

Abstract
Alzheimer’s disease (AD) is a current public health challenge and will remain until the development of an effective intervention. However, developing an effective treatment for the disease requires a thorough understanding of its etiology, which is currently lacking. Although several studies have shown the association between oxidative damage and AD, only a few have clarified the specific mechanisms involved. Herein, we reviewed recent preclinical and clinical studies that indicated the significance of oxidative damage in AD, as well as potential antioxidants. Although several factors regulate oxidative stress in AD, we centered our investigation on apolipoprotein E and the gut microbiome. Apolipoprotein E, particularly apolipoprotein E-ε4, can impair the structural facets of the mitochondria. This, in turn, can minimize the mitochondrial functionality and result in the progressive build-up of free radicals, eventually leading to oxidative stress. Similarly, the gut microbiome can influence oxidative stress to a significant degree via its metabolite, trimethylamine N-oxide. Given the various roles of these two factors in modulating oxidative stress, we also discuss the possible relationship between them and provide future research directions.
Keywords: Neurodegeneration, gut-brain axis, reactive oxygen species, antioxidants, mitochondria dysfunction
1.Introduction
A progressive cognitive impairment is a significant indication of Alzheimer’s disease (hereafter: AD). AD has three main stages: preclinical AD, mild cognitive impairment, and dementia [1, 2]. In the United States, one out of every ten people aged 65 and up has AD [2]. The vast number of people affected with AD globally places a considerable strain on health resources. For example, as the disease progresses, patients become reliant on close family members, healthcare professionals, and caregivers to carry out their daily activities. The current medications for mild-to-moderate AD patients are cholinesterase inhibitors (galantamine, rivastigmine, and donepezil). Moderate-to-severe AD patients use memantine. Memantine suppresses the toxic effects of excess glutamate while simultaneously controlling its activation. Also, Namzaric (memantine + donepezil) is used to treat symptoms of moderate-to-severe AD. Unfortunately, current treatments merely deal with the symptoms, and medications are used cautiously due to their numerous adverse effects. Moreover, the efficacy of these medications decreases as the disease progresses [3].
Although enormous efforts have been undertaken to determine AD’s underlying causative and pathophysiological factors, these investigations have been largely inconclusive. However, recent studies such as the potentiality of 40-Hz light flickers as an interventional therapy and early detection of the disease via blood plasma have shown promising results [4, 5]. The latter study is particularly appealing because it evaluated potential disease biomarkers in a large number of cognitively impaired and unimpaired individuals, with their aberrancy associated with neurodegeneration [5]. Given the limited availability of biological indicators for monitoring AD development, blood plasma could be beneficial in studying the disease trajectory in both preclinical and clinical settings.
Mitochondria produce the ATP (adenosine triphosphate) needed for various physiological functions. Endoplasmic reticulum cytochrome P450, nicotinamide adenine dinucleotide phosphate-oxidase (NOX), and xanthine oxidase (XO) are among the mitochondrial and non-mitochondrial enzymes implicated in the endogenous free radical production via the electron transport chain [6]. Besides, the mitochondria regulate intracellular Ca2+ homeostasis and produce molecules that influence cellular apoptosis [7]. The inositol 1,4,5-triphosphate receptor (IP3R) regulates Ca2+ homeostasis and promotes increased intracellular Ca2+ by releasing Ca2+ from the endoplasmic reticulum (ER). As intracellular [Ca2+] levels rise, mitochondria absorb it through the intracellular mitochondria-associated membranes (MAMs). Also, Ca2+ exchange between the ER and mitochondria modulates mitochondrial respiration [8, 9], with its aftereffect being ROS (reactive oxygen species) formation [10].
Reactive oxygen species (ROS) are unstable, highly reactive species formed predominantly in the cell’s mitochondria as by-products of cellular metabolism. Oxygen-derived ROS include reactive nitrogen species (RNS), hydroxyl radicals, and superoxide anions. Hydrogen peroxide (H2O2) is an example of non-free radical ROS [11]. ROS (such as H2O2) are essential for cellular signaling in moderate quantities. For instance, low amounts of H2O2 promote cell growth in cancer and non-transformed stem cells [12]. Moreover, H2O2 is a second messenger that modulates macrophagic functions during the respiratory burst, which is notable in inflammation [13]. Nonetheless, chronic inflammation causes the overproduction of free radical species and leads to oxidative stress, impairing the neuronal membrane [14, 15].
ROS can be produced both exogenously (through medicines) and endogenously by the brain. The thioredoxin (Trx) system, which includes thioredoxin reductase (TrxR) and nicotinamide adenine dinucleotide phosphate hydrogen (NADPH), is essential for regulating ROS levels in the brain. The counteractive measures of the TrxR system in controlling ROS may be due to the minimal catalase expression in the brain [16]. Catalase is an antioxidant that can neutralize aberrant amounts of ROS to prevent oxidative stress. The brain’s reduced catalase is unsurprising, given that it requires high glucose metabolism and oxygen consumption for physiological activities. Thus, it is predisposed to oxidative stress due to higher ROS levels concomitant with limited counter-regulatory measures. In addition, biomolecules such as lipids are copious in the brain, increasing their susceptibility to oxidation in the presence of ROS [17].
The fundamental pathological features of AD comprise accumulated β-amyloid and tau protein phosphorylation. β-amyloid, in particular, causes neuronal damage and oxidative stress by disrupting the oxidant-antioxidant system, reducing synaptic plasticity, impairing metabolism, and inducing neuroinflammation [18]. On the other hand, an in-vitro study showed oxidative stress to aggravate soluble β-amyloid precursor protein and coincided with increased vascular endothelial growth factor (VEGF) [19].
Suggestions that a combination of several factors may cause AD have been proposed. Therefore, we conducted a systematic review to ascertain the role of apolipoprotein E (APOE) and gut microbiome dyshomeostasis in oxidative stress in AD. Also, we examined several potential antioxidants currently being explored for AD. Although the various sections of our theme are narrow, we aim to shed light and expand the horizon on the current understanding of the relationship between APOE, gut microbiome, and oxidative damage in AD.
2.Methods
2.1. Data sources and search terms
Publication databases and academic search engines, such as PubMed, Web of Science, and Scopus, were scoured extensively for studies used in this report. We conducted the searches at two different times. Two independent reviewers performed the first search (Benson and Favour) between July and August 2020. One independent reviewer (William) did the second search in January 2021. Search terms used were “Alzheimer disease” or “Neuro-degenerative disease” or “Neurodegeneration” and “oxidative stress” or “oxidative damage” or “gut microbiome oxidative stress” or “gut microbiome Alzheimer’s disease” or “gut microbiome neuro-degeneration” or “APOE and gut microbiome”. In addition, we narrowed our search to publications in English, with great emphasis on preclinical and clinical studies published within the last five years. Also, we prioritized in-vivo investigations over in-vitro.
In section 6, we used the clinicaltrials.gov database with the search terms “antioxidants for Alzheimer disease” together with filters “completed” (under ‘Status’), “studies with results” (under ‘Study Results’), and “approved for marketing” (under ‘Expanded Access below Status’) that yielded eight results. After, we amended the search terms to include studies that were currently in the recruiting stages. Thus, the search terms “antioxidant” and “recruiting studies” (under status) and “Alzheimer’s disease” generated ten studies.
2.2. Selection criteria
To include an article in the review, we used three indicators: 1) the study had to be original, along with the employment of appropriate animal models (either transgenic or drug-induced) and human samples; 2) the study had to involve both control and experimental groups; and 3) the study had to address the pathophysiological alterations (either β-amyloid plaques or neurofibrillary tangles), together with oxidative stress biomarkers. We omitted studies having the following criteria: 1) review articles; 2) non-experimental studies; 3) those with unavailable full texts, and 4) studies published over a decade ago. We consulted one reviewer (Yili or Marong) to discuss any disputes regarding a study inclusion or exclusion (supplementary Fig. 1).
3.Mitochondrial dysfunction and oxidative stress
Several factors contribute to mitochondrial dysfunction (MtD). Sirtuins (SIRTs), specifically SIRT3, are one example. SIRT3, abundant in the brain, is associated with mitochondrial function, aging, and longevity. Moreover, they regulate glutathione (GSH) [20]. Mitochondrial complex 1, which contains mitochondrial-NADH dehydrogenases 2/4 (MT-ND2 and MT-ND4), controls intracellular ROS levels. Reduced SIRT3 activity resulted in downregulated MT-ND2 and MT-ND4 levels, with concomitant effects of ROS overproduction, cytochrome c release, and neurodegeneration [21]. Cytochrome C is a pro-apoptotic factor, and studies have thoroughly investigated and reviewed the association between apoptosis and neurodegeneration [22-24]. Furthermore, recent studies have shown that elevated Ca2+ levels within the mitochondria cause neuronal death via free radical formation [25-26]. These findings are intriguing because Ca2+ accumulation in the mitochondria induces cytochrome c release into the cytosol via a stimulated mitochondrial-permeability transition pore (mPTP), with cytochrome c activating the caspase cascade to promote apoptosis [8-9]. Moreover, oxidative stress causes continuous alterations in the mitochondrial structure. For instance, prolonged oxidative stress disrupts functional tubular mitochondrial networks, resulting in mitochondrial fragmentation and apoptosis, decreased ATP production, energy metabolism, and an inability to meet neuronal cell energy demands [7]. Neuronal death occurs as a result of the limited availability of energy.
In the same way that mitochondria generate ROS, they also have mechanisms to handle ROS formation. For example, GSH peroxidase (GPx) is a free radical scavenger that regulates H2O2 within the mitochondria [27]. Similarly, copper-zinc superoxide dismutase 1 (SOD1) and copper chaperone for SOD1 (CCS) neutralizes mitochondria-generated free radicals [28]. However, damage to these mitochondrial antioxidative mechanisms might result in free radical aberrancy, and eventually, oxidative stress. For example, in the Haber-Weiss reaction, sustained elevated H2O2 levels within the mitochondria react with superoxide ions to form an even more reactive hydroxyl radical. In such an event, the cell’s fate is oxidative insults to proteins, mitochondrial DNA, and mPTP activation, which results in MtD and eventual cell death [13, 29].
mPTP appears to be a vital element in MtD, yet its structure and components are not well understood. However, we speculate that mPTP activation caused by either Ca2+ overload, stress, or unknown factors may result in the release of antioxidant systems from the mitochondria. Because mitochondria are the primary source of free radical production, the limited number of antioxidants that remain will be gradually overwhelmed by the constant formation of free radicals, ultimately causing oxidative stress and expediting related diseases, such as AD.
3.1.Mitochondrial dysfunction, oxidative stress, and Alzheimer's disease
Oxidative stress due to MtD has been implicated in AD, given that MtD is the chief instigator of free radical formation. The loss of mitochondrial function affects APP expression and processing, resulting in β-amyloid accumulation [30]. The build-up of intracellular β-amyloid in the mitochondria leads to its dysfunction. Furthermore, β-amyloid interaction with various mitochondrial proteins, the inner mitochondrial membrane, and the matrix impairs oxidative phosphorylation, increasing ROS synthesis [31]. The observed early manifestation of oxidative stress in AD could underlie its role in the disease etiology [17, 32]. Increased β-amyloid levels have been long linked to high degrees of oxidative proteins, lipids, and nucleic acids in the AD hippocampus and cortex [33]. Moreover, there is a relationship between AD and nuclear and mitochondrial DNA oxidation, as higher oxidized base (such as 8-hydroxyguanine and 5-hydroxyuracil) levels have been observed in the frontal, parietal, and temporal lobes of the brain [34-35]. Besides, a report indicated an elevated 8-hydroxyguanine in the hippocampus of preclinical stage AD patients [36].
MtD and tau pathology have a positive feedback interaction. The delivery of mitochondria in neurons is critical due to the importance of the mitochondria in maintaining neuronal function. Microtubule-associated proteins, like tau, transport mitochondria across the axon into synapses [37]. Overexpression and tau hyper-phosphorylation affect its localization and distribution [38, 39], impairing axonal transport, and causing synaptic loss [37]. Furthermore, hyperphosphorylated tau undermines mitochondrial function by lowering antioxidant activity. The resultant effect of this is oxidative stress, which hinders ATP production and causes synaptic dysfunction [40]. A preclinical investigation showed MtD to result from apoptosis, oxidative stress and upregulated voltage-dependent anion channel 1 protein (VDAC1). Interestingly, an upregulated VDAC1 was also noted in post-mortem AD brains [41], indicating that it may play a role in MtD. The question as to whether prospective VDAC1 inhibitors could alleviate MtD in AD is a future research topic that warrants thorough investigations using multiple preclinical models of AD.
4.Apolipoprotein E: Function and role in AD
Cholesterol is an organic molecule that is required for life sustenance and is generated chiefly in the liver. It maintains cell wall integrity and acts as a building block for many hormones (such as testosterone, cortisol, and aldosterone). Lipoproteins comprise apolipoprotein and lipids and transport cholesterol through the bloodstream [1, 42]. Apolipoproteins include apolipoprotein A-1 (APOA-I), apolipoprotein B-48 (APOB-48), apolipoprotein C-II (APOC-II), and apolipoprotein E (APOE). Their differences correspond to diverse molecular weights and functions [42]. For instance, APOC-II functions in energy delivery and storage through its hydrolyzation of chylomicrons and very-low-density lipoprotein (VLDL) [43]. We have previously reported the multiple polymorphic alleles of APOE and their frequency in humans [1]. APOE-ε4, in particular, has been associated with late-onset AD [44-46]. Furthermore, several cardiovascular and neurological diseases have shown the involvement of APOE [47-50]. The different APOE confer variable risks to AD. Although the precise reason for this variation has yet to be determined, several studies have attempted to provide some clarifications. These clarifications span from dysfunctions of the insulin signaling and blood-brain barrier (BBB) to levels of APOE concentration, cholesterol, and β-amyloid secretion [51-57]. For example, Chan and colleagues discovered that APOEε4-APP mice have much higher hippocampal β-amyloid levels than APOE-ε3-APP mice, which correlated with impaired insulin signaling and poor spatial memory [53]. The impaired insulin signaling pathway could be related to IGF-1 (insulin-like growth factor 1), IDE (insulin-degrading enzyme), and GLUT-4 (Glucose transporter type 4) reduced in significant amounts in the brains of animals carrying the APOE-ε4 allele [54]. Furthermore, Shinohara and colleagues discovered that APOE-ε2-TR mice have higher APOE in their CSF, cortex, hippocampus, and plasma than APOE-ε3 & ε4-TR mice. When cholesterol was measured, the CSF and plasma showed a similar pattern, and the brain cortex exhibited opposing results. The previous findings resulted in better memory function in APOE-ε2-TR mice than their age-matched counterparts [52]. Thus, there is the likelihood that higher APOE concentrations in AD might require a protective functioning gene (like APOE-ε2) to control brain cholesterol effectively and improve cognition, which may reduce AD risk. The findings from Shinohara et al. study [52] are supported by a recent study that examined over 5000 post-mortem brain tissues and reported a lower risk of homozygous APOE-ε2 people developing AD [58].
The BBB impermeability acts as a protective mechanism for the brain, preventing circulating bacteria and viruses from entering the brain. In a neurological condition, this protective mechanism may hinder therapeutic drug delivery for favorable consequences. Hence, the BBB mechanism is a double-edged sword. Dysregulated BBB in AD and other neurological diseases has been widely studied [55-57]. Particularly in AD, the involvement of the APOE-ε4 allele in BBB impairment has been evidenced, with the stimulation of the CypA-MMP9 signaling pathway touted as the potential mediator [59]. Therefore, future studies targeting this signaling pathway as an early interventional approach in halting or reversing cognitive dysfunction in AD might be worthwhile.
Sortilin is an important APOE metabolic regulator, and its role in AD is ambiguous. For instance, the interplay of APOE-ε3 and sortilin resulted in the modulation of omega-3 lipids. This finding corresponded with minimized levels of pro-inflammatory (TNF-α) and astrocytic (GFAP) markers. Not surprising, the opposite was true in APOE-ε4’s interaction with sortilin [60]. Furthermore, several human investigations have found disparate single nucleotide polymorphisms of the SORT1 (sortilin 1) gene; for example, rs17646665 lowers AD risk, while rs1010159 appears to increase the risk of mild cognitive impairment (MCI) progressing to AD [61, 62]. Notwithstanding, the SNP rs1764665 finding from the above study should be interpreted with caution due to its lack of correlation with conventional AD biomarkers, like β-amyloid and phosphorylated tau in cerebrospinal fluid (CSF) [62]. More intriguingly, previous studies also found no link between this genetic mutation and AD [63, 64]. Thus, SNP rs1764665 lower risk of AD may be relevant to those who live in a specific region, specifically Scandinavia, and maybe Europe.
4.1.Apolipoprotein E and oxidative stress
Several reasons explain the link between APOE and oxidative stress, one being APOE’s unique thiol-mediated antioxidant actions. Thiols, such as cysteine and GSH, are free radical scavengers with well-studied functional roles [65, 66]. Thiol levels are significantly reduced in AD [67, 68], leading to disease instigation or exacerbation. While APOE-ε3 and APOE-ε2 have corresponding one and two thiol-free groups, APOE-ε4 has none. The thiol groups of APOE-ε2 occupy positions 112 and 158 of the N-terminus. Likewise, that of APOE-ε3 is at 112 of the N-terminus [69]. Because the number of thiol groups corresponds to its antioxidant capabilities, a compound with more free thiol groups will have more robust antioxidant activity. On that basis, the absence of thiol-mediated activities in APOE-ε4 might compel the oxidation of biomolecules and enhance β-amyloid aggregation [70]. Similarly, the presence of two free thiol groups in APOE-ε2 may underlie its protectiveness in AD.
Superoxide dismutase (SOD) is a potent antioxidant enzyme that comes in three types: SOD1, SOD2, and SOD3. SODs - 1, 2, and 3 are in the cytoplasm, mitochondria, and extracellular space, respectively, and have varying physiological roles. A study demonstrated a positive relationship between SOD level and longevity in species like Schizosaccharomyces pombe and Lasius niger [71, 72]. Several studies have reported dysregulated SOD enzymes in both MCI and AD. Various biological markers, such as advanced oxidation protein products (AOPP) and ferric reducing antioxidant power (FRAP), are used to assess the degree of oxidative insults in both preclinical and clinical studies. Chico and colleagues evaluated the oxidative damage in 119 patients with MCI and AD. Relative to healthy controls, FRAP was considerably lower in MCI and AD subjects. Contrarily, AOPP was detected in more significant quantities in patient groups than in healthy controls. When comparing the SOD levels of MCI and healthy patients, MCI patients showed an increased level [73]. Several factors could have accounted for the increased SOD in MCI patients. For instance, both patient and healthy groups were on the Mediterranean diet, and the patient group might have adhered to the plan better than their healthy counterparts. However, the more significant finding was that 74% of the MCI patients were non-APOE-ε4 carriers. The importance of this discovery is because the increased SOD level in MCI patients relative to healthy controls might reflect the higher number of non-APOE-ε4 carriers. As to whether this result would have changed as the disease progressed is anyone’s guess.
Several studies have implicated FoxO (forkhead box O) and PCG-1α (peroxisome proliferator-activated receptor gamma coactivator 1-alpha) in various diseases, such as AD and oxidative stress disorders [74-76]. The involvement of FoxO-3 in AD or oxidative damage is puzzling due to studies showing contrasting results. For example, impaired HDAC (histone deacetylase) caused FoxO-3α upregulation, resulting in oxidative stress inhibition [77]. Similarly, the beneficial effects of FoxO-3 in obviating axonal dysfunction and oxidative insult have been evidenced [74, 75]. However, the initial findings are complicated by a study that found FoxO-3α to cause neuronal apoptosis via impaired miR-132/212, with concomitant effect of neurodegeneration [78]. Junxiang et al. did a post-mortem brain tissue analysis of APOE-ε4 carriers and non-carriers. Compared to non-APOE-ε4 carriers, they found that APOE-ε4 carriers had impaired mitochondrial structure and function due to mitigated levels of PCG-1α and SIRT3. Moreover, the observed mitochondrial dysfunction coincided with synaptic loss and oxidative stress [79]. PCG-1α is closely related to SIRT3, and both are essential modulators of mitochondrial dynamics. Their disparate functions have been thoroughly examined [76, 80-82]. During physical activities, the levels of these proteins are elevated [83, 84], and their overexpression alleviates mitochondrial detriments [76, 80]. These findings are interesting because they may underlie the rationale behind the clinical trial study by the University of Calgary (elaborated in section 6) to determine the possibility of physical activities in preventing neurodegeneration.
We have previously stated the relationship between APOE and sortilin. Sortilin-related vacuolar protein sorting 10 (VSP10P) domain-containing receptor 2 (SorCS2) belongs to the same transmembrane protein family as SORT1 [85]. Malik et al. showed SorCS2 upregulation to obviate neuronal death and oxidative injury by promoting neuronal cysteine. The increase in neuronal cysteine was concomitant with enhanced GSH formation. Most importantly, the interplay between SorCS2 and the excitatory amino acid transporter 3 (EAAT3) mediated the previously mentioned findings [86]. In AD, this transporter is abnormally aggregated in the hippocampus and causes neuronal decline [87]. Based on the prior studies, SorCS2 inhibition may impair EAAT3 functionality, trigger oxidative stress, and cause neuronal loss.
Although SODs are potent antioxidants, structural changes can reduce their efficacy. For example, rs2070424 and rs4880 are corresponding SOD1 and SOD2 genetic polymorphisms that increase AD risk [88, 89]. In individuals possessing combined APOE-ε4 and rs4880-T alleles, the likelihood of developing amnestic MCI (aMCI) was greater than in healthy controls, and the probability of getting AD was higher when compared to aMCI individuals [90]. However, in the absence of the APOE-ε4 allele, there was no risk of developing either AD or aMCI [90], implying that having the rs-4880 allele alone is not an AD risk. It is worth noting that several investigations [88, 91] have corroborated the previous finding. Therefore, we speculate that having the APOE-ε4 allele may cause antioxidant dysfunctionality of the SOD gene, especially in those carrying the rs-4880 variant, and increase their risk of AD.
Given that MCI is an antecedent condition to AD, alterations in oxidative stress biomarkers may indicate the progression of MCI to AD. For instance, the recent study by Deng et al. reported decreased SOD activity and increased plasma protein-methionine sulfoxide in AD individuals than MCI patients, indicating that oxidative stress was more profound in AD [92]. However, drawing a definitive conclusion will require further studies.
In summary, APOE-ε4 plays a multifaceted role in oxidative stress. We hypothesize that its modulation of SOD activity may result in; 1) the disease progression from MCI to AD, and 2) the activation of oxidative stress and other AD-related pathological features, which may exacerbate the disease. The careful monitoring of conventional oxidative stress biomarkers (such as AOPP and FRAP), coupled with interventions targeting their dysregulation may potentially alter the disease course from MCI to AD.
5.The gut microbiome
Microbial species abound and are diverse in the human gastrointestinal system. The majority of these organisms are bacteria, fungi, and viruses. An evolving body of knowledge concerning the gut microbiome and its effects on human health continues to grow. The human gut microbiota influences brain function via immunological, humoral, and neurological pathways [93]. Each person has an extensive gut microbiota that can be affected by diet and lifestyle, making the gut microbiota more susceptible to dysfunctional changes [94].
In this section, we review the possible link between the gut microbiome, APOE, and neurodegeneration. We also examine the gut metabolite involved in oxidative stress.
5.1.The gut microbiome and Apolipoprotein E
Multitudinous bacteria harbor within the gut. It is therefore not surprising that studies have associated several of them with AD. For instance, investigations have linked Ruminococcaceae and Erysipelotrichaceae with APOE. Erysipelotrichaceae and Ruminococcaceae, the most dominant bacteria families in the gut microbiome, are prevalent in AD patients [95, 96]. Parikh et al. found a correlation between the increased abundance of Erysipelotrichaceae in 4 and 6 - month-old EFAD mice and APOE-ε4 [95]. Also, an increase in Erysipelotrichaceae species was reported in 18-month-old APOE-ε4 transgenic mice relative to age-matched APOE-ε3 counterparts [96]. The proliferated population of this bacteria family in the gut microbiome appears to be related to higher cholesterol levels [97]. In addition, there is a positive correlation between this bacteria family and TNF-α [98]. Although the relationship between TNF-α and cholesterol concerning Erysipelotrichaceae is unclear, a high cholesterol diet may likely cause an aggravated formation of Erysipelotrichaceae species within the gut, which in turn could enhance TNF-α production and lead to inflammation [99]. It is worth mentioning that APOE-ε4 has a predilection for binding to VLDL and causes its build-up [100]. Hence, the APOE-ε4 transgenic mice used in [95] and [96] studies may have had elevated cholesterol levels caused by several factors, such as diet, lack of exercise, or both. The elevated cholesterol levels may have then triggered the increased Erysipelotrichaceae. Tran and colleagues also found considerably higher Erysipelotrichaceae in 4-month-old APOE-ε3 transgenic mice than in the age-matched APOE-ε4 transgenic mice group, although the reverse was true in 18-month-old animals [96]. Several factors, such as different animal breeds, different purchasing vendors, and ages of mice, may have accounted for the discrepancy in results between the two studies [95, 96] concerning the young-aged animals.
Compared to both APOE-ε3 and APOE-ε4 groups of similar age, a significantly high number of Ruminococcaceae species were discerned in APOE-ε2 EFAD mice [95]. Furthermore, disease-free individuals showed comparable findings [96]. Ruminococcaceae species produces short-chain fatty acids (SCFA), such as butyrate [96]. The beneficial effects of butyrate include the inhibition of β-amyloid plaque and controlling inflammation [101, 102]. In major depression and Crohn’s disease, reduced levels of these species have been observed [103, 104]. Particularly in Crohn’s disease, the reduced quantity of these species may have enhanced inflammatory factors and exacerbated the condition. Because APOE-ε2 is a protective allele in AD, we believe its protectiveness could be mediated by the proliferation of the Ruminococcaceae species in the gastrointestinal tract. Although the underlying reason is speculative, SCFA (like butyrate) generated by Ruminococcaceae species may modulate inflammation and minimize AD risk. Noteworthy is that a study found an association between the inhibition of the NF-κB pathway and Ruminococcaceae species [105], which could indicate the potential signaling pathway by which these bacteria control inflammation.
There is a link between APOE and the gut microbiome. Nevertheless, how APOE modulates the gut microbiota is presently unknown. Several hypotheses indicate inflammation as a central facet [95, 96], and APOE-ε4 is associated with pro-inflammation [106, 107]. This might explain why butyrate-generating gut bacteria, which can regulate inflammation, are considerably lower in APOE-ε4 than in APOE-ε2 alleles [95, 96]. Intriguingly, whether the increased relative abundance of butyrate-producing gut bacteria (via fecal transplantation or probiotics) in homozygous or heterozygous APOE-ε4 individuals could decrease AD risk, halt AD progression, or repudiate AD remains to be elucidated. Future studies delineating the above quandary with relevant animal models and human samples could improve our understanding and open novel interventional avenues for AD.
5.2.The gut microbiome and neurodegeneration
The correlation between the gut microbiome and neurodegeneration has generated significant interest in recent times and is an evolving investigative area. Sampson and colleagues demonstrated gut microbes to cause the augmentation of α-synuclein, a pathological feature of Parkinson’s disease. In addition, gut microbes generated short-chain fatty acids that resulted in neuroinflammation and affected the motor function of Thy1-α-Syn mice [108]. Preclinical and clinical AD investigations have evidenced alterations in the gut microbiome. For instance, the daily transfer of fecal microbiota in ADLPAPT (AD-like pathology with amyloid and neurofibrillary tangles) mice alleviated a myriad of AD-related pathological features and signs, such as β-amyloid accumulation, gliosis, tau pathology, and cognitive impairment. In addition, intestinal microbiota alterations aggravated the gut permeability, further leading to intestinal and systemic inflammation. Besides, these changes preceded AD pathogenesis [109]. The latter finding is interesting considering that a clinical study reported gut microbiota alteration preceding dementia [110]. Similarly, the evaluation of stool and inflammatory indicators showed increased inflammation in cognitively impaired individuals than aged-matched controls, concomitant with a higher abundance of proinflammatory taxa (Escherichia/Shigella). As expected, the anti-inflammatory marker (IL-10) was decreased in cognitively impaired individuals, also coinciding with a lower abundance of the anti-inflammatory taxon (Eubacterium rectale) [111].
Given the above studies, peripheral factors may affect CNS disorders. We do concur with other studies [110, 112-114] in speculating that peripheral agent causing inflammation may disrupt the impermeable integrity of the BBB and gut. The resultant effect of this is the aggravation of harmful molecules (such as lipopolysaccharides; LPS) that could be passed through to the CNS to promote neuroinflammation and lead to neurodegeneration (Fig. 1).
Figure 1.
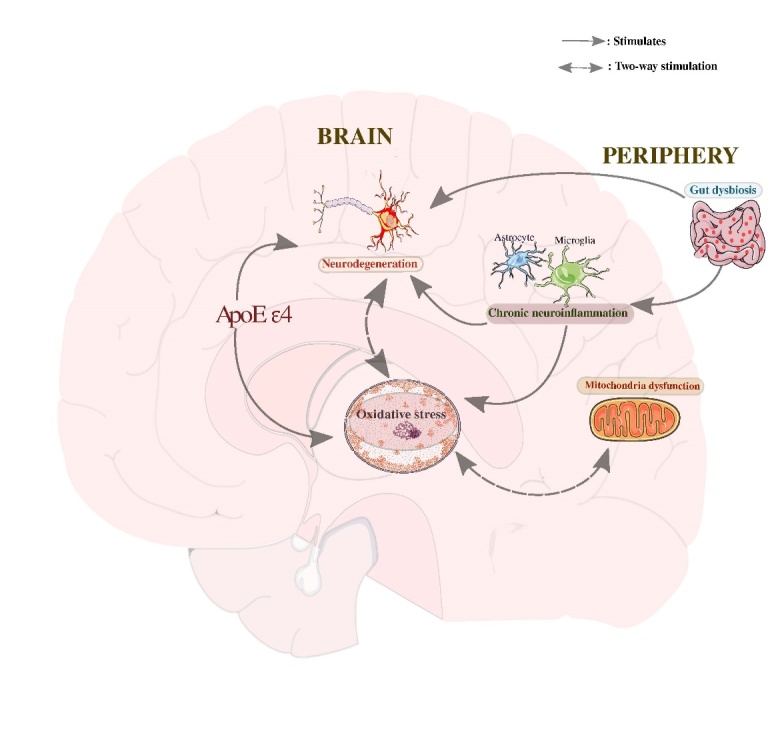
The various pathways resulting in oxidative stress and neurodegeneration. Lipopolysaccharides from gut dysbiosis impair the blood-brain barrier integrity, leading to neuroinflammation and neurodegeneration. During neuroinflammation, the activations of microglia and astrocytes enhance free radical synthesis that progressively increases the risk of oxidative stress. Furthermore, there is a higher risk of β-amyloid aggregation and oxidative stress in the absence of thiol-mediated antioxidant properties of APOE-ε4. β-amyloid plaques trigger neuronal damage and oxidative stress via the oxidant-antioxidant system disequilibrium. Chronic oxidative stress impairs mitochondrial function that exacerbates oxidative insults and causes neuronal dysfunction. Also, oxidative stress upregulates amyloid precursor protein, which may result in neurodegeneration. Relevant references are in the main text.
5.3.The gut microbiome and oxidative stress
Investigations have established the correlation between gut microbiome dysbiosis and resultant health effects. A significant component of the pathway by which this occurs is oxidative stress. As has been outlined, ROS and RNS are crucial in body regulatory functions, not least are microbial destruction by neutrophils and cytokines. The inability of established counterregulatory factors, including GSH, to limit the deleterious effect of excess oxidative species on normal cell architecture has already been determined as the cause of many human ailments [115]. Regarding neurodegenerative diseases, studies have shown oxidative stress as a contributory factor. Gram-negative gut bacteria have been singled out as a possible etiological factor via the destructive effects of their LPS. A positive correlation exists between LPS and NADPH oxidase 2 (NOD2), with the potential resultant effect of neuroinflammation [114]. This is supported by several human studies that have ascertained the role of LPS in AD pathogenesis. For instance, examined brain tissues of AD patients revealed a higher level of LPS than age-matched healthy controls. More importantly, these observations were noted in amyloid plaques and surrounding blood vessels [116]. Also, human AD brains showed a higher degree of LPS in the hippocampal and superior temporal lobe [117]. Despite unclear reasons, one possible route by which LPS induces oxidative stress in AD could be through its concerted interaction with CD-14 (cluster of differentiation-14) and TLR-4 (toll-like receptor-4), which then triggers both astrocytes and microglia. The instigation of these cells may cause the release of complement proteins, proinflammatory cytokines, and chemokines, resulting in oxidative stress. Both mitochondria damage and chronic inflammation are consequences of oxidative stress that can eventually lead to neuronal loss [118].
Trimethylamine N-oxide (TMAO) is a gut metabolite, and studies have associated it with several human diseases. Salmon, milk, and red meat consumption in humans undergo initial metabolization into trimethylamine (TMA) by the gut bacteria. Following its absorption via the intestinal walls, TMA is then conveyed to the liver. With oxygenation, the liver breaks it down to TMAO [119]. In analyzing AD and MCI patients, their CSF showed higher levels of TMAO. Concomitantly, neuronal degeneration and other AD-related pathological markers, such as aberrancy in the ratio of phosphorylated tau to β-amyloid were also discerned [120]. Several human and animal studies have pointed to TMAO as being involved in oxidative stress, with its exacerbated levels associated with aging. In APOE knock-out mice put on a diet that included TMAO, increased plasma levels of TNF-α, MCP-1, and IL-1β (implying inflammation), as well as corresponding higher and lower plasma concentration of malondialdehyde and GSH-Px/SOD activities (indicating oxidative stress) were detected [121]. Also, TMAO level was higher in aged Fisher-344 male rats than in younger controls. The above observation concurred with the exacerbated formation of superoxide, upregulated pro-inflammatory cytokines, and downregulated endothelial nitric oxide synthase [122]. These findings are supported by a more recent that showed TMAO to instigate oxidative stress and cause endothelial impairment in older individuals [123]. However, a study has reported contrary findings concerning TMAO’s association with oxidative stress. Intriguingly, this study used only healthy women (between 65 and 70 years) and detected no association between TMAO and oxidative stress following a 24-week supplementation of 1500-mg L-carnitine-L-tartrate [124]. To our knowledge, this is the only study to report the absence of any link between TMAO and oxidative stress.
We suspect that increased aging, particularly in AD, may cause elevated circulatory levels of TMAO that can trigger the overexpression of cytokines to promote oxidative stress and compromise endothelial function, eventually resulting in neurodegeneration. Nonetheless, a more precise delineation of the association between TMAO and oxidative stress warrants further studies using applicable human and animal models. Most importantly, data analysis from these studies should factor in the age, sex, and disease state of experimental models. Also, when assessing TMAO in AD, neurofibrillary tangles and β-amyloid plaques should be considered. Similarly, as certain foods are sources of TMAO, there is the likelihood that significant moderation of these foods may be beneficial for aged individuals, especially those at risk of AD. The above statement will corroborate our previous report where we suggested nutrition as a possible interventional agent for AD [125].
Complex interactions between microbiota, as previously reported, influence oxidative species and hinders the antioxidant counterregulatory mechanisms. Various metabolites of microbiota, including short-chain fatty acids, TMAO, and polyphenols, may regulate the oxidative state of the CNS.
6.Prospective interventional agents
AD’s complex pathophysiological mechanism partly accounts for the lack of an effective treatment. For almost two decades, no pharmaceutical agent had been approved to either reverse or improve disease symptoms until September 2019, when health regulators in China endorsed the use of sodium oligomannate for mild-to-moderate AD. Before its endorsement, Xinyu and colleagues used human and animal models (5xFAD (Tg), C57, and APP/PS1 mice), where they found sodium oligomannate to alter the gut microbiota, with concomitant effects of attenuated neuroinflammation and enhanced cognition [126]. The significance of this study lies in shedding light on the possibility that targeting the gut microbiota dysregulation and its metabolites could halt the progression of AD-related pathological indicators and ameliorate the disease.
Antioxidants, classified as endogenous (primarily enzymes) or exogenous (non-enzymes obtained from food products), can halt oxidative stress. The endogenous antioxidant includes SOD, catalase, and GSH. The GSH system comprises GSH peroxidase (GPx) and GSH reductase [127]. Given that antioxidants counteract the activity of free radicals, a thorough understanding of their usefulness for AD and other neurodegenerative diseases is paramount. Catalase and GPx convert H2O2 to less harmful substances (water and oxygen). However, the GPx-GSH system is limited by the availability of GSH, which has to be regenerated from GSSG (glutathione disulfide) by flavoenzyme, GSH reductase, and NADPH. This limitation decreases the efficiency of the GPx-GSH system in removing H2O2 [27]. Several antioxidants for AD have undergone clinical trials. For example, the use of resveratrol in mild-to-moderate AD patients showed increased and decreased plasma matrix metalloproteinase (MMP)-10 and CSF MMP-9, respectively. In addition, regulated neuroinflammation via elevated fibroblast growth factor-2, macrophage-derived chemokine, and IL-4, as well as reduced RANTES (Regulated on Activation, Normal T-Cell Expressed and Secreted) and IL12p40 was detected [128]. These findings corresponded with enhanced mini-mental status examination (MMSE) and Alzheimer's Disease Cooperative Study—Activities of Daily Living Inventory (ADCS-ADL) scores and reduced β-amyloid in the CSF. Concerning the inhibition of some ILs, particularly IL12p40, there have been suggestions that its impairment could reduce accumulated β-amyloid plaques while enhancing cognition [129, 130]. In phase 2 randomized controlled trial, the administration of 2-mg circadin (prolonged released melatonin) tablets showed considerable improvement in productive sleep and cognitive function. Noteworthy is that this medication was given along with anticholinesterase inhibitors, indicating the efficacy of a combinational therapy for AD. Besides ascertaining circadin’s potency, the study also showed the possible association between cognitive decline and sleep insufficiency [131]. Studies have evidenced the link between AD and sleep dysfunction. For instance, an 11.2-year observational study of non-demented subjects correlated premature light-out time and prolonged sleep latency with dementia risk, particularly AD. Premature light-out time and prolonged sleep latency are indicators of poor sleep quality [132]. Intriguingly, the study found the absence of a correlation between dementia risk and disruption to the 24-hr activity rhythm. This finding complicates the specific relationship between sleep disturbance and AD, given that rest-activity rhythm disturbance in AD has been linked to increased and decreased NREM (Non-rapid eye movement) sigma and delta power, respectively [133]. Regardless of these contrasting findings, the potential applicability of melatonin for AD as a preventive agent has given grounds for researchers from the University of Iowa to undertake a 9-month clinical trial study involving 230 participants. Study participants will be administered 5-mg melatonin and have their AD biological markers assessed at various time points. For instance, evaluations of CSF t-tau and the ratio of p-tau to β-amyloid will take place eight weeks before initiating treatment and weeks 16 and 44 after treatment. Also, neuropsychological examinations will be employed to evaluate cognition (from clinicaltrials.gov, NCT03954899). Aside from being antioxidants, melatonin and resveratrol are SIRT1 activators and can potentially mitigate AD pathological features. In particular, SIRT1’s role in AD has been extensively reviewed [134], implying that their applicability in AD could be promising. Therefore, it would be interesting if relevant AD models harboring both β-amyloid and tau pathological features can evaluate the efficacy of the combined usage of melatonin and resveratrol for the disease.
Vitamins can alleviate oxidative stress and modulate inflammation by neutralizing free radical activities, thus being potential adjuncts in AD treatment [125]. Compared to the placebo-administered group, a 5-year administration of α-tocopherol (vitamin E) showed remarkable retardation of activities of daily living in mild-to-moderate AD patients [135]. We noted two major factors from this study. Firstly, the study participants were already on ChEIs (donepezil and galantamine), suggesting that this study was ascertaining the effectiveness of a combinational therapy for AD. Secondly, α-tocopherol alone (i.e., α-tocopherol + ChEI) was more effective than the combined administration of α-tocopherol and memantine (i.e., α-tocopherol + memantine + ChEI). Contrastingly, a concluded clinical trial found the combined usage of omega-3 fatty acids and lipoic acid to decelerate both functional and cognitive deteriorations in AD patients [136]. We previously suggested the employment of combinational therapy as a probable intervention for AD [125]. As might be expected, along with the clinical evidence from this observational study [135], not all combinational therapies are likely to be effective. Nonetheless, we still believe that combined interventional agents will be the right course for AD due to the intricate pathophysiological mechanism of the disease, with extensive studies needed for better understanding before the employment of this course of action.
Finding an effective interventional agent for AD looks more promising than ever owing to a greater understanding of the disease trajectory over the last decades since its first report by Alois Alzheimer, together with the development of innovative diagnostic techniques. Table 1 summarizes clinical studies on antioxidants that have either been completed or are in the recruiting stage. Paul Rosenberg and colleagues from John Hopkins University and Icahn School of Medicine are undertaking a phase 1 clinical trial that will employ bioactive dietary polyphenol preparation (BDPP, comprising resveratrol and grape seed polyphenolic extract) to ascertain its efficacy against prediabetes or type 2 diabetes and MCI. Diabetes is a known AD risk factor [137, 138]. Assessed outcomes will include cognition, mood, and adverse effects (from clinicaltrials.gov, NCT02502253). Also, Marc Poulin and colleagues will conduct a non-invasive clinical study on the use of aerobic exercise (walking and jogging) for individuals at high risk of AD and associated dementia. The study concludes in January 2025 and will assess brain structure and function, coupled with sleep quality and cognition at different time points. In essence, the study seeks to demonstrate the link between physical activity and cognitive processes and its potential in halting age-related neurodegeneration (from clinicaltrials.gov, NCT03035851).
Table 1.
Summary of clinical studies of prospective antioxidants for AD (Compiled from clinicaltrials.gov)
| Clinical trial number | Interventional agents | Aim | Number of participants and age range (years) | Current status | Primary purpose | Estimated completion date |
|---|---|---|---|---|---|---|
| NCT03514875 | MitoQ | To evaluate the impact of this agent on cerebrovascular blood flow and carotid artery vasodilation | N = 12 Age: 50-85 |
Recruiting | Treatment |
October 2021 |
| NCT03841539 | Mediterranean diet Low fat diet |
To ascertain the effect of these interventions on brain volume and antioxidant level, as well as memory and cardiometabolic biological markers | N = 200 Age: ≥ 65 |
Recruiting | Prevention | February 2023 |
| NCT01982578 | Genistein | To establish the efficacy of this agent in AD | N= 50 Age: ≥ 18 |
Recruiting | Treatment | December 2020 |
| NCT03978052 | Epigallocatechin-gallate plus physical activity plus diet plus mental health promotion | To determine whether this multimodal action can halt cognitive decline | N = 200 Age: 60-80 |
Recruiting | Prevention | September 2021 |
| NCT04740580 | Glutathione (Glycine plus N-acetylcysteine) | To assess the impact of glutathione on cognition | N = 52 Age: 55- 85 |
Recruiting | Other | May 2025 |
| NCT03361410 | Grape powder | To evaluate the impact of this agent on neuropsychological behavior and cerebral metabolism | N = 32 Age: 65 - 85 |
Recruiting | Treatment | January 2021 |
| NCT04063124 | Dasatinib plus Quercetin | To ascertain the penetrance ability of this combined agents in AD individuals | N = 5 Age: ≥ 65 |
Recruiting | Treatment | August 2023 |
| NCT01780974 | Lipoic acid plus omega-3 fatty acids | To evaluate the efficacy of this combined agent in inhibiting AD | N = 42 Age: ≥ 55 |
Completed | Prevention | October 2015 |
| NCT01354444 | Carvedilol | To determine if this agent can ameliorate AD | N = 29 Age: 0 - 100 |
Completed | Treatment | January 2017 |
| NCT01058941 | Lipoic acid plus fish oil concentrate | To ascertain if the combined agent can halt AD progression | N = 67 Age: ≥ 55 |
Completed | Treatment | December 2014 |
| NCT00597376 | Cerefolin NAC | To determine the correlation between this dietary supplement and cognition | N=104 Age: ≥ 60 |
Completed | Prevention | May 2011 |
MitoQ: mitoquinone; AD: Alzheimer disease
7.Concluding remarks and research directions
The implication of oxidative stress in AD is multifactorial. However, whether it induces or is an aftereffect of AD, along with its precise time of emergence (either early or late stage of the disease), is presently open to question. There is the likelihood of oxidative stress being an instigator, accelerator, and aftermath of AD. Given that completed multitudinous AD studies are leaving us with unanswered queries, more thorough investigations need to address the specific position of oxidative stress in the pathophysiological context of AD, which could be instrumental in the development of effective interventions for the disease. Future studies on the subject of oxidative stress in AD could include:
-
•
Identifying the “driver” and “passenger” between β-amyloid and tau protein and oxidative damage. Is β-amyloid/tau protein the “driver” that spurs oxidative damage or vice versa?
-
•
Determining various gut bacteria family/phylum species involved in oxidative stress and their regulatory targets.
-
•
Thorough elucidation of oxidized lipids and proteins that may serve as AD biomarkers in the clinical setting.
-
•
Targeting oxidative damage in the early AD stage via gut microbiome modulation and ascertaining its likelihood of halting disease progression
-
•
Establishing the specific role of cell cycle dysfunction in AD and discovering innovative approaches to repairing this dysfunction.
-
•
Determining the specific features of mitochondria involved in AD and their potential targets for prospective pharmaceutical agents.
-
•
Establishing the possible relationship between mammalian vacuolar protein sorting 10 protein, APOE, and oxidative stress.
Supplementary Materials
The Supplementary data can be found online at: www.aginganddisease.org/EN/10.14336/AD.2021.0616.
{kind=link}
Acknowledgements
This study was supported by funds from the National Science Foundation of China (grant numbers 81871063).
Footnotes
Disclosure statement
No conflicts of interest exist.
References
- [1].Botchway BO, Iyer IC (2017). Alzheimer’s Disease - The Past, the Present and the Future. Sci J Clin Med, 6(1):1-19. [Google Scholar]
- [2].Alzheimer’s Association (2019). Alzheimer’s disease facts and figures. Alzheimer’s dementia, 15(3):321-87 [Google Scholar]
- [3].National Institute on Aging (2018). Treatment of Alzheimer’s Disease: How is Alzheimer’s Disease Treated. Available from: www.nia.nih.gov/health/how-alzheimers-disease-treated.
- [4].Iaccarino HF, Singer AC, Martorell AJ, Rudenko A, Gao F, Gillingham TZ, et al. (2016). Gamma frequency entrainment attenuates amyloid load and modifies microglia. Nature, 540(7632): 230-235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Moscoso A, Grothe MJ, Ashton NJ, Karikari TK, Lantero Rodríguez J, Snellman A, et al. (2021). Longitudinal Associations of Blood Phosphorylated Tau181 and Neurofilament Light Chain with Neurodegeneration in Alzheimer Disease. JAMA Neurol, 78(4): 396-406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Kim GH, Kim JE, Rhie SJ, and Yoon S (2015). The Role of Oxidative Stress in Neurodegenerative Diseases. Exp Neurobiol, 24(4), 325-340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Hung CH, Cheng SS, Cheung YT, Wuwongse S, Zhang NQ, Ho YS, et al. (2018). A reciprocal relationship between reactive oxygen species and mitochondrial dynamics in neurodegeneration. Redox Biol, 14:7-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Szymański J, Janikiewicz J, Michalska B, Patalas-Krawczyk P, Perrone M, Ziółkowski W, et al. (2017). Interaction of Mitochondria with the Endoplasmic Reticulum and Plasma Membrane in Calcium Homeostasis, Lipid Trafficking and Mitochondrial Structure. Int J Mol Sci, 18(7):1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Marchi S, Giorgi C, Oparka M, Duszynski J, Wieckowski MR, Pinton P (2014). Oncogenic and oncosuppressive signal transduction at mitochondria-associated endoplasmic reticulum membranes. Mol Cell Oncol, 1(2):e956469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Yetkin-Arik B, Vogels I, Nowak-Sliwinska P, Weiss A, Houtkooper RH, Van Noorden C, et al. (2019). The role of glycolysis and mitochondrial respiration in the formation and functioning of endothelial tip cells during angiogenesis. Sci Rep, 9(1):12608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Tönnies E, Trushina E (2017). Oxidative Stress, Synaptic Dysfunction, and Alzheimer's Disease. J Alzheimer's Dis, 57(4):1105-1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Liao X, Huang C, Zhang D, Wang J, Li J, Jin H, et al. (2017). Mitochondrial catalase induces cells transformation through nucleolin-dependent Cox-2 mRNA stabilization. Free Radic Biol Med, 113:478-486. [DOI] [PubMed] [Google Scholar]
- [13].Kanta J (2011). The role of hydrogen peroxide and other reactive oxygen species in wound healing. Acta medica, 54(3):97-101. [DOI] [PubMed] [Google Scholar]
- [14].Hansen DV, Hanson JE, Sheng M (2018). Microglia in Alzheimer's disease. J Cell Biol, 217(2):459-472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Cheignon C, Tomas M, Bonnefont-Rousselot D, Faller P, Hureau C, Collin F (2018). Oxidative stress and the amyloid beta peptide in Alzheimer's disease. Redox Biol, 14:450-464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ren X, Zou L, Zhang X, Branco V, Wang J, Carvalho C, et al. (2017). Redox Signaling Mediated by Thioredoxin and Glutathione Systems in the Central Nervous System. Antioxid. Redox Signal, 27(13):989-1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Wang X, Wang W, Li L, Perry G, Lee HG, Zhu X (2014). Oxidative stress and mitochondrial dysfunction in Alzheimer's disease. Biochim Biophys Acta, 1842(8):1240-1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Butterfield DA, Boyd-Kimball D (2019). Redox proteomics and amyloid β-peptide: insights into Alzheimer disease. J Neurochem, 151(4):459-487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Muche A, Arendt T, Schliebs R (2017). Oxidative stress affects processing of amyloid precursor protein in vascular endothelial cells. PLoS One, 12(6):e0178127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Someya S, Yu W, Hallows WC, Xu J, Vann JM, Leeuwenburgh C, et al. (2010). Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell, 143(5):802-812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Lee J, Kim Y, Liu T, Hwang YJ, Hyeon SJ, Im H, et al. (2018). SIRT3 deregulation is linked to mitochondrial dysfunction in Alzheimer's disease. Aging Cell, 17(1):e12679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kim YJ, Kim SH, Park Y, Park J, Lee JH, Kim BC, et al. (2020). miR-16-5p is upregulated by amyloid β deposition in Alzheimer's disease models and induces neuronal cell apoptosis through direct targeting and suppression of BCL-2. Exp Gerontol, 136:110954. [DOI] [PubMed] [Google Scholar]
- [23].Sun W, Zhao J, Li C (2020). Dexmedetomidine Provides Protection Against Hippocampal Neuron Apoptosis and Cognitive Impairment in Mice with Alzheimer's Disease by Mediating the miR-129/YAP1/JAG1 Axis. Mol Neurobiol, 57(12):5044-5055. [DOI] [PubMed] [Google Scholar]
- [24].Chen SY, Gao Y, Sun JY, Meng XL, Yang D, Fan LH, et al. (2020). Traditional Chinese Medicine: Role in Reducing β-Amyloid, Apoptosis, Autophagy, Neuroinflammation, Oxidative Stress, and Mitochondrial Dysfunction of Alzheimer's Disease. Front Pharmacol, 11:497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Jadiya P, Kolmetzky DW, Tomar D, Di Meco A, Lombardi AA, Lambert JP, et al. (2019). Impaired mitochondrial calcium efflux contributes to disease progression in models of Alzheimer's disease. Nat Commun, 10(1):3885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Calvo-Rodriguez M, Hou SS, Snyder AC, Kharitonova EK, Russ AN, Das S, et al. (2020). Increased mitochondrial calcium levels associated with neuronal death in a mouse model of Alzheimer's disease. Nat Commun, 11(1):2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Espinosa-Diez C, Miguel V, Mennerich D, Kietzmann T, Sánchez-Pérez P, Cadenas S, et al. (2015). Antioxidant responses and cellular adjustments to oxidative stress. Redox Biol, 6:183-197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Suzuki Y, Ali M, Fischer M, Riemer J (2013). Human copper chaperone for superoxide dismutase 1 mediates its own oxidation-dependent import into mitochondria. Nat Commun, 4:2430. [DOI] [PubMed] [Google Scholar]
- [29].Angelova PR, Abramov AY (2016). Functional role of mitochondrial reactive oxygen species in physiology. Free Radic Biol Med, 100:81-85. [DOI] [PubMed] [Google Scholar]
- [30].Sochocka M, Koutsouraki ES, Gasiorowski K, Leszek J (2013). Vascular oxidative stress and mitochondrial failure in the pathobiology of Alzheimer's disease: a new approach to therapy. CNS Neurol. Disord Drug Targets, 12(6):870-881. [DOI] [PubMed] [Google Scholar]
- [31].Pagani L, Eckert A (2011). Amyloid-Beta interaction with mitochondria. Int. [J]. Alzheimers Dis, 2011:925050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].de Leeuw FA, Peeters C, Kester MI, Harms AC, Struys EA, Hankemeier T, et al. (2017). Blood-based metabolic signatures in Alzheimer's disease. Alzheimers Dement, 8:196-207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Butterfield DA, Lauderback CM (2002). Lipid peroxidation and protein oxidation in Alzheimer's disease brain: potential causes and consequences involving amyloid beta-peptide-associated free radical oxidative stress. Free Radic Biol Med, 32(11):1050-1060. [DOI] [PubMed] [Google Scholar]
- [34].Mecocci P, MacGarvey U, Beal MF (1994). Oxidative damage to mitochondrial DNA is increased in Alzheimer's disease. Ann Neurol, 36(5):747-751. [DOI] [PubMed] [Google Scholar]
- [35].Gabbita SP, Lovell MA, Markesbery WR (1998). Increased nuclear DNA oxidation in the brain in Alzheimer's disease. J Neurochem, 71(5):2034-2040. [DOI] [PubMed] [Google Scholar]
- [36].Lovell MA, Soman S, Bradley MA (2011). Oxidatively modified nucleic acids in preclinical Alzheimer's disease (PCAD) brain. Mech. Ageing Dev, 132(8-9): 443-448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Wang ZX, Tan L, Yu JT (2015). Axonal transport defects in Alzheimer's disease. Mol Neurobiol, 51(3):1309-1321. [DOI] [PubMed] [Google Scholar]
- [38].Shahpasand K, Uemura I, Saito T, Asano T, Hata K, Shibata K, et al. (2012). Regulation of mitochondrial transport and inter-microtubule spacing by tau phosphorylation at the sites hyperphosphorylated in Alzheimer's disease. J Neurosci, 32(7):2430-2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Rodríguez-Martín T, Cuchillo-Ibáñez I, Noble W, Nyenya F, Anderton BH, Hanger DP (2013). Tau phosphorylation affects its axonal transport and degradation. Neurobiol. Aging, 34(9):2146-2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Li XC, Hu Y, Wang ZH, Luo Y, Zhang Y, Liu XP, et al. (2016). Human wild-type full-length tau accumulation disrupts mitochondrial dynamics and the functions via increasing mitofusins. Sci Rep, 6:24756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Manczak M, Reddy PH (2012). Abnormal interaction of VDAC1 with amyloid beta and phosphorylated tau causes mitochondrial dysfunction in Alzheimer's disease. Hum Mol Genet, 21(23):5131-5146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Feingold KR (2021). Introduction to Lipids and Lipoproteins. In Feingold KR (Eds.) et al., Endotext. MDText.com, Inc. [Google Scholar]
- [43].Wolska A, Dunbar RL, Freeman LA, Ueda M, Amar MJ, Sviridov DO, et al. (2017). Apolipoprotein C-II: New findings related to genetics, biochemistry, and role in triglyceride metabolism. Atherosclerosis, 267:49-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Wang HF, Yu JT, Zhang W, Wang W, Liu QY, Ma XY, et al. (2012). SORCS1 and APOE polymorphisms interact to confer risk for late-onset Alzheimer's disease in a Northern Han Chinese population. Brain Res, 1448:111-116. [DOI] [PubMed] [Google Scholar]
- [45].Lu RC, Wang H, Tan MS, Yu JT, Tan L (2014). TMEM106B and APOE polymorphisms interact to confer risk for late-onset Alzheimer's disease in Han Chinese. J Neural Transm,121(3):283-287. [DOI] [PubMed] [Google Scholar]
- [46].Lescai F, Chiamenti AM, Codemo A, Pirazzini C, D'Agostino G, Ruaro C, et al. (2011). An APOE haplotype associated with decreased ε4 expression increases the risk of late onset Alzheimer's disease. J Alzheimer's Dis, 24(2):235-245. [DOI] [PubMed] [Google Scholar]
- [47].Lagging C, Lorentzen E, Stanne TM, Pedersen A, Söderholm M, Cole JW, et al. (2019). APOE ε4 is associated with younger age at ischemic stroke onset but not with stroke outcome. Neurology, 93(19):849-853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Zhao LL, Su G, Chen LX, Yan Q, Wang XP, Yuan W, et al. (2017). Apolipoprotein E polymorphisms are associated with ischemic stroke susceptibility in a Northwest China Han population. Biosci Rep, 37(6): BSR20171088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Li Z, Ding C, Gong X, Wang X, Cui T (2016). Apolipoprotein E ε4 Allele was Associated with Nonlesional Mesial Temporal Lobe Epilepsy in Han Chinese Population. Medicine, 95(9):e2894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Morton AM, Koch M, Mendivil CO, Furtado JD, Tjønneland A, Overvad K, et al. (2018). Apolipoproteins E and CIII interact to regulate HDL metabolism and coronary heart disease risk. JCI Insight, 3(4):e98045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Huang YA, Zhou B, Wernig M, Südhof TC (2017). ApoE2, ApoE3, and ApoE4 Differentially Stimulate APP Transcription and Aβ Secretion. Cell, 168(3):427-441.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Shinohara M, Kanekiyo T, Yang L, Linthicum D, Shinohara M, Fu Y, et al. (2016). APOE2 eases cognitive decline during Aging: Clinical and preclinical evaluations. Ann Neurol, 79(5):758-774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Chan ES, Shetty MS, Sajikumar S, Chen C, Soong TW, Wong BS (2016) ApoE4 expression accelerates hippocampus-dependent cognitive deficits by enhancing Aβ impairment of insulin signaling in an Alzheimer's disease mouse model. Sci Rep, 6:26119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Keeney JT, Ibrahimi S, Zhao L (2015). Human ApoE isoforms differentially modulate glucose and amyloid metabolic pathways in female brain: evidence of the mechanism of neuroprotection by ApoE2 and implications for Alzheimer’s disease prevention and early intervention. [J]. Alzheimer’s Dis, 48(2):411-424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Shimizu F, Nishihara H, Kanda T (2018). Blood-brain barrier dysfunction in immuno-mediated neurological diseases. Immunol Med, 41(3):120-128. [DOI] [PubMed] [Google Scholar]
- [56].Profaci CP, Munji RN, Pulido RS, Daneman R (2020). The blood-brain barrier in health and disease: Important unanswered questions. J Exp Med, 217(4):e20190062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Sweeney MD, Sagare AP, Zlokovic BV (2018). Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat Rev Neurol, 14(3): 133-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Reiman EM, Arboleda-Velasquez JF, Quiroz YT, Huentelman MJ, Beach TG, Caselli RJ, et al. (2020). Exceptionally low likelihood of Alzheimer's dementia in APOE2 homozygotes from a 5,000-person neuropathological study. Nat Commun, 11(1):667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Montagne A, Nation DA, Sagare AP, Barisano G, Sweeney MD, Chakhoyan A, et al. (2020). APOE4 leads to blood-brain barrier dysfunction predicting cognitive decline. Nature, 581(7806):71-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Asaro A, Carlo-Spiewok AS, Malik AR, Rothe M, Schipke CG, Peters O, et al. (2020). Apolipoprotein E4 disrupts the neuroprotective action of sortilin in neuronal lipid metabolism and endocannabinoid signaling. Alzheimers Dement, 16(9):1248-1258. [DOI] [PubMed] [Google Scholar]
- [61].Piscopo P, Tosto G, Belli C, Talarico G, Galimberti D, Gasparini M, et al. (2015). SORL1 Gene is Associated with the Conversion from Mild Cognitive Impairment to Alzheimer's Disease. J Alzheimer's Dis, 46(3):771-776. [DOI] [PubMed] [Google Scholar]
- [62].Andersson CH, Hansson O, Minthon L, Andreasen N, Blennow K, Zetterberg H, et al. (2016). A Genetic Variant of the Sortilin 1 Gene is Associated with Reduced Risk of Alzheimer's Disease. J Alzheimer's Dis, 53(4):1353-1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Reitz C, Tosto G, Vardarajan B, Rogaeva E, Ghani M, Rogers RS, et al. (2013). Independent and epistatic effects of variants in VPS10-d receptors on Alzheimer disease risk and processing of the amyloid precursor protein (APP). Transl Psychiatry, 3(5):e256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Beecham GW, Hamilton K, Naj AC, Martin ER, Huentelman M, Myers AJ, et al. (2014). Genome-wide association meta-analysis of neuropathologic features of Alzheimer's disease and related dementias. PLoS Genet, 10(9):e1004606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Baba SP, Bhatnagar A (2018). Role of thiols in oxidative stress. Curr. Opin. Toxicol, 7:133-139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Ulrich K, Jakob U (2019). The role of thiols in antioxidant systems. Free Radic Biol Med, 140:14-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Persichilli S, Gervasoni J, Di Napoli A, Fuso A, Nicolia V, Giardina B, et al. (2015). Plasma thiols levels in Alzheimer's disease mice under diet-induced hyperhomocysteinemia: effect of S-adenosylmethionine and superoxide-dismutase supplementation. J Alzheimer's Dis, 44(4):1323-1331. [DOI] [PubMed] [Google Scholar]
- [68].Gumusyayla S, Vural G, Bektas H, Deniz O, Neselioglu S, Erel O (2016). A novel oxidative stress marker in patients with Alzheimer’s disease: Dynamic thiol-disulphide homeostasis. Acta Neuropsychiatr, 28(6):315-320. [DOI] [PubMed] [Google Scholar]
- [69].Muñoz SS, Garner B, Ooi L (2019). Understanding the Role of ApoE Fragments in Alzheimer's Disease. Neurochem Res, 44(6):1297-1305. [DOI] [PubMed] [Google Scholar]
- [70].Di Domenico F, Pupo G, Giraldo E, Badìa MC, Monllor P, Lloret A, et al. (2016). Oxidative signature of cerebrospinal fluid from mild cognitive impairment and Alzheimer disease patients. Free Radic Biol Med, 91:1-9. [DOI] [PubMed] [Google Scholar]
- [71].Ogata T, Senoo T, Kawano S, Ikeda S (2016). Mitochondrial superoxide dismutase deficiency accelerates chronological aging in the fission yeast Schizosaccharomyces pombe. Cell Biol Int, 40(1):100-106. [DOI] [PubMed] [Google Scholar]
- [72].Lucas ER, Keller L (2018). Elevated expression of ageing and immunity genes in queens of the black garden ant. Exp Gerontol, 108:92-98. [DOI] [PubMed] [Google Scholar]
- [73].Chico L, Simoncini C, Lo Gerfo A, Rocchi A, Petrozzi L, Carlesi C, et al. (2013). Oxidative stress and APO E polymorphisms in Alzheimer's disease and in mild cognitive impairment. Free Radic Res, 47(8):569-576. [DOI] [PubMed] [Google Scholar]
- [74].Hwang I, Oh H, Santo E, Kim DY, Chen JW, Bronson RT, et al. (2018). FOXO protects against age-progressive axonal degeneration. Aging Cells, 17(1): e12701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Kops GJ, Dansen TB, Polderman PE, Saarloos I, Wirtz KW, Coffer PJ, et al. (2002). Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature, 419(6904):316-321. [DOI] [PubMed] [Google Scholar]
- [76].Kang C, Goodman CA, Hornberger TA, Ji LL (2015). PGC-1α overexpression by in vivo transfection attenuates mitochondrial deterioration of skeletal muscle caused by immobilization. FASEB J, 29(10):4092-4106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Shimazu T, Hirschey MD, Newman J, He W, Shirakawa K, Le Moan N, et al. (2013). Suppression of oxidative stress by β-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science, 339(6116):211-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Wong HKA, Veremeyko T, Patel N, Lemere CA, Walsh DM, Esau C, et al. (2013). De-repression of FOXO3a death axis by microRNA-132 and-212 causes neuronal apoptosis in Alzheimer's disease. Hum Mol Genet, 22(15):3077-3092. [DOI] [PubMed] [Google Scholar]
- [79].Yin J, Reiman EM, Beach TG, Serrano GE, Sabbagh MN, Nielsen M, et al. (2020). Effect of ApoE isoforms on mitochondria in Alzheimer disease. Neurology, 94(23):e2404-e2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Kong X, Wang R, Xue Y, Liu X, Zhang H, Chen Y, et al. (2010). Sirtuin 3, a new target of PGC-1alpha, plays an important role in the suppression of ROS and mitochondrial biogenesis. PLoS One, 5(7):e11707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Zhang J, Xiang H, Liu J, Chen Y, He RR, Liu B (2020). Mitochondrial Sirtuin 3: New emerging biological function and therapeutic target. Theranostics, 10(18):8315-8342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Kanwal A (2018). Functional and therapeutic potential of mitochondrial SIRT3 deacetylase in disease conditions. Expert Rev. Clin Pharmacol, 11(12):1151-1155. [DOI] [PubMed] [Google Scholar]
- [83].Vargas-Ortiz K, Perez-Vazquez V, Diaz-Cisneros FJ, Figueroa A, Jiménez-Flores LM, Rodriguez-DelaRosa G, et al. (2015). Aerobic Training Increases Expression Levels of SIRT3 and PGC-1α in Skeletal Muscle of Overweight Adolescents Without Change in Caloric Intake. Pediatr. Exerc Sci, 27(2):177-184. [DOI] [PubMed] [Google Scholar]
- [84].Palacios OM, Carmona JJ, Michan S, Chen KY, Manabe Y, Ward JL, et al. (2009). Diet and exercise signals regulate SIRT3 and activate AMPK and PGC-1alpha in skeletal muscle. Aging, 1(9):771-783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Januliene D, Manavalan A, Ovesen PL, Pedersen KM, Thirup S, Nykjær A, et al. (2017). Hidden Twins: SorCS Neuroreceptors Form Stable Dimers. J Mol Biol, 429(19):2907-2917. [DOI] [PubMed] [Google Scholar]
- [86].Malik AR, Szydlowska K, Nizinska K, Asaro A, van Vliet EA, Popp O, et al. (2019). SorCS2 Controls Functional Expression of Amino Acid Transporter EAAT3 and Protects Neurons from Oxidative Stress and Epilepsy-Induced Pathology. Cell Rep, 26: 2792-2804.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Duerson K, Woltjer RL, Mookherjee P, Leverenz JB, Montine TJ, Bird TD, et al. (2009). Detergent-insoluble EAAC1/EAAT3 aberrantly accumulates in hippocampal neurons of Alzheimer's disease patients. Brain Pathol, 19(2):267-278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Wiener HW, Perry RT, Chen Z, Harrell LE, Go RC (2007). A polymorphism in SOD2 is associated with development of Alzheimer's disease. Genes Brain Behav, 6(8):770-775. [DOI] [PubMed] [Google Scholar]
- [89].Spisak K, Klimkowicz-Mrowiec A, Pera J, Dziedzic T, Aleksandra G, Slowik A (2014). rs2070424 of the SOD1 gene is associated with risk of Alzheimer's disease. Neurol Neurochir Pol, 48(5):342-345. [DOI] [PubMed] [Google Scholar]
- [90].Gamarra D, Elcoroaristizabal X, Fernández-Martínez M, De Pancorbo MM (2015). Association of the C47T Polymorphism in SOD2 with Amnestic Mild Cognitive Impairment and Alzheimer's Disease in Carriers of the APOEε4 Allele. Dis Markers, 2015:746329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Camporez D, Belcavello L, Almeida JFF, Silva-Sena GG, Pimassoni LHS, Morelato RL, et al. (2021). Positive association of a Sirt1 variant and parameters of oxidative stress on Alzheimer's disease. Neurol Sci, 42(5):1843-1851. [DOI] [PubMed] [Google Scholar]
- [92].Deng Y, Marsh BM, Moskovitz J (2019). Increased Levels of Protein-methionine Sulfoxide in Plasma Correlate with a Shift from a Mild Cognitive Impairment to an Alzheimer's Disease Stage. Innov. Clin Neurosci, 16(7-08):29-31. [PMC free article] [PubMed] [Google Scholar]
- [93].Cerovic M, Forloni G, Balducci C (2019) Neuroinflammation and the Gut Microbiota: Possible Alternative Therapeutic Targets to Counteract Alzheimer’s Disease? Front. Aging Neurosci, 11:284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Santoro A, Ostan R, Candela M, Biagi E, Brigidi P, Capri M, et al. (2018) Gut microbiota changes in the extreme decades of human life: a focus on centenarians. Cell Mol Life Sci. 75(1):129-148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Parikh IJ, Estus JL, Zajac DJ, Malik M, Maldonado Weng J, Tai LM, et al. (2020). Murine Gut Microbiome Association with APOE Alleles. Front Immunol, 11:200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Tran T, Corsini S, Kellingray L, Hegarty C, Le Gall G, Narbad A, et al. (2019). APOE genotype influences the gut microbiome structure and function in humans and mice: relevance for Alzheimer's disease pathophysiology. FASEB J, 33(7):8221-8231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Martínez I, Perdicaro DJ, Brown AW, Hammons S, Carden TJ, Carr TP, et al. (2013). Diet-induced alterations of host cholesterol metabolism are likely to affect the gut microbiota composition in hamsters. Appl Environ Microbiol, 79(2):516-524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Dinh DM, Volpe GE, Duffalo C, Bhalchandra S, Tai AK, Kane AV, et al. (2015). Intestinal microbiota, microbial translocation, and systemic inflammation in chronic HIV infection. J Infect Dis, 211(1):19-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Kaakoush NO (2015). Insights into the Role of Erysipelotrichaceae in the Human Host. Front. Cell Infect Microbiol, 5:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Mahley RW (2016). Apolipoprotein E: from cardiovascular disease to neurodegenerative disorders. J Mol Med, 94(7):739-746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Ho L, Ono K, Tsuji M, Mazzola P, Singh R, Pasinetti GM (2018). Protective roles of intestinal microbiota derived short chain fatty acids in Alzheimer's disease-type beta-amyloid neuropathological mechanisms. Expert Rev Neurother, 18(1):83-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Cleophas M, Ratter JM, Bekkering S, Quintin J, Schraa K, Stroes ES, et al. (2019). Effects of oral butyrate supplementation on inflammatory potential of circulating peripheral blood mononuclear cells in healthy and obese males. Sci Rep, 9(1):775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Jiang H, Ling Z, Zhang Y, Mao H, Ma Z, Yin Y, et al. (2015). Altered fecal microbiota composition in patients with major depressive disorder. Brain Behav. Immun, 48:186-194. [DOI] [PubMed] [Google Scholar]
- [104].Alam MT, Amos G, Murphy A, Murch S, Wellington E, Arasaradnam RP (2020). Microbial imbalance in inflammatory bowel disease patients at different taxonomic levels. Gut Pathog, 12:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Quévrain E, Maubert MA, Michon C, Chain F, Marquant R, Tailhades J, et al. (2016). Identification of an anti-inflammatory protein from Faecalibacterium prausnitzii, a commensal bacterium deficient in Crohn's disease. Gut, 65(3):415-425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Shi Y, Yamada K, Liddelow SA, Smith ST, Zhao L, Luo W, et al. (2017). ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature, 549(7673):523-527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Tao Q, Ang T, DeCarli C, Auerbach SH, Devine S, Stein TD, et al. (2018). Association of Chronic Low-grade Inflammation with Risk of Alzheimer Disease in ApoE4 Carriers. JAMA Netw Open, 1(6):e183597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Sampson TR, Debelius JW, Thron T, Janssen S, Shastri GG, Ilhan ZE, et al. (2016). Gut Microbiota Regulate Motor Deficits and Neuroinflammation in a Model of Parkinson's Disease. Cell, 167(6):1469-1480.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Kim MS, Kim Y, Choi H, Kim W, Park S, Lee D, et al. (2020). Transfer of a healthy microbiota reduces amyloid and tau pathology in an Alzheimer's disease animal model. Gut, 69(2):283-294. [DOI] [PubMed] [Google Scholar]
- [110].Li B, He Y, Ma J, Huang P, Du J, Cao L, et al. (2019). Mild cognitive impairment has similar alterations as Alzheimer's disease in gut microbiota. Alzheimers. Dement, 15(10):1357-1366. [DOI] [PubMed] [Google Scholar]
- [111].Cattaneo A, Cattane N, Galluzzi S, Provasi S, Lopizzo N, Festari C, et al. (2017) Association of brain amyloidosis with pro-inflammatory gut bacterial taxa and peripheral inflammation markers in cognitively impaired elderly. Neurobiol. Aging, 49:60-68. [DOI] [PubMed] [Google Scholar]
- [112].Pistollato F, Sumalla Cano S, Elio I, Masias Vergara M, Giampieri F, Battino M (2016). Role of gut microbiota and nutrients in amyloid formation and pathogenesis of Alzheimer disease. Nutr Rev, 74(10):624-634. [DOI] [PubMed] [Google Scholar]
- [113].Dinan TG, Cryan JF (2017). Gut instincts: microbiota as a key regulator of brain development, ageing and neurodegeneration. J Physiol, 595(2):489-503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Loffredo L, Ettorre E, Zicari AM, Inghilleri M, Nocella C, Perri L, et al. (2020). Oxidative Stress and Gut-Derived Lipopolysaccharides in Neurodegenerative Disease: Role of NOX2. Oxid. Med Cell Longev, 2020:8630275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Lynch SV, Pedersen O (2016). The Human Intestinal Microbiome in Health and Disease. N Engl J Med, 375(24):2369-2379. [DOI] [PubMed] [Google Scholar]
- [116].Zhan X, Stamova B, Jin LW, DeCarli C, Phinney B, Sharp FR (2016). Gram-negative bacterial molecules associate with Alzheimer disease pathology. Neurology, 87(22):2324-2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Zhao Y, Jaber V, Lukiw W J (2017). Secretory Products of the Human GI Tract Microbiome and Their Potential Impact on Alzheimer's Disease (AD): Detection of Lipopolysaccharide (LPS) in AD Hippocampus. Front Cell Infect Microbiol, 7:318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Zakaria R, Wan Yaacob WM, Othman Z, Long I, Ahmad AH, Al-Rahbi B (2017). Lipopolysaccharide-induced memory impairment in rats: a model of Alzheimer's disease. Physiol Res, 66(4):553-565. [DOI] [PubMed] [Google Scholar]
- [119].Chan C, Law B, Waye M, Chan J, So W, Chow KM (2019). Trimethylamine-N-oxide as One Hypothetical Link for the Relationship between Intestinal Microbiota and Cancer - Where We Are and Where Shall We Go? J Cancer, 10(23):5874-5882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Vogt NM, Romano KA, Darst BF, Engelman CD, Johnson SC, Carlsson CM, et al. (2018). The gut microbiota-derived metabolite trimethylamine N-oxide is elevated in Alzheimer's disease. Alzheimer's Res Ther, 10(1):124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].He Z, Kwek E, Hao W, Zhu H, Liu J, Ma KY, et al. (2021). Hawthorn fruit extract reduced trimethylamine-N-oxide (TMAO)-exacerbated atherogenesis in mice via anti-inflammation and anti-oxidation. Nutr Metab, 18:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Li T, Chen Y, Gua C, Li X (2017). Elevated Circulating Trimethylamine N-Oxide Levels Contribute to Endothelial Dysfunction in Aged Rats through Vascular Inflammation and Oxidative Stress. Front Physiol, 8:350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Brunt VE, Gioscia-Ryan RA, Casso AG, Van Dongen NS, Ziemba BP, Sapinsley ZJ, et al. (2020). Trimethylamine-N-Oxide Promotes Age-Related Vascular Oxidative Stress and Endothelial Dysfunction in Mice and Healthy Humans. Hypertension, 76(1):101-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Olek RA, Samulak JJ, Sawicka AK, Hartmane D, Grinberga S, Pugovics O, et al. (2019). Increased Trimethylamine N-Oxide Is Not Associated with Oxidative Stress Markers in Healthy Aged Women. Oxid. Med. Cell. Longev, 2019:6247169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Botchway BOA, Moore MK, Akinleye FO, Iyer IC, Fang M (2018). Nutrition: Review on the Possible Treatment for Alzheimer’s Disease. J Alzheimer’s Dis, 61(3):867-883. [DOI] [PubMed] [Google Scholar]
- [126].Wang X, Sun G, Feng T, Zhang J, Huang X, Wang T, et al. (2019). Sodium oligomannate therapeutically remodels gut microbiota and suppresses gut bacterial amino acids-shaped neuroinflammation to inhibit Alzheimer’s disease progression. Cell Res, 29(10):787-803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Collin F, Cheignon C, Hureau C (2018). Oxidative stress as a biomarker for Alzheimer's disease. Biomark Med, 12(3):201-203. [DOI] [PubMed] [Google Scholar]
- [128].Moussa C, Hebron M, Huang X, Ahn J, Rissman RA, Aisen PS, et al. (2017). Resveratrol regulates neuro-inflammation and induces adaptive immunity in Alzheimer's disease. J Neuroinflammation, 14(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Eede P, Obst J, Benke E, Yvon-Durocher G, Richard BC, Gimber N, et al. (2020). Interleukin-12/23 deficiency differentially affects pathology in male and female Alzheimer's disease-like mice. EMBO Rep, 21(3):e48530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Vom Berg J, Prokop S, Miller KR, Obst J, Kälin RE, Lopategui-Cabezas I, et al. (2012). Inhibition of IL-12/IL-23 signaling reduces Alzheimer's disease-like pathology and cognitive decline. Nat Med, 18(12): 1812-1819. [DOI] [PubMed] [Google Scholar]
- [131].Wade AG, Farmer M, Harari G, Fund N, Laudon M, Nir T, et al. (2014). Add-on prolonged-release melatonin for cognitive function and sleep in mild to moderate Alzheimer's disease: a 6-month, randomized, placebo-controlled, multicenter trial. Clin Interv Aging, 9:947-961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Lysen TS, Luik AI, Ikram MK, Tiemeier H, Ikram MA (2020). Actigraphy-estimated sleep and 24-hour activity rhythms and the risk of dementia. Alzheimers Dement, 16(9):1259-1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Filon MJ, Wallace E, Wright S, Douglas DJ, Steinberg LI, Verkuilen CL, et al. (2020). Sleep and diurnal rest-activity rhythm disturbances in a mouse model of Alzheimer's disease. Sleep, 43(11): zsaa087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Rizzi L, Roriz-Cruz M (2018). Sirtuin 1 and Alzheimer's disease: An up-to-date review. Neuropeptides, 71:54-60 [DOI] [PubMed] [Google Scholar]
- [135].Dysken MW, Sano M, Asthana S, Vertrees JE, Pallaki M, Llorente M, et al. (2014). Effect of vitamin E and memantine on functional decline in Alzheimer disease: the TEAM-AD VA cooperative randomized trial. JAMA, 311(1):33-44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Shinto L, Quinn J, Montine T, Dodge HH, Woodward W, Baldauf-Wagner S, et al. (2014). A randomized placebo-controlled pilot trial of omega-3 fatty acids and alpha lipoic acid in Alzheimer's disease. J Alzheimer's Dis, 38(1):111-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Jayaraj RL, Azimullah S, Beiram R (2020). Diabetes as a risk factor for Alzheimer’s disease in the Middle East and its shared pathological mediators. Saudi J Biol Sci, 27(2):736-750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Thomas KR, Bangen KJ, Weigand AJ, Edmonds EC, Sundermann E, Wong C, et al. (2020). Type 2 Diabetes Interacts with Alzheimer Disease Risk Factors to Predict Functional Decline. Alzheimer Dis Assoc Disord, 34(1):10-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.