

Abstract
Salt stress significantly reduces the productivity of crop plants including maize (Zea mays). miRNAs are major regulators of plant growth and stress responses, but few studies have examined the potential impacts of miRNAs on salt stress responses in maize. Here, we show that ZmmiR169q is responsive to stress-induced ROS signals. After detecting that salt stress and exogenous H2O2 treatment reduced the accumulation of ZmmiR169q, stress assays with transgenic materials showed that depleting ZmmiR169q increased seedling salt tolerance whereas overexpressing ZmmiR169q decreased salt tolerance. Helping explain these observations, we found that ZmmiR169q repressed the transcript abundance of its target NUCLEAR FACTOR YA8 (ZmNF-YA8), and overexpression of ZmNF-YA8 in maize improved salt tolerance, specifically by transcriptionally activating the expression of the efficient antioxidant enzyme PEROXIDASE1. Our study reveals a direct functional link between salt stress and a miR169q-NF-YA8 regulatory module that plants use to manage ROS stress and strongly suggests that ZmNF-YA8 can be harnessed as a resource for developing salt-tolerant crop varieties.
A regulatory module consisting of miR169q and NUCLEAR FACTOR YA8 is associated with reactive oxygen species levels and mediates salt tolerance by regulating the antioxidant enzyme system in maize roots.
Introduction
Salinity is one of the most decisive environmental factors limiting plant growth and productivity, with especially pronounced limitations for crop production in arid and semi-arid regions. Global annual losses from salt-affected land presently exceed US$12 billion, and are rising (Qadir et al., 2008; Flowers et al., 2010). Some of the known factors contributing to increases in soil salinity include poor irrigation practices, improper application of fertilizers, and industrial pollution (Ouhibi et al., 2014). For plants, salt accumulation in soil solution causes reductions in water and nutrient uptake and also induces ionic stress, osmotic stress, and secondary stresses like oxidative stress (Ismail et al., 2014; Yang and Guo, 2018).
Oxidative stress signaling and detoxification mechanisms for reactive oxygen species (ROS) are both known to be essential components of salinity stress tolerance physiology in plants. ROS bursts resulting from abiotic stress can function as secondary signals for salinity response pathways (Zhu et al., 2020), yet excessive ROS cause serious damage to cells (Munne-Bosch et al., 2013). Relatively little is known about how plants sense ROS or about how ROS specifically trigger activation/inactivation of downstream oxidative-stress-related genetic and physiological responses.
MicroRNAs (miRNAs) are now understood to regulate gene expression for multiple developmental and signaling pathways in plants, and recent studies have shown that miRNAs function in response to environmental stresses in a miRNA-, stress-, tissue-, and genotype-dependent manner (Weiberg et al., 2014; Yu et al., 2017). Thus, miRNAs are now accepted as potential new targets for genetically improving plant tolerance to various stresses. miRNAs can induce gene silencing via mechanisms including guiding post-transcriptional gene silencing through mRNA degradation and via translational inhibition (Yu et al., 2017). Multiple miRNAs have been reported to be induced by salt stress in several different plant species, including miR156 (Ma et al., 2021), miR159 (Wang et al., 2013), miR165 (Jia et al., 2015), miR167 (Ye et al., 2020), miR168 (Qin et al., 2015), miR169 (Luan et al., 2014), miR319 (Zhou et al., 2013), miR393 (Denver and Ullah, 2019; Gao et al., 2011; Busch and Montgomery, 2015), miR395 (Kim et al., 2010b), miR396 (Gao et al., 2010), miR398 (Sunkar et al., 2006; Jagadeeswaran et al., 2009), miR399 (Ma et al., 2010), and miR402 (Sunkar and Zhu, 2004; Kim et al., 2010a). Notably, almost all of these salt-stress-induced miRNAs are evolutionarily conserved.
Foundational studies in this area established for example that overexpression of rice (Oryza sativa) miR319 enhanced salinity tolerance in creeping bentgrass (Agrostis stolonifera) (Zhou et al., 2013), and overexpressing OsmiR393 and osa-MIR396c decreased salt tolerance in both Arabidopsis thaliana and rice (Gao et al., 2010, 2011). Overexpression of miR395 affects plant tolerance to salinity and drought stress in Arabidopsis (Kim et al., 2010b). In Arabidopsis, salt stress induces accumulation of miR402, and overexpression of miR402 promotes the growth of plants under salt stress (Sunkar and Zhu, 2004; Kim et al., 2010a). Although there are now multiple examples of miRNAs functioning in plant responses to salt stress, the specific mechanisms through which these miRNA molecules exert their effects are typically not well understood.
Previous studies have shown that multiple members of the miR169 family respond differentially to salt stress in various plants: for example, miR169g accumulates in response to high salinity in rice (Zhao et al., 2009) but the level of miR169g is greatly reduced by high salinity in Arabidopsis seedlings (Pegler et al., 2019). We previously identified maize (Zea mays) miR169 family members that are differentially accumulated upon high salinity treatment. We found that 125 mM NaCl treatment caused a dramatic reduction in the level of one of these miRNAs, ZmmiR169q (Luan et al., 2014), and therefore hypothesized that this miRNA may somehow function in maize responses to salinity stress. Here, we show that ZmmiR169q functions as an ROS responder which can rapidly perceive salinity-induced ROS accumulation in maize roots. After finding that both salt stress and exogenous H2O2 treatment significantly reduces ZmmiR169q accumulation, stress assays with transgenic materials showed that depleting ZmmiR169q increases seedling salt tolerance, whereas overexpressing ZmmiR169q decreased salt tolerance, exhibiting for example substantial increases in biomass. At the molecular level, we also discovered that ZmmiR169q targets mRNA transcripts encoding the nuclear factor Y subunit A 8 (NF-YA8, Zm00001d022109; GRMZM2G038303), which was named as ZmNF-YA14 in our previous report (Luan et al., 2014), and named ZmNF-YA8 in a recent report (Liu et al., 2021). So we chose the gene name of Zm00001d022109 as ZmNF-YA8, for consistency. ZmNF-YA8 specifically binds to two CCAAT boxes present in the promoter of the maize peroxidase 1 gene (ZmPER1), thereby positively regulating ZmPER1 transcription and promoting ZmPER1-catalyzed antioxidative metabolism to scavenge excessive ROS. Thus, beyond characterizing ZmmiR169q as a ROS responder and revealing a miR169q-NF-YA8 regulatory module that plants use to manage ROS stress, our study clearly suggests that genetic manipulation of ZmNF-YA8 should facilitate the development of salt-tolerant crop varieties.
Results
Salinity stress reduces the accumulation of ZmmiR169q, which negatively regulates salt tolerance in maize
We previously conducted an analysis of miR169 family expression based on stem-loop RT-qPCR and found that ZmmiR169q in roots was significantly down-regulated upon salt treatment (Luan et al., 2014), so we conducted a GUS-staining assay of pmiR169q:GUS. The results showed that the expression of ZmmiR169q was reduced by 1 h high salinity treatment (200 mM NaCl), then dropped to the bottom at 2 h treatment, followed by a gradual increase after 4 h (Supplemental Figure S1) suggesting the possibility that ZmmiR169q responds to salt stress. We further confirmed the consistency of expression patterns of ZmmiR169q between B73 and B104 under short-term and long-term salt stress conditions (Supplemental Figure S2).
To investigate the role of ZmmiR169q in the salt responses, we generated ZmmiR169 knockdown transgenic maize plants with miRNA short tandem target mimicry (STTM, these plants were named MIM169; the two independent lines were MIM169-1 and MIM169-2) (Figure 1, a) and ZmmiR169q overexpression plants (two lines were named miR169q OE2 and miR169q OE10) (Figure 1, b). Confirming the success of our transgenic approaches, we found that the miRNA abundance of ZmmiR169q was decreased in MIM169 plants and increased in miR169q OE plants (Figure 1, c and e). We then grew the transgenic lines and their corresponding wild-type (WT) plants under normal or salt stress (200 mM NaCl) conditions. Under control growth conditions, no visible differences were found between WT and MIM169 plants or between WT and miR169q OE plants (Figure 1, g and h). In contrast, the growth of MIM169 transgenic maize plants was faster than WT plants under salt stress conditions for 15 d (Figure 1, g), and the survival rates of the MIM169-1 and MIM169-2 lines were 40% and 47% greater than that of WT plants when treated with 200 mM NaCl for 40 d (Figure 1, d). Again, consistent with a potential role for ZmmiR169q in salt stress responses in maize, we found that miR169q OE2 and miR169q OE10 plants were smaller, exhibited more severe damage under salt stress conditions for 12 d (Figure 1, h), and had significantly reduced survival rates compared with WT plants under salt stress for 28 d (Figure 1, f). We also assessed the dry weight of these plants: the dry weight of MIM169 plants was similar to B73 plants (Figure 1, i). And there was no significant difference in dry weight between miR169q OE and B104 plants (Figure 1, k). After NaCl treatment, the dry weight of MIM169 plants was significantly higher than that of B73 plants (Figure 1, j). Consistently, the dry weight of miR169q OE plants was reduced significantly compared with B104 plants (Figure 1, l). Collectively, these results indicated that ZmmiR169q functions as a negative regulator of salt tolerance.
Figure 1.

ZmmiR169q negatively regulates salt tolerance in maize. a, A diagram of the ZmmiR169 STTM construct (MIM169). b, A diagram of the ZmmiR169q overexpression construct (miR169q OE). PUBI represents the Ubiquitin promoter and TNOS represents the NOS terminator. c and e, The expression levels of ZmmiR169q in MIM169 roots (c) and miR169q OE roots (e). d and g, MIM169 transgenic lines (MIM169-1 and MIM169-2) exhibit salt tolerance, survival was assayed by treatment in 200-mM NaCl for 40 d (d). f and h, ZmmiR169q overexpression transgenic maize lines (miR169q OE2 and miR169q OE10) showed increased sensitivity to salt, survival was assayed by treatment in 200 mM NaCl for 28 d (f). g, Image of seedling leaves after treatment with 200 mM NaCl for 15 d. h, 20-dy-old miR169q OE2, miR169q OE10, and WT plants grown under 200 mM NaCl conditions for 12 d. Similar results were observed for three independent experiments; B73 and B104 roots were examined as controls. The expression levels of ZmmiR169q were normalized to the level of U6. Values are means ± sd, n = 3. Scale bars = 5cm. i and j, Dry weight of B73 and MIM169 seedlings under normal and salt stress conditions. k and l, Dry weight of B104 and miR169q OE seedlings under normal and salt stress conditions. All asterisks denote a statistically significant difference from the WT, Student’s t test, **P < 0.01.
ZmmiR169q rapidly responds to stress-induced ROS
To investigate gene network(s) through which ZmmiR169q may impact the maize root, we harvested WT and miR169q OE2 roots at 21 d after germination and performed RNA-sequencing. More than 72% of the raw reads from each sample were successfully mapped to annotated gene-coding regions of the maize genome (Supplemental Table S1), and a total of 484 significantly differentially up-regulated and 851 down-regulated genes were detected in miR169q OE compared with WT roots (Figure 2, a). Notably, the term “tetrapyrrole binding” was the most strongly enriched term in the GO analysis of the differentially expressed genes (DEGs) (Figure 2, b); tetrapyrroles are widely understood as mediators of oxidative stress responses and the mitigation of ROS accumulation as cofactors in the antioxidant systems (Busch and Montgomery, 2015). There were also some DEGs, including many encoding peroxidases (POD), with functional annotations potentially related to oxidative stress metabolism and physiology (Figure 2, c). Thus, our transcriptome analysis supports our initial physiological findings from the salt stress growth assessment and again indicates the potential involvement of ZmmiR169q in salinity induced-oxidative stress. Specifically, we speculate that ZmmiR169q may rapidly sense stress-induced ROS and then trigger a cascade that reduces the ROS level. Pursuing this hypothesis, we used hydrogen peroxide (H2O2) treatments to examine how the expression of ZmmiR169q is affected by excess ROS stress in maize WT plants. Reverse transcription quantitative PCR (RT-qPCR) showed that ZmmiR169q expression decreased sharply at 1 h post exposure to 1 mM H2O2, bottomed at 2 h, and then increased after 4 h (Figure 2, d). In addition, the expression patterns of ZmmiR169q in both B73 and B104 were the same under H2O2 treatment (Supplemental Figure S3). To determine whether reduced ZmmiR169q accumulation was caused by downregulation of transcription of the ZmmiR169q precursor, we investigated the expression level of the ZmmiR169q precursor (pre-miR169q) in maize roots by RT-qPCR upon ROS exposure. Our results showed that the level of pre-miR169q and ZmmiR169q had similar patterns under 1 mM H2O2 treatment (Figure 2, d). Further GUS assays with transgenic maize plants expressing β-glucuronidase under the control of the ZmmiR169q native promoter (pmiR169q:GUS) indicated that exposure to 1 mM H2O2 resulted in decreased transcription from the ZmmiR169q promoter (Figure 2, e). In addition, we used the ROS-scavenger GSH to determine how ROS impact the expression levels of ZmmiR169q. Exogenous application of GSH obviously repressed ROS accumulation (Figure 2, f) and neutralized the expression reduction of ZmmiR169q under 1 mM H2O2 treatment (Figure 2, g), suggesting that ROS directly affect the expression of ZmmiR169q. Collectively, these results support that ZmmiR169q expression levels impact multiple pathways of oxidative metabolism in cells and show that increased oxidative stress rapidly reduces both the transcription and overall accumulation of ZmmiR169q in maize roots.
Figure 2.

RNA-seq of WT and miR169q OE roots implicate ZmmiR169q in salinity induced-oxidative stress. a, Genes differentially expressed in the miR169q OE transgenic (seedling roots) line compared with WT. A total of 484 up-regulated and 851 down-regulated genes were detected in miR169q OE roots compared with WT. b, Top 10 terms from a gene ontology (GO) enrichment analysis of the DEGs of the molecular function GO category: the GO term “tetrapyrrole binding” was the most strongly enriched. c, Heatmap illustrating the DEGs detected between miR169q OE and WT plants with functional annotations related to oxidative stress and antioxidant activity. d, ZmmiR169q and pre-miR169q expression patterns in WT roots under 1-mM H2O2 exposure were assessed by RT-qPCR. ZmmiR169q or pre-miR169q expression was normalized the U6 level. Values are means ± sd, n = 3. e, Histochemical staining of pmiR169q:GUS transgenic plants after 1-mM H2O2 treatment. Bars = 1 cm. Both the RT-qPCR and histochemical staining showed that the expression of ZmmiR169q decreased sharply at 1 h, bottomed at 2 h, and then increased after 4 h when maize seedlings were exposed to 1 mM H2O2. f, NBT and DAB staining of the root tip of miR169q OE after 1-mM H2O2 treatment or 1-mM H2O2 supplemented with 100 μM GSH. Bars = 2 mm. g, ZmmiR169q expression level in the roots of WT after 1 mM H2O2 treatment for 1 h or 1 mM H2O2 supplemented with 100 μM GSH for 1 h was assessed by RT-qPCR. ZmmiR169q expression was normalized the U6 level. Values are means ± sd, n = 3. All asterisks denote a statistically significant difference from the control, Student’s t test, **P < 0.01, *P < 0.05.
ZmmiR169q exerts its functions through its downstream target, Nuclear factor Y, Subunit A8
NF-Ys are heterotrimeric transcription factors comprising NF-YA, NF-YB, and NF-YC subunits. Functional NF-Y heterotrimers can regulate gene transcription by binding to CCAAT box regulatory elements present in the promoters of eukaryotic genes (Gnesutta et al., 2019). In plants, NF-Y subunits are encoded by multigene families whose members show structural and functional diversification, with demonstrated functions in the development of primary and lateral roots, in root nodule and arbuscular mycorrhizal symbioses, and in plant responses to biotic and abiotic stresses (Sorin et al., 2014). Our previous study employed RNA ligase-mediated RACE to show that ZmmiR169q targets seven NF-YA genes in maize (ZmNF-YA1, ZmNF-YA4, ZmNF-YA6, ZmNF-YA7, ZmNF-YA8/14, ZmNF-YA11, and ZmNF-YA13) (Luan et al., 2014). Here, we confirmed that ZmmiR169q can regulate the ZmNF-YA8 gene which has a ZmmiR169q target site by transient assay (Supplemental Figure S4). And our RNA-seq experiment showed that ZmNF-YA8 expression was significantly lower in miR169q OE2 roots than in WT roots. Further, RT-qPCR analysis showed that ZmNF-YA8 (Zm00001d022109) expression was significantly increased in MIM169q roots compared with the WT (Figure 3, a) and was dramatically decreased in roots of miR169q OE plants (Figure 3, b). These findings confirm our previous characterization of ZmNF-YA8 as a target of ZmmiR169q. Moreover, we investigated the tissue/cell specificity of ZmmiR169q and ZmNF-YA8 expression patterns and observed strong GUS activity in pmiR169:GUS transgenic plants and pNF-YA8:GUS transgenic plants in the root cortex and endodermis tissues of the mature zone (Figure 3, c). These findings suggest that the observed impacts of differential ZmmiR169q accumulation upon salt and oxidative stress in maize may be mediated via ZmNF-YA8, potentially via ZmmiR169q-mediated disruption of ZmNF-YA8 mRNA.
Figure 3.

ZmmiR169q regulates salt tolerance by repressing transcription of its downstream target ZmNF-YA8. a and b, ZmNF-YA8 transcript levels in MIM169 roots (a) and miR169q OE roots (b); WT roots were used for a control and ZmNF-YA8 expression was normalized to Actin1. Values are means ± sd, n = 3. Asterisks denote a statistically significant difference from the control, Student’s t test, **P < 0.01. ZmNF-YA8 expression was significantly increased in MIM169 than in WT roots, while ZmNF-YA8 expression was significantly reduced in miR169q OE roots. c, β-Glucuronidase staining of pmiR169q:GUS and pNF-YA8:GUS root tissues to examine tissue/cell specificity of ZmmiR169q and ZmNF-YA8 expression. Shown are cross-sections of unstained root tips (left) and the stained root tips (right) of the primary root. Similar results were observed for three independent experiments. Black scale bar, 50 µm. The results showed that both the GUS activity in pmiR169:GUS transgenic plants and pNF-YA8:GUS transgenic plants was predominantly evident in the root cortex and endodermis tissues of the mature root zone. d, ZmNF-YA8 expression in NF-YA8 OE lines. Values are expressed as means ± sd, n = 3, normalized to Actin1. e, The survival rate was determined after treatment in 200 mM NaCl for 40 d. Data are expressed as means ± sd. Similar results were observed for three independent experiments. f, NF-YA8 OE6 and NF-YA8 OE8 lines showed salt tolerance. White scale bars = 5 cm. g and h, Dry weight in NF-YA8 OE lines, measured under normal conditions and after treatment with 200 mM NaCl for 20 d. Values are means ± sd, n = 3. All asterisks denote a statistically significant difference from the control, Student’s t test, **P < 0.01.
To characterize any effects of ZmNF-YA8 on salt tolerance in maize, we generated ZmNF-YA8 overexpression transgenic lines (NF-YA8 OE) and investigated ZmNF-YA8 expression levels in the OE lines (Figure 3, d). Recalling our findings about ZmmiR169q as an apparent negative regulator of salt tolerance, we conducted salt stress assays with the NF-YA8 OE plants. No obvious differences in overall growth were detected between NF-YA8 OE and B104 plants under normal conditions (Figure 3, f and g); in contrast, the NF-YA8 OE lines showed significant increases in their survival rates and dry weight compared with B104 plants under salt stress (Figure 3, e, f, and h). The survival rates of NF-YA8 OE plants were approximately 45% and 24% higher than those of WT plants (12%), respectively (Figure 3, e). These results demonstrate that ZmNF-YA8 functions as a positive regulator to increase maize salt tolerance.
A ZmmiR169q/ZmNF-YA8 module regulates ROS accumulation
As salt stress can drive accumulation of ROS in plants, we next investigated ROS levels in the leaves and roots of different maize lines under normal conditions (0 mM NaCl) or 200 mM NaCl treatment (Figure 4, a–h) by monitoring the accumulation of superoxide (O2−; nitroblue tetrazolium staining) and H2O2 (3,3′-diaminobenzidine staining). As expected, the WT plants exhibited increased accumulation of both O2− and H2O2 upon exposure to the salt stress condition. Under both normal and salt stress conditions, when compared with the WT, miR169q OE transgenic roots had increased O2− and H2O2 levels whereas NF-YA8 OE transgenic roots had reduced levels (Figure 4, a–h). To determine whether the observed increases in ROS levels correspond with increased cell injury, we evaluated the malondialdehyde (MDA) content in the different maize lines upon salt stress. Upon salt stress, the miR169q OE transgenic lines (with higher ROS levels) accumulated more MDA than WT plants, whereas the NF-YA8 OE transgenic plants (lower ROS accumulation) accumulated less MDA than WT plans (Figure 4, i).
Figure 4.

A Zma-miR169q/ZmNF-YA8 module regulates ROS levels in maize. a and b, NBT staining of seedling leaves under normal or salt stress (200 mM NaCl) growth conditions. c and d, DAB staining of seedling leaves. e and f, NBT staining of seedling whole roots. g and h, DAB staining of seedling whole roots. The single-root image is partial enlarged view of root in box. i, MDA content in WT, miR169q OE, and NF-YA8 OE plants exposed to 200 mM NaCl for 1 h. j–l, SOD, POD, and CAT enzyme activities in WT, miR169q OE, and NF-YA8 OE plants exposed to 200 mM NaCl for 1 h. At least five leaves from six individual plants per line were used for each experiment. The data are presented as the mean ± sd of three independent experiments. Asterisks indicate significant difference compared with WT (Student’s t test, **P < 0.01, *P < 0.05).
Antioxidant enzymes function in ROS detoxification, contributing to ROS scavenging under abiotic stresses (Miller et al., 2010; Huang et al., 2013; Zhang et al., 2016). We then measured the activities of antioxidant enzymes including SOD, CAT, and POD in different maize lines after treatment with 200 mM NaCl for 96 h. The activities of these three enzymes in the NF-YA8 OE transgenic plants were significantly higher than those in the WT plants (Figure 4, j–l). In contrast, the activities of these three enzymes in the miR169q OE plants were significantly reduced (Figure 4, j–l). These results demonstrate that manipulation of ZmNF-YA8 levels—including via miR169q-mediated reduction and direct overexpression of ZmNF-YA8—affects plant salt tolerance by altering MDA levels in cells and by regulating the overall capacity for ROS scavenger enzyme activity. Thus, the miR169q-ZmNF-YA8 regulatory module that we have characterized is likely to interact with downstream target genes that function in these cellular defense processes.
ZmNF-YA8 activates transcription of the maize peroxidase 1 gene ZmPER1
To investigate ZmNF-YA8’s downstream gene regulatory network, roots of WT and NF-YA8 OE plants were harvested at post-germination day 21 for RNA-seq-based transcriptomics analysis. More than 82% of the raw reads from each sample were mapped to the annotated gene-coding regions (Supplemental Table S2), and a total of 948 up-regulated significantly DEGs and 1,371 down-regulated DEGs were detected in our comparison of NF-YA8 OE and WT plants (Supplemental Figure S5, a). We detected enrichment for GO terms associated with tetrapyrrole binding, oxidoreductase activity, catalytic activity, and antioxidant activity for the DEGs in NF-YA8 OE plants (Supplemental Figure S5, b), among which there were 34 genes predicted to encode POD enzymes (Supplemental Figure S5, c). We also analyzed DEGs in comparisons including WT versus miR169q OE and WT versus NF-YA8 OE. Relative to WT, we found that Zm00001d018618 was among the significantly down-regulated DEGs in the miR169q OE line but among the up-regulated DEGs in the NF-YA8 OE line (Figure 5, a and Supplemental Table S2). Zm00001d018618 is predicted to encode a POD 1 protein named ZmPER1. Orthologs of ZmPER1 have been identified in many plant species (Supplemental Figure S6, a), but their function(s) remain unknown. Analysis of an expression profile for ZmPER1 obtained from gene expression atlas for maize development (Walley et al., 2016) indicated that ZmPER1 is prominently expressed in roots (Supplemental Figure S6, b). The RT-qPCR of ZmPER1 confirmed our findings from the RNA-seq analysis: the abundance of ZmPER1 increases upon ZmNF-YA8 mRNA accumulation (Figure 5, b), supporting the hypothesis that ZmPER1 transcription may be positively regulated by ZmNF-YA8. We further examined the expression levels of ZmNF-YA8 and ZmPER1 in the roots of WT B104 under 200 mM NaCl treatment. These two genes were up-regulated at 2 h, then slowly down-regulated at 48 h of treatment, followed by a gradual increase till 11 d after treatment (Supplemental Figure S7). These results suggest that ZmNF-YA8 and ZmPER1 are salt responsive, and their kinetics fit with that of ZmmiR169q (Supplemental Figure S2, a).
Figure 5.

ZmNF-YA8 directly binds to the ZmPER1 promoter. a, Venn diagram exhibiting one downregulated DEG in miR169q OE overlapped with the upregulated DEG in NF-YA8 OE line. b, RT-qPCR validation of RNA-seq analysis for ZmPER1 transcripts. Black bars indicate log2 (fold change) calculated from RT-qPCR analysis, the expression level of ZmNF-YA8 was normalized to that of Actin1; gray bars indicate fold change calculated from RNA-seq analysis. c, Schematic diagrams of the promoters of ZmPER1, in which the CCAAT-box elements are denoted using orange rectangles. The segments marked with C2, C4, and C5 represent the promoter fragments used in the Y1H assay. ChIP-F and ChIP-R mean primers used in ChIP–qPCR assay. The segments marked with P1, P2, P3, and P4 represent the truncated promoter fragments containing different CCAAT-boxes used in the dual-LUC transient expression assays. P1 comprises three CCAAT-boxes; P2 comprises only the C5 CCAAT-box; P3 comprises the C2 and C4 CCAAT-boxes; and P4 comprises only the C2 CCAAT-box. d, Growth of yeast cells of co-transformants (bait + prey) on SD/-Leu/-Ura medium supplemented without (left panels) or with (right panels) AbA. Y1H assay showing direct binding of ZmNF-YA8 to the CCAAT-box in the ZmPER1 promoter. C2m, C4m, and C5m means the C2, C4, and C5 CCAAT box was mutated, respectively. e, ChIP assay of ZmPER1 in 3-week-old NF-YA8-flag OE seedlings grown under 200 mM NaCl. Values given are mean ± sd (n = 3). **P < 0.01 by the Student’s t test. f, Schematic diagram showing the effectors and reporter used in the transient transcriptional activity assays, REN means Renilla LUC, LUC means firefly LUC. g, ZmNF-YA8 activates ZmPER1 expression. The data are presented as the mean ± sd (n = 3), Student’s t test, **P < 0.01.
ZmNF-YA8 directly binds to CCAAT-boxes in the ZmPER1 promoter
We analyzed the cis-elements in the predicted promoter region (∼2 kb upstream of the translation initiation site) of the ZmPER1 locus seeking to identify potential binding sites of ZmNF-YA8, operating under the assumption that this protein may target similar sequences to previously characterized members of this transcription factor family (Xu et al., 2014). Interestingly, three typical CCAAT-boxes were found at –896 nt, –416 nt, and –354 nt from the ATG site in the sense strand of the ZmPER1 promoter, named as C2, C4, and C5, respectively (Figure 5, c). To test if these three motifs were functional for ZmNF-YA8 binding, we conducted a yeast one-hybrid (Y1H) assay: the full-length CDS of ZmNF-YA8 was fused with GAL4AD to generate the prey vector, while bait vectors were constructed using six fragments containing the three tandem CCAAT boxes (C2, C4, and C5) and three tandem mutant CCAAT boxes (C2m, C4m, and C5m), respectively. The Y1H assays showed that the ZmNF-YA8 fusion protein specifically interacts with C4 and C5, the two CCAAT-boxes closest to the transcription start site (TSS) of ZmPER1. Mutations of the cis-element sequence in the C4 and C5 fragments abolished ZmNF-YA8 binding, suggesting that this interaction is authentic (Figure 5, d). To further confirm this binding, we conducted a chromatin immunoprecipitation (ChIP)-qPCR assay using the NF-YA8-flag OE transgenic plants, which revealed significant enrichment for occupancy of the NF-YA8-flag fusion protein at the ZmPER1 promoter region containing the CCAAT boxes (Figure 5, e).
Next, we examined whether ZmNF-YA8 could directly regulate the transcription of the target ZmPER1 using a dual-LUC reporter assay in maize protoplasts, in which the coding sequence of ZmNF-YA8 (driven by the 35S promoter) was used as the effectors and the luciferase (LUC) gene driven by different truncation fragments from the ZmPER1 promoter were used as reporters (Figure 5, f). We found that the P1-PER1::LUC, P2-PER1::LUC, and P3-PER1::LUC reporters were activated in maize protoplasts in the absence of ZmNF-YA8 (Figure 5, g). In addition, such an enhancement is dependent on the singleness of the C5 box (Figure 5, g), indicating that the presence of a single CCAAT box (at -354 nt from the TSS, C5) was sufficient for ZmNF-YA8 binding. We thus concluded that ZmNF-YA8 directly binds to the C5 CCAAT box in the ZmPER1 promoter and enhances its expression.
Discussion
MiRNAs function extensively in plant responses to biotic and abiotic stresses, as well as in mediating cross-talk among these stress pathways (Weiberg et al., 2014; Cai et al., 2018). The miR169 family was the first miRNA family characterized in the plant kingdom, and miR169 family members are known to regulate biotic stress and abiotic stress-associated molecular pattern-triggered responses in Arabidopsis (Li et al., 2008b; Xu et al., 2014; Hanemian et al., 2016; Serivichyaswat et al., 2017). How miR169 members regulate salt tolerance is largely unknown in maize. Because the root system is the first organ to undergo salt stress, we focused on the functions of ZmmiR169 mainly in roots. In the present study, based on transcriptomics profiling and subsequent functional analyses, we revealed a full loop—from salt-induced ROS accumulation to ROS scavenging—that is mediated by the ZmmiR169q/NF-YA8 module (Figure 6). Salt stress induces ROS accumulation, which in turn decreases ZmmiR169q levels in maize roots.
Figure 6.
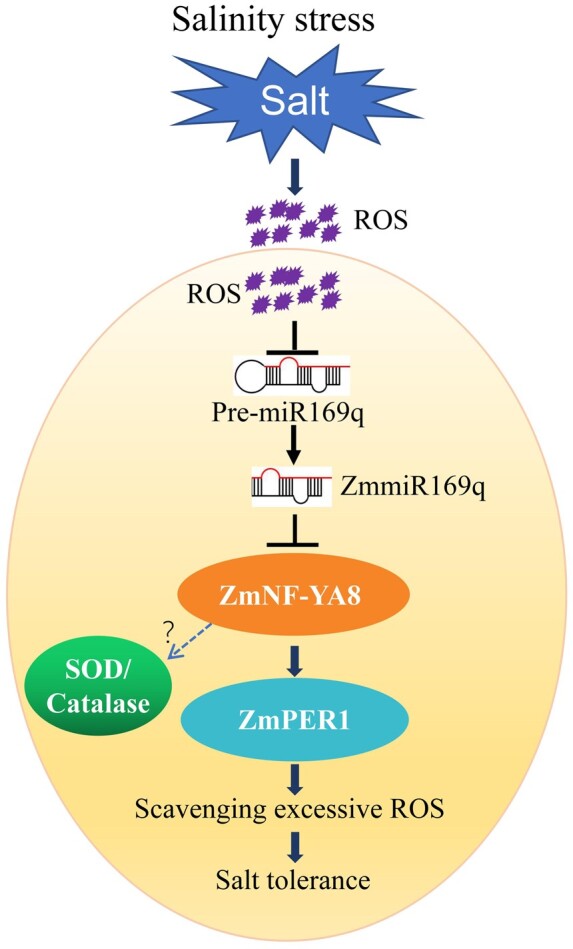
A proposed model of ZmmiR169q-mediated plant salt tolerance. Without salinity stress, ZmmiR169q regulated the expression of ZmNF-YA8 to maintain the ROS levels for normal metabolism in maize cells. When the plants were exposed to salinity stress, accumulated ROS suppressed the transcription of pre-miR169q, and the repressed ZmmiR169q accumulation elevated the expression of ZmNF-YA8. The up-regulated ZmNF-YA8 promoted the expression level of ZmPERs, resulting in higher POD enzyme activity. Besides activated ZmPERs transcriptional programs, NF-YA8 also could target SOD/CAT systems to modulate cellular redox homeostasis, forming a complex network between ROS and miRNA/target regulation. As a result, the enhanced total enzyme activity contributed to scavenge the excessive salinity-induced ROS in plant cells, which protected against cell membrane injury, thus response to salinity stress. The size of each round indicates the expression level. Arrows mean positive regulation and blunt-ended bars indicate inhibition.
Based on our observations of increased tolerance in MIM169 plants and increased susceptibility in miR169q OE plants, we conclude that ZmmiR169q is a negative regulator in response to salt stress. ZmNF-YA8 transcripts are targeted by ZmmiR169q in maize roots: salt stress causes significant increases in ZmNF-YA8 transcript abundance, and overexpression of ZmNF-YA8 conferred salt stress tolerance, yielding a phenotype similar to that of miR169 mimicry maize. The ZmNF-YA8 protein transcriptionally activates the expression level of the POD gene ZmPER1, and this antioxidant enzyme represents the first line of defense against salt stress-induced ROS accumulation, there are also other antioxidant enzymes that work together with ZmPER1 (Supplemental Figure S5, c). Our work thus supports the following model for the situation in WT plants: under normal conditions, the presence of ZmmiR169q functions in repressing ZmNF-YA8-mediated responses to excessive ROS levels. When the ROS levels in cells are elevated during salt stress, the accumulation of ZmmiR169q decreases sharply, which de-represses translation of ZmNF-YA8 transcripts, thereby enabling promoting ZmNF-YA8’s binding and transcriptional activation of the highly efficient antioxidant enzyme ZmPER1 or other ZmPERs (Figure 6). Thus, our study suggests that manipulating ZmNF-YA8 can be understood as a promising strategy for developing salt-tolerant crop varieties.
To date, NF-YA-encoding genes are the only demonstrated targets for miR169 in plants (Rhoades et al., 2002). NF‐YA recognizes the CCAAT motif frequently observed in eukaryotic promoters, and this motif has been repeatedly implicated in developmental and stress-responsive processes in plants (Petroni et al., 2012; Laloum et al., 2013). An AtmiR169/NF-YA5 module was shown to regulate drought stress response in Arabidopsis (Li et al., 2008). The bZIP28 is mobilized to nucleus by proteolysis and recruits NF-Y subunits to form a transcriptional complex that upregulates the transcripts of endoplasmic reticulum (ER) stress-induced genes in the response to ER stress (Liu and Howell, 2010a, 2010b). Our work demonstrates a miR169q-NF-YA8 regulatory module that plants use to manage ROS under salt stress, which expands our understanding of miR169-mediated regulation of NF-YA impacts on plant stress responses.
A study of cotton reported that plants exposed to salinity stress exhibited elevated ROS production whereas reduced accumulation of ghr-miR414c (Wang et al., 2019). In Arabidopsis, miR398 expression is transcriptionally downregulated by oxidative stress, and this downregulation affects the accumulation of CSD1 and CSD2 mRNA and then alters oxidative stress tolerance (Sunkar et al., 2006; Jagadeeswaran et al., 2009). Studies in animals have functionally linked oxidative stress signaling and the induction of miRNAs (Kerrigan, 1990; Sorescu and Griendling, 2002; Lin et al., 2009). Emerging evidence has implied that ROS can directly exert profound effects on miRNA transcription and miRNAs may regulate the expression of redox sensors and other ROS modulators such as ROS scavenger, then turnover to regulate cell fate, but these reports specifically concern tumorigenesis (Lan et al., 2018; Ebrahimi et al., 2020). However, the roles of miRNAs in sensing stress-induced ROS and the nature of the interplay between ROS and miRNAs in plants remain largely unknown, here we found increased ROS levels resulting from salt stress cause the down-regulation of ZmmiR169q, our work illuminates an excellent experimental model—maize roots and the miR169q-NF-YA8 regulatory module—for elucidating these biochemical (and/or chemical) mechanisms. So our discovery of a ROS effector in roots that exerts downstream effects on salt tolerance at the whole-plant level represents a substantial deepening in our understanding ROS–miRNA processes in plants. Moreover, the agronomically attractive phenotypes we observed for NF-YA8 OE and MIM169 plants clearly support the idea that traditional, transgenic, or genome-editing-based genetic manipulations of the ZmNF-YA8 gene or ZmmiR169 should enable the development of excellent yielding yet salt-tolerance crop varieties.
Materials and methods
Plant materials
Maize (Z. mays L.) varieties used in this work included B73, B104, and Hi (II).
Plant growth and salt treatments
Root samples were prepared using a paper roll culturing technique. Kernels were surface-sterilized with 6% (v/v) sodium hypochlorite, placed on a double layer of brown germination papers (Anchor Paper Company, Cat SD3836S), and wrapped into rolls. Rolls were placed in 2-L beakers with 400 mL of half-strength Hoagland nutrient solution and held vertically for 3 weeks in a plant growth chamber under the following conditions: 28°C/20°C day/night temperature; 16-/8-h light/dark photoperiod; 3,300 lux light intensity; and a relative humidity of 70%. The culture solution was replaced every 3 d until the V1 stage, then 200 mM NaCl solutions were added to the cultures. To analyze abundance of pre-miR169q and mature miR169q, the treated roots of miR169q OE seedlings were harvested at 0, 1, 2, and 4 h. To analyze GUS activities, the roots of treated pmiR169q:GUS transgenic plants were harvested at 0, 1, 2, and 4 h. For determining ROS levels, the roots of B104 and homozygous lines of miR169q OE, NF-YA8 OE were harvested at 0 and 1 h. For determining MDA contents and ROS scavenging enzyme activities, the roots of B104 and homozygous lines of miR169q OE, NF-YA8 OE were harvested at 0 and 96 h. Treated roots were harvested at 0 h for transcript analysis and all samples were frozen immediately in liquid nitrogen and stored at −80°C until analysis. For survival analysis, maize plants were cultivated in pots (diameter of 8.5 cm) filled with uniformly mixed substrate (www.pindstrup.com). Ten plants were planted in each pot, and grown in a greenhouse at 26°C, with a 16-h/8-h light/dark photoperiod. Three-week-old seedlings were treated with 200 mM NaCl or water (control) for 35 d, and then the surviving plants were counted. The dry weights of the aerial parts of plants were also measured after treatment with 200 mM NaCl for 20 d.
H2O2 treatment
Maize roots for the H2O2 sensitivity assay of plants were grown based on the paper roll culture method described above. Three-week-old inbred line B104 and pmiR169q:GUS transgenic seedlings were treated with 1 mM H2O2. Roots of B104 were harvested at the given time points (0, 1, 2, and 4 h) after treatment and frozen in liquid nitrogen for total RNA extraction. Then to detect abundance of pre-miR169q and mature miR169q by RT-qPCR and stem-loop qPCR. Roots of pmiR169q:GUS transgenic lines were harvested at the given time points (0, 1, 2, and 4 h) to detect GUS activities.
Constructs for genetic transformation
To construct pmiR169q:GUS and pNF-YA8:GUS, a region of approximately 2.0 kb of DNA upstream of ZmmiR169q and ZmNF-YA8 was amplified from the genomic DNA of B73 using the primers P-miR169q-F and P-miR169q-R, P-YA8-F, and P-YA8-R, respectively (Supplemental Table S2). Two fragments were separately cloned into the binary vector pCAMBIA3301 to generate the pmiR169q:GUS construct and pNF-YA8:GUS construct. To prepare the ZmmiR169 target mimic construct (MIM169), a 21-nucleotide motif complementary to the miR165/166 in STTM165/166-48 (Tang et al., 2012) was replaced with a sequence complementary to ZmmiR169. The STTM165/166-48 fragment with the miR169q-complementary motif was inserted into the BamHI and BstEII sites downstream of the Ubi promoter in the pCAMBIA3301-Ubi vector (constructed by our laboratory). pmiR169q:GUS, pNF-YA8:GUS and MIM169 constructs were then introduced into Agrobacterium tumefaciens strain EHA105, and then transformed into the immature embryos of HiII to regenerate seedlings, respectively. MIM169/HiII transgenic lines were continuously backcrossed to B73, and the homozygous BC6F3 transgenic maize plants were used in experiments.
To generate the ZmmiR169q-overexpression construct (miR169q OE), the genomic DNA sequence surrounding the pre-miR169q was amplified using PCR. The amplicon was sequenced and subcloned into the BamHI and BstEII sites downstream of the Ubi promoter in the pCAMBIA3301-Ubi vector (constructed by our laboratory). For the ZmNF-YA8-flag-overexpressing construct (NF-YA8-flag OE), full-length 3× flag DNA sequence was amplified with PCR and then inserted between BstBI and BstEII restriction sites of the pCAMBIA3301-Ubi vector (pCAMBIA3301-Ubi-flag). The maize ZmNF-YA8 CDS sequence of B73 was then amplified using PCR with deletion of the translation termination codon (TAG) and an oligonucleotide sequence containing a BstBI site. The fragment was fused and cloned into the pCAMBIA3301-Ubi-flag vector digested by BamHI and BstBI using the ClonExpress Entry One Step Cloning Kit (C114-01, Vazyme). In addition, we detected the ZmNF-YA8 3′-UTR sequence of B73 and B104 inbred lines using PCR-sequencing, the results showed that the ZmmiR169q target site was conserved between B73 and B104 (Supplemental Figure S8). The two OE recombinant vectors were transformed into A. tumefaciens strain EHA105 and were then transformed into B104. The homozygous T4 transgenic maize plants were used in subsequent experiments. Details of the primers used for constructing vectors are listed in Supplemental Table S2.
GUS assay
Histochemical staining of transgenic GUS plants was conducted as follows: the whole roots of 3-week-old transgenic plants were vacuum infiltrated in staining solution containing 1 mM X-Gluc (5-bromo-4-chloro-3-indolyl-β-glucuronide; Clontech), 1% N,N-dimethylformamide, 0.5 mM potassium ferricyanide, 0.5 mM potassium ferrocyanide, 0.1% Triton X-100 and 50 mM potassium phosphate buffer (pH 7.0) for 30 min, and incubated overnight at 37°C in the dark. Afterward, the roots were decolorized and fixed in 70% (v/v) ethanol and photographed using a scanner (Microtek, ZhongJing Limited Company, Shanghai). Sections of 500 microns were cut using a Leica microtome VT1000S and photographed using an Invitrogen EVOS XL microscope. The GUS activity assay was carried out as described previously (Jefferson et al., 1987).
RNA isolation and RT-qPCR analysis
Total RNA (including miRNA) was extracted using the miRcute miRNA kit (Tiangen, Beijing, China). To analyze pre-miR169q and ZmmiR169q expression, miRNA first-strand cDNA was synthesized using the miRcute miRNA First-Strand cDNA Synthesis kit (Tiangen). The expression of pre-miR169q and ZmmiR169q was then quantified by RT-qPCR using the SYBR PrimeScript miRNA RT-PCR kit (Tiangen) with U6 as the internal control, respectively. To analyze target gene expression of ZmmiR169q, ZmNF-YA8, and other downstream genes, approximately 2 μg total RNA for each sample was reverse transcribed using the FastQuant Super Mix kit (Tiangen). The resulting cDNA was used as the template for a qPCR assay, which was completed with the SYBR Green SuperReal PreMix Plus kit (Tiangen). The actin1 gene (GRMZM2G126010) was used for normalization. The qPCR analysis involved three biological replicates for each miRNA and gene. The PCR program consisted of an initial denaturation step at 95°C for 5 min, followed by 40 cycles of 95°C for 15 s, 60°C for 20 s, and 72°C for 20 s. Relative expression levels were calculated according to the delta–delta threshold cycle (2−ΔΔCT) relative quantification method. All qPCR reactions were performed in an ABI 7500 real-time instrument. Values shown are means ± sd of three biological replicates. In some cases, means were compared with Student’s t tests. Details regarding the qPCR primers are listed in Supplemental Table S2.
Transcriptome sequencing and informatics analyses
Roots of WT, WT treated with 200 mM NaCl, miR169q OE and NF-YA8 OE from 3-week-old seedlings grown in paper rolls in a plant growth chamber (three biological replicates) were harvested and flash-frozen in liquid nitrogen. Total RNA from the materials was isolated as described above. RNA-seq libraries were constructed using the QiaQuick PCR extraction kit, end repaired, poly (A) added, and ligated to Illumina sequencing adapters. Libraries were sequenced on an Illumina HiSeq2500 instrument by Gene Denovo Co. (Guangzhou, China).
Raw RNA-seq libraries were processed using a pipeline to trim adapter sequences and filter out low-quality reads as described before (Zhu et al., 2017). TopHat version 2.0.3.12 (Kim et al., 2013) was used to map reads to the maize genome (AGPv3 sequences; http://ensembl.gramene.org/Zea_mays/Info/Index). Genome-mapped reads were assembled, and transcripts were reconstructed using Cufflinks (Trapnell et al., 2012). Gene abundances were quantified in RESM (Li and Dewey, 2011). Comparisons of WT and OE transgenic lines were used to calculate differential expression in the edgeR package (http://www.rproject.org/). Genes with a fold change ≥2 and a false discovery rate (FDR) < 0.05 were identified as significantly DEGs. DEGs were then subjected to enrichment analysis of GO functions and KEGG pathways.
Histochemical staining of O2− and H2O2
Histochemical staining of O2− and H2O2 was performed using NBT and DAB, respectively. Briefly, whole maize leaves were infiltrated with 50 mM sodium phosphate (pH 7.5) containing 0.2% NBT (Sigma) or 10 mM sodium phosphate (pH 6.5) containing 1 mg mL−1 DAB (Sigma), followed by 10 min vacuuming, then incubation at 37°C in the dark for 5 and 10 h, respectively. Whole maize roots were infiltrated with same NBT staining solution or DAB staining solution, followed by incubation at 37°C in the dark for 2 and 6 h, respectively. Then the leaves and roots were washed with ethanol to bleach out the chlorophyll in boiled water for 10 min, then transferred into fresh ethanol. Finally, the leaves were photographed using a camera and whole roots were scanned by a micro computed scanner (WinRHIZO-VT1000S), root tips were photographed using a stereo microscope (LEICA-M165-FC) under uniform lighting.
Determination of MDA concentration and antioxidant enzyme activities
For the MDA concentration assay, fresh roots of WT, miR169q OE, or NF-YA8 OE from 0 mM NaCl treatment were homogenized in 5 mL of 10% trichloroacetic acid with a pestle and mortar. Homogenates were centrifuged at 4,000 × g for 20 min. To each 2-mL aliquot of the supernatant, 2 mL of 0.6% thiobarbituric acid in 10% TCA was added. The mixtures were heated at 100°C for 15 min and then quickly cooled in an ice bath. After centrifugation at 10,000 × g for 20 min, the absorbance of the supernatant was recorded at 532 and 450 nm. Lipid peroxidation was expressed as the MDA content in nmol per g FW. Details of the procedures for determining activities of POD, superoxide dismutase (SOD), and catalase (CAT) were described previously (Xu et al., 2019). Briefly, roots excised from the seedlings after treatment with 0 or 200 mM NaCl for 96 h were immediately frozen in liquid nitrogen and finely ground into powder with a pestle. Then, the activities of CAT, SOD, and POD were determined according to the protocols of the plant POD assay kit (A084-3, Nanjing Jiancheng Bioengineering Institute, Nanjing, China), total SOD (T-SOD) assay kit (A001-1-1, Jiancheng), and the CAT assay kit (A007-1-1, Jiancheng), respectively.
Y1H assay
To test the binding of ZmNF-YA8 to ZmPER1 promoter in yeast, a Y1H assay was completed with the Matchmaker Gold Yeast One-Hybrid Library Screening System (Clontech Laboratories, Mountain View, CA, USA). Three DNA fragments containing CCAAT-box elements in the sense strand of the ZmPER1 promoter, named C2, C4, and C5 were separately synthesized (Supplemental Table S2) and three tandem repeats were inserted into the reporter vector pAbAi to obtain pPER3C2-AbAi, pPER3C4-AbAi, and pPER3C5-AbAi plasmids, respectively. To test the specificity of the binding sites, three fragments carrying the same flanking regions but with mutated CCAAT-box elements (i.e. replaced with 5′-CTAGT-3′) (Supplemental Table S2), were synthesized and inserted into the pAbAi reporter vector to generate the pPER3C2m-AbAi, pPER3C4m-AbAi, and pPER3C5m-AbAi plasmids. The full-length ZmNF-YA8 coding sequence was amplified using PCR and inserted into the pGADT7 vector. The recombinant pGADT7-NF-YA8 plasmid was used to transform Y1H Gold yeast strain cells carrying the linearized pPER3C2-AbAi, pPER3C4-AbAi, pPER3C5-AbAi, pPER3C4m-AbAi, and pPER3C5m-AbAi. Transformed yeast cells were detected by spotting serial dilutions (1:10, 1:100, and 1:1,000) of yeast onto agar-solidified synthetic dextrose (SD)/−Leu medium supplemented with 100 ng/mL aureobasidin A (AbA). The pGADT7-Rec-p53 and p53-AbAi plasmids were used as positive controls. Details regarding the primers used for this assay are listed in Supplemental Table S2.
Transient expression assays in maize protoplasts and in N. benthamiana
For the dual-LUC transient expression assays, a 1,208-bp ZmPER1 promoter segment containing three CCAAT boxes (C2, C4, and C5), a 845-bp ZmPER1 promoter segment containing two CCAAT boxes (C2 and C4), a 698-bp ZmPER1 promoter segment containing one CCAAT box (C2), and a 387-bp ZmPER1 promoter segment containing one CCAAT box (C5) were separately amplified from inbred line B73 and recombined into BamHI and SalI sites of pGreenII 0800-LUC vector, generating the pZmPER1-P1:LUC, pZmPER1-P2:LUC, and pZmPER1-P3:LUC plasmids as reporters. The Renilla LUC (REN) gene driven by the 35S promoter in the pGreenII 0800-LUC vector was used as the internal control. The full-length CDS of ZmNF-YA8 was amplified and recombined into BamH1 and Xho1 sites of pGreenII 62-SK vector driven by the 35S promoter, forming the ZmNF-YA8 effectors. The empty pGreenII 62-SK vector was used as a control. The transient dual-LUC assays were performed in maize protoplasts collected from the leaves of 2-week-old etiolated seedlings of inbred line B73 (Gao et al., 2019). The LUC signal was detected using dual-LUC assay reagents (vazyme) following the manufacturer’s instructions. Relative LUC activity was calculated by normalizing LUC activity to REN activity. The constructs pUBI:miR169q, pUBI:NF-YA8-CDS, pUBI:NF-YA8-CDS-3′UTR (normal 3′-UTR), and pUBI:NF-YA8-CDS-mutant 3′UTR were kept in our laboratory. Transient expression experiments in N. benthamiana were performed according to the previous report (Luan et al., 2015). Nicotiana benthamiana Actin1 expression was used as an internal standard for normalization. The sequences of primers used in the transient expression assay are listed in Supplemental Table S2.
ChIP and qPCR analysis
A ChIP–qPCR assay was performed as previously described (Xu et al., 2014). Briefly, 3-week-old seedlings of NF-YA8 OE were treated with 200 mM NaCl for 96 h, then the seedlings were cross-linked with 1% formaldehyde and the chromatin complexes were sonicated at 4°C to generate 200–500-bp fragments. The sheared chromatin was immunoprecipitated, washed, and reverse cross-linked. A polyclonal anti-FLAG M2 antibody (F1804, Sigma–Aldrich, St. Louis, MO, USA) was used, with IgG as a negative control. The purified precipitated DNA was dissolved in water for qPCR analysis. The values were standardized to the input DNA to obtain the fold enrichment. GH3 was used as an internal control. Details regarding the primers used for this assay are listed in Supplemental Table S2.
Statistical analyses
All experiments were performed at least three times. All RT-qPCR reactions and other quantitative analysis were repeated at least three times. The Student’s t test was used to evaluate the significant differences among the various genotypes, treated or untreated with salt or H2O2.
Accession numbers
Sequence data from this article can be found in the GenBank/EMBL data libraries under accession numbers: POD 1(NM_001154199), NF-YA8 (NM_001153839), and miR169q (NR_121140).
Supplemental data
Supplemental Figure S1. Salt stress rapidly reduces ZmmiR169q expression in maize roots.
Supplemental Figure S2. The expression profiles of ZmmiR169q in B73 and B104 under 200 mM NaCl treatment.
Supplemental Figure S3. The expression profiles of ZmmiR169q in B73 and B104 under 1 mM H2O2 treatment.
Supplemental Figure S4. Coexpression of zmmiR169q and ZmNF-YA8 gene in a transient expression system in N. benthamiana cells.
Supplemental Figure S5. ZmNF-YA8 was involved in antioxidant pathway.
Supplemental Figure S6. Gene tree of Zm00001d018618 and expression profiles in different tissues.
Supplemental Figure S7. Expression profiles of ZmNF-YA8 and ZmPER1 in root of B104 under 200 mM NaCl treatment.
Supplemental Figure S8. Aligment of ZmmiR169q target sites in the 3′-UTR of ZmNF-YA8 between B73 and B104.
Supplemental Table S1. Mapping ratio of reference genome.
Supplemental Table S2. List of primers used in this study.
Supplementary Material
Acknowledgments
We thank Dr. Rongfeng Huang (Biotechnology Research Institute, Chinese Academy of Agricultural Sciences) for kindly providing the pCAMBIA1300–nLUC/cLUC vectors; we appreciated Prof. Haiyang Wang and Wen-Xue Li provided valuable comments to improve paper.
Funding
This work was supported by the National Key Research and development Program of China (2016YFD0101002), the National Natural Science Foundation of China (30272068), Agricultural Science and Technology Innovation Program of CAAS (CAAS-XTCX20190025), and Central Public-Interest Scientific Institution Basal Research Fund (1610392020001).
Conflict of interest statement. Authors declare no conflict of interest.
M.X. and L.W. designed the research. L.X. performed the survival and biomass detection, Y1H, RT-qPCR, H2O2 treatment, and malondialdehyde and antioxidant enzyme activities determination. M.Zhu. performed the statistical analysis, ChIP-qPCR, histochemical staining. M. Zhang performed transcriptome sequencing, and analyzed the data. M.L. generated the ZmmiR169- and ZmNF-YA8-related transgenic plants. L.X. and L.J. performed the dual luciferase experiments. Y.L. performed the plant culture and salt treatment. J.Z. designed some of the experiments. M.X. wrote the manuscript.
The author responsible for distribution of materials integral to the findings presented in this article in accordance with the policy described in the Instructions for Authors (https://academic.oup.com/plphys/pages/General-Instructions) is Miaoyun Xu (xumiaoyun@caas.cn).
References
- Busch AWU, Montgomery BL (2015) Interdependence of tetrapyrrole metabolism, the generation of oxidative stress and the mitigative oxidative stress response. Redox Biol 4: 260–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Q, He B, Kogel KH, Jin H (2018) Cross-kingdom RNA trafficking and environmental RNAi-nature’s blueprint for modern crop protection strategies. Cur Opin Microbiol 46: 58–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denver B, Ullah H (2019) miR393s regulate salt stress response pathway in Arabidopsis thaliana through scaffold protein RACK1A mediated ABA signaling pathways. Plant SignalBehav 14: 1600394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebrahimi SO, Reiisi S, Shareef S (2020) miRNAs, oxidative stress, and cancer: A comprehensive and updated review. J Cell Physiol 235: 8812–8825 [DOI] [PubMed] [Google Scholar]
- Flowers TJ, Galal HK, Bromham L (2010) Evolution of halophytes: multiple origins of salt tolerance in land plants. Funct Plant Biol 37: 604–612 [Google Scholar]
- Gao L, Shen G, Zhang L, Qi J, Zhang C, Ma C, Li J, Wang L, Malook SU, Wu J (2019) An efficient system composed of maize protoplast transfection and HPLC-MS for studying the biosynthesis and regulation of maize benzoxazinoids. Plant Methods 15: 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao P, Bai X, Yang L, Lv DK, Li Y, Cai H, Ji W, Guo DJ, Zhu YM (2010) Over-expression of osa-MIR396c decreases salt and alkali stress tolerance. Planta 231: 991–1001 [DOI] [PubMed] [Google Scholar]
- Gao P, Bai X, Yang LA, Lv DK, Pan X, Li Y, Cai H, Ji W, Chen Q, Zhu YM (2011) osa-MIR393: a salinity- and alkaline stress-related microRNA gene. Mol Biol Rep 38: 237–242 [DOI] [PubMed] [Google Scholar]
- Gnesutta N, Chiara M, Bernardini A, Balestra M, Horner DS, Mantovani R (2019) The plant NF-Y DNA matrix in vitro and in vivo. Plants (Basel) 8: 406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanemian M, Barlet X, Sorin C, Yadeta KA, Keller H, Favery B, Simon R, Thomma BPHJ, Hartmann C, Crespi M, et al. (2016) Arabidopsis CLAVATA1 and CLAVATA2 receptors contribute to Ralstonia solanacearum pathogenicity through a miR169-dependent pathway. New Phytol 211: 502–515 [DOI] [PubMed] [Google Scholar]
- Huang J, Levine A, Wang ZF (2013) Plant Abiotic Stress. Sci World J 2013: 432836 [Google Scholar]
- Ismail A, Takeda S, Nick P (2014) Life and death under salt stress: same players, different timing? J Exp Bot 65: 2963–2979 [DOI] [PubMed] [Google Scholar]
- Jagadeeswaran G, Saini A, Sunkar R (2009) Biotic and abiotic stress down-regulate miR398 expression in Arabidopsis. Planta 229: 1009–1014 [DOI] [PubMed] [Google Scholar]
- Jefferson AR, Kavanagh AT, Bevan WM (1987) GUS fusions: beta-glucuronidase as a sensitive and versatile gene fusion marker in higher plants. EMBO J 6: 3901–3907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia X, Ding N, Fan W, Yan J, Gu Y, Tang X, Li R, Tang G (2015) Functional plasticity of miR165/166 in plant development revealed by small tandem target mimic. Plant Sci 233: 11–21 [DOI] [PubMed] [Google Scholar]
- Kerrigan P (1990) Effective and appropriate prescribing. Practitioner 234: 193. [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL (2013) TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14: R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, Kwak KJ, Jung HJ, Lee HJ, Kang H (2010a) MicroRNA402 affects seed germination of Arabidopsis thaliana under stress conditions via targeting DEMETER-LIKE Protein3 mRNA. Plant Cell Physiol 51: 1079–1083 [DOI] [PubMed] [Google Scholar]
- Kim JY, Lee HJ, Jung HJ, Maruyama K, Suzuki N, Kang H (2010b) Overexpression of microRNA395c or 395e affects differently the seed germination of Arabidopsis thaliana under stress conditions. Planta 232: 1447–1454 [DOI] [PubMed] [Google Scholar]
- Laloum T, De Mita S, Gamas P, Baudin M, Niebel A (2013) CCAAT-box binding transcription factors in plants: Y so many? Trends Plant Sci 18: 594–595 [DOI] [PubMed] [Google Scholar]
- Lan J, Huang Z, Han J, Shao J, Huang C (2018) Redox regulation of microRNAs in cancer. Cancer Lett 418: 250–259 [DOI] [PubMed] [Google Scholar]
- Li B, Dewey CN (2011) RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12: 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li WX, Oono Y, Zhu J, He XJ, Wu JM, Iida K, Lu XY, Cui X, Jin H, Zhu JK (2008a) The Arabidopsis NFYA5 transcription factor is regulated transcriptionally and posttranscriptionally to promote drought resistance. Plant Cell 20: 2238–2251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Zhu YM, Liu Y, Shu YJ, Meng FJ, Lu YM, Bai X, Liu B, Guo DJ (2008b) Genome-wide identification of osmotic stress response gene in Arabidopsis thaliana. Genomics 92: 488–493 [DOI] [PubMed] [Google Scholar]
- Lin Y, Liu X, Cheng Y, Yang J, Huo Y, Zhang C (2009) Involvement of microRNAs in hydrogen peroxide-mediated gene regulation and cellular injury response in vascular smooth muscle cells. J Biol Phys Chem 284: 7903–7913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu BX, Zhang B, Yang ZR, Liu Y, Yang SP, Shi YL, Jiang CF, Qin F (2021) Manipulating ZmEXPA4 expression ameliorates the drought-induced prolonged anthesis and silking interval in maize. Plant Cell 33: 2058–2071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JX, Howell SH (2010a) bZIP28 and NF-Y transcription factors are activated by ER stress and assemble into a transcriptional complex to regulate stress response genes in Arabidopsis. Plant Cell 22: 782–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JX, Howell SH (2010b) Endoplasmic reticulum protein quality control and its relationship to environmental stress responses in plants. Plant Cell 22: 2930–2942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luan MD, Xu MY, Lu YM, Zhang QX, Zhang L, Zhang CY, Fan YL, Lang ZH, Wang L (2014) Family-wide survey of miR169s and NF-YAs and their expression profiles response to abiotic stress in maize roots. PLoS ONE 9: e91369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luan MD, Xu MY, Lu YM, Zhang L, Fan YL, Wang L (2015) Expression of zma-miR169 miRNAs and their target ZmNF-YA genes in response to abiotic stress in maize leaves. Gene 555: 178–185 [DOI] [PubMed] [Google Scholar]
- Ma HS, Liang D, Shuai P, Xia XL, Yin WL (2010) The salt- and drought-inducible poplar GRAS protein SCL7 confers salt and drought tolerance in Arabidopsis thaliana. J Exp Bot 61: 4011–4019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Xue H, Zhang F, Jiang Q, Yang S, Yue P, Wang F, Zhang Y, Li L, He P, et al. (2021) The miR156/SPL module regulates apple salt stress tolerance by activating MdWRKY100 expression. Plant Biotechnol J 19: 311–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller G, Suzuki N, Ciftci-Yilmaz S, Mittler R (2010) Reactive oxygen species homeostasis and signalling during drought and salinity stresses. Plant Cell Environ 33: 453–467 [DOI] [PubMed] [Google Scholar]
- Munne-Bosch S, Queval G, Foyer CH (2013) The impact of global change factors on redox signaling underpinning stress tolerance. Plant Physiol 161: 5–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouhibi C, Attia H, Rebah F, Msilini N, Chebbi M, Aarrouf J, Urban L, Lachaal M (2014) Salt stress mitigation by seed priming with UV-C in lettuce plants: Growth, antioxidant activity and phenolic compounds. Plant Physiol Biochem 83: 126–133 [DOI] [PubMed] [Google Scholar]
- Pegler JL, Oultram JMJ, Grof CPL, Eamens AL (2019) Profiling the abiotic stress responsive microRNA landscape of Arabidopsis thaliana. Plants (Basel) 8: 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petroni K, Kumimoto RW, Gnesutta N, Calvenzani V, Fornari M, Tonelli C, Holt BF, Mantovani R (2012) The promiscuous life of plant NUCLEAR FACTOR Y transcription factors. Plant Cell 24: 4777–4792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qadir M, Tubeileh A, Akhtar J, Larbi A, Minhas PS, Khan MA (2008) Productivity enhancement of salt-affected environments through crop diversification. Land Degrad Dev 19: 429–453 [Google Scholar]
- Qin Z, Chen J, Jin L, Duns GJ, Ouyang P (2015) Differential expression of miRNAs under salt stress in Spartina alterniflora leaf tissues. J Nanosci Nanotechnol 15: 1554–1561 [DOI] [PubMed] [Google Scholar]
- Rhoades MW, Reinhart BJ, Lim LP, Burge CB, Bartel B, Bartel DP (2002) Prediction of plant microRNA targets. Cell 110: 513–520 [DOI] [PubMed] [Google Scholar]
- Serivichyaswat PT, Susila H, Ahn JH (2017) Elongated hypocotyl 5-Homolog (HYH) negatively regulates expression of the ambient temperature-responsive microRNA gene MIR169. Front Plant Sci 8: 2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorescu D, Griendling KK (2002) Reactive oxygen species, mitochondria, and NAD(P)H oxidases in the development and progression of heart failure. Congest Heart Fail 8: 132–140 [DOI] [PubMed] [Google Scholar]
- Sorin C, Declerck M, Christ A, Blein T, Ma LN, Lelandais-Briere C, Njo MF, Beeckman T, Crespi M, Hartmann C (2014) A miR169 isoform regulates specific NF-YA targets and root architecture in Arabidopsis. New Phytol 202: 1197–1211 [DOI] [PubMed] [Google Scholar]
- Sunkar R, Kapoor A, Zhu JK (2006) Posttranscriptional induction of two Cu/Zn superoxide dismutase genes in Arabidopsis is mediated by downregulation of miR398 and important for oxidative stress tolerance. Plant Cell 18: 2415–2415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunkar R, Zhu JK (2004) Novel and stress-regulated microRNAs and other small RNAs from Arabidopsis. Plant Cell 16: 2001–2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang G, Yan J, Gu Y, Qiao M, Fan R, Mao Y, Tang X (2012) Construction of short tandem target mimic (STTM) to block the functions of plant and animal microRNAs. Methods 58: 118–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L (2012) Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7: 562–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walley JW, Sartor RC, Shen ZX, Schmitz RJ, Wu KJ, Urich MA, Nery JR, Smith LG, Schnable JC, Ecker JR, et al. (2016) Integration of omic networks in a developmental atlas of maize. Science 353: 814–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Wang Q, Zhang B (2013) Response of miRNAs and their targets to salt and drought stresses in cotton (Gossypium hirsutum L.). Gene 530: 26–32 [DOI] [PubMed] [Google Scholar]
- Wang W, Liu D, Chen DD, Cheng YY, Zhang XP, Song LR, Hu MJ, Dong J, Shen FF (2019) MicroRNA414c affects salt tolerance of cotton by regulating reactive oxygen species metabolism under salinity stress. RNA Biol 16: 362–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiberg A, Wang M, Bellinger M, Jin H (2014) Small RNAs: a new paradigm in plant–microbe interactions. Annu Rev Phytopathol 52: 495–516 [DOI] [PubMed] [Google Scholar]
- Xu MY, Zhang L, Li WW, Hu XL, Wang MB, Fan YL, Zhang CY, Wang L (2014) Stress-induced early flowering is mediated by miR169 in Arabidopsis thaliana. J Exp Bot 65: 89–101 [DOI] [PubMed] [Google Scholar]
- Xu Y, Zou JJ, Zheng HY, Xu MY, Zong XF, Wang L (2019) RNA-Seq transcriptome analysis of rice primary roots reveals the role of flavonoids in regulating the rice primary root growth. Genes 10: 213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Guo Y (2018) Elucidating the molecular mechanisms mediating plant salt-stress responses. New Phytol 217: 523–539 [DOI] [PubMed] [Google Scholar]
- Ye Y, Wang J, Wang W, Xu LA (2020) ARF family identification in Tamarix chinensis reveals the salt responsive expression of TcARF6 targeted by miR167. PeerJ 8: e8829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Jia T, Chen X (2017) The ‘how’ and ‘where’ of plant microRNAs. New Phytol 216: 1002–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Zhu GZ, Du L, Shang XG, Cheng CZ, Yang B, Hu Y, Cai CP, Guo WZ (2016) Genetic regulation of salt stress tolerance revealed by RNA-Seq in cotton diploid wild species, Gossypium davidsonii. Sci Rep 6: 20582. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Zhao B, Ge L, Liang R, Li W, Ruan K, Lin H, Jin Y (2009) Members of miR-169 family are induced by high salinity and transiently inhibit the NF-YA transcription factor. BMC Mol Biol 10: 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M, Li DY, Li ZG, Hu Q, Yang CH, Zhu LH, Luo H (2013) Constitutive expression of a miR319 gene alters plant development and enhances salt and drought tolerance in transgenic Creeping bentgrass. Plant Physiol 161: 1375–1391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Chen CJ, Zeng J, Yun Z, Liu YL, Qu HX, Jiang YM, Duan XW, Xia R (2020) MicroRNA528, a hub regulator modulating ROS homeostasis via targeting of a diverse set of genes encoding copper-containing proteins in monocots. New Phytol 225: 385–399 [DOI] [PubMed] [Google Scholar]
- Zhu M, Zhang M, Xing L, Li W, Jiang H, Wang L, Xu M (2017) Transcriptomic analysis of long non-coding RNAs and coding genes uncovers a complex regulatory network that is involved in maize seed development. Genes (Basel) 8: 274. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.