

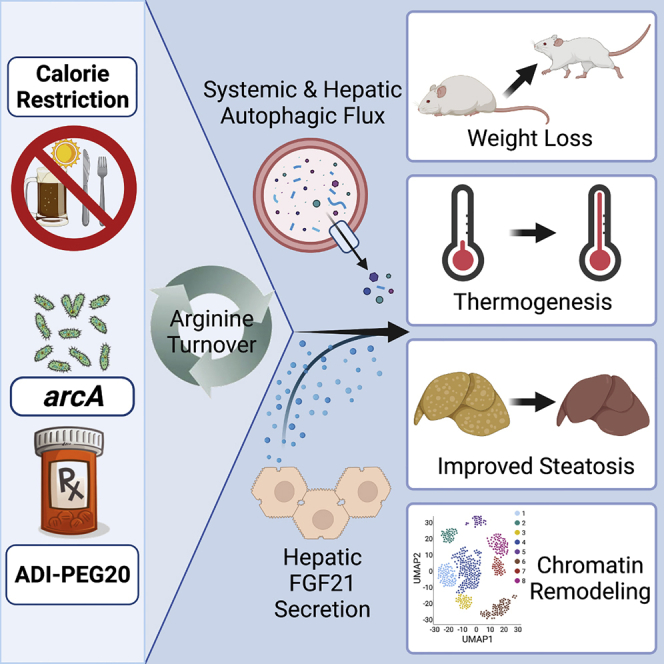
Summary
Obesity is a multi-systemic disorder of energy balance. Despite intense investigation, the determinants of energy homeostasis remain incompletely understood, and efficacious treatments against obesity and its complications are lacking. Here, we demonstrate that conferred arginine iminohydrolysis by the bacterial virulence factor and arginine deiminase, arcA, promotes mammalian energy expenditure and insulin sensitivity and reverses dyslipidemia, hepatic steatosis, and inflammation in obese mice. Extending this, pharmacological arginine catabolism via pegylated arginine deiminase (ADI-PEG 20) recapitulates these metabolic effects in dietary and genetically obese models. These effects require hepatic and whole-body expression of the autophagy complex protein BECN1 and hepatocyte-specific FGF21 secretion. Single-cell ATAC sequencing further reveals BECN1-dependent hepatocyte chromatin accessibility changes in response to ADI-PEG 20. The data thus reveal an unexpected therapeutic utility for arginine catabolism in modulating energy metabolism by activating systemic autophagy, which is now exploitable through readily available pharmacotherapy.
Keywords: liver, ADI-PEG 20, arginine, arginine deiminase, arcA, arginase, autophagy, Beclin-1, fasting, caloric restriction, energy metabolism, insulin resistance, glucose transport, GLUT, non-alcoholic fatty liver disease, thermogenesis, diabetes, obesity, FGF21
Graphical abstract
Highlights
-
•
Pegylated arginine deiminase (ADI-PEG 20) is currently used to treat liver tumors
-
•
ADI-PEG 20 improves insulin sensitivity, dyslipidemia, and liver fat in obese mice
-
•
ADI-PEG 20 improves energy homeostasis by driving systemic and hepatocyte autophagy
-
•
Arginine catabolism is a tractable pathway to treat obesity and related disorders
Zhang et al. show that promoting systemic arginine catabolism by expressing hepatocyte arginine deiminase—or by treating mice with the drug ADI-PEG 20—induces systemic and hepatic autophagic flux to ameliorate obesity and its complications in mice.
Introduction
Obesity is a disorder of energy balance afflicting an estimated 1 in 5 individuals worldwide.1 It is associated with multiple morbidities, including metabolic syndrome, cardiovascular death, type 2 diabetes mellitus, and non-alcoholic steatohepatitis.2 Yet, despite decades of investigation into the determinants of energy homeostasis, the incidence of overweight, obesity, and their complications continue to rise worldwide. Currently available therapies that modulate energy homeostasis as a root cause to these complex disorders are limited in number, efficacy, and mechanistic action.
Intermittent fasting and caloric restriction (IF and CR) are effective therapies against obesity and its complications, including non-alcoholic fatty liver disease (NAFLD), dyslipidemia, and insulin resistance, in mice and in humans.3, 4, 5, 6, 7, 8 However, intensive lifestyle modifications are rarely sustainable in real-world settings.9 We previously found that the hepatocyte response to glucose deprivation is sufficient to mimic several key therapeutic effects of generalized IF and CR on hepatic steatosis, hepatic inflammation, and insulin resistance,10, 11, 12, 13, 14, 15, 16, 17, 18, 19 in part by inducing hepatocyte autophagic flux and secretion of the anti-diabetic hepatokine, FGF21.20 We thus set out here to leverage this pathway against metabolic disease. Clinically, this approach is of particular interest, because hepatocyte glucose transport and its downstream pathways are amenable to pharmacological therapy.
We previously identified the arginine ureahydrolase, arginase 2 (ARG2), as a hepatocyte glucose withdrawal-induced factor. Induction of ARG2 is sufficient to exert part of the therapeutic metabolic sequelae of caloric restriction.16 Subsequent data further demonstrated that arginase 1 and 2 polymorphisms determine circulating arginine levels in arginine-supplemented and unsupplemented dietary contexts. Together, the data initiated the hypothesis that augmenting arginine catabolism can modulate host arginine status—and thereby therapeutically direct energy metabolism.
Whereas mammalian ARG2 is a low-affinity (Km ∼2mM), moderate-capacity arginine ureahydrolase,21 we turned to the therapeutic potential of a naturally occurring, high-affinity arginine iminohydrolase, arcA, as a potentially more efficacious means by which to modify host arginine status. arcA is a bacterial virulence factor that is evolutionarily primed for this duty, because its high binding affinity for ARG2 (Km = 34.5 μM) permits bacterial niche establishment by competing arginine away from host inflammatory nitric oxide synthases and toward bacterial ATP production.22 The favorable enzymatic properties of arcA and the ready availability of a pharmacotherapy that mimics these properties led us to examine efficacy and mechanisms of therapeutic arginine catabolism against obesity and its complications.
Here, we demonstrate that hepatocyte and systemic arginine status is therapeutically modifiable, particularly through the bacterial arginine deiminase, arcA. Hepatocyte-directed arcA expression increased basal caloric expenditure and improved glucose and insulin tolerance in genetically diabetic mice. We then provide evidence corroborating the insulin-sensitizing and thermic effects of a arginine deiminase-based pharmacological agent, ADI-PEG 20. Mechanistically, we show enhanced arginine catabolism exerts its therapeutic effects on host metabolism by activating systemic autophagic flux and hepatocyte-specific FGF21 secretion. We conclude that hepatocyte and systemic arginine catabolism play a canonical role in dictating peripheral energy and insulin homeostasis, which is pharmacologically modifiable using readily available agents.
Results
Ectopic expression of the arginine deiminase, arcA, induces thermogenesis and insulin sensitivity
The mammalian arginine ureahydrolases, arginase 1 and arginase 2 (ARG1 and ARG2), mediate arginine hydrolysis to ornithine and urea with low substrate affinity. We reported that fasting induces hepatocyte arginase 2 (Arg2) expression, and that Arg2 upregulation in the absence of caloric restriction is sufficient to induce peripheral thermogenesis and insulin sensitization in genetic and diet-induced obese animals. This suggested that either or both arginine catabolism per se and ARG2 scaffolding and signaling mediate these effects. To test the hypothesis that hepatocyte arginine catabolism enhances hepatic and peripheral energy metabolism apart from ARG2 upregulation, and apart from ARG2 products ornithine and urea, we expressed arcA in hepatocytes. arcA encodes a high-affinity arginine deiminase23 that differs from the arginases in its enzymatic products, citrulline and ammonia.22 We delivered arcA or eGFP (an ectopic expression control vector) via tail-vein injection of AAV8 encoding arcA or eGFP under hepatocyte-specific thyroxine binding globulin promoter control in 5-week-old db/db mice. Five weeks following gene delivery, we verified arcA expression (Figure S1A) and subjected all mice to a battery of metabolic assays and tissue collection (Figure 1A). Hepatocyte-directed arcA increased body mass-by-time interaction with modest but significant increases in fat mass percentage and lower lean mass percentage (Figures 1B and 1C), as quantified by echoMRI analysis. Nonetheless, hepatocyte arcA increased VO2, VCO2 and caloric expenditure throughout both dark and light cycles (Figures 1D and 1E) without altering respiration exchange rate (RER), total locomotion (Figures S1B and S1C), or food consumption (Figure S1D). Analysis of covariance (ANCOVA) further confirmed significantly different heat versus body-weight regression relationships (Figure 1F). arcA reduced fasting serum glucose and improved glucose and insulin tolerance testing (Figures 1G, 1H, and 1I). In contrast, fasting serum low-density lipoprotein cholesterol (LDL-C) and NEFA were significantly decreased, whereas fasting triglycerides (TGs) and total cholesterol (TC) were trended toward and significantly elevated in arcA-expressing diabetic mice, respectively (Figures 1J–1M).
Figure 1.

Hepatic AAV8-mediated arcA increases thermogenesis and insulin sensitivity in db/db mice
(A) Schematic of experimental design used to test the role of AAV8-mediated mouse codon optimized arcA overexpression in db/db mice.
(B and C) Body weight (B) and body fat and lean mass (C) percentage of composition of AAV8-eGFP or AAV8-arcA-injected db/db mice (n = 8 mice per group).
(D) Whole-body oxygen consumption (VO2), carbon dioxide (VCO2), and energy expenditure during light and dark cycle (shaded area) in AAV8-injected db/db mice.
(E) Quantified VO2, VCO2, and energy expenditure during light and dark cycle.
(F) Body weight to energy expenditure regression test during light and dark cycle.
(G) Serum glucose in AAV8-eGFP and AAV8-arcA-treated db/db mice.
(H and I) Intraperitoneal tolerance tests for insulin (ITT, H) and for glucose (GTT, I).
(J–M) Serum non-esterified fatty acid (FFA, J), low-density lipoprotein cholesterol (LDL-C, K), triglycerides (TGs, L), and cholesterol (TC, M) in AAV8-eGFP and AAV8-arcA-treated db/db mice.
Data represented in mean ± SEM. Each data point represents an individual animal. Exact p values are shown. Statistical significance was determined using two-way ANOVA in (B), (D), (H), and (I). Unpaired two-tailed Student’s t test was used in (C), (E), (G), and (J)–(M).
Hepatic metabolic analysis revealed that serum alanine aminotransferase and aspartate aminotransferases (ALT and AST), markers of hepatocellular lysis and enzymatic excursion, were lower in arcA-expressing mice, whereas hepatocyte synthetic function, as quantified by serum albumin, was unchanged in ADI-expressing versus GFP-expressing db/db mice (Figures 2A–2C). This hepatoprotective effect of ADI expression correlated with reduced percentage steatotic area on histologic quantification (Figure 2D) and reduced intrahepatic TGs, TC, and non-esterified fatty acids (NEFA, Figures 2E–2G), but without changes in liver mass, or liver-weight-to-body-weight ratio (Figures S1E and S1F). Targeted hepatic metabolomics revealed significantly lower intrahepatic ornithine, citrulline, asparagine, and alanine, to suggest an arginine shunt away from urea cycle flux (Figure 2H). Liver transcriptomic analysis revealed clear separation in gene expression profiling secondary to arcA expression when compared with GFP-expressing controls (Figures 2I, 2J, and 2K). Although arcA expression increased fatty acid import and decreased fatty acid export gene expression of Cd36 and Mttp, respectively, via quantitative real-time PCR (Figure S1G), pathway analysis revealed significant upregulation in multiple metabolic processes upon arcA overexpression, including organic and fatty acid metabolism, arachidonic acid metabolism,14 and fatty acid oxidation (Figures 2L, left panel, and S1H). Concomitantly, we observed suppression in pro-inflammatory pathways, including adhesion, locomotion, cell migration, immune regulation, and immune effector responses (Figure 2L, right panel). Transcriptomic suggestions of suppressed inflammation were confirmed by quantitative real-time PCR, demonstrating decreased hepatic expression of Il-1β, Il-6, Tnf-α, Ccl2, and Cxcl9 (Figure S1I). Markers of macrophage infiltration, Cd68 and Mmp2, along with markers of fibrosis development, Col1a1 and Timp1, are also suppressed by arcA overexpression in the liver (Figures S1J and S1K). Consistent with the reduction in inflammatory response, hepatocyte-specific overexpression of arcA also led to significant increases in expression of gene encoding urea cycle enzymes like Ass1, Otc, and Cps1 (Figures S1L), and a significant decrease in Glul expression (Figures S1M). These findings mirrored the increased energy expenditure and anti-inflammatory phenotypes observed previously in Arg2-overexpressing liver.16 Moreover, these hepatic metabolic and inflammatory improvements associated with enhanced arcA-induced LC3B-II and FGF21 protein accumulation in liver and serum—each serving as biomarkers of hepatocyte fasting- and stress-response activation (Figures 2M–2O).20,24 Cellular alterations in fasting signaling correlated with enhanced cellular respiration, as quantified by Seahorse respirometry upon ADI overexpression in isolated mouse hepatocyte cell line, AML12 (Figure 2P). Indeed, ADI expression significantly induced time-by-treatment analysis of overall respiration, as well as non-mitochondrial oxygen consumption and spare respiratory capacity, whereas we observed trends toward increased basal and maximal capacity that did not reach statistical significance (Figure 2Q).
Figure 2.

Hepatic AAV8-mediated arcA attenuates hepatic steatosis and inflammation in db/db mice
(A–C) Serum ALT (A), AST (B), and albumin (C) contents.
(D) Liver sections stained with hematoxylin and eosin (H&E) with steatotic area (e.g., aparenchymal space) quantified (right). Scale bars, 100 μm.
(E–G) Triglyceride (E), cholesterol (F), and non-esterified fatty acid (G) contents in the livers of AAV8-eGFP and AAV8-arcA mice.
(H) Targeted metabolomic analysis of serum amino acids and urea cycle intermediaries from db/db AAV8-eGFP and AAV8-arcA mice.
(I) Principal component analysis (PCA) plot of bulk liver RNA-seq samples from AAV8-eGFP and AAAV8-arcA-injected db/db mice. The first two principal components (PCs) are plotted. Variance proportions are shown along each component axis. The plot model 56% of the total data variance.
(J) Volcano plot of the distribution of all differentially expressed genes between AAV8-eGFP and AAV8-arcA-injected db/db mice, mapping the 28 upregulated genes (red), 255 downregulated genes (blue), and non-significant genes (gray). Black vertical lines highlight log fold changes (FC) of −2 and 2, while the black horizontal line represents a padj of 0.05.
(K) Heatmap showing all significantly expressed genes in the livers of AAV8-arcA-injected db/db mice.
(L) Differentially up- (left) and downregulated (right) genes enriched and identified by Gene Ontology enrichment analysis of biological processes between the livers of AAV8-eGFP and AAV8-arcA-injected db/db mice.
(M) Western blot analysis of FGF21 and LC3B in liver samples from db/db AAV8-eGFP and AAV8-arcA mice. β-actin was used as a loading control.
(N) Western blot quantifications of LC3B (left) and FGF21 (right) (n = 8 mice per group).
(O) Serum FGF21 contents.
(P and Q) From left to right, mitochondrial respiration (P), non-mitochondrial oxygen consumption, spare respiration capacity, basal respiration, and maximal respiration (Q) in vitro AML12 treated with Ad-eGFP (n = 4) or Ad-arcA (n = 5).
Data represented in mean ± SEM. Each data point represents an individual animal. Exact p values are shown. Statistical significance was determined using two-way ANOVA in (P). Statistical significance was determined using unpaired two-tailed Student’s t test in (A)–(G), (N), (O), and (Q).
Pharmacological arginine catabolism induces thermogenesis and insulin tolerance in genetically diabetic mice
Hepatocyte-specific arcA expression ameliorated insulin resistance, glucose intolerance, and dyslipidemia in genetically diabetic mice. ADI-PEG 20 is a stabilized, pegylated arginine deiminase conjugate that has been used against various cancers, including hepatocellular carcinoma.25,26 We tested the hypothesis that systemically administered arginine catabolism recapitulates the metabolic effects of hepatocyte-directed arcA/ADI expression. To that end, we treated db/db diabetic mice with vehicle or ADI-PEG 20 (5 IU/week intraperitoneally) for 5 weeks prior to metabolic testing and tissue analysis (Figure 3A). In contrast with hepatocyte arcA, ADI-PEG 20 attenuated weight gain (Figure 3B), reduced endpoint fat mass percentage and increased lean mass percentage (Figure 3C) relative to vehicle-treated db/db mice. No changes were observed in ghrelin and leptin gene expression and protein abundance (Figures S2A and S2B). Total daily food consumption was significantly reduced with ADI-PEG 20 treatment (Figure S2C). However, temporal alignment of food intake and body weight indicates rapid, statistically significant weight loss several weeks prior to detection of changes in food consumption (Figure S2D). Together, the data suggested that mechanisms apart from food intake are more likely to drive at least the acute therapeutic effects of ADI-PEG 20 on body mass (Figures S2A–S2D). These favorable changes in body weight and composition were observed alongside light and dark cycle increases in VO2, VCO2, and caloric expenditure (Figure 3D), in the absence of changes in respiratory exchange ratio or locomotion (Figures S2E and S2F). In light of observed body-weight-attenuating effects of ADI-PEG 20, we performed linear regression analyses and analysis of covariance comparing heat:body weight relationships in vehicle- and drug-treated animals. This confirmed significant differences in heat:weight regression curves (Figure 3E). Dissected liver mass was lower, whereas no difference in liver mass: body-weight ratio was observed. However, we observed a decrease in WAT mass and WAT mass:body-weight ratio, and increased iBAT mass:body-weight ratio in ADI-PEG 20-treated mice (Figures S2G and S2H). This associated with ADI-PEG 20-induced cellular oxidative respiration, as quantified by Seahorse respirometry in murine hepatocyte cell line AML12 treated with 0.5 μg/mL ADI-PEG 20 (72 h) versus vehicle (Figure 3F). Upon examining the parameters of oxidative respiration, ADI-PEG 20 treatment increased basal oxygen consumption rate (OCR), maximal respiration, spare respiratory capacity, and ATP-coupled respiration (Figure 3G). Concordant with hepatocyte arcA effects, ADI-PEG 20 reduced fasting glucose and glucose and insulin tolerance (Figures 3H–3J) and fasting serum TGs, TC, NEFA, and LDL-C in ADI-PEG 20-treated mice relative to vehicle-treated mice (Figure 3K).
Figure 3.

ADI-PEG 20 treatment improves whole-body metabolism and insulin sensitivity
Five-week-old male db/db mice and their male db/db littermates were randomly grouped (n = 12 per group) and treated with saline (control) or ADI-PEG 20 for 4 weeks.
(A) Schematic of experimental design used to test the metabolic effects of ADI-PEG 20 in db/db mice.
(B) Body weight.
(C) Whole-body fat and lean mass percentage of composition of db/db mice after 5 weeks of ADI-PEG 20 treatment.
(D) Whole-body oxygen consumption (VO2), carbon dioxide (VCO2), and energy expenditure with quantification during light and dark cycles in vehicle- and ADI-PEG 20-treated mice.
(E) Body weight to energy expenditure regression test during light cycle.
(F and G) Mitochondrial respiration (F) and OCR parameters of basal respiration, maximal respiration, spare respiration capacity, and ATP-production coupled respiration (G) of vehicle- and ADI-PEG 20-treated AML12 cells in vitro (n = 12–14 per group).
(H) Serum glucose.
(I and J) Intraperitoneal insulin tolerance test (ITT, I) and glucose tolerance test (GTT, J).
(K) Serum triglyceride, cholesterol, FFA, and LDL-C.
(L) Serum ALT (left), AST (middle), and albumin (right) contents.
(M) Liver triglyceride, cholesterol, and FFA.
(N) Liver sections stained with hematoxylin and eosin (H&E) or oil red O. Scale bars, 100 μm. Liver sections were stained with H&E with steatotic area (e.g., aparenchymal space) quantified. Scale bars, 100 μm.
(O) Western blot analysis of FGF21, LC3B, and SQSTM1/p62 in liver samples from db/db AAV8-eGFP and AAV8-arcA mice. β-actin was used as a loading control.
(P) Western blot quantifications of FGF21 (left), LC3B (middle), and SQSTM1/p62 (right) (n = 4 mice per group).
(Q) Serum FGF21.
Data represented in mean ± SEM. Each data point represents an individual animal. Exact p values are shown. Statistical significance was determined using two-way ANOVA in (B), (D), (F), (I), and (J). Unpaired two-tailed Student’s t test was used in (C), (G), (H), (K)–(N), (P), and (Q).
Hepatic metabolic characterization revealed lower serum ALT and albumin (Figure 3L), with a trend toward improved intrahepatic TGs (−22%, p = 0.0656), and significantly lower intrahepatic cholesterol and NEFA after ADI-PEG 20 treatment (Figure 3M). Interestingly, in contrast to arcA overexpression, we observed increased Il-1β, Il-6, Tnf-α, Ccl2, and Cxcl9 (Figure S2I) along with Cd68, but not Mmp2 (Figure S2J). However, no changes were observed in fibrotic gene expression of Col1a1 and Timp1 (Figure S2K). To assess gene expression related to hepatic ammonia production and scavenging, we measured the expression of urea cycle genes and glutamine synthetase (Glul). We observed significant decrease in urea cycle gene expression including Ass1, Slc25a15, Otc, and Cps1 (Figure S2L), and we observed an increase in Glul expression (Figure S2M). Nevertheless, biochemical improvements were corroborated by percentage of steatotic area, which was significantly reduced in ADI-PEG 20-treated mice without evidence of cellular inflammatory infiltrate in treated or untreated liver (Figure 3N). Intrahepatic Fgf21 mRNA (Figure S2N) and FGF21 protein, LC3BII, and circulating FGF21 were greater in drug-treated mice, whereas hepatic p62/SQSTM1 was unchanged (Figures 3O, 3P, and 3Q). Because p62/SQSTM1 is also transcriptionally upregulated in response to inflammation, oxidative, and other cellular stressors,27,28 we quantified hepatic p62/Sqstm1 mRNA. This revealed a significant p62/Sqstm1 mRNA downregulation in ADI-PEG 20-treated mouse liver (Figure S2O). Interrogation of hepatic mTORC signaling in ADI-PEG 20-treated mice revealed significantly lower phosphorylated p70S6K (threonine 389) and trends toward reduced mTOR (serine 2448) phosphorylation. However, suppression of this arm of the pathway was associated with unaltered ULK1 (serine 757) phosphorylation and with increased 4E-BP1 (threonine 36/47) (Figures S2P and S2Q). We observed enhanced hepatocyte fasting-like and fatty acid oxidation-associated gene expression via Fgf21 and Ucp1, increased fat oxidation gene expression in Cpt1β, Ucp2, and Ucp3, and reduced hepatic gluconeogenic gene expression of Pck1, G6pc, and Fbp1 (Figures S2N, S2R, and S2S),13 without changes in lipid transporters Cd36 or Mttp or cell stress markers Grp78 or Atf4/Chop (Figures S2T and S2U). Overall, ADI-PEG 20 reduced histologic and biochemical hepatic steatosis in contexts of suppressed hepatic fatty acid oxidation and gluconeogenic gene expression.
Hepatocyte FGF21 links systemic arginine catabolism to the thermic and glucose homeostatic effects of ADI-PEG 20 in a western-diet-fed model of metabolic disease
Our data in genetically obese models previously16 and herein indicate a protective effect of arginine hydrolysis in the progression of diabetes in a genetically predisposed model. We showed the downstream signaling effectors of GLUT blockade, induce FGF21 secretion.13 To more completely define and provide mechanistic insights into the action of ADI-PEG 20, we relied on a western diet (WD)-fed model of obesity to examine (1) the extent to which arginine catabolic effects are generalizable across obese models, (2) the degree to which ADI-PEG 20 can reverse, as opposed to attenuate, progression of the deleterious effects of obesity, and (3) the interaction between ADI-induced hepatic FGF21 secretion and its therapeutic effects. We therefore placed wild-type (WT, e.g., Fgf21fl/fl genotype) male mice and their hepatocyte-specific FGF21-deficient littermates (FGF21 LKO, e.g., Fgf21fl/fl × albumin-Cre+) on a 12-week WD (Figure 4A). Thereafter, mice were treated with ADI-PEG 20 (5 IU/week i.p.) for 4 weeks while continuously on diet for a total of 16 weeks. In both WT and FGF21 LKO mice, ADI-PEG 20 reversed WD-induced weight gain (Figures 4B–4D) without altering food consumption (Figure S3A). EchoMRI-based body composition analysis demonstrated decreased fat accumulation and concomitantly increased lean mass:total mass ratio in both WD-fed WT and FGF21 LKO mice (Figure 4E). ADI-PEG 20 trended to increased O2-CO2 exchange and caloric expenditure (Figure 4F) in the absence of changes to locomotion in WD-fed WT mice (Figures S4B and S4C), and these effects were abrogated in FGF21 LKO mice, as quantified by linear analysis of body heat:body weight regression curves (Figure 4G). In contrast, fasting serum insulin levels and glucose tolerance were improved in WD-fed WT and FGF21 LKO groups (Figures 4H and 4I). Interestingly, however, ADI-PEG 20 improved insulin tolerance modestly, and yet this improvement was significantly reversed in ADI-PEG 20-treated FGF21 LKO mice (Figure 4J). Moreover, ADI-PEG 20 decreased fasting serum TGs, TC, and NEFA in WD-fed WT mice. However, only the anti-dyslipidemic effect on TGs, but not on TC, LDL-C, and NEFA, was reversed in FGF21-deficient mice (Figures 4K–4N).
Figure 4.

Hepatic-specific Fgf21 knockout partially abolishes ADI-PEG 20-mediated therapeutic effects
(A) Schematic of experimental design used to test the role of ADI-PEG 20 in Fgf21 LKO WD-fed mice.
(B–E) Body weight over time (B), end point body weight (C), and change in body weight (D) of vehicle- and ADI-PEG 20-treated Fgf21 LKO mice (vehicle-treated Fgf21fl/fl mice, n = 5. ADI-PEG 20-treated Fgf21fl/fl mice, n = 10. ADI-PEG 20-treated Fgf21 LKO mice, n = 7).
(E) Change in body fat and lean composition.
(F) Whole-body oxygen consumption (VO2), carbon dioxide (VCO2), and energy expenditure during light and dark cycle (shaded area) in vehicle- and ADI-PEG 20-treated Fgf21 LKO mice.
(G) Body weight to energy expenditure regression test during light and dark cycle.
(H) Serum insulin in vehicle- and ADI-PEG 20-treated Fgf21 LKO mice.
(I and J) Intraperitoneal tolerance tests for glucose (GTT, I) and for glucose (ITT, J).
(K–N) Serum triglyceride (K), cholesterol (L), non-esterified fatty acid (M), and low-density lipoprotein cholesterol (N) in vehicle- and ADI-PEG 20-treated Fgf21 LKO mice.
(O and P) Serum ALT (O) and serum albumin (P) contents.
(Q and R) Liver sections stained with hematoxylin and eosin (H&E, Q) with steatotic area (e.g., aparenchymal space) quantified (R). Scale bars, 100 μm.
(S–U) Triglyceride (S), cholesterol (T), and non-esterified fatty acid (U) contents in the livers of vehicle- and ADI-PEG 20-treated Fgf21 LKO mice.
(V) Serum FGF21 content.
Data represented in mean ± SEM. Each data point represents an individual animal. Exact p values are shown. Statistical significance was determined using two-way ANOVA in (B), (E), (F), (I), and (J). Unpaired two-tailed Student’s t test was used in (C), (D), (H), (K)–(P), and (R)–(V).
Hepatic analysis in this diet-induced obese model revealed that ADI-PEG 20 did not alter liver weight:body mass ratios (Figures S3D and S3E), serum transaminases, or albumin in any group (Figures 4O, 4P, and 4Q). ADI-PEG 20 reduced the percentage steatotic area histologically (Figure 4R), and this was FGF21 dependent. Biochemical analysis of hepatic lipids confirmed reduction of intrahepatic TGs, cholesterol, and NEFA by ADI-PEG 20. The therapeutic effect of this agent on intrahepatic cholesterol—but not on TGs or NEFA—was reversed in ADI-PEG 20-treated FGF21 LKO mice (Figures 4S, 4T, and 4U). Notably, ADI-PEG 20 increased FGF21 peptide, and FGF21 LKO mice had circulating FGF21 levels that matched basal FGF21 levels (Figure 4V). Each of these ADI-PEG 20-induced changes was associated with reductions in Pck1, G6pc, and Fbp1, although these reductions were not FGF21 dependent (Figure S3F). Together, the data indicate that hepatocyte FGF21 mediates the thermogenic, insulin/glucose-sensitizing, and anti-dyslipidemic effects of ADI-PEG 20 but is dispensable for the body mass, composition, and anti-steatotic effects of FGF21.
Hepatocyte-specific Beclin 1 mediates the anti-dyslipidemic effect of ADI-PEG 20
Autophagic flux data indicate robust hepatic activation of autophagy in conjunction with FGF21 secretion. We directly tested the hypothesis that the metabolic actions of ADI-PEG 20 require hepatocyte-specific autophagic flux through BECN1. To that extent, we subjected WT (e.g., Becn1fl/fl) and hepatocyte-specific Becn1-deficient mice (BECN1 LKO, e.g., Becn1fl/fl × albumin-Cre+) to WD for 12 weeks. This was followed by 4-week ADI-PEG 20 treatment at 5 IU/week i.p. (Figure 5A). We first confirmed knockout of BECN1 in liver by gene expression analysis (Figure S4A). ADI-PEG 20 again decreased total body mass and fat percentage, increased lean mass percentage, and trended to increase O2-CO2 exchange and caloric expenditure in both WT and BECN1 LKO mice (Figures 5B–5E) without changes in respiratory exchange ratios or locomotion (Figures S4B and S4C). ADI-PEG 20 significantly decreased glucose and insulin tolerance and fasting insulin in WT mice (Figures 5F–5H). ADI-PEG 20-mediated improvements in glucose tolerance occurred independently of BECN1 (Figure 5F), but BECN1 deficiency abrogated ADI-PEG 20 effects on insulin tolerance and fasting insulin levels (Figures 5G and 5H). Similarly, serum TGs, TC, LDL-C, and NEFA were decreased in drug-treated WT mice, and this effect was attenuated in BECN1 LKO mice (Figures 5I–5L).
Figure 5.

Hepatic autophagy is necessary for ADI-PEG 20-mediated therapeutic effects in WD-fed mice
(A) Schematic of experimental design used to test the role of ADI-PEG 20 in WD-fed Becn1 LKO mice.
(B–D) Body weight (B), body fat (C), and lean mass (D) composition in vehicle- and ADI-PEG 20-treated Becn1 LKO mice (n = 9, 10, 5, 5 mice per group).
(E) Whole-body oxygen consumption (VO2), carbon dioxide (VCO2), and energy expenditure during light and dark cycle (shaded area) in vehicle- and ADI-PEG 20-treated Becn1 LKO mice.
(F and G) Intraperitoneal tolerance tests for insulin (ITT, F) and for glucose (GTT, G).
(H) Serum insulin in vehicle- and ADI-PEG 20-treated Becn1 LKO mice.
(I–L) Serum triglyceride (I), cholesterol (J), non-esterified fatty acid (K), and low-density lipoprotein cholesterol (L) in vehicle- and ADI-PEG 20-treated Becn1 LKO mice.
(M) Serum ALT in vehicle- and ADI-PEG 20-treated Becn1 LKO mice.
(N–P) Triglyceride (N), cholesterol (O), and non-esterified fatty acid (P) contents in the livers of vehicle- and ADI-PEG 20-treated Becn1 LKO mice.
(Q and R) Liver sections stained with hematoxylin and eosin (H&E, Q) with steatotic area (e.g., aparenchymal space) quantified (R). Scale bars, 100 μm.
(S) Serum FGF21 content in vehicle- and ADI-PEG 20-treated Becn1 LKO mice.
Data represented in mean ± SEM. Each data point represents an individual animal. Exact p values are shown. Statistical significance was determined using two-way ANOVA in (F) and (G). Unpaired two-tailed Student’s t test was used in (B)–(D), (H)–(P), (R), and (S).
Hepatic analysis revealed improved liver TGs, TC, and transaminases AST and ALT in WT but not BECN1 LKO liver (Figures 5M–5P). Similarly, BECN1 was dispensable for ADI-PEG 20-induced reductions in liver-weight-to-body-weight ratio (Figures S4D and S4E). This was not a ubiquitous lipid effect, however, as hepatic FFA content was suppressed in both ADI-PEG 20-treated WT and BECN1 LKO mice (Figure 5Q), independent of genotype. Hepatic lipid effects were corroborated by histologic analysis. ADI-PEG 20 reduced steatotic percentage area in WT but not BECN1 LKO mice (Figure 5R). Surprisingly, BECN1 deficiency reversed ADI-PEG 20 effects on serum and hepatic lipids, even though serum FGF21 was significantly elevated in ADI-PEG 20-treated BECN1 LKO mice relative to similarly treated WT mice (Figure 5S). No changes in hepatic stress markers GRP78 or ATF4/CHOP expression were observed (Figure S4F), confirming that the drug was not simply inducing autophagic flux and FGF21 release due to a somewhat non-specific ER stress response. Together with FGF21 loss-of-function data, data in BECN1-deficient mice indicate overall that FGF21 is necessary but not sufficient to exert the pleiotropic therapeutic actions of hepatocyte and systemic arginine catabolism.
Systemic autophagy mediates the metabolic effects of ADI-PEG 20
Arginine is sensed through the SLC38A9-CASTOR1/2 signaling complexes to activate mTORC1 and block autophagic flux during nutrient-replete conditions.29,30 On that basis, we demonstrated that arginine iminohydrolysis via ADI-PEG 20 and ADI expression induce hepatic LC3BII accumulation (Figures 2M and 3N). Yet ADI-PEG 20 retained multiple therapeutic effects in both WT and BECN1 LKO mice. This prompted the hypothesis that systemic autophagic flux mediates the breadth of ADI-PEG 20 metabolic action. To directly test the participation of hepatic and extrahepatic autophagic flux in mediating the effects of ADI-PEG 20, we subjected WT and BECN1-haploinsufficient mice to 12 weeks of WD, followed by 4-week vehicle or ADI-PEG 20 treatment (Figure 6A). Again, ADI-PEG 20 reduced body mass and fat mass and increased percentage lean mass in both WT and Becn1+/− mice (Figures 6B–6D). In contrast, ADI-PEG 20 increased insulin tolerance and reduced fasting insulin and glucose (Figures 6E–6G). Reductions in insulin tolerance and fasting glucose were attenuated in the absence of a haploid Becn1, although ADI-PEG 20-mediated reductions in fasting insulin remained BECN1 independent (Figure 6F). ADI-PEG 20 reduced fasting TGs, TC, NEFA, LDL-C, and glucose in WT mice (Figures 6H–6K). These effects were significantly reversed in ADI-PEG 20-treated Becn1+/− mice.
Figure 6.

Whole-body Becn1 Het abolishes the therapeutic effects of ADI-PEG 20 in WD-fed mice
(A) Schematic of experimental design used to test the role of ADI-PEG 20 in Becn1Het WD-fed mice.
(B–D) Body weight (B), body fat (C), and lean mass (D) percentage of composition of vehicle- and ADI-PEG 20-treated Becn1 Het mice (n = 7, 3, 7, and 9 mice per group).
(E) Intraperitoneal tolerance tests for insulin (ITT).
(F and G) Serum insulin (F) and serum glucose (G) in vehicle- and ADI-PEG 20-treated Becn1Het mice.
(H–K) Serum non-esterified fatty acid (H), low-density lipoprotein cholesterol (I), triglyceride (J), and cholesterol (K) in vehicle- and ADI-PEG 20-treated Becn1 Het mice.
(L and M) Serum ALT (L) and serum albumin (M) contents in vehicle- and ADI-PEG 20-treated Becn1 Het mice.
(N–P) Triglyceride (N), cholesterol (O), and non-esterified fatty acid (P) contents in the livers of vehicle- and ADI-PEG 20-treated Becn1 Het mice.
(Q and R) Liver sections stained with hematoxylin and eosin (H&E, Q) with steatotic (e.g., aparenchymal space) quantified (R). Scale bars, 100 μm.
(S and T) Targeted metabolomic analysis of liver (S) and serum (T) amino acids and urea cycle intermediaries from vehicle- and ADI-PEG 20-treated Becn1Het mice.
Data represented in mean ± SEM. Each data point represents an individual animal. Exact p values are shown. Statistical significance was determined using two-way ANOVA in (E). Unpaired two-tailed Student’s t test was used in (B)–(D) and (F)–(R).
In liver, ADI-PEG 20 again reduced hepatic TGs, TC, and NEFA as well as circulating ALT and AST (Figures 6L–6Q) with trends toward decreased liver weight and liver-body-weight ratio (Figures S5A and S5B). However, all effects required a full complement of Becn1, and ADI-PEG 20 failed to reduce any of these outcomes in Becn1+/− mice. We confirmed this histologically, as ADI-PEG 20 reduced percentage steatotic area in both WT and Becn1+/− mice, yet ADI-PEG 20-treated Becn1 haploinsufficient mice nevertheless had higher mean percentage steatotic area when compared with drug-treated WT controls (Figure 6R).
Similarly, ADI-PEG 20 broadly increased hepatic amino acid content and ornithine, urea, and citrulline content. These increases were uniformly muted in ADI-PEG 20-treated Becn1+/− mice (Figure 6S). In contrast, we did not observe consistent changes in serum amino acid levels (Figure 6T).
arcA and ADI-PEG 20 induced FGF21 and other gene-expression alterations. We thus tested the hypothesis that the therapeutic effects of ADI-PEG 20 associated with hepatocellular population-specific epigenetic alterations in chromatin accessibility. We performed single-cell ATAC sequencing in livers from WT and Becn1+/− mice on a 12-week WD followed by 4-week ADI-PEG 20 treatment. Of the nine distinct clusters defined, we differentiated hepatocyte populations from endothelial, Ito, leukocyte, and cholangiocyte cell types based on key hepatocyte markers. This included albumin, Hnf4a, Hnf1a, and FoxA1 (Figures 7A–7C). Based on this separation, we noted that ADI-PEG 20 reduced the inflammatory macrophage population in WT but not in Becn1+/− mice (Figure 7D). Overall, pseudo-bulk PCA analysis revealed broad separation of accessibility in the chromatin landscape of WT mice treated with ADI-PEG 20, whereas untreated WT mice clustered indistinguishably with treated and untreated Becn1+/− mice (Figure 7E). UMAP analysis further demonstrated most prominent separation of hepatocyte chromatin accessibility in WT but not Becn1+/− mice hepatocytes (Figures 7F and 7G). Chromatin changes induced by ADI-PEG 20 were thus almost completely dependent on BECN1, particularly in the hepatocyte population. More detailed comparison of vehicle- and ADI-PEG 20-treated hepatocyte populations demonstrated significant reductions in peaks in encoding regions along chromosome 18 (Figure 7H, Cidea), chromosome 11 (Figure 7I, Fasn), chromosome 19 (Figure 7J, Gpam), chromosome 4 (Figure 7K, Mtor), and chromosome 11 (Figure 7L, Gpx3). In contrast, increased chromatin accessibility was demonstrated in ADI-PEG 20-treated WT mice along chromosome 1 near the ureagenic gene Cps1 (Figure 7M). Each of these changes in chromatin structure was BECN1 dependent. Together, in vivo genetic data indicate that systemic autophagic flux via BECN1 mediate the insulin-sensitizing, dyslipidemic, chromatin-restructuring, and anti-steatotic effects of ADI-PEG 20.
Figure 7.

Single-cell ATAC sequencing reveals alterations in the hepatocyte-selective chromatin accessibility landscape upon systemic arginine deprivation
(A) UMAP projection of 40773 liver cells from scATAC-seq where cells that share similar chromatin accessibility landscape are grouped through unsupervised clustering. Each point represents a single cell captured.
(B) Dot-plot analysis of known cell-specific marker gene expression used to assign identity to the clusters.
(C) UMAP visualization of the clusters showing the assigned identity for each cell type identified.
(D) The proportion of cells that contributes to each cell type by each liver sample.
(E) Psuedobulk principle component analysis of all scATAC-seq samples. The first two principal components (PCs) are plotted. Variance proportions are shown along each component axis. The plot model 77% of the total data variance.
(F) UMAP visualization of cell clusters split by genotype between wild-type (WT) and Becn1+/− (Becn1Het).
(G) UMAP visualization of cell clusters split by treatment of vehicle and ADI-PEG 20 between genotypes.
(H–M) Hepatocyte-specific chromatin landscapes are shown for genes Cidea (H), Fasn (I), Gpam (J), Mtor (K), Gpx3 (I), and Cps1 (M).
Discussion
Treatments for obesity and its broad sequelae currently fall short of ideal in their number and breadth of mechanism. Here, we demonstrate a target process, arginine catabolism, and commandeer a naturally occurring bacterial virulence factor to leverage this process. Specifically, we show that the high-affinity arginine deiminase,22 arcA, enhances host arginine catabolism to activate key hepatocyte fasting-like signals and systemic autophagic flux to ameliorate multiple metabolic complications in obese mice. We then nominate ADI-PEG 20 to drive these processes exogenously. Finally, we combine in vivo pharmacology, mouse genetics, and advanced single-cell-based approaches to delineate an autophagic flux- and FGF21-dependent mechanism that shares common intermediaries with responses generalized fasting, and the canonical hepatocyte glucose fasting responses.7,17,18,31, 32, 33, 34
Arginine “deprivation” generally, and ADI-PEG 20 specifically, have demonstrated utility and safety in clinical35, 36, 37 (NCT03922880) and pre-clinical38, 39, 40, 41, 42 contexts targeting tumor metabolism. Indeed, the majority of reports indicate that arginine deprivation attenuates growth in multiple tumor types,43 including among others, breast,41 prostate,44 pancreatic,45 and liver46,47 tumors that specifically lack the rate-limiting arginine biosynthetic enzyme, argininosuccinate synthetase 1 (ASS1). Yet recent evidence surprisingly suggests that ASS1 deficiency is common also to livers from patients with obesity, simple steatosis, and non-alcoholic steatohepatitis.48 Consistent with these findings, experimental NASH models also exhibit impaired hepatic ureagenesis.49 Therefore, the steatotic and inflamed liver and the hepatic tumor share a degree of arginine dependence to fuel distinct energy-dependent pathophysiologies. A key difference, however, is that tumors have high proliferative energy requirements due to increased pyrimidine biosynthesis, fueled by aspartate shunting in the absence of ASS1.50 This sensitizes ASS1-deficient tumors to apoptotic death upon arginine deprivation.42 On the other hand, the non-proliferative, steatotic hepatocyte exhibits greater substrate plasticity, which renders it amenable to non-lethal arginine deprivation. This instead results in hepatocyte-directed adaptations that coordinate hepatic and extrahepatic compensation. This is possible, in large part, because hepatocytes possess compensatory autophagic and fasting autocrine and paracrine signaling to rectify nutrient deficiencies cell-autonomously, and direct extrahepatic tissues to aid in this compensation. For example, we show that hepatocytes secrete FGF21 in response to ADI-PEG 20 treatment, the overall purpose of which is to enhance peripheral insulin sensitivity and drive lipid catabolism in response to an apparent fasting-like state.20,51,52
These concepts of arginine-dependent metabolism may, in part, explain the exquisite and targeted sensitivity of the steatotic liver to both hepatocyte-directed and systemic arginine perturbations. This both clinically important and expedient for at least two reasons. First, these data demonstrate that the intersection of its arginine sensitivity and the homeostatic plasticity of the liver during arginine deprivation may facilitate the broad therapeutic effects of ADI-PEG 20. Second, nutrient sensing functions of the liver permit systemic arginine targeting with effects that extend those observed in hepatocyte-specific targeting.
L-arginine is a semi-essential amino acid and is one of the most versatile amino acids with multiple competing metabolic fates in the liver. This raises the possibility that long-term perturbation could negatively impact other aspects of metabolic function. We cannot definitively rule out this possibility. Although precise time span over which arginine depletion exerts metabolic benefits remains unclear, our prior studies indicate quite durable 12- to 14-week efficacy of hepatocyte-specific Arg2 overexpression in vivo.16 In the current study, mice were treated with ADI-PEG 20 for up to 5 weeks without apparent adverse effects. Similarly, two human trials treated patients with ADI-PEG 20 between 8 and 41 weeks. All of these patients were evaluated for ADI-PEG 20-treatment-induced toxicities. Overall, the toxicity was found to be <5%.37 A more recent clinical study in patients with advanced hepatocellular carcinoma and other gastrointestinal malignancies, 24 weeks of arginine depletion was sustained in patients with no predominant safety signal observed other than hematologic toxicity. All cases were resolved and manageable.53 Moreover, human studies indicate rapid and persistent arginine depletion up to 18 weeks after initial dosing,54 and an ongoing phase 3 trial will define ADI-PEG 20 durability through up to 103 weeks after initial dosing (NCT03449901). The current evidence does not suggest obvious deleterious metabolic effects of chronic arginine deprivation. Similarly, data on effects of long-term L-arginine supplementation are conflicting in both pre-clinical and clinical studies,23,55, 56, 57, 58 Taken together, the data do not reveal negative metabolic sequelae from chronic modulation of nitrogen status. What is clear is the net metabolic benefit at least of short-term arginine manipulation. Nevertheless, we anticipate that the hepatocyte epigenomic alterations we observed in association with the metabolic effects of ADI-PEG 20 extend the effective durability of arginine depletion beyond dosing of the drug. The precedent for this phenomenon is the well-known epigenetic and durable effects of IF and CR on host metabolism.59, 60, 61 Yet independent of its durability, the clinical utility of even short-term arginine-targeting therapy remains. Here, we directly modeled arcA efficacy in a leptin-melanocortin pathway signaling-deficient db/db diabetic model.62 This by itself may address an unmet need that exists in treating monogenic and other refractory obesity, wherein acute weight loss preceding bariatric surgery optimizes surgical outcomes. This currently represents one unique challenge that even short-term metabolic therapy could address.
A second potential limitation of this approach is that ADI-PEG 20 does not clearly invoke a single, linear cascade to produce its effects. It appears that cell-autonomous autophagic, epigenetic, and autocrine/endocrine effects of this therapy are all contributory to its effect, akin to a broad stimulus, such as fasting, caloric restriction, or carbohydrate restriction. Furthermore, current data demonstrate that BECN1, a canonical autophagic mediator protein, may have pleiotropic functions that go beyond autophagic flux. This includes functions such as vesicular sorting, autophagy-dependent cell death, centrosome functions, cytokinesis, and vision cycle.63, 64, 65 On this basis, we acknowledge the potentially much broader functions of BECN1 that may indicate as yet unidentified processes mediating the therapeutic effects of forced arginine catabolism.
In contrast, more targeted therapies, such as melanocortin receptor agonists, act on a cognate receptor with well-defined action.62,66 Regardless of mechanism, however, this pleiotropism may ultimately prove to be a primary strength on two bases. First, it is likely that most obesity is a common final manifestation of multiple signaling, carbon flux, and autophagic defects that culminate in a common, gross phenotype. Second, perturbations in linear pathways, at least historically using leptin as the exemplar, have proved to permit homeostatic compensation to attenuate these therapeutic effects.67 We postulate that adaptation to the broader perturbation of arginine deprivation may attenuate the degree to which compensatory mechanisms can attenuate its therapeutic effects. And whereas we do not yet directly demonstrate the connection between arginine turnover and the requirement for fasting-like signals (FGF21, autophagic flux) in the ADI-PEG 20 response, we hope to highlight in this work the concept of arginine deprivation as a tractable therapeutic pathway that mediates its effects through canonical fasting-like response pathways.
Thus, overall, we have demonstrated that systemic and hepatocyte-directed arginine deprivation is sufficient to induce adaptive hepatocyte fasting-like responses, and we introduce a readily available pharmacotherapy that leverages this pathway. The data justify arginine catabolism as a target pathway in treating metabolic disease, and in light of precedent safety and efficacy data in patients over the course of a decade in clinical use, the data justify the use of ADI-PEG 20 therapy to drive therapeutic arginine catabolism in human clinical trials against obesity and its metabolic sequelae.
Limitations of the study
There are two major limitations of the current study. First, the long-term effects of exogenous systemic arginine catabolism remain incompletely characterized. Second, the current study is performed in murine models, and thus further examination must define the extent to which the mechanisms of arginine catabolism translate to human therapy.
STAR★Methods
Key resources table
| Reagent or resource | Source | Identifier |
|---|---|---|
| Antibodies | ||
| FGF21 | Abcam | Cat# ab171941; RRID:AB_2629460 |
| β-ACTIN | Cell Signaling Technology | Cat# 3700S; RRID:AB_2242334 |
| LC3B | Novus Biologicals | Cat# NB100-2220; RRID:AB_10003146 |
| p62/SQSTM1 | Abcam | Cat# ab56416; RRID:AB_945626 |
| GHRL | Santa Cruz | Cat# sc-517596; RRID: NA |
| phospho-mTOR (Ser2448) | Cell Signaling Technology | Cat# 5536S; RRID:AB_10691552 |
| mTOR | Cell Signaling Technology | Cat# 2983S; RRID:AB_2105622 |
| phospho-ULK1 (Ser757) | Cell Signaling Technology | Cat# 14202S; RRID:AB_2665508 |
| ULK1 | Cell Signaling Technology | Cat# 8054S; RRID:AB_11178668 |
| phospho-p70 S6 Kinase (Thr389) | Cell Signaling Technology | Cat# 9234S; RRID:AB_2269803 |
| p70 S6 Kinase | Cell Signaling Technology | Cat# 2708S; RRID:AB_390722 |
| phospho-4E-BP1 (Thr37/46) | Cell Signaling Technology | Cat# 2855S; RRID:AB_560835 |
| 4E-BP1 | Cell Signaling Technology | Cat# 9644S; RRID:AB_2097841 |
| Horse Anti-Mouse IgG-HRP | Cell Signaling Technology | Cat# 7076S; RRID:AB_330924 |
| Goat Anti-Rabbit IgG-HRP | Cell Signaling Technology | Cat# 7074S; RRID:AB_2099233 |
| Bacterial and virus strains | ||
| AAV8-eGFP | Vector Biolabs | Cat# 1060 |
| AAV8-TGB-arcA | Vector Biolabs | NA |
| Ad-CMV-eGFP | Vector Biolabs | NA |
| Ad-CMV-arcA-eGFP | Vector Biolabs | NA |
| Biological samples | ||
| Fetal Bovine Serum (FBS) | GIBCO | Cat# 26140-079 |
| Chemicals, peptides, and recombinant proteins | ||
| ADI-PEG 20 | Polaris Pharmaceuticals Inc. | Kit# 36386 |
| TRIzol | Invitrogen | Cat# 15596018 |
| 0.9% Sodium Chloride Injection, USP | Hospira | |
| Fast SYBR Green Master Mix | Applied Biosystems | Cat# 4385612 |
| Pierce Protease and Phosphatase Inhibitor Mini Tablets, EDTA-Free | Thermo Scientific | Cat# A32961 |
| 10X TBST | EZ BioResearch | Cat# S-1012 |
| 10X Tris/Glycine Buffer | Bio-Rad | Cat# 1610734 |
| 10X Tris/Glycine/SDS Buffer | Bio-Rad | Cat# 1610732 |
| Mini-PROTEAN TGX Stain-Free Gels | Bio-Rad | Cat# 4568094 |
| Clarity Western ECL Substrate | Bio-Rad | Cat# 1705060 |
| Humulin R | Lilly USA, LLC | NDC 0002-8215-17 |
| Ultra Sensitive Mouse Insulin ELISA Kit | Crystal Chem | Cat# 90080 |
| Mouse/Rat Fibroblast Growth Factor 21 ELISA | BioVendor | Cat# RD291108200R |
| Glucose Colorimetric Assay Kit | Cayman Chemical | Cat# 10009582 |
| Triglycerides Standard | Pointe Scientific | Cat# T7531-STD |
| Infinity Triglycerides | Thermo Scientific | Cat# TR22421 |
| Cholesterol Standard | Stanbio | Cat# 1012-030 |
| Infinity Cholesterol | Thermo Scientific | Cat# TR13421 |
| NEFA Standard Solution | Fujifilm | Cat# 276-76491 |
| HR Series NEFA-HR(2) Color Reagent A | Fujifilm | Cat# 999-34691 |
| HR Series NEFA-HR(2) Solvent A | Fujifilm | Cat# 995-34791 |
| HR Series NEFA-HR(2) Color Reagent B | Fujifilm | Cat# 991-34891 |
| HR Series NEFA-HR(2) Solvent B | Fujifilm | Cat# 993-35191 |
| HDL-C/LDL-C Calibrator | Fujifilm | Cat# 990-28011 |
| L-Type LDL-C Reagent 1 | Fujifilm | Cat# 993-00404 |
| L-Type LDL-C Reagent 2 | Fujifilm | Cat# 999-00504 |
| Albumin from Bovine Serum | Sigma-Aldrich | SKU A3983-50G |
| 2-Propanol | Sigma-Aldrich | SKU 190764-500ML |
| Methanol | Sigma-Aldrich | SKU 179337-4L-PB |
| Chloroform | Sigma-Aldrich | SKU 319988-500ML |
| Ethanol | Decon Laboratories, Inc. | Cat# 2701 |
| D-(+)-Glucose | Sigma-Aldrich | SKU G8270-100G |
| DMEM/F12(1:1) (1X) | GIBCO | Cat# 11330-032 |
| Insulin-Transferrin-Selenium 100X | GIBCO | Cat# 41400-045 |
| Dexamethasone | Sigma-Aldrich | SKU D4902-100MG |
| Pen Strep | GIBCO | Cat# 15140-122 |
| Rodent Diet: Adjusted Calories Diet (42% from fat) | Envigo Teklad Diets | Cat# TD.88137 |
| Nuclease-Free Water | Invitrogen | Cat# AM9937 |
| Critical commercial assays | ||
| Seahorse XF Cell Mito Stress Test Kit | Agilent | Cat# 103015-100 |
| QuantiTect Reverse Transcription Kit | QIAGEN | Cat# 205314 |
| Deposited data | ||
| Gene Expression RNA-seq data | This paper | GEO: GSE191295 |
| Single-cell ATAC-seq data | This paper | GEO: GSE192413 |
| Experimental models: Cell lines | ||
| Mouse: AML12 | ATCC | Cat# CRL-2254; RRID:CVCL_0140 |
| Experimental models: Organisms/strains | ||
| Mouse: C57B/J6 | Jackson Laboratory | RRID: IMSR_JAX:000664 |
| Mouse: db/db | Jackson Laboratory | RRID: IMSR_JAX:000642 |
| Mouse: Becn1+/− | Jackson Laboratory | RRID: IMSR_JAX:018429 |
| Mouse: Becn1 flox | Jackson Laboratory | RRID: IMSR_JAX:028794 |
| Mouse: Alb1-cre | Jackson Laboratory | RRID: IMSR_JAX:016832 |
| Mouse: Fgf21 flox | Jackson Laboratory | RRID: IMSR_JAX:022361 |
| Oligonucleotides | ||
| qPCR Primers | This paper | Table S1 |
| Software and algorithms | ||
| GraphPad Prism 7 | GraphPad Software Inc. | http://www.graphpad.com; RRID:SCR_002798 |
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/; RRID:SCR_003070 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Brian DeBosch (deboschb@wustl.edu).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Mice, diets, and treatments
All animal protocols were approved by the Washington University School of Medicine Animal Studies Committee. Male C57B/6J mice, db/db mice, Becn1 heterozygous (Becn1+/−) mice, Becn1fl/fl mice, and Fgf21fl/fl mice (mouse strains 000664, 000642, 018429, 028794, and 022361 respectively) were purchased directly from the Jackson Laboratory (Bar Harbor, ME). Liver specific knockout mice of Becn1−/− and Fgf21−/− were generated from Becn1fl/fl mice and Fgf21fl/fl mice which were bred with Alb1-Cre transgenic mice from Jackson Laboratory (mouse strain 016832).
All strains of genetically altered mice were on a C57BL/6J background. Control mice were negative for Cre recombinase and matched by genetic background, age, and sex. All animals were housed at the Washington University Medical School in St. Louis in a 12-h alternating light-dark, temperature-controlled, specific pathogen-free barrier facility prior to and throughout experimentation.
All animals received humane care and procedures were performed in accordance with the approved guidelines by the Animal Studies Committee at Washington University School of Medicine. All animal studies were performed in accordance with the criteria and ethical regulations outlined by the Institutional Animal Care and Use Committee (IACUC).
Five-week-old mice were fed ad libitum: a normal chow diet (NCD) or a Western diet (WD) (TD.88137: 42% kcal fat; Envigo Teklad Diets, Madison, WI, USA) for 16 weeks. All animals received non-supplemented drinking water.
For ADI-PEG 20 studies, five-week-old male mice were fed a normal chow diet (NCD) or a Western diet (TD.88137, Envigo Teklad Diets, Madison, WI, USA) for 12 weeks prior to treatment and then ADI-PEG 20 (Polaris Pharmaceuticals, Inc., San Francisco, CA, USA) treatment started for 4 weeks while continuously on the same diet. Once per week, 5IU/mouse of ADI-PEG 20 was administered through intraperitoneal injection. At the end of the experiment, animals were sacrificed, and the liver, fat and serum were harvested for subsequent analysis.
Cell cultures and treatment
α mouse liver 12 (AML12) cells were purchased from American Type Culture Collection (ATCC) [CRL-2254; Research Resource Identifier (RRID): CVCL_0140] and maintained per American Type Culture Collection guidelines. AML12 cells were cultured in Dulbecco’s Modified Eagle Medium high glucose/Ham’s F12 (DMEM-F12 (1:1) (1X), GIBCO) and supplemented with 10% fetal bovine serum (FBS, GIBCO), 40 ng/mL dexamethasone (Sigma Aldrich), 10 μg/mL insulin, 5.5 μg/mL transferrin, and 5 ng/mL selenium (GIBCO), and 1% penicillin/streptomycin/fungizone (GIBCO). AML12 cells were propagated in tissue culture treated 10 cm dishes (TPP). All cell lines were seeded at > 95% viability.
Method details
AAV8- and adenovirus-mediated overexpression
Serotype 8 AAV (AAV8) was administered via tail vein as we previously reported.16 The arcA viral vectors (AAV8-TBG-arcA and Ad-arcA) were obtained directly from Vector Biolabs Inc (Malvern, PA, USA).
Intraperitoneal glucose tolerance test
Intraperitoneal glucose tolerance tests were carried out on mice fasted for 6 hours on aspen bedding. Basal blood glucose concentrations were determined for each mouse prior to glucose administration using a hand-held glucose meter (Arkray USA, Inc., Minneapolis, MN, USA). Each mouse then received 2g per kg body weight of glucose, except for db/db mice, which received 1 g per kg body weight of glucose through intraperitoneal injection and blood glucose concentrations were subsequently measured at 30, 60, 90 and 120 minutes post glucose administration.
Intraperitoneal insulin tolerance test
Intraperitoneal insulin tolerance tests were carried out on mice fasted for 4 hours on aspen bedding. Basal blood glucose concentrations were determined for each mouse prior to insulin administration using a hand-held glucose meter (Arkray USA, Inc., Minneapolis, MN, USA). Each mouse then received 0.75 IU per kg body weight of insulin (Lilly USA, LLC Indianapolis, IN, USA) through intraperitoneal injection and blood glucose concentrations were subsequently measured at 30, 60, 90 and 120 minutes post insulin administration.
Clinical chemistry measurements and hepatic lipid analyses
For all other serum analyses, submandibular blood collection was performed immediately prior to sacrifice and serum was separated. Insulin ELISA (Millipore #EZRMI-13K), triglycerides (Thermo Fisher Scientific #TR22421), cholesterol (Thermo Fisher Scientific #TR13421), and free fatty acids (Wako Diagnostics #999-34691, #995-34791, #991-34891, #993-35191) quantification were performed using commercially available reagents according to manufacturer’s directions. Albumin levels were quantified using an AMS LIASYS Chemistry Analyzer.
Measurement of liver triglycerides
Liver-specific lipids were extracted and analyzed from snap frozen liver tissue samples. ∼50 mg hepatic tissue samples were homogenized in 2:1 chloroform:methanol. In total, 0.25%–0.5% of each extract was evaporated overnight prior to biochemical quantification of triglycerides, cholesterol, and free fatty acids (FFA) using reagents described above, precisely according to manufacturer’s directions.
Body composition analysis
Body composition analysis was carried out in unanesthetized mice using an EchoMRI 3-1 device (Echo Medical Systems) via the Washington University Diabetic Mouse Models Phenotyping Core Facility.
Indirect calorimetry and food intake measurement
All measurements were performed in a PhenoMaster System (TSE systems) via the Washington University Diabetic Mouse Models Phenotyping Core Facility, which allowed metabolic performance measurement and activity monitoring by an infrared light = beam frame. Mice were placed at room temperature (22–24 °C) in separate chambers of the PhenoMaster open-circuit calorimetry. Mice were allowed to acclimatize in the chambers for 4 h. Food and water were provided ad libitum in the appropriate devices. The parameters of indirect calorimetry (VO2, VCO2, respiratory exchange ratio (RER), heat and movement) were measured for at least 24 h for a minimum of one light cycle (6:01 am to 6:00 pm) and one dark cycle (6:01 pm to 6:00 am). Presented data are average values obtained in these recordings.
Quantitative real-time RT-PCR
Total RNA was prepared by homogenizing snap-frozen livers or cultured hepatocytes in Trizol reagent (Invitrogen #15596026) according to the manufacturer’s protocol. cDNA was prepared using QIAGEN Quantitect reverse transcriptase kit (QIAGEN #205310). Real-time qPCR was performed with Step-One Plus Real-Time PCR System (Applied Biosystems) using SYBR Green master Mix Reagent (Applied Biosystems) and specific primer pairs. Relative gene expression was calculated by a comparative method using values normalized to the expression of an internal control gene.
Immunoblotting
Tissues were homogenized in RIPA lysis buffer (50mM Tris, 1% NP-40, 0.1% SDS, 0.5% Sodium Deoxycholate, 150 mM NaCl, pH 8.0) supplemented with protease and phosphatase inhibitors (Thermo Scientific). After homogenization, lysate was centrifuged at 18,000 g for 15 min at 4C, and the supernatant was recovered. Protein concentration was determined by BCA Assay Kit (Thermo Scientific) and was adjusted to 2mg/mL. Samples for western blotting were prepared by adding Laemmli buffer at a ratio of 1:1 and heating at 95 C for 5 min. The prepared samples were subjected to 10% or 13% SDS-PAGE, followed by electrical transfer onto a nitrocellulose membrane using the Trans-Blot Turbo system (Bio-Rad). After blocking the membrane with 5% milk in TBST, the membrane was incubated in primary antibody at 4°C overnight. The blot was developed after secondary antibody incubation using Pierce ECL Western Blotting Substrate (Thermo Scientific). Blots were developed according to the manufacturer’s instructions. Protein expression levels were quantified with ImageJ Lab software and normalized to the levels of β-Actin.
Antibodies
Antibodies against FGF21 (Abcam Cat. # ab171941), SQSTM1/p62 (Abcam Cat. # ab56416), LC3B (Novus Biologicals Cat. # NB100-2220), and β-Actin (Cell Signaling Cat. # 3700S). The dilution ratio for all primary antibodies was 1:1,000. The secondary antibodies used in this study were peroxidase-conjugated anti-rabbit IgG (Cell Signaling Cat. # 7074S) and anti-mouse IgG (Cell Signaling Cat. # 7076S) were purchased from Cell Signaling Technology (CST) (Beverly, MA, USA), in which were used at a 1:5,000 dilution.
Histological analysis
Formalin-fixed paraffin-embedded liver sections were stained by H&E via the Washington University Digestive Diseases Research Core Center. OCT-embedded frozen liver sections were stained by Oil Red O according to standard protocols flowered by microscopic examination. Three liver sections were examined and evaluated for each animal. For Oil red-O staining, ice-cold methanol-fixed frozen sections from mice were stained according to described protocols.11,12,68
RNA-seq
RNA-seq was performed by the Washington University Genome Technology Access Center (GTAC). Library preparation was performed with 10uG of total RNA with a Bioanalyzer RIN score greater than 8.0. Ribosomal RNA was removed by poly-A selection using Oligo-dT beads (mRNA Direct kit, Life Technologies). mRNA was then fragmented in buffer containing 40mM Tris Acetate pH 8.2, 100mM Potassium Acetate and 30mM Magnesium Acetate and heating to 94 degrees for 150 s. mRNA was reverse transcribed to yield cNDA using SuperScript III RT enzyme (Life Technologies, per manufacturer’s instructions) and random hexamers. A second strand reaction was performed to yield ds-cDNA. cDNA was blunt ended, had an A base added to the 3′ ends, and then had Illumina sequencing adapters ligated to the ends. Ligated fragments were then amplified for 12 cycles using primers incorporating unique index tags. Fragments were sequenced on an Illumina HiSeq-3000 using single reads extending 50 bases.
RNA-seq reads were aligned to the Ensembl release 76 top-level assembly with STAR version 2.0.4b. Gene counts were derived from the number of uniquely aligned unambiguous reads by Subread:featureCount version 1.4.5. Transcript counts were produced by Sailfish version 0.6.3. Sequencing performance was assessed for total number of aligned reads, total number of uniquely aligned reads, genes and transcripts detected, ribosomal fraction known junction saturation and read distribution over known gene models with RSeQC version 2.3.
To enhance the biological interpretation of the large set of transcripts, grouping of genes/transcripts based on functional similarity was achieved using the R/Bioconductor packages GAGE and Pathview. GAGE and Pathview were also used to generate pathway maps on known signaling and metabolism pathways curated by KEGG.
SC-ATAC sequencing
Tissues were harvested and frozen samples were sent to Active Motif to perform the scATAC-seq assay. Tissues were prepared as described by 10X Genomics Demonstrated Protocol – Nuclei Isolation from Mouse Brain Tissue for Single Cell ATAC Sequencing Rev B with some modifications. Briefly, tissues were minced in ice cold lysis buffer followed by dounce homogenization and incubated on ice for 10 minutes. Lysate was strained, washed, and nuclei were resuspended and counted using a Countess II FL Automated Cell Counter. Isolated nuclei were then used as input following the 10X Genomics Chromium Next GEM Single Cell ATAC Reagent Kits v1.1 manual. Targeting a 5,000 nuclei recovery, samples were added to the tagmentation reaction, loaded into the Chromium Controller for nuclei barcoding, and prepared for library construction following manufacturer’s protocol (10X Genomics PN-1000175). Resulting libraries were quantified using the KAPA Library Quantification Kit for Illumina platforms (KAPA Biosystems), and sequenced with PE34 sequencing on the NextSeq 500/550 sequencer (Illumina).
Sequenced data were processed with the Cell Ranger ATAC software, with alignment to the mouse (mm10) genome. The Cell Ranger output files were used as input to Active Motif’s proprietary analysis program, which creates Excel tables containing detailed information on cluster-specific peak locations, gene annotations, and motif enrichment.
The alignment files generated by Cell Ranger were also processed as pseudo-bulk ATAC-Seq samples. Duplicate reads were removed, only reads mapping as matched pairs and only uniquely mapped reads (mapping quality ≥ 1) were used for further analysis. Alignments were extended in silico at their 3′ ends to a length of 200 bp and assigned to 32-nt bins along the genome. The resulting histograms (genomic “signal maps”) were stored in bigWig files. Peaks were identified using the MACS 2.1.0 algorithm at a cutoff of p value 10−7, without control file, and with the –nomodel option. Peaks that were on the ENCODE blacklist of known false ChIP-Seq peaks were removed. Signal maps and peak locations were used as input data to Active Motif’s proprietary analysis program, which creates Excel tables containing detailed information on sample comparison, peak metrics, peak locations, and gene annotations.
Targeted metabolomics
We performed targeted metabolomics as reported with minor modifications.69 Briefly, the liver samples were homogenized in water (4 mL/g liver). The amino acids in 20 μL of mouse serum or liver homogenate were extracted with protein precipitation in the presence of internal standards (13C6,15N-Ile, d3-Leu, d8-Lys, d8-Phe, d8-Trp, d4-Tyr, d8-Val, d7-Pro, 13C4-Thr, d3-Met, d2-Gly, 15N2-Asn, d4-Cit, d3-Asp, 13C5-Gln, 13C6-His, d3-Glu, d4-Ala, d3-Ser, 13C5-Orn, and 13C6-Arg). Quality control (QC) samples for livers and sera were prepared from pooled partial study samples and injected every 5 study samples to monitor intra-batch precision. Only the lipid species with CV% < 15% for QC injections are reported. The Ile, Leu, Lys, Phe, Trp, Tyr, Val, Pro, Thr, Met, Gly, Asn, Cit, Asp, Gln, His, Glu, Ala, Ser, Orn, and Arg were analyzed on 4000 QTRAP mass spectrometer coupled with a Prominence LC-20AD HPLC system. Data processing was conducted with Analyst 1.5.1 (Applied Biosystems).
Extracellular flux analysis
In vitro respiration measurements were performed using the Seahorse xFE96 Analyzer (Agilent) with the AML12 immortalized mouse hepatocyte cell line. Cells were seeded to near confluency. Cells were treated with adenoviruses, Ad-eGFP (Control) or Ad-arcA, for 24 hours in regular media, and subjected to fresh media for an additional 24 hours prior to analysis. The Seahorse Mito Stress Test kit (Agilent) was used according to manufacturer instructions.
Quantification and statistical analysis
Statistical analyses
Data were analyzed using GraphPad Prism version 7.05 (RRID:SCR_015807). p < 0.05 was defined as statistically significant. Data shown are as mean ± SEM. Unpaired 2-tailed homoscedastic t tests with Bonferroni post hoc correction for multiple comparisons were used for all analyses unless otherwise noted in the Figure Legends. Two-way ANOVA was also used for analyses with two independent variables.
Acknowledgments
This work was supported by grants from the NIDDK (1R01DK126622-01A1), NHLBI (1R01HL147968-01A1), AASLD (Pilot Research Award), NCCIH (1R21AT010520-01), NIH/National Center for Advancing Translational Sciences (NCATS, #UL1TR002345), NIH R56 (DK115764), AGA-Gilead Sciences Research Scholar Award in Liver Disease, the AGA-Allergan Foundation Pilot Research Award in Non-Alcoholic Fatty Liver Disease, the Washington University Digestive Disease Research Core Center (P30DK52574), Washington University Diabetes Research Center (P30DK020579), the Nutrition & Obesity Research Center (P30DK056341), The Association for Aging Research Junior Faculty Award, the Robert Wood Johnson Foundation, Washington University Center for Autophagy Therapeutics Research, and the Longer Life Foundation. Y.Z. is a predoctoral student supported by the Washington University School of Medicine Pediatric Gastroenterology Research Training Grant (NIDDK, T32DK077653).
Author contributions
B.J.D. conceived and coordinated the study. B.J.D. and Y.Z. wrote the paper. Y.Z., C.B.H., and B.J.D. designed, performed, and analyzed the experiments. B.V.T. coordinated, performed, and analyzed metabolomics experiments. J.S.B. coordinated ADI-PEG 20 experiments and analyzed the data. All authors reviewed the results and approved the final version of the manuscript.
Declaration of interests
Part of this study was funded by a sponsored research agreement awarded by Polaris Pharmaceuticals (to B.J.D.). B.J.D. is the lead inventor on US Patent Application #17/050,318. Relevant US Patent Publication #US2021/0077598, toward which the presented data are material. J.S.B. is an employee of Polaris Pharmaceuticals, Inc.
Published: January 18, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2021.100499.
Supplemental information
Data and code availability
-
•
Bulk RNA-seq data and single-cell ATAC-seq data have been deposited at the Gene Expression Omnibus (GEO) with accession codes GSE191295 and GSE192413, respectively, and are publicly available as of the date of publication. Accession numbers are also listed in the key resources table.
-
•
This paper does not generate custom code.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.
References
- 1.Blüher M. Obesity: global epidemiology and pathogenesis. Nat. Rev. Endocrinol. 2019;15:288–298. doi: 10.1038/s41574-019-0176-8. [DOI] [PubMed] [Google Scholar]
- 2.Saklayen M.G. The Global Epidemic of the Metabolic Syndrome. Curr. Hypertens. Rep. 2018;20:12. doi: 10.1007/s11906-018-0812-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wan R., Camandola S., Mattson M.P. Intermittent fasting and dietary supplementation with 2-deoxy-d-glucose improve functional and metabolic cardiovascular risk factors in rats. FASEB J. 2003;17:1133–1134. doi: 10.1096/fj.02-0996fje. [DOI] [PubMed] [Google Scholar]
- 4.Patterson R.E., Laughlin G.A., LaCroix A.Z., Hartman S.J., Natarajan L., Senger C.M., Martínez M.E., Villaseñor A., Sears D.D., Marinac C.R., Gallo L.C. Intermittent Fasting and Human Metabolic Health. J. Acad. Nutr. Diet. 2015;115:1203–1212. doi: 10.1016/j.jand.2015.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mattson M.P., Longo V.D., Harvie M. Impact of intermittent fasting on health and disease processes. Ageing Res. Rev. 2017;39:46–58. doi: 10.1016/j.arr.2016.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Cabo R., Mattson M.P. Effects of Intermittent Fasting on Health, Aging, and Disease. N. Engl. J. Med. 2019;381:2541–2551. doi: 10.1056/NEJMra1905136. [DOI] [PubMed] [Google Scholar]
- 7.Longo V.D., Panda S. Fasting, Circadian Rhythms, and Time-Restricted Feeding in Healthy Lifespan. Cell Metab. 2016;23:1048–1059. doi: 10.1016/j.cmet.2016.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wei M., Brandhorst S., Shelehchi M., Mirzaei H., Cheng C.W., Budniak J., Groshen S., Mack W.J., Guen E., Di Biase S., et al. Fasting-mimicking diet and markers/risk factors for aging, diabetes, cancer, and cardiovascular disease. Sci. Transl. Med. 2017;9:eaai8700. doi: 10.1126/scitranslmed.aai8700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mauro M., Taylor V., Wharton S., Sharma A.M. Barriers to obesity treatment. Eur. J. Intern. Med. 2008;19:173–180. doi: 10.1016/j.ejim.2007.09.011. [DOI] [PubMed] [Google Scholar]
- 10.DeBosch B.J., Chen Z., Finck B.N., Chi M., Moley K.H. Glucose transporter-8 (GLUT8) mediates glucose intolerance and dyslipidemia in high-fructose diet-fed male mice. Mol. Endocrinol. 2013;27:1887–1896. doi: 10.1210/me.2013-1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DeBosch B.J., Chen Z., Saben J.L., Finck B.N., Moley K.H. Glucose transporter 8 (GLUT8) mediates fructose-induced de novo lipogenesis and macrosteatosis. J. Biol. Chem. 2014;289:10989–10998. doi: 10.1074/jbc.M113.527002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DeBosch B.J., Heitmeier M.R., Mayer A.L., Higgins C.B., Crowley J.R., Kraft T.E., Chi M., Newberry E.P., Chen Z., Finck B.N., et al. Trehalose inhibits solute carrier 2A (SLC2A) proteins to induce autophagy and prevent hepatic steatosis. Sci. Signal. 2016;9:ra21. doi: 10.1126/scisignal.aac5472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang Y., Higgins C.B., Mayer A.L., Mysorekar I.U., Razani B., Graham M.J., Hruz P.W., DeBosch B.J. TFEB-dependent induction of thermogenesis by the hepatocyte SLC2A inhibitor trehalose. Autophagy. 2018;14:1959–1975. doi: 10.1080/15548627.2018.1493044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Higgins C.B., Zhang Y., Mayer A.L., Fujiwara H., Stothard A.I., Graham M.J., Swarts B.M., DeBosch B.J. Hepatocyte ALOXE3 is induced during adaptive fasting and enhances insulin sensitivity by activating hepatic PPARγ. JCI Insight. 2018;3:e120794. doi: 10.1172/jci.insight.120794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mayer A.L., Zhang Y., Feng E.H., Higgins C.B., Adenekan O., Pietka T.A., Beatty W.L., DeBosch B.J. Enhanced Hepatic PPARα Activity Links GLUT8 Deficiency to Augmented Peripheral Fasting Responses in Male Mice. Endocrinology. 2018;159:2110–2126. doi: 10.1210/en.2017-03150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang Y., Higgins C.B., Fortune H.M., Chen P., Stothard A.I., Mayer A.L., Swarts B.M., DeBosch B.J. Hepatic arginase 2 (Arg2) is sufficient to convey the therapeutic metabolic effects of fasting. Nat. Commun. 2019;10:1587. doi: 10.1038/s41467-019-09642-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang Y., DeBosch B.J. Using trehalose to prevent and treat metabolic function: effectiveness and mechanisms. Curr. Opin. Clin. Nutr. Metab. Care. 2019;22:303–310. doi: 10.1097/MCO.0000000000000568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang Y., Shaikh N., Ferey J.L., Wankhade U.D., Chintapalli S.V., Higgins C.B., Crowley J.R., Heitmeier M.R., Stothard A.I., Mihi B., et al. Lactotrehalose, an Analog of Trehalose, Increases Energy Metabolism Without Promoting Clostridioides difficile Infection in Mice. Gastroenterology. 2020;158:1402–1416. doi: 10.1053/j.gastro.2019.11.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Helsley R.N., Moreau F., Gupta M.K., Radulescu A., DeBosch B., Softic S. Tissue-Specific Fructose Metabolism in Obesity and Diabetes. Curr. Diab. Rep. 2020;20:64. doi: 10.1007/s11892-020-01342-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Flippo K.H., Potthoff M.J. Metabolic Messengers: FGF21. Nat. Metab. 2021;3:309–317. doi: 10.1038/s42255-021-00354-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Caldwell R.B., Toque H.A., Narayanan S.P., Caldwell R.W. Arginase: an old enzyme with new tricks. Trends Pharmacol. Sci. 2015;36:395–405. doi: 10.1016/j.tips.2015.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pols T., Singh S., Deelman-Driessen C., Gaastra B.F., Poolman B. Enzymology of the pathway for ATP production by arginine breakdown. FEBS J. 2021;288:293–309. doi: 10.1111/febs.15337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hadi A., Arab A., Moradi S., Pantovic A., Clark C.C.T., Ghaedi E. The effect of l-arginine supplementation on lipid profile: a systematic review and meta-analysis of randomised controlled trials. Br. J. Nutr. 2019;122:1021–1032. doi: 10.1017/S0007114519001855. [DOI] [PubMed] [Google Scholar]
- 24.Madrigal-Matute J., Cuervo A.M. Regulation of Liver Metabolism by Autophagy. Gastroenterology. 2016;150:328–339. doi: 10.1053/j.gastro.2015.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feun L., Savaraj N. Pegylated arginine deiminase: a novel anticancer enzyme agent. Expert Opin. Investig. Drugs. 2006;15:815–822. doi: 10.1517/13543784.15.7.815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Abou-Alfa G.K., Qin S., Ryoo B.Y., Lu S.N., Yen C.J., Feng Y.H., Lim H.Y., Izzo F., Colombo M., Sarker D., et al. Phase III randomized study of second line ADI-PEG 20 plus best supportive care versus placebo plus best supportive care in patients with advanced hepatocellular carcinoma. Ann. Oncol. 2018;29:1402–1408. doi: 10.1093/annonc/mdy101. [DOI] [PubMed] [Google Scholar]
- 27.Johansen T., Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy. 2011;7:279–296. doi: 10.4161/auto.7.3.14487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lamark T., Svenning S., Johansen T. Regulation of selective autophagy: the p62/SQSTM1 paradigm. Essays Biochem. 2017;61:609–624. doi: 10.1042/EBC20170035. [DOI] [PubMed] [Google Scholar]
- 29.Saxton R.A., Chantranupong L., Knockenhauer K.E., Schwartz T.U., Sabatini D.M. Mechanism of arginine sensing by CASTOR1 upstream of mTORC1. Nature. 2016;536:229–233. doi: 10.1038/nature19079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chantranupong L., Scaria S.M., Saxton R.A., Gygi M.P., Shen K., Wyant G.A., Wang T., Harper J.W., Gygi S.P., Sabatini D.M. The CASTOR Proteins Are Arginine Sensors for the mTORC1 Pathway. Cell. 2016;165:153–164. doi: 10.1016/j.cell.2016.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kading J., Finck B.N., DeBosch B.J. Targeting hepatocyte carbohydrate transport to mimic fasting and calorie restriction. FEBS J. 2021;288:3784–3798. doi: 10.1111/febs.15482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Madeo F., Carmona-Gutierrez D., Hofer S.J., Kroemer G. Caloric Restriction Mimetics against Age-Associated Disease: Targets, Mechanisms, and Therapeutic Potential. Cell Metab. 2019;29:592–610. doi: 10.1016/j.cmet.2019.01.018. [DOI] [PubMed] [Google Scholar]
- 33.Mardones P., Rubinsztein D.C., Hetz C. Mystery solved: Trehalose kickstarts autophagy by blocking glucose transport. Sci. Signal. 2016;9:fs2. doi: 10.1126/scisignal.aaf1937. [DOI] [PubMed] [Google Scholar]
- 34.Zhang Y., DeBosch B.J. Microbial and metabolic impacts of trehalose and trehalose analogues. Gut Microbes. 2020;11:1475–1482. doi: 10.1080/19490976.2020.1750273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Szlosarek P.W., Steele J.P., Nolan L., Gilligan D., Taylor P., Spicer J., Lind M., Mitra S., Shamash J., Phillips M.M., et al. Arginine Deprivation With Pegylated Arginine Deiminase in Patients With Argininosuccinate Synthetase 1-Deficient Malignant Pleural Mesothelioma: A Randomized Clinical Trial. JAMA Oncol. 2017;3:58–66. doi: 10.1001/jamaoncol.2016.3049. [DOI] [PubMed] [Google Scholar]
- 36.Synakiewicz A., Stachowicz-Stencel T., Adamkiewicz-Drozynska E. The role of arginine and the modified arginine deiminase enzyme ADI-PEG 20 in cancer therapy with special emphasis on Phase I/II clinical trials. Expert Opin. Investig. Drugs. 2014;23:1517–1529. doi: 10.1517/13543784.2014.934808. [DOI] [PubMed] [Google Scholar]
- 37.Tsai H.-J., Jiang S.S., Hung W.-C., Borthakur G., Lin S.-F., Pemmaraju N., Jabbour E., Bomalaski J.S., Chen Y.-P., Hsiao H.-H., et al. A Phase II Study of Arginine Deiminase (ADI-PEG20) in Relapsed/Refractory or Poor-Risk Acute Myeloid Leukemia Patients. Sci. Rep. 2017;7:11253. doi: 10.1038/s41598-017-10542-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kremer J.C., Prudner B.C., Lange S.E.S., Bean G.R., Schultze M.B., Brashears C.B., Radyk M.D., Redlich N., Tzeng S.-C., Kami K., et al. Arginine Deprivation Inhibits the Warburg Effect and Upregulates Glutamine Anaplerosis and Serine Biosynthesis in ASS1-Deficient Cancers. Cell Rep. 2017;18:991–1004. doi: 10.1016/j.celrep.2016.12.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miraki-Moud F., Ghazaly E., Ariza-McNaughton L., Hodby K.A., Clear A., Anjos-Afonso F., Liapis K., Grantham M., Sohrabi F., Cavenagh J., et al. Arginine deprivation using pegylated arginine deiminase has activity against primary acute myeloid leukemia cells in vivo. Blood. 2015;125:4060–4068. doi: 10.1182/blood-2014-10-608133. [DOI] [PubMed] [Google Scholar]
- 40.Locke M., Ghazaly E., Freitas M.O., Mitsinga M., Lattanzio L., Lo Nigro C., Nagano A., Wang J., Chelala C., Szlosarek P., Martin S.A. Inhibition of the Polyamine Synthesis Pathway Is Synthetically Lethal with Loss of Argininosuccinate Synthase 1. Cell Rep. 2016;16:1604–1613. doi: 10.1016/j.celrep.2016.06.097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Qiu F., Chen Y.R., Liu X., Chu C.Y., Shen L.J., Xu J., Gaur S., Forman H.J., Zhang H., Zheng S., et al. Arginine starvation impairs mitochondrial respiratory function in ASS1-deficient breast cancer cells. Sci. Signal. 2014;7:ra31. doi: 10.1126/scisignal.2004761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Changou C.A., Chen Y.R., Xing L., Yen Y., Chuang F.Y., Cheng R.H., Bold R.J., Ann D.K., Kung H.J. Arginine starvation-associated atypical cellular death involves mitochondrial dysfunction, nuclear DNA leakage, and chromatin autophagy. Proc. Natl. Acad. Sci. USA. 2014;111:14147–14152. doi: 10.1073/pnas.1404171111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fultang L., Vardon A., De Santo C., Mussai F. Molecular basis and current strategies of therapeutic arginine depletion for cancer. Int. J. Cancer. 2016;139:501–509. doi: 10.1002/ijc.30051. [DOI] [PubMed] [Google Scholar]
- 44.Kim R.H., Coates J.M., Bowles T.L., McNerney G.P., Sutcliffe J., Jung J.U., Gandour-Edwards R., Chuang F.Y., Bold R.J., Kung H.J. Arginine deiminase as a novel therapy for prostate cancer induces autophagy and caspase-independent apoptosis. Cancer Res. 2009;69:700–708. doi: 10.1158/0008-5472.CAN-08-3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lowery M.A., Yu K.H., Kelsen D.P., Harding J.J., Bomalaski J.S., Glassman D.C., Covington C.M., Brenner R., Hollywood E., Barba A., et al. A phase 1/1B trial of ADI-PEG 20 plus nab-paclitaxel and gemcitabine in patients with advanced pancreatic adenocarcinoma. Cancer. 2017;123:4556–4565. doi: 10.1002/cncr.30897. [DOI] [PubMed] [Google Scholar]
- 46.McAlpine J.A., Lu H.T., Wu K.C., Knowles S.K., Thomson J.A. Down-regulation of argininosuccinate synthetase is associated with cisplatin resistance in hepatocellular carcinoma cell lines: implications for PEGylated arginine deiminase combination therapy. BMC Cancer. 2014;14:621. doi: 10.1186/1471-2407-14-621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thongkum A., Wu C., Li Y.-Y., Wangpaichitr M., Navasumrit P., Parnlob V., Sricharunrat T., Bhudhisawasdi V., Ruchirawat M., Savaraj N. The Combination of Arginine Deprivation and 5-Fluorouracil Improves Therapeutic Efficacy in Argininosuccinate Synthetase Negative Hepatocellular Carcinoma. Int. J. Mol. Sci. 2017;18:1175. doi: 10.3390/ijms18061175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eriksen P.L., Vilstrup H., Rigbolt K., Suppli M.P., Sørensen M., Heebøll S., Veidal S.S., Knop F.K., Thomsen K.L. Non-alcoholic fatty liver disease alters expression of genes governing hepatic nitrogen conversion. Liver Int. 2019;39:2094–2101. doi: 10.1111/liv.14205. [DOI] [PubMed] [Google Scholar]
- 49.Thomsen K.L., Grønbæk H., Glavind E., Hebbard L., Jessen N., Clouston A., George J., Vilstrup H. Experimental nonalcoholic steatohepatitis compromises ureagenesis, an essential hepatic metabolic function. Am. J. Physiol. Gastrointest. Liver Physiol. 2014;307:G295–G301. doi: 10.1152/ajpgi.00036.2014. [DOI] [PubMed] [Google Scholar]
- 50.Rabinovich S., Adler L., Yizhak K., Sarver A., Silberman A., Agron S., Stettner N., Sun Q., Brandis A., Helbling D., et al. Diversion of aspartate in ASS1-deficient tumours fosters de novo pyrimidine synthesis. Nature. 2015;527:379–383. doi: 10.1038/nature15529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Potthoff M.J., Inagaki T., Satapati S., Ding X., He T., Goetz R., Mohammadi M., Finck B.N., Mangelsdorf D.J., Kliewer S.A., Burgess S.C. FGF21 induces PGC-1alpha and regulates carbohydrate and fatty acid metabolism during the adaptive starvation response. Proc. Natl. Acad. Sci. USA. 2009;106:10853–10858. doi: 10.1073/pnas.0904187106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Markan K.R., Naber M.C., Ameka M.K., Anderegg M.D., Mangelsdorf D.J., Kliewer S.A., Mohammadi M., Potthoff M.J. Circulating FGF21 is liver derived and enhances glucose uptake during refeeding and overfeeding. Diabetes. 2014;63:4057–4063. doi: 10.2337/db14-0595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Harding J.J., Do R.K., Dika I.E., Hollywood E., Uhlitskykh K., Valentino E., Wan P., Hamilton C., Feng X., Johnston A., et al. A phase 1 study of ADI-PEG 20 and modified FOLFOX6 in patients with advanced hepatocellular carcinoma and other gastrointestinal malignancies. Cancer Chemother. Pharmacol. 2018;82:429–440. doi: 10.1007/s00280-018-3635-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Beddowes E., Spicer J., Chan P.Y., Khadeir R., Corbacho J.G., Repana D., Steele J.P., Schmid P., Szyszko T., Cook G., et al. Phase 1 Dose-Escalation Study of Pegylated Arginine Deiminase, Cisplatin, and Pemetrexed in Patients With Argininosuccinate Synthetase 1-Deficient Thoracic Cancers. J. Clin. Oncol. 2017;35:1778–1785. doi: 10.1200/JCO.2016.71.3230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tomé L.A., Yu L., de Castro I., Campos S.B., Seguro A.C. Beneficial and harmful effects of L-arginine on renal ischaemia. Nephrol. Dial. Transplant. 1999;14:1139–1145. doi: 10.1093/ndt/14.5.1139. [DOI] [PubMed] [Google Scholar]
- 56.Dioguardi F.S. To give or not to give? Lessons from the arginine paradox. J. Nutrigenet. Nutrigenomics. 2011;4:90–98. doi: 10.1159/000327777. [DOI] [PubMed] [Google Scholar]
- 57.Nogiec C.D., Kasif S. To supplement or not to supplement: a metabolic network framework for human nutritional supplements. PLoS ONE. 2013;8:e68751. doi: 10.1371/journal.pone.0068751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nitz K., Lacy M., Atzler D. Amino Acids and Their Metabolism in Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2019;39:319–330. doi: 10.1161/ATVBAHA.118.311572. [DOI] [PubMed] [Google Scholar]
- 59.Taylor R. Calorie restriction for long-term remission of type 2 diabetes. Clin. Med. (Lond.) 2019;19:37–42. doi: 10.7861/clinmedicine.19-1-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fontana L., Meyer T.E., Klein S., Holloszy J.O. Long-term calorie restriction is highly effective in reducing the risk for atherosclerosis in humans. Proc. Natl. Acad. Sci. USA. 2004;101:6659–6663. doi: 10.1073/pnas.0308291101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fontana L. Interventions to promote cardiometabolic health and slow cardiovascular ageing. Nat. Rev. Cardiol. 2018;15:566–577. doi: 10.1038/s41569-018-0026-8. [DOI] [PubMed] [Google Scholar]
- 62.Quarta C., Claret M., Zeltser L.M., Williams K.W., Yeo G.S.H., Tschöp M.H., Diano S., Brüning J.C., Cota D. POMC neuronal heterogeneity in energy balance and beyond: an integrated view. Nat. Metab. 2021;3:299–308. doi: 10.1038/s42255-021-00345-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Itakura E., Kishi C., Inoue K., Mizushima N. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol. Biol. Cell. 2008;19:5360–5372. doi: 10.1091/mbc.E08-01-0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Galluzzi L., Green D.R. Autophagy-Independent Functions of the Autophagy Machinery. Cell. 2019;177:1682–1699. doi: 10.1016/j.cell.2019.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Funderburk S.F., Wang Q.J., Yue Z. The Beclin 1-VPS34 complex--at the crossroads of autophagy and beyond. Trends Cell Biol. 2010;20:355–362. doi: 10.1016/j.tcb.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Clément K., van den Akker E., Argente J., Bahm A., Chung W.K., Connors H., De Waele K., Farooqi I.S., Gonneau-Lejeune J., Gordon G., et al. Setmelanotide POMC and LEPR Phase 3 Trial Investigators Efficacy and safety of setmelanotide, an MC4R agonist, in individuals with severe obesity due to LEPR or POMC deficiency: single-arm, open-label, multicentre, phase 3 trials. Lancet Diabetes Endocrinol. 2020;8:960–970. doi: 10.1016/S2213-8587(20)30364-8. [DOI] [PubMed] [Google Scholar]
- 67.Mantzoros C.S., Flier J.S. Editorial: leptin as a therapeutic agent--trials and tribulations. J. Clin. Endocrinol. Metab. 2000;85:4000–4002. doi: 10.1210/jcem.85.11.7062. [DOI] [PubMed] [Google Scholar]
- 68.Mayer A.L., Higgins C.B., Heitmeier M.R., Kraft T.E., Qian X., Crowley J.R., Hyrc K.L., Beatty W.L., Yarasheski K.E., Hruz P.W., DeBosch B.J. SLC2A8 (GLUT8) is a mammalian trehalose transporter required for trehalose-induced autophagy. Sci. Rep. 2016;6:38586. doi: 10.1038/srep38586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Prinsen H.C.M.T., Schiebergen-Bronkhorst B.G.M., Roeleveld M.W., Jans J.J.M., de Sain-van der Velden M.G.M., Visser G., van Hasselt P.M., Verhoeven-Duif N.M. Rapid quantification of underivatized amino acids in plasma by hydrophilic interaction liquid chromatography (HILIC) coupled with tandem mass-spectrometry. J. Inherit. Metab. Dis. 2016;39:651–660. doi: 10.1007/s10545-016-9935-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Bulk RNA-seq data and single-cell ATAC-seq data have been deposited at the Gene Expression Omnibus (GEO) with accession codes GSE191295 and GSE192413, respectively, and are publicly available as of the date of publication. Accession numbers are also listed in the key resources table.
-
•
This paper does not generate custom code.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.