

Abstract
Congenital disorders resulting in pathological protein deficiencies are most often treated postnatally with protein or enzyme replacement therapies. However, treatment of these disorders in utero before irreversible disease onset could significantly minimize disease burden, morbidity, and mortality. One possible strategy for the prenatal treatment of congenital disorders is the in utero delivery of messenger RNA (mRNA). mRNA is a gene therapeutic that has previously been investigated for protein replacement therapies and gene editing technologies. While viral vectors have been explored to induce intracellular expression of mRNA, they are limited in their clinical application due to concerns of immunogenicity and genomic integration. As an alternative to viral vectors, safe and efficient in utero mRNA delivery can be achieved using ionizable lipid nanoparticles (LNPs). While LNPs have demonstrated potent in vivo mRNA delivery to the liver following intravenous administration, intra-amniotic delivery has the potential to deliver mRNA to cells and tissues beyond those in the liver, such as in the skin, lung, and digestive tract. However, LNP stability in fetal amniotic fluid and how this stability affects mRNA delivery has not been previously investigated. Here, we engineered a library of LNPs using orthogonal design of experiments (DOE) to evaluate how LNP structure affects ex utero stability in amniotic fluid, and whether a lead candidate identified from these stability measurements enables intra-amniotic mRNA delivery in utero. We used a combination of techniques including dynamic light scattering (DLS), transmission electron microscopy (TEM), and chromatography followed by protein content quantification to screen ex vivo LNP stability in amniotic fluids. These results identified multiple lead LNP formulations that are highly stable in amniotic fluids ranging from small animals to humans, including mouse, sheep, pig, and human amniotic fluid samples. We then demonstrate that stable LNPs from the ex utero screen in mouse amniotic fluid enabled potent mRNA delivery in vitro and in utero following intra-amniotic injection in a murine model. This exploration of ex utero stability in amniotic fluids demonstrates a means by which to identify novel LNP formulations for prenatal treatment of congenital disorders via in utero mRNA delivery.
Keywords: Gene therapy, lipid nanoparticles, in utero, mRNA, nucleic acid therapeutics
1. Introduction
Recent advances in prenatal care and genetic medicine have led to improvements in fetal diagnostics including fetal whole exome sequencing and non-invasive fetal genetic testing via detection of cell-free fetal DNA in maternal serum [1–3]. This progress has enabled prenatal diagnosis of many genetic diseases such as β-thalassemia, cystic fibrosis, and glycogen storage disorders [4–6]. Although many genetic diseases can be treated after birth, postnatal treatments have limited efficacy in diseases where the onset of irreversible pathology begins in utero. Instead, prenatal gene therapies, including protein replacement and gene editing therapeutics, allow for the treatment of congenital disorders prior to or in the early stages of pathology to reduce disease burden, morbidity, and mortality [7,8]. Additionally, there are a number of ontological properties of the developing fetus that result in practical and therapeutic advantages for prenatal gene therapy. First, the small size of the fetus allows for maximum dosing per fetal weight, therefore minimizing the challenges associated with the large-scale manufacture of gene therapies [9,10]. Additionally, progenitor cells, which are an ideal target for genetic correction, are more abundant and accessible in utero, and physical barriers to delivery such as mucus membranes and the glycocalyx are less developed in the fetus [5,6]. With these notable advantages, some congenital diseases that are currently treated postnatally with protein or enzyme replacement therapeutics may be ideal candidates for prenatal gene therapy [11,12].
Two factors are critical to the delivery of gene therapies before and after birth – the delivery route and delivery vehicle. Multiple studies in small and large animal models have demonstrated the ability to target a number of different fetal organs by administering viral vectors via different injection routes [5,13–17]. First, intravenous injection of adenoviruses and adeno-associated viruses (AAVs) via the vitelline vein has demonstrated robust targeting to the fetal liver [13]. Intramuscular injection of the fetal hindlimb resulted in efficient delivery of AAVs to the skeletal muscle [14]. Intra-amniotic delivery of lentiviral vectors have been shown to target stem cells of a number of different organs in the developing fetus depending on the gestational age at which the vector was delivered [15,16]. For example, late gestation intra-amniotic injection of viral vectors has been shown to target the fetal lungs and gastrointestinal tract by taking advantage of normal fetal breathing and swallowing movements [5,17]. Alternatively, in large animal models, fetal intra-tracheal injections can also directly target the lungs while avoiding technical difficulties that exist in small animal models and minimizing the dilutional effect of the large amniotic fluid volume on the therapeutic cargo [18].
Prenatal protein and enzyme replacement gene therapies can be administered via various delivery strategies. One option is the direct administration of whole proteins in utero, but these therapeutics are limited by the in vitro synthesis of proteins with the correct post-translational modifications [11]. This challenge can be overcome by viral or non-viral mediated delivery of nucleic acids which are instead translated endogenously in the host. Viral vectors for the delivery of nucleic acids have shown promise in prenatal applications [5,13,19], but pose the risk of genomic integration [20,21]. Additional challenges of viral vectors such as immunogenicity and limitations regarding repeat dosing can be addressed with non-viral nucleic acid delivery [20,22]. Non-viral nucleic acid delivery includes the administration of therapeutic messenger RNA (mRNA) which initiates transient protein expression in the cytosol and therefore avoids nuclear transport and the risk of genomic integration [11,20]. However, mRNA faces similar delivery challenges in utero as it does in adults, including rapid degradation by nucleases present in the body and inefficient transport across the cell membrane due to its large size and negative charge [23]. These challenges have limited the broad clinical use of mRNA therapeutics and necessitate the development of mRNA delivery technologies for in vivo delivery [20,23].
Numerous delivery platforms have been investigated for the delivery of mRNA in vivo such as polymeric and lipid-based nanoparticle (NP) systems [20,22,24]. One polymeric system, specifically poly(lactic-co-glycolic acid) (PLGA) NPs, has shown efficient delivery of gene editing nucleic acids to fetal hematopoietic stem cells for the prenatal treatment of β-thalassemia [6]. However, other polymeric NPs for gene delivery that use highly cationic molecules such as polyethyleneimine (PEI) have been found to be highly toxic, therefore limiting their clinical application [22,25]. Instead, ionizable lipid nanoparticle (LNP) platforms can be used for the therapeutic delivery of mRNA, and they are more clinically advanced than polymeric systems following the recent Food and Drug Administration (FDA) approval of Alnylam’s Onpattro siRNA LNP therapeutic [22,26] and emergency use authorization of Moderna and Pfizer/BioNTech’s mRNA vaccines against COVID-19 [27,28]. Ionizable LNPs benefit from high nucleic acid encapsulation efficiencies and small sizes (<100 nm) making them ideal vectors for in utero intracellular delivery [29]. Additionally, LNPs contain an ionizable lipid component that remains neutral at physiological pH, but after cellular uptake, becomes charged in the acidic endosomal environment allowing for enhanced endosomal escape and potent intracellular mRNA delivery [24,30–32]. A final advantage of LNPs is their modular design; the excipients and their respective molar ratios, the ionizable lipid, and the lipid to nucleic acid ratio can all be individually optimized to improve biodistribution and intracellular delivery for a particular application [33–36]. As LNP technology advances and the number of possible formulations continues to grow, there is a need for assays to evaluate and predict LNP performance for in vivo, including prenatal applications.
Recent work has demonstrated the substantial effect of the biological environment on NP stability, biodistribution, and delivery [37–39], yet these works have primarily focused on the effect of blood, serum, and simulated interstitial fluid biological environments. Fetal amniotic fluid, the biological environment for intra-amniotically administered in utero gene therapeutics, is a similar protein-rich environment as serum and likely will influence LNP stability and delivery. However to our knowledge, the effects of amniotic fluids on LNP stability have not been previously investigated. Therefore, we hypothesized that evaluating LNP stability in amniotic fluid could identify novel LNPs that are well-suited for prenatal gene therapies. To this end, we sought to explore the stability of a library of LNPs in mouse, sheep, pig, and human amniotic fluids, and determine if these measures of stability correlate with in vitro and in utero mRNA delivery.
Here, we orthogonally designed a library of 16 ionizable LNPs with varying excipient molar ratios and developed a minimal resource ex vivo stability assay using dynamic light scattering (DLS). Orthogonal design of experiments (DOE) was used to screen a space of 256 possible LNP formulations by combining four molar ratios of each of four excipients (ionizable lipid, DOPE, cholesterol, and lipid-PEG) with 16 formulations. We explored the stability of this library in a group of amniotic fluids and identified stable and unstable LNPs in each of the fluids tested. Also, by screening each LNP in the same fluid type across species, we identified correlations of LNP stability in amniotic fluids across species. Next, the LNP library was screened for luciferase mRNA delivery in vitro. These results demonstrated correlations between ex vivo stability in mouse amniotic fluid and in vitro luciferase mRNA delivery. We then tested a stable and unstable LNP from ex vivo screening in a mouse model of intra-amniotic fetal delivery which showed that the more stable LNP had higher in utero mRNA delivery than an unstable LNP. Finally, we demonstrate the structure function relationships of LNP excipients on ex utero amniotic fluid stability and in vitro mRNA delivery. Taken together, we have explored ex utero LNP stability in amniotic fluids as a means to identify LNP formulations for prenatal mRNA delivery with potential applications in treating congenital diseases.
2. Materials and Methods
2.1. Ionizable lipid Synthesis and mRNA Production
Ionizable lipid B-4 and C12–200 were prepared via Michael addition chemistry as previously described [40]. Briefly, the polyamine core (Enamine Inc., Monmouth Junction, NJ) was combined with excess lipid epoxide (Sigma-Aldrich, St. Louis, MO) in a 4 mL glass scintillation vial under gentle stirring with a magnetic stir bar for 2 days at 80 °C. The reaction mixture was dried using a Rotovap R-300 (Buchi, New Castle, DE) and used for LNP formulation.
The firefly luciferase gene sequence was codon optimized, synthetized, and cloned into our proprietary mRNA production plasmid. The m1Ψ UTP nucleoside modified Fluc mRNA was co-transcriptionally capped using the trinucleotide cap1 analogue (TriLink, San Diego, CA), and engineered to contain 101 nucleotide-long poly(A) tail. Transcription was performed using MegaScript T7 RNA polymerase (Invitrogen, Waltham, MA) and mRNA was precipitated using lithium chloride and purified by cellulose chromatography as previously described [41]. The produced mRNAs were analyzed by agarose gel electrophoresis, sequenced, subjected to a standard J2 dot blot, assayed for INF induction in human monocyte derived dendritic cells, and stored frozen at −80 °C for future use.
2.2. LNP Formulation
LNPs were formulated using a 10:1 weight ratio of ionizable lipid B-4 to luciferase mRNA [40]. First, ionizable lipid B-4 was combined in an ethanol phase with cholesterol (Sigma-Aldrich), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE, Avanti, Alabaster, AL), and 1,2-dimyristoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (ammonium salt) (C14-PEG2000, Avanti) at varying molar ratios (Table 1) to a total volume of 112.5 μL. A separate aqueous phase was prepared with 25 μg of luciferase mRNA in 10 mM citrate buffer (pH = 3) to a total volume of 337.5 μL. With a syringe pump, the ethanol and aqueous phases were combined to form LNPs via chaotic mixing using a microfluidic device designed with herringbone features as previously described [42]. LNPs were dialyzed against 1X PBS with a molecular weight cutoff of 20 kDa for 2 h, filtered using a 0.22 μm filter, and stored at 4 °C for later use. All materials were prepared and handled ribonuclease-free throughout synthesis, formulation, and characterization steps.
Table 1.
LNP library formulations including the molar ratio and molar percentage of excipients.
| Molar Ratios | Molar Percentage (%) | |||||||
|---|---|---|---|---|---|---|---|---|
| Name | B-4 | DOPE | Chol | PEG | B-4 | DOPE | Chol | PEG |
| A1 | 15 | 10 | 5 | 0.5 | 49.18 | 32.79 | 16.39 | 1.64 |
| A2 | 15 | 20 | 20 | 4.5 | 25.21 | 33.61 | 33.61 | 7.56 |
| A3 | 15 | 30 | 35 | 8.5 | 16.95 | 33.90 | 39.55 | 9.60 |
| A4 | 15 | 40 | 50 | 12.5 | 12.77 | 34.04 | 42.55 | 10.64 |
| A5 | 25 | 10 | 20 | 8.5 | 39.37 | 15.75 | 31.50 | 13.39 |
| A6 | 25 | 20 | 5 | 12.5 | 40.00 | 32.00 | 8.00 | 20.00 |
| A7 | 25 | 30 | 50 | 0.5 | 23.70 | 28.44 | 47.39 | 0.47 |
| A8 | 25 | 40 | 35 | 4.5 | 23.92 | 38.28 | 33.49 | 4.31 |
| A9 | 35 | 10 | 35 | 12.5 | 37.84 | 10.81 | 37.84 | 13.51 |
| A10 | 35 | 20 | 50 | 8.5 | 30.84 | 17.62 | 44.05 | 7.49 |
| A11 | 35 | 30 | 5 | 4.5 | 46.98 | 40.27 | 6.71 | 6.04 |
| A12 | 35 | 40 | 20 | 0.5 | 36.65 | 41.88 | 20.94 | 0.52 |
| A13 | 45 | 10 | 50 | 4.5 | 41.10 | 9.13 | 45.66 | 4.11 |
| A14 | 45 | 20 | 35 | 0.5 | 44.78 | 19.90 | 34.83 | 0.50 |
| A15 | 45 | 30 | 20 | 12.5 | 41.86 | 27.91 | 18.60 | 11.63 |
| A16 | 45 | 40 | 5 | 8.5 | 45.69 | 40.61 | 5.08 | 8.63 |
2.3. Dynamic Light Scattering and Zeta Potential
For initial dynamic light scattering (DLS) measurements, 10 μL of each LNP solution was diluted 100X in 1X PBS in 4 mL disposable cuvettes. For baseline zeta potential measurements, 20 μL of each LNP solution was diluted 50X in deionized water in DTS1070 zeta potential cuvettes (Malvern Panalytical, Malvern, UK). Four measurements each with at least 10 runs were recorded for each sample using a Zetasizer Nano (Malvern Instruments, Malvern, UK). Data are reported as mean ± standard deviation (n = 3 to 4 measurements).
2.4. LNP pKa Measurements
Surface ionization measurements to calculate the pKa of each LNP formulation were performed as previously described [43]. Briefly, buffered solution containing 150 mM sodium chloride, 20 mM sodium phosphate, 20 mM ammonium acetate, and 25 mM ammonium citrate was adjusted to pH 2 to 12 in increments of 0.5. 125 μL of each pH-adjusted solution and 5 μL of each LNP formulation were plated in triplicate in black 96-well plates. 2-(p-toluidinyl)naphthalene-6-sulfonic acid (TNS) was then added to each well to a final TNS concentration of 6 μM. The fluorescence intensity was read on an Infinite 200 Pro plate reader (Tecan, Morrisville, NC) at an excitation wavelength of 322 nm and an emission wavelength of 431 nm. Using least squares regression, the pKa was taken as the pH corresponding to half-maximum fluorescence intensity, i.e., 50% protonation.
2.5. LNP Encapsulation Efficiency
mRNA encapsulation efficiencies of each LNP formulation were calculated using the Quant-iT-RiboGreen (Thermo Fisher Scientific, Waltham, MA) assay as previously described [44]. Each LNP sample was diluted to approximately 2 ng/μL in two microcentrifuge tubes containing 1X TE buffer or 0.1% (v/v) Triton X-100 (Sigma-Aldrich). LNPs in Triton-X were left to lyse for 20 minutes. After incubation, LNPs in TE buffer and Triton X-100 as well as mRNA standards were plated in triplicate in black 96-well plates and the fluorescent RiboGreen reagent was added per manufacturer’s instructions. Fluorescence intensity was read on an Infinite 200 Pro plate reader (Tecan) at an excitation wavelength of 480 nm and an emission wavelength of 520 nm. RNA content was estimated by comparison to a standard curve estimated using least squares linear regression (LSLR). Encapsulation efficiency was calculated as where A is the RNA content in TE buffer and B is the RNA content in Triton X-100. Encapsulation efficiencies are reported as mean ± standard deviation (n = 3).
2.6. Animal Experiments
All animal use and experimental protocols were approved by the Institutional Animal Care and Use Committees (IACUC) at the Children’s Hospital of Philadelphia’s (CHOP) and the University of Pennsylvania and followed guidelines set forth in the National Institutes of Health’s Guide for the Care and Use of Laboratory Animals. Balb/c (stock #000651) mice were purchased from The Jackson Laboratory (Bar Harbor, ME) and housed in the Laboratory Animal Facility of the Colket Translational Research Building at CHOP. Females of breeding age were paired with males and separated at 24 h to achieve time-dated pregnant dams for amniotic fluid collections or in utero LNP injections as described below. Time-dated Suffolk ewes were obtained from MacCauley Suffolks (Atglen, PA) and time-dated miniature Yucatan swine were obtained from Sinclair Bio Resources (Auxvasse, MO).
2.7. Fluid Collection
For murine serum collections, 8-week-old female C57BL/6 mice (Jackson Laboratory, 18–21 g) were subjected to tail vein blood collections. Blood was centrifuged at 10,000 g for 10 min and the serum supernatant was collected and the cell pellet discarded.
For murine amniotic fluid collections, time-dated Balb/c dams were sacrificed at gestational day 16 (E16), and under sterile conditions a midline laparotomy was performed and the uterine horn was removed. A 27 gauge needle was used to aspirate amniotic fluid from each individual amniotic sac and amniotic fluid was stored at −80 °C. For one biological replicate, amniotic fluid was pooled from several fetuses; the volume of fluid obtained from the amniotic cavity of a single fetus was approximately 100 μL.
For sheep amniotic fluid collection, a time-dated ewe at gestational day 110 (term is approximately 145 days) was anesthetized with 15 mg/kg of intramuscular ketamine with maintenance of general anesthesia using inhaled isoflurane (2–4% in O2) and propofol (0.2 to 1 mg/kg/min). Intraoperative monitoring included pulse oximetry and constant infusion of isotonic saline administered via a central venous line placed in a jugular vein. Under sterile conditions, a lower midline laparotomy was performed and the uterus was exposed. A small hysterotomy was then performed and the amniotic fluid was aspirated using a 60 mL syringe and stored at −80 °C until use.
For pig amniotic fluid collection, a time-dated sow at gestational day 100 (term is approximately 114 days) was anesthetized with intramuscular ketamine and acepromazine with maintenance of general anesthesia using inhaled isoflurane and propofol. Intraoperative monitoring included pulse oximetry and constant infusion of isotonic saline administered via a central venous line placed in a jugular vein. Under sterile conditions, a midline laparotomy was performed and the uterus was exposed and amniotic fluid was aspirated using a 60 mL syringe via a small hysterotomy.
For human specimens, amniotic fluid was collected from the amniotic cavity of an approximately 24 week gestation fetus undergoing an open fetal surgical procedure. Specifically, at the time of hysterotomy, amniotic fluid was aspirated in a sterile manner in a 60 mL syringe and subsequently stored at −80 °C until use. Amniotic fluid collection was approved by the Children’s Hospital of Philadelphia Institutional Review Board (IRB #14–010958).
2.8. Amniotic Fluid Characterization
To characterize the fluids used in this study, pH and protein concentration of all five fluids (mouse serum, mouse amniotic fluid, sheep amniotic fluid, pig amniotic fluid, and human amniotic fluid) were measured. Three pH measurements of each fluid were recorded using a glass combination microelectrode (Thermo Fisher Scientific). Using a NanoQuant Plate (Tecan), protein concentration was estimated by measuring absorbance at excitation wavelengths of 260 nm and 280 nm on an Infinite 200 Pro plate reader (Tecan). pH values and protein concentrations are reported as mean ± standard deviation (n = 3).
2.9. LNP Stability in Mouse Amniotic Fluid
DLS was used to assess LNP stability in mouse amniotic fluid based on previously described assessments of nanoparticle behavior in human serum albumin and fetal bovine serum (FBS) [45,46]. Briefly, a range of mouse amniotic fluid percentages were selected – 0%, 25%, 50%, 75%, and 100% (v/v). DLS measurements of the LNP alone and the fluid alone were used for the 0% and 100% fluid percentages, respectively. The other fluid percentages were calculated using the equation
where A is the volume of fluid and B is the volume of LNP in 1X PBS. The LNP volume was held constant at 10 μL for all DLS stability measurements. For all fluid percentages, LNPs were incubated in E16 mouse amniotic fluid for 30 min at 37 °C under gentle agitation at 300 rpm. After 30 min, the entire incubation volume of each sample was diluted 100X in PBS and transferred to a cuvette for DLS measurement.
A range of incubation timepoints was also selected – 0 min, 5 min, 15 min, 30 min, 60 min, 120 min, and 240 min. For all time points, LNPs were incubated in 50% (v/v) mouse amniotic fluid at 37 °C under gentle agitation at 300 rpm. Following incubation, the LNP and fluid samples were prepared for DLS as described above.
Both experiments were repeated in triplicate using three biological replicates of E16 mouse amniotic fluid. All DLS readings in the present study involved four independent measurements, each the average of 10 runs. The LNP size described throughout this study is the mean peak intensity diameter (nm) of intensity distribution measurements from DLS. Intensity curves are shown as the mean intensity (n = 3 to 4) for each data point as a function of size (nm). Size and polydispersity index (PDI) are reported as the mean ± standard deviation (n = 3 to 4 measurements per biological replicate).
2.10. LNP Library Stability Screen in Mouse Serum and Mouse, Pig, Sheep and Human Amniotic Fluid
All 16 LNP formulations were evaluated in the five fluids listed above. Due to the precious nature of many of these samples and the reasonable standard deviations of measurements collected with three biological replicates of mouse amniotic fluid (Figure 2b), only one biological replicate of each fluid was used in the library screen. Following incubation in 50% (v/v) fluid for 30 min at 37 °C with gentle agitation at 300 rpm, the entire incubation volume was diluted 100X in 1X PBS and transferred to a cuvette for DLS measurement.
Figure 2 -. Ex utero LNP stability in mouse amniotic fluid.
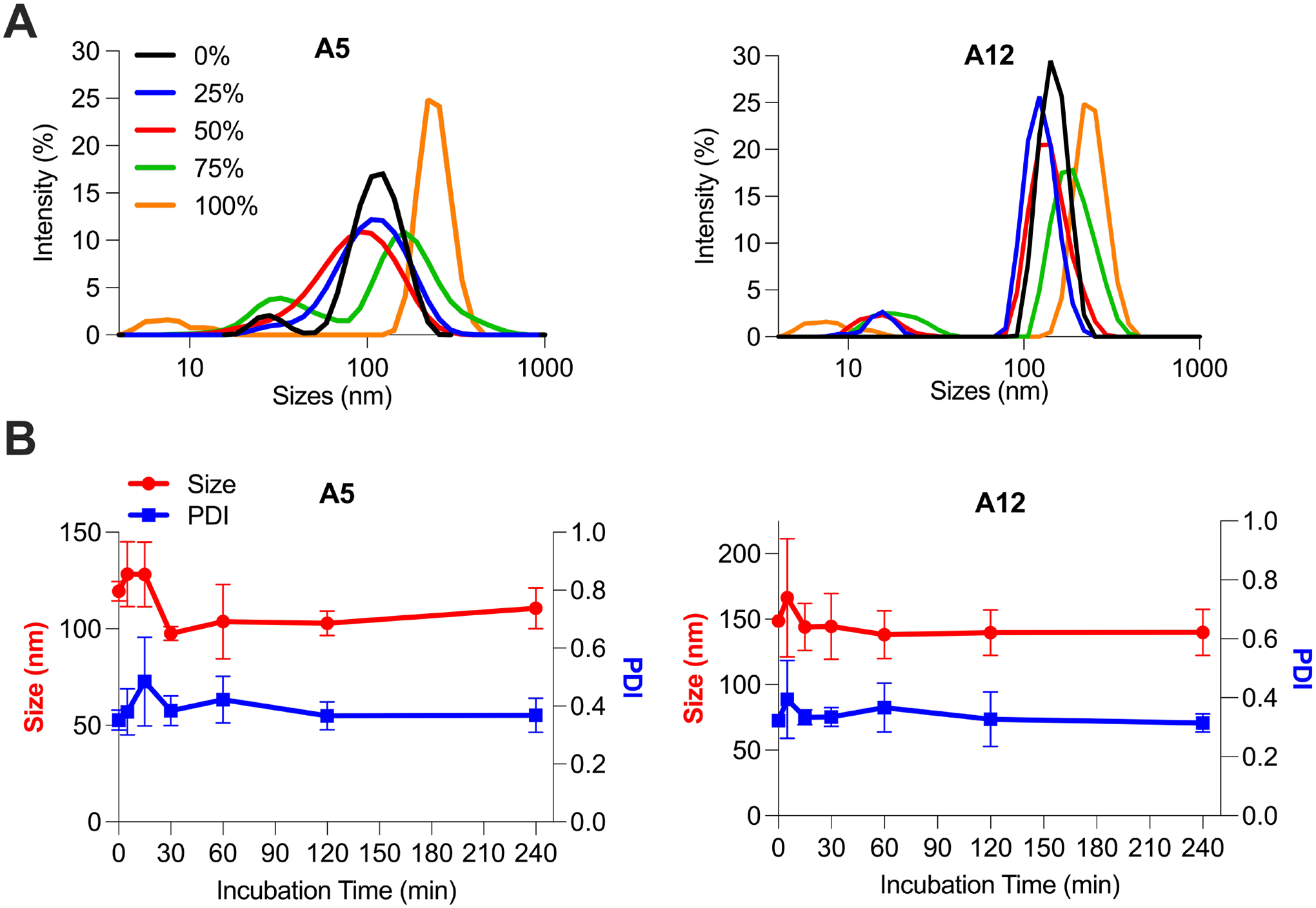
(A) Stability assessment of LNPs A5 (34 ng/μL) and A12 (41 ng/μL) in mouse amniotic fluid with varying fluid percentages and incubation times. LNPs A5 and A12 were incubated in five percentages of mouse amniotic fluid – 0%, 25%, 50%, 75%, and 100% (volume mouse amniotic fluid/total volume) – for 30 minutes. Intensity curves were recorded by DLS for both formulations across fluid percentages to demonstrate size and PDI changes. (B) LNPs A5 and A12 were incubated for seven time points – 0 min, 5 min, 15 min, 30 min, 60 min, 120 min, and 240 min – in 50% (v/v) mouse amniotic fluid. Size and PDI were measured by DLS for both formulations across timepoints. Data points are presented as means and standard deviations among n = 3 to 4 technical replicates for each of three biological replicates.
The percent change in size for each LNP and fluid combination was calculated as
where A is the mean LNP size in PBS and B is the LNP size in fluid. Similarly, the percent change in PDI for each LNP and fluid combination was calculated as
where C is the mean PDI of the LNP in PBS and D is the PDI of the LNP in fluid.
Percent change in size and percent change in PDI measurements were compared by 2-way ANOVA across fluid type and formulation with the Tukey-Kramer correction for multiple comparisons. Hits were identified as LNP and fluid combinations that had significantly (p < α = 0.05) lower percent change in size or percent change in PDI measurements than the same LNP in mouse serum.
An instability parameter was defined to concurrently evaluate the effect of both percent change in size and percent change in PDI on overall LNP stability; it is defined as the mean of the two measurements. LSLR was used to compare mean instability parameter measurements for the LNP library across species for amniotic fluids with goodness of fit quantified by the coefficient of determination R2.
2.11. TEM and Zeta Potential Characterization of LNPs in Mouse Amniotic Fluid
Transmission electron microscopy (TEM) images were obtained using a JEOL 1010 electron microscope system (Jeol, Tokyo, Japan) operated at 80 kV. LNP samples were deposited on thin carbon films (Ted Pella Inc., Redding, CA) supported by nickel grids and were stained with 2% uranyl acetate (Electron Microscopy Sciences, Hatfield, PA) before observation. For LNP formulations in PBS and with mouse amniotic fluid, the shortest edge to edge diameter of 20 particles was manually measured with ImageJ. The reported diameter is the mean ± standard deviation (n = 20).
For zeta potential measurements of LNPs in mouse amniotic fluid, LNPs A12 and A1 were incubated for 30 min at 37 °C with gentle agitation at 300 rpm in six percentages of mouse amniotic fluid – 0%, 10%, 25%, 50%, 75%, and 100% (v/v). Samples were immediately diluted 50X in deionized water and loaded into DTS1070 zeta potential cuvettes (Malvern Panalytical). Zeta potential was measured using a Zetasizer Nano (Malvern Instruments) and measurements are reported as mean ± standard deviation (n = 3).
2.12. Chromatography and Protein Quantification of LNPs in Mouse Amniotic Fluid
Based on a previous study [47], the most (A12) and least stable (A1) LNPs from the mouse amniotic fluid stability screen were incubated in mouse amniotic fluid and isolated from unbound fluid proteins via a Sepharose CL-6B affinity chromatography column. 32 fractions, each equal and approximately 100 μL in volume, were collected following loading of (i) A12 with mouse amniotic fluid, (ii) A1 with mouse amniotic fluid, or (iii) free mouse amniotic fluid. For all three samples, protein concentration was evaluated in each fraction using a NanoQuant Plate (Tecan) and read on an Infinite 200 Pro plate reader (Tecan). Fractions with non-zero protein concentration readings that did not overlap with free mouse amniotic fluid fractions were identified by plotting protein concentration versus fraction number. These identified fractions were pooled and further evaluated for protein content using a micro bicinchoninic acid (BCA) protein assay kit (Thermo Fisher). Pooled fractions from column separation, LNPs A12 and A1 in PBS, and standard curve samples were incubated at a 1:1 ratio of sample to working reagent at 60 °C for 60 min. Following incubation, samples were allowed to cool and plated in triplicate on a 96-well plate. Absorbance at a wavelength of 562 nm was immediately read on an Infinite 200 Pro plate reader (Tecan). The protein concentration was quantified by comparing sample absorbances to a standard curve using LSLR. A paired t test was used to determine significant differences in protein concentration between LNPs in PBS and with mouse amniotic fluid. Protein concentrations are reported as mean ± standard deviation (n = 3).
2.13. In Vitro LNP-mediated Luciferase mRNA Delivery to HeLa Cells
HeLa cells (ATCC no. CCL-2) were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) with L-glutamine (Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (FBS) (Gibco, Dublin, Ireland) and 1% penicillin-streptomycin (Gibco). Cells were plated at 10,000 cells per well in 100 μL of medium in tissue culture treated 96-well plates and were left to adhere overnight. All 16 LNP formulations were incubated with 50% (v/v) mouse amniotic fluid for 30 min at 37 °C under gentle agitation at 300 rpm. LNP formulations pre-incubated in mouse amniotic fluid or LNPs in PBS were used to treat cells at a dose of 50 ng of mRNA per 10,000 cells. As a positive control, the transfection reagent lipofectamine MessengerMAX (Thermo Fisher Scientific) was combined with luciferase mRNA for 10 min as per the manufacturer’s protocol and was used to treat cells at a dose of 50 ng of mRNA per 10,000 cells. 24 h after treatment with LNPs and lipofectamine, cells were centrifuged at 300 g for 5 min and excess medium was removed. 50 μL of 1X lysis buffer (Promega, Madison, WI) followed by 100 μL of luciferase assay substrate (Promega) was added to each well. After 10 minutes of incubation, luminescence was quantified using an Infinite 200 Pro plate reader (Tecan). The luminescence signal for each condition was normalized by dividing by the luminescence signal of untreated control cells. To evaluate cytotoxicity, additional plates were prepared as described above. After 24 h, 100 μL of CellTiter-Glo (Promega) was added to each well and the luminescence corresponding to ATP production was quantified using a plate reader following 10 min of incubation. Luminescence for each group was normalized by dividing by the luminescence signal of untreated control cells.
Luciferase expression and percent cell viability are reported as mean ± standard deviation (n = 3 biological replicates and at least 2 technical replicates per plate). GraphPad Prism’s ROUT method [48] with Q = 5% was used to identify outliers across treatment conditions and subsequently remove them from mean and standard deviation calculations. For both luciferase expression and percent viability, 2-way ANOVA with the Tukey-Kramer correction for multiple comparisons was used to compare means across formulation and treatment condition.
2.14. In Vitro LNP-mediated Luciferase mRNA Delivery to Primary Fetal Lung Cells
Fetal lung cells were harvested from a single pregnant Balb/c female mouse (stock #000651) that was time-dated at gestational day E16. The pregnant dam was euthanized and a laparotomy was performed to expose the uterine horns. Six fetuses were removed and a dissection microscope was used to perform a thoracotomy and isolate the fetal lung cells. This lung tissue was digested mechanically and filtered through a 100 μM cell strainer to isolate cells. These cells were washed with 1X PBS and then cultured in DMEM supplemented with 15% FBS and 1% penicillin-streptomycin. Cells were incubated at 37°C in a humidified 5% CO2 atmosphere.
Primary fetal lung cells were plated in a 96-well plate at a density of 20,000 cells per well. Cells were treated with either A4 or A12 LNPs containing luciferase mRNA at doses ranging from 10 to 100 ng per 20,000 cells. Luciferase expression and cell viability experiments were performed as described above. Luciferase expression and percent cell viability are reported as mean ± standard deviation (n = 6). Unpaired t tests with the Holm-Sídák correction for multiple comparisons was used to evaluate differences in luciferase expression between LNPs A12 and A4 for tested each dose.
2.15. In Utero Studies
In utero intra-amniotic injections were performed as previously described [5]. Briefly, under isoflurane anesthesia and after providing local anesthetic with 0.25% bupivacaine subcutaneously, a midline laparotomy was performed and the uterine horn was exposed. Under a dissecting microscope, 30 μL of PBS or LNPs concentrated to 325 ng/μL was injected into the amniotic cavity of each fetus using a custom made 80 μm beveled glass micropipette and an automated microinjector (Narishige IM-400 Electric Microinjector, Narishige International USA Inc., Amityville, NY). After successful injection, the uterus was returned to the peritoneal cavity and the abdomen was closed with a single layer of absorbable 4–0 polyglactin 910 suture. A group size of n = 5 was used for each of the three treatment groups (LNP A12, LNP A4, and PBS injections).
2.16. Luciferase Imaging and Quantification
We sought to assess luciferase signal in treated fetuses as well as individual fetal organs following in utero intra-amniotic injection of LNPs containing luciferase mRNA using methods previously described [34]. Specifically, mice were imaged 4 h after intra-amniotic injection of LNPs or PBS. Luciferase imaging was performed using an in vivo imaging system (IVIS, PerkinElmer, Waltham, MA). 10 min before sacrifice and imaging, dams were injected intraperitoneally with D-luciferin and potassium salt at 150 mg/kg (Biotium, Fremont, CA). Pregnant dams were then placed supine into the IVIS, and luminescence signal was detected with a 60 s exposure time. Next, a midline laparotomy was performed to expose the uterine horn, and luciferase imaging was repeated. Following imaging of the dam with the uterine horn exposed, fetuses were removed and individually imaged using IVIS with 60 s exposure times. The fetal liver, intestines, lungs, and brain were subsequently removed and imaged by IVIS. Image analysis was conducted using the Living Image software (PerkinElmer). To quantify luminescence flux, a rectangular region of interest (ROI) was placed in an area without any luminescence signal in the same image. Normalized flux was calculated by dividing the total flux from the ROI over the fetus or organ by the total flux from the background ROI. For each treatment group, the ROUT outlier test with Q = 1% was used to identify and remove outliers. Reported fetal and organ bioluminescence represent the mean ± standard error of the mean (SEM) (n ≥ 4). The representative organ IVIS images shown are those that have the highest luminescence values for each treatment condition.
3. Results
3.1. LNP Library Design, Formulation, and Characterization
To engineer and identify stable LNPs in each amniotic fluid of interest, a library of 16 LNP formulations was designed using an orthogonal design of experiments (DOE) approach. Orthogonal DOE was chosen because it is a well-defined methodology for screening nanoparticles, while minimizing the total number of formulations tested [33,40,49]. Theoretically, 256 combinations are possible when varying four molar ratios of each of four excipients. However, by using orthogonal design, the effects of the four excipients and their four molar ratios can be evaluated using only 16 formulations (Table 1). Therefore, four excipients at varying molar ratios (Figure 1) were used to formulate LNPs: (i) an ionizable lipid, (ii) 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), (iii) cholesterol, and (iv) lipid-anchored polyethylene glycol (lipid-PEG) (Supplementary Figure 1). The ionizable lipid B-4 was selected based on previously published work from our group demonstrating that LNPs formulated with the B-4 ionizable lipid had the highest fetal lung delivery following vitelline vein injection in gestational age E16 fetuses [34]. Fetal lungs are often one main organ target for intra-amniotic administration of gene therapies [5]. DOPE, cholesterol, and lipid-PEG were selected based on previous work indicating their inclusion in LNPs enables efficient delivery of mRNA in adult mice [50]. The phospholipid DOPE promotes LNP membrane formation and endosomal escape, cholesterol enhances membrane stability, and lipid-PEG limits immune system recognition and rapid clearance [50]. Due to the structural impact of these LNP excipients, we hypothesized that screening a range of molar ratios for each of these lipid excipients would impact ex utero LNP stability in amniotic fluids. The molar ratio ranges for each of these lipid excipients were selected by expanding the ranges used in previous LNP excipient optimization work [33] to create a library with substantial excipient deviations from traditional LNP formulations.
Figure 1 -. Overview of LNP library design, formulation, and ex utero screening in amniotic fluids to predict intra-amniotic delivery.
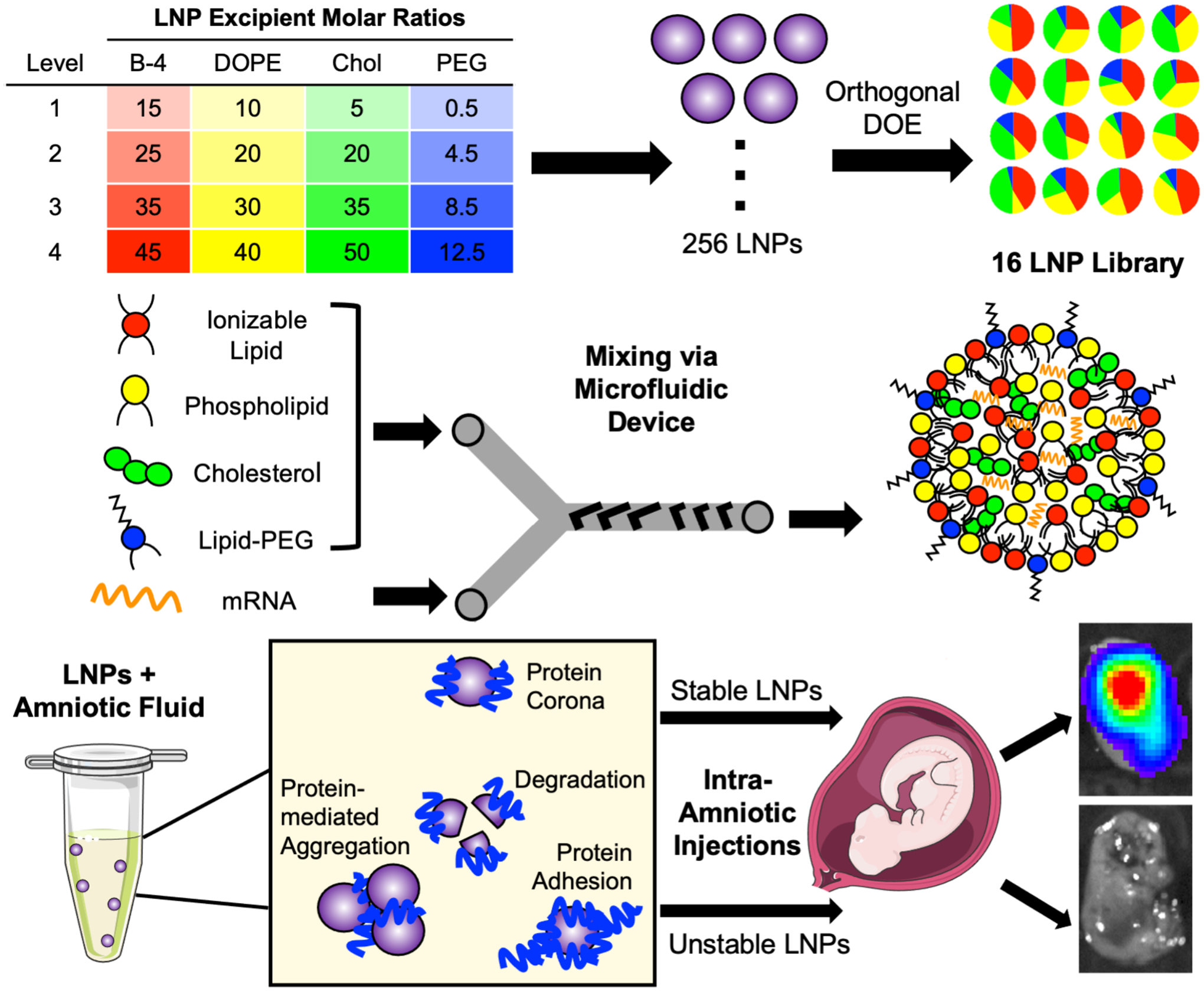
A library of 16 LNP formulations was generated using orthogonal design of experiments (DOE) to explore four molar ratios of each of four excipients. Next, each LNP was synthesized by combining an ethanol phase of lipid excipients – including ionizable lipid, DOPE phospholipid, cholesterol, and lipid-PEG – with an aqueous phase containing luciferase mRNA. The two phases were mixed at controlled flow rates in a microfluidic device to form LNPs. Then, LNPs were screened ex utero in fetal fluids to identify stable and unstable LNPs for intra-amniotic delivery.
Following library design, all 16 LNPs were formulated using the ionizable lipid B-4. As previously described, the ionizable lipid was synthesized using Michael addition chemistry where the polyamine core was reacted with 14-carbon alkyl tails [40]. B-4 was then mixed in an ethanol phase with the remaining lipid excipients – DOPE, cholesterol, and lipid-PEG – and combined with an aqueous phase of luciferase mRNA via chaotic mixing in a microfluidic device (Figure 1) [42].
LNPs were characterized by hydrodynamic diameter, polydispersity index (PDI), encapsulation efficiency, pKa, and zeta potential (Table 2). Using intensity measurements from dynamic light scattering (DLS), LNP size ranged from 46 to 153 nm and six out of 16 LNPs had PDIs > 0.3. A RiboGreen assay was used to characterize mRNA encapsulation efficiency, and seven out of 16 LNPs had encapsulation efficiencies ≤ 75%. These results indicate that the wide range of molar ratios selected for library design conferred large LNP size, high polydispersity, and low encapsulation efficiency for some formulations. Next, LNPs were characterized by their pKa, or the pH at which the LNP is 50% protonated. The pKa of an LNP indicates its ability to escape the acidic environment of the endosome following endocytosis [51]. In the endosome, LNPs with pKa values < 7 will become protonated causing their membrane lipids to fuse with the anionic lipid of the endosome, and release their mRNA cargo into the cytosol [51,52]. Typically, ionizable LNPs with pKa values from 6 to 7 enable potent delivery of nucleic acids [34,43,51,52]. The measured pKa values for our 16 LNP library ranged from 6.03 to 6.63 indicating that all LNPs were in the optimal range to enable endosomal escape. Finally, zeta potential measurements ranged from −7.4 to 25 mV, and 13 out of 16 LNPs had neutral to positive zeta potential values as expected due to the cationic nature of the B-4 ionizable lipid and DOPE. Interestingly, a trend between the molar ratio of PEG in the LNP formulations and zeta potential was observed. Namely, increasing the molar ratio of PEG resulted in decreasing zeta potential measurements (Supplementary Figure 2). There were no notable trends between zeta potential and the molar ratios of the B-4 ionizable lipid, DOPE, or cholesterol.
Table 2.
Characterization of LNP library including size and PDI in PBS, mRNA concentration, encapsulation efficiency, pKa, and zeta potential.
| Size (nm) | PDI | mRNA (ng/μL) | Encapsulation Efficiency (%) | pKa | Zeta Potential (mV) | |
|---|---|---|---|---|---|---|
| A1 | 137 ± 15 | 0.28 ± 0.03 | 33 ± 3 | 75 ± 2 | 6.63 | 25.0 ± 2.0 |
| A2 | 105 ± 7 | 0.24 ± 0.02 | 31 ± 9 | 84 ± 6 | 6.17 | −0.01 ± 0.8 |
| A3 | 62 ± 11 | 0.37 ± 0.02 | 46 ± 11 | 93 ± 1 | 6.53 | 1.5 ± 0.3 |
| A4 | 95 ± 10 | 0.28 ± 0.02 | 44 ± 26 | 89 ± 7 | 6.52 | 2.4 ± 0.5 |
| A5 | 100 ± 6 | 0.19 ± 0.03 | 34 ± 4 | 53 ± 4 | 6.57 | −7.4 ± 0.6 |
| A6 | 89 ± 3 | 0.18 ± 0.02 | 14 ± 3 | 38 ± 9 | 6.28 | 4.6 ± 0.9 |
| A7 | 88 ± 6 | 0.20 ± 0.01 | 21 ± 0.3 | 93 ± 0.2 | 6.51 | 19.6 ± 0.2 |
| A8 | 46 ± 3 | 0.61 ± 0.05 | 35 ± 3 | 96 ± 0.1 | 6.48 | 15.2 ± 0.8 |
| A9 | 153 ± 6 | 0.22 ± 0.03 | 23 ± 0.1 | 63 ± 0.2 | 6.46 | 7.7 ± 0.3 |
| A10 | 110 ± 13 | 0.30 ± 0.10 | 30 ± 1 | 94 ± 0.2 | 6.46 | 10.2 ± 0.5 |
| A11 | 137 ± 11 | 0.41 ± 0.05 | 31 ± 2 | 69 ± 2 | 6.53 | 6.0 ± 1.0 |
| A12 | 89 ± 5 | 0.25 ± 0.01 | 41 ± 7 | 93 ± 1 | 6.21 | 14.5 ± 0.8 |
| A13 | 148 ± 10 | 0.33 ± 0.07 | 37 ± 6 | 95 ± 0.9 | 6.45 | 11.7 ± 0.7 |
| A14 | 136 ± 9 | 0.28 ± 0.02 | 18 ± 4 | 89 ± 2 | 6.03 | 18.9 ± 0.5 |
| A15 | 108 ± 2 | 0.31 ± 0.03 | 13 ± 3 | 31 ± 5 | 6.52 | −0.9 ± 0.7 |
| A16 | 142 ± 10 | 0.31 ± 0.02 | 7 ± 4 | 16 ± 6 | 6.44 | 3.2 ± 0.9 |
3.2. Ex Utero LNP Stability in Mouse Amniotic Fluid
Nanoparticles can undergo a variety of changes in biological fluids including aggregation [38,46], protein corona formation [47,53], and degradation [38,54] (Figure 1), all of which impact the in vitro or in vivo stability of the drug delivery system. Previous work has studied the stability of lipid-based nanoparticle systems in well-characterized fluids such as serum using DLS [37,46,53]. DLS is a minimal resource, quantitative assay for measuring the size distribution and polydispersity of a nanoparticle sample, and the stability of LNPs following ex vivo incubation in a variety of fluids can be assessed using this technique. For example, more stable LNPs will exhibit smaller size and polydispersity changes upon incubation in fluid [46], therefore facilitating the identification of highly stable and unstable LNPs in each fluid of interest. To determine the DLS incubation parameters for the ex vivo screening of the LNP library, we selected a range of mouse amniotic fluid percentages – 0%, 25%, 50%, 75%, and 100% (v/v) – and a range of incubation times – 0 min, 5 min, 15 min, 30 min, 60 min, 120 min, and 240 min – for evaluation. We selected two LNPs – A5 and A12 – each with different excipient molar ratios for this preliminary investigation, and utilized mouse amniotic fluid collected from gestational day 16 (E16) fetuses. E16 amniotic fluid is representative of the biological environment in the amniotic sac at the onset of fetal breathing and was selected as it represents the timeframe during which intra-amniotic gene therapies could be administered to take advantage of fetal inhalation and ingestion of LNPs from the amniotic fluid for lung and digestive tract delivery [5].
Across a range of amniotic fluid percentages, LNP A5 exhibited broadening of the DLS intensity curve along the x-axis as amniotic fluid percentage increased, until ultimately becoming bimodal in 75% amniotic fluid (Figure 2A). This suggests that the A5 LNP population became more heterogenous in size with increasing polydispersity as the amniotic fluid percentage increased. In contrast, as the amniotic fluid percentage increased, the A12 intensity curves shifted right along the size axis and began overlapping with the intensity curve for 100% mouse amniotic fluid. This suggests that as the mouse amniotic fluid percentage increased, the mean A12 LNP size increased with little change in polydispersity. In general, for both LNPs, there were less substantial effects on size and polydispersity in 25% mouse amniotic fluid. Additionally, this low fluid percentage is less physiologically relevant for applications of intra-amniotic injection of LNPs where the particles would be exposed to 100% amniotic fluid in the sac. Alternatively, in 75% mouse amniotic fluid, both LNPs were unstable with either a very high polydispersity or a large increase in size. Therefore, to ensure resolution between stable and unstable LNPs in each of the fluids of interest, 50% (v/v) amniotic fluid percentage was selected for the subsequent library stability screen.
Previous work has evaluated lipid-based nanoparticle stability following incubation in protein-rich fluid for times comparable to 5 and 15 min [46]. However, results in the present study indicate that size and PDI measurements from these incubation times had large standard deviations and did not represent longer-term LNP stability in mouse amniotic fluid (Figure 2B). Instead, there were minimal changes in size and PDI measurements for incubation times greater than or equal to 30 min. Therefore, while the 240 min incubation timepoint was selected to represent the maximum LNP residence time in amniotic fluid before evaluation of in utero delivery, we selected 30 min as our incubation time as it sufficiently represents longer-term behavior of LNPs in mouse amniotic fluid.
3.3. LNP Stability in Mouse Serum and Mouse, Large Animal, and Human Amniotic Fluids
After determining the appropriate incubation parameters above (30 min in 50% (v/v) fluid) using only two LNPs and one fluid of interest, DLS was used to evaluate the ex vivo stability of all 16 LNPs in the following five fluids: mouse serum, mouse amniotic fluid, sheep amniotic fluid, pig amniotic fluid, and human amniotic fluid. Mouse serum was selected as one fluid of interest as numerous prior studies have characterized the stability of lipid-based nanoparticle systems in various blood and serum fluids, including human plasma and fetal bovine serum (FBS) [37,46,53]. Additionally, as some of these studies report significant lipid nanoparticle instability at low concentrations (1% or 2% v/v) of FBS [37,46], we hypothesized that mouse serum could serve as a positive control for this screen (Figure 3A).
Figure 3 – LNP library stability in mouse serum and amniotic fluids.
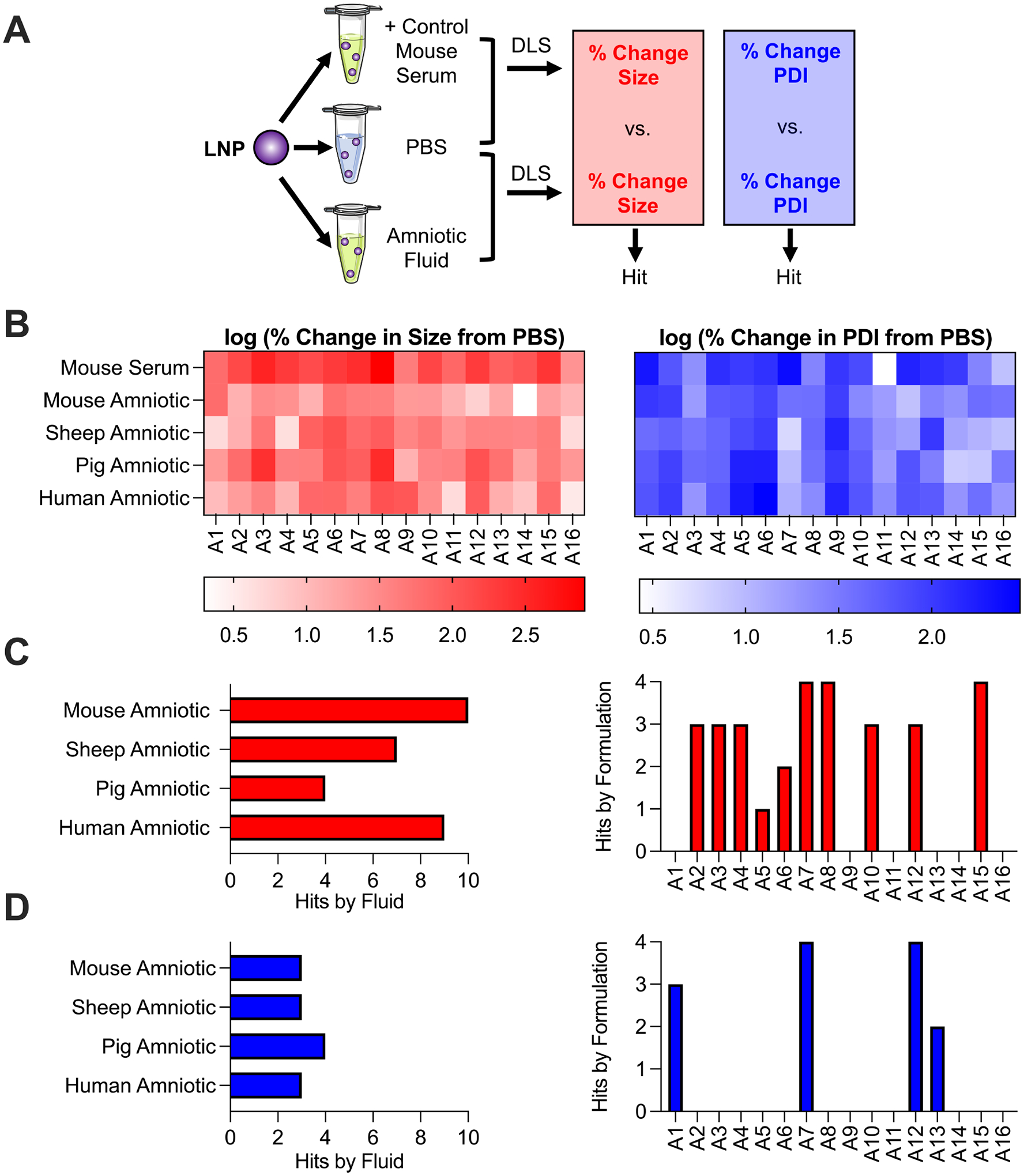
(A) Schematic depicting LNP library screen using DLS where percent change in LNP size or PDI in each amniotic fluid is calculated from the LNP size or PDI in PBS alone. These percent change measurements are compared to those in mouse serum as a positive control to identify hits. (B) Heatmaps depicting log transforms of LNP percent change in size and percent change in PDI (from PBS) in each fluid. Red – darker colors represent larger percent changes in size from the LNP in PBS alone. Blue – darker colors represent larger percent changes in PDI from the LNP in PBS alone. (C and D) 2-way ANOVA results indicating hits across amniotic fluids and formulations for percent change in size (C) and percent change in PDI (D) measurements. A hit is defined as an LNP in a given amniotic fluid with a significantly smaller (p < 0.05) percent change in size or PDI measurement than the LNP in mouse serum as determined from 2-way ANOVA.
Results of the screen were quantified via percent change in size and percent change in PDI (both from the LNP in PBS alone) following incubation in fluid. These percent change parameters were selected to take into account the initial size and PDI of the LNP before incubation, as they varied widely across the library (Table 2). Also, these parameters allow for an intuitive understanding of stability; stable LNPs have low percent change in size or PDI measurements following incubation in a given fluid. Therefore, results were presented in a heatmap with the log transforms of the percent change in size and percent change in PDI measurements (Figure 3B). Log transformation allows a larger range of values to be presented in a color gradated scale, without having very large measurements diminish the resolution present between smaller measurements.
As hypothesized, mouse serum served as a positive control for this library screen since many LNPs performed substantially worse in serum than the other fetal fluids (Figure 3B). To aid in visualization of the library screen findings, a 2-way ANOVA was performed to define hits, or LNPs in a given fluid whose percent change in size or percent change in PDI measurements were significantly smaller (p < 0.05) than the same LNP in mouse serum (Figures 3C and 3D). First, for percent change in size measurements (Figure 3C), there were ten and nine LNP hits in mouse amniotic and human amniotic fluids, respectively, while there were only four LNP hits in pig amniotic fluid. Taken together, these results suggest that more LNPs from the library were stable in mouse and human amniotic fluids than in pig amniotic fluid. However, for percent change in PDI measurements (Figure 3D), all fluids had only between three and four LNP hits, suggesting that the LNP library performed similarly across all fluids.
Percent change in size measurements identified that ten out of 16 LNPs in the library were a hit in at least one fluid (Figure 3C). Yet, percent change in PDI measurements identified that only four out of 16 LNPs in the library were a hit in at least one fluid (Figure 3D). Collectively, these results suggest that there were substantially fewer hits for percent change in PDI measurements than percent change in size measurements when compared to mouse serum. In other words, LNPs in a given fluid are more likely to have significantly smaller percent change in size measurements compared to mouse serum than percent change in PDI measurements. It is important to note that LNPs such as A9 and A16 were not identified as hits in any of the fetal fluids. However, this is likely because these LNPs appeared to be stable in mouse serum, as they exhibited low percent change in size and PDI measurements. Therefore, no significant improvements compared mouse serum in any of the fetal fluids could be identified. While these results are intriguing, LNPs A9 and A16 also had low encapsulation efficiencies (≤ 75%), therefore limiting their application for mRNA delivery and future exploration in this study.
The majority of LNPs presented both substantial percent change in size and percent change in PDI measurements following incubation in fluid. However, some LNPs appeared to demonstrate mainly high percent change in size measurements (A8), while others presented mainly high percent change in PDI measurements (A9) (Figure 3B). Therefore, we rationalized that both parameters should be considered when evaluating overall LNP stability. To determine the most and least stable LNP in each of the fluids, we averaged both stability measurements – percent change in size and percent change in PDI – for each LNP in the library to determine an overall lowest and highest LNP instability parameter, respectively. The top LNPs in each amniotic fluid evaluated were as follows: A12 for mouse amniotic, A14 for pig amniotic, and A16 for sheep and human amniotic. A12 and A14 LNPs had several commonalities: a moderate to high molar ratio of B-4 ionizable lipid (35 – 45), a low molar ratio of cholesterol (20 – 35), and a low molar ratio of lipid-PEG (0.5) compared to traditional LNP formulations for mRNA delivery. However, as mentioned above, LNP A16 had an encapsulation efficiency of less than 75%, so this formulation likely would require further optimization for sheep and human intra-amniotic delivery.
Representative DLS intensity curves (Supplementary Figure 3) of the most stable LNPs often showed little to no size or PDI change in the presence of amniotic fluid compared to the intensity curve of the LNP in PBS alone. Instead, intensity curves of the least stable LNPs in each of the fluids showed increased PDI, bimodal behavior, and substantial size increases as curves shifted right and sometimes completely overlapped with the intensity curve of the amniotic fluid background.
3.4. Stability Correlations in Amniotic Fluid Across Species
Next, we sought to identify any correlations between LNP stability in amniotic fetal fluids across species using the above defined instability parameter (Figure 4). First, LNP instability parameters in mouse and pig amniotic fluids only mildly correlated with those in human amniotic fluid (R2 = 0.2314 and R2 = 0.2868, respectively). However, there was a moderate correlation of the instability parameter measurements between sheep amniotic and human amniotic fluids (R2 = 0.7099). In terms of stability, these results suggest that LNPs performed most similarly in our sample of sheep amniotic fluid as they did in human amniotic fluid, more so than in our samples of mouse and pig amniotic fluids. Next, LNP instability parameters in pig and sheep amniotic fluids had little to no correlation with those in mouse amniotic fluid (R2 = 0.0423 and R2 = 0.1450, respectively). These results suggest that mouse amniotic LNP stability may not accurately correlate with stability in our amniotic fluid samples of larger species, specifically pig and sheep. Taken together, these results demonstrate differences in LNP stability in amniotic fluids between small animal models and large animal models or humans, perhaps due to gestational age differences at the time of amniotic fluid collection and total length of gestational periods. Finally, fluids used in this screen were characterized in terms of their pH and protein concentration (Supplementary Table 1). Specifically, the pH of the four amniotic fluids and mouse serum ranged from 7.58 to 8.45. Also, the protein concentration in the amniotic fluids ranged from 140 to 515 ng/μL, while the protein concentration in mouse serum was 1507 ng/μL. This is consistent with previous work which has found total protein concentration in human amniotic fluid to be up to 12.5-fold lower than in human serum [55]. While LNPs generally performed worse in mouse serum, there were no notable trends between stability measurements of the LNP library and either fluid pH or protein concentration.
Figure 4 – LNP instability parameter correlations for amniotic fluids across species.
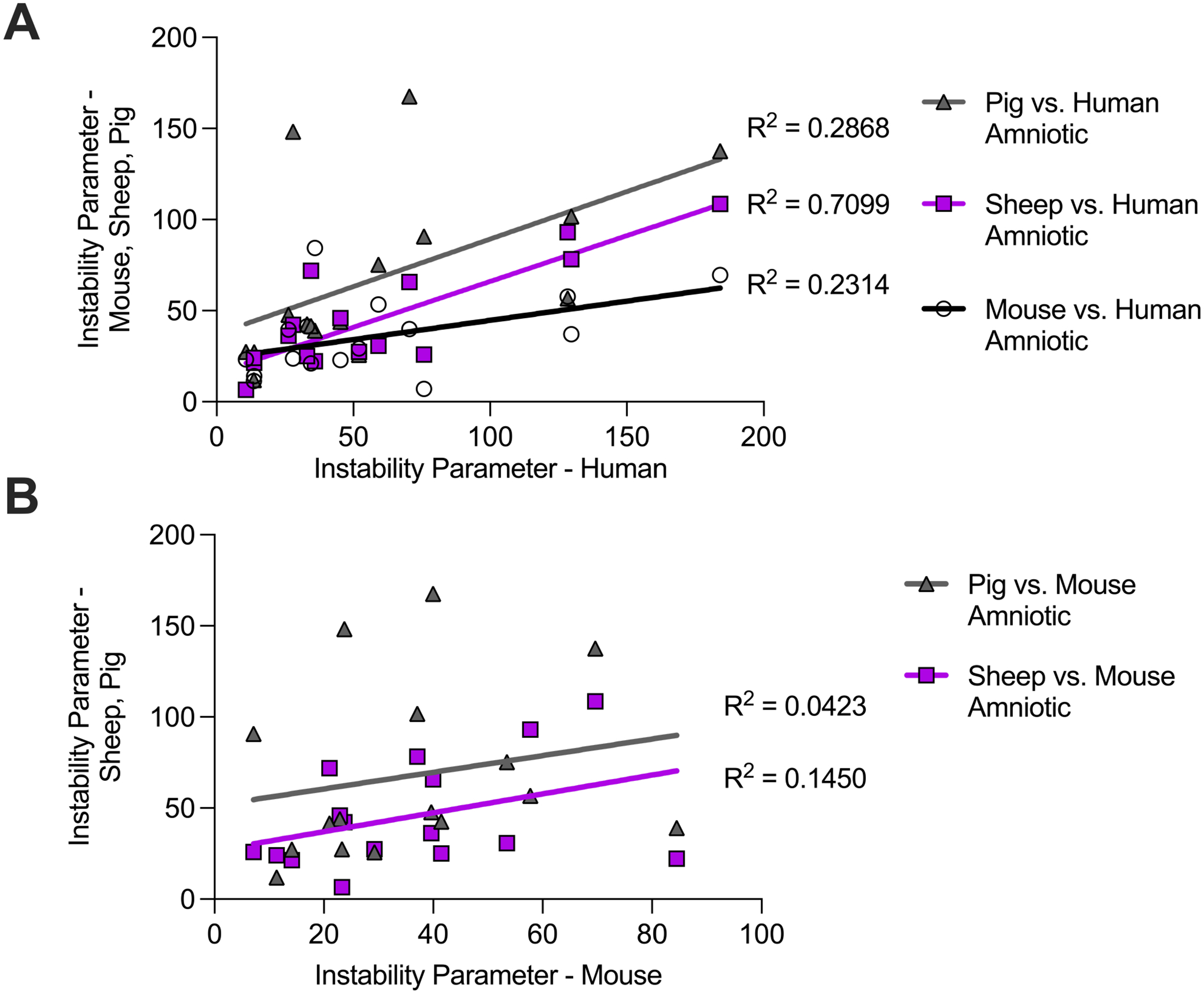
(A) Instability parameter measurements of the LNP library in mouse, sheep, and pig amniotic fluids (y axis) correlated with human amniotic fluid (x axis). (B) Instability parameter measurements of the LNP library in sheep and pig amniotic fluids (y axis) correlated with mouse amniotic fluid (x axis). The coefficient of determination R2 of the least squares linear regressions indicate the goodness of fit for the instability parameter correlations.
3.5. Effect of Ionizable Lipid on LNP Stability
To evaluate the generalizable nature of this ex vivo stability assay across different ionizable lipids, we looked at the effect of changing the ionizable lipid in an LNP formulation on stability measurements. To this end, we selected excipient formulation A5 and replaced the B-4 ionizable lipid with C12–200, a well characterized ionizable lipid for mRNA delivery [40,56,57]. There were no significant (*p < 0.05) differences in the measured size of the B-4 and C12–200 LNPs in any of the fluids evaluated (Supplementary Figure 4A). However, in two of the amniotic fluids evaluated, the C12–200 LNP had significantly (*p < 0.05 and ***p<0.001) higher PDI measurements than the B-4 LNP (Supplementary Figure 4B). We hypothesize this difference was due to the significantly higher initial PDI of the C12–200 LNP in PBS alone compared to the B-4 formulation also in PBS. These results suggest the reproducibility of ex vivo stability measurements for formulations with different ionizable lipids.
3.6. LNP Morphology and Zeta Potential Effects in Mouse Amniotic Fluid
To visualize LNP morphological changes following incubation in mouse amniotic fluid, transmission electron microscopy (TEM) was used to visualize the morphology of the most stable (A12) and least stable (A1) LNPs from the above ex utero stability screen in mouse amniotic fluid. As stated above, the most and least stable LNPs in mouse amniotic fluid were determined by averaging percent change in size and percent change in PDI stability measurements of each formulation in the library for an overall lowest and highest instability parameter, respectively. First, TEM images of LNPs A12 and A1 in PBS showed primarily spherical and monodisperse particles (Figure 5A). Particle analysis of TEM images indicated A12 had a mean size of 97 ± 17 nm and A1 had a mean size of 71 ± 12 nm. Upon incubation in mouse amniotic fluid, TEM images of the most stable LNP (A12) showed little shape or size changes. However, the least stable LNP (A1) showed substantial aggregation and clustering in mouse amniotic fluid. These qualitative morphological changes are confirmed with TEM image particle analysis where the LNP size was 118 ± 43 nm for A12 and 176 ± 110 nm for A1 following incubation in mouse amniotic fluid.
Figure 5 – LNP morphology and protein interactions in mouse amniotic fluid.
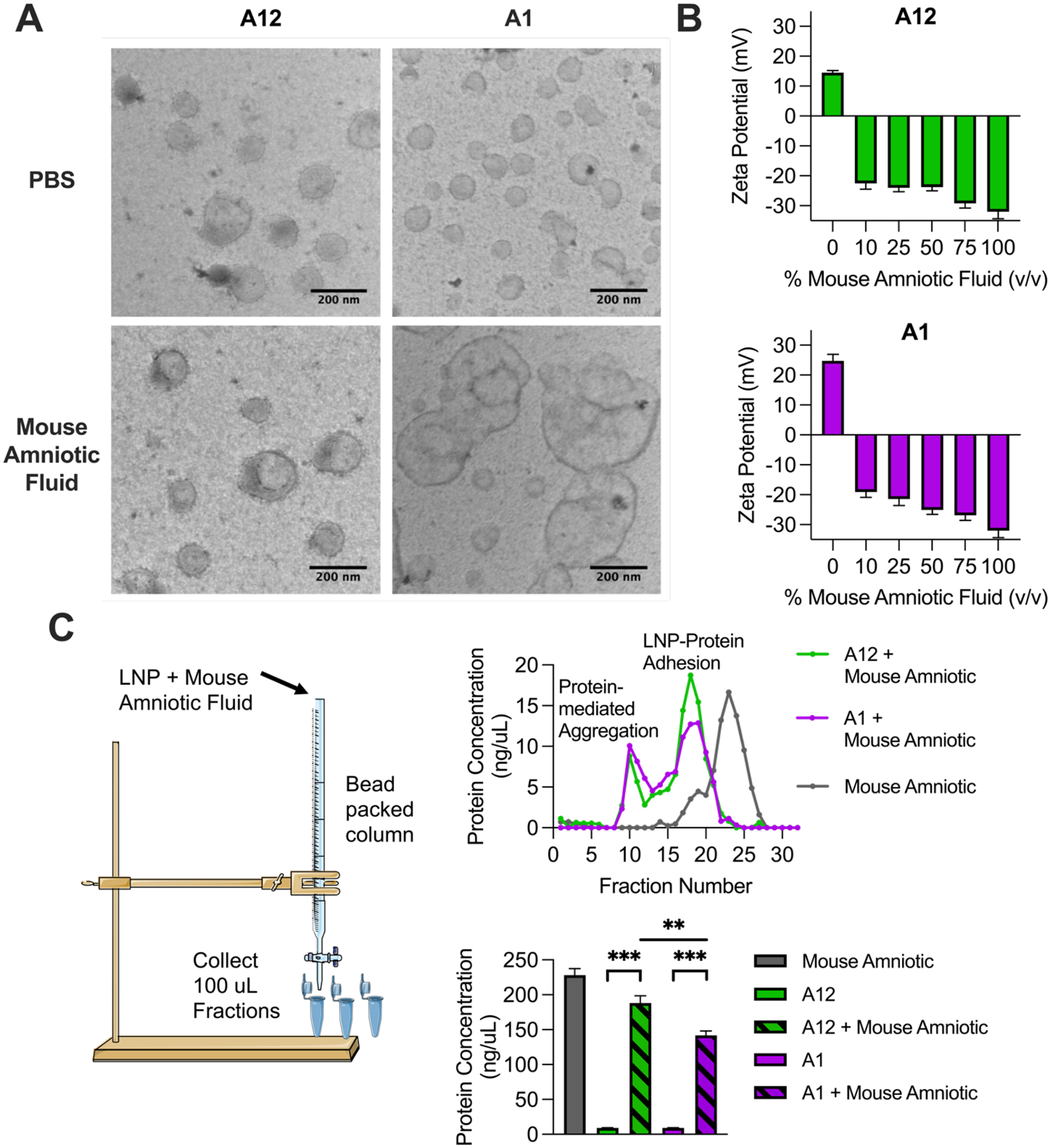
(A) TEM images of the most stable (A12) and least stable (A1) LNPs from ex utero mouse amniotic fluid screen. (B) Zeta potential of A12 and A1 LNPs with increasing percentages (v/v) of mouse amniotic fluid. (C) BCA assay identifying protein content bound to LNPs following LNP incubation in mouse amniotic fluid and chromatographic separation of LNPs from unbound mouse amniotic fluid. Data is presented as means with standard deviations of n = 3 to 4 measurements.
To further characterize LNP-protein effects following incubation in mouse amniotic fluid, we measured the zeta potential of LNPs A12 and A1 following incubation in increasing fluids percentages of mouse amniotic fluid (Figure 5B). Previous findings report that zeta potential measurements became more negative as NPs were incubated in increasing concentrations of protein-rich fluid [58]. Here, both LNPs A12 and A1 alone had positive zeta potential measurements that immediately became negative upon addition of mouse amniotic fluid. The zeta potential measurements became increasingly more negative as fluid percentage increased, as is consistent with previous findings, due to what we hypothesize is increased protein adhesion to the particle.
3.7. Chromatography and Protein Quantification of LNPs in Mouse Amniotic Fluid
As mouse serum and the amniotic fluids evaluated in this study are protein-rich biological environments, we sought to identify the presence of bound proteins on LNPs A12 and A1 following incubation in mouse amniotic fluid. To do so, we expanded on a previously reported methodology of Sepharose column separation to isolate LNPs from unbound protein-rich fluid [47]. Using this protocol, free mouse amniotic fluid and LNPs A12 and A1 pre-incubated in mouse amniotic fluid were each individually passed through a Sepharose column (Figure 5C). 32 chromatographic fractions were collected for each of the three samples, and the protein concentration of each fraction was measured using Tecan’s NanoQuant Plate. Plots of protein concentration as a function of chromatographic fraction indicate the presence of two peaks for LNP samples. We hypothesize that the first peaks represent LNP aggregates which would be larger in size and elute from the separation column first before smaller LNPs with bound proteins on their surfaces, representing the second peaks detected in the chromatographic fractions.
Fractions with non-zero protein readings and no overlap with the elution of free mouse amniotic fluid were pooled and used to measure protein content via BCA assay. The BCA assay indicated significant (***p < 0.0002) protein content on LNPs A12 and A1 that were pre-incubated in mouse amniotic fluid compared to the same LNPs in PBS alone. Interestingly, the most stable LNP (A12) from the ex utero mouse amniotic stability screen had significantly (***p < 0.0021) higher protein content bound to the surface than the least stable (A1) LNP from the stability screen. This assay confirms that proteins derived from mouse amniotic fluid are bound to LNPs A12 and A1 following incubation, and we hypothesize that these LNP-protein interactions likely contribute to the previous stability findings.
3.8. In Vitro LNP-mediated mRNA Delivery
To establish trends between ex vivo LNP stability and mRNA delivery, we evaluated LNP-mediated luciferase mRNA delivery and toxicity of the library in vitro in HeLa cells (Figure 6). HeLa cells were selected for this in vitro library screen as epithelial cells are found in several major organ targets for intra-amniotic injection of LNPs, including the skin, pulmonary and digestive tract organs. Treatment conditions included LNPs alone and LNPs pre-incubated in mouse amniotic fluid. HeLa cells were dosed with LNPs or lipofectamine at a concentration of 50 ng per 10,000 cells. Lipofectamine is a commonly used transfection agent and is often considered the gold standard for in vitro nucleic acid delivery [59,60]. To evaluate LNP delivery, 24 h after treatment, luciferase expression was quantified using bioluminescence measurements (Figure 6A). Seven LNPs – A2, A3, A4, A7, A8, A12, and A14 – with mouse amniotic fluid had significantly (*p < 0.05) higher luciferase expression than lipofectamine. For 15 of 16 LNPs in the library, there were no significant differences in luciferase expression between the LNP alone and the LNP with mouse amniotic fluid treatment conditions, except for A7 which demonstrated significantly (p < 0.05) better delivery in the presence of mouse amniotic fluid. Percent cell viability was also evaluated 24 h following treatment with LNPs or lipofectamine. LNPs A1, A7, and A8 with mouse amniotic fluid had significantly lower cell viability compared to lipofectamine (Figure 6B). Notably, three LNPs – A7, A13, and A14 – had significantly better cell viability in the presence of mouse amniotic fluid than in PBS alone.
Figure 6 – In vitro LNP-mediated luciferase mRNA delivery.
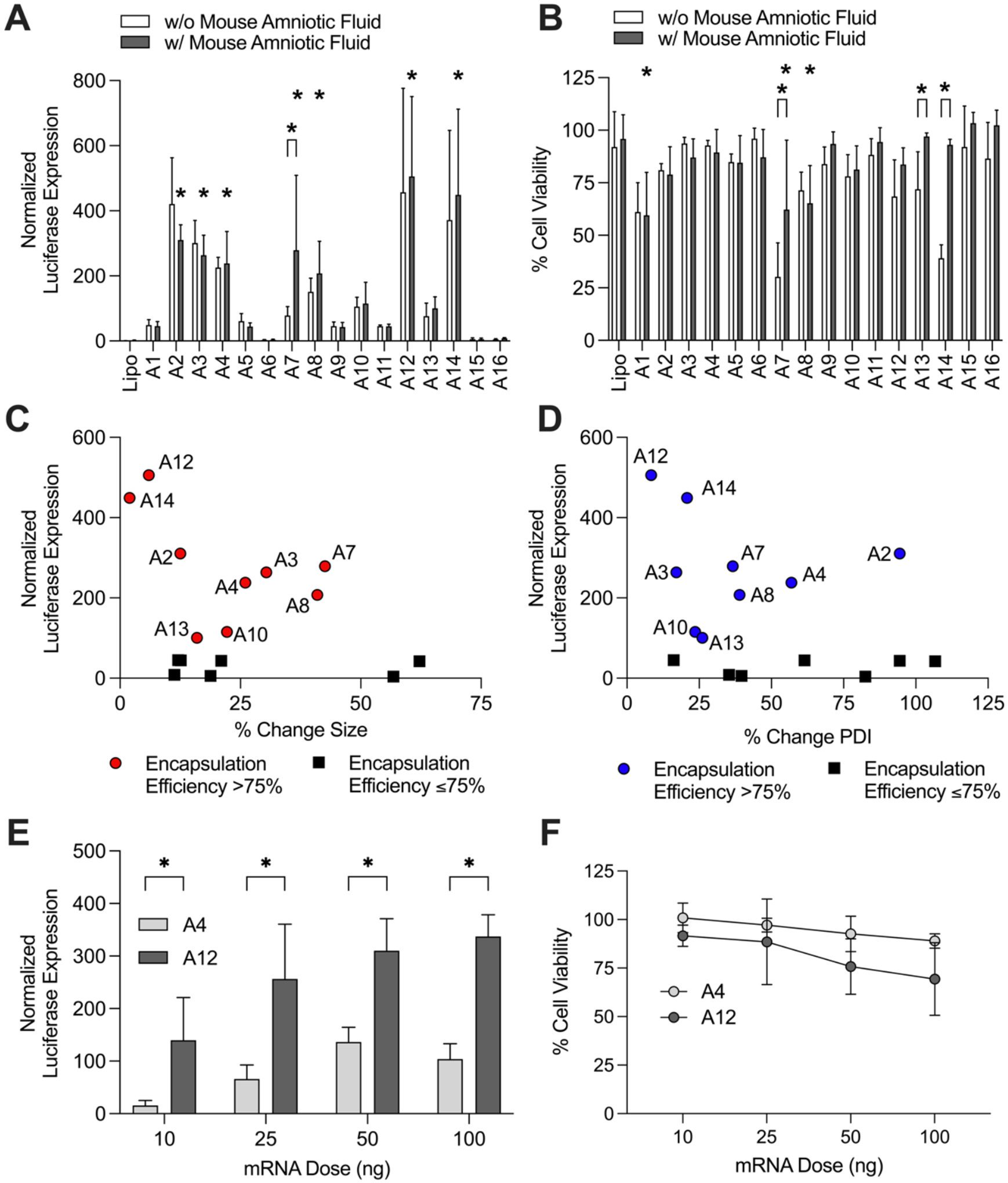
(A) LNP-mediated luciferase mRNA delivery in HeLa cells. Cells were treated with the 16 LNP library in PBS alone and pre-incubated in mouse amniotic fluid. Luciferase expression for each treatment condition was normalized to untreated cells and compared to lipofectamine MessengerMAX delivery using 2-way ANOVA for significance. Seven LNPs with mouse amniotic fluid had significant (*p < 0.05) delivery compared to lipofectamine. Only LNP A7 had significantly different delivery with mouse amniotic fluid compared to the same formulation in PBS alone. (B) Cell viability following treatment with the LNP library in PBS alone or pre-incubated in mouse amniotic fluid. LNPs A1, A7, and A8 had significantly (*p < 0.05) lower cell viability compared to lipofectamine. LNPs A7, A13, and A14 had significantly better cell viability after pre-incubation in mouse amniotic fluid compared to the same formulation in PBS alone. (C and D) Inverse correlation between luciferase expression and percent change in size (C) and percent change in PDI (D) in mouse amniotic fluid. Particles with encapsulation efficiencies ≤75% excluded from correlation. (E) LNP-mediated luciferase mRNA delivery in primary mouse fetal lung cells with stable LNP A12 and unstable LNP A4. LNP A12 had significantly higher luciferase expression (*p < 0.05 via Welch’s t test) at all doses compared to LNP A4. (F) Cell viability in primary mouse fetal lung cells with LNP A12 and LNP A4.
As expected, luciferase expression demonstrated a strong correlation with encapsulation efficiency, as LNPs with less than or equal to 75% encapsulation had little to no delivery (Supplementary Figure 5). Focusing on LNPs with encapsulation efficiencies greater than 75%, we assessed correlations between luciferase expression and percent change in size or percent change in PDI ex utero stability measurements in mouse amniotic fluid (Figure 6C and 6D). We noted a general inverse correlation between luciferase expression and both percent change in size and percent change in PDI. A12, the most stable LNP in mouse amniotic fluid, had the highest luciferase expression of all LNPs evaluated in the library. These results demonstrate the ability of the stability measurements to predict top in vitro performers such as LNPs A12 or A14. Unlike these stability measurements, there were no clear correlations between zeta potential measurements and in vitro luciferase mRNA delivery of the LNP library (Supplementary Figure 6).
To validate these in vitro results in primary cells, we isolated lungs from fetuses removed from time-dated pregnant mice at gestational age E16. Single cell suspensions were created from these fetal lung tissues and seeded in a well plate for LNP-mediated luciferase mRNA delivery and cell viability. LNP A12 – the most stable LNP in amniotic fluid – and LNP A4 – with substantially lower stability and in vitro mRNA delivery in HeLa cells – were used to treat cells at doses of 10, 25, 50, and 100 ng of mRNA per 20,000 cells. At all four doses, LNP A12 had significantly (*p < 0.05) higher luciferase expression than LNP A4 (Figure 6E). This is consistent with the results in HeLa cells where LNP A12 demonstrated approximately two to three-fold higher delivery than LNP A4. Cell viability in the primary fetal lung cells indicated some decline in cell viability for LNP A12 at increasing doses, likely due to the high luciferase expression at the 100 ng dose and increased fragility of the primary cells compared to the immortalized HeLa cells (Figure 6F). These results further demonstrate the potential for higher mRNA delivery with stable LNPs such as A12 over unstable LNPs such as A4.
3.9. LNP Structure Function Relationships with Ex Utero Stability and In Vitro Delivery
As the LNP library was designed with four molar ratio levels of each of four excipients, we sought to investigate LNP structure function relationships with ex utero stability measurements and in vitro delivery (Figure 7). For ionizable lipid B-4, we found that percent change in size and percent change in PDI decreased as molar ratio increased, yet there was no noticeable trend for delivery (Figure 7A). For both DOPE and cholesterol, percent change in PDI decreased and luciferase expression increased as the excipient molar ratio increased (Figures 7B and 7C). If our stability measurements are accurate predictors of mRNA delivery, we would expect trends such as these where percent change in size and percent change in PDI stability measurements should get smaller as delivery improves. Finally, for PEG, percent change in PDI increased and luciferase expression decreased as the molar ratio of PEG increased (Figure 7D). Again, we notice an inverse trend between our percent change in PDI stability measurements and luciferase delivery. As it appears that the percent change in PDI measurements track as expected with luciferase delivery, these results suggest that the PDI stability measurements might better predict in vitro delivery than percent change in size measurements. Additionally, these results help identify certain molar ratios (10 for DOPE, 5 for cholesterol, 12.5 for PEG) that play a role in LNP instability as seen by high percent change in PDI measurements and low luciferase delivery.
Figure 7 – LNP structure function relationships with ex utero stability and in vitro delivery.
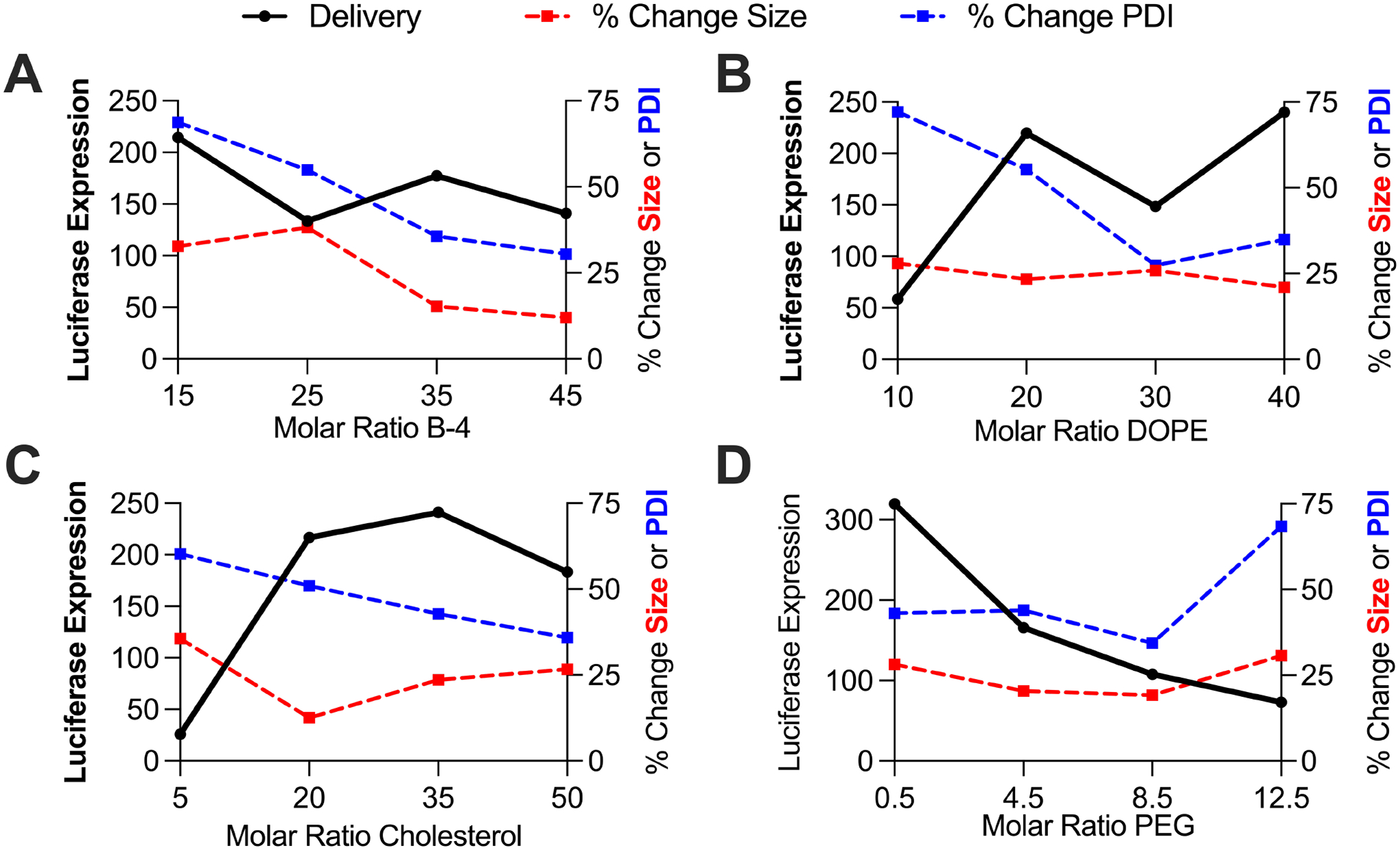
Each data point represents an average of the stability and luciferase expression measurements of the four LNPs with the given excipient molar ratio. (A) Percent change in size and PDI decrease as ionizable lipid B-4 increases. (B) & (C) Percent change in PDI decreases and luciferase expression increases as the molar ratio of DOPE and cholesterol increases. (D) Percent change in PDI increases and luciferase expression decreases as the molar ratio of PEG increases.
3.10. LNP Mediated Intra-Amniotic Luciferase mRNA Delivery
Two LNPs were selected to evaluate the correlation between ex utero stability measurements in mouse amniotic fluid and intra-amniotic luciferase mRNA delivery to E16 fetal mice. Gestational age E16 fetuses were selected for intra-amniotic injection as E16 amniotic fluid was used in the previous ex utero stability measurements. Additionally, gestational age E16 represents the biological environment at the onset of fetal breathing for inhalation and ingestion of LNPs from the amniotic fluid [5]. LNP A12 was selected as it was the most stable LNP in mouse amniotic fluid and had the highest in vitro luciferase mRNA delivery. LNP A4 was selected for its poorer ex utero stability in mouse amniotic fluid and lower in vitro delivery, while still having a suitable encapsulation efficiency of mRNA. Other formulations such as A1 and A9 were less stable in mouse amniotic fluid, yet their encapsulation efficiencies are less than 75% and would not allow for accurate comparisons of in utero mRNA delivery with LNP A12.
LNPs A12 and A4 were concentrated to 325 ng/μL and 30 μL of LNP or PBS was injected into five individual fetal amniotic sacs of E16 pregnant dams for each test condition (Figure 8A). Four hours after injection, luciferin was administered to the dams and IVIS imaging was used to quantify luciferase expression. IVIS images of the dams and exposed uterine horns showed no luciferase delivery for sacs receiving PBS and A4 injections (Figure 8B). In contrast, there was clear luminescence in sacs receiving A12 injections. Fetuses were removed and individually assessed by IVIS imaging. When quantified and averaged, fetal bioluminescence was significantly higher (*p < 0.05) for fetuses receiving LNP A12 injections compared to both LNP A4 and PBS injections (Figure 8C). There was no significant delivery of the A4 LNP compared to PBS control.
Figure 8 – LNP-mediated intra-amniotic luciferase mRNA delivery.
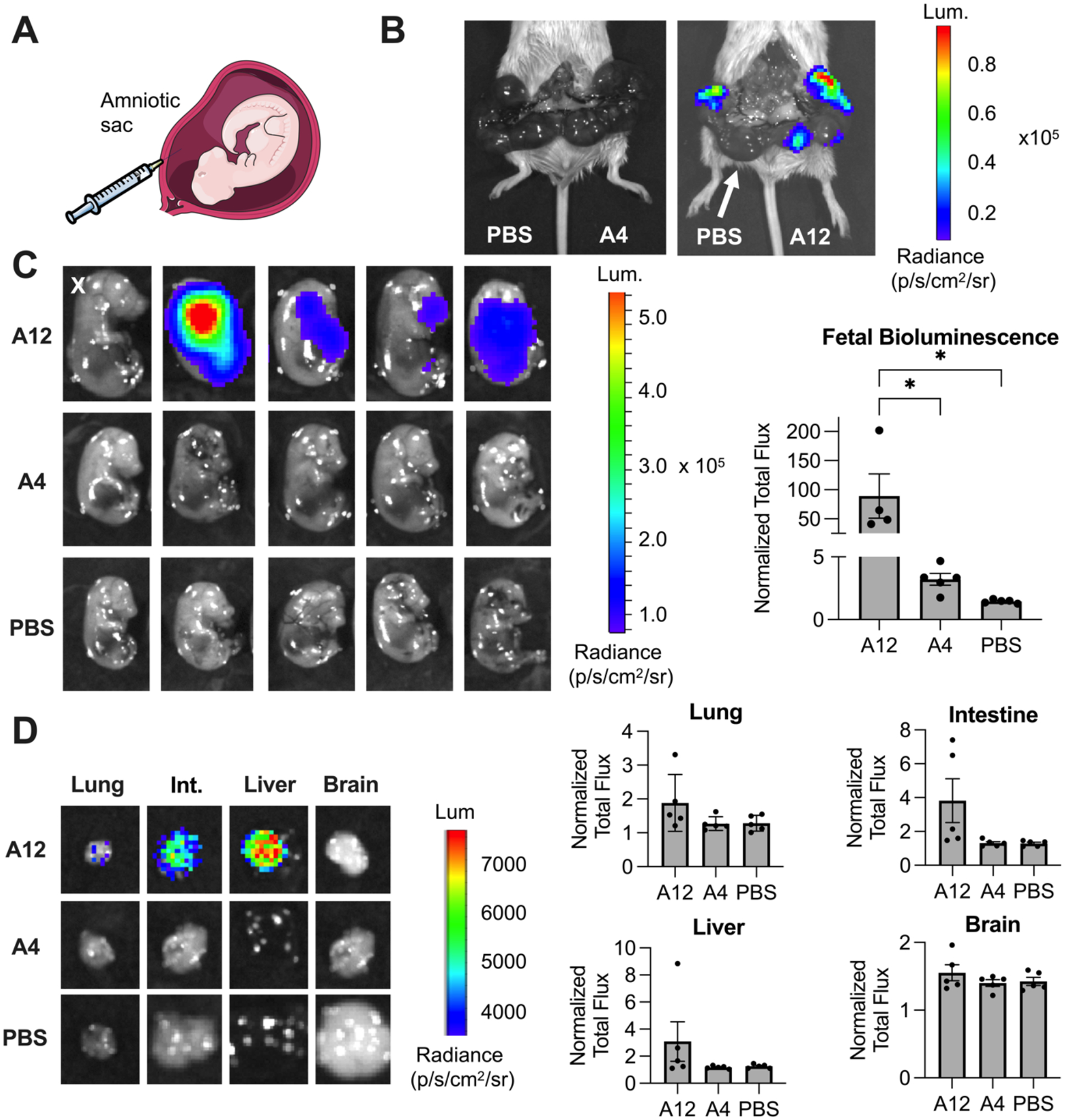
Two LNPs – A12 and A4 – were selected to evaluate in utero luciferase mRNA delivery. (A) Schematic of intra-amniotic injection. (B) Left - IVIS image of dam and exposed uterine horn with pups in the four left sacs receiving PBS control and pups in the five right sacs receiving A4 LNP injections. Right - strong luciferase expression in the uterine horn where pups received A12 LNP injections, other than one sac (denoted with white arrow) that instead received PBS as a control injection. (C) IVIS images (left) and quantification (right) of fetal bioluminescence after surgical removal from the dams. IVIS images indicate variability in luciferase expression for A12 LNP condition, with the luciferase expression from one fetus identified as an outlier (denoted with an X) and removed from analysis. A12 LNP had significantly higher fetal luciferase expression, as quantified by normalized total flux, compared to both A4 LNP and PBS control injections. (D) IVIS images (left) of the highest luciferase expression in each organ for all conditions. Quantification (right) of fetal organ bioluminescence following dissection. There was no significant difference in the normalized total flux for A12 LNP compared to A4 LNP or PBS control across four organs shown.
Finally, fetal organs including the lung, intestine, liver, and brain were isolated and assessed by IVIS for bioluminescence. Fetuses undergoing intra-amniotic injection with LNP A12 demonstrated luminescence in the lung and intestines as well as the liver, consistent with fetal swallowing and inhalation of the amniotic fluid containing LNP A12. In contrast, no luminescence was detected in any organs of those fetuses in which LNP A4 or PBS was injected into the amniotic sac. (Figure 8D). Overall, this data demonstrates proof-of-concept that our ex utero stability measurements in mouse amniotic fluid correlate with in utero intra-amniotic luciferase mRNA delivery.
4. Discussion
In the present work, we explored ex utero LNP stability in various fetal fluid biological environments and demonstrated correlations between stability measurements and LNP-mediated in vitro and in utero luciferase mRNA delivery. By using DLS and measured changes in LNP size and PDI following incubation in fluid, this stability assay requires minimal LNP and fluid resources. As a result, a larger number of formulations could be evaluated using this ex vivo approach than in other labor intensive and expensive in vivo screening experiments. For example, here, the ex vivo screening of our 16 LNP library identified excipient formulations in mouse, sheep, pig, and human amniotic fluids that were highly stable. Interestingly, LNPs behaved differently in each of these amniotic fluids. This could be explained by differences in the gestational periods of each of these species; the gestational periods of these species range from 20 to 280 days for mice and humans, respectively. Therefore, substantial developmental changes that alter amniotic fluid composition may occur over the span of hours in mice and over the span of days or weeks in humans. These results justify the need to optimize LNP formulations for gestational age and species-specific delivery. Future ex vivo screening could include design of a second generation library for each of the amniotic fluids of interest to further assess LNP stability across gestational age.
Library screening and establishing structure function relationships between LNP formulation and delivery are increasingly valuable as the number of possible formulations continues to grow with research on new modular LNP components. For example, recent work has shown that varying the molar ratio of cationic lipids such as DOTAP in LNP formulations can shift organ biodistribution in an effort to improve delivery to a target of interest [36]. Previous work has also demonstrated that different phospholipids such as DOPE and DSPC, or different ratios of lipid to nucleic acid cargo can improve encapsulation and delivery of one or multiple nucleic acids [35]. While we have demonstrated the reproducible nature of this assay with different ionizable lipids, we hypothesize that many of these modular changes in LNP formulation could impact ex vivo LNP stability, helping to further the understanding of LNP structure function relationships.
Like in this present study, mice are often used to evaluate in utero therapeutic delivery due to their short gestational period (approximately 20 days) and ability to simultaneously carry multiple fetuses per dam [61]. However, the small size of the mouse fetus presents technical challenges with respect to evaluating delivery approaches that would be possible in humans. For example, direct intra-tracheal or intra-esophageal injections of LNPs may be optimal delivery routes to target the lungs and gastrointestinal tract in humans, respectively [18]. However, intra-amniotic injections have more established safety profiles with groups demonstrating their safety for both fetus and dam in mouse models [5,15,16]. Additionally, amniocentesis is a similar procedure to intra-amniotic injections that is performed regularly in the clinic for sampling human amniotic fluid. When considering these unique injection routes, preclinical, large animal models may provide valuable information. These larger animal models, including time-dated pregnant pigs and sheep, are labor and cost prohibitive and are only used for well-characterized and clinically translatable technologies, therefore limiting in vivo LNP optimization in these species. However, the longer gestational period of sheep (approximately 145 days) in contrast to that of the mouse (approximately 20 days) more closely mimics the development period of the human fetus. This may be one explanation for the stronger correlation of LNP stability measurements in sheep and human amniotic fluids in contrast to mouse amniotic fluid. To combat the challenges associated with differences between small and large animal models, similar ex utero stability screening as performed in in this study could enable identification of novel LNP formulations specifically for larger species using unique biological environments such as fetal amniotic fluid.
Beyond using DLS to characterize size and PDI changes upon incubation in fluid, we sought to characterize morphological and protein effects to further understand ex utero LNP stability. Upon incubation in mouse amniotic fluid, the least stable LNP (A1) from the ex utero mouse amniotic fluid stability screen exhibited substantial morphological changes, including aggregation and increased size. Instead, the most stable LNP (A12) in mouse amniotic fluid presented little morphological changes, with only a small increase in size. These morphological differences visualized in TEM images confirmed what we found in the ex utero stability screen where more stable LNPs had little size or PDI changes upon incubation in fluids, but less stable particles exhibited substantial size and PDI changes from the LNPs in PBS alone.
Next, we hypothesized these differences in stability might be due to differences in the amount of bound protein on the LNP surface, as the fluids we evaluated in this study are protein-rich biological environments. However, opposite of what we hypothesized, the more stable LNP in mouse amniotic fluid (A12) had significantly higher protein content bound to the surface than the least stable LNP (A1) from the ex utero stability screen. This finding suggests some LNP structure function relationship that makes certain formulations better suited to resist conformational changes in protein rich biological environments. We hypothesize these findings could be due to different types of protein coronas that form on the surface of nanoparticles in biological fluids. In general, protein coronas are considered to be the sum of all the proteins that adsorb on the surface of nanoparticles such as LNPs when they come in contact with a protein-rich biological environment such as serum or amniotic fluid [38]. Types of protein coronas include “hard” coronas which represent proteins that bind directly to the LNP surface with high affinity, while “soft” coronas are considered a looser, more dynamic protein layer that interacts more freely with the biological environment [38]. Less stable LNPs are potentially forming primarily hard protein coronas that more substantially impact LNP conformation and resulting size and PDI measurements using DLS. Instead, more stable LNPs may be forming primarily soft protein coronas with reversibly bound proteins, consequently resulting in less substantial conformational changes as measured by size and PDI measurements [62]. Future work could further characterize the formation of these fetal fluid protein coronas, including identifying specific proteins, their relative quantities, and correlations between DLS stability measurements and protein corona formation. For example, previous work has demonstrated differences in the proteome of human amniotic fluid versus serum and noted that proteins commonly found in serum such as albumin, globulin, lipoprotein, and apoproteins are found in significantly lower quantities in human amniotic fluid than serum [55,63]. Therefore, a robust proteomic analysis of a variety of LNPs in amniotic fluid could provide valuable insights compared to those for serum and blood.
Besides LNP stability, the biological environment can also impact in vitro delivery. Previously, it has been proposed that protein coronas can have paradoxical effects on in vitro cellular uptake and LNP delivery [62]. For example, while proteins on the LNP surface can trigger and enhance cellular uptake, specifically via protein-receptor interactions, proteins bound to the LNP surface can also decrease LNP adhesion to the cell membrane due to surface free-energy limitations, therefore decreasing LNP uptake [62]. Here we found no significant difference in LNP mediated luciferase mRNA delivery for 15 of the 16 LNPs in the presence of mouse amniotic fluid compared to each LNP in PBS alone. Interestingly though, we did identify improved cell viability for three LNPs in the presence of mouse amniotic fluid compared to the LNP in PBS alone. Perhaps the presence of bound proteins on the LNP surface reduces some of cellular toxicity associated with the LNP itself.
We evaluated structure function relationships between LNP excipient molar ratios and their ex utero stability and in vitro luciferase mRNA delivery in mouse amniotic fluid. These relationships indicated that the percent change in PDI stability measurements more closely tracked as expected with mRNA delivery than percent change in size stability measurements. In other words, LNP percent change in PDI measurements and LNP-mediated luciferase mRNA delivery were inversely related as expected. As changes in PDI are representative of changes in size distribution, it is likely that large PDIs indicate the presence of both large LNP aggregates and small broken down LNPs due to high protein content on the LNP surface. Perhaps, these LNP distribution changes are more indicative of functional delivery in a protein-rich environment than increases in LNP size. Additionally, the inverse relationship between percent change in PDI measurements and mRNA delivery was especially strong when observing variations in molar ratio of PEG. This is an interesting observation as PEG is often included in LNP formulations to reduce immune system recognition and rapid clearance that is often initiated by protein adhesion to the LNP surface [50]. However, we found that increased PEG appears to be detrimental to LNP stability and functional delivery in mouse amniotic fluid. Also, the most (A12) and least (A1) stable LNPs in mouse amniotic fluid both had a PEG molar ratio of 0.5. Yet, the formulations differed in the other three lipid components – the more stable LNP had higher molar ratios of ionizable lipid, DOPE, and cholesterol than the least stable LNP. These results suggest that the ionizable lipid, DOPE, and cholesterol which aid in membrane formation, cargo complexation, and rigidity likely play an essential role in the stability of LNPs in biological environments.
Finally, intra-amniotic delivery of a highly stable and less stable LNP from the ex utero mouse amniotic stability screen demonstrated significantly increased in utero luciferase mRNA delivery for the more stable LNP compared to the less stable LNP. As hypothesized based on the timing of injection during mouse development when fetal breathing and swallowing movements are active, the intra-amniotically injected LNPs demonstrated some signal in the intestine and lung. We also observed LNP signal in the fetal liver, likely due to the soft nature of LNPs and their ability to escape the fetal lung and intestinal tissue and enter circulation. These results are consistent with previous work in adult mouse models where lipid-based nanoparticles are found in the liver following inhalation [64,65]. Additional bioluminescent signal in the fetal images could represent some LNP-mediated luciferase mRNA delivery to fetal membranes within the amniotic sac or the fetal skin. We also noted some luciferase expression variability amongst the fetuses receiving the most stable LNP treatment which also justifies future investigation in alternative delivery routes such as intra-tracheal injections which are likely to be more translatable in the clinic. Ultimately, these in vivo results demonstrate the ability of ex utero LNP stability to predict LNP mediated in utero luciferase mRNA delivery.
In conclusion, here we have explored ex utero LNP stability in a series of amniotic fluids to identify highly stable LNP formulations for in utero mRNA delivery. This work can be continued with second generation libraries to further optimize formulations for the non-viral treatment of congenital diseases in utero, or explored with other protein-rich biological fluids for different organ and disease target applications. Overall, this proof-of-concept study demonstrates correlations between ex utero stability measurements and in utero luciferase mRNA delivery, therefore indicating the potential of similar stability measurements to identify lead LNP formulations for prenatal gene therapy technologies.
Supplementary Material
7. Acknowledgements
We acknowledge the Children’s Hospital of Philadelphia Small Animal Imaging Facility for the maintenance of the IVIS. Research reported in this publication was supported in part by the Institute for Translational Medicine and Therapeutics (ITMAT) Transdisciplinary Program in Translational Medicine and Therapeutics. This work was also supported by the U.S. National Institutes of Health (NIH) Director’s New Innovator Awards (DP2 TR002776 to M.J.M. and DP2HL152427 to W.H.P.), NIH 1R01DK123049-01 to W.H.P. and M.J.M., and a Burroughs Wellcome Fund Career Award at the Scientific Interface (CASI) to M.J.M. K.L.S. was supported by a National Science Foundation (NSF) Graduate Research Fellowship.
Footnotes
5. Author Credit
K.L.S. – Conceptualization, Methodology, Investigation, Formal Analysis, Visualization, Writing (Original Draft), Writing (Review and Editing); M.M.B. – Conceptualization, Investigation, Writing (Review and Editing); B.W., S.K.B., R.P., A.D., N.G., and S.P. – Investigation, Methodology, Formal Analysis; A.G.H. – Formal Analysis and Writing (Review and Editing); M.G.A. and D.W. – Resources; W.H.P. – Conceptualization, Writing (Review and Editing), Resources, Supervision, and Funding Acquisition; M.J.M. – Conceptualization, Writing (Review and Editing), Resources, Supervision, Project Administration, and Funding Acquisition.
6. Declaration of Competing Interests
D.W. is an inventor on several patents related to this work filed by the Trustees of the University of Pennsylvania (11/990,646; 13/585,517; 13/839,023; 13/839,155; 14/456,302; 15/339,363; 16/299,202). M.J.M. is an inventor on a patent related to this work filed by the Trustees of the University of Pennsylvania (PCT/US20/56252). The authors declare that they have no other competing interests.
9. Data Availability
The data that support the findings of this report are available upon reasonable request to the corresponding authors.
10. References
- [1].Yang Y, Muzny DM, Reid JG, Bainbridge MN, Willis A, Ward PA, Braxton A, Beuten J, Xia F, Niu Z, Hardison M, Person R, Bekheirnia MR, Leduc MS, Kirby A, Pham P, Scull J, Wang M, Ding Y, Plon SE, Lupski JR, Beaudet AL, Gibbs RA, Eng CM, Clinical Whole-Exome Sequencing for the Diagnosis of Mendelian Disorders, 10.1056/NEJMoa1306555. (2013). 10.1056/NEJMoa1306555. [DOI] [PMC free article] [PubMed]
- [2].Lord J, McMullan DJ, Eberhardt RY, Rinck G, Hamilton SJ, Quinlan-Jones E, Prigmore E, Keelagher R, Best SK, Carey GK, Mellis R, Robart S, Berry IR, Chandler KE, Cilliers D, Cresswell L, Edwards SL, Gardiner C, Henderson A, Holden ST, Homfray T, Lester T, Lewis RA, Newbury-Ecob R, Prescott K, Quarrell OW, Ramsden SC, Roberts E, Tapon D, Tooley MJ, Vasudevan PC, Weber AP, Wellesley DG, Westwood P, White H, Parker M, Williams D, Jenkins L, Scott RH, Kilby MD, Chitty LS, Hurles ME, Maher ER, Bateman M, Berry IR, Best SK, Campbell C, Campbell J, Carey G, Chandler KE, Chitty LS, Cilliers D, Cohen K, Collingwood E, Constantinou P, Cresswell L, Delmege C, Eberhardt RY, Edwards SL, Ellis R, Evans J, Everett T, Pinto CF, Forrester N, Fowler E, Gardiner C, Hamilton S, Healey K, Henderson A, Holden ST, Homfray T, Hudson R, Hurles ME, Jenkins L, Keelagher R, Kilby MD, Lester T, Lewis R, Lord J, Maher ER, Marton T, McMullan DJ, Mehta S, Mellis R, Newbury-Ecob R, Park S-M, Parker M, Prescott K, Prigmore E, Quarrell OW, Quinlan-Jones E, Ramsden SC, Rinck G, Robart S, Roberts E, Rowland J, Scott RH, Steer J, Tapon D, Taylor EJ, Tooley MJ, Vasudevan PC, Weber AP, Wellesley DG, Westwood P, White H, Williams D, Wilson E, Prenatal exome sequencing analysis in fetal structural anomalies detected by ultrasonography (PAGE): a cohort study, The Lancet. 393 (2019) 747–757. 10.1016/S0140-6736(18)31940-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Lo YMD, Tein MSC, Lau TK, Haines CJ, Leung TN, Poon PMK, Wainscoat JS, Johnson PJ, Chang AMZ, Hjelm NM, Quantitative Analysis of Fetal DNA in Maternal Plasma and Serum: Implications for Noninvasive Prenatal Diagnosis, Am. J. Hum. Genet 62 (1998) 768–775. 10.1086/301800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Roseman DS, Khan T, Rajas F, Jun LS, Asrani KH, Isaacs C, Farelli JD, Subramanian RR, G6PC mRNA Therapy Positively Regulates Fasting Blood Glucose and Decreases Liver Abnormalities in a Mouse Model of Glycogen Storage Disease 1a, Mol. Ther 26 (2018) 814–821. 10.1016/j.ymthe.2018.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Alapati D, Zacharias WJ, Hartman HA, Rossidis AC, Stratigis JD, Ahn NJ, Coons B, Zhou S, Li H, Singh K, Katzen J, Tomer Y, Chadwick AC, Musunuru K, Beers MF, Morrisey EE, Peranteau WH, In utero gene editing for monogenic lung disease, Sci. Transl. Med 11 (2019). 10.1126/scitranslmed.aav8375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Ricciardi AS, Bahal R, Farrelly JS, Quijano E, Bianchi AH, Luks VL, Putman R, López-Giráldez F, Coşkun S, Song E, Liu Y, Hsieh W-C, Ly DH, Stitelman DH, Glazer PM, Saltzman WM, In utero nanoparticle delivery for site-specific genome editing, Nat. Commun 9 (2018) 2481. 10.1038/s41467-018-04894-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Palanki R, Peranteau WH, Mitchell MJ, Delivery technologies for in utero gene therapy, Adv. Drug Deliv. Rev 169 (2021) 51–62. 10.1016/j.addr.2020.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Almeida-Porada G, Atala A, Porada CD, In utero stem cell transplantation and gene therapy: rationale, history, and recent advances toward clinical application, Mol. Ther. Methods Clin. Dev 5 (2016) 16020. 10.1038/mtm.2016.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Peranteau WH, Flake AW, The Future of In Utero Gene Therapy, Mol. Diagn. Ther 24 (2020) 135–142. 10.1007/s40291-020-00445-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].DeWeerdt S, Prenatal gene therapy offers the earliest possible cure, Nature 564 (2018) S6–S8. 10.1038/d41586-018-07643-z. [DOI] [PubMed] [Google Scholar]
- [11].Trepotec Z, Lichtenegger E, Plank C, Aneja MK, Rudolph C, Delivery of mRNA Therapeutics for the Treatment of Hepatic Diseases, Mol. Ther 27 (2019) 794–802. 10.1016/j.ymthe.2018.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Connolly B, Isaacs C, Cheng L, Asrani KH, Subramanian RR, SERPINA1 mRNA as a Treatment for Alpha-1 Antitrypsin Deficiency, J. Nucleic Acids. 2018 (2018) e8247935. 10.1155/2018/8247935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Rossidis AC, Stratigis JD, Chadwick AC, Hartman HA, Ahn NJ, Li H, Singh K, Coons BE, Li L, Lv W, Zoltick PW, Alapati D, Zacharias W, Jain R, Morrisey EE, Musunuru K, Peranteau WH, In utero CRISPR-mediated therapeutic editing of metabolic genes, Nat. Med 24 (2018) 1513–1518. 10.1038/s41591-018-0184-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Bouchard S, MacKenzie TC, Radu AP, Hayashi S, Peranteau WH, Chirmule N, Flake AW, Long-term transgene expression in cardiac and skeletal muscle following fetal administration of adenoviral or adeno-associated viral vectors in mice, J. Gene Med 5 (2003) 941–950. 10.1002/jgm.421. [DOI] [PubMed] [Google Scholar]
- [15].Stitelman DH, Endo M, Bora A, Muvarak N, Zoltick PW, Flake AW, Brazelton TR, Robust In Vivo Transduction of Nervous System and Neural Stem Cells by Early Gestational Intra Amniotic Gene Transfer Using Lentiviral Vector, Mol. Ther 18 (2010) 1615–1623. 10.1038/mt.2010.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Endo M, Zoltick PW, Peranteau WH, Radu A, Muvarak N, Ito M, Yang Z, Cotsarelis G, Flake AW, Efficient In Vivo Targeting of Epidermal Stem Cells by Early Gestational Intraamniotic Injection of Lentiviral Vector Driven by the Keratin 5 Promoter, Mol. Ther 16 (2008) 131–137. 10.1038/sj.mt.6300332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Boyle MP, Enke RA, Adams RJ, Guggino WB, Zeitlin PL, In Utero AAV-Mediated Gene Transfer to Rabbit Pulmonary Epithelium, Mol. Ther 4 (2001) 115–121. 10.1006/mthe.2001.0428. [DOI] [PubMed] [Google Scholar]
- [18].David AL, Peebles DM, Gregory L, Themis M, Cook T, Coutelle C, Rodeck CH, Percutaneous Ultrasound-Guided Injection of the Trachea in Fetal Sheep: A Novel Technique to Target the Fetal Airways, Fetal Diagn. Ther 18 (2003) 385–390. 10.1159/000071984. [DOI] [PubMed] [Google Scholar]
- [19].M. G, M. Cnz, W. Ams, S. E, B. Smk, H. Br, K. S, P. Dp, B. D, H. S, R.-L. A, B. S, H. M, P. Da, P. Fm, M. K, B. A, C. Jd, C. Jky, C. Sh, W. Sn, R. Aa, Fetal gene therapy for neurodegenerative disease of infants., Nat. Med 24 (2018) 1317–1323. 10.1038/s41591-018-0106-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Yin H, Kanasty RL, Eltoukhy AA, Vegas AJ, Dorkin JR, Anderson DG, Non-viral vectors for gene-based therapy, Nat. Rev. Genet 15 (2014) 541–555. 10.1038/nrg3763. [DOI] [PubMed] [Google Scholar]
- [21].Howe SJ, Mansour MR, Schwarzwaelder K, Bartholomae C, Hubank M, Kempski H, Brugman MH, Pike-Overzet K, Chatters SJ, de Ridder D, Gilmour KC, Adams S, Thornhill SI, Parsley KL, Staal FJT, Gale RE, Linch DC, Bayford J, Brown L, Quaye M, Kinnon C, Ancliff P, Webb DK, Schmidt M, von Kalle C, Gaspar HB, Thrasher AJ, Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients, J. Clin. Invest 118 (2008) 3143–3150. 10.1172/JCI35798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Hajj KA, Whitehead KA, Tools for translation: non-viral materials for therapeutic mRNA delivery, Nat. Rev. Mater 2 (2017) 1–17. 10.1038/natrevmats.2017.56. [DOI] [Google Scholar]
- [23].Kowalski PS, Rudra A, Miao L, Anderson DG, Delivering the Messenger: Advances in Technologies for Therapeutic mRNA Delivery, Mol. Ther 27 (2019) 710–728. 10.1016/j.ymthe.2019.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kauffman KJ, Webber MJ, Anderson DG, Materials for non-viral intracellular delivery of messenger RNA therapeutics, J. Controlled Release. 240 (2016) 227–234. 10.1016/j.jconrel.2015.12.032. [DOI] [PubMed] [Google Scholar]
- [25].Lv H, Zhang S, Wang B, Cui S, Yan J, Toxicity of cationic lipids and cationic polymers in gene delivery, J. Controlled Release. 114 (2006) 100–109. 10.1016/j.jconrel.2006.04.014. [DOI] [PubMed] [Google Scholar]
- [26].Garber K, Alnylam launches era of RNAi drugs, Nat. Biotechnol 36 (2018) 777–778. 10.1038/nbt0918-777. [DOI] [PubMed] [Google Scholar]
- [27].Jackson LA, Anderson EJ, Rouphael NG, Roberts PC, Makhene M, Coler RN, McCullough MP, Chappell JD, Denison MR, Stevens LJ, Pruijssers AJ, McDermott A, Flach B, Doria-Rose NA, Corbett KS, Morabito KM, O’Dell S, Schmidt SD, Swanson PA, Padilla M, Mascola JR, Neuzil KM, Bennett H, Sun W, Peters E, Makowski M, Albert J, Cross K, Buchanan W, Pikaart-Tautges R, Ledgerwood JE, Graham BS, Beigel JH, An mRNA Vaccine against SARS-CoV-2 — Preliminary Report, N. Engl. J. Med 383 (2020) 1920–1931. 10.1056/NEJMoa2022483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Vogel AB, Kanevsky I, Che Y, Swanson KA, Muik A, Vormehr M, Kranz LM, Walzer KC, Hein S, Güler A, Loschko J, Maddur MS, Ota-Setlik A, Tompkins K, Cole J, Lui BG, Ziegenhals T, Plaschke A, Eisel D, Dany SC, Fesser S, Erbar S, Bates F, Schneider D, Jesionek B, Sänger B, Wallisch A-K, Feuchter Y, Junginger H, Krumm SA, Heinen AP, Adams-Quack P, Schlereth J, Schille S, Kröner C, de la Caridad Güimil Garcia R, Hiller T, Fischer L, Sellers RS, Choudhary S, Gonzalez O, Vascotto F, Gutman MR, Fontenot JA, Hall-Ursone S, Brasky K, Griffor MC, Han S, Su AAH, Lees JA, Nedoma NL, Mashalidis EH, Sahasrabudhe PV, Tan CY, Pavliakova D, Singh G, Fontes-Garfias C, Pride M, Scully IL, Ciolino T, Obregon J, Gazi M, Carrion R, Alfson KJ, Kalina WV, Kaushal D, Shi P-Y, Klamp T, Rosenbaum C, Kuhn AN, Türeci Ö, Dormitzer PR, Jansen KU, Sahin U, Immunogenic BNT162b vaccines protect rhesus macaques from SARS-CoV-2, Nature. (2021) 1–10. 10.1038/s41586-021-03275-y. [DOI] [PubMed] [Google Scholar]
- [29].Kulkarni JA, Cullis PR, van der Meel R, Lipid Nanoparticles Enabling Gene Therapies: From Concepts to Clinical Utility, Nucleic Acid Ther 28 (2018) 146–157. 10.1089/nat.2018.0721. [DOI] [PubMed] [Google Scholar]
- [30].Riley RS, June CH, Langer R, Mitchell MJ, Delivery technologies for cancer immunotherapy, Nat. Rev. Drug Discov 18 (2019) 175–196. 10.1038/s41573-018-0006-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Patel S, Ashwanikumar N, Robinson E, DuRoss A, Sun C, Murphy-Benenato KE, Mihai C, Almarsson Ö, Sahay G, Boosting Intracellular Delivery of Lipid Nanoparticle-Encapsulated mRNA, Nano Lett 17 (2017) 5711–5718. 10.1021/acs.nanolett.7b02664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Gilleron J, Querbes W, Zeigerer A, Borodovsky A, Marsico G, Schubert U, Manygoats K, Seifert S, Andree C, Stöter M, Epstein-Barash H, Zhang L, Koteliansky V, Fitzgerald K, Fava E, Bickle M, Kalaidzidis Y, Akinc A, Maier M, Zerial M, Image-based analysis of lipid nanoparticle-mediated siRNA delivery, intracellular trafficking and endosomal escape, Nat. Biotechnol 31 (2013) 638–646. 10.1038/nbt.2612. [DOI] [PubMed] [Google Scholar]
- [33].Cheng Q, Wei T, Jia Y, Farbiak L, Zhou K, Zhang S, Wei Y, Zhu H, Siegwart DJ, Dendrimer-Based Lipid Nanoparticles Deliver Therapeutic FAH mRNA to Normalize Liver Function and Extend Survival in a Mouse Model of Hepatorenal Tyrosinemia Type I, Adv. Mater 30 (2018) 1805308. 10.1002/adma.201805308. [DOI] [PubMed] [Google Scholar]
- [34].Riley RS, Kashyap MV, Billingsley MM, White B, Alameh M-G, Bose SK, Zoltick PW, Li H, Zhang R, Cheng AY, Weissman D, Peranteau WH, Mitchell MJ, Ionizable lipid nanoparticles for in utero mRNA delivery, Sci. Adv 7 (2021) eaba1028. 10.1126/sciadv.aba1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Ball RL, Hajj KA, Vizelman J, Bajaj P, Whitehead KA, Lipid Nanoparticle Formulations for Enhanced Co-delivery of siRNA and mRNA, Nano Lett (2018) 9. [DOI] [PubMed] [Google Scholar]
- [36].Cheng Q, Wei T, Farbiak L, Johnson LT, Dilliard SA, Siegwart DJ, Selective organ targeting (SORT) nanoparticles for tissue-specific mRNA delivery and CRISPR–Cas gene editing, Nat. Nanotechnol 15 (2020) 313–320. 10.1038/s41565-020-0669-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Chen D, Ganesh S, Wang W, Amiji M, The role of surface chemistry in serum protein corona-mediated cellular delivery and gene silencing with lipid nanoparticles, Nanoscale. 11 (2019) 8760–8775. 10.1039/C8NR09855G. [DOI] [PubMed] [Google Scholar]
- [38].Francia V, Schiffelers RM, Cullis PR, Witzigmann D, The Biomolecular Corona of Lipid Nanoparticles for Gene Therapy, Bioconjug. Chem 31 (2020) 2046–2059. 10.1021/acs.bioconjchem.0c00366. [DOI] [PubMed] [Google Scholar]
- [39].Stebounova LV, Guio E, Grassian VH, Silver nanoparticles in simulated biological media: a study of aggregation, sedimentation, and dissolution, J. Nanoparticle Res 13 (2011) 233–244. 10.1007/s11051-010-0022-3. [DOI] [Google Scholar]
- [40].Kauffman KJ, Dorkin JR, Yang JH, Heartlein MW, DeRosa F, Mir FF, Fenton OS, Anderson DG, Optimization of Lipid Nanoparticle Formulations for mRNA Delivery in Vivo with Fractional Factorial and Definitive Screening Designs, Nano Lett 15 (2015) 7300–7306. 10.1021/acs.nanolett.5b02497. [DOI] [PubMed] [Google Scholar]
- [41].Baiersdörfer M, Boros G, Muramatsu H, Mahiny A, Vlatkovic I, Sahin U, Karikó K, A Facile Method for the Removal of dsRNA Contaminant from In Vitro-Transcribed mRNA, Mol. Ther. Nucleic Acids. 15 (2019) 26–35. 10.1016/j.omtn.2019.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Chen D, Love KT, Chen Y, Eltoukhy AA, Kastrup C, Sahay G, Jeon A, Dong Y, Whitehead KA, Anderson DG, Rapid Discovery of Potent siRNA-Containing Lipid Nanoparticles Enabled by Controlled Microfluidic Formulation, J Am Chem Soc (2012) 4. [DOI] [PubMed] [Google Scholar]
- [43].Hajj KA, Ball RL, Deluty SB, Singh SR, Strelkova D, Knapp CM, Whitehead KA, Branched-Tail Lipid Nanoparticles Potently Deliver mRNA In Vivo due to Enhanced Ionization at Endosomal pH, Small. 15 (2019) 1805097. 10.1002/smll.201805097. [DOI] [PubMed] [Google Scholar]
- [44].Heyes J, Palmer L, Bremner K, MacLachlan I, Cationic lipid saturation influences intracellular delivery of encapsulated nucleic acids, J. Controlled Release. 107 (2005) 276–287. 10.1016/j.jconrel.2005.06.014. [DOI] [PubMed] [Google Scholar]
- [45].Schroffenegger M, Leitner NS, Morgese G, Ramakrishna SN, Willinger M, Benetti EM, Reimhult E, Polymer Topology Determines the Formation of Protein Corona on Core–Shell Nanoparticles, ACS Nano. 14 (2020) 12708–12718. 10.1021/acsnano.0c02358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Malcolm DW, Varghese JJ, Sorrells JE, Ovitt CE, Benoit DSW, The Effects of Biological Fluids on Colloidal Stability and siRNA Delivery of a pH-Responsive Micellar Nanoparticle Delivery System, ACS Nano. 12 (2018) 187–197. 10.1021/acsnano.7b05528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Amici A, Caracciolo G, Digiacomo L, Gambini V, Marchini C, Tilio M, Capriotti AL, Colapicchioni V, Matassa R, Familiari G, Palchetti S, Pozzi D, Mahmoudi M, Laganà A, In vivo protein corona patterns of lipid nanoparticles, RSC Adv 7 (2017) 1137–1145. 10.1039/C6RA25493D. [DOI] [Google Scholar]
- [48].Motulsky HJ, Brown RE, Detecting outliers when fitting data with nonlinear regression – a new method based on robust nonlinear regression and the false discovery rate, BMC Bioinformatics. 7 (2006) 123. 10.1186/1471-2105-7-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Li B, Luo X, Deng B, Wang J, McComb DW, Shi Y, Gaensler KML, Tan X, Dunn AL, Kerlin BA, Dong Y, An Orthogonal Array Optimization of Lipid-like Nanoparticles for mRNA Delivery in Vivo, Nano Lett 15 (2015) 8099–8107. 10.1021/acs.nanolett.5b03528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Oberli MA, Reichmuth AM, Dorkin JR, Mitchell MJ, Fenton OS, Jaklenec A, Anderson DG, Langer R, Blankschtein D, Lipid Nanoparticle Assisted mRNA Delivery for Potent Cancer Immunotherapy, Nano Lett 17 (2017) 1326–1335. 10.1021/acs.nanolett.6b03329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Jayaraman M, Ansell SM, Mui BL, Tam YK, Chen J, Du X, Butler D, Eltepu L, Matsuda S, Narayanannair JK, Rajeev KG, Hafez IM, Akinc A, Maier MA, Tracy MA, Cullis PR, Madden TD, Manoharan M, Hope MJ, Maximizing the Potency of siRNA Lipid Nanoparticles for Hepatic Gene Silencing In Vivo, Angew. Chem 124 (2012) 8657–8661. 10.1002/ange.201203263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Whitehead KA, Dorkin JR, Vegas AJ, Chang PH, Veiseh O, Matthews J, Fenton OS, Zhang Y, Olejnik KT, Yesilyurt V, Chen D, Barros S, Klebanov B, Novobrantseva T, Langer R, Anderson DG, Degradable lipid nanoparticles with predictable in vivo siRNA delivery activity, Nat. Commun 5 (2014) 4277. 10.1038/ncomms5277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Colapicchioni V, Tilio M, Digiacomo L, Gambini V, Palchetti S, Marchini C, Pozzi D, Occhipinti S, Amici A, Caracciolo G, Personalized liposome–protein corona in the blood of breast, gastric and pancreatic cancer patients, Int. J. Biochem. Cell Biol 75 (2016) 180–187. 10.1016/j.biocel.2015.09.002. [DOI] [PubMed] [Google Scholar]
- [54].Chen S, Tam YYC, Lin PJC, Sung MMH, Tam YK, Cullis PR, Influence of particle size on the in vivo potency of lipid nanoparticle formulations of siRNA, J. Controlled Release. 235 (2016) 236–244. 10.1016/j.jconrel.2016.05.059. [DOI] [PubMed] [Google Scholar]
- [55].Tong X-L, Wang L, Gao T-B, Qin Y-G, Qi Y-Q, Xu Y-P, Potential Function of Amniotic Fluid in Fetal Development—Novel Insights by Comparing the Composition of Human Amniotic Fluid with Umbilical Cord and Maternal Serum at Mid and Late Gestation, J. Chin. Med. Assoc 72 (2009) 368–373. 10.1016/S1726-4901(09)70389-2. [DOI] [PubMed] [Google Scholar]
- [56].Love KT, Mahon KP, Levins CG, Whitehead KA, Querbes W, Dorkin JR, Qin J, Cantley W, Qin LL, Racie T, Frank-Kamenetsky M, Yip KN, Alvarez R, Sah DWY, de Fougerolles A, Fitzgerald K, Koteliansky V, Akinc A, Langer R, Anderson DG, Lipid-like materials for low-dose, in vivo gene silencing, Proc. Natl. Acad. Sci 107 (2010) 1864–1869. 10.1073/pnas.0910603106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Guimaraes PPG, Zhang R, Spektor R, Tan M, Chung A, Billingsley MM, El-Mayta R, Riley RS, Wang L, Wilson JM, Mitchell MJ, Ionizable lipid nanoparticles encapsulating barcoded mRNA for accelerated in vivo delivery screening, J. Controlled Release. 316 (2019) 404–417. 10.1016/j.jconrel.2019.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Gräfe C, Weidner A, Lühe M. v. d., Bergemann C, Schacher FH, Clement JH, Dutz S, Intentional formation of a protein corona on nanoparticles: Serum concentration affects protein corona mass, surface charge, and nanoparticle–cell interaction, Int. J. Biochem. Cell Biol 75 (2016) 196–202. 10.1016/j.biocel.2015.11.005. [DOI] [PubMed] [Google Scholar]
- [59].Cardarelli F, Digiacomo L, Marchini C, Amici A, Salomone F, Fiume G, Rossetta A, Gratton E, Pozzi D, Caracciolo G, The intracellular trafficking mechanism of Lipofectamine-based transfection reagents and its implication for gene delivery, Sci. Rep 6 (2016) 25879. 10.1038/srep25879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Wang T, Larcher LM, Ma L, Veedu RN, Systematic Screening of Commonly Used Commercial Transfection Reagents towards Efficient Transfection of Single-Stranded Oligonucleotides, Mol. J. Synth. Chem. Nat. Prod. Chem 23 (2018). 10.3390/molecules23102564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Figueroa-Espada CG, Hofbauer S, Mitchell MJ, Riley RS, Exploiting the placenta for nanoparticle-mediated drug delivery during pregnancy, Adv. Drug Deliv. Rev 160 (2020) 244–261. 10.1016/j.addr.2020.09.006. [DOI] [PubMed] [Google Scholar]
- [62].Chen D, Ganesh S, Wang W, Amiji M, Plasma protein adsorption and biological identity of systemically administered nanoparticles, Nanomed. 12 (2017) 2113–2135. 10.2217/nnm-2017-0178. [DOI] [PubMed] [Google Scholar]
- [63].Cho C-KJ, Shan SJ, Winsor EJ, Diamandis EP, Proteomics Analysis of Human Amniotic Fluid *, Mol. Cell. Proteomics. 6 (2007) 1406–1415. 10.1074/mcp.M700090-MCP200. [DOI] [PubMed] [Google Scholar]
- [64].Truzzi E, Nascimento TL, Iannuccelli V, Costantino L, Lima EM, Leo E, Siligardi C, Gualtieri ML, Maretti E, In Vivo Biodistribution of Respirable Solid Lipid Nanoparticles Surface-Decorated with a Mannose-Based Surfactant: A Promising Tool for Pulmonary Tuberculosis Treatment?, Nanomaterials. 10 (2020) 568. 10.3390/nano10030568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Gaspar DP, Gaspar MM, Eleutério CV, Grenha A, Blanco M, Gonçalves LMD, Taboada P, Almeida AJ, Remuñán-López C, Microencapsulated Solid Lipid Nanoparticles as a Hybrid Platform for Pulmonary Antibiotic Delivery, Mol. Pharm 14 (2017) 2977–2990. 10.1021/acs.molpharmaceut.7b00169. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this report are available upon reasonable request to the corresponding authors.