

Abstract
Impaired white adipose tissue (WAT) function has been recognized as a critical early event in obesity-driven disorders, but high buoyancy, fragility, and heterogeneity of primary adipocytes have largely prevented their use in drug discovery efforts highlighting the need for human stem cell based approaches. Here, we utilized human stem cells to derive metabolically functional 3D adipose tissue (iADIPO) in a micro-physiological system (MPS). Surprisingly, previously reported WAT differentiation approaches created insulin resistant WAT ill-suited for T2DM drug discovery. Using three independent insulin sensitivity assays, i.e. glucose and fatty acid uptake and suppression of lipolysis, as our functional readouts we derived new differentiation conditions yielding hormonally responsive iADIPO. Through concomitant optimization of an iADIPO-MPS we are able to obtain WAT with more unilocular and significantly larger (~40%) lipid droplets compared to iADIPO in 2D culture, increased insulin responsiveness of glucose uptake (2~3 fold), fatty acid uptake (3~6 fold), and ~40% suppressing of stimulated lipolysis giving us a dynamic range that is competent to current in vivo and ex vivo models, allowing us to identify both insulin sensitizers and desensitizers.
Keywords: organ-on-a-chip, microphysiological system, human induced pluripotent stem cells, white adipose tissue, insulin sensitivity
Graphical Abstract
This study used human stem cells to derive metabolically functional 3D adipose tissue in a micro-physiological system. Through concomitant optimization, the adipose system showed mostly unilocular and significantly larger lipid droplets, increased insulin responsiveness of glucose uptake, fatty acid uptake and suppressing of stimulated lipolysis compared to 2D culture, providing a good dynamic range to identify insulin sensitizers and desensitizers.
1. Introduction
Obesity-related disorders, particularly type-2 diabetes mellitus (T2DM), continue to increase in the US and worldwide[1]. One of the early events in the pathogenesis of T2DM is insulin resistance in white adipose tissue (WAT), which disrupts the normal postprandial response to insulin[2]. Insulin resistance causes dysregulated lipolysis and impaired uptake of glucose and fatty acid resulting in hyperglycemia and elevated serum free fatty acid levels that lead to ectopic lipid deposition and impaired organ function such as nonalcoholic fatty liver disease (NAFLD) and glucolipotoxicity in β-cells ultimately resulting in overt diabetes[3].
Given its central role in energy homeostasis[4], WAT has been recognized as a critical target for T2DM investigation and pharmacotherapeutic interventions. Many previous studies have utilized murine preadipocytes cell lines and animal models to investigate WAT. However, their usefulness for translational applications is limited by significant species differences between the lipid metabolism in humans and rodents[5]. Human primary adipocytes, though readily obtainable from obese subjects, are often heterogeneous, highly fragile, too buoyant to be maintained in standard tissue culture (TC) conditions, and often limited for high-throughput screening approaches[6]. Alternatively WAT can be derived from human stem cells however only with varying and often low rates of adipogenesis, such as 30%−85% differentiation potential for adipose-derived stromal cells[7], 30%−50% for human mesenchymal stem cells (hMSCs)[8] and 3%−10% potential for induced pluripotent stem cells (iPSCs)[9]. Adipogenesis of human stem cells can be greatly increased via the forced expression of the adipogenic master regulator PPARγ[9–10]. Human iPSCs offer many advantages such as indefinite propagation, easy derivatization, reduced ethical concerns, and ability to derive multiple tissues from an isogenic source. Previous studies of human stem cell derived WAT typically focus on achieving robust lipid droplet formation and adipogenic gene expression profiles but only few of these studies have addressed functional competence [11] and some of these studies reported a lack of insulin responsiveness[12]. Indeed, in our hands existing iPSC to WAT differentiation protocols[13] result in completely insulin resistant WAT (Figure S1), limiting their usefulness for drug discovery.
Further, standard 2D tissue culture (TC) models do not fully replicate the physiological environment of human WAT and frequently fail to reproduce key features such as unilocular lipid droplets. TC conditions often lack physiological circulation that continuously delivers nutrients, removes wastes, and transfers hormones and cytokines. Additionally, the 2D environment of standard TC fails to provide physiological cell-to-cell and cell-to-matrix interactions found in WAT, which are essential for their differentiation and function[14]. 3D models were therefore developed. Adipocyte progenitors can directly form 3D spheroid on a chemically tethered surface or grow on a 3D scaffold. Nonetheless, the mature adipocytes become fragile and are subject to disassociation and necrosis as similar as 2D condition. Encapsulation of adipocyte progenitors in 3D addresses the concern by isolating cells from environmental impacts[15], and increases adipogenesis compared to 2D culture[16]. However, the requirement of cell numbers and materials increased greatly[17]. Organ-on-a-chip system, also known as the micro-physiological system (MPS), is an emerging platform that can address many of these concerns. Derived from well-established microfluidic technology, MPS allow for the circulating of culture medium in a well-controlled manner, provide a stable encapsulated microenvironment for differentiation and long-term culturing, and minimizes the requirement for cells and materials[18]. Thus, there is a clear need to create a MPS platform that uses human iPSCs to reconstruct competent metabolic functions of WAT, which has not been well-developed until now[19].
To address these issues, we created a hormonally responsive iADIPO in a MPS using human mesenchymal stem cells (hMSCs) and induced pluripotent stem cells (iPSCs) that in proof-of-concept screens allowed us to detect both insulin sensitizing and desensitizing drugs. To the best of our knowledge, this is the first study that reconstructs the key hormonal responses of 3D remodeled iADIPO-MPS using hMSCs and iPSCs. Our study provides a pre-clinical model that may facilitate drug development, support profound investigation of obesity and T2DM, and climb a stair toward the ultimate “human-on-a-chip” goal.
2. Results
2.1. Investigation of hMSCs differentiation toward metabolically competent WAT
Initial studies by us demonstrated that following existing published differentiation protocols[13], hMSCs-derived adipocytes accumulated lipid droplets but were insulin resistant (Figure. S1). Thus, we decided to further optimize differentiation conditions with physiological insulin responses as our primary criterion in addition to lipid droplet accumulation. While increasing PPARγ agonist rosiglitazone led to a dose dependent increase in lipid accumulation, insulin sensitivity was only observed in a narrow range (Figure S2a). The expressions of multiple adipocyte-specific genes were all increased with increasing concentration of rosiglitazone (ANOVA: p=0.0007 for HSL, and p <0.0001 for other genes, Figure 1a). Higher rosiglitazone concentration also led to higher basal nutrient uptake (ANOVA: p<0.0001 for fatty acid, and p=0.0003 for glucose, Figure 1b&c). However, differentiated adipocytes were desensitized to insulin under some conditions. Robust insulin-induced uptakes were only obtained in a range of rosiglitazone between 20~100 nM for free fatty acid uptake (Figure 1b), and 20~1000 nM for glucose uptake (Figure 1c). 100 nM was identified as the acceptable concentration yielding both robust adipogenesis and highly sensitive insulin responses. Based on this protocol, the lipolysis of differentiated adipocytes was characterized. As shown in Figure 1d, lipolysis, as measured by glycerol release, was robustly stimulated by 1 μM isoproterenol establishing a ß-adrenergic activated baseline. Additional inclusion of 1 μM insulin significantly inhibited catecholamine stimulated lipolysis. Therefore, the key physiological functions of WAT, i.e. insulin-induced nutrient uptake and suppression of β-adrenergically stimulated lipolysis, were well-recapitulated by using hMSCs-derived adipocytes obtaining similar results to previous in vivo or ex vivo studies[20].
Figure 1: optimization of hMSCs differentiation.
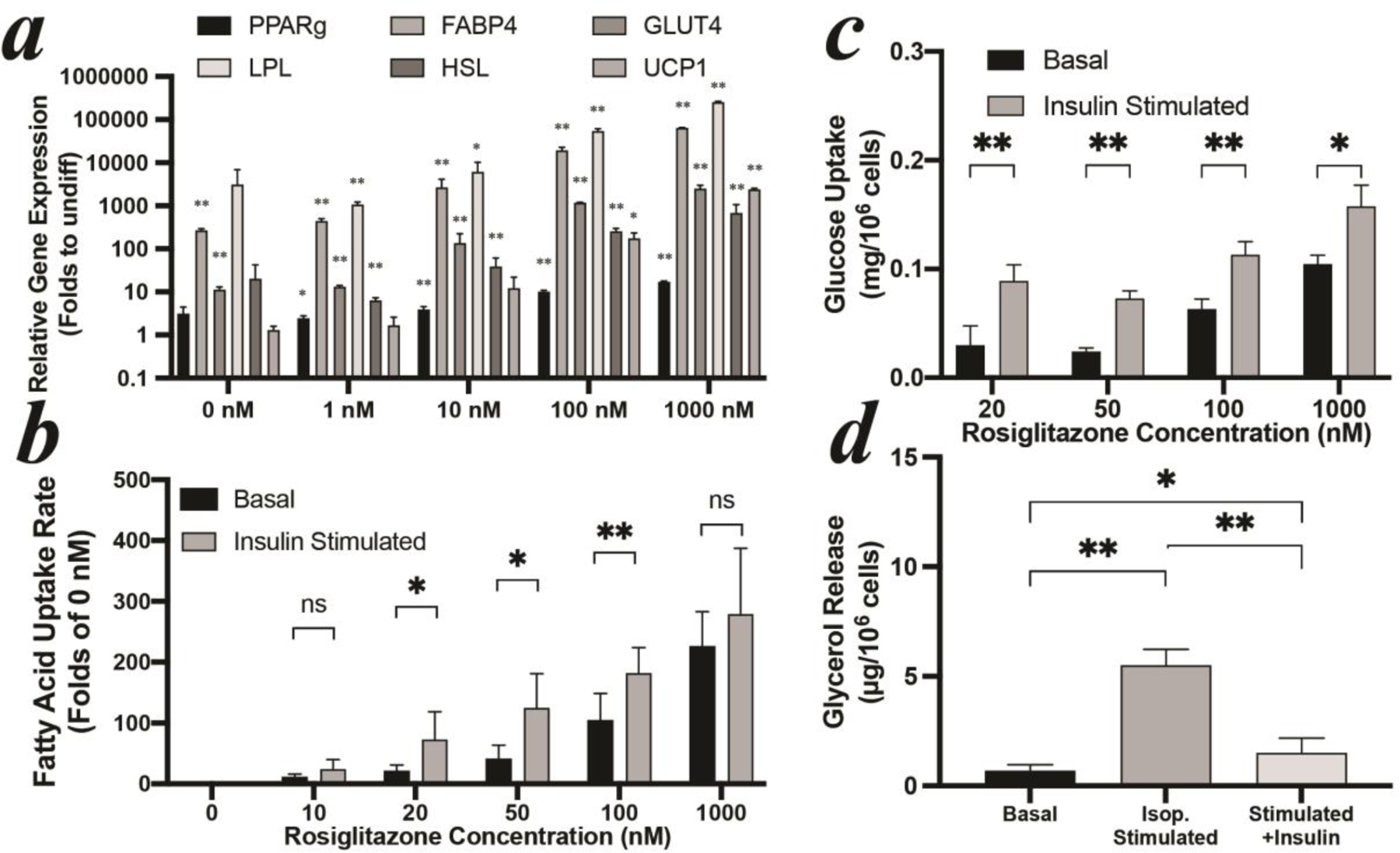
(a) gene expression, (b) free fatty acid uptake, and (c) glucose uptake of hMSCs-derived adipocytes with different rosiglitazone concentrations. (d) lipolysis of hMSCs-derived adipocytes with 100 nM rosiglitazone. All hMSCs were differentiated for 7 days. Expression values in (a) are shown relative to the mean of undifferentiated hMSCs set as 1. Stars label significant difference compared to undifferentiated hMSCs.
2.2. Investigation of iPSCs differentiation toward metabolically competent WAT
WTC11 iPSCs were firstly differentiated to mesenchymal level (iPSC-MSCs) and lentivirally transfected with PPARγ, the master gene to regulate adipogenesis[9]. The differentiation cocktail of iPSC-MSCs was slightly modified by adding SB431542, to suppress chondrogenesis[21], and doxycycline to induce exogenous PPARγ in transfected iPSC-MSCs. The supportive role of rosiglitazone in the adipogenesis of iPSC-MSCs is in line with that of hMSCs (Figure S2a). Higher rosiglitazone dose led to an increased expression of adipogenic specific hallmark genes: relatively 5~20 folds increase of all genes once rosiglitazone dose above 10 nM (Figure 2a). However, insulin desensitization also occurred once rosiglitazone dose was above 1000 nM (Figure 2b). There is a window that allows for robust insulin responsiveness in both glucose and fatty acid uptakes by keeping rosiglitazone concentrations at 100 nM (Figure 2b&c). Under this condition, lipolysis can be activated by isoproterenol, releasing ~3 folds more glycerol into the surrounding medium; and can be suppressed by insulin (Figure 2d). Thus, the anabolic and catabolic functions of WAT were reconstructed using iPSCs. Importantly, this optimized differentiation protocol is not specific to one iPSC line (WTC11 in this case), as we were able to obtain similar results in terms of generating insulin sensitive WAT from a total of 4 human iPSC lines including 1023–5, K3, and G15AO (Figure S3)
Figure 2: optimization of iPSC-MSCs differentiation.
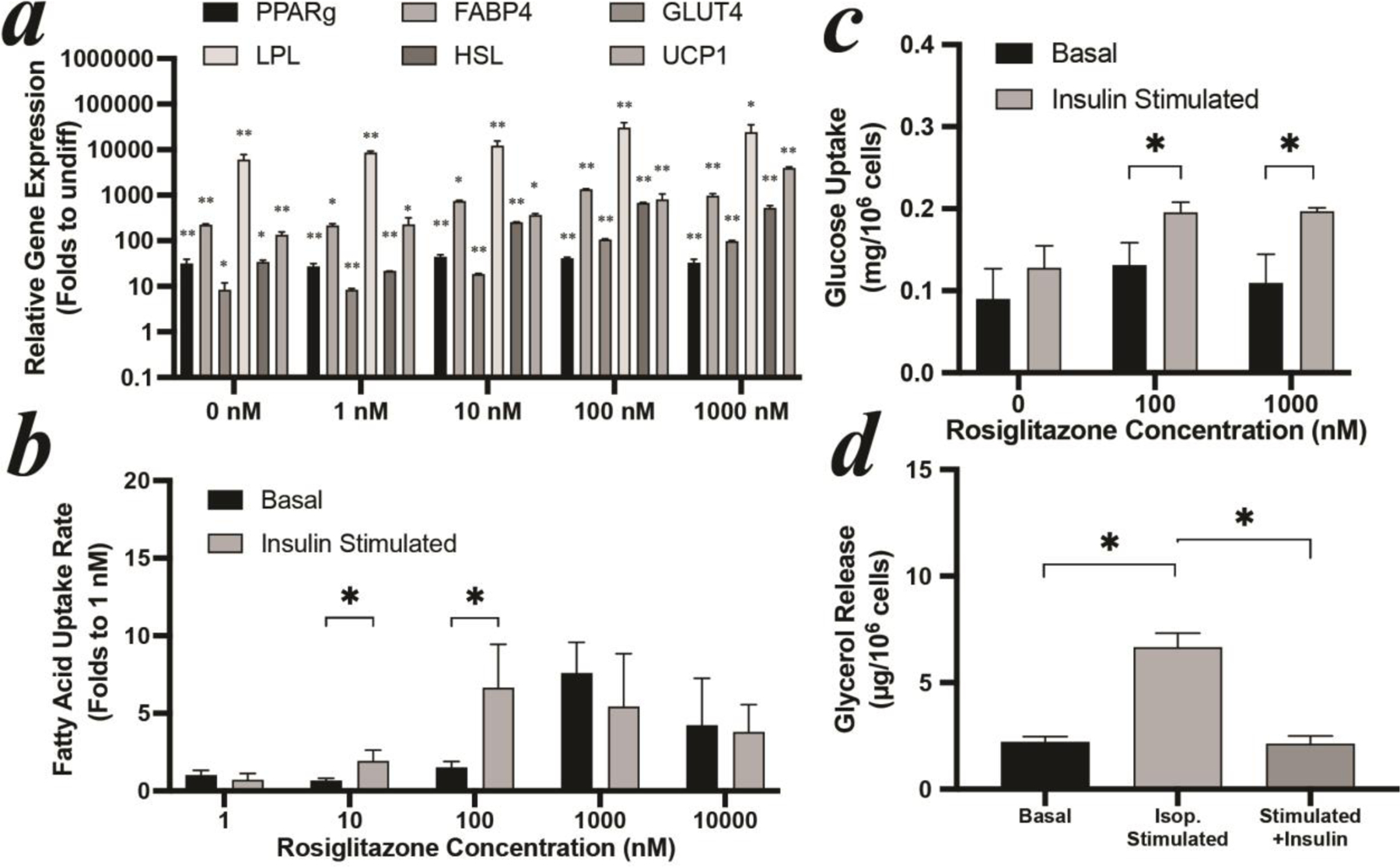
(a) gene expression, (b) free fatty acid uptake, and (c) glucose uptake of iPSCs-derived adipocytes with different rosiglitazone concentrations. (d) lipolysis of iPSCs-derived adipocytes with 100 nM rosiglitazone. All iPSCs-derived adipocytes were differentiated for 14 days. Expression values in (a) are shown relative to the mean of undifferentiated iPSC-MSCs set as 1. Stars label significant difference compared to undifferentiated iPSC-MSCs.
2.3. Adipogenic gene expression and function of iADIPO
The adipogenesis of our stem cell-derived adipocytes (iADIPO) was compared to commercially pooled human subcutaneous mature adipocytes originated from liposuction. The hMSCs-derived adipocytes were further matured to Day 14 to meet the same differentiation period of iPSCs-derived adipocytes. Both showed more lipid droplet coverage and larger droplet size than the commercial adipocytes (Figure S2b–d), matching previous reports[9]. Hallmark adipogenic genes expression shows that hMSCs-derived adipocytes were not significantly different to commercial primary adipocytes except for lower UCP1 expression (p=0.0002, Figure 3a). Further, iPSCs-derived adipocytes expressed significantly higher PPARγ (p<0.0001), FABP4 (p<0.0001), HSL (p=0.0105) compared to the commercial primary adipocytes while demonstrating lower UCP1 expression (p=0.0032). Compared to the adipocytes from human biopsies, both stem cell-derived adipocytes show no significant differences in expression of all WAT specific hallmark genes, excepting for the PPARγ in iPSCs-derived adipocytes (p<0.0001) due to the exogeneous induction. The hormonal responsiveness of our stem cell-derived adipocytes and that of commercial adipocytes were also compared. While the relative changes in lipolysis brought about by ß-adrenergic activation and insulin-induced suppression were comparable between hMSCs-derived, iPSCs-derived and commercial adipocytes (Figure 3b), absolute glycerol release rates under stimulated conditions were higher in iADIPO. The insulin-induced glucose uptakes between iADIPO and commercial adipocytes were significantly different (Figure 3c). Insulin was able to robustly induce glucose uptake in both stem cell derived adipocytes. In particular, the hMSCs-derived adipocytes can uptake ~3 folds more glucose upon insulin stimulation, following the similar response shown in Day 7 (Figure 1c). The commercial mature adipocytes, however, failed to display insulin stimulated glucose uptake, which suggests severe insulin resistance.
Figure 3: Comparison to commercial human adipocytes.
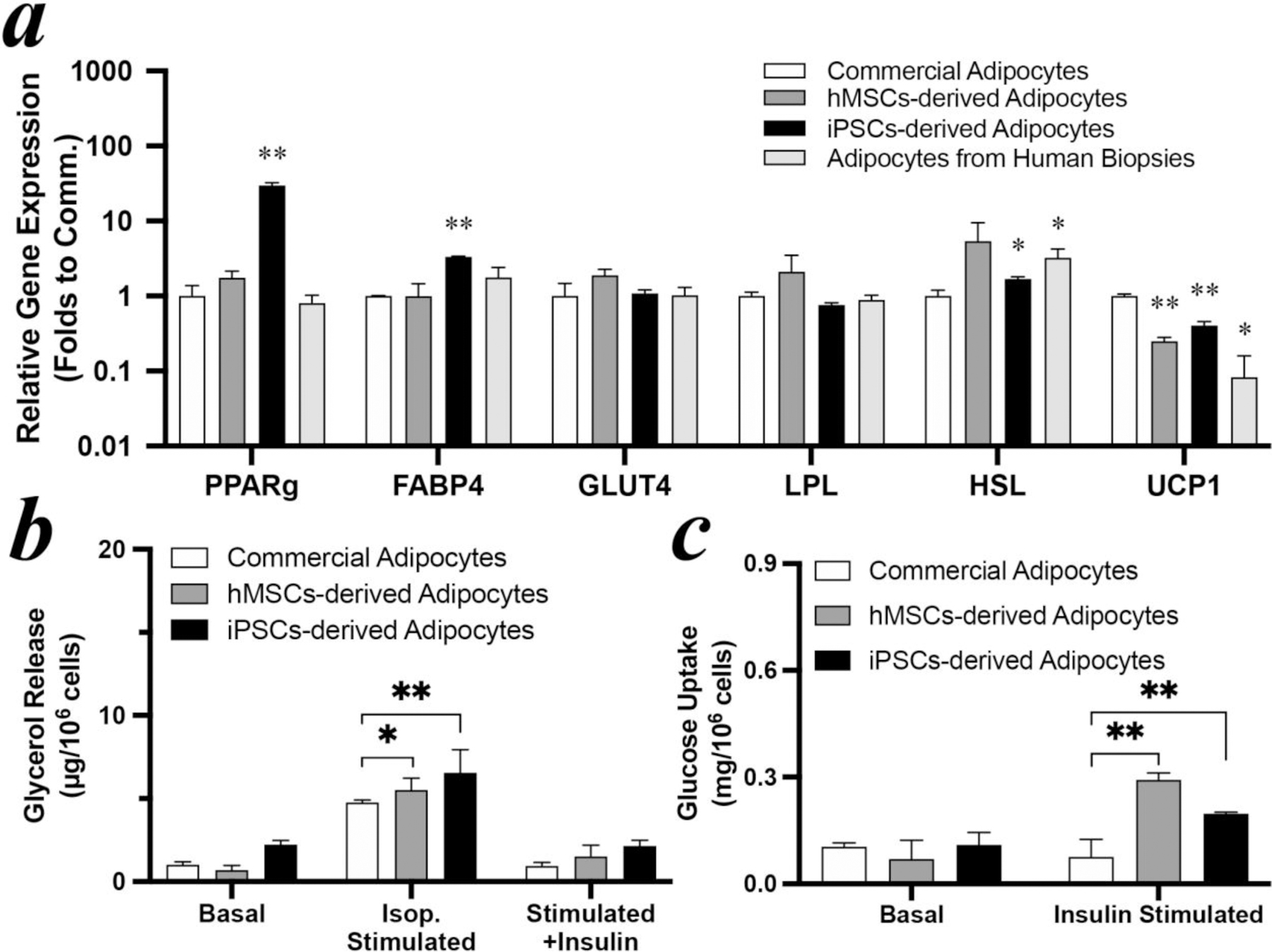
(a) gene expression, (b) lipolysis, and (c) glucose uptake of hMSCs- and iPSCs-derived adipocytes after 14 days differentiation with 100 nM rosiglitazone and commercial adipocytes. Expression values in (a) are shown relative to the mean of commercial adipocytes set as 1. Stars label significant difference compared to commercial mature adipocytes.
2.4. MPS design and optimization
To enhance the differentiation and the survival of adipocyte progenitors in a microfluidic environment, MPS layout and culture conditions were investigated. Since geometric cues may affect adipogenesis[22], varied sizes of cell chambers were examined for effects on adipocyte development (Figure 4a). The diameter of cell chambers ranged from 500, 1000, 1500 to 2000 μm to cover the common sizes used in microfluidic-based MPS[23]. Differentiating hMSCs on Day 4, the stage that has been initiated to adipogenesis commitment, were dissociated from tissue culture flasks as pre-induced stem cells. Adhesion peptide modified HA hydrogel, which was synthesized based on our previously study, was mixed with dissociated cells and loaded in MPS. This hydrogel can assist 3D culture and gradually degrade with tissue remodeling process during differentiation. Initial cell density was selected as 4x107 cells/mL according to previous 3D models [15d, 15e, 16, 24]. After 3 days, the cells aggregated and clustered at the center (Figure 4b), implying an active WAT remodelling process that is commonly observed during adipogenesis differentiation[25]. As shown in the schematic (Figure 4b), there is a ring-shape area with sparsely distributed cells (defined as cell lack area) between cell cluster edge and cell chamber sidewall. The adipocytes within this area are not expected to differentiate as homogenous as the cluster area due to the low cell confluence. Based on statistical analysis (Figure 4c), cell chambers with diameters of 1500 μm showed minimal cell lack area and were used for subsequent MPS generations.
Figure 4: Investigation of cell chamber size of MPS.
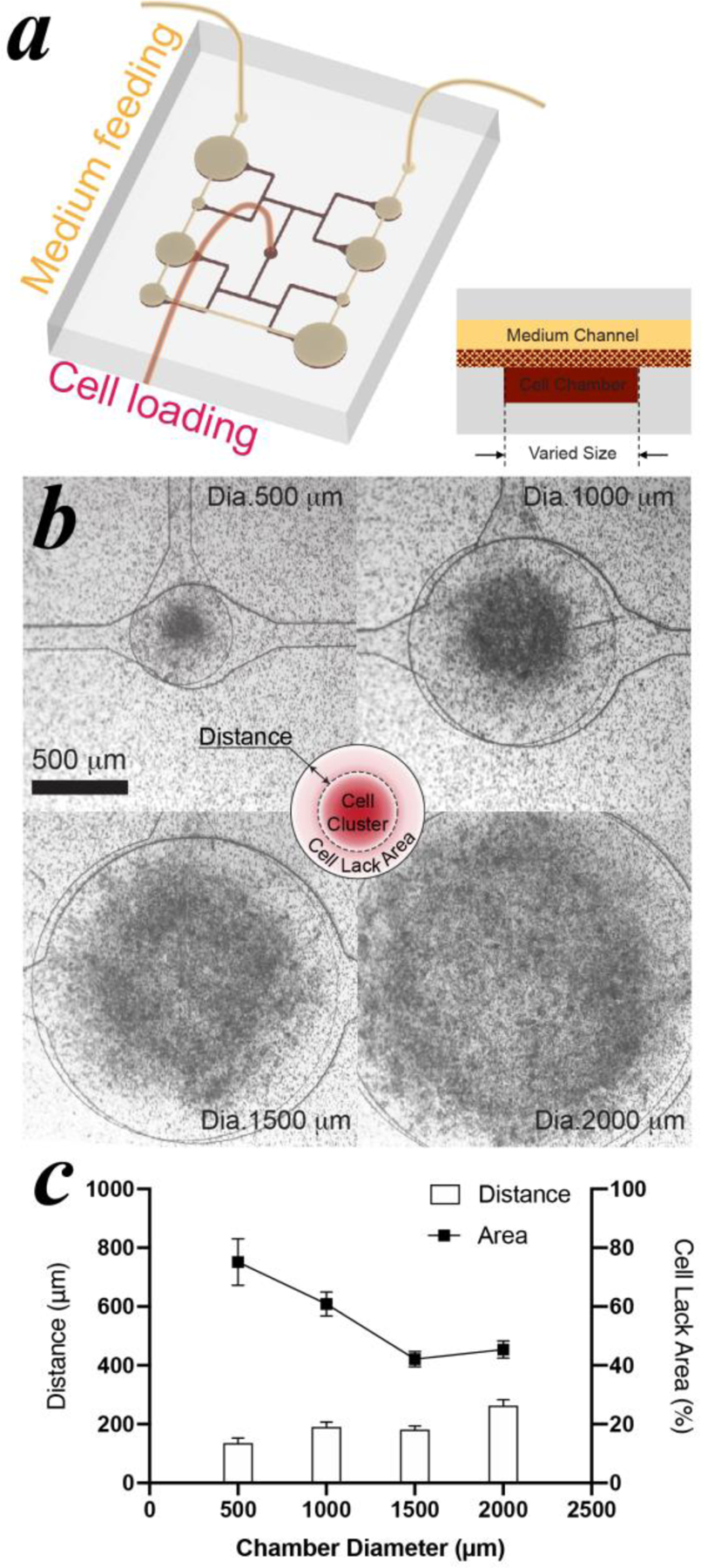
(a) schematic of MPS design. (b) cell clustering in vary-sized cell chambers after 3 days loading. (c) statistical analysis of the distance between the cell cluster edge and the cell chamber sidewall, and the calculated area ratio of cell lack area to chamber area. Pre-induced hMSCs were loaded in the MPS at a cell density of 4×107 cells/mL.
Next, the effect of cell loading density on adipogenesis was investigated. Pre-induced cell-hydrogel slurries with densities from 1×107 to 8×107 cells/mL were loaded into MPS with identical cell chamber sizes (Figure 5a). After 6 days (i.e. Day 10 of differentiation), cell clusters formed in all groups. The cluster with the highest cell density, 8×107 cells/mL, showed the smallest cell lack area than other groups (Figure 5b). As a result, more adipocytes containing lipid droplets were distributed the entire cell cluster. On the other end, there were only a few lipid droplets formed at the center in the chamber loaded with 2×107 cells/mL (Figure 5b and S4) and nearly no observable lipid droplets in the chamber loaded with 1×107 cells/mL.
Figure 5: Investigation of cell loading density in MPS.
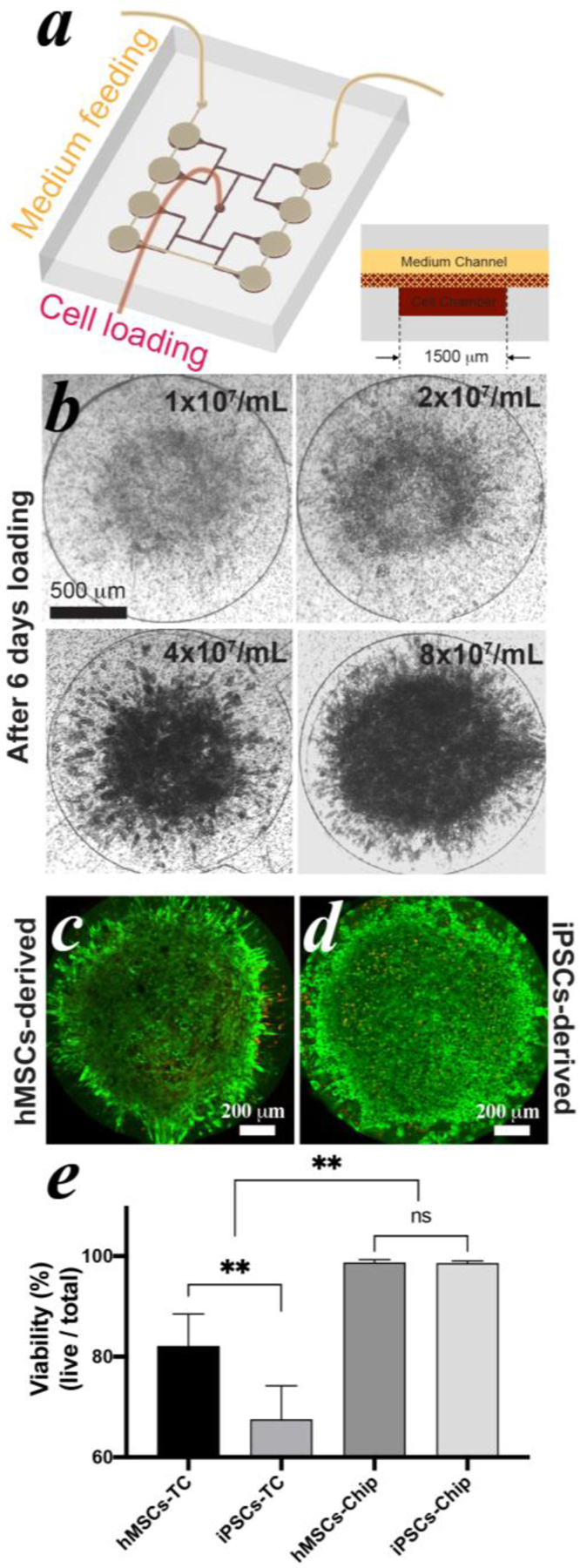
(a) schematic of MPS design. (b) hMSCs-derived cell clustering in MPS after 6 days loading with varied cell density. The live and dead cells of hMSCs- (c) and iPSCs-derived adipocytes (d) were stained after 30 days since differentiation with a cell loading density of 8×107 cells/mL. Live cells were stained in green and dead cells in red. (e) cell viability analysis of both adipocytes cultured in MPS and in TC condition after 30 days since differentiation.
To investigate our ability to establish long-term culture, pre-induced hMSCs and iPSC-MSCs with a density of 8×107 cells/mL were loaded, differentiated, and maintained in the MPS. After 30 days, cell viability was determined using a live/dead cell staining assay (Figure 5c&d). The fluorescent images show that the viability of both adipocytes was highly maintained with nearly no dead cells, even at the central area. Both stem cell derived adipocytes kept overall viability >98% without apparent loss of morphology (Figure 5e). For the purpose of comparison, the same batches of hMSCs and iPSC-MSCs were also plated, differentiated, and maintained under standard TC condition for the same period of time (Figure S5a&b). The overall viability of the hMSCs-derived adipocytes was ~82.1% and of iPSCs-derived was ~67.5% in TC conditions, which were significantly lower than that in the MPS. In addition, the buoyant lipid droplets started to break the fragile matured adipocytes and float in medium after ~24 days since differentiation in spite of careful handling and minimized disruption during media exchanges (Figure S5c).
2.5. Engineering of iADIPO-MPS
Given the promising viability of both stem cell-derived adipocytes in the MPS, the morphology and the physiological functions of iADIPO in MPS was examined. Nuclei, lipid droplets, and F-actin of hMSCs-derived adipocytes were stained as blue, green and red, respectively after 10 days loading (Figure 6a). The adipocytes self-remodeled in the MPS, developed lipid droplets over the whole cluster and were distributed stereoscopically, as the sideview observed in the 3D reconstruction. Most of the lipid droplets sized from 10 μm to 40 μm (Figure 6b). Importantly, the cells in the MPS displayed significantly larger lipid droplets with lower locularity compared to the TC condition (Figure 6c&d), indicating the formation of more unilocular and larger adipocytes in MPS.
Figure 6: Reconstitution of hMSCs-derived iADIPO-MPS.
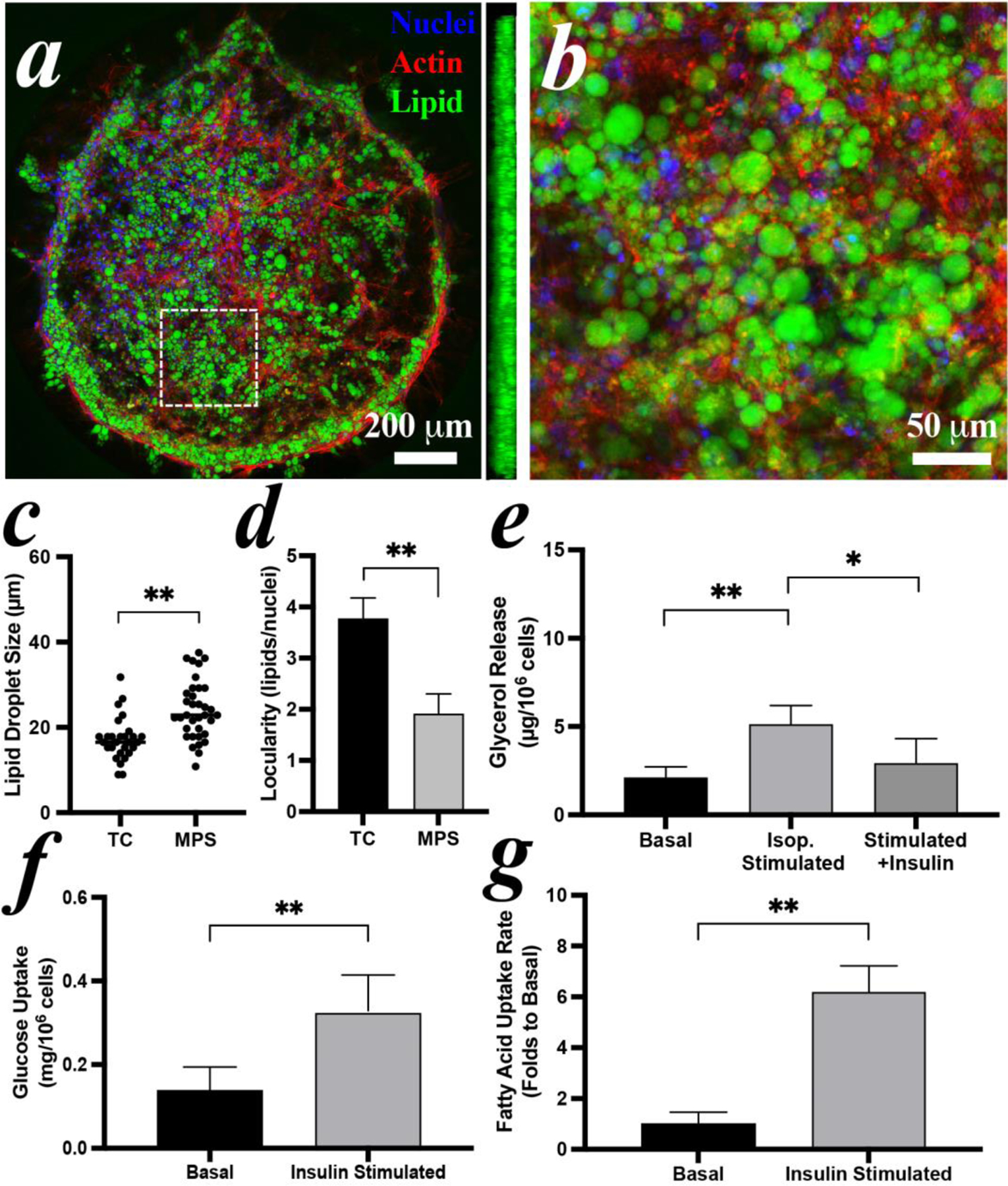
(a) morphology of adipocytes in MPS and (b) zoomed view of white dash-lined area in (a). A sideview of 3D reconstruction in (a) is inserted aside. (c) lipid droplet size and (d) locularity of the adipocytes differentiated in TC condition and in MPS. Average diameters are 16.5 μm in TC and 22.9 μm in MPS. (e) lipolysis and (f) glucose uptake of the adipocytes in the MPS. (g) analysis of fluorescent fatty acid uptake by adipocytes in MPS with or without insulin stimulation. Pre-induced hMSCs were loaded in MPS at Day 4 and imaged or assayed at Day 14.
To investigate the hormonal responses of the hMSCs-derived iADIPO-MPS lipolysis was examined by measuring the glycerol in the retained assay solution in MPS. The iADIPO-MPS released significantly more glycerol upon isoproterenol stimulation than the basal condition (Figure 6e). The lipolysis was decreased ~43% upon suppression by insulin. The glucose uptake of iADIPO-MPS was determined by measuring the glucose clearance of retained medium in the MPS. Upon insulin stimulation, ~0.33 mg glucose was consumed per 106 adipocytes, significantly higher than the basal (Figure 6f). The fatty acid uptake of iADIPO-MPS was characterized by measuring fluorescent fatty acid accumulation in adipocytes. With insulin stimulation, most of the adipocytes emitted an intensive fluorescence after 30 mins (Figure S6a), indicating a significantly active uptake of the fluorescent fatty acid (Figure 6g).
2.6. Engineering of iPSC based iADIPO-MPS
The morphology of iPSCs-derived adipocytes in MPS was also characterized by staining nuclei, lipid droplets, and cytoskeleton. The confocal image of the entire MPS showed clear clustering of adipocytes at the center with 3D distributed lipid droplets as the sideview observed (Figure 7a). The adipocytes developed lipid droplets sized from 20~30 μm (Figure 7b). Again, the adipocytes in the MPS formed significantly larger lipid droplets with lower locularity compared to TC conditions (Figure 7c&d).
Figure 7: Reconstitution of iPSCs-derived iADIPO-MPS.
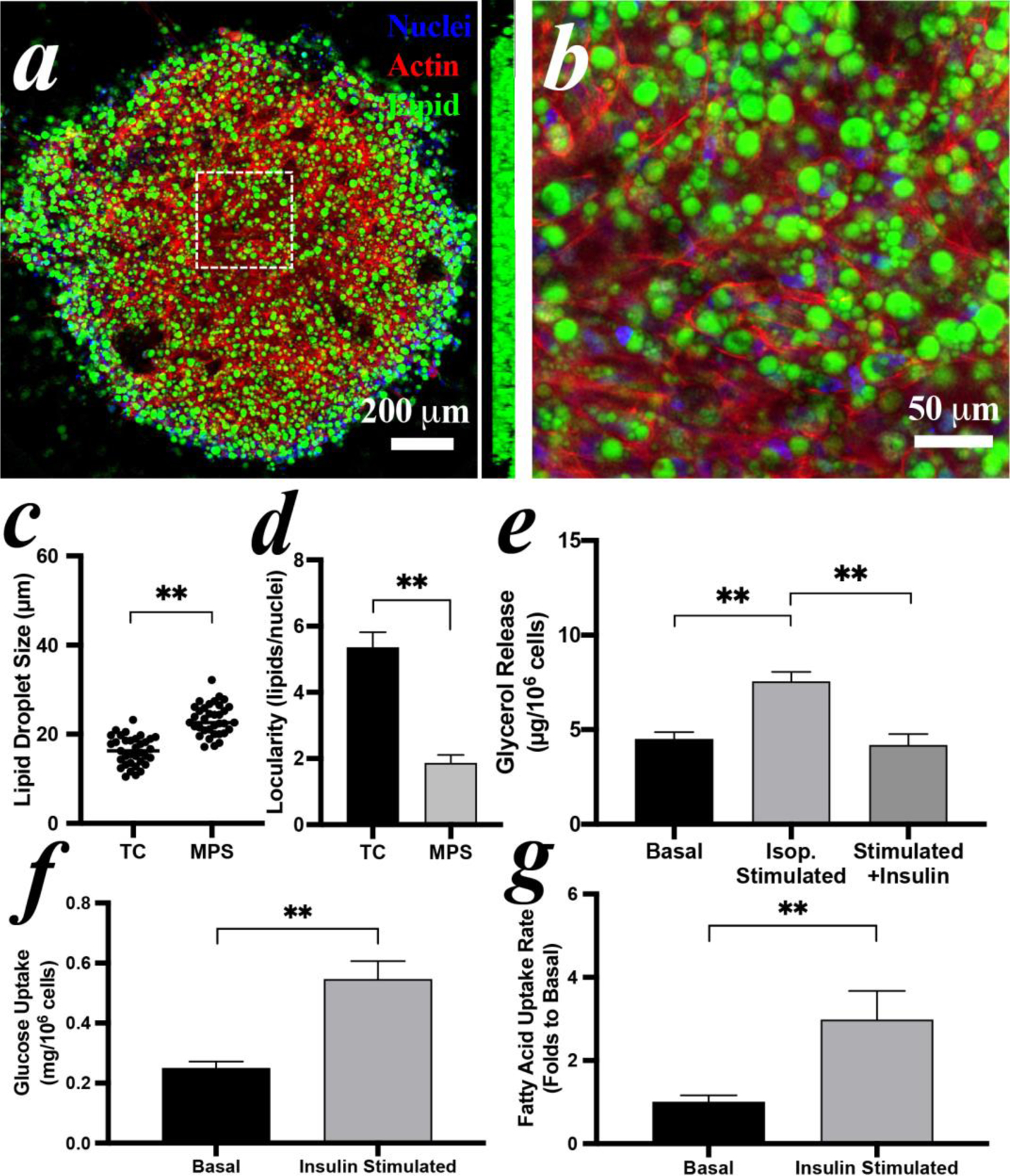
(a) morphology adipocytes in MPS and (b) zoomed view of white dash-lined area in (a). A sideview of 3D reconstruction in (a) is inserted aside. (c) lipid droplet size and (d) locularity of the adipocytes differentiated in TC condition and in MPS. Average diameters of adipocytes are 16.3 μm in TC and 22.6 μm in MPS. (e) lipolysis and (f) glucose uptake of the adipocytes in the MPS. (g) analysis of fluorescent fatty acid uptake by adipocytes in MPS with or without insulin stimulation. Pre-induced iPSC-MSCs were loaded in MPS at Day 4 and imaged or assayed at Day 14.
The hormonal responses of the iPSCs-derived iADIPO-MPS were examined following the same strategies as hMSCs-derived ones. The lipolysis released significantly more glycerol by isoproterenol stimulation than the basal condition and can be suppressed by insulin (Figure 7e). The glucose clearance of retained medium in the MPS is ~0.55 mg/106 cell in the MPS, which is significantly higher than the basal (Figure 7f). The accumulation of fluorescent fatty acid in the iADIPO-MPS was also intensively increased upon stimulation of insulin (Figure S6b), indicating a significantly active uptake and robust insulin responsiveness (Figure 7g).
2.7. Drug screening in iADIPO-MPS
To show the utility of iADIPO-MPS for the pharmaceutical interrogation of insulin sensitivity, several potential insulin sensitizing or desensitizing compounds were assessed. Four iADIPO-MPSs were connected in series to assure similar conditions during differentiation (Figure 8a). There are ~0.03 ×106 cells/MPS with 10 µL/hours medium flow rate, totally 480 µL flow through in 48 hours. As a comparison, there are ~0.05 ×106 cells/well of a 96-well plate with 100 µL medium refresh in 48 hours. Thus, medium in a MPS is refreshed about 8 times more than in TC leading us to assume that formation of nutrient gradients in our system are minimal, which is supported by the fact that we did not find obvious morphological or functional differences between up- and down-steam chambers. On Day 14, iPSCs-derived iADIPO-MPSs were switched to individual circuits for drug administration (Figure 8b). The four iADIPO-MPSs were infused with media containing either 10 mM metformin, 5 μM hydrocortisone, or 10 μM atorvastatin, according to their common dosages in previous studies[26]. After three days of drug treatment, fluorescent fatty acid uptakes of eight chambers on each iADIPO-MPS were microscopically measured showing that metformin-treated iADIPO-MPS maintained high insulin sensitivity while hydrocortisone and atorvastatin treatments resulted in insulin resistance (Figure 8c). To further quantify drug effectiveness, the uptake indexes are calculated based on the folds of insulin-induced uptake over basal in each cell chamber. Intriguingly, the insulin-induced uptake index of metformin-treated iADIPO-MPS is significantly higher than that of the non-treated group, confirming the well-known insulin-sensitizing effects of metformin (Figure 8d) and thus demonstrating that the iADIPO-MPS platform can reliably identify both insulin sensitizers and desensitizers.
Figure 8: Drug Screening in iADIPO-MPS.
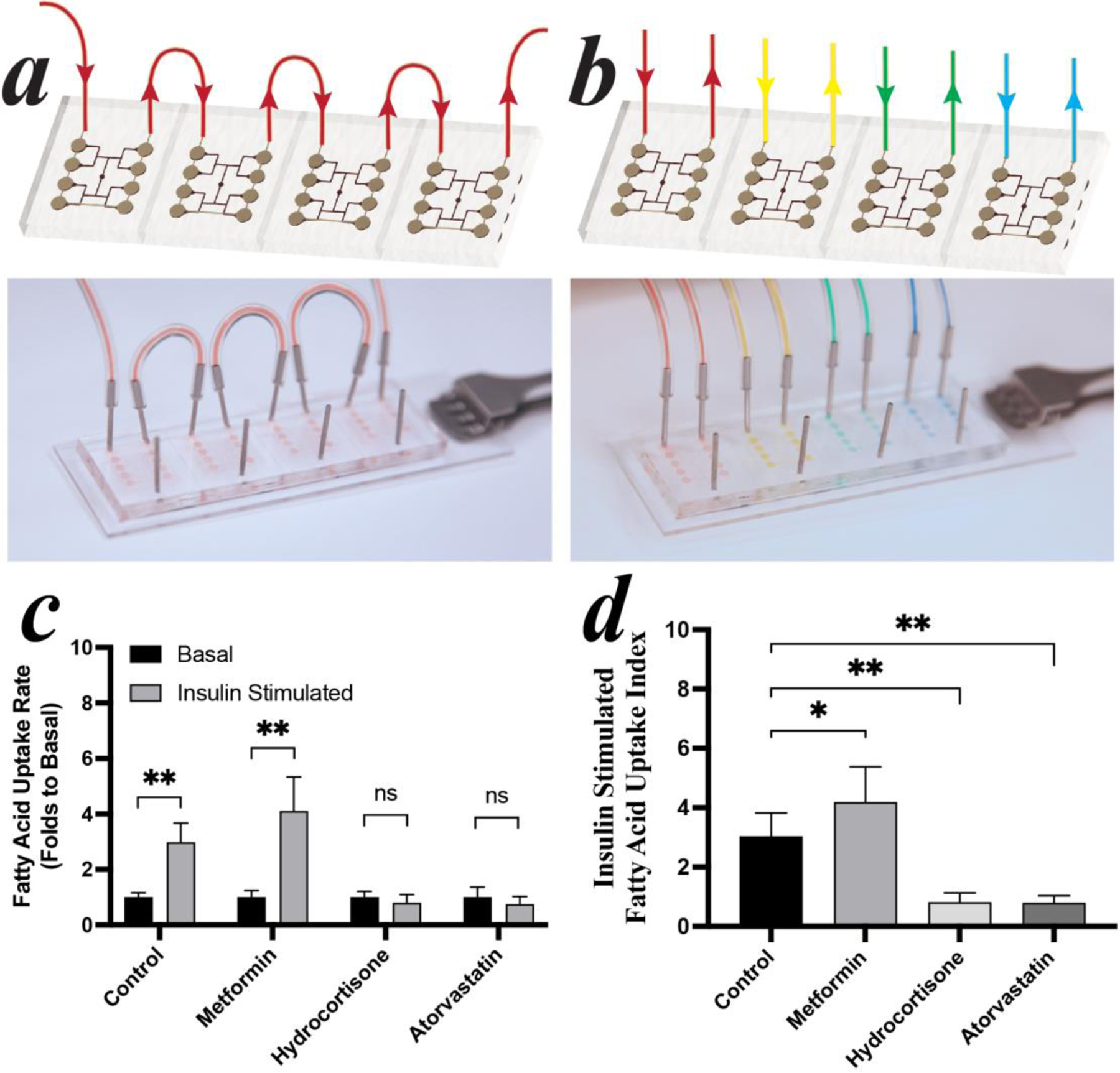
(a) In-series culture in iADIPO-MPS array. (b) Different drugs administrated in each iADIPO-MPS. (c) fluorescent fatty acid uptake by iADIPO-MPS with or without insulin stimulation. (d) uptake indexes of fatty acid uptake determined by the folds of the insulin stimulated uptakes over the basal under each condition.
3. Discussion
Taken together, our findings show that hormonally responsive and metabolically fully competent human iADIPO-MPS can be created and utilized for drug discovery. We were surprised to find that standard differentiation protocols[13] resulted in insulin resistant cells and that even commercially available adipocytes were unable to demonstrate insulin induced glucose uptake, a hallmark of insulin sensitivity[2b]. Through careful adjustment of differentiation conditions, we were able to identify key variables that allowed for both robust adipogenic differentiation of human stem cells as well as hormonal sensitivity. The three classic factors for adipogenesis[27]: insulin, dexamethasone, IBMX were orthogonally investigated to ensure that selected doses can meet the best initial induction (Figure S7a–c). We also examined two adipogenic inducers, rosiglitazone[13a, 13b, 13d] and omega-3 fatty acid[28]. Based on our orthogonal tests the effectiveness of rosiglitazone was much higher than that of DHA (Figure. S7d). DHA may also lead to insulin resistant adipocytes (Figure S7e–f). Thus, rosiglitazone emerged as a key factor both in terms of differentiation efficiency and insulin sensitivity. Although rosiglitazone has been extensively used in the clinic as an insulin sensitizer[29] we found that concentrations over 1000 nM may result in insulin resistant cells. Particularly, high rosiglitazone doses led to insulin insensitive cells while very low levels of rosiglitazone led to insulin sensitive cells but low lipid droplet accumulation (Figure 1&2, Figure S1&2). The mechanism behind this insulin desensitizing effect is unclear but here are many examples of adipocyte differentiation factors, such as insulin, corticosteroid, free fatty acids, that show a biphasic response. On one hand, they are critical to initiate the commitment of adipogenesis, further increase maturation and lipid accumulation[13]. On the other hand, they may cause mature adipocytes to be insulin resistance[26b]. Similarly, rosiglitazone shows different effects on preadipocytes and mature adipocytes in previous studies[30]. Due to its biphasic roles, the rosiglitazone dose needs to be precisely controlled to promote adipogenesis while generating insulin sensitivity adipocytes. The narrow working range of rosiglitazone was also observed in other studies[31]. In addition, rosiglitazone can induce browning of the adipocytes resulting in a more than hundreds-fold induction of UCP1[32]. Brown and beige adipocytes show decreased reliance on the insulin sensitive GLUT4 but enhance GLUT1 expression[33], which may also decrease insulin sensitivity[34]. This may explain the cause of reduced insulin sensitivity in the 1000 nM rosiglitazone treated adipocytes, which expressed ~14 folds (hMSCs) and ~5 folds (iPSCs) higher UCP1 comparing to 100 nM rosiglitazone treated adipocytes (Figure 1a&2a). Interestingly, the commercial pooled human subcutaneous adipocytes clearly contained a population of UCP1-positive brown/beige adipocytes (~2 folds higher than our adipocytes) that may be due to isolation and/or culture conditions when the cells were generated.
While WAT in MPSs have been reported previously using murine[19b, 35] and human pre-adipocytes[12, 36] as well as primary adipocytes[23, 37], we further optimized them for use with human stem cells by adjusting MPS geometry and culture environment from 2D to 3D. We assessed cell viability and differentiation with the optimized MPS design and culture conditions and compared them to standard TC conditions. Cell viability is highly maintained in MPS and better than in TC, even at such high cell loading density (Figure. 5e). This result indicates that our MPS facilitated nutrient supply and waste removal very well. Besides, many of the dead cells located at the cell lack area rather than the expected stereoscopic center. It may suggest that confluent cells with restricted cell spreading in a limited space is essential to support adipocytes for long-term culturing[38]. As such, higher cell density is preferable in respect to cell viability in our iADIPO-MPS. We also believe that such outcome is due to the isoporous membrane, which isolates the cells from medium disturbance. This design is especially supportive on cell viability for fragile cells, such as mature adipocytes. Recently, other groups also showed that a similar design can maintain the viability of primary human adipocytes[23d]. Regarding the remodeling capability and the presence of lipid droplets in Figure 4b and Figure 5b, adipogenesis in 3D matrix seems not obviously affected by cell chamber size, but highly relied on cell loading density. The ineffectiveness of geometry size may be due to the 3D environment, where most of the cells cannot sense the geometric cues provided by the cell chamber. Instead, they are surrounded by the hydrogel matrix and other cells. As such, the cell confluence in 3D space, which correlates to cell loading density, becomes dominated for adipogenesis at a high loading density. Given the diameter of hMSCs is ~20 μm[39], the cells only occupy below ~10% of the entire cell-hydrogel suspension volume at ~1×107 cells/mL. Thus, the cells are most likely encapsulated by hydrogel with negligible chance of contacting other cells, resulting in limited differentiation. The cell confluence increases with the increase of loading density, promoting overall adipogenic differentiation. Once hMSCs are loaded at ~8×107 cells/mL, they occupy above ~50% of the cell-hydrogel volume, which is close to the fraction of densely packed monodispersed spheres (50%~70%)[40]. In other words, nearly all the cells contact to each other that creates a fully confluent and even mechanically compacted microenvironment in limited 3D space, where round hMSCs with restricted cell spreading are preferable to differentiate toward adipogenic lineage, prolong culture longevity and support hypertrophy[22, 38b, 41]. The void between packed cells is filled with the biodegradable hydrogel that provides initial support but can be digested once cells start remodeling. We want to point out that other 3D culture strategies, such as hanging drop method[15c, 42] or extrusion-based techniques[16, 43], require significantly more cells and materials (~106 cells/sample) compared to our MPS (~104 cells/sample) to achieve similar outcomes[44].
Even when comparing adipocytes generated with our optimized protocol, iADIPO-MPS exhibited similar and even higher hormonal response levels (i.e. lipolysis and uptakes normalized by cell number) compared to TC conditions (Figure 6&7) and much better than commercially pooled human mature adipocytes. Particularly, insulin was able to robustly induce glucose uptake in both stem cell-derived adipocytes by 2~3 folds, matching the responses seen in murine cell line-derived adipocytes such as 3T3L1 adipocytes or human preadipocytes[20, 36b, 38b, 45]. Our data also show that the MPS environment is suitable for long-term culture and analysis. Further, iADIPO-MPS not only formed significantly larger lipid droplets, but also displayed many unilocular lipid droplets (Figure 6&7), a hallmark of WAT in vivo that is not normally reproduced in TC[19b, 23d, 46]. Remarkably, insulin responsiveness based on indexes of fold stimulation of glucose and fatty acid uptake over basal, increased from 1.5~2 to 2~3 folds for glucose uptake, from 1.5~4 to 3~6 folds for fatty acid uptake. Also, experimental variance in the iADIPO-MPS was smaller resulting in improved significance levels (p=0.0026, 0.0002, 0.0028) compared to standard TC conditions (p=0.01~0.05).
Conclusion
In summary, we report here the development of metabolically competent white adipocyte MPS derived from human mesenchymal and induced pluripotent stem cells and demonstrate their utility for the discovery of insulin sensitizing and desensitizing compounds based on three insulin sensitivity indexes. This system leverages the advantages of utilizing human cells while enabling the reduction of necessary animal experimentation. Thus, the iADIPO-MPS system should lend itself both to drug discovery in the diabetes space as well as the detection of environmental obesogens.
Methods
MPS fabrication
Patterned master templates for the medium channel and the cell chambers were fabricated with a thickness of 60 μm by standard photolithography using SU-8 (MicroChem Corp). The chamber was designed as a circular shape to match the preference for adipogenic differentiation[41]. The microfluidic patterns were replica molded from the master templates to polydimethylsiloxane (PDMS, Sylgard 184, Dow) slabs by soft lithography. The inlet/outlets were holed on the medium channel slab using a 0.75 mm biopsy punch (World Precision Instruments LLC). A polyethylene terephthalate (PET) isoporous membrane (TRAKETCH, SABEU GmbH & Co. KG) was activated by oxygen plasma (Plasma Equipment Technical Services) at 60 W under ~0.6 Torr for 60 s and then chemically decorated at 80°C for 30 mins in a solution of 97% isopropyl alcohol, 2% bis(3-(trimethoxysilyl)propyl)anime, and 1% Milli-Q water. After decoration, the membrane was rinsed in pure isopropyl alcohol and stored in anhydrous ethanol solution until further use. For MPS assembling, the cell chamber and medium channel PDMS slabs were activated by oxygen plasma at 60 W under ~0.6 Torr for 30 s and immediately sandwiched a size-trimmed decorated PET membrane with proper alignment. The device was then baked at 110 °C for 30 mins for ethanol removal, bonding stabilization, and device sterilization.
Adhesion peptide modified hyaluronic acid synthesis
Modification of hyaluronic acid bases on the previous study of our group[47]. Briefly, hyaluronic acid (65 kDa, Lifecore Biomedical) carrying hydrazide groups was synthesized and then reacted to acryloxysuccinimide to generate acrylate groups. The collagen I short peptide sequence C1[48] (CGGGF(HYP)GER, GenScript), was reacted with the acrylate-HA at room temperature to form adhesion side chains. After synthesis, the adhesion peptide modified HA precursor was lyophilized until further use.
Cell culture and differentiation
All iPSCs were firstly differentiated into mesenchymal level (iPSC-MSCs) following the standard procedure (Stem Cell Technologies). The fidelity of our subcutaneous human mesenchymal stem cells (Catalog#: SP-F-SL, Lot#: SL0060; 0064, pooled donors with BMI 25.0–29.99, Zenbio Inc.) and WTC11 iPSC-MSCs was identified by examining typical mesenchymal stem cell surface markers[49], including positive expression of the cluster of differentiation 73 (CD 73), CD 90, CD 105, and negative expression of CD45 (Figure S8a&b). Mesenchymal potency was verified by differentiating them into chondrocytes and osteoblasts (Figure S8c–e). For proliferation, hMSCs were cultivated in the complete medium (DMEM/F12 medium with 10% fetal bovine serum, 1% HEPES, 1% penicillin/streptomycin) with supplements of 5 ng/mL human fibroblast growth factor and 5 ng/mL human epidermal growth factor (all from ThermoFisher). While iPSC-MSCs were cultivated in a commercial complete medium (MesenCult-ACF Plus, Stem Cell Technologies). The sub-culture of both cells was below 70% of confluency to maintain proliferation and differentiation capacities. For the purpose of differentiation, hMSCs (within 9th generation) were cultured to 100% confluency (Day −2) with additional 2 days post-confluence culture. Differentiation was initiated by the complete medium with supplements of 500 nM insulin (Humulin R, Eli Lilly), 0.25 μM dexamethasone (Sigma-Aldrich), 0.25 mM 3-isobutyl-1-methylxanthine (Sigma-Aldrich) and rosiglitazone (Sigma-Aldrich) at designated concentrations for 4 days (Day 0 to 4). The rest of the differentiation was followed by culturing in complete medium with supplements of 500 nM insulin and rosiglitazone at the same concentration for additional 4 days (Day 4 to 8). After Day 8, the major differentiation was finished, while an extensive period of culturing in the complete medium with 500 nM insulin allows further adipocyte maturation and lipid accumulation. Following the same protocol, differentiation of iPSC-MSCs (within 13th generation) showed low efficiency. Exogenous PPARγ was thus lentivirally transfected into iPSC-MSCs, which were then sub-cultured 3 passages to stabilize cell proliferation. The differentiation of transfected iPSC-MSCs shared the similar differentiation protocol with addition of 1 μg/mL doxycycline to turn on the exogeneous PPARγ expression until Day 14. A TGF-β pathway inhibitor, SB431542 (abcam), was added into media from Day −2 to Day 14 to prevent chondrogenesis[21]. To compare our hMSCs derived adipocytes with a gold standard, commercially pooled human subcutaneous mature adipocytes (Catalog#: SA-1024-SL, pooled donor lot, ZenBio Inc.) in 24-well culture plate were purchased and maintained in the recommended commercial medium (ZenBio Inc.) for 2 days prior to assays. Special measures were implemented to reduce any disturbance to commercial and long-term cultured stem cell-derived adipocytes (>14 days), including careful plate handling, minimizing exposure out of the incubator, and gently refreshing medium by half-volume every two days.
Human subjects
All the study participants were part of Inflammation, Diabetes, Ethnicity, and Obesity (IDEO) cohort, which has been previously described[50]. Briefly, IDEO consists of 25–65 year-old men and women of multiple ethnicities and across a wide BMI range (18.5–52 kg/m2) living in the San Francisco Bay Area; exclusion factors include smoking, unstable weight within the last 3 months (>3% weight gain or loss), a diagnosed inflammatory or infectious disease, liver failure, renal dysfunction, cancer, and reported alcohol consumption of >20 grams per day. Each participant consented to take part in the study, which was approved by the University of California San Francisco (UCSF) Committee on Human Research. Participants were recruited from medical and surgical clinics at the University of California san Francisco (UCSF) and the Zuckerberg San Francisco General Hospital, or through local public advertisements (NCT03022682).
Subcutaneous white adipose tissue (WAT) biopsies sampling
Subcutaneous WAT samples were obtained from the subjects by 2 different methods as described previously[50]. In most cases, samples were collected using a 2.1 mm blunt, side-ported liposuction catheter (Tulip CellFriendly™ GEMS system Miller Harvester, Tulip Medical Products) from the peri-umbilical area under local anesthesia. Some of the samples were obtained during elective abdominal or bariatric surgery. WAT samples were freed of visible connective tissue and rinsed to remove blood and clots, after which they were further washed with Krebs-Ringer bicarbonate (KRB) buffer supplemented with 1% BSA and stored at −80°C. The study involved samples from four female participants aged 31–51 years with BMI ranging from 25–30 kg/m2. Detailed information about the study participants was listed in Table S1.
RNA extraction, cDNA synthesis and real-time qPCR.
Total RNA from the tested cells was isolated with Trizol (Invitrogen) and purified using organic extraction method by alcohol precipitation and rehydration. RNA from the human biopsies was extract and purified using column-based strategy (RNeasy lipid tissue mini kit, QIAGEN) according to manufacturer’s instructions. The RNA yield was determined using the NanoDrop One UV-Vis spectrophotometer (Thermo Scientific). Isolated RNA (1000 ng) was converted to cDNA using the Maxima First-Strand cDNA kit (Thermo Scientific) on a thermo cycler (Bio-Rad). 10 ng cDNA was used for qPCR with TaqMan Universal Master Mix (Applied Biosystems) and validated PrimeTime primer probe sets (Integrated DNA Technologies) on a QuantStudio 5 Real-Time PCR machine (Applied Biosystems). Each sample contains three replicates for qPCR to avoid artificial error. Reference gene, PPIA, was used as an internal normalization control in all samples. Expression of a certain gene between samples were determined by comparative Ct method (or so-called delta-delta Ct): the difference in Ct values (delta Ct) between a target gene and PPIA was calculated in each sample, then was directly compared to that of other samples. Primer sequences are listed in Supplementary Table. S1.
Culture in MPS
To ensure the adipogenesis of loaded stem cells, the initial stage of differentiation was pre-induced in a tissue culture flask, where tiny lipid droplets would present in >80% stem cells at Day 4. Pre-induced stem cells were dissociated (TrypLE Express, Gibco) and centrifuged as cell pellets. Adhesion peptide modified HA precursor was dissolved in triethanolamine-buffer (TEOA; 0.3 M, pH 8) at a concentration of 3 wt%. Prior to cell loading, 10 wt% MMP cleavable peptides (CQPQGLAKC, GenScript) were dissolved in TEOA and directly added to the cell pellet with dissolved HA precursor under a peptide-to-precursor volume ratio of 1:10. The hydrogel-cell mixture was injected into the cell chamber of the device and incubate at 37 °C for crosslinking. After 1 hour, the medium channel of the device was connected with catheter couplers (Instech Laboratories) and tubes (Cole-Parmer). Culture medium was infused at a flow rate of 10 μL/hour using a syringe pump (Harvard Apparatus). Metformin, hydrocortisone or atorvastatin (all from Sigma-Aldrich) was added into medium during drug administration.
Hormone-stimulated WAT function assays
Before all assays, adipocytes were subject to serum-free starvation with low glucose DMEM (1g/L glucose, phenol free, gibco) overnight. For lipolysis assay, adipocytes were incubated at 37°C in HBSS solution with 0.1% fatty acid free bovine serum albumin (BSA, ThermoFisher) with proper stimulation factors: 1 μM isoproterenol (Sigma-Aldrich), or 1 μM isoproterenol plus 1 μM insulin (Sigma-Aldrich). After 90 mins, the assay solution was collected. The lipolysis progress was determined by measuring glycerol concentration in the collected solution using a commercial reagent (Free Glycerol Reagent, Sigma-Aldrich), which enzymatically reacted with glycerol and yielded a colorimetric signal to be read in absorbance at 540 nm. For glucose uptake assay, adipocytes were glucose-free starved using KRPH buffer with 2% BSA prior to examination. After 45 mins, adipocytes were assayed in low glucose DMEM without or with 1 μM insulin stimulation at 37°C for 3 hours. The uptake was measured by the clearance of glucose in the medium using Amplex Red kit (ThermoFisher Scientific). For the fatty acid uptake assay in TC condition, adipocytes were immersed in HBSS solution with 0.1% BSA, 2 mM trypan blue (Sigma), additional 3.5 g/mL glucose (Sigma), and 1 μM fluorescent fatty acid (Bodipy 3823, Invitrogen) at 37°C for 90 mins. The dynamic uptake of fatty acid was measured by the fluorescence of cell layer at excitation/emission of 488/515 nm. For the fatty acid assay in the device, HBSS solution with 0.1% BSA, additional 3.5 g/mL glucose, and 1 μM Bodipy 3823 were infused into the medium channel at a flow rate of 20 μL/min. The entire system was placed in an incubation tank (Zeiss) with 5% CO2 supply and 37°C heating. The dynamic uptake was monitored using a fluorescence microscope at designated time points up to 30 mins. In both TC and MPS conditions, fatty acid uptake was calculated based on the increase of fluorescence within adipocytes after a certain period, in unit of relative fluorescence unit per time, and normalize to the basal uptake of a designated condition as 1 for comparison purpose.
Fluorescence staining
Cell viability was justified by staining live cells with 2 μM calcein AM and dead cells with 4 μM ethidium homodimer-1 (Invitrogen) in HBSS for 15 mins. All images were captured within 1 hour with proper incubation. For the purpose of morphology investigation, adipocytes were firstly fixed by 4% paraformaldehyde for 30 mins in TC conditions, or for 2 hours in the MPS. Nuclei, lipid droplets, and F-actin, were respectively stained by 300 nM DAPI (ThermoFisher), 1 μM Bodipy D3922 (ThermoFisher), and 1 × phalloidim-iFluor 647 (abcam) in PBS with 1% BSA for 1 hour in TC conditions, or overnight in the MPS. Samples were mounted (Diamond Antifade, Invitrogen) after staining and rinsing.
Characterizations
Bright-field images were taken by a wide-field inverted microscope (Axiovert 100 TV, Zeiss). Fluorescent images were captured by a wide-field fluorescence microscope (AxioObserver Z1, Zeiss) and confocal fluorescence microscopes (LSM710, LSM880, Zeiss). To measure time-lapsed fluorescence change, the look-up-table and other imaging settings were kept consistent. Images were analyzed and processed by Zen (Zeiss) or ImageJ (NIH). Absorbance and fluorescence of assay solutions were read by a microplate reader (SpectraMax i3x, Molecular Devices).
Statistics
All results from the TC condition were obtained from at least 3 replication groups. Lipid size was determined by analyzing at least 4 samples in each group, where the largest 7 lipids were measured in each image. For the in-device glucose and lipolysis assays, the results were obtained from 3 repeating groups, where each group combines the assay solution collected from 2 chips, and each chip contained 8 cell chambers. For the in-device fatty acid uptake assay, the images of each condition were taken from 8 cell chambers with same initiation time. In experiments that need to quantify stimulation-induced indexes, basal and stimulated conditions were sequentially assayed and sampled from the same chip to reduce variance. Cell number in TC (Figure 1c&d, 2c&d, 3b&c) were counted based on nuclei per randomly selected views (1mm × 1mm) then multiply cell culture area. The cell number in MPS (6d-e, 7d-e) is estimated based on cell density in every loading then multiply cell chamber volume. All results were statistically analyzed by JMP 11 (SAS Institute). Two-tailed student t-test was used to compare the difference between two groups. ANOVA was performed to assess the change of gene expression and function over rosiglitazone concentration. P-value <0.05 was evaluated as significant (*), <0.005 as highly significant (**). Error bars represent standard derivation in each group.
Supplementary Material
Acknowledgements
This study is supported by NIH grant 1UG3DK120004. L. Qi was supported by a postdoctoral fellowship from the Siebel Stem Cell Institute. We appreciate generous gift of WTC11 iPSCs from Prof. Kevin Healy group at UC Berkeley, 1023-5 and K3 iPSCs from Prof. Holger Willenbring group at UCSF, and G15AO iPSCs from Barcelona Stem Cell Bank. We thank Prof. Kevin Healy group for suggestions in device fabrication. Confocal imagines were captured at the CRL Molecular Imaging Center, RRID:SCR_017852, supported by Gordon and Betty Moore Foundation.
Footnotes
Competing interests
There is no competing interest to declare.
Contributor Information
Lin Qi, Department of Nutritional Science and Toxicology, College of Natural Resources, University of California, Berkeley, Berkeley, California, 94720, USA.
Peter James Zushin, Department of Nutritional Science and Toxicology, College of Natural Resources, University of California, Berkeley, Berkeley, California, 94720, USA.
Ching-Fang Chang, Department of Nutritional Science and Toxicology, College of Natural Resources, University of California, Berkeley, Berkeley, California, 94720, USA.
Yue Tung Lee, Department of Nutritional Science and Toxicology, College of Natural Resources, University of California, Berkeley, Berkeley, California, 94720, USA.
Diana L. Alba, Division of Endocrinology, Diabetes, and Metabolism, Department of Medicine, University of California, San Francisco; Diabetes Center, University of California, San Francisco, San Francisco, California 94143, USA
Suneil Koliwad, Division of Endocrinology, Diabetes, and Metabolism, Department of Medicine, University of California, San Francisco; Diabetes Center, University of California, San Francisco, San Francisco, California 94143, USA.
Andreas Stahl, Department of Nutritional Science and Toxicology, College of Natural Resources, University of California, Berkeley, Berkeley, California, 94720, USA.
References
- [1].WHO, Obesity and overweight, https://http://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight, accessed.
- [2].a) Haffner SM, Diabetes research and clinical practice 2003, 61 Suppl 1, S9; [DOI] [PubMed] [Google Scholar]; b) Santoro A, McGraw TE, Kahn BB, Cell metabolism 2021, 33, 748. [DOI] [PubMed] [Google Scholar]
- [3].a) Birkenfeld AL, Shulman GI, Hepatology 2014, 59, 713; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Cusi K, Current diabetes reports 2010, 10, 306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Rosen ED, Spiegelman BM, Cell 2014, 156, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Chusyd DE, Wang D, Huffman DM, Nagy TR, Frontiers in nutrition 2016, 3, 10; [DOI] [PMC free article] [PubMed] [Google Scholar]; Hauner H, Skurk T, Wabitsch M, in Adipose tissue protocols, Springer; 2001, p. 239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].a) Zhang H, Kumar S, Barnett A, Eggo M, Journal of Endocrinology 2000, 164, 119; [DOI] [PubMed] [Google Scholar]; b) Harms MJ, Li Q, Lee S, Zhang C, Kull B, Hallen S, Thorell A, Alexandersson I, Hagberg CE, Peng X-R, Cell reports 2019, 27, 213. [DOI] [PubMed] [Google Scholar]
- [7].Sen A, Lea-Currie YR, Sujkowska D, Franklin DM, Wilkison WO, Halvorsen YDC, Gimble JM, J Cell Biochem 2001, 81, 312. [DOI] [PubMed] [Google Scholar]
- [8].Zuk PA, Zhu M, Mizuno H, Huang J, Futrell JW, Katz AJ, Benhaim P, Lorenz HP, Hedrick MH, Tissue engineering 2001, 7, 211. [DOI] [PubMed] [Google Scholar]
- [9].Ahfeldt T, Schinzel RT, Lee Y-K, Hendrickson D, Kaplan A, Lum DH, Camahort R, Xia F, Shay J, Rhee EP, Nature cell biology 2012, 14, 209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Elefanty AG, Stanley EG, Nature cell biology 2012, 14, 126. [DOI] [PubMed] [Google Scholar]
- [11].a) Unser AM, Tian Y, Xie Y, Biotechnology advances 2015, 33, 962; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Mohsen-Kanson T, Hafner AL, Wdziekonski B, Takashima Y, Villageois P, Carrière A, Svensson M, Bagnis C, Chignon-Sicard B, Svensson PA, Stem Cells 2014, 32, 1459; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Warren CR, O’Sullivan JF, Friesen M, Becker CE, Zhang X, Liu P, Wakabayashi Y, Morningstar JE, Shi X, Choi J, Xia F, Peters DT, Florido MHC, Tsankov AM, Duberow E, Comisar L, Shay J, Jiang X, Meissner A, Musunuru K, Kathiresan S, Daheron L, Zhu J, Gerszten RE, Deo RC, Vasan RS, O’Donnell CJ, Cowan CA, Cell stem cell 2017, 20, 547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Liu Y, Kongsuphol P, Gourikutty SBN, Ramadan Q, Biomed Microdevices 2017, 19, 18. [DOI] [PubMed] [Google Scholar]
- [13].a) Merlin J, Sato M, Nowell C, Pakzad M, Fahey R, Gao J, Dehvari N, Summers RJ, Bengtsson T, Evans BA, Hutchinson DS, Cellular signalling 2018, 42, 54; [DOI] [PubMed] [Google Scholar]; b) Revittser A, Neguliaev YA, Cell and Tissue Biology 2018, 12, 367; [DOI] [PubMed] [Google Scholar]; c) Contador D, Ezquer F, Espinosa M, Arango-Rodriguez M, Puebla C, Sobrevia L, Conget P, Experimental Biology and Medicine 2015, 240, 1235; [DOI] [PubMed] [Google Scholar]; d) Pu Y, Veiga-Lopez A, Cellular & molecular biology letters 2017, 22, 6. [DOI] [PubMed] [Google Scholar]
- [14].a) Kelly JL, Findlay MW, Knight KR, Penington A, Thompson EW, Messina A, Morrison WA, Tissue engineering 2006, 12, 2041; [DOI] [PubMed] [Google Scholar]; b) Wang W, Itaka K, Ohba S, Nishiyama N, Chung UI, Yamasaki Y, Kataoka K, Biomaterials 2009, 30, 2705; [DOI] [PubMed] [Google Scholar]; c) Yang YI, Kim HI, Choi MY, Son SH, Seo MJ, Seo JY, Jang WH, Youn YC, Choi KJ, Cheong SH, J Cell Physiol 2010, 224, 807. [DOI] [PubMed] [Google Scholar]
- [15].a) Hassan W, Dong Y, Wang W, Stem cell research & therapy 2013, 4, 1; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Gerlach JC, Lin Y-C, Brayfield CA, Minteer DM, Li H, Rubin JP, Marra KG, Tissue Engineering Part C: Methods 2012, 18, 54; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Gwon K, Kim E, Tae G, Acta biomaterialia 2017, 49, 284; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Kuss M, Kim J, Qi D, Wu S, Lei Y, Chung S, Duan B, Acta biomaterialia 2018, 71, 486; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Yang JP, Anderson AE, McCartney A, Ory X, Ma G, Pappalardo E, Bader J, Elisseeff JH, Tissue Engineering Part A 2017, 23, 253; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Louis F, Kitano S, Mano JF, Matsusaki M, Acta biomaterialia 2019, 84, 194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Hsiao AY, Okitsu T, Teramae H, Takeuchi S, Advanced healthcare materials 2016, 5, 548. [DOI] [PubMed] [Google Scholar]
- [17].Azizipour N, Avazpour R, Rosenzweig DH, Sawan M, Ajji A, Micromachines 2020, 11, 599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].a) Huh D, Kim HJ, Fraser JP, Shea DE, Khan M, Bahinski A, Hamilton GA, Ingber DE, Nature protocols 2013, 8, 2135; [DOI] [PubMed] [Google Scholar]; b) Caplin JD, Granados NG, James MR, Montazami R, Hashemi N, Adv Healthc Mater 2015, 4, 1426. [DOI] [PubMed] [Google Scholar]
- [19].a) Wu X, Schneider N, Platen A, Mitra I, Blazek M, Zengerle R, Schule R, Meier M, Proc Natl Acad Sci U S A 2016, 113, E4143; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Li X, Easley CJ, Analytical and bioanalytical chemistry 2018, 410, 791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].a) Stolic M, Russell A, Hutley L, Fielding G, Hay J, MacDonald G, Whitehead J, Prins J, International journal of obesity 2002, 26, 17; [DOI] [PubMed] [Google Scholar]; b) Westergren H, Danielsson A, Nystrom FH, Strålfors P, Metabolism 2005, 54, 781; [DOI] [PubMed] [Google Scholar]; c) Lundgren M, Burén J, Ruge T, Myrnas T, Eriksson JW, The Journal of Clinical Endocrinology & Metabolism 2004, 89, 2989; [DOI] [PubMed] [Google Scholar]; d) Giorgino F, Laviola L, Eriksson JW, Acta Physiologica Scandinavica 2005, 183, 13; [DOI] [PubMed] [Google Scholar]; e) Varlamov O, Somwar R, Cornea A, Kievit P, Grove KL, Roberts CT Jr, American Journal of Physiology-Endocrinology and Metabolism 2010, 299, E486; [DOI] [PubMed] [Google Scholar]; f) Arner P, Kriegholm E, Engfeldt P, Bolinder J, The Journal of clinical investigation 1990, 85, 893; [DOI] [PubMed] [Google Scholar]; g) Stich V, Berlan M, Proceedings of the Nutrition Society 2004, 63, 369; [DOI] [PubMed] [Google Scholar]; h) Barbe P, Millet L, Galitzky J, Lafontan M, Berlan M, British journal of pharmacology 1996, 117, 907; [DOI] [PubMed] [Google Scholar]; i) Campbell PJ, Carlson MG, Hill J, Nurjhan N, American Journal of Physiology-Endocrinology And Metabolism 2006, 263, E1063; [DOI] [PubMed] [Google Scholar]; j) Meek SE, Nair KS, Jensen MD, Diabetes 1999, 48, 10. [DOI] [PubMed] [Google Scholar]
- [21].Pfeifer CG, Karl A, Kerschbaum M, Berner A, Lang S, Schupfner R, Koch M, Angele P, Nerlich M, Mueller MB, International journal of stem cells 2019, 12, 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].McBeath R, Pirone DM, Nelson CM, Bhadriraju K, Chen CS, Developmental cell 2004, 6, 483. [DOI] [PubMed] [Google Scholar]
- [23].Wu X, Schneider N, Platen A, Mitra I, Blazek M, Zengerle R, Schüle R, Meier M, Proceedings of the National Academy of Sciences 2016, 113, E4143; [DOI] [PMC free article] [PubMed] [Google Scholar]; a) Zambon A, Zoso A, Gagliano O, Magrofuoco E, Fadini GP, Avogaro A, Foletto M, Quake S, Elvassore N, Anal Chem 2015, 87, 6535; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Li X, Brooks JC, Hu J, Ford KI, Easley CJ, Lab Chip 2017, 17, 341; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Rogal J, Binder C, Kromidas E, Roosz J, Probst C, Schneider S, Schenke-Layland K, Loskill P, Sci Rep 2020, 10, 6666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Mauney JR, Nguyen T, Gillen K, Kirker-Head C, Gimble JM, Kaplan DL, Biomaterials 2007, 28, 5280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].a) Mariman EC, Wang P, Cellular and molecular life sciences 2010, 67, 1277; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chun T-H, Adipocyte 2012, 1, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].a) Zulian A, Cancello R, Girola A, Gilardini L, Alberti L, Croci M, Micheletto G, Danelli P, Invitti C, Obesity facts 2011, 4, 27; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lo KA, Labadorf A, Kennedy NJ, Han MS, Yap YS, Matthews B, Xin X, Sun L, Davis RJ, Lodish HF, Cell reports 2013, 5, 259; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Henriksbo BD, Tamrakar AK, Xu J, Duggan BM, Cavallari JF, Phulka J, Stampfli MR, Ashkar AA, Schertzer JD, Diabetes 2019, 68, 1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Skurk T, Hauner H, in Human cell culture protocols, Springer; 2012, p. 215. [Google Scholar]
- [28].Hilgendorf KI, Johnson CT, Mezger A, Rice SL, Norris AM, Demeter J, Greenleaf WJ, Reiter JF, Kopinke D, Jackson PK, Cell 2019, 179, 1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].a) Kintscher U, Law RE, American journal of physiology. Endocrinology and metabolism 2005, 288, E287; [DOI] [PubMed] [Google Scholar]; b) Albrektsen T, Frederiksen KS, Holmes WE, Boel E, Taylor K, Fleckner J, Diabetes 2002, 51, 1042. [DOI] [PubMed] [Google Scholar]
- [30].Wang P, Renes J, Bouwman F, Bunschoten A, Mariman E, Keijer J, Diabetologia 2007, 50, 654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Wang Y, Zhu W, Zhang H, Journal of Reproduction and Contraception 2014, 25, 199. [Google Scholar]
- [32].Fayyad AM, Khan AA, Abdallah SH, Alomran SS, Bajou K, Khattak MNK, International journal of molecular sciences 2019, 20, 1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Czech MP, Molecular metabolism 2020, 34, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Ebeling P, Koistinen HA, Koivisto VA, FEBS letters 1998, 436, 301. [DOI] [PubMed] [Google Scholar]
- [35].a) Loskill P, Sezhian T, Tharp KM, Lee-Montiel FT, Jeeawoody S, Reese WM, Zushin P-JH, Stahl A, Healy KE, Lab Chip 2017, 17, 1645; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tanataweethum N, Zelaya A, Yang F, Cohen RN, Brey EM, Bhushan A, Biotechnol Bioeng 2018, 115, 1979; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Brooks JC, Judd RL, Easley CJ, in Thermogenic Fat, Springer; 2017, p. 185; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Zhu J, He J, Verano M, Brimmo AT, Glia A, Qasaimeh MA, Chen P, Aleman JO, Chen W, Lab Chip 2018, 18, 3550; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Dugan CE, Kennedy RT, Methods in enzymology 2014, 538, 195; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Clark AM, Sousa KM, Jennings C, MacDougald OA, Kennedy RT, Anal Chem 2009, 81, 2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].a) Liu Y, Kongsuphol P, Chiam SY, Zhang QX, Gourikutty SBN, Saha S, Biswas SK, Ramadan Q, Lab Chip 2019, 19, 241; [DOI] [PubMed] [Google Scholar]; b) Kongsuphol P, Gupta S, Liu Y, Gourikutty SBN, Biswas SK, Ramadan Q, Scientific reports 2019, 9, 1. [DOI] [PubMed] [Google Scholar]
- [37].Lau FH, Vogel K, Luckett JP, Hunt M, Meyer A, Rogers CL, Tessler O, Dupin CL, Hilaire H, Islam KN, Tissue Engineering Part C: Methods 2018, 24, 135. [DOI] [PubMed] [Google Scholar]
- [38].a) Nelson CM, Chen CS, FEBS letters 2002, 514, 238; [DOI] [PubMed] [Google Scholar]; b) Pope BD, Warren CR, Dahl MO, Pizza CV, Henze DE, Sinatra NR, Gonzalez GM, Chang H, Liu Q, Glieberman AL, Lab Chip 2020, 20, 4152. [DOI] [PubMed] [Google Scholar]
- [39].Krueger TEG, Thorek DLJ, Denmeade SR, Isaacs JT, Brennen WN, Stem cells translational medicine 2018, 7, 651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Dullien FA, Porous media: fluid transport and pore structure, Academic press, 2012. [Google Scholar]
- [41].Kilian KA, Bugarija B, Lahn BT, Mrksich M, Proc Natl Acad Sci U S A 2010, 107, 4872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Klingelhutz AJ, Gourronc FA, Chaly A, Wadkins DA, Burand AJ, Markan KR, Idiga SO, Wu M, Potthoff MJ, Ankrum JA, Scientific reports 2018, 8, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Tytgat L, Van Damme L, Arevalo M. d. P. O., Declercq H, Thienpont H, Otteveare H, Blondeel P, Dubruel P, Van Vlierberghe S, International journal of biological macromolecules 2019, 140, 929. [DOI] [PubMed] [Google Scholar]
- [44].Cui P, Wang S, Journal of pharmaceutical analysis 2019, 9, 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].a) Lee MJ, Wu Y, Fried SK, Obesity 2012, 20, 2334; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Brannmark C, Paul A, Ribeiro D, Magnusson B, Brolen G, Enejder A, Forslow A, PloS one 2014, 9, e113620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Volz A-C, Omengo B, Gehrke S, Kluger PJ, Differentiation 2019, 110, 19. [DOI] [PubMed] [Google Scholar]
- [47].Jha AK, Tharp KM, Browne S, Ye J, Stahl A, Yeghiazarians Y, Healy KE, Biomaterials 2016, 89, 136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Knight CG, Morton LF, Peachey AR, Tuckwell DS, Farndale RW, Barnes MJ, J Biol Chem 2000, 275, 35. [DOI] [PubMed] [Google Scholar]
- [49].Louwen F, Ritter A, Kreis N, Yuan J, Obesity Reviews 2018, 19, 888. [DOI] [PubMed] [Google Scholar]
- [50].a) Alba DL, Farooq JA, Lin MY, Schafer AL, Shepherd J, Koliwad SK, The Journal of Clinical Endocrinology & Metabolism 2018, 103, 3194; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Oguri Y, Shinoda K, Kim H, Alba DL, Bolus WR, Wang Q, Brown Z, Pradhan RN, Tajima K, Yoneshiro T, Cell 2020, 182, 563. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.