

Abstract
The gut microbiome plays a critical role in various inflammatory conditions, and its modulation is a potential treatment option for these conditions. The role of the gut microbiome in the pathogenesis of thromboembolism has not been fully elucidated. In this review, we summarize the evidence linking the gut microbiome to the pathogenesis of arterial and venous thrombosis. In a human host, potentially pathogenic bacteria are normal residents of the human gut microbiome, but significantly outnumbered by commensal anaerobic bacteria. Several disease states with an increased risk of venous thromboembolism (VTE) are associated with an imbalance in the gut microbiome characterized by a decrease in commensal anaerobic bacteria and an increase in the abundance of pathogenic bacteria of which the most common is the gram-negative Enterobacteriaceae (ENTERO) family. Bacterial lipopolysaccharides (LPS), the glycolipids found on the outer membrane of gram-negative bacteria, is one of the links between the microbiome and hypercoagulability. LPS binds to toll-like receptors to activate endothelial cells and platelets, leading to activation of the coagulation cascade. Bacteria in the microbiome can also metabolite compounds in the diet to produce important metabolites like trimethylamine-N-oxide (TMAO). TMAO causes platelet hyperreactivity, promotes thrombus formation and is associated with cardiovascular disease. Modulating the gut microbiome to target LPS and TMAO levels may be an innovative approach for decreasing the risk of thrombosis.
Keywords: Thrombosis, Microbiota, Lipopolysaccharides, Dysbiosis
Introduction
Venous and arterial thromboembolic events are rapidly growing clinical problems. The rates of venous thromboembolism (VTE) in pediatric patients have increased by almost 70% based on a retrospective cohort from 2001 to 2007[1, 2]. The incidence of VTE continues to rise rapidly in adulthood after the fourth decade of life to 5–6 per 1000 people annually [3]. The morbidity associated with VTE – such as bleeding events from anticoagulation, recurrence, lack of thrombus resolution and post-thrombotic syndrome (PTS) -- is substantial. Similarly, cardiovascular disease (CVD) leading to arterial thromboembolic events is an emerging problem in both children and adults[4]. The pathogenesis of thrombosis is complex and occurs from the additive effects of genetic and environmental risk factors[5]. Hence, there is a strong need to identify modifiable risk factors to prevent thrombotic events and by extension, adverse long-term outcomes.
The role of the gut microbiome in various aspects of human health (ranging from obesity and CVD to autoimmune phenomena) is becoming increasingly recognized as having a significant impact on host physiology [6–10]. The gut microbiome is a modifiable host factor for several chronic diseases and is the focus of novel therapies such as fecal microbial transplant (FMT), probiotics, dietary changes, selective antibiotic use, and targeted inhibition of microbial enzymes [6, 11–15]. Perturbations of the gut microbiome from various environmental or genetic factors can lead to activation of inflammatory pathways in vascular endothelial cells, platelets, and innate immune cells resulting in release of various coagulation proteins leading to a prothrombotic state [16–21]. This review discusses current data on the association of gut microbiome perturbations and thrombosis and its potential as a preventive treatment option for VTE.
Overview of the gut microbiome
The human body is colonized by trillions of resident microorganisms that inhabit numerous host body niches with the highest density residing in the gastrointestinal tract[22]. The gut microbiota refers to all the microorganisms in the intestinal tract, and the microbiome is a collective term that includes all the genes of the microbiota and their by-products. Colonization of the gastrointestinal tract begins at birth and rapidly gains diversity during the first few years of life as diet, genetics, and environmental insults, such as antibiotics, come into play[23–25]. The microbiome composition can vary significantly from person to person and within the same person as they age, but in general, family members tend to have similar gut microbiome signatures, which is likely a combination of both genetic and environmental (e.g., diet) factors[6]. The microbiome maintains a complex balance with environmental and host genetic factors, the disruption of which leads to the development of various disease states[6, 7]. The gut epithelial barrier typically restricts the microbiomes to the intestinal lumen, but its function can be impaired by various factors, including inflammation, nutrition, and antibiotic use[26]. This “leaky” epithelial barrier allows for the translocation of gut microbial products and metabolites into the portal and then systemic circulation leading to multiple pathologies including potentially thrombosis [26].
Microbiome composition
Microbial composition and density vary along the gastrointestinal tract with a limited number and lower burden of bacterial species in the small intestine (< 104 cells per gram feces) as compared to the colon with ~ 1011 to 1012 cells per gram feces [6, 27, 28]. Usually, the gut microbiota is diverse with an assortment of gram-positive, gram-negative, obligate and facultative anaerobic bacteria [25]. In a healthy human adult, the gut microbiota is comprised of an abundance (>95%) of commensal obligate anaerobic bacteria, of which the majority is the phyla Firmicutes and Bacteroidetes[25]. Potentially pathogenic bacteria also referred to as pathobionts (e.g., facultative gram-negative bacteria, such as Escherichia coli), are normal residents of the human gut microbiome, but are greatly outnumbered (<1% relative abundance) by commensal anaerobic bacteria[29]. Dysbiosis is defined as a microbial imbalance that typically manifests as a decrease in microbial diversity: for example, a decrease in commensal anaerobic gut bacteria and an overgrowth of pathobionts such as the bacterial family Enterobacteriaceae (ENTERO)[30, 31]. ENTERO are gram-negative facultative anaerobes able to thrive with or without oxygen and are recognized as harmful, pathogenic, and proinflammatory bacteria[32]. The microbiome promotes immune system development. On the contrary, dysbiosis and ENTERO expansion is associated with obesity, diabetes, inflammatory diseases, and other chronic disease states [6].
Metabolite production
Another mechanism by which the gut microbiome induces host response is via gut microbiota derived metabolites. Western diets are rich in trimethylamine (TMA) containing nutrients such as choline, carnitine, and phosphatidylcholine. The gut microbiota metabolizes these nutrients from the host’s diet into metabolites such as trimethylamine-N-oxide (TMAO)[33]. Western diets promote enrichment of a gut microbiome that is associated with increased TMAO production (figure 1a)[19]. Higher plasma TMAO levels are associated with adverse outcomes of CVD and thrombotic events and TMAO is currently being studied as a therapeutic option for CVD prevention[19, 34, 35].
Figure 1:
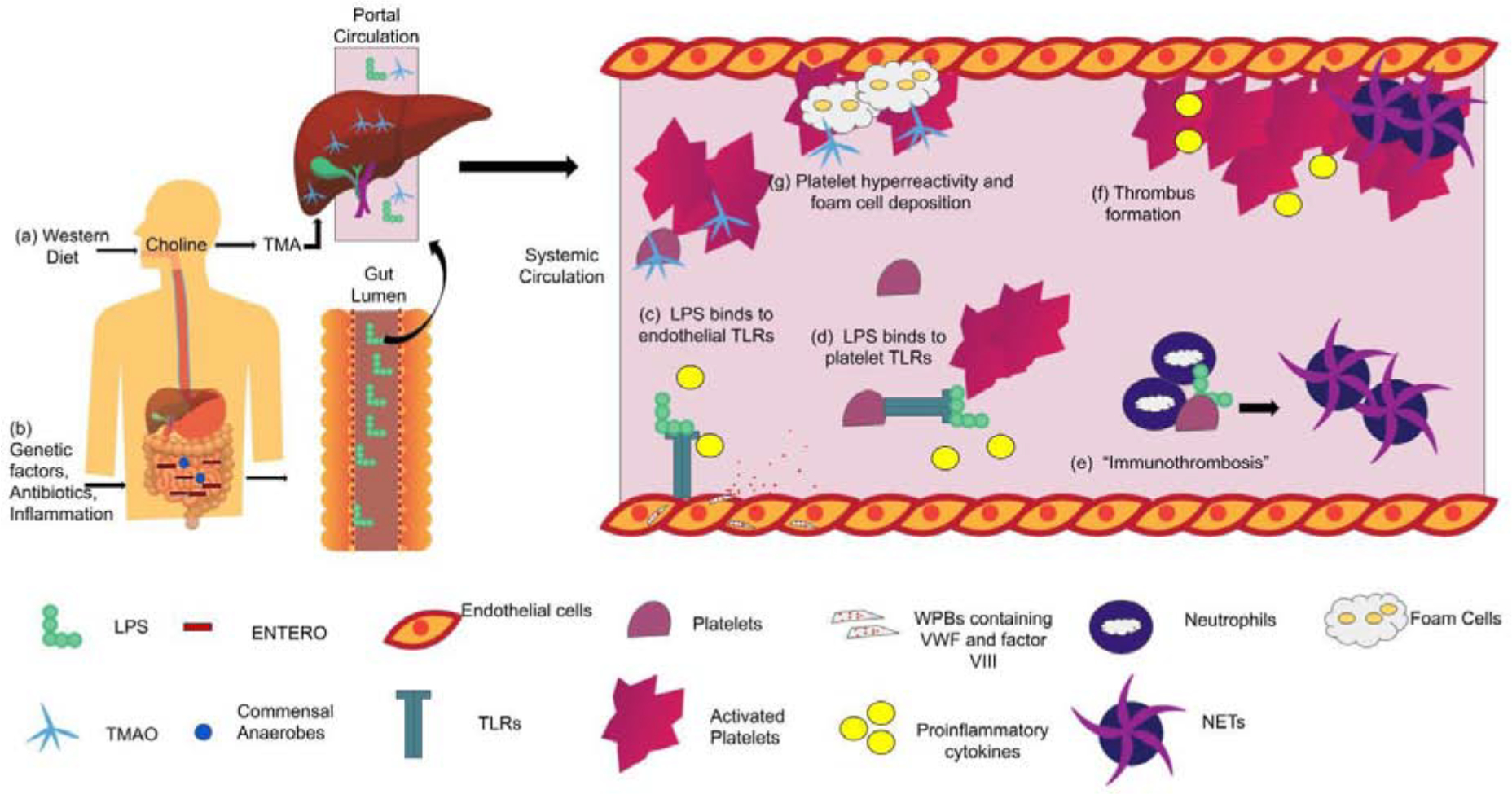
Proposed mechanisms of how the microbiome induces thrombosis
(a) Western diet are choline-rich which is metabolized to TMA by TMA lyases in the gut. TMA enter portal circulation in the liver where it is converted to TMAO and released into circulation.
(b) Presence of genetic and environmental factors lead to dysbiosis with expansion of ENTERO and decrease in microbial diversity. Increase in ENTERO leads to increase in LPS production in the gut lumen. LPS translocates through an impaired gut epithelial barrier into portal and then systemic circulation.
(c) LPS leads to activation of TLR4 on endothelial cells which leads to release of proinflammatory cytokines and exocytosis of Weibel palade bodies to release factor VIII and vWF.
(d) LPS binds to platelet TLRs leading to activation of platelets, release of alpha and dense granules and release of cytokines.
(e) TLRs also mediate platelet-neutrophil interactions and “immunothrombosis” by binding to neutrophils to increase formation of NETs, which can activate the contact factor pathway and contribute to aggregation.
(f) Platelet hyperreactivity, release of coagulation factors, NET formation and proinflammatory cytokines lead to thrombus formation.
(g) Upon entering systemic circulation, TMAO leads to platelet hyperresponsiveness and increases deposition of foam cells in the early steps in atherosclerosis.
Profiling the microbiome
In the past, culture-based approaches have not accurately characterized the composition of the gut microbiome, given that many obligate anaerobic species are difficult or impossible to culture[36]. Quantitative polymerase chain reaction (qPCR) allows for determining absolute levels of specific species or groups of gut microbiota but cannot provide community microbiome taxonomic composition data (Table 1). With the advent of next-generation sequencing approaches, investigators focused on the bacterial 16S ribosomal RNA (16S rRNA) gene, which contains genetic sequences common to all bacteria but also variable regions that are unique for specific groups or species of bacteria[37]. Hence, by taking genomic DNA recovered from a complex bacterial community (e.g. human fecal sample), the variable regions of all the bacterial 16S rRNA genes could be simultaneously amplified, sequenced, and then analyzed (by interrogation of a 16S rRNA sequence database) to produce a readout of the relative abundances of various gut microbiota in that community[25, 36, 38]. More recently, metagenomic shotgun sequencing (MSS), which directly sequences all the genomic DNA and forgoes amplicon libraries, has led to higher taxonomic resolution and an unbiased approach to profiling the gut microbiome[36]. Thus, there is great interest in identifying a distinct microbiome signature associated with CVD or increased TMAO production. For example, 16s RNA sequencing of atherosclerotic plaques shows an increased density of certain bacterial species also present in the oral cavity and gut of these patients and correlated with higher levels of cholesterol[39].
Table 1:
Comparison of various gut microbiome profiling approaches
| Technique | Population | Method | Pros | Cons |
|---|---|---|---|---|
| Quantitative polymerase chain reaction (qPCR) | Bacteria, viruses, fungi | Group specific primers are used to clone segment of 16S rRNA gene | Low cost and labor | Offers no metagenomic insight |
| Only method that determines absolute concentration | Can only detect one bacterial group per experiment (i.e. 16S rRNA for E. coli to determine ENTERO) | |||
| Can be performed within a day | ||||
| 16S ribosomal RNA gene sequencing (16S) | Bacteria only | Amplifies the V1-V9 region of 16S rRNA gene and clusters sequences into operational taxonomic units (OTUs) | More affordable | Can have taxonomic bias (primer design, amplification conditions) |
| Less complex informatics than MSS | ||||
| OTUs are references against database of 16S rRNA sequences to determine taxa | Determines relative abundance | |||
| Can simultaneously detect thousands of different taxa | ||||
| Metagenomic Shotgun Sequencing (MSS) | Bacteria, viruses, fungi | Unrestricted sequencing genome of all microorganisms in a sample | Can detect low abundance organisms | High cost |
| Can pair with metabolomic profiling to detect metabolites such as TMAO | Complex bioinformatics analysis | |||
| Uses marker genes for taxonomic identification | Can simultaneously detect thousands of different taxa | Takes weeks to months to perform |
rRNA, ribosomal RNA; ENTERO, Enterobacteriaceae, V, variable; ENTERO, Enterobacteriaceae; TMAO, trimethylamine-N-oxide;
Yet both 16S rRNA and MSS are time-consuming, laborious, and not suited for real-time gut microbiota monitoring in patients (Table 1). A major limitation of these methods is they only allow for determining the relative abundance of microorganisms, not the absolute levels, which can be misleading as an increase in relative abundance may not always correlate with an increase in absolute abundance . As such, novel methods are needed to provide point of care gut microbiota profiling in the future.
Inflammation and dysbiosis associated VTE
Inflammation is an important risk factor for VTE. Inflammation activates endothelial cells, platelets, and leukocytes to initiate coagulation [40]. It also leads to a consumptive coagulopathy and increase in pro-inflammatory cytokines, chemokines and various leukocyte subtypes [41, 42]. Several inflammatory states such as obesity, sepsis/infection, inflammatory bowel disease (IBD) and intestinal failure (IF) are associated with both dysbiosis and higher incidence of VTE (Table 2)[19, 30, 31, 43–55].
Table 2:
Disease states associated with high risk of VTE and dysbiosis
| Disease State: | VTE Risk: | Study: | Microbiome Changes: | Study: |
|---|---|---|---|---|
| ICU patients |
|
Kaplan, 2015 [43] |
|
McDonald, 2016 [31] |
| IBD |
|
Bernstein 2001 [44] Kappelman 2011 [45] Miehsler 2004 [46] |
|
Manichanh, 2006 [47] Frank, 2011 [48] Pastorelli, 2013 [49] Pastorelli, 2015 [50] |
| Obesity |
|
Ageno 2008 [51] |
|
Zhu, 2016 [19] Karlsson, 2012 [52] Ley, 2005 [53] |
| Intestinal Failure |
|
Journeycake 2015[54] Gonzalez-Hernandez 2016 [55] |
|
Piper, 2017 [30] |
ENTERO, Enterobacteriaceae; TMAO, trimethylamine-N-oxide;
Dysbiosis, LPS, and hypercoagulability
One of the links between dysbiosis and thrombosis is lipopolysaccharides (LPS), the glycolipid found on the outer membrane of gram-negative bacteria [56]. LPS is a heat-stable endotoxin that can enter the blood either by local or systemic infection [57]. In sepsis, patients have LPS levels a hundred-fold higher than healthy controls, which leads to overproduction of inflammatory cytokines and the clinical findings of gram-negative septic shock (organ failure, hypotension, respiratory distress, and disseminated intravascular coagulation (DIC) from dysregulation of coagulation cascade) [58]. Metabolic endotoxemia is a condition with chronically elevated plasma levels of LPS that are 10–50 times lower than sepsis resulting in a chronic inflammatory state [57]. Metabolic endotoxemia results from bacteria and their endotoxins, such as LPS, entering systemic circulation by translocating across an impaired intestinal lumen due to inflammation and injury [59]. LPS can translocate across different regions of GI tract from the lymphatic system in the oral cavity to the defective mucosal barrier in the intestinal mucosa (figure 1b)[49, 50, 60, 61].
LPS and metabolic endotoxemia
There has been difficulty finding a reliable LPS assay to measure these low levels of LPS in metabolic endotoxemia. The horseshoe crab (Limulus Polyphemus or Tachypleus species) has blood that is very sensitive to and will clot in the presence of LPS. One of the first assays used for LPS detections was a limulus amebocyte lysate (LAL) assays that would determine the presence of LPS in the patient’s plasma by measuring activation of the coagulation cascade in a horshecrab’s blood[62]. Since then, further assays have been developed including ELISA and Endotoxin activity assay (EAA), however, their reliability and interpretation are inconsistent and levels can fluctuance quickly within the bloodstream [62]. Another major limitation, is that not all LPS is the same as it can have differing structures and immunogenecity depending on the species to induce a host response. For example, Bacteriodes are commensal gram negative obligate anaerobes which also produce LPS but their LPS is structurally distinct and is beneficial as it inihibits innate immune signaling compared to LPS from E. Coli which is a potent immune activator[63, 64].
Despite these limitations, metabolic endotoxemia has been shown to be present in a variety of disease states such as atherosclerosis, autoimmune diseases, metabolic syndrome, cirrhosis, depression, type 2 diabetes, obesity, traumatic brain injury, multi-organ failure, depression and HIV [62]. To further highlight how dysbiosis plays a critical role in metabolic endotoxemia, in IBD, abundance of ENTERO in the microbiome of patients is associated with increased serum LPS levels compared to healthy controls[50]. Patients with cirrhosis similarly have higher levels of circulating systemic LPS secondary to translocation of bacteria and their by-products from the intestinal lumen into the portal and systemic circulation[65, 66]. Much higher levels of LPS are found in portal circulation than systemic circulation, suggesting a mechanistic process of spillover from the gut lumen into the portal and then systemic circulation[66–68]. These findings have also been recapitualated in a mouse model as high-fat diets in mice (as model of obesity) induce a change in gut microbiota populations towards an increase in abundance of gram-negative bacteria and increased intestinal permeability, which leads to LPS translocating into the systemic circulation with resultant metabolic endotoxemia[69].
Evidence for LPS-induced hypercoagulability
How increased LPS may lead to hypercoagulability has been evidenced in studies across various disease states and in mouse models(Table 3). In cirrhosis patients, elevated serum LPS levels positively correlate with factor (F) VIII levels, von Willebrand factor (VWF), and thrombin generation (as measured by prothrombin fragments F1+2 and D-dimer levels) [67, 68, 70]. Administration of enteral non-absorbable antibiotics led to a decrease in LPS levels and a concomitant decrease in thrombin generation [70]. Similarly, in IBD patients, LPS levels correlated with D-dimer and prothrombin fragment levels, and this association was stronger in patients with colonic disease where there is a higher bacterial burden than the rest of the GI tract [50].
Table 3:
LPS Mediated hypercoagulability
| Pathways | Finding: | References: |
|---|---|---|
| General | Administration of LPS potentiated thrombus formation in murine models | Wang, 2008 [71] |
| LPS levels correlated with elevated levels of vWF and factor VIII | Carnavale, 2017 [68] | |
| Platelet activation | Platelets exposed to LPS have increase aggregation | Raparelli, 2017 [78] |
| TLR4 mediated platelet activation | Degranulation of alpha and dense granules | Andonegui, 2005 [17] |
| Release of inflammatory cytokines | Zhang, 2009 [80] | |
| Release vWF and potentiates thrombin | Rivadeneyra, 2014 [85] | |
| Mediate platelet-neutrophil interactions and NET formation | D’Atri and Schattner, 2017 [73] | |
| TLR4 mediated activation of endothelial cells | Release of vWF, Factor VIII and P-selectin from Weibel Palade bodies | Carnevale, 2017 [68] Pan, 2016 [79] |
| TLR 2 mediated platelet activation | Platelet activation, aggregation with leukocytes, release of alpha and dense granules | Blair, 2009 [83] Rex, 2009 [84] Rivadeneyra, 2014 [85] |
| TLR 2 mediated activation of endothelial cells | Increase vWF, adhesion and arterial thrombus growth | Jäckel, 2017 [21] |
| Increase tissue factor, PAI-1 and decrease TPA and TFPI | Shin, 2011 [82] |
LPS, lipopolysaccharides; VWF, von Willebrand factor; TL4, toll-like receptor 4 ; NET, neutrophil endothelial traps ; PAI-1, plasminogen activator inhibitor-1; TPA, tissue plasminogen activator; TFPI, tissue factor pathway inhibitor
Wang et al. have studied the effect of LPS on both arterial and venous thrombosis in a murine model [71]. In this model, high levels of ferric chloride (FeCl3) are used to induce vessel injury either to the carotid artery or vena cava[71]. Dose-dependent thrombosis; time to thrombosis and thrombus size are used as metrics of hypercoagulability[71]. Wang et al. discovered that administration of LPS potentiated thrombus formation even at lower ferric chloride concentrations where vessel injury was not induced[71].
Toll-like receptor pathways in hypercoagulability
LPS is a key activator of the innate immune system via toll-like receptors (TLRs) expressed on both immune and non-immune cells, including macrophages, lymphocytes, endothelial cells, dendritic cells and platelets[72]. TLRs are key activators of the innate immune system that recognize pathogen-associated molecular patterns (PAMPs) found in bacteria, viruses, and fungi and play a key role in the activation of the innate immune system[72, 73]. Over 10 different TLRs have been identified of which the best studied are TLR2 and TLR4, which are expressed on nucleated endothelial cells, white blood cells, and platelets[50, 74, 75]. Platelet TLRs are the bridge in “thromboinflammation” with their role in inflammation, platelet adhesion and aggregation and the formation of neutrophil extracellular traps (NETs)[73]. There have also been studies to demonstrate that higher levels of TLR transcript expression can be associated with cardiovascular risk and inflammatory biomarkers to further highlight the association with thrombosis[76].
TLR4
TLR4 is the primary receptor for LPS on nucleated cells and platelets[77]. In clinical studies of IBD patients, LPS levels correlate positively with TLR4 concentrations to highlight the role of LPS-TLR4 pathway in triggering coagulation in these patients [50]. Ex vivo studies of platelets from cirrhosis patients with elevated LPS levels show platelet aggregation is increased in response to subthreshold concentrations of common agonists (such as Adenine di-Phosphate or collagen) compared to healthy controls and regardless of platelet count [78]. This response is blunted in the presence of TLR4 inhibition to further highlight the importance of this pathway [78].
TLR4 is found on endothelial cells which contain Weibel-Palade bodies (WPBs) that store and release modulators of vascular inflammation, including VWF, factor VIII and P-selectin [68, 79]. In vitro studies of human umbilical vein endothelial cells (HUVEC) incubation with LPS at concentrations observed in cirrhosis patients show that TLR4 activation induces VWF and factor VIII expression from endothelial cells (figure 1c)[68]. When these cells were incubated with a TLR4 inhibitor, this effect was mitigated suggesting that TLR4 pathways mediates exocytosis of Weibel-Palade bodies. Binding of LPS to TLR4 leads to activation of a MyD88-dependent pathway which causes the release of proinflammatory cytokines[80].
On platelets, TLR4 mediates platelet-neutrophil interactions by increasing binding to neutrophils, formation of NETs, and histone-mediated platelet responses that increase thrombin generation[73].
LPS has also been shown to activate platelets directly through aggregation, dense/alpha granule release and fibrinogen binding (figure 1d)[17]. However this role is controversial in other studies that show LPS may inhibit platelets or not induce any response either directly or through potentiation [81]. This discordant data may be due to a variety of factors including platelet preparation, insufficient amounts of soluble CD14 (needed for LPS mediated platelet response), the strain and concentration of LPS, stimulation time and biphasic response to cGMP signaling[73].
TLR2
TLR2 is also found on endothelial cells and platelets. TLR2 normally forms a heterodimer with either TLR1 or TLR6 and recognizes the PAMPs on a wide range of peptidoglycans on mainly gram-positive and some gram-negative bacteria[73, 77].
TLR2 pathway is vital for endothelial cells as TLR2 receptors are found on the hepatic endothelial cells and their activation will lead to an increase in VWF and an increase in arterial thrombus growth [21]. Germ-free mice (mice without microbiota) and TLR2 deficient mice have reduced thrombus formation highlighting the importance of the microbiota for TLR2 activation. Furthermore, treatment of in vitro HUVEC with TLR2 agonists leads to procoagulant effects with increased production of tissue factor, plasminogen activator inhibitor-1 (PAI-1), and decreased production of tissue plasminogen activator and tissue factor pathway inhibitor (TFPI) [82].
In platelets, activation of the TLR2 pathway leads to platelet activation, aggregation with leukocytes, and release of alpha and dense granules [83–85]. There are some conflicting studies suggest TLR2/6 may inhibit platelet activation but further studies are needed to understand the true pathophysiology [86].
Understanding immunothrombosis
“Immunothrombosis” is a term coined by Englemann and Massberg on the role of thrombosis in preventing intravascular antimicrobial spread through the formation of thrombi in microvessels by the recruitment of innate immune cells and platelets [87]. Neutrophils, monocytes, and dendritic cells work together to inhibit pathogen dissemination in the vasculature by initiating thrombosis through fibrin formation and platelet activation [87]. “Immunothrombosis” is distinct from the process of primary and secondary hemostasis as it occurs mostly in the intravascular compartment and could potentially be a novel treatment option for preventing pathological thrombosis [87]. Hemostasis and immunothrombosis work as a continuum for preventing blood loss and intravascular antimicrobial dissemination, but aberrant activation can lead to pathologic thrombosis seen in CVD and DVTs [87].
Neutrophils
Neutrophils are the major effectors of innate immunity and plan a dual function in host defense and thrombosis (Table 4). They aggregate rapidly at the site of vessel injury with platelets and can contribute to the formation of large-vessel thrombosis in cardiovascular diseases [88]. Upon activation, neutrophils will release NETs that are a matrix of DNA and histones projected from the neutrophils. NETs are protective against infections as they work as scaffolds to enhance neutrophil-mediated trapping and killing of pathogens[87, 89]. However, they can also induce a strong procoagulation response and be injurious to tissues by leading to pathologic consequences such as inflammatory diseases and transfusion-related acute lung injury (TRALI)[90].
Table 4:
Immunothrombosis in hypercoagulability
| Pathway/ Cell: | Key Finding: | Reference |
|---|---|---|
| Neutrophils | NETs illicit a strong procoagulant response | Fuchs, 2012 [89] |
| LPS induces expression of adhesion VCAM-1 and ICAM1 | Sawa, 2008 [95] | |
| Breakdown of NETs with DNAse will lead prevent thrombus formation | Brill, 2012 [97] | |
| Platelets | Bind to LPS and present it to neutrophils and immune cells | Semple, 2007 [98] |
| Bind to neutrophils and endothelial cells to foster additional immune cell recruitment | von Brühl, 2012[94] | |
| Contact activation (intrinsic) pathway | Elevated LPS levels activate KKS | Wu, 2015 [100] |
| LPS increased FXII release of Interleukins | Toossi, 1992 [101] Gobel, 2016 [102] | |
| Tissue factor (extrinsic) pathway | LPS leads to externalization of phosphatidylserines that lead to increase TF activation | Yang, 2019 [105] |
| Inhibition of TF prevents DIC findings in endotoxemia | Pawlinski, 2010 [106] |
NETS, neutrophil endothelial traps; VCAM, vascular cell adhesion molecule; ICAM, intracellular adhesion molecules; LPS, lipopolysaccharides; DNAase, deoxyribonuclease; KKS, Kallikrein-Kinin system
NET formation in “immunothrombosis” leads to platelet-neutrophil interactions that activate the contact factor and contribute to aggregation (figure 1e). Histone components of the extracellular nucleosomes in NETs can activate TLR2 and TLR4[91]. Activated neutrophils will deposit serine proteases that inactivate important anticoagulants like TFPI and thrombomodulin, which leads to resultant hypercoagulability[92, 93]. NETs also activate the contact factor pathway by activating factor XII and bind to VWF to facilitate platelet recruitment and activation[94]. TLR4 pathway also induces NET formation by increasing endothelial expression of important adhesion molecules, including intercellular adhesion molecule-1 (ICAM-1) [20, 95, 96]. Furthermore, blocking NET formation in preclinical models by treatment of mice with deoxyribonuclease (DNase) that breakdown NETs, will prevent thrombus formation [97].
Platelets
Platelets play a critical supportive role in immunothrombosis through pathogen recognition and augmentation of prothrombotic pathways involved in innate immune cells. Platelet will bind to LPS on circulating bacteria and present these pathogens to neutrophils and other innate immune cells[98]. In arterial thrombus, they are the first cells recruited to the sites of endothelial disruption and lead to the expression of adhesion molecules and release of chemokines to recruit innate immune cells[99]. In DVTs, platelets accumulate at the inflammatory sites and participate in receptor-dependent binding to neutrophils and monocytes and direct platelet-endothelial cell interactions that foster additional immune cell recruitment [94].
LPS and the coagulation cascade
Contact activation (intrinsic) pathway
Elevated LPS levels in endotoxemia can directly activate kallikrein-kinin system (KKS)[100]. Generated kallikrein reciprocally activates factor XII which leads to activation of the intrinsic coagulation system and cleaves high molecular weight kininogen (HK) to lead to the liberation of the proinflammatory mediator, bradykinin[100]. HK is also a critical LPS carrier as mice lacking HK are resistant to LPS-induced mortality, and replenishing HK results in restored susceptibility of animals [12]. In mononuclear cell cultures, FXII-induced release of interleukin (IL)-1 is augmented in the presence of LPS[101]. Furthermore, FXII-treated splenic dendritic cells exposed to LPS, produce higher amounts of IL-6 and IL-23 further corroborating that FXII and the KKS are instrumental in mediating host immune responses[102].
6.2. Tissue factor (extrinsic) pathway
LPS can also affect the extrinsic coagualation cascade. Phosphatidylserines enhance activity of tissue factor (TF) by increasing TF binding to FVII and formation of cofactor-protease complexes in the coagulation cascade[103]. Phosphatidylserines are normally found in the inner leaflet of the plasma membrane and will externalize in the presence of LPS to the outer leaflet of the plasma membrane leading to TF activation[103, 104]. The mechanism of LPS induced phosphatidylserine expression is through LPS activation of an intracellular cytosolic LPS receptor, Caspase-11 (Casp11) which triggers gastrodermin D (GSDMD) pore formation leads to calcium influx and activation of transmebrane proteins that mediate phosphatidylserine expression[105]. Deletion of Casp11 will lead to decrease LPS-induced thrombin generation and platelet aggregation in mice[105]. Prior studies have also suggested an important link between extrinsic pathway and endotoxemia as inhibition of TF prevents endotoxemia-induced DIC[106]. This combination of platelet hyperreactivity, release of coagulation factors, cytokine release and NET formation can lead to hypercoagulablity and possibly thrombus formation (figure 1f). This supports an important role of immunothrombosis in the pathogenesis of VTE and as a possible therapeutic target for future studies. Further details on mechanisms of LPS-induced hypercoagulability are extensively reviewed elsewhere[85, 107–110].
Microbiome metabolites and thrombosis risk
The gut microbiome processes the undigested dietary polysaccharides from the host’s diet and can alter metabolite production in the host [24]. A western diet, high in fat and sugar, has increased content of phosphatidylcholine, choline and carnitine[24]. TMA lyases in the gut microbiome metabolize choline from the diet into TMA, which then goes through the portal circulation to the liver where it is converted into TMAO[24, 34, 111]. Microorganisms associated with increase TMAO production were difficult and inconsistently identified with 16s rRNA sequencing as production of a specific metabolite like TMAO in the abudance a certain bacterial taxa does not necessarily imply causality[112–115]. Reference genomic studies have been more helpful in identify TMAO producing species in the phylum Actinobacteria, Firmicutes and Proteobacteria through sceening for species with pontential to be involved in metabolic pathway of TMA production[115, 116]. TMAO has gained importance because it enhances cardiovascular and thrombosis risk by atherosclerosis progression through vascular inflammation, endothelial cell dysfunction, foam cell formation, and platelet hyperreactivity[19, 117–120].
Other microbiome metabolites that correlate with the risk of CVD are bile acids (BA) and short-chain fatty acids (SCFA)[121]. BA are synthesized from cholesterol in the liver and are vital for cholesterol excretion in the feces. Changes in the microbiome can decrease BA synthesis, leading to decreased excretion of cholesterol and increased risk for CVD[122]. SCFA result from fermentation of indigestible fibers by the microbiota, and while elevated levels are thought to generally have an anti-inflammatory effect, uncontrolled levels can be a risk factor for CVD[123–125]. While there have been advances in our ability to detect these metabolites and study correlations, our understanding of the exact mechanisms remains unclear.
Mechanisms of TMAO
Atherosclerotic diseases, such as myocardial infarction and stroke, are multifactorial in origin however, there is increasing evidence that changes in the microbiome may influence the progression of atherosclerosis[125]. Atherosclerotic disease has been shown to be associated with increased metabolism of dietary lipid phosphatidylcholine to TMAO [24]. Plasma TMAO levels are thought to correlate with coronary atherosclerotic plaque burden and are an independent predictor of CVD disease in humans (Table 5) [19, 24, 114]. TMAO increases the deposition of cholesterol-laden macrophage foam cells onto vessels, which is one of the earliest steps in the development of atherosclerotic disease (figure 1g)[24]. The microbiome plays an important role in this as a choline-rich diet will augment atherosclerosis while the depletion of gut flora with antibiotics, even in the presence of a choline-rich diet, will suppress this response[24].
Table 5:
TMAO mediated hypercoagulability
| Finding: | References: |
|---|---|
| TMAO levels are independent predictors of CVD | Wang, 2011 [24] |
| Intestinal microbiota is needed to produce TMAO and accelerate atherosclerosis | Koeth, 2013 [114] |
| Dietary choline and elevated TMAO has a direct prothrombotic effect in humans | Zhu, 2016 [19] |
| Use of DMB to lower TMAO levels leads to reduced size of atherosclerotic lesions | Wang, 2015 [15] |
CVD, cardiovascular disease; TMAO, trimethylamine-N-oxide; DMB, 3,3-dimethyl-1-butanol
In mouse models of thrombosis, higher levels of TMAO in choline-supplemented mice are associated with a shorter time to cessation of blood flow after FeCl3 injury[19]. Furthermore, when germ-free mice with no gut colonization are supplemented with choline alone, there is no effect on the time to cessation of blood flow, underscoring the critical role of the gut microbiome in the prothrombotic phenotype with TMAO[19]. A choline-rich diet induces dysbiosis by an increase in the phyla of Firmicutes and Proteobacteria and transplantation of this microbiome composition to germ free mice results in increased platelet responsiveness (as measured by ex vivo platelet aggregometry) and in vivo thrombosis potential (as measured by the FeCl3 carotid artery injury model)[19, 126]. TMAO does not stimulate platelets directly but enhances the release of Ca2+ and activation to platelet agonists to show a TMAO-dependent enhancement [19].
Unfortunately, these findings are not consistent as other studies have shown that TMAO correlated inversely with atherosclerotic lesion size, and further studies are needed to fully understand its role in thrombosis[127]. Jonsson et al. determined that while choline supplementation did increase TMAO levels, the size of aortic lesions was not affected by choline supplementation and did not correlate with TMAO levels[113]. However these studies differed significantly in methods as Jonsson et al. used germ free mice as microbiota depleted controls and supplemented choline at an older age of 8 weeks (when atherosclerotic would have already developed in mice) as compared to Wang et al. who used antibiotic to deplete the gut microbiota and started choline supplementation at only 4 weeks[24]. TMAO is also not as clear of a predictor for VTE as a cross-sectional study of patients with acute VTE, higher levels of TMAO correlated with mortality but not with VTE recurrence[128].
Treatment options
Dysbiosis
Many treatment options to modulate dysbiosis in the gut microbiome are currently being explored (Table 6). Prebiotics, foods, or dietary supplements that “induce” the growth or activity of beneficial gut commensal anaerobes , and probiotics, live organisms that are ingested, have been studied as treatment options in IBD and Clostridium difficile (C. difficile) infection with mixed reports on efficacy[11, 129, 130]. The use of certain broad-spectrum antibiotics, such as vancomycin and cephalexin, can lead to long term changes in the composition of the microbiome with the expansion of pathobionts, such as ENTERO, which can potentially lead to opportunistic infections[13, 131]. The use of clindamycin in the hematopoietic stem cell transplantation (HSCT) population leads to depletion of commensal anaerobes (Firmicutes), which is associated with adverse outcomes of graft-versus-host disease (GVHD)[7]. How modulating the gut microbiome with antibiotics can lead to ENTERO expansion, and increased thrombotic risk has not been studied.
Table 6:
Treatment Options
| Options: | Mechanism: | |
|---|---|---|
| Dysbiosis | Prebiotics, probiotics | Ingestion of live organisms or supplements to induce growth of certain commensal anaerobes |
| Antibiotics | Depletion of pathogenic microbiota leading to overgrowth of healthy gut flora | |
| Fecal microbial transplant | Transplantation of donor fecal samples | |
| LPS | C11C1 | Monoclonal antibody that blocks LPS binding site |
| CD14 antibodies | Lead to TLR4 internalization and decrease LPS response | |
| Statins | Lead to increase expression of LDL-R and increase clearance of LPS | |
| PCSK9 | Inhibit degradation of LDL-R leading to increase LPS clearance | |
| IL-1B inhibitors | Block downstream TLR4 pathway leading to decrease in CVD | |
| TMAO | FMO inhibitors | Block conversion of TMA into TMAO |
| DMB | Inhibit microbial TMA lyases to inhibit production of TMA | |
| CutC/D | Target microbial proteins |
C11C1, HMW Kininogen antibody; LPS, lipopolysaccharides; PCSK9, proprotein convertase subtilisin/ kexin type 9; LDL-R, low density lipoprotein receptor; IL, interleukin; TLR4,toll-like receptor 4; CVD, cardiovascular disease; FMO, Flavin monooxygenases; DMB, 3,3-dimethyl-1-butanol;
FMT is the transplantation of a donor’s fecal samples by either oral or rectal delivery to restore gut microbiota homeostasis. FMT is a safe and effective treatment option for recurrent C. difficile infection with remission rates of over 80% compared to antibiotics alone[14, 132]. The role of FMT in metabolic syndrome has been studied in animal models and humans with favorable results[133, 134]. Autologous FMT also has promising potential as a therapeutic option in HSCT, where the loss of microbial diversity during transplant is associated with systemic infections, C. difficile infections and GVHD[135]. The expansion of commensal anaerobes, specifically phyla Firmicutes, in successfully transplanted patients is thought to have an important role in restoring the microbiome and reducing C. difficile infections[14, 136]. Healthy donor FMT can also affect hypercoagulabilty in metabolic syndrome patients by leading to a prolonged lag time on thrombin generation and downregulation of coagulation related proteins[137].
FMT, however, is not without risks. Recently in 2019, there was an FDA warning about the use of FMT after two immunocompromised patients developed invasive extended spectrum-beta lactamase Escherichia coli infections after transplants from the same donor (https://www.fda.gov/vaccines-blood-biologics/safety-availability-biologics/important-safety-alert-regarding-use-fecal-microbiota-transplantation-and-risk-serious-adverse). The donor had not been tested for multi-drug resistant organisms, which is typically performed by many groups administering FMT.
LPS:
Treatment options to target LPS have also long been explored. One option is to block LPS with therapies such as a HMW Kininogen antibody (C11C1), a monoclonal antibody directed to the LPS binding site on domain 5 of HK [138]. C11C1 has been shown to reduce LPS-induced systemic inflammation and circulating LPS levels in mice[138]. Several TLR4 antagonists have been developed and investigated in clinical trials but with little efficacy[138, 139].
Another option is to increase the clearance of LPS. CD14 is the LPS sensing receptors on monocytes that binds to LPS and transfer it to the TLR4 complex[140]. Anti-CD 14 antibodies can lead to TLR4 internalization, decrease LPS response and may be a novel therapeutic option[141]. LPS is also cleared through the cholesterol pathway, including the low density lipoprotein receptor (LDLR)[142]. Statins increase expression of LDLR and have general anti-inflammatory effect but in clinical trials of sepsis do not shown a survival benefit against sepsis. The use of proprotein convertase subtilisin/ kexin type 9 (PCSK9) inhibitors to block degradation of LDL receptors and lead to increase LPS clearance can also be considered[143, 144]
Targeting the downstream effectors of the TLR-4 signaling pathway are yet another therapeutic possibility. Activator protein 1,signal transducers and activators of transcription family of transcription factors and interferon regulatory factors are currently being explored as potential options for modulating inflammation and sepsis[145]. The Canakinumab Anti-inflammatory Thrombosis Outcome Study (CANTOS) trial has shown that IL-1B inhibitors, downstream of the TLR4 pathway, lead to a lower incidence of cardiovascular events[146]. Another option that has yet to be explored is using DNase, which inhibits NET formation as a possible therapeutic drug for thrombolysis[89]. Whether these therapies will be effective in decreasing thrombosis risk in dysbiosis has not been explored.
TMAO
The discovery of TMAO as a risk factor for arterial thrombosis has led to research targeting this pathway as a treatment modality to decrease the risk of CVD. Flavin monooxygenases (FMOs), mainly FMO3, convert TMA into TMAO [34]. There is a genetic disorder of FMO3 deficiency known as fish odor syndrome, but recreating this in humans is clinically limited as it has the unfortunate side effect of a strong door from the increase in TMA secretion through the sweat glands, urine, and breath[147]. A structural analog of choline, 3,3-dimethyl-1-butanol (DMB), is found naturally in food products and decreases TMAO production by inhibiting microbial TMA lyases to lower TMA levels [15]. CutC/D inhibitors that target microbial proteins in choline metabolism are another treatment option. These inhibitors would accumulate only in the intestinal microbes and block TMA and TMAO production to reduce thrombosis potential [35].
Conclusion
The role of the gut microbiome in various aspects of human health is becoming increasingly recognized. The etiology of thrombosis is a complex interplay between the coagulation system, innate immune system, and inflammation. Inflammation is further associated with dysbiosis, increase in intestinal permeability and production of specific metabolites. Identification of important metabolites related to the gut microbiome and cardiovascular risk, such as TMAO, has led to research in the development of targeted therapies [19, 35]. The role of gut microbiota derived LPS in hypercoagulability and VTE continues to be studied and treatment options are yet to be explored. The added inflammatory effect of dysbiosis that activates the innate immune system is yet another pathway that could be targeted. The authors are currently leading an ongoing single institutional prospective pilot study on new pediatric VTE with the primary objectives of determining if patients who develop VTE have a distinct gut microbiome signature with expansion of ENTERO. Furthermore, they are determining whether these patients have elevated plasma LPS levels and if these levels correlate with hypercoagulability as determined by thrombin generation assay. The primary goal is to establish a gut microbiome signature for VTE and elevated LPS levels as a possible biomarker of VTE in high-risk patients. Causality could then be tested by utilizing a murine model of thrombosis to gain mechanistic insight into how dysbiosis leads to hypercoagulability. Our ability to understand the microbiome and its role in thrombosis is critical in optimizing treatment and developing new preventative and therapeutic strategies with limited bleeding risk.
Highlights:
Several disease states with high risk of thrombosis are associated with dysbiosis
Bacterial LPS from the gut can bind to TLRs and activates the coagulation cascade
The microbiome can affect the innate immune system and lead to “immunothrombosis”
Gut metabolites like TMAO can promote cardiovascular disease and thrombosis
Acknowledgement
We would like to thank the Hemostasis and Thrombosis Research Society (HTRS), HTRS Publication Committee, and Canadian Venous Thromboembolism Clinical Trials and Outcomes Research (CanVECTOR) for providing helpful feedback and comments on the manuscript.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors. A.Z is supported by a grant from the National Institutes of Health (1K23HL132054-01). A.Y.K is supported by the Roberta I. and Norman L. Pollock Fund, the US National Institutes of Health (NIH) grants R01AI123163 and R01CA231303, and Centers for Disease Control/National Center for Emerging and Zoonotic Infectious Diseases. These funding sources were not involved in the writing of the report or the decision to submit the article for publication.
Abbreviations:
- VTE
Venous thromboembolism
- ENTERO
Enterobacteriaceae
- LPS
Lipopolysaccharides
- TMAO
Trimethylamine-N-oxide
- PTS
Post-thrombotic syndrome
- CVD
Cardiovascular Disease
- FMT
Fecal microbial transplant
- TMA
Trimethylamine
- qPCR
Quantitative polymerase chain reaction
- rRNA
Ribosomal RNA
- MSS
Metagenomic shotgun sequencing
- IBD
Inflammatory bowel disease
- IF
Intestinal failure
- DIC
Disseminated intravascular coagulation
- LAL
Limulus amebocyte lysate
- EAA
Endotoxin activity assay
- F
Factor
- VWF
von Willebrand factor
- FeCl3
Ferric chloride
- TLRs
Toll-like receptors
- PAMPs
Pathogen-associated molecular patterns
- NETs
Neutrophil extracellular traps
- WBP
Weibel-Palade bodies
- HUVEC
Human umbilical vein endothelial cells
- PAI-1
Plasminogen activator inhibitor-1
- TFPI
Tissue factor pathway inhibitor
- TRALI
Transfusion-related acute lung injury
- ICAM-1
Intercellular adhesion molecule-1
- DNase
Deoxyribonuclease
- KKS
Kallikrein-kinin system
- HK
High molecular weight kininogen
- IL
Interleukin
- TF
Tissue factor
- Casp11
Caspase-11
- GSDMD
Gastrodermin D
- BA
Bile acids
- SCFA
Short-chain fatty acids
- HSCT
Hematopoietic stem cell transplantation
- GVHD
Graft-versus-host disease
- C11C1
HMW Kininogen antibody
- LDLR
Low density lipoprotein receptor
- PCSK9
Proprotein convertase subtilisin/ kexin type 9
- CANTOS
Canakinumab Anti-inflammatory Thrombosis Outcome Study
- FMOs
Flavin monooxygenases
- DMB
3,3-dimethyl-1-butanol
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest
None
References
- [1].Raffini L, Huang YS, Witmer C, Feudtner C, Dramatic increase in venous thromboembolism in children’s hospitals in the United States from 2001 to 2007, Pediatrics 124(4) (2009) 1001–8. [DOI] [PubMed] [Google Scholar]
- [2].Chalmers EA, Epidemiology of venous thromboembolism in neonates and children, Thromb Res 118(1) (2006) 3–12. [DOI] [PubMed] [Google Scholar]
- [3].Silverstein MD, Heit JA, Mohr DN, Petterson TM, O’Fallon WM, Melton LJ, Trends in the incidence of deep vein thrombosis and pulmonary embolism: a 25-year population-based study, Arch Intern Med 158(6) (1998) 585–93. [DOI] [PubMed] [Google Scholar]
- [4].May AL, Kuklina EV, Yoon PW, Prevalence of cardiovascular disease risk factors among US adolescents, 1999–2008, Pediatrics 129(6) (2012) 1035–41. [DOI] [PubMed] [Google Scholar]
- [5].Cushman M, Epidemiology and risk factors for venous thrombosis, Semin Hematol 44(2) (2007) 62–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Spor A, Koren O, Ley R, Unravelling the effects of the environment and host genotype on the gut microbiome, Nat Rev Microbiol 9(4) (2011) 279–90. [DOI] [PubMed] [Google Scholar]
- [7].Simms-Waldrip TR, Sunkersett G, Coughlin LA, Savani MR, Arana C, Kim J, Kim M, Zhan X, Greenberg DE, Xie Y, Davies SM, Koh AY, Antibiotic-Induced Depletion of Anti-inflammatory Clostridia Is Associated with the Development of Graft-versus-Host Disease in Pediatric Stem Cell Transplantation Patients, Biol Blood Marrow Transplant 23(5) (2017) 820–829. [DOI] [PubMed] [Google Scholar]
- [8].Frankel AE, Coughlin LA, Kim J, Froehlich TW, Xie Y, Frenkel EP, Koh AY, Metagenomic Shotgun Sequencing and Unbiased Metabolomic Profiling Identify Specific Human Gut Microbiota and Metabolites Associated with Immune Checkpoint Therapy Efficacy in Melanoma Patients, Neoplasia 19(10) (2017) 848–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Aron-Wisnewsky J, Clément K, The gut microbiome, diet, and links to cardiometabolic and chronic disorders, Nat Rev Nephrol 12(3) (2016) 169–81. [DOI] [PubMed] [Google Scholar]
- [10].Fan D, Coughlin LA, Neubauer MM, Kim J, Kim MS, Zhan X, Simms-Waldrip TR, Xie Y, Hooper LV, Koh AY, Activation of HIF-1α and LL-37 by commensal bacteria inhibits Candida albicans colonization, Nat Med 21(7) (2015) 808–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Qiao YQ, Cai CW, Ran ZH, Therapeutic modulation of gut microbiota in inflammatory bowel disease: More questions to be answered, J Dig Dis 17(12) (2016) 800–810. [DOI] [PubMed] [Google Scholar]
- [12].Yang A, Xie Z, Wang B, Colman RW, Dai J, Wu Y, An essential role of high-molecular-weight kininogen in endotoxemia, J Exp Med 214(9) (2017) 2649–2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Tanaka S, Kobayashi T, Songjinda P, Tateyama A, Tsubouchi M, Kiyohara C, Shirakawa T, Sonomoto K, Nakayama J, Influence of antibiotic exposure in the early postnatal period on the development of intestinal microbiota, FEMS immunology and medical microbiology 56(1) (2009) 80–7. [DOI] [PubMed] [Google Scholar]
- [14].Leffler DA, Lamont JT, Clostridium difficile infection, The New England journal of medicine 372(16) (2015) 1539–48. [DOI] [PubMed] [Google Scholar]
- [15].Wang Z, Roberts AB, Buffa JA, Levison BS, Zhu W, Org E, Gu X, Huang Y, Zamanian-Daryoush M, Culley MK, DiDonato AJ, Fu X, Hazen JE, Krajcik D, DiDonato JA, Lusis AJ, Hazen SL, Non-lethal Inhibition of Gut Microbial Trimethylamine Production for the Treatment of Atherosclerosis, Cell 163(7) (2015) 1585–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Dunzendorfer S, Lee HK, Tobias PS, Flow-dependent regulation of endothelial Toll-like receptor 2 expression through inhibition of SP1 activity, Circ Res 95(7) (2004) 684–91. [DOI] [PubMed] [Google Scholar]
- [17].Andonegui G, Kerfoot SM, McNagny K, Ebbert KV, Patel KD, Kubes P, Platelets express functional Toll-like receptor-4, Blood 106(7) (2005) 2417–23. [DOI] [PubMed] [Google Scholar]
- [18].Dauphinee SM, Karsan A, Lipopolysaccharide signaling in endothelial cells, Lab Invest 86(1) (2006) 9–22. [DOI] [PubMed] [Google Scholar]
- [19].Zhu W, Gregory JC, Org E, Buffa JA, Gupta N, Wang Z, Li L, Fu X, Wu Y, Mehrabian M, Sartor RB, McIntyre TM, Silverstein RL, Tang WHW, DiDonato JA, Brown JM, Lusis AJ, Hazen SL, Gut Microbial Metabolite TMAO Enhances Platelet Hyperreactivity and Thrombosis Risk, Cell 165(1) (2016) 111–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Komatsu S, Berg RD, Russell JM, Nimura Y, Granger DN, Enteric microflora contribute to constitutive ICAM-1 expression on vascular endothelial cells, American journal of physiology. Gastrointestinal and liver physiology 279(1) (2000) G186–91. [DOI] [PubMed] [Google Scholar]
- [21].Jäckel S, Kiouptsi K, Lillich M, Hendrikx T, Khandagale A, Kollar B, Hörmann N, Reiss C, Subramaniam S, Wilms E, Ebner K, Brühl MV, Rausch P, Baines JF, Haberichter S, Lämmle B, Binder CJ, Jurk K, Ruggeri ZM, Massberg S, Walter U, Ruf W, Reinhardt C, Gut microbiota regulate hepatic von Willebrand factor synthesis and arterial thrombus formation via Toll-like receptor-2, Blood 130(4) (2017) 542–553. [DOI] [PubMed] [Google Scholar]
- [22].Sender R, Fuchs S, Milo R, Revised Estimates for the Number of Human and Bacteria Cells in the Body, PLoS biology 14(8) (2016) e1002533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Singh RK, Chang HW, Yan D, Lee KM, Ucmak D, Wong K, Abrouk M, Farahnik B, Nakamura M, Zhu TH, Bhutani T, Liao W, Influence of diet on the gut microbiome and implications for human health, J Transl Med 15(1) (2017) 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, Dugar B, Feldstein AE, Britt EB, Fu X, Chung YM, Wu Y, Schauer P, Smith JD, Allayee H, Tang WH, DiDonato JA, Lusis AJ, Hazen SL, Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease, Nature 472(7341) (2011) 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Jandhyala SM, Talukdar R, Subramanyam C, Vuyyuru H, Sasikala M, Nageshwar Reddy D, Role of the normal gut microbiota, World J Gastroenterol 21(29) (2015) 8787–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kiouptsi K, Reinhardt C, Contribution of the commensal microbiota to atherosclerosis and arterial thrombosis, British journal of pharmacology 175(24) (2018) 4439–4449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Dave M, Higgins PD, Middha S, Rioux KP, The human gut microbiome: current knowledge, challenges, and future directions, Transl Res 160(4) (2012) 246–57. [DOI] [PubMed] [Google Scholar]
- [28].Xu J, Gordon JI, Honor thy symbionts, Proc Natl Acad Sci U S A 100(18) (2003) 10452–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Fouhy F, Ross RP, Fitzgerald GF, Stanton C, Cotter PD, Composition of the early intestinal microbiota: knowledge, knowledge gaps and the use of high-throughput sequencing to address these gaps, Gut Microbes 3(3) (2012) 203–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Piper HG, Fan D, Coughlin LA, Ho EX, McDaniel MM, Channabasappa N, Kim J, Kim M, Zhan X, Xie Y, Koh AY, Severe Gut Microbiota Dysbiosis Is Associated With Poor Growth in Patients With Short Bowel Syndrome, JPEN J Parenter Enteral Nutr 41(7) (2017) 1202–1212. [DOI] [PubMed] [Google Scholar]
- [31].McDonald D, Ackermann G, Khailova L, Baird C, Heyland D, Kozar R, Lemieux M, Derenski K, King J, Vis-Kampen C, Knight R, Wischmeyer PE, Extreme Dysbiosis of the Microbiome in Critical Illness, mSphere 1(4) (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Lupp C, Robertson ML, Wickham ME, Sekirov I, Champion OL, Gaynor EC, Finlay BB, Host-mediated inflammation disrupts the intestinal microbiota and promotes the overgrowth of Enterobacteriaceae, Cell Host Microbe 2(3) (2007) 204. [DOI] [PubMed] [Google Scholar]
- [33].Peng J, Xiao X, Hu M, Zhang X, Interaction between gut microbiome and cardiovascular disease, Life Sci 214 (2018) 153–157. [DOI] [PubMed] [Google Scholar]
- [34].Bennett BJ, de Aguiar Vallim TQ, Wang Z, Shih DM, Meng Y, Gregory J, Allayee H, Lee R, Graham M, Crooke R, Edwards PA, Hazen SL, Lusis AJ, Trimethylamine-N-oxide, a metabolite associated with atherosclerosis, exhibits complex genetic and dietary regulation, Cell Metab 17(1) (2013) 49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Roberts AB, Gu X, Buffa JA, Hurd AG, Wang Z, Zhu W, Gupta N, Skye SM, Cody DB, Levison BS, Barrington WT, Russell MW, Reed JM, Duzan A, Lang JM, Fu X, Li L, Myers AJ, Rachakonda S, DiDonato JA, Brown JM, Gogonea V, Lusis AJ, Garcia-Garcia JC, Hazen SL, Development of a gut microbe-targeted nonlethal therapeutic to inhibit thrombosis potential, Nat Med 24(9) (2018) 1407–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Koh AY, Potential for Monitoring Gut Microbiota for Diagnosing Infections and Graft-versus-Host Disease in Cancer and Stem Cell Transplant Patients, Clin Chem 63(11) (2017) 1685–1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, Fierer N, Knight R, Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample, Proc Natl Acad Sci U S A 108 Suppl 1 (2011) 4516–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Kim J, Kim MS, Koh AY, Xie Y, Zhan X, FMAP: Functional Mapping and Analysis Pipeline for metagenomics and metatranscriptomics studies, BMC Bioinformatics 17(1) (2016) 420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Koren O, Spor A, Felin J, Fåk F, Stombaugh J, Tremaroli V, Behre CJ, Knight R, Fagerberg B, Ley RE, Bäckhed F, Human oral, gut, and plaque microbiota in patients with atherosclerosis, Proc Natl Acad Sci U S A 108 Suppl 1 (2011) 4592–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Branchford BR, Carpenter SL, The Role of Inflammation in Venous Thromboembolism, Front Pediatr 6 (2018) 142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Saghazadeh A, Rezaei N, Inflammation as a cause of venous thromboembolism, Crit Rev Oncol Hematol 99 (2016) 272–85. [DOI] [PubMed] [Google Scholar]
- [42].Lentz SR, Thrombosis in the setting of obesity or inflammatory bowel disease, Blood 128(20) (2016) 2388–2394. [DOI] [PubMed] [Google Scholar]
- [43].Kaplan D, Casper TC, Elliott CG, Men S, Pendleton RC, Kraiss LW, Weyrich AS, Grissom CK, Zimmerman GA, Rondina MT, VTE Incidence and Risk Factors in Patients With Severe Sepsis and Septic Shock, Chest 148(5) (2015) 1224–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Bernstein CN, Blanchard JF, Houston DS, Wajda A, The incidence of deep venous thrombosis and pulmonary embolism among patients with inflammatory bowel disease: a population-based cohort study, Thromb Haemost 85(3) (2001) 430–4. [PubMed] [Google Scholar]
- [45].Kappelman MD, Horvath-Puho E, Sandler RS, Rubin DT, Ullman TA, Pedersen L, Baron JA, Sorensen HT, Thromboembolic risk among Danish children and adults with inflammatory bowel diseases: a population-based nationwide study, Gut 60(7) (2011) 937–43. [DOI] [PubMed] [Google Scholar]
- [46].Miehsler W, Reinisch W, Valic E, Osterode W, Tillinger W, Feichtenschlager T, Grisar J, Machold K, Scholz S, Vogelsang H, Novacek G, Is inflammatory bowel disease an independent and disease specific risk factor for thromboembolism?, Gut 53(4) (2004) 542–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Manichanh C, Rigottier-Gois L, Bonnaud E, Gloux K, Pelletier E, Frangeul L, Nalin R, Jarrin C, Chardon P, Marteau P, Roca J, Dore J, Reduced diversity of faecal microbiota in Crohn’s disease revealed by a metagenomic approach, Gut 55(2) (2006) 205–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Frank DN, Robertson CE, Hamm CM, Kpadeh Z, Zhang T, Chen H, Zhu W, Sartor RB, Boedeker EC, Harpaz N, Pace NR, Li E, Disease phenotype and genotype are associated with shifts in intestinal-associated microbiota in inflammatory bowel diseases, Inflamm Bowel Dis 17(1) (2011) 179–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Pastorelli L, De Salvo C, Mercado JR, Vecchi M, Pizarro TT, Central role of the gut epithelial barrier in the pathogenesis of chronic intestinal inflammation: lessons learned from animal models and human genetics, Front Immunol 4 (2013) 280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Pastorelli L, Dozio E, Pisani LF, Boscolo-Anzoletti M, Vianello E, Munizio N, Spina L, Tontini GE, Peyvandi F, Corsi Romanelli MM, Vecchi M, Procoagulatory state in inflammatory bowel diseases is promoted by impaired intestinal barrier function, Gastroenterol Res Pract 2015 (2015) 189341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Ageno W, Becattini C, Brighton T, Selby R, Kamphuisen PW, Cardiovascular risk factors and venous thromboembolism: a meta-analysis, Circulation 117(1) (2008) 93–102. [DOI] [PubMed] [Google Scholar]
- [52].Karlsson FH, Fåk F, Nookaew I, Tremaroli V, Fagerberg B, Petranovic D, Bäckhed F, Nielsen J, Symptomatic atherosclerosis is associated with an altered gut metagenome, Nat Commun 3 (2012) 1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Ley RE, Backhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI, Obesity alters gut microbial ecology, Proc Natl Acad Sci U S A 102(31) (2005) 11070–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Journeycake JM, Buchanan GR, Catheter-related deep venous thrombosis and other catheter complications in children with cancer, J Clin Oncol 24(28) (2006) 4575–80. [DOI] [PubMed] [Google Scholar]
- [55].Gonzalez-Hernandez J, Daoud Y, Styers J, Journeycake JM, Channabasappa N, Piper HG, Central venous thrombosis in children with intestinal failure on long-term parenteral nutrition, J Pediatr Surg 51(5) (2016) 790–3. [DOI] [PubMed] [Google Scholar]
- [56].Whitfield C, Trent MS, Biosynthesis and export of bacterial lipopolysaccharides, Annu Rev Biochem 83 (2014) 99–128. [DOI] [PubMed] [Google Scholar]
- [57].Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, Neyrinck AM, Fava F, Tuohy KM, Chabo C, Waget A, Delmée E, Cousin B, Sulpice T, Chamontin B, Ferrières J, Tanti JF, Gibson GR, Casteilla L, Delzenne NM, Alessi MC, Burcelin R, Metabolic endotoxemia initiates obesity and insulin resistance, Diabetes 56(7) (2007) 1761–72. [DOI] [PubMed] [Google Scholar]
- [58].Opal SM, Scannon PJ, Vincent JL, White M, Carroll SF, Palardy JE, Parejo NA, Pribble JP, Lemke JH, Relationship between plasma levels of lipopolysaccharide (LPS) and LPS-binding protein in patients with severe sepsis and septic shock, J Infect Dis 180(5) (1999) 1584–9. [DOI] [PubMed] [Google Scholar]
- [59].Fukui H, Gut-liver axis in liver cirrhosis: How to manage leaky gut and endotoxemia, World journal of hepatology 7(3) (2015) 425–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].van Deventer SJ, ten Cate JW, Tytgat GN, Intestinal endotoxemia. Clinical significance, Gastroenterology 94(3) (1988) 825–31. [DOI] [PubMed] [Google Scholar]
- [61].Spadoni I, Zagato E, Bertocchi A, Paolinelli R, Hot E, Di Sabatino A, Caprioli F, Bottiglieri L, Oldani A, Viale G, Penna G, Dejana E, Rescigno M, A gut-vascular barrier controls the systemic dissemination of bacteria, Science 350(6262) (2015) 830–4. [DOI] [PubMed] [Google Scholar]
- [62].Munford RS, Endotoxemia-menace, marker, or mistake?, J Leukoc Biol 100(4) (2016) 687–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Vatanen T, Kostic AD, d’Hennezel E, Siljander H, Franzosa EA, Yassour M, Kolde R, Vlamakis H, Arthur TD, Hamalainen AM, Peet A, Tillmann V, Uibo R, Mokurov S, Dorshakova N, Ilonen J, Virtanen SM, Szabo SJ, Porter JA, Lahdesmaki H, Huttenhower C, Gevers D, Cullen TW, Knip M, Xavier RJ, Variation in Microbiome LPS Immunogenicity Contributes to Autoimmunity in Humans, Cell 165(4) (2016) 842–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].d’Hennezel E, Abubucker S, Murphy LO, Cullen TW, Total Lipopolysaccharide from the Human Gut Microbiome Silences Toll-Like Receptor Signaling, mSystems 2(6) (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Nolan JP, The role of intestinal endotoxin in liver injury: a long and evolving history, Hepatology 52(5) (2010) 1829–35. [DOI] [PubMed] [Google Scholar]
- [66].Lumsden AB, Henderson JM, Kutner MH, Endotoxin levels measured by a chromogenic assay in portal, hepatic and peripheral venous blood in patients with cirrhosis, Hepatology 8(2) (1988) 232–6. [DOI] [PubMed] [Google Scholar]
- [67].Violi F, Ferro D, Clotting activation and hyperfibrinolysis in cirrhosis: implication for bleeding and thrombosis, Semin Thromb Hemost 39(4) (2013) 426–33. [DOI] [PubMed] [Google Scholar]
- [68].Carnevale R, Raparelli V, Nocella C, Bartimoccia S, Novo M, Severino A, De Falco E, Cammisotto V, Pasquale C, Crescioli C, Scavalli AS, Riggio O, Basili S, Violi F, Gut-derived endotoxin stimulates factor VIII secretion from endothelial cells. Implications for hypercoagulability in cirrhosis, J Hepatol 67(5) (2017) 950–956. [DOI] [PubMed] [Google Scholar]
- [69].Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, Burcelin R, Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice, Diabetes 57(6) (2008) 1470–81. [DOI] [PubMed] [Google Scholar]
- [70].Ferro D, Quintarelli C, Lattuada A, Leo R, Alessandroni M, Mannucci PM, Violi F, High plasma levels of von Willebrand factor as a marker of endothelial perturbation in cirrhosis: relationship to endotoxemia, Hepatology 23(6) (1996) 1377–83. [DOI] [PubMed] [Google Scholar]
- [71].Wang X, Lipopolysaccharide augments venous and arterial thrombosis in the mouse, Thromb Res 123(2) (2008) 355–60. [DOI] [PubMed] [Google Scholar]
- [72].Mogensen TH, Pathogen recognition and inflammatory signaling in innate immune defenses, Clin Microbiol Rev 22(2) (2009) 240–73, Table of Contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].LP DA, Schattner M, Platelet toll-like receptors in thromboinflammation, Frontiers in bioscience (Landmark edition) 22 (2017) 1867–1883. [DOI] [PubMed] [Google Scholar]
- [74].Cognasse F, Hamzeh H, Chavarin P, Acquart S, Genin C, Garraud O, Evidence of Toll-like receptor molecules on human platelets, Immunol Cell Biol 83(2) (2005) 196–8. [DOI] [PubMed] [Google Scholar]
- [75].O’Neill LA, Golenbock D, Bowie AG, The history of Toll-like receptors - redefining innate immunity, Nat Rev Immunol 13(6) (2013) 453–60. [DOI] [PubMed] [Google Scholar]
- [76].Koupenova M, Mick E, Mikhalev E, Benjamin EJ, Tanriverdi K, Freedman JE, Sex differences in platelet toll-like receptors and their association with cardiovascular risk factors, Arteriosclerosis, thrombosis, and vascular biology 35(4) (2015) 1030–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Kawai T, Akira S, The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors, Nat Immunol 11(5) (2010) 373–84. [DOI] [PubMed] [Google Scholar]
- [78].Raparelli V, Basili S, Carnevale R, Napoleone L, Del Ben M, Nocella C, Bartimoccia S, Lucidi C, Talerico G, Riggio O, Violi F, Low-grade endotoxemia and platelet activation in cirrhosis, Hepatology 65(2) (2017) 571–581. [DOI] [PubMed] [Google Scholar]
- [79].Pan J, Dinh TT, Rajaraman A, Lee M, Scholz A, Czupalla CJ, Kiefel H, Zhu L, Xia L, Morser J, Jiang H, Santambrogio L, Butcher EC, Patterns of expression of factor VIII and von Willebrand factor by endothelial cell subsets in vivo, Blood 128(1) (2016) 104–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Zhang G, Han J, Welch EJ, Ye RD, Voyno-Yasenetskaya TA, Malik AB, Du X, Li Z, Lipopolysaccharide stimulates platelet secretion and potentiates platelet aggregation via TLR4/MyD88 and the cGMP-dependent protein kinase pathway, J Immunol 182(12) (2009) 7997–8004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Ward JR, Bingle L, Judge HM, Brown SB, Storey RF, Whyte MK, Dower SK, Buttle DJ, Sabroe I, Agonists of toll-like receptor (TLR)2 and TLR4 are unable to modulate platelet activation by adenosine diphosphate and platelet activating factor, Thromb Haemost 94(4) (2005) 831–8. [PubMed] [Google Scholar]
- [82].Shin HS, Xu F, Bagchi A, Herrup E, Prakash A, Valentine C, Kulkarni H, Wilhelmsen K, Warren S, Hellman J, Bacterial lipoprotein TLR2 agonists broadly modulate endothelial function and coagulation pathways in vitro and in vivo, J Immunol 186(2) (2011) 1119–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Blair P, Rex S, Vitseva O, Beaulieu L, Tanriverdi K, Chakrabarti S, Hayashi C, Genco CA, Iafrati M, Freedman JE, Stimulation of Toll-like receptor 2 in human platelets induces a thromboinflammatory response through activation of phosphoinositide 3-kinase, Circ Res 104(3) (2009) 346–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Rex S, Beaulieu LM, Perlman DH, Vitseva O, Blair PS, McComb ME, Costello CE, Freedman JE, Immune versus thrombotic stimulation of platelets differentially regulates signalling pathways, intracellular protein-protein interactions, and alpha-granule release, Thromb Haemost 102(1) (2009) 97–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Rivadeneyra L, Carestia A, Etulain J, Pozner RG, Fondevila C, Negrotto S, Schattner M, Regulation of platelet responses triggered by Toll-like receptor 2 and 4 ligands is another non-genomic role of nuclear factor-kappaB, Thromb Res 133(2) (2014) 235–43. [DOI] [PubMed] [Google Scholar]
- [86].Kälvegren H, Skoglund C, Helldahl C, Lerm M, Grenegård M, Bengtsson T, Toll-like receptor 2 stimulation of platelets is mediated by purinergic P2X1-dependent Ca2+ mobilisation, cyclooxygenase and purinergic P2Y1 and P2Y12 receptor activation, Thromb Haemost 103(2) (2010) 398–407. [DOI] [PubMed] [Google Scholar]
- [87].Engelmann B, Massberg S, Thrombosis as an intravascular effector of innate immunity, Nat Rev Immunol 13(1) (2013) 34–45. [DOI] [PubMed] [Google Scholar]
- [88].Horne BD, Anderson JL, John JM, Weaver A, Bair TL, Jensen KR, Renlund DG, Muhlestein JB, Which white blood cell subtypes predict increased cardiovascular risk?, Journal of the American College of Cardiology 45(10) (2005) 1638–43. [DOI] [PubMed] [Google Scholar]
- [89].Fuchs TA, Brill A, Wagner DD, Neutrophil extracellular trap (NET) impact on deep vein thrombosis, Arteriosclerosis, thrombosis, and vascular biology 32(8) (2012) 1777–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Thomas GM, Carbo C, Curtis BR, Martinod K, Mazo IB, Schatzberg D, Cifuni SM, Fuchs TA, von Andrian UH, Hartwig JH, Aster RH, Wagner DD, Extracellular DNA traps are associated with the pathogenesis of TRALI in humans and mice, Blood 119(26) (2012) 6335–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Semeraro F, Ammollo CT, Morrissey JH, Dale GL, Friese P, Esmon NL, Esmon CT, Extracellular histones promote thrombin generation through platelet-dependent mechanisms: involvement of platelet TLR2 and TLR4, Blood 118(7) (2011) 1952–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Massberg S, Grahl L, von Bruehl ML, Manukyan D, Pfeiler S, Goosmann C, Brinkmann V, Lorenz M, Bidzhekov K, Khandagale AB, Konrad I, Kennerknecht E, Reges K, Holdenrieder S, Braun S, Reinhardt C, Spannagl M, Preissner KT, Engelmann B, Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases, Nat Med 16(8) (2010) 887–96. [DOI] [PubMed] [Google Scholar]
- [93].Pham CT, Neutrophil serine proteases: specific regulators of inflammation, Nat Rev Immunol 6(7) (2006) 541–50. [DOI] [PubMed] [Google Scholar]
- [94].von Brühl ML, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, Khandoga A, Tirniceriu A, Coletti R, Köllnberger M, Byrne RA, Laitinen I, Walch A, Brill A, Pfeiler S, Manukyan D, Braun S, Lange P, Riegger J, Ware J, Eckart A, Haidari S, Rudelius M, Schulz C, Echtler K, Brinkmann V, Schwaiger M, Preissner KT, Wagner DD, Mackman N, Engelmann B, Massberg S, Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo, J Exp Med 209(4) (2012) 819–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Sawa Y, Ueki T, Hata M, Iwasawa K, Tsuruga E, Kojima H, Ishikawa H, Yoshida S, LPS-induced IL-6, IL-8, VCAM-1, and ICAM-1 expression in human lymphatic endothelium, The journal of histochemistry and cytochemistry : official journal of the Histochemistry Society 56(2) (2008) 97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Obi AT, Andraska E, Kanthi Y, Kessinger CW, Elfline M, Luke C, Siahaan TJ, Jaffer FA, Wakefield TW, Henke PK, Endotoxaemia-augmented murine venous thrombosis is dependent on TLR-4 and ICAM-1, and potentiated by neutropenia, Thromb Haemost 117(2) (2017) 339–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Brill A, Fuchs TA, Savchenko AS, Thomas GM, Martinod K, De Meyer SF, Bhandari AA, Wagner DD, Neutrophil extracellular traps promote deep vein thrombosis in mice, J Thromb Haemost 10(1) (2012) 136–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Semple JW, Aslam R, Kim M, Speck ER, Freedman J, Platelet-bound lipopolysaccharide enhances Fc receptor-mediated phagocytosis of IgG-opsonized platelets, Blood 109(11) (2007) 4803–5. [DOI] [PubMed] [Google Scholar]
- [99].Furie B, Furie BC, Mechanisms of thrombus formation, The New England journal of medicine 359(9) (2008) 938–49. [DOI] [PubMed] [Google Scholar]
- [100].Wu Y, Contact pathway of coagulation and inflammation, Thromb J 13 (2015) 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Toossi Z, Sedor JR, Mettler MA, Everson B, Young T, Ratnoff OD, Induction of expression of monocyte interleukin 1 by Hageman factor (factor XII), Proc Natl Acad Sci U S A 89(24) (1992) 11969–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Gobel K, Pankratz S, Asaridou CM, Herrmann AM, Bittner S, Merker M, Ruck T, Glumm S, Langhauser F, Kraft P, Krug TF, Breuer J, Herold M, Gross CC, Beckmann D, Korb-Pap A, Schuhmann MK, Kuerten S, Mitroulis I, Ruppert C, Nolte MW, Panousis C, Klotz L, Kehrel B, Korn T, Langer HF, Pap T, Nieswandt B, Wiendl H, Chavakis T, Kleinschnitz C, Meuth SG, Blood coagulation factor XII drives adaptive immunity during neuroinflammation via CD87-mediated modulation of dendritic cells, Nat Commun 7 (2016) 11626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Grover SP, Mackman N, Tissue Factor: An Essential Mediator of Hemostasis and Trigger of Thrombosis, Arteriosclerosis, thrombosis, and vascular biology 38(4) (2018) 709–725. [DOI] [PubMed] [Google Scholar]
- [104].Nagata S, Suzuki J, Segawa K, Fujii T, Exposure of phosphatidylserine on the cell surface, Cell Death Differ 23(6) (2016) 952–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Yang X, Cheng X, Tang Y, Qiu X, Wang Y, Kang H, Wu J, Wang Z, Liu Y, Chen F, Xiao X, Mackman N, Billiar TR, Han J, Lu B, Bacterial Endotoxin Activates the Coagulation Cascade through Gasdermin D-Dependent Phosphatidylserine Exposure, Immunity 51(6) (2019) 983–996.e6. [DOI] [PubMed] [Google Scholar]
- [106].Pawlinski R, Wang J-G, Owens AP 3rd, Williams J, Antoniak S, Tencati M, Luther T, Rowley JW, Low EN, Weyrich AS, Mackman N, Hematopoietic and nonhematopoietic cell tissue factor activates the coagulation cascade in endotoxemic mice, Blood 116(5) (2010) 806–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Zarbock A, Ley K, McEver RP, Hidalgo A, Leukocyte ligands for endothelial selectins: specialized glycoconjugates that mediate rolling and signaling under flow, Blood 118(26) (2011) 6743–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Carestia A, Kaufman T, Rivadeneyra L, Landoni VI, Pozner RG, Negrotto S, D’Atri LP, Gomez RM, Schattner M, Mediators and molecular pathways involved in the regulation of neutrophil extracellular trap formation mediated by activated platelets, J Leukoc Biol 99(1) (2016) 153–62. [DOI] [PubMed] [Google Scholar]
- [109].Ao L, Song Y, Fullerton DA, Dinarello CA, Meng X, The interaction between myocardial depressant factors in endotoxemic cardiac dysfunction: role of TNF-alpha in TLR4-mediated ICAM-1 expression, Cytokine 38(3) (2007) 124–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Schattner M, Platelet TLR4 at the crossroads of thrombosis and the innate immune response, J Leukoc Biol (2018). [DOI] [PubMed] [Google Scholar]
- [111].Nicholls AW, Mortishire-Smith RJ, Nicholson JK, NMR spectroscopic-based metabonomic studies of urinary metabolite variation in acclimatizing germ-free rats, Chemical research in toxicology 16(11) (2003) 1395–404. [DOI] [PubMed] [Google Scholar]
- [112].Zhu Y, Jameson E, Crosatti M, Schafer H, Rajakumar K, Bugg TD, Chen Y, Carnitine metabolism to trimethylamine by an unusual Rieske-type oxygenase from human microbiota, Proc Natl Acad Sci U S A 111(11) (2014) 4268–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Lindskog Jonsson A, Caesar R, Akrami R, Reinhardt C, Fak Hallenius F, Boren J, Backhed F, Impact of Gut Microbiota and Diet on the Development of Atherosclerosis in Apoe(−/−) Mice, Arteriosclerosis, thrombosis, and vascular biology 38(10) (2018) 2318–2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Koeth RA, Wang Z, Levison BS, Buffa JA, Org E, Sheehy BT, Britt EB, Fu X, Wu Y, Li L, Smith JD, DiDonato JA, Chen J, Li H, Wu GD, Lewis JD, Warrier M, Brown JM, Krauss RM, Tang WH, Bushman FD, Lusis AJ, Hazen SL, Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis, Nat Med 19(5) (2013) 576–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Falony G, Vieira-Silva S, Raes J, Microbiology Meets Big Data: The Case of Gut Microbiota-Derived Trimethylamine, Annu Rev Microbiol 69 (2015) 305–321. [DOI] [PubMed] [Google Scholar]
- [116].Craciun S, Balskus EP, Microbial conversion of choline to trimethylamine requires a glycyl radical enzyme, Proceedings of the National Academy of Sciences of the United States of America 109(52) (2012) 21307–21312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Ma G, Pan B, Chen Y, Guo C, Zhao M, Zheng L, Chen B, Trimethylamine N-oxide in atherogenesis: impairing endothelial self-repair capacity and enhancing monocyte adhesion, Bioscience reports 37(2) (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Seldin MM, Meng Y, Qi H, Zhu W, Wang Z, Hazen SL, Lusis AJ, Shih DM, Trimethylamine N-Oxide Promotes Vascular Inflammation Through Signaling of Mitogen-Activated Protein Kinase and Nuclear Factor-kappaB, Journal of the American Heart Association 5(2) (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Sun X, Jiao X, Ma Y, Liu Y, Zhang L, He Y, Chen Y, Trimethylamine N-oxide induces inflammation and endothelial dysfunction in human umbilical vein endothelial cells via activating ROS-TXNIP-NLRP3 inflammasome, Biochem Biophys Res Commun 481(1–2) (2016) 63–70. [DOI] [PubMed] [Google Scholar]
- [120].Tang WH, Wang Z, Levison BS, Koeth RA, Britt EB, Fu X, Wu Y, Hazen SL, Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk, The New England journal of medicine 368(17) (2013) 1575–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Smolinska A, Tedjo DI, Blanchet L, Bodelier A, Pierik MJ, Masclee AAM, Dallinga J, Savelkoul PHM, Jonkers D, Penders J, van Schooten FJ, Volatile metabolites in breath strongly correlate with gut microbiome in CD patients, Analytica chimica acta 1025 (2018) 1–11. [DOI] [PubMed] [Google Scholar]
- [122].Sayin SI, Wahlstrom A, Felin J, Jantti S, Marschall HU, Bamberg K, Angelin B, Hyotylainen T, Oresic M, Backhed F, Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist, Cell Metab 17(2) (2013) 225–35. [DOI] [PubMed] [Google Scholar]
- [123].Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, Nakanishi Y, Uetake C, Kato K, Kato T, Takahashi M, Fukuda NN, Murakami S, Miyauchi E, Hino S, Atarashi K, Onawa S, Fujimura Y, Lockett T, Clarke JM, Topping DL, Tomita M, Hori S, Ohara O, Morita T, Koseki H, Kikuchi J, Honda K, Hase K, Ohno H, Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells, Nature 504(7480) (2013) 446–50. [DOI] [PubMed] [Google Scholar]
- [124].Singh V, Chassaing B, Zhang L, San Yeoh B, Xiao X, Kumar M, Baker MT, Cai J, Walker R, Borkowski K, Harvatine KJ, Singh N, Shearer GC, Ntambi JM, Joe B, Patterson AD, Gewirtz AT, Vijay-Kumar M, Microbiota-Dependent Hepatic Lipogenesis Mediated by Stearoyl CoA Desaturase 1 (SCD1) Promotes Metabolic Syndrome in TLR5-Deficient Mice, Cell Metab 22(6) (2015) 983–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Ahmad A, Dwivedi G, O’Gara F, Caparros-Martin J, Ward N, The gut microbiome and atherosclerosis: current knowledge and clinical potential, Am J Physiol Heart Circ Physiol (2019). [DOI] [PubMed] [Google Scholar]
- [126].Romano KA, Vivas EI, Amador-Noguez D, Rey FE, Intestinal microbiota composition modulates choline bioavailability from diet and accumulation of the proatherogenic metabolite trimethylamine-N-oxide, mBio 6(2) (2015) e02481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Collins HL, Drazul-Schrader D, Sulpizio AC, Koster PD, Williamson Y, Adelman SJ, Owen K, Sanli T, Bellamine A, L-Carnitine intake and high trimethylamine N-oxide plasma levels correlate with low aortic lesions in ApoE(−/−) transgenic mice expressing CETP, Atherosclerosis 244 (2016) 29–37. [DOI] [PubMed] [Google Scholar]
- [128].Reiner MF, Muller D, Gobbato S, Stalder O, Limacher A, Bonetti NR, Pasterk L, Mean M, Rodondi N, Aujesky D, Angelillo-Scherrer A, Matter CM, Luscher TF, Camici GG, von Eckardstein A, Beer JH, Gut microbiota-dependent trimethylamine-N-oxide (TMAO) shows a U-shaped association with mortality but not with recurrent venous thromboembolism, Thromb Res 174 (2018) 40–47. [DOI] [PubMed] [Google Scholar]
- [129].Hickson M, D’Souza AL, Muthu N, Rogers TR, Want S, Rajkumar C, Bulpitt CJ, Use of probiotic Lactobacillus preparation to prevent diarrhoea associated with antibiotics: randomised double blind placebo controlled trial, BMJ (Clinical research ed.) 335(7610) (2007) 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Allen SJ, Wareham K, Wang D, Bradley C, Hutchings H, Harris W, Dhar A, Brown H, Foden A, Gravenor MB, Mack D, Lactobacilli and bifidobacteria in the prevention of antibiotic-associated diarrhoea and Clostridium difficile diarrhoea in older inpatients (PLACIDE): a randomised, double-blind, placebo-controlled, multicentre trial, Lancet (London, England) 382(9900) (2013) 1249–57. [DOI] [PubMed] [Google Scholar]
- [131].Ubeda C, Taur Y, Jenq RR, Equinda MJ, Son T, Samstein M, Viale A, Socci ND, van den Brink MR, Kamboj M, Pamer EG, Vancomycin-resistant Enterococcus domination of intestinal microbiota is enabled by antibiotic treatment in mice and precedes bloodstream invasion in humans, The Journal of clinical investigation 120(12) (2010) 4332–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].van Nood E, Vrieze A, Nieuwdorp M, Fuentes S, Zoetendal EG, de Vos WM, Visser CE, Kuijper EJ, Bartelsman JF, Tijssen JG, Speelman P, Dijkgraaf MG, Keller JJ, Duodenal infusion of donor feces for recurrent Clostridium difficile, The New England journal of medicine 368(5) (2013) 407–15. [DOI] [PubMed] [Google Scholar]
- [133].de Groot PF, Frissen MN, de Clercq NC, Nieuwdorp M, Fecal microbiota transplantation in metabolic syndrome: History, present and future, Gut Microbes 8(3) (2017) 253–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Vrieze A, Van Nood E, Holleman F, Salojarvi J, Kootte RS, Bartelsman JF, Dallinga-Thie GM, Ackermans MT, Serlie MJ, Oozeer R, Derrien M, Druesne A, Van Hylckama Vlieg JE, Bloks VW, Groen AK, Heilig HG, Zoetendal EG, Stroes ES, de Vos WM, Hoekstra JB, Nieuwdorp M, Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome, Gastroenterology 143(4) (2012) 913–6.e7 [DOI] [PubMed] [Google Scholar]
- [135].Taur Y, Coyte K, Schluter J, Robilotti E, Figueroa C, Gjonbalaj M, Littmann ER, Ling L, Miller L, Gyaltshen Y, Fontana E, Morjaria S, Gyurkocza B, Perales MA, Castro-Malaspina H, Tamari R, Ponce D, Koehne G, Barker J, Jakubowski A, Papadopoulos E, Dahi P, Sauter C, Shaffer B, Young JW, Peled J, Meagher RC, Jenq RR, van den Brink MRM, Giralt SA, Pamer EG, Xavier JB, Reconstitution of the gut microbiota of antibiotic-treated patients by autologous fecal microbiota transplant, Sci Transl Med 10(460) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Song Y, Garg S, Girotra M, Maddox C, von Rosenvinge EC, Dutta A, Dutta S, Fricke WF, Microbiota dynamics in patients treated with fecal microbiota transplantation for recurrent Clostridium difficile infection, PLoS One 8(11) (2013) e81330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Mohammed Y, Kootte RS, Kopatz WF, Borchers CH, Büller HR, Versteeg HH, Nieuwdorp M, van Mens TE, The intestinal microbiome potentially affects thrombin generation in human subjects, Journal of thrombosis and haemostasis : JTH (2019) 10.1111/jth.14699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Tidswell M, Tillis W, Larosa SP, Lynn M, Wittek AE, Kao R, Wheeler J, Gogate J, Opal SM, Phase 2 trial of eritoran tetrasodium (E5564), a toll-like receptor 4 antagonist, in patients with severe sepsis, Critical care medicine 38(1) (2010) 72–83. [DOI] [PubMed] [Google Scholar]
- [139].Rice TW, Wheeler AP, Bernard GR, Vincent JL, Angus DC, Aikawa N, Demeyer I, Sainati S, Amlot N, Cao C, Ii M, Matsuda H, Mouri K, Cohen J, A randomized, double-blind, placebo-controlled trial of TAK-242 for the treatment of severe sepsis, Critical care medicine 38(8) (2010) 1685–94. [DOI] [PubMed] [Google Scholar]
- [140].Gioannini TL, Teghanemt A, Zhang D, Coussens NP, Dockstader W, Ramaswamy S, Weiss JP, Isolation of an endotoxin-MD-2 complex that produces Toll-like receptor 4-dependent cell activation at picomolar concentrations, Proc Natl Acad Sci U S A 101(12) (2004) 4186–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Kim D, Kim JY, Anti-CD14 antibody reduces LPS responsiveness via TLR4 internalization in human monocytes, Molecular immunology 57(2) (2014) 210–5. [DOI] [PubMed] [Google Scholar]
- [142].Feingold KR, Grunfeld C, The acute phase response inhibits reverse cholesterol transport, J Lipid Res 51(4) (2010) 682–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Almog Y, Shefer A, Novack V, Maimon N, Barski L, Eizinger M, Friger M, Zeller L, Danon A, Prior statin therapy is associated with a decreased rate of severe sepsis, Circulation 110(7) (2004) 880–5. [DOI] [PubMed] [Google Scholar]
- [144].Kruger P, Bailey M, Bellomo R, Cooper DJ, Harward M, Higgins A, Howe B, Jones D, Joyce C, Kostner K, McNeil J, Nichol A, Roberts MS, Syres G, Venkatesh B, A multicenter randomized trial of atorvastatin therapy in intensive care patients with severe sepsis, American journal of respiratory and critical care medicine 187(7) (2013) 743–50. [DOI] [PubMed] [Google Scholar]
- [145].Roy A, Srivastava M, Saqib U, Liu D, Faisal SM, Sugathan S, Bishnoi S, Baig MS, Potential therapeutic targets for inflammation in toll-like receptor 4 (TLR4)-mediated signaling pathways, International immunopharmacology 40 (2016) 79–89. [DOI] [PubMed] [Google Scholar]
- [146].Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, Kastelein JJP, Cornel JH, Pais P, Pella D, Genest J, Cifkova R, Lorenzatti A, Forster T, Kobalava Z, Vida-Simiti L, Flather M, Shimokawa H, Ogawa H, Dellborg M, Rossi PRF, Troquay RPT, Libby P, Glynn RJ, Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease, The New England journal of medicine 377(12) (2017) 1119–1131. [DOI] [PubMed] [Google Scholar]
- [147].Dolphin CT, Janmohamed A, Smith RL, Shephard EA, Phillips IR, Missense mutation in flavin-containing mono-oxygenase 3 gene, FMO3, underlies fish-odour syndrome, Nature genetics 17(4) (1997) 491–4. [DOI] [PubMed] [Google Scholar]