

Abstract
DNA mismatch repair (MMR) corrects non-Watson-Crick basepairs generated by replication errors, recombination intermediates, and some forms of chemical damage to DNA. In MutS and MutL homolog-dependent MMR, damaged bases do not identify the error-containing daughter strand that must be excised and resynthesized. In organisms like Escherichia coli that use methyl-directed MMR, transient undermethylation identifies the daughter strand. For other organisms, growing in vitro and in vivo evidence suggest that strand discrimination is mediated by DNA replication-associated daughter strand nicks that direct asymmetric loading of the replicative clamp (the β-clamp in bacteria and the proliferating cell nuclear antigen, PCNA, in eukaryotes). Structural modeling suggests that replicative clamps mediate strand specificity either through the ability of MutL homologs to recognize the fixed orientation of the daughter strand relative to one face of the replicative clamps or through parental strand-specific diffusion of replicative clamps on DNA, which places the daughter strand in the MutL homolog endonuclease active site. Finally, identification of bacteria that appear to lack strand discrimination mediated by a replicative clamp and a pre-existing nick suggest that other strand discrimination mechanisms exist or that these organisms perform MMR by generating a double-stranded DNA break intermediate, which may be analogous to NucS-mediated MMR.
Keywords: DNA mismatch repair, strand discrimination, DNA mispair
1. Introduction
DNA mismatch repair (MMR) must recognize and properly repair a small number of mispairs within a large background of normally base-paired DNA (1-3). Mispairs are base pairs that violate the Watson-Crick base-paring rules or small insertions or deletions present on only one strand of DNA, which most commonly arise due to misincorporation errors by DNA polymerases. MMR mediated by homologs of Escherichia coli MutS and MutL use an excision-repair mechanism in which a portion of the daughter (newly synthesized) strand including the mispair is excised and resynthesized by DNA polymerases using information from the parental (template) strand. The ability of MMR to discriminate between the parental and daughter strands is essential; if excision and resynthesis are inappropriately targeted to the parental strand, the mispair is converted to a mutation (Fig. 1). In principle, mutations disrupting strand discrimination but not mispair-provoked excision would be expected to cause an increased mutation rate; this appears to be true in E. coli dam mutants. These mutants have increased mutations rates due to the lack of strand discrimination signals and MMR protein-mediated excision that likely occurs on both strands, as evidenced by the accumulation of double-strand breaks and a hyper-recombination phenotype (4-7).
Figure 1. MMR must target the daughter DNA strand.
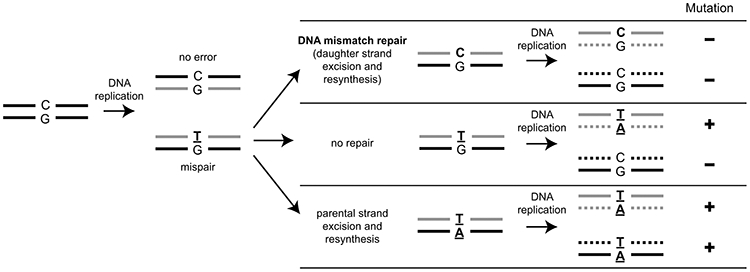
Hypothetical processing of a T:G mispair arising due to a DNA replication error can lead to multiple genetic outcomes. In MMR, excision and resynthesis target the daughter strand (gray), which eliminates the mispair prior to DNA replication and no mutant progeny are produced. If the mispair is unrepaired, then DNA replication will generate one wild-type progeny and one mutant progeny. If the parental strand (black) were to be targeted by excision and resynthesis, then the mispair would be converted into a mutation and propagated in all subsequent rounds of DNA replication.
2. Strand discrimination in methyl-directed MMR
Methyl-directed MMR is present in a subset of gamma-proteobacteria, including the orders Enterobacteriales (containing E. coli), Pasteurellales, Vibrionales, Aeromonadales, and a subset of the Alteromonadales as well as some bacteria that appear to have obtained methyl-directed MMR by horizontal gene transfer (8). In these bacteria, the DNA adenine methyltransferase Dam modifies the N6 position of adenosines in the palindromic sequence d(GATC) (9). After DNA replication, Dam sites are transiently hemi-methylated with a methylated parental strand and an unmethylated daughter strand. Transient hemi-methylation is the strand-discrimination signal for methyl-directed MMR (10), as was proposed based on the analysis of repair tracts of heteroduplexes introduced into E. coli (11). The requirement for the hemi-methylation explains why both loss of Dam methylation and increased Dam methylation cause mutator phenotypes (5, 12). Hemi-methylated d(GATC) sites also have additional roles, such as preventing re-replication of DNA and organizing the nucleoid (13, 14). Hemi-methylated sites are fully methylated by Dam within seconds, as measured for plasmids, to minutes, as measured for chromosomes under slow growth conditions (15-17). The speed of methylation likely stems from the processivity of the Dam methyltransferase, which modifies over 50 d(GATC) sites on one strand after each DNA binding event (18). Rapid loss of the strand-discrimination signals by chromosome maturation processes likely require the prompt recognition of mispairs and may explain why localization of MutS homologs to the replication fork is conserved throughout evolution (19-23), even if the strand-discrimination signals are not.
Mispair recognition by the MutS homodimeric ATPase initiates methyl-directed MMR (Fig. 2). Finding mispairs in vitro involves three-dimensional collisions between MutS and DNA followed by one-dimensional diffusion involving rotation-coupled translation of a MutS “mispair-searching complex” along the DNA backbone (24, 25) that in vivo likely occurs by MutS homodimers tethered to the replication fork (19-23). The cryo-electron microscopy (cryo-EM) structure of the one-dimensional scanning complex shows that the bound homoduplex DNA is partially bent, likely indicating an attempt by MutS to identify mispairs through DNA distortion (26). Upon mispair recognition, the DNA is bent by MutS is ~45-60° at the mispair, allowing one base of the mispair to be recognized by π-stacking with a highly conserved phenylalanine side chain in the MutS mispair-recognition domain (26-28). Mispair recognition stabilizes MutS on DNA (24, 25) and also aligns the domains of MutS so that subsequent conversion to the “sliding clamp” conformation by ATP binding is no longer sterically blocked (26). The MutS sliding clamp releases the mispair, thereby eliminating the DNA bend, and likely has kinked lever domains to more extensively wrap around the unbent DNA; the MutS sliding clamp rapidly migrates bidirectionally along the DNA backbone, and falls off of DNAs at free ends (26, 29, 30). This sliding clamp form of MutS and/or an intermediate “pre-mobile” state observed kinetically and by cryo-EM appear to be the conformations of MutS that recruit the MutL homodimer (26, 31-33). MutS homologs recognize mispairs in an asymmetric way: one subunit recognizes the mispair, the other does not. This asymmetry, however, does not mediate strand specificity. The same base, such as a T in a G:T mispair, is always recognized by the mispair-recognizing subunit, regardless of whether the T is in the parental or daughter strand. In addition, the sliding clamp form of MutS diffuses along the DNA with limited contact with the DNA backbone (24, 25), suggesting that any strand asymmetry in the MutS mispair-recognition complex is lost upon formation of the MutS sliding clamp.
Figure 2. Methyl-directed MMR.
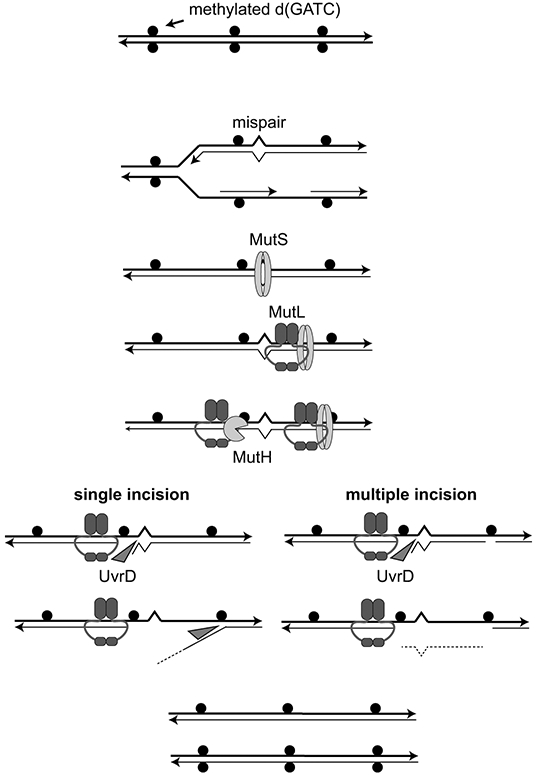
Replication of d(GATC) sites that are methylated on both strands (solid circles) generates hemi-methylated sites that distinguish the parental and daughter strands. Mispairs generated during DNA replication are recognized by the MutS homodimer that then recruits the MutL homodimer. MutL complexes (or MutS-MutL complexes) can then migrate along DNA to activate the MutH single-stranded endonuclease at hemi-methylated d(GATC) either 5’ or 3’ to the mispair. These nicks become entry sites for the recruitment and activation of the UvrD helicase that either generates a single-stranded daughter strand flap (single MutH incision) or a gap in which the mispair-proximal region of the daughter strand is removed (multiple MutH incision). Gap filling by DNA polymerase and DNA ligase completes the repair of the daughter strand.
Loading of the MutL homodimeric ATPase onto DNA by MutS generates MutL sliding clamps (Fig. 2), which migrate bidirectionally along DNA and can pass over protein-DNA complexes (29, 34, 35). These MutL sliding clamps (or potentially MutS-MutL sliding clamps) play roles in mediating downstream steps in MMR. Single molecule studies of E. coli proteins have demonstrated that MutL loading occurs randomly on DNA after MutS sliding clamp formation at mispairs (34). This random loading, combined with the inability of MutS sliding clamps to distinguish between the strands, argues against strand-specific loading of MutL complexes.
MutL activates the latent MutH endonuclease at hemi-methylated d(GATC) sites to make a single-stranded cleavage (nick) 5’ of the G on the unmethylated daughter strand (36-38) (Fig. 2). MutH is a monomeric endonuclease in the same family as the Sau3AI restriction enzyme and recognizes the unmethylated adenosine of the daughter strand using a tyrosine residue (E. coli Y212), which is required for cleavage (39-41). Thus, MutH bridges the mispair-recognition components of MMR with Dam methylase-directed strand discrimination. Migration of MutL (or MutS-MutL) sliding clamps on DNA allows MutH to be activated at d(GATC) sites up to 1-2 kb away from the mispair in either direction in vitro (42). In vivo, the average distance between d(GATC) sites in E. coli is far shorter, around 240 bp, and only 2% of these sites are separated by more than 1 kb. Although bidirectional utilization of hemi-methylated d(GATC) sites has been observed in vitro, analysis of MMR in vivo suggests that the first d(GATC) site between the mispair and the progressing replication fork is used and that removal of the daughter strand occurs in the opposite direction as replication (43). Thus E. coli MMR is bidirectional with regards to the mispair, but predominantly unidirectional with regard to DNA replication, which may allow the MMR system to use the most recently replicated GATC sites that are the most likely to retain their strand-discriminating hemi-methylated status.
MutL also recruits and activates the UvrD DNA helicase (DNA helicase II) in a mechanism that is independent of ATP hydrolysis (44) (Fig. 2). UvrD uses the MutH-generated single-stranded DNA nicks as entry sites to displace the daughter strand regardless of whether the nick where it is loaded is 5’ or 3’ to the mispair (45, 46); to do this, UvrD translocates on the parental strand for 5’ nicks and on the daughter strand for 3’ nicks, as it is a dedicated 3’-5’ DNA helicase. UvrD has limited processivity as a dsDNA helicase (40-50 bp in one study and ~250 bp in another) but substantial processivity (~2400 nt) as a ssDNA translocase (47-49), which has led to a model suggesting that MutL loads multiple UvrD helicases, each of which only unwinds a portion of DNA before dissociating and is replaced by another helicase that translocates along the growing ssDNA gap (50). Although the UvrD-generated single-stranded DNA can be degraded by single-stranded nucleases in vitro (51), it is unclear if this is an important processing pathway in vivo. Moreover, repair is stimulated by the presence of multiple daughter strand nicks that flank the mispair, which would allow UvrD to directly remove the daughter strand to generate a gap (52). Termination of strand displacement on substrates that only have a single hemi-methylated d(GATC) site ends about 100 nucleotides past the mispair (53). The resulting gap can be filled by the action of DNA polymerase III and DNA ligase in vitro (54), which appears to depend on the ability of MutS to recruit the replicative β-clamp (55).
3. Strand discrimination in organisms with methyl-independent MMR
How organisms with methyl-independent MMR perform strand discrimination in vivo is controversial, although pre-existing DNA nicks suffice for strand discrimination in vitro. Methyl-independent MMR share many initial steps with methyl-dependent MMR. First, mispairs are recognized by MutS homologs; in eukaryotes these are the partially redundant eukaryotic heterodimers MSH2-MSH6 (MutSα) and MSH2-MSH3 (MutSβ). MSH2-MSH6 preferentially recognizes base-base mispairs and short insertion/deletion mispairs, whereas MSH2-MSH3 only recognizes a subset of base-base mispairs and has greater affinity for larger insertion/deletion mispairs (56, 57). Second, MutS homologs recruit MutL homologs to DNA. In eukaryotes, the MutL homologs involved in mitotic MMR are MLH1-PMS2 (MutLα, called Mlh1-Pms1 in Saccharomyces cerevisiae), which has a major role in MMR, and MLH1-MLH3 (MutLγ), which has an important role in meiotic crossing over but only a minor role in MMR (58, 59). Two major differences from methyl-dependent MMR are the lack of Dam or MutH homologs and the presence of a single-stranded DNA endonuclease activity in the MutL homologs (present in the eukaryotic PMS2 and MLH3 subunits but not MLH1), which is activated by replicative sliding clamps: the β-clamp for bacteria and proliferating cell nuclear antigen (PCNA) for eukaryotes (60-72). Consistent with this, disruption of the MutL-β-clamp interaction causes a weak mutator phenotype in E. coli but completely eliminates methyl-independent MMR in Bacillus subtilis (70). In bacterial methyl-independent MMR, the UvrD helicase appears to acts in MMR, as uvrD mutants cause increased mutation rates (73), and UvrD activity in these organisms is stimulated by MutL (65). Eukaryotes, however, appear to primarily target the daughter strand with nucleases.
3.1. Early experimental evidence for eukaryotic strand-discrimination in vitro
Initial efforts to investigate eukaryotic MMR in vitro focused on reactions involving cell-free extracts and plasmid substrates that typically contained a mispair and a nick present either 5’ or 3’ of the mispair (74-78) (Fig. 3). In systems used to follow mismatch-provoked excision, the mispair-containing substrates were more extensively resected than fully base-paired substrates (79). Excision targeted the nicked strand and excised from the nick to sites that were generally 100 nucleotides or further past the mispair (79, 80). The lack of a pre-existing nick in the substrate eliminated repair in Drosophila nuclear extracts and gave rise to products that showed no strand bias in mammalian nuclear extracts or Xenopus egg extracts (74, 77, 81). Thus, strand discrimination in cell-free extracts is dictated by the presence of a pre-existing nick on one of the strands, though these results do not reveal if the nick directly or indirectly mediates strand discrimination.
Figure 3. In vitro reconstituted eukaryotic MMR reactions.
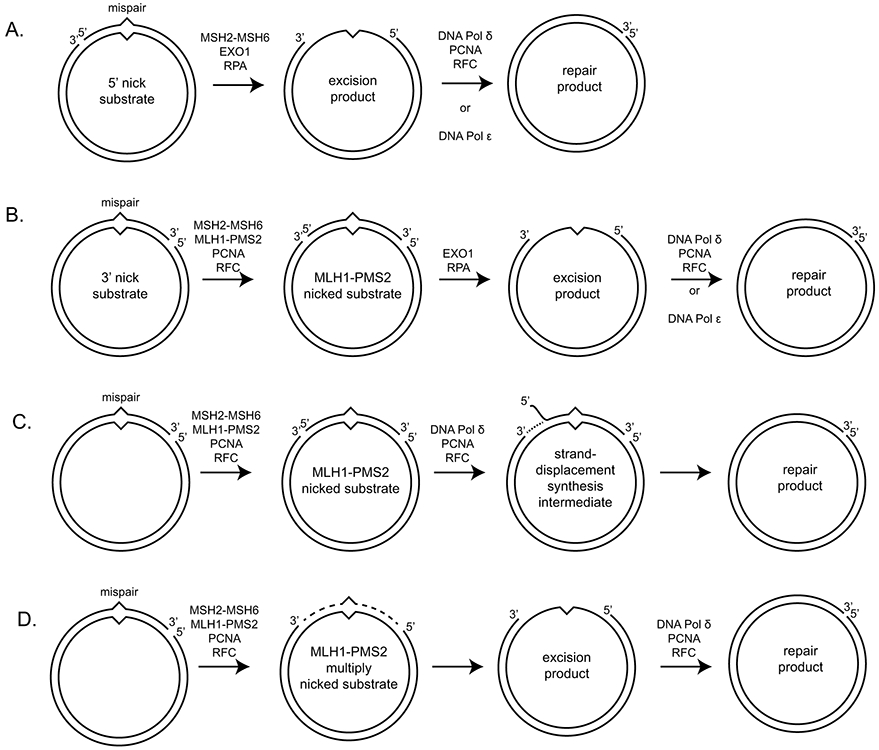
A. EXO1-dependent repair of a mispair-containing substrate with a 5’ nick can proceed by mispair recognition by MSH2-MSH6 and EXO1 recruitment. Nicked strand degradation uses the 5’-3’ exonuclease activity of EXO1. B. EXO1-dependent repair of a substrate with a 3’ nick requires the MLH1-PMS2 endonuclease and its PCNA activator to generate a 5’ nick. Repair then occurs for the 5’ nick containing substrate. C. One possible repair reaction of a 3’ nicked substrate in the absence of EXO1 repair can proceed by generation of a 5’ nick by MLH1-PMS2 followed by strand displacement synthesis by DNA polymerase δ. D. Another possible repair reaction in the absence of EXO1 can proceed by the generation of multiple nicks by MLH1-PMS2 that can give rise to a gap through daughter fragment dissociation, which can then be repaired by gap filling using a DNA polymerase.
3.2. Direct utilization of nicks for eukaryotic strand discrimination in vitro
In vitro reconstitution of MMR reactions with purified human and S. cerevisiae proteins revealed that a nick could mediate strand-specificity if the nick was 5’ to the mispair (82-84) (Fig. 3A). This reaction only required MSH2-MSH6, Exonuclease 1 (EXO1), which was implicated as a MMR factor on the basis of its interaction with MSH2 (85), and the single-strand binding protein RPA, which improves excision by EXO1. This reaction is, in essence, the simplest strand-specific reaction. However, this nicked-strand specificity is driven by: [1] the direct recognition of the nick by EXO1, [2] EXO1’s obligatory 5’-3’ exonuclease directionality, and [3] the ability of MSH2-MSH6 to recruit and promote EXO1 processivity. The increased processivity is most likely due to the interaction of MSH2 with the of the MSH2-interaction peptide sequences (SHIP boxes) in the unstructured C-terminal tail of EXO1 (86). Complete repair reactions, which additionally require gap-filling, can be performed upon the addition of DNA polymerase δ with Replication Factor C (RFC) and PCNA or DNA polymerase ε (83, 84, 87, 88). In the reconstituted reaction, the presence of MLH1-PMS2 improves the mismatch specificity of the reaction (82).
3.3. Strand discrimination at a distance in eukaryotic reconstituted reactions in vitro
Reconstituted MMR reactions using substrates with 3’ nicks have also been performed (87, 89, 90) (Fig. 3B). In addition to the proteins required for 5’ nick-directed excision, these reactions also require MLH1-PMS2, PCNA, and RFC. Repair of 3’ nicked substrates depends upon: [1] the ability of MSH2-MSH6 to recognize mispairs and recruit MLH1-PMS2, but not form sliding clamps or bind PCNA, and [2] a functional MLH1-PMS2 nuclease active site (60, 61, 89, 91). The new protein requirements for the 3’ nicked substrates can be explained if MLH1-PMS2, PCNA, and RFC act to: [1] generate new nicks 5’ of the mispair, which allows a 5’ nick-directed reaction to proceed, and [2] retain strand discrimination. The RFC and PCNA requirements can be explained by MLH1-PMS2 activity: PCNA activates the MLH1-PMS2 endonuclease (60, 61), and RFC is known to be required only for loading PCNA in these reactions (92). Consistently, the PCNA inhibitor protein p21 completely block 3’ nick-directed excision in nuclear extracts but only partially block 5’ nick-directed excision (90). A striking feature of this retention of strand discrimination is that new strand-specific nicks can be generated hundreds of nucleotides away from the initial 3’ nick (60, 61, 89). Thus, the interactions of MLH1-PMS2, PCNA, and RFC with each other and the substrate DNA appears to be mediate strand discrimination at a distance.
An EXO1-independent reconstituted reaction can be performed in which DNA polymerase δ uses strand-displacement synthesis from a 5’ nick on substrates where MLH1-PMS2 is present to make nicks bracketing the mispair (Fig. 3C; (93)). Like the strand specificity of EXO1 in reconstituted reactions, strand specificity of the DNA polymerase is entirely due to the direct use of the 3’ end at a 5’ nick as a primer; the strand discrimination at a distance is again mediated by MLH1-PMS2, PCNA, and RFC. Although this reconstituted reaction is EXO1-independent, it is unclear if it plays a major role in EXO1-independent reactions in vivo. Loss of the gene encoding EXO1 in both mice and S. cerevisiae does not cause a substantial loss of MMR (78, 85). Genetic analyses to define the EXO1-independent pathway in in exo1Δ S. cerevisiae strains have revealed an accumulation of Pms1-GFP foci (20) and a hypersensitivity to defects that decrease the nuclease activity of Mlh1-Pms1 (human MLH1-PMS2; (89, 94, 95)), suggesting that at least one EXO1-independent pathway involves sequential cleavage events by Mlh1-Pms1 in a strand-specific manner (Fig. 3D; (96)).
3.4. The MutL-β-clamp/PMS2-PCNA interaction: the lynchpin of strand discrimination?
In eukaryotic reconstituted reactions in vitro, reactions where nick-directed strand discrimination occurs over a distance require both MLH1-PMS2 and PCNA. As for methyl-directed MMR, neither the recruitment of MutS homologs nor recruitment of MutL homologs seem likely mediate strand discrimination, as described above. Thus, strand discrimination as observed in vitro probably involve interactions between the pre-existing nick, the migrating MutL homolog clamp, and/or the replicative sliding clamp.
MutL homologs are comprised of N- and C-terminal domains that are separated by an unstructured interdomain linker (Fig. 4A). The N-terminal domain belongs to the GHKL-family of ATPases and dimerizes upon ATP binding to form the MutL sliding clamp (97, 98). The C-terminal domain in most organisms is also comprised of two subdomains: a “dimerization” subdomain that mediates constitutive dimerization and a “regulatory” subdomain insertion into the dimerization subdomain that contains a sequence that mediates interaction with PCNA or the β-clamp (99, 100). The dimerization subdomain contains the nuclease active site. In MutL homodimers, both subunits contain active sites; however, the nuclease active site and the PCNA interaction motifs in eukaryotic heterodimers exist on the same subunit (Fig. 4B) (60, 61, 64). Remarkably, engineered B. subtilis MutL C-terminal domain dimers possessing only one endonuclease active site are functional, and the subunit with the endonuclease active site requires the ability to bind the β-clamp for full activity, suggesting that bacterial MutL, like bacterial MutS, is functionally asymmetric (101) and that the core aspects of this functional asymmetry are reflected in the duplication and specialization of the MLH1-PMS2 and MLH1-MLH3 eukaryotic homologs.
Figure 4. Features of the C-terminal domains of MutL homologs.

A. Ribbon diagram of E. coli MutL homolog comprised of N-terminal ATPase domains (top; PDB ID 1b62 (98)) and a C-terminal dimerization domain made up of a dimerization/nuclease subdomain and a regulatory subdomain (bottom; PDB ID 1x9z (99)). B. Comparison of the C-terminal domain structures of E. coli MutL (a methyl-directed MutL without an endonuclease active site; PDB ID 1x9z (99)), B. subtilis MutL (a methyl-independent MutL with an endonuclease active site; PDB ID 3kdk (64)), Aquifex aeolicus MutL (containing an endonuclease active site but not a regulatory subdomain; PDB ID 5b42 (151)), and S. cerevisiae Mlh1-Pms1 (containing an endonuclease active site and a regulatory subdomain in the Pms1 protein; PDB ID 4e4w (154)). C. Diagram of MutLs from methyl-dependent and methyl-independent subfamilies I-III. Endonuclease domains are missing from the methyl-dependent MutL proteins, but present in the methyl-independent MutLs. The methyl-independent subfamilies II and III lack the regulatory domain involved in interaction with the β-clamp through mutation (subfamily II) or deletion (subfamily III).
PCNA and the β-clamp are preferentially loaded onto the 3’ end of nicks and gaps in double-stranded DNA by the eukaryotic RFC clamp loader and the bacterial clamp loader (γ-complex), respectively (102). The replicative sliding clamps have “front” and “back” faces along the DNA axis; however, this asymmetry alone cannot explain strand specific cleavage, particularly for plasmid substrates where these clamps can diffuse around the entire ring and present both the “front” and “back” faces towards the site of the mispair. Thus, strand-specificity by the replication clamp/MutL homolog complex must involve additional asymmetry introduced by the bound DNA. The available data are consistent with two possible mechanisms: [1] a “MutL-directed strand specificity” mechanism and [2] a “strand-specific clamp diffusion” mechanism.
In the “MutL-directed strand specificity” mechanism, the asymmetry of the parental and daughter strands is read out using the relative orientations of the strands relative to the front face of the sliding clamp. Here, the asymmetric loading of the replicative clamps (Fig. 5A) allows the strands to be distinguished based on their orientation relative to the clamp faces. Using a definition of the “front” face of the replicative clamp as the face where the replicative clamp loader and MutL homologs bind (100, 103), the daughter strand always emerges in a 5’-3’ direction from the front face and the parental strand always emerges in a 3’-5’ direction. This asymmetry is tolerant of non-strand-specific diffusion of the replicative clamps along the DNA (Fig. 5B). However, the mechanism requires that MutL homologs must specifically cleave the strand emerging in the 5’-3’ direction from the “front” face of the replicative clamp (Fig. 5C).
Figure 5. The MutL-directed strand specificity model for strand-specific cleavage.

A. The bacteria β-clamp (green) is loaded by the clamp loader (subunits black to grey) so that the “front” face is oriented towards the 3’ end of the discontinuous (daughter) strand. The model was generated using the E. coli β-clamp DNA complex (PDB ID 3bep (111)), the E. coli clamp loader bound to a primer-template DNA (grey; PDB ID 3glf (102)) and the β-clamp complex with clamp loader δ subunit (not shown; PDB ID 1jqj (103)). B. The orientation of the faces of the replicative clamps relative to the two strands are preserved during potentially non-strand-specific diffusion along DNA. C. To mediate daughter strand specific cleavage, clamp-bound MutL homologs (blue) must specifically cleave the strand emerging in a 5’ to 3’ direction from the “front” face of the replicative clamp (red arrows). Image generated with the B. subtilis β-clamp fused to the MutL regulatory subdomain (PDB ID 6e8d (100)).
In the “strand-specific clamp diffusion” mechanism, the strand specificity is imparted to the replicative sliding clamps during loading and retained by strand-specific diffusion. This model posits that only the replicative clamps mediate strand specificity; the MutL homologs cleavage is directed to the strand presented by the clamp. The key requirement of this mechanism is that the replicative clamps must diffuse in a strand-specific way along DNA after asymmetric loading. Growing evidence indicates that both PCNA and the β-clamp asymmetrically interact with DNA that passes through the center of the clamps (104-107). Additionally, single molecule studies have revealed that PCNA migrates along DNA primarily through a translation mechanism that is coupled with the DNA helical pitch, consistent with tracking along a single strand (108). The mechanism underlying strand-specific tracking of PCNA has been revealed by the crystal structure and molecular dynamics simulation of PCNA in complex with a double-stranded DNA fragment; PCNA contains a path of DNA phosphate-interacting residues that readily track along a single strand as the protein rotates (Fig. 6A,B; (106, 107)). Strand-specific tracking of the replicative clamps bound to DNA polymerases is more equivocal; the 8 Å resolution cryo-EM structure of the E. coli beta clamp with DNA polymerase III and DNA did not show asymmetric DNA recognition by the beta clamp (109), but a recent 3 Å cryo-EM structure of the human DNA polymerase δ-PCNA revealed asymmetric PCNA-DNA contacts biased to the template strand in the processive conformation (110). Despite this, it is not clear that the replicative clamps associated with the processive DNA polymerases are the replicative clamps used in MMR, particularly given that MutL homolog-associated repair foci do not localize with replication forks, unlike MutS homologs (20).
Figure 6. The strand-specific clamp diffusion model for strand-specific cleavage.

A. Structure of the human PCNA trimer (green surface and cartoon) bound to a double-stranded DNA fragment with the tracked strand in orange (PDB ID 6gis (106)). B. Detail of the interactions of the phosphates of the tracked DNA strand with positively charged residues primarily in one PCNA subunit. C. The bacteria β-clamp (green) is loaded onto the continuous (parental; orange) strand of a DNA duplex by the clamp loader (subunits black to grey) as shown in Figure 5A, orienting the clamps to track along the parental strand by strand-specific diffusion. D. The β-clamp (green) interaction with the regulatory subdomain of the MutL C-terminus (light blue) places the active site of the molecule (dark blue) in an appropriate position to cleave the non-tracked, daughter DNA strand (red). This model, together with strand-specific loading shown in panel C, explains how nicks, the replicative sliding clamp, and MutL homologs can mediate strand specificity. The model was generated using the structure of the B. subtilis β-clamp fused to the MutL regulatory subdomain (PDB ID 6e8d (100)), the B. subtilis MutL C-terminal domain (PDB ID 3kdk (64)), and the E. coli β-clamp DNA complex (PDB ID 3bep (111)).
A structural model for the “strand-specific clamp diffusion” mechanism can be built following the work of McNally et al. (105) in modeling an RFC-PCNA-DNA loading intermediate through superimposition of the E. coli clamp loader (γ-complex) bound to a primer-template DNA (102), the E. coli β-clamp bound to DNA (111), and B-form DNA (Fig. 6C). This model indicates that the β-clamp is loaded so that it makes extensive contacts with the continuous DNA strand, suggesting that DNA backbone-coupled diffusion acts along this continuous (parental) strand. Note that this model has the same δ-subunit/β-clamp interaction as the isolated β-δ complex (103); however, the angles between the subunits are different, as the β-clamp orientation in the isolated structure is sterically precluded by the other subunits in the γ-complex. Similarly, superimposition of the structure of the B. subtilis β-clamp with the regulatory C-terminal subdomain of MutL (100), the structure of the B. subtilis C-terminal domain (64), and the structure of the E. coli β-clamp bound asymmetrically to DNA (111) can be used to generate a model for DNA cleavage (Fig. 6D). This second model suggests that the MutL subunit that makes contact with the β-clamp is the only one positioned so that the active site can interact with DNA, consistent with results from engineered heterodimeric B. subtilis MutL variants (101) and the conservation of a PCNA interaction for the active site-containing PMS2 (S. cerevisiae Pms1) and MLH3 subunits, but not MLH1 (69). This second model also suggests that the strand that comes into the vicinity of the MutL endonuclease active site is not the strand that the β-clamp appears to track along. Combining both models in the “strand-specific clamp diffusion” mechanism indicates that the clamp loader loads replicative sliding clamps so that they track along the parental strand and that interaction with MutL homologs specifically positions the daughter strand for cleavage.
Regardless of how the replicative clamp-MutL homolog complexes mediate strand-specificity, this specificity is consistent with multiple observations. First, under conditions of very low ionic strength, MLH1-PMS2 can nick substrates independent of loading by MSH2-MSH6. In the absence of PCNA and RFC, cleavage of the nicked and continuous strands of a plasmid substrate were similar, but addition of PCNA and RFC increased the nicking of the already nicked DNA strand (61). Second, there is no bias in strand cleavage on supercoiled or bubble-containing heteroduplex-containing substrates lacking nicks where PCNA can be loaded and MLH1-PMS2 can be activated independently of MSH2-MSH6 and where the loading of both proteins is not directed to a specific strand (92). Third, pre-loading of PCNA onto a nicked plasmid followed by DNA ligation prior is sufficient to mediate nicked strand-specific MMR in Xenopus egg nucleoplasmic extracts (112). The ability of PCNA to bind MSH2-MSH6 through the N-terminal PIP box (113-116) was required for preventing PCNA unloading in these extracts; however, loss of the PIP box interaction only causes a modest MMR defect in S. cerevisiae (113, 114), suggesting that PCNA retention is not required for most MMR in vivo. Strand-specific repair mediated by pre-loaded PCNA on substrates without nicks, however, strongly argues that PCNA loading asymmetry is sufficient to retain strand-specificity, even though it cannot rule out potential roles of MLH1-PMS2 in the recognition of pre-existing nicks, which should resemble products of the endonuclease reaction.
3.5. Experimental evidence for nicks in eukaryotic strand discrimination in vivo.
If pre-existing nicks and strand-specific translocation of replicative sliding clamps are also fundamental components of strand discrimination in vivo, then one obvious source of these pre-existing nicks is DNA replication. A role for some form of DNA replication intermediate in mediating strand discrimination, such as daughter-strand discontinuities, would explain the narrow post-replication temporal window for MMR proficiency in S. cerevisiae (117, 118) and the failure of screens for mutator mutants in S. cerevisiae to identify novel dedicated strand-discrimination components acting in MMR (95, 119, 120).
Several lines of evidence are consistent with the roles of both DNA-bound PCNA and daughter strand nicks and gaps in mediating strand discrimination in vivo. First, S. cerevisiae PCNA mutations give rise to MMR defects in vivo, and many of these mutant alleles encode proteins with defects in PCNA trimer stability and in DNA loading (94, 95, 121-125). Second, overexpression of the replicative DNA ligase, Cdc9, causes a MMR defect in S. cerevisiae that synergizes with defects in both the Exo1-dependent and Exo1-independent MMR pathways (118). Interestingly, Cdc9 overexpression is not associated with loss of DNA-bound PCNA, despite the fact that PCNA unloading by the Elg1 alternative RFC complex occurs after ligation of Okazaki fragments (126). Third, delaying the expression of Cdc9 until the G2/M phase of the cell cycle is sufficient to rescue the ability of G2/M-expressed Mlh1-Pms1 to mediate MMR at loci that replicate in S-phase (118). Together these results indicate that the lifetime of replication-associated nicks controls the temporal window in which strand discrimination can occur in vivo.
Inappropriately increased levels of DNA-bound PCNA, caused by deletion of ELG1 or by PCNA mutations, cause a modest MMR defect that is epistatic to the MMR-defective msh6Δ msh3Δ double mutation (127). This result has been interpreted to be due to over-recruitment of Msh2-Msh6 (127). An intriguing possibility, however, is that PCNA molecules, at some rate, lose strand-specific diffusion along DNA that is required in the “strand-specific clamp diffusion” mechanism. Single-molecule experiment suggest that PCNA spends 97-99% of its time helically sliding along the DNA and the rest in a non-helical mode that does not track along the backbone (108). Thus, timely removal of PCNA by the Elg1 alternative RFC complex may prevent the accumulation of PCNA molecules that have lost strand-specific diffusion. This model would predict that the elg1Δ mutation would cause increased mutation rates due to a loss of strand-discrimination but not mispair-provoked excision, similar to the effect of dam mutations in E. coli (4-7).
3.6. Sources of daughter-strand nicks in eukaryotes in vivo.
Nuclear DNA replication involves the action of three major DNA polymerases at the replication fork (128). DNA polymerase α adds 20-30 nucleotides to a ~10 base RNA primer generated by DNA primase. This occurs during both leading and lagging strand replication at replication origins and at the initiation of each Okazaki fragment during lagging strand replication (every ~200 nt in eukaryotes). DNA polymerase δ plays a major role in extending the RNA primers in lagging strand replication, whereas DNA polymerase ε plays the major role in leading strand replication (129). Both DNA polymerases δ and ε contain intrinsic proofreading 3’-5’ exonuclease activities that improve their fidelity, whereas DNA polymerase α does not and has an estimated error rate of 10−4 errors per base synthesized (130). Thus, lagging strand replication is both the most obvious source of transient nicks as well as a likely source of most of the replication errors (10-20% of the nucleotides in Okazaki fragments are synthesized by DNA polymerase α). Genetic and biochemical results suggest that errors generated by DNA polymerase α are repaired using MMR or using strand displacement synthesis by DNA polymerase δ combined with 5’ flap cleavage by flap-endonuclease 1 (FEN1) (131-133). Additionally, mutator variants of DNA polymerases, single-stranded oligonucleotide transformation, and 8-oxoguanine-induced mutagenesis in ogg1Δ strains of S. cerevisiae suggest that MMR suppresses lagging strand mutations at a somewhat higher rate than the leading strand (134-136). Thus, there is substantial evidence of the use of daughter-strand nicks during lagging strand replication.
Despite the fact that leading strand replication lacks Okazaki fragment-like formation of regular daughter strand nicks, there is direct evidence that MMR acts on both leading and lagging DNA strand replication. Proofreading-defective versions of both DNA polymerases δ and ε are synthetically lethal with loss of MMR in haploid S. cerevisiae cells due to “replication error-induced extinction” (137, 138). Additionally, MMR acts on errors made by mutator variants of all three replicative DNA polymerases (131). Thus, eukaryotic MMR can correct mispairs on daughter strands generated during both leading and lagging strand replication.
It has been proposed that misincorporated ribonucleotides could be a major strand discrimination signal during leading strand replication after these ribonucleotides are converted to gaps by the action of RNase H2 (139, 140). Single ribonucleotides support strand-specific MMR in nuclear extracts and reconstituted biochemical reactions in vitro (139), but the single ribonucleotides in these reactions are a source of strand discontinuities, and discontinuities generated by the excision of other lesions, such as O6-methylguanine (141), can also work. The in vivo evidence for a major role of ribonucleotides is more equivocal. RNase H2 mutants do not cause a strong mutator phenotype even in S. cerevisiae strains with polymerase mutations that increase the ribonucleotide misincorporation rate by 10-fold (120, 139, 140, 142, 143). In addition, the spectrum of mutations in RNase H2-deficient strains are dominated by rather unusual deletions of short tandem repeats that depend on the action of topoisomerase I on ribonucleotides (142, 143) and are very different than those caused by loss of MMR (144). Thus, while it is probable that ribonucleotides can be used as a source of strand discontinuities like O6-methylguanine (141), the available evidence suggests that they are unlikely to be the major contributor in vivo.
Recent high-throughput sequencing strategies have been used to map nicks in both human and S. cerevisiae cells have identified nicks formed during both leading and lagging strand replication (145). Ablation of the Cdc9 DNA ligase led to a substantial accumulation of unligated Okazaki fragments formed during lagging strand replication. In contrast, most of the nicks detected in wild-type cells expressing Cdc9 result from leading strand replication. These results suggest that leading strand replication may be less perfectly continuous in vivo than predicted. Thus, it seems likely that nicks can be strand discrimination signals during both leading and lagging strand synthesis and that these signals are transient, particularly on the lagging strand, consistent with in vivo results for MMR proficiency (117, 118).
3.7. Maintenance of strand-discrimination signals in vivo?
Several lines of evidence indicate that the presence of pre-existing nicks is insufficient for mediating MMR in vivo and that MutL homologs maintain strand-discrimination signals. First, despite the presence of MLH1-PMS2-independent in vitro reactions (82, 83), both humans and S. cerevisiae require the endonuclease activity MLH1-PMS2 for MMR in vivo (59, 146). Second, nuclear extracts of the human colorectal cancer cell line HCT116, which lack MLH1, do not support excision from a 5’ nick (147). This MutL homolog endonuclease requirement would be consistent with a competition between nick utilization by MMR and DNA ligation in vivo (118) and with a requirement for gaps and not nicks in MMR mediated by Xenopus egg nucleoplasmic extracts (112). Thus, daughter strand-specific cleavage might not only direct strand-specific MMR, but might also maintain the proficiency of MMR via ongoing daughter strand cleavage to generate additional single-stranded nicks and/or gaps. Loss of the mispair during MMR-mediated excision and subsequent loss of ongoing cleavage by MutL homologs would then allow the competing gap-filling and nick-ligation reactions to reseal the daughter strand and complete repair.
4.0. Mismatch repair without strand discrimination?
A MMR reaction that lacks a requirement for strand discrimination was proposed nearly 40 years ago (148). This repair was envisioned to involve the formation of double-stranded DNA breaks (DSBs) at mispairs combined with homologous recombination with the sister chromatid. Remarkably NucS-mediated MMR, which makes double-strand breaks at mispairs (149, 150), may use this type of mechanism. NucS prevents mutations and suppresses homeologous recombination in vivo in the bacterium Mycobacterium smegmatis, and NucS homologs are primarily found in Actinobacteria, Euryarchaeota, Thaumarchaeota, Aigarchaeota, Crenarchaeota, and Korarchaeota both individually and alongside a MutS/L-dependent MMR system (149).
Intriguingly, a recombination-mediated MMR mechanism might act in a subset of MutS/L-containing bacteria. A number of bacteria with methyl-independent MMR, including those in the phyla Aquificae, Thermotogae, Cyanobacteria, and Nitrospirae, contain MutL homologs lacking the β-clamp interaction motif either through substitutions in the motif (subfamily II) or through deletion (subfamily III) (Fig. 4C; (151)). Subfamily II and III MutL proteins also lack the negatively charged patch on the C-terminal domain, which is neutralized by the β-clamp interaction in subfamily I. Consistent with these structural features, these MutL proteins have β-clamp-independent endonuclease activities (151), and reconstituted MMR reactions with Thermus thermophilus proteins (subfamily II) were mispair-specific but not strand specific (152). Subfamily II and III MutL proteins also have short or non-existent interdomain linkers between the N- and C-terminal domains, which play key roles in the ability of E. coli and eukaryotic MutL homologs to migrate along DNA (29, 35, 153). Thus, subfamily II and III MutL proteins may use an unidentified strand-discrimination strategy or use the lack of strand discrimination (and lack of migrating MutL clamps) to generate mispair-proximal DSBs to drive recombination-mediated MMR.
5.0. Conclusions
Although the details of strand-discrimination differ between methyl-directed and methyl-independent MMR, the general strategy is the same. MMR is initiated at daughter-specific nicks strand, taking advantage of a transient replication-associated signal that distinguishes the daughter and parental strands. The methyl-independent excision-repair system, as present in eukaryotes and bacteria including B. subtilis, is simpler than the more derived methyl-directed MMR system present in bacteria like E. coli. Strand discrimination at a distance in these systems seems directed by details of the asymmetric replicative clamp loading at replication-generated nicks and the interactions of the replicative clamp to activate the MutL homolog. It remains to be seen, however, if subfamily II and III MutL homologs, which lack strand discrimination in the presence of a nicked plasmid and the β-clamp, mediate MMR without strand discrimination. If so, it raises the question of whether these HR-mediated MMR systems were ancestral to or were derived from the nick-directed MMR system found in organisms like eukaryotes.
Acknowledgements.
This review only exists due to the gentle encouragement of Dr. Sam Wilson and is dedicated to the memory of his scientific contributions both as a researcher and as a tireless proponent of excellence in the DNA repair field. I also want to thank the anonymous reviewer whose arguments outlined the basis of the “MutL-directed strand specificity” mechanism.
Funding.
This work was supported by the Ludwig Institute for Cancer Research and the NIH grant R01 GM50006.
References
- 1.Fishel R, Mismatch repair. J Biol Chem 290, 26395–26403 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Iyer RR, Pluciennik A, Burdett V, Modrich PL, DNA mismatch repair: functions and mechanisms. Chem Rev 106, 302–323 (2006). [DOI] [PubMed] [Google Scholar]
- 3.Reyes GX, Schmidt TT, Kolodner RD, Hombauer H, New insights into the mechanism of DNA mismatch repair. Chromosoma 124, 443–462 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marinus MG, Morris NR, Pleiotropic effects of a DNA adenine methylation mutation (dam-3) in Escherichia coli K12. Mutat Res 28, 15–26 (1975). [DOI] [PubMed] [Google Scholar]
- 5.Marinus MG, Morris NR, Biological function for 6-methyladenine residues in the DNA of Escherichia coli K12. J Mol Biol 85, 309–322 (1974). [DOI] [PubMed] [Google Scholar]
- 6.Glickman B, van den Elsen P, Radman M, Induced mutagenesis in dam- mutants of Escherichia coli: a role for 6-methyladenine residues in mutation avoidance. Mol Gen Genet 163, 307–312 (1978). [DOI] [PubMed] [Google Scholar]
- 7.Marinus MG, Recombination is essential for viability of an Escherichia coli dam (DNA adenine methyltransferase) mutant. J Bacteriol 182, 463–468 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Putnam CD, Evolution of the methyl directed mismatch repair system in Escherichia coli. DNA Repair (Amst) 38, 32–41 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hattman S, Brooks JE, Masurekar M, Sequence specificity of the P1 modification methylase (M.Eco P1) and the DNA methylase (M.Eco dam) controlled by the Escherichia coli dam gene. J Mol Biol 126, 367–380 (1978). [DOI] [PubMed] [Google Scholar]
- 10.Pukkila PJ, Peterson J, Herman G, Modrich P, Meselson M, Effects of high levels of DNA adenine methylation on methyl-directed mismatch repair in Escherichia coli. Genetics 104, 571–582 (1983). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wagner R Jr., Meselson M, Repair tracts in mismatched DNA heteroduplexes. Proc Natl Acad Sci U S A 73, 4135–4139 (1976). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Herman GE, Modrich P, Escherichia coli K-12 clones that overproduce dam methylase are hypermutable. J Bacteriol 145, 644–646 (1981). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Campbell JL, Kleckner N, coli oriC E and the dnaA gene promoter are sequestered from dam methyltransferase following the passage of the chromosomal replication fork. Cell 62, 967–979 (1990). [DOI] [PubMed] [Google Scholar]
- 14.Weitao T, Nordstrom K, Dasgupta S, Escherichia coli cell cycle control genes affect chromosome superhelicity. EMBO Rep 1, 494–499 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Campbell JL, Kleckner N, The rate of Dam-mediated DNA adenine methylation in Escherichia coli. Gene 74, 189–190 (1988). [DOI] [PubMed] [Google Scholar]
- 16.Stancheva I, Koller T, Sogo JM, Asymmetry of Dam remethylation on the leading and lagging arms of plasmid replicative intermediates. EMBO J 18, 6542–6551 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ogden GB, Pratt MJ, Schaechter M, The replicative origin of the E. coli chromosome binds to cell membranes only when hemimethylated. Cell 54, 127–135 (1988). [DOI] [PubMed] [Google Scholar]
- 18.Urig S et al. , The Escherichia coli dam DNA methyltransferase modifies DNA in a highly processive reaction. J Mol Biol 319, 1085–1096 (2002). [DOI] [PubMed] [Google Scholar]
- 19.Liao Y, Schroeder JW, Gao B, Simmons LA, Biteen JS, Single-molecule motions and interactions in live cells reveal target search dynamics in mismatch repair. Proc Natl Acad Sci U S A 112, E6898–6906 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hombauer H, Campbell CS, Smith CE, Desai A, Kolodner RD, Visualization of eukaryotic DNA mismatch repair reveals distinct recognition and repair intermediates. Cell 147, 1040–1053 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Simmons LA, Davies BW, Grossman AD, Walker GC, Beta clamp directs localization of mismatch repair in Bacillus subtilis. Mol Cell 29, 291–301 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smith BT, Grossman AD, Walker GC, Visualization of mismatch repair in bacterial cells. Mol Cell 8, 1197–1206 (2001). [DOI] [PubMed] [Google Scholar]
- 23.Lenhart JS, Sharma A, Hingorani MM, Simmons LA, DnaN clamp zones provide a platform for spatiotemporal coupling of mismatch detection to DNA replication. Mol Microbiol 87, 553–568 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cho WK et al. , ATP alters the diffusion mechanics of MutS on mismatched DNA. Structure 20, 1264–1274 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gorman J et al. , Single-molecule imaging reveals target-search mechanisms during DNA mismatch repair. Proc Natl Acad Sci U S A 109, E3074–3083 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fernandez-Leiro R et al. , The selection process of licensing a DNA mismatch for repair. Nat Struct Mol Biol 28, 373–381 (2021). [DOI] [PubMed] [Google Scholar]
- 27.Obmolova G, Ban C, Hsieh P, Yang W, Crystal structures of mismatch repair protein MutS and its complex with a substrate DNA. Nature 407, 703–710 (2000). [DOI] [PubMed] [Google Scholar]
- 28.Lamers MH et al. , The crystal structure of DNA mismatch repair protein MutS binding to a G x T mismatch. Nature 407, 711–717 (2000). [DOI] [PubMed] [Google Scholar]
- 29.Acharya S, Foster PL, Brooks P, Fishel R, The coordinated functions of the E. coli MutS and MutL proteins in mismatch repair. Mol Cell 12, 233–246 (2003). [DOI] [PubMed] [Google Scholar]
- 30.Hura GL et al. , DNA conformations in mismatch repair probed in solution by X-ray scattering from gold nanocrystals. Proc Natl Acad Sci U S A 110, 17308–17313 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Groothuizen FS et al. , MutS/MutL crystal structure reveals that the MutS sliding clamp loads MutL onto DNA. Elife 4, e06744 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.LeBlanc SJ et al. , Coordinated protein and DNA conformational changes govern mismatch repair initiation by MutS. Nucleic Acids Res 46, 10782–10795 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Qiu R et al. , MutL traps MutS at a DNA mismatch. Proc Natl Acad Sci U S A 112, 10914–10919 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu J et al. , Cascading MutS and MutL sliding clamps control DNA diffusion to activate mismatch repair. Nature 539, 583–587 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mardenborough YSN et al. , The unstructured linker arms of MutL enable GATC site incision beyond roadblocks during initiation of DNA mismatch repair. Nucleic Acids Res 47, 11667–11680 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ahrends R et al. , Identifying an interaction site between MutH and the C-terminal domain of MutL by crosslinking, affinity purification, chemical coding and mass spectrometry. Nucleic Acids Res 34, 3169–3180 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hall MC, Matson SW, The Escherichia coli MutL protein physically interacts with MutH and stimulates the MutH-associated endonuclease activity. J Biol Chem 274, 1306–1312 (1999). [DOI] [PubMed] [Google Scholar]
- 38.Welsh KM, Lu AL, Clark S, Modrich P, Isolation and characterization of the Escherichia coli mutH gene product. J Biol Chem 262, 15624–15629 (1987). [PubMed] [Google Scholar]
- 39.Ban C, Yang W, Structural basis for MutH activation in E.coli mismatch repair and relationship of MutH to restriction endonucleases. EMBO J 17, 1526–1534 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee JY et al. , MutH complexed with hemi- and unmethylated DNAs: coupling base recognition and DNA cleavage. Mol Cell 20, 155–166 (2005). [DOI] [PubMed] [Google Scholar]
- 41.Friedhoff P, Thomas E, Pingoud A, Tyr212: a key residue involved in strand discrimination by the DNA mismatch repair endonuclease MutH. J Mol Biol 325, 285–297 (2003). [DOI] [PubMed] [Google Scholar]
- 42.Au KG, Welsh K, Modrich P, Initiation of methyl-directed mismatch repair. J Biol Chem 267, 12142–12148 (1992). [PubMed] [Google Scholar]
- 43.Hasan AM, Leach DR, Chromosomal directionality of DNA mismatch repair in Escherichia coli. Proc Natl Acad Sci U S A 112, 9388–9393 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Robertson AB, Pattishall SR, Gibbons EA, Matson SW, MutL-catalyzed ATP hydrolysis is required at a post-UvrD loading step in methyl-directed mismatch repair. J Biol Chem 281, 19949–19959 (2006). [DOI] [PubMed] [Google Scholar]
- 45.Hall MC, Jordan JR, Matson SW, Evidence for a physical interaction between the Escherichia coli methyl-directed mismatch repair proteins MutL and UvrD. EMBO J 17, 1535–1541 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yamaguchi M, Dao V, Modrich P, MutS and MutL activate DNA helicase II in a mismatch-dependent manner. J Biol Chem 273, 9197–9201 (1998). [DOI] [PubMed] [Google Scholar]
- 47.Ali JA, Lohman TM, Kinetic measurement of the step size of DNA unwinding by Escherichia coli UvrD helicase. Science 275, 377–380 (1997). [DOI] [PubMed] [Google Scholar]
- 48.Fischer CJ, Maluf NK, Lohman TM, Mechanism of ATP-dependent translocation of E.coli UvrD monomers along single-stranded DNA. J Mol Biol 344, 1287–1309 (2004). [DOI] [PubMed] [Google Scholar]
- 49.Dessinges MN, Lionnet T, Xi XG, Bensimon D, Croquette V, Single-molecule assay reveals strand switching and enhanced processivity of UvrD. Proc Natl Acad Sci U S A 101, 6439–6444 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Matson SW, Robertson AB, The UvrD helicase and its modulation by the mismatch repair protein MutL. Nucleic Acids Res 34, 4089–4097 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Burdett V, Baitinger C, Viswanathan M, Lovett ST, Modrich P, In vivo requirement for RecJ, ExoVII, ExoI, and ExoX in methyl-directed mismatch repair. Proc Natl Acad Sci U S A 98, 6765–6770 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hermans N et al. , Dual daughter strand incision is processive and increases the efficiency of DNA mismatch repair. Nucleic Acids Res 44, 6770–6786 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Grilley M, Griffith J, Modrich P, Bidirectional excision in methyl-directed mismatch repair. J Biol Chem 268, 11830–11837 (1993). [PubMed] [Google Scholar]
- 54.Lahue RS, Au KG, Modrich P, DNA mismatch correction in a defined system. Science 245, 160–164 (1989). [DOI] [PubMed] [Google Scholar]
- 55.Pluciennik A, Burdett V, Lukianova O, O'Donnell M, Modrich P, Involvement of the beta clamp in methyl-directed mismatch repair in vitro. J Biol Chem 284, 32782–32791 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Marsischky GT, Filosi N, Kane MF, Kolodner R, Redundancy of Saccharomyces cerevisiae MSH3 and MSH6 in MSH2-dependent mismatch repair. Genes Dev 10, 407–420 (1996). [DOI] [PubMed] [Google Scholar]
- 57.Dowen JM, Putnam CD, Kolodner RD, Functional studies and homology modeling of Msh2-Msh3 predict that mispair recognition involves DNA bending and strand separation. Mol Cell Biol 30, 3321–3328 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang TF, Kleckner N, Hunter N, Functional specificity of MutL homologs in yeast: evidence for three Mlh1-based heterocomplexes with distinct roles during meiosis in recombination and mismatch correction. Proc Natl Acad Sci U S A 96, 13914–13919 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Flores-Rozas H, Kolodner RD, The Saccharomyces cerevisiae MLH3 gene functions in MSH3-dependent suppression of frameshift mutations. Proc Natl Acad Sci U S A 95, 12404–12409 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kadyrov FA, Dzantiev L, Constantin N, Modrich P, Endonucleolytic function of MutLalpha in human mismatch repair. Cell 126, 297–308 (2006). [DOI] [PubMed] [Google Scholar]
- 61.Kadyrov FA et al. , Saccharomyces cerevisiae MutLalpha is a mismatch repair endonuclease. J Biol Chem 282, 37181–37190 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Correa EM, Martina MA, De Tullio L, Argarana CE, Barra JL, Some amino acids of the Pseudomonas aeruginosa MutL D(Q/M)HA(X)(2)E(X)(4)E conserved motif are essential for the in vivo function of the protein but not for the in vitro endonuclease activity. DNA Repair (Amst) 10, 1106–1113 (2011). [DOI] [PubMed] [Google Scholar]
- 63.Iino H et al. , Characterization of C- and N-terminal domains of Aquifex aeolicus MutL endonuclease: N-terminal domain stimulates the endonuclease activity of C-terminal domain in a zinc-dependent manner. Biosci Rep 31, 309–322 (2011). [DOI] [PubMed] [Google Scholar]
- 64.Pillon MC et al. , Structure of the endonuclease domain of MutL: unlicensed to cut. Mol Cell 39, 145–151 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mauris J, Evans TC Jr., A human PMS2 homologue from Aquifex aeolicus stimulates an ATP-dependent DNA helicase. J Biol Chem 285, 11087–11092 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Duppatla V, Bodda C, Urbanke C, Friedhoff P, Rao DN, The C-terminal domain is sufficient for endonuclease activity of Neisseria gonorrhoeae MutL. Biochem J 423, 265–277 (2009). [DOI] [PubMed] [Google Scholar]
- 67.Mauris J, Evans TC, Adenosine triphosphate stimulates Aquifex aeolicus MutL endonuclease activity. PLoS One 4, e7175 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fukui K, Nishida M, Nakagawa N, Masui R, Kuramitsu S, Bound nucleotide controls the endonuclease activity of mismatch repair enzyme MutL. J Biol Chem 283, 12136–12145 (2008). [DOI] [PubMed] [Google Scholar]
- 69.Genschel J et al. , Interaction of proliferating cell nuclear antigen with PMS2 is required for MutLalpha activation and function in mismatch repair. Proc Natl Acad Sci U S A 114, 4930–4935 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pillon MC et al. , The sliding clamp tethers the endonuclease domain of MutL to DNA. Nucleic Acids Res 43, 10746–10759 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cannavo E et al. , Regulation of the MLH1-MLH3 endonuclease in meiosis. Nature 10.1038/s41586-020-2592-2 (2020). [DOI] [PubMed] [Google Scholar]
- 72.Kulkarni DS et al. , PCNA activates the MutLgamma endonuclease to promote meiotic crossing over. Nature 10.1038/s41586-020-2645-6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Oliver A, Baquero F, Blazquez J, The mismatch repair system (mutS, mutL and uvrD genes) in Pseudomonas aeruginosa: molecular characterization of naturally occurring mutants. Mol Microbiol 43, 1641–1650 (2002). [DOI] [PubMed] [Google Scholar]
- 74.Holmes J Jr., Clark S, Modrich P, Strand-specific mismatch correction in nuclear extracts of human and Drosophila melanogaster cell lines. Proc Natl Acad Sci U S A 87, 5837–5841 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Thomas DC, Roberts JD, Kunkel TA, Heteroduplex repair in extracts of human HeLa cells. J Biol Chem 266, 3744–3751 (1991). [PubMed] [Google Scholar]
- 76.Muster-Nassal C, Kolodner R, Mismatch correction catalyzed by cell-free extracts of Saccharomyces cerevisiae. Proc Natl Acad Sci U S A 83, 7618–7622 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Varlet I, Radman M, Brooks P, DNA mismatch repair in Xenopus egg extracts: repair efficiency and DNA repair synthesis for all single base-pair mismatches. Proc Natl Acad Sci U S A 87, 7883–7887 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wei K et al. , Inactivation of Exonuclease 1 in mice results in DNA mismatch repair defects, increased cancer susceptibility, and male and female sterility. Genes Dev 17, 603–614 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang H, Hays JB, Mismatch repair in human nuclear extracts. Quantitative analyses of excision of nicked circular mismatched DNA substrates, constructed by a new technique employing synthetic oligonucleotides. J Biol Chem 277, 26136–26142 (2002). [DOI] [PubMed] [Google Scholar]
- 80.Fang WH, Modrich P, Human strand-specific mismatch repair occurs by a bidirectional mechanism similar to that of the bacterial reaction. J Biol Chem 268, 11838–11844 (1993). [PubMed] [Google Scholar]
- 81.Brown TC, Jiricny J, Different base/base mispairs are corrected with different efficiencies and specificities in monkey kidney cells. Cell 54, 705–711 (1988). [DOI] [PubMed] [Google Scholar]
- 82.Genschel J, Modrich P, Mechanism of 5'-directed excision in human mismatch repair. Mol Cell 12, 1077–1086 (2003). [DOI] [PubMed] [Google Scholar]
- 83.Bowen N et al. , Reconstitution of long and short patch mismatch repair reactions using Saccharomyces cerevisiae proteins. Proc Natl Acad Sci U S A 110, 18472–18477 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhang Y et al. , Reconstitution of 5'-directed human mismatch repair in a purified system. Cell 122, 693–705 (2005). [DOI] [PubMed] [Google Scholar]
- 85.Tishkoff DX et al. , Identification and characterization of Saccharomyces cerevisiae EXO1, a gene encoding an exonuclease that interacts with MSH2. Proc Natl Acad Sci U S A 94, 7487–7492 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Goellner EM et al. , Identification of Exo1-Msh2 interaction motifs in DNA mismatch repair and new Msh2-binding partners. Nat Struct Mol Biol 25, 650–659 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dzantiev L et al. , A defined human system that supports bidirectional mismatch-provoked excision. Mol Cell 15, 31–41 (2004). [DOI] [PubMed] [Google Scholar]
- 88.Bowen N, Kolodner RD, Reconstitution of Saccharomyces cerevisiae DNA polymerase epsilon-dependent mismatch repair with purified proteins. Proc Natl Acad Sci U S A 114, 3607–3612 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Smith CE et al. , Activation of Saccharomyces cerevisiae Mlh1-Pms1 Endonuclease in a Reconstituted Mismatch Repair System. J Biol Chem 290, 21580–21590 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Guo S et al. , Differential requirement for proliferating cell nuclear antigen in 5' and 3' nick-directed excision in human mismatch repair. J Biol Chem 279, 16912–16917 (2004). [DOI] [PubMed] [Google Scholar]
- 91.Iyer RR et al. , The MutSalpha-proliferating cell nuclear antigen interaction in human DNA mismatch repair. J Biol Chem 283, 13310–13319 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pluciennik A et al. , PCNA function in the activation and strand direction of MutLalpha endonuclease in mismatch repair. Proc Natl Acad Sci U S A 107, 16066–16071 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kadyrov FA et al. , A possible mechanism for exonuclease 1-independent eukaryotic mismatch repair. Proc Natl Acad Sci U S A 106, 8495–8500 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Goellner EM et al. , PCNA and Msh2-Msh6 activate an Mlh1-Pms1 endonuclease pathway required for Exo1-independent mismatch repair. Mol Cell 55, 291–304 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Amin NS, Nguyen MN, Oh S, Kolodner RD, exo1-Dependent mutator mutations: model system for studying functional interactions in mismatch repair. Mol Cell Biol 21, 5142–5155 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Goellner EM, Putnam CD, Kolodner RD, Exonuclease 1-dependent and independent mismatch repair. DNA Repair (Amst) 32, 24–32 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ban C, Junop M, Yang W, Transformation of MutL by ATP binding and hydrolysis: a switch in DNA mismatch repair. Cell 97, 85–97 (1999). [DOI] [PubMed] [Google Scholar]
- 98.Ban C, Yang W, Crystal structure and ATPase activity of MutL: implications for DNA repair and mutagenesis. Cell 95, 541–552 (1998). [DOI] [PubMed] [Google Scholar]
- 99.Guarne A et al. , Structure of the MutL C-terminal domain: a model of intact MutL and its roles in mismatch repair. EMBO J 23, 4134–4145 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Almawi AW et al. , Binding of the regulatory domain of MutL to the sliding beta-clamp is species specific. Nucleic Acids Res 47, 4831–4842 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Liu L, Ortiz Castro MC, Rodriguez Gonzalez J, Pillon MC, Guarne A, The endonuclease domain of Bacillus subtilis MutL is functionally asymmetric. DNA Repair (Amst) 73, 1–6 (2019). [DOI] [PubMed] [Google Scholar]
- 102.Simonetta KR et al. , The mechanism of ATP-dependent primer-template recognition by a clamp loader complex. Cell 137, 659–671 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Jeruzalmi D et al. , Mechanism of processivity clamp opening by the delta subunit wrench of the clamp loader complex of E. coli DNA polymerase III. Cell 106, 417–428 (2001). [PubMed] [Google Scholar]
- 104.Mayanagi K et al. , Mechanism of replication machinery assembly as revealed by the DNA ligase-PCNA-DNA complex architecture. Proc Natl Acad Sci U S A 106, 4647–4652 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.McNally R, Bowman GD, Goedken ER, O'Donnell M, Kuriyan J, Analysis of the role of PCNA-DNA contacts during clamp loading. BMC Struct Biol 10, 3 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.De March M et al. , Structural basis of human PCNA sliding on DNA. Nat Commun 8, 13935 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ivanov I, Chapados BR, McCammon JA, Tainer JA, Proliferating cell nuclear antigen loaded onto double-stranded DNA: dynamics, minor groove interactions and functional implications. Nucleic Acids Res 34, 6023–6033 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kochaniak AB et al. , Proliferating cell nuclear antigen uses two distinct modes to move along DNA. J Biol Chem 284, 17700–17710 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Fernandez-Leiro R, Conrad J, Scheres SH, Lamers MH, cryo-EM structures of the E. coli replicative DNA polymerase reveal its dynamic interactions with the DNA sliding clamp, exonuclease and tau. Elife 4 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lancey C et al. , Structure of the processive human Pol delta holoenzyme. Nat Commun 11, 1109 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Georgescu RE et al. , Structure of a sliding clamp on DNA. Cell 132, 43–54 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kawasoe Y, Tsurimoto T, Nakagawa T, Masukata H, Takahashi TS, MutSalpha maintains the mismatch repair capability by inhibiting PCNA unloading. Elife 5 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Shell SS, Putnam CD, Kolodner RD, The N terminus of Saccharomyces cerevisiae Msh6 is an unstructured tether to PCNA. Mol Cell 26, 565–578 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Flores-Rozas H, Clark D, Kolodner RD, Proliferating cell nuclear antigen and Msh2p-Msh6p interact to form an active mispair recognition complex. Nat Genet 26, 375–378 (2000). [DOI] [PubMed] [Google Scholar]
- 115.Clark AB, Valle F, Drotschmann K, Gary RK, Kunkel TA, Functional interaction of proliferating cell nuclear antigen with MSH2-MSH6 and MSH2-MSH3 complexes. J Biol Chem 275, 36498–36501 (2000). [DOI] [PubMed] [Google Scholar]
- 116.Kleczkowska HE, Marra G, Lettieri T, Jiricny J, hMSH3 and hMSH6 interact with PCNA and colocalize with it to replication foci. Genes Dev 15, 724–736 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hombauer H, Srivatsan A, Putnam CD, Kolodner RD, Mismatch repair, but not heteroduplex rejection, is temporally coupled to DNA replication. Science 334, 1713–1716 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Reyes GX et al. , Ligation of newly replicated DNA controls the timing of DNA mismatch repair. Curr Biol 31, 1268–1276 e1266 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Schmidt TT et al. , Alterations in cellular metabolism triggered by URA7 or GLN3 inactivation cause imbalanced dNTP pools and increased mutagenesis. Proc Natl Acad Sci U S A 114, E4442–E4451 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Huang ME, Rio AG, Nicolas A, Kolodner RD, A genomewide screen in Saccharomyces cerevisiae for genes that suppress the accumulation of mutations. Proc Natl Acad Sci U S A 100, 11529–11534 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Umar A et al. , Requirement for PCNA in DNA mismatch repair at a step preceding DNA resynthesis. Cell 87, 65–73 (1996). [DOI] [PubMed] [Google Scholar]
- 122.Johnson RE et al. , Evidence for involvement of yeast proliferating cell nuclear antigen in DNA mismatch repair. J Biol Chem 271, 27987–27990 (1996). [DOI] [PubMed] [Google Scholar]
- 123.Chen C, Merrill BJ, Lau PJ, Holm C, Kolodner RD, Saccharomyces cerevisiae pol30 (proliferating cell nuclear antigen) mutations impair replication fidelity and mismatch repair. Mol Cell Biol 19, 7801–7815 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lau PJ, Flores-Rozas H, Kolodner RD, Isolation and characterization of new proliferating cell nuclear antigen (POL30) mutator mutants that are defective in DNA mismatch repair. Mol Cell Biol 22, 6669–6680 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Zhou Y, Hingorani MM, Impact of individual proliferating cell nuclear antigen-DNA contacts on clamp loading and function on DNA. J Biol Chem 287, 35370–35381 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kubota T, Katou Y, Nakato R, Shirahige K, Donaldson AD, Replication-Coupled PCNA Unloading by the Elg1 Complex Occurs Genome-wide and Requires Okazaki Fragment Ligation. Cell Rep 12, 774–787 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Paul Solomon Devakumar LJ, Gaubitz C, Lundblad V, Kelch BA, Kubota T, Effective mismatch repair depends on timely control of PCNA retention on DNA by the Elg1 complex. Nucleic Acids Res 47, 6826–6841 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Burgers PM, Polymerase dynamics at the eukaryotic DNA replication fork. J Biol Chem 284, 4041–4045 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kunkel TA, Burgers PM, Dividing the workload at a eukaryotic replication fork. Trends Cell Biol 18, 521–527 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kunkel TA, Hamatake RK, Motto-Fox J, Fitzgerald MP, Sugino A, Fidelity of DNA polymerase I and the DNA polymerase I-DNA primase complex from Saccharomyces cerevisiae. Mol Cell Biol 9, 4447–4458 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lujan SA et al. , Mismatch repair balances leading and lagging strand DNA replication fidelity. PLoS Genet 8, e1003016 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Pavlov YI et al. , Evidence that errors made by DNA polymerase alpha are corrected by DNA polymerase delta. Curr Biol 16, 202–207 (2006). [DOI] [PubMed] [Google Scholar]
- 133.Liu S et al. , Okazaki fragment maturation involves alpha-segment error editing by the mammalian FEN1/MutSalpha functional complex. EMBO J 34, 1829–1843 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Pavlov YI, Mian IM, Kunkel TA, Evidence for preferential mismatch repair of lagging strand DNA replication errors in yeast. Curr Biol 13, 744–748 (2003). [DOI] [PubMed] [Google Scholar]
- 135.Kow YW, Bao G, Reeves JW, Jinks-Robertson S, Crouse GF, Oligonucleotide transformation of yeast reveals mismatch repair complexes to be differentially active on DNA replication strands. Proc Natl Acad Sci U S A 104, 11352–11357 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Nick McElhinny SA, Kissling GE, Kunkel TA, Differential correction of lagging-strand replication errors made by DNA polymerases {alpha} and {delta}. Proc Natl Acad Sci U S A 107, 21070–21075 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Williams LN, Herr AJ, Preston BD, Emergence of DNA polymerase epsilon antimutators that escape error-induced extinction in yeast. Genetics 193, 751–770 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Morrison A, Johnson AL, Johnston LH, Sugino A, Pathway correcting DNA replication errors in Saccharomyces cerevisiae. EMBO J 12, 1467–1473 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Ghodgaonkar MM et al. , Ribonucleotides misincorporated into DNA act as strand-discrimination signals in eukaryotic mismatch repair. Mol Cell 50, 323–332 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Lujan SA, Williams JS, Clausen AR, Clark AB, Kunkel TA, Ribonucleotides are signals for mismatch repair of leading-strand replication errors. Mol Cell 50, 437–443 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Olivera Harris M et al. , Mismatch repair-dependent metabolism of O6-methylguanine-containing DNA in Xenopus laevis egg extracts. DNA Repair (Amst) 28, 1–7 (2015). [DOI] [PubMed] [Google Scholar]
- 142.Allen-Soltero S, Martinez SL, Putnam CD, Kolodner RD, A saccharomyces cerevisiae RNase H2 interaction network functions to suppress genome instability. Mol Cell Biol 34, 1521–1534 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kim N et al. , Mutagenic processing of ribonucleotides in DNA by yeast topoisomerase I. Science 332, 1561–1564 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Tishkoff DX, Filosi N, Gaida GM, Kolodner RD, A novel mutation avoidance mechanism dependent on S. cerevisiae RAD27 is distinct from DNA mismatch repair. Cell 88, 253–263 (1997). [DOI] [PubMed] [Google Scholar]
- 145.Sriramachandran AM et al. , Genome-wide Nucleotide-Resolution Mapping of DNA Replication Patterns, Single-Strand Breaks, and Lesions by GLOE-Seq. Mol Cell 78, 975–985 e977 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Peltomaki P, Lynch syndrome genes. Fam Cancer 4, 227–232 (2005). [DOI] [PubMed] [Google Scholar]
- 147.Papadopoulos N et al. , Mutation of a mutL homolog in hereditary colon cancer. Science 263, 1625–1629 (1994). [DOI] [PubMed] [Google Scholar]
- 148.Wagner R et al. , Involvement of Escherichia coli mismatch repair in DNA replication and recombination. Cold Spring Harb Symp Quant Biol 49, 611–615 (1984). [DOI] [PubMed] [Google Scholar]
- 149.Castaneda-Garcia A et al. , A non-canonical mismatch repair pathway in prokaryotes. Nat Commun 8, 14246 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Ishino S et al. , Identification of a mismatch-specific endonuclease in hyperthermophilic Archaea. Nucleic Acids Res 44, 2977–2986 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Fukui K, Baba S, Kumasaka T, Yano T, Structural Features and Functional Dependency on beta-Clamp Define Distinct Subfamilies of Bacterial Mismatch Repair Endonuclease MutL. J Biol Chem 291, 16990–17000 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Shimada A et al. , MutS stimulates the endonuclease activity of MutL in an ATP-hydrolysis-dependent manner. FEBS J 280, 3467–3479 (2013). [DOI] [PubMed] [Google Scholar]
- 153.Kim Y, Furman CM, Manhart CM, Alani E, Finkelstein IJ, Intrinsically disordered regions regulate both catalytic and non-catalytic activities of the MutLalpha mismatch repair complex. Nucleic Acids Res 47, 1823–1835 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Gueneau E et al. , Structure of the MutLalpha C-terminal domain reveals how Mlh1 contributes to Pms1 endonuclease site. Nat Struct Mol Biol 20, 461–468 (2013). [DOI] [PubMed] [Google Scholar]