

Abstract
Recent genetic studies of neurodevelopmental disorders point to synaptic proteins and ion channels as key contributors to disease pathogenesis. While many of these proteins, such as the L-type calcium channel Cav1.2 or the postsynaptic scaffolding protein SHANK3, have well-studied functions in mature neurons, new evidence indicates that they may subserve novel, distinct roles in immature cells as the nervous system is assembled in prenatal development. Emerging tools and technologies, including single cell sequencing and human cellular models of disease, are illuminating differential isoform utilization, spatiotemporal expression, and subcellular localization of ion channels and synaptic proteins in the developing brain compared to the adult, providing new insights into the regulation of developmental processes. We propose that it is essential to consider the temporally distinct and cell-specific roles of these proteins during development and maturity in our framework for understanding neuropsychiatric disorders.
In Brief
In this Perspective, Panagiotakos and Pașca highlight mechanisms conferring unique developmental roles to disease-associated ion channels and synaptic proteins, arguing that incorporating spatiotemporally-distinct and cell type-specific functions is essential for our emerging framework for understanding neuropsychiatric disorders.
Introduction
The nervous system builds itself through a series of cellular processes that emerge from an elaborate set of genetic programs, encompassing stem and progenitor cell proliferation to neuronal migration and terminal differentiation (Di Bella et al., 2021; Greig et al., 2013; Leone et al., 2008; Miyata et al., 2010; Rubenstein and Rakic, 1999; Shibata et al., 2015). The coordinated cellular choreography that underlies neural development generates immense cell diversity, incorporating features such as the distant migration of multiple cell types from their place of origin into their final circuit, the elimination of certain cells even after they have been integrated into circuits, and the conserved timing of specific cellular events across different species (Bonnefont and Vanderhaeghen, 2021; Jabaudon, 2017; Kwan et al., 2012; Wamsley and Fishell, 2017; Wong and Marín, 2019). This dynamic selforganization of the developing nervous system often involves utilizing the same gene, gene network, signaling pathway or cellular process in different contexts and time periods. In this perspective, we discuss novel, surprising roles for proteins associated with neurodevelopmental disorders, placing special emphasis on dynamic changes in their expression and regulation that may critically impact disease-related cellular phenotypes. Our focus is specifically on ion channels and synaptic proteins, as they have been strongly linked to neurodevelopmental disorders and proposed as convergence points for disease etiology (De Rubeis et al., 2014; Gilman et al., 2011; Iossifov et al., 2014; Jamain et al., 2003; Levy et al., 2011; Sanders et al., 2011; Zoghbi, 2003). We present selected examples illustrating that temporally regulated and cell-specific changes in gene transcription, alternative splicing and subcellular localization result in the emergence of unique developmental functions of these disease-relevant proteins. Incorporating these roles into efforts towards a systems-level understanding of neurodevelopmental disorders is likely to provide insights into both the proper assembly of the nervous system, as well as the consequences of disease risk-conferring gene variants on neuronal identity and mature circuit function.
Temporally-regulated and cell type-specific enrichment of ion channel subtypes
Precise spatiotemporal regulation of gene expression is a hallmark of normal nervous system development (Miller et al., 2014; Nowakowski et al., 2017; Pletikos et al., 2014; Polioudakis et al., 2019; Telley et al., 2016; Thompson et al., 2014; Trevino et al., 2020; Zhu et al., 2018b), and a number of studies in recent years have brought to light the developmental dynamics of transcripts encoding ion channel subunits, ion pumps and synaptic components (Gordon et al., 2021; Mayer et al., 2019; Smith et al., 2018, 2021; Vitali et al., 2018). Single cell RNA sequencing efforts are also providing unprecedented transcriptomic resolution of neural cell types (Hodge et al., 2019; Yao et al., 2021), allowing for the interrogation of disease-relevant gene expression in specific neural populations (Trevino et al., 2021; Velmeshev et al., 2019; Zuccaro et al., 2021). As ion channels and synaptic proteins have emerged as significant players in neurodevelopmental disorders (De Rubeis et al., 2014; Gilman et al., 2011; Iossifov et al., 2014; Jamain et al., 2003; Levy et al., 2011; Sanders et al., 2011; Zoghbi, 2003), understanding how their expression is regulated across development both temporally and spatially is essential for our ability to define the temporal windows and candidate cell types that are altered in the context of disease.
A prime example of the phenotypic consequences of temporally-regulated enrichment of disease-associated ion channels is the voltage-gated sodium channels Nav1.1, Nav1.2, and Nav1.3, encoded by SCN1A, SCN2A, and SCN3A, respectively (Smith and Walsh, 2020). Both gain- and loss-of-function mutations have been identified in all three genes encoding these sodium channels and are associated with different developmental phenotypes (Meisler and Kearney, 2005; Meisler et al., 2021; Sanders et al., 2018; Zaman et al., 2020). SCN3A expression is elevated in immature progenitors and neurons of the fetal brain, giving way to postnatal expression of SCN1A and SCN2A (Beckh et al., 1989; Smith et al., 2018). Consequently, emerging data indicates that mutations in SCN3A can lead to developmental brain malformations like polymicrogyria (Smith et al., 2018; Zaman et al., 2020), whereas SCN1A and SCN2A variants typically affect neurophysiology and are more commonly associated with infantile epilepsies (Meisler and Kearney, 2005; Sanders et al., 2018). In line with this, a recent study demonstrated that expression of disease-associated SCN3A variants in immature neurons of the developing human and ferret cerebral cortex results in deficits in neuronal migration and placement, in addition to malformed gyri (Smith et al., 2018).
In addition to temporal expression dynamics, Nav channels have been reported to display cell type-specific enrichment in the developing brain, which may also contribute to distinct phenotypic outcomes when they are mutated. Nav1.1 channels, for example, are predominantly expressed in parvalbumin (PV) cortical interneurons (Yu et al., 2006). Consequently, Scn1a loss of function selectively reduces sodium current density in inhibitory interneurons (Yu et al., 2006), and behavioral deficits in an Scn1a haploinsufficiency mouse model of Dravet syndrome can be rescued by augmenting GABAergic activity with clonazepam (Han et al., 2012). A knock-in Scn1a loss-of-function mouse model also displays early postnatal epilepsy due to a transient impairment of PV interneuron activity (Favero et al., 2018; Ogiwara et al., 2007). Intriguingly, the output of PV interneurons appears to normalize in adulthood, owing to compensatory changes in the distribution of sodium channels in these cells (Favero et al., 2018). This observation underscores another important idea: it is critical to distinguish between initial mechanistic drivers of disease onset and secondary cellular and molecular mechanisms contributing to chronic disease states.
In contrast to Nav1.1, Nav1.2 is enriched in cortical pyramidal neurons (Hu et al., 2009; Spratt et al., 2019), and several studies have described electrophysiological impairments in both developing and mature excitatory neurons resulting from Scn2a inactivation (Ben-Shalom et al., 2017; Spratt et al., 2019, 2021; Zhang et al., 2021). A recent study also demonstrated that transcripts encoding Nav1.2 are specifically expressed in both the mouse and baboon in a differentiating population of spiking oligodendrocyte progenitors (Gould and Kim, 2021). This is consistent with previous work suggesting that oligodendrocyte spiking is dependent on Nav1.2 currents (Berret et al., 2017; Jiang et al., 2013). SCN2A inactivation disrupts oligodendrocyte maturation (Gould and Kim, 2021), highlighting an emerging, unconventional role for Nav1.2 channels in the differentiation of a cell type not traditionally thought of as “excitable”. Future studies exploring the functional consequences of individual mutations, as well as their impact on the expression and functions of these channels in specific developing cell populations, are an essential next step towards unraveling the contributions of cell type-specific dysfunction to disease etiology. Understanding how disease-associated changes in electrical signals and ionic influx alter developmental events in human-specific cell types will be important. Towards this goal, approaches functionally characterizing multiple disease-relevant ion channel mutations, at scale and in human cells (e.g. using automated patching, Optopatch), and connecting gene expression with cell function (Patch-seq) will be particularly informative.
Other disease-associated ion channels display dynamic patterns of subunit composition during different developmental windows, including GABA receptor subunit switching during differentiation (Mayer et al., 2019), AMPA receptor subunit switching during neuronal maturation (Kumar et al., 2002), and shifting ratios of voltage-gated potassium channel subunits encoded by KCNQ2 and KCNQ3 in early postnatal life (Kanaumi et al., 2008; Tinel et al., 1998). Here we would like to highlight another important developmental switch in ion channel subunit composition: the temporal utilization of NMDA receptor subunits GluN2A and GluN2B, encoded by GRIN2A and GRIN2B, respectively. Early studies in the embryonic turtle cortex demonstrate that NMDA receptor expression and functional NMDA currents precede the formation of synapses in differentiating neurons (Blanton et al., 1990), and it has been reported that a switch in subunit composition from GluN2B to GluN2A begins in rodent cortical neurons at birth (Sheng et al., 1994; Watanabe et al., 1992). Similarly, studies on human fetal tissue report declining expression of GRIN2B as cortical development proceeds (Bagasrawala et al., 2017). This key developmental transition can be recapitulated in human induced pluripotent stem (hiPS) cell-derived cortical organoids that were allowed to mature for extensive periods in vitro (Gordon et al., 2021).
Compared to their GluN2B-containing counterparts, GluN2A-expressing NMDA receptors have distinct electrophysiological properties, including more rapid desensitization and different responses to allosteric modulators (Paoletti et al., 2013; Wyllie et al., 2013). As a consequence of the enrichment pattern of channels containing these subunits, GRIN2B gene variants are often associated with earlier neurodevelopmental phenotypes, including cortical malformations and autism spectrum disorders (ASD) (Endele et al., 2010; O’Roak et al., 2012). In line with this, inactivation of Grin2b in rodent models points to roles for GluN2B-containing NMDA receptors in developmental events occurring during late prenatal and early postnatal life, including migration, morphological maturation and circuit integration (De Marco García et al., 2015; Espinosa and Luo, 2008; Jiang et al., 2015). For example, knocking down GluN2B, but not GluN2A, in the embryonic rodent cortex results in selective deficits in radial migration (Jiang et al., 2015). In developing mouse hippocampal neurons and layer IV cortical neurons, cell-autonomous GluN2B function is also required for proper pruning of primary dendrites and dendritic patterning, respectively, but appears unnecessary for initial dendritic elaboration (Espinosa and Luo, 2008). In the neonatal somatosensory cortex, GluN2B is essential for morphological maturation and circuit integration of specific developing cortical interneuron subtypes (De Marco García et al., 2015). Intriguingly, GluN2B-containing NMDA receptors in these populations are specifically activated by projections from the thalamus, suggesting a potential mechanism by which distinct circuit components can be selectively affected by disease-associated mutations. Several studies are also illuminating, however, that the dynamics of GluN2 subunit expression may differ in individual cell types (Matta et al., 2013). For instance, whereas medial ganglionic eminence (MGE)-derived interneurons display an activity-dependent GluN2 subunit switch, a subpopulation of caudal ganglionic eminence (CGE)derived hippocampal interneurons retains GluN2B-containing NMDA receptors throughout development, well into the third postnatal week in the mouse (Matta et al., 2013). GluN2B also appears to play a critical role in adult-born neurons as they integrate into olfactory circuits (Kelsch et al., 2012). These findings support the notion that, based on cell type-specific differences in the dynamics of ion channel subunit expression, disease-associated mutations may impinge on different cell types at distinct developmental stages to contribute to the emergence of clinically distinct phenotypes.
Intriguingly, a recent study has implicated rare truncating and damaging missense variants in GRIN2A in schizophrenia risk (Singh et al., 2020), highlighting that mutations in different developmentally regulated ion channel subunits may also lead to distinct neuropsychiatric conditions with differing age of onset. Single cell RNA sequencing analysis on human fetal cortical specimens has demonstrated expression of GRIN2A in human cortical neural stem cells (Mayer et al., 2019), suggesting a more dynamic pattern of GRIN2A regulation through cortical lineage progression. This underscores, once again, the importance of exploring functional consequences of disease-associated GRIN2 variants at different developmental stages, in different cell types, and in humans as well as other species. Indeed, ion channels are becoming increasingly implicated in normal neural progenitor cell function. Temporally regulated progenitor hyperpolarization, driven in large part by dynamic changes in the expression of potassium channels during embryonic development, is essential for the normal sequential progression of cortical neurogenesis (Vitali et al., 2018). More recently, consistent with a dynamic role for ion channels in neural progenitor cell proliferation, neurodevelopmental disease-associated hyperpolarization activated cyclic nucleotide-gated cation (HCN) channels were shown to regulate progenitor cell cycle dynamics, with HCN loss of function resulting in G1 accumulation and microcephaly (Schlusche et al., 2021). Taken together, these studies and others are defining new roles for ion channels, which have been canonically associated with mature neuronal function, whereby they can regulate progenitor proliferation and other earlier stages of brain assembly.
Another area of growing interest centers around the understanding of evolutionary conservation and divergence of genetic networks underlying neurodevelopmental processes. An extensive transcriptomic study conducted across 16 regions of the developing human and macaque brain recently found that heterochronic processes between the two species include molecular pathways related to synaptic activity (Zhu et al., 2018b). Of note, the genes encoding the ASD-associated synaptic proteins SHANK2 and SHANK3 were expressed earlier in the macaque neocortex and other brain regions relative to humans, whereas the schizophrenia-associated GRIA1, encoding the AMPA receptor subunit GluR1, is expressed earlier in the developing human brain (Zhu et al., 2018b). Such studies are beginning to illuminate dynamic species-specific differences in the expression timing of disease-associated transcripts involved in modulating neuronal activity during development, shedding light both on evolutionary changes in the coordination of developmental programs, as well as on human-specific features of disease-associated gene regulation.
Temporally regulated splicing of ion channels and synaptic proteins
In comparison to other tissues, the brain exhibits substantial levels of alternative splicing (Yeo et al., 2004). In the embryonic brain, dynamic changes in splicing contribute to key developmental transitions (Boutz et al., 2007; Makeyev et al., 2007; Zheng and Black, 2013), including the process of neuronal differentiation in the cerebral cortex (Zhang et al., 2016), and several studies have illuminated developmental splicing switches in genes encoding ion channels or related to synaptic transmission (Flaherty et al., 2019; Gazina et al., 2010; Liang et al., 2021; Panagiotakos et al., 2019; Tang et al., 2009, 2011; Weyn-Vanhentenryck et al., 2018; Zheng et al., 2012). Not surprisingly, given the importance of cell type-specific splicing in dictating key steps in neural development, genome-wide changes in alternative splicing and activity-dependent splicing networks have been reported in neuropsychiatric disorders like ASD, schizophrenia and bipolar disorder (Gandal et al., 2018a; Irimia et al., 2014; Parikshak et al., 2016; Quesnel-Vallières et al., 2016; Sanders et al., 2020; Weyn-Vanhentenryck et al., 2014).
Mutations in individual ion channels or synaptic proteins that impact alternative splicing also play essential roles in the emergence of disease-relevant cellular phenotypes (Flaherty et al., 2019; Panagiotakos et al., 2019). Whereas significant overlap is observed in differential gene expression across neuropsychiatric disorders, changes in cell type-specific isoform expression show a high degree of disease specificity (Gandal et al., 2018a). Disease-specific alterations in splicing machinery (e.g. RBFOX1) that impinge on transcripts encoding ion channels and synaptic proteins have also been reported (De Rubeis et al., 2014; Gandal et al., 2018a; Lee et al., 2016; Tang et al., 2009; Weyn-Vanhentenryck et al., 2014). Importantly, activity-dependent splicing factors like Rbfox1 have been shown to direct cell-type specific splicing programs in developing cortical circuits (Wamsley et al., 2018), suggesting another possible molecular substrate for the emergence of distinct cellular phenotypes. These findings demonstrate the necessity of understanding spatiotemporally regulated and cell type-specific splicing patterns in the brain across the lifespan to define cell- and circuit-specific mechanisms underpinning different neuropsychiatric disorders.
The impact of altered splicing in the context of neurodevelopmental disease is exemplified by the monogenic, syndromic ASD Timothy Syndrome (TS). Classical TS is caused by a point mutation in the alternatively spliced exon 8A of CACNA1C, which encodes the pore-forming subunit of the neuropsychiatric disease-associated calcium channel Cav1.2 (Bhat et al., 2012; Cross-Disorder Group of the Psychiatric Genomics Consortium, 2013; Panagiotakos et al., 2019; Paşca et al., 2011; Splawski et al., 2004). CACNA1C isoforms containing exon 8A are enriched in immature cortical progenitors and young neurons in both the mouse and human, and neuronal differentiation is accompanied by a switch in CACNA1C exon utilization from exon 8A to its mutually exclusive counterpart exon 8 (Figure 1) (Panagiotakos et al., 2019; Tang et al., 2011). Intriguingly, in addition to increasing calcium influx by impairing voltage-dependent channel inactivation (Paşca et al., 2011; Splawski et al., 2004), the TS mutation prevents the splicing switch in CACNA1C from exon 8A to exon 8 in differentiating human cortical excitatory neurons from TS patients (Panagiotakos et al., 2019). Consequently, TS cortical neurons derived from hiPS cells continue to express exon 8A transcripts containing the mutation at increased levels. This prolonged developmental misexpression of mutant channels later in development and the ensuing reduction in wild type exon 8-expressing channels contribute to impairments in the differentiation of deep layer cortical projection neurons (Panagiotakos et al., 2019; Paşca et al., 2011), which have been implicated in the pathogenesis of ASD (Willsey et al., 2013).
Figure 1:
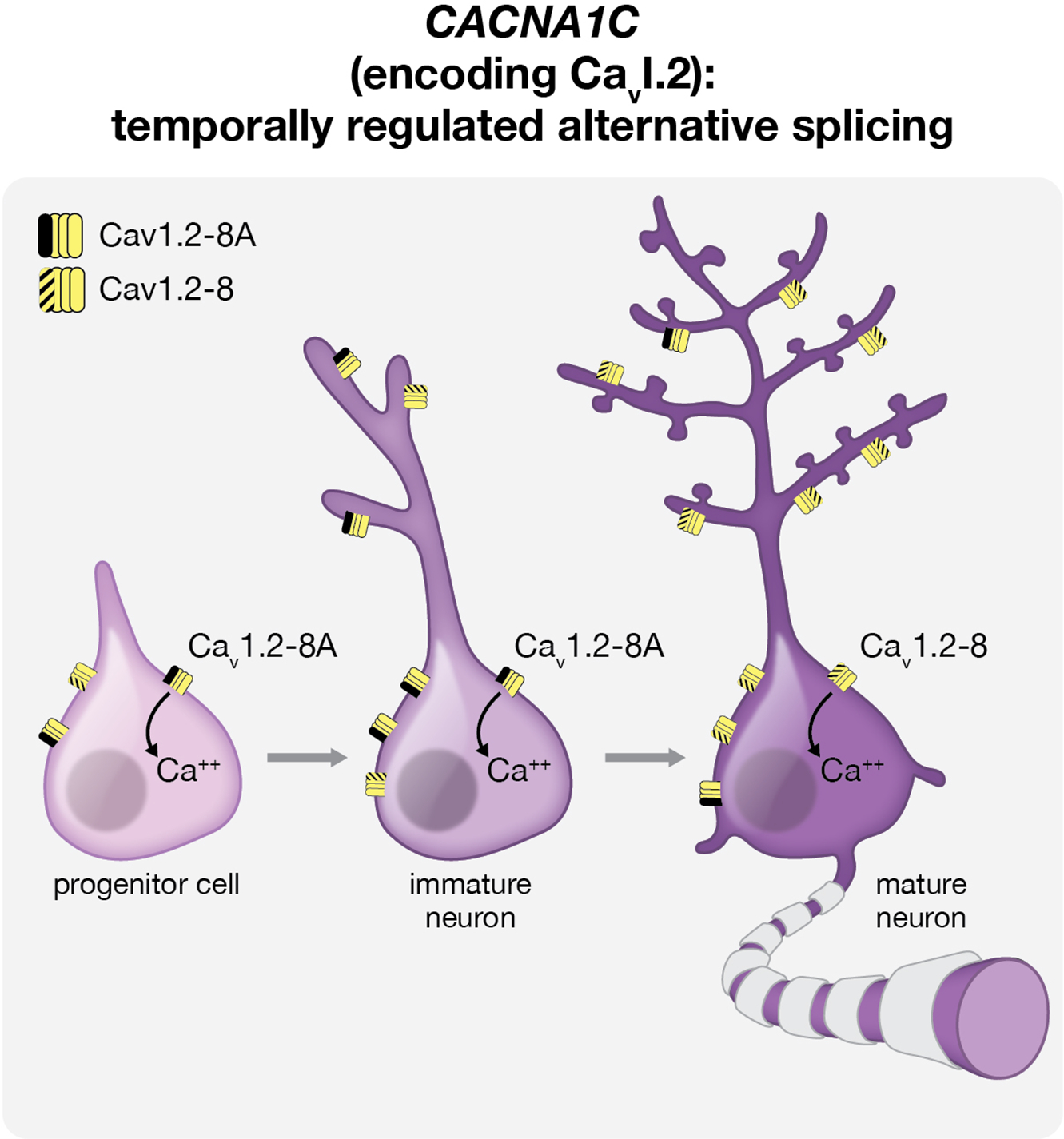
Dynamic changes in CACNA1C splicing may underlie novel roles for Cav1.2 channels in neuronal differentiation. Utilization of the Timothy syndrome-associated exons 8 and 8a of CACNA1C in the developing brain is temporally regulated. Exon 8a-expressing transcripts are enriched in neural progenitors and immature neurons (left, center), whereas channels encoded by exon 8-containing transcripts are more highly expressed in mature neurons (right). The TS mutation promotes persistently increased expression of exon 8A transcripts containing the mutation during neuronal differentiation and maturation.
In addition to differentiation deficits, TS channels have been linked to phenotypes in different cell types and at different stages of cortical neuron maturation, including impaired interneuron migration (Birey et al., 2017), deficits in radial migration in developing excitatory neurons (Kamijo et al., 2018)dendritic retraction in cortical pyramidal neurons (Krey et al., 2013). Investigating how altered splicing contributes to these developmental phenotypes underlying TS in different neural cell types is thus of great interest. It should be noted here that since CACNA1C transcripts differing only in exon 8 or exon 8A expression generate Cav1.2 channels with only subtle differences in channel physiology (Welling et al., 1997; Zühlke et al., 1998), it is possible that swapping these exons alone may predominantly impact channel conformation or downstream signaling interactions to affect neuronal development, as opposed to simply changing ionic flux. Further supporting a role for the TS mutation in promoting splicing deficits in CACNA1C, we recently discovered using hiPS cell-derived forebrain assembloids that the splicing switch from exon 8A to exon 8 is also impaired in interneuron populations from TS patients (Birey et al., 2021).
Developmental, cell- and tissue-specific regulation of other CACNA1C exons has been reported in the human, as well as the rodent, (Diebold et al., 1992; Soldatov, 1994; Tang et al., 2007, 2009; Welling et al., 1997), with a recent study identifying multiple fulllength region-specific CACNA1C splice isoforms in the adult human brain (Clark et al., 2020). As inclusion of some exons can significantly impact channel properties (Tang et al., 2009), it is likely that alternative splicing plays a key role in directing the developmental functions of these channels by generating isoforms with distinct biophysical and/or signaling properties. Perhaps disease-causing mutations cause specific deficits by influencing splicing to generate specific isoforms during discrete developmental windows. Incorporating the expression patterns of different channel splice variants into our framework for understanding CACNA1C-associated neuropsychiatric disorders will illuminate the cell types or developmental stages that drive the emergence of specific disease-relevant cellular phenotypes.
Other disease-associated ion channels also display developmentally regulated splice isoform utilization (Gazina et al., 2015; Liang et al., 2021; Liu et al., 2019; Plummer et al., 1997; Smith et al., 2001). For example, SCN2A, the gene encoding the voltage-gated sodium channel Nav1.2, is strongly associated with ASD, intellectual disability (ID) and infantile seizures (Sanders et al., 2018). SCN2A contains two mutually exclusive exons, 5N and 5A, whose utilization is dynamically controlled during development and into adulthood (Gazina et al., 2015; Liang et al., 2021). The “neonatal” 5N exon is enriched in SCN2A transcripts in the embryonic and early postnatal brain (Gazina et al., 2015; Liang et al., 2021). In rodents, 5N inclusion declines postnatally as Scn2a mRNA levels increase, resulting in “adult” exon 5A-containing isoforms encoding the majority of Nav1.2 channels in cortical neurons after the second postnatal week (Gazina et al., 2010, 2015). These splicing dynamics also differ across brain regions in the mouse (Gazina et al., 2010), and different patterns of exon 5A and 5N splicing have been reported in the genes coding for other voltage-gated sodium channel alpha subunits in the mouse and human brain, including the neurodevelopmental disease-associated SCN8A (encoding Nav1.6) (Gazina et al., 2010; Liang et al., 2021).
Expression of exon 5A or 5N-containing SCN2A variants in cortical excitatory neurons is predicted to differentially impact channel properties and thereby neural excitability. For instance, neonatal isoforms result in reduced neuronal excitability (Gazina et al., 2015; Xu et al., 2007). A recent report also suggests that cell type-specific SCN2A splice variants may generate channels that are optimized for the electrophysiological properties of the neurons in which they are expressed (Lignani et al., 2020). Notably, studies have found that multiple SCN2A mutations associated with infantile epilepsy preferentially or more strongly impact channels encoded by neonatal 5N isoforms, rendering neonatal channels more functionally similar to adult channels (Thompson et al., 2020; Xu et al., 2007). Other data indicate that splice isoforms of sodium channels associated with epilepsy disorders, such as Nav1.1 (encoded by SCN1A), are differentially responsive to anti-epileptic drugs (Tate et al., 2005; Thompson et al., 2011). This suggests that alternative splicing also has important consequences for developing therapies. Collectively, these findings underscore that it is essential to profile the expression of specific ion channel splice variants across development and to functionally characterize their impact on different developmental processes. This will enable the field to better predict the consequences of disease-causing mutations and the effects of therapeutic interventions.
Looking ahead, as with Timothy Syndrome, the use of hiPS cell-derived organoid and assembloid platforms will be especially powerful to probe the functional consequences of SCN2A splice isoforms and disease-associated mutations in human cells (Paşca, 2018). Human neurons lacking one or both copies of SCN2A have been generated from control hiPS cells or human embryonic stem (hES) cell lines using CRISPR approaches and have been shown to phenocopy some of the electrophysiological phenotypes reported in Scn2a mouse models (Deneault et al., 2018; Lu et al., 2019; Ogiwara et al., 2018; Planells-Cases et al., 2000). To date, hiPS cell derivatives have been successfully used to probe cellular phenotypes resulting from SCN1A mutations in Dravet syndrome patients (Jiao et al., 2013; Liu et al., 2013; Sun et al., 2016).
In addition to ion channels, synaptic proteins are known to undergo alternative splicing to generate cell type-specific isoforms, as exemplified by the presynaptic neurexins (Chih et al., 2006; Dai et al., 2019; Fuccillo et al., 2015; Jenkins et al., 2016; Schreiner et al., 2014; Treutlein et al., 2014). Nrxn1, for example, is alternatively spliced to yield an array of cell type-specific neurexins in the mouse cerebral cortex that specify individual synapses by interacting selectively with isoforms of different post-synaptic partners (most notably the neuroligins) (Fuccillo et al., 2015; Schreiner et al., 2014). In humans, deletions in NRXN1 (encoding neurexin-1) have been strongly associated with various neuropsychiatric disorders, including ASD and schizophrenia (Ching et al., 2010; De Rubeis et al., 2014; Kim et al., 2008), and differential splicing of NRXN1 has been reported in ASD, schizophrenia and bipolar disorder (Gandal et al., 2018a). NRXN1 expression and NRXN1α and NRXN1β isoform levels have been shown to increase during fetal development and peak at birth in post-mortem human cortical specimens (Jenkins et al., 2016), and a recent study reports that NRXN1α isoform diversity is developmentally regulated, with fetal prefrontal cortex samples displaying substantially higher isoform diversity than both adult prefrontal cortex and hiPS cell-derived control neurons (Flaherty et al., 2019). This latter study goes on to show that the abundance of wild type NRXN1α isoforms is markedly reduced in patient-derived neurons bearing heterozygous loss-of-function mutations in NRXN1, accompanied by increased expression of NRXN1β isoforms and, in some genotypes, aberrant expression of novel “mutant” NRXN1α splice variants (Flaherty et al., 2019). Changes in NRXN1 splice isoform expression resulting from NRXN1 mutations were correlated with deficits in neuronal activity, morphology and maturation (Flaherty et al., 2019). Intriguingly, overexpression of wild type NRXN1α isoforms was not able to rescue patient neurons that expressed dominant negative “mutant” NRXN1α isoforms, highlighting the significance of understanding the precise impact of different disease-causing mutations on the splice variant repertoire of synaptic proteins and the pathological consequences of altered splicing on neuronal function. Functional studies in rodents indicate that combinatorial transsynaptic interactions between specific pairs of presynaptic neurexin and postsynaptic neuroligin isoforms are critical for synaptic assembly, plasticity and cell-specific connectivity (Singh et al., 2016; Uchigashima et al., 2020). Disease-associated mutations in NRXN1 could potentially eliminate NRXN1 isoforms that specify individual synaptic types, or, as described by Flaherty et al., could introduce “mutant” isoforms that alter the repertoire of NRXN1 isoforms to ultimately influence neuronal activity (Flaherty et al., 2019). How such “mutant” isoforms interfere with synaptic function or whether they take on novel roles remains unclear. Mutations in genes encoding astrocyte-secreted linker proteins that enable interactions between “incompatible” neurexin-neuroligin isoform pairs (Singh et al., 2016) could also indirectly impinge on neurexin signaling. One such protein, encoded by SPARCL1, has been associated with ASD (De Rubeis et al., 2014). Given the cell type specificity of neurexins, it will be especially important to interrogate the impact of NRXN1 mutations in different cell types and at different developmental stages.
In summary, two key ideas are essential to consider in our discussion of mechanisms that contribute to the emergence of specific cellular phenotypes in neurodevelopmental disorders. Our knowledge of the spatiotemporal expression profiles and unique functions of ion channel and synaptic protein isoforms will enable a deeper understanding of how mutations that confer risk for neurodevelopmental disorders can lead to circuit dysfunction. At the same time, some disease-relevant mutations may promote misexpression of splice variants at inappropriate times or in the incorrect cell types, or alternately, aberrant expression of “mutant” splice isoforms, to drive cellular phenotypes that underlie disease.
Developmentally regulated subcellular localization
In addition to developmentally regulated, cell-specific gene expression and alternative splicing events, temporal changes in subcellular localization may represent another key contributor to the unique developmental functions of disease-related ion channels and synaptic proteins. As mentioned above, loss-of-function mutations in SCN2A have been associated with ASD and ID, whereas gain-of-function SCN2A variants have been linked to infantile epilepsies (Sanders et al., 2018). While only a small fraction of SCN2A disease-associated mutations have been functionally characterized, a recent study demonstrated that 11 different ASD-associated SCN2A variants inhibited or completely abrogated Nav1.2 channel function in HEK cells. Moreover, these mutations had more pronounced effects on the excitability of immature cortical neurons as compared to mature pyramidal cells (BenShalom et al., 2017). While it is possible that the temporal switch from 5N-expressing SCN2A variants to adult 5A isoforms contributes to the selective consequences of these ASD mutations in immature neurons, the specificity of these deficits likely also results from the developmental regulation of Nav1.2 subcellular localization during early postnatal life.
In immature cortical neurons, Nav1.2 channels are expressed throughout the axon and in the axon initial segment (AIS), making them critical for action potential generation and propagation (Figure 2) (Boiko et al., 2003; Gazina et al., 2015; Spratt et al., 2019). In mature cortical neurons, however, Nav1.2 channels are replaced by Nav1.6 at the distal AIS and nodes of Ranvier, and the restriction of Nav1.2 expression to the proximal AIS, soma and dendrites changes its contribution to mature neuronal function (Figure 2) (Boiko et al., 2003; Gazina et al., 2015; Hu et al., 2009; Spratt et al., 2019). As a consequence of this developmental reorganization of Nav1.2 channel distribution, Nav1.2 loss of function has differential effects on cortical neurons at different stages of maturation: whereas Scn2a haploinsufficiency in immature neurons during the first postnatal week reduces neuronal excitability and impairs spiking, mature Scn2a+/– cortical neurons instead display deficits in dendritic action potential backpropagation and synaptic function, as well as structurally immature dendritic spines (Spratt et al., 2019). Two recent studies further interrogated the effects of complete Scn2a loss of function on mature neuronal activity, discovering unexpected cellular phenotypes upon complete inactivation of Scn2a (Scn2a−/−) (Spratt et al., 2021; Zhang et al., 2021). Notably, adult mice lacking both copies of Scn2a in mature cortical neurons display increased neuronal excitability due to compensatory changes in potassium channel expression (Spratt et al., 2021; Zhang et al., 2021). These tantalizing data set the stage for future studies exploring: how different SCN2A variants impact dendritic structure, neuronal excitability and synaptic function at specific stages of neuronal maturation; how early changes in neuronal maturation contribute to circuit assembly and function; how compensatory changes in ion channel expression or distribution can result in paradoxical phenotypes; and how the developmental change in Nav1.2 channel localization contributes to the phenotypic heterogeneity of SCNA-associated disorders.
Figure 2:
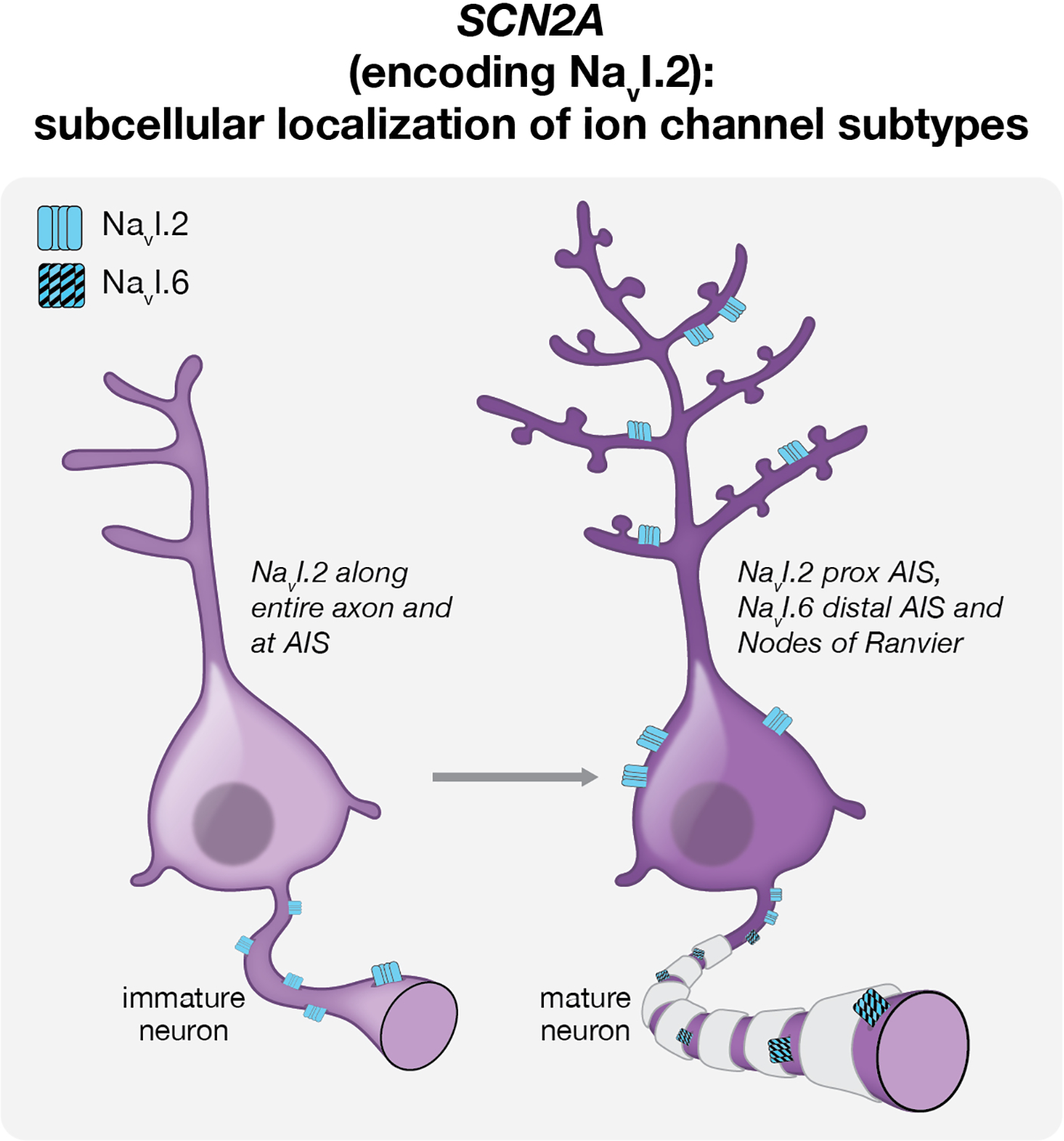
Developmental reorganization of Nav1.2 channels contributes to distinct functions at different stages of neuronal maturation. In immature neurons (left), Nav1.2 channels decorate the axon initial segment (AIS) and entire length of the axon, underpinning their indispensable role in action potential generation and propagation at early postnatal ages. In mature neurons (right), Nav1.2 channels display somatodendritic localization and are restricted to the proximal AIS. They are replaced in the distal AIS and Nodes of Ranvier by Nav1.6 channels. As a consequence of this dynamic subcellular regulation of channel distribution, Scn2a inactivation yields distinct electrophysiological phenotypes in immature and mature cortical projection neurons, and SCN2A loss-of-function variants preferentially affect immature neuron excitability.
Recent studies have also shed light on spatiotemporal regulation of synaptic protein distribution as a potential driver of their novel developmental functions. In one study, the ASD and Phelan-McDermid (PMD) Syndrome-associated synaptic protein SHANK3 (Bonaglia et al., 2001; De Rubeis et al., 2014; Durand et al., 2007; Sanders et al., 2015; Satterstrom et al., 2020) was found to play a critical but unexpected developmental role in regulating intrinsic neuronal excitability in human neurons (Yi et al., 2016). SHANK3 is enriched in neuronal post-synaptic densities, where it acts as a scaffold that orchestrates post-synaptic signaling (Naisbitt et al., 1999; Sheng and Kim, 2000). Consequently, it has been thought that mutations affecting SHANK3 expression or function cause neurodevelopmental deficits primarily by impairing synaptic activity (Bozdagi et al., 2010; Kouser et al., 2013; Monteiro and Feng, 2017; Wang et al., 2011). Interestingly, however, PMD patient neurons derived from hiPS cells display increased input resistance in addition to altered synaptic transmission (Kathuria et al., 2018; Shcheglovitov et al., 2013), supporting the possibility that SHANK3 may subserve a distinct earlier role separate from its conventional function as a scaffolding protein at the synapse. Using homologous recombination to conditionally inactivate SHANK3 in hES cell lines, Yi et al. generated matched isogenic control and SHANK3 mutant neurons and found that SHANK3 loss of function, in addition to causing synaptic deficits, resulted in preceding changes: impaired dendritic arborization and increased neuronal input resistance and excitability (Yi et al., 2016). The authors show that this effect on neuronal excitability is mediated by a direct interaction between SHANK3 and voltage gated HCN channels. They also demonstrate that persistent pharmacological suppression of Ih currents phenocopies the deficits in neuronal morphology and synaptic function observed in SHANK3 mutant neurons. This novel, earlier developmental function of SHANK3 may thus underlie the constellation of cellular phenotypes resulting from SHANK3 disease-associated loss-of-function mutations.
The role of SHANK3 in regulating neuronal excitability is also highlighted by a recent assembloid model of the cortico-striatal circuit (Miura et al., 2020). Striatal organoids derived from patients with PMD did not display changes in neuronal activity when grown in isolation, as measured by calcium imaging, but following assembly with cortical glutamatergic neurons, striatal neurons from PMD patients displayed an increased number of calcium spike events (Miura et al., 2020). These data in human cells are consistent with a study in rodents demonstrating that early increases in cortical activity in Shank3B−/− mutant animals drive premature maturation of striatal projection neurons and corticostriatal connectivity (Peixoto et al., 2016). A subsequent study performed in Shank3B−/− mice revealed that cortical hyperactivity is the result of impaired interneuron function, and selective disruption of Shank3 in cortical interneurons promotes pyramidal neuron hyperexcitability (Chen et al., 2020). These findings reinforce the importance of interrogating the function of neuropsychiatric disease risk genes in different cell types to uncover disease mechanisms.
Notably, transcripts encoding proteins in the SHANK family are alternatively spliced to generate isoforms with distinct subcellular localization and temporal expression patterns in the mouse (Lim et al., 1999; Peça et al., 2011; Wang et al., 2014) suggests that differential expression of SHANK3 isoforms may also contribute to diverse subcellular functions of SHANK3 proteins. While Shank3B−/− mice completely lack two of the major Shank3 isoforms and express markedly reduced levels of a third isoform (Peça et al. 2011), mouse models in which different Shank3 isoforms are deleted have been generated by targeting regions corresponding to specific protein interaction domains (Zhu et al. 2018). Such models also support the idea that Shank3 deficiency impacts neuronal excitability (Zhu et al., 2018a). However, only disruption of specific Shank3 isoforms is associated with HCN channelopathy, whereas disrupting a domain that spares one isoform does not impact Ih currents (Zhu et al., 2018a). Understanding how neurodevelopmental disease-associated mutations in SHANK3 impinge on the subcellular localization and function of individual SHANK3 isoforms in different neuronal subtypes will therefore be essential to shed light on the unique cellular phenotypes resulting from different mutations (Wang et al., 2014).
In a similar vein, the gene encoding the ASD and ID-related synaptic protein SynGAP, SYNGAP1, is alternatively spliced to generate proteins that display unique spatiotemporal localization in immature neurons (Araki et al., 2020). SynGAP is a postsynaptic protein localized to dendritic spines that has been shown to interact with PSD-95 and regulate synaptic plasticity (Chen et al., 1998; Kim et al., 1998). Multiple ASD- and ID-associated de novo splice site mutations in SYNGAP1 have been identified (Vlaskamp et al., 2019), and loss-of-function SYNGAP1 variants have been strongly associated with a variety of neurodevelopmental disorders (Deciphering Developmental Disorders Study, 2015; Hamdan et al., 2009; Parker et al., 2015; Rauch et al., 2012). A recent study by Araki et al. demonstrates differential subcellular localization and biochemical properties of distinct SynGAP isoforms. Expression of the shorter SynGAP-β isoform, which is diffusely cytoplasmically localized in immature neurons, precedes expression of synaptically targeted SynGAP isoforms and plays a specific role in the elaboration of dendritic arbors (Figure 3) (Araki et al., 2020). In contrast, the most well studied SynGAP isoform, SynGAP-α1, is concentrated at the postsynaptic density by virtue of two structural domains, both of which SynGAP-β lacks: a coiled-coiled domain enabling phase separation and a PDZ binding domain enabling tight association with PSD-95 (Araki et al., 2020). Mutations in SynGAP-α1 preventing phase separation or PSD-95 binding render it more similar to SynGAP-β, disrupting synaptic targeting and redistributing SynGAP-α1 to the dendrite, where it then contributes to dendritic arborization (Araki et al., 2020). This work underscores the importance of characterizing the spatiotemporal expression patterns and sub-cellular functions of different isoforms in order to inform our understanding of neurodevelopmental disease-associated SYNGAP1 variants.
Figure 3:
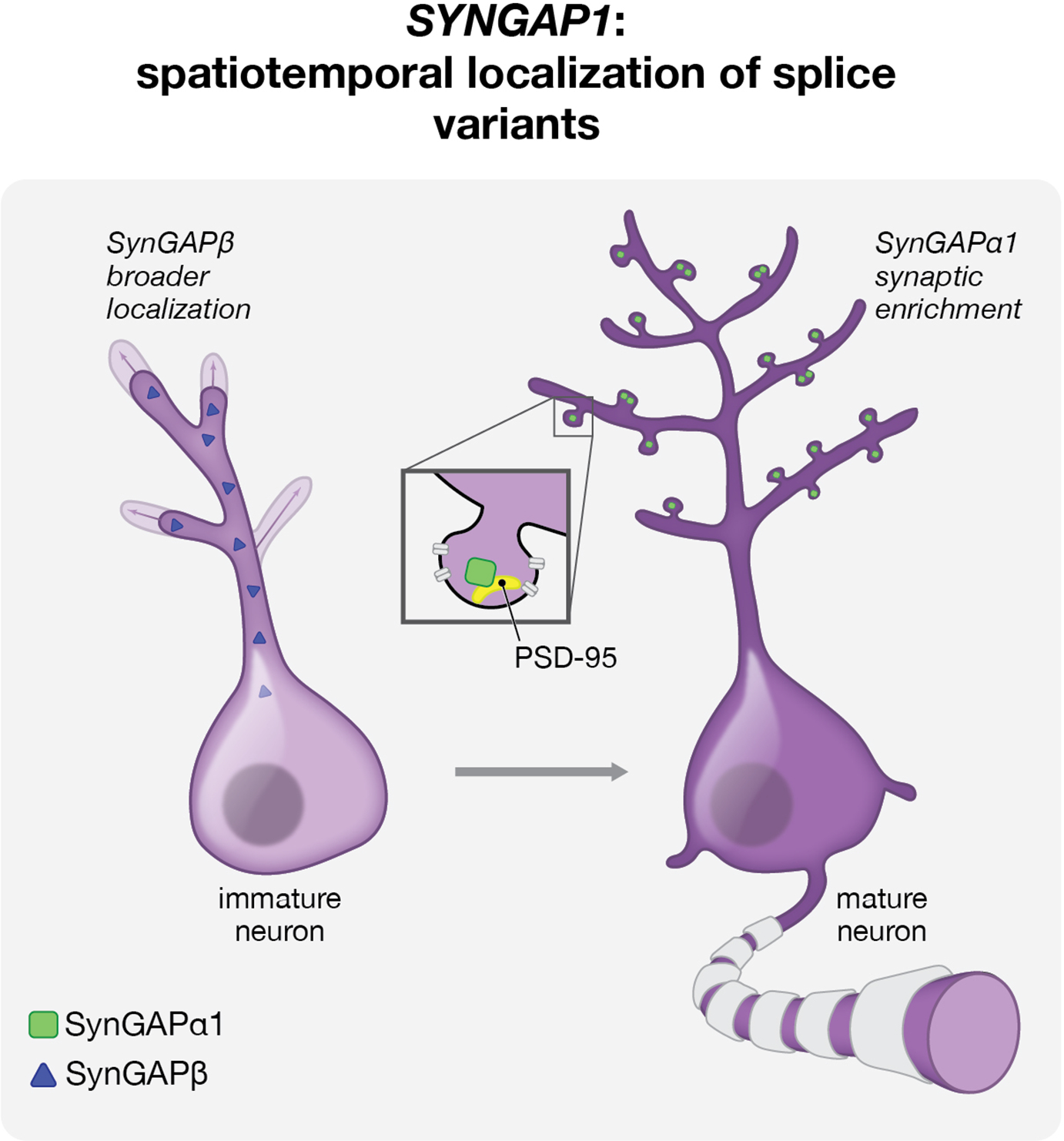
Alternative splicing of Syngap1 gives rise to SynGAP isoforms with distinct, temporally-regulated subcellullar localization in immature and mature neurons. Earlier and less synaptically-enriched expression of the β isoform of SynGAP in immature neurons (left) results in a specific role for this classical synaptic protein in dendritic arborization. In mature neurons (right), SynGAP-α1 is enriched at the postsynaptic density, where it directly interacts with PSD-95 and regulates synaptic plasticity. Dendritic arborization is altered in human neurons lacking all SYNGAP1 isoforms, but it is not yet clear in human cells how this developmental reorganization of SynGAP isoforms might be influenced by SYNGAP1 mutations.
hiPS cell-derived neurons engineered to lack all SynGAP isoforms display alterations in dendritic morphogenesis and synaptic transmission (Llamosas et al., 2020) It remains unclear, however, how different SynGAP isoforms are localized to regulate distinct aspects of neuronal maturation in human cells and how these isoforms might be disrupted by disease-associated SYNGAP1 mutations. Together, these examples draw attention to dynamic changes in the subcellular localization of disease-relevant ion channels and synaptic proteins that can give rise to distinct functions in different cellular compartments and at different stages of development. They reinforce the need to conduct parallel investigations in model organisms and human cells using single cell and spatial “omics” approaches to probe the dynamic spatiotemporal and cell type-specific changes that occur at critical developmental transitions.
Looking ahead
A burgeoning of genetic studies has identified many neuropsychiatric disease-associated risk variants. Substantial evidence has also emerged pointing to genetic overlap across multiple neuropsychiatric disorders (Cross-Disorder Group of the Psychiatric Genomics Consortium, 2013; Cross-Disorder Group of the Psychiatric Genomics Consortium et al., 2013; Cross-Disorder Group of the Psychiatric Genomics Consortium. Electronic address: plee0@mgh.harvard.edu and Cross-Disorder Group of the Psychiatric Genomics Consortium, 2019; Gandal et al., 2018b). The use of integrative analyses on genetic and proteomic datasets in the past decade has defined splicing networks, protein interaction maps, and neurodevelopmental programs onto which genetic risk converges (Neale et al., 2012; O’Roak et al., 2012; Sanders et al., 2012; Sullivan and Geschwind, 2019). More recently, bulk and single cell interrogation of chromatin dynamics and gene regulatory networks in the developing human cortex has enabled the identification of novel ASD-associated gene variants and the mapping of non-coding mutations in an ASD patient cohort onto specific developing cell types (Markenscoff-Papadimitriou et al., 2020; Trevino et al., 2021). Stemming from such studies, it has been proposed that specific cellular processes, developmental windows or cell types (Gilman et al., 2011; Gulsuner et al., 2013; Li et al., 2018; Parikshak et al., 2016; State and Šestan, 2012; Tebbenkamp et al., 2014; Willsey et al., 2013; Zoghbi, 2003) in the prenatal brain are preferentially vulnerable to neuropsychiatric disease-associated mutations. These ideas have spurred exciting, largescale collaborative efforts aimed at massively parallel interrogations of disease risk genes (Sanders et al., 2019; Willsey et al., 2018, 2021). The ultimate goal of such endeavors is to define gene and protein interaction networks and identify points of convergence at the cellular or circuit level in the context of disease.
It has become increasingly clear, however, that disease-associated risk genes can display dynamic expression trajectories across development (Gulsuner et al., 2013; Li et al., 2018; Tebbenkamp et al., 2014; Willsey and State, 2015; Zhu et al., 2018b). More recent studies are also revealing a complexity beyond the level of gene transcription. As we discuss in this perspective, mechanisms including splice isoform selection and differential subcellular localization confer non-canonical, temporally distinct roles to proteins encoded by disease risk genes. We presented individual examples of spatiotemporal changes in gene expression, isoform utilization and subcellular localization as three axes of dynamic change that bestow novel developmental functions on individual well-studied, disease-associated ion channels and synaptic proteins. These snapshots highlight the possibility that preventing or altering such spatiotemporally regulated events is another way in which disease-associated mutations might disrupt specific cellular behaviors in the developing brain.
As described above, rigorous functional interrogation of disease-associated ion channels and synaptic proteins in differentiating and/or maturing cells is also uncovering unexpected disease-relevant phenotypes. Such cellular phenotypes appear to result as a consequence of the complex spatial, temporal and cell type-specific regulation of these proteins, in some cases secondary to homeostatic mechanisms. To fully understand the contribution of neuropsychiatric disorder risk genes to cellular phenotypes underlying disease, we must extend our emerging systems-level framework of neuropsychiatric disorders. We propose that this should include a more refined mechanistic understanding of how the protein products of disease risk genes dynamically change in space and time within specific cell types to impart unique functions. How do these molecular dynamics in turn impact the association of these proteins with proteins encoded by other risk genes in different neural cell types and at different developmental stages? Moving forward, we must consolidate a more complete understanding of the full repertoire of developmental changes in ion channel and synaptic protein expression and localization, not just individual changes across a single axis as those described above. How are these dynamics regulated across neuronal maturation? In graded or restricted areal- or laminar-specific patterns? With cell type type-specific enrichment? In distinct brain regions? Across lineage progression? How can we map cellular phenotypes and candidate disease-relevant molecular pathways onto multiple changing trajectories across space and time? Moreover, we must begin to incorporate how extrinsic features, including electrical activity, immune state, and metabolic function, can intersect with these cellular and subcellular properties to promote phenotypic heterogeneity.
ACKNOWLEDGEMENTS
We thank T. Palmer, M. State and J. Rubenstein for critical feedback on our manuscript. We also thank the members of our laboratories for important discussions surrounding disease mechanisms and cell type specificity.
This work was supported by the UCSF Program for Breakthrough Biomedical Research, Sandler Foundation (Faculty Fellows Program), NIH R01 MH125004 and R56 MH127075 (to G.P.), and the Stanford Brain Organogenesis (Wu Tsai Neurosciences Institute), Bio-X, a New York Stem Cell Foundation–Robertson Investigator Award and Chan Zuckerberg Ben Barres Investigator Award, NIH R01 MH115012 (to S.P.P.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Araki Y, Hong I, Gamache TR, Ju S, Collado-Torres L, Shin JH, and Huganir RL (2020). SynGAP isoforms differentially regulate synaptic plasticity and dendritic development. Elife 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagasrawala I, Memi F, Radonjic N, V., and Zecevic N (2017). N-Methyl d-Aspartate Receptor Expression Patterns in the Human Fetal Cerebral Cortex. Cereb. Cortex 27, 5041–5053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckh S, Noda M, Lübbert H, and Numa S (1989). Differential regulation of three sodium channel messenger RNAs in the rat central nervous system during development. EMBO J. 8, 3611–3616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Shalom R, Keeshen CM, Berrios KN, An JY, Sanders SJ, and Bender KJ (2017). Opposing effects on NaV1.2 function underlie differences between SCN2A variants observed in individuals with autism spectrum disorder or infantile seizures. Biol. Psychiatry 82, 224–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berret E, Barron T, Xu J, Debner E, Kim EJ, and Kim JH (2017). Oligodendroglial excitability mediated by glutamatergic inputs and Nav1.2 activation. Nat. Commun 8, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat S, Dao DT, Terrillion CE, Arad M, Smith RJ, Soldatov NM, and Gould TD (2012). CACNA1C (Cav1.2) in the pathophysiology of psychiatric disease. Prog. Neurobiol 99, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birey F, Andersen J, Makinson CD, Islam S, Wei W, Huber N, Fan HC, Metzler KRC, Panagiotakos G, Thom N, et al. (2017). Assembly of functionally integrated human forebrain spheroids. Nature 545, 54–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birey F, Li M-Y, Gordon A, Thete MV, Valencia AM, Revah O, Pașca AM, Geschwind DH, and Pașca SP (2021). Dissecting the molecular basis of human interneuron migration in forebrain assembloids from Timothy syndrome. [DOI] [PubMed] [Google Scholar]
- Blanton MG, Lo Turco JJ, and Kriegstein AR (1990). Endogenous neurotransmitter activates N-methyl-D-aspartate receptors on differentiating neurons in embryonic cortex. Proc. Natl. Acad. Sci. U. S. A 87, 8027–8030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boiko T, Van Wart A, Caldwell JH, Levinson SR, Trimmer JS, and Matthews G (2003). Functional specialization of the axon initial segment by isoform-specific sodium channel targeting. J. Neurosci 23, 2306–2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonaglia MC, Giorda R, Borgatti R, Felisari G, Gagliardi C, Selicorni A, and Zuffardi O (2001). Disruption of the ProSAP2 gene in a t(12;22)(q24.1;q13.3) is associated with the 22q13.3 deletion syndrome. Am. J. Hum. Genet 69, 261–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnefont J, and Vanderhaeghen P (2021). Neuronal fate acquisition and specification: time for a change. Curr. Opin. Neurobiol 66, 195–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutz PL, Stoilov P, Li Q, Lin C-H, Chawla G, Ostrow K, Shiue L, Ares M Jr, and Black DL (2007). A post-transcriptional regulatory switch in polypyrimidine tract-binding proteins reprograms alternative splicing in developing neurons. Genes Dev. 21, 1636–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozdagi O, Sakurai T, Papapetrou D, Wang X, Dickstein DL, Takahashi N, Kajiwara Y, Yang M, Katz AM, Scattoni ML, et al. (2010). Haploinsufficiency of the autism-associated Shank3 gene leads to deficits in synaptic function, social interaction, and social communication. Mol. Autism 1, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HJ, Rojas-Soto M, Oguni A, and Kennedy MB (1998). A synaptic Ras-GTPase activating protein (p135 SynGAP) inhibited by CaM kinase II. Neuron 20, 895–904. [DOI] [PubMed] [Google Scholar]
- Chen Q, Deister CA, Gao X, Guo B, Lynn-Jones T, Chen N, Wells MF, Liu R, Goard MJ, Dimidschstein J, et al. (2020). Dysfunction of cortical GABAergic neurons leads to sensory hyper-reactivity in a Shank3 mouse model of ASD. Nat. Neurosci 23, 520–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chih B, Gollan L, and Scheiffele P (2006). Alternative splicing controls selective transsynaptic interactions of the neuroligin-neurexin complex. Neuron 51, 171–178. [DOI] [PubMed] [Google Scholar]
- Ching MSL, Shen Y, Tan W-H, Jeste SS, Morrow EM, Chen X, Mukaddes NM, Yoo S-Y, Hanson E, Hundley R, et al. (2010). Deletions of NRXN1 (neurexin-1) predispose to a wide spectrum of developmental disorders. Am. J. Med. Genet. B Neuropsychiatr. Genet 153B, 937–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark MB, Wrzesinski T, Garcia AB, Hall NAL, Kleinman JE, Hyde T, Weinberger DR, Harrison PJ, Haerty W, and Tunbridge EM (2020). Long-read sequencing reveals the complex splicing profile of the psychiatric risk gene CACNA1C in human brain. Mol. Psychiatry 25, 37–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross-Disorder Group of the Psychiatric Genomics Consortium (2013). Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet 381, 1371–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross-Disorder Group of the Psychiatric Genomics Consortium, Lee SH, Ripke S, Neale BM, Faraone SV, Purcell SM, Perlis RH, Mowry BJ, Thapar A, Goddard ME, et al. (2013). Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat. Genet 45, 984–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross-Disorder Group of the Psychiatric Genomics Consortium. Electronic address: plee0@mgh.harvard.edu, and Cross-Disorder Group of the Psychiatric Genomics Consortium (2019). Genomic Relationships, Novel Loci, and Pleiotropic Mechanisms across Eight Psychiatric Disorders. Cell 179, 1469–1482.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai J, Aoto J, and Südhof TC (2019). Alternative Splicing of Presynaptic Neurexins Differentially Controls Postsynaptic NMDA and AMPA Receptor Responses. Neuron 102, 993–1008.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deciphering Developmental Disorders Study (2015). Large-scale discovery of novel genetic causes of developmental disorders. Nature 519, 223–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Marco García NV, Priya R, Tuncdemir SN, Fishell G, and Karayannis T (2015). Sensory inputs control the integration of neurogliaform interneurons into cortical circuits. Nat. Neurosci 18, 393–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deneault E, White SH, Rodrigues DC, Ross PJ, Faheem M, Zaslavsky K, Wang Z, Alexandrova R, Pellecchia G, Wei W, et al. (2018). Complete Disruption of AutismSusceptibility Genes by Gene Editing Predominantly Reduces Functional Connectivity of Isogenic Human Neurons. Stem Cell Reports 11, 1211–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, Kou Y, Liu L, Fromer M, Walker S, et al. (2014). Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Bella DJ, Habibi E, Stickels RR, Scalia G, Brown J, Yadollahpour P, Yang SM, Abbate C, Biancalani T, Macosko EZ, et al. (2021). Molecular logic of cellular diversification in the mouse cerebral cortex. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diebold RJ, Koch WJ, Ellinor PT, Wang JJ, Muthuchamy M, Wieczorek DF, and Schwartz A (1992). Mutually exclusive exon splicing of the cardiac calcium channel alpha 1 subunit gene generates developmentally regulated isoforms in the rat heart. Proc. Natl. Acad. Sci. U. S. A 89, 1497–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand CM, Betancur C, Boeckers TM, Bockmann J, Chaste P, Fauchereau F, Nygren G, Rastam M, Gillberg IC, Anckarsäter H, et al. (2007). Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat. Genet 39, 25–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endele S, Rosenberger G, Geider K, Popp B, Tamer C, Stefanova I, Milh M, Kortüm F, Fritsch A, Pientka FK, et al. (2010). Mutations in GRIN2A and GRIN2B encoding regulatory subunits of NMDA receptors cause variable neurodevelopmental phenotypes. Nat. Genet 42, 1021–1026. [DOI] [PubMed] [Google Scholar]
- Espinosa JS, and Luo L (2008). Timing neurogenesis and differentiation: insights from quantitative clonal analyses of cerebellar granule cells. J. Neurosci 28, 2301–2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favero M, Sotuyo NP, Lopez E, Kearney JA, and Goldberg EM (2018). A transient developmental window of fast-spiking interneuron dysfunction in a mouse model of Dravet syndrome. J. Neurosci [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaherty E, Zhu S, Barretto N, Cheng E, Deans PJM, Fernando MB, Schrode N, Francoeur N, Antoine A, Alganem K, et al. (2019). Neuronal impact of patient-specific aberrant NRXN1α splicing. Nat. Genet 51, 1679–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuccillo MV, Földy C, Gökce Ö, Rothwell PE, Sun GL, Malenka RC, and Südhof TC (2015). Single-Cell mRNA Profiling Reveals Cell-Type-Specific Expression of Neurexin Isoforms. Neuron 87, 326–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandal MJ, Zhang P, Hadjimichael E, Walker RL, Chen C, Liu S, Won H, van Bakel H, Varghese M, Wang Y, et al. (2018a). Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandal MJ, Haney JR, Parikshak NN, Leppa V, Ramaswami G, Hartl C, Schork AJ, Appadurai V, Buil A, Werge TM, et al. (2018b). Shared molecular neuropathology across major psychiatric disorders parallels polygenic overlap. Science 359, 693–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazina EV, Richards KL, Mokhtar MBC, Thomas EA, Reid CA, and Petrou S (2010). Differential expression of exon 5 splice variants of sodium channel alpha subunit mRNAs in the developing mouse brain. Neuroscience 166, 195–200. [DOI] [PubMed] [Google Scholar]
- Gazina EV, Leaw BTW, Richards KL, Wimmer VC, Kim TH, Aumann TD, Featherby TJ, Churilov L, Hammond VE, Reid CA, et al. (2015). “Neonatal” Nav1.2 reduces neuronal excitability and affects seizure susceptibility and behaviour. Hum. Mol. Genet 24, 1457–1468. [DOI] [PubMed] [Google Scholar]
- Gilman SR, Iossifov I, Levy D, Ronemus M, Wigler M, and Vitkup D (2011). Rare de novo variants associated with autism implicate a large functional network of genes involved in formation and function of synapses. Neuron 70, 898–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon A, Yoon S-J, Tran SS, Makinson CD, Park JY, Andersen J, Valencia AM, Horvath S, Xiao X, Huguenard JR, et al. (2021). Long-term maturation of human cortical organoids matches key early postnatal transitions. Nat. Neurosci 24, 331–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould E, and Kim JH (2021). SCN2A contributes to oligodendroglia excitability and development in the mammalian brain. Cell Rep. 36, 109653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greig LC, Woodworth MB, Galazo MJ, Padmanabhan H, and Macklis JD (2013). Molecular logic of neocortical projection neuron specification, development and diversity. Nat. Rev. Neurosci 14, 755–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulsuner S, Walsh T, Watts AC, Lee MK, Thornton AM, Casadei S, Rippey C, Shahin H, Consortium on the Genetics of Schizophrenia (COGS), PAARTNERS Study Group, et al. (2013). Spatial and temporal mapping of de novo mutations in schizophrenia to a fetal prefrontal cortical network. Cell 154, 518–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamdan FF, Gauthier J, Spiegelman D, Noreau A, Yang Y, Pellerin S, Dobrzeniecka S, Côté M, Perreau-Linck E, Carmant L, et al. (2009). Mutations in SYNGAP1 in autosomal nonsyndromic mental retardation. N. Engl. J. Med 360, 599–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S, Tai C, Westenbroek RE, Yu FH, Cheah CS, Potter GB, Rubenstein JL, Scheuer T, de la Iglesia HO, and Catterall WA (2012). Autistic-like behaviour in Scn1a+/− mice and rescue by enhanced GABA-mediated neurotransmission. Nature 489, 385–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodge RD, Bakken TE, Miller JA, Smith KA, Barkan ER, Graybuck LT, Close JL, Long B, Johansen N, Penn O, et al. (2019). Conserved cell types with divergent features in human versus mouse cortex. Nature 573, 61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W, Tian C, Li T, Yang M, Hou H, and Shu Y (2009). Distinct contributions of Na(v)1.6 and Na(v)1.2 in action potential initiation and backpropagation. Nat. Neurosci 12, 996–1002. [DOI] [PubMed] [Google Scholar]
- Iossifov I, O’Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, Stessman HA, Witherspoon KT, Vives L, Patterson KE, et al. (2014). The contribution of de novo coding mutations to autism spectrum disorder. Nature 515, 216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irimia M, Weatheritt RJ, Ellis JD, Parikshak NN, Gonatopoulos-Pournatzis T, Babor M, Quesnel-Vallières M, Tapial J, Raj B, O’Hanlon D, et al. (2014). A highly conserved program of neuronal microexons is misregulated in autistic brains. Cell 159, 1511–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jabaudon D (2017). Fate and freedom in developing neocortical circuits. Nat. Commun 8, 16042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamain S, Quach H, Betancur C, Råstam M, Colineaux C, Gillberg IC, Soderstrom H, Giros B, Leboyer M, Gillberg C, et al. (2003). Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat. Genet 34, 27–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins AK, Paterson C, Wang Y, Hyde TM, Kleinman JE, and Law AJ (2016). Neurexin 1 (NRXN1) splice isoform expression during human neocortical development and aging. Mol. Psychiatry 21, 701–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Jiang W, Zou J, Wang B, Yu M, Pan Y, Lin Y, Mao Y, and Wang Y (2015). The GluN2B subunit of N-methy-D-asparate receptor regulates the radial migration of cortical neurons in vivo. Brain Res. 1610, 20–32. [DOI] [PubMed] [Google Scholar]
- Jiang P, Chen C, Liu X-B, Selvaraj V, Liu W, Feldman DH, Liu Y, Pleasure DE, Li RA, and Deng W (2013). Generation and characterization of spiking and nonspiking oligodendroglial progenitor cells from embryonic stem cells. Stem Cells 31, 2620–2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao J, Yang Y, Shi Y, Chen J, Gao R, Fan Y, Yao H, Liao W, Sun X-F, and Gao S (2013). Modeling Dravet syndrome using induced pluripotent stem cells (iPSCs) and directly converted neurons. Hum. Mol. Genet 22, 4241–4252. [DOI] [PubMed] [Google Scholar]
- Kamijo S, Ishii Y, Horigane S-I, Suzuki K, Ohkura M, Nakai J, Fujii H, TakemotoKimura S, and Bito H (2018). A Critical Neurodevelopmental Role for L-Type Voltage-Gated Calcium Channels in Neurite Extension and Radial Migration. J. Neurosci 38, 5551–5566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanaumi T, Takashima S, Iwasaki H, Itoh M, Mitsudome A, and Hirose S (2008). Developmental changes in KCNQ2 and KCNQ3 expression in human brain: possible contribution to the age-dependent etiology of benign familial neonatal convulsions. Brain Dev. 30, 362–369. [DOI] [PubMed] [Google Scholar]
- Kathuria A, Nowosiad P, Jagasia R, Aigner S, Taylor RD, Andreae LC, Gatford NJF, Lucchesi W, Srivastava DP, and Price J (2018). Stem cell-derived neurons from autistic individuals with SHANK3 mutation show morphogenetic abnormalities during early development. Mol. Psychiatry 23, 735–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelsch W, Li Z, Eliava M, Goengrich C, and Monyer H (2012). GluN2B-Containing NMDA Receptors Promote Wiring of Adult-Born Neurons into Olfactory Bulb Circuits. J. Neurosci 32, 12603–12611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H-G, Kishikawa S, Higgins AW, Seong I-S, Donovan DJ, Shen Y, Lally E, Weiss LA, Najm J, Kutsche K, et al. (2008). Disruption of neurexin 1 associated with autism spectrum disorder. Am. J. Hum. Genet 82, 199–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Liao D, Lau LF, and Huganir RL (1998). SynGAP: a synaptic RasGAP that associates with the PSD-95/SAP90 protein family. Neuron 20, 683–691. [DOI] [PubMed] [Google Scholar]
- Kouser M, Speed HE, Dewey CM, Reimers JM, Widman AJ, Gupta N, Liu S, Jaramillo TC, Bangash M, Xiao B, et al. (2013). Loss of predominant Shank3 isoforms results in hippocampus-dependent impairments in behavior and synaptic transmission. J. Neurosci 33, 18448–18468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krey JF, Paşca SP, Shcheglovitov A, Yazawa M, Schwemberger R, Rasmusson R, and Dolmetsch RE (2013). Timothy syndrome is associated with activity-dependent dendritic retraction in rodent and human neurons. Nat. Neurosci 16, 201–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar SS, Bacci A, Kharazia V, and Huguenard JR (2002). A developmental switch of AMPA receptor subunits in neocortical pyramidal neurons. J. Neurosci 22, 3005–3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan KY, Sestan N, and Anton ES (2012). Transcriptional co-regulation of neuronal migration and laminar identity in the neocortex. Development 139, 1535–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J-A, Damianov A, Lin C-H, Fontes M, Parikshak NN, Anderson ES, Geschwind DH, Black DL, and Martin KC (2016). Cytoplasmic Rbfox1 Regulates the Expression of Synaptic and Autism-Related Genes. Neuron 89, 113–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leone DP, Srinivasan K, Chen B, Alcamo E, and McConnell SK (2008). The determination of projection neuron identity in the developing cerebral cortex. Curr. Opin. Neurobiol 18, 28–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy D, Ronemus M, Yamrom B, Lee Y-H, Leotta A, Kendall J, Marks S, Lakshmi B, Pai D, Ye K, et al. (2011). Rare de novo and transmitted copy-number variation in autistic spectrum disorders. Neuron 70, 886–897. [DOI] [PubMed] [Google Scholar]
- Li M, Santpere G, Imamura Kawasawa Y, Evgrafov OV, Gulden FO, Pochareddy S, Sunkin SM, Li Z, Shin Y, Zhu Y, et al. (2018). Integrative functional genomic analysis of human brain development and neuropsychiatric risks. Science 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang L, Fazel Darbandi S, Pochareddy S, Gulden FO, Gilson MC, Sheppard BK, Sahagun A, An J-Y, Werling DM, Rubenstein JLR, et al. (2021). Developmental dynamics of voltage-gated sodium channel isoform expression in the human and mouse brain. Genome Med. 13, 135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lignani G, Liavas A, Kullmann DM, and Schorge S (2020). Alternative splicing tunes sodium channels to support channel- and neuron-specific effects.
- Lim S, Naisbitt S, Yoon J, Hwang JI, Suh PG, Sheng M, and Kim E (1999). Characterization of the Shank family of synaptic proteins. Multiple genes, alternative splicing, and differential expression in brain and development. J. Biol. Chem 274, 29510–29518. [DOI] [PubMed] [Google Scholar]
- Liu H, Wang H, Peterson M, Zhang W, Hou G, and Zhang Z-W (2019). N-terminal alternative splicing of GluN1 regulates the maturation of excitatory synapses and seizure susceptibility. Proc. Natl. Acad. Sci. U. S. A 116, 21207–21212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Lopez-Santiago LF, Yuan Y, Jones JM, Zhang H, O’Malley HA, Patino GA, O’Brien JE, Rusconi R, Gupta A, et al. (2013). Dravet syndrome patient-derived neurons suggest a novel epilepsy mechanism. Ann. Neurol 74, 128–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llamosas N, Arora V, Vij R, Kilinc M, Bijoch L, Rojas C, Reich A, Sridharan B, Willems E, Piper DR, et al. (2020). SYNGAP1 Controls the Maturation of Dendrites, Synaptic Function, and Network Activity in Developing Human Neurons. J. Neurosci 40, 7980–7994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C, Shi X, Allen A, Baez-Nieto D, Nikish A, Sanjana NE, and Pan JQ (2019). Overexpression of NEUROG2 and NEUROG1 in human embryonic stem cells produces a network of excitatory and inhibitory neurons. FASEB J. 33, 5287–5299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makeyev EV, Zhang J, Carrasco MA, and Maniatis T (2007). The MicroRNA miR-124 promotes neuronal differentiation by triggering brain-specific alternative pre-mRNA splicing. Mol. Cell 27, 435–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markenscoff-Papadimitriou E, Whalen S, Przytycki P, Thomas R, Binyameen F, Nowakowski TJ, Kriegstein AR, Sanders SJ, State MW, Pollard KS, et al. (2020). A Chromatin Accessibility Atlas of the Developing Human Telencephalon. Cell 182, 754–769.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matta JA, Pelkey KA, Craig MT, Chittajallu R, Jeffries BW, and McBain CJ (2013). Developmental origin dictates interneuron AMPA and NMDA receptor subunit composition and plasticity. Nat. Neurosci 16, 1032–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer S, Chen J, Velmeshev D, Mayer A, Eze UC, Bhaduri A, Cunha CE, Jung D, Arjun A, Li E, et al. (2019). Multimodal Single-Cell Analysis Reveals Physiological Maturation in the Developing Human Neocortex. Neuron 102, 143–158.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisler MH, and Kearney JA (2005). Sodium channel mutations in epilepsy and other neurological disorders. J. Clin. Invest 115, 2010–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisler MH, Hill SF, and Yu W (2021). Sodium channelopathies in neurodevelopmental disorders. Nat. Rev. Neurosci 22, 152–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JA, Ding S-L, Sunkin SM, Smith KA, Ng L, Szafer A, Ebbert A, Riley ZL, Royall JJ, Aiona K, et al. (2014). Transcriptional landscape of the prenatal human brain. Nature 508, 199–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura Y, Li M-Y, Birey F, Ikeda K, Revah O, Thete MV, Park J-Y, Puno A, Lee SH, Porteus MH, et al. (2020). Generation of human striatal organoids and cortico-striatal assembloids from human pluripotent stem cells. Nat. Biotechnol 38, 1421–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyata T, Kawaguchi D, Kawaguchi A, and Gotoh Y (2010). Mechanisms that regulate the number of neurons during mouse neocortical development. Curr. Opin. Neurobiol 20, 22–28. [DOI] [PubMed] [Google Scholar]
- Monteiro P, and Feng G (2017). SHANK proteins: roles at the synapse and in autism spectrum disorder. Nat. Rev. Neurosci 18, 147–157. [DOI] [PubMed] [Google Scholar]
- Naisbitt S, Kim E, Tu JC, Xiao B, Sala C, Valtschanoff J, Weinberg RJ, Worley PF, and Sheng M (1999). Shank, a novel family of postsynaptic density proteins that binds to the NMDA receptor/PSD-95/GKAP complex and cortactin. Neuron 23, 569–582. [DOI] [PubMed] [Google Scholar]
- Neale BM, Kou Y, Liu L, Ma’ayan A, Samocha KE, Sabo A, Lin C-F, Stevens C, Wang L-S, Makarov V, et al. (2012). Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 485, 242–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowakowski TJ, Bhaduri A, Pollen AA, Alvarado B, Mostajo-Radji MA, Di Lullo E, Haeussler M, Sandoval-Espinosa C, Liu SJ, Velmeshev D, et al. (2017). Spatiotemporal gene expression trajectories reveal developmental hierarchies of the human cortex. Science 358, 1318–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogiwara I, Miyamoto H, Morita N, Atapour N, Mazaki E, Inoue I, Takeuchi T, Itohara S, Yanagawa Y, Obata K, et al. (2007). Nav1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: a circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation. J. Neurosci 27, 5903–5914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogiwara I, Miyamoto H, Tatsukawa T, Yamagata T, Nakayama T, Atapour N, Miura E, Mazaki E, Ernst SJ, Cao D, et al. (2018). Nav1.2 haplodeficiency in excitatory neurons causes absence-like seizures in mice. Commun Biol 1, 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, Levy R, Ko A, Lee C, Smith JD, et al. (2012). Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485, 246–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panagiotakos G, Haveles C, Arjun A, Petrova R, Rana A, Portmann T, Paşca SP, Palmer TD, and Dolmetsch RE (2019). Aberrant calcium channel splicing drives defects in cortical differentiation in Timothy syndrome. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoletti P, Bellone C, and Zhou Q (2013). NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci 14, 383–400. [DOI] [PubMed] [Google Scholar]
- Parikshak NN, Swarup V, Belgard TG, Irimia M, Ramaswami G, Gandal MJ, Hartl C, Leppa V, Ubieta L de la T, Huang J, et al. (2016). Genome-wide changes in lncRNA, splicing, and regional gene expression patterns in autism. Nature 540, 423–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker MJ, Fryer AE, Shears DJ, Lachlan KL, McKee SA, Magee AC, Mohammed S, Vasudevan PC, Park S-M, Benoit V, et al. (2015). De novo, heterozygous, loss-offunction mutations in SYNGAP1 cause a syndromic form of intellectual disability. Am. J. Med. Genet. A 167A, 2231–2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paşca SP (2018). Building three-dimensional human brain organoids. Nat. Neurosci [DOI] [PubMed] [Google Scholar]
- Paşca SP, Portmann T, Voineagu I, Yazawa M, Shcheglovitov A, Paşca AM, Cord B, Palmer TD, Chikahisa S, Nishino S, et al. (2011). Using iPSC-derived neurons to uncover cellular phenotypes associated with Timothy syndrome. Nat. Med 17, 1657–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peça J, Feliciano C, Ting JT, Wang W, Wells MF, Venkatraman TN, Lascola CD, Fu Z, and Feng G (2011). Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature 472, 437–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peixoto RT, Wang W, Croney DM, Kozorovitskiy Y, and Sabatini BL (2016). Early hyperactivity and precocious maturation of corticostriatal circuits in Shank3B(−/−) mice. Nat. Neurosci 19, 716–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planells-Cases R, Caprini M, Zhang J, Rockenstein EM, Rivera RR, Murre C, Masliah E, and Montal M (2000). Neuronal death and perinatal lethality in voltage-gated sodium channel alpha(II)-deficient mice. Biophys. J 78, 2878–2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pletikos M, Sousa AMM, Sedmak G, Meyer KA, Zhu Y, Cheng F, Li M, Kawasawa YI, and Sestan N (2014). Temporal specification and bilaterality of human neocortical topographic gene expression. Neuron 81, 321–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plummer NW, McBurney MW, and Meisler MH (1997). Alternative splicing of the sodium channel SCN8A predicts a truncated two-domain protein in fetal brain and non-neuronal cells. J. Biol. Chem 272, 24008–24015. [DOI] [PubMed] [Google Scholar]
- Polioudakis D, de la Torre-Ubieta L, Langerman J, Elkins AG, Shi X, Stein JL, Vuong CK, Nichterwitz S, Gevorgian M, Opland CK, et al. (2019). A Single-Cell Transcriptomic Atlas of Human Neocortical Development during Mid-gestation. Neuron 103, 785–801.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quesnel-Vallières M, Dargaei Z, Irimia M, Gonatopoulos-Pournatzis T, Ip JY, Wu M, Sterne-Weiler T, Nakagawa S, Woodin MA, Blencowe BJ, et al. (2016). Misregulation of an Activity-Dependent Splicing Network as a Common Mechanism Underlying Autism Spectrum Disorders. Mol. Cell 64, 1023–1034. [DOI] [PubMed] [Google Scholar]
- Rauch A, Wieczorek D, Graf E, Wieland T, Endele S, Schwarzmayr T, Albrecht B, Bartholdi D, Beygo J, Di Donato N, et al. (2012). Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet 380, 1674–1682. [DOI] [PubMed] [Google Scholar]
- Rubenstein JL, and Rakic P (1999). Genetic control of cortical development. Cereb. Cortex 9, 521–523. [DOI] [PubMed] [Google Scholar]
- Sanders SJ, Ercan-Sencicek AG, Hus V, Luo R, Murtha MT, Moreno-De-Luca D, Chu SH, Moreau MP, Gupta AR, Thomson SA, et al. (2011). Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron 70, 863–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, Ercan-Sencicek AG, DiLullo NM, Parikshak NN, Stein JL, et al. (2012). De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 485, 237–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SJ, He X, Willsey AJ, Ercan-Sencicek AG, Samocha KE, Cicek AE, Murtha MT, Bal VH, Bishop SL, Dong S, et al. (2015). Insights into Autism Spectrum Disorder Genomic Architecture and Biology from 71 Risk Loci. Neuron 87, 1215–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SJ, Campbell AJ, Cottrell JR, Moller RS, Wagner FF, Auldridge AL, Bernier RA, Catterall WA, Chung WK, Empfield JR, et al. (2018). Progress in Understanding and Treating SCN2A-Mediated Disorders. Trends Neurosci. 41, 442–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SJ, Sahin M, Hostyk J, Thurm A, Jacquemont S, Avillach P, Douard E, Martin CL, Modi ME, Moreno-De-Luca A, et al. (2019). A framework for the investigation of rare genetic disorders in neuropsychiatry. Nat. Med 25, 1477–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SJ, Schwartz GB, and Farh KK-H (2020). Clinical impact of splicing in neurodevelopmental disorders. Genome Med. 12, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satterstrom FK, Kosmicki JA, Wang J, Breen MS, De Rubeis S, An J-Y, Peng M, Collins R, Grove J, Klei L, et al. (2020). Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell 180, 568–584.e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlusche AK, Vay SU, Kleinenkuhnen N, Sandke S, Campos-Martin R, Florio M, Huttner W, Tresch A, Roeper J, Rueger MA, et al. (2021). Developmental HCN channelopathy results in decreased neural progenitor proliferation and microcephaly in mice. [DOI] [PMC free article] [PubMed]
- Schreiner D, Nguyen T-M, Russo G, Heber S, Patrignani A, Ahrné E, and Scheiffele P (2014). Targeted combinatorial alternative splicing generates brain region-specific repertoires of neurexins. Neuron 84, 386–398. [DOI] [PubMed] [Google Scholar]
- Shcheglovitov A, Shcheglovitova O, Yazawa M, Portmann T, Shu R, Sebastiano V, Krawisz A, Froehlich W, Bernstein JA, Hallmayer JF, et al. (2013). SHANK3 and IGF1 restore synaptic deficits in neurons from 22q13 deletion syndrome patients. Nature 503, 267–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M, and Kim E (2000). The Shank family of scaffold proteins. J. Cell Sci 113 (Pt 11), 1851–1856. [DOI] [PubMed] [Google Scholar]
- Sheng M, Cummings J, Roldan LA, Jan YN, and Jan LY (1994). Changing subunit composition of heteromeric NMDA receptors during development of rat cortex. Nature 368, 144–147. [DOI] [PubMed] [Google Scholar]
- Shibata M, Gulden FO, and Sestan N (2015). From trans to cis: transcriptional regulatory networks in neocortical development. Trends Genet. 31, 77–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SK, Stogsdill JA, Pulimood NS, Dingsdale H, Kim YH, Pilaz L-J, Kim IH, Manhaes AC, Rodrigues WS Jr, Pamukcu A, et al. (2016). Astrocytes Assemble Thalamocortical Synapses by Bridging NRX1α and NL1 via Hevin. Cell 164, 183–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh T, Neale BM, Daly MJ, and on behalf of the Schizophrenia Exome Meta-Analysis (SCHEMA) Consortium (2020). Exome sequencing identifies rare coding variants in 10 genes which confer substantial risk for schizophrenia. medRxiv 2020.09.18.20192815. [Google Scholar]
- Smith RS, and Walsh CA (2020). Ion Channel Functions in Early Brain Development. Trends Neurosci. 43, 103–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JS, Iannotti CA, Dargis P, Christian EP, and Aiyar J (2001). Differential expression of kcnq2 splice variants: implications to m current function during neuronal development. J. Neurosci 21, 1096–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RS, Kenny CJ, Ganesh V, Jang A, Borges-Monroy R, Partlow JN, Hill RS, Shin T, Chen AY, Doan RN, et al. (2018). Sodium Channel SCN3A (NaV1.3) Regulation of Human Cerebral Cortical Folding and Oral Motor Development. Neuron 99, 905–913.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RS, Florio M, Akula SK, Neil JE, Wang Y, Hill RS, Goldman M, Mullally CD, Reed N, Bello-Espinosa L, et al. (2021). Early role for a Na+,K+-ATPase (ATP1A3) in brain development. Proc. Natl. Acad. Sci. U. S. A 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soldatov NM (1994). Genomic structure of human L-type Ca2+ channel. Genomics 22, 77–87. [DOI] [PubMed] [Google Scholar]
- Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, Napolitano C, Schwartz PJ, Joseph RM, Condouris K, et al. (2004). Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 119, 19–31. [DOI] [PubMed] [Google Scholar]
- Spratt PWE, Ben-Shalom R, Keeshen CM, Burke KJ Jr, Clarkson RL, Sanders SJ, and Bender KJ (2019). The autism-associated gene Scn2a contributes to dendritic excitability and synaptic function in the prefrontal cortex. Neuron 103, 673–685.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spratt PWE, Alexander RPD, Ben-Shalom R, Sahagun A, Kyoung H, Keeshen CM, Sanders SJ, and Bender KJ (2021). Paradoxical hyperexcitability from NaV1.2 sodium channel loss in neocortical pyramidal cells. Cell Rep. 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- State MW, and Šestan N (2012). Neuroscience. The emerging biology of autism spectrum disorders. Science 337, 1301–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan PF, and Geschwind DH (2019). Defining the Genetic, Genomic, Cellular, and Diagnostic Architectures of Psychiatric Disorders. Cell 177, 162–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Paşca SP, Portmann T, Goold C, Worringer KA, Guan W, Chan KC, Gai H, Vogt D, Chen Y-JJ, et al. (2016). A deleterious Nav1.1 mutation selectively impairs telencephalic inhibitory neurons derived from Dravet Syndrome patients. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang ZZ, Hong X, Wang J, and Soong TW (2007). Signature combinatorial splicing profiles of rat cardiac- and smooth-muscle Cav1.2 channels with distinct biophysical properties. Cell Calcium 41, 417–428. [DOI] [PubMed] [Google Scholar]
- Tang ZZ, Zheng S, Nikolic J, and Black DL (2009). Developmental control of CaV1.2 L-type calcium channel splicing by Fox proteins. Mol. Cell. Biol 29, 4757–4765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang ZZ, Sharma S, Zheng S, Chawla G, Nikolic J, and Black DL (2011). Regulation of the mutually exclusive exons 8a and 8 in the CaV1.2 calcium channel transcript by polypyrimidine tract-binding protein. J. Biol. Chem 286, 10007–10016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tate SK, Depondt C, Sisodiya SM, Cavalleri GL, Schorge S, Soranzo N, Thom M, Sen A, Shorvon SD, Sander JW, et al. (2005). Genetic predictors of the maximum doses patients receive during clinical use of the anti-epileptic drugs carbamazepine and phenytoin. Proc. Natl. Acad. Sci. U. S. A 102, 5507–5512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tebbenkamp ATN, Willsey AJ, State MW, and Sestan N (2014). The developmental transcriptome of the human brain: implications for neurodevelopmental disorders. Curr. Opin. Neurol 27, 149–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telley L, Govindan S, Prados J, Stevant I, Nef S, Dermitzakis E, Dayer A, and Jabaudon D (2016). Sequential transcriptional waves direct the differentiation of newborn neurons in the mouse neocortex. Science 351, 1443–1446. [DOI] [PubMed] [Google Scholar]
- Thompson CH, Kahlig KM, and George AL Jr (2011). SCN1A splice variants exhibit divergent sensitivity to commonly used antiepileptic drugs. Epilepsia 52, 1000–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson CH, Ben-Shalom R, Bender KJ, and George AL (2020). Alternative splicing potentiates dysfunction of early-onset epileptic encephalopathy SCN2A variants. J. Gen. Physiol 152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson CL, Ng L, Menon V, Martinez S, Lee C-K, Glattfelder K, Sunkin SM, Henry A, Lau C, Dang C, et al. (2014). A high-resolution spatiotemporal atlas of gene expression of the developing mouse brain. Neuron 83, 309–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinel N, Lauritzen I, Chouabe C, Lazdunski M, and Borsotto M (1998). The KCNQ2 potassium channel: splice variants, functional and developmental expression. Brain localization and comparison with KCNQ3. FEBS Lett. 438, 171–176. [DOI] [PubMed] [Google Scholar]
- Treutlein B, Gokce O, Quake SR, and Südhof TC (2014). Cartography of neurexin alternative splicing mapped by single-molecule long-read mRNA sequencing. Proc. Natl. Acad. Sci. U. S. A 111, E1291–E1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trevino AE, Sinnott-Armstrong N, Andersen J, Yoon S-J, Huber N, Pritchard JK, Chang HY, Greenleaf WJ, and Pașca SP (2020). Chromatin accessibility dynamics in a model of human forebrain development. Science 367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trevino AE, Müller F, Andersen J, Sundaram L, Kathiria A, Shcherbina A, Farh K, Chang HY, Pașca AM, Kundaje A, et al. (2021). Chromatin and gene-regulatory dynamics of the developing human cerebral cortex at single-cell resolution. Cell. [DOI] [PubMed] [Google Scholar]
- Uchigashima M, Konno K, Demchak E, Cheung A, Watanabe T, Keener DG, Abe M, Le T, Sakimura K, Sasaoka T, et al. (2020). Specific Neuroligin3–αNeurexin1 signaling regulates GABAergic synaptic function in mouse hippocampus. Elife 9, e59545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velmeshev D, Schirmer L, Jung D, Haeussler M, Perez Y, Mayer S, Bhaduri A, Goyal N, Rowitch DH, and Kriegstein AR (2019). Single-cell genomics identifies cell type-specific molecular changes in autism. Science 364, 685–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitali I, Fièvre S, Telley L, Oberst P, Bariselli S, Frangeul L, Baumann N, McMahon JJ, Klingler E, Bocchi R, et al. (2018). Progenitor Hyperpolarization Regulates the Sequential Generation of Neuronal Subtypes in the Developing Neocortex. Cell 174, 1264–1276.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlaskamp DRM, Shaw BJ, Burgess R, Mei D, Montomoli M, Xie H, Myers CT, Bennett MF, XiangWei W, Williams D, et al. (2019). SYNGAP1 encephalopathy: A distinctive generalized developmental and epileptic encephalopathy. Neurology 92, e96–e107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wamsley B, and Fishell G (2017). Genetic and activity-dependent mechanisms underlying interneuron diversity. Nat. Rev. Neurosci 18, 299–309. [DOI] [PubMed] [Google Scholar]
- Wamsley B, Jaglin XH, Favuzzi E, Quattrocolo G, Nigro MJ, Yusuf N, KhodadadiJamayran A, Rudy B, and Fishell G (2018). Rbfox1 Mediates Cell-type-Specific Splicing in Cortical Interneurons. Neuron 100, 846–859.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, McCoy PA, Rodriguiz RM, Pan Y, Je HS, Roberts AC, Kim CJ, Berrios J, Colvin JS, Bousquet-Moore D, et al. (2011). Synaptic dysfunction and abnormal behaviors in mice lacking major isoforms of Shank3. Hum. Mol. Genet 20, 3093–3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Xu Q, Bey AL, Lee Y, and Jiang Y-H (2014). Transcriptional and functional complexity of Shank3 provides a molecular framework to understand the phenotypic heterogeneity of SHANK3 causing autism and Shank3 mutant mice. Mol. Autism 5, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M, Inoue Y, Sakimura K, and Mishina M (1992). Developmental changes in distribution of NMDA receptor channel subunit mRNAs. Neuroreport 3, 1138–1140. [DOI] [PubMed] [Google Scholar]
- Welling A, Ludwig A, Zimmer S, Klugbauer N, Flockerzi V, and Hofmann F (1997). Alternatively spliced IS6 segments of the alpha 1C gene determine the tissue-specific dihydropyridine sensitivity of cardiac and vascular smooth muscle L-type Ca2+ channels. Circ. Res 81, 526–532. [DOI] [PubMed] [Google Scholar]
- Weyn-Vanhentenryck SM, Mele A, Yan Q, Sun S, Farny N, Zhang Z, Xue C, Herre M, Silver PA, Zhang MQ, et al. (2014). HITS-CLIP and integrative modeling define the Rbfox splicing-regulatory network linked to brain development and autism. Cell Rep. 6, 1139–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyn-Vanhentenryck SM, Feng H, Ustianenko D, Duffié R, Yan Q, Jacko M, Martinez JC, Goodwin M, Zhang X, Hengst U, et al. (2018). Precise temporal regulation of alternative splicing during neural development. Nat. Commun 9, 2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willsey AJ, and State MW (2015). Autism spectrum disorders: from genes to neurobiology. Curr. Opin. Neurobiol 30, 92–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willsey AJ, Sanders SJ, Li M, Dong S, Tebbenkamp AT, Muhle RA, Reilly SK, Lin L, Fertuzinhos S, Miller JA, et al. (2013). Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell 155, 997–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willsey AJ, Morris MT, Wang S, Willsey HR, Sun N, Teerikorpi N, Baum TB, Cagney G, Bender KJ, Desai TA, et al. (2018). The Psychiatric Cell Map Initiative: A Convergent Systems Biological Approach to Illuminating Key Molecular Pathways in Neuropsychiatric Disorders. Cell 174, 505–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willsey HR, Exner CRT, Xu Y, Everitt A, Sun N, Wang B, Dea J, Schmunk G, Zaltsman Y, Teerikorpi N, et al. (2021). Parallel in vivo analysis of large-effect autism genes implicates cortical neurogenesis and estrogen in risk and resilience. Neuron 109, 788–804.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong FK, and Marín O (2019). Developmental Cell Death in the Cerebral Cortex. Annu. Rev. Cell Dev. Biol 35, 523–542. [DOI] [PubMed] [Google Scholar]
- Wyllie DJA, Livesey MR, and Hardingham GE (2013). Influence of GluN2 subunit identity on NMDA receptor function. Neuropharmacology 74, 4–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu R, Thomas EA, Jenkins M, Gazina EV, Chiu C, Heron SE, Mulley JC, Scheffer IE, Berkovic SF, and Petrou S (2007). A childhood epilepsy mutation reveals a role for developmentally regulated splicing of a sodium channel. Mol. Cell. Neurosci 35, 292–301. [DOI] [PubMed] [Google Scholar]
- Yao Z, van Velthoven CTJ, Nguyen TN, Goldy J, Sedeno-Cortes AE, Baftizadeh F, Bertagnolli D, Casper T, Chiang M, Crichton K, et al. (2021). A taxonomy of transcriptomic cell types across the isocortex and hippocampal formation. Cell 184, 3222–3241.e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo G, Holste D, Kreiman G, and Burge CB (2004). Variation in alternative splicing across human tissues. Genome Biol. 5, R74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi F, Danko T, Botelho SC, Patzke C, Pak C, Wernig M, and Südhof TC (2016). Autism-associated SHANK3 haploinsufficiency causes Ih channelopathy in human neurons. Science 352, aaf2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu FH, Mantegazza M, Westenbroek RE, Robbins CA, Kalume F, Burton KA, Spain WJ, McKnight GS, Scheuer T, and Catterall WA (2006). Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat. Neurosci 9, 1142–1149. [DOI] [PubMed] [Google Scholar]
- Zaman T, Helbig KL, Clatot J, Thompson CH, Kang SK, Stouffs K, Jansen AE, Verstraete L, Jacquinet A, Parrini E, et al. (2020). SCN3A-Related Neurodevelopmental Disorder: A Spectrum of Epilepsy and Brain Malformation. Ann. Neurol 88, 348–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Chen X, Eaton M, Wu J, Ma Z, Lai S, Park A, Ahmad TS, Que Z, Lee JH, et al. (2021). Severe deficiency of the voltage-gated sodium channel NaV1.2 elevates neuronal excitability in adult mice. Cell Rep. 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Chen MH, Wu X, Kodani A, Fan J, Doan R, Ozawa M, Ma J, Yoshida N, Reiter JF, et al. (2016). Cell-Type-Specific Alternative Splicing Governs Cell Fate in the Developing Cerebral Cortex. Cell 166, 1147–1162.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S, and Black DL (2013). Alternative pre-mRNA splicing in neurons: growing up and extending its reach. Trends Genet. 29, 442–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S, Gray EE, Chawla G, Porse BT, O’Dell TJ, and Black DL (2012). PSD-95 is post-transcriptionally repressed during early neural development by PTBP1 and PTBP2. Nat. Neurosci 15, 381–388, S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu M, Idikuda VK, Wang J, Wei F, Kumar V, Shah N, Waite CB, Liu Q, and Zhou L (2018a). Shank3-deficient thalamocortical neurons show HCN channelopathy and alterations in intrinsic electrical properties. J. Physiol 596, 1259–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Sousa AMM, Gao T, Skarica M, Li M, Santpere G, Esteller-Cucala P, Juan D, Ferrández-Peral L, Gulden FO, et al. (2018b). Spatiotemporal transcriptomic divergence across human and macaque brain development. Science 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoghbi HY (2003). Postnatal neurodevelopmental disorders: meeting at the synapse? Science 302, 826–830. [DOI] [PubMed] [Google Scholar]
- Zuccaro E, Murek V, Kim K, Chen H-H, Mancinelli S, Oyler-Castrillo P, Jiménez-Barrón LT, Gerhardinger C, Brown JR, Byrnes A, et al. (2021). Human-specific enrichment of schizophrenia risk-genes in callosal neurons of the developing neocortex. [Google Scholar]
- Zühlke RD, Bouron A, Soldatov NM, and Reuter H (1998). Ca2+ channel sensitivity towards the blocker isradipine is affected by alternative splicing of the human alpha1C subunit gene. FEBS Lett. 427, 220–224. [DOI] [PubMed] [Google Scholar]