

Abstract
Candida parapsilosis accounts for a significant number of nosocomial fungemias, but in fact, no effective and verified genetic fingerprinting method has emerged for assessing the relatedness of independent isolates for epidemiological studies. A complex 15-kb DNA fingerprinting probe, Cp3-13, was therefore isolated from a library of C. parapsilosis genomic DNA fragments. The efficacy of Cp3-13 for DNA fingerprinting was verified by a comparison of its clustering capacity with those of randomly amplified polymorphic DNA analysis and internally transcribed spacer region sequencing, by testing species specificity, and by assessing its capacity to identify microevolutionary changes both in vitro and in vivo. Southern blot hybridization of EcoRI/SalI-digested DNA with Cp3-13 provides a fingerprinting system that (i) identifies the same strain in independent isolates, (ii) discriminates between unrelated isolates, (iii) separates independent isolates into valid groups in a dendrogram, (iv) identifies microevolution in infecting populations, and (v) is amenable to automatic computer-assisted DNA fingerprint analysis. This probe is now available for epidemiological studies.
Genetic fingerprinting has emerged as an indispensable tool for studying the incidence, distribution, and origin of fungal disease in human populations. A number of different genetic fingerprinting methods have been used, including multilocus enzyme electrophoresis (MLEE), restriction fragment length polymorphisms (RFLP) visualized by ethidium bromide staining, RFLP with hybridization probes, randomly amplified polymorphic DNA (RAPD) analysis, a variety of additional PCR-based methods, electrophoretic karyotyping, and sequencing (35). All of these methods have been demonstrated to be useful, but some are more effective than others at different levels of genetic resolution. As these different genetic fingerprinting methods have been applied and their efficacies assessed, a number of desirable, and in some cases essential, characteristics have been identified. An effective method should be relatively easy to perform, highly reproducible between experiments and laboratories, and amenable to automated, computer-assisted analysis. An effective method should also be capable of identifying the same strain in different isolates, distinguishing between completely unrelated isolates, clustering moderately related isolates, and resolving microevolution within a strain. Recently, we demonstrated that three methods, MLEE, RFLP with the complex probe Ca3, and RAPD analysis, fulfilled these requirements for fingerprinting Candida albicans strains (26). However, we concluded that DNA fingerprinting with the complex probe Ca3 (1, 18, 28) was our method of choice for large epidemiological studies because of its high level of reproducibility, speed, amenability to automatic computer-assisted analysis, species specificity, clustering capacity, and sensitivity in distinguishing microevolutionary change (26, 35). Its capacity to handle all levels of relatedness results from the following combination of sequences it includes: invariant sequences, low variability sequences, moderately variable sequences, and hypervariable sequences. We have used Ca3 as a paradigm for cloning and characterizing species-specific complex DNA fingerprinting probes for additional infectious fungi, including Candida dubliniensis (10), Candida glabrata (19), Candida tropicalis (11), and Aspergillus fumigatus (9). Here we describe the cloning and characterization of a complex probe for Candida parapsilosis.
C. parapsilosis accounts for a significant proportion of nosocomial fungemias (39, 40). Although it was originally believed to be a very weak commensal organism, colonizing only 4 to 5% of hospitalized individuals (5, 23), it has recently been demonstrated that C. parapsilosis is selectively carried in the oral cavity of children (12, 15). More importantly, C. parapsilosis has been repeatedly associated with intravascular catheter infections (14), presumably because of its enhanced capacity to adhere to prosthetic materials (4, 40) in biofilms (22) formed through the production of extracellular slime (2, 23). To develop a complex DNA fingerprinting probe for C. parapsilosis, we screened a library of C. parapsilosis genomic DNA fragments ranging between 9 and 23 kb for complex sequences that contained one or more repetitive elements. The efficacy of the selected probe, Cp3-13, was verified (i) by a comparison of its clustering capacity with those of RAPD analysis and internally transcribed spacer region (ITS) sequencing, (ii) by testing species specificity, and (iii) by assessing its capacity to identify microevolutionary changes in vitro and in vivo.
MATERIALS AND METHODS
Strain maintenance and growth conditions.
The date, anatomical location, and geographical location of isolation of each strain used in this study are presented in Table 1. All strains were originally typed by sugar assimilation profiles and stored in sterile 20% glycerol at −70°C. In select cases (Pf6, Pf9, J920296, J920297, p191, p203, Y-12969, Y-7681, Y-17456, and Y-543), strains were retyped with the ID32C kit from bioMérieux (Lyon, France). For standard DNA fingerprinting experiments, each isolate was grown on YPD agar plates (2% glucose, 2% Bacto Peptone, 1% yeast extract, 2% agar) at 25°C. For experiments in which the stability of the Southern blot hybridization pattern was assessed over time, 5 × 106 cells from a single colony derived from a single cell from each of four strains were inoculated into 25 ml of YPD liquid medium and grown to stationary phase at 37°C. Cells from this flask (5 × 106) were inoculated into a second flask, and the process was repeated. After 200 generations, cells were plated at low density on YPD agar plates, and nine colonies of each original strain were prepared for DNA fingerprinting.
TABLE 1.
Isolates used in this study
| Isolate | Year | Site | Location |
|---|---|---|---|
| Pf1 | 1995 | Blood | Pittsburgh, Pa. |
| Pf2 | 1995 | Blood | Miami, Fla. |
| Pf3 | 1995 | Blood | Miami, Fla. |
| Pf4 | 1995 | Blood | Ft. Lauderdale, Fla. |
| Pf5 | 1995 | Blood | Ft. Lauderdale, Fla. |
| Pf6 | 1995 | Blood | Savannah, Ga. |
| Pf7 | 1995 | Blood | Sioux Falls, S. Dak. |
| Pf8 | 1996 | Blood | Pittsburgh, Pa. |
| Pf9 | 1996 | Blood | Miami, Fla. |
| Pf10 | 1996 | Blood | Miami, Fla. |
| Pf11 | 1996 | Blood | New York, N.Y. |
| Pf12 | 1996 | Blood | New York, N.Y. |
| Pf13 | 1996 | Blood | Richmond, Va. |
| Pf14 | 1996 | Blood | Richmond, Va. |
| Pf15 | 1996 | Blood | Richmond, Va. |
| Pf16 | 1997 | Blood | Ankara, Turkey |
| Pf17 | 1997 | Blood | Coimbra, Portugal |
| Pf18 | 1997 | Blood | Coimbra, Portugal |
| Pf19 | 1997 | Blood | Coimbra, Portugal |
| Pf20 | 1997 | Blood | Coimbra, Portugal |
| Pf21 | 1997 | Blood | Genoa, Italy |
| Pf22 | 1997 | Blood | Genoa, Italy |
| Pf23 | 1997 | Blood | Phoenix, Ariz. |
| Pf24 | 1997 | Blood | New Hyde Park, N.Y. |
| Pf25 | 1997 | Blood | New Hyde Park, N.Y. |
| Pf26 | 1997 | Blood | New Hyde Park, N.Y. |
| Pf27 | 1997 | Blood | Spokane, Wash. |
| Pf28 | 1997 | Blood | Spokane, Wash. |
| Pf29 | 1997 | Blood | St. Louis, Mo. |
| Pf30 | 1997 | Blood | St. Louis, Mo. |
| Pf31 | 1997 | Blood | Galveston, Tex. |
| Pf32 | 1998 | Blood | Indianapolis, Ind. |
| Pf33 | 1998 | Urine | Iowa City, Iowa |
| Pf35 | 1998 | Blood | New York, N.Y. |
| Pf36 | 1998 | Blood | Charlotte, N.C. |
| Pf37 | 1998 | Blood | Charlotte, N.C. |
| Pf38 | 1998 | Blood | Charlotte, N.C. |
| Pf39 | 1998 | Blood | Charlottesville, Va. |
| Pf40 | 1998 | Blood | Charlottesville, Va. |
| Pf41 | 1998 | Blood | Charlottesville, Va. |
| Pf45 | 1996 | NAd | Westchester, N.Y. |
| Pf46 | 1996 | NA | Westchester, N.Y. |
| Pf47 | 1996 | NA | Westchester, N.Y. |
| Pf48 | 1996 | NA | Westchester, N.Y. |
| Pf49 | NA | NA | NA |
| 79Tw1 | 1997 | Oral cavity | Iowa City, Iowa |
| 82T1 | 1997 | Oral cavity | Iowa City, Iowa |
| 1480.21 | NA | Pharynx | Detroit, Mich. |
| 1480.22 | NA | Pharynx | Detroit, Mich. |
| 1480.23 | NA | Blood | Royal Oak, Mich. |
| 1480.24 | NA | Rectum | Detroit, Mich. |
| 1480.25 | NA | HCWc | Detroit, Mich. |
| 1480.26 | NA | Wound | Detroit, Mich. |
| 1480.27 | NA | HCWc | Detroit, Mich. |
| 1480.28 | NA | HCWc | Detroit, Mich. |
| 1480.29a | NA | Sprue | Puerto Rico |
| 1480.30 | NA | Stool | Detroit, Mich. |
| B66126 | 1991 | Nail | Belgium |
| J920296 | 1992 | Vagina | Belgium |
| J941226/2 | 1994 | Tinea pedis | Belgium |
| J941818 | 1994 | Surveillance | The Netherlands |
| J941824 | 1994 | Surveillance | The Netherlands |
| J941830 | 1994 | Surveillance | The Netherlands |
| J932072 | 1994 | Nail | Belgium |
| J940043 | 1993 | Nail | Belgium |
| J941074 | 1994 | Oral cavity | Belgium |
| J940889 | 1994 | Oral cavity | Belgium |
| J940360 | 1994 | Fingernail | Belgium |
| J940723 | 1994 | Fingernail | Belgium |
| J941340P | 1994 | Toenail | Belgium |
| J941130 | 1994 | NA | Philippines |
| J941221P | 1994 | Nail | Belgium |
| J941253 | 1994 | Fingernail | Belgium |
| J941335 | 1994 | Nail | Greece |
| J941350 | 1994 | Nail | Belgium |
| p190b | 1994 | Oral cavity | Iowa City, Iowa |
| p191b | 1994 | Oral cavity | Iowa City, Iowa |
| p192b | 1994 | Oral cavity | Iowa City, Iowa |
| p193b | 1994 | Oral cavity | Iowa City, Iowa |
| p196b | 1994 | Oral cavity | Iowa City, Iowa |
| p201b | 1994 | Oral cavity | Iowa City, Iowa |
| p203b | 1994 | Oral cavity | Iowa City, Iowa |
| p205b | 1994 | Oral cavity | Iowa City, Iowa |
| p206b | 1994 | Oral cavity | Iowa City, Iowa |
| p218b | 1994 | Oral cavity | Iowa City, Iowa |
| Y-543a | NA | NA | NA |
| Y-17456a | NA | NA | NA |
| Y-12969a | NA | Sprue | Puerto Rico |
Strains 1480.29 and Y-12969 are both ATCC 22019 obtained from different laboratories. Y-543, Y-17456, Y-12969, and Y-7681 were obtained from C Kurtzman. Y-543 and Y-12969 are C. parapsilosis. Y-17456 is a Candida sp.
Represent isolates taken from one individual during a single episode.
HCW, health care worker.
NA, not available.
Cloning a complex DNA fingerprinting probe.
The general methods for cloning a complex DNA fingerprinting probe were similar to those previously described for C. albicans (28, 35), C. tropicalis (11), C. dubliniensis (10), and A. fumigatus (9). In brief, a Sau3AI partial digest of genomic DNA of C. parapsilosis strain J940889 was separated in a sucrose gradient. Fractions were run in a 0.8% agarose gel to determine molecular size. The fractions containing fragments between 9 and 23 kb were pooled and cloned into λEMBL3 by methods described by the manufacturer (Stratagene, La Jolla, Calif.). The resultant genomic library was amplified and used to infect Escherichia coli strain P2392. E. coli, infected at a density of 10,000 plaques per 150-mm-diameter petri dish, was then transferred to duplicate nitrocellulose filters. Each filter was prehybridized at 65°C for 20 min in a solution containing 1% bovine serum albumin, 7% sodium dodecyl sulfate (SDS), 0.5 M NaH2PO4 (pH 7.0), and 1 mM EDTA (3). One filter was hybridized overnight at 65°C with randomly primed [32P]dCTP-labeled TaqI-digested genomic DNA of C. parapsilosis J940889, and the other filter was hybridized with randomly primed [32P]dCTP-labeled ribosomal DNA (rDNA) of C. albicans. The filters were then washed with a solution containing 1% SDS, 40 mM NaH2PO4, and 1 mM EDTA for 20 min and autoradiographed. The two autoradiograms were aligned, and intense spots were identified. Intense spots that were present in the filter hybridized with genomic DNA but absent from the filter hybridized with rDNA were picked and resuspended in 500 μl of a solution containing 0.1 M NaCl, 0.02 M MgSO4 · 7H2O, 50 mM Tris-HCl(pH 7.5), and 0.01% gelatin plus 20 μl of chloroform and then screened a second time in a similar fashion. Clones of putative complex probes were amplified in E. coli P2392.
Southern blot hybridization.
Southern blot hybridization with DNA fingerprinting probes was performed according to the methods of Schmid et al. (32). In brief, DNA was extracted from each test isolate according to the method of Scherer and Stevens (30). Three micrograms of the DNA preparation was digested with EcoRI or a combination of EcoRI and SalI (4 U per μg of DNA) for 16 h at 37°C. Digested DNA preparations were then electrophoresed at 55 V in a 0.75% agarose gel until the bromophenol blue dye front was 50 mm from the bottom edge of a 25-cm gel. DNA was then transferred to a nylon membrane (29). C. parapsilosis strain J940043 was used in all studies as a reference strain, and its DNA was run in the far right and far left lanes of each fingerprinting gel. The membrane was prehybridized with sheared calf thymus DNA, hybridized with a randomly primed [32P]dCTP-labeled probe, and finally autoradiographed as previously described (10, 11, 19, 32).
Computer-assisted analysis of Southern blot hybridization patterns.
Autoradiogram images were imported into the DENDRON software program (Solltech Inc., Oakdale, Iowa) using an HP Scanjet IIcx flatbed scanner with transparency accessory (Hewlett Packard, Palo Alto, Calif.). Distortions in the image were corrected using the unwarping and straightening options of DENDRON. Lanes and bands were automatically identified and analyzed (35). Patterns of different C. parapsilosis isolates were compared by computing the similarity coefficient (SAB) between every pair according to the following formula:
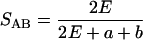 |
where E is the number of bands shared by patterns A and B, a is the number of bands unique to pattern A, and b is the number of bands unique to pattern B. The SAB can vary from 0.0 (no bands common) to 1.0 (all bands common). The unweighted pair-group method (33) was then used to generate dendrograms based on the SABs.
RAPD analysis.
DNA was prepared for RAPD analysis by mixing 5 × 109 cells per ml in 400 μl of 1× TE (10 mM Tris-HCl, 1 mM EDTA, pH 8.0) with 300 μl of glass beads (0.45-mm diameter) and agitating with a bead beater (Biospec Products, Bartlesville, Okla.) to disrupt cells. SDS was added to a final concentration of 2% (wt/vol), and the preparation was extracted twice with phenol-chloroform and twice with chloroform alone. DNA was precipitated with ethanol and suspended in 500 μl of water. PCRs were performed in 0.5-ml microcentrifuge tubes containing 25 μl of the following reaction mixture: 1 ng of C. parapsilosis DNA, 2.5 μl of 10 × buffer (Roche/Boehringer Mannheim, Indianapolis, Ind.), 1.5 U of Taq DNA polymerase (Roche/Boehringer Mannheim), 200 μM (each) dATP, dCTP, dGTP, and dTTP (Roche/Boehringer Mannheim), and a 0.4 mM concentration of one of the primers listed below. The amplification was performed in a thermal cycler (Lab Line, Melrose Park, Ill.) under the following conditions: 45 cycles of 1 min at 94°C, 2 min at 36°C, and 2 min at 73°C. PCR products were separated by electrophoresis in a 1.3% (wt/vol) agarose gel and stained with ethidium bromide. Forty primers from kits A and E of Operon Technologies (Alameda, Calif.) were screened for the ability to create reproducible bands and to discriminate between test isolates. The following 10-base primers fulfilled these requisites: OPA-1 (5′-CAGGCCCTTC-3′), OPA-6 (5′-GGTCCCTGAC-3′), OPA-10 (5′-GTGATCGCAG-3′), OPA-20 (5′-GTTGCGATCC-3′), OPE-10 (5′-CACCAGGTGA-3′), and OPE-13 (5′-CCCGATTCGG-3′). SABs were computed between pairs of isolates by manually entering the patterns to band data files in DENDRON, which calculated the SABs based upon the combined proportions of matches (26). The dendrogram was then created by the unweighted pair-group method (33).
CHEF electrophoresis.
To prepare chromosomal DNA for contour-clamped homogeneous electric field (CHEF) electrophoresis, cells were grown overnight in YPD medium at 25°C, pelleted by centrifugation, washed twice with sterile water, and washed once with 1 M sorbitol. The cells were resuspended in a solution of 1 M sorbitol, 25 mM EDTA, and 50 mM dithiothreitol and incubated for 30 min at room temperature. The cells were then pelleted and resuspended at a concentration of 109 cells per ml in a solution containing 1 M sorbitol, 0.1 M sodium citrate (pH 5.8), 10 mM EDTA, and 0.4 mg of Zymolyase 100T (Seikagaku America, Rockville, Md.) per ml and incubated at 37°C for 30 min. Spheroplast formation was monitored microscopically. When digestion had achieved greater than 90% spheroplast formation, the cells were pelleted, washed twice in a solution containing 1 M sorbitol and 250 mM EDTA, and resuspended in the same solution at a density of 109 per ml. The spheroplast suspension was mixed in a one-to-one ratio with 1× TE (10 mM Tris-HCl, 1 mM EDTA, pH 8.0) containing 1% agarose (low-melt preparative grade; Bio-Rad Laboratories, Hercules, Calif.) and pipetted into plug molds. When the plugs solidified, they were suspended in a solution of 0.5 mM EDTA, 1% Sarkosyl, and 5 mg of proteinase K per ml for 72 h at 37°C. Plugs were washed five times with 1× TE (pH 8.0) and stored at 4°C. For electrophoresis, plugs were loaded on a separation gel containing 22.5 mM Tris-HCl (pH 8.3), 22.5 mM boric acid, 1 mM EDTA, and 1% (wt/vol) agarose. Separation of chromosomes in the CHEF mapper (Bio-Rad Laboratories) was performed at 14°C using the multistate protocol of a 120° angle at a gradient of 4.5 V/cm, with pulse intervals of 120 s for the first 24 h and 240 s for the next 36 h. The final gel was stained with ethidium bromide, photographed, Southern blotted, and hybridized with probe.
ITS sequence analysis.
To compare ITS rDNA sequences, genomic DNA was amplified using the primers ITS1 (5′-TCCGTAGGTGAACCTGCGC-3′) and ITS4 (5′TCCTCCGCTTATTGATATGC-3′) (16). The PCR products were cloned into pGEM-T Easy Vector (Promega Corp., Madison, Wis.). The nucleotide sequences of inserts were determined with an ABI Model 1373A auto sequencing system (Perkin-Elmer/Applied Biosystems, Foster City, Calif.) using the PCR cycle sequencing protocol and fluorescent dye terminator dideoxynucleotides (Perkin-Elmer/Applied Biosystems).
Homology and alignment of nucleotide sequences were performed with MacDNASIS pro, v3.6, software (Hitachi Software Engineering, San Bruno, Calif.). Pairwise comparison between the ITS sequences was performed with the DNAdist program of the PHYLIP package, version 3.5, using Jukes and Cantor distances (http://evolution.genetics.washington.edu/phylip.html). Trees were inferred with the KITSCH program of the PHYLIP package. The data set was bootstrapped (1,000 replicates) by sequential use of the SEQboot, DNAdist, KITSCH, and Consense programs of the PHYLIP package.
RESULTS
Cloning a DNA fingerprinting probe.
A genomic library was constructed in λEMBL3 from a Sau3AI partial digest of C. parapsilosis strain J940889. Partially digested fragments of 9 to 23 kb, representing no less than five genomic equivalents, were cloned into λEMBL3. The library was plated out on 30 150-mm-diameter petri dishes. Each petri dish contained 10,000 plaques. The library on each plate was transferred in duplicate to nitrocellulose membranes and hybridized in parallel with either radiolabeled C. parapsilosis genomic DNA or radiolabeled C. albicans rDNA. The hybridization conditions were nonsaturating, which allowed discrimination between clones of unique sequences (low intensity hybridization) and clones containing moderately to highly repetitive sequences (high intensity hybridization). Of the 300,000 plaques screened, 60 gave intense signals with radiolabeled genomic DNA but not with radiolabeled rDNA. Putative clones were screened a second time under identical conditions as the first screen. Clones were then radiolabeled and used to probe Southern blots of EcoRI-digested DNA of two unrelated test isolates of C. parapsilosis. Of the 60 initial test clones, only three generated Southern blot hybridization patterns that differed between the two test isolates. Two of the clones, however, generated a pattern with only a single polymorphic band and were thus deemed insufficient for DNA fingerprinting. The final 15-kb clone, Cp3-13, generated multiple polymorphic bands.
Cp3-13 hybridization is species specific.
To test whether the hybridization pattern it generates is species specific, Cp3-13 was used to probe a Southern blot that contained EcoRI-digested DNA of C. parapsilosis and 13 related yeast species. Cp3-13 hybridized strongly only to C. parapsilosis, demonstrating that Cp3-13 was species specific (Fig. 1).
FIG. 1.

Species specificity of the probe Cp3-13. Southern blots of EcoRI-digested DNA of the 14 noted yeast species were probed with Cp3-13.
Developing more complexity in the Cp3-13 DNA fingerprinting pattern.
Although Cp3-13 could distinguish between the two unrelated test isolates, the hybridization patterns with EcoRI-digested genomic DNA lacked sufficient complexity, as is evident in the patterns of four test isolates in Fig. 2A. The patterns consisted of between eight and nine bands of varying intensities (Fig. 2A). Each included five monomorphic bands and three to four polymorphic bands. To increase the complexity of the pattern (i.e., the total number of bands) and, more importantly, the number of polymorphic bands, genomic DNA was digested with five different restriction enzymes alone and with 11 different combinations of two restriction enzymes. Of the 16 restriction digests, the combination of EcoRI plus SalI provided the greatest pattern complexity (i.e., the greatest number of band differences) between test isolates (Fig. 2B). The EcoRI/SalI patterns contained bands of relatively similar intensities over a wide range of molecular weights. The number of bands in the EcoRI/SalI patterns of the four test isolates averaged 14.3, ranging between 12 and 19. The patterns contained three monomorphic bands. We therefore selected the EcoRI/SalI combination for digesting C. parapsilosis DNA in all subsequent fingerprinting studies with the probe Cp3-13.
FIG. 2.

Southern blot hybridization patterns of EcoRI-digested DNA (A) and EcoRI- and SalI-digested DNA (B) of four unrelated strains probed with Cp3-13. Digested DNA was electrophoresed in a 0.75% agarose gel, Southern blotted, and hybridized with Cp3-13. The arrowheads to the right of each gel represent invariant (monomorphic) bands. Molecular sizes are noted in kilobases. The origins of the strains are described in Table 1.
Cp3-13 sequences reside on multiple chromosomes.
To assess the distribution of sequences homologous to Cp3-13 in the C. parapsilosis genome, chromosomes of four unrelated C. parapsilosis isolates separated by CHEF electrophoresis were probed with the Cp3-13 probe. The number of bands in the ethidium bromide-stained CHEF patterns was similar for the four isolates. Ten chromosomes were resolved for isolates J941074, J940043, and Pf19, and 12 were resolved for isolate 1480.26 (Fig. 3A). The bands in the pattern of strain J941074, our reference strain, were arbitrarily numbered 1 through 10, beginning with the highest-molecular-weight band (Fig. 3A). The molecular sizes of the high-molecular-weight chromosomes 1, 2, and 3 were similar between the pair-test strains, but those of the mid- and low-molecular-size chromosomes varied between strains (Fig. 3A). Fernando and Samaranayake (6) also noted chromosome length polymorphisms, especially in the mid- and low-molecular-size chromosomes. Cp3-13 hybridized intensively to chromosomes 4, 5, 9, and 10 (Fig. 3B). Similar hybridization patterns were evident for isolates J940043, 1480.26, and Pf19 (Fig. 3B).
FIG. 3.

Hybridization of CHEF-separated chromosomes of four unrelated C. parapsilosis isolates with Cp3-13. Chromosomes of J941074, J940043, 1480.26, and Pf19 were separated by CHEF and then the gel was stained with ethidium bromide (EtBr) (A). Numbers to the left of the gel represent chromosomal bands of strain J941074. The gel was Southern blotted and probed with Cp3-13 (B).
Cp3-13 discriminates between unrelated isolates.
To test its resolving power, Southern blots of 89 C. parapsilosis isolates were probed with Cp3-13, and the resulting Southern blot patterns were compared in a dendrogram based on SAB values computed between all pairs. The collection included 78 isolates that were presumed unrelated (Table 1). In addition, the collection contained 10 isolates, p190, p191, p192, p193, p196, p201, p203, p205, p206, and p218, that were isolated from one individual during a single episode of oral candidiasis (Table 1). The collection included isolates collected in several geographical locations, including 14 states in the United States, six European countries, and one Asian country (Table 1). In Fig. 4A, the original banding patterns are presented for 11 representative isolates. In Fig. 4B, the gel image has been unwarped using computer-assisted methods (35). In the two outermost lanes, the banding patterns are presented for the reference strain J940043. Note that the two reference strain patterns are identical both for band positions and intensities. The pattern of strain J940043 contained 16 bands varying in intensity. The patterns of the additional isolates contained between 12 and 16 bands. Three monomorphic bands were evident at 3.4, 3.2, and 1.3 kb. The majority of bands were adequately separated in all patterns for automated computer-assisted analysis.
FIG. 4.

Examples of the Southern blot hybridization patterns of isolates probed with Cp3-13. The original pattern image is presented in panel A and the pattern image unwarped with DENDRON software is presented in panel B. Samples of C. parapsilosis DNA were digested with EcoRI and SalI and electrophoresed in a 0.75% agarose gel. Molecular sizes in kilobases of select bands are indicated to the left of the gels. Arrow heads represent invariant bands. The reference strain J940043 was analyzed in the outer two lanes to normalize the gel during computer-assisted analysis.
The average SAB, based on band position alone, for the entire collection was 0.45 ± 0.22. In the dendrogram generated for the entire collection, the isolates separated into three deep-rooted groups, with nodes below an SAB of 0.30 (Fig. 5). The three groups contained 79, 8, and 1 isolate, respectively. Group I isolates exhibited an average SAB of 0.53 ± 0.15, and group II isolates exhibited an average SAB of 0.38 ± 0.13. Group I isolates could be further subdivided into groups Ia and Ib at a node of 0.37. Group Ia isolates exhibited an average SAB of 0.54 ± 0.15, and group Ib isolates exhibited an average SAB of 0.58 ± 0.15. Isolate Y-17456, which was previously distinguished by Kurtzman and Robnett (13) as a potential species distinct from C. parapsilosis, resided in group II. The fingerprinting patterns of group II isolates had far less complexity than those of group I isolates. While the number of bands in the patterns of group I isolates ranged between 12 and 16, the number of bands in the patterns of group II isolates ranged between 4 and 7. Group II isolates shared one monomorphic band that was absent from the group I patterns. The difference in the patterns is demonstrated in a comparison of group I, group II, and group III patterns in Fig. 6.
FIG. 5.

Dendrogram generated from SABs computed for pair-wise comparisons of 87 unrelated isolates collected worldwide and fingerprinted with Cp3-13. SABs were calculated utilizing band position alone (see Materials and Methods). Prominent clusters are indicated with vertical bars to the right of the dendrogram. Isolates clustered in the p1 and p2 groups are from the same patient. Isolate origins are presented in Table 1.
FIG. 6.

Representative Cp3-13 Southern blot hybridization patterns of isolates that fell outside the major group I cluster formed by Cp3-13 fingerprinting. Representative patterns for group I (J940043), group II (Pf6, Pf9, p203, and Y17456) and group III (J920296) isolates are presented.
In order to identify highly related isolates, we selected an arbitrary SAB threshold of 0.90. That threshold was used to identify groups of highly related isolates in C. albicans using a comparable complex probe, Ca3. For comparable sets of C. albicans isolates DNA fingerprinted with Ca3, the average SAB ranged between 0.65 and 0.69 (21, 26, 35). Since the average SAB of the C. parapsilosis collection fingerprinted with Cp3-13 was 0.45 ± 0.22, the use of 0.90 as a threshold for high relatedness was even more stringent when applied to C. parapsilosis. At this threshold, 10 clusters of highly related isolates formed (Fig. 5). Two of the clusters, labeled p1 and p2 (Fig. 5), included five isolates each of the 10 isolates collected from a single episode of oral candidiasis (Table 1). The clusters connected in the dendrogram at a node of 0.18, demonstrating that they represented two coinfecting, unrelated strains of C. parapsilosis. A third cluster included six isolates collected exclusively from the Detroit, Mich., area (1480.24, 1480.30, 1480.23, 1480.21, 1480.22, and 1480.25). Five of these six isolates were collected from patients or health care workers in the same hospital, suggesting a nosocomial outbreak of a single endemic strain. Two additional Detroit isolates (1480.27 and 1480.28) clustered with these six isolates when the SAB threshold was reduced to 0.88 (Fig. 5). Five clusters included two isolates each, from the following locations (Fig. 5): Belgium (J940723) and The Netherlands (J941818); Greece (J941335) and Belgium (J941340P); Richmond, Va. (Pf12 and Pf13); Miami, Fla. (Pf10), and Genoa, Italy (Pf21); and (1480.29 and Y-12969) (these isolates proved to be American Type Culture Collection (ATCC) strain 22019 obtained independently from two laboratories (Table 1). Finally, there was one cluster of four isolates, two from Detroit (1480.28 and 1480.27) and two from Belgium (J941253 and J941226), that coclustered with six Detroit isolates (1480.21, 1480.22, 1480.23, 1480.24, 1480.25, and 1480.30) at a reduced SAB threshold of 0.88 (Fig. 5). These results provide examples of geographical specificity as well as geographical mixing.
Verification of Cp3-13 clustering by comparison with RAPD analysis.
The most direct way of verifying the efficacy of a fingerprinting probe is to compare its clustering capabilities with those of an unrelated fingerprinting method (10, 11, 19, 26, 35). To verify the efficacy of Cp3-13, we compared its clustering capacity with that of the RAPD method for 21 isolates of C. parapsilosis that included representatives of groups Ia, Ib, II, and III. To obtain the necessary level of discrimination by the RAPD method, 42 primers were tested. Thirty-one primers identified differences among group II isolates. Six of these also identified differences among group I isolates and were used for further analysis. Examples of RAPD analyses performed on group I and group II isolates are presented in Fig. 7 for primers OPA-17, OPE-2, and OPE-13. Because OPA-17 and OPE-2 identified variability among group II but not group I isolates (Fig. 7), they were excluded. Because OPE-13 identified variability among isolates of both groups (Fig. 7), it was included. Amplification with each of the six selected primers was repeated twice to assure reproducibility, and only the major bands were used in the analysis. Band data were entered directly into band data files in the DENDRON program for subsequent calculations of SABs. A total of 68 band positions were scored, of which 24 were present in group I isolates and 41 were present in group II isolates. Of the 24 in group I, 10 were polymorphic (42%), and of the 41 in group II, 25 were polymorphic (61%). Bands were entered into the DENDRON band data file manually, SABs were computed based on band positions alone to generate a matrix of SAB values, and a dendrogram was generated from the matrix in the same manner that a dendrogram was generated with Cp3-13 hybridization patterns (26). The dendrograms generated by the RAPD and Cp3-13 methods are shown in Fig. 8A and B, respectively. The structures of the dendrograms were remarkably similar. Both methods separated the 21 isolates into the same three groups as follows: 16 group I isolates, 5 group II isolates, and 1 group III isolate. Both methods separated group I isolates further into two subgroups, Ia and Ib. All of the isolates which were clustered into group Ib by Cp3-13 fingerprinting were also clustered into that group by RAPD fingerprinting. However, two additional isolates placed in group Ib by Cp3-13 fingerprinting were placed in group Ia by RAPD fingerprinting. These results demonstrate a reasonably high degree of parity in the clustering capabilities of Cp3-13 and RAPD fingerprinting and serve to cross-verify the two methods.
FIG. 7.

Three examples of RAPD patterns produced for 10 strains amplified with the primers OPA-17, OPE-2, or OPE-13. Although OPA-17 and OPE-2 discriminated between group II isolates, they did not discriminate between group I isolates and were therefore eliminated. OPE-13 was included in the RAPD primers used since it discriminated between strains in groups I and II.
FIG. 8.

A comparison of the dendrograms generated by the RAPD method (A) and Cp3-13 fingerprinting (B) for 21 independent C. parapsilosis isolates. Asterisks represent strains clustered into group Ib by the RAPD method that were clustered into group Ia by Cp3-13.
Group I isolates exhibit reduced genetic diversity.
The preceding comparison of genetic relatedness both by Cp3-13 fingerprinting and by RAPD fingerprinting revealed that group I isolates were genetically less diverse than group II isolates. This difference was evident in the average SAB values as well as the proportion of polymorphic bands in the RAPD analysis (42% for group I isolates and 61% for group II isolates). A comparison of the data obtained with RAPD analysis of group I isolates of C. parapsilosis and those obtained with RAPD analysis of other Candida species suggests that group I isolates also exhibit less genetic diversity than other species. To test this suggestion, we compared a “polymorphic index,” computed as the number of polymorphic bands divided by the number of primers screened, amongst a number of species in which the same array of primers was tested. In this case, relative genetic diversity is proportional to the ratio. The ratios were 0.25 for C. parapsilosis group I isolates, 0.37 for C. dubliniensis group I isolates (10), 0.46 for C. glabrata isolates (19), 0.53 for C. albicans isolates (26), and 0.64 for C. tropicalis isolates (11). These results support earlier observations using RAPD analysis (20), MLEE analysis (16), electrophoretic karyotyping (20), and mitochondrial DNA analysis (20) that group I isolates exhibited decreased genetic variability.
Further verification of grouping by ITS sequencing.
Lin et al. (16) distinguished three distinct groups of C. parapsilosis, not only by MLEE, but also by sequencing the 5.8S rDNA and adjacent ITS regions. To further verify that the three groups identified by Cp3-13 represented the three groups distinguished in this previous study, we cloned and sequenced the rDNA regions that included ITS1 and ITS2 for three group I isolates (J940889, 1480.21, and J940723), three group II isolates (Pf9, p203, and Y-17456), and the one group III isolate (J920296). These sequences were compared to those previously analyzed (ITS-GI, ITS-GII, and ITS-GIII) (16). The tree generated with the combined data is presented in Fig. 9. Our group I, group II, and group III isolates clustered with the group I, group II, and group III isolates previously identified by Lin et al. (16) (Fig. 9). In addition, J940723, a member of the subgroup Ib, was least related to the other group I isolates, J940889 and 1480.21, which were members of subgroup Ia. These results further validate Cp3-13 groupings.
FIG. 9.

Dendrogram based on ITS sequences. Representative isolates of C. parapsilosis groups were used for ITS sequencing and compared to published sequences ITS-GI, ITS-GII, and ITS-GIII of groups I, II and III, respectively (16). The tree was generated using pair-wise comparisons of ITS sequences of ∼490 bp. Bootstrap values collected from 1,000 replicates are given at the respective nodes.
In vitro and in vivo stability of Cp3-13 fingerprint patterns.
For a DNA fingerprinting method to be effective, the DNA fingerprint must be relatively stable over time (35). On the other hand, one attribute of complex probes is that they sometimes function as indicators of microevolution through hybridization with hypervariable sequences like the RPS elements in C. albicans (25). To test both pattern stability and the capacity to identify microevolutionary changes in C. parapsilosis in vitro, clones of four isolates, Pf16, 1480.23, J940043, and J941253, were grown for 200 generations. At the end of this growth regimen, cells were plated at low density and nine single colonies of each isolate were DNA fingerprinted with Cp3-13. The fingerprints were compared with those of the original isolates. In every case, the general Cp3-13 pattern was maintained over 200 generations (Fig. 10). However, in the case of two of the four isolates, Pf16 (Fig. 10B) and 1490.23, microevolution was evident after 200 generations. No changes were evident in the pattern of isolate J940043 (Fig. 10A) or J941253.
FIG. 10.

In vitro analysis of the stability of the pattern generated by Cp3-13 and the capacity of Cp3-13 to discriminate microevolutionary changes. Southern blots of EcoRI/SalI-digested DNA of C. parapsilosis J940043 (A) and Pf16 (B) at 0 generations (original) and after 200 generations. Ten individual clones were selected randomly for analysis at 200 generations for J940043, and nine individual clones were selected randomly for analysis at 200 generations for Pf16. Variant patterns are numbered at the bottom of each blot (variations in patterns are demarcated by asterisks).
To investigate both stability and microevolution in vivo, C. parapsilosis isolates were obtained from the oral cavity of a human immunodeficiency virus-positive individual presenting with his first case of oral thrush. Thirteen C. parapsilosis clones were picked from a single primary culture and DNA fingerprinted with Cp3-13. Two unrelated strains were evident in the 13 isolates. The first was a group I strain and included isolates p190, p191, p192, p193, and p196. These isolates formed cluster p1 in the general dendrogram in Fig. 5. These isolates exhibited microevolutionary changes, separating them into two subgroups with an SAB node of 0.97. The second was a group II strain and included p201, p203, p205, p206, and p218. These isolates exhibited no microevolution identified by Cp3-13 (Fig. 5). These results suggest that the Cp3-13 pattern is relatively stable in vivo and that Cp3-13 can identify microevolutionary changes occurring in infecting populations.
Phenotypic variation and Cp3-13 pattern stability.
When stored cultures of clones of C. parapsilosis isolates were plated, we frequently observed variant colony morphologies at relatively high frequencies (10−2 to 10−4). Switching was first reported in C. parapsilosis by Lott et al. (20). The differences in colony morphologies were associated with differences in the proportions of budding cells and pseudohyphae in the colony domes (Fig. 11A). To test whether variant phenotypes were associated with changes in the Cp3-13 fingerprint, a stored culture of C. parapsilosis isolate J941226, a member of group I (Fig. 5), was plated at low density, and representatives of the five colony phenotypes, “smooth,” “rough,” “snowball,” “concentric,” and “crepe” (Fig. 11A), were picked and DNA fingerprinted with Cp3-13. The five phenotypes in the switching repertoire exhibited identical Cp3-13 hybridization patterns (Fig. 11B).
FIG. 11.

Phenotypic switching in C. parapsilosis is not associated with changes in Cp3-13 pattern. (A) Colony and cellular phenotypes of strain J941226. (B) Cp3-13 fingerprinting patterns of the five phenotypes.
DISCUSSION
C. parapsilosis represents the second most prevalent Candida species causing bloodstream infections (24). In order to understand its distribution, incidence, and modes of transmission, we must have as a tool an effective genetic fingerprinting system. In the past, C. parapsilosis has been fingerprinted by electrophoretic karyotyping (27, 38), MLEE (16), restriction enzyme analysis of genomic DNA (27, 38), RAPD analysis (20), and sequencing of ITS sequences flanking the 5.8S rDNA gene (16). For reasons articulated in a recent review on DNA fingerprinting (35), Southern blot hybridization with complex DNA fingerprinting probes represents one of the most effective methods for preparing large-scale epidemiological studies. Because the DNA fingerprint patterns they produce are amenable to computer-assisted analysis and storage, they can also provide databases that can be used effectively in retrospective analyses and for comparison of data collected from different laboratories (34). For this reason, we set out to develop a complex probe for C. parapsilosis.
Cloning Cp3-13.
Using a procedure effectively used in the past to clone complex probes for a variety of fungal species (9–11, 19, 28, 31), we cloned a 15-kb fragment, Cp3-13, that generated a complex Southern blot hybridization pattern when used to probe EcoRI/SalI-digested genomic DNA. The number of total bands, variable bands, and monomorphic bands in the pattern generated by Cp3-13 was similar to that of the patterns generated by the best-characterized complex probe, Ca3, with EcoRI-digested C. albicans DNA (32). Cp3-13 hybridized intensely with three chromosome-sized bands in a CHEF gel and less intensely with two additional chromosomes, suggesting it is represented at different copy numbers on 5 of the 10 chromosomes separated by CHEF. Interestingly, this distribution held true for four different, presumably unrelated test isolates of C. parapsilosis.
Verification of the discriminatory ability of Cp3-13 fingerprinting.
The discriminatory capacity of Cp3-13 was quite good. Cp3-13 was able to identify identical strains collected in different samples, discriminate between presumably unrelated isolates, and identify microevolution. To verify these levels of discrimination, we compared it to the RAPD method of DNA fingerprinting by a cluster analysis. Both methods separated a test collection of 21 isolates into three major groups with identical members. The two methods further separated the group I isolates into subgroups Ia and Ib with similar members. Of the 16 group I members, only 2 (13%) were not separated by the two methods into the same subgroups. These results demonstrate a relatively good level of parity between the discriminatory capacity of the two methods. We further verified the discriminatory power of Cp3-13 by ITS sequencing. ITS sequencing separated test isolates into the same three groups, and furthermore, cogrouped the test isolates with group I, group II, and group III isolates previously discriminated by Lin et al. (16) using ITS sequencing. These results further validate the discriminatory capacity of Cp3-13 as well as reinforce the separation of C. parapsilosis strains into three definable groups.
Cp3-13 identifies microevolution.
Perhaps one of the most important but neglected topics in medical mycology is microevolution in commensal and pathogenic populations (35). Pathogenic microorganisms, especially those that are also commensals, must rapidly adapt to changes in host physiology, the host immune response, drug therapy, and environmental changes such as anatomical location. Several methods have been demonstrated to measure microevolution within infecting populations (7, 8, 10, 17, 18, 26, 30, 36, 37, 41). In a comparison between MLEE, RAPD analysis, and fingerprinting with the complex probe Ca3, the last method proved the most sensitive for discriminating microevolution (26). It was therefore suggested that one desirable feature of a complex probe is the capability of identifying microevolution (35). The results presented have demonstrated that Cp3-13 not only discriminates deep-rooted differences between strains in groups and subgroups but also can identify changes in a clonal population within a few hundred generations in vitro and changes that have occurred in vivo in an infecting population. Since one can obtain rates of spontaneous change in vitro (25), Cp3-13 can potentially be used to estimate the length of time a particular strain has infected a host, although our limited data suggest that the rate of high-frequency reorganization may be strain dependent.
The regulation of Cp3-13 is not the basis of switching in C. parapsilosis.
Lott et al. (20) first reported high-frequency switching in C. parapsilosis. To test whether the reorganization of sequences homologous to Cp3-13 was the basis of variant phenotypes in stored C. parapsilosis cultures, we compared the Cp3-13 Southern blot hybridization patterns amongst five switch phenotypes. No differences in pattern were observed. A similar lack of involvement has been demonstrated for Ca3 reorganization in C. albicans switching (34).
Summary.
We have developed an effective complex DNA fingerprinting probe for C. parapsilosis that will (i) identify the same strain in independent isolates, (ii) discriminate unrelated isolates, (iii) separate isolates into valid groups in a dendrogram, (iv) identify microevolution in infecting populations, and (v) be amenable to automatic computer-assisted DNA fingerprint analysis. This probe is now available to researchers for use in epidemiological studies of C. parapsilosis.
ACKNOWLEDGMENT
This research was funded by Public Health Service grants AI39735 and DEI0758 to D.R.S. from the National Institutes of Health.
REFERENCES
- 1.Anderson J, Srikantha T, Morrow B, Miyasaki S H, White T C, Agabian N, Schmid J, Soll D R. Characterization and partial sequence of the fingerprinting probe Ca3 of Candida albicans. J Clin Microbiol. 1993;31:1472–1480. doi: 10.1128/jcm.31.6.1472-1480.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Branchini M L, Pfaller M A, Rhine-Chalberg J, Frempong T, Isenberg H D. Genotypic variation and slime production among blood and catheter isolates of Candida parapsilosis. J Clin Microbiol. 1994;32:452–456. doi: 10.1128/jcm.32.2.452-456.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Church G M, Gilbert W. Genomic sequencing. Proc Natl Acad Sci USA. 1984;81:1991–1995. doi: 10.1073/pnas.81.7.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Critchley I A, Douglas L J. Differential adhesion of pathogenic Candida species to epithelial and inert surfaces. FEMS Microbiol Lett. 1985;28:199–203. [Google Scholar]
- 5.Dick J D, Merz W G, Saral R. Incidence of polyene-resistant yeasts recovered from clinical specimens. Antimicrob Agents Chemother. 1980;18:158–163. doi: 10.1128/aac.18.1.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fernando P H, Samaranayake L P. Chromosome length polymorphism in clinical isolates of Candida parapsilosis. APMIS. 1998;106:941–946. [PubMed] [Google Scholar]
- 7.Franzot S P, Mukherjee J, Cherniak R, Chen L C, Hamdan J S, Casadevall A. Microevolution of a standard strain of Cryptococcus neoformans resulting in differences in virulence and other phenotypes. Infect Immun. 1998;66:89–97. doi: 10.1128/iai.66.1.89-97.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fries B C, Chen F, Currie B P, Casadevall A. Karyotype instability in Cryptococcus neoformans infection. J Clin Microbiol. 1996;34:1531–1534. doi: 10.1128/jcm.34.6.1531-1534.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Girardin H, Latgé J-P, Srikantha T, Morrow B, Soll D R. Development of DNA probes for fingerprinting Aspergillus fumigatus. J Clin Microbiol. 1993;31:1547–1554. doi: 10.1128/jcm.31.6.1547-1554.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Joly S, Pujol C, Rysz M, Vargas K, Soll D R. Development and characterization of complex DNA fingerprinting probes for the infectious yeast Candida dubliniensis. J Clin Microbiol. 1999;37:1035–1044. doi: 10.1128/jcm.37.4.1035-1044.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Joly S, Pujol C, Schröppel K, Soll D R. Development of two species-specific fingerprinting probes for broad computer-assisted epidemiological studies of Candida tropicalis. J Clin Microbiol. 1996;34:3063–3071. doi: 10.1128/jcm.34.12.3063-3071.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kleinegger C, Lockhart S R, Vargas K, Soll D R. Frequency, intensity, species, and strains of oral yeast vary as a function of host age. J Clin Microbiol. 1996;34:2246–2254. doi: 10.1128/jcm.34.9.2246-2254.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kurtzman C P, Robnett C J. Identification of clinically important ascomycetous yeasts based on nucleotide divergence in the 5′ end of the large-subunit (26S) ribosomal DNA gene. J Clin Microbiol. 1997;35:1216–1223. doi: 10.1128/jcm.35.5.1216-1223.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Levin A S, Costa S F, Mussi N S, Basso M, Sinto S L, Machado C, Geiger D C, Villares M C, Schreiber A Z, Barone A A, Branchini M L. Candida parapsilosis fungemia associated with implantable and semi-implantable central venous catheters and the hands of healthcare workers. Diagn Microbiol Infect Dis. 1998;30:243–249. doi: 10.1016/s0732-8893(98)00006-6. [DOI] [PubMed] [Google Scholar]
- 15.Levy I, Rubin L G, Vasishtha S, Tucci V, Sood S K. Emergence of Candida parapsilosis as the predominant species causing candidemia in children. Clin Infect Dis. 1998;26:1086–1088. doi: 10.1086/520277. [DOI] [PubMed] [Google Scholar]
- 16.Lin D, Wu L-C, Rinaldi M G, Lehmann P F. Three distinct genotypes within C. parapsilosis from clinical sources. J Med Vet Mycol. 1995;33:1815–1821. doi: 10.1128/jcm.33.7.1815-1821.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lockhart S, Fritch J J, Meier A S, Schröppel K, Srikantha T, Galask R, Soll D R. Colonizing populations of Candida albicans are clonal in origin but undergo microevolution through C1 fragment reorganization as demonstrated by DNA fingerprinting and C1 sequencing. J Clin Microbiol. 1995;33:1501–1509. doi: 10.1128/jcm.33.6.1501-1509.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lockhart S R, Reed B D, Pierson C L, Soll D R. Most frequent scenario for recurrent Candida vaginitis is strain maintenance with “substrain shuffling”: demonstration by sequential DNA fingerprinting with probes Ca3, C1, and CARE2. J Clin Microbiol. 1996;34:767–777. doi: 10.1128/jcm.34.4.767-777.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lockhart S R, Joly S, Pujol C, Sobel J D, Pfaller M A, Soll D R. Development and verification of fingerprinting probes for Candida glabrata. Microbiology. 1997;143:3733–3746. doi: 10.1099/00221287-143-12-3733. [DOI] [PubMed] [Google Scholar]
- 20.Lott T J, Keykendall R J, Welbel S F, Pramanik A, Lasker B A. Genomic heterogeneity in the yeast Candida parapsilosis. Curr Genet. 1993;23:463–467. doi: 10.1007/BF00312635. [DOI] [PubMed] [Google Scholar]
- 21.Marco F, Lockhart S, Pfaller M, Pujol C, Rangel-Frausto M S, Wiblin T, Blumberg H M, Edwards J E, Jarvis W, Saiman L, Patterson J E, Rinaldi M G, Wenzel R P, Soll D R the NEMIS Study Group. Elucidating the origins of nosocomial infections with Candida albicans by DNA fingerprinting with the complex probe Ca3. J Clin Microbiol. 1999;37:2817–2828. doi: 10.1128/jcm.37.9.2817-2828.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marrie T J, Cooper J H, Costerton J W. Ultrastructure of Candida parapsilosis endocarditis. Infect Immun. 1984;45:390–398. doi: 10.1128/iai.45.2.390-398.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pfaller M A. Epidemiology of candidiasis. J Hosp Infect. 1995;30:329–338. doi: 10.1016/0195-6701(95)90036-5. [DOI] [PubMed] [Google Scholar]
- 24.Pfaller M A, Messer S A, Houston A, Rangel-Frausto M S, Wiblin T, Blumberg H M, Edwards J E, Jarvis W, Martin M A, Neu H C, Saiman L, Patterson J E, Dibb J C, Roldan C M, Rinaldi M G, Wenzel R P. National Epidemiology of Mycoses Survey: a multicenter study of strain variation and antifungal susceptibility among isolates of Candida species. Diagn Microbiol Infect Dis. 1998;31:289–296. doi: 10.1016/s0732-8893(97)00245-9. [DOI] [PubMed] [Google Scholar]
- 25.Pujol C, Joly S, Nolan B, Srikantha T, Soll D R. Microevolutionary changes in Candida albicans identified by the complex Ca3 probe involve insertions and deletions of RPS units at specific genomic sites. Microbiology. 1999;145:2635–2646. doi: 10.1099/00221287-145-10-2635. [DOI] [PubMed] [Google Scholar]
- 26.Pujol C, Joly S, Lockhart S, Noel S, Tibayrenc M, Soll D R. Parity among the randomly amplified polymorphic DNA method, multilocus enzyme electrophoresis, and Southern blot hybridization with the moderately repetitive DNA probe Ca3 for fingerprinting Candida albicans. J Clin Microbiol. 1997;35:2348–2358. doi: 10.1128/jcm.35.9.2348-2358.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Riederer K, Fozo P, Khatib R. Typing of Candida albicans and Candida parapsilosis: species-related limitations of electrophoretic karyotyping and restriction endonuclease analysis of genomic DNA. Mycoses. 1998;41:397–402. doi: 10.1111/j.1439-0507.1998.tb00360.x. [DOI] [PubMed] [Google Scholar]
- 28.Sadhu C, McEachern M J, Rustchenko-Bulgac E P, Schmid J, Soll D R, Hicks J. Telomeric and dispersed repeat sequences in Candida yeasts and their use in strain identification. J Bacteriol. 1991;173:842–850. doi: 10.1128/jb.173.2.842-850.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sambrook J, Fritsch E F, Maniatis T. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 30.Scherer S, Stevens D A. Application of DNA typing methods to epidemiology and taxonomy of Candida species. J Clin Microbiol. 1987;25:675–679. doi: 10.1128/jcm.25.4.675-679.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Scherer S, Stevens D A. A Candida albicans dispersed, repeated gene family and its epidemiological applications. Proc Natl Acad Sci USA. 1988;85:1452–1456. doi: 10.1073/pnas.85.5.1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schmid J, Voss E, Soll D R. Computer-assisted methods for assessing Candida albicans strain relatedness by Southern blot hybridization with repetitive sequence Ca3. J Clin Microbiol. 1990;28:1236–1243. doi: 10.1128/jcm.28.6.1236-1243.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sneath P H A, Sokal R R. Numerical taxonomy. W. H. San Francisco, Calif: Freeman and Co.; 1973. [Google Scholar]
- 34.Soll D R. High-frequency switching in Candida albicans. Clin Microbiol Rev. 1992;5:183–203. doi: 10.1128/cmr.5.2.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Soll D R. The ins and outs of DNA fingerprinting the infectious fungi. Clin Microbiol Rev. 2000;13:332–370. doi: 10.1128/cmr.13.2.332-370.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Soll D R, Galask R, Schmid J, Hanna C, Mac K, Morrow B. Genetic dissimilarity of commensal strains carried in different anatomical locations of the same healthy women. J Clin Microbiol. 1991;29:1702–1710. doi: 10.1128/jcm.29.8.1702-1710.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sullivan D, Haynes K, Morgan G, Shanley D, Coleman D. Persistence, replacement, and microevolution of Cryptococcus neoformans strains in recurrent meningitis in AIDS patients. J Clin Microbiol. 1996;34:1739–1744. doi: 10.1128/jcm.34.7.1739-1744.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Waggoner-Fountain L A, Walker M W, Hollis R J, Pfaller M A, Ferguson J E, Wenzel R P, Donowitz L G. Vertical and horizontal transmission of unique Candida species to premature newborns. Clin Infect Dis. 1996;22:803–808. doi: 10.1093/clinids/22.5.803. [DOI] [PubMed] [Google Scholar]
- 39.Weems J J., Jr Candida parapsilosis: epidemiology, pathogenicity, clinical manifestations, and antimicrobial susceptibility. Clin Infect Dis. 1992;14:756–766. doi: 10.1093/clinids/14.3.756. [DOI] [PubMed] [Google Scholar]
- 40.Weems J J, Jr, Chamberland M E, Ward J, Willy M, Padhye A A, Solomon S L. Candida parapsilosis fungemia associated with parenteral nutrition and contaminated blood pressure transducers. J Clin Microbiol. 1987;25:1029–1032. doi: 10.1128/jcm.25.6.1029-1032.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.White T C, Pfaller M A, Rinaldi M G, Smith J, Redding S W. Stable azole drug resistance associated with a substrain of Candida albicans from an HIV-infected patient. Oral Dis. 1997;3:S102–S109. doi: 10.1111/j.1601-0825.1997.tb00336.x. [DOI] [PubMed] [Google Scholar]