

Summary
The Columbia Cancer Target Discovery and Development (CTD2) Center is developing PANACEA, a resource comprising dose-responses and RNA sequencing (RNA-seq) profiles of 25 cell lines perturbed with ∼400 clinical oncology drugs, to study a tumor-specific drug mechanism of action. Here, this resource serves as the basis for a DREAM Challenge assessing the accuracy and sensitivity of computational algorithms for de novo drug polypharmacology predictions. Dose-response and perturbational profiles for 32 kinase inhibitors are provided to 21 teams who are blind to the identity of the compounds. The teams are asked to predict high-affinity binding targets of each compound among ∼1,300 targets cataloged in DrugBank. The best performing methods leverage gene expression profile similarity analysis as well as deep-learning methodologies trained on individual datasets. This study lays the foundation for future integrative analyses of pharmacogenomic data, reconciliation of polypharmacology effects in different tumor contexts, and insights into network-based assessments of drug mechanisms of action.
Keywords: polypharmacology, pharmacogenomics, DREAM challenge, community challenge
Graphical abstract
Highlights
-
•
Drug-perturbed RNA sequencing data can be used to identify drug targets
-
•
Technology-based drug-target definitions often subsume literature definitions
-
•
Literature and screening datasets provide complementary information on drug mechanisms
Douglass et al. report the results of a crowdsourced challenge to develop machine-learning algorithms that use drug-perturbed transcriptome data to rapidly predict drug targets on a proteomic scale. Winning methods effectively predicted off-target binding of clinical kinase inhibitors and clarified disparate literature on these drugs’ mechanisms of action.
Introduction
Non-canonical drug targets are known to contribute to clinical toxicity due to off-target effects. More recent work, however, suggests that off targets may contribute to clinical efficacy.1,2 Systematic, de novo elucidation of compound mechanisms of action (MoAs), including polypharmacology, is thus emerging as a critical, yet still highly elusive, problem in clinical oncology. Availability of methodologies for the comprehensive assessment of on- and off-target drug binding could help discriminate between targets driving efficacy or toxicity and those producing non-relevant clinical effects.3
Traditionally, the molecular targets of a drug that comprise its MoA have been defined by detailed thermodynamic (binding strength) and crystallographic (binding structure) characterization of a drug’s interaction with individual proteins.4 This approach is quite effective, as it directly facilitates structure-based drug design. Unfortunately, such a “one-drug/one-target” paradigm is often insufficient to mechanistically elucidate clinical phenotypes induced by even classical drugs, such as aspirin.5,6 As a result, there is an urgent need to systematically re-assess drug MoAs in terms of their proteome-wide polypharmacology, which is defined as their ability to inhibit or activate proteins across a comprehensive, proteome-wide landscape.7
An increasing number of efforts have emerged to leverage large-scale perturbational profiles—e.g., mRNA profiles of cell lines and tissues before and after perturbation with a small compound—to predict both high-affinity binding targets and context-specific effectors.8, 9, 10, 11 The key assumption behind the use of perturbational profiles for this purpose is that differential gene expression is controlled by transcription factors and co-factors that represent the key downstream effectors of a compound’s high-affinity binding targets (Figure 1A).12,13 For example, the drug lapatinib inhibits EGFR (Epidermal Growth Factor Receptor), which induces gene expression changes via downstream transcription factors, including MYC and E2F family proteins (effectors).14,15 As a result, drug-induced differential expression of MYC and E2F transcriptional targets may help distinguish EGFR inhibitors from inhibitors with a different downstream effector repertoire (Figure 1A). By extension, compounds targeting the same proteins should induce similar transcriptional signatures, which in turn can shed insight into its MoA (Figure S1).
Figure 1.

Underlying data and structure of polypharmacology community challenge
(A) Drug mechanism can be divided into direct binding targets and downstream effectors.
(B) The PANACEA-database-given transcriptional profiles of cell lines perturbed by clinical oncology drugs.
(C) Kinome-binding profiles of 32 kinase inhibitors.
(D) Transcriptional Hallmark programs induced by 32 kinase inhibitors (this data represents the average of two technical replicates where the same cell line was perturbed and sequenced on 2 different days).
(E) Challenge structure: participants are given perturbed RNA-seq and dose response data and asked to predict protein targets.
(F) Challenge evaluation: participant predictions are evaluated based on the enrichment of <μM binders within each drug target prediction vector.
The availability of compound- and tissue-specific dose-response curves (DRCs) further improves target assessments. First, it allows perturbational profile generation at high, yet sub-lethal, concentrations, thus preventing an emergence of cell-mediated responses, such as apoptosis or cellular stress, that would confound the true MoA. Second, the availability of differential cell viability in multiple molecularly distinct tissues further informs on compound activity based on distinct cellular and pathway architectures.16
Protein kinases represent one of the most thoroughly studied drug target classes. Protein kinase inhibitors are designed to target some of the most frequently mutated oncogenes, whose inhibition has been the hallmark of the oncogene addiction hypothesis.17 Moreover, ATP-competitive pull-down assays enable effective and systematic binding affinity measurements across comprehensive protein kinase repertoires. The most comprehensive such evaluation to date, the Kinome-Binding Resource (KBR), measured the affinity of 230 clinically relevant kinase inhibitors across 255 kinases.18 While restricted to this protein class, this dataset is well-suited to benchmarking methods aimed at predicting drug polypharmacology by providing criteria for the evaluation of systems pharmacology approaches (Figure S2).
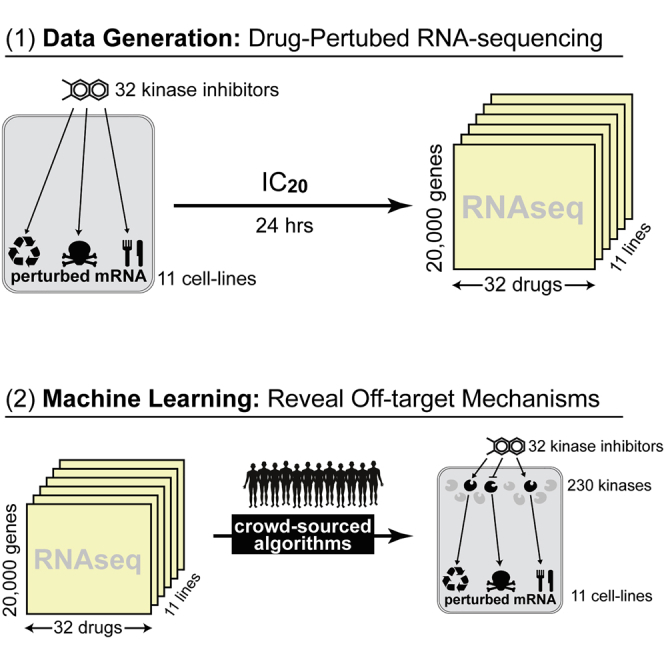
To assess the research community’s ability to predict kinase inhibitors’ MoAs from drug perturbation profiles, we designed a DREAM Challenge19,20 using KBR to provide ground-truth compound MoAs and PANACEA (Pan-cancer Analysis of Chemical Entity Activity)—a large-scale resource comprising genome-wide RNA sequencing (RNA-seq) profiles and matched DRCs of multiple cell lines following perturbation with hundreds of clinically relevant compounds—to provide data that may be used to predict compound MoAs. This significantly extends previous computational and systems pharmacology DREAM challenges by shifting the question from drug sensitivity to MoA prediction. The PANACEA data used in this challenge includes matched DRCs and perturbational RNA-seq profiles representing 11 cell lines following perturbation with approximately 400 clinical oncology drugs in replicate—including US Food and Drug Administration (FDA)-approved and late-stage (phase 2 and 3) compounds (Figure 1B). From the challenge, we specifically selected a subset of 32 kinase inhibitors that were also represented in the KBR (Figures 1C and 1D).
Challenge participants were provided with perturbational profiles and DRCs for each drug (blinded) and cell line (Figure 1E) and were asked to predict high-affinity binding targets for the 32 selected drugs by developing and training machine-learning algorithms using these data. Teams were further encouraged to use public data sources, such as the Cancer Cell Line Encyclopedia,21 the Genomics of Drug Sensitivity in Cancer database,22 and the CMap L1000 database,23 and to leverage insights and models developed in previous DREAM Challenges.8,19,24,25
Previous projects, such as the IDG-DREAM Drug Kinase Binding Challenge25 and the Multi-targeting Drug DREAM Challenge,26 challenged the community to develop computational methods that leveraged publicly available chemical (e.g., chemical fingerprints, protein structures) and kinase binding data to predict drug-target interactions without using compound treatment data in a biological context. In contrast, this challenge asked the community to develop methods that could rank the proteins most affected by a compound using publicly available biochemical data. In order to make the challenge realistic, participants were blinded to the compound identity and to the fact that they were selected from the KBR collection. The challenge operated from December 2019 to February 2020 and led to the development and assessment of state-of-the-art approaches for inferring drug MoAs from perturbational profiles, as described herein.
Results
Challenge requirements and data
In the CTD2 Pancancer Drug Activity DREAM Challenge, participants were asked to use DREAM-provided and publicly available pharmacogenomic datasets—including cell-line-matched DRCs and gene expression profiles of drug-naive and -perturbed cells (perturbational profiles)—to predict compound binding proteins (high-affinity targets) of 32 anonymized drugs (Figure 1A, Table S1). The DREAM-provided dataset comprised 704 DRCs and matched perturbational profiles of these 32 drugs (Table S2) in 11 cell lines representing molecularly distinct tumor subtypes in replicate (Figure 1E). All drugs used in the challenge had perturbational profiling in PANACEA and high-affinity binding characterization in the Kinome-Binding Resource (KBR).18 While the full PANACEA manuscript is being published independently, all data related to this challenge is made contextually available with the publication of this manuscript (see Data and code availability).
Participants were encouraged to combine these data with additional publicly available resources to infer high-affinity binding targets of the 32 drugs from a repertoire of ∼1,300 potential drug targets, defined as the union of all DrugBank-reported targets and the 255 kinases profiled in the KBR. Drug names were obfuscated to prevent trivial training of the algorithm on the KBR data (Figure 1F), and participants were not aware that the KBR data would be used as a gold standard for performance assessment.
Consistent with past DREAM studies, the challenge included a leaderboard round followed by a final validation round.20 During the former, teams were allowed to submit up to five predictions for the 32 compounds, which were scored and posted to a public leaderboard. The purpose of this round was to enable experimentation and conceptual flexibility in model development by providing rapid feedback on the accuracy of the model while also encouraging competition among participants. A limit of 5 submissions was chosen to allow model refinement without compromising the statistical independence of the training and testing model, thus minimizing the potential for over-fitting. In the final validation round, participants were asked to submit their final model’s predictions with the accompanying source code, thus allowing for objective validation of their methodology. Model performance was evaluated according to each team’s ability to prioritize bona fide targets of the 32 drugs, with the latter defined as having a dissociation constant Kd < 1 μM in the KBR, according to two complementary metrics, which were summarized by two sub-challenges:
Sub-challenge 1 (SC1) was designed to assess the ability of each submitted prediction to identify high-affinity binding targets (Kd < 1 μM) of each of the 32 compounds among the top 10 highest-scoring predicted targets. The rationale for selecting the top 10 targets was to represent the number of predictions that could be realistically validated using experimental assays. For each submitted drug prediction, a p value was calculated by filtering the prediction list to consider only targets in the KBR and comparing the number of bona fide targets (Kd < 1 μM in the KBR) in the top 10 predicted targets to a null model generated from all possible targets and was similarly filtered to consider only targets in the KBR. A final integrated score was computed by averaging the -log2(p value) for each drug across all 32 drugs.
Sub-challenge 2 (SC2) was designed to assess the ability of each submitted prediction to accurately rank all the (for the participants) unknown bona fide targets (Kd < 1 μM in the KBR) of each of the 32 compounds by computing their enrichment—and associated p value—within the ranked list of predicted targets. The rationale for this second metric was to provide a more comprehensive and fine-grained comparison of the different methodologies (Figure 1F). Similar to SC1, a final integrated score was computed by averaging the -log2(p value) for each drug across all 32 drugs.
Challenge results
During the leaderboard phase, 21 teams contributed 86 prediction matrices of which 39 (45%) showed a geometric mean (across drugs for each team) p value of <0.01 for both SC1 and SC2. Interestingly, SC1 and SC2 scores revealed distinct distribution profiles: on average, most predictions were statistically significantly enriched on the top 10 target metric (SC1) but not on the entire list enrichment (SC2) (Figures S3A and S3B)
Consistent with previous DREAM Challenges, we assessed whether the performances across teams were statistically different for both sub-challenges by estimating a Bayes factor using a bootstrap analysis (see STAR Methods). The Bayes factor is a metric used to compare two (or more) statistical models; a model with a Bayes factor BF ≤3 indicates that the model is statistically indistinguishable from the top-ranked model. Figures S3C and 3D summarize the results of this analysis, with each box showing a team’s bootstrapped scores, and the color of the box indicating the Bayes factor relative to the top performer. Using this criteria, Team Atom and Team Netphar were confirmed as the top performers in SC1 and SC2, respectively (Figures S3C and S3D), while team SBNB was a close second in SC2 (Bayes factor 3–5). A description of the algorithms from teams Atom, Netphar, and SBNB is provided in the STAR Methods.
When scoring the algorithms for the challenge, we filtered the predictions to the 255 kinases in the gold-standard dataset (i.e., KBR compendium). However, it would be possible in principle for challenge participants to rank kinase targets in the correct order but below incorrect targets not included in the KBR, such that this filtering step would boost their performance relative to participants who had ranked kinase targets in the correct order and above non-kinase targets. To address this issue, SC1 and SC2 results were re-scored considering the full list of 1,259 targets. As with the scoring for the main challenge, SC1 evaluated the top 10 predictions to assess whether any given top 10 overlapped with the gold-standard dataset. SC2 looked at the rank of the gold-standard targets within the submitted predictions. This analysis (Figures S3E and S3F) changed the ranking of some teams in SC1, most notably Team Theragen, whose original second place fell to ninth place when non-KBR targets were not pre-filtered. In addition, Team Netphar’s SC1 performance substantially increased, moving from 5th to 1st position.
To better understand the models and the difference in their performances, we examined sub-challenge scores on an individual drug basis (Figures S3G and S3H). Two clusters emerged, which separated teams based on whether they had used additional training datasets to train their algorithms. (Table 1). In general, consistent with prior results on the value of evidence integration,25 overall performance was positively correlated with the number of additional databases utilized in the analysis, accounting for 27% of the variance in SC1 and a remarkable 82% of the variance in SC2.
Table 1.
Number of additional datasets used by participants for training and algorithm class
| Team | SC1 | SC2 | No. of drug-AUC datasetsa | No. of drug-mRNA datasetsb | No. of drug-target datasetsc | Total training datasets | Algorithm class |
|---|---|---|---|---|---|---|---|
| Netphar | 12.6 | 70.9 | 6 | 1 | 4 | 11 | similarity |
| SBNB | 11.7 | 59.2 | 6 | 3 | 2 | 11 | similarity |
| Xielab | 13.8 | 50.3 | 6 | 2 | 1 | 9 | similarity |
| Atom | 17.4 | 49.3 | – | 2 | 4 | 6 | NN |
| DMIS_PDA | 13.8 | 35.2 | – | 2 | 1 | 3 | NN |
| Theragen | 15.1 | 17.3 | – | 2 | 1 | 3 | similarity |
| Signal | 6.3 | 6.1 | – | 1 | 2 | 4 | regression |
| TeamAxolotl | 6.2 | 1.1 | – | – | 2 | 3 | NN |
| AMbeRland | 3.3 | 1.1 | – | – | – | 0 | unsup. |
| SenthamizhaV | 7.4 | 0.9 | – | – | – | 0 | unsup. |
Training data source contribution to model performances
Both SC2 winning teams, Netphar and SBNB, employed multiple highly curated datasets for training their algorithm. Netphar relied on the multi-database resources DrugComb (cytotoxicity)35 and DrugTargetCommons (drug targets),36 and SBNB relied on the multi-modality ChemicalChecker database.37 Figure 2 provides a high-level conceptual summary of the types of data sources included in these meta-databases organized by data type and source.
Figure 2.

The universe of training data used in this challenge
Drug-perturbation datasets can be divided into two major categories: technology-based and literature-based, each with distinct limitations.
Overall, the datasets used to train the algorithms could be divided into two main categories: experimental screening-based and literature curation-based (Figure 2). Screening approaches have the advantages of providing measurements that are quantitative, directly comparable, and systematic (i.e., low sparsity). However, they may suffer from technological platform bias. Literature curation has the advantage of reflecting a multi-laboratory consensus but suffers from the disparate, ad hoc nature of the measurements and from lack of systematic assessment (high sparsity) (Figure 2). Team performance was further stratified based on whether they relied on (1) drug-target databases, (2) drug-perturbational databases, and/or (3) cytotoxicity databases. As further discussed below, drug-target and -perturbational databases provided the greatest accuracy boost across all drugs.
Critically, all teams chose to use literature-based datasets for identifying candidate drug targets (Figure 2). This is an important detail because while methods were trained on literature-based “drug-target” definitions, they were eventually evaluated based on objective, high-accuracy ATP-competitive assays (Figure 1F). To better understand the overlap between literature- and ATP-based drug targets, we evaluated the overlap between DrugBank and KBR targets (Figure 3). Specifically, we measured the number of DrugBank-reported protein kinase targets that were recovered across a range of affinity thresholds from 1 nM to 10 μM in the KBR (Figure 3A). Encouragingly, almost 80% of them were identified in the KBR using a Kd < 1 μM threshold (Figure 3A), consistent with a common “rule-of-thumb” for drug-lead development.4
Figure 3.

Comparison of DrugBank and kinome drug target definitions
(A) An affinity threshold of 1 μM within the kinome database successfully recovered almost 80% of the kinase targets within DrugBank.
(B) The kinome-defined drug targets appear to reveal a large number of new drug-targets (in red) in addition to the canonical drug targets (in black).
(C) Drug target pairs overlap across four drug target universes.
(D) Drug target pairs not detected in the kinome database used for PANACEA evaluation.
(E) Number of successful top 10 predictions for each drug and team across the different drug target universes.
Interestingly, while a 1 μM threshold identified the majority of DrugBank kinase targets, it also revealed the presence of a significant number of new targets not reported in DrugBank (Figure 3B). Overall, this shows that while DrugBank is mostly recapitulated by the KBR, the reverse is not true, suggesting that DrugBank may not contain all high-affinity targets of a drug. A key question raised by this comparison is whether the winning method’s performance may have been driven entirely by canonical DrugBank targets. To address this question, we evaluated the ratio between the scores of the top three winning teams when either DrugBank or KBR targets were used as bona fide high-affinity targets of the 32 drugs used in the challenge (Figure S4). While the scores based on DrugBank targets were consistently higher (Netphar, 3:2; SBNB, 4:2; and Atom, 1.7:1.4), they all showed positive enrichment within the prediction vector (Figure S4). This result implies that literature-curated drug targets can be successfully used to bootstrap the polypharmacology analysis of otherwise uncharacterized drugs, thus further supporting the value of these resources. However, this may reduce algorithm performance for new compounds that are not yet included in any database.
In addition to DrugBank, two additional drug target databases—ChEMBL32 and DrugTargetCommons36—were used by the top-performing teams. Plotting the overlap of all drug-target pairs across all four drug-target databases, only 121 targets (34%) were found to be unique to the KBR (Figure 3C). Taken together, these databases provided up to 2,386 additional drug-target interactions, of which 520 (21%) were evaluated in the KBR but were found to have affinities >1 μM, suggesting that they are false positive drug-target interactions (Figure 3D).
We compared the ranked performance of each prediction using these various databases as gold standards (KBR/Kinome, ChEMBL, DrugTargetCommons, KinomeScan, and DrugBank) to evaluate the stability of the predictions with different ground truths (Figure S5). As is expected, different ground-truth datasets have a substantial effect on team ranking, though the SC2 metric (rank across all targets for which data are available in the gold-standard dataset) is more stable than the SC1 metric (rank across top ten targets only).
Interestingly, when comparing the overlap of the top 10 targets predicted by the winning teams in each database, the observed differences strongly reflect the training datasets used by each team (Figure 3E). For instance, as one would expect, SBNB and Netphar results were biased toward DrugBank and DrugTargetCommons targets, respectively.
Kinase groups have distinct transcriptional programs
We next explored drivers of model performance by examining prediction accuracy for individual kinase inhibitor groups (Figures 4A and 4B).38 Significant heterogeneity in methods performance across individual drugs was observed, suggesting that differences in modeling strategies (see the next section) may be leveraged to predict different drug classes. For instance, all winning methods performed better on the tyrosine kinase inhibitors group than on any other kinase group (Figure 4C).
Figure 4.

Different kinase pathways show distinct mRNA signatures when inhibited
(A and B) Across all models, tyrosine kinase (TK)-targeting drugs performed the best.
(C) Distribution of kinases profiled across the Human Kinome annotated by kinase group.
(D) Correlation of kinase-binding data with transcriptional program.
(E and F) KEGG pathway transformation of kinase space from (C) revealed pathway-specific transcriptional signatures
Based on this observation, we hypothesized that specific kinase groups and families may be associated with distinct transcriptional programs. To evaluate the general relationships between kinase targets and mRNA programs, we assessed the correlation of the KBR-reported Kd with transcriptional hallmarks (as detailed in Supplemental information) across 84 drugs present in both databases (Figure 4D). This correlation matrix is plotted with phylogenetic tree-based kinase groups annotated on the top bars. Examining the protein kinase mRNA program matrix, no strong kinase group clustering was observed, indicating that kinase class is not generally sufficient to predict downstream transcriptional effects (Figure 4D, columns), although tyrosine kinases showed weak clustering with proliferation programs (Figure 4D, rows, bottom cluster): E2F targets, MYC targets, G2M checkpoint, oxidative phosphorylation, and mTOR-signaling.
To better understand the nature of the biological pathways underlying this association, we projected kinases into the KEGG pathway space39 (see Method details for figures), which yielded a matrix of associations between kinase-signaling pathways and downstream transcriptional programs (Figure 4E). This analysis revealed a distinct pattern of transcriptional signatures that distinguished tyrosine kinase inhibitors from cell-cycle inhibitors and TGFβ inhibitors (Figure 4E), consistent with the known hierarchical structure of these signaling pathways, e.g., MYC and cell-cycle suppression via RTK-inhibition, in contrast to cell-cycle but not MYC suppression via CDK-inhibition (Figure 4F).14
Methodological summary
Overall, the methods submitted to the final validation round could be broken into three general categories:
-
1.
Methods relying on a weighted average of differential gene expression and area under the curve (AUC)-based DRC similarity across drugs and drug targets. These included Netphar, SBNB, Xielab, and Theragen.
-
2.
Methods relying on neural networks trained on prior information relating differential gene expression to drug-targets. These included Atom, DMIS_PDA, and TeamAxolotl
-
3.
Methods based on fully unsupervised data transformation combining differential gene expression and DRC data. These included AMbeRland, SenthamizhamV, and Signal.
Generally, similarity-weighted average methodologies performed best in SC2 (Netphar 1st, SBNB 2nd, and Theragen 3rd)—i.e., they were better at predicting the entire range of targets— while Neural Network-based methodologies performed best in SC1 (Atom 1st and DMIS_PDA 3rd)—i.e., they were better at predicting targets in the range that could lead to realistic experimental validation. Fully unsupervised methods showed the worst performance. Nonetheless, they achieved statistical significance without leveraging any prior knowledge, suggesting the potential for mechanistic insight that could be combined with prior knowledge in future approaches.
In addition, there were differences in the training datasets used by algorithms in the first two categories. While weighted similarity methods used both transcriptional and cytotoxicity data (Figures 1E and 5A), neural network methods were trained exclusively with transcriptional profile data (see Contribution of drug sensitivity data). Intriguingly, the winning neural network method (Atom) used protein sequence data to further train their neural network (Figure 5B). This particular prior knowledge is worth noting, as it underlies several traditional approaches to structure-based drug design (e.g., ligand docking to homology models) and off-target discovery (e.g., BLAST searches in DrugBank31). Unfortunately, while such an approach may eventually help distinguish high-affinity binding targets from key downstream effectors, the use of protein sequence information improved Atom’s performance only by a small, non-statistically significant amount.
Figure 5.

Comparison of the two winning strategies: weighted similarity and neural networks
(A) Team Netphar (who won SC2) used a simple matrix manipulation procedure to predict drug targets.
(B) Team Atom (who won SC1) used a protein-sequence-trained neural network.
Contribution of drug sensitivity data
Previous work has shown that training on drug sensitivity profile data can provide a comparable prediction performance to training on transcriptional signatures.40 As such, we sought to investigate the contributions of drug sensitivity and drug transcriptional data to the performance of the winning Netphar model (which utilized both). Drug sensitivity training data was obtained from DrugComb, a curated database that includes batch-corrected drug sensitivities for both single drugs and drug combinations.35 In addition to the commonly used IC50 (half maximum inhibitory concenration), DrugComb provides an AUC-based relative inhibition (RI) metric,41 which captures both the potency and efficacy of drug responses (Figure S6A).
Examining correlations between predicted and gold-standard targets, we found that adding drug sensitivity data significantly improved prediction accuracy relative to transcriptional data alone (Figure S6B). Performance improvements were driven by several individual drugs whose targets were poorly predicted based on perturbational profile data only, including sunitinib, crizotinib, and crenolanib (Figure S6C). Finally, we tested whether the additional efficacy information provided by the RI metric improved model performance. Indeed, use of the RI metric in the predictive algorithms produced statistically significant, albeit marginal, overall improvement (median 0.18 compared to 0.19, paired Wilcoxon test p value = 0.025), highlighting the potential value of this metric in modeling drug properties.
Discussion
MoA elucidation is a critical, yet time-consuming, step in the drug development process,42 as it helps to identify on- and off-target effects supporting the activity of the compound (polypharmacology) as well as off-target effects that may cause unwanted toxicity. This addresses two major reasons for clinical trial failures: lack of safety and efficacy.43,44 Failure rates may be substantially reduced if compound MoAs could be assessed more accurately and comprehensively (Figures S1A and S1B).
A drug MoA is defined as the set of biochemical interactors and effectors through which the drug produces its pharmacological effects, both positive and negative. These are almost invariably cell-context-specific. Despite its relevance, MoA characterization still represents a significant challenge, which is only partially addressed by experimental and computational strategies. Most of the experimental approaches rely on direct binding assays, such as ATP competitive pull-down,18 affinity purification45,46 or affinity chromatography assays.47 These labor-intensive methods are generally limited to the identification of high-affinity binding targets rather than the full protein repertoire responsible for compound activity in a tissue and are often restricted to a specific protein family, such as protein kinases (Figure S1A). Thus, critically relevant targets outside of these relatively narrow confines may be missed, as shown by the recent reclassification of the MET tyrosine receptor kinase inhibitor tivantinib as a microtubule inhibitor.48 Indeed, drug polypharmacology is emerging as a critical concept that increasingly impacts the mechanistic understanding of a drug’s disease-specific impact, for instance via a field effect mediated by multiple targets rather than by their primary, high-affinity binding target (Figure S1B). For example, OTS964 is a compound originally developed as a MELK inhibitor and was recently shown to manifest its antitumoral activity via an entirely different target, CDK11, which had originally been missed in its MoA characterization.1
A few computational approaches have also been developed to infer MoA,49, 50, 51 including using structural and/or genomic information,52 text-mining algorithms,53 or data mining.54,55 As such, they rely on detailed three-dimensional structures of both the drug molecules and the target proteins or on prior knowledge of related compounds. More recently, systematic gene expression profiling (GEP) following compound perturbations in cell lines8,11,23,56 has furthered the development of computational methods for MoA analysis (Figure S1B).
To address these issues, we hosted a DREAM community challenge to assess computational approaches for drug MoA inference from drug perturbational profiles using a comprehensive, experimental protein kinase binding affinity benchmark. The objective benchmark used in this challenge is the Kinome-Binding Resource (KBR), a systematic set of ATP-competitive binding assays assessing the ability of 230 candidate kinase inhibitor molecules to bind to one of 255 protein kinases (Figure S2).
A critical issue emerging from the evaluation of individual prediction performance and individual databases is that the concept of drug target is still poorly defined and inconsistent (Figure S2). For instance, even when restricting the comparison strictly to protein kinases, the comparison of targets defined in DrugBank versus KBR shows that the former may be missing data and may contain false positive targets whose binding affinity is >1 μM (Figure 3). Yet, it is unclear whether there may be false negatives in the KBR, for example, if allosteric binding or protein degradation occurred upon drug binding, as it would be missed by an ATP-competitive binding assay. More critically, it is unclear whether the targets reported in one database but not the other may play a relevant pharmacological role either in disease treatment or in the emergence of undesirable side effects.
While this was not the main objective of the DREAM Challenge, the study also provides significant insights on the network of effector proteins downstream of high-affinity binding targets. Indeed, the fact that the perturbational signature significantly contributed to correct target inference suggests that downstream transcriptional regulators represent a valuable reporter assay that can distinguish the MoA of different compounds (Figures 4 and S6). Furthermore, the analysis shows that availability of matched DRCs and perturbational profile data for each drug provided a significant contribution to the quality of the prediction. For instance, drugs such as sunitinib, crizotinib, and crenolanib produced significantly worse performances when the analysis was restricted to perturbational profiles but performed significantly better when DRCs and perturbational profile data were integrated.
An interesting observation that emerged from this challenge is that tyrosine kinase inhibitors were predicted with higher accuracy by all methods (Figures 4A–4C). Examining correlations between binding constants and transcriptional profiles, we found that tyrosine kinases inhibitors were mostly associated with suppression of proliferation signatures (Figure 4D). This is perhaps unsurprising, as growth factor control of the cell cycle is typically mediated by receptor tyrosine kinases. Looking at enrichment of KEGG pathways within Figure 4D’s correlation matrix, we were able to identify a decoupling in the effects of MYC and the cell cycle (Figure 4E) that was consistent with the hierarchy of known proliferation pathways (Figure 4F). These results provide evidence that drug-perturbed transcriptional signatures can retain information on the signaling pathways directly downstream of molecular drug targets.
While we did not observe major differences between model performances based on modeling strategy, generally, similarity-weighted average methodologies performed best in SC2 (Figures S7 and S8), while neural-network-based methodologies performed best in SC1 (Figure S9). An important insight arising from the challenge is that computational methods for MoA inference are best at identifying similarities between unknown compounds and compounds already reported in existing databases rather than at elucidating compound MoAs de novo. Indeed, all of the methodologies that did not rely on prior databases underperformed when compared with those that did. The fact that all the proposed methodologies produced statistically significant results suggests that genome-wide perturbational profiles bring de novo predictions of compound MoAs a step closer to being effectively useful in drug discovery.
For the best-performing drug classes, differential transcriptional signals could be traced to specific patterns of co-regulated transcriptional gene sets or hallmarks (Figures 1D, 4D, and 4E). These patterns can be directly explained by the hierarchical structure of kinase signaling cascades in canonical pathways (Figure 4F). Critically, this insight highlights the strengths and weaknesses of mRNA-based target inference where:
-
•
Targets within the same pathway can be difficult to differentiate (e.g., EGFR, RAS, RAF, and MEK inhibitors) due to transcriptional phenocopying.
-
•
Targets at pathway branch points are easier to predict due to the differential transcriptional effects they induce (e.g., RTK versus CDK versus TGFβ inhibitors).
For example, while RTK inhibitors could be effectively distinguished from CDK inhibitors, distinguishing the more subtle differences between drugs within each class should prove more challenging.
Overall, this work suggests that predictive models can leverage perturbational data to effectively infer the MoA of small molecules and to reveal biological and clinical insights about druggable pathways. Future studies using computational modeling to tackle this problem will be critical to the successful application of these methods. Specifically, developing a more systematic knowledge of drug targets, particularly for non-kinase targets, may improve the ability of the community to develop accurate models. Additional development and benchmarking of unsupervised prediction methods may also be required for the accurate prediction of targets of novel molecules. Finally, future work will be necessary to elucidate the best practices, limitations, and general applicability of these methods as a step in the drug discovery pipeline.
Limitations of the study
It is important to note that these finding are most applicable to kinases due to the focus of our drug library on kinase inhibitors. While kinase inhibitors form the largest class of targeted therapy (which often assume specificity), care should be taken in extending the results to other oncology drugs such as cell-cycle inhibitors and DNA-damaging agents. In addition, it should be noted that our perturbation data are collected on only 11 cell lines and so may not recapitulate the transcriptional effects of these 32 kinase inhibitors across all cancer types. Finally, while several different methods were evaluated in this challenge, other methods not evaluated in the present study may also be performant when applied to this machine-learning problem.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| AEE788 | SelleckChem | S1486 |
| Afatinib | SelleckChem | S1011 |
| AZD5363 | SelleckChem | S8019 |
| Bafetinib | SelleckChem | S1369 |
| Bosutinib | SelleckChem | S1014 |
| Cabozantinib | SelleckChem | S1119 |
| Cediranib | SelleckChem | S1017 |
| Crenolanib | SelleckChem | S2730 |
| Crizotinib | SelleckChem | S1068 |
| Dacomitinib | SelleckChem | S2727 |
| Dasatinib | SelleckChem | S1021 |
| Dovitinib | SelleckChem | S1018 |
| Foretinib | SelleckChem | S1111 |
| Gefitinib | SelleckChem | S1025 |
| Icotinib | SelleckChem | S2922 |
| Imatinib | SelleckChem | S2475 |
| KW2449 | SelleckChem | S2158 |
| Lapatinib | SelleckChem | S2111 |
| Linifanib | SelleckChem | S1003 |
| MGCD365 | SelleckChem | S1361 |
| MK2206 | SelleckChem | S1078 |
| Neratinib | SelleckChem | S2150 |
| Nilotinib | SelleckChem | S1033 |
| Osimertinib | SelleckChem | S7297 |
| Ponatinib | SelleckChem | S1490 |
| Quizartinib | SelleckChem | S1526 |
| Regorafenib | SelleckChem | S1178 |
| Sorafenib | SelleckChem | S1040 |
| Sunitinib | SelleckChem | S1042 |
| Tivantinib | SelleckChem | S2753 |
| Vandetanib | SelleckChem | S1046 |
| Varlitinib | SelleckChem | S2755 |
| Critical commercial assays | ||
| CellTiter-Glo Luminescent Viability Assay | Promega | G7570 |
| Deposited data | ||
| PANACEA gene expression profiles. | This paper | GEO: GSE186341 |
| Experimental models: Cell lines | ||
| AsPC-1 | ATCC | ATCC Cat# CRL-1682; RRID:CVCL_0152 |
| DU 145 | ATCC | ATCC Cat# HTB-81; RRID:CVCL_0105 |
| EFO-21 | DSMZ | DSMZ Cat# ACC-235; RRID:CVCL_0029 |
| HCC1143 | ATCC | ATCC Cat# CRL-2321; RRID:CVCL_1245 |
| HF2597 | Henry Ford | N/A |
| HSTS | Broad | RRID:CVCL_L296 |
| KRJ1 | Califano Lab | RRID:CVCL_8886 |
| LNCaP | ATCC | ATCC Cat# CRL-1740; RRID:CVCL_1379 |
| NCI-H1793 | ATCC | ATCC Cat# CRL-5896; RRID:CVCL_1496 |
| PANC-1 | ATCC | ATCC Cat# CRL-1469; RRID:CVCL_0480 |
| U-87 MG | ATCC | ATCC Cat# HTB-14; RRID:CVCL_0022 |
| Software and algorithms | ||
| STAR aligner, 2.5.2b | Dobin et al.57 | https://github.com/alexdobin/STAR |
| Limma 3.48.1 | Ritchie et al.58 | https://bioconductor.org/packages/release/bioc/html/limma.html |
| DESeq2 | Love et al.59 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| ComBat | Johnson et al.60 | https://bioconductor.org/packages/release/bioc/html/sva.html |
| Analysis code | This paper | https://github.com/Sage-Bionetworks-Challenges/CTD2-Panacea-Challenge |
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Andrea Califano (ac2248@cumc.columbia.edu).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Cell line viability
Cell-lines were obtained from ATCC and cultured using prescribed conditions. To determine optimal seeding density for compound titrations (i.e., cell-growth is linear for the duration of experiment), 3.2 million cells of each cell line were plated and viability measured using CelTiter Glo (Promega Corp.) at 24, 48, 72 and 96 hours. Briefly, 10 mL of 320,000 cells/mL cell-solution was added to column 11 of a 12w deep-well plate. 5mL from column 11 was then serially diluted 1:1 from column 11 through column 2. The Hamilton MicroLab automated liquid handling system’s Cell Line Optimization protocol was used to split the 12 w plates between 4 384 well plates for incubation. 384 well plates were stored in the incubator and at 24, 48, 72 and 96 hours 1 plate was removed and allowed to sit for 15 minutes at room temperature. 25 uL of Cell Titer Glo was added to each well and shaken at 800rpm for 5 min. Finally luminescence was read using the EnVision Multi-Label Reader (Perkin Elmer Inc.). The seeding density which resulted in linear increase of the cells was used for the perturbation experiments.
Method details
Collaborative methods overview
The PANACEA database was developed in collaboration between Columbia University Irving Medical Centers (CUIMC)’s High Throughput Screening Center (HTS), Sulzberger Genome Center and the Califano Laboratory in the Department of Systems Biology. Briefly, HTS handled cell-culture, cell-perturbation experiments and RNA extraction; the Genome Center performed RNA sequencing and the Califano laboratory performed data normalization, quality control, benchmarking and scientific and statistical analysis.
Compound titration curves
To determine the 48h ED20 of each drug, cell lines were plated into 96-well tissue culture plates, in 100 μL total volume, and incubated at 37°C. After 16 hours the plates were removed from the incubator and compounds were transferred into assay wells (1 μL) in triplicate. Plates were then returned to the incubator. After 48 hours the assay plates were removed from the incubator and allowed to cool to room temperature prior to the addition of 100 μL of CellTiter-Glo (Promega Inc.) per well. The plates were then mechanically shaken for 5 minutes prior to readout on the EnVision Multi-Label Reader (Perkin Elmer Inc.) using the enhanced luminescence module. Relative cell viability was computed using matched DMSO control wells as reference. ED20 was estimated by fitting a four-parameter sigmoid model to the titration results.
Perturbational profile generation
Using the previously described plating and perturbation procedure we perturbed each cell-line with each drug at its 48h ED20 value (measured above) or its CMax concentration. In order to optimize the clinical translation potential of the perturbation databases, we used the CMax, defined as the maximum plasma concentration after the administration of the drug at the maximum tolerated dose in patients, (whenever available from published pharmacokinetic studies), as an upper bound for the perturbation studies (Table S1). The mRNA from these cells was isolated and profiled by PLATESeq (Nat. Commun. 2017, 8, 105) at 24h after each perturbation.
Quantification and statistical analysis
Profile normalization
RNASeq reads were mapped for each well to the human reference genome assembly 38 using the STAR aligner,57 version 2.5.2b. Individual plates counts files were then combined, normalized and corrected for batch effects. First, individual counts files were combined across genes and ERCC2 spike-in counts removed, yielding the raw counts file for each cell-line experiment. Second, raw counts were quantile normalized and variance stabilized based on the negative binomial distribution with the DESeq R system package.59 To account for plate-based batch effects (which are common with drug-perturbed transcriptomic data) normalized expression was batch corrected using ComBat.60
Kinome and PANACEA data formatting
Kinome-binding data from Klaeger et al.18 was downloaded at https://www.proteomicsdb.org/#projects/4257 via “Supplementary Table 3 Drug Matrices.” Raw data was transformed to -log10 scale and NA’s replaced with the matrix maximum -log10(Kd) of ∼4.3 to represent the limit of detection of the technology. PANACEA differential gene expression data was calculated using a moderated Student’s t test as implemented in the limma package58 from Bioconductor (version 3.48.1) with respect to pooled DMSO controls across all cell-line plates.
Baseline model
For the baseline model, we used drug perturbation gene expression data from the LINCS-L1000 project23 and drug-target information from the Drug Repurposing Hub.61 We calculated consensus signatures40 for each drug with known target molecules. The DREAM-PANACEA gene expression dataset was standardized using the control measurements, and consensus signature (average across cell lines) was calculated for each DREAM-PANACEA drug. We calculated the similarity (Spearman’s correlation) matrix between the LINCS and DREAM-PANACEA drug signatures, using only the measured (landmark) genes of LINCS-L1000. For each DREAM-PANACEA drug, we performed target enrichment (including the mode of action (i.e., activation or inhibitor), using the viper R package12) using the drug similarity vector and the known targets of the LINCS drugs. The normalized enrichment scores from target enrichment were further rank transformed for each drug, and submitted as baseline prediction.
Scoring algorithms
Participants submitted predictions for a list of 1259 “druggable” targets and 30 drugs, with each prediction being a confidence score between 0 and 1 (where one is most confident that the target is a true target of a drug). We then filtered each submission to only consider the 255 targets in the gold standard dataset. For the purposes of calculating p values, we created 1000 null models by generating 1000 random prediction sets. These random predictions were generated by sampling (without replacement) the 255 gold standard targets using the dplyr “sample_frac” function to obtain a randomly-ranked set of targets (this procedure was repeated 1000 times).62
For each submission, we filtered the predictions to the 255 kinases being evaluated. For SC1, we scored teams by evaluating the enrichment of their top 10 predictions for each drug in the gold standard dataset, as well as for one null model prediction, performing a paired Wilcoxon rank sum test (Mann-Whitney test) to generate a p value for each prediction. We repeated this each null model to generate a distribution of 1000 p values for each submission, and calculated the mean p value as the participants’ score. For SC2, the methodology and null models were identical, but instead of evaluating the enrichment of the top 10 predicted targets in the gold standard dataset, we assessed the ranks of the true targets within the full vector of 255 predicted targets for each drug. We again performed a paired Wilcoxon rank sum test (Mann-Whitney test) to generate a p value for each submission. We repeated this for each null model to generate a distribution of 1000 p values for each submission and calculated the mean p value as the participants’ score. In the post-challenge phase of this study, we re-evaluated the performance of each team (Figure S3) by repeating this analysis but omitting the kinase filtering step described above.
Alternate gold standard evaluation
Data from ChEMBL, DrugTargetCommons, KinomeScan (generated by HMS LINCS consortium), and DrugBank for the 1259 “druggable” targets used in this challenge were collected and formatted in the same manner as the KBR dataset used in the challenge. A set of 1000 null models was generated using the 1259 “druggable” targets. The scoring was performed for the SC1 and SC2 metrics as described in the previous section. Due to the very different target universes and completeness of each dataset, we converted the absolute scores to ranks to make it easier to compare the relative differences between the different datasets.
Determination of top performers and data leak
Winners were determined by calculating a Bayes factor relative to the top-ranked submission in each category. In this context, we used the Bayes factor, a likelihood ratio, to compare the difference between the top-ranked model and all other models in each subchallenge. The Bayes factor indicates the relative difference between the predictive power of the two models; with larger Bayes factor values corresponding an larger difference between the models. Ties were defined as models with a Bayes factor ≤ 3 relative to the top-ranked model.
We calculated Bayes factors by bootstrapping all of the submissions that qualified for final scoring by performing 10000 iterations of sampling with replacement for each submission. For each bootstrap, we calculated the p values as described above to generate a distribution of scores for each submission. Using this distribution of p values, Bayes factors were calculated for each submission relative to the top-scoring team using the challengescoring R package (https://github.com/sage-bionetworks/challengescoring). Ties were defined as submissions with a Bayes factor ≤ 3 relative to the top submission. During the scoring of the final round, we discovered that a portion of the Challenge dose-response data had been revealed to the public via a preprint. Upon reviewing the writeups, we saw that Team netphar (not knowing that this was the challenge data) described using this information to fine-tune some of the compound predictions for better performance. To ensure a level playing field and to ensure that this team’s model was generalizable and did not use the preprint data, we worked with Team netphar to remove this fine-tuning step and rescore the prediction. Importantly, the analyses presented in this manuscript to determine the top performers used the new prediction file that omits the fine-tuning step and leaked data.
Detailed computational procedure Figures 1C and 1D
PANACEA differential gene expression data were transformed into “Transcriptional Hallmarks” based on definitions of 50 transcriptional signatures defined in63. Briefly, an average z-score was calculated for each signature by averaging the z-scores of the individual genes for each signature. PanACEA cell-lines were then averaged to yield a single 32x50 matrix reflecting the relationships of 32 drugs and 50 transcriptional hallmarks. PANACEA and Kinome-binding matrices were then processed for visualization by (1) filtering for the top 30 kinases and signatures by variance and (2) clustering rows and column based on pearson correlation. Filtered data were then visualized using the heatmap.2 function of the gplots package in R. Sidebar annotations of canonical drug-targets were defined based on the DrugBank definitions of drug-targets as detailed in Table S1.
Detailed computational procedure Figure 3A
To assess the agreement between DrugBank-Literature and Kinome-binding data, we first defined our reference as: all the kinase-targets defined by DrugBank for our 32-drug library. As the Kinome-binding data give continuous measurements, it is necessary to define a Kd-threshold to binarize the Kinome-data to compare with DrugBank. For each Kd-threshold, we then calculated the coverage of DrugBank by counting the number of drug-kinase edges identified in the Kinome data and divided by the total number of drug-kinase edges in DrugBank. (B) To visualize the new-targets defined in the Kinome-data (but not in DrugBank) we plotted the number of overlapping drug-targets in black (defined in Figure 3A) and newly identified drug-targets in red (defined as NOT being present within DrugBank) for each drug. We then sorted based on total number of targets to aid assessment of polypharmacology.
Detailed computational procedure Figures 4A and 4B
To better understand the performance of the three winning models on individual drugs we recalculated team-scores for each drug as z-scores for enrichment (in red) or depletion (in blue) for < uM targets within each drug-vector for both SC1 and SC2. Recalculated scores were sorted by the rank of the average performance across all three teams to identify the drugs which all models performed well on. To better understand the type of inhibitors that models performed the best on we calculated the enrichment of each drugbank target kinases (as defined in Table S1) over the ranked 32-drug vector in Figures 4A and 4B using the aREA algorithm in the viper package in R.12 (C) To visualize the kinases sampled by the Klaeger et. al18 definitions of drug-targets we color-coded individual kinase-nodes within the Human Kinome phylogenetic tree obtained from the CORAL tool.64 Kinases measured in the Kinome-dataset were color coded based the Kinase group that they were a member of as defined in Manning et al.38 (D) To better understand the relationships between individual kinases and down-stream transcriptional programs we calculated the correlation matrix between the Kinome Kd’s (250 kinases x 84 drugs) and the pan-cancer transcriptional signature PANACEA-data (50 signatures x 84 drugs) across 84 overlapping drugs that occurred in both datasets. Correlation matrix was then clustered based on correlation and visualized using the heatmap.2 function in the gplots package in R. Top side-bars were color-coded by kinase groups as defined in Manning et al.38 and colors were chosen to match the Kinome-coverage-phylogenetic tree in Figure 4C. (E) To better understand the signaling pathways involved in the kinase-mRNA correlations in Figure 4D, we transformed the individual kinase columns in Figure 4D’s correlation matrix into KEGG-defined signaling pathways. This was done by calculating the enrichment of pathway-specific kinases within each transcriptional program vector using the aREA algorithm in the viper package in R. This generated a normalized enrichment score (NES) for each kinase-pathway/mRNA-program pair that is equivalent to a z-score. The visualization on Figure 4E was obtained from the raw pathway-program matrix by slicing top columns associated with receptor tyrosine kinase controlled pathways.
Detailed computational procedure Figure S4
To compare the relative scores of DrugBank-defined targets and New-Kinome-Data-set defined targets within the winning teams predictions we first normalized all scores by the average-score. The purpose of this was to assure that a random-selection of drug-targets would have a normalized score of 1. For each winning team, and for each drug, we then calculated the average scores of Drug-Bank defined targets and Kinome-defined targets. Encouraging, while DrugBank Targets were consistently higher than Kinome-defined targets, both sets consistently scored better than random sets of drug-targets.
Detailed algorithm for winning team “Netphar”
The Netphar team collected three types of data related to the compounds: 1) Drug sensitivity data; 2) Drug induced gene signature data and 3) Drug target interaction data. For drug sensitivity data, we utilized the DrugComb database, which is a crowd-sourcing database to collect comprehensive drug sensitivity screen data, including both monotherapy drug screens and drug combination screens.35 DrugComb currently consists of drug sensitivity data for 466k combination and 710k monotherapy drug screenings. From DrugComb, we found n = 116 drugs that have dose-response data on at least 7 of the 11 cell lines. Furthermore, for each compound-cell pair, we determined IC20 and RI (relative inhibition, which is based on area under the log10-scaled dose-response curves41) score, as more robust measures for drug sensitivity.
For drug-target interaction data, we utilized the DrugTargetCommons which is a crowdsourcing-based database to collectively and manually curate the comprehensive drug-target bioactivity values.36 The bioactivity values were transformed into a confidence score between 0 and 1 to indicate the binding affinity potential.
To determine the best machine learning models to predict the drug targets, we considered two classes of methods including weighted averaging and regression (Figure below). For weighted averaging, the prediction was made based on the multiplication of the Pearson correlation matrix and the drug-target interaction matrix; while for regression, we considered standard machine learning algorithms including ElasticNet, RandomForest and GBM (Gradient Boosting Machine), for which the model was trained on the n = 116 compounds that were found in DrugComb, and then tested on the n = 32 Challenge compounds. We have utilized the LINCS-L1000 data23 to evaluate the methods, and determined the weighted averaging approach that performed better than regression based on 10-fold cross validation.
Detailed algorithm for winning team “SBNB” (Figure S8):
As SBNB team, we approached the challenge as a data integration exercise, where we first adapted the transcriptional and sensitivity signatures of the DREAM Challenge compounds to the format of the Chemical Checker (CC).37 The CC is a resource that provides processed, harmonized, and ready-to-use bioactivity signatures for about 1M compounds, offering a rich portrait of the small molecule data available in the public domain, and opening an opportunity for making queries that would be otherwise impossible using chemical information alone. The CC expresses bioactivity data as numerical vectors, making them suitable for similarity measurements, clustering, visualization and prediction tasks. Among others, the CC contains cell line sensitivity (Sens) and differential gene expression (DGEx) bioactivity signatures for tens of thousands of compounds (CC compounds), being thus possible to relate this data to the DREAM compounds. To integrate DREAM compounds with CC compounds, we built six different signature types from those bioactivity spaces similar to the ones provided by the DREAM challenge. In three of them, we used growth-inhibition (GI) data of eight cell lines common to the Cancer Therapeutics Response Portal (CTRP28) and the DREAM panel. We then used GI data as features to train a classifier to infer the expected CTRP sensitivity (Sens) profile of DREAM compounds, as well as biomarkers and annotations from the PharmacoDB resource.65 Thus, we could connect the DREAM compounds to the hundreds of drugs available in the public drug sensitivity panels. Likewise, DREAM DGEx data were integrated with LINCS DGEx (Level 5) signatures,23 along with the Touchstone reference collection of perturbational profiles. Additionally, we mapped the DREAM and LINCS DGEx to a collection of manually curated expression signatures from Gene Expression Omnibus (CREEDS30), in order to capture cell-unspecific profiles, since only one cell line was shared between the DREAM and LINCS L1000 resources. We moved from individual gene expression to global expression signatures with the aim of capturing possible transcriptional regulatory programs shared among the compounds, enabling thus a more comprehensive integration of the DREAM and LINCS datasets.
Once we contextualised DREAM compounds within the larger CC compounds collection, we used the Sens/DiffGEx signatures as input for conventional target prediction methods, based on previously known ligand-binding profiles. In brief, to prevent overfitting due to the limited number of CC-exp compounds, we first used CC signatures to train a k-nearest neighbors (kNN) classifier to identify the most probable targets for each DREAM compound.
We simply looked in DrugBank for CC compounds having similar signatures to the DREAM compounds, and suggested the CC annotated targets as putative targets for the DREAM compounds. Then, in a second step, we used a much larger set of over 100k bioactive compounds in the Chemical Checker, for which we inferred their gene expression and cell line sensitivity signatures to train a multitask, quality-aware artificial neural network (ANN) primarily based on chemogenomics data (i.e., compound interactions) from ChEMBL66 and refined it with DrugBank drug-target data.31 More specifically, we trained a deep neural network (implemented with Tensorflow v1.12.) with 2 hidden layers (of 256 and 128 units, both using RELU activation and 20% dropout for regularization) and a last multitask classification output layer (with sigmoid activation and 1 unit for each annotated target). Given the gene expression and cell line sensitivity signatures of a drug, this architecture returns a vector of probabilities for each annotated target. We first trained the model using the ChEMBL universe of targets (456 proteins, 87904 different compounds) for 50 epochs with a high learning rate (1e-3). We used the trained network as a starting point to fine-tune the network (transfer learning on the whole network with a low learning rate of 1e-5) with Drugbank targets (456 proteins, 3409 compounds). In both cases we used the normalized average of compound signature confidence (obtained from the CC pipeline) to weight the sample, hence, making the network predictions aware of the input quality (quality-aware). To obtain the final ranking we first computed the closest 1, 5 and 10 nearest neighbors, assembling the results and using them to rank each target accordingly, as previous attempts showed a good performance for the challenge SC2. Then, to improve the challenge SC1, we reordered the top 10 targets for each drug according to the ANN prediction (i.e., we placed in the top 10 the top 10 targets with higher probability scores according to the ANN). Finally, those protein targets of the challenge not annotated in Drugbank were placed at the end, ranked according to the drug counts in ChEMBL (thus, sorted by their prior probability of being a target).
Detailed algorithm for winning team “ATOM” (Figure S9):
For each compound, its compound-perturbed gene expression feature was calculated from Level 5 data of the LINCS L1000 platform of phase I (GSE92742) and phase II (GSE70138). To obtain a consensus feature for each compound without considering other conditions like cell line, dose and time, all the Level 5 signatures corresponding to the same compound were selected and averaged using MODZ algorithm introduced in L1000 paper.23 In order to suit for the challenge, we compared the RNA-seq data with the L1000 data and selected 973 overlapping genes as input features.
During the model training, a graph-based multi-task constraint was used to train our model (described below). The target similarity graph was constructed by using two types of metrics, including a sequence similarity from protein primary sequences as well as a genomic similarity from gene knockdown perturbed gene expression profiles. The protein primary sequences were first obtained from UniProt database according to their gene IDs. Then, the Smith-Waterman sequence alignment scores were computed by an alignment tool (https://github.com/mengyao/Complete-Striped-Smith-Waterman-Library). The sequence similarity between two proteins was then defined as the normalized alignment scores, that is, , where stands for the alignment score between protein sequences and . The gene knockdown perturbed gene expression profiles were obtained from the L1000 database and processed using the same protocol as drug features described above. The genomic similarity between two targets was defined as , where stands for Pearson’s correlation coefficient between gene expression profiles and . Finally, we averaged these two matrices and constructed a K-nearest neighbor (KNN) (K = 10) graph as our final target similarity graph.
As the problem is to predict the potential targets for a compound/drug of interest, we formulate this problem as a multi-label classification problem, where the input of a compound is the compound-perturbed gene expression feature derived from LINCS L1000 platform, and the output is a binary vector indicating the binding probabilities to 769 pre-defined protein targets. We used an ensemble of neural networks to make predictions.
We use an ensemble of single-layer neural networks to model the relationship between and . The model architecture of each base learner (i.e., a single-layer neural network) is shown in Figure 5. For each base learner, three losses are used to train its parameters. The first one is Bayesian Personalized Ranking (BPR) loss.67 Specifically, let denote the predicted score between drug and protein produced by our model. Then, BPR loss is defined as: , where protein is the known target of drug while protein is not. During neural network training, we sampled a batch (batch size = 256) of drugs, and for each drug , we sampled pairs (, ) and (, ) to perform forward and backward propagation. The second loss is a multi-task constraint loss (Zhou et al., 2011). The multi-task constraint loss is defined as: , where is the learnable parameter of the last layer of the neural network, is the normalized graph laplacian of the target similarity graph defined above. This loss encourages the similar targets to have similar classifiers. The last loss is the weight decay (i.e., L2_regularization) for controlling the model complexity. The combined loss is defined as BPR_Loss + + L2_regularization, where and are used to balance different losses. This combined loss was optimized by Adam optimizer with learning rate = 0.001.
We used 10-fold cross validation to train models. For each fold, 1/10 of the drugs were used as test data. Among the remaining 9/10 drugs, 1/10 of drugs were left out as validation data and the rest drugs were used as training data. This strategy was used to perform hyperparameter selection (i.e., dropout rate, , , hidden size of the neural network and training epoch). During training, early stopping was used to prevent overfitting. For each epoch, we compared the model performance on validation data with the best performance. The training process would be stopped as long as the performance on the validation data no longer improves in consecutive 100 epochs.
We used ensemble learning approach to further boost the performance. We constructed 100 different neural network models from { = 0.0001, 0.00001} × { = 0.0001} × {hidden size of neural network = 256, 512, 1024, 2048, 4096} × {10 different folds}. These hyperparameter ranges produced decent prediction performance during our hyperparameter selection. We then averaged the prediction scores from these models to produce the final scores.
Consortia
The members of the CTD2 Drug Activity DREAM Challenge Community Consortium are: Renata Retkute, Alidivinas Prusokas, Augustinas Prusokas, Andrea Degasperi, Yasin Memari, João M. L. Dias, Guillermo de Anda-Jáuregui, Santiago Castro-Dau, Cristóbal Fresno, Laura Gómez-Romero, Humberto Gutiérrez-González, Enrique Hernández-Lemus], Soledad Ochoa, José María Zamora-Fuentes, Yue Qiu, Di He, Lei Xie, Gwanghoon Jang, Jungsoo Park, Sungjoon Park, Buru Chang, Sunkyu Kim, Jaewoo Kang, Eugene F. Douglass Jr., Robert Allaway, Bence Szalai, Ron Realubit, Charles Karan, Wenyu Wang, Tingzhong Tian, Adrià Fernández-Torras, Jing Tang, Shuyu Zheng, Alberto Pessia, Ziaurrehman Tanoli, Mohieddin Jafari, Fangping Wan, Shuya Li, Yuanpeng Xiong, Jianyang Zeng, Miquel Duran-Frigola, Martino Bertoni, Pau Badia-i-Mompel, Lídia Mateo, Oriol Guitart-Pla, Patrick Aloy, Verena Chung, Julio Saez-Rodriguez, Justin Guinney, Daniela Gerhard, and Andrea Califano (see Data S1 for consortium author affiliations).
Acknowledgments
This work was supported by the National Cancer Institute’s Office of Cancer Genomics Cancer Target Discovery and Development (CTD2) initiative. The results published here are based in whole or in part upon data generated by CTD2 Network (https://ocg.cancer.gov/programs/ctd2/data-portal), established by the National Cancer Institute’s Office of Cancer Genomics. P.A. acknowledges the support of the Generalitat de Catalunya (RIS3CAT Emergents CECH: 001-P-001682 and VEIS: 001-P-001647), the Spanish Ministerio de Economía y Competitividad (BIO2016-77038-R), the European Research Council (SysPharmAD: 614944), and the European Commission (RiPCoN: 101003633). B.S. was supported by the Premium Postdoctoral Fellowship Program of the Hungarian Academy of Sciences (460044). A.C. was supported by a Cancer Target Discovery and Development Center grant (U01-CA168426) and by NIH shared instrumentation grants S10-OD012351 and S10-OD021764. A.C., J.G., R.J.A., and E.F.D. were supported through supplemental funding from the CTD2 program (U01-CA217862). J.T. acknowledges the support of European Research Council (ERC) starting grant DrugComb (no. 716063) and the Academy of Finland (no. 317680). J.T. and Z.T. were supported by European Commission H2020 EOSC-life (no. 824087). W.W. was funded by the Doctoral Program of Biomedicine, University of Helsinki, and K. Albin Johanssons stiftelse. M.J. was supported by the Academy of Finland (no. 332454). The Challenge team NetPhar acknowledges their colleagues Bulat Zagidullin and Jehad Aldahdooh for technical support, as well as the CSC – IT Center for Science, Finland, for computational resources.
Author contributions
E.F.D., R.J.A., and B.S. contributed equally to this work and have the right to list their names first on their CVs. Conceptualization, E.F.D., A.C., D.S.G., A.F.-T., M.D.-F., P.A., W.W., J.T., and T.T.; data curation, E.F.D., R.R., C.K., A.F.-T., M.D.-F., L.M., W.W., S.Z., A.P., Z.T., M.J., T.T., and S.L.; formal analysis, E.F.D., A.C., A.F.-T., M.D.-F., M.B., P.B.-i.-M., L.M., W.W., J.T., S.Z., M.J., A.P., T.T., S.L., R.J.A., and B.S.; funding acquisition, A.C., P.A., J.T., W.W., J.Z., and J.G.; investigation, E.F.D., A.C., D.G., A.F.-T., M.D.-F., M.B., P.B.-i.-M., L.M., P.A., J.T., W.W., A.P., M.J., T.T., F.W., S.L., Y.X., J.Z., R.J.A., B.S., and J.S.-R.; methodology, E.F.D., A.C., A.F.-T., M.D.-F., M.B., P.B.-i.-M., L.M., J.T., W.W., A.P., M.J., T.T., F.W., S.L., R.J.A., B.S., and J.S.-R.; project administration, E.F.D., A.C., D.G., M.D.-F., P.A., J.T., W.W., T.T., J.Z., R.J.A., and J.G.; resources, P.A., J.T., J.Z., V.C., and R.J.A., software, E.F.D., A.F.-T., M.D.-F., M.B., P.B.-i.-M., L.M., O.G.-P., W.W., J.T., S.Z., M.J., A.P., T.T., F.W., S.L., R.J.A., V.C., and B.S.; supervision, A.C., D.S.G., M.D.-F., P.A., J.T., J.Z., J.G., and J.S.-R.; validation, E.F.D., A.F.-T., M.D.-F., M.B., P.B.-i.-M., W.W., J.T., S.Z., T.T., F.W., R.J.A., and B.S.; visualization, E.F.D., A.F.-T., M.D.-F., W.W., F.W., T.T., R.J.A, and V.C.; writing – original draft, E.F.D., A.C., D.S.G., A.F.-T., M.D.-F., P.A., W.W., T.T., J.G., R.J.A., and B.S.; writing – review & editing, E.F.D., A.C., D.S.G., A.F.-T., M.D.-F., P.A., W.W., J.T., S.Z., M.J., Z.T., A.P., T.T., F.W., J.G., R.J.A., V.C., and J.S.-R.
Declaration of interests
A.C. is founder, equity holder, and consultant of DarwinHealth, Inc., a company that has licensed the PANACEA database used in this manuscript from Columbia University. Columbia University is also an equity holder in DarwinHealth, Inc. J.S.-R. has received funding from GSK and Sanofi and personal fees from Travere Therapeutics. B.S. received consultation fees from Turbine, Ltd. All other authors declare no competing interests.
Published: January 18, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2021.100492.
Supplemental information
This table includes Drug Name, identifier codes, disease-indication, pathway information, FDA approval status and maximum concentration in serum.
Data and code availability
Data used in the challenge, submission writeups, and other Challenge resources can be found at https://www.doi.org/10.7303/syn20968331.
Raw data is also available through the Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/) under accession number GEO: GSE186341. Code for scoring the predictions and for generating the null models is available here: https://github.com/Sage-Bionetworks-Challenges/CTD2-Panacea-Challenge, and a Docker container that was used to deploy the scoring algorithm in this challenge is available to all registered Synapse users via the Synapse Docker registry (https://www.synapse.org/#!Synapse:syn20968331/wiki/597042). Links to all submitted writeups, Docker containers containing method source code, and Docker documentation for this challenge can be found on the Challenge wiki: https://www.synapse.org/#!Synapse:syn20968331/wiki/607259.
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.
References
- 1.Lin A., Giuliano C.J., Palladino A., John K.M., Abramowicz C., Yuan M.L., Sausville E.L., Lukow D.A., Liu L., Chait A.R., et al. Off-target toxicity is a common mechanism of action of cancer drugs undergoing clinical trials. Sci. Transl. Med. 2019;11:eaaw8412. doi: 10.1126/scitranslmed.aaw8412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dar A.C., Das T.K., Shokat K.M., Cagan R.L. Chemical genetic discovery of targets and anti-targets for cancer polypharmacology. Nature. 2012;486:80–84. doi: 10.1038/nature11127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hopkins A.L. Network pharmacology: the next paradigm in drug discovery. Nat. Chem. Biol. 2008;4:682–690. doi: 10.1038/nchembio.118. [DOI] [PubMed] [Google Scholar]
- 4.Anderson A.C. The process of structure-based drug design. Chem. Biol. 2003;10:787–797. doi: 10.1016/j.chembiol.2003.09.002. [DOI] [PubMed] [Google Scholar]
- 5.Bedard P.L., Hyman D.M., Davids M.S., Siu L.L. Small molecules, big impact: 20 years of targeted therapy in oncology. Lancet. 2020;395:1078–1088. doi: 10.1016/S0140-6736(20)30164-1. [DOI] [PubMed] [Google Scholar]
- 6.Proschak E., Stark H., Merk D. Polypharmacology by Design: A Medicinal Chemist’s Perspective on Multitargeting Compounds. J. Med. Chem. 2019;62:420–444. doi: 10.1021/acs.jmedchem.8b00760. [DOI] [PubMed] [Google Scholar]
- 7.Milletti F., Vulpetti A. Predicting polypharmacology by binding site similarity: from kinases to the protein universe. J. Chem. Inf. Model. 2010;50:1418–1431. doi: 10.1021/ci1001263. [DOI] [PubMed] [Google Scholar]
- 8.Bansal M., Yang J., Karan C., Menden M.P., Costello J.C., Tang H., Xiao G., Li Y., Allen J., Zhong R., et al. NCI-DREAM Community. NCI-DREAM Community A community computational challenge to predict the activity of pairs of compounds. Nat. Biotechnol. 2014;32:1213–1222. doi: 10.1038/nbt.3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Iorio F., Bosotti R., Scacheri E., Belcastro V., Mithbaokar P., Ferriero R., Murino L., Tagliaferri R., Brunetti-Pierri N., Isacchi A., di Bernardo D. Discovery of drug mode of action and drug repositioning from transcriptional responses. Proc. Natl. Acad. Sci. USA. 2010;107:14621–14626. doi: 10.1073/pnas.1000138107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shen Y., Alvarez M.J., Bisikirska B., Lachmann A., Realubit R., Pampou S., Coku J., Karan C., Califano A. Systematic, network-based characterization of therapeutic target inhibitors. PLoS Comput. Biol. 2017;13:e1005599. doi: 10.1371/journal.pcbi.1005599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Woo J.H., Shimoni Y., Yang W.S., Subramaniam P., Iyer A., Nicoletti P., Rodríguez Martínez M., López G., Mattioli M., Realubit R., et al. Elucidating Compound Mechanism of Action by Network Perturbation Analysis. Cell. 2015;162:441–451. doi: 10.1016/j.cell.2015.05.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alvarez M.J., Shen Y., Giorgi F.M., Lachmann A., Ding B.B., Ye B.H., Califano A. Functional characterization of somatic mutations in cancer using network-based inference of protein activity. Nat. Genet. 2016;48:838–847. doi: 10.1038/ng.3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alvarez M.J., Subramaniam P.S., Tang L.H., Grunn A., Aburi M., Rieckhof G., Komissarova E.V., Hagan E.A., Bodei L., Clemons P.A., et al. A precision oncology approach to the pharmacological targeting of mechanistic dependencies in neuroendocrine tumors. Nat. Genet. 2018;50:979–989. doi: 10.1038/s41588-018-0138-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kolch W., Halasz M., Granovskaya M., Kholodenko B.N. The dynamic control of signal transduction networks in cancer cells. Nat. Rev. Cancer. 2015;15:515–527. doi: 10.1038/nrc3983. [DOI] [PubMed] [Google Scholar]
- 15.Blumer K.J., Johnson G.L. Diversity in function and regulation of MAP kinase pathways. Trends Biochem. Sci. 1994;19:236–240. doi: 10.1016/0968-0004(94)90147-3. [DOI] [PubMed] [Google Scholar]
- 16.Schenone M., Dančík V., Wagner B.K., Clemons P.A. Target identification and mechanism of action in chemical biology and drug discovery. Nat. Chem. Biol. 2013;9:232–240. doi: 10.1038/nchembio.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weinstein I.B. Cancer. Addiction to oncogenes–the Achilles heel of cancer. Science. 2002;297:63–64. doi: 10.1126/science.1073096. [DOI] [PubMed] [Google Scholar]
- 18.Klaeger S., Heinzlmeir S., Wilhelm M., Polzer H., Vick B., Koenig P.-A., Reinecke M., Ruprecht B., Petzoldt S., Meng C., et al. The target landscape of clinical kinase drugs. Science. 2017;358:eaan4368. doi: 10.1126/science.aan4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Costello J.C., Heiser L.M., Georgii E., Gönen M., Menden M.P., Wang N.J., Bansal M., Ammad-ud-din M., Hintsanen P., Khan S.A., et al. NCI DREAM Community A community effort to assess and improve drug sensitivity prediction algorithms. Nat. Biotechnol. 2014;32:1202–1212. doi: 10.1038/nbt.2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saez-Rodriguez J., Costello J.C., Friend S.H., Kellen M.R., Mangravite L., Meyer P., Norman T., Stolovitzky G. Crowdsourcing biomedical research: leveraging communities as innovation engines. Nat. Rev. Genet. 2016;17:470–486. doi: 10.1038/nrg.2016.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barretina J., Caponigro G., Stransky N., Venkatesan K., Margolin A.A., Kim S., Wilson C.J., Lehár J., Kryukov G.V., Sonkin D., et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iorio F., Knijnenburg T.A., Vis D.J., Bignell G.R., Menden M.P., Schubert M., Aben N., Gonçalves E., Barthorpe S., Lightfoot H., et al. A Landscape of Pharmacogenomic Interactions in Cancer. Cell. 2016;166:740–754. doi: 10.1016/j.cell.2016.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Subramanian A., Narayan R., Corsello S.M., Peck D.D., Natoli T.E., Lu X., Gould J., Davis J.F., Tubelli A.A., Asiedu J.K., et al. A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell. 2017;171:1437–1452.e17. doi: 10.1016/j.cell.2017.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Menden M.P., Wang D., Mason M.J., Szalai B., Bulusu K.C., Guan Y., Yu T., Kang J., Jeon M., Wolfinger R., et al. AstraZeneca-Sanger Drug Combination DREAM Consortium Community assessment to advance computational prediction of cancer drug combinations in a pharmacogenomic screen. Nat. Commun. 2019;10:2674. doi: 10.1038/s41467-019-09799-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cichońska A., Ravikumar B., Allaway R.J., Wan F., Park S., Isayev O., Li S., Mason M., Lamb A., Tanoli Z., et al. IDG-DREAM Drug-Kinase Binding Prediction Challenge Consortium Crowdsourced mapping of unexplored target space of kinase inhibitors. Nat. Commun. 2021;12:3307. doi: 10.1038/s41467-021-23165-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xiong Z., Jeon M., Allaway R.J., Kang J., Park D., Lee J., Jeon H., Ko M., Jiang H., Zheng M., et al. Crowdsourced identification of multi-target kinase inhibitors for RET- and TAU-based disease: the Multi-Targeting Drug DREAM Challenge. PLoS Comput. Biol. 2021;17:e1009302. doi: 10.1371/journal.pcbi.1009302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shoemaker R.H. The NCI60 human tumour cell line anticancer drug screen. Nat. Rev. Cancer. 2006;6:813–823. doi: 10.1038/nrc1951. [DOI] [PubMed] [Google Scholar]
- 28.Seashore-Ludlow B., Rees M.G., Cheah J.H., Cokol M., Price E.V., Coletti M.E., Jones V., Bodycombe N.E., Soule C.K., Gould J., et al. Harnessing Connectivity in a Large-Scale Small-Molecule Sensitivity Dataset. Cancer Discov. 2015;5:1210–1223. doi: 10.1158/2159-8290.CD-15-0235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Haverty P.M., Lin E., Tan J., Yu Y., Lam B., Lianoglou S., Neve R.M., Martin S., Settleman J., Yauch R.L., Bourgon R. Reproducible pharmacogenomic profiling of cancer cell line panels. Nature. 2016;533:333–337. doi: 10.1038/nature17987. [DOI] [PubMed] [Google Scholar]
- 30.Wang Z., Monteiro C.D., Jagodnik K.M., Fernandez N.F., Gundersen G.W., Rouillard A.D., Jenkins S.L., Feldmann A.S., Hu K.S., McDermott M.G., et al. Extraction and analysis of signatures from the Gene Expression Omnibus by the crowd. Nat. Commun. 2016;7:12846. doi: 10.1038/ncomms12846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wishart D.S., Feunang Y.D., Guo A.C., Lo E.J., Marcu A., Grant J.R., Sajed T., Johnson D., Li C., Sayeeda Z., et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 2018;46(D1):D1074–D1082. doi: 10.1093/nar/gkx1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mendez D., Gaulton A., Bento A.P., Chambers J., De Veij M., Félix E., Magariños M.P., Mosquera J.F., Mutowo P., Nowotka M., et al. ChEMBL: towards direct deposition of bioassay data. Nucleic Acids Res. 2019;47(D1):D930–D940. doi: 10.1093/nar/gky1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kanehisa M., Sato Y., Furumichi M., Morishima K., Tanabe M. New approach for understanding genome variations in KEGG. Nucleic Acids Res. 2019;47(D1):D590–D595. doi: 10.1093/nar/gky962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Günther S., Kuhn M., Dunkel M., Campillos M., Senger C., Petsalaki E., Ahmed J., Urdiales E.G., Gewiess A., Jensen L.J., et al. SuperTarget and Matador: resources for exploring drug-target relationships. Nucleic Acids Res. 2008;36:D919–D922. doi: 10.1093/nar/gkm862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zagidullin B., Aldahdooh J., Zheng S., Wang W., Wang Y., Saad J., Malyutina A., Jafari M., Tanoli Z., Pessia A., Tang J. DrugComb: an integrative cancer drug combination data portal. Nucleic Acids Res. 2019;47(W1):W43–W51. doi: 10.1093/nar/gkz337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tang J., Tanoli Z.-U.-R., Ravikumar B., Alam Z., Rebane A., Vähä-Koskela M., Peddinti G., van Adrichem A.J., Wakkinen J., Jaiswal A., et al. Drug Target Commons: A Community Effort to Build a Consensus Knowledge Base for Drug-Target Interactions. Cell Chem. Biol. 2018;25:224–229.e2. doi: 10.1016/j.chembiol.2017.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Duran-Frigola M., Pauls E., Guitart-Pla O., Bertoni M., Alcalde V., Amat D., Juan-Blanco T., Aloy P. Extending the small-molecule similarity principle to all levels of biology with the Chemical Checker. Nat. Biotechnol. 2020;38:1087–1096. doi: 10.1038/s41587-020-0502-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Manning G., Whyte D.B., Martinez R., Hunter T., Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 39.Kanehisa M., Furumichi M., Tanabe M., Sato Y., Morishima K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017;45(D1):D353–D361. doi: 10.1093/nar/gkw1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Szalai B., Subramanian V., Holland C.H., Alföldi R., Puskás L.G., Saez-Rodriguez J. Signatures of cell death and proliferation in perturbation transcriptomics data-from confounding factor to effective prediction. Nucleic Acids Res. 2019;47:10010–10026. doi: 10.1093/nar/gkz805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Malyutina A., Majumder M.M., Wang W., Pessia A., Heckman C.A., Tang J. Drug combination sensitivity scoring facilitates the discovery of synergistic and efficacious drug combinations in cancer. PLoS Comput. Biol. 2019;15:e1006752. doi: 10.1371/journal.pcbi.1006752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Scannell J.W., Blanckley A., Boldon H., Warrington B. Diagnosing the decline in pharmaceutical R&D efficiency. Nat. Rev. Drug Discov. 2012;11:191–200. doi: 10.1038/nrd3681. [DOI] [PubMed] [Google Scholar]
- 43.Kola I., Landis J. Can the pharmaceutical industry reduce attrition rates? Nat. Rev. Drug Discov. 2004;3:711–715. doi: 10.1038/nrd1470. [DOI] [PubMed] [Google Scholar]
- 44.Wehling M. Assessing the translatability of drug projects: what needs to be scored to predict success? Nat. Rev. Drug Discov. 2009;8:541–546. doi: 10.1038/nrd2898. [DOI] [PubMed] [Google Scholar]
- 45.Hirota T., Lee J.W., St John P.C., Sawa M., Iwaisako K., Noguchi T., Pongsawakul P.Y., Sonntag T., Welsh D.K., Brenner D.A., et al. Identification of small molecule activators of cryptochrome. Science. 2012;337:1094–1097. doi: 10.1126/science.1223710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ito T., Ando H., Suzuki T., Ogura T., Hotta K., Imamura Y., Yamaguchi Y., Handa H. Identification of a primary target of thalidomide teratogenicity. Science. 2010;327:1345–1350. doi: 10.1126/science.1177319. [DOI] [PubMed] [Google Scholar]
- 47.Aebersold R., Mann M. Mass spectrometry-based proteomics. Nature. 2003;422:198–207. doi: 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- 48.Basilico C., Pennacchietti S., Vigna E., Chiriaco C., Arena S., Bardelli A., Valdembri D., Serini G., Michieli P. Tivantinib (ARQ197) displays cytotoxic activity that is independent of its ability to bind MET. Clin. Cancer Res. 2013;19:2381–2392. doi: 10.1158/1078-0432.CCR-12-3459. [DOI] [PubMed] [Google Scholar]
- 49.Keiser M.J., Setola V., Irwin J.J., Laggner C., Abbas A.I., Hufeisen S.J., Jensen N.H., Kuijer M.B., Matos R.C., Tran T.B., et al. Predicting new molecular targets for known drugs. Nature. 2009;462:175–181. doi: 10.1038/nature08506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lomenick B., Hao R., Jonai N., Chin R.M., Aghajan M., Warburton S., Wang J., Wu R.P., Gomez F., Loo J.A., et al. Target identification using drug affinity responsive target stability (DARTS) Proc. Natl. Acad. Sci. USA. 2009;106:21984–21989. doi: 10.1073/pnas.0910040106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Miller W.H., Jr., Schipper H.M., Lee J.S., Singer J., Waxman S. Mechanisms of action of arsenic trioxide. Cancer Res. 2002;62:3893–3903. [PubMed] [Google Scholar]
- 52.Yamanishi Y., Araki M., Gutteridge A., Honda W., Kanehisa M. Prediction of drug-target interaction networks from the integration of chemical and genomic spaces. Bioinformatics. 2008;24:i232–i240. doi: 10.1093/bioinformatics/btn162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li J., Zhu X., Chen J.Y. Building disease-specific drug-protein connectivity maps from molecular interaction networks and PubMed abstracts. PLoS Comput. Biol. 2009;5:e1000450. doi: 10.1371/journal.pcbi.1000450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hansen N.T., Brunak S., Altman R.B. Generating genome-scale candidate gene lists for pharmacogenomics. Clin. Pharmacol. Ther. 2009;86:183–189. doi: 10.1038/clpt.2009.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Perlman L., Gottlieb A., Atias N., Ruppin E., Sharan R. Combining drug and gene similarity measures for drug-target elucidation. J. Comput. Biol. 2011;18:133–145. doi: 10.1089/cmb.2010.0213. [DOI] [PubMed] [Google Scholar]
- 56.Lamb J., Crawford E.D., Peck D., Modell J.W., Blat I.C., Wrobel M.J., Lerner J., Brunet J.-P., Subramanian A., Ross K.N., et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006;313:1929–1935. doi: 10.1126/science.1132939. [DOI] [PubMed] [Google Scholar]
- 57.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Johnson W.E., Li C., Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8:118–127. doi: 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- 61.Corsello S.M., Bittker J.A., Liu Z., Gould J., McCarren P., Hirschman J.E., Johnston S.E., Vrcic A., Wong B., Khan M., et al. The Drug Repurposing Hub: a next-generation drug library and information resource. Nat. Med. 2017;23:405–408. doi: 10.1038/nm.4306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wickham H., Averick M., Bryan J., Chang W., McGowan L., François R., Grolemund G., Hayes A., Henry L., Hester J., et al. Welcome to the tidyverse. J. Open Source Softw. 2019;4:1686. [Google Scholar]
- 63.Liberzon A., Birger C., Thorvaldsdóttir H., Ghandi M., Mesirov J.P., Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1:417–425. doi: 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Metz K.S., Deoudes E.M., Berginski M.E., Jimenez-Ruiz I., Aksoy B.A., Hammerbacher J., Gomez S.M., Phanstiel D.H. Coral: Clear and Customizable Visualization of Human Kinome Data. Cell Syst. 2018;7:347–350.e1. doi: 10.1016/j.cels.2018.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Smirnov P., Kofia V., Maru A., Freeman M., Ho C., El-Hachem N., Adam G.-A., Ba-Alawi W., Safikhani Z., Haibe-Kains B. PharmacoDB: an integrative database for mining in vitro anticancer drug screening studies. Nucleic Acids Res. 2018;46(D1):D994–D1002. doi: 10.1093/nar/gkx911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gaulton A., Hersey A., Nowotka M., Bento A.P., Chambers J., Mendez D., Mutowo P., Atkinson F., Bellis L.J., Cibrián-Uhalte E., et al. The ChEMBL database in 2017. Nucleic Acids Res. 2017;45(D1):D945–D954. doi: 10.1093/nar/gkw1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rendle S., Freudenthaler C., Gantner Z., Schmidt-Thieme L. BPR: Bayesian Personalized Ranking from Implicit Feedback. arXiv. 2012 https://arxiv.org/abs/1205.2618 1205.2618v1. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This table includes Drug Name, identifier codes, disease-indication, pathway information, FDA approval status and maximum concentration in serum.
Data Availability Statement
Data used in the challenge, submission writeups, and other Challenge resources can be found at https://www.doi.org/10.7303/syn20968331.
Raw data is also available through the Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/) under accession number GEO: GSE186341. Code for scoring the predictions and for generating the null models is available here: https://github.com/Sage-Bionetworks-Challenges/CTD2-Panacea-Challenge, and a Docker container that was used to deploy the scoring algorithm in this challenge is available to all registered Synapse users via the Synapse Docker registry (https://www.synapse.org/#!Synapse:syn20968331/wiki/597042). Links to all submitted writeups, Docker containers containing method source code, and Docker documentation for this challenge can be found on the Challenge wiki: https://www.synapse.org/#!Synapse:syn20968331/wiki/607259.
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.