

Abstract
Maternal gestational exposures to traffic and urban air pollutant particulates have been linked to increased risk and/or worsening asthma in children; however, mechanisms underlying this vertical transmission are not entirely understood. It was postulated that gestational particle exposure might affect the ability to elicit specialized pro-resolving mediator (SPM) responses upon allergen encounter in neonates. Lipidomic profiling of 50 SPMs was performed in lungs of neonates born to mice exposed to concentrated urban air particles (CAP), diesel exhaust particles (DEP) or less immunotoxic titanium dioxide particles (TiO2). While asthma-like phenotype was induced with identical eosinophilia intensity across neonates of all particle-exposed mothers, levels of LXA4, HEPE and HETE isoforms, and HDoHe were only decreased by CAP and DEP only but not by TiO2. However, RvE2 and RvD1 were inhibited by all particles. In contrast, isomers of Maresin1 and Protectin D1 were variably elevated by CAP and DEP, whereas Protectin DX, PGE2 and TxB2 were increased in all groups. Only Protectin D1/DX, MaR1(n-3,DPA), 5(S),15(S)-DiHETE, PGE2 and RvE3 correlated with eosinophilia but the majority of other analytes, elevated or inhibited, showed no marked correlation with inflammation intensity. Evidence indicates that gestational particle exposure leads to both particle-specific and non-specific effects on the SPM network.
Introduction
Exposure to environmental pollutant particles contributes to asthma susceptibility and may explain in part the alarming rise of allergic asthma. Worldwide, an estimated 300 million people suffer from asthma which accounts for 1% of disability-adjusted life years lost (Masoli et al. 2004). The prevalence of asthma is expected to continue to rise with increasing global urbanization (Masoli et al. 2004), especially in industrialized countries, including the United States (ISAAC 1998; Mannino et al. 1998; Lai et al. 2009). Enhanced asthma risk and worsened course of the disease have been extensively linked to exposure to air pollutants including especially urban air contaminants and traffic exhaust (Venn et al. 2001; Miyake et al 2002; Cheng et al 2014; Khreis et al. 2017; Akinbami et al. 2010; Gasana et al. 2012; Nishimura et al 2013; Li et al. 2011; Patel et al. 2011; Juhn et al. 2010). Although there is some discrepancy on which components of air pollution are associated with asthma development (Orellano et al. 2017; Bowatte et al. 2015; Sbihi et al. 2016; Ma et al. 2002), this issue merits further study. Particulate matter (PM) is thought to help trigger or enhance asthma (Hamada et al. 1999; 2000; 2002; Goldsmith et al. 1999; Goldsmith and Kobzik 1999) and may also be associated with sustained inflammation (Dostert et al. 2008; Pope et al. 2016; Castaneda et al 2017), however more studies are needed to determine the mechanisms underlying PM-mediated triggered or sustained inflammation in order to investigate potential strategies for mitigation of PM health effects. (Flesher, Herbert and Kumar 2014; Rogerio et al. 2012; Aoki et al. 2008; Hisada, Aoki-Saito and Koga 2017).
In our model, in utero exposure of mice to titanium dioxide (TiO2), diesel exhaust (DEP) and concentrated urban air particles (CAP) leads to abnormal, enhanced susceptibility to allergens (Fedulov et al. 2008; Fedulov and Kobzik 2008; Gregory et al. 2017). Hence, maternal gestational exposure to PM aberrantly polarizes immune regulation in the neonates in a way that predisposes these animals to allergic airway inflammation but the nature of this effect remained largely unknown. Our model recapitulates the link between maternal particle exposure and childhood asthma in humans (Wright et al. 2021; Hazlehurst et al. 2021) which simulates maternal asthma linkage to childhood asthma (Lim, Kobzik and Dahl 2010). One might speculate the mechanisms underlying maternal particle-induced effects might exhibit similarities noted in maternal allergy. There are data to support that such maternal effects may be in part mediated by transplacental cytokines (Hamada et al. 2003), epigenetic mechanisms (Gregory et al. 2017; Fedulov and Kobzik 2011) and by microbiome (Vuillermin et al. 2017). Previously Hamada et al (2003) and Fedulov and Kobzik 2011 determined using maternal allergy model ovalbumin (OVA) rather than particle exposure to demonstrate that maternal effects are allergen-independent. Other investigators also showed that maternal particle exposure promoted responses to other allergens (Qian et al. 2020). The link between air pollutants and early origins of respiratory diseases is a field of active study (Kim et al. 2018; Benedikter et al 2018).
Normal resolution of inflammation is increasingly recognized as an active process (Barnig, Frossard and Levy 2018). Specialized pro-resolving mediators (SPMs) are signaling molecules of lipid nature formed in cells by metabolism of polyunsaturated fatty acids by lipoxygenases, cyclooxygenase, and cytochrome P450 monooxygenase enzymes and include resolvins, protectins, maresins and lipoxins. Together these components orchestrate resolution of inflammation by signaling through specific receptors (Serhan, Chiang and Dalli 2015; Barnig and Levy 2015; Pirault and Bäck 2018). Evidence suggests this signaling may be impaired in asthma as shown for some of the SPMs (Barnig, Frossard and Levy 2018; Barnig and Levy 2015; Duvall, Bruggemann and Levy 2017; Planaguma and Levy 2008) but for other substances it is less well characterized. Because of the lipid nature of these mediators, these agents cannot be measured by transcriptomic or proteomic methods which may account for paucity of experimental reports on the SPM ‘landscape’. Most studies investigating levels of SPMs in tissues of model animals focus on one or a few of SPM molecules. Lack of a comprehensive analysis of the SPM ‘landscape’ in asthmatic airways limits the ability to identify potential therapeutic targets. Further, the influence of PM on SPM production and signaling have not apparently been examined. Anecdotal reports to support the feasibility of such link exists (Nordgren et al. 2013; Lu et al 2018; Beck-Speier et al. 2012), which fueled our enthusiasm.
The aim of this study was to determine the levels of SPMs in the lungs of neonates born to mothers exposed during gestation to particulates. Previously a model was optimized such that low-dose allergen OVA induced no (or minimal) response in control neonates, but produced an asthma-like phenotype in offspring of exposed mothers (Fedulov et al. 2005; 2007; 2008; Fedulov and Kobzik 2008; Gregory et al. 2017; Hamada et al. 2003). It was hypothesized that in utero particle exposure that predisposes to asthma affects the ability to elicit SPM production in neonatal lung upon exposure to a low dose of allergen. A second aim was to determine whether 3 types of particulates that induce allergen ‘responsiveness’ to the same extent might also affect SPM levels similarly or differently. The third aim was to test how useful is the LC/MS lipidomic tissue profiling approach for studies of SPM pathway effects linked to particulate exposure and to obtain a characteristic signature snapshot of SPM levels altered by gestational particle exposure at the start of the resolution process.
Materials and Methods
Animals.
BALB/c mice were obtained from Charles River Laboratories as time-pregnant dams (gestational day E13). Animals were maintained in the barrier facility of Rhode Island Hospital, fed commercial pelleted mouse feed, and provided water ad libitum. The facility maintains 22–24°C temperature with a 12-hr light/dark cycle. All mice were exposed to particles at gestational days E14-E20. After the pups were born, each received 5 ug OVA with 1 mg alum in 0.1 ml of PBS intraperitoneally (ip) at postnatal day 3 (P3). On days 12–14 of life (P12-P14), these neonates were exposed to aerosolized OVA (3% (w/v) OVA (grade V; Sigma-Aldrich) in PBS, pH7.4) for 10 min on 3 consecutive days. The aerosol exposure was performed within individual compartments of a mouse pie chamber (Braintree Scientific) using a Pari IS2 nebulizer (Sun Medical Supply) connected to an air compressor (PulmoAID; DeVilbiss).
All studies comply with ARRIVE guidelines were performed in compliance with the National Institutes of Health guide for the care and use of laboratory animals, and were approved by the IACUC of Rhode Island Hospital.
Particles.
Titanium dioxide (TiO2), CAS Registry Number 13463-67-7, were a gift from Dr. L. Kobzik (Harvard School of Public Health, Boston, MA) and used previously in our studies (Fedulov et al. 2008; Zhang et al. 2015; Lamoureux et al. 2010). Concentrated urban air particles (CAP) were obtained via Harvard School of Public Health particle concentrator (batch #816) and represent urban contaminants typically present in Boston air (Demokritou et al. 2003; Lawrence et al. 2004; Savage et al. 2003) and well characterized in our prior studies (Gregory et al. 2017; Imrich, Ning and Kobzik 2000; Sigaud et al. 2007; Zhou and Kobzik 2006; Mandarino et al. 2020). Diesel exhaust particles (DEP), CAS Number 1333-86-4, were generously provided by Dr. Ian Gilmour at the U.S. Environmental Protection Agency and used by us in earlier studies (Fedulov et al. 2008; Gregory et al. 2017; Mandarino et al. 2020). All particles were of comparable “fine” size of the PM2.5 class with mean particle size of approximately 1 μm although not identical; see micrograph in (Mandarino et al. 2020). All particles were sonicated on ice to break up clumps using Qsonica Q55 probe sonicator prior to instillation.
Exposure
Gestational maternal exposures were performed daily for 6 days at gestational days E14–20 via intranasal insufflations of 8.3 μg/mouse/day particle suspensions in 50 μl PBS under light isoflurane anesthesia. While particle deposition after aerosols differs slightly vs. instillations (Brain et al. 1976), the instillation of particle suspensions is a useful and established method (Fedulov et al. 2008; Gregory et al. 2017; Ichinose, Furuyama and Sagai 1995; Miller et al. 2013). The exposures were performed inside a fume hood in a procedure room in the barrier facility.
Analysis.
At postnatal day 16 (P16) bronchoalveolar lavage (BAL) and lungs were harvested from the neonates after an injection of sodium pentobarbital. BAL was performed 5 times with 0.3 ml sterile PBS without Ca2+ or Mg2+ (Lonza) instilled into the trachea which was cannulated via a neck incision. Lavage fluid (recovery volume was approximately 90% of instilled) was collected and centrifuged at 300 g for 10 min, and the cell pellet resuspended in 0.1 ml PBS. The total cell yield was quantified in a Goryaev chamber. BAL differential cell counts were performed on cytocentrifuge slides prepared by centrifugation of samples at 200g for 5 min (Cytospin 2; Shandon). These slides were fixed in 95% methanol and stained with Diff-Quick (VWR), a modified Wright-Giemsa stain, and cells counted for each sample under a light microscope with enumeration of macrophages, eosinophils and lymphocytes. The lungs were removed from the chest immediately after lavage, placed on ice in Eppendorf tubes for the duration of experiment and then frozen to −80 °C. The lungs were not perfused prior to harvest.
Lipidomics was performed at the Wayne State University Lipidomic core. The lipidomics panel is capable of detecting a comprehensive list of 50 analytes that included these SPMs, isoforms and precursors: 5(S),15(S)-DiHEPE, 5(S),15(S)-DiHETE, 5(S),12(S)-DiHETE, 5(S),6(R)-DiHETE, LTB4, 20-hydroxy LTB4, 20-COOH LTB4, LXA5, LXA4, LXB4, 15-epi LXA4, 7(S)-Maresin1, Maresin1, MaR1(n-3,DPA), 5-HEPE, 11-HEPE, 12-HEPE, 15(S)-HEPE, 18-HEPE, 5-HETE, 11-HETE, 12-HETE, 15-HETE, 4-HDoHE, 7-HDoHE, 14-HDoHE, 13-HDoHE, 17-HDoHE, 10S,17S-DiHDoHE, PD1, AT-PD1, PD1(n-3,DPA), 22-OH-PD1, 12(S)-HHTrE, PGE2, PGD2, PGF2a, TXB2, RvE3, RvE2, RvE1, RvD6, RvD5, RvD5(n-3,DPA), RvD1, AT-RvD1, RvD2, AT-RvD3, RvD3, RvD4. (HETE = hydroxyeicosatetraenoic acid; HEPE = hydroxyicosapentaenoic acid; LX=lipoxin; TX = thromboxane; PG = prostaglandin; PD = protectin, Rv=resolvin). They are grouped by biochemical categories in Table 1.
Table 1.
SPM analytes grouped by biochemical nature.
| DHA metabolome | EPA metabolome | AA metabolome | DHA pathway markers |
|---|---|---|---|
| RvD1 | RvE1 | LXA4 | 4-HDoHE |
| AT-RvD1 | RvE2 | AT-LXA4 | 7-HDoHE |
| RvD2 | RvE3 | LXB4 | 14-HDoHE |
| RvD3 | 5S,15S-diHEPE | AT-LXB4 (15-epi LXA4) | 13-HDoHE |
| RvD4 | LXA5 | 5S,15S-diHETE | 17-HDoHE |
| AT-RvD3 | LTB4 | EPA pathway markers | |
| RvD5 and RvD5(n-3,DPA) | 20-OH-LTB4 | 18-HEPE | |
| RvD6 | 20-COOH-LTB4 | 15-HEPE | |
| PD1 and PD1(n-3,DPA) | 5S,12S-diHETE | 12-HEPE | |
| AT-PD1 | 5(S),6(R)-DiHETE | 11-HEPE | |
| 10S,17S-DiHDoHE (PDx) | PGD2 | 5-HEPE | |
| 22-OH-PD1 | PGE2 | AA pathway markers | |
| Maresin 1 | PGF2α | 15-HETE | |
| 7(S)-Maresin1 | TXB2 | 12-HETE | |
| MaR1(n-3,DPA) | 12(S)-HHTrE | 11-HETE | |
| 5-HETE |
Homogenized lung samples were spiked with 5 ng each (in 150 μl methanol) of 15(S)-HETE-d8,14(15)-EpETrE-d8, Resolvin D2-d5, Leukotriene B4-d4, and Prostaglandin E1-d4 as internal standards for recovery and quantitation and mixed thoroughly. The samples were then extracted for PUFA metabolites using C18 extraction columns as described in (Maddipati and Zhou 2011; Markworth et al. 2013; Maddipati et al. 2014; Norris et al. 2018). Briefly, the internal standard spiked samples were applied to conditioned C18 cartridges, washed with 15% methanol in water followed by hexane and dried under vacuum. The cartridges were eluted with 0.5 ml methanol. The eluate was dried under a gentle stream of nitrogen. The residue was redissolved in 50 μl methanol-25 mM aqueous ammonium acetate (1:1) and subjected to LC/MS analysis.
Mass-spectrometry-based lipidomic profiling of a panel of 50 analytes including key SPMs in lungs from neonate mice responding to low-dose allergen regimen HPLC was conducted using a Prominence XR system (Shimadzu) using Luna C18 (3μ, 2.1×150 mm) column. The mobile phase consisted of a gradient between A: methanol-water-acetonitrile (10:85:5 v/v) and B: methanol-water-acetonitrile (90:5:5 v/v), both containing 0.1% ammonium acetate. The gradient program with respect to the composition of B was as follows: 0–1 min, 50%; 1–8 min, 50–80%; 8–15 min, 80–95%; and 15–17 min, 95%. The flow rate was 0.2 ml/min. The HPLC eluate was directly introduced to ESI source of QTRAP5500 mass analyzer (ABSCIEX) in the negative ion mode with following conditions: Curtain gas: 35 psi, GS1: 35 psi, GS2: 65 psi, Temperature: 600 °C, Ion Spray Voltage: −1500 V, Collision gas: low, Declustering Potential: −60 V, and Entrance Potential: −7 V. The eluate was monitored by Multiple Reaction Monitoring (MRM) method to detect unique molecular ion – daughter ion combinations for each of the lipid mediators using a scheduled MRM around the expected retention time for each compound. Optimized Collisional Energies (18 – 35 eV) and Collision Cell Exit Potentials (7 – 10 V) were used for each MRM transition. Spectra of each peak detected in the scheduled MRM were recorded using Enhanced Product Ion scan to confirm the structural identity. Data were collected using Analyst 1.6.2 software and the MRM transition chromatograms were quantitated by MultiQuant software (both from ABSCIEX). The internal standard signals in each chromatogram were used for normalization, recovery, as well as relative quantitation of each analyte. Analyte values were normalized to total protein amount (in mg) in each sample to determine the concentration in lung tissue.
Data analysis.
Data from 3 repeat experiments tested over 2 separate LC/MS lipidomic measurements were assembled into a matrix table in Excel 2010 (Microsoft), values below the threshold of detection were assigned a value of 0.001 and the data were analyzed and plotted using Prism 7.02 (GraphPad). To make pooling possible the data were normalized to an average of the negative control group (Neg). The data passed the D’Agostino-Pearson, Shapiro-Wilk and Kolmogorov-Smirnov normality tests, and the Brown-Forsythe test indicated the variances allow using parametric criteria. To determine significance of differences between means we used ANOVA with Fisher’s LSD test. Our rationale is based upon (Rothman 1990; Saville 1990; Keppel and Wickens 2004; Chen, Feng and Yi 2017; Motulsky 2021; Perneger 1998). It is noteworthy that in contrast to a typical search for significant effects in a data matrix, a data mining approach was not used for all analytes at once as often done in ‘omics’ studies, but rather comparisons were performed across groups for each analyte separately. In addition, the focus was on a few scientifically sensible comparisons rather than every possible comparison. Each particle group was compared to either a negative or vehicle control and not negative controls to each other, or particles groups to each other. Therefore it is conceivable that Fisher’s LSD test is appropriate to prevent a high level of false-negatives (Perneger 1998). For an added level of scrutiny adjusted q-values were calculated using an improved adaptive method of Benjamini et al (2006) which is based upon the original Benjamini and Hochberg (1995) method but has more power. All significant comparisons (Fisher p-values and Benjamini q-values) were assembled into Supplementary Table 1. The differences were accepted as significant when p was less than 0.05. Comparisons were identified where a mean in a treatment group (TiO2, CAP or DEP) was significantly different from either negative control (Neg or PBS), and these are highlighted on the charts with an asterisk (if the difference was significant only against Neg but not against PBS, this is separately noted).
Results
Neonates of mice exposed to TiO2, CAP or DEP, but not control neonates born to mothers receiving vehicle (PBS) or to intact negative control mothers (Neg), exhibited significant allergic airway inflammation as evidenced by elevated BAL eosinophil counts (Figure 1). Two days after the last aerosol challenge this inflammation was maximal which is when resolution of inflammation starts (Yi et al. 2018; Koltsida et al. 2013).
Figure 1. Schematic of the protocol and airway eosinophilia.
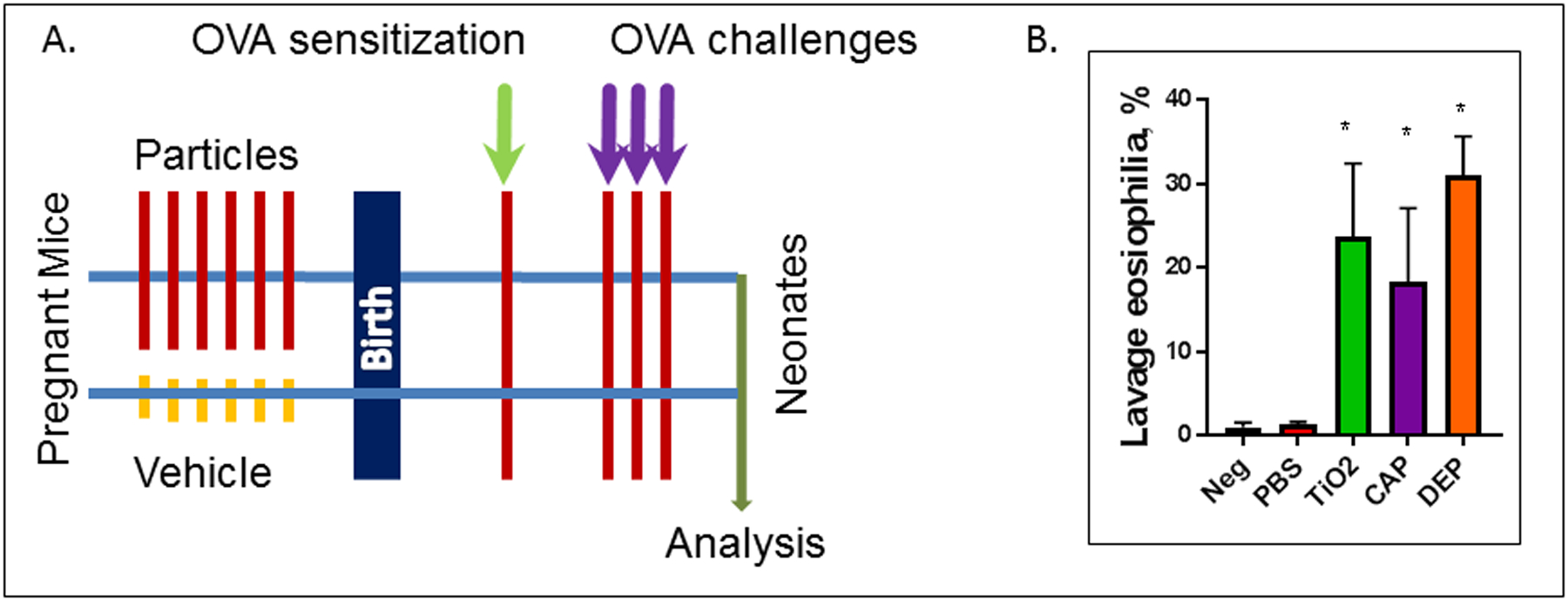
(A). Pregnant BALB/c mice received 6 consecutive daily instillations (E14–20) of TiO2, CAP or DEP particles, or PBS vehicle control, or remained untreated (‘Neg’). Neonates received a single sensitization injection of OVA+alum at P3 and a low-dose set of 3 OVA aerosol challenges at P12–14 which induces allergic airway inflammation seen as BAL eosinophilia at P16 (B). * p<0.05 (ANOVA).
Previously our OVA model demonstrated characteristically the presence of BAL eosinophilia consistent with lung tissue infiltration, elevated airway hyperresponsiveness in a methacholine test and increased BAL cytokine levels (Fedulov et al. 2005; 2007; 2008, Gregory et al. 2017; Hamada et al. 2003); hence measurements of these phenomena were not measured solely for the purpose of this report. This investigation focused on the utility of LC/MS lipidomic approach in providing an overview of SPM profiles. In lung tissue specimens tested, this measurement detected 41 out of 50 analytes; 9 were not detected: 20-hydroxy LTB4, 20-COOH LTB4, LXA5, AT-PD1, PD1(n-3,DPA), 22-OH-PD1, RvE1, RvD2, and AT-RvD3. Data for 21analytes which differed are presented in Figure 2. These were subdivided into those inhibited in the particles’ groups (Figure 2A) and those elevated (Figure 2B).
Figure 2. Lipidomic profiling of neonatal lung tissue.
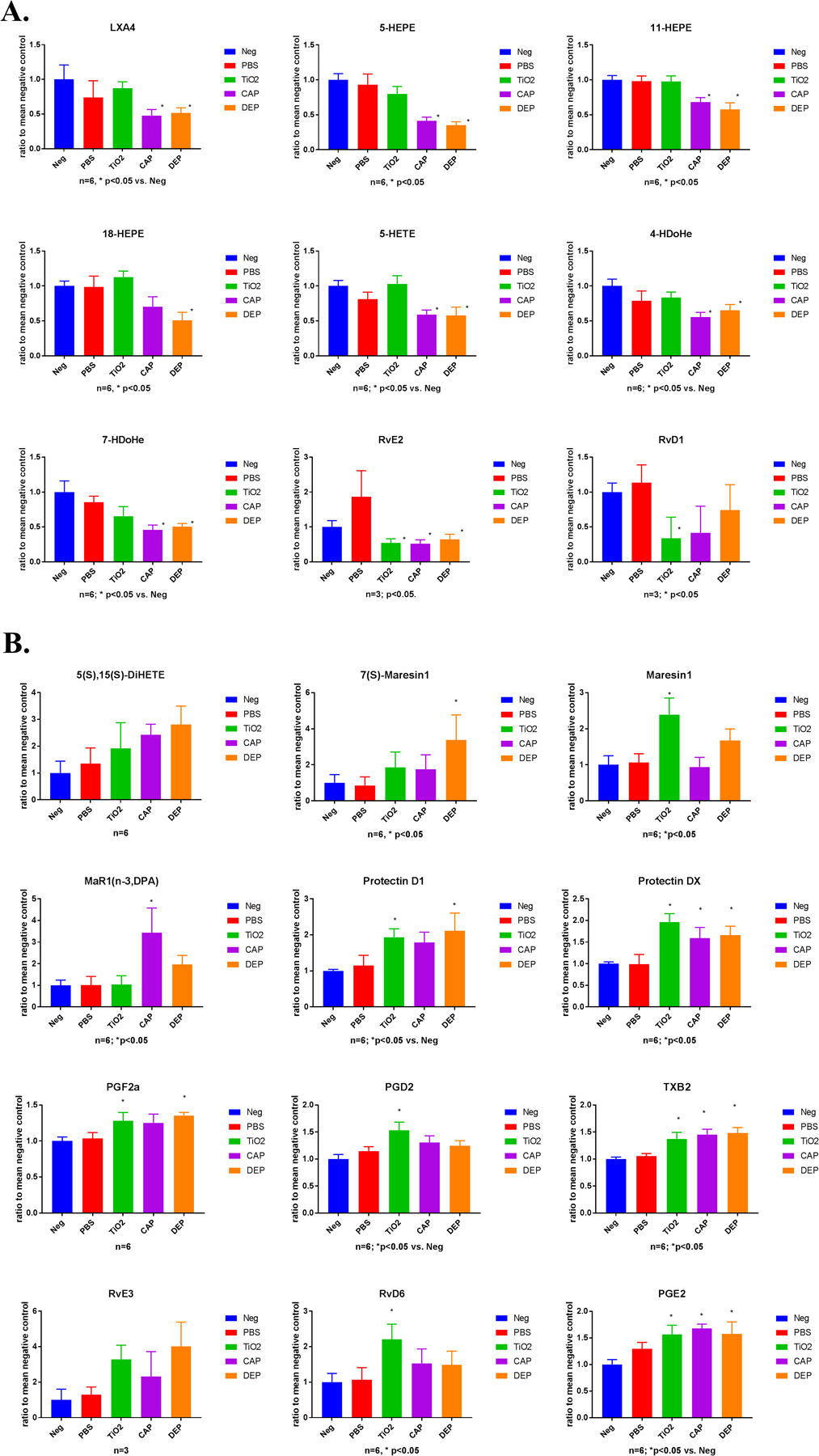
A). SPMs inhibited, and B) SPMs elevated in particle exposure groups. (n)=6 per group unless otherwise noted on the chart. Asterisk (*) denotes p<0.05 (ANOVA) for a particle group comparison versus either PBS control or negative control (the latter is separately noted on the chart). See explanation and rationale in Methods; detailed p-values and comparisons are included in Supplementary Table 1.
Lipoxin A4 (LXA4) the only lipoxin detected was significantly lower in CAP and DEP groups compared to either Neg or PBS controls. Surprisingly, there was no marked change in the TiO2 group which may in part be attributed to diminished immunotoxicity of TiO2 particles. A similar pattern was noted with most other inhibited analytes: 5-HEPE, 11-HEPE, 18-HEPE, 5-HETE, 4-HDoHE, and 7-HDoHE. Resolvins E2 and D1 showed a departure from this pattern which were inhibited in all three particles groups albeit with variable statistical significance. It should also be noted that in one of the LC/MS runs RvE2 and D1 were below the level of detection (LOD) in all groups.
Among those analytes elevated in the particle groups, prostaglandins E2, F2a and D2 and thromboxane B2 were increased with significantly either in all three particle groups, in two or only in one, but in cases where there were no significant alterations the mean values still displayed a consistent trend towards elevation. Given the pro-inflammatory nature of prostaglandins and their role in asthmatic inflammation it is not surprising that these substances were present in essentially all inflamed groups albeit with variable significance.
Maresin 1 epimers were variably elevated in TiO2 (mostly Maresin 1), in DEP (mostly 7(S)-Maresin 1) and CAP (mostly n-3 DPA form) samples. Evidence thus indicates that elevation of Maresin 1 class molecules occurred in particle groups.
Protectin 1 showed a marked rise in the TiO2 and DEP particle groups but only numerically higher for CAP. 10S,17S-DiHDoHE (an isomer of protectin D1, known as protectin DX) was significantly increased in all particle groups, whereas 5(S),15(S)-DiHETE exhibited a rising trend that did not reach ssignificance. Finally, resolvins D6 and E3 trended towards elevation in TiO2, CAP and DEP groups. RvE3 was only detectable in the first run, but not in the second run.
Twenty analytes were not markedly affected: 5(S),15(S)-DiHEPE, 5(S),12(S)-DiHETE, 5(S),6(R)-DiHETE, LTB4, LXB4, 15-epi LXA4, 12-HEPE, 15(S)-HEPE, 11-HETE, 12-HETE, 15-HETE, 14-HDoHE, 13-HDoHE, 17-HDoHE, 12(S)-HHTrE, RvD5(n-3,DPA), AT-RvD1, RvD3, RvD4, RvD5.
Correlation analysis
For those analytes altered significantly a linear correlation analysis was performed to determine whether there was a link between airway (BAL) eosinophils and analyte levels (Figure 3). It is of intertest that none of the analytes inhibited in utero by TiO2, CAP or DEP exposure correlated with eosinophil counts. Of the elevated analytes, only 6 showed a mild (R2 = 0.2 – 0.3) significant correlation, the exception was 10S,17S-DiHDoHE which correlated strongly (R2 = 0.5). Overall, while most SPM levels were affected by in utero particle exposure, few were directly linked to individual eosinophilia.
Figure 3. Correlation analysis.
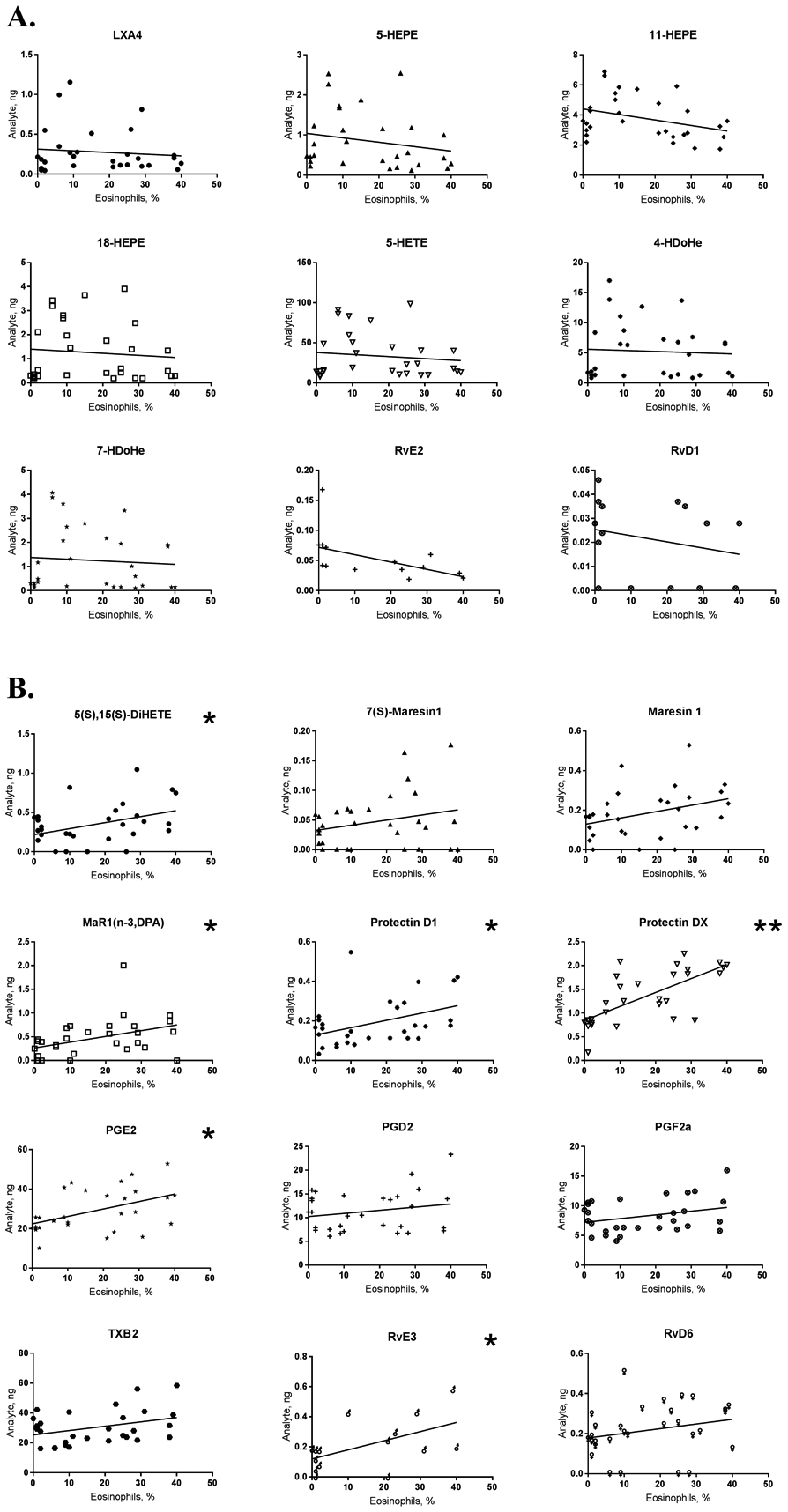
Analyte concentration and eosinophil percentage were correlated individually across all neonatal samples for inhibited (A) and elevated (B) SPMs. (n) = 30; * P<0.05, ** P<0.0001 (Pearson).
Discussion
Maternal more so than paternal asthma (Lim, Kobzik and Dahl 2010; Martinez et al. 1995; Barrett 2008) and maternal exposures to environmental contaminants (Murdzoska et al. 2010; Breton et al. 2009; Shankardass et al. 2009; Blacquiere et al. 2009; Wang and Pinkerton 2007) are strongly linked to asthma in a child (Wright et al. 2021; Hazlehurst et al. 2021). Although the mechanism underlying this maternal effect remains to be determined one possibility is that the maternal immune system acts to influence the developing immune system in utero or shortly after birth either through epigenetic mechanisms or by polarizing immune regulation (Gregory et al 2017; Fedulov and Kobzik 2011; Prescott and Clifton 2009).
In our model maternal gestational exposure to particles induces increased asthma ‘preparedness’ in the neonates. This effect is not specific to the nature of particles and was noted for TiO2, CAP, DEP and black carbon (Goldsmith et al. 1999; Gregory et al 2017). The model generally mimics the human phenomenon of increased asthma incidence in families living in polluted, PM2.5 −rich environments (Wright et al. 2021; Hazlehurst et al. 2021).
Concentrated urban air particles (CAP)
Concentrated urban air particles (CAP) are perhaps the most relevant in our study as these components simulate exposures of a city dweller and serve as a helpful surrogate for urban air pollutants (Ghio et al 2012). Human exposures to CAP are ubiquitous in the urban setting and have been associated with worsening of asthma symptoms (Kelly and Fussell 2015; Alexis et al. 2014). Since CAPs comprise a variety of particle sizes and compositions these produce a large variety of deleterious effects which always include inflammation (Godleski et al. 2002).
Mouse exposure is typically modeled through either CAP suspensions (Harkema et al. 2004; Imrich et al. 2007) or inside urban air particle concentrator chamber (Mauad et al. 2008). Co-exposure of young mice to air pollutant aerosols results in allergic sensitization to inhaled OVA, which produces no marked effect in the absence of the pollutant (Hamada et al. 2000). The CAPs used were obtained using Harvard Ambient Particle Concentrator (Demokritou et al. 2003; Lawrence et al. 2004) and are well characterized (Savage et al. 2003; Imrich, Ning and Kobzik 2000; Sigaud et al. 2007; Zhou and Kobzik 2006; Mandarino et al. 2020; Harkema et al. 2004).
Diesel exhaust particles (DEP)
Diesel exhaust particles (DEP) are known to be pro-inflammatory and pro-allergic particles (Suzuki et al 1996), possibly because these substances contain soluble components, including pyrene, a known stimulant of IL-4 expression (Bommel et al. 2000). Human exposure occurs through the exhaust of diesel engines and is postulated to be a component of higher asthma risk in high-traffic areas (McConnell et al. 2006; Boehmer et al. 2010). The significance of DEP is highlighted by the recent “dieselgate” scandal which revealed that auto manufacturers bypassed regulations to allow prohibited levels of diesel emissions.
Based upon the finding reported here that CAP and DEP impair the ability to elicit production of a number of SPMs it is conceivable that SPM network may mediate the maternally transmitted ‘asthma preparedness’ effect of the particles, which renders a hypothesis that Lipoxin A4 and Resolvins E2 and D1 are deficient and may be considered for therapeutic purposes.
Titanium dioxide (TiO2)
Titanium dioxide (TiO2) fine-sized particles are considered immunologically ‘inert’ and often used as a negative control in particle exposure experiments (van Maanen et al. 1999), in part because these agents do not contain soluble components. Interestingly, TiO2 are not completely innocuous and may initiate pulmonary inflammation with activation of antigen-presenting cells and production of certain chemokines (Warheit et al. 2005; Renwick et al. 2004; Drumm et al. 1999) and IL-13 (Ahn et al. 2005). Fedulov et al (2008) showed that airway TiO2 exposure during pregnancy leads to the same (enhanced) inflammation in lungs of pregnant dams as that initiated by DEP and results in increased asthma susceptibility in the neonates (Figure 1). Human exposure to TiO2 aerosol is rare and linked to paint manufacturing process (IARC 2010); however human exposure to TiO2 suspensions is almost ubiquitous albeit not in an airborne way as it is a component of many foods, cosmetic and hygienic products (Davidson 2020; Rompelberg et al. 2016; Wu and Hicks 2020). Our findings suggest that indirect (maternal) exposure to TiO2 particle suspension affects the elicitation of SPM production in response to allergic inflammation, which might be important in illustrating the potential adverse health effects of TiO2-containing products.
While CAP and DEP are known to trigger or exacerbate asthma in humans and mice, previously Zhang et al (2015) reported that a relatively ‘inert’ TiO2 also produced similar effects. It is of interest that other investigations noted this effect is linked to pregnancy hormones which predispose the airway macrophages to elicit inflammatory response to TiO2 particles that are normally innocuous (Fedulov et al. 2008). In agreement with these observations all three particle types elicited identically intense responsiveness to OVA (Figure 1B). It should be noted that two negative controls – offspring of intact mice (Neg) and newborns from mice who received only vehicle (PBS) – partly to ensure the instillation procedure is innocuous and partly to have additional duplication of the negative control group.
A unique benefit of our model is that the lungs of the neonates are not directly exposed to particles, which enables dissecting the allergic inflammation in the lung from any potential co-effect of PM. It needs to be emphasized that all neonates including both negative control groups received the same OVA sensitization and aerosol challenge protocol.
While airway eosinophilia was similar across all PM exposure groups, a dichotomy of the inhibitory effect of particles on SPM levels was detected between TiO2 and other more toxic particulates CAP and DEP. The finding that LXA4, forms of hydroxyeicosapentaenoic acid (HEPE) and hydroxyeicosatetraenoic acid (HETE) as well as hydroxydocosahexaenoic acid (HDoHE) are only inhibited in CAP and DEP groups, but not in TiO2, suggests that changes identified were in fact linked to the nature of the particle exposure, and not only to the intensity of eosinophilic inflammation which was identical. This is also evident from lack of correlation of airway eosinophilia to levels of most SPMs. It is possible that generally lower immunotoxicity of TiO2 particles in contrast to CAP or DEP contributes to this dichotomous effect; however, further studies are needed to determine whether the mechanisms by which TiO2 induce neonatal asthma ‘preparedness’ and those that alter SPM levels are different.
5-HETE and 5-HEPE are major metabolites produced by 5-lipoxygenase from arachidonic acid (AA) and eicosapentaenoic acid (EPA). 5-HETE is a precursor of leukotrienes, and 5-HEPE might exert its own anti-inflammatory/pro-resolving effects (Onodera et al. 2017). 15-HETE is a precursor of LXA4 and B4 (Halade, Black and Verma 2018). It is interesting that 15-HETE along with LXB4 was not markedly affected, but LXA4 was significantly decreased by CAP and DEP. It is conceivable that this might be attributed to differences in activity of LXA4 and LXB4 hydrolases or in the relative functioning of 12-LOX and 5-LOX. It may also be related to uneven consumption or decay of LXA4.
When analyzing the subset of SPMs and precursors that were elevated, a rise in prostaglandins E2, F2a and D2 and thromboxane B2 was found. This elevation is not surprising considering what is known regarding the generally pro-inflammatory nature of these molecules and their role in asthmatic inflammation (Claar, Hartert and Peebles 2015). It is possible, as noted here, that an inflamed lung might exhibit higher levels of these molecules than non-inflamed lung. PGE2 is pleiotropic and its elevation in eosinophilic lung may be viewed as part of a compensatory/protective response (Sastre and del Pozo 2012).
Protectin D1 (PD1) is produced (elevated) in asthmatic lung in humans and mice and is endogenously generated in allergic lung (Levy et al. 2007) which is consistent with findings that this substance it was elevated in our model. 10S,17S-DiHDoHE is a protectin isomer (protectin DX) and changed similarly to PD1. Notably, exogenous (therapeutic) PD1 attenuates asthmatic airway inflammation (Levy et al. 2007) and hence this increase may be associated with a component of the resolution response. Similarly, therapeutic Maresin 1 ameliorates asthmatic inflammation (Ou et al. 2021; Krishnamoorthy 2015); however, less is known regarding whether Maresin 1 is elevated endogenously and/or at which time point during an asthmatic inflammatory response. There are reports that Maresin 1 might be unchanged or inhibited (Miyata et al. 2021). The same is true for RvE3 and a few other metabolites tested (Miyata et al. 2021); however, data indicate that the asthma model used in that study was entirely different. Even less information was found relating to 5(S),15(S)-DiHETE levels in asthmatic lung but, given that 5(S)15(S)-DiHETE is a product of eosinophils and chemotactic for them (Morita, Schröder and Christophers 1990) it appears that it was elevated in correlation with eosinophil counts.
E-series resolvins are synthetized somewhat divergently. Unlike RvE1, RvE2 and RvE3 which are biosynthesized from 18-HEPE via the 12/15-LOX pathway in eosinophils, RvE1 is produced from 5S (6)-epoxy-18R-HEPE in activated neutrophils (Lee 2021). E-resolvins occur at different timepoints in the resolution process (Lee 2021), hence our ‘snapshot’ of the lipidomic profiling detected a decrease of RvE2 consistent with reduced 18-HEPE levels accompanied by rise of RvE3 and absence of RvE1 in asthmatic inflamed lungs.
Resolvins D-series are primarily synthetized in neutrophils and macrophages (Serhan et al. 2002), while RvD1 is produced rather rapidly upon an inflammatory trigger (Dalli, Colas and Serhan 2014. RvD6 kinetics have not apparently been reported (Lee 2021); however, it is possible that this might explain why the former RvD1was inhibited while the latter RvD6 was elevated in our asthma samples. Without knowing time kinetic differences one might expect these SPM to be altered in a similar manner. Findings such as this emphasize the need to perform time-course studies for a comprehensive understanding of the kinetics of these molecules in asthma (Levy et al. 2001). While our study is a one-timepoint ‘snapshot’ of the SPM landscape and further studies are needed to determine how changes in this landscape unfold dynamically, a ‘proof-of-concept’ report is provided that LC/MS -based lipidomics approach might be useful for better understanding of the SPM network in asthmatic lung and in resolution of PM–induced effects. Our study emphasizes the previously underappreciated toxicity mechanism attributed to PM and may help expand on the potential of SPM in therapeutics. For instance, other investigators showed a link of direct exposure to PM2.5 and ultrafine particles to LXA4 and 15(S)-HETE (Lu et al 2018; Beck-Speier et al. 2012), or an effect of Maresin-1 reducing inflammatory responses to organic dusts (Nordgren et al. 2013); however, a more comprehensive SPM profiling connecting PM effects on SPM responses is required. Similarly, in non-PM scenarios therapeutic approaches using synthetic SPMs in animal models have succeeded (Flesher, Herbert and Kumar 2014; Rogerio et al. 2012; Aoki et al. 2008; Hisada, Aoki-Saito and Koga 2017) without a comprehensive analysis that would detail the ‘landscape’ of SPMs in asthmatic airways, which may be of utility to identify lacking molecules that would serve as most beneficial therapeutic targets.
Usefulness of an ‘omics’ approach is that it provides added value in a modern tool box for toxicological studies (Miller et al 2014; Bonvallot et al 2014). Our findings suggested that all particles, whether (1) multi- or single-component, (2) soluble (digestible by phagocytes) or insoluble, (3) organic or metal, when administered during gestation might trigger increased asthma ‘preparedness’ in neonates. If true, this would suggest that it is the maternal airway that leads to vertical transmission of the phenotype, and not a potential direct fetotoxic effect of the particulates. Gregory et al (2017) pursued this hypothesis where it was found that the phenotype is inherited for 2–3 generations indicating an epigenetic mechanism of inheritance. Thus, it is conceivable that neonatal asthmatic inflammation is worsened by impairments in SPM network. With respect to transgenerational findings one might postulate that epigenetic changes in the enzymes of lipid metabolism such as cyclo- and lipoxygenases may be involved in this phenomenon. However, lack of correlation for most SPM with the main metric of allergic airway inflammation–eosinophil counts suggests that SPM mechanisms are more complex than a mere enhanced response to an inflammatory stimulus. Our findings also suggest that the altered SPMs might serve as biomarkers of prolonged/delayed resolution of asthmatic inflammation where mechanistic involvement of SPMs in resolution makes them valuable candidates. Finally, alterations in tissue levels of SPMs may be linked to cellular content changes. Our data demonstrated that the link of eosinophilia to SPMs was seen only for a few analytes, but it is feasible to postulate that other cells, especially their content in lung tissue and not BAL, might be linked. Although many analytes were at the lower margin of detection, an entire pair of lungs was used for SPM profiling making detailed immunohistological correlations not possible. This hypothesis may fuel future studies.
Particle dosage used in our study is a factor that needs to be considered. The dose of particles of 8.3 μg/mouse/day multiplied by 6 days of sub-chronic exposure is likely on a larger side when compared to real-life human exposure. However, the following justification is proposed. A ‘subchronic’ low-dose regimen of PM was selected to mimic human exposure. The complexities of inhalation toxicology studies are well-known when it comes to calculating doses (Reiprich et al. 2013; Phillips 2017; Chen and Lippmann 2015). Urban PM2.5 concentration ranges from 8.4 (median) and 38.3 (maximum) μg/m3 in (Penttinen et al. 2001) to 33.6 and up to 114.8 μg/m3 in some traffic-related scenarios (Zuurbier et al. 2010); ranges from 19.7 μg/m3 to 110 μg/m3 can occur as daily variations (Lippmann, Gordon and Chen 2005). Recalculating for ventilation, these values might result in 45.6 to 100.4 μg/hr during outdoor activity (Zuurbier et al. 2010). Concentration indoors might be lower than outdoors but without special filtration this reduction is approximately 36% (Cui et al. 2020). From this, a typical daily dose of PM2.5 in a city can reach mg ranges (but will vary widely). The following were taken into consideration body weight, surface area of the lung and ventilation volume (Brain et al. 1976; Ichinose, Furuyama and Sagai 1995; Warheit et al. 2005; Osier and Oberdörster 1997; Driscoll et al. 2000; Oberdorster 1996; Brown et al. 2005; Phillips 2017), the volume to be instilled, body position and anesthesia (Southam 2002); In addition taken into account was nasal clearance of a fraction of particles (Brain et al. 1976), that rodents are less susceptible to particles than humans (Bendtsen et al. 2020) and considerations of excessive dosages used in some studies (Curbani et al. 2019). Another basis for estimation was that one-time doses of 5–15 μg per mouse produced mild inflammatory responses and are considered ‘low’ (Riva et al. 2011), whereas doses of 20–50 μg enable more prominent and statistically significant effects to occur (Stoeger et al. 2006). In our prior studies 50 μg/mouse was employed for an acute exposure (Fedulov et al. 2008, Gregory et al. 2017); therefore, based upon published data and prior experience 1/6th of that (= 8.3 μg/mouse/day) was selected for a 6-day exposure. Of note, an experimental model needs to induce a phenotype in a feasibly short time. Further (1) since it is the gestational exposure that was the focus of this investigation, and (2) mouse pregnancy is 21 days, this timeframe for exposure is especially limited and prompts for a stronger dose stimulus. It is acceptable and a common and reasonable practice to exaggerate exposure stimuli in animal model studies. Doses in this range or higher were successfully used by other investigators (Reiprich et al. 2013; Lippmann, Gordon and Chen 2005; Bendtsen et al. 2020; Curbani et al. 2019; Stoeger et al. 2006). It is noteworthy that whether lower doses would produce the same effects was not examined. Evidence indicates similar to other animal models, our results should not be blindly extrapolated to human effects.
It should be noted that lungs were lavaged prior to harvesting to provide counts of inflammatory cells in order to correlate SPM levels to eosinophilia. BAL fluid was similarly tested for SPM levels, but most analytes were either not detected or on the lower margin of detection even though some SPMs were found in human lavage samples. This suggests that most SPMs do not get washed out with lavage. It is noteworthy that the lungs were not perfused at harvest and hence a small amount of blood retained in the vessels may have contributed to the measured concentration.
In Supplementary Table 1 the p-values are provided from Fisher’s LSD post-test and q-values from the Benjamini-Krieger-Yekutieli –adjusted post-ANOVA test. While both tests returned identical p-values, the adjusted q-values are in a few cases different and because of the higher stringency of the adjustment do not allow the same conclusion of statistical significance. Because Fisher’s test allows conclusions of significance in hypothesis-driven pre-planned comparisons of specific groups within each analyte, while Benjamini-Krieger-Yekutieli enables conclusions of significance for comparisons that were not pre-planned and implicit to comparing all the groups, both are reported to provide a helpful hypothesis-generating approach.
Conclusions
Maternal gestational exposures to TiO2, CAP or DEP particles that induced a similar allergen ‘responsiveness’ in neonates to a variable extent stimulated the ability to elicit SPM production in neonatal lung upon allergen challenge. While many SPMs were altered similarly by all three particle types, there were distinct patterns seen with CAP and DEP but not with TiO2. Nine analytes inhibited and 12 analytes elevated by maternal particles exposure were found in our lung samples. While neither of the 9 inhibited analytes nor 6 of the elevated correlated directly with eosinophil counts, a few, specifically those directly linked to eosinophils as a source, demonstrated significant correlations. Closely related SPMs such as maresin epimers and protectin changed congruently whereas some SPMs seemingly of the same class such as resolvins E and D showed divergent responses.
Supplementary Material
Supplemental Figure 1. Schematic of SPM biosynthesis.
{kind=link}
Acknowledgements
This study was supported by a National Institutes of Environmental Health Sciences (NIEHS) grant R01ES030227 to AVF, and by Brown Physicians Incorporated Academic Assessment grant funds to AVF.
The Lipidomics Core at WSU is supported in part by National Center for Research Resources, National Institutes of Health Grant S10RR027926. We thank Dr. Rao Maddipati for his guidance in LC/MS lipidomics.
Abbreviations
- 5S,15S-diHETE
5S,15S-dihydroxy-eicosa-6E, 8Z, 11Z, 13E-tetraenoic acid
- 10S,17S-DiHDoHE
10,17-dihydroxydocosahexaenoic acid
- HDoHE
hydroxydocosahexaenoic acid
- HETE
hydroxyeicosatetraenoic acid
- HEPE
hydroxyeicosapentaenoic acid
- LX
lipoxin
- TX
thromboxane
- PG
prostaglandin
- PD
protectin
- Rv
resolvin
- LT
leukotriene
- TiO2
titanium dioxide particles
- DEP
diesel exhaust particles
- CAP
concentrated urban air particles
- SPM
specialized pro-resolving mediators
- OVA
ovalbumin
- PM
particulate matter
- MRM
Multiple Reaction Monitoring
- HPLC
high-performance liquid chromatography
- LC/MS
liquid chromatography–mass spectrometry
- BAL
bronchoalveolar lavage
- PBS
phosphate-buffered saline
Footnotes
Disclosure statement.
The authors declare no conflict of interest.
Data availability statement
Raw data can be available from AVF by a reasonable request through the editorial office.
References
- Ahn M-H, Kang C-M, Park C-S, Park S-J, Rhim T, Yoon P-O, Chang HS, Kim S-H, Kyono H, and Kim KC. 2005. Titanium dioxide particle–induced goblet cell hyperplasia : Association with mast cells and IL-13. Respiratory Research 6:34. doi: 10.1186/1465-9921-6-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akinbami LJ, Lynch CD, Parker JD, and Woodruff TJ. 2010. The association between childhood asthma prevalence and monitored air pollutants in metropolitan areas, United States, 2001–2004. Environmental Research 110: 294–301. doi: 10.1016/j.envres.2010.01.001 [DOI] [PubMed] [Google Scholar]
- Alexis NE, Huang YCT, Rappold AG, Kehrl H, Devlin R, and Peden DB. 2014. Patients with asthma demonstrate airway inflammation after exposure to concentrated ambient particulate matter. American Journal of Respiratory and Critical Care Medicine 190: 235–237. doi: 10.1164/rccm.201401-0126LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki H, Hisada T, Ishizuka T, Utsugi M, Kawata T, Shimizu Y, Okajima F, Dobashi K, and Mori M. 2008. Resolvin E1 dampens airway inflammation and hyperresponsiveness in a murine model of asthma. Biochemical and Biophysical Research Communications 367: 509–515. doi: 10.1016/j.bbrc.2008.01.012. [DOI] [PubMed] [Google Scholar]
- Barnig C, Frossard N, and Levy BD. 2018. Towards targeting resolution pathways of airway inflammation in asthma. Pharmacology & Therapeutics 186: 98–113. doi: 10.1016/j.pharmthera.2018.01.004. [DOI] [PubMed] [Google Scholar]
- Barnig C, and Levy BD. 2015. Innate immunity is a key factor for the resolution of inflammation in asthma. European Respiratory Review 24: 141–153. doi: 10.1183/09059180.00012514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett EG 2008. Maternal influence in the transmission of asthma susceptibility. Pulmonary Pharmacology & Therapeutics 21: 474–484. doi: 10.1016/j.pupt.2007.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck-Speier I, Karg E, Behrendt H, Stoeger T, and Alessandrini F. 2012. Ultrafine particles affect the balance of endogenous pro- and anti-inflammatory lipid mediators in the lung: In-vitro and in-vivo studies. Particle and Fibre Toxicology 9. 10.1186/1743-8977-9-27. doi: 10.1186/1743-8977-9-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendtsen KM, Gren L, Malmborg VB, Shukla PC, Tunér M, Essig YJ, Krais AM, et al. 2020. Particle characterization and toxicity in C57BL/6 mice following instillation of five different diesel exhaust particles designed to differ in physicochemical properties. Particle and Fibre Toxicology 17. 10.1186/s12989-020-00369-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedikter BJ, Wouters EMF, Savelkoul PHM, Rohde GGU and Stassen FRM 2018. Extravascular vesicles released in response to respiratory exposures: Implications for chronic disease. Journal of Toxicology and Environmental Health B 21: 142–160. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, and Hochberg Y. 1995. Controlling the false discovery rate: A practical and powerful approach to multiple testing. Journal of the Royal Statistical Society 57: 289–300. [Google Scholar]
- Benjamini Y, Krieger AM, and Yekutieli D. 2006. Adaptive linear step-up procedures that control the false discovery rate. Biometrika 93: 491–507. [Google Scholar]
- Blacquiere MJ, Timens W, Melgert BN, Geerlings M, Postma DS, and Hylkema MN. 2009. Maternal smoking during pregnancy induces airway remodelling in mice offspring. European Respiratory Journal 33:1133–1140: 00129608. doi: 10.1183/09031936.00129608. [DOI] [PubMed] [Google Scholar]
- Boehmer TK, Foster SL, Henry JR, Woghiren-Akinnifesi EL, Yip FY, and Centers for Disease Control and Prevention (CDC). 2013. Residential proximity to major highways - United States, 2010. Morbidity and Mortality Weekly Report Supplements 62: 46–50. [PubMed] [Google Scholar]
- Bömmel H, Li-Weber M, Serfling E, and Duschl A. 2000. The environmental pollutant pyrene induces the production of IL-4. Journal of Allergy and Clinical Immunology 105: 796–802. doi: 10.1067/mai.2000.105124. [DOI] [PubMed] [Google Scholar]
- Bonvallot N, Tremblay-Franco M, Chevrier C, Canlet C, Debrauwer L, Cravedi J-P and Cordier S. 2014. Potential input from metabolomics for exploring and understanding the links between environment and health. Journal of Toxicology and Environmental Health B 17: 21–44. [DOI] [PubMed] [Google Scholar]
- Bowatte G, Lodge C, Lowe AJ, Erbas B, Perret J, Abramson MJ, Matheson M, and Dharmage SC. 2015. The influence of childhood traffic-related air pollution exposure on asthma, allergy and sensitization: A systematic review and a meta-analysis of birth cohort studies. Allergy 70: 245–256. doi: 10.1111/all.12561. [DOI] [PubMed] [Google Scholar]
- Brain JD, Knudson DE, Sorokin SP, and Davis MA. 1976. Pulmonary distribution of particles given by intratracheal instillation or by aerosol inhalation. Environmental Research 11: 13–33. doi: 10.1016/0013-9351(76)90107-9. [DOI] [PubMed] [Google Scholar]
- Breton CV, Byun H-M, Wenten M, Pan F, Yang A, and Gilliland FD. 2009. Prenatal tobacco smoke exposure affects global and gene-specific DNA methylation. American Journal of Respiratory and Critical Care Medicine 180: 462–467. doi: 10.1164/rccm.200901-0135OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JS, Wilson WE, and Grant LD. 2005. Dosimetric comparisons of particle deposition and retention in rats and humans. Inhalation Toxicology 17: 355–385. doi: 10.1080/08958370590929475. [DOI] [PubMed] [Google Scholar]
- Castaneda AR, Bein KJ, Smiley-Jewell S, and Pinkerton KE. 2017. Fine particulate matter (PM2.5) enhances allergic sensitization in BALB/c mice. Journal of Toxicology and Environmental Health A 80: 197–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, and Lippmann M. 2015. Inhalation toxicology methods: The generation and characterization of exposure atmospheres and inhalational exposures. Current Protocols in Toxicology 63.: 10.1002/0471140856.tx2404s63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S-Y, Feng Z, and Yi X. 2017. A general introduction to adjustment for multiple comparisons. Journal of Thoracic Disease 9: 1725–1729. doi: 10.21037/jtd.2017.05.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng M-H, Chen C-C, Chiu H-F, and Yang C-Y 2014. Fine particulate air pollution and hospital admissions for asthma: A case-crossover study in Taipei. Journal of Toxicology and Environmental Health A 77: 1075–1083. [DOI] [PubMed] [Google Scholar]
- Claar D, Hartert TV, and Peebles RS Jr. 2015. The role of prostaglandins in allergic lung inflammation and asthma. Expert Review of Respiratory Medicine 9: 55–72. doi: 10.1586/17476348.2015.992783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui X, Li Z, Teng Y, Barkjohn KK, Norris CL, Fang L, Daniel GN, et al. 2020. Association between bedroom particulate matter filtration and changes in airway pathophysiology in children with asthma. JAMA Pediatrics 174: 533. doi: 10.1001/jamapediatrics.2020.0140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curbani F, de Oliveira Busato F, Marcarini do Nascimento M, Olivieri DN, and Tadokoro CE. 2019. Inhale, exhale: Why particulate atter exposure in animal models are so acute? Data and facts behind the history. Data in Brief 25: 104237. doi: 10.1016/j.dib.2019.104237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalli J, Colas RA, and Serhan CN. 2013. Novel N-3 Immunoresolvents: Structures and actions. Scientific Reports 3. 10.1038/srep01940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson K 2020. Titanium dioxide in food — Should you be concerned? Healthline. www.healthline.com/nutrition/titanium-dioxide-in-fooduses. Accessed on August 30, 2021. [Google Scholar]
- Demokritou P, Gupta T, Ferguson S, and Koutrakis P. 2003. Development of a high-volume concentrated ambient particles system (CAPS) for human and animal inhalation toxicological studies. Inhalation Toxicology 15 : 111–129. doi: 10.1080/08958370304475. [DOI] [PubMed] [Google Scholar]
- Keppel G, and Wickens TD. 2004. Design and Analysis: A Researcher’s Handbook. IBSN:0135159415.
- Dostert C, Pétrilli V, Van Bruggen R, Steele C, Mossman BT, and Tschopp J. 2008. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 320: 674–677. doi: 10.1126/science.1156995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driscoll KE 2000. Intratracheal instillation as an exposure technique for the evaluation of respiratory tract toxicity: Uses and limitations. Toxicological Sciences 55: 24–35. doi: 10.1093/toxsci/55.1.24. [DOI] [PubMed] [Google Scholar]
- Drumm K, Schindler H, Buhl R, Küstner E, Smolarski R, and Kienast K. 1999. Indoor air pollutants stimulate interleukin-8-specific MRNA expression and protein secretion of alveolar macrophages. Lung 177: 9–19. [DOI] [PubMed] [Google Scholar]
- Duvall MG, Bruggemann TR, and Levy BD. 2017. Bronchoprotective mechanisms for specialized pro-resolving mediators in the resolution of lung inflammation. Molecular Aspects of Medicine 58: 44–56. doi: 10.1016/j.mam.2017.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedulov A, Silverman E, Xiang Y, Leme A, and Kobzik L. 2005. Immunostimulatory CpG oligonucleotides abrogate allergic susceptibility in a murine model of maternal asthma transmission. Journal of Immunology 175: 4292–4300. [DOI] [PubMed] [Google Scholar]
- Fedulov AV, and Kobzik L. 2011. Allergy risk is mediated by dendritic cells with congenital epigenetic changes. American Journal of Respiratory Cell and Molecular Biology 44: 285–292. doi: 10.1165/rcmb.2009-0400OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedulov AV, and Kobzik L. 2008. Immunotoxicologic analysis of maternal transmission of asthma risk. Journal of Immunotoxicology 5: 445–452. doi: 10.1080/15476910802481765. [DOI] [PubMed] [Google Scholar]
- Fedulov AV, Leme A, Yang Z, Dahl M, Lim R, Mariani TJ, and Kobzik L. 2008. Pulmonary exposure to particles during pregnancy causes increased neonatal asthma susceptibility. American Journal of Respiratory Cell and Molecular Biology 38: 57–67. doi: 10.1165/rcmb.2007-0124OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedulov AV, Leme AS, and Kobzik L. 2007. Duration of allergic susceptibility in maternal transmission of asthma risk. American Journal of Reproductive Immunology 58: 120–128. doi: 10.1111/j.1600-0897.2007.00496.x. [DOI] [PubMed] [Google Scholar]
- Flesher RP, Herbert C, and Kumar RK. 2014. Resolvin E1 promotes resolution of inflammation in a mouse model of an acute exacerbation of allergic asthma. Clinical Science 126: 805–818. doi: 10.1042/CS20130623. [DOI] [PubMed] [Google Scholar]
- Gasana J, Dillikar D, Mendy A, Forno E, and Ramos Vieira E. 2012. Motor vehicle air pollution and asthma in children: A meta-analysis. Environmental Research 117: 36–45. doi: 10.1016/j.envres.2012.05.001. [DOI] [PubMed] [Google Scholar]
- Ghio AJ, Carraway MS and Madden MC. 2012. Composition of air pollution particles and oxidative stress in cells tissues and living systems. Journal of Toxicology and Environmental Health B 15: 1–21. [DOI] [PubMed] [Google Scholar]
- Godleski JJ, Clarke RW, Coull BA, Saldiva PH, Jiang NF, Lawrence J, and Koutrakis P. 2002. Composition of inhaled urban air particles determines acute pulmonary responses. Annals of Occupational Hygiene 46 (Suppl_1): 419–424. [Google Scholar]
- Goldsmith CA, Hamada K, Ning Y, Qin G, Catalano P, Krishna Murthy GG, Lawrence J, and Kobzik L. 1999. Effects of environmental aerosols on airway hyperresponsiveness in a murine model of asthma. Inhalation Toxicology 11: 981–998. doi: 10.1080/089583799196646. [DOI] [PubMed] [Google Scholar]
- Goldsmith CA, and Kobzik L. 1999. Particulate air pollution and asthma: A review of epidemiological and biological studies. Reviews on Environmental Health 14: 121–134. [DOI] [PubMed] [Google Scholar]
- Gregory DJ, Kobzik L, Yang Z, McGuire CC, and Fedulov AV. 2017. Transgenerational transmission of asthma risk after exposure to environmental particles during pregnancy. American Journal of Physiology. Lung Cellular and Molecular Physiology 313: L395–L405. doi: 10.1152/ajplung.00035.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halade GV, Black LM, and Verma MK. 2018. Paradigm shift – Metabolic transformation of docosahexaenoic and eicosapentaenoic acids to bioactives exemplify the promise of fatty acid drug discovery. Biotechnology Advances 36: 935–953. doi: 10.1016/j.biotechadv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamada K, Goldsmith C-A, Goldman A, and Kobzik L. 2000. Resistance of very young mice to inhaled allergen sensitization is overcome by co-exposure to an air-pollutant aerosol. American Journal of Respiratory and Critical Care Medicine 161: 1285–1293. doi: 10.1164/ajrccm.161.4.9906137. [DOI] [PubMed] [Google Scholar]
- Hamada K, Goldsmith CA, and Kobzik L. 1999. Increased airway hyperresponsiveness and inflammation in a juvenile mouse model of asthma exposed to air-pollutant aerosol. Journal of Toxicology and Environmental Health A 58: 129–143. [DOI] [PubMed] [Google Scholar]
- Hamada K, Goldsmith CA, Suzaki Y, Goldman A, and Kobzik L. 2002. Airway hyperresponsiveness caused by aerosol exposure to residual oil fly ash leachate in mice. Journal of Toxicology and Environmental Health A 65:1351–1365. doi: 10.1080/00984100290071586. [DOI] [PubMed] [Google Scholar]
- Hamada K, Suzaki Y, Goldman A, Ning YY, Goldsmith C, Palecanda A, Coull B, Hubeau C, and Kobzik L. 2003. Allergen-independent maternal transmission of asthma susceptibility. Journal of Immunology 170: 1683–1689. doi: 10.4049/jimmunol.170.4.1683. [DOI] [PubMed] [Google Scholar]
- Harkema JR, Keeler G, Wagner J, Morishita M, Timm E, Hotchkiss J, Marsik F, Dvonch T, Kaminski N, and Barr E. 2004. Effects of concentrated ambient particles on normal and hypersecretory airways in rats. Health Effects Research Institute 120: 1–68; discussion 69–79. [PubMed] [Google Scholar]
- Hazlehurst MF, Carroll KN, Loftus CT, Szpiro AA, Moore PE, Kaufman JD, Kirwa K, et al. 2021. Maternal exposure to PM2.5 during pregnancy and asthma risk in early childhood: Consideration of phases of fetal lung development. Environmental Epidemiology 5: e130. doi: 10.1097/ee9.0000000000000130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hisada T, Aoki-Saito H, and Koga Y. 2017. Are specialized pro-resolving mediators promising therapeutic agents for severe bronchial asthma? Journal of Thoracic Disease 9: 4266–4269. doi: 10.21037/jtd.2017.10.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans.; International Agency for Research on Cancer. “Carbon Black, Titanium Dioxide, and Talc.” Lyon, France: International Agency for Research on Cancer, 2010. Lyon, France : Distributed by WHO Press; 201. MMS ID 9916672343406676; ISBN 9283212932 (pbk.); NLM ID 101667234. [PMC free article] [PubMed] [Google Scholar]
- Ichinose T, Furuyama A, and Sagai M. 1995. Biological effects of diesel exhaust particles (DEP). II. Acute toxicity of DEP introduced into lung by intratracheal instillation. Toxicology 99: 153–167. doi: 10.1016/0300-483x(94)03013-r. [DOI] [PubMed] [Google Scholar]
- Imrich A, Ning Y, and Kobzik L. 2000. Insoluble components of concentrated air particles mediate alveolar macrophage responses in vitro. Toxicology and Applied Pharmacology 167 140–150. doi: 10.1006/taap.2000.9002. [DOI] [PubMed] [Google Scholar]
- Imrich A, Ning Y, Lawrence J, Coull B, Gitin E, Knutson M, and Kobzik L. 2007. Alveolar macrophage cytokine response to air pollution particles: Oxidant mechanisms. Toxicology and Applied Pharmacology 218: 256–264: 033. doi: 10.1016/j.taap.2006.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juhn YJ, Qin R, Urm S, Katusic S, and Vargas-Chanes D. 2010. The influence of neighborhood environment on the incidence of childhood asthma: A propensity score approach. Journal of Allergy and Clinical Immunology 125: 838–843.e2. doi: 10.1016/j.jaci.2009.12.998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly FJ, and Fussell JC. 2015. Air pollution and public health: Emerging hazards and improved understanding of risk. Environmental Geochemistry and Health 37: 631–649. doi: 10.1007/s10653-015-9720-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khreis H, Kelly C, Tate J, Parslow R, Lucas K, and Nieuwenhuijsen M. 2017. Exposure to traffic-related air pollution and risk of development of childhood asthma: A systematic review and meta-analysis. Environment International. 100:1–31. doi: 10.1016/j.envint.2016.11.012. [DOI] [PubMed] [Google Scholar]
- Kim D, Chen Z, Zhou L-F, and Huang S-X. 2018. Air pollutants and early origins of respiratory diseases. Chronic Diseases and Translational Medicine 4: 75–94. doi: 10.1016/j.cdtm.2018.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koltsida O, Karamnov S, Pyrillou K, Vickery T, Chairakaki A, Tamvakopoulos C, Sideras P, Serhan CN, and Andreakos E. 2013. Toll‐like receptor 7 stimulates production of specialized pro‐resolving lipid mediators and promotes resolution of airway inflammation. EMBO Molecular Medicine 5: 762–775. doi: 10.1002/emmm.201201891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamoorthy N, Burkett PR, Dalli J, Abdulnour R-EE, Colas R, Ramon S, Phipps RP, et al. 2015. Cutting edge: Maresin-1 engages regulatory T cells to limit Type 2 innate lymphoid cell activation and promote resolution of lung inflammation. Journal of Immunology. 194: 863–867. doi: 10.4049/jimmunol.1402534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai CKW, Beasley R, Crane J, Foliaki S, Shah J, Weiland S, and the ISAAC Phase Three Study Group. 2009. Global variation in the prevalence and severity of asthma symptoms: Phase three of the International Study of Asthma and Allergies in Childhood (ISAAC). Thorax. 64: 476–483. doi: 10.1136/thx.2008.106609. [DOI] [PubMed] [Google Scholar]
- Lamoureux DP, Kobzik L, and Fedulov AV. 2010. Customized PCR-Array analysis informed by gene-chip microarray and biological hypothesis reveals pathways involved in lung inflammatory response to titanium dioxide in pregnancy. Journal of Toxicology and Environmental Health A 73: 596–606. doi: 10.1080/15287390903566641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence J, Wolfson JM, Ferguson S, Koutrakis P, and Godleski J. 2004. Performance stability of the Harvard ambient particle concentrator. Aerosol Science and Technology 38: 219–227. DOI: 10.1080/02786820490261735. [DOI] [Google Scholar]
- Lee CH 2021. Role of specialized pro-resolving lipid mediators and their receptors in virus infection: A promising therapeutic strategy for SARS-CoV-2 cytokine storm. Archives of Pharmacal Research. 44: 84–98. doi: 10.1007/s12272-020-01299-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy BD, Clish CB, Schmidt B, Gronert K, and Serhan CN. 2001. Lipid mediator class switching during acute inflammation: Signals in resolution. Nature Immunology. 2: 612–619. doi: 10.1038/89759. [DOI] [PubMed] [Google Scholar]
- Levy BD, Kohli P, Gotlinger K, Haworth O, Hong S, Kazani S, Israel E, Haley KJ, and Serhan CN. 2007. Protectin D1 is generated in asthma and dampens airway inflammation and hyperresponsiveness. Journal of Immunology 178: 496–502. doi: 10.4049/jimmunol.178.1.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Batterman S, Wasilevich E, Elasaad H, Wahl R, and Mukherjee B. 2011. Asthma exacerbation and proximity of residence to major roads: A population-based matched case-control study among the pediatric Medicaid population in Detroit, Michigan. Environmental Health 10. doi: 10.1186/1476-069X-10-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim RH, Kobzik L, and Dahl M. 2010. Risk for asthma in offspring of asthmatic mothers versus fathers: A meta-analysis. PloS One 5: e10134. doi: 10.1371/journal.pone.0010134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippmann M, Gordon T, and Chen LC. 2005. Effects of subchronic exposures to concentrated ambient particles (CAPs) in mice: I. Introduction, objectives, and experimental plan. Inhalation Toxicology 17: 177–187. doi: 10.1080/08958370590912716. [DOI] [PubMed] [Google Scholar]
- Lu X, Fu H, Han F, Fang Y, Xu J, Zhang L, and Du Q. 2018. Lipoxin A4 regulates PM2.5-induced severe allergic asthma in mice via the Th1/Th2 balance of group 2 innate lymphoid cells. Journal of Thoracic Disease 10: 1449–1459. doi: 10.21037/jtd.2018.03.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma JYC, and Ma JKH. 2002. The dual effect of the particulate and organic components of diesel exhaust particles on the alteration of pulmonary immune/inflammatory responses and metabolic enzymes. Journal of Environmental Science and Health. Part C, Environmental Carcinogenesis & Ecotoxicology Reviews 20: 117–147. doi: 10.1081/GNC-120016202. [DOI] [PubMed] [Google Scholar]
- van Maanen JM, Borm PJ, Knaapen A, van Herwijnen M, Schilderman PA, Smith KR, Aust AE, Tomatis M, Fubini B. 1999. In vitro effects of coal fly ashes: Hydroxyl radical generation, iron release, and DNA damage and toxicity in rat lung epithelial cells. Inhalation Toxicology 11:1123–1141. doi: 10.1080/089583799196628. [DOI] [PubMed] [Google Scholar]
- Maddipati KR, and Zhou S-L. 2011. Stability and analysis of eicosanoids and docosanoids in tissue culture media. Prostaglandins & Other Lipid Mediators 94: 59–72. [DOI] [PubMed] [Google Scholar]
- Maddipati KR, Romero R, Chaiworapongsa T, Zhou S-L, Xu Z, Tarca AL, Kusanovic JP, Munoz H, and Honn KV. 2014. Eicosanomic profiling reveals dominance of the epoxygenase pathway in human amniotic fluid at term in spontaneous labor. Federation of American Societies for Experimental Biology Journal 28: 4835–4846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandarino A, Gregory DJ, McGuire CC, Leblanc BW, Witt H, Rivera LM, Godleski JJ, and Fedulov AV. 2020. The effect of talc particles on phagocytes in co-culture with ovarian cancer cells. Environmental Research 180: 108676. doi: 10.1016/j.envres.2019.108676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannino DM, Homa DM, Pertowski CA, Ashizawa A, Nixon LL, Johnson CA, Ball LB, Jack E, and Kang DS. 1998. Surveillance for asthma--United States, 1960–1995. CDC Surveillance Summaries: Morbidity and Mortality Weekly Report 47: 1–27. [PubMed] [Google Scholar]
- Markworth JF, Vella L, Lingard BS, Tull DL, Rupasinghe TW, Sinclair AJ, Maddipati KR, and Cameron-Smith D. 2013. Human inflammatory and resolving lipid mediator responses to resistance exercise and ibuprofen treatment. American Journal of Physiology. Regulatory, Integrative and Comparative Physiology 305: R1281–R1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez FD, Wright AL, Taussig LM, Holberg CJ, Halonen M, and Morgan WJ. 1995. Asthma and wheezing in the first six years of life. The Group Health Medical Associates. New England Journal of Medicine 332:133–138. doi: 10.1056/NEJM199501193320301. [DOI] [PubMed] [Google Scholar]
- Masoli M, Fabian D, Holt S, Beasley R, and Global Initiative for Asthma (GINA) Program. 2004. The global burden of asthma: Executive Summary of the GINA Dissemination Committee Report. Allergy 59: 469–478. doi: 10.1111/j.1398-9995.2004.00526.x. [DOI] [PubMed] [Google Scholar]
- Mauad T, Rivero DHRF, de Oliveira RC, de Faria Coimbra Lichtenfels AJ, Guimarães ET, de Andre PA, Kasahara DI, de Siqueira Bueno HM, and Saldiva PHN. 2008. Chronic exposure to ambient levels of urban particles affects mouse lung development. American Journal of Respiratory and Critical Care Medicine 178: 721–728. doi: 10.1164/rccm.200803-436OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConnell R, Berhane K, Yao L, Jerrett M, Lurmann F, Gilliland F, Künzli N, et al. 2006. Traffic, susceptibility, and childhood asthma. Environmental Health Perspectives 114: 766–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller I, Serchi T, Murk AJ and Gutleb AC. 2014. The added value of proteomics for toxicological studies. Journal of Toxicology and Environmental Health B 17: 225–246. [DOI] [PubMed] [Google Scholar]
- Miller MR, McLean SG, Duffin R, Lawal AO, Araujo JA, Shaw CA, Mills NL, Donaldson K, Newby DE, and Hadoke PWF. 2013. Diesel exhaust particulate increases the size and complexity of lesions in atherosclerotic mice. Particle and Fibre Toxicology 10: 61. doi: 10.1186/1743-8977-10-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyake Y, Yura A, and Iki M. 2002. Relationship between distance from major roads and adolescent health in Japan. Journal of Epidemiology 12: 418–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyata J, Yokokura Y, Moro K, Arai H, Fukunaga K, and Arita M. 2021. 12/15-Lipoxygenase regulates IL-33-induced eosinophilic airway inflammation in mice. Frontiers in Immunology 12: 687192. doi: 10.3389/fimmu.2021.687192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita E, Schröder JM, and Christophers E. 1990. Identification of a novel and highly potent eosinophil chemotactic lipid in human eosinophils treated with arachidonic a cid. Journal of Immunology 144: 1893–1900. [PubMed] [Google Scholar]
- Murdzoska J, Devadason SG, Khoo S-K, Landau LI, Young S, Goldblatt J, Zhang G, Le Souëf PN, and Hayden CM. 2010. In utero smoke exposure and role of maternal and infant glutathione S-transferase genes on airway responsiveness and lung function in infancy. American Journal of Respiratory and Critical Care Medicine 181: 64–71. doi: 10.1164/rccm.200812-1887OC. [DOI] [PubMed] [Google Scholar]
- Motulsky HJ GraphPad Statistics Guide. GraphPad Prism Statistics Guide. Chapter “When it makes sense to not correct for multiple comparisons”) accessed on October 1, 2021 at: https://www.graphpad.com/guides/prism/latest/statistics/stat_when_to_not_correct_for_2.htm
- Nishimura KK, Galanter JM, Roth LA, Oh SS, Thakur N, Nguyen EA, Thyne S, et al. 2013. Early-life air pollution and asthma risk in minority children. The GALA II and SAGE II Studies. American Journal of Respiratory and Critical Care Medicine 188: 309–318. doi: 10.1164/rccm.201302-0264OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordgren TM, Heires AJ, Wyatt TA, Poole JA, LeVan TD, Cerutis DR, and Romberger DJ. 2013. Maresin-1 reduces the pro-inflammatory response of bronchial epithelial cells to organic dust. Respiratory Research 14. 10.1186/1465-9921-14-51.. doi: 10.1186/1465-9921-14-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris PC, Skulas-Ray AC, Riley I, Richter CK, Kris-Etherton PM, Jensen GL, Serhan CN, and Maddipati KR. 2018. Identification of specialized pro-resolving mediator clusters from healthy adults after intravenous low-dose endotoxin and omega-3 supplementation: A methodological validation. Scientific Reports 8: 18050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberdorster G 1996. Significance of particle parameters in the evaluation of exposure-dose-response relationships of inhaled particles. Inhalation Toxicology 8 Suppl: 73–89. [PubMed] [Google Scholar]
- Onodera T, Fukuhara A, Shin J, Hayakawa T, Otsuki M, and Shimomura I. 2017. Eicosapentaenoic acid and 5-HEPE enhance macrophage-mediated TREG induction in mice. Scientific Reports 7. doi: 10.1038/s41598-017-04474-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orellano P, Quaranta N, Reynoso J, Balbi B, and Vasquez J. 2017. Effect of outdoor air pollution on asthma exacerbations in children and adults: Systematic review and multi-level meta-analysis. PloS One 12: e0174050. doi: 10.1371/journal.pone.0174050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osier M 1997. Intratracheal inhalation vs intratracheal instillation: Differences in particle effects. Fundamental and Applied Toxicology 40: 220–227. doi: 10.1006/faat.1997.2390. [DOI] [PubMed] [Google Scholar]
- Ou G, Liu Q, Yu C, Chen X, Zhang W, Chen Y, Wang T, et al. 2021. The protective effects of Maresin 1 in the OVA-induced asthma mouse model. Mediators of Inflammation 2021: 1–11. doi: 10.1155/2021/4131420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel MM, Quinn JW, Jung KH, Hoepner L, Diaz D, Perzanowski M, Rundle A, Kinney PL, Perera FP, and Miller RL. 2011. Traffic density and stationary sources of air pollution associated with wheeze, asthma, and immunoglobulin E from birth to age 5 years among New York City children. Environmental Research 111: 1222–1229. doi: 10.1016/j.envres.2011.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penttinen P, Timonen KL, Tiittanen P, Mirme A, Ruuskanen J, and Pekkanen J. 2001. Number concentration and size of particles in urban air: Effects on spirometric lung function in adult asthmatic subjects. Environmental Health Perspectives 109: 319–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perneger TV 1998. What’s wrong with Bonferroni adjustments. British Medical Journal 316: 1236–1238. doi: 10.1136/bmj.316.7139.1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips JE 2017. Inhaled efficacious dose translation from rodent to human: A retrospective analysis of clinical standards for respiratory diseases. Pharmacology & Therapeutics 178: 141–147. doi: 10.1016/j.pharmthera.2017.04.003. [DOI] [PubMed] [Google Scholar]
- Pirault J, and Bäck M. 2018. Lipoxin and resolvin receptors transducing the resolution of inflammation in cardiovascular disease. Frontiers in Pharmacology 9. 10.3389/fphar.2018.01273. doi: 10.3389/fphar.2018.01273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planaguma A, and Levy BD. 2008. Uncontrolled airway inflammation in lung disease represents a defect in counter-regulatory signaling. Future Lipidology 3: 697–704. doi: 10.2217/17460875.3.6.697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pope CA, Bhatnagar A, McCracken JP, Abplanalp W, Conklin DJ, and O’Toole T. 2016. Exposure to fine particulate air pollution is associated with endothelial injury and systemic inflammation. Circulation Research. 119: 1204–1214. doi: 10.1161/CIRCRESAHA.116.309279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prescott SL, and Clifton V. 2009. Asthma and pregnancy: Emerging evidence of epigenetic interactions in utero. Current Opinion in Allergy and Clinical Immunology 9: 417–426. doi: 10.1097/ACI.0b013e328330634f. [DOI] [PubMed] [Google Scholar]
- Qian Q, Chowdhury BP, Sun Z, Lenberg J, Alam R, Vivier E, and Gorska MM. 2020. Maternal diesel particle exposure promotes offspring asthma through NK cell–derived granzyme B. Journal of Clinical Investigation. 130:4133–4151. doi: 10.1172/JCI130324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiprich M, Rudzok S, Schütze N, Simon JC, Lehmann I, Trump S, and Polte T. 2013. Inhibition of endotoxin-induced perinatal asthma protection by pollutants in an experimental mouse model. Allergy 68: 481–489. doi: 10.1111/all.12121. [DOI] [PubMed] [Google Scholar]
- Renwick LC, Brown D, Clouter A, and Donaldson K. 2004. Increased inflammation and altered macrophage chemotactic responses caused by two ultrafine particle types. Occupational and Environmental Medicine 61: 442–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riva DR, Magalhães CB, Lopes AA, Lanças T, Mauad T, Malm O, Valença SS, Saldiva PH, Faffe DS, and Zin WA. 2011. Low dose of fine particulate matter (PM2.5) can induce acute oxidative stress, inflammation and pulmonary impairment in healthy mice. Inhalation Toxicology 23: 257–267. doi: 10.3109/08958378.2011.566290. [DOI] [PubMed] [Google Scholar]
- Rogerio AP, Haworth O, Croze R, Oh SF, Uddin M, Carlo T, Pfeffer MA, Priluck R, Serhan CN, and Levy BD. 2012. Resolvin D1 and aspirin-triggered Resolvin D1 promote resolution of allergic airways responses. Journal of Immunology 189: 1983–1991. doi: 10.4049/jimmunol.1101665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rompelberg C, Heringa MB, van Donkersgoed G, Drijvers J, Roos A, Westenbrink S, Peters R, van Bemmel G, Brand W, and Oomen AG. 2016. Oral intake of added titanium dioxide and its nanofraction from food products, food supplements and toothpaste by the Dutch population. Nanotoxicology 10: 1404–1414. doi: 10.1080/17435390.2016.1222457. [DOI] [PubMed] [Google Scholar]
- Rothman KJ 1990. No adjustments are needed for multiple comparisons. nnEpidemiology 1: 43–46. [PubMed] [Google Scholar]
- Sastre B, and del Pozo V. 2012. Role of in asthma and nonasthmatic eosinophilic bronchitis. Mediators of Inflammation 2012: 1–9. doi: 10.1155/2012/645383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage ST, Lawrence J, Katz T, Stearns RC, Coull BA, and Godleski JJ. 2003. Does the Harvard/U.S Environmental Protection Agency ambient particle concentrator change the toxic potential of particles? Journal of the Air & Waste Management Association 53: 1088–1097. doi: 10.1080/10473289.2003.10466267. [DOI] [PubMed] [Google Scholar]
- Saville DJ 1990. Multiple comparison procedures: The practical solution. American Statistician 44: 174–180. [Google Scholar]
- Sbihi H, Tamburic L, Koehoorn M, and Brauer M. 2016. Perinatal air pollution exposure and development of asthma from birth to age 10 years. European Respiratory Journal 47: 1062–1071. doi: 10.1183/13993003.00746-2015. [DOI] [PubMed] [Google Scholar]
- Serhan CN, Chiang N, and Dalli J. 2015. The resolution code of acute inflammation: Novel pro-resolving lipid mediators in resolution. Seminars in Immunology 27: 200–215. doi: 10.1016/j.smim.2015.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serhan CN, Hong S, Gronert K, Colgan SP, Devchand PR, Mirick G, and Moussignac R-L. 2002. Resolvins. Journal of Experimental Medicine 196: 1025–1037. doi: 10.1084/jem.20020760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankardass K, McConnell R, Jerrett M, Milam J, Richardson J, and Berhane K. 2009. Parental stress increases the effect of traffic-related air pollution on childhood asthma incidence. Proceedings of the National Academy of Sciences of the United States of America 106: 12406–12411. doi: 10.1073/pnas.0812910106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigaud S, Goldsmith C-AW, Zhou H, Yang Z, Fedulov A, Imrich A, and Kobzik L. 2007. Air pollution particles diminish bacterial clearance in the primed lungs of m ice. Toxicology and Applied Pharmacology 223: 1–9. doi: 10.1016/j.taap.2007.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southam DS, Dolovich M, O’Byrne PM, and Inman MD. 2002. Distribution of intranasal instillations in mice: Effects of volume, time, body position, and anesthesia. American Journal of Physiology. Lung Cellular and Molecular Physiology 282: L833–L839. doi: 10.1152/ajplung.00173.2001. [DOI] [PubMed] [Google Scholar]
- Stoeger T, Reinhard C, Takenaka S, Schroeppel A, Karg E, Ritter B, Heyder J, and Schulz H. 2006. Instillation of six different ultrafine carbon particles indicates a surface area threshold dose for acute lung inflammation in mice. Environmental Health Perspectives 114: 328–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Kanoh T, Ishimori M, Ikeda S, and Ohkuni H. 1996. Adjuvant activity of diesel exhaust particulates (DEP) in production of anti-IgE and anti-IgG1 antibodies to mite allergen in mice. Journal of Clinical & Laboratory Immunology 48: 187–199. [PubMed] [Google Scholar]
- Venn AJ, Lewis SA, Cooper M, Hubbard R, and Britton J. 2001. Living near a main road and the risk of wheezing illness in children. American Journal of Respiratory and Critical Care Medicine 164: 2177–2180. doi: 10.1164/ajrccm.164.12.2106126. [DOI] [PubMed] [Google Scholar]
- Vuillermin PJ, Macia L, Nanan R, Tang MLK, Collier F, and Brix S. 2017. The maternal microbiome during pregnancy and allergic disease in the offspring. Seminars in Immunopathology 39: 669–675. doi: 10.1007/s00281-017-0652-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, and Pinkerton KE. 2007. Air pollutant effects on fetal and early postnatal development. Birth Defects Research. Part C, Embryo Today: Reviews 81: 144–154. doi: 10.1002/bdrc.20097. [DOI] [PubMed] [Google Scholar]
- Warheit DB, Brock WJ, Lee KP, Webb TR, and Reed KL. 2005. Comparative pulmonary toxicity inhalation and instillation studies with different TiO2 particle formulations: Impact of surface treatments on particle toxicity. Toxicological Sciences 88 : 514–524. doi: 10.1093/toxsci/kfi331. [DOI] [PubMed] [Google Scholar]
- Worldwide variations in the prevalence of asthma symptoms: the International Study of Asthma and Allergies in Childhood (ISAAC). Eur Respir J. 1998;12(2):315–35. [DOI] [PubMed] [Google Scholar]
- Wright RJ, Hsu H-HL, Chiu Y-HM, Coull BA, Simon MC, Hudda N, Schwartz J, Kloog I, and Durant JL. 2021. Prenatal ambient ultrafine particle exposure and childhood asthma in the Northeastern United States. American Journal of Respiratory and Critical Care Medicine 204: 788–796. doi: 10.1164/rccm.202010-3743OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu F, and Hicks AL. 2020. Estimating human exposure to titanium dioxide from personal care products through a social survey approach. Integrated Environmental Assessment and Management 16: 10–16. doi: 10.1002/ieam.4197. [DOI] [PubMed] [Google Scholar]
- Yi S, Zhai J, Niu R, Zhu G, Wang M, Liu J, Huang H, et al. 2018. Eosinophil recruitment is dynamically regulated by interplay among lung dendritic cell subsets after allergen challenge. Nature Communications 9. 10.1038/s41467-018-06316-9. doi: 10.1038/s41467-018-06316-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Mikhaylova L, Kobzik L, and Fedulov AV. 2015. Estrogen-mediated impairment of macrophageal uptake of environmental TiO2particles to explain inflammatory effect of TiO2 on airways during pregnancy. Journal of Immunotoxicology 12: 81–91. . doi: 10.3109/1547691X.2014.899411. [DOI] [PubMed] [Google Scholar]
- Zhou H, and Kobzik L. 2007. Effect of concentrated ambient particles on macrophage phagocytosis and killing of Streptococcus pneumoniae. American Journal of Respiratory Cell and Molecular Biology 36: 460–465. doi: 10.1165/rcmb.2006-0293OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuurbier M, Hoek G, Oldenwening M, h Lenters V, Meliefste K, van den Hazel P, and Brunekreef B. 2010. Commuters’ exposure to particulate matter air pollution is affected by mode of transport, fuel type, and route. Environmental Health Perspectives 118: 783–789. doi: 10.1289/ehp.0901622. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Schematic of SPM biosynthesis.
Data Availability Statement
Raw data can be available from AVF by a reasonable request through the editorial office.