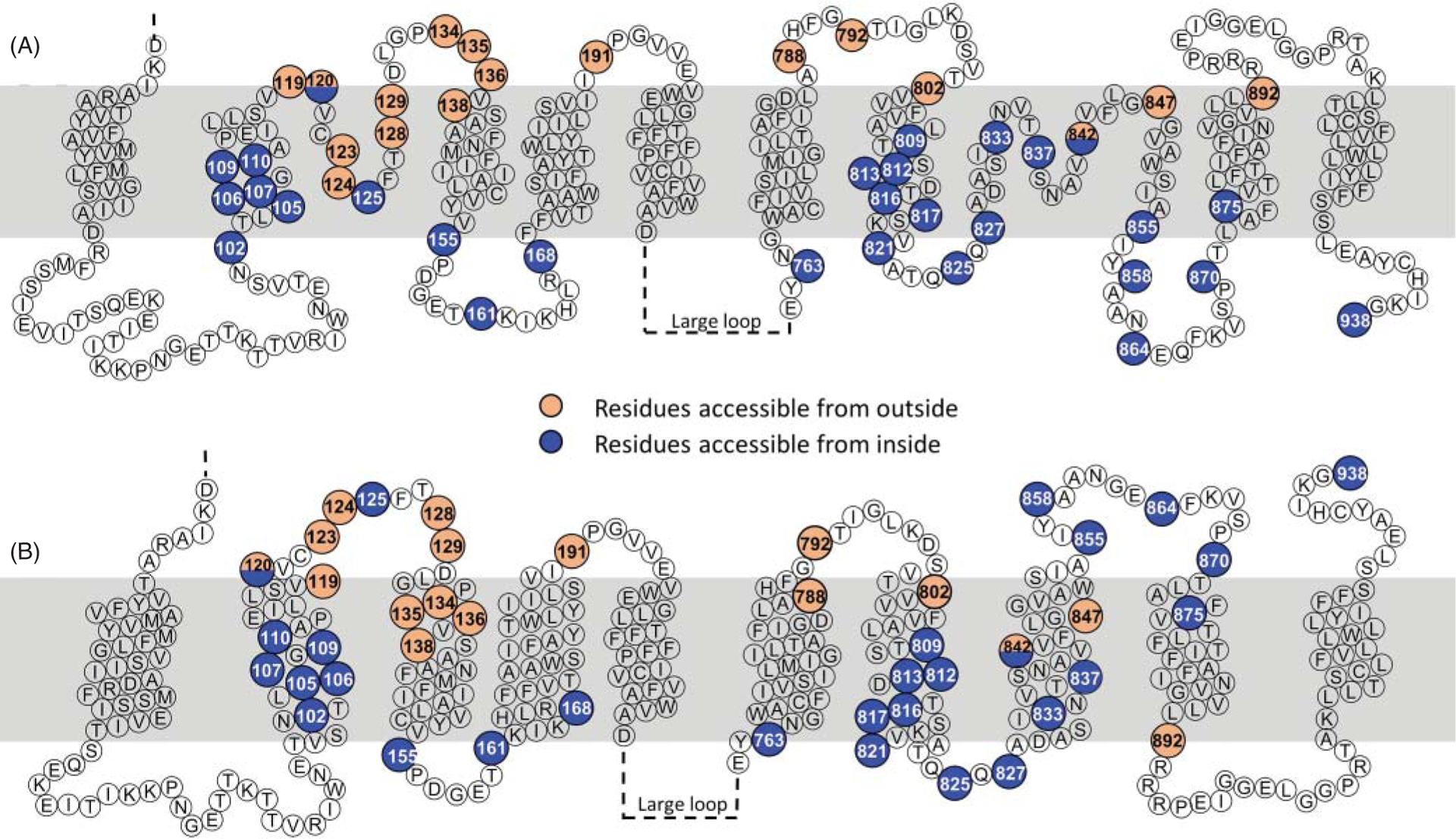
(A) Cysteine scanning mutagenesis and epitope mapping studies have been instrumental in deciphering the topology of the cardiac exchanger (35, 50, 112, 114, 186, 187, 212). The figure shows the amino acids that were replaced with cysteine and found accessible to membrane-impermeable sulfhydryl agents either from the cytoplasm (blue circles) or the external side of the membrane (orange circles). Based on these investigations, NCX was modeled to have an extracellular N-terminus, and a cytoplasmic C-terminus. Moreover, residues 120 and 125 within the α1-repeat were found to be exposed to the intracellular environment as well as several residues within the α2-repeat, constraining these regions into reentrant loops. This organization corresponds to the one shown in Figure 4C. (B) With the atomic structure of the archaebacterial exchanger at hand (150), NCX1.1 is now assumed to be organized in 10 transmembrane segments (228, 265), as shown in Figure 4E. However, this new arrangement places several residues previously found accessible to the cytoplasmic application of sulfhydryl reagents (blue circles), now facing the extracellular environment, including the C-terminus. Note that the new architecture does not include reentrant loops. More investigations are needed to fully elucidate the topology of the mammalian exchanger.
Information garnered from such experiments revealed three important features. First, the loop connecting TMSs 2 and 3 invaginated into the plasma membrane without fully crossing it (Figures 4C and 5A) (112, 114), resembling the P-loops found in ion channels. Second, TMS 6 was not a transmembrane region but was part of the large intracellular loop (Figures 4C) (186). Third, TMS 9 was also arranged as a P-loop (112, 114, 186, 187). The rearrangement of TMSs 6 and 9 as nonspanning helices downsized NCX1 to nine TMSs, with an extracellular N-terminus and an intracellular C-terminus (Figures 4C and 5A).
Even though the reorganization of NCX into 9 helices had been validated by several laboratories (112, 114, 186, 187), its accuracy came into question with the availability of the atomic structure of the homologous archaebacterial exchanger, NCX_Mj (150) which revealed 10 TMSs (Figure 4D). Among these helices, TMSs 2, 3, 7, and 8 are nestled in the center of the protein harboring 12 ion coordinating residues, as detailed in section titled “Structural Organization of the α-Repeats”. This transport machinery is surrounded by the remaining 6 TMSs that form an outer scaffold. The reader interested in learning more about the archaebacterial exchanger structural and functional details is referred to excellent reviews published on this subject (73, 74). Herein, the archaebacterial exchanger will be marginally discussed with the goal to learn about the key elements of its mammalian counterpart and how it has shaped the research related to the cardiac exchanger. For example, the presence of an additional TMS and an extracellular C-terminus in NCX_Mj (Figure 4A) (150) clearly diverges from the predicted and experimentally derived topology of NCX (compare Figure 4C with Figure 4D). These newly revealed differences gave a push to reassess the orientation of NCX’s C-terminus using various mapping approaches such as cross-linking and epitope-tagging. Results from these investigations support NCX’s C-terminus exposure to the extracellular milieu (228, 265), suggesting that the mammalian NCX follows an arrangement similar to NCX_Mj by being arranged into 10 TMSs, in spite of their low overall sequence identity. This new organization (Figures 4E and 5B) has important implications for the distal end of NCX, as it leaves little room for the reentrant loop previously modeled after TMS 7, as further detailed in the following section. Also, this new topological arrangement places several residues previously found accessible from the intracellular milieu now exposed to the extracellular environment (Figure 5), calling for further investigations.
Structural organization of the α-repeats
A defining feature of the NCX family is the presence of two conserved regions known as α-repeats (Figure 6A). These motifs were first demonstrated in the Drosophila exchanger (also known as CALX) due to their intramolecular homology (Figure 6B) (245). Their presence as a conserved tandem repeat suggests that the exchanger arose from a gene duplication event. As these α-repeat regions are present in all members of the Ca2+/cation exchanger superfamily (158), they have become the signature sequence of this class of proteins.
Figure 6. Sequence alignment of the α-repeats.
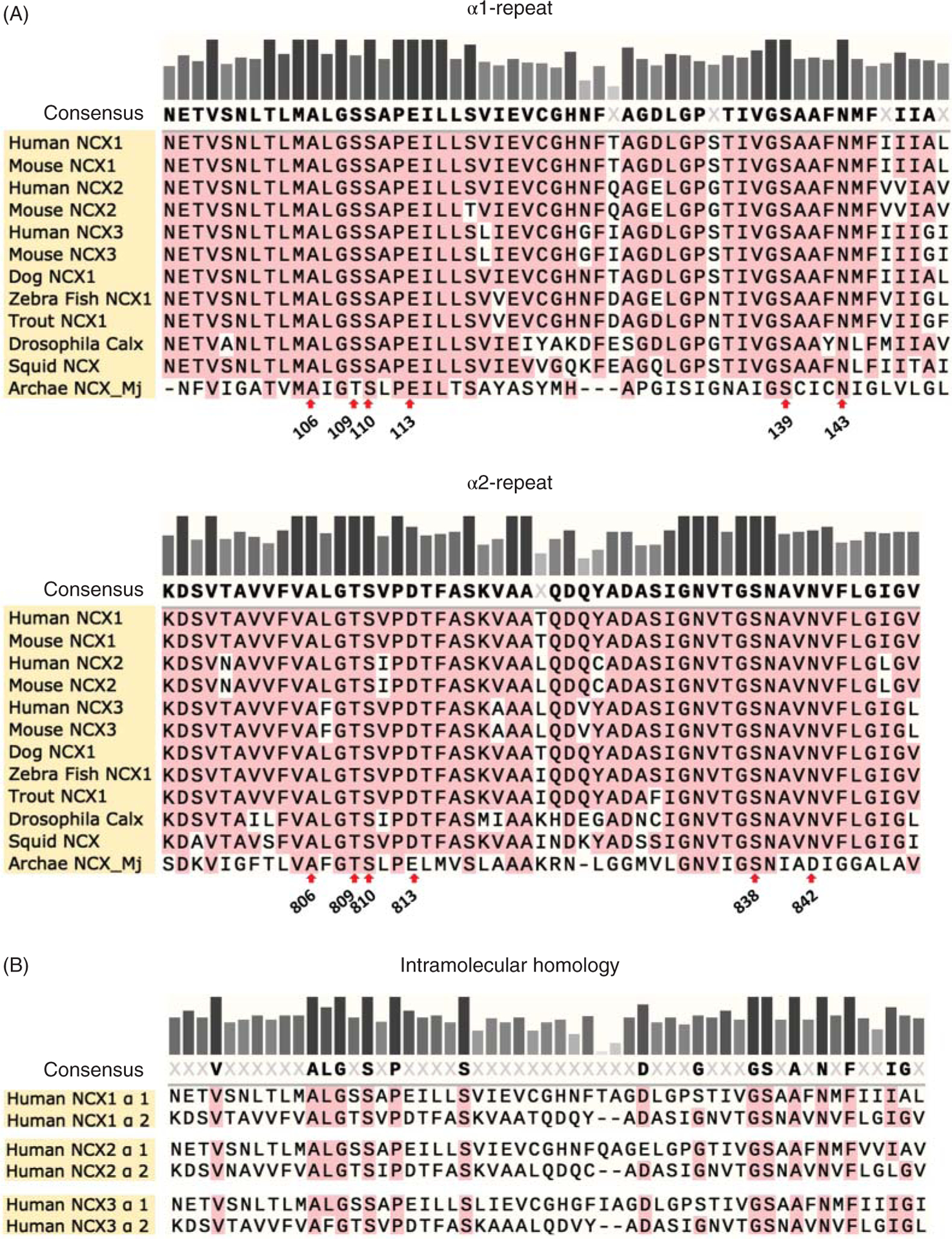
(A) The sequence alignment illustrates the conservation of the α repeat regions within the Na+-Ca2+ exchanger family. This is consistent with a broad range of evidence underlying their essential role in ion transport (125, 183, 184, 195). The α1-repeat encompasses residues 96 to 150 of NCX1.1, while the α2-repeat stretches from 797 to 849 (amino acid numbering of NCX1.1 without the signal sequence). These boundaries were selected as previously described in Ref. 245. The residues that coordinate Na+ and Ca2+ ions, as revealed by the atomic structure of the archaebacterial exchanger (150), are indicated with a red arrow. (B) In addition to the conservation between species, the α-repeats show intramolecular homology. This forms the foundation for the idea that exchangers are a result of gene duplication.
In the canine cardiac exchanger isoform, the α1-repeat consists of residues 96 to 150 and α2-repeat encompasses residues 797 to 849 (this numbering excludes the 32 amino acids encoding for the signal peptide). The boundaries of these motifs are based on the sequence similarity with the Drosophila exchanger, as first described by Schwarz and Benzer (Figure 6) (245). The first repeat includes TMS 2 and a portion of TMS 3 (Figure 4). The extracellular loop linking these two helices has been predicted to dip into the membrane from the extracellular side without fully crossing it (Figures 4C and 5A). This model stems from studies showing that two cysteines introduced within this loop via mutagenesis (N125C and E120C, Figure 5) reacted with cytoplasmically applied sulfhydryl agents (112, 114, 187). In contrast, the corresponding loop in the archaebacterial exchanger is a shorter extracellular link lacking any invagination into the lipid bilayer (Figure 4D) (150), a result that may raise concerns about the fine organizational details of the mammalian isoforms.
The architecture of the α2-repeat is also uncertain. While there is consensus that the N-terminal section of the α2-repeat forms TMS 7 both in the mammalian and archaebacterial exchanger, biochemical studies conducted on the cardiac NCX1.1 isoform and the atomic structure of NCX_Mj depict a different scenario for the remaining portion of the α2-repeat and the following amino acids. Indeed, residues 825 to 870 were found to form a second cytoplasmic invagination into the plasma membrane in the cardiac exchanger (112, 187) (Figures 4C and 5A), whereas they define a new TMS, TMS 8, and the following extracellular linker in the archaebacterial exchanger (residues 226 to 256) (Figure 4D) (150). An important observation is that TMS 8 of NCX_Mj carries residues essential for coordinating transported ions (Figure 11) (150). Since the archaea and eukaryotic exchangers transport Na+ and Ca2+ with the same 3:1 stoichiometry (225, 254), the current assumption is that the mammalian isoform follows the NCX_Mj architecture, with the C-terminal end of the α2-repeat forming an extra helix carrying conserved residues for ion coordination. The reasoning behind the topological discrepancies of the α-repeats as predicted by the cysteine scanning mutagenesis and the structure of the archaebacterial exchanger remains puzzling and requires investigation to reexamine the fine structural organization of these essential domains of the mammalian exchangers.
Figure 11. The ion coordinating residues are conserved between the archaebacterial and mammalian exchangers.
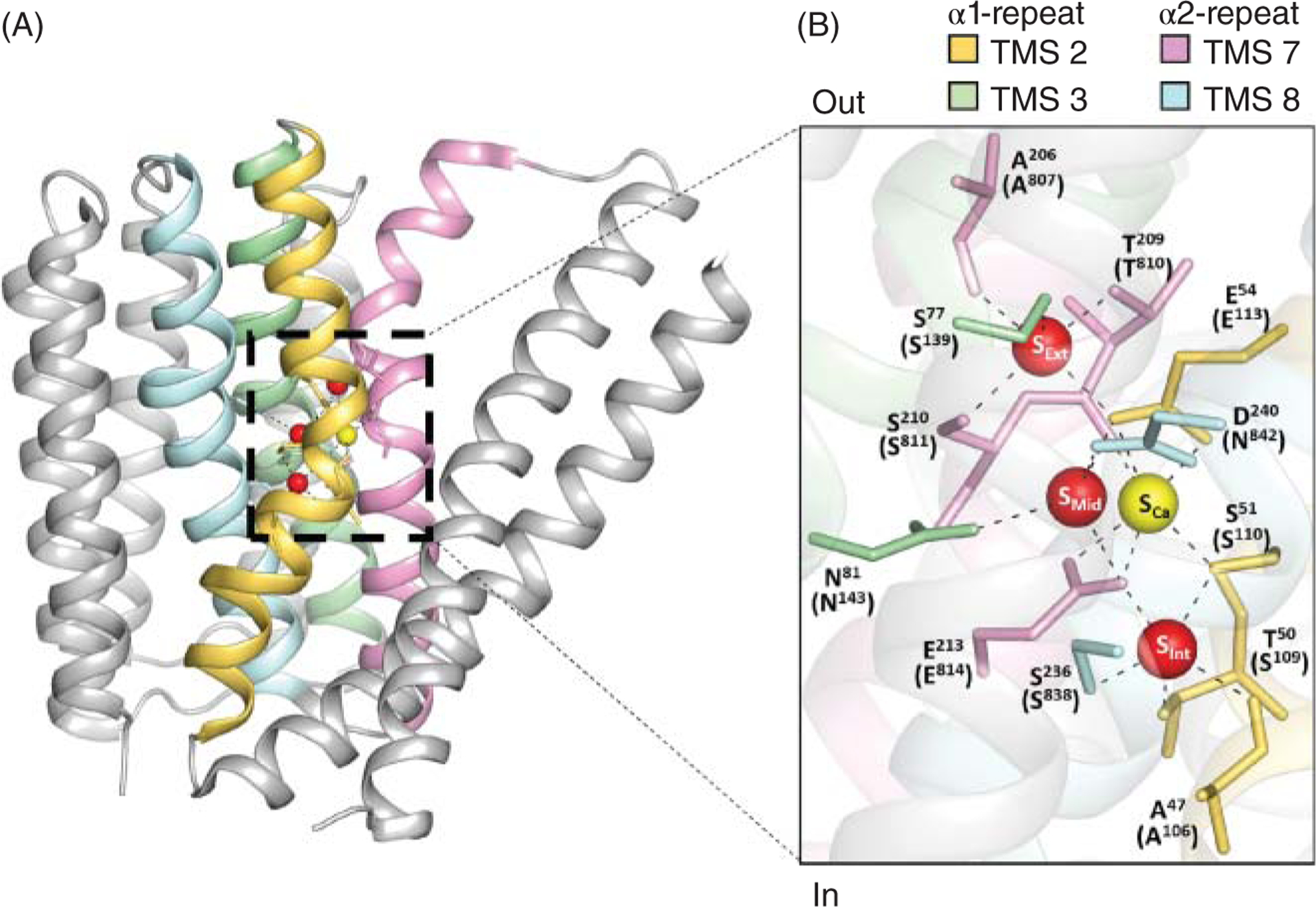
(A) The atomic structure of the archaebacterial exchanger revealed 12 amino acids within the α-repeats (TMS 2 yellow, TMS 3 green, TMS 7 pink, and TMS 8 cyan) that coordinate Na+ and Ca2+ ions (PDB code 3V5U) (150). (B) The residues involved in ion coordination in NCX_Mj are shown. In parenthesis are reported the homologous residues in the mammalian NCX1.1 (numbered without taking into account the signal peptide). The three sodium coordinating sites are identified as SExt, SMid, and SInt. The Ca2+ ion binding site is referred to as SCa (150). New evidence indicates that SMid coordinates water instead of Na+ (151, 162).
Organization of NCX cytoplasmic regions
The cardiac exchanger is modeled to have five cytoplasmic loops (Figures 4 and 5). These loop regions lack any sequence similarity with NCX_Mj and remain largely unexplored with the important exception of the large loop linking TMS 5 and 6. Herein, we discuss the various cytoplasmic loops progressing from the N to C terminal region of the protein, following the topology as predicted by the archaebacterial exchanger. Differences between the architecture of the mammalian exchanger, as determined by biochemical studies, and the atomic structure of the homologous archaebacterial exchanger will be discussed to highlight potential discrepancies in their organization.
Cytoplasmic loop between TMSs 1 and 2
The first cytoplasmic loop (~22 amino acid) connects TMSs 1 to 2, based on the structural similarity with NCX_Mj (Figures 4D, 4E, and 5). As it links two helices vital for ion transport (see section titled “Organization of the Mammalian Na+-Ca2+ Exchanger”), this uncharted domain may be relevant for controlling NCX1.1 activity. While further investigations are needed to establish its relevancy, preliminary evidence supports its potential importance as insertion of a Flag epitope (8 a.a.) within TMSs 1 and 2 linker region significantly reduced the activity of the rat brain exchanger (35).
Cytoplasmic loop between TMSs 3 and 4
The second cytoplasmic loop located between helices 3 and 4 is shorter in size, encompassing approximately 6 amino acids, as dictated by the structure of NCX_Mj. Mutation of residues Val 155 and Arg 161 within this loop to a cysteine did not affect activity but rendered the cardiac exchanger responsive to cytoplasmic application of sulfhydryl reagents, confirming the intracellular location of this domain (Figure 5) (112).
Cytoplasmic loop between TMSs 5 and 6
The third intracellular loop linking TMS 5 to 6 is the largest, encompassing approximately 500 amino acids (Figure 7), and is the most extensively investigated. This loop is not essential for function as its deletion did not prevent transport activity (168), however, it harbors several domains involved in regulatory processes. Below we will review the “hot spots” found within this large cytoplasmic domain.
Figure 7. Domains and residues relevant for NCX1.1 regulation.
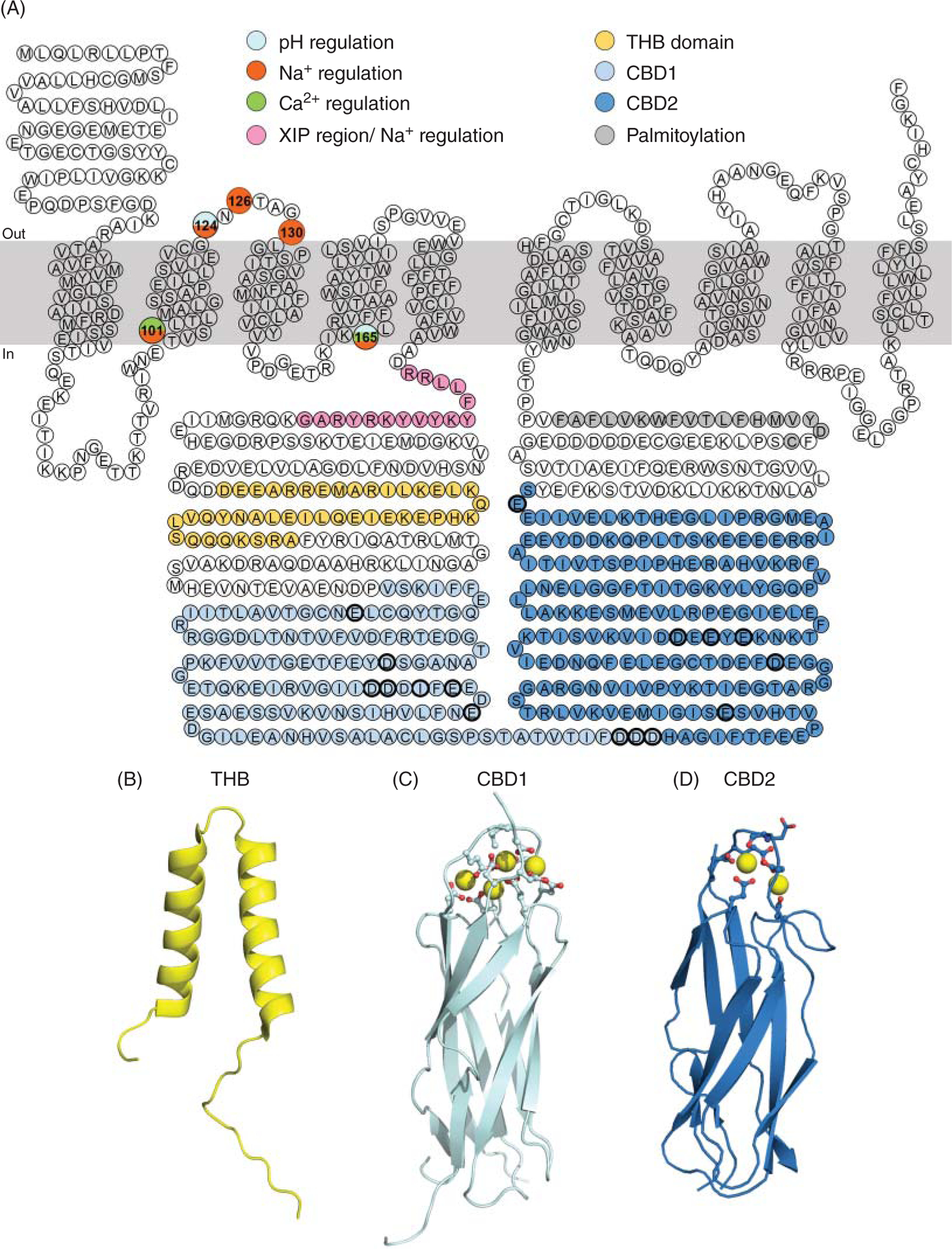
(A) Highlighted are the residues of the canine cardiac Na+-Ca2+ exchanger found important for its regulation. For a detailed role of these regions in controlling NCX transport activity please see the main text. Briefly, the residues shown in pink constitute the XIP region, which is intimately involved with the Na+-dependent inactivation (166). The two helix bundle domain (THB) follows in yellow and its structure is shown in (B) (PDB# 6BV7) (295). The role of this domain remains to be determined. The residues encompassing the two Ca2+ regulatory domains are shown in light blue (Ca2+-binding domain 1, CBD1) and dark blue (Ca2+-binding domain 2, CBD2), their atomic structure is shown in (C) and (D), respectively [CBD1 PDB# 2PDK (189) and CBD2 PDB# 2QVM (16)]. At the distal end of the large cytoplasmic loop, there is a stretch of residues (in dark grey) forming an amphipathic helix, required for palmitoylation of cysteine at position 739 (207). Single point mutations that affect Na+, Ca2+, and pH regulation are indicated.
XIP region
The XIP region is composed of a stretch of hydrophobic and positive residues (a.a. 219–238) (149), located immediately after the C-terminal end of TMS 5 (Figures 7 and 8). The name of this region, XIP, originates from a synthetic peptide with the same sequence as the endogenous XIP (XIP, eXchanger Inhibitory Peptide) which, when applied exogenously from the cytoplasmic side, was found to be a potent inhibitor of NCX activity (149). The structural architecture of the XIP region remains unknown but its sequence resembles a putative calmodulin-binding site (149). Accordingly, a synthetic peptide corresponding to the XIP region has been found to bind calmodulin with an affinity of approximately 200 nM (149). Whether XIP interacts with calmodulin in the full length exchanger remains to be established. More recent evidence, suggests that calmodulin may mediate its functional effects on NCX by interacting with an alternative site. This conclusion stems from a study showing that deletion of residues 684 to 703, in a distal region of the large cytoplasmic loop, prevents calmodulin enhancing NCX transport activity and promoting its trafficking to the plasma membrane (33).
Figure 8. The XIP region is essential for Na+-dependent inactivation.
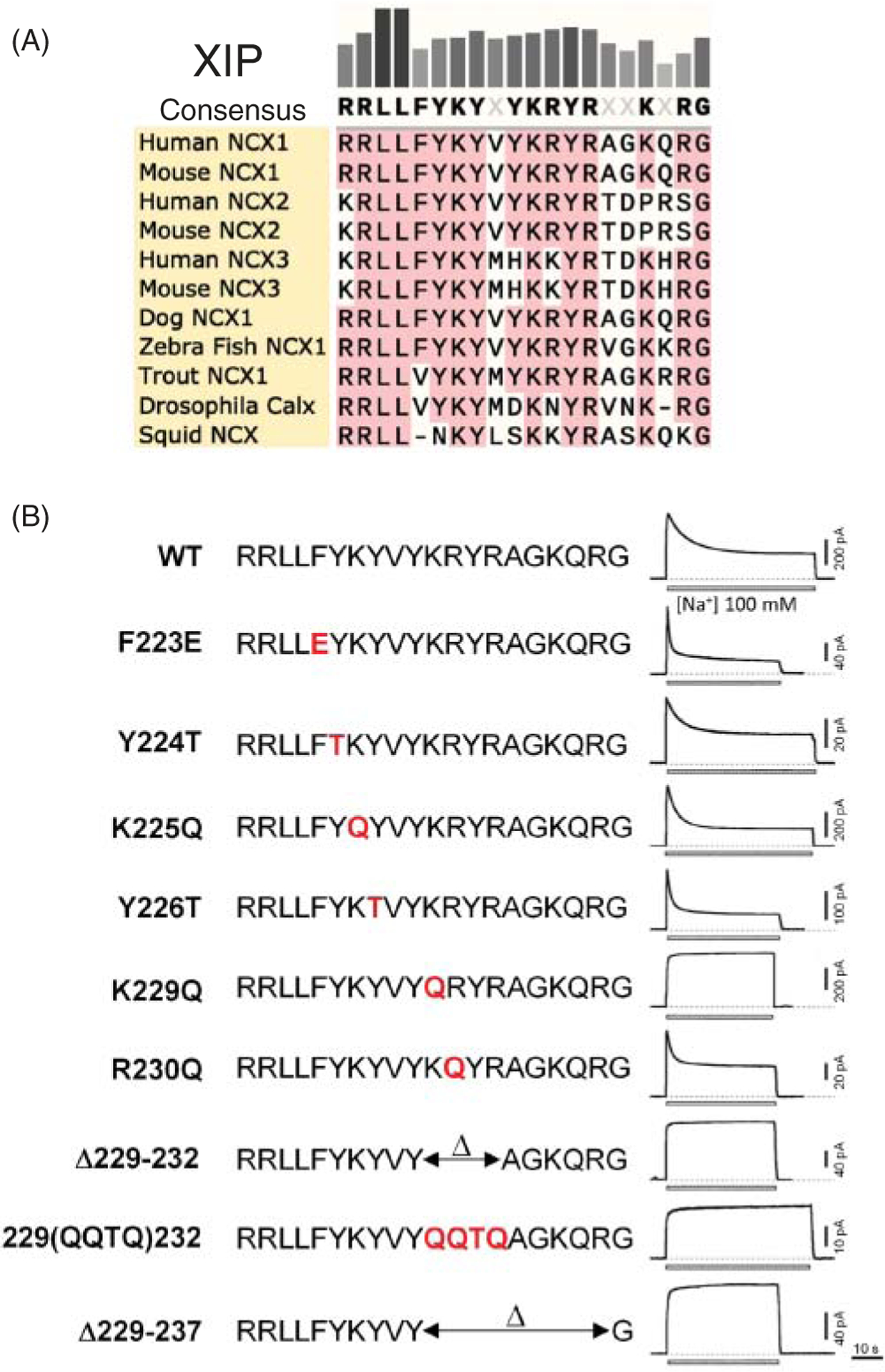
(A) Sequence alignment of the XIP region (amino acids 219 to 238, note NCX_Mj has no XIP region). (B) Mutations within this region either accelerate or abrogate the time course of current decay due to raised intracellular Na+, so-called “Na+-dependent inactivation”. Modified, with permission, from Matsuoka S, et al., 1997 (166).
More well determined is the role that the XIP region plays in the regulatory process called Na+-dependent inactivation (Figure 8), which is discussed in detail in section titled “Inactivation by Intracellular Na+”.
Catenin-like domain 1
The region (a.a. 238 to 370) following the XIP sequence has been designated as a putative catenin-like domain due to its sequence similarity with α-catenin (95), Accordingly, this domain has been assumed to have a structure similar to α-catenin with five antiparallel helices (95). Recent nuclear magnetic resonance (NMR) studies, however, reveal that this region is largely unstructured with the exception of two antiparallel α helices, encompassing residues 281 to 301 and 307 to 323 (295) (Figure 7). This motif is highly conserved among the exchangers and has been designated as the “two helix bundle” (THB) (295).
The functional role of THB and the remaining portion of the catenin-like domain 1, has yet to be established. Electrophysiological studies show that introduction of the fluorescent protein YFP about 20 amino acids upstream of the THB domain (a.a. 266) did not dramatically alter the biophysical properties of NCX as compared to the wild type (193). Also, splitting of NCX into two halves, at position 266, did not affect either the reassembling of the protein in the plasma membrane after co-expression of the two domains or its biophysical properties (192). Together, these findings suggest that this region is not essential for activity.
Ca2+-binding domains
Following the THB domain, two distinct structural regions that coordinate Ca2+ ions are found and are referred to as calcium-binding domains or CBD (Figure 7). These two Ca2+ regulatory domains are not present in the archaebacterial exchanger NCX_Mj, suggesting that eukaryotic exchangers have evolved a sophisticated way to regulate their transport activity in a cell-specific manner to finely control Ca2+ homeostasis.
Calcium ions bound to these sites are not transported but trigger conformational changes that increase NCX turn over rate and antagonize the Na+-dependent inactivation, resulting in enhanced activity (103, 167, 196). NMR and X-ray structural studies show that these regulatory domains share a common architecture consisting of seven antiparallel β-strands (Figure 7B) (16, 93, 95, 189). Despite their general structural similarity, the two CBDs display important functional differences. The first Ca2+-binding domain (CBD1, residues 301–500) is conserved between the various isoforms and coordinates 4 Ca2+ ions (Figure 9). In contrast, the second Ca2+-binding domain (CBD2, residues 501–689 in NCX1.1) is subjected to alternative splicing (Figures 2 and 10A), generating CBD2s which vary in the number of Ca2+ ions they can coordinate (Figure 10B) (93, 132). Generally, excitable cells possess a Ca2+-coordinating CBD2 (exon A), while exchangers in nonexcitable cells express a CBD2 lacking any Ca2+ coordination site (exon B) (Figure 3) (220).
Figure 9. Properties of the exchanger Ca2+-binding domain 1 (CBD1).
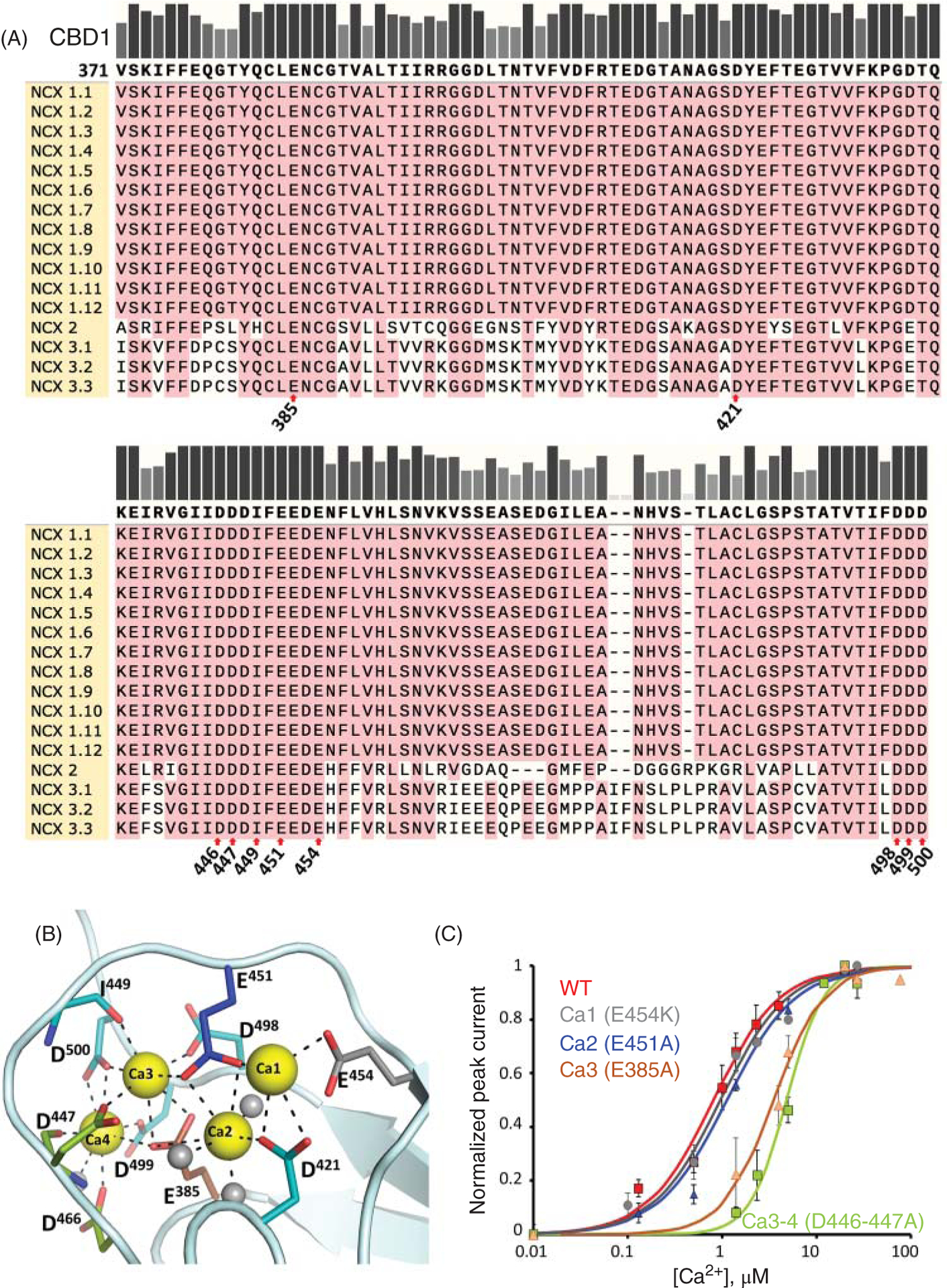
(A) Protein sequence alignment of the CBD1 region among human exchanger isoforms. Numbering is reported according to NCX1.1 sequence, excluding the signal peptide. Residues involved in ion coordination are highlighted with red arrows. (B) This domain coordinates 4 Ca2+ ions (PDB code 2DPK, Ca2+ ions are shown in yellow) (189) but only the ion coordinating sites Ca3 and Ca4 have been shown to be relevant for regulation of the cardiac exchanger (196). (C) The ionic current generated by the cardiac exchanger can be evaluated using the giant patch technique (100). Shown is the Ca2+ dependency of WT NCX1.1 ionic currents and the indicated mutants (196). Mutations E385A and D446A-D447A disrupt the binding of Ca2+ from sites 3 and 4 respectively and decreases the sensitivity of NCX1.1 to cytoplasmic Ca2+ of approximately 10-fold.
Figure 10. Properties of the exchanger Ca2+-binding domain 2 (CBD2).
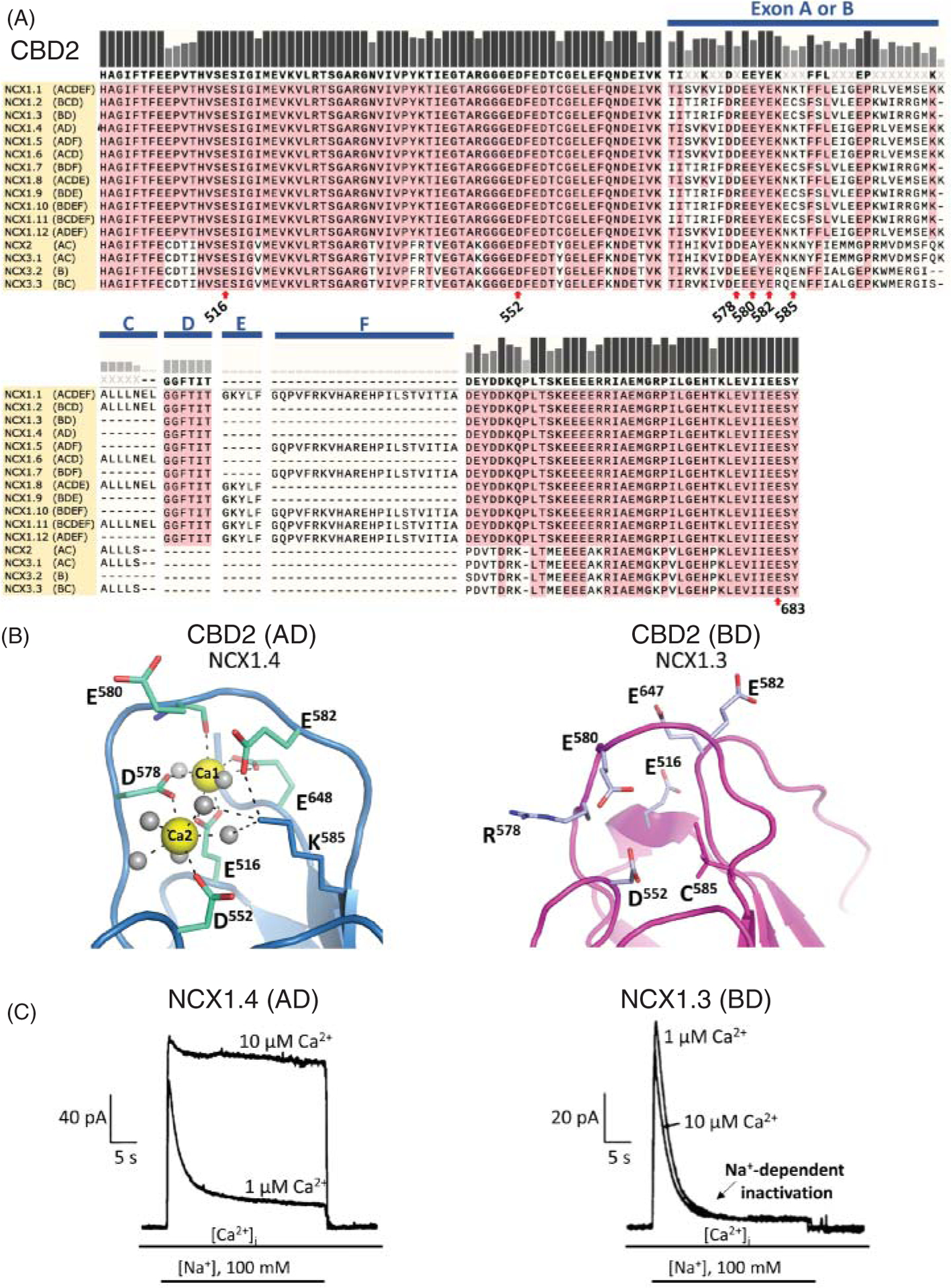
(A) Protein sequence alignment of CBD2 regions among human exchanger isoforms. The protein regions generated by alternative splicing (see Figure 2) are shown. Key residues for Ca2+ coordination are indicated. The lysine at position 585 does not directly coordinate Ca2+ ions but maintains CBD2 structural integrity by coordinating Asp 552 and Glu 648 in the absence of Ca2+ (16, 93). Residue 683 (Glu) corresponds to Glu 648 in isoforms expressing exons AD and to Glu 647 in exon BD expressing exchangers. Numbering is reported according to NCX1.1 sequence, excluding the signal peptide. (B) The alternative splicing within CBD2 affects Ca2+ coordination. NMR and X-ray structures show that isoforms expressing exon A allows coordination of two Ca2+ ions (PDB code 2QVM) (16, 93, 95), while those expressing exon B (PDB code 2KLT) (93) are unable to coordinate Ca2+. The presence of a positive charge at position 578 and a cysteine at position 585 in exon B expressing CBD2s prevents Ca2+ coordination (16, 93, 95). (C) Example of ionic currents recorded from exchangers expressing either exon A or B is shown. While the steady state current of the brain isoform NCX1.4 (AD exon) augmented in the presence of 10 μM cytoplasmic Ca2+, the kidney NCX1.3 (BD exon) steady state ionic current was insensitive to changes in intracellular Ca2+ concentration. The binding of Ca2+ to CBD2 enables exon A expressing isoforms to overcome the Na+-dependent inactivation with elevation in intracellular Ca2+ (49). In contrast, exon B expressing exchangers lack this important regulatory feature. Modified, with permission, from Dunn J, et al., 2002 (49).
Both domains respond to Ca2+ binding with conformational changes. While CBD1 requires nanomolar levels of cytosolic Ca2+ (Kh ~ 200 nM) to modify its configuration, CBD2 has a lower sensitivity to Ca2+ (~12 μM), and it shows more restricted movements than CBD1 (16, 23, 69, 93, 126). It has to be noted that, when conjoined, the two domains display properties that do not resemble those observed in the isolated CBD1 and CBD2, suggesting synergist interactions between them that lead to enhanced Ca2+ sensitivity and slower movements (69, 126). How these movements relate to enhanced transport activity will be discussed in section titled “Regulation by Cytosolic Ca2+”.
Catenin-like domain 2
This region, also referred to as cytoplasmic segment 2-Cyto2 (295), spans residues 651 to 705 (95) and, as seen for the catenin-like domain 1 (a.a. 238–370), it shares some sequence similarity with α-catenin and suggested to follow a similar organization with oppositely oriented α-helices (95). Though, NMR analysis of residues 657 to 702 indicates that this section of the loop is mostly disordered in the canine NCX1.4 (exons AD) (295). As previously mentioned, this region may include a potential binding site for calmodulin in correspondence of residues 684 to 703 (33). Binding of calmodulin to this site seems to modulate both NCX membrane trafficking and transport activity when expressed in HEK cells (33). If this regulation occurs in other tissues remains to be established.
Palmitoylation domain
The region that follows the catenin-like domain 2 has gained increased attention due to its multifactorial effects on NCX1.1 activity. Previously modeled as TMS 6 (a.a. 738–757) but then reassigned as an intracellular segment (186), this region includes an amphipathic α-helix (a.a. 740–756), which is required for the addition of palmitate at cysteine 738 (Figure 7) (226). Palmitoylation of NCX appears to influence its trafficking and oligomerization (see section titled “S-palmitoylation”). Residues within this helix (750KVXF) have also been implicated in interacting with phosphatases, which together with two protein kinases (PKA and PKC), and the anchoring protein mAKAP, form a macromolecular complex within the C terminal region of NCX cytoplasmic loop (244). The presence of this macromolecular complex has yet to be confirmed by other studies and the functional effects of phosphorylation on NCX activity remain controversial (see section titled “Phosphorylation”) (177).
Cytoplasmic loop between TMSs 7 and 8
As predicted by the atomic structure of the archaebacterial NCX_Mj, the loop between TMSs 7 and 8 is a short linker encompassing residues 824 to 827 (Figure 5B). This region was initially modeled as part of the reentrant loop within the α2-repeat (Figures 4C and 5A) (112, 114, 186). Single cysteine residues introduced at positions 825 and 827 were found to react with cytoplasmically applied sulfhydryl agents, confirming the cytoplasmic orientation of this loop. Furthermore, the introduction of cysteine at position 825 (D825C) decreased the sensitivity of NCX to both external Ca2+ and Ni2+ (a known blocker of NCX), when assessed using Na+-dependent 45Ca2+ uptake assay (114). Interestingly, this residue is highly conserved among the mammalian exchanger isoforms but is absent in NCX_Mj (Figure 6).
Cytoplasmic loop between TMSs 9 and 10
While cysteine scanning mutagenesis studies initially determined that this loop was extracellularly located (Figure 5A) (a.a. ~892 to 909) (112, 114, 187), it is now predicted to be exposed to the cytoplasm, as dictated by the NCX_Mj structure (Figure 5B) (150). Information is lacking regarding the role of this region in NCX activity.
Oligomerization
The cardiac NCX can exist at least as a dimer in the plasma membrane as assessed by optical and biochemical studies (126, 227). Cross-linking investigations suggest TMSs 1 and 2 as the potential contact surfaces between two exchangers (227). Moreover, fluorescence resonance energy transfer (FRET) studies show that the N terminal portions of the large cytoplasmic loop are in close contact (<10 nm) between two adjacent exchangers and that this distance narrows upon binding of Ca2+ to the CBDs (126). Recent biochemical evidence suggests that NCX1.1 palmitoylation also restructures existing dimers (79).
Despite this evidence, the functional effects of NCX oligomerization remain to be determined, especially in light of the fact that the archaebacterial homolog NCX_Mj exchanger was crystallized as a monomer. Examples of multimeric assembly of exchangers belonging to the same superfamily can be found in the literature. For example, the Na+/Ca2+-K+ exchanger dimerizes via interactions mediated by transmembrane domains (129) and the Ca2+/H+ antiporter protein YfkE from Bacillus subtilis (290), which shares structural similarity with NCX_Mj (290), exists as a trimer in the membrane (290). Perhaps, these solute carriers proteins form oligomers to regulate their transport activity, as seen in other transporters (11, 56, 57, 61, 91, 108, 199, 234, 235, 256, 300) or ion channels (44, 105, 255, 268).
Mechanisms of Ion Exchange
NCX is a bidirectional electrogenic transporter
The exchanger harnesses the energy of the electrochemical gradient of Na+ to move Ca2+ against its concentration gradient. It is generally accepted that NCX moves three Na+ ions in exchange for one Ca2+ ion, although other transport stoichiometries have been reported (130). The net positive charge moved across the membrane generates an ionic current that can be recorded using electrophysiological techniques (169). Normally, NCX extrudes Ca2+ from the cell. This is the most recognized operational mode of NCX and it is referred to as “forward mode” (Figure 1). In the heart, during the plateau and repolarization phases of the cardiac action potential, Ca2+ extrusion via NCX helps restore diastolic Ca2+ levels (40, 285, 286), while the coupled Na+ influx may contribute to membrane depolarization (3, 223).
As NCX is a bidirectional transporter (Figure 1), under certain membrane potentials and Na+ and Ca2+ concentrations, it can reverse, leading to Ca2+ influx and extrusion of Na+ from the cell. The perfect scenario for this to occur is in correspondence of the peak of the cardiac action potential (285, 286), when membrane depolarization and high intracellular Na+ levels allow NCX to reverse briefly. The resulting Ca2+ influx elevates Ca2+ within the dyadic cleft (140, 271), and consequently, less Ca2+ influx via the voltage-dependent Ca2+ channels will be required to open RyRs and initiate contraction (140, 271). Ca2+ influx via NCX reverse mode is, however, mainly linked to pathologies accompanied by elevation in intracellular Na+, such as during heart failure (40, 286) and ischemia/reperfusion (110). If persistent, the resulting Ca2+ overload leads to cell death, making NCX a direct mediator of ischemic injury (213).
Independently of its transport mode, the molecular mechanisms that allow NCX to move ions across the membrane remain vague. NCX is expected to follow an alternating access mechanism (119) (Figure 1) in which the binding pockets are exposed either to the extracellular environment (outward-facing state) or the cytoplasm (inward-facing state). During this process, NCX transitions through states in which the ions are trapped within the protein (occluded states) (Figure 1). The resolved structure of the homologous archaebacterial exchanger (150) has given a glimpse into this cycle by showing the protein in the outward state configuration. This milestone has certainly pushed forward the field, but atomic structures of the protein in different configurations are needed to fully unlock all the steps involved in ion transport. Yet, important information has emerged from the extensive investigations conducted on the mammalian exchangers and its archaebacterial homologue, and below we will summarize the current understanding of NCX mechanisms of transport.
Regions relevant to ion transport
Residues coordinating Na+ and Ca2+ ions
Since the cloning of NCX, a considerable number of mutagenesis studies have highlighted the α-repeats as essential domains for ion translocation (112, 114, 125, 184, 186, 195). These experimental observations have been validated by the crystal structure of the archaebacterial homolog NCX_Mj by revealing that the 12 amino acids that coordinate three Na+ and one Ca2+ ions are localized within the α-repeats. Specifically, four residues (A47, T50, S51, E54) are found in TMS 2, four (A206, T209, S210, E213) in TMS 7 and two each in TMS 3 (S77, N81) and TMS 8 (S236, D240) (Figure 11A) (150). These residues are arranged in a diamond shape, forming four coordinating sites which are referred to as Sext, Smid, Sint, and SCa (Figure 11B). The initial assumption was that the Sext, Smid, and Sint sites coordinated Na+, while Ca+2 ion occupied the SCa site (150). New evidence suggests that Smid is in fact occupied by a water molecule rather than Na+ (151, 162). By forming hydrogen bonds with the E54 (TMS 2) and E213 (TMS 7) residues, it appears that this bound water molecule stabilizes the remaining Na+ ion binding sites (162). This hypothesis has been corroborated by X-ray diffraction studies of NCX_Mj crystals soaked in the presence of different extracellular Na+ concentrations in the absence of Ca2+ (151). The resultant crystal structures also revealed that SCa can coordinate Na+ ions in addition of being the functional Ca2+-binding site, although with lower affinity.
A sequence comparison between NCX_MJ and NCX1.1 α-repeats (Figures 6 and 11B) shows that of the 12 ion-coordinating amino acids of NCX_Mj, nine are identical, two show conservative substitutions and one differs. In spite of this high conservation, several of these residues contribute differently to the transport activity of the mammalian and archaea exchanger, as detailed below.
Identical residues
Among the nine identical coordinating residues between the two exchangers, five positions show functional conservancy (S51, E54, S77, T209, S210 in NCX_Mj, which correspond to S110, E113, S139, T810, and S811 in the cardiac NCX1.1, Figure 11B) (273) as their replacement leads to either inactivity or drastic decreases in the apparent affinity for transported ions in both proteins (Figure 12) (125, 184, 273). In contrast, the asparagine found at position 81 in NCX_Mj and 143 in NCX1.1 appears to play a different role in the two exchangers. Initially described as part of the low affinity Na+ binding site SMid, this residue is now modeled to coordinate a water molecule in NCX_Mj (151, 162). Mutagenesis at this site does not impact NCX_Mj activity (273), whereas replacement of the equivalent residue within the cardiac exchanger abolishes transport function. The striking effects that mutation of N143 has on the transport properties of the mammalian exchanger hints at the possibility of the Smid site playing a more prominent role in the mammalian isoform than in the archaea transporter. Similarly, the serine residue at position 236 in NCX_Mj and 838 in NCX1.1, although conserved between the two proteins, seems to contribute to ion transport differently. Replacement of S838 with an alanine in NCX1.1 results in an inactive exchanger (Figure 12) (184), while the analogous mutation in NCX_Mj has little effect on its activity (273). This serine, together with A47, T50, S51, and E213, coordinates Na+ at its most intracellular site in NCX_Mj (Figure 11A). It appears that the homologous site in the cardiac exchanger, which is made of residues A106, S109, S110, E814, and S838 (Figure 11B), is less tolerant to mutagenesis, as replacement of any of these residues either drastically shifts Na+ and Ca2+ affinity (Figures 12A and 12B, respectively) or abolishes activity entirely (125, 184).
Figure 12. Residues relevant for NCX transport activity.
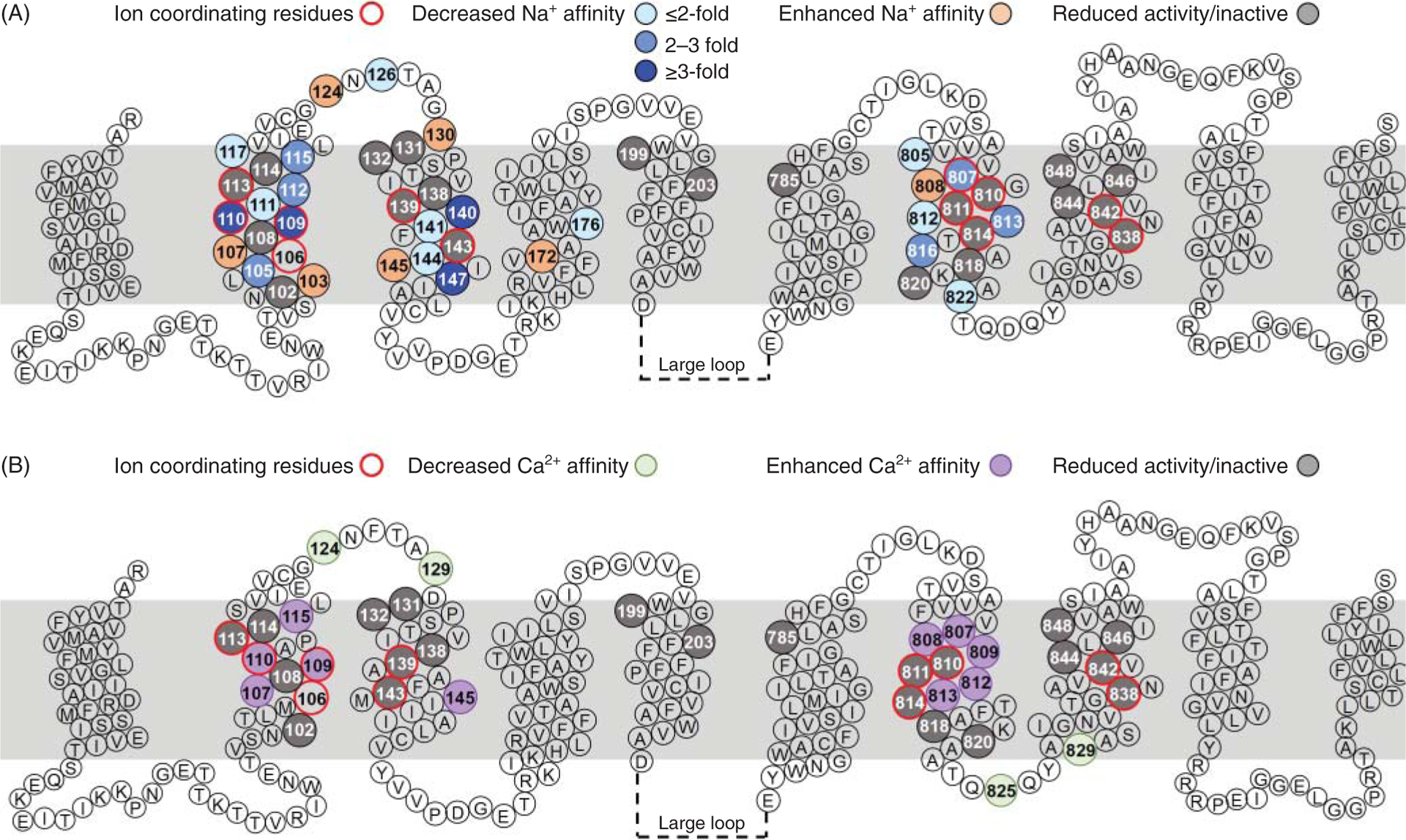
The location of residues whose replacement perturbed NCX1.1 transport activity is shown. Amino acid numbering is based on the sequence of the canine NCX1.1 exchanger, excluding the signal peptide. The indicated residues were mutated one at a time and the activity of the resulting exchanger was analyzed either electrophysiologically or as Na-dependent 45Ca2+ uptake (47, 112, 114, 125, 183, 184, 195). Mutations within the α-repeats drastically affect transport activity. This is consistent with these regions harboring the residues involved in ion coordination, as revealed by the crystal structure of NCX_Mj (150) (see Figure 11). The amino acids coordinating the transported ions (marked with a red circle) are well conserved between NCX_Mj and the mammalian exchanger (see Figure 11) and their replacement impairs activity (grey circle).
Conserved residues
The first ion-coordinating residue that shows conservation between the two exchangers contributes to the SMid site and is T50 in NCX_Mj and S109 in NCX1.1 (Figures 6 and 11B). Prior to the crystallization studies, the impact of this residue on NCX1.1 activity was investigated by combining mutagenesis with Na+-dependent Ca2+ fluxes (184). Using this approach, it was demonstrated that replacement of serine at position 109 with either an Ala or Gly resulted in inactivity, demonstrating for the first time a crucial role of this residue in transport activity. Subsequent electrophysiological studies established that the exchanger mutant S109C could generate ionic currents, although less efficiently when compared to wild-type exchanger. Importantly, the study revealed that mutations at this site significantly decreased the sensitivity of NCX1.1 to intracellular Na+ (Figure 12A) while enhancing its response to intracellular Ca2+ (Figure 12B) (125), consistent with a strategic role of this residue in the transport activity of the mammalian exchanger. It is striking that replacement of the homologous residue in NCX_Mj has little effect on transport rates (273), suggesting possible functional divergence between the two proteins at this site.
The second conservative substitution is the negatively charged glutamic acid at position 213 in NCX_Mj, which is an aspartic acid in NCX1.1 (D814) (Figures 6 and 11B). This residue is essential to coordinate both Na+ and Ca2+ ions at the SCa site in NCX_Mj. There is functional conservancy at this position as mutagenesis at this site strongly impairs the activity of both exchangers (Figure 12) (184, 273).
Non-conserved residues
The only ion coordinating residue not conserved between NCX_Mj and NCX1.1 is an aspartate at position 240 in NCX_Mj, which is replaced by an asparagine (N842) in the cardiac isoform (Figures 6 and 11B). In the archaebacterial exchanger, this residue is protonated and contributes to the SMid site occupied by a water molecule (151, 162). Replacement of this negatively charged residue in NCX_Mj (D240N), to mimic the mammalian exchanger, accelerates both Na+/Ca2+ and Ca2+/Ca2+ exchange rates (162). Although NCX_Mj D240N mutant does not fully resemble the mammalian exchanger in terms of its higher turnover rate, the phenotype observed in D240N underlines a role of this residue in setting the transport cycle rate in the archaebacterial isoform (162). Conversely, mutation N842D in the cardiac isoform significantly reduces the activity when assessed via Na+-dependent Ca45 uptake in oocytes (184). A detailed electrophysiological analysis of the effects of this mutation will have to be performed to determine its biophysical effect in the mammalian exchanger.
Further investigations analyzing the similarities and differences in the ion coordinating sites and their respective response/susceptibility to mutagenesis may help understand how the mammalian exchanger achieves a faster turnover rate than NCX_Mj (73).
Non-coordinating residues relevant for ion transport within the α-repeats
Mutagenesis of residues not directly involved in ion coordination has revealed effects on transport activity in both the mammalian and archaebacterial exchanger, although their relative impact on function differs between the two. As an example, replacement of two conserved prolines (P53 and P212) in NCX_Mj, which are near the coordinating residues in TMS 2 and TMS 7, drastically affects its transport properties (273). Comparative mutations in NCX1.1 (P112 and P813) had limited effects on its activity (125, 184). Similarly, mutation of residues C78 and C80, adjacent to the ion coordinating sites Ser77 and Asn81 in TMS3 of NCX_Mj, drastically lessens the transport capabilities of the protein (273), whereas the equivalent mutations in the mammalian exchanger, A140C and F142C, evoke the milder phenotypes of either a decreased affinity for transported Na+ (A140C) (Figure 12A) or unaltered properties (F142C) (195).
Within the α1-repeat, the extracellular linker between TMSs 2 and 3 (residues 119 to 136), previously modeled to form a reentrant loop in NCX1.1 (Figure 5A), shows limited similarity with NCX_Mj (Figure 4A). While more investigations are needed to define the architecture of this domain in NCX1.1, evidence indicates that this loop is not essential for transport, but it can shape NCX transport properties (195). Replacement of histidine at position 124 and phenylalanine at 126 elicited a modest but significant effect on the affinity of the transporter for cytosolic Na+, while also altering the cooperativity of binding (Figure 12A) (195). Furthermore, point mutations in this region reduce the apparent affinity for extracellular Ni2+, which is routinely used as an exchanger blocker (115). These observations underscore a role of this region in the transport function of NCX.
Residues outside the α-repeats relevant for ion transport
Following the newly modeled TMS 8, NCX1.1 has a stretch of 14 extracellular residues not present in NCX_Mj (a.a. 859 to 871) (Figure 5B). Iwamoto and colleagues replaced three of these residues (Asn861, Q864, and Gly870) with cysteine to assess the role of this region in transport. The biophysical properties of the resulting exchangers resembled those observed in the WT exchanger, suggesting that these residues are not essential for transport (112, 114). Three adjacent positions (855, 858, and 864) were found accessible to sulfhydryl agents when applied cytoplasmically (252) (Figure 5), in contrast to their predicted extracellular location in NCX_Mj. This disagreement stresses the need for further investigation of this region.
Several residues within TMSs 5, 6, and 9 have been shown to be essential for NCX1.1 activity as single point mutations within these helices result in inactive exchangers (Figure 12) (114, 184). Among them is residue Glu 199 (TMS 5). The corresponding residue in NCX_Mj is Asp 127, as determined by X-ray crystallography, and it appears to coordinate an extracellular Ca2+ ion (150). The functional relevance of this Ca2+-binding site is unclear for NCX_Mj and its particular relevance for NCX activity is otherwise unexplored. In the mammalian exchanger, this negatively charged amino acid is conserved among the three isoforms and is found within a region that shares some similarity to the Na+/K+ ATPase (NKA) (183, 184). Whether replacement of this residue impairs trafficking or impedes transport activity remains to be established.
The remaining TMSs are unexplored. Mutagenesis analysis of TMSs 1 and 6 should be prioritized as they are considered to be the gating helices that allow the transition from the inward to outward facing configurations, as detailed below.
Mechanisms of transport
During transport, NCX changes conformations to allow the ion-binding sites to be alternatively exposed to either the cytoplasm (inward-facing state) or the outside of the cell (outward-facing state) (Figure 1). How the mammalian NCX transitions between these states remain largely unresolved, albeit analysis of several outward-facing structures of NCX_Mj obtained in the presence of various concentrations of extracellular Na+ has revealed important steps into this process (Figure 13) (151). In the absence of bound Na+, there are two water filled pathways that connect the extracellular milieu to the ion-binding sites located in the core of the protein. The first water filled pathway is lined by TMSs 3 and 7 while the second is surrounded by TMSs 7 and 2. In this Na+ unbounded state, TMS 7 forms hydrophobic contacts with TMS 6, which is intimately bundled with TMS 1, both positioned toward the center of the membrane. Upon binding of Na+ to SExt, the N-terminal half of TMS 7 bends, breaking TMS 7 into two short helices (151). This bending/breaking triggers two events: it closes access to the water filled pathways, thereby occluding the ion coordinating sites from the external milieu; and, it disengages the hydrophobic contacts between TMSs 7 and 6. This allows the bundled TMS 6 and TMS 1 helices to slide toward the extracellular side (151), making the ion binding pockets accessible to the cytoplasm.
Figure 13. Molecular mechanisms of ion transport.
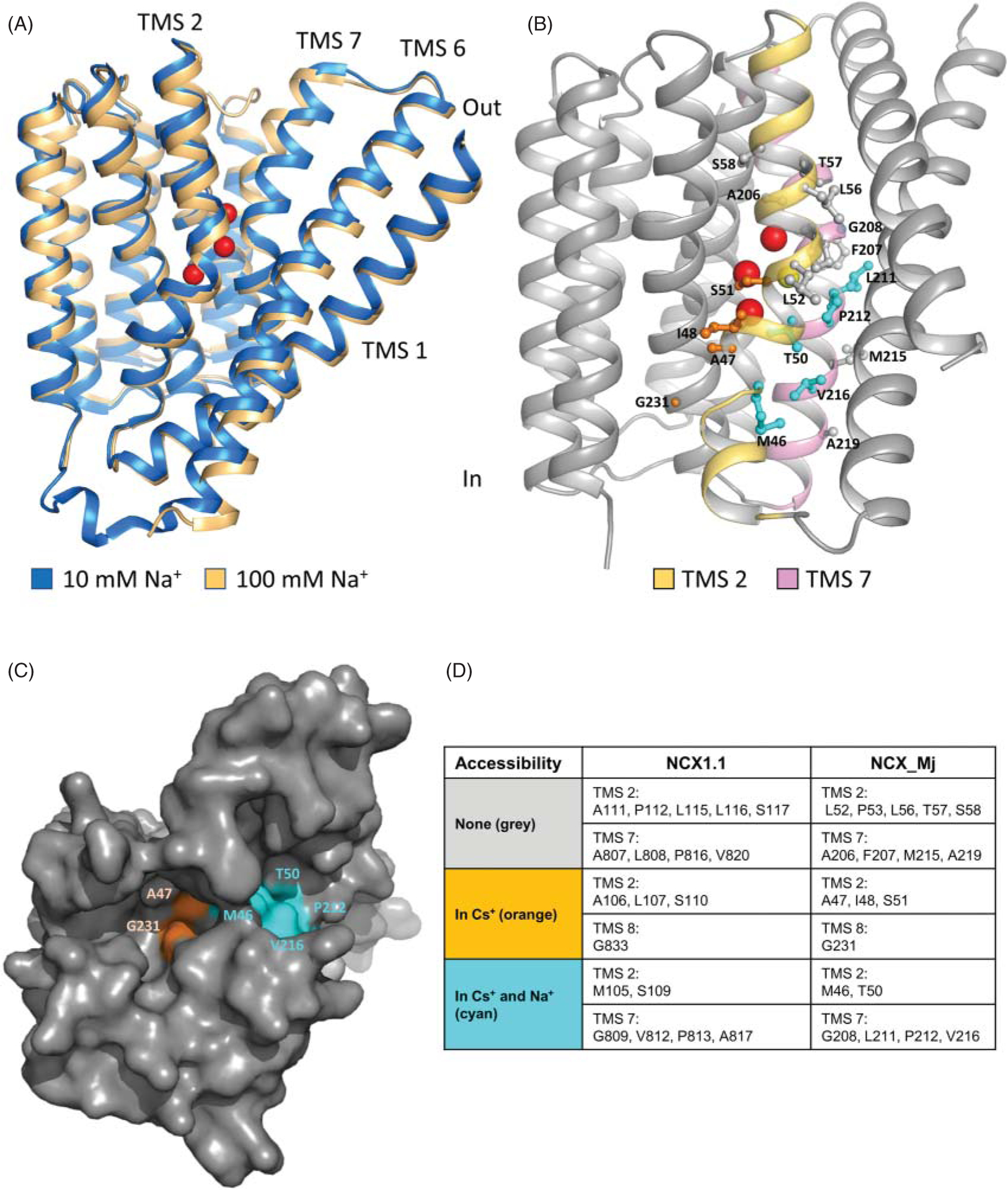
(A) Superimposed structures of NCX_Mj obtained in the presence of high Na+ (100 mM, gold, PDB code 5HXE) or low Na+ (10 mM, blue, PDB code 5HWY), Ca2+ ions were absent (151). Binding of Na+ (red spheres) elicits conformational changes throughout the protein. The movements of the external half of TMS 7 govern the transition from outward facing state to the occluded configuration (151). In the absence of Na+, TMS 7 is a contiguous “straight” helix. Upon binding of Na+ to SExt, TMS 7 bends (forming two helices) resulting in its movement away from TMS 6. This allows TMSs 1 and 6 to slide perpendicular to the membrane and transition the protein through occluded state (not shown) to the inward state (not shown). (B) Cysteines scanning mutagenesis allowed the mapping of the residues of the canine cardiac exchanger (NCX1.1) accessible from the cytoplasmic side. Shown are the homologous NCX_Mj residues mapped onto the inward model (125, 150). Cysteine introduced at positions highlighted in cyan were accessible to cytoplasmically applied membrane-impermeable sulfhydryl agent independently of NCX1.1 conformational state of. Residues labeled in orange were accessible to intracellular sulfhydryl agents only when the exchanger was constrained to the inward state. Residues labeled in grey were unaffected by sulfhydryl agents as assessed by ionic current inhibition. (C) Surface representation of NCX_Mj viewed from the cytoplasm. The inward-facing model predicts two separate cytoplasmic pathways for ions to reach their binding sites (150) and cysteine scanning mutagenesis conducted in the mammalian exchanger support this hypothesis (125). Residues are color coded as described in (B). (D) NCX_Mj residues corresponding to those of NCX1.1 tested for reactivity to sulfhydryl reagents are highlighted and indicated in the table. (B–D) Modified, with permission, from John SA, et al., 2013 (125), National Academy of Sciences.
Two independent studies support this model. First, hydrogen-deuterium exchange mass spectrometry (HDX-MS) studies confirmed flexibility of the extracellular end of TMS 7 upon Na+ or Ca2+ binding/occlusion in NCX_Mj (75). Second, a comparison between the NCX_Mj structure in the outward facing configuration and the related Ca2+/H+ exchanger structure in the cytosolic facing conformation (277) supports the idea that the transition between the inward and outward facing states is associated with a sliding motion of the similarly bundled TMSs 1 and 6. Intriguingly, in all the mammalian exchangers TMS 6 is directly connected to the large cytoplasmic loop, the site of numerous allosteric regulatory sites (see section titled “Regulation by Cytosolic Factors”). Perhaps, this large regulatory loop tunes the movements of TMS 6 and therefore controls NCX transport cycle.
The steps that govern the transitions from inward to outward facing in the mammalian exchanger remain to be determined. The current hypothesis is that the archaea and eukaryotic exchangers will follow similar steps. Electrophysiological studies combined with cysteine scanning mutagenesis support the idea that eukaryotic exchanger has also two cytoplasmic aqueous cavities flanked by TMSs 2 and 7 (Figure 13), which were predominately accessible when the transporter was constrained into the inward-facing state (125). This discovery is consistent with the idea that these two helices, in addition to flanking the extracellular ion conducting pathways, also delimit the intracellular ion/aqueous passageway(s). Moreover, the state-dependent access of these pathways (Figure 13) is indicative of conformational changes at these sites. Rapid advancements have been recently accomplished in deciphering the structure of transporters via Cryo-EM; the hope is that this information will be soon available for the mammalian exchanger allowing a deeper understanding of its mechanisms of transport.
Regulation by Cytosolic Factors
Evolution has equipped NCX with a variety of regulatory mechanisms to suit its surroundings. This is particularly needed in the heart where a fine-tuning of NCX activity is crucial to maintain proper function. For example, Na+ and Ca2+, in addition to being transported, play key regulatory roles. Increased levels of Ca2+ enhance NCX activity to favor Ca2+ extrusion, while elevations in intracellular Na+ inhibit transport, possibly to preclude NCX from reversing and thereby preventing Ca2+ influx. This duality of function, working as both modulators and substrates has proven burdensome in determining their molecular mechanisms of action. Nevertheless, much progress has been achieved and important molecular steps involved in these two intertwined ionic modulatory mechanisms has been deciphered. Moreover, new regulatory pathways have emerged which control NCX, some of which may play important roles in cardiac physiology, and/or pathology. We will summarize the advancements toward the understanding of the many and varied NCX regulatory mechanisms below.
Inactivation by intracellular Na+
Molecular mechanisms
The Na+-dependent inactivation describes a time-dependent decay of exchanger current due to increased levels of intracellular Na+ (Figures 8 and 10). This regulatory mechanism seems to originate from the occupancy of the transport site(s) by cytosolic Na+. Such an occurrence drives the protein into a new conformational state that prevents activity (so-called-inactivated state) (103). This hypothesis is founded on the observation that the sensitivity of both the exchanger current and its inactivation to intracellular Na+ are nearly identical, suggesting that they originate from the same mechanism (103). How the binding of Na+ to its intracellular transport sites drives NCX into the inactivated state has yet to be defined, but evidence clearly supports the involvement of the XIP region in this process (166) (see section titled “Organization of NCX Cytoplasmic Regions”). The current model predicts that XIP is tethered to PIP2 (or other anionic phospholipids), which by preventing the development of Na+-dependent inactivation allows for exchange activity. Following increases in cytoplasmic Na+ and the occupancy of the transport sites, a conformational change enables XIP to be released from PIP2 (86). The unbound XIP subsequently binds to a docking site on the exchanger to initiate the inactivation process. Several regions have been postulated to be the XIP binding site. A stretch of negatively charged residues (a.a. 445–455) within CBD1 has been proposed to interact with XIP via electrostatic interactions (84). This is consistent with evidence showing that positively charged residues within the XIP region are essential for the development of the Na+-dependent inactivation (87, 166). Another region implicated in XIP mooring is within the region of amino acids 562 to 688, in the C-terminal end of CBD2 (159, 168, 176). Electrophysiological recordings obtained from an exchanger split in the large cytoplasmic loop and lacking residues 266 to 672, demonstrated that absence of these residues did not ablate XIP sensitivity or the Na+-dependent inactivation (192). This observation suggests that the remaining region 672 to 688 is sufficient to retain XIP binding to allow for Na+ inactivation. Consistent with this hypothesis, deletion of residues 680 to 685 from the full-length exchanger NCX1.1 abrogates Na+ modulation (170), but it also removes Ca2+ sensitivity. More recently, it has been shown that a peptide corresponding to residues 709 to 728 interacts with the purified XIP and that deletion of the corresponding residues within NCX1.1 disrupts the binding of XIP to the exchanger as detected by co-immunoprecipitation (79).
These apparent divergent results may be explained by assuming that several domains of NCX form the docking site for XIP. This hypothesis seems supported by mutagenesis analysis combined with electrophysiological studies showing that the residues that impact this process are spread out within the protein. For example, two residues located at the cytoplasmic ends of TMSs 2 and 4 (Asn101 and His165, respectively) (Figure 7), play an important role in this allosteric mechanism as their replacement abolishes NCX Na+-dependent inactivation (47, 124). Single point mutations within the XIP regions can either abolish or enhance this process (Figure 8) (166); while Cys 739 controls the extent of Na+-dependent inactivation via its palmitoylation (226). Finally, residues close to the extracellular end of TMS2 (His124, Phe126, and Asp130) appear to control the kinetics of Na+ inactivation (195). This region is close to the Na+ binding sites and may influence the kinetics by an allosteric effect on Na+ binding.
The involvement of multiple disparate locations of NCX provides evidence that a complex system governs the modulation of NCX by cytoplasmic Na+ and a clear picture of how any or all of these domains interact to inactivate NCX will likely be revealed only after resolving the atomic structure of the mammalian exchanger.
Physiological relevance
The extent of Na+-dependent inactivation that NCX undergoes in native cells remains to be established. Electrophysiological investigations indicate that approximately 15 to 20 mM of intracellular Na+ is required to inactivate about 50% of the exchanger population (103, 165, 166). As these intracellular Na+ levels are considered not physiologically relevant, this regulation is often assumed to be inconsequential under physiological conditions (15). Moreover, the extent of inactivation is highly sensitive to secondary factors such as cytoplasmic Ca2+, PIP2, and protons, complicating the investigations of this regulatory mechanisms in vivo. In excitable cells (52, 95, 144), increases in intracellular Ca2+ rescue NCX from the inactive state (101, 167, 196). Similarly, PIP2 reduces the extent of NCX Na+ inactivation, and new evidence indicates that Cys739, located at the C-terminus of the large cytoplasmic loop, is involved in this process. It appears that palmitoylation of this residue enhances the affinity of XIP for anionic lipids, preventing this domain from inactivating the transporter (59, 79, 226).
In contrast to intracellular Ca2+ and PIP2, cytoplasmic acidification facilitates inactivation of NCX by Na+ (45, 46, 124). This synergy may be relevant during ischemic events when both Na+ and protons are elevated, which will lead to increased inactivation and less Ca2+ removal from the cytoplasm.
The observation that NCX Na+-dependent inactivation is highly conserved among the family of exchangers suggests that this regulation is of evolutionary advantage. NCX isoforms such as NCX3 (153), the Drosophila (CalX) (51), and squid exchangers (88), all show inhibition of their currents by elevated Na+ levels. An important exception is NCX2, which lacks this modulatory mechanism (153). NCX2 XIP region diverges the most from its homologs, perhaps altering its structural organization and autoregulatory properties (Figure 8). This is consistent with results showing that a peptide with a sequence analogous to NCX2 XIP region is a weak inhibitor of NCX1 activity (87). NCX2 is mainly found in the brain and perhaps here this regulatory mechanism would impair neuronal function.
The uncertainty about the relevance of NCX Na+-dependent inactivation in cardiac physiology calls for an intensive effort from the field, a nontrivial task due to the numerous technical challenges in studying this regulation in intact myocytes, isolated heart and in vivo. A possible methodology to address this issue has recently been reported with the generation of a new mouse line in which NCX is mutated at the genomic level via CRISPR to have this regulation abrogated. Such an approach seems promising (246, 260) to investigate how the Na+-dependent inactivation of NCX1.1 or lack thereof impacts cardiac function.
Regulation by cytosolic Ca2+
Molecular mechanisms
The modulation of NCX activity by cytoplasmic Ca2+ is well documented with the first report published more than 30 years ago (42). Key advancements were then achieved after the cloning of NCX in 1990 (185), which allowed heterologous expression of this transporter and its electrophysiological characterization (98). These investigations showed that cytoplasmic Ca2+ enhances transport by increasing the turnover rate of NCX and counteracting the extent of the Na+-dependent inactivation (101, 167, 196). As removal of the large cytoplasmic loop abolished Ca2+ regulation but not transport activity (168), it was concluded that cytoplasmic Ca2+ was binding to a domain separate from the transport site. Because of this important distinction, Ca2+ involved in the modulation of NCX is referred to as “regulatory Ca2+.” A central portion of the loop was found to form a high affinity Ca2+ regulatory site (residues 371–508) (145). A peptide including these residues was shown to undergo Ca2+-dependent conformational changes in physiological settings (197, 292). In 2006, NMR structural studies provided the first atomic view of this modulatory site and to the surprise of the field, revealed a second domain (Figures 7, 9, and 10) (95). This milestone and the following structural investigations (16, 92–95, 144, 145, 189, 288, 289) confirmed that the first Calcium-Binding Domain 1 (CBD1) encompasses residue 371 to 500 (145). This domain coordinates four Ca2+ ions (Figure 9B, see section titled “Organization of NCX Cytoplasmic Regions”) with an affinity for Ca2+ of ~0.2 μM (72, 126). Electrophysiological and biochemical studies corroborate the essential role of this region in NCX modulation by cytoplasmic Ca2+ (70, 72, 92, 101, 126, 145, 167, 168, 194, 196, 197, 288, 289, 292) and showed that occupancy of sites 3 and 4 is sufficient to confer Ca2+ regulation to the full-length exchanger (Figure 9C), as mutations of strategic residues that coordinate Ca2+ at sites 1 and 2 do not affect the response of NCX to Ca2+ (196).
As mentioned above, a second CBD2 was later discovered (95). CBD2 is less sensitive to intracellular Ca2+ (23, 95, 126, 196) with an affinity of about 12 μM and coordinates Ca2+ at only two sites (Figures 10), which are not functionally equivalent. Disruption of Ca2+ site 1 completely abolishes the sensitivity of the cardiac NCX to regulatory Ca2+, while site 2 does not contribute significantly to the Ca2+ regulatory properties of NCX ionic currents (16, 196). In the cardiac isoform, coordination of two Ca2+ ions by CBD2 is conferred by the presence of exon A. Exon B expressing exchangers, which are found in nonexcitable cells, lack the Ca2+ ion coordination capability (49, 52, 71, 92–94). This has important functional implications as expression of exon A allows intracellular Ca2+ to alleviate the inhibitory effects of the Na+-dependent inactivation (Figure 10B), while exon B expressing isoforms lack this ability as detailed in section titled “Influence of Cytoplasmic Ca2+ on the Na+ Dependent Inactivation”.
In addition to exon A, CBD2 of the cardiac exchanger is composed of exons CDEF which encode for regions at the opposite end of the Ca2+ ion coordinating sites. This arrangement puts them in close proximity to the CBD1’s Ca2+ ion coordinating region. Studies conducted with the conjoint CBDs show that expression of exons C, E, and F enhances CBD1’s affinity for Ca2+ (68, 93). Deletion of the equivalent regions from the full-length exchanger minimally impacts its biophysical features (196). This leaves the role of regions encoded by CDEF in controlling Ca2+ regulation uncertain.
The presence of multiple Ca2+ coordinating sites raises the question of how their occupancy leads to enhanced exchanger activity. The expectation is that Ca2+-dependent conformational changes in the CBDs are then transmitted to the membranous region of NCX leading to enhanced activity. Indeed, biochemical, structural, and optical studies have established that binding of Ca2+ to the isolated domains triggers conformational changes (25, 94, 145, 197, 292), which are more conspicuous in CBD1 than in CBD2 (16, 93, 95, 126, 197). Ca2+ driven movements have also been detected in the full-length exchanger, mainly due to the unbinding and binding of Ca2+ to CBD1 (126).
To better decipher the movements that drive NCX transport activity, numerous studies have been conducted using the two domains conjoined together but as separate entities from the full-length protein. Small-angle X-Ray scattering analysis indicates that binding of Ca2+ to the purified conjoined CBD1 and CBD2 domains leads to a more compact conformation (93), while NMR and Fluorescence Resonance studies suggest a more elongated (126, 197, 240) and rigid conformation (72, 240). In spite of these apparent discrepancies, a consistent observation is that, when in tandem, the two domains elicit movements not observed in the two separate domains due to inter-domain connections. It has been proposed that the arginine at position 532 of CBD2 is essential for this cross talk by contributing to Ca2+ coordination within CBD1 via a salt bridges with two negatively charged aspartate residues found at sites 3 and 4 of CBD1 (D499 and D500) (72). This electrostatic interaction stabilizes Ca2+ in CBD1 and makes the conformation of the conjoined domains more rigid (72). Conversely, in the absence of Ca2+, the linker between the two domains gains flexibility, allowing the two CBDs to move freely with respect to each other (72). This observation suggests that the more “tense” (Ca2+ bound) structure activates NCX, while the more “relaxed” (Ca2+ unbound) structure leads to decreased exchanger activity.
While, the electrostatic interactions between the two CBDs may offer an explanation for the movements seen in the conjoined domains (69, 126, 197), they do not appear as the main driving force underlying the response of the full-length exchanger to changes in intracellular Ca2+. This is because replacement of Arg 532 with Cys in the full-length exchanger only modestly affected the sensitivity of NCX1.1 to cytosolic Ca2+ (194). Likewise by inserting a spacer of 7 Ala between the two CBDs, to potentially uncouple them, had minimal impact on NCX Ca2+ regulation (194).
More effort needs to be invested to fully disclose how the full-length protein responds to changes in intracellular Ca2+ due to its physiological implications. For example, there is no information on how the movements of the CBDs are coupled to NCX enhanced activity. A possibility is that the catenin-like domains flanking the CBDs act as springs that drive the exchanger into a more active state. Alternatively, the CBDs may anchor to another domain of the protein or the transmembrane region, modulating its movement. Interestingly, the exchanger CBDs are structurally homologous to the C2 domain, whose plasma membrane localization is driven by Ca2+ occupancy (29). To date, only a few residues outside the cytoplasmic loop have been shown to influence Ca2+ regulation. Replacement of the Asn 101 or the His 165, which are found at the cytoplasmic ends of TMS 2 and 4 respectively, renders NCX insensitive to intracellular Ca2+ (194). Perhaps these regions are a contact point between the transport machinery of the membranous NCX and the large cytoplasmic loop possibly translating its movements into changes in affinity for the transported ions (43).
Physiological relevance
The amount of Ca2+ needed to activate NCX in native cells remains vague, as a wide range of apparent affinities have been reported in the literature (20 to 800 nM) (100, 101, 171, 196). In some species, evidence indicates that this regulation takes place under physiological conditions (197, 284, 292). Enhancement of transport activity by elevations of intracellular Ca2+ has been observed in both ferret and rabbit cardiomyocytes (77, 284), while NCX seems already fully activated under resting conditions when investigated in murine cardiac myocytes (284). This diverse response may be related to different cytoplasmic environments, or alternatively due to changes in the molecular properties of the exchanger found in these species. Interestingly, Kim and Lee cloned the cardiac murine exchanger (134) and reported the presence of a glutamine at position 682 instead of a glutamic acid as seen in the canine (185), ferret, rabbit, and human exchanger. This glutamine is next to the negatively charged glutamic acid 683, an essential component of CBD2 Ca2+ coordination site 1 and its replacement abolishes NCX Ca2+ regulation (196). The substitution of a negative charge at this position in the murine sequence may affect the ability of NCX to respond to intracellular Ca2+. It should be noted, however, that the latest NCBI Databases (RefSeq, NM_011406.3, CCDS 37706.1) reports the presence of a glutamic acid at position 682, as is seen in other species such as human and dog. Since the mouse model is extensively used for investigating the role of NCX and in general the process of excitation contraction coupling, the amino acid sequence of both CBD1 and CBD2 should be confirmed. (Sequencing in the authors’ laboratory of C57BL/6J mice indicate the presence of glutamic acids at positions 682 and 683.)
Influence of cytoplasmic Ca2+ on the Na+ dependent inactivation
In addition to increasing the turnover rate of NCX, cytoplasmic Ca2+ can relieve NCX from the Na+-dependent inactivation process, resulting in enhanced transport activity. This effect is only observable in exchangers expressing exon A, while NCX1 splice variants expressing exon B, are unable to overcome the Na+-dependent inactivation with increased levels of intracellular Ca2+ (49, 52, 93). This striking difference is readily observed by comparing the biophysical properties of the brain (NCX1.4, exon AD expression) and kidney (NCX1.3, exon BD expression) exchanger isoforms (Figure 10C) (49). While steady-state ionic currents of the brain isoform NCX1.4, were augmented by cytoplasmic Ca2+, steady-state currents recorded from the kidney isoform NCX1.3 were insensitive to changes in intracellular Ca2+ (Figure 10C) (49, 52). Sequence comparison between the exons shows a negative charge, Asp 578, in exon A and a positive charge, Arg 578, in exon B (Figure 10). This difference prevents Ca2+ coordination in exon B expressing NCX1 isoforms (Figure 10B) (93). Consistently, the substitution of Asp 578 in NCX1.4 with Arg, making it like exon B, was sufficient to prevent Ca2+ antagonizing the Na+-dependent inactivation (49). Similarly, mutation of Asp 578 in the cardiac exchanger NCX1.1 (exon ACDEF) abrogates its sensitivity to increasing intracellular Ca2+ levels, including its effects on Na+-dependent inactivation (16).
Even though CBD2 is the main domain that appears to be responsible for affecting Na+ dependent inactivation, mutations within CBD1 have also been shown to interfere in this process (196). This observation suggests a complex network of interactions between these domains and other allosteric regulatory regions of NCX. Deciphering the mechanisms by which Ca2+ and Na+ communicate is not an easy task, and resolution may require a crystal or cryo-electron structure of the mammalian isoform.
Regulation by cytosolic protons
The exchanger transport function is highly sensitive to changes in intracellular protons. The first evidence of this allosteric modulation was reported in 1982 by Philipson and colleagues (201, 205). The authors investigated the effects of changes in pH on NCX activity measured as Na+-dependent 45Ca2+ uptake from isolated canine cardiac sarcolemmal vesicles (201, 205). They found that acidic pH was a strong inhibitor of transport function, while alkaline pH enhanced its activity. Insights into the molecular mechanisms that govern this modulation started emerging a decade later when the giant patch technique (100) was applied to record the small ionic currents generated by NCX from cardiac myocytes. Several important observations stemmed from these studies (45, 46, 103). First, 50% of NCX current was inhibited at a pH of approximately of 7.3, well within physiological pH range. Second, alkaline pH strongly enhanced NCX currents and eliminated its sensitivity to both cytoplasmic Ca2+ and Na+, leaving the transporter in a constitutively active state (Figure 14). Third, proton inhibition was enhanced by intracellular Na+, suggesting a link between the Na+-dependent inactivation process and proton regulation. Fourth, proteolytic degradation of the cytoplasmic components of NCX with chymotrypsin abolished the regulation of NCX by cytosolic protons. As chymotrypsin also eliminates Na+ and Ca2+ regulation (96), it was implied that protons, Na+ and Ca2+ act on a common domain within the large cytoplasmic loop (45, 46). This hypothesis was explored in more depth by Boyman and colleagues (22). Their studies revealed that the Ca2+-binding domains could also coordinate protons which, by displacing the Ca2+ from their binding sites, inhibit transport function (22). Electrophysiological investigations, however, do not support this hypothesis by showing that a full-length exchanger with the Ca2+-binding domains mutated to prevent Ca2+ coordination still responds to changes in cytoplasmic levels of protons in a manner similar to the native exchanger (Figure 14) (124). Rather than the CBDs being responsible, it was shown that two histidine amino acids, His124 and His165, located at the extracellular end of TMS 2 and the cytoplasmic end of TMS 4, respectively, were essential for NCX pH modulation (Figures 7 and 14) (124). Indeed, their replacement decreased the sensitivity of NCX to cytoplasmic protons and in the case of the histidine at position 165, it also decreased the cooperativity of proton binding (Figure 14) (124).
Figure 14. pH regulation of NCX1.1.
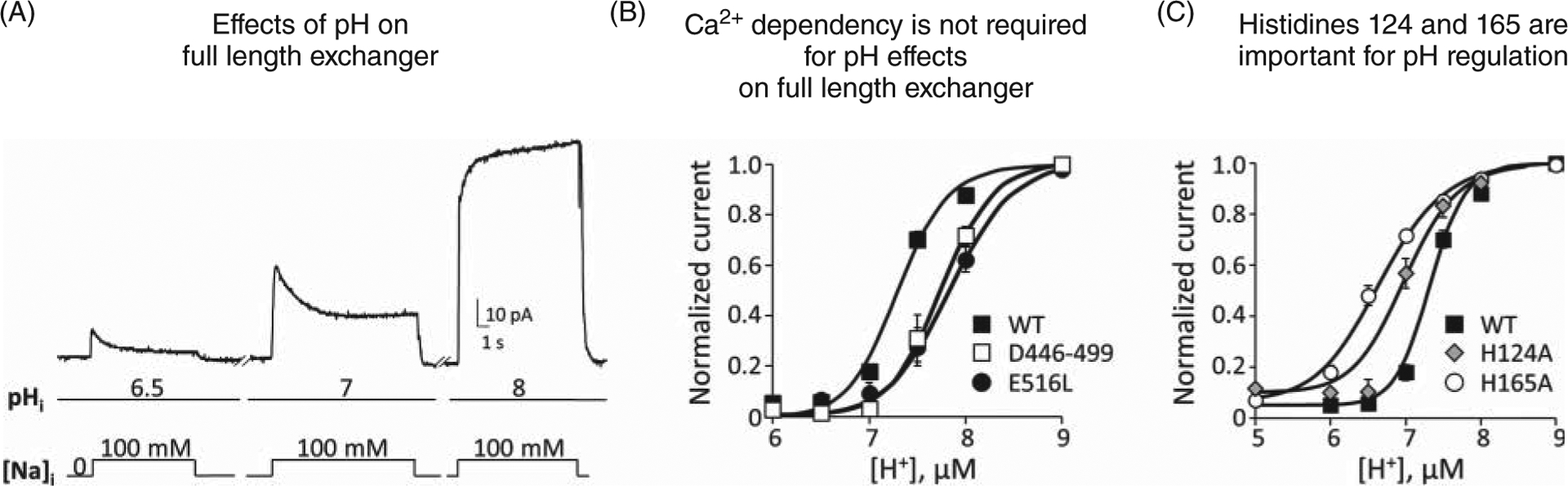
(A) Cytoplasmic acidification inhibits NCX activity; conversely, alkaline pH enhances NCX ionic currents and renders NCX insensitive to both Ca2+ and Na+ (not shown) (45, 46, 124). Outward exchanger currents were recorded from oocytes expressing the canine cardiac exchanger NCX1.1 using the giant patch technique. (B) Detailed electrophysiological studies show that mutations that disrupt binding of Ca2+ either to CBD1 (Δ446–499) or CBD2 (E516L) do not decrease NCX pH sensitivity (124). This result suggests that displacement of Ca2+ from the regulatory sites is not essential to confer pH sensitivity to the full-length exchanger. This contrasts with experiments conducted with the isolated Ca2+-binding domains (CBD1 and CBD2) showing that protons can displace Ca2+ from their binding sites (22). (C) Replacement of histidine 125 and 165 with alanine drastically decreases the sensitivity of the cardiac exchanger to cytosolic protons and in the case of position 165 also affected the cooperativity of the binding. Measurements were conducted on the canine cardiac exchanger expressed in Xenopus laevis oocytes as detailed in (124). (A,C) Modified, with permission, from John S, et al., 2018 (124), © 2018, Rockfeller University Press.
The high sensitivity of NCX to cytoplasmic pH underscores the impact that this regulation may have in cardiac function under conditions in which the cytoplasmic pH changes: dropping in ischemia and rising during metabolic alkalosis. By further elucidating the molecular mechanisms of NCX pH regulation, we will have a better understanding of how this regulation tunes NCX activity in its native environment.
Regulation by lipids
The role of lipids in controlling NCX activity was first shown in 1983 with studies demonstrating an enhancement of exchange activity measured from highly purified canine cardiac sarcolemmal vesicles subjected to phospholipid hydrolysis (202). Today, it is well demonstrated that a broad spectrum of lipids are capable of modifying NCX activity including anionic phospholipids (204, 205), anionic amphiphiles (99, 200), fatty acids (206), phosphatidyl serine, cholesterol (276), and long-chain acyl CoA (233). All act to enhance the activity of NCX.
The mechanism of action of lipids on NCX seems complex and multifaceted. There is general consensus that these lipids target NCX Na+-dependent inactivation and, by antagonizing it, enhance exchange activity (86, 98, 250, 294). The current hypothesis is that XIP is anchored to PIP2 (86) but is released from it when Na+ is bound to the transport sites. The change in conformation following the unbinding of XIP from the membrane leads to exchanger inactivation (see section titled “Inactivation by Intracellular Na+”) and therefore transport inhibition. High levels of PIP2 or other anionic lipids act to limit the release of XIP, preventing the Na+-dependent inactivation process. Several lines of evidence support this hypothesis. For example, a peptide with a sequence identical to the XIP region binds both to PIP2 (6, 14, 86) and different types of lipids (248). Moreover, mutations within the XIP region abolish NCX activation by PIP2 (86) or long-chain acyl CoA esters (233). It must be pointed out that high levels of PIP2 can lead to opposing effects. Indeed, if too elevated, PIP2 decreases transport activity by reducing the number of exchangers at the plasma membrane via endocytosis (294).
Inhibition of NCX activity has been also observed upon application of amphiphiles (131) and n-3 polyunsaturated fatty acid (PUFAs) from fish oil (2, 291). It is not clear why certain lipids produce contrasting effects on NCX. Perhaps the experimental conditions used to investigate NCX properties influence the ultimate output. Another factor contributing to these divergent responses may be the alternative splicing of NCX. Fatty acids seem to inhibit more effectively isoforms expressing exons B and D (NCX1.3), found in smooth muscle and kidney, than the cardiac exchanger, which expresses exons ACDEF (NCX1.1) (2). As these alternative splice isoforms have CBD2 with different sequence and properties, the results suggest that this domain may be an alternative target for lipid regulation, but further studies are required. This is relevant as changes in lipid and fatty acid composition of plasma membranes are observed both during exercise and ischemia (233, 272) and their action on NCX may influence cardiac function.
Post-Transational Modifications
NCX is subjected to a variety of posttranslational modifications. Not all have been investigated with respect to their effects on NCX activity, biophysical characterization, or modulation of trafficking.
Glycosylation
Cloning and sequence analysis of the cardiac NCX1.1 revealed 6 potential N-linked glycosylation sites (106). A series of deletions combined with site-directed mutagenesis identified Asn 9 of the extracellular N-terminal region of the cardiac NCX1.1 as the only site at which glycosylation actually occurs (Figure 4) (106). In the cardiac exchanger NCX1.1, glycosylation does not have apparent functional consequences (106), while glycosylation of NCX3 at site Asn 45 seems relevant for targeting the protein to the plasma membrane (156).
Disulfide bond
The cardiac NCX1.1 possesses one extracellular disulfide bond between Cys 792 and either one of the two cysteines in the N terminus (Cys 14 or 20) (Figure 4E). While the presence of the disulfide bond is not essential for NCX activity, evidence suggests that it may affect the protein’s expression levels as an exchanger with either cysteine 20 or 792 mutated to alanine showed a significant decrease in expression (241). Of note, the presence of the disulfide bond results in a different migration pattern on a SDS-PAGE, a 160-kDa band under nonreducing conditions, and a 120 kDa band under reducing conditions (241).
S-palmitoylation
Protein palmitoylation consists in the linkage of a 16-carbon palmitate moiety to intracellular cysteine. This modulation is known to control numerous signaling pathways of ion channels such as their spatial organization in the membrane, trafficking, and turnover pattern (253). NCX1.1 was recently found to undergo this posttranslational modification and its impact on exchanger activity or physiological function is still developing (59, 79, 207, 226). NCX1.1 palmitoylation occurs in the Golgi and involves the single cysteine at position 739 (Figure 7). A short helix that follows this residue (a.a. 740–756) is essential for this posttranslational modification to occur, possibly by being the docking site for the S-acylation enzyme (207). Both the Cys 739 and this short α-helix are found at the C-terminus of the large cytoplasmic loop, just prior to TMS 6. As this TMS is considered essential for the ion translocation cycle (150, 151), this position may be strategic for modulation of NCX1.1 activity.
Palmitoylation of NCX seems to affect several of its features: it modifies NCX1.1’s dimer structure (79); it is essential for the development of the Na+-dependent inactivation (226); regulates the lifetime of NCX in the plasma membrane during stress conditions (226), and alters the affinity between the XIP domain and its binding site made of residues 709 to 728 (79). As about 60% of the exchangers in cardiac myocytes are palmitoylated (226), this posttranslational modification may represent a new emerging mechanism to control heart function by influencing exchanger activity and expression. An important physiological role of NCX palmitoylation regulation in general, may be supported by the observation that Cys 739 and the α-helix which follows are evolutionarily conserved among the mammalian isoforms, NCX2 and NCX3.
Phosphorylation
Due to the impact of β-adrenergic stimulation on cardiac function, efforts have been undertaken to determine whether NCX activity is itself influenced by phosphorylation. Evidence shows that both PKA and PKC can enhance the activity of the cardiac exchanger (113, 116, 153). It appears that PKA, two phosphatases PP1 and PP2 and A-kinase anchoring protein mAKAP, are tethered to the C-terminus of NCX forming a macromolecular complex (244), although the presence of this complex has not been confirmed by other studies. Also, whether it allows direct phosphorylation of NCX by PKA in the heart remains controversial, with conflicting reports suggesting that NCX may not be the direct target of PKA (76, 152, 238, 244, 282). The sites of phosphorylation within NCX are also contentious. Threonine at position 371 within the large cytoplasmic loop has been proposed as a putative site, as a short identical peptide carrying this residue could be phosphorylated by PKA (282). However, this residue is not accessible in the full-length exchanger, supporting the hypothesis that NCX is not directly phosphorylated in native tissues (282). Accordingly, it has been postulated that an accessory subunit may be subjected to phosphorylation/dephosphorylation to regulate NCX activity. This theory is based on evidence showing that phospholemman (PLM) associates with NCX and inhibits its activity when phosphorylated (31, 83, 299).
Cleavage by Ca2+-dependent proteases μ-Calpain
The cardiac NCX1.1 is a substrate for the Ca2+-dependent proteases μ-Calpain but the functional implications of its cleavage are contradictory. Electrophysiological recordings from Xenopus oocytes expressing the cardiac exchanger demonstrated that proteolysis of NCX1.1 by μ-Calpain removes the requirement of intracellular Ca2+ for activity, which thereby enhances transport function (104). In contrast, NCX1.1 ionic currents were inhibited upon cleavage by calpain after expression of the exchanger in human embryonic kidney cells (281). The authors showed that calpain cleaves NCX at Met 337 after directly binding to the protein (281). Similarly, proteolysis of NCX3 by calpain at sites 504, 510, and 512, which are not well conserved within NCX1, has been shown to inhibit transport activity (9). μ-Calpain controls several essential physiological processes such as muscle contraction, cell proliferation, differentiation, and apoptosis (28, 259). Its activity is also upregulated in ischemia-reperfusion injury in the heart (111). Given these physiological and pathological effects of μ-Calpain, more effort should be funneled into understanding how NCX is affected by this endogenous protease.
Physiological Role of the Na+-Ca2+ Exchanger in the Heart
General function
The primary role of NCX1.1 in the heart is to extrude Ca2+ out of the cell, countering the Ca2+ that enters the cytoplasm during systole through L-type Ca2+ channels (LTCC) (26). This is an essential step for relaxation and thus normal heart function.
There is a large body of evidence that demonstrates the essential role of NCX in excitation-contraction (EC) coupling and the reader is referred to several reviews about this topic (15, 17, 80, 198). These investigations have analyzed the cellular localization of NCX within cardiac cells and its proximity to other proteins relevant for EC coupling. Moreover, genetically modified mice engineered to either overexpress or knockout NCX have been instrumental in consolidating the important role of this protein in normal and diseased hearts. We will elaborate on these aspects of this essential protein.
Expression and distribution of NCX1.1 in mammalian cardiac myocytes
Expression of NCX1.1 peaks near birth and then declines during maturation to adulthood. It appears that the density of NCX1.1 is about 2.5- to 6-fold greater in neonatal myocytes when compared to adult cells, as assessed by RNA levels and ionic current recordings (4, 5, 18). In neonatal cells, NCX1.1 is homogenously distributed on the cell surface (82), although clusters of exchangers spaced at approximately 0.7 μm have been reported (38).
As the T-tubule develops, NCX1.1 localizes within these highly specialized structures (38, 58, 247). It has been estimated that in adult myocytes more than 60% of the population of exchangers is within the T-tubule (38, 39), although recent super-resolution imaging microscopy investigations suggest that this may be an overestimation (161). As seen in neonatal myocytes, adult myocytes show exchanger clusters both in the T tubules and the remaining sarcolemma (120).
Functional coupling with other cardiac proteins
The knowledge of how NCX functionally and spatially relates to other cardiac proteins is crucial to understand how this protein influences cell excitability and contractility. Below, we will summarize how NCX interacts with other proteins important for EC coupling.
Colocalization with voltage-dependent Na+ channels
Voltage-dependent Na+ channels are an essential component of EC coupling by causing the rapid upstroke of the action potential and controlling SR release (141). By elevating the submembrane Na+ levels and depolarizing the plasma membrane, they create the perfect scenario for NCX1.1 to transiently reverse at the peak of the action potential (283, 285). The Ca2+ entry via reversed NCX1.1 then influences Ca2+ release from the SR (140, 141, 155, 180, 271) (Figure 15, panel 1) by acting in synergy with the Ca2+ entering via the voltage-dependent Ca2+ channels to trigger the opening of RyRs (140, 271). A requisite for this cross talk is the close proximity between NCX and the voltage-dependent Na+ channels, creating a functional microdomain that allows NCX to sense elevations in cytosolic Na+ (Figure 15, panel 1). The presence of this close interaction remains controversial as immunocytochemistry studies show NCX either clustered with neuronal-type Na+-channels in the T-tubule (24, 67, 222), or in a T tubule domain that does not include either Na+ or Ca2+ channels (247). This controversy has emerged also from electrophysiological investigations. Studies conducted in mice showed that low concentrations of TTX (~100 nM), which preferentially inhibit the neuronal-type Na+ channels, significantly reduced Ca2+ release from the SR (271) by preventing NCX to reverse. In contrast, investigations conducted in rabbits suggest that Ca2+ entry via NCX due to the opening of the Na+ channels is minimal (283).
Figure 15. NCX1.1 distribution in cardiac cells.
The cartoon summarizes the distribution of NCX within adult cardiomyocytes and its coupling to other proteins essential for excitation-contraction coupling. (A) The exchanger has been localized in proximity to voltage-dependent Na+ (Nav,). The influx of Na+ via voltage-dependent Na+ channels drives NCX to function in reverse mode and therefore prime the dyadic cleft with Ca2+ (140, 271). (B) Changes in NCX1.1 expression (overexpression or knockout) regulate the extent of the voltage-dependent Ca2+ channels (LTCCs) Ca2+-dependent inactivation process, suggesting that these two proteins are in close proximity within the dyadic cleft. (C) NCX forms a macromolecular complex with the Na+/K+ ATPase (NKA), inositol triphosphate receptors (IP3R) and ankyrin (ANKB) (172). Genetic disorders due to defective expression of ankyrin result in disruption of this macromolecular complex leading to cardiac arrhythmia (173). (D) NCX appears to be in tight connection with the transient receptor potential channel (TRPC3) channels. The current model predicts that the influx of Na+ via TRPC3 tunes NCX transport activity by changing the Na+ levels in its proximity. If elevated enough, Na+ may reverse NCX augmenting intracellular Ca2+ levels (53, 54).
These results underscore the relevance of addressing the spatial organization of transporters and channels within cardiac myocytes to unravel how proteins in close proximity to NCX can affect cardiac contractility by influencing its transport.
Coupling with Ryanodine receptors
NCX1.1 has been found to form clusters both in neonatal and adult myocytes, some of which can be found in close proximity to RyRs (Figure 15, panel 2) (38). This spatial co-localization seems more prominent in neonatal myocytes, where NCX plays an essential role in initiating contraction. Indeed, in neonatal cardiac myocytes, the release of Ca2+ through RyRs seems triggered mainly by Ca2+ influx driven by NCX functioning in the reverse mode (38, 107, 287), and not by the Ca2+ entering via the voltage-dependent Ca2+ channels (LTCCs). This is in sharp contrast to that seen in adult cardiac cells. Consistent with this, in adult myocytes, only a small percentage of exchangers (5%–27%) are adjacent to the RyRs within the T-tubule (38, 120, 243, 247, 267). This reflects upon the preeminent role that LTCCs play in activating Ca2+ release from the RyR in adult myocytes (Figure 15).
Macromolecular complex with Ankyrin and the Na+-K+ ATPase
A physical interaction between NCX1.1 and ankyrin was first reported in 1993 (148). The ankyrin family consists of three proteins that link plasma membrane proteins to cytoskeletal elements. Ankyrin B is the isoform known to interact with NCX1.1. This coupling seems mediated by the ankyrin repetitive β hairpin loops of repeats 16 to 18 (37), which binds to an undisclosed site within NCX. In the heart, ankyrin B tethers together the Na+-K+ ATPase (NKA), the inositol triphosphate receptor (IP3R), and NCX1.1, creating a molecular complex in cardiac myocytes (Figure 15, panel 3) (172). Loss of function of ankyrin B, and thus of this macromolecular complex, causes type 4 long-QT cardiac arrhythmia and sudden death (173). This has been confirmed in a mouse model in which haploinsufficiency of ankyrin B elicits a similar phenotype and disrupts both the trafficking and expression of NCX1.1, NKA, and IP3R (173). Evidence shows that NKA α2 is the predominant isoform functionally coupled to NCX1.1 (39, 118, 257, 264) and the main pump involved in Na+ handling (118, 264, 298). The close proximity between the NKA and NCX1.1 enforced by ankyrin may attenuate any localized Na+ accumulation in the proximity of NCX1.1. This, in turn, may facilitate Ca2+ extrusion by limiting the extent of Na+-dependent inactivation and/or favoring NCX1.1 forward mode. The cross talk between NKA and NCX1.1 may underline the ability of NKA to modulate cardiac contractility (41, 257, 263). This hypothesis has been bolstered through experiments conducted in cardiac myocytes which show reduced Ca2+ extrusion via NCX1.1 after uncoupling NKA from ankyrin (257).
Transient receptor potential protein (TRPC) and NCX signaling complex
Immunocytochemistry studies combined with co-immunoprecipitation and pulldown experiments have highlighted a close association between the TRPC3 ion channel and NCX1.1 in HEK cells (237). These studies also revealed that the C-terminus of TRPC3 channel directly interacts with NCX1.1 (237). A signaling complex composed of TRPC3 channel and NCX has also been demonstrated in isolated cardiac myocytes (48, 54, 78). Although not a direct player in EC coupling, the nonselective cation channel TRPC3 has been shown to influence cardiac contractility via its interaction with NCX1.1 (48). The influx of Na+ via TRPC3 is thought to enhance intracellular Ca2+ either by slowing down Ca2+ extrusion via NCX1, due to a shift in its reversal potential, or by reversing NCX1.1, leading to Ca2+ influx (53, 54) (Figure 15, panel 4).
Mouse Models with Genetically Modified Na+-Ca2+ Exchangers
Mouse models that genetically alter the expression of NCX have been invaluable tools in advancing our understanding of the physiological roles of NCX1.1 in the heart. These models range from ablation (knockout), overexpression, conditional, and inducible conditional (Figure 16). This section will review the most relevant accomplishments related to these investigations. The interested reader is referred to (80, 175, 198, 214, 249) for more in-depth discussion of this developing field.
Figure 16. NCX1 genetically modified mouse lines.
(A) The cartoon shows a timeline of the mouse lines generated with altered expression levels of NCX. (B,C) Overexpression or ablation of NCX affects the cardiac action potential duration. The effects on the action potential waveforms have been connected to changes in K+ currents density (219), although both Ca2+ channels and NCX may contribute to this phenotype. Additional studies are needed to fully elucidate the impact of NCX on both the contractile and electrical properties of the heart. (B,C) Modified, with permission, from Bögeholz N, et al., 2016 (20), © 2016, Elsevier.
NCX1.1 overexpressing transgenic mice
The first transgenic mouse line generated to address the physiological impact of NCX1.1 relied on its overexpression. Utilizing the α-MHC promoter, heterozygous and homozygous overexpressing mice were produced that generated 2.3-and 3.1-fold increased levels of exchanger protein in the heart, respectively (1, 215, 229, 280, 293). There are two commonalities between the heterozygous and homozygous overexpressors: firstly is the absence of adaptation in the expression levels of other cardiac proteins, especially those intimately involved in Ca2+ handling; secondly is the presence of an altered action potential waveform showing a decreased peak amplitude and prolonged duration (Figure 16).
While the increase in the expression level of NCX1.1 in heterozygous mice (~2.3-fold) appeared to have minimal effects on Ca2+ handling (1, 280, 293), the higher levels of NCX expression seen in homozygous mice (~3.1) led to aberrant EC coupling. The enhanced Ca2+ efflux via NCX resulted in faster Ca2+ transients with smaller amplitude, despite unchanged Ca2+ levels in the SR (229). A different scenario has been reported in rabbit cardiomyocytes, where acute overexpression of NCX by adenoviral-mediated gene transfer leads to both decreased SR Ca2+ content and diastolic Ca2+ levels (224).
Another key feature observed in adult ventricular cardiac myocytes isolated from overexpressing NCX1.1 mice is the enhancement of the ionic currents generated by LTCC. The robust Ca2+ efflux due to the increased number of exchangers in the sarcolemma decreases the amount of Ca2+ in the dyadic cleft, which in turn enhances LTCC ionic currents by limiting the extent of Ca2+ dependent inactivation (215). The presence of smaller amplitude Ca2+ transients despite larger L-type Ca2+ currents is a characteristic of defective EC coupling (215).
A lasting controversy in the field was whether NCX1.1 reverse-mode (Ca2+ entry/Na+ efflux) could trigger release of Ca2+ from the SR in adult cardiac myocytes (141). The homozygous overexpressing mouse cardiac myocytes showed Ca2+ transients in the presence of the Ca2+ channel blocker Nifedipine (229, 293). This observation supports the hypothesis that the influx of Ca2+, in the setting of a 3-fold increase in NCX, can drive the release of Ca2+ from the SR.
As enhanced expression levels of NCX1.1 are a common feature of heart failure and hypertrophy (209), studies conducted with these mice have helped define the role of this protein in the genesis of these conditions. For example, overexpression of NCX1.1 evokes hypertrophy, blunted response to β-adrenergic stimulation, intolerance to exercise, and increased mortality due to heart failure (236). This phenotype has been attributed to enhanced Ca2+ entry via reverse exchange at the end of contraction and during rest (236, 266), which in turn contributes to the onset of hypertrophy and heart failure (229, 236, 266). Higher levels of NCX1.1 also lead to delayed afterdepolarizations (DADs) resulting in spontaneous action potentials (20, 217). This is consistent with reports linking NCX1 to arrhythmia (210, 211) due to its ability to influence cell excitability via its electrogenic transport.
Finally, studies have exploited NCX1.1 overexpressing mice to gain more insights into how NCX1.1 participates in the injury associated with an ischemic insult (36, 85) via its functional coupling with the cardiac Na+-H+ exchanger (NHE). NHE slows NCX1.1 forward mode by raising intracellular Na+ in the process of extruding excess protons (66). If sufficiently elevated, intracellular Na+ can actually reverse NCX1.1 resulting in excessive Ca2+ influx and ultimately cell death (21, 110, 275). Conflicting results have been reported showing that overexpression of NCX1.1 can either protect (85) or increase susceptibility to ischemic injury (36). This may be due to differences in either experimental protocols (ischemia only vs. ischemia-reperfusion) or differences across genders, as data show that only male overexpressing NCX1.1 mice displayed enhanced sensitivity to ischemia-reperfusion injury (36, 262). This result implies that female-specific hormones may have a protective effect preventing injury in spite of the overexpression of NCX1.1 (36). This hypothesis is consistent with estrogens having a significant inhibitory effect on the rise in intracellular Ca2+ and Na+ during ischemia (262) and a protective role during metabolic stress (128). These observations raise important questions about gender differences in relation to the role of NCX1.1 in cardiac function both in health and disease.
Global NCX1 knockout mice
Ablation of NCX1 in mice results in death at the embryonic stage (between days 9.5 and 11 days post coitum) (32, 138, 230, 279). Histological analysis of the embryonic heart showed thinning of the ventricle walls and a reduction in the number of cardiac cells (279), while at the cellular level myocytes showed enlarged and irregular mitochondria, disorganized myofibrils, nuclear condensation and cell shrinkage, indicative of apoptosis (230, 278, 279). At the functional level, heart tubes isolated from homozygous NCX1 knockout mice displayed normal Ca2+ transients upon stimulation but whether they can spontaneously contract remains controversial (32, 138, 230).
Although Ca2+ handling was not drastically affected by NCX1 ablation when investigated in physiological conditions, the absence of NCX1 rendered the heart tubes more susceptible to stress such as increased stimulus frequency or application of isoproterenol (138, 230). One explanation is that the rapid and elevated changes in intracellular Ca2+ cannot be accommodated by the remaining Ca2+ extrusion mechanism, namely the PMCA, due to its low transport capacity.
Investigations conducted with heart tubes from embryonic mice lacking NCX1 reveal two important pharmacological observations related to this transporter. First, NCX is essential for the effects exerted by cardiac glycosides (231). By blocking the NKA, ouabain increases cytosolic Na+ levels, which in turn leads to decreased Ca2+ efflux via NCX and therefore a surplus of Ca2+ entering the SR. The increased Ca2+ load results in more forceful contractions, revealing the mechanism of the positive inotropic effect of cardiac glycosides. The second observation was that two compounds thought to be selective blockers for NCX1.1, KB-R7943, and SEA0400, instead lack specificity since they both depressed calcium transients in the absence of NCX1.1 (231).
Unlike the homozygous mouse model, NCX1 heterozygous knockout mice live to adulthood with little cardiac remodeling or dysfunction, despite decreased NCX1 expression of approximately 50% (19). The electrical and contractile properties of cardiac myocytes isolated from this mouse model resemble those found in the conditional cardiac-specific knockout, which are detailed below. Here we only briefly mention that heterozygous global NCX1 KO showed a significant shortening of the action potential (19) and Ca2+ transients with a slower decay. The occurrence of early and delayed afterdepolarizations was also reduced in the presence of decreased exchanger protein levels (19). Cumulatively, these findings underline the fundamental role NCX1 plays in influencing both contractility and excitability.
Ventricular-specific NCX1 knockout mice
The early lethality of the global exchanger knockout drove the development of a ventricular-specific knockout of NCX1 (89). With their survivability, this mouse line allowed investigation of the role of NCX1 in cardiac physiology without confounding effects from other tissues. Ablation of NCX1 from the ventricles resulted in a modest reduction in contractility (89) and increased ventricular fibrosis (127). Of particular note is that, despite the conditional system, NCX1 is ablated only in approximately 80% to 90% of cardiomyocytes (89). Although it seems unlikely, it is conceivable that the remaining 10% to 20% of cells would suffice for cardiac function. At the cellular level, ablation of NCX1 did not affect the expression of other Ca2+ handling proteins and had little impact on Ca2+ dynamics as both diastolic Ca2+ levels and Ca2+ transients were comparable to those of WT mice (89). To account for the ability to control Ca2+ homeostasis in the absence of NCX, attribution has been given to the voltage-dependent Ca2+ channels (LTCC) (218). Although their level of expression was unchanged (89), a reduction in Ca2+ current of 50% was recorded in the absence of NCX1 (218), allowing PMCA to manage alone the increase in intracellular Ca2+. Moreover, the absence of NCX1 leads to increased Ca2+ levels in the dyadic cleft, which in turn facilitates Ca2+ release from the SR via activation of RyR and promotes LTCC Ca2+-dependent inactivation (89, 216, 218). This is consistent with a broad range of evidence supporting a role of NCX in tuning Ca2+ levels within the restricted space of the dyadic cleft (140, 180, 271).
Genetic ablation of ventricular NCX1 results in a shortening of the action potential duration (219). Based on a mouse-specific action potential computer model as well as experimental findings, this has been attributed to an upregulation of the expression of the voltage-dependent transient outward current (Ito), mediated in part by the potassium channel Kv4.2, and the repolarizing effects of the enhanced efflux of K+ (219). However, this is unlikely the only contributing factor to the shortening of the action potential duration as heterozygous KO mice show a similar phenotype but with unchanged K+-currents (19, 20) (Figure 16C). Further studies are needed to detail the molecular mechanisms that lead to the changes in action potential duration seen both in the overexpressor and knockout NCX cardiac myocytes.
Conditional (ventricle specific) NCX knockout mice have further defined the contribution of this transporter to pathophysiological conditions such as ischemia/reperfusion (I/R). In the absence of NCX1, the heart is less susceptible to this insult, exhibiting reduced maximum contracture, delayed ischemic contracture, and a smaller decline in ATP levels during ischemia (110). With reperfusion, these conditional mouse hearts have less necrosis, better recovery of phosphocreatine, and improved left ventricular developed pressure compared to WT hearts (110). These results support NCX1 being a major player in the pathological and detrimental effects linked to ischemia (117, 142, 242). While removal of NCX1 in mice is beneficial in the setting of ischemia/reperfusion, its absence makes the heart less tolerant to stresses such as transaortic constriction and chronic β-adrenergic stimulation (e.g., isoproterenol infusion). NCX1 knockout mice also have a blunted inotropic response to dobutamine (127). A fine balance of NCX activity is essential for the well-being of the heart both in physiological settings and during stress conditions.
NCX1 knockout in sinoatrial node
Heart rhythm is generated by dedicated pacemaker cells. These highly specialized cells within the sinoatrial node (SAN), located in the right atrium, generate spontaneous depolarizations which lead to action potentials that drive the heart rate. This self-generating stimulus is driven by two cellular pathways working in concert. The first, the membrane clock, relies on the orchestrated operation of voltage-sensitive membrane currents. Primarily, it is the onset of the hyperpolarization-activated pacemaker current (If or funny current) that brings the membrane potential to its threshold value triggering an action potential (12, 27, 261). The second, the Ca2+ clock, relies on spontaneous Ca2+ release from the SR which, by activating the electrogenic NCX, depolarizes the membrane (160). The reader is directed to more comprehensive reviews focusing on this topic (34, 137, 139).
To illuminate the role of NCX1 in the pacemaking activity of the heart, four mouse models have been generated. One model entailed ablation of NCX1 from the entire atria (including pacemaking cells) in a constitutive manner using sarcolipin Cre mice (81). The absence of NCX1 led to hearts with increased size and fibrosis and altered electrical properties including a slower heart rate, the absence of P waves on electrocardiogram (ECG), and the presence of a slow junctional escape rhythm (81). Moreover, optical recordings of Ca2+ transients and action potentials from SAN/atrial tissue isolated from this mouse line revealed abnormal pacemaker activity with bursts of spontaneous depolarizations and Ca2+ transients interrupted by long pauses associated with nondepolarizing Ca2+ waves. This abnormal rhythm was alleviated either by Ca2+ buffering with BAPTA-AM (270) or application of apamin (269), a specific blocker of the small conductance Ca2+-activated potassium channels. These findings imply that the high Ca2+ levels due to the absence of NCX1 inactivate LTCCs and activate small conductance Ca2+-activated potassium channels, thus inhibiting plasma membrane depolarization (269, 270).
The three additional mouse models created to address the role of NCX in SAN activity involved a more targeted ablation of NCX and/or an inducible system. Gao et al. used gene painting to ablate NCX1 in the SAN or a tamoxifen-inducible knockout of NCX in all atrial and ventricular cells using the MerCreMer promoter (65), while Herrmann et al used a tamoxifen-inducible Cre to target NCX1 in nodal and conduction system cells containing HCN4 channels (90). These models retained their P wave on ECG. The only major change in the MerCreMer and gene painting models was a blunted response to isoproterenol, whereas the HCN4 Cre animals showed progressively slower heart rate with SAN pauses, SAN dysrhythmias, atrioventricular block, and sinus arrest (65, 90).
At the cellular level, the constitutive (81) and inducible knockout models (65, 90) showed some notable differences. For example, irregular spontaneous action potentials and their associated Ca2+ transients were recorded from SAN cells after inducible knockout (65, 90), while constitutive ablation of NCX resulted in electrically silent cells (81). Moreover, LTCC ionic current amplitude was significantly decreased in the constitutive atrial-specific NCX1 knockout model (81) but not in the inducible conduction system knockout. The residual expression of NCX in pacemaker cells detected in the inducible NCX1 knockout cardiac pacemaker cells (65, 90) may underlie this difference by preventing Ca2+ accumulation, and therefore LTCC Ca2+-dependent inactivation.
The different phenotypes observed in these NCX1 knockout models imply a complex molecular system designed to guarantee pacemaker automaticity, involving both the voltage and Ca2+ clocks. The synergy between the two coupled clocks ensures proper cardiac function and therefore survival (139).
Atrial specific NCX1 knockout
To date, there are no detailed published investigations regarding the electrical and Ca2+ handling properties of atrial cells in the absence of NCX1. NCX1 is highly expressed in atrial tissue and its specific ablation produces important abnormalities in atrial myocyte morphology and EC coupling. Most notably, atrial myocytes isolated from atrial-specific NCX1 knockout mouse hearts exhibit loss of transverse-axial tubules (TATs) (297). These results were surprising for two reasons. First, they demonstrated the presence of an unexpectedly robust TAT system in atrial cells from control mice, allowing these cells to generate synchronous Ca2+ transients. Prior to this investigation, the common assumption was that atrial cells from small mammals exhibit a reduced density of T-tubules in atrial cardiomyocytes leading to a nonuniform Ca2+ release pattern (109, 135). Second, this complex structure was lost in the absence of NCX1 leading to changes in the kinetics of Ca2+ transients (296, 297). This observation reveals a new role of NCX in the structural arrangements of cardiac myocytes.
Transgenic mice expressing NCX1 with altered allosteric properties
In spite of the progress achieved toward the understanding of the molecular mechanisms that govern NCX modulation by cytosolic signaling factors, how the allosteric control of NCX1 activity impacts cardiac function remains largely elusive. The technical difficulties in investigating this process in native cells have limited such investigations. A step forward toward addressing this important knowledge gap was the development of a transgenic mouse line overexpressing an exchanger insensitive to the effects that cytosolic Ca2+ and Na+ exert on its activity, while maintaining transport properties intact. This was accomplished by overexpressing an exchanger with residues 680 to 685 deleted in cardiac tissue (170). Removal of these amino acids abolishes both Na+ and Ca2+ secondary regulation of NCX1 (168) and electrophysiological studies conducted on cardiac myocytes isolated from the transgenic mice confirmed that these regulations were largely eliminated (170), in spite of the presence of the native exchanger. More importantly, the ionic currents recorded from cardiac myocytes overexpressing the Δ680 to 685 exchanger failed to augment with increasing Ca2+ concentrations as seen in control cells (284), suggesting that the exchanger Ca2+ regulation is a physiologically relevant process. This was further corroborated by evidence showing that papillary muscles isolated from Δ680 to 685 show enhanced postrest papillary muscle force compared to WT hearts (170).
With the advent and application of CRISPR technology, we await the future investigation of NCX in mouse models that can rely on normal expression profiles but with defined phenotypes.
Concluding Remarks and Future Directions
In the last decade, striking advancements have been achieved toward the understanding of the structure and function of NCX1.1. Structures of isolated components of NCX1.1 have been resolved as well as the atomic organization of a full-length archaebacterial homologue. These milestones have advanced our mechanistic knowledge of how this protein exchanges ions, and how it is modulated. The development of new transgenic mice, with NCX1 either ablated or over-expressed, have helped to define the physiological impact of this protein in cardiac function. The knockout studies also highlight the remarkable plasticity of the mammalian heart to survive and function, ostensibly near normal, until early adulthood despite the near total ablation of NCX in cardiomyocytes. These studies demonstrate the need for the development of a highly selective NCX blocker, which would allow acute inhibition and prevent adaptation in experimental setups at the various stages of mouse development.
There is urgency to further understand how the mammalian exchanger behaves in its native environment and especially how it responds to stimuli within cells or its neighboring proteins. Such investigations were technically challenging if not impossible until a few years ago but new methodologies are now in place to resolve these questions. For example, there is a strong hope that in the next decade we will see the structure of the mammalian exchanger resolved thanks to cryo-electron microscopy. Alongside this, the CRISPR/Cas9 technique has opened new avenues to generate knockin mice in which the allosteric regulation of NCX1 is selectively modified or eliminated, making it possible to address its specific physiological and pathophysiological impact without changes in its expression profile. Such studies will help in the development of new targeted therapies as this transporter has been implicated in numerous cardiac and neurological conditions.
Didactic Synopsis.
Major teaching points
The Na+-Ca2+ exchangers are a family of proteins that use the electrochemical gradient of Na+ to move Ca2+ out of the cell, against its concentration gradient.
The electrogenic Na+-Ca2+ exchanger is a bidirectional transporter, therefore under certain membrane potentials and Na+ and Ca2+ concentrations it can reverse and bring Ca2+ into the cell.
Understanding the mode of operation of the Na+-Ca2+ exchanger is necessary to comprehend how cells maintain optimal intracellular Ca2+ levels, vital for numerous physiological processes.
Intracellular Na+, Ca2+ and H+ are essential regulators of Na+-Ca2+ exchanger activity.
The Na+-Ca2+ exchanger is abundant in cardiac myocytes where it controls intracellular Ca2+ to modulate cardiac contractility.
Impairments of Na+-Ca2+ exchanger activity have been linked to cardiac diseases, such as arrhythmia, heart failure, and ischemia-reperfusion injury.
Acknowledgements
Grants
This work was supported by the following NIH/NHLBI grants: R01 HL130308 (M. Ottolia), R01 HL152296 (M. Ottolia) and MPI R01 HL147569 (J. I. Goldhaber/M. Ottolia).
Footnotes
Disclosures
No conflicts of interest, financial or otherwise, are declared by the authors.
References
- 1.Adachi-Akahane S, Lu L, Li Z, Frank JS, Philipson KD, Morad M. Calcium signaling in transgenic mice overexpressing cardiac Na+-Ca2+2+ exchanger. J Gen Physiol 109: 717–729, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ander BP, Hurtado C, Raposo CS, Maddaford TG, Deniset JF, Hryshko LV, Pierce GN, Lukas A. Differential sensitivities of the NCX1.1 and NCX1.3 isoforms of the Na+-Ca2+ exchanger to alpha-linolenic acid. Cardiovasc Res 73: 395–403, 2007. [DOI] [PubMed] [Google Scholar]
- 3.Armoundas AA, Hobai IA, Tomaselli GF, Winslow RL, O’Rourke B. Role of sodium-calcium exchanger in modulating the action potential of ventricular myocytes from normal and failing hearts. Circ Res 93: 46–53, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Artman M Sarcolemmal Na+-Ca2+ exchange activity and exchanger immunoreactivity in developing rabbit hearts. Am J Physiol 263: H1506–H1513, 1992. [DOI] [PubMed] [Google Scholar]
- 5.Artman M, Ichikawa H, Avkiran M, Coetzee WA. Na+/Ca2+ exchange current density in cardiac myocytes from rabbits and guinea pigs during postnatal development. Am J Physiol 268: H1714–H1722, 1995. [DOI] [PubMed] [Google Scholar]
- 6.Asteggiano C, Berberian G, Beauge L. Phosphatidyl inositol-4,5-bisphosphate bound to bovine cardiac Na+/Ca2+ exchanger displays a MgATP regulation similar to that of the exchange fluxes. Eur J Biochem 268: 437–442, 2001. [DOI] [PubMed] [Google Scholar]
- 7.Baker PF, Blaustein MP. Sodium-dependent uptake of calcium by crab nerve. Biochim Biophys Acta 150: 167–170, 1968. [DOI] [PubMed] [Google Scholar]
- 8.Baker PF, Blaustein MP, Hodgkin AL, Steinhardt RA. The influence of calcium on sodium efflux in squid axons. J Physiol 200: 431–458, 1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bano D, Young KW, Guerin CJ, Lefeuvre R, Rothwell NJ, Naldini L, Rizzuto R, Carafoli E, Nicotera P. Cleavage of the plasma membrane Na+/Ca2+ exchanger in excitotoxicity. Cell 120: 275–285, 2005. [DOI] [PubMed] [Google Scholar]
- 10.Barnes KV, Cheng G, Dawson MM, Menick DR. Cloning of cardiac, kidney, and brain promoters of the feline ncx1 gene. J Biol Chem 272: 11510–11517, 1997. [PubMed] [Google Scholar]
- 11.Bartholomaus I, Milan-Lobo L, Nicke A, Dutertre S, Hastrup H, Jha A, Gether U, Sitte HH, Betz H, Eulenburg V. Glycine transporter dimers: Evidence for occurrence in the plasma membrane. J Biol Chem 283: 10978–10991, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baruscotti M, Bucchi A, Viscomi C, Mandelli G, Consalez G, Gnecchi-Rusconi T, Montano N, Casali KR, Micheloni S, Barbuti A, DiFrancesco D. Deep bradycardia and heart block caused by inducible cardiac-specific knockout of the pacemaker channel gene Hcn4. Proc Natl Acad Sci U S A 108: 1705–1710, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bassani RA, Bassani JW, Bers DM. Mitochondrial and sarcolemmal Ca2+ transport reduce [Ca2+]i during caffeine contractures in rabbit cardiac myocytes. J Physiol 453: 591–608, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beauge L, Asteggiano C, Berberian G. Regulation of phosphatidylinositol-4,5-biphosphate bound to the bovine cardiac Na+/Ca2+ exchanger. Ann N Y Acad Sci 976: 288–299, 2002. [DOI] [PubMed] [Google Scholar]
- 15.Bers DM. Cardiac excitation-contraction coupling. Nature 415: 198–205, 2002. [DOI] [PubMed] [Google Scholar]
- 16.Besserer GM, Ottolia M, Nicoll DA, Chaptal V, Cascio D, Philipson KD, Abramson J. The second Ca2+-binding domain of the Na+ Ca2+ exchanger is essential for regulation: Crystal structures and mutational analysis. Proc Natl Acad Sci U S A 104: 18467–18472, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blaustein MP, Lederer WJ. Sodium/calcium exchange: Its physiological implications. Physiol Rev 79: 763–854, 1999. [DOI] [PubMed] [Google Scholar]
- 18.Boerth SR, Zimmer DB, Artman M. Steady-state mRNA levels of the sarcolemmal Na+-Ca2+ exchanger peak near birth in developing rabbit and rat hearts. Circ Res 74: 354–359, 1994. [DOI] [PubMed] [Google Scholar]
- 19.Bogeholz N, Pauls P, Bauer BK, Schulte JS, Dechering DG, Frommeyer G, Kirchhefer U, Goldhaber JI, Muller FU, Eckardt L, Pott C. Suppression of early and late afterdepolarizations by heterozygous knockout of the Na+/Ca2+ exchanger in a murine model. Circ Arrhythm Electrophysiol 8: 1210–1218, 2015. [DOI] [PubMed] [Google Scholar]
- 20.Bogeholz N, Pauls P, Kaese S, Schulte JS, Lemoine MD, Dechering DG, Frommeyer G, Goldhaber JI, Seidl MD, Kirchhefer U, Eckardt L, Muller FU, Pott C. Triggered activity in atrial myocytes is influenced by Na+/Ca2+ exchanger activity in genetically altered mice. J Mol Cell Cardiol 101: 106–115, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bountra C, Vaughan-Jones RD. Effect of intracellular and extracellular pH on contraction in isolated, mammalian cardiac muscle. J Physiol 418: 163–187, 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boyman L, Hagen BM, Giladi M, Hiller R, Lederer WJ, Khananshvili D. Proton-sensing Ca2+ binding domains regulate the cardiac Na+/Ca2+ exchanger. J Biol Chem 286: 28811–28820, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boyman L, Mikhasenko H, Hiller R, Khananshvili D. Kinetic and equilibrium properties of regulatory calcium sensors of NCX1 protein. J Biol Chem 284: 6185–6193, 2009. [DOI] [PubMed] [Google Scholar]
- 24.Brette F, Orchard CH. Density and sub-cellular distribution of cardiac and neuronal sodium channel isoforms in rat ventricular myocytes. Biochem Biophys Res Commun 348: 1163–1166, 2006. [DOI] [PubMed] [Google Scholar]
- 25.Breukels V, Vuister GW. Binding of calcium is sensed structurally and dynamically throughout the second calcium-binding domain of the sodium/calcium exchanger. Proteins 78: 1813–1824, 2010. [DOI] [PubMed] [Google Scholar]
- 26.Bridge JH, Smolley JR, Spitzer KW. The relationship between charge movements associated with ICa and INa-Ca in cardiac myocytes. Science 248: 376–378, 1990. [DOI] [PubMed] [Google Scholar]
- 27.Bucchi A, Barbuti A, Difrancesco D, Baruscotti M. Funny current and cardiac rhythm: Insights from HCN knockout and transgenic mouse models. Front Physiol 3: 240, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bukowska A, Lendeckel U, Bode-Boger SM, Goette A. Physiologic and pathophysiologic role of calpain: Implications for the occurrence of atrial fibrillation. Cardiovasc Ther 30: e115–127, 2012. [DOI] [PubMed] [Google Scholar]
- 29.Chaptal V, Besserer GM, Ottolia M, Nicoll DA, Cascio D, Philipson KD, Abramson J. How does regulatory Ca2+ regulate the Na+-Ca2+ exchanger? Channels (Austin) 1: 397–399, 2007. [DOI] [PubMed] [Google Scholar]
- 30.Cheng G, Hagen TP, Dawson ML, Barnes KV, Menick DR. The role of GATA, CArG, E-box, and a novel element in the regulation of cardiac expression of the Na+-Ca2+ exchanger gene. J Biol Chem 274: 12819–12826, 1999. [DOI] [PubMed] [Google Scholar]
- 31.Cheung JY, Rothblum LI, Moorman JR, Tucker AL, Song J, Ahlers BA, Carl LL, Wang J, Zhang XQ. Regulation of cardiac Na+/Ca2+ exchanger by phospholemman. Ann N Y Acad Sci 1099: 119–134, 2007. [DOI] [PubMed] [Google Scholar]
- 32.Cho CH, Kim SS, Jeong MJ, Lee CO, Shin HS. The Na+ -Ca2+ exchanger is essential for embryonic heart development in mice. Mol Cells 10: 712–722, 2000. [DOI] [PubMed] [Google Scholar]
- 33.Chou AC, Ju YT, Pan CY. Calmodulin interacts with the sodium/calcium exchanger NCX1 to regulate activity. PLoS One 10: e0138856, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cingolani E, Goldhaber JI, Marban E. Next-generation pacemakers: From small devices to biological pacemakers. Nat Rev Cardiol 15: 139–150, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cook O, Low W, Rahamimoff H. Membrane topology of the rat brain Na+-Ca2+ exchanger. Biochim Biophys Acta 1371: 40–52, 1998. [DOI] [PubMed] [Google Scholar]
- 36.Cross HR, Lu L, Steenbergen C, Philipson KD, Murphy E. Overexpression of the cardiac Na+/Ca2+ exchanger increases susceptibility to ischemia/reperfusion injury in male, but not female, transgenic mice. Circ Res 83: 1215–1223, 1998. [DOI] [PubMed] [Google Scholar]
- 37.Cunha SR, Bhasin N, Mohler PJ. Targeting and stability of Na+/Ca2+ exchanger 1 in cardiomyocytes requires direct interaction with the membrane adaptor ankyrin-B. J Biol Chem 282: 4875–4883, 2007. [DOI] [PubMed] [Google Scholar]
- 38.Dan P, Lin E, Huang J, Biln P, Tibbits GF. Three-dimensional distribution of cardiac Na+-Ca2+ exchanger and ryanodine receptor during development. Biophys J 93: 2504–2518, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Despa S, Brette F, Orchard CH, Bers DM. Na/Ca exchange and Na/KATPase function are equally concentrated in transverse tubules of rat ventricular myocytes. Biophys J 85: 3388–3396, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Despa S, Islam MA, Weber CR, Pogwizd SM, Bers DM. Intracellular Na+ concentration is elevated in heart failure but Na/K pump function is unchanged. Circulation 105: 2543–2548, 2002. [DOI] [PubMed] [Google Scholar]
- 41.Despa S, Lingrel JB, Bers DM. Na+/K+-ATPase alpha2-isoform preferentially modulates Ca2+ transients and sarcoplasmic reticulum Ca2+ release in cardiac myocytes. Cardiovasc Res 95: 480–486, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.DiPolo R Calcium influx in internally dialyzed squid giant axons. J Gen Physiol 73: 91–113, 1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.DiPolo R, Beauge L. In the squid axon Na+/Ca2+ exchanger the state of the Ca i-regulatory site influences the affinities of the intra- and extracellular transport sites for Na+ and Ca2+. Pflugers Arch 456: 623–633, 2008. [DOI] [PubMed] [Google Scholar]
- 44.Dixon RE, Yuan C, Cheng EP, Navedo MF, Santana LF. Ca2+ signaling amplification by oligomerization of L-type Cav1.2 channels. Proc Natl Acad Sci U S A 109: 1749–1754, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Doering AE, Lederer WJ. The mechanism by which cytoplasmic protons inhibit the sodium-calcium exchanger in guinea-pig heart cells. J Physiol 466: 481–499, 1993. [PMC free article] [PubMed] [Google Scholar]
- 46.Doering AE, Lederer WJ. The action of Na+ as a cofactor in the inhibition by cytoplasmic protons of the cardiac Na+-Ca2+ exchanger in the guinea-pig. J Physiol 480 (Pt 1): 9–20, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Doering AE, Nicoll DA, Lu Y, Lu L, Weiss JN, Philipson KD. Topology of a functionally important region of the cardiac Na+/Ca2+ exchanger. J Biol Chem 273: 778–783, 1998. [DOI] [PubMed] [Google Scholar]
- 48.Doleschal B, Primessnig U, Wolkart G, Wolf S, Schernthaner M, Lichtenegger M, Glasnov TN, Kappe CO, Mayer B, Antoons G, Heinzel F, Poteser M, Groschner K. TRPC3 contributes to regulation of cardiac contractility and arrhythmogenesis by dynamic interaction with NCX1. Cardiovasc Res 106: 163–173, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dunn J, Elias CL, Le HD, Omelchenko A, Hryshko LV, Lytton J. The molecular determinants of ionic regulatory differences between brain and kidney Na+/Ca2+ exchanger NCX1 isoforms. J Biol Chem 277: 33957–33962, 2002. [DOI] [PubMed] [Google Scholar]
- 50.Durkin JT, Ahrens DC, Pan YC, Reeves JP. Purification and amino-terminal sequence of the bovine cardiac sodium-calcium exchanger: Evidence for the presence of a signal sequence. Arch Biochem Biophys 290: 369–375, 1991. [DOI] [PubMed] [Google Scholar]
- 51.Dyck C, Maxwell K, Buchko J, Trac M, Omelchenko A, Hnatowich M, Hryshko LV. Structure-function analysis of CALX1.1, a Na+-Ca2+ exchanger from Drosophila. Mutagenesis of ionic regulatory sites. J Biol Chem 273: 12981–12987, 1998. [DOI] [PubMed] [Google Scholar]
- 52.Dyck C, Omelchenko A, Elias CL, Quednau BD, Philipson KD, Hnatowich M, Hryshko LV. Ionic regulatory properties of brain and kidney splice variants of the NCX1 Na+-Ca2+ exchanger. J Gen Physiol 114: 701–711, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Eder P, Poteser M, Romanin C, Groschner K. Na+ entry and modulation of Na+/Ca2+ exchange as a key mechanism of TRPC signaling. Pflugers Arch 451: 99–104, 2005. [DOI] [PubMed] [Google Scholar]
- 54.Eder P, Probst D, Rosker C, Poteser M, Wolinski H, Kohlwein SD, Romanin C, Groschner K. Phospholipase C-dependent control of cardiac calcium homeostasis involves a TRPC3-NCX1 signaling complex. Cardiovasc Res 73: 111–119, 2007. [DOI] [PubMed] [Google Scholar]
- 55.Eisner DA, Caldwell JL, Kistamas K, Trafford AW. Calcium and excitation-contraction coupling in the heart. Circ Res 121: 181–195, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Faham S, Watanabe A, Besserer GM, Cascio D, Specht A, Hirayama BA, Wright EM, Abramson J. The crystal structure of a sodium galactose transporter reveals mechanistic insights into Na+/sugar symport. Science 321: 810–814, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Farhan H, Freissmuth M, Sitte HH. Oligomerization of neurotransmitter transporters: A ticket from the endoplasmic reticulum to the plasma membrane. Handb Exp Pharmacol: 233–249, 2006. [DOI] [PubMed] [Google Scholar]
- 58.Frank JS, Mottino G, Reid D, Molday RS, Philipson KD. Distribution of the Na+-Ca2+ exchange protein in mammalian cardiac myocytes: An immunofluorescence and immunocolloidal gold-labeling study. J Cell Biol 117: 337–345, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fuller W, Reilly L, Hilgemann DW. S-palmitoylation and the regulation of NCX1. Channels Austin 10: 75–77, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Furman I, Cook O, Kasir J, Low W, Rahamimoff H. The putative amino-terminal signal peptide of the cloned rat brain Na+-Ca2+ exchanger gene RBE-1 is not mandatory for functional expression. J Biol Chem 270: 19120–19127, 1995. [DOI] [PubMed] [Google Scholar]
- 61.Fuster D, Moe OW, Hilgemann DW. Steady-state function of the ubiquitous mammalian Na/H exchanger NHE1 in relation to dimer coupling models with 2Na/2H stoichiometry. J Gen Physiol 132: 465–480, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gabellini N, Bortoluzzi S, Danieli GA, Carafoli E. The gene promoter of human Na+/Ca2+ exchanger isoform 3 SLC8A3 is controlled by cAMP and calcium. Ann N Y Acad Sci 976: 282–284, 2002. [DOI] [PubMed] [Google Scholar]
- 63.Gabellini N, Bortoluzzi S, Danieli GA, Carafoli E. The human SLC8A3 gene and the tissue-specific Na+/Ca2+ exchanger 3 isoforms. Gene 298: 1–7, 2002. [DOI] [PubMed] [Google Scholar]
- 64.Gabellini N, Bortoluzzi S, Danieli GA, Carafoli E. Control of the Na+/Ca2+ exchanger 3 promoter by cyclic adenosine monophosphate and Ca2+ in differentiating neurons. J Neurochem 84: 282–293, 2003. [DOI] [PubMed] [Google Scholar]
- 65.Gao Z, Rasmussen TP, Li Y, Kutschke W, Koval OM, Wu Y, Wu Y, Hall DD, Joiner ML, Wu XQ, Swaminathan PD, Purohit A, Zimmerman K, Weiss RM, Philipson KD, Song LS, Hund TJ, Anderson ME. Genetic inhibition of Na+-Ca2+ exchanger current disables fight or flight sinoatrial node activity without affecting resting heart rate. Circ Res 112: 309–317, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Garciarena CD, Youm JB, Swietach P, Vaughan-Jones RD. H+-activated Na+ influx in the ventricular myocyte couples Ca2+-signalling to intracellular pH. J Mol Cell Cardiol 61: 51–59, 2013. [DOI] [PubMed] [Google Scholar]
- 67.Gershome C, Lin E, Kashihara H, Hove-Madsen L, Tibbits GF. Colocalization of voltage-gated Na+ channels with the Na+/Ca2+ exchanger in rabbit cardiomyocytes during development. Am J Physiol Heart Circ Physiol 300: H300–H311, 2011. [DOI] [PubMed] [Google Scholar]
- 68.Giladi M, Bohbot H, Buki T, Schulze DH, Hiller R, Khananshvili D. Dynamic features of allosteric Ca2+ sensor in tissue-specific NCX variants. Cell Calcium 51: 478–485, 2012. [DOI] [PubMed] [Google Scholar]
- 69.Giladi M, Boyman L, Mikhasenko H, Hiller R, Khananshvili D. Essential role of the CBD1-CBD2 linker in slow dissociation of Ca2+ from the regulatory two-domain tandem of NCX1. J Biol Chem 285: 28117–28125, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Giladi M, Hiller R, Hirsch JA, Khananshvili D. Population shift underlies Ca2+-induced regulatory transitions in the sodium-calcium exchanger NCX. J Biol Chem 288: 23141–23149, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Giladi M, Khananshvili D. Molecular determinants of allosteric regulation in NCX proteins. Adv Exp Med Biol 961: 35–48, 2013. [DOI] [PubMed] [Google Scholar]
- 72.Giladi M, Sasson Y, Fang X, Hiller R, Buki T, Wang YX, Hirsch JA, Khananshvili D. A common Ca2+-driven interdomain module governs eukaryotic NCX regulation. PLoS One 7: e39985, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Giladi M, Shor R, Lisnyansky M, Khananshvili D. Structure-functional basis of ion transport in sodium-calcium exchanger NCX proteins. Int J Mol Sci 17: 1949, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Giladi M, Tal I, Khananshvili D. Structural features of ion transport and allosteric regulation in sodium-calcium exchanger NCX proteins. Front Physiol 7: 30, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Giladi M, van Dijk L, Refaeli B, Almagor L, Hiller R, Man P, Forest E, Khananshvili D. Dynamic distinctions in the Na+/Ca2+ exchanger adopting the inward- and outward-facing conformational states. J Biol Chem 292: 12311–12323, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ginsburg KS, Bers DM. Isoproterenol does not enhance Ca-dependent Na/Ca exchange current in intact rabbit ventricular myocytes. J Mol Cell Cardiol 39: 972–981, 2005. [DOI] [PubMed] [Google Scholar]
- 77.Ginsburg KS, Weber CR, Bers DM. Cardiac Na+-Ca2+ exchanger: Dynamics of Ca2+-dependent activation and deactivation in intact myocytes. J Physiol 591: 2067–2086, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Goel M, Zuo CD, Sinkins WG, Schilling WP. TRPC3 channels colocalize with Na+/Ca2+ exchanger and Na+ pump in axial component of transverse-axial tubular system of rat ventricle. Am J Physiol Heart Circ Physiol 292: H874–H883, 2007. [DOI] [PubMed] [Google Scholar]
- 79.Gok C, Plain F, Robertson AD, Howie J, Baillie GS, Fraser NJ, Fuller W. Dynamic palmitoylation of the sodium-calcium exchanger modulates its structure, affinity for lipid-ordered domains, and inhibition by XIP. Cell Rep 31: 107697, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Goldhaber JI, Philipson KD. Cardiac sodium-calcium exchange and efficient excitation-contraction coupling: Implications for heart disease. Adv Exp Med Biol 961: 355–364, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Groenke S, Larson ED, Alber S, Zhang R, Lamp ST, Ren X, Nakano H, Jordan MC, Karagueuzian HS, Roos KP, Nakano A, Proenza C, Philipson KD, Goldhaber JI. Complete atrial-specific knockout of sodium-calcium exchange eliminates sinoatrial node pacemaker activity. PLoS One 8: e81633, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Haddock PS, Coetzee WA, Cho E, Porter L, Katoh H, Bers DM, Jafri MS, Artman M. Subcellular [Ca2+]i gradients during excitation-contraction coupling in newborn rabbit ventricular myocytes. Circ Res 85: 415–427, 1999. [DOI] [PubMed] [Google Scholar]
- 83.Hafver TL, Hodne K, Wanichawan P, Aronsen JM, Dalhus B, Lunde PK, Lunde M, Martinsen M, Enger UH, Fuller W, Sjaastad I, Louch WE, Sejersted OM, Carlson CR. Protein phosphatase 1c associated with the cardiac sodium calcium exchanger 1 regulates its activity by dephosphorylating Serine 68-phosphorylated phospholemman. J Biol Chem 291: 4561–4579, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hale CC, Bliler S, Quinn TP, Peletskaya EN. Localization of an exchange inhibitory peptide XIP binding site on the cardiac sodium-calcium exchanger. Biochem Biophys Res Commun 236: 113–117, 1997. [DOI] [PubMed] [Google Scholar]
- 85.Hampton TG, Wang JF, DeAngelis J, Amende I, Philipson KD, Morgan JP. Enhanced gene expression of Na+/Ca2+ exchanger attenuates ischemic and hypoxic contractile dysfunction. Am J Physiol Heart Circ Physiol 279: H2846–H2854, 2000. [DOI] [PubMed] [Google Scholar]
- 86.He Z, Feng S, Tong Q, Hilgemann DW, Philipson KD. Interaction of PIP2 with the XIP region of the cardiac Na/Ca exchanger. Am J Physiol Cell Physiol 278: C661–C666, 2000. [DOI] [PubMed] [Google Scholar]
- 87.He Z, Petesch N, Voges K, Roben W, Philipson KD. Identification of important amino acid residues of the Na+-Ca2+ exchanger inhibitory peptide, XIP. J Membr Biol 156: 149–156, 1997. [DOI] [PubMed] [Google Scholar]
- 88.He Z, Tong Q, Quednau BD, Philipson KD, Hilgemann DW. Cloning, expression, and characterization of the squid Na+-Ca2+ exchanger NCX-SQ1. J Gen Physiol 111: 857–873, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Henderson SA, Goldhaber JI, So JM, Han T, Motter C, Ngo A, Chantawansri C, Ritter MR, Friedlander M, Nicoll DA, Frank JS, Jordan MC, Roos KP, Ross RS, Philipson KD. Functional adult myocardium in the absence of Na+-Ca2+ exchange: Cardiac-specific knockout of NCX1. Circ Res 95: 604–611, 2004. [DOI] [PubMed] [Google Scholar]
- 90.Herrmann S, Lipp P, Wiesen K, Stieber J, Nguyen H, Kaiser E, Ludwig A. The cardiac sodium-calcium exchanger NCX1 is a key player in the initiation and maintenance of a stable heart rhythm. Cardiovasc Res 99: 780–788, 2013. [DOI] [PubMed] [Google Scholar]
- 91.Herz K, Rimon A, Jeschke G, Padan E. Beta-sheet-dependent dimerization is essential for the stability of NhaA Na+/H+ antiporter. J Biol Chem 284: 6337–6347, 2009. [DOI] [PubMed] [Google Scholar]
- 92.Hilge M Ca2+ regulation of ion transport in the Na+/Ca2+ exchanger. J Biol Chem 287: 31641–31649, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hilge M, Aelen J, Foarce A, Perrakis A, Vuister GW. Ca2+ regulation in the Na+/Ca2+ exchanger features a dual electrostatic switch mechanism. Proc Natl Acad Sci U S A 106: 14333–14338, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hilge M, Aelen J, Perrakis A, Vuister GW. Structural basis for Ca2+ regulation in the Na+/Ca2+ exchanger. Ann N Y Acad Sci 1099: 7–15, 2007. [DOI] [PubMed] [Google Scholar]
- 95.Hilge M, Aelen J, Vuister GW. Ca2+ regulation in the Na+/Ca2+ exchanger involves two markedly different Ca2+ sensors. Mol Cell 22: 15–25, 2006. [DOI] [PubMed] [Google Scholar]
- 96.Hilgemann DW. Regulation and deregulation of cardiac Na+-Ca2+ exchange in giant excised sarcolemmal membrane patches. Nature 344: 242–245, 1990. [DOI] [PubMed] [Google Scholar]
- 97.Hilgemann DW. Unitary cardiac Na+, Ca2+ exchange current magnitudes determined from channel-like noise and charge movements of ion transport. Biophys J 71: 759–768, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hilgemann DW, Ball R. Regulation of cardiac Na+,Ca2+ exchange and KATP potassium channels by PIP2. Science 273: 956–959, 1996. [DOI] [PubMed] [Google Scholar]
- 99.Hilgemann DW, Collins A. Mechanism of cardiac Na+-Ca2+ exchange current stimulation by MgATP: Possible involvement of aminophospholipid translocase. J Physiol 454: 59–82, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hilgemann DW, Collins A, Cash DP, Nagel GA. Cardiac Na+-Ca2+ exchange system in giant membrane patches. Ann N Y Acad Sci 639: 126–139, 1991. [DOI] [PubMed] [Google Scholar]
- 101.Hilgemann DW, Collins A, Matsuoka S. Steady-state and dynamic properties of cardiac sodium-calcium exchange. Secondary modulation by cytoplasmic calcium and ATP. J Gen Physiol 100: 933–961, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hilgemann DW, Lu CC. Giant membrane patches: Improvements and applications. Methods Enzymol 293: 267–280, 1998. [DOI] [PubMed] [Google Scholar]
- 103.Hilgemann DW, Matsuoka S, Nagel GA, Collins A. Steady-state and dynamic properties of cardiac sodium-calcium exchange. Sodium-dependent inactivation. J Gen Physiol 100: 905–932, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hnatowich M, Le HD, DeMoissac D, Ranson K, Yurkov V, Gilchrist JS, Omelchenko A. mu-Calpain-mediated deregulation of cardiac, brain, and kidney NCX1 splice variants. Cell Calcium 51: 164–170, 2012. [DOI] [PubMed] [Google Scholar]
- 105.Hoshi M, Du XX, Shinlapawittayatorn K, Liu H, Chai S, Wan X, Ficker E, Deschenes I. Brugada syndrome disease phenotype explained in apparently benign sodium channel mutations. Cir Cardiovasc Genet 7: 123–131, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hryshko LV, Nicoll DA, Weiss JN, Philipson KD. Biosynthesis and initial processing of the cardiac sarcolemmal Na+-Ca2+ exchanger. Biochim Biophys Acta 1151: 35–42, 1993. [DOI] [PubMed] [Google Scholar]
- 107.Huang J, Hove-Madsen L, Tibbits GF. Na+/Ca2+ exchange activity in neonatal rabbit ventricular myocytes. Am J Physiol Cell Physiol 288: C195–C203, 2005. [DOI] [PubMed] [Google Scholar]
- 108.Hunte C, Screpanti E, Venturi M, Rimon A, Padan E, Michel H. Structure of a Na+/H+ antiporter and insights into mechanism of action and regulation by pH. Nature 435: 1197–1202, 2005. [DOI] [PubMed] [Google Scholar]
- 109.Huser J, Lipsius SL, Blatter LA. Calcium gradients during excitation-contraction coupling in cat atrial myocytes. J Physiol 494 (Pt 3): 641–651, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Imahashi K, Pott C, Goldhaber JI, Steenbergen C, Philipson KD, Murphy E. Cardiac-specific ablation of the Na+-Ca2+ exchanger confers protection against ischemia/reperfusion injury. Circ Res 97: 916–921, 2005. [DOI] [PubMed] [Google Scholar]
- 111.Inserte J, Hernando V, Garcia-Dorado D. Contribution of calpains to myocardial ischaemia/reperfusion injury. Cardiovasc Res 96: 23–31, 2012. [DOI] [PubMed] [Google Scholar]
- 112.Iwamoto T, Nakamura TY, Pan Y, Uehara A, Imanaga I, Shigekawa M. Unique topology of the internal repeats in the cardiac Na+/Ca2+ exchanger. Febs Letters 446: 264–268, 1999. [DOI] [PubMed] [Google Scholar]
- 113.Iwamoto T, Pan Y, Wakabayashi S, Imagawa T, Yamanaka HI, Shigekawa M. Phosphorylation-dependent regulation of cardiac Na+/Ca2+ exchanger via protein kinase C. J Biol Chem 271: 13609–13615, 1996. [DOI] [PubMed] [Google Scholar]
- 114.Iwamoto T, Uehara A, Imanaga I, Shigekawa M. The Na+/Ca2+ exchanger NCX1 has oppositely oriented reentrant loop domains that contain conserved aspartic acids whose mutation alters its apparent Ca2+ affinity. J Biol Chem 275: 38571–38580, 2000. [DOI] [PubMed] [Google Scholar]
- 115.Iwamoto T, Uehara A, Nakamura TY, Imanaga I, Shigekawa M. Chimeric analysis of Na+/Ca2+ exchangers NCX1 and NCX3 reveals structural domains important for differential sensitivity to external Ni2+ or Li+. J Biol Chem 274: 23094–23102, 1999. [DOI] [PubMed] [Google Scholar]
- 116.Iwamoto T, Wakabayashi S, Shigekawa M. Growth factor-induced phosphorylation and activation of aortic smooth muscle Na+/Ca2+ exchanger. J Biol Chem 270: 8996–9001, 1995. [DOI] [PubMed] [Google Scholar]
- 117.Iwamoto T, Watanabe Y, Kita S, Blaustein MP. Na+/Ca2+ exchange inhibitors: A new class of calcium regulators. Cardiovasc Hematol Disord Drug Targets 7: 188–198, 2007. [DOI] [PubMed] [Google Scholar]
- 118.James PF, Grupp IL, Grupp G, Woo AL, Askew GR, Croyle ML, Walsh RA, Lingrel JB. Identification of a specific role for the Na,KATPase alpha 2 isoform as a regulator of calcium in the heart. Mol Cell 3: 555–563, 1999. [DOI] [PubMed] [Google Scholar]
- 119.Jardetzky O Simple allosteric model for membrane pumps. Nature 211: 969–970, 1966. [DOI] [PubMed] [Google Scholar]
- 120.Jayasinghe ID, Cannell MB, Soeller C. Organization of ryanodine receptors, transverse tubules, and sodium-calcium exchanger in rat myocytes. Biophys J 97: 2664–2673, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Jeffs GJ, Meloni BP, Bakker AJ, Knuckey NW. The role of the Na+/Ca2+ exchanger NCX in neurons following ischaemia. J Clin Neurosci 14: 507–514, 2007. [DOI] [PubMed] [Google Scholar]
- 122.Jeffs GJ, Meloni BP, Sokolow S, Herchuelz A, Schurmans S, Knuckey NW. NCX3 knockout mice exhibit increased hippocampal CA1 and CA2 neuronal damage compared to wild-type mice following global cerebral ischemia. Exp Neurol 210: 268–273, 2008. [DOI] [PubMed] [Google Scholar]
- 123.Jeon D, Chu K, Jung KH, Kim M, Yoon BW, Lee CJ, Oh U, Shin HS. Na+/Ca2+ exchanger 2 is neuroprotective by exporting Ca2+ during a transient focal cerebral ischemia in the mouse. Cell Calcium 43: 482–491, 2008. [DOI] [PubMed] [Google Scholar]
- 124.John S, Kim B, Olcese R, Goldhaber JI, Ottolia M. Molecular determinants of pH regulation in the cardiac Na+-Ca2+ exchanger. J Gen Physiol 150: 245–257, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.John SA, Liao J, Jiang Y, Ottolia M. The cardiac Na+-Ca2+ exchanger has two cytoplasmic ion permeation pathways. Proc Natl Acad Sci U S A 110: 7500–7505, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.John SA, Ribalet B, Weiss JN, Philipson KD, Ottolia M. Ca2+-dependent structural rearrangements within Na+-Ca2+ exchanger dimers. Proc Natl Acad Sci U S A 108: 1699–1704, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Jordan MC, Henderson SA, Han T, Fishbein MC, Philipson KD, Roos KP. Myocardial function with reduced expression of the sodium-calcium exchanger. J Card Fail 16: 786–796, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Jovanovic S, Jovanovic A, Shen WK, Terzic A. Low concentrations of 17beta-estradiol protect single cardiac cells against metabolic stress-induced Ca2+ loading. J Am Coll Cardiol 36: 948–952, 2000. [DOI] [PubMed] [Google Scholar]
- 129.Kang K, Bauer PJ, Kinjo TG, Szerencsei RT, Bonigk W, Winkfein RJ, Schnetkamp PP. Assembly of retinal rod or cone Na+/Ca2+-K+ exchanger oligomers with cGMP-gated channel subunits as probed with heterologously expressed cDNAs. Biochemistry 42: 4593–4600, 2003. [DOI] [PubMed] [Google Scholar]
- 130.Kang TM, Hilgemann DW. Multiple transport modes of the cardiac Na+/Ca2+ exchanger. Nature 427: 544–548, 2004. [DOI] [PubMed] [Google Scholar]
- 131.Keller M, Pignier C, Niggli E, Egger M. Mechanisms of Na+-Ca2+ exchange inhibition by amphiphiles in cardiac myocytes: Importance of transbilayer movement. J Membr Biol 198: 159–175, 2004. [DOI] [PubMed] [Google Scholar]
- 132.Khananshvili D Structure-dynamic coupling through Ca2+-binding regulatory domains of mammalian NCX isoform/splice variants. Adv Exp Med Biol 981: 41–58, 2017. [DOI] [PubMed] [Google Scholar]
- 133.Kim I, Koh GY, Lee CO. Identification of alternatively spliced Na+-Ca2+ exchanger isoforms expressed in the heart. Comp Biochem Physiol B Biochem Mol Biol 119: 157–161, 1998. [DOI] [PubMed] [Google Scholar]
- 134.Kim I, Lee CO. Cloning of the mouse cardiac Na+-Ca2+ exchanger and functional expression in Xenopus oocytes. Ann N Y Acad Sci 779: 126–128, 1996. [DOI] [PubMed] [Google Scholar]
- 135.Kockskamper J, Sheehan KA, Bare DJ, Lipsius SL, Mignery GA, Blatter LA. Activation and propagation of Ca2+ release during excitation-contraction coupling in atrial myocytes. Biophys J 81: 2590–2605, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kofuji P, Lederer WJ, Schulze DH. Mutually exclusive and cassette exons underlie alternatively spliced isoforms of the Na/Ca exchanger. J Biol Chem 269: 5145–5149, 1994. [PubMed] [Google Scholar]
- 137.Kohajda Z, Loewe A, Toth N, Varro A, Nagy N. The cardiac pacemaker story-fundamental role of the Na+/Ca2+ exchanger in spontaneous automaticity. Front Pharmacol 11: 516, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Koushik SV, Wang J, Rogers R, Moskophidis D, Lambert NA, Creazzo TL, Conway SJ. Targeted inactivation of the sodium-calcium exchanger Ncx1 results in the lack of a heartbeat and abnormal myofibrillar organization. FASEB J 15: 1209–1211, 2001. [DOI] [PubMed] [Google Scholar]
- 139.Lakatta EG, Maltsev VA, Vinogradova TM. A coupled SYSTEM of intracellular Ca2+ clocks and surface membrane voltage clocks controls the timekeeping mechanism of the heart’s pacemaker. Circ Res 106: 659–673, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Larbig R, Torres N, Bridge JH, Goldhaber JI, Philipson KD. Activation of reverse Na+-Ca2+ exchange by the Na+ current augments the cardiac Ca2+ transient: Evidence from NCX knockout mice. J Physiol 588: 3267–3276, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Leblanc N, Hume JR. Sodium current-induced release of calcium from cardiac sarcoplasmic reticulum. Science 248: 372–376, 1990. [DOI] [PubMed] [Google Scholar]
- 142.Lee C, Dhalla NS, Hryshko LV. Therapeutic potential of novel Na+-Ca2+ exchange inhibitors in attenuating ischemia-reperfusion injury. Can J Cardiol 21: 509–516, 2005. [PubMed] [Google Scholar]
- 143.Lee SL, Yu AS, Lytton J. Tissue-specific expression of Na+-Ca2+ exchanger isoforms. J Biol Chem 269: 14849–14852, 1994. [PubMed] [Google Scholar]
- 144.Lee SY, Giladi M, Bohbot H, Hiller R, Chung KY, Khananshvili D. Structure-dynamic basis of splicing-dependent regulation in tissue-specific variants of the sodium-calcium exchanger. FASEB J 30: 1356–1366, 2016. [DOI] [PubMed] [Google Scholar]
- 145.Levitsky DO, Nicoll DA, Philipson KD. Identification of the high affinity Ca2+-binding domain of the cardiac Na+-Ca2+ exchanger. J Biol Chem 269: 22847–22852, 1994. [PubMed] [Google Scholar]
- 146.Li XF, Lytton J. A circularized sodium-calcium exchanger exon 2 transcript. J Biol Chem 274: 8153–8160, 1999. [DOI] [PubMed] [Google Scholar]
- 147.Li Z, Matsuoka S, Hryshko LV, Nicoll DA, Bersohn MM, Burke EP, Lifton RP, Philipson KD. Cloning of the NCX2 isoform of the plasma membrane Na+-Ca2+ exchanger. J Biol Chem 269: 17434–17439, 1994. [PubMed] [Google Scholar]
- 148.Li ZP, Burke EP, Frank JS, Bennett V, Philipson KD. The cardiac Na+-Ca2+ exchanger binds to the cytoskeletal protein ankyrin. J Biol Chem 268: 11489–11491, 1993. [PubMed] [Google Scholar]
- 149.Li ZP, Nicoll DA, Collins A, Hilgemann DW, Filoteo AG, Penniston JT, Weiss JN, Tomich JM, Philipson KD. Identification of a peptide inhibitor of the cardiac sarcolemmal Na+-Ca2+ exchanger. J Biol Chem 266: 1014–1020, 1991. [PubMed] [Google Scholar]
- 150.Liao J, Li H, Zeng W, Sauer DB, Belmares R, Jiang Y. Structural insight into the ion-exchange mechanism of the sodium/calcium exchanger. Science 335: 686–690, 2012. [DOI] [PubMed] [Google Scholar]
- 151.Liao J, Marinelli F, Lee C, Huang Y, Faraldo-Gomez JD, Jiang Y. Mechanism of extracellular ion exchange and binding-site occlusion in a sodium/calcium exchanger. Nat Struct Mol Biol 23: 590–599, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Lin X, Jo H, Sakakibara Y, Tambara K, Kim B, Komeda M, Matsuoka S. Beta-adrenergic stimulation does not activate Na+/Ca2+ exchange current in guinea pig, mouse, and rat ventricular myocytes. Am J Physiol Cell Physiol 290: C601–C608, 2006. [DOI] [PubMed] [Google Scholar]
- 153.Linck B, Qiu Z, He Z, Tong Q, Hilgemann DW, Philipson KD. Functional comparison of the three isoforms of the Na+/Ca2+ exchanger NCX1, NCX2, NCX3. Am J Physiol 274: C415–C423, 1998. [DOI] [PubMed] [Google Scholar]
- 154.Lindgren RM, Zhao J, Heller S, Berglind H, Nister M. Molecular cloning and characterization of two novel truncated isoforms of human Na+/Ca2+ exchanger 3, expressed in fetal brain. Gene 348: 143–155, 2005. [DOI] [PubMed] [Google Scholar]
- 155.Lipp P, Niggli E. Sodium current-induced calcium signals in isolated guinea-pig ventricular myocytes. J Physiol 474: 439–446, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Liu T, Zhao J, Ibarra C, Garcia MU, Uhlen P, Nister M. Glycosylation controls sodium-calcium exchanger 3 sub-cellular localization during cell cycle. Eur J Cell Biol 97: 190–203, 2018. [DOI] [PubMed] [Google Scholar]
- 157.Lytton J Na+/Ca2+ Exchangers and Ca2+ Transport in Neurons. Boston, MA: Springer, 2007. [Google Scholar]
- 158.Lytton J Na+/Ca2+ exchangers: Three mammalian gene families control Ca2+ transport. Biochem J 406: 365–382, 2007. [DOI] [PubMed] [Google Scholar]
- 159.Maack C, Ganesan A, Sidor A, O’Rourke B. Cardiac sodium-calcium exchanger is regulated by allosteric calcium and exchanger inhibitory peptide at distinct sites. Circ Res 96: 91–99, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Maltsev VA, Lakatta EG. Normal heart rhythm is initiated and regulated by an intracellular calcium clock within pacemaker cells. Heart Lung Circ 16: 335–348, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Manfra O, Shen X, Hell JW, Edward Louch W. Super-resolution dSTORM imaging of calcium handling proteins in cardiomyocytes. Biophysical Journal 114: 620a, 2018. [Google Scholar]
- 162.Marinelli F, Almagor L, Hiller R, Giladi M, Khananshvili D, Faraldo-Gomez JD. Sodium recognition by the Na+/Ca2+ exchanger in the outward-facing conformation. Proc Natl Acad Sci U S A 111: E5354–E5362, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Marshall CR, Fox JA, Butland SL, Ouellette BF, Brinkman FS, Tibbits GF. Phylogeny of Na+/Ca2+ exchanger NCX genes from genomic data identifies new gene duplications and a new family member in fish species. Physiol Genomics 21: 161–173, 2005. [DOI] [PubMed] [Google Scholar]
- 164.Matsuoka S, Hilgemann DW. Steady-state and dynamic properties of cardiac sodium-calcium exchange. Ion and voltage dependencies of the transport cycle. J Gen Physiol 100: 963–1001, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Matsuoka S, Hilgemann DW. Inactivation of outward Na+-Ca2+ exchange current in guinea-pig ventricular myocytes. J Physiol 476: 443–458, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Matsuoka S, Nicoll DA, He Z, Philipson KD. Regulation of cardiac Na+-Ca2+ exchanger by the endogenous XIP region. J Gen Physiol 109: 273–286, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Matsuoka S, Nicoll DA, Hryshko LV, Levitsky DO, Weiss JN, Philipson KD. Regulation of the cardiac Na+-Ca2+ exchanger by Ca2+. Mutational analysis of the Ca2+-binding domain. J Gen Physiol 105: 403–420, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Matsuoka S, Nicoll DA, Reilly RF, Hilgemann DW, Philipson KD. Initial localization of regulatory regions of the cardiac sarcolemmal Na+-Ca2+ exchanger. Proc Natl Acad Sci U S A 90: 3870–3874, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Matsuoka S, Philipson KD, Hilgemann DW. Multiple functional states of the cardiac Na+-Ca2+ exchanger. Whole-cell, native-excised, and cloned-excised properties. Ann N Y Acad Sci 779: 159–170, 1996. [DOI] [PubMed] [Google Scholar]
- 170.Maxwell K, Scott J, Omelchenko A, Lukas A, Lu L, Lu Y, Hnatowich M, Philipson KD, Hryshko LV. Functional role of ionic regulation of Na+/Ca2+ exchange assessed in transgenic mouse hearts. Am J Physiol 277: H2212–H2221, 1999. [DOI] [PubMed] [Google Scholar]
- 171.Miura Y, Kimura J. Sodium-calcium exchange current. Dependence on internal Ca and Na and competitive binding of external Na and Ca. J Gen Physiol 93: 1129–1145, 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Mohler PJ, Davis JQ, Bennett V. Ankyrin-B coordinates the Na/K ATPase, Na/Ca exchanger, and InsP3 receptor in a cardiac T-tubule/SR microdomain. PLoS Biol 3: e423, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Mohler PJ, Schott JJ, Gramolini AO, Dilly KW, Guatimosim S, duBell WH, Song LS, Haurogne K, Kyndt F, Ali ME, Rogers TB, Lederer WJ, Escande D, Le Marec H, Bennett V. Ankyrin-B mutation causes type 4 long-QT cardiac arrhythmia and sudden cardiac death. Nature 421: 634–639, 2003. [DOI] [PubMed] [Google Scholar]
- 174.Molinaro P, Cuomo O, Pignataro G, Boscia F, Sirabella R, Pannaccione A, Secondo A, Scorziello A, Adornetto A, Gala R, Viggiano D, Sokolow S, Herchuelz A, Schurmans S, Di Renzo G, Annunziato L. Targeted disruption of Na+/Ca2+ exchanger 3 NCX3 gene leads to a worsening of ischemic brain damage. J Neurosci 28: 1179–1184, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Molinaro P, Natale S, Serani A, Calabrese L, Secondo A, Tedeschi V, Valsecchi V, Pannaccione A, Scorziello A, Annunziato L. Genetically modified mice to unravel physiological and pathophysiological roles played by NCX isoforms. Cell Calcium 87: 102189, 2020. [DOI] [PubMed] [Google Scholar]
- 176.Molinaro P, Pannaccione A, Sisalli MJ, Secondo A, Cuomo O, Sirabella R, Cantile M, Ciccone R, Scorziello A, di Renzo G, Annunziato L. A new cell-penetrating peptide that blocks the autoinhibitory XIP domain of NCX1 and enhances antiporter activity. Mol Ther 23: 465–476, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Morad M, Cleemann L, Menick DR. NCX1 phosphorylation dilemma: A little closer to resolution. Focus on “Full-length cardiac Na+/Ca2+ exchanger 1 protein is not phosphorylated by protein kinase A”. Am J Physiol Cell Physiol 300: C970–C973, 2011. [DOI] [PubMed] [Google Scholar]
- 178.Muller JG, Isomatsu Y, Koushik SV, O’Quinn M, Xu L, Kappler CS, Hapke E, Zile MR, Conway SJ, Menick DR. Cardiac-specific expression and hypertrophic upregulation of the feline Na+-Ca2+ exchanger gene H1-promoter in a transgenic mouse model. Circ Res 90: 158–164, 2002. [DOI] [PubMed] [Google Scholar]
- 179.Nakasaki Y, Iwamoto T, Hanada H, Imagawa T, Shigekawa M. Cloning of the rat aortic smooth muscle Na+/Ca2+ exchanger and tissue-specific expression of isoforms. J Biochem 114: 528–534, 1993. [DOI] [PubMed] [Google Scholar]
- 180.Neco P, Rose B, Huynh N, Zhang R, Bridge JH, Philipson KD, Goldhaber JI. Sodium-calcium exchange is essential for effective triggering of calcium release in mouse heart. Biophys J 99: 755–764, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Nicholas SB, Philipson KD. Cardiac expression of the Na+/Ca2+ exchanger NCX1 is GATA factor dependent. Am J Physiol 277: H324–H330, 1999. [DOI] [PubMed] [Google Scholar]
- 182.Nicholas SB, Yang W, Lee SL, Zhu H, Philipson KD, Lytton J. Alternative promoters and cardiac muscle cell-specific expression of the Na+/Ca2+ exchanger gene. Am J Physiol 274: H217–H232, 1998. [DOI] [PubMed] [Google Scholar]
- 183.Nicoll DA, Hryshko LV, Matsuoka S, Frank JS, Philipson KD. Mutagenesis studies of the cardiac Na+-Ca2+ exchanger. Ann N Y Acad Sci 779: 86–92, 1996. [DOI] [PubMed] [Google Scholar]
- 184.Nicoll DA, Hryshko LV, Matsuoka S, Frank JS, Philipson KD. Mutation of amino acid residues in the putative transmembrane segments of the cardiac sarcolemmal Na+-Ca2+ exchanger. J Biol Chem 271: 13385–13391, 1996. [DOI] [PubMed] [Google Scholar]
- 185.Nicoll DA, Longoni S, Philipson KD. Molecular cloning and functional expression of the cardiac sarcolemmal Na+-Ca2+ exchanger. Science 250: 562–565, 1990. [DOI] [PubMed] [Google Scholar]
- 186.Nicoll DA, Ottolia M, Lu L, Lu Y, Philipson KD. A new topological model of the cardiac sarcolemmal Na+-Ca2+ exchanger. J Biol Chem 274: 910–917, 1999. [DOI] [PubMed] [Google Scholar]
- 187.Nicoll DA, Ottolia M, Philipson KD. Toward a topological model of the NCX1 exchanger. Ann N Y Acad Sci 976: 11–18, 2002. [DOI] [PubMed] [Google Scholar]
- 188.Nicoll DA, Quednau BD, Qui Z, Xia YR, Lusis AJ, Philipson KD. Cloning of a third mammalian Na+-Ca2+ exchanger, NCX3. J Biol Chem 271: 24914–24921, 1996. [DOI] [PubMed] [Google Scholar]
- 189.Nicoll DA, Sawaya MR, Kwon S, Cascio D, Philipson KD, Abramson J. The crystal structure of the primary Ca2+ sensor of the Na+/Ca2+ exchanger reveals a novel Ca2+ binding motif. J Biol Chem 281: 21577–21581, 2006. [DOI] [PubMed] [Google Scholar]
- 190.Niggli E, Lederer WJ. Molecular operations of the sodium-calcium exchanger revealed by conformation currents. Nature 349: 621–624, 1991. [DOI] [PubMed] [Google Scholar]
- 191.On C, Marshall CR, Chen N, Moyes CD, Tibbits GF. Gene structure evolution of the Na+-Ca2+ exchanger NCX family. BMC Evol Biol 8: 127, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Ottolia M, John S, Qiu Z, Philipson KD. Split Na+-Ca2+ exchangers. Implications for function and expression. J Biol Chem 276: 19603–19609, 2001. [DOI] [PubMed] [Google Scholar]
- 193.Ottolia M, John S, Ren X, Philipson KD. Fluorescent Na+-Ca+ exchangers: Electrophysiological and optical characterization. J Biol Chem 282: 3695–3701, 2007. [DOI] [PubMed] [Google Scholar]
- 194.Ottolia M, Nicoll DA, John S, Philipson KD. Interactions between Ca2+ binding domains of the Na+-Ca2+ exchanger and secondary regulation. Channels Austin 4: 159–162, 2010. [DOI] [PubMed] [Google Scholar]
- 195.Ottolia M, Nicoll DA, Philipson KD. Mutational analysis of the alpha-1 repeat of the cardiac Na+-Ca2+ exchanger. J Biol Chem 280: 1061–1069, 2005. [DOI] [PubMed] [Google Scholar]
- 196.Ottolia M, Nicoll DA, Philipson KD. Roles of two Ca2+-binding domains in regulation of the cardiac Na+-Ca2+ exchanger. J Biol Chem 284: 32735–32741, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Ottolia M, Philipson KD, John S. Conformational changes of the Ca2+ regulatory site of the Na+-Ca2+ exchanger detected by FRET. Biophys J 87: 899–906, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Ottolia M, Torres N, Bridge JH, Philipson KD, Goldhaber JI. Na/Ca exchange and contraction of the heart. J Mol Cell Cardiol 61: 28–33, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Perez C, Khafizov K, Forrest LR, Kramer R, Ziegler C. The role of trimerization in the osmoregulated betaine transporter BetP. EMBO Rep 12: 804–810, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Philipson KD. Interaction of charged amphiphiles with Na+-Ca2+ exchange in cardiac sarcolemmal vesicles. J Biol Chem 259: 13999–14002, 1984. [PubMed] [Google Scholar]
- 201.Philipson KD, Bersohn MM, Nishimoto AY. Effects of pH on Na+-Ca2+ exchange in canine cardiac sarcolemmal vesicles. Circ Res 50: 287–293, 1982. [DOI] [PubMed] [Google Scholar]
- 202.Philipson KD, Frank JS, Nishimoto AY. Effects of phospholipase C on the Na+-Ca2+ exchange and Ca2+ permeability of cardiac sarcolemmal vesicles. J Biol Chem 258: 5905–5910, 1983. [PubMed] [Google Scholar]
- 203.Philipson KD, Longoni S, Ward R. Purification of the cardiac Na+-Ca2+ exchange protein. Biochim Biophys Acta 945: 298–306, 1988. [DOI] [PubMed] [Google Scholar]
- 204.Philipson KD, Nishimoto AY. Na+-Ca2+ exchange in inside-out cardiac sarcolemmal vesicles. J Biol Chem 257: 5111–5117, 1982. [PubMed] [Google Scholar]
- 205.Philipson KD, Nishimoto AY. Stimulation of Na+-Ca2+ exchange in cardiac sarcolemmal vesicles by phospholipase D. J Biol Chem 259: 16–19, 1984. [PubMed] [Google Scholar]
- 206.Philipson KD, Ward R. Effects of fatty acids on Na+-Ca2+ exchange and Ca2+ permeability of cardiac sarcolemmal vesicles. J Biol Chem 260: 9666–9671, 1985. [PubMed] [Google Scholar]
- 207.Plain F, Congreve SD, Yee RSZ, Kennedy J, Howie J, Kuo CW, Fraser NJ, Fuller W. An amphipathic alpha-helix directs palmitoylation of the large intracellular loop of the sodium/calcium exchanger. J Biol Chem 292: 10745–10752, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Pogwizd SM. Clinical potential of sodium-calcium exchanger inhibitors as antiarrhythmic agents. Drugs 63: 439–452, 2003. [DOI] [PubMed] [Google Scholar]
- 209.Pogwizd SM, Bers DM. Na/Ca exchange in heart failure: Contractile dysfunction and arrhythmogenesis. Ann N Y Acad Sci 976: 454–465, 2002. [DOI] [PubMed] [Google Scholar]
- 210.Pogwizd SM, Bers DM. Cellular basis of triggered arrhythmias in heart failure. Trends Cardiovasc Med 14: 61–66, 2004. [DOI] [PubMed] [Google Scholar]
- 211.Pogwizd SM, Qi M, Yuan W, Samarel AM, Bers DM. Upregulation of Na+/Ca2+ exchanger expression and function in an arrhythmogenic rabbit model of heart failure. Circ Res 85: 1009–1019, 1999. [DOI] [PubMed] [Google Scholar]
- 212.Porzig H, Li Z, Nicoll DA, Philipson KD. Mapping of the cardiac sodium-calcium exchanger with monoclonal antibodies. Am J Physiol 265: C748–C756, 1993. [DOI] [PubMed] [Google Scholar]
- 213.Pott C, Eckardt L, Goldhaber JI. Triple threat: The Na+/Ca2+ exchanger in the pathophysiology of cardiac arrhythmia, ischemia and heart failure. Curr Drug Targets 12: 737–747, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Pott C, Goldhaber JI, Philipson KD. Genetic manipulation of cardiac Na+/Ca2+ exchange expression. Biochem Biophys Res Commun 322: 1336–1340, 2004. [DOI] [PubMed] [Google Scholar]
- 215.Pott C, Goldhaber JI, Philipson KD. Homozygous overexpression of the Na+-Ca2+ exchanger in mice: Evidence for increased transsarcolemmal Ca2+ fluxes. Ann N Y Acad Sci 1099: 310–314, 2007. [DOI] [PubMed] [Google Scholar]
- 216.Pott C, Henderson SA, Goldhaber JI, Philipson KD. Na+/Ca2+ exchanger knockout mice: Plasticity of cardiac excitation-contraction coupling. Ann N Y Acad Sci 1099: 270–275, 2007. [DOI] [PubMed] [Google Scholar]
- 217.Pott C, Muszynski A, Ruhe M, Bogeholz N, Schulte JS, Milberg P, Monnig G, Fabritz L, Goldhaber JI, Breithardt G, Schmitz W, Philipson KD, Eckardt L, Kirchhof P, Muller FU. Proarrhythmia in a non-failing murine model of cardiac-specific Na+/Ca 2+ exchanger over-expression: Whole heart and cellular mechanisms. Basic Res Cardiol 107: 247, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Pott C, Philipson KD, Goldhaber JI. Excitation-contraction coupling in Na+-Ca2+ exchanger knockout mice: Reduced transsarcolemmal Ca2+ flux. Circ Res 97: 1288–1295, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Pott C, Ren X, Tran DX, Yang MJ, Henderson S, Jordan MC, Roos KP, Garfinkel A, Philipson KD, Goldhaber JI. Mechanism of shortened action potential duration in Na+-Ca2+ exchanger knockout mice. Am J Physiol Cell Physiol 292: C968–C973, 2007. [DOI] [PubMed] [Google Scholar]
- 220.Quednau BD, Nicoll DA, Philipson KD. Tissue specificity and alternative splicing of the Na+/Ca2+ exchanger isoforms NCX1, NCX2, and NCX3 in rat. Am J Physiol 272: C1250–C1261, 1997. [DOI] [PubMed] [Google Scholar]
- 221.Quednau BD, Nicoll DA, Philipson KD. The sodium/calcium exchanger family-SLC8. Pflugers Arch 447: 543–548, 2004. [DOI] [PubMed] [Google Scholar]
- 222.Radwanski PB, Brunello L, Veeraraghavan R, Ho HT, Lou Q, Makara MA, Belevych AE, Anghelescu M, Priori SG, Volpe P, Hund TJ, Janssen PM, Mohler PJ, Bridge JH, Poelzing S, Gyorke S. Neuronal Na+ channel blockade suppresses arrhythmogenic diastolic Ca2+ release. Cardiovasc Res 106: 143–152, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Ramos-Franco J, Aguilar-Sanchez Y, Escobar AL. Intact heart loose patch photolysis reveals ionic current kinetics during ventricular action potentials. Circ Res 118: 203–215, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Ranu HK, Terracciano CM, Davia K, Bernobich E, Chaudhri B, Robinson SE, Bin Kang Z, Hajjar RJ, MacLeod KT, Harding SE. Effects of Na+/Ca2+-exchanger overexpression on excitation-contraction coupling in adult rabbit ventricular myocytes. J Mol Cell Cardiol 34: 389–400, 2002. [DOI] [PubMed] [Google Scholar]
- 225.Reeves JP, Hale CC. The stoichiometry of the cardiac sodium-calcium exchange system. J Biol Chem 259: 7733–7739, 1984. [PubMed] [Google Scholar]
- 226.Reilly L, Howie J, Wypijewski K, Ashford ML, Hilgemann DW, Fuller W. Palmitoylation of the Na/Ca exchanger cytoplasmic loop controls its inactivation and internalization during stress signaling. FASEB J 29: 4532–4543, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Ren X, Nicoll DA, Galang G, Philipson KD. Intermolecular cross-linking of Na+-Ca2+ exchanger proteins: Evidence for dimer formation. Biochemistry 47: 6081–6087, 2008. [DOI] [PubMed] [Google Scholar]
- 228.Ren X, Philipson KD. The topology of the cardiac Na+/Ca2+ exchanger, NCX1. J Mol Cell Cardiol 57: 68–71, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Reuter H, Han T, Motter C, Philipson KD, Goldhaber JI. Mice overexpressing the cardiac sodium-calcium exchanger: Defects in excitation-contraction coupling. J Physiol 554: 779–789, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Reuter H, Henderson SA, Han T, Mottino GA, Frank JS, Ross RS, Goldhaber JI, Philipson KD. Cardiac excitation-contraction coupling in the absence of Na+ - Ca2+ exchange. Cell Calcium 34: 19–26, 2003. [DOI] [PubMed] [Google Scholar]
- 231.Reuter H, Henderson SA, Han T, Ross RS, Goldhaber JI, Philipson KD. The Na+-Ca2+ exchanger is essential for the action of cardiac glycosides. Circ Res 90: 305–308, 2002. [DOI] [PubMed] [Google Scholar]
- 232.Reuter H, Seitz N. The dependence of calcium efflux from cardiac muscle on temperature and external ion composition. J Physiol 195: 451–470, 1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Riedel MJ, Baczko I, Searle GJ, Webster N, Fercho M, Jones L, Lang J, Lytton J, Dyck JR, Light PE. Metabolic regulation of sodium-calcium exchange by intracellular acyl CoAs. Embo J 25: 4605–4614, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Rimon A, Tzubery T, Padan E. Monomers of the NhaA Na+/H+ antiporter of Escherichia coli are fully functional yet dimers are beneficial under extreme stress conditions at alkaline pH in the presence of Na+ or Li+. J Biol Chem 282: 26810–26821, 2007. [DOI] [PubMed] [Google Scholar]
- 235.Robertson JL, Kolmakova-Partensky L, Miller C. Design, function and structure of a monomeric ClC transporter. Nature 468: 844–847, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Roos KP, Jordan MC, Fishbein MC, Ritter MR, Friedlander M, Chang HC, Rahgozar P, Han T, Garcia AJ, Maclellan WR, Ross RS, Philipson KD. Hypertrophy and heart failure in mice overexpressing the cardiac sodium-calcium exchanger. J Card Fail 13: 318–329, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Rosker C, Graziani A, Lukas M, Eder P, Zhu MX, Romanin C, Groschner K. Ca2+ signaling by TRPC3 involves Na+ entry and local coupling to the Na+/Ca2+ exchanger. J Biol Chem 279: 13696–13704, 2004. [DOI] [PubMed] [Google Scholar]
- 238.Ruknudin AM, Wei SK, Haigney MC, Lederer WJ, Schulze DH. Phosphorylation and other conundrums of Na/Ca exchanger, NCX1. Ann N Y Acad Sci 1099: 103–118, 2007. [DOI] [PubMed] [Google Scholar]
- 239.Sahin-Toth M, Nicoll DA, Frank JS, Philipson KD, Friedlander M. The cleaved N-terminal signal sequence of the cardiac Na+-Ca2+ exchanger is not required for functional membrane integration. Biochem Biophys Res Commun 212: 968–974, 1995. [DOI] [PubMed] [Google Scholar]
- 240.Salinas RK, Bruschweiler-Li L, Johnson E, Bruschweiler R. Ca2+ binding alters the interdomain flexibility between the two cytoplasmic calcium-binding domains in the Na+/Ca2+ exchanger. J Biol Chem 286: 32123–32131, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Santacruz-Toloza L, Ottolia M, Nicoll DA, Philipson KD. Functional analysis of a disulfide bond in the cardiac Na+-Ca2+ exchanger. J Biol Chem 275: 182–188, 2000. [DOI] [PubMed] [Google Scholar]
- 242.Schafer C, Ladilov Y, Inserte J, Schafer M, Haffner S, Garcia-Dorado D, Piper HM. Role of the reverse mode of the Na+/Ca2+ exchanger in reoxygenation-induced cardiomyocyte injury. Cardiovasc Res 51: 241–250, 2001. [DOI] [PubMed] [Google Scholar]
- 243.Schulson MN, Scriven DR, Fletcher P, Moore ED. Couplons in rat atria form distinct subgroups defined by their molecular partners. J Cell Sci 124: 1167–1174, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Schulze DH, Muqhal M, Lederer WJ, Ruknudin AM. Sodium/calcium exchanger NCX1 macromolecular complex. J Biol Chem 278: 28849–28855, 2003. [DOI] [PubMed] [Google Scholar]
- 245.Schwarz EM, Benzer S. Calx, a Na-Ca exchanger gene of Drosophila melanogaster. Proc Natl Acad Sci U S A 94: 10249–10254, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Scranton K, Umar S, Calmettes G, Eghbali M, Goldhaber JI, John SA, Olcese R, Escobar AL, Ottolia M. The Na+-dependent inactivation of NCX1.1 is physiologically relevant to cardiac function. Biophys J 118: 100a–101a, 2020. [Google Scholar]
- 247.Scriven DR, Dan P, Moore ED. Distribution of proteins implicated in excitation-contraction coupling in rat ventricular myocytes. Biophys J 79: 2682–2691, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Shannon TR, Hale CC, Milanick MA. Interaction of cardiac Na-Ca exchanger and exchange inhibitory peptide with membrane phospholipids. Am J Physiol 266: C1350–C1356, 1994. [DOI] [PubMed] [Google Scholar]
- 249.Shattock MJ, Ottolia M, Bers DM, Blaustein MP, Boguslavskyi A, Bossuyt J, Bridge JH, Chen-Izu Y, Clancy CE, Edwards A, Goldhaber J, Kaplan J, Lingrel JB, Pavlovic D, Philipson K, Sipido KR, Xie ZJ. Na+/Ca2+ exchange and Na+/K+-ATPase in the heart. J Physiol 593: 1361–1382, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Shen C, Lin MJ, Yaradanakul A, Lariccia V, Hill JA, Hilgemann DW. Dual control of cardiac Na+ Ca2+ exchange by PIP2: Analysis of the surface membrane fraction by extracellular cysteine PEGylation. J Physiol 582: 1011–1026, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Shieh BH, Xia Y, Sparkes RS, Klisak I, Lusis AJ, Nicoll DA, Philipson KD. Mapping of the gene for the cardiac sarcolemmal Na+-Ca2+ exchanger to human chromosome 2p21-p23. Genomics 12: 616–617, 1992. [DOI] [PubMed] [Google Scholar]
- 252.Shigekawa M, Iwamoto T, Uehara A, Kita S. Probing ion binding sites in the Na+/Ca2+ exchanger. Ann N Y Acad Sci 976: 19–30, 2002. [DOI] [PubMed] [Google Scholar]
- 253.Shipston MJ. Ion channel regulation by protein palmitoylation. J Biol Chem 286: 8709–8716, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Shlosman I, Marinelli F, Faraldo-Gomez JD, Mindell JA. The prokaryotic Na+/Ca2+ exchanger NCX_Mj transports Na+ and Ca2+ in a 3:1 stoichiometry. J Gen Physiol 150: 51–65, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Shuart NG, Haitin Y, Camp SS, Black KD, Zagotta WN. Molecular mechanism for 3:1 subunit stoichiometry of rod cyclic nucleotide-gated ion channels. Nat Commun 2: 457, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Sitte HH, Farhan H, Javitch JA. Sodium-dependent neurotransmitter transporters: Oligomerization as a determinant of transporter function and trafficking. Mol Interv 4: 38–47, 2004. [DOI] [PubMed] [Google Scholar]
- 257.Skogestad J, Aronsen JM, Tovsrud N, Wanichawan P, Hougen K, Stokke MK, Carlson CR, Sjaastad I, Sejersted OM, Swift F. Coupling of the Na+/K+-ATPase to Ankyrin B controls Na+/Ca2+ exchanger activity in cardiomyocytes. Cardiovasc Res 116: 78–90, 2020. [DOI] [PubMed] [Google Scholar]
- 258.Sokolow S, Manto M, Gailly P, Molgo J, Vandebrouck C, Vanderwinden JM, Herchuelz A, Schurmans S. Impaired neuromuscular transmission and skeletal muscle fiber necrosis in mice lacking Na/Ca exchanger 3. J Clin Invest 113: 265–273, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Sorimachi H, Ono Y. Regulation and physiological roles of the calpain system in muscular disorders. Cardiovasc Res 96: 11–22, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Steccanella F, Scranton K, Panday N, Angelini M, Zhang R, Lotteau S, John SA, Olcese R, Goldhaber JI, Ottolia M. Genetic ablation of NCX1.1 Na+-dependent inactivation impacts cardiac action potential and Ca2+ transient. Biophys J 118: 100a, 2020. [Google Scholar]
- 261.Stieber J, Herrmann S, Feil S, Loster J, Feil R, Biel M, Hofmann F, Ludwig A. The hyperpolarization-activated channel HCN4 is required for the generation of pacemaker action potentials in the embryonic heart. Proc Natl Acad Sci U S A 100: 15235–15240, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Sugishita K, Su Z, Li F, Philipson KD, Barry WH. Gender influences [Ca2+]i during metabolic inhibition in myocytes overexpressing the Na+-Ca2+ exchanger. Circulation 104: 2101–2106, 2001. [DOI] [PubMed] [Google Scholar]
- 263.Swift F, Birkeland JA, Tovsrud N, Enger UH, Aronsen JM, Louch WE, Sjaastad I, Sejersted OM. Altered Na+/Ca2+-exchanger activity due to downregulation of Na+/K+-ATPase alpha2-isoform in heart failure. Cardiovasc Res 78: 71–78, 2008. [DOI] [PubMed] [Google Scholar]
- 264.Swift F, Tovsrud N, Enger UH, Sjaastad I, Sejersted OM. The Na+/K+-ATPase alpha2-isoform regulates cardiac contractility in rat cardiomyocytes. Cardiovasc Res 75: 109–117, 2007. [DOI] [PubMed] [Google Scholar]
- 265.Szerencsei RT, Kinjo TG, Schnetkamp PP. The topology of the C-terminal sections of the NCX1 Na +/Ca 2+ exchanger and the NCKX2 Na +/Ca 2+ -K + exchanger. Channels Austin 7: 109–114, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Terracciano CM, Souza AI, Philipson KD, MacLeod KT. Na+-Ca2+ exchange and sarcoplasmic reticular Ca2+ regulation in ventricular myocytes from transgenic mice overexpressing the Na+-Ca2+ exchanger. J Physiol 512 (Pt 3): 651–667, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Thomas MJ, Sjaastad I, Andersen K, Helm PJ, Wasserstrom JA, Sejersted OM, Ottersen OP. Localization and function of the Na+/Ca2+-exchanger in normal and detubulated rat cardiomyocytes. J Mol Cell Cardiol 35: 1325–1337, 2003. [DOI] [PubMed] [Google Scholar]
- 268.Tombola F, Ulbrich MH, Kohout SC, Isacoff EY. The opening of the two pores of the Hv1 voltage-gated proton channel is tuned by cooperativity. Nat Struct Mol Biol 17: 44–50, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Torrente AG, Zhang R, Wang H, Zaini A, Kim B, Yue X, Philipson KD, Goldhaber JI. Contribution of small conductance K+ channels to sinoatrial node pacemaker activity: Insights from atrial-specific Na+/Ca2+ exchange knockout mice. J Physiol 595: 3847–3865, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Torrente AG, Zhang R, Zaini A, Giani JF, Kang J, Lamp ST, Philipson KD, Goldhaber JI. Burst pacemaker activity of the sinoatrial node in sodium-calcium exchanger knockout mice. Proc Natl Acad Sci U S A 112: 9769–9774, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Torres NS, Larbig R, Rock A, Goldhaber JI, Bridge JH. Na+ currents are required for efficient excitation-contraction coupling in rabbit ventricular myocytes: A possible contribution of neuronal Na+ channels. J Physiol 588: 4249–4260, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.van der Vusse GJ, van Bilsen M, Reneman RS. Ischemia and reperfusion induced alterations in membrane phospholipids: An overview. Ann N Y Acad Sci 723: 1–14, 1994. [PubMed] [Google Scholar]
- 273.van Dijk L, Giladi M, Refaeli B, Hiller R, Cheng MH, Bahar I, Khananshvili D. Key residues controlling bidirectional ion movements in Na+/Ca2+ exchanger. Cell Calcium 76: 10–22, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Van Eylen F, Kamagate A, Herchuelz A. A new Na/Ca exchanger splicing pattern identified in situ leads to a functionally active 70kDa NH2-terminal protein. Cell Calcium 30: 191–198, 2001. [DOI] [PubMed] [Google Scholar]
- 275.Vaughan-Jones RD, Spitzer KW, Swietach P. Intracellular pH regulation in heart. J Mol Cell Cardiol 46: 318–331, 2009. [DOI] [PubMed] [Google Scholar]
- 276.Vemuri R, Philipson KD. Phospholipid composition modulates the Na+-Ca2+ exchange activity of cardiac sarcolemma in reconstituted vesicles. Biochim Biophys Acta 937: 258–268, 1988. [DOI] [PubMed] [Google Scholar]
- 277.Waight AB, Pedersen BP, Schlessinger A, Bonomi M, Chau BH, Roe-Zurz Z, Risenmay AJ, Sali A, Stroud RM. Structural basis for alternating access of a eukaryotic calcium/proton exchanger. Nature 499: 107–110, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Wakimoto K, Fujimura H, Iwamoto T, Oka T, Kobayashi K, Kita S, Kudoh S, Kuro-o M, Nabeshima Y, Shigekawa M, Imai Y, Komuro I. Na+/Ca2+ exchanger-deficient mice have disorganized myofibrils and swollen mitochondria in cardiomyocytes. Comp Biochem Physiol B Biochem Mol Biol 135: 9–15, 2003. [DOI] [PubMed] [Google Scholar]
- 279.Wakimoto K, Kobayashi K, Kuro OM, Yao A, Iwamoto T, Yanaka N, Kita S, Nishida A, Azuma S, Toyoda Y, Omori K, Imahie H, Oka T, Kudoh S, Kohmoto O, Yazaki Y, Shigekawa M, Imai Y, Nabeshima Y, Komuro I. Targeted disruption of Na+/Ca2+ exchanger gene leads to cardiomyocyte apoptosis and defects in heartbeat. J Biol Chem 275: 36991–36998, 2000. [DOI] [PubMed] [Google Scholar]
- 280.Wang J, Chan TO, Zhang XQ, Gao E, Song J, Koch WJ, Feldman AM, Cheung JY. Induced overexpression of Na+/Ca2+ exchanger transgene: Altered myocyte contractility, [Ca2+]i transients, SR Ca2+ contents, and action potential duration. Am J Physiol Heart Circ Physiol 297: H590–H601, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Wanichawan P, Hafver TL, Hodne K, Aronsen JM, Lunde IG, Dalhus B, Lunde M, Kvaloy H, Louch WE, Tonnessen T, Sjaastad I, Sejersted OM, Carlson CR. Molecular basis of calpain cleavage and inactivation of the sodium-calcium exchanger 1 in heart failure. J Biol Chem 289: 33984–33998, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Wanichawan P, Louch WE, Hortemo KH, Austbo B, Lunde PK, Scott JD, Sejersted OM, Carlson CR. Full-length cardiac Na+/Ca2+ exchanger 1 protein is not phosphorylated by protein kinase A. Am J Physiol Cell Physiol 300: C989–C997, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Weber CR, Ginsburg KS, Bers DM. Cardiac submembrane [Na+] transients sensed by Na+-Ca2+ exchange current. Circ Res 92: 950–952, 2003. [DOI] [PubMed] [Google Scholar]
- 284.Weber CR, Ginsburg KS, Philipson KD, Shannon TR, Bers DM. Allosteric regulation of Na/Ca exchange current by cytosolic Ca in intact cardiac myocytes. J Gen Physiol 117: 119–131, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Weber CR, Piacentino V 3rd, Ginsburg KS, Houser SR, Bers DM. Na+-Ca2+ exchange current and submembrane [Ca2+] during the cardiac action potential. Circ Res 90: 182–189, 2002. [DOI] [PubMed] [Google Scholar]
- 286.Weber CR, Piacentino V 3rd, Houser SR, Bers DM. Dynamic regulation of sodium/calcium exchange function in human heart failure. Circulation 108: 2224–2229, 2003. [DOI] [PubMed] [Google Scholar]
- 287.Wetzel GT, Chen F, Klitzner TS. Na+/Ca2+ exchange and cell contraction in isolated neonatal and adult rabbit cardiac myocytes. Am J Physiol 268: H1723–H1733, 1995. [DOI] [PubMed] [Google Scholar]
- 288.Wu M, Le HD, Wang M, Yurkov V, Omelchenko A, Hnatowich M, Nix J, Hryshko LV, Zheng L. Crystal structures of progressive Ca2+ binding states of the Ca2+ sensor Ca2+ binding domain 1 CBD1 from the CALX Na+/Ca2+ exchanger reveal incremental conformational transitions. J Biol Chem 285: 2554–2561, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Wu M, Tong S, Gonzalez J, Jayaraman V, Spudich JL, Zheng L. Structural basis of the Ca2+ inhibitory mechanism of Drosophila Na+/Ca2+ exchanger CALX and its modification by alternative splicing. Structure 19: 1509–1517, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Wu M, Tong S, Waltersperger S, Diederichs K, Wang M, Zheng L. Crystal structure of Ca2+/H+ antiporter protein YfkE reveals the mechanisms of Ca2+ efflux and its pH regulation. Proc Natl Acad Sci U S A 110: 11367–11372, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Xiao YF, Ke Q, Chen Y, Morgan JP, Leaf A. Inhibitory effect of n-3 fish oil fatty acids on cardiac Na+/Ca2+ exchange currents in HEK293t cells. Biochem Biophys Res Commun 321: 116–123, 2004. [DOI] [PubMed] [Google Scholar]
- 292.Xie Y, Ottolia M, John SA, Chen JN, Philipson KD. Conformational changes of a Ca2+-binding domain of the Na+/Ca2+ exchanger monitored by FRET in transgenic zebrafish heart. Am J Physiol Cell Physiol 295: C388–C393, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Yao A, Su Z, Nonaka A, Zubair I, Lu L, Philipson KD, Bridge JH, Barry WH. Effects of overexpression of the Na+-Ca2+ exchanger on [Ca2+]i transients in murine ventricular myocytes. Circ Res 82: 657–665, 1998. [DOI] [PubMed] [Google Scholar]
- 294.Yaradanakul A, Feng S, Shen C, Lariccia V, Lin MJ, Yang J, Kang TM, Dong P, Yin HL, Albanesi JP, Hilgemann DW. Dual control of cardiac Na+ Ca2+ exchange by PIP2: Electrophysiological analysis of direct and indirect mechanisms. J Physiol 582: 991–1010, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Yuan J, Yuan C, Xie M, Yu L, Bruschweiler-Li L, Bruschweiler R. The intracellular loop of the Na+/Ca2+ exchanger contains an “Awareness Ribbon”-shaped two-helix bundle domain. Biochemistry 57: 5096–5104, 2018. [DOI] [PubMed] [Google Scholar]
- 296.Yue X, Hazan A, Lotteau S, Zhang R, Torrente AG, Philipson KD, Ottolia M, Goldhaber JI. Na/Ca exchange in the atrium: Role in sinoatrial node pacemaking and excitation-contraction coupling. Cell Calcium 87: 102167, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Yue X, Zhang R, Kim B, Ma A, Philipson KD, Goldhaber JI. Heterogeneity of transverse-axial tubule system in mouse atria: Remodeling in atrial-specific Na+-Ca2+ exchanger knockout mice. J Mol Cell Cardiol 108: 50–60, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Zhang J, Lee MY, Cavalli M, Chen L, Berra-Romani R, Balke CW, Bianchi G, Ferrari P, Hamlyn JM, Iwamoto T, Lingrel JB, Matteson DR, Wier WG, Blaustein MP. Sodium pump alpha2 subunits control myogenic tone and blood pressure in mice. J Physiol 569: 243–256, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Zhang XQ, Ahlers BA, Tucker AL, Song J, Wang J, Moorman JR, Mounsey JP, Carl LL, Rothblum LI, Cheung JY. Phospholemman inhibition of the cardiac Na+/Ca2+ exchanger. Role of phosphorylation. J Biol Chem 281: 7784–7792, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Zhao J, Shigaki T, Mei H, Guo YQ, Cheng NH, Hirschi KD. Interaction between Arabidopsis Ca2+/H+ exchangers CAX1 and CAX3. J Biol Chem 284: 4605–4615, 2009. [DOI] [PubMed] [Google Scholar]