

Abstract
Diffuse midline glioma (DMG) is a deadly pediatric and adolescent central nervous system (CNS) tumor localized along the midline structures of the brain atop the spinal cord. With a median overall survival (OS) of just 9–11-months, DMG is characterized by global hypomethylation of histone H3 at lysine 27 (H3K27me3), driven by recurring somatic mutations in H3 genes including, HIST1H3B/C (H3.1K27M) or H3F3A (H3.3K27M), or through overexpression of EZHIP in patients harboring wildtype H3. The recent World Health Organization’s 5th Classification of CNS Tumors now designates DMG as, ‘H3 K27-altered’, suggesting that global H3K27me3 hypomethylation is a ubiquitous feature of DMG and drives devastating transcriptional programs for which there are no treatments. H3-alterations co-segregate with various other somatic driver mutations, highlighting the high-level of intertumoral heterogeneity of DMG. Furthermore, DMG is also characterized by very high-level intratumoral diversity with tumors harboring multiple subclones within each primary tumor. Each subclone contains their own combinations of driver and passenger lesions that continually evolve, making precision-based medicine challenging to successful execute. Whilst the intertumoral heterogeneity of DMG has been extensively investigated, this is yet to translate to an increase in patient survival. Conversely, our understanding of the non-genomic factors that drive the rapid growth and fatal nature of DMG, including endogenous and exogenous microenvironmental influences, neurological cues, and the posttranscriptional and posttranslational architecture of DMG remains enigmatic or at best, immature. However, these factors are likely to play a significant role in the complex biological sequelae that drives the disease. Here we summarize the heterogeneity of DMG and emphasize how analysis of the posttranslational architecture may improve treatment paradigms. We describe factors that contribute to treatment response and disease progression, as well as highlight the potential for pharmaco-proteogenomics (i.e., the integration of genomics, proteomics and pharmacology) in the management of this uniformly fatal cancer.
Subject terms: CNS cancer, Paediatric cancer
Introduction
Diffuse midline glioma
Diffuse midline glioma (DMG) is a devastating high-grade glioma (HGG) responsible for 50% of all childhood HGGs [1]. DMG is most frequently diagnosed in the brainstem (especially the pons, where it has historically been called diffuse intrinsic pontine glioma - DIPG), and less frequently in the midbrain, thalamus, and spine with patients presenting neurological symptoms such as cranial nerve deficits (facial asymmetry and diplopia), cerebellar signs (ataxia and dysarthria) and long tract signs (hyperreflexia and decreased strength) [2]. DMG patients face a very poor median overall survival (OS) of just 9–11-months, with <10% of patients with pontine tumors surviving two years post-diagnosis [3]. Due to the location within the critical structures of the brain or spine, and the diffuse and infiltrative growth characteristics of the tumor, significant surgical resection is extremely challenging to execute and most often impossible. This leaves radiotherapy as the only standard treatment; however, benefits are temporary for those who respond [4].
Investigations into the multitude of complex biological sequelae that underpin tumor formation and disease progression are key priorities in DMG. Landmark molecular profiling studies have identified numerous key genetic and epigenetic alterations, many co-segregating with age of onset, anatomical location, clinical outcome, histopathological and radiological features [1, 5, 6]. Investigations into the molecular pathogenesis of DMG has led to the classification of several molecular subtypes [1, 7]. The World Health Organization’s (WHO) 5th Classification of Central Nervous System (CNS) Tumors, designates DMG as “diffuse midline glioma, H3 K27-altered” representing the majority of DIPGs as well as tumors found along the midline (e.g., brainstem, midbrain, thalamus, and spine). This classification encompasses molecular subtypes categorized according to alterations to lysine 27 in histone H3 (H3 K27-altered) [8], as well as patients harboring wildtype H3 and concomitant overexpression of the EZH inhibitory protein (EZHIP) [9].
DMG histone H3 modifications
The devastating transcriptional programs influenced by H3-alterations in DMG are fundamentally controlled by posttranslational modification (PTMs) of the 59 amino acid long N-terminal tail of H3, in both histone 3 isoform 1 (H3.1) and histone 3 isoform 3 (H3.3). These PTMs include acetylation, methylation, phosphorylation and ubiquitination and dictate protein structure, stability and accessibility, hence promotes or represses the activation of the transcriptional machinery complexes [10]. H3.1 variants are encoded by a cluster of intronless genes (HIST1 cluster), expressed in a replication dependent manner during the S-phase of the cell cycle [11]. Comparatively, H3.3 variants are independently encoded by two, intron-possessing genes (H3F3A and H3F3B) and are expressed throughout the cell cycle, however, are enriched at various stages of differentiation [12]. Of the genes encoding H3 histone variants, HIST1H3B (H3.1) and H3F3A (H3.3) harbor recurring mutations in DMG leading to the establishment of the molecular subtypes [9]. Between these two histone H3 genes, there are significant structural differences in both the presence of exon and introns, and nucleotide length (Fig. 1A, B), with only 3.4% genomic homogeny. However, the resultant histone proteins show 98.5% homogeny (pairwise sequence alignment using the EMBOSS needle alignment tool [13]). Key lysine residues such as K27 and K36 are conserved and play pivotal roles in the epigenetic regulation of transcription.
Fig. 1. Gene-RNA-protein alignments of the mutant histone H3 genes that give rise to diffuse midline glioma.
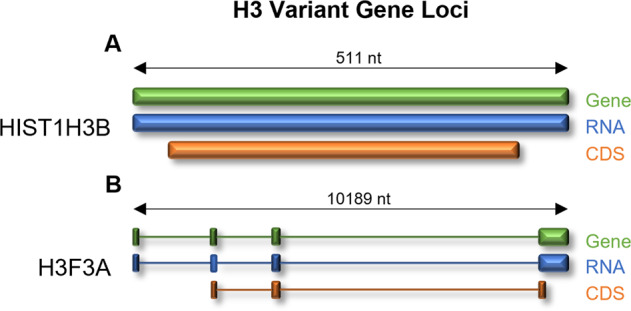
A The HIST1H3B (H3.1) gene is a short (511 nt) intronless gene, translated into a 136 amino acid, 15,404 (Da) protein. B Comparatively, the H3F3A gene is a long (10,189 nt), intron-containing gene, translated into a 136 amino acid, 15,328 (Da) protein.
Diffuse midline glioma, H3 K27-altered
H3.1K27M- and H3.3K27M- DMG
Histone 3 lysine 27 to methionine (H3K27M) mutations occur in both H3.1 and H3.3 histone variants and are mutually exclusive. H3.1K27M is identified in 12–19% of DMG cases, with a median OS rate of 15 months, while H3.3K27M is identified in 65% of cases, with a median OS of 9 months [14]. H3K27M mutations are only translated into 3–17% of the total H3 protein pool [15], however, cause global loss of histone 3 lysine 27 trimethylation (H3K27me3) in the remaining wildtype H3 protein, leading to gene silencing normally regulated by the polycomb repressive complex 2 (PRC2) complex [16]. In healthy cells, the PRC2 complex auto-methylates its core subunits including the Suppressor of zeste 12 protein homolog (SUZ12) and two mutually exclusive and interchangeable catalytic subunits, Enhancer of zeste homolog 1/2 (EZH1/EZH2). Auto-methylation of PRC2 subunits increase histone methyltransferase activity by promoting accessibility to H3 tails where the catalytic pocket of PRC2 can methylate H3K27. Importantly, auto-methylation of EZH2 at K514me3 is reduced in cells transduced with H3K27M with a concomitant reduction in H3K27me2/me3 [17]. DMG harbor even more of a profound loss of EZH2-K514me3 and H3K27me2/me3, indicative of the reduced intrinsic activity of PRC2 [17]. Although the mechanistic basis for the loss of EZH2 methylation and hence PRC methyltransferase activity is not unequivocally resolved, structural studies indicate that the H3K27M mutant peptide shows affinity for EZH2 which may sequester or alter the conformation of the complex [18, 19]. Alternatively, H3K27M may impair the spread of repressive marks, that are preferentially retained at unmethylated CpG islands, affecting lowly-expressed genes influencing neurogenesis [15]. Nevertheless, global hypomethylation of H3K27 inhibits gene silencing and cell differentiation while promoting proliferation and is accompanied by synchronous co-enrichment in elevated H3K36me2, methylated by the Nuclear receptor binding SET domain protein 1/2 (NSD1/2) [20].
As the dominant molecular feature, treatment strategies targeting H3K27M are a priority, however, it has remained undruggable to-date. Given the associated loss of trimethylation and hence increased H3K27 acetylation (H3K27ac), it is of some surprise that most research has focused on the use of histone deacetylase (HDAC) inhibitors (HDACis), such as panobinostat, rather than inhibitors of histone acetyltransferases (HATs). Nevertheless, HDACis show low nanomolar cytotoxicity against DMG cell cultures and are effective in DMG patient-derived xenograft (PDX) mouse models [21, 22]. The HDACis, valproic acid, panobinostat, quisinostat and romidepsin, induce a dose-dependent global increase in H3K27ac and H3K27me3 [21–23], suggestive of a partial rescue of global H3K27 hypomethylation. This is consistent with findings that polyacetylation at residues both proximal and distal to K27M can greatly diminish PRC2 inhibition. Unfortunately, HDACi-induced partial rescue of global hypomethylation is transient [24], encouraging exploration into chemicals that synergize with HDACi. One promising candidate that is in preclinical development is the proteasome inhibitor marizomib which invokes acute toxicity through uncoupling of respiration and inhibition of glycolysis leading to metabolic catastrophe in DMG cells [25].
Wildtype H3 K27 EZHIP-DMG
DMG cases harboring wildtype H3 is seen in approximately 10–15% of cases, with a median OS of 15 months, similar to H3.1K27M DMG [5]. Characterized by the overexpression of the CXorf67 gene which encodes EZHIP [26], EZHIP-overexpressing DMG are a recent addition to the new WHO DMG subtype classification system [27]. EZHIP overexpression occurs in most wildtype H3 DMG cases [26], showing the ubiquitous nature of global H3K27me3 hypomethylation in DMG. A C-terminal peptide in EZHIP mimics the amino acid sequence of H3K27M, sequestering or altering the conformation of PRC2, reducing its histone methyltransferase activity [28]. Inhibition of PRC2 activity leads to an aberrant enrichment of H3K27ac marks and transcriptional programs remarkably analogous to H3K27M [19]. While direct inhibitors of EZHIP are yet to be developed, HDACis, such as panobinostat and quisinostat, show preclinical activity in wildtype H3 DMG, specifically those found in the pontine regions of the brain and that harbor a high mutational burden [22].
DMG somatic and clonal heterogeneity
We are gaining a greater understanding of the somatic heterogeneity of DMG [1], however, what is less understood is the impact of distinct genomic subclonal populations [6], potentially underpinning the lack of effective treatments. While recurring genetic H3-alterations are hallmark features of DMG, multiple co-segregating mutations are patient-specific, conferring their own varying level of poor prognoses and midline localization (Table 1, Fig. 2). The genomic landscapes of DMG have been comprehensively characterized [1], and in most cases, highlight the alterations that harbor potential for therapeutic targeting [29]. Even though we are yet to translate these discoveries into improved outcomes, these sophisticated real-time studies are providing us with increased knowledge of the co-occurring somatic events that underpin the genomic heterogeneity of DMG, critical information for the design of effective combination treatment strategies. To aid in the development of such combination strategies, in the following sections we summarize recurring somatic mutations linked with each DMG a subtype, list the recurrent midline localizations of these driver gene alterations (Fig. 2), and highlight potential therapies and research priorities that we hope will help to increase the durability and effectiveness of strategies targeting these mutations.
Table 1.
Recurring genomic and proteomic alterations in diffuse midline glioma.
| Genetic Alteration | Mutated Sites | Molecular Subtype | Prevalence | Location | Treatment | Reference | Section |
|---|---|---|---|---|---|---|---|
| ACVR1 | R206H, G328V, G328W | H3.1K27M, EZHIP | 32% | Pons, thalamus | LDN212854 | [27, 46, 92, 93] | Activin receptor type-1 (ACVR1) |
| ATM | G2342V, L2877F | H3.3K27M, EZHIP | 6% | Pons | AZD1390 | [27, 43, 94] | ND |
| ATRX | H2254R, R2197L, L1357fs | H3.3K27M, EZHIP | 10% | Pons, thalamus | Pyridostatin | [27, 49, 93] | ND |
| BCOR | C1363fs, A535V, G101fs | H3.1K27M, EZHIP | 8% | Pons | [27, 93–95] | ND | |
| BCORL1 | S425I, R21H | H3.1K27M, H3.3K27M | 7% | Pons | [1, 95] | ND | |
| CCND1/2/3 | Amplification | H3.3K27M | 15% | Pons, thalamus | Palbociclib, ribociclib, abemaciclib | [43] | G1/S-specific cyclin-D2 (CCND2) / Cyclin-dependent kinases 4 and 6 (CDK4, CDK6) |
| CDK4/6 | Amplification, L185V | H3.3K27M | 15% | Pons | Palbociclib, ribociclib, abemaciclib | [1] | G1/S-specific cyclin-D2 (CCND2) / Cyclin-dependent kinases 4 and 6 (CDK4, CDK6) |
| DDX11 | R186W, R167T | H3.3K27M | 6% | Pons | Irinotecan | [49] | ND |
| EGFR | R108K, Amplification, CNG | H3.1K27M, H3.3K27M, EZHIP | 4% | Pons, thalamus | Gefitinib, erlotinib | [1, 5, 14, 49, 96] | ND |
| FGFR1 | K697E, N98S, N546K, K656E | H3.3K27M, H3.1K27M, EZHIP | 12.5% | Pons, thalamus, midbrain | AZ4547, dovatinib, PD173074, ponatinib | [27, 93, 97] | ND |
| GNAQ | T96S | H3.3K27M | 6% | Pons | Tris DBA palladium | [49] | ND |
| IGF2R | K162R, D1830E | H3.1K27M | 8% | Pons | GSK1838705A | [68] | ND |
| KDM6A | Deletion, CNL | H3.3K27M | 6% | Pons | [49, 94] | ND | |
| KDR | S1154P, Amplification, CNG | H3.3K27M | 4.8% | Pons | Mebendazole | [1, 49, 98] | ND |
| KIT | T96P, Amplification, CNG | H3.3K27M | 4.8% | Pons | Mebendazole | [1, 49, 98] | ND |
| KMT5B | R187*, M646fs | H3.3K27M, EZHIP | 1% | Pons | Olaparib, talazoparib | [6] | ND |
| MET | P664P, Amplification | H3.3K27M | 10% | Pons, midbrain | Cabozantinib | [5, 49] | ND |
| MTOR | A1971V | H3.3K27M | 1% | Pons | Everolimus, fimepinostat, AZD2014 | [1, 55, 99] | Phosphatidylinositol-4,5-bisphosphate 3-kinase signaling cascade (PIK3CA/PIK3R1/PTEN/MTOR) |
| MYC | R33C, Amplification | H3.3K27M | 12% | Pons | Omomyc | [1, 57] | MYC proto-oncogene protein (MYC)/ MYCN proto-oncogene protein (MYCN) |
| MYCN | Amplification, CNG | EZHIP | 8% | Pons | Bromodomain inhibitors | [14, 68] | MYC proto-oncogene protein (MYC)/ MYCN proto-oncogene protein (MYCN) |
| NF1 | G295R, R1204W, Deletion | H3.3K27M | 10% | Pons | Binimetinib, trametinib | [1, 14, 95, 99] | ND |
| NTRK1/2/3 | TPM3_NTRK1 VCL_NTRK2 ETV6_NTRK3 | H3.3K27M | 3.7% | Pons, midbrain | Larotrectinib | [5, 98] | ND |
| PDGFRA | Y288C, C235Y, Amplification | H3.3K27M | 30% | Pons | Crenolanib, dasatinib | [1, 42, 93] | Platelet derived growth factor receptor alpha (PDGFRA) |
| PIK3CA | E545K, I391M, H1047R | H3.3K27M, H3.1K27M, EZHIP | 12% | Pons, thalamus, midbrain | Paxalisib, fimepinostat | [43, 55, 93] | Phosphatidylinositol-4,5-bisphosphate 3-kinase signaling cascade (PIK3CA/PIK3R1/PTEN/MTOR) |
| PIK3R1 | K567E, G376R | H3.3K27M, EZHIP | 18% | Pons, thalamus, midbrain | Paxalisib, everolimus, fimepinostat | [27, 43, 49, 55] | Phosphatidylinositol-4,5-bisphosphate 3-kinase signaling cascade (PIK3CA/PIK3R1/PTEN/MTOR) |
| PPM1D | W427*, E525X, Q404X, E405X, 428 fs | H3.1K27M, H3.3K27M, EZHIP | 25% | Pons, thalamus, midbrain | CCT007093, GSK2830371, olaparib | [27, 53, 70] | Protein phosphatase, Mg2+/Mn2+dependent 1D (PPM1D) |
| PTEN | A126S, R130X, Deletion | H3.1K27M, H3.3K27M, EZHIP | 19% | Pons, thalamus | Fimepinostat | [7, 27, 43, 49, 53, 55] | Phosphatidylinositol-4,5-bisphosphate 3-kinase signaling cascade (PIK3CA/PIK3R1/PTEN/MTOR) |
| RB1 | Amplification, Deletion | H3.1K27M, H3.3K27M | 16% | Pons, thalamus | [14, 97] | ND | |
| RPTOR | D857N | H3.3K27M | 1% | Pons | [1, 43] | ND | |
| TERT | C228T, C250T | H3.3K27M | 2% | Pons | Imetelstat | [5] | ND |
| TOP3A | C633Y | H3.3K27M | 3–4% | Pons, midbrain | PIP-199 | [1] | ND |
| TP53 | G245S, R175H, R248Q, R248W, R273C, R273H, S241F, V157F | H3.1K27M, H3.3K27M, EZHIP | 60–80% | Pons, thalamus, midbrain | APR-246, GSK-J4 | [1, 27, 30] | Cellular tumor antigen p53 (TP53) |
| TSC2 | D1587V, Q1035* | EZHIP | 2% | Pons | Rapamycin | [1] | ND |
ND not determined.
Fig. 2. H3-altered diffuse midline glioma recurrent somatic mutations associated with each midline localization.

A Most frequently, diffuse midline glioma (DMG) is localized in the pons (green), midbrain (pink) and thalamus (orange). B Venn diagram of recurrent somatic mutations seen in each H3-altered subtype, H3.1K27M (purple), H3.3K27M (light blue) and EZHIP (sky blue). Identity of recurrent somatic mutations in C H3.1K27M, D H3.3K27M and E EZHIP DMG H3-altered. Genomic information obtained by examining the comprehensive data published by whole-exome and whole-genome studies, references are including in Table 1.
Cellular tumor antigen p53 (TP53)
Cellular tumor antigen p53 (TP53) is the second most recurring lesion in H3.3K27M DMG (60-80%) [30] (Table 1, Fig. 2B). TP53 mutations are also seen in H3.1K27M and EZHIP DMG, however, considerably less frequently (13.3% and 11.1% respectively) [1, 27]. TP53 is a tumor suppressor, encoding the p53 protein which transmits a variety of stress-inducing signals to different antiproliferative cellular responses including apoptosis, senescence, and cell-cycle arrest [31]. Mutations in the TP53 gene are known to lead to tumor immortality through epigenetic dysregulation and elimination of H3K27me3-driven oncogene repression [32]. These mutations are also the main driver of increased radiotherapy-resistance in DMG, both in patients and corresponding cellular models [33]. The introduction of loss of function (LoF) mutations in the TP53 gene and knock-in of H3.3K27M mutations are enough to induce neural stem cell self-renewal in mice. This occurs via transcriptional and epigenetic control of the proliferative genes necessary to drive DMG formation in vivo and are further exacerbated by knock-in of activating mutations in platelet derived growth factor receptor alpha (PDGFRA, discussed in “Platelet derived growth factor receptor alpha (PDGFRA)”) [34].
The recurrent nature of TP53 mutations highlight the importance of therapies that act as surrogate regulators of apoptosis and cell cycle arrest; however, this has been notoriously difficult to achieve. Examining the functional outcomes of TP53 mutations which result in either a partial or full distortion to the DNA-binding domain, has led to the development of the prodrug, APR-246. Upon activation, APR-246 binds to and stabilizes mutant p53, reactivating the protein and driving tumor suppression in preclinical models [35]. In addition to p53 reactivation, APR-246 also elevates reactive oxygen species (ROS) production through dysregulation of redox systems. This propels apoptotic elements and drives oxidative DNA damage, increasing genotoxic stress [36]. The efficacy of APR-246 is further enhanced by radiotherapy (~50%), and when combined with the Jumonji demethylase inhibitor GSK-J4 [30], increased survival of preclinical models [37, 38].
As TP53 LoF mutations are challenging to treat, targeting cell proliferation/survival signaling pathways activated uniquely in response to TP53 mutations, may be an alternative therapeutic approach. Tyrosine phosphoproteomic profiling of TP53-mutant mouse tumor models, revealed unique up-regulation of the proto-oncogene, receptor tyrosine kinase (RTK) Mesenchymal–epithelial transition factor (MET) [39]. Treatment of cultured p53-null cells exhibiting MET amplification with a selective MET tyrosine kinase inhibitor (TKI) (PHA-665752) abrogated aberrant tyrosine phosphorylation and blocked cell proliferation. MET inhibition has also shown preclinical efficacy in TP53 mutant glioblastoma (GBM) cell lines and patient-derived GBM cells, particularly when combined with inhibitors of epidermal growth factor receptor (EGFR) [40]. These observations highlight a possible treatment option for TP53 mutant DMG.
Platelet derived growth factor receptor alpha (PDGFRA)
Activation of PDGFRA accelerates DMG formation in mice, with recurring mutations seen in 14.4% of DMG patients [41] and gene amplification in 30% of DMG, primarily H3.3K27M tumors [42] (Table 1, Fig. 2B). Platelet derived growth factor receptors (PDGFRs) influence cell migration, proliferation and survival with ligand induced receptor dimerization driving auto-phosphorylation and activation [43]. Transduction of downstream signals is regulated by a multitude of pathways, but predominately PI3K/Akt/mTOR [41, 43] (discussed in “Phosphatidylinositol-4,5-bisphosphate 3-kinase signaling cascade (PIK3CA/PIK3R1/PTEN/MTOR)”). Co-segregating with H3.3K27M, amplified PDGFRA strongly promotes glioma formation in vivo, resulting in a clinically aggressive form of DMG [41] (Fig. 2B). Tyrosine kinase inhibitors (TKIs) approved by the US Food and Drug Administration (FDA), such as dasatinib and crenolanib, target PDGFRA, however are cytostatic, not cytotoxic, and hence do not extend the survival of DMG patients [44] (Table 2). Deep proteomic, phosphoproteomic and transcriptomic profiling of PDGFRA-mutant HGG mouse models identified PI3K/Akt signaling as responsible for driving MYC (discussed in “MYC proto-oncogene protein (MYC)/ MYCN proto-oncogene protein (MYCN)”) and JUN (Transcription factor AP-1) activity [45], to create a positive-feedback loop increasing expression of multiple other RTKs leading to oncogene addiction. Importantly, the activity of oncogenic signaling pathways activated downstream of PDGFRA mutations were only visible via assessment of the phosphoproteome, highlighting the importance of the evaluation of the posttranslational architecture of DMG in the design effective treatment strategies.
Table 2.
Clinical trials involving the use of targeted therapy drugs in DMG and other pediatric gliomas.
| Treatment | Clinical Trial Identifier | Phase | Description | Posted results |
|---|---|---|---|---|
| Abemaciclib | NCT02644460 | Phase I [Recruiting] | Clinical trial evaluating abemaciclib in patients with newly diagnosed or relapsed/refractory DMG aged 2–25-years. | |
| NCT04238819 | Phase I [Recruiting] | Study to determine the safety and efficacy of abemaciclib in combination with temozolomide and irinotecan in patients with relapsed/refractory solid tumors aged up to 18-years. | ||
| AZD1390 | NCT03215381 | Phase I [Completed] | Study to analyze the PK of AZD1390 in healthy adult males aged between 20–65-years. | No results posted |
| NCT03423628 | Phase I [Recruiting] | Study to test safety, tolerability, and PK of AZD1390 and radiotherapy for the treatment of glioblastoma in patients aged18–130-years. | ||
| AZD2014 | NCT02619864 | Phase I [Completed] | Clinical trial to determine MTD of AZD2014 in combination with temozolomide in glioblastoma patients 18- years and older. | No results posted |
| Binimetinib | NCT02285439 | Phase I/II [Active, not recruiting] | Clinical trial to determine MTD of binimetinib (MEK162) in patients with low grade glioma aged 1–18-years. | |
| BMS-986158 | NCT03936465 | Phase I [Recruiting] | Study investigating the bromodomain inhibitor, BMS-986158, for brain tumors in patients aged 1-21-years. | |
| Cabozantinib | NCT02885324 | Phase II [Recruiting] | Clinical trial to test cabozantinib for HGG patients aged 2–21-years. | |
| Crenolanib | NCT01393912 | Phase I [Completed] | Phase I clinical evaluated crenolanib in patients with newly diagnosed DMG or in recurrent, progressive, or refractory HGG aged 18 months-21-years. | No results posted |
| Dasatinib | NCT01644773 | Phase I [Completed] | Clinical trial to determine MTD of crizotinib and dasatinib for patients with DMG and other HGG aged 2–21-years. | No results posted |
| NCT00996723 | Phase I [Completed] | Clinical trial to evaluate the combination of vandetanib and dasatinib during and after radiotherapy in patients with DMG 18 months–21-years. | No results posted | |
| Erlotinib | NCT01182350 | Phase II [Terminated] | Clinical trial tested combinations of FDA-approved agents (including erlotinib) in patients with DMG aged 3–18-years based on specific biologic targets. | 64.4% of patients had a 9-month overall survival rate following treatment |
| Erlotinib | NCT02233049 | Phase II [Unknown] | Clinical trial comparing response of DMG patient to erlotinib, everolimus and/or dasatinib depending on biological abnormalities, aged 6 months–25-years. | |
| Everolimus | NCT03387020 | Phase I [Completed] | This study examined the side effects and best dose of ribociclib and everolimus as well as how well they work in treating patients, aged 1-21-years, with recurrent/refractory DMG. | MTD for ribociclib and everolimus was determined to be 120 and 1.2 mg/m2/day respectively. |
| NCT03355794 | Phase I [Active, not recruiting] | Clinical trial examining the safety of ribociclib and everolimus, when administered to DMG patients aged 1-30-years following radiation therapy. | ||
| NCT03632317 | Phase II [Withdrawn] | Clinical trial evaluated the activity of panobinostat in combination with everolimus for patients aged 2 to 30 years with newly diagnosed HGG or DMG after radiotherapy. | Low accrual | |
| Fimepinostat | NCT03893487 | Early Phase I [Recruiting] | Clinical trial studying the efficacy of fimepinostat in treating patients aged 3 to 39 years with newly diagnosed DMG | |
| Gefitinib | NCT00042991 | Phase I/II [Completed] | Clinical trial studied the efficacy of gefitinib, in combination with radiation therapy, in treating patients aged 3-21 years with brainstem gliomas and glioblastoma. | This trial found median progression -free survival to be 7.43 months on average while overall survival was 12.12 months |
| NCT00052208 | Phase I/II [Completed] | Study investigated side effects and best dose of gefitinib when administered in conjunction with radiotherapy as well as its effectiveness in treating patients of all ages with glioblastoma. | No results posted | |
| Imetelstat | NCT01836549 | Phase II [Terminated] | Phase II clinical trial studied the efficacy of imetelstat in treating patients aged 12 months to 21 years with recurrent or refractory brain tumors. | Terminated due to several intracranial hemorrhages and recommendation by the PBTC DSMB. |
| Larotrectinib | NCT04655404 | Early Phase I [Recruiting] | Clinical trial evaluating the disease status in patients aged up to 21 years with HGG with TRK fusion following larotrectinib treatment. | |
| Mebendazole | NCT01837862 | Phase I/II [Recruiting] | Study determining the safety and efficacy of mebendazole in combination with chemotherapy drugs for the treatment of DMG in patients between 1 and 21 years of age. | |
| NCT02644291 | Phase I [Recruiting] | Clinical trial investigating the safety and side effects of mebendazole in patients aged 1-21 years for recurrent brain cancers that are no longer responsive to standard therapies. | ||
| Olaparib | NCT03233204 | Phase II [Recruiting] | Study observing the effectiveness of olaparib in treating patients (12 months to 21 years) with relapsed/refractory solid tumors, which possess defects in DNA damage repair genes. | |
| NCT03155620 | Phase II [Recruiting] | Phase II trial studying the efficacy of genetic testing-directed treatment (including the drug olaparib) in patients, between aged 12 months to 21 years, with advanced solid tumors. | ||
| ONC201 | NCT03416530 | Phase I [Recruiting] | Multicenter, seven arm, dose escalation, clinical trial studying ONC201 in patients aged 2 to 18 years with DMG and recurrent/refractory H3 K27M gliomas. | |
| Palbociclib | NCT03709680 | Phase I [Recruiting] | Study evaluating palbociclib in combination with chemotherapy in patents aged 2-20 years with medulloblastoma or DMG. | |
| Ponatinib | NCT02478164 | Phase II [Completed] | Clinical trial studied ponatinib as a potential treatment for recurrent glioblastoma unresponsive to bevacizumab in patients aged 18 years and older. | This clinical trial had no patients with a 3-month progression free survival and an average overall survival of 98 days. |
| Rapamycin | NCT02420613 | Phase I [Active, not recruiting] | Phase I trial studying the side effects and best dose of rapamycin (temsirolimus) when given together with vorinostat and with or without radiation therapy in patients aged 7 months to 21 years with DMG | |
| Ribociclib | NCT03355794 | Phase I [Active, not recruiting] | Clinical trial examining the safety of ribociclib and everolimus, when administered to patients aged 12 months to 30 years with DMG following radiation therapy. | |
| NCT02607124 | Phase I/II [Terminated] | Study investigated the effects of ribociclib when given after radiation therapy in DMG patients aged 12 months to 30 years. | Terminated due to a competing trial opened for patient population with combination of ribociclib and everolimus. | |
| NCT03387020 | Phase I [Completed] | Clinical trial examined the side effects and best dose of ribociclib and everolimus and their efficacy in treating patients aged 1-21 years with treatment resistant or relapsed malignant brain tumors, including DMG. | No results posted | |
| Talazoparib | NCT04740190 | Phase II [Recruiting] | Study testing the effectiveness of talazoparib in recurrent glioblastoma in patients aged 18 years and older. | |
| Trametinib | NCT03919071 | Phase II [Recruiting] | Clinical trial examining how well dabrafenib and trametinib works after radiation therapy in HGG patients aged 3- 25 years. |
Activin receptor type-1 (ACVR1)
Activin receptor type I (ACVR1) is mutated in approximately 32% of all DMG [32], 87% of H3.1K27M [1] and 72% (13/18 cases) of EZHIP [27] (Table 1, Fig. 2A–C). ACVR1 encodes the serine/threonine protein kinase, activin receptor-like kinase-2 (ALK2), belonging to bone morphogenetic protein (BMP) signaling pathway, transforming growth factor-beta (TGF-β) superfamily. Activation of ALK2 regulates morphogenesis, differentiation, proliferation, and apoptosis during embryonic development [43]. ALK2 phosphorylation activates BMP signaling and leads to the phosphorylation and activation of the SMAD (mothers against decapentaplegic family) transcription factors [43]. ACVR1 mutations lead to constitutive activation of BMP signaling, thus, activation of SMAD, driving expression of DNA-binding protein inhibitors, ID1 and ID2. This expression promotes tumor initiation and accelerates gliomagenesis whilst repressing differentiation [43, 46]. Somatic mutations in ACVR1 are unique to DMG, with analogous germline mutations seen in fibrodysplasia ossificans progressiva (FOP). Currently, there are no curative treatments for ACVR1 mutations in FOP, thus treatments have instead focused on the inhibition of BMP [43]. Preclinical compounds such as LDN-212184, have been developed to inhibit ALK2 and hence phosphorylation of SMAD, with pharmacokinetic (PK) studies showing sufficient brain penetration [46]. Although LDN-212184 extends survival of orthotopic DMG PDX mouse models, ALK2 inhibitors, with improved specificity and potency, are necessary if these treatments are to translate to better outcomes for DMG patients.
Given that the intracellular activity of SMADs is reliant on their phosphorylation, numerous protein phosphatases are known to downregulate their activity and may serve as targets for therapies to enhance ALK2 inhibition [47]. The serine/threonine protein phosphatase, PP2A is a SMAD-associated phosphatase frequently showing reduced activity in cancer, with activity increased using fingolimod (FTY720), an FDA approved drug used in the treatment of multiple sclerosis, and hence has excellent brain penetration [48]. This encourages preclinical assessment of FTY720 in combination with therapies targeting ALK2 for ACVR1 mutant DMG.
Phosphatidylinositol-4,5-bisphosphate 3-kinase signaling cascade (PIK3CA/PIK3R1/PTEN/MTOR)
Mutations in the components of the Phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K) signaling axis are recognized drivers of gliomagenesis in DMG [43]. Mutations in the PI3K catalytic subunit alpha (PIK3CA) are seen in 12% of DMG (Fig. 2), while PI3K regulatory subunit 1 (PIK3R1) mutations are present in 18%, most commonly in H3.3K27M and EZHIP subtypes [43, 49] (Table 1, Figs. 2B, C). PIK3CA mutations lead to constitutive lipid kinase activity thereby driving cellular transformation [50], while PIK3R1 mutations activate wildtype PIK3CA or PI3K signaling [51]. Phosphatase and tensin homolog (PTEN) is the well-established negative regulator of this signaling cascade, and an important tyrosine kinase tumor suppressor [52]. PTEN is mutated in 4% of H3.1K27M [53], 6% of H3.3K27M [49], and 6% of EZHIP DMG [27] (Table 1, Fig. 2). Loss of PTEN can occur through a chromosomal deletion of 10q and is determined to be an early event in DMG development [52].
Paxalisib is an FDA approved, PI3K/Akt inhibitor developed to penetrate the brain and decrease activity of signaling cascades [54]. Recently, paxalisib was shown to reduce growth and proliferation in PI3K-mutant and wildtype DMG cell lines [54] and has recently entered combination clinical trials for DMG (NCT05009992). Fimepinostat (CUDC-907), is a dual PI3K/HDAC inhibitor (Table 2), that inhibits radiation-induced DNA repair pathways including homologous recombination and nonhomologous end-joining, leading to G1 cell-cycle arrest and apoptosis [55] and is also in DMG clinical trials (NCT03893487). Despite PI3K alterations being some of the common events in DMG and other cancers, pharmacological inhibition of PI3K has resulted in variable clinical responses. This raises the possibility of an inherent mechanism of resistance. Indeed, mouse tumor models show insulin feedback is induced by PI3K inhibitors, reactivating PI3K signaling, thus compromising their efficacy [56]. Insulin-feedback is effectively controlled using anti-glycemic approaches, which greatly enhances the therapeutic effectiveness of PI3K inhibitors, an approach that warrants rigorous testing in DMG.
MYC proto-oncogene protein (MYC) / MYCN proto-oncogene protein (MYCN)
The MYC proto-oncogene transcription factor family, including MYCL proto-oncogene (MYCL), Cellular myelocytomatosis oncogene (c-MYC) and MYCN proto-oncogene (MYCN), are important mediators of many growth-promoting signal transduction pathways. MYC alterations are common in human cancers including DMG, reported in 20% of the H3.3K27M subtypes [57] (Table 1, Fig. 2B). Interestingly, the MYCN gene has been used to define a subset of DMG characterized by CpG hypermethylation, high-grade histology, and chromothripsis on chromosome 2p in tumors, leading to recurrent amplification of MYCN [7]. This subtype is seen in 8% of DMG, and predominately associated with EZHIP [7]. High MYC activity is correlated with poor outcomes, but it is very difficult to target owing to its ‘undruggable’ protein structure [57]. To overcome this, Omomyc, a peptide-based dominant negative inhibitor, was developed to outcompete MYC/MAX dimers for binding to E-box DNA sequences. MYC dimerizes with MAX to activate transcription and promote cell proliferation. Omomyc binds to MAX, blocking MYC binding, repressing MYC-mediated gene expression profiles in H3.3K27M DMG preclinical models [57, 58].
Hyperacetylation of H3K27 in DMG is driven by the activity of acetyl-binding, bromodomain and extraterminal (BET) proteins, particularly bromodomain-containing protein 4 (BRD4) which are implicated in tumor progression and aggressiveness [59]. MYC is highly occupied by H3.3K27M and H3K27ac super-enhancers and hence highly expressed in DMG. This is slightly at odds with what has been previously shown, where MYC/MYCN amplification was only observed in EZHIP DMG [7]. Panobinostat decreased oncogenic MYC target gene expression causing cell death in DMG preclinical models [21].
Targeting BRD4-driven MYC activity, using the BBB-penetrant bromodomain inhibitor, JQ1, is effective as a monotherapy in EZHIP DMG models when used at high-doses, however, H3K27M DMG are less sensitive [23]. Combinations of JQ1 and MRK003, a gamma-secretase inhibitor that reduces NOTCH1 expression, reduced H3K27M DMG growth and survival [60], highlighting this as a potential strategy for DMG.
G1/S-specific cyclin-D2 (CCND2) / cyclin-dependent kinases 4 and 6 (CDK4, CDK6)
G1/S-specific cyclin-D2 (CCND2) functions as a regulator of Cyclin-dependent kinase 4 and 6 (CDK4/CDK6) which contributes to the temporal coordination of the cell cycle [61], and typically altered in H3.3K27M DMG (Table 1, Fig. 2B). Activation of the cyclin/CDK complex leads to the hyperphosphorylation of the Retinoblastoma (RB) tumor suppressor protein and dissociation from the transcription factor E2F1. The recruitment of HATs and subsequent transcription of E2F1 target genes advance mitotic progression through the G1/S phase [61]. Modulation of cyclins such as CCND2 (G1/S-specific cyclin-D2) occurs in nearly all tumors with the amplification of CCND2 being a frequent genomic event in DMG and predominates in tumors of the pontine region [1]. CDK4/6 mutations are also common in DMG, highlighting the potential of targeting the cyclin signaling axis. In melanoma, activating mutations in CDK4 abolish interactions with the tumor suppressor Cyclin dependent kinase inhibitor 2 A’s (CDKN2A) / p16INK4A, rendering the protein constitutively active, leading to uncontrolled cell cycling [62].
Given the importance of cyclin proteins in mitosis, several CDK4/6 inhibitors have been developed and are under clinical evaluation in DMG [63] (Table 2). Palbociclib has completed dose escalation studies, while two other trials are testing ribociclib. Abemaciclib is suggested to be the most effective CCND2-CDK4/6 inhibitor due to higher CDK4/6 affinity/potency compared to palbociclib and ribociclib [63]. While these CDK4/6 inhibitors have shown some efficacy in tumors with deregulated cell-cycle control, resistance to these therapies is common. Phosphoproteomic profiling of cancer models resistant to CDK4/6 inhibitors revealed enhanced Mitogen-activated protein kinase (MAPK) signaling, therapeutically exposed using the FDA approved MEK inhibitor, trametinib [64] (Table 2). Targeting MEK is a treatment paradigm that has recently come into clinical thinking particularly for HGGs and DIPGs harboring germline or somatic mutations in Neurofibromatosis type 1 (NF1) [65, 66]. Furthermore, the combination of MEK1/2 and CDK4/6 inhibition showed therapeutic synergy across a broad panel of high-risk neuroblastoma preclinical models [67], a treatment paradigm that warrants exploration in DMG.
Protein phosphatase, Mg2+/Mn2+ dependent 1D (PPM1D)
Activating mutations in the Protein phosphatase, Mg2+/Mn2+ dependent 1D (PPM1D) are seen in 15% of H3.3K27M [68], 3.8% of H3.1K27M [68], and 11.1% of EZHIP [27] DMG (Table 1, Fig. 2). PPM1D mutations inactivate ATM- (Ataxia telangiectasia mutated protein) and ATR- (Ataxia telangiectasia and Rad3-related protein) mediated DNA damage response (DDR) and dephosphorylate H2A.X and p53 in response to radiation [69]. Knockdown of PPM1D is almost curative in DMG PDX models [70]. Hence an allosteric, non-competitive inhibitor of PPM1D, GSK2830371, has been developed, which drives p53-dependent inhibition of DMG tumor growth [70]. Additionally, GSK2830371 sensitized PPM1D-mutant DMG to PARP inhibition using olaparib (Table 2). Although olaparib showed no brain penetration against intact BBB, tumor penetration was detected in orthotopic GBM xenografts, penetrating both tumor core and tumor margins of GBM patients with recurrent tumors [71]. Whether GBM’s tumor vasculature characteristics (leaky vasculature) translate to olaparib uptake in DMG is yet to be reported.
Intertumoral and intratumoral heterogeneity
The somatic alterations summarized above offer some biological insight into the roles these recurring mutations play in DMG and present us with a suite of therapeutic vulnerabilities. Despite the number of currently used targeted therapies developed to combat the various co-occurring mutations in DMG, most are still undergoing early-stage clinical trials to establish toxicity profiles. More recent studies are now using them in combination, however, the maximum tolerated doses for these combinations are yet to be determined, and efficacy signals are unknown (Table 2).
The complexity of these clonal cancer genomes emphasizes why a one-drug-fits-all model has unequivocally failed patients. Furthermore, tumors typically have multiple mutations of unknown significance, making it challenging to ascertain which of these genetic lesions are primary oncogenic drivers. Investigation into the effectiveness of drug combinations as means to combat the somatic heterogeneity of DMG has been performed in multiple DMG models using high-throughput drug screening, and comprehensive molecular analysis to correlate drug sensitivities with genomic influences [21, 25]. While the intertumoral heterogeneity of DMG is well established and exploited to develop treatments (Table 1, Table 2), the intratumoral diversity of this cancer represents a relatively novel field in which to better understand DMG. Analysis of nonsilent mutations in DMG revealed high intratumor (clonal) heterogeneity as a proportion of nonslient mutations [53], the extent of which is greater than or equal to all other cancers [72]. In both DMG and pediatric GBMs, multiple somatic subclones co-exist, both spatially and temporally [6, 44]. These co-existing tumor subclones are suspected to play important tumorigenic roles in response to CNS active therapies and may enhance proliferative, tissue invasion and oncogenic signaling, to promote tumor cell dissemination. Indeed, DMG subclones co-cultured or transplanted together, enhanced the invasion into surrounding tissues of less-mobile colonies [44]. These tumor subclones, populated by varying co-segregating driver and passenger mutations, continually evolve alongside tumor burden during a patient’s clinical journey (Fig. 3). Targeting of one of these subclonal driver mutations may show success in combating that oncogenic colony, however, genomic diversity between subclones encourages their spread and indeed diversification, ultimately leading to resistance. Hence targeted therapies provide limited therapeutic benefits [73]. The growth and diversification of these subclones influence, and are influenced by, non-genomic factors including the tumor microenvironment (TME), concurrent corticosteroid therapy, growth factors, stress hormones and electrical signals [74] that increase invasiveness and provide a survival advantage, as well as potentiate intratumoral heterogeneity [75] (discussed further in “Phosphoproteomic and tumor microenvironmental influences”).
Fig. 3. Clonal evolution, tumor burden and non-genomic contributions to diffuse midline gliomas development and progression.
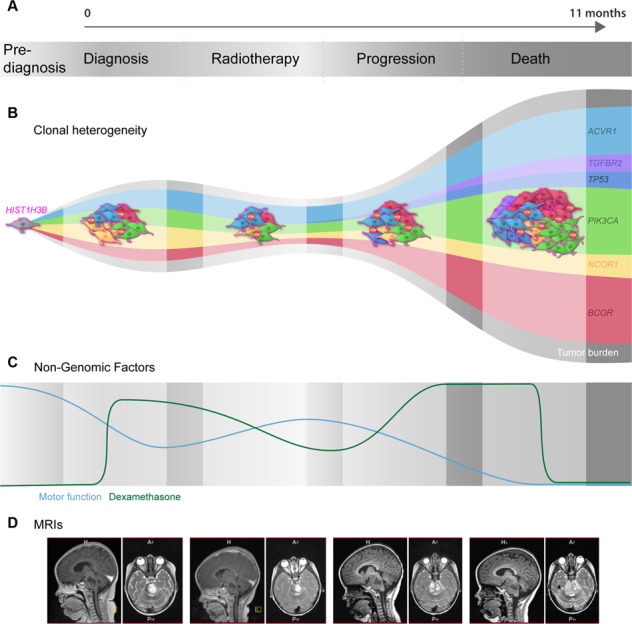
A Diffuse midline glioma (DMG) tumor burden continually increases following diagnosis. Similarly, the clonal heterogeneity of DMG evolves throughout a patient’s disease, potentially influenced by endogenous and exogeneous factors, including microsatellite instability, treatment, and steroids. B Representation of tumor evolution beginning with a single tumor cell harboring a HIST1H3B mutation, cell outlined in pink. As the tumor grows and diversifies, it gains new driver and passenger mutations. Driver mutations in this example include ACVR1 in light blue, TGFBR2 in purple, TP53 in dark blue, PIK3CA in green, NCOR1 in yellow and BCOR in red. C In addition to the increased clonal heterogeneity and tumor burden, non-genomic factors fluctuate throughout disease progression and likely contribute to growth and survival. The patient’s degree of motor function, represented by a blue line, is inversely proportional to tumor burden, whereas corticosteroids anti-inflammatory medications (dexamethasone), represent by a green line, is relatively proportional to tumor burden beginning with a sharp increase at diagnosis, sustained then decreased during radiotherapy, and adjusted to meet the patient’s symptoms [100]. D Representative magnetic resonance imaging (MRI) of tumor development and progression throughout a DMG patient’s journey.
Moving towards systems biological approach
Germline analysis
To progress treatment and improve outcomes we need to take a systems-wide view of DMG and employ multi-omics approaches to develop treatments, starting with the analysis of both the somatic and germline mutations. Characterization of a patient’s germline and somatic variants, coupled with understanding of the systems-wide response to therapies, will potentiate the personalization of drug-dose and -timing, as well as the selection of the most appropriate therapy for each patient. Germline DNA analysis is increasingly becoming as important as somatic evaluation. Not only does it aid in distinguishing key somatic events, but also identifies any potential inherited influences that may play an important role in drug PK and pharmacodynamic (PD) disposition. For example, patients carrying Uridine 5’-diphospho-glucuronosyltransferase (UGT) polymorphisms have impaired ability to inactivate 7-ethyl-10-hydroxycamptothecin (SN-38), the active metabolite of irinotecan, and hence face significant toxicities. If polymorphisms are known, patients receive reduced dose [76] which may affect response. This may be an important consideration in the case of DMG response to radiotherapy, as common TP53 germline mutations may influence response [33].
It is important to consider that gene expression profiles rarely correlate with the abundance of the corresponding protein, influenced by a range of post -transcriptional / -translational effects. These effects include amino acid composition which impacts the rate of translation elongation [77], miRNA expression, and the battery of posttranslational modification (>200) that characterize the mammalian proteome [78]. This may provide us with some insight into why genomically-targeted, precision therapies are yet to provide a therapeutic benefit for DMG patients. A quantitative trait loci (QTL) is a region of DNA associated with a particular phenotypic trait that varies dependent on polygenic effects such as expression of multiple genes, and the environment in which they are expressed; while protein quantitative trait loci (pQTLs) provide a functional readout to identify how genetic variants regulate protein expression; necessary to reveal the proteome’s role in disease causation [79]. QTL mapping using somatic mutations, germline variants and sensitivity to 265 drugs, in 993 cancer cell lines, identified 78 drugs with at least one significant genetic association (drug response QTL). Remarkably, nine of these genetic associations involved germline variants, comparable with effects related to somatic variants, highlighting that germline variants contribute to protein abundance, a key factor in modeling drug sensitivity and response [80]. Given germline mutations in cancer-predisposing genes are seen in 9% of non-DMG pediatric cancers [81], it is likely that these mutations have a yet-to-be-determined role in the gliomagenesis and treatment resistant characteristics of DMG. While genomic sequencing provides a static snapshot of the cellular environment, these studies are limited in their ability to interpret the contribution of genetic features to the biology of patient’s tumor, leaving clinicians with therapies that offer little hope of efficacy. The cancer proteome, however, can narrow the gap between genotype and phenotype, providing a more appropriate platform for studying the kinetics of drug response, as it accounts for the plasticity and dynamic nature of cancer cells and, when assessed using tumors treated in vivo, can help to reveal the important contribution of the TME [82].
Phosphoproteomic and tumor microenvironmental influences
Global assessment of the PTMs that influence the activity of oncoproteins that ultimately drive gliomagenesis in DMG is now necessary. Coupled with molecular and pharmacological information, assessment of the proteome and posttranslational architecture of DMG reveal the proteomic heterogeneity of DMG in response to treatment. While some DMG harbor constitutively activated RAS/MAPK pathways [57], not all cells within the tumor will show this signal-type. Proteomic heterogeneity is not always a simple consequence of the heterogeneity of the genome and is always affected by endogenous factors, including metabolic and neurological cues, and exogenous stressors such as steroids, experimental treatments and radiotherapy [57]. The TME itself is a complex network of cells, organelles and structures such as blood vessels, neurons, astrocytes, microglia, and oligodendrocytes and filled with endogenous factors, including catecholamines such as dopamine, insulin, growth hormones, factors likely to influence tumor development and progression [57] (Fig. 4).
Fig. 4. Non-genomic contributions to diffuse midline glioma growth and progression.

Diffuse midline gliomas (DMG) are vastly complex tumors localized in the midline structures of the brain. At diagnosis (primary tumor) DMG harbor numerous driver mutations, (highlighted by tumor cells of varying color), that contribute to drive the evolution of the cancer. Surrounding cells and structures of the midline of the brain such as blood vessels, neurons, astrocytes, microglia, and oligodendrocytes contribute to the gliomagenesis of DMG, whether through direct physical connections or by more indirect mechanisms, such as electrical signals (yellow lightening blots) between glioma and normal cells or the contribution of growth hormones, NGF, VEGF, TGF-β, and prolactin, or even, endogenous factors, such as hypoxia, dopamine, insulin, catecholamines. The extracellular cues drive posttranslational modifications (PTMs) that influence the activity of oncoproteins that contribute to the aggressive nature of the disease. It is likely that exogenous factors, such as radiotherapy (radioactive symbol) and corticosteroids (dexamethasone), also contribute to the disease, the impact on tumor growth and treatment resistance yet to be fully understood. The diffuse and infiltrative growth of this cancer also leads to dissemination throughout the brain. Disseminated subclones, however, can differ in genomic and proteomic characteristics to that of the primary tumor, influenced by clonal selection supported by non-genomic factors from each different region of the brain, and lead to distinct survival and proliferative advantages, highlighting the challenge we face in developing treatment strategies that will lead to long-term survival for patients diagnosed with DMG.
Interestingly, but perhaps not surprisingly, another microenvironmental influence on DMG tumor progression is neuronal activity. Electrochemical communications occur between gliomas and neurons through synapses and drive proliferation, differentiation, and survival (Fig. 4). Neuronal excitability and thus the release of growth factors promote glioma propagation [74]. A common characteristic of the TME is the infiltration of immune cells which influence the behavior of tumor cells. In adult GBMs, infiltrating immune cells stimulate tumor proliferation and invasion. Tumor-associated macrophages originate from microglia and/or bone marrow-derived monocytes to drive cellular migration and metastasis and release pro-inflammatory cytokines to promote a glioma stem-like state [83]. However, primary DMG display little immune cell infiltration when compared to GBM and other low-grade gliomas (LGG). These findings reveal minimal immune activation in the DMG TME, the impact of which is currently unclear [44]. These microenvironmental influences are vital to tumor development and, when acting in conjunction proteomic heterogeneity, aids in the natural selection of aggressive tumor subclones [72] (Fig. 4). For instance, this can occur by changing the milieu of certain conditions and/or growth factors following therapy. The hypoxic microenvironment that remains following radiotherapy typically favors tumor cells harboring activated HIF1α, aiding survival and poor outcomes [84].
While these microenvironmental factors likely influence tumor progression and the effectiveness of treatments, systems-wide analysis can be used to predict drug efficacy. Indeed, proteomics has been acknowledged as one of the most effective predictors of drug cytotoxicity compared to gene copy number, mutations, and methylation analysis, which provide poor predictors of the sensitivity of human cancer cell lines to an extensive list of oncology and non-oncology drugs [85].
Pharmaco-proteogenomics
There are clear benefits to integrating complementary pharmacogenomic and pharmacoproteomic research when investigating new treatment paradigms for patients. Techniques such as single nucleotide polymorphism (SNP) arrays, whole genome sequencing (WGS) and RNA-sequencing (RNA-seq) should be implemented in conjunction with high-throughput quantitative proteomics [86, 87], validated via high-throughput multiplex immunohistochemical imaging and TME analysis using limited tissue from biopsy. This will provide us with a more effective target prediction platform, and lead to more durable responses [88].
Large-scale, comprehensive proteogenomic analysis in pediatric brain cancers has recently been performed using 218 tumors across 7 histological types with thousands of proteins and phosphoproteins, correlated with mutations and copy number variations (CNVs). Of these pediatric CNS tumors, 25 were HGGs, 10 of which were localized to the midline [89]. Proteomic and phosphoproteomic profiling analyzed by consensus clustering revealed eight distinct subtypes, used to predict survival outcomes, proliferation indices and pathway activation, that spanned histological boundaries. Of the 10 HGG localized to midline structures, 7 subtyped within the HGG-rich cluster, while 3 were grouped in LGG BRAFFusion-rich, Ganglioglioma-rich and Ependymoma clusters. Kinase enrichment analysis using HGG phosphoproteomes, revealed an elevated abundance of phosphorylated substrates of CDK1 and CDK2. CDK2 shared kinase-substrate association with Minichromosome maintenance complex component 2 (MCM2) at Ser 139 and Nucleophosmin (NPM1) at Ser 70, with MCM2 and NPM1 vital for the regulation of cell proliferation, suggesting CDK2 plays a dominant role in promoting DMG cell proliferation, and hence a target of worthy pursuit. An independent signaling node in HGG was the Calcium/calmodulin dependent protein kinase II alpha (CAMK2A) also shown to be the most abundant protein in gangliogliomas; but in HGG a higher correlation between kinase activity and protein abundance was evident [89], highlighting the importance of simultaneous analysis of the transcriptome/proteome and the phosphoproteome. CAMK2A in association with Gap junction protein alpha 1 (GJA1) phosphorylated at Ser 325 and Ser 314, plays an important role in metastatic invasion, promoting gap junction assembly between glioma cells and surrounding astrocytes [90] as well as increasing synaptic transmission through the phosphorylation of Synapsin-1 (SYN1) at Ser 605 by CAMK2A [89]. Given the limited number of DMG samples profiled in this study, it is currently unknown whether these two mutually exclusive HGG proliferation (CKD2-MCM2-NPM1) and invasion (CAMK2A-GJA1-SYN1) oncogenic signaling pathways are associated with specific DMG H3-subtypes, highlighting the necessity for similar studies assessing the phospho-proteogenomic characteristics of DMG, studies that are ongoing in our laboratory. Nevertheless, the observation that the phosphorylation status of these pathways dictates their oncogenic function, which remains invisible to genomic approaches, highlights the importance of a systems-wide view to aid in the development of effective treatment strategies for DMG patients.
Importantly, not only can pharmaco-proteogenomics offer a functional molecular readout of the potential of therapeutic targets but can also be used to refine molecular subtypes based on drug response. Quantitative proteomics coupled with RNA sequencing led to the development of a distinct binary classification system for Isocitrate dehydrogenase 1/2 (IDH) wildtype GBM. These tumors were clustered based on elevated protein expression of either FKBP9 (Prolyl isomerase 9), or PHGDH (Phosphoglycerate dehydrogenase) and RFTN2 (Raftlin family member 2) [91]. Tumors with high expression of FKBP9 correlated with poorer prognosis IDH wildtype GBMs, while high expression of PHGDH and RFTN2 provided a more favorable prognosis. Integration of these proteomic subtypes with pharmacological profiles using matched patient-derived GBM cells showed mTORC1/2 inhibition (AZD2014) as an efficient strategy for patients harboring poor prognostic biomarkers. This highlights the power of pharmaco-proteogenomics to uncover treatment targets that guide therapeutic interventions that could possibly be used to provide more of a functional readout than simply subtyping DMG based on H3 K27-alterations [8].
Naturally, there are challenges associated with employing ‘multi-omics’ techniques in DMG. Genomics is challenged by poor drug responses, defining candidate genes, reproducibility, statistical analysis and the requirement to interrogate large datasets of polymorphisms in large numbers of patients [87], while proteomics is challenged by the need for large amounts of starting material, which is almost impossible to obtain at biopsy, complex protocols and study design, with clinical sites needing appropriate infrastructure, expertise and robust analytical tools to ensure successful execution [88].
It is now a key challenge to try and integrate CNS pharmacokinetics into chemotherapeutics and pharmaco-proteogenomics to design regimens that will benefit patients. Treatments based solely on genomic prediction are limited due to the posttranscriptional and posttranslational architecture of DMG, which are yet to be fully elucidated. Genomics has become an essential element of sophisticated clinical trials [29] with genome data from tumor biopsies and/or blood plasma being used to assign the tumor to a molecular subtype and to detect genetically-distinct tumor subclones. Despite this, genomic data alone does not always provide the required insights into a patient prognosis or treatment options that deliver improved patient outcomes. Coupling phosphoproteomic approaches with in-depth genomic analyses will help to identify recurrent genetic alterations and their associated protein-controlled functional outcomes, the therapeutic potential of which can be furthered by the integration of pharmacological studies to accelerate novel clinical trials with biomarkers of prognostic and predictive value. In summary, sophisticated imaging and nuclear medicine should guide resection of multiple biopsy samples from representative regions of the tumor to help overcome regional clonal heterogeneity which will aid in the prediction of beneficial therapies, particularly when genomics is coupled with proteomics which will additionally aid in the assessment of regional contributions of the TME and immune system.
Conclusion
Despite extensive developments in novel targeted therapies and precision medicines, the prognosis and outcomes of patients diagnosed with DMG remain unacceptably poor. The recent 5th Edition of the WHO Classification of Tumors of the CNS, subtypes DMG based on H3 K27-alterations and facilitates the categorization of patients according to distinct clinicopathological and molecular features. It is important to note that the hallmark H3-alterations that give rise to DMG are somewhat unique to these tumors; therefore, novel modalities targeting these alterations herald our greatest chance to improve treatment. However, long-term successful outcomes will require treatments that take into appreciation the yet-to-be-characterized proteomic heterogeneity of DMG, including the assessment of the posttranslational architecture. Furthermore, future studies focused on regional contributions to tumor growth and survival are also needed as are studies to determine the mechanisms that influence immune system avoidance. Until genomics-based treatment target identification is integrated with pharmacogenomics and pharmacoproteomics research, the success of trials will remain low, with little hope of patients achieving long-term survival. Coupled evaluation of the DMG genome with the respective proteome, will enhance treatment selection/development, refine the evaluation of patient prognosis, and lead to the development, we hope, of approaches that improve outcomes for those diagnosed with the most aggressive, and poorly survived pediatric cancer.
Author contributions
Contributions: IJF and MDD conceived and designed the study. IJF, SMW and MDD analyzed the data. All authors wrote the manuscript. IJF and MDD designed the figures. Finishing of the figures was performed by Somersault18:24. All authors edited the manuscript and approved the final version.
Funding
MDD is supported by a Defeat DIPG ChadTough New Investigator Fellowship and an NHMRC Investigator Grant – GNT1173892. The contents of the published material are solely the responsibility of the research institutions involved or individual authors and do not reflect the views of NHMRC. This project was supported by the DIPG Collaborative, RUN DIPG Ltd, Strategic Group, McDonald Jones Foundation, Vinva Foundation, PNOC Foundation, Yuvaan Tiwari Foundation, Kiriwina Investments, Kids Cancer Project, and the Hunter Medical Research Institute. ERJ is supported via the Josephine Dun Scholarship from the Isabella and Marcus Foundation Scholarship, a sub fund of Australian Communities Foundation.
Competing interests
MDD is a parent of a child lost to DIPG and the Founder and a Director of the non-for-profit charity RUN DIPG Ltd. The remaining authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Mackay A, Burford A, Carvalho D, Izquierdo E, Fazal-Salom J, Taylor KR, et al. Integrated molecular meta-analysis of 1,000 pediatric high-grade and diffuse intrinsic pontine glioma. Cancer Cell. 2017;32:520–.e525. doi: 10.1016/j.ccell.2017.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hong S, Kim IH, Wang KC. Outcome and prognostic factors of childhood diffuse brainstem glioma. Cancer Res Treat. 2005;37:109–13. doi: 10.4143/crt.2005.37.2.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hoffman LM, Veldhuijzen van Zanten SEM, Colditz N, Baugh J, Chaney B, Hoffmann M, et al. Clinical, radiologic, pathologic, and molecular characteristics of long-term survivors of diffuse intrinsic pontine glioma (DIPG): a collaborative report from the International and European Society for Pediatric Oncology DIPG Registries. J Clin Oncol. 2018;36:1963–72. doi: 10.1200/JCO.2017.75.9308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sulman EP, Eisenstat DD. World Cancer Day 2021 - perspectives in pediatric and adult neuro-oncology. Front Oncol. 2021;11:659800. doi: 10.3389/fonc.2021.659800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu G, Diaz AK, Paugh BS, Rankin SL, Ju B, Li Y, et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat Genet. 2014;46:444–50. doi: 10.1038/ng.2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vinci M, Burford A, Molinari V, Kessler K, Popov S, Clarke M, et al. Functional diversity and cooperativity between subclonal populations of pediatric glioblastoma and diffuse intrinsic pontine glioma cells. Nat Med. 2018;24:1204–15. doi: 10.1038/s41591-018-0086-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Buczkowicz P, Hoeman C, Rakopoulos P, Pajovic S, Letourneau L, Dzamba M, et al. Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nat Genet. 2014;46:451–6. doi: 10.1038/ng.2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, et al. The 2021 WHO Classification of tumors of the central nervous system: a summary. Neuro-Oncology 2021. [DOI] [PMC free article] [PubMed]
- 9.Rushing EJ WHO classification of tumors of the nervous system: preview of the upcoming 5th edition. Magazine of European Medical Oncology 2021.
- 10.Martin DDO, Kay C, Collins JA, Nguyen YT, Slama RA, Hayden MR. A human huntingtin SNP alters post-translational modification and pathogenic proteolysis of the protein causing Huntington disease. Sci Rep. 2018;8:8096. doi: 10.1038/s41598-018-25903-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marzluff WF, Gongidi P, Woods KR, Jin J, Maltais LJ. The human and mouse replication-dependent histone genes. Genomics. 2002;80:487–98. [PubMed] [Google Scholar]
- 12.Szenker E, Ray-Gallet D, Almouzni G. The double face of the histone variant H3.3. Cell Res. 2011;21:421–34. doi: 10.1038/cr.2011.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Madeira F, Park YM, Lee J, Buso N, Gur T, Madhusoodanan N, et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019;47:W636–W641. doi: 10.1093/nar/gkz268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Buczkowicz P, Hawkins C. Pathology, molecular genetics, and epigenetics of diffuse intrinsic pontine glioma. Front Oncol. 2015;5:147. doi: 10.3389/fonc.2015.00147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harutyunyan AS, Krug B, Chen H, Papillon-Cavanagh S, Zeinieh M, De Jay N, et al. H3K27M induces defective chromatin spread of PRC2-mediated repressive H3K27me2/me3 and is essential for glioma tumorigenesis. Nat Commun. 2019;10:1262. doi: 10.1038/s41467-019-09140-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Silveira AB, Kasper LH, Fan Y, Jin H, Wu G, Shaw TI, et al. H3.3 K27M depletion increases differentiation and extends latency of diffuse intrinsic pontine glioma growth in vivo. Acta Neuropathol. 2019;137:637–55. doi: 10.1007/s00401-019-01975-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee CH, Yu JR, Granat J, Saldaña-Meyer R, Andrade J, LeRoy G, et al. Automethylation of PRC2 promotes H3K27 methylation and is impaired in H3K27M pediatric glioma. Genes Dev. 2019;33:1428–40. doi: 10.1101/gad.328773.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lewis PW, Muller MM, Koletsky MS, Cordero F, Lin S, Banaszynski LA, et al. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science. 2013;340:857–61. doi: 10.1126/science.1232245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chan KM, Fang D, Gan H, Hashizume R, Yu C, Schroeder M, et al. The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes Dev. 2013;27:985–90. doi: 10.1101/gad.217778.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yu JR, LeRoy G, Bready D, Frenster JD, Saldana-Meyer R, Jin Y, et al. The H3K36me2 writer-reader dependency in H3K27M-DIPG. Sci Adv. 2021; 7. [DOI] [PMC free article] [PubMed]
- 21.Grasso CS, Tang Y, Truffaux N, Berlow NE, Liu L, Debily MA, et al. Functionally defined therapeutic targets in diffuse intrinsic pontine glioma. Nat Med. 2015;21:555–9. doi: 10.1038/nm.3855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vitanza NA, Biery MC, Myers C, Ferguson E, Zheng Y, Girard EJ, et al. Optimal therapeutic targeting by HDAC inhibition in biopsy-derived treatment-naïve diffuse midline glioma models. Neuro-Oncol. 2021;23:376–86. doi: 10.1093/neuonc/noaa249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nagaraja S, Vitanza NA, Woo PJ, Taylor KR, Liu F, Zhang L, et al. Transcriptional dependencies in diffuse intrinsic pontine glioma. Cancer Cell. 2017;31:635–52 e636. doi: 10.1016/j.ccell.2017.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brown ZZ, Muller MM, Jain SU, Allis CD, Lewis PW, Muir TW. Strategy for “detoxification” of a cancer-derived histone mutant based on mapping its interaction with the methyltransferase PRC2. J Am Chem Soc. 2014;136:13498–501. doi: 10.1021/ja5060934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin GL, Wilson KM, Ceribelli M, Stanton BZ, Woo PJ, Kreimer S, et al. Therapeutic strategies for diffuse midline glioma from high-throughput combination drug screening. Sci Transl Med. 2019;11:eaaw0064. doi: 10.1126/scitranslmed.aaw0064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Antin C, Tauziède-Espariat A, Debily MA, Castel D, Grill J, Pagès M, et al. EZHIP is a specific diagnostic biomarker for posterior fossa ependymomas, group PFA and diffuse midline gliomas H3-WT with EZHIP overexpression. Acta Neuropathol Commun. 2020;8:183. doi: 10.1186/s40478-020-01056-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Castel D, Kergrohen T, Tauziède-Espariat A, Mackay A, Ghermaoui S, Lechapt E, et al. Histone H3 wild-type DIPG/DMG overexpressing EZHIP extend the spectrum diffuse midline gliomas with PRC2 inhibition beyond H3-K27M mutation. Acta Neuropathologica. 2020;139:1109–13. doi: 10.1007/s00401-020-02142-w. [DOI] [PubMed] [Google Scholar]
- 28.Jain SU, Rashoff AQ, Krabbenhoft SD, Hoelper D, Do TJ, Gibson TJ, et al. H3 K27M and EZHIP Impede H3K27-Methylation Spreading by Inhibiting Allosterically Stimulated PRC2. Mol Cell. 2020;80:726–.e727. doi: 10.1016/j.molcel.2020.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wong M, Mayoh C, Lau LMS, Khuong-Quang D-A, Pinese M, Kumar A, et al. Whole genome, transcriptome and methylome profiling enhances actionable target discovery in high-risk pediatric cancer. Nat Med. 2020;26:1742–53. doi: 10.1038/s41591-020-1072-4. [DOI] [PubMed] [Google Scholar]
- 30.Nikolaev A, Fiveash JB, Yang ES. Combined targeting of mutant p53 and Jumonji family histone demethylase augments therapeutic efficacy of radiation in H3K27M DIPG. Int J Mol Sci. 2020;21:490.. doi: 10.3390/ijms21020490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yin Y, Stephen CW, Luciani MG, Fåhraeus R. p53 Stability and activity is regulated by Mdm2-mediated induction of alternative p53 translation products. Nat Cell Biol. 2002;4:462–7. doi: 10.1038/ncb801. [DOI] [PubMed] [Google Scholar]
- 32.Lapin DH, Tsoli M, Ziegler DS. Genomic insights into diffuse intrinsic pontine glioma. Front Oncol. 2017;7:57. doi: 10.3389/fonc.2017.00057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Werbrouck C, Evangelista CCS, Lobón-Iglesias M-J, Barret E, Le Teuff G, Merlevede J, et al. TP53 pathway alterations drive radioresistance in diffuse intrinsic pontine gliomas (DIPG) Clin Cancer Res. 2019;25:6788. doi: 10.1158/1078-0432.CCR-19-0126. [DOI] [PubMed] [Google Scholar]
- 34.Larson JD, Kasper LH, Paugh BS, Jin H, Wu G, Kwon CH, et al. Histone H3.3 K27M accelerates spontaneous brainstem glioma and drives restricted changes in bivalent gene expression. Cancer Cell. 2019;35:140–.e147. doi: 10.1016/j.ccell.2018.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Q, Bykov VJN, Wiman KG, Zawacka-Pankau J. APR-246 reactivates mutant p53 by targeting cysteines 124 and 277. Cell Death Dis. 2018;9:439. doi: 10.1038/s41419-018-0463-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sillar JR, Germon ZP, DeIuliis GN, Dun MD. The role of reactive oxygen species in acute myeloid leukaemia. Int J Mol Sci. 2019;20:6003.. doi: 10.3390/ijms20236003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mu M-D, Qian Z-M, Yang S-X, Rong K-L, Yung W-H, Ke Y. Therapeutic effect of a histone demethylase inhibitor in Parkinson’s disease. Cell Death Dis. 2020;11:927. doi: 10.1038/s41419-020-03105-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Omar SI, Lepre MG, Morbiducci U, Deriu MA, Tuszynski JA. Virtual screening using covalent docking to find activators for G245S mutant p53. PLoS ONE. 2018;13:e0200769. doi: 10.1371/journal.pone.0200769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ali NA, Wu J, Hochgräfe F, Chan H, Nair R, Ye S, et al. Profiling the tyrosine phosphoproteome of different mouse mammary tumour models reveals distinct, model-specific signalling networks and conserved oncogenic pathways. Breast Cancer Res. 2014;16:437. doi: 10.1186/s13058-014-0437-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Velpula KK, Dasari VR, Asuthkar S, Gorantla B, Tsung AJ. EGFR and c-Met cross talk in glioblastoma and its regulation by human cord blood stem cells. Transl Oncol. 2012;5:379–92. doi: 10.1593/tlo.12235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Paugh BS, Zhu X, Qu C, Endersby R, Diaz AK, Zhang J, et al. Novel oncogenic PDGFRA mutations in pediatric high-grade gliomas. Cancer Res. 2013;73:6219–29. doi: 10.1158/0008-5472.CAN-13-1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Castel D, Philippe C, Calmon R, Le Dret L, Truffaux N, Boddaert N, et al. Histone H3F3A and HIST1H3B K27M mutations define two subgroups of diffuse intrinsic pontine gliomas with different prognosis and phenotypes. Acta Neuropathol. 2015;130:815–27. doi: 10.1007/s00401-015-1478-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Duchatel RJ, Jackson ER, Alvaro F, Nixon B, Hondermarck H, Dun MD. Signal transduction in diffuse intrinsic pontine glioma. Proteomics. 2019;19:e1800479. doi: 10.1002/pmic.201800479. [DOI] [PubMed] [Google Scholar]
- 44.Kluiver TA, Alieva M, van Vuurden DG, Wehrens EJ, Rios AC. Invaders exposed: understanding and targeting tumor cell invasion in diffuse intrinsic pontine glioma. Front Oncol. 2020;10:92. doi: 10.3389/fonc.2020.00092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang H, Diaz AK, Shaw TI, Li Y, Niu M, Cho JH, et al. Deep multiomics profiling of brain tumors identifies signaling networks downstream of cancer driver genes. Nat Commun. 2019;10:3718. doi: 10.1038/s41467-019-11661-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hoeman CM, Cordero FJ, Hu G, Misuraca K, Romero MM, Cardona HJ, et al. ACVR1 R206H cooperates with H3.1K27M in promoting diffuse intrinsic pontine glioma pathogenesis. Nat Commun. 2019;10:1023. doi: 10.1038/s41467-019-08823-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bruce DL, Sapkota GP. Phosphatases in SMAD regulation. FEBS Lett. 2012;586:1897–905. doi: 10.1016/j.febslet.2012.02.001. [DOI] [PubMed] [Google Scholar]
- 48.Garner EF, Williams AP, Stafman LL, Aye JM, Mroczek-Musulman E, Moore BP, et al. FTY720 decreases tumorigenesis in group 3 medulloblastoma patient-derived xenografts. Sci Rep. 2018;8:6913. doi: 10.1038/s41598-018-25263-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mueller S, Jain P, Liang WS, Kilburn L, Kline C, Gupta N, et al. A pilot precision medicine trial for children with diffuse intrinsic pontine glioma-PNOC003: a report from the pacific pediatric Neuro-Oncology Consortium. Int J Cancer. 2019;145:1889–901. doi: 10.1002/ijc.32258. [DOI] [PubMed] [Google Scholar]
- 50.Gymnopoulos M, Elsliger MA, Vogt PK. Rare cancer-specific mutations in PIK3CA show gain of function. Proc Natl Acad Sci USA. 2007;104:5569–74. doi: 10.1073/pnas.0701005104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Oliver MD, Fernández-Acero T, Luna S, Rodríguez-Escudero I, Molina M, Pulido R, et al. Insights into the pathological mechanisms of p85α mutations using a yeast-based phosphatidylinositol 3-kinase model. Biosci Rep. 2017;37:2.. doi: 10.1042/BSR20160258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Koschmann C, Farooqui Z, Kasaian K, Cao X, Zamler D, Stallard S, et al. Multi-focal sequencing of a diffuse intrinsic pontine glioma establishes PTEN loss as an early event. npj Precis Oncol. 2017;1:32. doi: 10.1038/s41698-017-0033-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nikbakht H, Panditharatna E, Mikael LG, Li R, Gayden T, Osmond M, et al. Spatial and temporal homogeneity of driver mutations in diffuse intrinsic pontine glioma. Nat Commun. 2016;7:11185. doi: 10.1038/ncomms11185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wen PY, Cloughesy TF, Olivero AG, Morrissey KM, Wilson TR, Lu X, et al. First-in-human phase I study to evaluate the brain-penetrant PI3K/mTOR inhibitor GDC-0084 in patients with progressive or recurrent high-grade glioma. Clin Cancer Res. 2020;26:1820. doi: 10.1158/1078-0432.CCR-19-2808. [DOI] [PubMed] [Google Scholar]
- 55.Pal S, Kozono D, Yang X, Fendler W, Fitts W, Ni J, et al. Dual HDAC and PI3K inhibition abrogates NFκB- and FOXM1-mediated DNA damage response to radiosensitize pediatric high-grade gliomas. Cancer Res. 2018;78:4007–21. doi: 10.1158/0008-5472.CAN-17-3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hopkins BD, Pauli C, Du X, Wang DG, Li X, Wu D, et al. Suppression of insulin feedback enhances the efficacy of PI3K inhibitors. Nature. 2018;560:499–503. doi: 10.1038/s41586-018-0343-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pajovic S, Siddaway R, Bridge T, Sheth J, Rakopoulos P, Kim B, et al. Epigenetic activation of a RAS/MYC axis in H3.3K27M-driven cancer. Nat Commun. 2020;11:6216. doi: 10.1038/s41467-020-19972-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Demma MJ, Mapelli C, Sun A, Bodea S, Ruprecht B, Javaid S, et al. Omomyc reveals new mechanisms to inhibit the MYC oncogene. Mol Cell Biol. 2019;39:e00248–19. doi: 10.1128/MCB.00248-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wiese M, Hamdan FH, Kubiak K, Diederichs C, Gielen GH, Nussbaumer G, et al. Combined treatment with CBP and BET inhibitors reverses inadvertent activation of detrimental super enhancer programs in DIPG cells. Cell Death Dis. 2020;11:673. doi: 10.1038/s41419-020-02800-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Taylor IC, Hütt-Cabezas M, Brandt WD, Kambhampati M, Nazarian J, Chang HT, et al. Disrupting NOTCH slows diffuse intrinsic pontine glioma growth, enhances radiation sensitivity, and shows combinatorial efficacy with bromodomain inhibition. J Neuropathol Exp Neurol. 2015;74:778–90. doi: 10.1097/NEN.0000000000000216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang L, Cui Y, Zhang L, Sheng J, Yang Y, Kuang G, et al. The silencing of CCND2 by promoter aberrant methylation in renal cell cancer and analysis of the correlation between CCND2 methylation status and clinical features. PLoS ONE. 2016;11:e0161859. doi: 10.1371/journal.pone.0161859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sheppard KE, McArthur GA. The cell-cycle regulator CDK4: an emerging therapeutic target in melanoma. Clin Cancer Res. 2013;19:5320. doi: 10.1158/1078-0432.CCR-13-0259. [DOI] [PubMed] [Google Scholar]
- 63.Schettini F, De Santo I, Rea CG, De Placido P, Formisano L, Giuliano M, et al. CDK 4/6 inhibitors as single agent in advanced solid tumors. Front Oncol. 2018;8:608. doi: 10.3389/fonc.2018.00608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.de Leeuw R, McNair C, Schiewer MJ, Neupane NP, Brand LJ, Augello MA, et al. MAPK reliance via acquired CDK4/6 inhibitor resistance in cancer. Clin Cancer Res. 2018;24:4201. doi: 10.1158/1078-0432.CCR-18-0410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lobbous M, Bernstock JD, Coffee E, Friedman GK, Metrock LK, Chagoya G, et al. An update on neurofibromatosis type 1-associated gliomas. Cancers. 2020;12:114.. doi: 10.3390/cancers12010114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vitanza NA, Khalatbari H, Ermoian R, Sarthy J, Lockwood CM, Cole BL, et al. Molecularly targeted treatments for NF1-mutant diffuse intrinsic pontine glioma. J Appl Lab Med. 2021;6:550–3. doi: 10.1093/jalm/jfaa086. [DOI] [PubMed] [Google Scholar]
- 67.Hart LS, Rader J, Raman P, Batra V, Russell MR, Tsang M, et al. Preclinical therapeutic synergy of MEK1/2 and CDK4/6 inhibition in neuroblastoma. Clin Cancer Res. 2017;23:1785–96. doi: 10.1158/1078-0432.CCR-16-1131. [DOI] [PubMed] [Google Scholar]
- 68.Taylor KR, Mackay A, Truffaux N, Butterfield Y, Morozova O, Philippe C, et al. Recurrent activating ACVR1 mutations in diffuse intrinsic pontine glioma. Nat Genet. 2014;46:457–61. doi: 10.1038/ng.2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ruark E, Snape K, Humburg P, Loveday C, Bajrami I, Brough R, et al. Mosaic PPM1D mutations are associated with predisposition to breast and ovarian cancer. Nature. 2013;493:406–10. doi: 10.1038/nature11725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Akamandisa MP, Nie K, Nahta R, Hambardzumyan D, Castellino RC. Inhibition of mutant PPM1D enhances DNA damage response and growth suppressive effects of ionizing radiation in diffuse intrinsic pontine glioma. Neuro Oncol. 2019;21:786–99. doi: 10.1093/neuonc/noz053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hanna C, Kurian KM, Williams K, Watts C, Jackson A, Carruthers R, et al. Pharmacokinetics, safety, and tolerability of olaparib and temozolomide for recurrent glioblastoma: results of the phase I OPARATIC trial. Neuro-Oncol. 2020;22:1840–50. doi: 10.1093/neuonc/noaa104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.McGranahan N, Swanton C. Clonal heterogeneity and tumor evolution: past, present, and the future. Cell. 2017;168:613–28. doi: 10.1016/j.cell.2017.01.018. [DOI] [PubMed] [Google Scholar]
- 73.Ma X, Edmonson M, Yergeau D, Muzny DM, Hampton OA, Rusch M, et al. Rise and fall of subclones from diagnosis to relapse in pediatric B-acute lymphoblastic leukaemia. Nat Commun. 2015;6:6604. doi: 10.1038/ncomms7604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Venkatesh HS, Morishita W, Geraghty AC, Silverbush D, Gillespie SM, Arzt M, et al. Electrical and synaptic integration of glioma into neural circuits. Nature. 2019;573:539–45. doi: 10.1038/s41586-019-1563-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Whiteside TL. The tumor microenvironment and its role in promoting tumor growth. Oncogene. 2008;27:5904–12. doi: 10.1038/onc.2008.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gillis NK, Patel JN, Innocenti F. Clinical implementation of germ line cancer pharmacogenetic variants during the next-generation sequencing era. Clin Pharm Ther. 2014;95:269–80. doi: 10.1038/clpt.2013.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Riba A, Di Nanni N, Mittal N, Arhne E, Schmidt A, Zavolan M. Protein synthesis rates and ribosome occupancies reveal determinants of translation elongation rates. Proc Natl Acad Sci USA. 2019;116:15023–32. doi: 10.1073/pnas.1817299116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dun MD, Chalkley RJ, Faulkner S, Keene S, Avery-Kiejda KA, Scott RJ, et al. Proteotranscriptomic profiling of 231-BR breast cancer cells: identification of potential biomarkers and therapeutic targets for brain metastasis. Mol Cell Proteom. 2015;14:2316–30. doi: 10.1074/mcp.M114.046110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.He B, Shi J, Wang X, Jiang H, Zhu H-J. Genome-wide pQTL analysis of protein expression regulatory networks in the human liver. BMC Biol. 2020;18:97. doi: 10.1186/s12915-020-00830-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Menden MP, Casale FP, Stephan J, Bignell GR, Iorio F, McDermott U, et al. The germline genetic component of drug sensitivity in cancer cell lines. Nat Commun. 2018;9:3385. doi: 10.1038/s41467-018-05811-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhang J, Walsh MF, Wu G, Edmonson MN, Gruber TA, Easton J, et al. Germline mutations in predisposition genes in pediatric cancer. N. Engl J Med. 2015;373:2336–46. doi: 10.1056/NEJMoa1508054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chambliss AB, Chan DW. Precision medicine: from pharmacogenomics to pharmacoproteomics. Clin Proteom. 2016;13:25. doi: 10.1186/s12014-016-9127-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chen Z, Hambardzumyan D. Immune microenvironment in glioblastoma subtypes. Front Immunol. 2018;9:1004. doi: 10.3389/fimmu.2018.01004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hirata E, Sahai E. Tumor microenvironment and differential responses to therapy. Cold Spring Harb Perspect Med. 2017;7:a026781.. doi: 10.1101/cshperspect.a026781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Corsello SM, Nagari RT, Spangler RD, Rossen J, Kocak M, Bryan JG, et al. Discovering the anti-cancer potential of non-oncology drugs by systematic viability profiling. Nat Cancer. 2020;1:235–48. doi: 10.1038/s43018-019-0018-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Murray HC, Enjeti AK, Kahl RGS, Flanagan HM, Sillar J, Skerrett-Byrne DA, et al. Quantitative phosphoproteomics uncovers synergy between DNA-PK and FLT3 inhibitors in acute myeloid leukaemia. Leukemia. 2021;35:1782–7. doi: 10.1038/s41375-020-01050-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Roden DM, Altman RB, Benowitz NL, Flockhart DA, Giacomini KM, Johnson JA, et al. Pharmacogenomics: challenges and opportunities. Ann Intern Med. 2006;145:749–57. doi: 10.7326/0003-4819-145-10-200611210-00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Nandal S, Burt T. Integrating pharmacoproteomics into early-phase clinical development: state-of-the-art, challenges, and recommendations. Int J Mol Sci. 2017;18:448.. doi: 10.3390/ijms18020448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Petralia F, Tignor N, Reva B, Koptyra M, Chowdhury S, Rykunov D, et al. Integrated proteogenomic characterization across major histological types of pediatric brain cancer. Cell. 2020;183:1962–.e1931. doi: 10.1016/j.cell.2020.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cooper CD, Lampe PD. Casein kinase 1 regulates connexin-43 gap junction assembly. J Biol Chem. 2002;277:44962–8. doi: 10.1074/jbc.M209427200. [DOI] [PubMed] [Google Scholar]
- 91.Oh S, Yeom J, Cho HJ, Kim J-H, Yoon S-J, Kim H, et al. Integrated pharmaco-proteogenomics defines two subgroups in isocitrate dehydrogenase wild-type glioblastoma with prognostic and therapeutic opportunities. Nat Commun. 2020;11:3288. doi: 10.1038/s41467-020-17139-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Baker SJ, Ellison DW, Gutmann DH. Pediatric gliomas as neurodevelopmental disorders. Glia. 2016;64:879–95. doi: 10.1002/glia.22945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hoffman LM, DeWire M, Ryall S, Buczkowicz P, Leach J, Miles L, et al. Spatial genomic heterogeneity in diffuse intrinsic pontine and midline high-grade glioma: implications for diagnostic biopsy and targeted therapeutics. Acta Neuropathol Commun. 2016;4:1. doi: 10.1186/s40478-015-0269-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Georgescu MM, Islam MZ, Li Y, Circu ML, Traylor J, Notarianni CM, et al. Global activation of oncogenic pathways underlies therapy resistance in diffuse midline glioma. Acta Neuropathol Commun. 2020;8:111. doi: 10.1186/s40478-020-00992-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhu X, Lazow MA, Schafer A, Bartlett A, Senthil Kumar S, Mishra DK, et al. A pilot radiogenomic study of DIPG reveals distinct subgroups with unique clinical trajectories and therapeutic targets. Acta Neuropathol Commun. 2021;9:14. doi: 10.1186/s40478-020-01107-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sievers P, Sill M, Schrimpf D, Stichel D, Reuss DE, Sturm D, et al. A subset of pediatric-type thalamic gliomas share a distinct DNA methylation profile, H3K27me3 loss and frequent alteration of EGFR. Neuro-Oncol. 2021;23:34–43. doi: 10.1093/neuonc/noaa251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Fontebasso AM, Papillon-Cavanagh S, Schwartzentruber J, Nikbakht H, Gerges N, Fiset PO, et al. Recurrent somatic mutations in ACVR1 in pediatric midline high-grade astrocytoma. Nat Genet. 2014;46:462–6. doi: 10.1038/ng.2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Pfaff E, El Damaty A, Balasubramanian GP, Blattner-Johnson M, Worst BC, Stark S, et al. Brainstem biopsy in pediatric diffuse intrinsic pontine glioma in the era of precision medicine: the INFORM study experience. Eur J Cancer. 2019;114:27–35. doi: 10.1016/j.ejca.2019.03.019. [DOI] [PubMed] [Google Scholar]
- 99.Walker JA, Upadhyaya M. Emerging therapeutic targets for neurofibromatosis type 1. Expert Opin Ther Targets. 2018;22:419–37. doi: 10.1080/14728222.2018.1465931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kostaras X, Cusano F, Kline GA, Roa W, Easaw J. Use of dexamethasone in patients with high-grade glioma: a clinical practice guideline. Curr Oncol. 2014;21:e493–503. doi: 10.3747/co.21.1769. [DOI] [PMC free article] [PubMed] [Google Scholar]