

Abstract
MECP2 gene transfer has been shown to extend the survival of Mecp2−/y knockout mice modelling Rett syndrome, an X-linked neurodevelopmental disorder. However, controlling deleterious overexpression of MECP2 remains the critical unmet obstacle towards a safe and effective gene therapy approach for Rett syndrome. A recently developed truncated miniMECP2 gene has also been shown to be therapeutic after AAV9-mediated gene transfer in knockout neonates.
We show that AAV9/miniMECP2 has a similar dose-dependent toxicity profile to that of a published second-generation AAV9/MECP2 vector after treatment in adolescent mice. To overcome that toxicity, we developed a risk-driven viral genome design strategy rooted in high-throughput profiling and genome mining to rationally develop a compact, synthetic microRNA target panel (miR-responsive auto-regulatory element, ‘miRARE’) to minimize the possibility of miniMECP2 transgene overexpression in the context of Rett syndrome gene therapy. The goal of miRARE is to have a built-in inhibitory element responsive to MECP2 overexpression. The data provided herein show that insertion of miRARE into the miniMECP2 gene expression cassette greatly improved the safety of miniMECP2 gene transfer without compromising efficacy. Importantly, this built-in regulation system does not require any additional exogenous drug application, and no miRNAs are expressed from the transgene cassette.
Although broad applications of miRARE have yet to be determined, the design of miRARE suggests a potential use in gene therapy approaches for other dose-sensitive genes.
Keywords: MECP2, AAV, Rett, intrathecal, microRNA
Sinnett et al. show that miniMECP2 gene therapy for Rett syndrome has similar dose-dependent toxicity to full-length MECP2 gene therapy. By developing a novel regulatory element to limit exogenous miniMECP2 expression, they demonstrate that the safety of miniMECP2 gene therapy can be improved without compromising efficacy.
Introduction
Rett syndrome is an X-linked neurodevelopmental disorder mediated by inactivating mutations in the transcription regulator methyl-CpG binding protein 2 (MECP2).1-3 Although Rett syndrome is primarily diagnosed in females, the male knockout mouse model is often used to assess new preclinical gene therapies.4–9 Knockout mouse phenotypes include decreased survival, poor motor coordination, abnormal gait, hindlimb clasping and decreased body weight.4–8,10 Although MECP2 is a ubiquitously expressed protein, global and peripheral mouse knockout models indicate that MECP2 expression in the CNS is sufficient to protect against many Rett syndrome phenotypes.10
Adeno-associated viral vector serotype 9 (AAV9) and AAV9-derived variants (i.e. PHP.B and PHP.eB) can cross the blood–brain barrier and transduce neurons, making them well-suited for gene replacement therapy in mice modelling CNS disorders.11,12 Although the more efficient variants PHP.B and PHP.eB are especially useful for proof-of-concept studies (e.g. the systemic PHP.eB/MECP2 study in Luoni et al.9), it is important to note that the LY6A receptor mediating their efficient blood–brain barrier transport is not expressed in non-human primates.13,14 Thus, AAV9 remains an important tool for full-factorial assessments of MECP2 gene therapy treatment paradigms.
One strategy to improve the efficiency of AAV9-mediated gene transfer is to pair the AAV9 capsid with a self-complementary viral genome. Compared to single-stranded AAV (ssAAV), self-complementary AAV (scAAV) has a smaller viral genome packaging capacity (∼2.2 kb excluding inverted terminal repeats), but permits more efficient transduction due to its ability to bypass the rate-limiting second-strand synthesis in host cells.15 The reduced packaging capacity of scAAV is critical for guiding MECP2 viral genome design as the ∼1.5 kb MECP2 gene limits the size of 5ʹ and 3ʹ regulatory elements included in the viral genome cassette. In contrast, a more recently developed therapeutic miniMECP2 gene (∼0.5 kb)7 frees up additional space within the self-complementary viral genome for inserting novel regulatory elements designed to improve the therapeutic index of Rett syndrome gene therapy.
Our interest in pairing a novel regulatory element with the self-complementary miniMECP2 viral genome derives from previously published studies showing dose-dependent toxicity after intracerebrospinal fluid administration of AAV9/MECP2 in juvenile mice.5 To be clear, all published AAV9/MECP2 and miniMECP2 gene therapies have been shown to extend knockout mouse survival.4–9 In knockout mice dosed at the neonatal age, survival and behavioural benefits have been realized with less pronounced side effects.4–7 However, when mice are treated at 4–5 weeks of age, a time more relevant for human translation, survival benefits have come with significant side effects (including death) and a lack of clear behavioural rescue.4,5 Moreover, this lack of clear behavioural rescue (within the context of AAV9-mediated gene transfer) has been observed across both knockout and T158M MECP2-expressing Rett syndrome mice treated during adolescence.4 Because a high intracerebrospinal fluid dose of AAV9/EGFP—but not AAV9/MECP2—is well-tolerated in wild-type mice,5 these dose-dependent side effects are most probably directly due to MECP2 overexpression. These side effects are, admittedly, not surprising in light of the symptoms seen in mouse models of MECP2 overexpression as well as in human MECP2 duplication syndrome.16–21 After years of iterative, full-factorial assessments of candidate MECP2 vectors, the field is wrestling with the same dilemma anticipated from MECP2-overexpression studies dating back 16 years.17,21 High doses of MECP2-expressing vectors may be harmful; low doses may not be effective. Clearly, this persistent dilemma warrants innovative viral genome design strategies that permit efficacy without compromising safety.
To further complicate the feasibility of gene therapy, the target human population is primarily females heterozygous for MECP2 mutations, which due to random X-inactivation have cellular mosaicism for MECP2 expression with a roughly equal mixture of dysfunctional cells and wild-type cells.22 Conceptually, if one delivers a corrected version of MECP2 equally to all cells, there would be the potential to rescue the dysfunctional cells while overexpressing MECP2 in the wild-type cells. One strategy to prevent gene overexpression-related toxicity is to insert microRNA targets into the 3ʹ untranslated region (UTR) of viral genomes. Endogenous miRNAs can base-pair with targets in viral genome-encoded mRNAs and ultimately decrease protein expression levels through RNA interference (RNAi).23 The established miR-122 target panel, for example, can constitutively decrease exogenous protein expression in the liver while permitting expression elsewhere in the body.24,25 Our goal for Rett syndrome gene therapy was to develop an miRNA target panel that conditionally regulates exogenous MECP2 in the same system we are trying to treat (i.e. the CNS). Our hypothesis is that such a target panel would buffer against deleterious overexpression of miniMECP2, while permitting sufficient transgene expression to exert a therapeutic effect similar to or greater than that of published MECP2 or miniMECP2 control vectors.7 We used a risk-driven viral genome design strategy rooted in high-throughput profiling and genome mining to develop a novel miRNA target panel (named the miR-responsive auto-regulatory element or ‘miRARE’) for regulating miniMECP2 expression. The objective was to create a feedback mechanism for negative transgene regulation that would be responsive to MECP2 overexpression. Data described herein show that miRARE improves the safety of scAAV9/miniMECP2 gene therapy without compromising efficacy following intracerebrospinal fluid injection in juvenile mice.
Materials and methods
Animals
Wild-type and knockout (B6.129P2(C)-Mecp2tm1.1Bird/J) mice were provided chow and water ad libitum and were housed on a 12-h light-dark cycle. Animal studies were conducted in accordance with a protocol approved by the Institutional Animal Care and Use Committee at the University of North Carolina (UNC) at Chapel Hill (Fig. 1) and the University of Texas Southwestern (UTSW) Medical Center (all other figures). All mice were weaned at postnatal Day 28 according to previously published methods.5 Mice at UTSW were housed and evaluated within a barrier facility.
Figure 1.
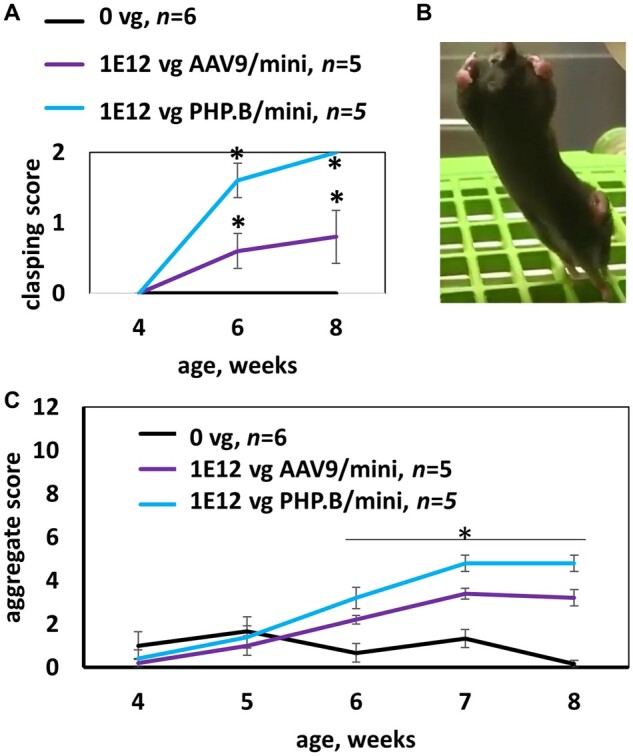
MiniMECP2 vectors cause side effects in wild-type mice. (A) In an acute toxicity study, overexpression of miniMECP2 caused significant increases in abnormal clasping scores. n = 5–6 mice per group. Saline versus virus treatments; and pre- versus post-injection: #P < 0.05. (B) Severe hindlimb clasping observed 3 days after treatment with PHP.B/miniMECP2 [1 × 1012 vg/mouse, intracisterna magna (ICM) administration]. In healthy mice, hindlimbs extend outwards. (C) Both AAV9/ and PHP.B/miniMECP2 significantly increase aggregate phenotype severity scores within 2 weeks post-injection (P < 0.05). Figure 1 shows data for mice housed and treated at UNC-Chapel Hill. All other mice in this paper were tested at UTSW. (A and C) Data are means ± SEM.
Vectors
AAV9/CBH-EGFP, AAV9/MeP426-hMECP2-myc-RDH1pA (denoted throughout as AAV9/MECP2), AAV9-MeP426-miniMECP2-myc-RDH1pA (denoted throughout as AAV9/miniMECP2) and AAV9-MeP426-miniMECP2-myc-miRARE-RDH1pA (denoted throughout as AAV9/miniMECP2-miRARE), PHP.B/miniMECP2-myc, PHP.B/miniMECP2-myc-miRARE and PHP.B/CBH-EGFP vectors were produced by UNC Vector Core.26 Vector certificates of analysis are provided in the Supplementary material. These are all self-complementary genomes, and all were prepared in a formulation buffer of 350 mM PBS containing 5% d-sorbitol. The full-length and miniMECP2 viral genome cassettes have been previously published.4,5,7 The six-target miRARE panel was inserted between two subcomponents of RDH1pA. Those subcomponents are a literature-based three-target panel for miR-19, miR-22 and miR-132 and a 110 bp fragment of the conserved distal polyadenylation signal.4,5 The sequence for miRARE is 5ʹ-CTGTTCTAGCCCCCAAAGAGTTTTCTGTGCTTGCTTTTGAAACTTGAAGTCTTGAAAACCAAAGACATAGATGTGAAAATTTTAGGCAGTGTAAGCTGATAGCACAAGTTCTGGCGACTCACAATTATGCTGTGAATTTTACAAAAAGAAGCAGTAATCTACCTCAGCCGATAAC-3′. Underlined sequences indicate seed matches for miR-9-5p, miR-26-5p miR-23-3p, miR-218-5p, miR-27-3p and let-7-5p, respectively. These targets and flanking sequences meet a number of criteria: (i) the bolded mutation was introduced to create a T1A anchor27; (ii) the spacers between each underlined sequence are within recommended ranges for cooperative repression28,29; and (iii) most of the targets permit a T9A or T9U.30
Treatments
Surgical intracisternal or percutaneous intrathecal injections (10 µl) were performed on 4–5-week-old mice. Previously published injection methods were used with the following exception: for surgeries, carprofen (5 mg/kg) and lidocaine (5 µl, 2% solution) were administered subcutaneously [instead of intraperitoneal tribromoethanol (Avertin®)] before inhalational isoflurane anaesthesia.5,31 Intrathecal injections were performed by personnel blinded to treatment. Personnel performing intrathecal injections did not have access to the list of treatment descriptions (concentration, capsid, viral genome design) and their corresponding identification numbers, which were used to blind those treatments. To ensure that the most severely affected knockout mice were randomly assigned to either saline or virus treatment groups, treatments were assigned blind to pre-injection weight and behaviour scores. Mice from multiple litters were assigned to each treatment group to prevent bias. Blinded saline treatments were interspersed with blinded virus treatments so that age-matched control mice could be evaluated in parallel with experimental mice. The number of mice evaluated per treatment group was based in part on literature,4,5 with the following exceptions: (i) group sizes were small (n = 5–6 mice per group) in Fig. 1 for humane reasons; and (ii) group sizes for miRNA profiling (n = 3 mice per treatment) were kept small with the expectation of using a secondary approach (e.g. bioinformatics) to strengthen the microarray dataset.
RNA purification
Three mice per group were treated with either saline, 1 × 1012 vg AAV9/MECP2 or 1 × 1012 vg AAV9/EGFP. Two to three weeks after injection, mice were euthanized with a lethal dose of intraperitoneal Avertin® and decapitated. Cervical cord, cerebellum and medulla were rapidly dissected, frozen on dry ice and immediately transferred to −80°C. Qiagen miRNeasy Mini kits were used to purify total RNA from thawed, lysed tissue. RNA samples were stored at −80°C until they were shipped on dry ice to LC Sciences for screening. Each sample was assigned an identification number to permit blind miRNA profiling. A list of identification numbers per blinded treatment group was provided to LC Sciences to permit statistical analyses.
MiRNA profiling
LC Sciences profiled mouse miRNAs from blinded RNA samples and conducted statistical analyses (microarray part number MRA-1002; miRBase version 21; n = three mice per group per tissue type with two screening replicates). See the Supplementary material for unblinded profile data. LC Sciences defined normalized signal intensities below 500 as low. Significantly increased miRNA expression levels described herein are limited to those having a mean signal intensity exceeding 500.
Bioinformatics
Throughout this paper, any discussion of endogenous targets is based solely on annotations listed in www.targetscan.org (last accessed October 2021). Excel files for 21 3ʹ UTRs were downloaded from www.targetscan.org Release 7.2 (March 2018).32 When two 3ʹ UTR sequences were available for a single gene, the most prevalent transcript was selected for analysis. For MECP2, for example, the 8-kb transcript from which a portion of RDH1pA is derived was used for analysis. Targets from downloaded files were merged into a single master file used to create a frequency table. All uniquely annotated targets were considered regardless of their context++ percentile scores,32 as these scores do not account for viral genome context. Targets appearing two or more times within a single transcript were counted only once per 3ʹ UTR. The exact number of putative targets may change over time if or when new versions of TargetScan are released. Supplementary Table 1 matches individual targets from miRARE to the endogenous mouse and human 3ʹ UTRs for 11 genes mediating intellectual disability.
Weekly weighing and aggregate behaviour scoring
An aggregate phenotype severity scale was used to assess behaviour in treated mice and is based on a previously published scale.33 This aggregate scale includes six subscales for abnormal mobility, abnormal gait, hindlimb clasping, tremors, abnormal breathing and abnormal general appearance. Hindlimb clasping was assessed by lifting the mouse by the tail so that hindlimbs were suspended. Scorers were blind to treatment and genotype but may have been able to infer genotype through visual assessment. Mice were weighed and scored pre-injection and weekly thereafter. Mice were assessed in a random order.
Survival
The date of death recorded for each mouse was the natural date of death with the following exceptions: mice that lost at least 20% of their peak body weight were euthanized, according to previously published methods4 or on the recommendation of UT Southwestern Animal Resource Center veterinary staff. Early euthanasias for severe health problems (prolapse with complications, tail lesions resulting in necrosis or followed by self-injury) are indicated in the uncensored survival curves.
Immunofluorescence and confocal analyses
Perfused brains were sectioned at 40-µm thickness and distributed serially among wells in a multi-well plate so that each well had a representative distribution of sagittal sections. Immunofluorescence analyses were conducted on fixed tissue according to previously published methods5 with the following exception: antigen retrieval was not performed. In our experience, antigen retrieval is necessary before blocking and incubating with anti-MECP2 primary antibodies, but not before blocking and incubating with anti-myc antibody. Immunolabelled sections were imaged with a Zeiss 880 confocal microscope using Zen software at UT Southwestern’s Live Cell Imaging Facility. All images used to calculate the percentage of myc-positive cells (myc+) (versus all DAPI+ cells) were taken at the same magnification with the same gain settings. For spinal cord segments, medulla and hippocampus, images were sampled from multiple (three to six) tissue sections. For the midbrain, we noted that the relatively high percentage of myc+ cells (for mice treated with unregulated vector) appeared within a narrow range along the x-axis of the brain (from medial to lateral). This range roughly corresponds to illustrated images 9–13 of the mouse sagittal Allen Brain Atlas.34,35 Therefore, multiple images were taken from one or two sections per microscope slide. Images for the pons and midbrain were typically taken from the same tissue sections.
Biodistribution and gene expression analyses
End-of-life tissue samples from saline- and virus-treated knockout mice were used for biodistribution and gene expression analyses. An AllPrep DNA/RNA/Protein Mini Kit (Cat no. 80004; Qiagen) was used to prepare genomic DNA and RNA. A Transcriptor First Strand complementary DNA Synthesis Kit (Cat no. 04897030001; Roche) was used to prepare cDNA from RNA. MiniMECP2 primer sequences were: 5′-CGGAAGCTTAAGCAAAGGAAATC-3′ (forward) and 5′-ACGCAATCAACTCCACTTTAGA-3′ (reverse). Cycling conditions were 98°C for 3 min followed by 40 cycles of the following: 98°C for 10 s (denature); 55°C for 30 s (anneal) and 60°C for 30 s (extend). Primer sequences and cycling conditions were kindly provided by Dr Ralph Hector at the University of Edinburgh. LMNB2 and ACTB were used as housekeeping controls for biodistribution and gene expression analyses, respectively.
Statistical analysis
The alpha-level used to determine significance was P < 0.05. LC Sciences performed statistical analyses of transformed signal intensities from miRNA microarray according to methods described in their technical bulletin.36 Analyses between groups were performed as requested (e.g. group A versus B). Because of limitations of the screening technique, P-values were interpreted with caution, and both screening and bioinformatics results were merged to justify miRNA targets selected for miRARE. The Gehan–Breslow–Wilcoxon test was used to calculate statistical significance between pairs of groups in Kaplan–Meier plots generated by GraphPad Prism. Except where otherwise noted, GraphPad was used to conduct two-way ANOVA followed by Tukey’s post hoc test for weight and behaviour data. For immunofluorescence analyses, we conducted two types of analysis. First, we conducted a two-way ANOVA followed by Tukey’s post hoc test. Conceptually, it could be possible for injection volume error to confound interpretation of expression data. Thus, we did a second analysis to address this conceptual confounding factor. Specifically, we used the medulla as an internal reference brain region and conducted a pair-wise t-test comparing the relative percentage of myc+ cells between the medulla and pons. For each host, the data-points for medulla and pons were paired, and the test was performed for each treatment group. In theory, this relative difference in the percentage of myc+ cells should be preserved even if a mouse is slightly underdosed. This allowed us to see whether relative differences between brain regions were reproducible for each host, or if those relative differences were mitigated by miRARE regulation in the pons.
Data availability
The raw data that support the findings of this study are available from the corresponding author, on reasonable request. Microarray data from LC Sciences are provided within the Supplementary material.
Results
MiniMECP2 vectors can cause side effects in wild-type mice
AAV9/MeP426-hMECP2-myc-RDH1pA (hereafter referred to as AAV9/MECP2) has been shown to cause dose-dependent side effects (e.g. weight loss and limb clasping abnormalities) after intracisternal (ICM) administration in adolescent mice.5 In contrast, relatively little has been published regarding the safety of AAV9/miniMECP2.7 Because the published MECP2 and miniMECP2 viral genomes feature the same promoter and 3ʹ UTR, we hypothesized that miniMECP2 viral vectors may recapitulate the same dose-dependent side effects as those observed for AAV9/MECP2.5,7 To quickly assess toxicity from miniMECP2 gene transfer, we injected wild-type adolescent mice with 1 × 1012 vg of AAV9/or PHP.B/miniMECP2 (ICM) and scored behaviour weekly. While these doses are extremely high, our ultimate goal is to develop a treatment that permits widespread MECP2 protein expression in the CNS, and may therefore require a high dose, without causing side effects. AAV9/miniMECP2 increased mean clasping scores within 2 weeks after injection (P < 0.05; Fig. 1A). PHP.B/miniMECP2 resulted in bilateral clasping as early as three days after injection (Fig. 1B). Both AAV9/and PHP.B/miniMECP2 increased aggregate phenotype severity scores within 2 weeks after injection (P < 0.05; Fig. 1C). Although transgenic mice expressing endogenous miniMECP2 exhibit mild clasping, the rapidly developing severe phenotypes that we observed seemed consistent with toxic overexpression and therefore warranted further optimization of the miniMECP2 viral genome.7
MicroRNA expression data can be used to rank targets from a bioinformatics analysis
Our goal for MECP2 gene therapy was to develop an miRNA target panel that conditionally regulates exogenous MECP2 in the same system we are trying to treat (i.e. the CNS; Fig. 2). Our hypothesis is that such a target panel would buffer against deleterious overexpression of MECP2, while permitting sufficient transgene expression to exert a therapeutic effect similar to or greater than that of a published MECP2 or miniMECP2 control vector.7 To identify miRNAs upregulated by toxic MECP2 gene therapy, we injected wild-type and knockout mice with saline, AAV9/EGFP or AAV9/MECP2 at 4–5 weeks of age (1 × 1012 vg/mouse, ICM). Two to three weeks after injection, we dissected three types of tissue that are close to the injection site: cervical cord, cerebellum and medulla. We purified total RNA from these samples, and then quantified the expression of 1900 mouse miRNAs via microarray (Supplementary material). This approach enabled us to identify MECP2-responsive miRNAs that are relevant within the CNS while forgoing the possibility of more granular, cell-type specific miRNA identification. Given our goal of finding miRNAs that are broadly applicable within the CNS and the immense technical hurdles involved in generating cell-type specific data we felt comfortable with the trade-off. We identified miRNAs whose expression levels were significantly increased at the tissue level in correlation with MECP2 (Fig. 3); however, these changes were too small in scale (many were <1.5-fold) for secondary confirmation with reverse transcription quantitative PCR. These significant but subtle changes in normalized fluorescence intensity, along with the general noisiness of the data, warranted an alternative secondary approach to justify the selection of miRNA targets for a new regulatory panel.
Figure 2.
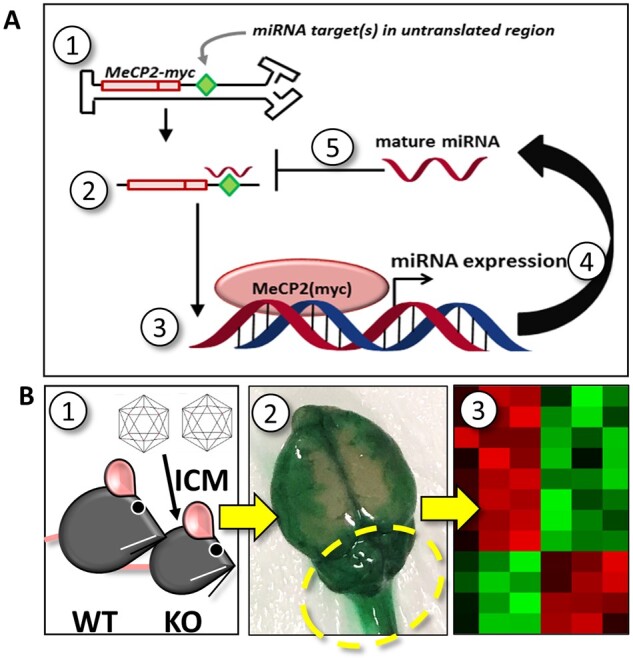
Schematic of strategy used to rationally synthesize a miRNA target panel tailored for Rett syndrome. (A) The first five steps of a conceptual negative feedback loop are indicated: (1) viral genome in nucleus; (2) exogenous MECP2-myc mRNA in cytoplasm; (3) translation and nuclear localization of MECP2-myc; (4) exogenous MECP2-myc upregulates miRNA expression; (5) mature miRNAs bind to non-coding targets in the 3ʹ UTR of an exogenous MECP2-myc transcript. After mature miRNAs bind to targets in the exogenous MECP2 mRNA, endogenous RNAi machinery (not shown) may conditionally silence exogenous MECP2 expression. The targets inserted into the viral genome may match targets found in the endogenous Mecp2 mRNA. Conceptually, this model could be adapted for other potentially toxic transcription factors that drive the expression of miRNAs in vivo. Alternatively, insertion of miRNA targets into the viral genome could improve the safety of gene therapies encoding potentially toxic cytoplasmic proteins, provided that those proteins indirectly upregulate miRNAs. (B) To identify translationally relevant miRNAs upregulated by toxic MECP2 gene transfer, (1) adolescent wild-type (WT) and knockout (KO) mice were treated intracisternally with saline, 1 × 1012 vg AAV9/MECP2, or 1 × 1012 vg AAV9/EGFP. (2) Two to three weeks later, CNS tissue nearest to the injection site (cerebellum, medulla and cervical cord; see dashed circle) was harvested from treated mice. The image shown in step 2 illustrates the spread of dye after ICM administration. The relatively concentrated dye localization near the cerebellum indicates how vulnerable tissue near the intracisternal site may be after treatment with high titre, toxic virus. (3) RNA was purified from dissected tissue, frozen and shipped to LC Sciences for miRNA profiling. Data from this screen were subsequently used to rank candidate miRNA targets from a secondary bioinformatics approach.
Figure 3.

Putative positive hits upregulated in correlation with endogenous or exogenous MECP2 according to miRNA microarray. (A–F) Significant differences between saline-treated knockout (KO) and saline-treated wild-type (WT) mice. (G–L) Significant differences between saline-treated and AAV9/MECP2-treated mice. (A–L) None of the 12 miRNAs listed here were significantly upregulated by AAV9/EGFP (versus saline-treated knockout or wild-type mice; P > 0.05). Each data-point represents the average of two screening replicates; error bars for each mean are not shown. n = 3 mice per group. P < 0.05 between the groups boxed in red. *A ‘reg1’ miRNA target panel featuring binding sites for miR-451a, miR-690, and let-7e-5p failed to improve the safety of miniMECP2 gene transfer (data not shown). As discussed in the main text, negative results (P > 0.05) should be interpreted with caution, as miRNAs upregulated at the cell-type level may be masked by noise from other cell types in the same tissue. Furthermore, low normalized fluorescence intensities may reflect low miRNA expression levels in a tissue sample but may fail to reveal high expression levels within a single cell type within said tissue. In general, many of the significant differences in normalized fluorescence intensity are for relatively small increases in mean signal between the indicated treatment groups. These subtle changes, along with the general noisiness of the data, warranted a secondary technique to justify the use of specific miRNA targets within a new target panel design. Finally, miRNAs significantly upregulated by AAV9/EGFP (versus saline-treated knockout mice) are miR-99a-5p, miR-107-3p and let-7f-5p in cervical cord (CC; data not shown). MiRNAs upregulated by AAV9/EGFP (versus saline-treated wild-type mice) are miR-669a-3p in cervical cord; let-7j in cerebellum; and miR-669f-3p, miR-669p-3p, miR-30c-5p and miR-669a-3p in medulla (data not shown).
We therefore decided to merge our microarray data with a new bioinformatics approach. We hypothesized that the 3ʹ UTRs of dose-sensitive CNS genes mediating phenotypically related and developmentally concurrent neurodevelopmental disorders may have a number of miRNA targets in common (Tables 1 and 2). If this hypothesis is supported, then we can synthesize a compact, multi-purpose target panel that is primarily intended for Rett syndrome gene therapy but may also be relevant for other diseases. We quantified the number of times annotated miRNA targets appeared among 11 3ʹ UTRs for a curated list of genes mediating intellectual disability (Tables 1, 2 and Fig. 4). A potential caveat to this secondary approach could relate to the observation that many dose-sensitive genes have relatively long 3ʹ UTRs (versus those of housekeeping or randomly selected genes). In other words, any apparent conservation of targets across our curated list of 3ʹ UTRs may simply be an artefact of these extremely long mRNA sequences. To address this concern, we conducted the same analysis for a random selection of human genes and saw that: (i) the 3ʹ UTRs for randomly selected genes had few miRNA targets in common (Supplementary Fig. 1); and (ii) the number of shared targets per 100-bp length of 3ʹ UTR sequence was lower for the randomly selected gene set than for our curated gene set (Supplementary Figs 2 and 3).
Table 1.
Curated list of confirmed or putative dose-sensitive genes mediating intellectual disability
| Gene | Role | Loss-of-function syndrome | Overexpression-related abnormality or syndrome |
Human Ensembl transcript numbera | |
|---|---|---|---|---|---|
| Monogenic mouse model | Clinical polygenic syndrome | ||||
| MECP2 b | Transcription | Rett syndrome | Yes | Yes | ENST00000303391.6 |
| TCF4 c | Transcription | Pitt-Hopkins | Yes | Yes | ENST00000354452.3 |
| MEF2C d | Transcription | MEF2C Haploinsufficiency | Noe (Drosophila data available) | Yes | ENST00000340208.5 |
| NSD1 f | Transcription | Sotos | Nog (Drosophila data available) | Yes | ENST00000439151.2 |
| ATRX h | Transcription | ATRX | Yes | Yes | ENST00000373344.5 |
| MBD5 i | Transcription | MAND | Noj (Drosophila data available) | Yes | ENST00000407073.1 |
| ZEB2 k | Transcription | Mowat-Wilson | Nol (Drosophila data available) | Yes | ENST00000558170.2 |
| UBE3A m | Degradation and transcription | Angelman | Yes | Yes | ENST00000232165.3 |
| DYRK1A n | Phosphorylation | DYRK1A | Yes | Yes | ENST00000339659.4 |
| RPS6KA3 o | Phosphorylation | Coffin-Lowry | Nop (Drosophila data available) | Yes | ENST00000379565.3 |
| SLC6A1 q | Transporter | Doose | Yes | Yes | ENST00000287766.4 |
Inactivating mutations in the genes listed here have been shown to mediate neurodevelopmental disorders characterized by intellectual disability as well as other phenotypes, such as seizures, stereotypies, abnormal speech and/or abnormal head size. Although the exact age of onset varies across loss-of-function syndromes, many of these syndromes are evident by 2 years of age. Reciprocal (overexpression-related) disorders may be mediated wholly or in part by the same genes. The contribution of a specific gene to specific phenotypes associated with a human chromosomal duplication may not always be known. To be clear, patients with supernumerary protein expression typically have duplications spanning larger chromosomal regions that encompass, but are not limited to, the genes indicated in this table. Therefore, Table 1 includes monogenic duplication mouse models that underscore the potential dose-sensitivity of these genes. Clinical vignettes describing intragenic duplications (which may yield truncated protein) are not considered in this table.
3ʹ UTR sequences are accessible at www.targetscan.org. The Ensembl transcript numbers listed here were used for analyses.32
TCF4 (transcription factor 4) references.39–45
MEF2C (myocyte enhancer factor 2C) references.46–49
Overexpression of the Drosophila orthologue MEF2 has been shown to cause lethality or abnormal wing morphology.48
NSD1 (nuclear receptor binding SET domain protein 1) references.50–53
Overexpression of the Drosophila orthologue NSD has been shown to cause development delay, early lethality, decreased wing size and disrupted organization of the eye (consequences of overexpression vary depending on the spatial extent of overexpression).54
ATRX (alpha-thalassemia/mental retardation, X-linked) references.55–58
MBD5 (methyl-CpG binding domain protein 5) references.59–62
Overexpression of the Drosophila orthologue sba has been shown to cause wing abnormalities.61
Overexpression of the Drosophila orthologue ZFH1 causes lethality.48
UBE3A (ubiquitin-protein ligase E3A) references.65–72
DYRK1A (dual specificity tyrosine-phosphorylation-regulated kinase 1A) references.73–79
RPS6KA3 (ribosomal protein S6 kinase A3) references.80–86
Overexpression of the Drosophila orthologue S6KII has been shown to exert a dominant negative effect in operant place learning.86
SLC6A1 references.87–90
Table 2.
Phenotypic overlap among selected loss-of-function disorders and reciprocal duplication disorders that encompass the same genes
| Gene | Intellectual disability | Speech abnormalities | Seizures | Microcephaly | Stereotypy |
|---|---|---|---|---|---|
| TCF4 | ✓/✓ | ✓/− | ✓/✓ | ✓/✓ | ✓/− |
| MECP2 | ✓/✓ | ✓/✓ | ✓/✓ | ✓/✓ | ✓/✓ |
| UBE3A | ✓/✓ | ✓/✓ | ✓/✓ | ✓/✓ | ✓/− |
| DYRK1A | ✓/✓ | ✓/✓ | ✓/✓ | ✓/✓ | ✓/✓ |
| MEF2C | ✓/✓ | ✓/✓ | ✓/- | −/✓ | ✓/− |
| NSD1 | ✓/✓ | ✓/✓ | ✓/✓ | −/✓ | |
| ATRX | ✓/✓ | ✓/✓ | ✓/✓ | ✓/− | ✓/− |
| RPS6KA3 | ✓/✓ | ✓/✓ | ✓/✓ | ✓/− | |
| MBD5 | ✓/✓ | ✓/✓ | ✓/✓ | ✓/− | |
| ZEB2 | ✓/✓ | ✓/✓ | ✓/− | ✓/✓a | |
| SLC6A1 | ✓/✓ | ✓/✓ | ✓/− | ✓/✓ | ✓/− |
The table mouse monogenic duplication models, human polygenic duplications and human trisomies. See Table 1 for references. Mouse and human phenotypes are aggregated together in Table 2. Speech abnormalities may encompass multiple phenotypes, such as lack of speech, babbling, dysprosody or delayed speech. The number and severity of symptoms may vary among patients with the same diagnosis. Early lethality in humans with duplication disorders may preclude documentation of specific phenotypes. A double tick (✓/✓) indicates phenotypes observed in loss-of-function and reciprocal duplication disorders or models. A single tick (✓/−) indicates phenotypes observed in loss-of-function disorders or models only.
Patient with 2q22.3 triplication had microcephaly at birth, but not at later age.64
Figure 4.
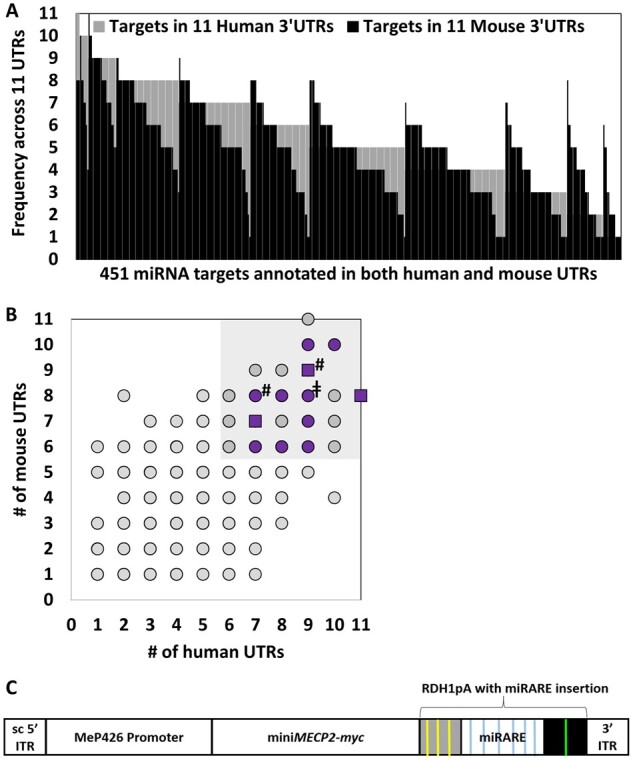
Microarray expression data were used to rank targets that appear frequently across a curated list of 3ʹ UTRs for genes mediating intellectual disability. (A) Many miRNA targets appear frequently across the curated list of 3ʹ UTRs. Among 2491 human targets and 1831 mouse targets, 451 targets had identical annotation across both mouse and human 3ʹ UTR datasets. (B) The scatter plot shows the same data as those shown in A. Targets that are annotated in over half of the examined 3ʹ UTR sequences for both species were prioritized for target panel design (shaded area). Within the shaded area, purple data-points indicate targets corresponding to miRNAs expressed at moderate to high levels in dissected cervical cord, cerebellar, and/or medullar tissue (signal intensity >500). Square data-points indicate targets for putative MECP2-responsive miRNAs indicated in Fig. 3A, C, D and G–J as well as two additional miRNAs (miR-9-5p and miR-27a-3p; indicated by a hash symbol), which showed trends in increased expression in cervical cord when data for MECP2− and MECP2+ treatment groups were aggregated together (data not shown). MECP2− groups, for example, would include both saline-treated and AAV9/EGFP-treated knockout mice. For clarity, we have limited notation (purple and square data-points) to the shaded area only. The double dagger symbol (‡) indicates the target for miR-218-5p included in the new panel design (‘miRARE’) for the purpose of making miRARE more broadly applicable for multiple gene therapy applications. Expression of miR-218-5p did not appear to be MECP2-responsive in our high throughput screening data. (C) Cartoon of the miniMECP2-miRARE viral genome cassette. Yellow and green lines indicate miRNA targets that are part of the previously published RDH1pA.4 Blue lines indicate miRARE targets that were inserted into RDH1pA. ITR = inverted terminal repeat; sc = mutated self-complementary ITR sequence.
After identifying miRNA targets that appear across most (≥6) of our 11 3ʹ UTRs (in both mouse and human datasets), we used our microarray data to prioritize targets for miRNAs expressed in tissue near the intracerebrospinal fluid injection site (Figs 3, 4 and Supplementary material). We then narrowed the list of the candidate targets to include those for miRNAs whose expression levels appeared to increase in correlation with MECP2. Finally, we sought to maximize regulatory potential while minimizing DNA sequence length by including a let-7-5p binding site predicted to base-pair with multiple potentially MECP2-responsive let-7-5p miRNAs. Ultimately, this two-pronged approach yielded a 175 bp six-target panel (miRARE), which was inserted into the miniMECP2 viral genome (Fig. 4). The self-complementary AAV9/MeP426-miniMECP2-myc-miRARE-RDH1pA viral genome is hereafter referred to as AAV9/miniMECP2-miRARE. Importantly, each of these miRARE targets are predicted to bind miRNAs that have been shown to be expressed in human brain tissue, from infancy through adolescence.37 Furthermore, the relative expression levels of six female human cerebellar miRNAs (miR-9-5p, miR-26b-5p miR-23a-3p, miR-218-5p, miR-27a-3p and let-7e-5p) roughly mirrors the relative expression levels in our miRNA profile, with miR-9-5p and miR-23a-3p being expressed at high and low levels, respectively.37
MiRARE improves the safety of miniMECP2 gene transfer in treated wild-type mice
We evaluated the safety of regulated miniMECP2 gene transfer in two treatment paradigms for adolescent mice: (i) ICM PHP.B-mediated gene transfer (Supplementary Figs 4 and 5); and (ii) intrathecal AAV9-mediated gene transfer. The ICM experiment with PHP.B provides data for highly efficient gene transfer and bridges our new miRARE data with Fig. 1, which compares ICM administration of unregulated AAV9/and PHP.B/miniMECP2 vectors. The PHP.B dataset is summarized as follows: miRARE provided robust regulation of miniMECP2-myc protein expression in wild-type brains (Supplementary Fig. 4), prevented weight loss in virus-treated wild-type mice (P < 0.05 versus unregulated vector and versus saline; Supplementary Fig. 5A), normalized aggregate phenotype scores for virus-treated wild-type mice (P < 0.05 versus unregulated vector; Supplementary Fig. 5B), delayed the onset of severe gait (P < 0.05 versus unregulated vector; Supplementary Fig. 5C) and almost completely eliminated the occurrence of severe hindlimb clasping in virus-treated wild-type mice (P < 0.05 versus unregulated vector; Supplementary Fig. 5D). The significant difference in the frequency of onset as well as the age of onset for severe gait and hindlimb clasping is particularly striking (Supplementary Fig. 5C and D). Across weight and behaviour readouts, there were no differences between saline- and PHP.B/miniMECP2-miRARE-treated wild-type mice.
Next, to align our miRARE assessment with methods relevant for human translation, we evaluated miRARE within the context of intrathecally administered AAV9. MiRARE provided several safety benefits. Specifically, miRARE prevented acute miniMECP2-mediated weight loss in wild-type mice at the highest dose tested (1 × 1012 vg/mouse; Fig. 5A). AAV9/miniMECP2-treated wild-type mice had significantly lower weight than that of saline-treated wild-type mice beginning at 15 weeks of age (P < 0.05). No significant difference was observed between saline- and AAV9/miniMECP2-miRARE-treated wild-type mice (1 × 1012 vg/mouse).
Figure 5.
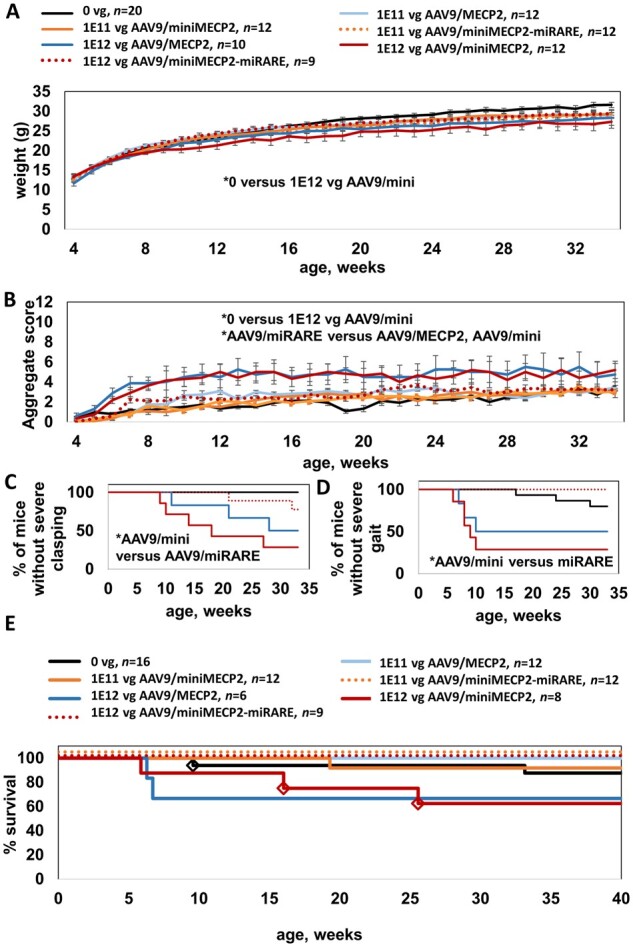
AAV9/miniMECP2-miRARE is well-tolerated after intrathecal administration in wild-type adolescents. Throughout this study, all treatments were administered between 4 and 5 weeks of age. (A) AAV9/miniMECP2-treated wild-type mice (1 × 1012 vg/mouse) had significantly lower weight than that of saline-treated wild-type mice beginning at 15 weeks of age (P < 0.05). No significant difference was observed between saline- and AAV9/miniMECP2-miRARE-treated wild-type mice (1 × 1012 vg/mouse). The same legend also applies to B. (B) AAV9/MECP2- and AAV9/miniMECP2-treated wild-type mice had a significantly higher mean aggregate behavioural severity score versus that observed for saline-treated mice (P < 0.05; at 6–30 and 7–27 weeks of age, respectively). AAV9/miniMECP2-miRARE-treated wild-type mice had a significantly lower mean aggregate severity score versus those of AAV9/MECP2- and AAV9/miniMECP2-treated mice at most time points from 11–19 and 9–20 weeks of age, respectively. No significant difference was observed between saline- and AAV9/miniMECP2-miRARE-treated wild-type mice (1 × 1012 vg/mouse). (C) Most wild-type mice treated with AAV9/miniMECP2-miRARE did not develop severe clasping abnormalities (severe clasping for n = 2/9 mice). (D) Wild-type mice treated with AAV9/miniMECP2-miRARE did not develop severe gait abnormalities (n = 0/9 mice). (C and D) Severe abnormal gait and severe clasping are each scored as a 2 on a scale of 0–2.33 All vectors were administered at 1 × 1012 vg/mouse. Mice that were euthanized early before developing severe gait or clasping scores (E) were excluded from data in C and D. (C and D) *P < 0.05 using Gehan–Breslow–Wilcoxon test, which can be used to evaluate significance for Kaplan–Meier plots. A one-way ANOVA of mean age of onset for D is not encouraged, as there are no time points listed here at which AAV9/miniMECP2-miRARE-treated wild-type mice develop severe gait abnormalities. (E) No early deaths have been observed for AAV9/miniMECP2-miRARE-treated wild-type mice. MiRARE groups are offset for clarity. Diamonds indicate veterinarian-requested euthanasias for bullying-related injuries (saline-treated) and for prolapses (AAV9/miniMECP2-treated). Prolapses were observed in 8–17% of AAV9/miniMECP2-treated wild-type mice (1 × 1011 to 1 × 1012 vg, respectively). Four AAV9/MECP2-treated mice were dissected at 8 weeks of age and were therefore excluded from the survival plot. The survival data for saline-treated wild-type mice also appears in Fig. 6D and Supplementary Fig. 9, as these mice were evaluated in parallel. (A and B) For simplicity only statistical differences between saline- and high-dose-treated wild-type mice were analysed. Data are means ± SEM. *P < 0.05.
MiRARE attenuated miniMECP2-mediated aggravation in wild-type aggregate phenotype severity scores (Fig. 5B). AAV9/MECP2- and AAV9/miniMECP2-treated wild-type mice had a significantly higher mean aggregate behavioural severity score versus that observed for saline-treated mice (P < 0.05; at 6–30 and 7–27 weeks of age, respectively). AAV9/miniMECP2-miRARE-treated wild-type mice had a significantly lower mean aggregate severity score versus those of AAV9/MECP2- and AAV9/miniMECP2-treated mice at most time points from 11–19 and 9–20 weeks of age, respectively. No significant difference was observed between saline- and AAV9/miniMECP2-miRARE-treated wild-type mice (1 × 1012 vg/mouse).
MiRARE attenuated miniMECP2-mediated aggravation in aggregate severity scores by reducing the frequency of miniMECP2-mediated severe hindlimb clasping and by preventing miniMECP2-mediated severely abnormal gait (Fig. 5C and D). In contrast, at least half of the wild-type mice treated with AAV9/MECP2 or AAV9/miniMECP2 developed severely abnormal gait or severe hindlimb abnormalities.
No early deaths were observed among AAV9/miniMECP2-miRARE-treated wild-type mice. Early veterinarian-requested euthanasias among AAV9/miniMECP2-treated wild-type mice were for complications from prolapses (Fig. 5E). Finally, no tail lesions were observed among AAV9/miniMECP2-miRARE-treated wild-type mice (0% of 21 mice). In contrast, lesions were observed in 8–17% of unregulated AAV9/miniMECP2-treated wild-type mice (1 of 12 mice treated with 1 × 1011 vg; 2 of 12 mice treated with 1 × 1012 vg). A tail lesion was observed in 1 of 12 wild-type mice treated with 1 × 1011 vg AAV9/MECP2.
MiniMECP2-miRARE gene therapy extends knockout survival despite robust downregulation
After intracisternal administration (1 × 1011 vg/mouse), PHP.B/miniMECP2 and PHP.B/miniMECP2-miRARE extended median survival by 21 and 29%, respectively. The extension in median survival for PHP.B/miniMECP2-miRARE-treated knockout mice was significant (P < 0.05) (Supplementary Fig. 6A). Biodistribution analyses in end-of-life tissue confirmed that both vectors were dosed equivalently in knockout mice (Supplementary Fig. 6B). To assess regulation globally across the knockout CNS, we quantified normalized miniMECP2 cDNA expression in the brain and spinal cord. MiRARE decreased miniMECP2 mRNA expression significantly in the brain, cervical cord, thoracic cord, and lumbar cord of treated knockout mice (P < 0.05) (Supplementary Fig. 6C). This is consistent with the miRARE-mediated regulation of protein expression observed in wild-type brain sections (Supplementary Fig. 4).
As a follow-up to the broad expression assessments described before, we decided to pursue a more granular multifactorial analysis—comparing two vectors and two genotypes in parallel—of miRARE-mediated expression. Importantly, we wanted to maximize spatial data. We suspected that a more rigorous immunofluorescence survey would permit identification of miRARE-mediated genotype-dependent regulation, even if that regulation were restricted to specific subregions within the brain. Because such a discovery would have huge translational value, we decided to conduct this analysis in tissue sections from wild-type and knockout mice treated with AAV9 vectors instead of PHP.B vectors (PHP.B is useful for proof-of-concept studies). We treated wild-type and knockout mice with 1 × 31012 vg/mouse of either regulated or unregulated vector (injected intrathecally). We imaged expression 3 weeks post-injection in multiple brain regions (hippocampus, pons, midbrain and medulla), as well as the spinal cord (cervical cord, thoracic cord and lumbar cord). In the spinal cord and medulla, miRARE-mediated regulation was robust across both genotypes (Fig. 6A and B). In the pons and midbrain, however, miRARE appeared to inhibit mean expression in a genotype-dependent manner, with significantly fewer myc+ cells observed in wild-type mice versus knockout mice (P < 0.05; Fig. 6B). Because of the translational value of a putative feedback-enabled viral genome, it is important that these early results be conveyed as transparently as possible. Thus, we included individual mouse data in Supplementary Fig. 7 so that the group size and the variability across hosts is clear, with representative images shown in Fig. 6C. In each wild-type host treated with unregulated vector, the pons had a relatively higher percentage of myc+ cells versus that of the medulla from the same host (P < 0.05, paired t-test; Supplementary Fig. 7). However, most miRARE-treated wild-type hosts did not have higher expression in the pons (versus the internal medulla control). This observation suggests that miRARE was strongly regulating expression in wild-type pons. In contrast, most knockout mice treated with either unregulated or regulated vectors had a relatively higher percentage of myc+ cells in the pons relative to the medulla (P < 0.05 for unregulated and regulated vectors).
Figure 6.

AAV9/miniMECP2-miRARE extends the survival of knockout mice despite regulation. (A) Percentage of myc+ cells is shown for cervical cord (CC), thoracic cord (TC) and lumbar cord (LC). MiRARE provided robust regulation across host genotypes. n = 3–5 mice per group. (B) Percentage of myc+ cells is shown. MiRARE significantly decreased expression in the pons and midbrain of AAV9/miniMECP2-miRARE-treated wild-type mice. P < 0.05 versus all other groups. The same legend applies for A and B. (C) Representative pons images. Scale bars = 20 µm. (D) Median survivals for saline- and AAV9/miniMECP2-miRARE-treated knockout mice are 9.6 and 15.0 weeks (P < 0.02). Compare to lower doses shown in Supplementary Fig. 9. Diamonds indicate veterinarian-requested euthanasias, which were primarily for severe tail lesions or tail self-amputations. (E) None of the treatments affected knockout weight. Any trends in increased weight for virus-treated knockout mice (versus saline-treated knockout mice) are insignificant because few mice are alive at later time points. n per group in E: wild-type, 0 vg (20); knockout, 0 vg (18); knockout, 1 × 1012 vg AAV9/MECP2 (12); knockout, 1 × 1012 vg AAV9/miniMECP2 (12); and knockout, 1 × 1012 vg AAV9/miniMECP2-miRARE (12). (D and E) The same wild-type control data appear in Fig. 5 as these mice were treated in parallel. (F) The frequency of achieving severe gait (score = 2) among saline-, AAV9/MECP2-, AAV9/miniMECP2- and AAV9/miniMECP2-miRARE-treated mice were 28%, 50%, 33% and 17%. Each mouse was administered 1 × 1012 vg. Black bars indicate the mean age of onset for severe clasping. Each data-point represents one mouse. Treatment groups are colour-coded as shown in D and E. The fraction of mice (subset versus entire group) is listed for each treatment group. AAV9/miniMECP2-miRARE-treated mice developing severe gait (n = 2 of 12) did so 4–5 weeks after other groups. Although this delay is significantly different, the number of data-points are too few to draw firm conclusions. Figure 5D and F show the same type of data. In general, Kaplan–Meier plots are the clearest way to visually communicate age-of-onset data (Fig. 5D), but these plots lend themselves to the assumption that all mice retaining normal gait live for the entire time period specified on the x-axis. This is true for wild-type mice (Fig. 5D) but not for knockout mice (F). Importantly, few AAV9/miniMECP2-miRARE-treated knockout mice developed severe gait, despite having an extended survival. For a graphical comparison, see Supplementary Fig. 8 for wild-type scatter plot data (also presented in Fig. 5D). (A, B and E) Data are mean ± SEM.
Given this initial evidence of potential genotype-dependent regulation, we expected miRARE to couple our two main goals of improving safety without compromising survival efficacy. Indeed, AAV9/miniMECP2-miRARE extended knockout survival by 56% (1 × 1012 vg/mouse, intrathecal; Fig. 6D). In contrast, the unregulated miniMECP2 gene transfer failed to extend knockout survival at either dose tested. Although there is an apparent trend for increased survival of knockout mice treated with AAV9/MECP2, this increase was not significant (P = 0.1). It is worth noting that although there is a strong trend towards increased survival with AAV9/MECP2, studies in wild-type mice showed unacceptable toxicity with this vector design at this dose (Fig. 5).
No treatment affected knockout weight (Fig. 6E).
AAV9/miniMECP2-miRARE may delay the age of onset of severely abnormal gait
Weekly behaviour data were used to tabulate the approximate age of onset for severely abnormal gait in knockout mice (score = 2 on a scale of 0–2). AAV9/miniMECP2-miRARE delayed the approximate age of onset of severely abnormal gait by 4–5 weeks (P < 0.05 versus all other groups, one-way ANOVA followed by Tukey’s post hoc test), with a similar or lower frequency of occurrence (versus all other knockout groups; Fig. 6F). Although this delay is statistically significant, the number of AAV9/miniMECP2-miRARE-treated knockout mice achieving severely abnormal gait are too few to draw firm conclusions. Note that the knockout gait data shown in Fig. 6F can be compared to the analogous wild-type data provided in Kaplan–Meier and scatter plot formats (Fig. 5D and Supplementary Fig. 8, respectively).
Adverse events were noted in some AAV-treated knockout mice
Tail lesions were observed across all treatment groups, including saline-treated knockout mice. The frequency of lesions among knockout mice treated with saline was 11% (2/18). The frequency of lesions among mice treated with 1 × 1011 vg of AAV9/MECP2, AAV9/miniMECP2 and AAV9/miniMECP2-miRARE were 25% (3/12), 42% (5/12) and 17% (2/12), respectively. The frequency of lesions among knockout mice treated with 1 × 1012 vg of AAV9/MECP2, AAV9/miniMECP2 and AAV9/miniMECP2-miRARE were 33% (4/12), 25% (3/12) and 17% (2/12), respectively. The inverse relationship between frequency of lesions and dose for AAV9/miniMECP2-treated knockout mice may be an artefact of early death. The respective median survivals for knockout mice treated with 1 × 1011 or 1 × 1012 vg AAV9/miniMECP2 are 10.4 and 9.6 weeks, respectively (Fig. 6D and Supplementary Fig. 9). It is worth noting that Luoni et al.9 showed that tail lesions in virus-treated knockout mice may be caused by an immune response to exogenous MECP2, which would not necessarily be influenced by miRARE.9
Discussion
This study is the first to quantify the dose-dependent side effects of unregulated AAV9/miniMECP2 in wild-type mice (Figs 1 and 5). These side effects recapitulate those previously observed for AAV9/MECP2 and therefore warranted further improvements to the viral genome.5
We developed a new target panel design strategy to create a regulated miniMECP2 viral genome featuring an ‘miRARE’ miRNA target panel. The intention was to use endogenous MECP2-responsive miRNAs to downregulate the miniMECP2 transgene in the event of MECP2 overexpression. Essentially, we wanted to create a safety valve to block transgene overexpression in any transduced cell. MiRARE meets the following criteria: (i) most of the targets are predicted to bind putative MECP2-responsive miRNAs; (ii) each target appears among 7–11 endogenous human 3ʹ UTRs from a curated list of dose-sensitive genes mediating intellectual disability (and may therefore be useful for other dose-sensitive CNS gene therapy applications); and (iii) the targets are predicted to bind miRNAs that have been shown to be expressed in human CNS tissue across developmental stages.37 Considering the abundance of many of these miRNA binding sites in endogenous 3ʹ UTRs, one might question why endogenous miRNA targets fail to prevent duplication syndromes in humans. We might speculate that endogenous miRNA targets may exert protective effects, and that the absence of those miRNA targets would make duplication syndromes more severe. However, this hypothesis is challenging to directly test and is beyond the scope of this study. By extension, one could then ask how inclusion of these miRNA targets in a vector genome could exert a stronger effect to protect against MECP2 (or miniMECP2) transgene overexpression. Ultimately, our strategy created a regulated viral genome that improved safety without compromising efficacy (versus that of the unregulated control vector) in two different treatment models. However, we acknowledge that the mechanistic underpinnings of this success are not fully elucidated. Conceptually, it may be possible for our selected miRNA targets to function more efficiently within the context of a small viral genome-encoded miniMECP2 mRNA versus that of a larger endogenous MECP2 mRNA, simply because smaller mRNAs in general are less resistant to RNAi (see Agarwal et al.32).
The construction of miRARE was motivated by the desire to create a negative feedback loop to control MECP2 expression. Our immunofluorescence data comparing expression between wild-type and knockout mice suggests that we have achieved a feedback loop (Fig. 6 and Supplementary Fig. 7). However, our observations need to be validated in future studies using an orthogonal approach. For example, one could administer the same dose intrathecally to mosaic females that are heterozygous-null for MECP2-EGFP and then quantify the percentage of myc+ cells in MECP2-EGFP+ and null cells within the pons and midbrain. The decrease in the percentage of myc+ pons and midbrain cells in AAV9/miniMECP2-miRARE-treated wild-type mice (versus AAV9/miniMECP2-miRARE-treated knockout mice and AAV9/miniMECP2-treated wild-type mice) is striking and may direct researchers towards meaningful efficacy readouts that are more granular and objective than the Bird scoring method.33 For example, the relatively high expression in knockout pons (versus wild-type pons) may direct researchers to analyse respiratory patterns via whole-body plethysmography.91
Other mechanism studies that may be helpful include elucidating miRARE function in neurons versus glia. However, these studies will face challenges. First, miRARE-RDH1pA features 10 miRNA targets (six for miRARE; four for RDH1pA) that may mediate different regulatory effects (constitutive and/or conditional) across different cell types.4,5 Second, there is no guarantee that positive cell culture-based documentation of negative feedback loops (such as the MECP2-miR-132 loop documented by Klein et al.92) will always be reproduced in the CNS in vivo. Third, physiologically meaningful changes in MECP2-responsive miRNAs within specific cell types (in vivo) may not be easily detected in RNA extracted from discrete, dissectable brain regions. Finally, literature describing miRNAs expressed in neuronal or glial cell types may not be informative for predicting miRARE function across cell types.93,94 Whereas published studies provide baseline cell-type specific miRNA profiles, our primary screen was designed to identify miRNAs that may be upregulated (at the tissue level) in a gene therapy overdose model.
Despite the currently limited mechanistic insights for miRARE, it is important to note that the design strategy for miRARE seeks to address limitations of conventional approaches for regulating transgene expression within the context of CNS gene therapies. Published approaches for regulating expression include strategies to impart cell-type specificity (e.g. neuronal promoters), eliminate peripheral expression (e.g. miR-122 target panels to constitutively inhibit hepatic expression), destabilize mRNA (in a manner that is not responsive to total MECP2 levels) and destabilize exogenous protein (e.g. fused degron domains).9,24,25,95,96 These published approaches, however, do not intend to cap exogenous protein expression levels in CNS cells receiving hundreds of vector genome copies versus those of CNS cells receiving few genome copies. Moreover, conventional regulatory approaches are not intended to control expression in response to MECP2 mosaicism, in which transduced cells express either wild-type endogenous MECP2 or mutant MECP2. This variability in vector copy number and endogenous wild-type MECP2 expression across transduced cells creates a need for true feedback-enabled regulation of MECP2. At the organismal level, such a feedback loop should maintain efficacy while improving safety. Ultimately, miRARE has thus far demonstrated promising expression data suggestive of a true feedback regulatory loop, and attenuates overexpression-related side effects in wild-type mice without compromising efficacy in knockout mice.
The knockout survival extension mediated by regulated miniMECP2 gene transfer (intrathecally) in knockout mice is modest compared to those published for neonatal gene transfer (within the context of intracerebrospinal fluid gene transfer mediated by AAV9).4,7,97 However, neonatal administration is not translationally relevant for Rett syndrome. Furthermore, published treatment paradigms mediating greater survival extensions in treated adolescent mice have limitations: (i) the viral genome cannot fully package based on predicted size6; and (ii) the selected capsid does not translate.9 Importantly, the modest AAV9/miniMECP2-miRARE-mediated extension in knockout survival was achieved while overcoming the primary barrier to successful MECP2 gene therapy: attenuating side effects in wild-type mice without compromising efficacy (versus that of an unregulated vector) in knockout mice at a translationally relevant age using a capsid that can be used in humans. Since AAV9/miniMECP2-miRARE did not reach an unacceptably toxic threshold, we speculate that improved vector technology permitting more efficient CNS gene transfer could improve the efficacy of this genome design while still maintaining safe regulation of MECP2 levels.
In future studies, it will be insightful to evaluate the relative performance of AAV9/miniMECP2-miRARE and control vectors (e.g. unregulated vectors or other published vectors) in MECP2-expressing Rett syndrome mouse models for two reasons: (i) it is unclear whether any given MECP2 vector will always achieve the same toxicity threshold across Rett syndrome mouse models; and (ii) endogenous MECP2-expressing mouse models may eliminate the bulk of the immune response recently shown to cause tail lesions in MECP2 virus-treated knockout mice.9 During the course of their study, Luoni et al.9 showed that tail lesions in virus-treated knockout mice may be caused by an immune response to exogenous MECP2. Our data also show an increased frequency of tail lesions among virus-treated knockout mice. We hypothesize that the low frequency of lesions observed in virus-treated wild-type mice (versus virus-treated knockout mice) may indicate a greatly attenuated immune response; the tail lesions in saline-treated knockout mice may have alternative mechanistic explanations (e.g. perhaps an aggressive biting or grooming behaviour that is independent of an immune response). Notably, other laboratories have previously observed skin sores or tail lesions in untreated mouse models of Rett syndrome.98,99 Although daily injectable cyclosporine A has been shown to reduce an immune response in virus-treated knockout mice,9 we do not expect this immunosuppressant to resolve the dose-dependent behavioural abnormalities observed in virus-treated wild-type mice, as these mice already express endogenous MECP2 (Fig. 5). Furthermore, the effect on any potential immune response on quality of life is currently unclear, as Gadalla et al.4 have previously shown that a first-generation MECP2 vector yielded similar behaviour and survival results across both knockout and T158M MECP2-expressing Rett syndrome mice. Importantly, miRARE has improved both safety and efficacy of intrathecal miniMECP2 gene transfer (in wild-type and knockout mice, respectively) without requiring a secondary intervention (Figs 5 and 6). Because the miRARE vector was well-tolerated in wild-type mice, we expect the vector to be well-tolerated in allele-specific Rett syndrome models. Overall, miRARE greatly attenuated these wild-type behavioural side effects while improving knockout survival in our lumbar intrathecal treatment model.
MiRARE may have practical use beyond Rett syndrome. It was designed using a combination of empirical data gathered from MECP2 overexpression studies, but also integrated knowledge gained from the analysis of several developmentally regulated, dose-sensitive genes. Thus, we recognize the possibility that miRARE could be used to provide feedback regulation of other dose-sensitive genes in the context of gene transfer, including genes beyond those analysed in the creation of miRARE. The design strategy of miRARE is of further significance because the target panel design strategy described herein is built on molecular data from an intentional overdose. Ultimately, risk-driven viral genome modification presents the field of gene therapy with a unique advantage that dose-dependent transgene-related side effects could be resolved within the encoded therapeutic product. Furthermore, it may be possible for researchers to adapt our risk-driven design strategy to apply miRNA-mediated feedback regulation to other groups of phenotypically related disorders.
Finally, we would like to acknowledge an unpublished call-to-action that concluded a recent cross-sector research conference for Rett syndrome: the Rett syndrome community should push the development of an MECP2 gene therapy that permits widespread but tightly regulated expression (Sarah Sinnett, summary of personal communication from general meeting discussion).100 In light of clearly documented dose-dependent side effects for MECP2, and now unregulated miniMECP2, gene therapy,5 it is important that Rett syndrome gene therapy trials move forward with great caution. We hope this study addresses widespread expectations for safer preclinical Rett syndrome gene therapy, and encourages researchers and clinicians to advocate for rigorous, transparent safety testing.
Supplementary Material
Acknowledgements
The authors would like to thank Mary Watkinson and Dr Xin Chen for providing technical assistance. The authors would like to thank Dr Erik Lykken and Melissa Hyatt for assisting with biodistribution and gene expression analyses. The authors would also like to thank Dr Erik Lykken for proofreading this paper.
Funding
A grant from Rett Syndrome Research Trust (RSRT) to S.J.G., a donation from Abeona Therapeutics to S.J.G. and general laboratory funds to S.J.G. (UTSW Department of Pediatrics) funded most of the project. A Mentored Training Fellowship (#3517) from Rettsyndrome.org to S.E.S. provided partial salary support. A grant from Taysha Gene Therapies to S.E.S. provided additional support. The authors would like to acknowledge the assistance of the UT Southwestern Live Cell Imaging Facility, a Shared Resources of the Harold C. Simmons Cancer Center, supported in part by an NCI Cancer Center Support Grant no, 1P30 CA142543-01.
Competing interests
S.J.G. declares a conflict of interest with Asklepios Biopharma, from which he has received patent royalties for IP that are not used in this study. S.J.G and S.E.S. declare a conflict of interest with Abeona Therapeutics, from which they have received royalties for miRARE. Additional IP related to the miniMECP2-miRARE vector, for which S.J.G. and S.E.S. are inventors, has been licensed to Taysha Gene Therapies.
Supplementary material
Supplementary material is available at Brain online.
Glossary
- AAV9
adeno-associated viral vector serotype 9
- miRARE
miR-responsive auto-regulatory element
Contributor Information
Sarah E Sinnett, Department of Pediatrics, University of Texas Southwestern Medical Center, Dallas, TX 75390, USA; Eugene McDermott Center for Human Growth and Development, University of Texas Southwestern Medical Center, Dallas, TX 75390, USA.
Emily Boyle, Department of Pediatrics, University of Texas Southwestern Medical Center, Dallas, TX 75390, USA.
Christopher Lyons, Department of Pediatrics, University of Texas Southwestern Medical Center, Dallas, TX 75390, USA.
Steven J Gray, Department of Pediatrics, University of Texas Southwestern Medical Center, Dallas, TX 75390, USA; Eugene McDermott Center for Human Growth and Development, University of Texas Southwestern Medical Center, Dallas, TX 75390, USA.
References
- 1. Chahil G, Bollu PC. Rett syndrome. In: StatPearls [Internet]. StatPearls Publishing; 2019. [PubMed] [Google Scholar]
- 2. Van den Veyver IB, Zoghbi HY. Genetic basis of Rett syndrome. Ment Retard Dev Disabil Res Rev . 2002;8(2):82–86. [DOI] [PubMed] [Google Scholar]
- 3. Mnatzakanian GN, Lohi H, Munteanu I, et al. A previously unidentified MECP2 open reading frame defines a new protein isoform relevant to Rett syndrome. Nat Genet . 2004;36(4):339–341. [DOI] [PubMed] [Google Scholar]
- 4. Gadalla KKE, Vudhironarit T, Hector RD, et al. Development of a novel AAV gene therapy cassette with improved safety features and efficacy in a mouse model of Rett syndrome. Mol Ther Methods Clin Dev . 2017;5:180–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sinnett SE, Gray SJ. Recent endeavors in MECP2 gene transfer for gene therapy of Rett syndrome. Discov Med . 2017;24(132):153–159. [PubMed] [Google Scholar]
- 6. Garg SK, Lioy DT, Cheval H, et al. Systemic delivery of MeCP2 rescues behavioral and cellular deficits in female mouse models of Rett syndrome. J Neurosci . 2013;33(34):13612–13620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tillotson R, Selfridge J, Koerner MV, et al. Radically truncated MeCP2 rescues Rett syndrome-like neurological defects. Nature . 2017;550(7676):398–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Matagne V, Ehinger Y, Saidi L, et al. A codon-optimized Mecp2 transgene corrects breathing deficits and improves survival in a mouse model of Rett syndrome. Neurobiol Dis . 2017;99:1–11. [DOI] [PubMed] [Google Scholar]
- 9. Luoni M, Giannelli S, Indrigo MT, et al. Whole brain delivery of an instability-prone Mecp2 transgene improves behavioral and molecular pathological defects in mouse models of Rett syndrome. Elife . 2020;9:e52629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ross PD, Guy J, Selfridge J, et al. Exclusive expression of MeCP2 in the nervous system distinguishes between brain and peripheral Rett syndrome-like phenotypes. Hum Mol Genet . 2016;25(20):4389–4404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gray SJ, Matagne V, Bachaboina L, Yadav S, Ojeda SR, Samulski RJ. Preclinical differences of intravascular AAV9 delivery to neurons and glia: a comparative study of adult mice and nonhuman primates. Mol Ther . 2011;19(6):1058–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Saraiva J, Nobre RJ, Pereira de Almeida L. Gene therapy for the CNS using AAVs: The impact of systemic delivery by AAV9. J Control Release . 2016;241:94–109. [DOI] [PubMed] [Google Scholar]
- 13. Matsuzaki Y, Tanaka M, Hakoda S, et al. Neurotropic properties of AAV-PHP.B are shared among diverse inbred strains of mice. Mol Ther . 2019;27(4):700–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Huang Q, Chan KY, Tobey IG, et al. Delivering genes across the blood-brain barrier: LY6A, a novel cellular receptor for AAV-PHP.B capsids. PLoS ONE . 2019;14(11):e0225206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. McCarty DM. Self-complementary AAV vectors; advances and applications. Mol Ther . 2008;16(10):1648–1656. [DOI] [PubMed] [Google Scholar]
- 16. Bodda C, Tantra M, Mollajew R, et al. Mild overexpression of MeCP2 in mice causes a higher susceptibility toward seizures. Am J Pathol . 2013;183(1):195–210. [DOI] [PubMed] [Google Scholar]
- 17. Collins AL, Levenson JM, Vilaythong AP, et al. Mild overexpression of MeCP2 causes a progressive neurological disorder in mice. Hum Mol Genet . 2004;13(21):2679–2689. [DOI] [PubMed] [Google Scholar]
- 18. Na ES, Nelson ED, Kavalali ET, Monteggia LM. The impact of MeCP2 loss- or gain-of-function on synaptic plasticity. Neuropsychopharmacology . 2013;38(1):212–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miguet M, Faivre L, Amiel J, et al. Further delineation of the MECP2 duplication syndrome phenotype in 59 French male patients, with a particular focus on morphological and neurological features. J Med Genet. 2018;55(6):359–371. [DOI] [PubMed] [Google Scholar]
- 20. Lim Z, Downs J, Wong K, Ellaway C, Leonard H. Expanding the clinical picture of the MECP2 duplication syndrome. Clin Genet . 2017;91(4):557–563. [DOI] [PubMed] [Google Scholar]
- 21. Luikenhuis S, Giacometti E, Beard CF, Jaenisch R. Expression of MeCP2 in postmitotic neurons rescues Rett syndrome in mice. Proc Natl Acad Sci U S A . 2004;101(16):6033–6038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Takagi N. The role of X-chromosome inactivation in the manifestation of Rett syndrome. Brain Dev . 2001;23 Suppl 1:S182–185. [DOI] [PubMed] [Google Scholar]
- 23. Dhungel B, Ramlogan-Steel CA, Steel JC. MicroRNA-regulated gene delivery systems for research and therapeutic purposes. Molecules . 2018;23(7):1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Qiao C, Yuan Z, Li J, et al. Liver-specific microRNA-122 target sequences incorporated in AAV vectors efficiently inhibits transgene expression in the liver. Gene Ther . 2011;18(4):403–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Geisler A, Jungmann A, Kurreck J, et al. microRNA122-regulated transgene expression increases specificity of cardiac gene transfer upon intravenous delivery of AAV9 vectors. Gene Ther . 2011;18(2):199–209. [DOI] [PubMed] [Google Scholar]
- 26. Clement N, Grieger JC. Manufacturing of recombinant adeno-associated viral vectors for clinical trials. Mol Ther Methods Clin Dev . 2016;3:16002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schirle NT, Sheu-Gruttadauria J, MacRae IJ. Structural basis for microRNA targeting. Science . 2014;346(6209):608–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Grimson A, Farh KK, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell . 2007;27(1):91–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Saetrom P, Heale BS, Snove O Jr, Aagaard L, Alluin J, Rossi JJ. Distance constraints between microRNA target sites dictate efficacy and cooperativity. Nucleic Acids Res . 2007;35(7):2333–2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell . 2005;120(1):15–20. [DOI] [PubMed] [Google Scholar]
- 31. Gray SJ, Choi VW, Asokan A, Haberman RA, McCown TJ, Samulski RJ. Production of recombinant adeno-associated viral vectors and use in in vitro and in vivo administration. Curr Protoc Neurosci . 2011;57:4.17.1–4.17.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Agarwal V, Bell GW, Nam JW, Bartel DP. Predicting effective microRNA target sites in mammalian mRNAs. Elife . 2015;4:e05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Guy J, Gan J, Selfridge J, Cobb S, Bird A. Reversal of neurological defects in a mouse model of Rett syndrome. Science . 2007;315(5815):1143–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lein ES, Hawrylycz MJ, Ao N, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature . 2007;445(7124):168–176. [DOI] [PubMed] [Google Scholar]
- 35.Allen Mouse Brain Atlas. Accessed 7 October 2021. https://mouse.brain-map.org/experiment/thumbnails/100042147?image_type=atlas
- 36.LC Sciences. microRNA Microarray Data Analysis. Accessed 7 October 2021. https://www.lcsciences.com/documents/application-notes/Tech-Bull-MicroRNA-Microarray-Data-Analysis.pdf
- 37. Ziats MN, Rennert OM. Identification of differentially expressed microRNAs across the developing human brain. Mol Psychiatry . 2014;19(7):848–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Leonard H, Cobb S, Downs J. Clinical and biological progress over 50 years in Rett syndrome. Nat Rev Neurol . 2017;13(1):37–51. [DOI] [PubMed] [Google Scholar]
- 39. de Winter CF, Baas M, Bijlsma EK, van Heukelingen J, Routledge S, Hennekam RC. Phenotype and natural history in 101 individuals with Pitt-Hopkins syndrome through an internet questionnaire system. Orphanet J Rare Dis . 2016;11:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dean L. Pitt-Hopkins syndrome. In: Pratt V, McLeod H, Rubinstein W, Dean L, Kattman B, Malheiro A, eds. Medical genetics summaries;2012. [Google Scholar]
- 41. Sweetser DA, Elsharkawi I, Yonker L, Steeves M, Parkin K, Thibert R. Pitt-Hopkins Syndrome. In: Adam MP, Ardinger HH, Pagon RA, et al. , eds. GeneReviews®;1993. [PubMed] [Google Scholar]
- 42. Brzozka MM, Radyushkin K, Wichert SP, Ehrenreich H, Rossner MJ. Cognitive and sensorimotor gating impairments in transgenic mice overexpressing the schizophrenia susceptibility gene Tcf4 in the brain. Biol Psychiatry . 2010;68(1):33–40. [DOI] [PubMed] [Google Scholar]
- 43. de Queiroz AM, Raffaini MS, de Camargo LM, de Pina Neto JM, Melo DG, Silva RA. Orofacial findings and dental treatment in an 8-year-old patient with trisomy 18: a case report. J Dent Child (Chic) . 2007;74(1):67–72. [PubMed] [Google Scholar]
- 44. Seltzer LE, Paciorkowski AR. Genetic disorders associated with postnatal microcephaly. Am J Med Genet C Semin Med Genet . 2014;166C(2):140–155. [DOI] [PubMed] [Google Scholar]
- 45. Matricardi S, Spalice A, Salpietro V, et al. Epilepsy in the setting of full trisomy 18: A multicenter study on 18 affected children with and without structural brain abnormalities. Am J Med Genet C Semin Med Genet . 2016;172(3):288–295. [DOI] [PubMed] [Google Scholar]
- 46. Vrecar I, Innes J, Jones EA, et al. Further clinical delineation of the MEF2C haploinsufficiency syndrome: report on new cases and literature review of severe neurodevelopmental disorders presenting with seizures, absent speech, and involuntary movements. J Pediatr Genet . 2017;6(3):129–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cesaretti C, Spaccini L, Righini A, et al. Prenatal detection of 5q14.3 duplication including MEF2C and brain phenotype. Am J Med Genet A . 2016;170A(5):1352–1357. [DOI] [PubMed] [Google Scholar]
- 48. Straub J, Gregor A, Sauerer T, et al. Genetic interaction screen for severe neurodevelopmental disorders reveals a functional link between Ube3a and Mef2 in Drosophila melanogaster. Sci Rep . 2020;10(1):1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Novara F, Rizzo A, Bedini G, et al. MEF2C deletions and mutations versus duplications: a clinical comparison. Eur J Med Genet . 2013;56(5):260–265. [DOI] [PubMed] [Google Scholar]
- 50. Tatton-Brown K, Rahman N. Sotos syndrome. Eur J Hum Genet . 2007;15(3):264–271. [DOI] [PubMed] [Google Scholar]
- 51. Rosenfeld JA, Kim KH, Angle B, et al. Further evidence of contrasting phenotypes caused by reciprocal deletions and duplications: Duplication of NSD1 causes growth retardation and microcephaly. Mol Syndromol . 2013;3(6):247–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Franco LM, de Ravel T, Graham BH, et al. A syndrome of short stature, microcephaly and speech delay is associated with duplications reciprocal to the common Sotos syndrome deletion. Eur J Hum Genet . 2010;18(2):258–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ball LJ, Sullivan MD, Dulany S, Stading K, Schaefer GB. Speech-language characteristics of children with Sotos syndrome. Am J Med Genet A . 2005;136A(4):363–367. [DOI] [PubMed] [Google Scholar]
- 54. Jeong Y, Kim T, Kim S, Hong YK, Cho KS, Lee IS. Overexpression of histone methyltransferase NSD in Drosophila induces apoptotic cell death via the Jun-N-terminal kinase pathway. Biochem Biophys Res Commun . 2018;496(4):1134–1140. [DOI] [PubMed] [Google Scholar]
- 55. Bouazzi H, Thakur S, Trujillo C, Alwasiyah MK, Munnich A. Novel ATRX gene damaging missense mutation c.6740A>C segregates with profound to severe intellectual deficiency without alpha thalassaemia. Indian J Med Res . 2016;143(1):43–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Stevenson RE. Alpha-thalassemia X-linked intellectual disability syndrome. In: Adam MP, Ardinger HH, Pagon RA, et al. , eds. GeneReviews®;1993. [Google Scholar]
- 57. Berube NG, Jagla M, Smeenk C, De Repentigny Y, Kothary R, Picketts DJ. Neurodevelopmental defects resulting from ATRX overexpression in transgenic mice. Hum Mol Genet . 2002;11(3):253–261. [DOI] [PubMed] [Google Scholar]
- 58. Lugtenberg D, de Brouwer AP, Oudakker AR, et al. Xq13.2q21.1 duplication encompassing the ATRX gene in a man with mental retardation, minor facial and genital anomalies, short stature and broad thorax. Am J Med Genet A . 2009;149A(4):760–766. [DOI] [PubMed] [Google Scholar]
- 59. Talkowski ME, Mullegama SV, Rosenfeld JA, et al. Assessment of 2q23.1 microdeletion syndrome implicates MBD5 as a single causal locus of intellectual disability, epilepsy, and autism spectrum disorder. Am J Hum Genet . 2011;89(4):551–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Mullegama SV, Rosenfeld JA, Orellana C, et al. Reciprocal deletion and duplication at 2q23.1 indicates a role for MBD5 in autism spectrum disorder. Eur J Hum Genet . 2014;22(1):57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kleefstra T, Kramer JM, Neveling K, et al. Disruption of an EHMT1-associated chromatin-modification module causes intellectual disability. Am J Hum Genet . 2012;91(1):73–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Chung BH, Mullegama S, Marshall CR, et al. Severe intellectual disability and autistic features associated with microduplication 2q23.1. Eur J Hum Genet . 2012;20(4):398–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hegarty SV, Sullivan AM, O'Keeffe GW. Zeb2: A multifunctional regulator of nervous system development. Prog Neurobiol . 2015;132:81–95. [DOI] [PubMed] [Google Scholar]
- 64. Yuan H, Zhang L, Chen M, Zhu J, Meng Z, Liang L. A de novo triplication on 2q22.3 including the entire ZEB2 gene associated with global developmental delay, multiple congenital anomalies and behavioral abnormalities. Mol Cytogenet . 2015;8:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Dagli AI, Mueller J, Williams CA. Angelman syndrome. In: Adam MP, Ardinger HH, Pagon RA, et al. , eds. GeneReviews®;1993. [Google Scholar]
- 66. Pelc K, Cheron G, Boyd SG, Dan B. Are there distinctive sleep problems in Angelman syndrome? Sleep Med . 2008;9(4):434–441. [DOI] [PubMed] [Google Scholar]
- 67. Pelc K, Cheron G, Dan B. Behavior and neuropsychiatric manifestations in Angelman syndrome. Neuropsychiatr Dis Treat . 2008;4(3):577–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Copping NA, Christian SGB, Ritter DJ, et al. Neuronal overexpression of Ube3a isoform 2 causes behavioral impairments and neuroanatomical pathology relevant to 15q11.2-q13.3 duplication syndrome. Hum Mol Genet . 2017;26(20):3995–4010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Finucane BM, Lusk L, Arkilo D, et al. 15q duplication syndrome and related disorders. In: Adam MP, Ardinger HH, Pagon RA, et al. , eds. GeneReviews®; 1993. [Google Scholar]
- 70. Wegiel J, Schanen NC, Cook EH, et al. Differences between the pattern of developmental abnormalities in autism associated with duplications 15q11.2-q13 and idiopathic autism. J Neuropathol Exp Neurol . 2012;71(5):382–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kim S, Chahrour M, Ben-Shachar S, Lim J. Ube3a/E6AP is involved in a subset of MeCP2 functions. Biochem Bioph Res Co . 2013;437(1):67–73. [DOI] [PubMed] [Google Scholar]
- 72. Noor A, Dupuis L, Mittal K, et al. 15q11.2 duplication encompassing only the UBE3A gene is associated with developmental delay and neuropsychiatric phenotypes. Hum Mutat . 2015;36(7):689–693. [DOI] [PubMed] [Google Scholar]
- 73. Luco SM, Pohl D, Sell E, Wagner JD, Dyment DA, Daoud H. Case report of novel DYRK1A mutations in 2 individuals with syndromic intellectual disability and a review of the literature. BMC Med Genet . 2016;17:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Araujo BH, Torres LB, Guilhoto LM. Cerebal overinhibition could be the basis for the high prevalence of epilepsy in persons with Down syndrome. Epilepsy Behav . 2015;53:120–125. [DOI] [PubMed] [Google Scholar]
- 75. Carter JC, Capone GT, Kaufmann WE. Neuroanatomic correlates of autism and stereotypy in children with Down syndrome. Neuroreport . 2008;19(6):653–656. [DOI] [PubMed] [Google Scholar]
- 76. Duchon A, Herault Y. DYRK1A, a dosage-sensitive gene involved in neurodevelopmental disorders, is a target for drug development in down syndrome. Front Behav Neurosci . 2016;10:104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Guedj F, Pereira PL, Najas S, et al. DYRK1A: A master regulatory protein controlling brain growth. Neurobiol Dis . 2012;46(1):190–203. [DOI] [PubMed] [Google Scholar]
- 78. Kent RD, Vorperian HK. Speech impairment in Down syndrome: A review. J Speech Lang Hear Res . 2013;56(1):178–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. De Toma I, Ortega M, Aloy P, Sabido E, Dierssen M. DYRK1A overexpression alters cognition and neural-related proteomic pathways in the hippocampus that are rescued by green tea extract and/or environmental enrichment. Front Mol Neurosci . 2019;12:272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Miyata Y, Saida K, Kumada S, et al. Periventricular small cystic lesions in a patient with Coffin-Lowry syndrome who exhibited a novel mutation in the RPS6KA3 gene. Brain Dev . 2018;40(7):566–569. [DOI] [PubMed] [Google Scholar]
- 81. Morino T, Ogata T, Horiuchi H, Yamaoka S, Fukuda M, Miura H. Eight years of follow-up after laminectomy of calcium pyrophosphate crystal deposition in the cervical yellow ligament of patient with Coffin-Lowry syndrome: A case report. Medicine (Baltimore). 2016;95(31):e4468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Tos T, Alp MY, Aksoy A, Ceylaner S, Hanauer A. A familial case of Coffin-Lowry syndrome caused by RPS6KA3 C.898C>T mutation associated with multiple abnormal brain imaging findings. Genet Couns . 2015;26(1):47–52. [PubMed] [Google Scholar]
- 83. Touraine RL, Zeniou M, Hanauer A. A syndromic form of X-linked mental retardation: the Coffin-Lowry syndrome. Eur J Pediatr . 2002;161(4):179–187. [DOI] [PubMed] [Google Scholar]
- 84. Matsumoto A, Kuwajima M, Miyake K, et al. An Xp22.12 microduplication including RPS6KA3 identified in a family with variably affected intellectual and behavioral disabilities. J Hum Genet . 2013;58(11):755–757. [DOI] [PubMed] [Google Scholar]
- 85. Tejada MI, Martinez-Bouzas C, Garcia-Ribes A, et al. A child with mild X-linked intellectual disability and a microduplication at Xp22.12 including RPS6KA3. Pediatrics . 2011;128(4):e1029-1033. [DOI] [PubMed] [Google Scholar]
- 86. Putz G, Bertolucci F, Raabe T, Zars T, Heisenberg M. The S6KII (rsk) gene of Drosophila melanogaster differentially affects an operant and a classical learning task. J Neurosci . 2004;24(44):9745–9751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Bittel DC, Kibiryeva N, Dasouki M, Knoll JH, Butler MG. A 9-year-old male with a duplication of chromosome 3p25.3p26.2: Clinical report and gene expression analysis. Am J Med Genet A . 2006;140(6):573–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Ma Y, Hu JH, Zhao WJ, et al. Overexpression of gamma-aminobutyric acid transporter subtype I leads to susceptibility to kainic acid-induced seizure in transgenic mice. Cell Res . 2001;11(1):61–67. [DOI] [PubMed] [Google Scholar]
- 89. Johannesen KM, Gardella E, Linnankivi T, et al. Defining the phenotypic spectrum of SLC6A1 mutations. Epilepsia . 2018;59(2):389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Natera-de Benito D, García-Pérez MA, Martínez-Granero MÁ, Izquierdo-López L. A patient with a duplication of chromosome 3p (p24.1p26.2): A comparison with other partial 3p trisomies. Am J Med Genet 2013;164A(2):548–550. [DOI] [PubMed] [Google Scholar]
- 91. Ramirez JM, Ward CS, Neul JL. Breathing challenges in Rett syndrome: Lessons learned from humans and animal models. Respir Physiol Neurobiol . 2013;189(2):280–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Klein ME, Lioy DT, Ma L, Impey S, Mandel G, Goodman RH. Homeostatic regulation of MeCP2 expression by a CREB-induced microRNA. Nat Neurosci . 2007;10(12):1513–1514. [DOI] [PubMed] [Google Scholar]
- 93. Jovicic A, Roshan R, Moisoi N, et al. Comprehensive expression analyses of neural cell-type-specific miRNAs identify new determinants of the specification and maintenance of neuronal phenotypes. J Neurosci . 2013;33(12):5127–5137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. He M, Liu Y, Wang X, Zhang MQ, Hannon GJ, Huang ZJ. Cell-type-based analysis of microRNA profiles in the mouse brain. Neuron . 2012;73(1):35–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Gray SJ, Foti SB, Schwartz JW, et al. Optimizing promoters for recombinant adeno-associated virus-mediated gene expression in the peripheral and central nervous system using self-complementary vectors. Hum Gene Ther . 2011;22(9):1143–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Quintino L, Namislo A, Davidsson M, et al. Destabilizing domains enable long-term and inert regulation of GDNF expression in the brain. Mol Ther Methods Clin Dev . 2018;11:29–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Gadalla KK, Bailey ME, Spike RC, et al. Improved survival and reduced phenotypic severity following AAV9/MECP2 gene transfer to neonatal and juvenile male Mecp2 knockout mice. Mol Ther . 2013;21(1):18–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Yasui DH, Gonzales ML, Aflatooni JO, et al. Mice with an isoform-ablating Mecp2 exon 1 mutation recapitulate the neurologic deficits of Rett syndrome. Hum Mol Genet . 2014;23(9):2447–2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Carrette LLG, Blum R, Ma W, Kelleher RJ 3rd, Lee JT. Tsix-Mecp2 female mouse model for Rett syndrome reveals that low-level MECP2 expression extends life and improves neuromotor function. Proc Natl Acad Sci USA . 2018;115(32):8185–8190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Bajikar S. Rett Syndrome Symposium and Workshop. Accessed 7 October 2021. https://nri.texaschildrens.org/event/rett-syndrome-symposium-and-workshop
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw data that support the findings of this study are available from the corresponding author, on reasonable request. Microarray data from LC Sciences are provided within the Supplementary material.