

Abstract
The molecular-scale morphology and topology of polyamide composite membranes determine the performance characteristics of these materials. However, molecular-scale simulations are computationally expensive and morphological and topological characterization of molecular structures are not well developed. Molecular dynamics simulation and analysis methods for the polymerization, hydration, and quantification of polyamide membrane structures were developed and compared to elucidate efficient approaches for producing and analyzing the polyamide structure. Polymerization simulations that omitted the reaction-phase solvent did not change the observed hydration, pore-size distribution, or water permeability, while improving the simulation efficiency. Pre-insertion of water into the aggregate pores (radius ≈ 4 Å) of dry domains enabled shorter hydration simulations and improved simulation scaling, without altering pore structure, properties, or performance. Medial axis and Minkowski functional methods were implemented to identify permeation pathways and quantify the polyamide morphology and topology, respectively. Better agreement between simulations and experimentally observed systems was accomplished by increasing the domain size rather than increasing the number of ensemble realizations of smaller systems. The largest domain hydrated was an order of magnitude larger by volume than the largest domain previously reported. This work identifies methods that can enable more efficient and meaningful fundamental modeling of membrane materials.
Keywords: molecular dynamics, polymerization, hydration methods, pore morphology and topology, Minkowski functionals
Graphical Abstract
1. Introduction
Reverse osmosis (RO) membrane treatment is a widely utilized and relatively energy efficient method of water purification for non-selective, near-complete removal of dissolved contaminants [1, 2]. Most commercial membranes are manufactured as thin-film composite (TFC) membranes, which have a 20–200 nm thick crosslinked, aromatic, polyamide layer that serves as the main barrier to dissolved species transport [3, 4].
Since the late 1970’s, improvements to active layer processing conditions identified via a build-and-test approach—i.e. new active layer chemistries, or processing methods, are identified via empirical observation, then individually synthesized, characterized, and tested—have resulted in significant incremental improvements to active layer performance, increasing water production while rejecting a greater fraction of contaminants [4]. However, while the build-and-test paradigm has yielded TFC membranes of improved performance, many design challenges remain that can be better addressed by improving the understanding of the relationship between fundamental properties and the resultant membrane performance [2, 5–7].
A pathway to determine fundamental property-performance relationships is via analysis of active layers at the molecular scale [2, 8, 9]. All transported molecules must travel through the molecular free volume of the active layer [7, 10–12]. Available pathways through the free volume are determined by the size of the transported molecule and the morphology and topology of the molecular potential field of the active layer [6, 7]. However, due to limitations of experimental characterization and restrictive configuration of RO membranes, it has proven difficult to produce in-lab observations of transport through polyamide active layers capable of informing fundamental property-performance relationships [6, 9, 13–15]. Accordingly, molecular dynamics (MD) simulations can be utilized to access the spatial and temporal scales required to quantify the interactions between the active layer structure and transported molecules [8, 9, 16], and analyze the effects of novel active layer compositions, such as those used in mixed-matrix membranes [17].
MD simulations of polyamide membranes consist of four general steps: polymerization, equilibration, hydration, and operation [9]. Each step is designed to mimic the corresponding in-lab step as closely as possible to provide realistic modeling of membrane structure and performance [9, 18, 19]. However, realistic MD simulations are computationally expensive, therefore modifications are made to these steps to reduce computational cost, often at the expense of the realism of the simulation [13, 18–21].
While some of the fundamentals and promise of MD methods for the analysis of membrane systems have appeared in the literature [8, 9], several open questions must be resolved to close the gap between promise and more routine application of these methods to advance membrane science. These open questions involve the identification and comparison of approaches to reduce the computational burden [19, 21], to quantify and reduce the uncertainty of predictions made [9, 16, 18, 21, 22], and to provide a means to more completely analyze the morphology and topology of the free volume structure and link these properties to the predicted performance [18].
Accordingly, the overall goal of this work is to advance critical aspects of methods for MD simulation and analysis of polyamide active layers. Said methods can then be used in the future to advance fundamental understanding of the relationship between active layer structure and performance. The specific objectives are: (1) to examine and compare polymerization techniques to model active layer processing; (2) to evaluate active layer hydration approaches for reducing computational cost; (3) to advance methods for evaluating active layer structure and linking structural characteristics to performance; and (4) to quantify the uncertainty in molecular-scale simulations and evaluate approaches for reducing the uncertainty.
2. Methods
2.1. Overview
MD simulations of crosslinked aromatic polyamide must be initiated ab initio since the molecular structure of these materials is not yet accessible via in-lab characterization. As a result, the simulation workflow includes polymerization, equilibration, hydration, and operation steps, which is intended to generate polyamide domains representative of TFC RO active layers, prepare them for operation, and observe their properties and performance. Performing simulations presented in the workflow in a completely realistic manner, however, is impractical due to the small time- and length-scales accessible via atomistic MD. Time steps for atomistic MD simulations are typically in the 10−15 s (1 femtosecond, fs) scale, and realistic length scales of hundreds of nanometers (nm) results in intractable levels of computational work.
Methods exist to mitigate the computational cost of long run time and large length-scale simulations required to adequately model polyamide active layers. For the purposes of this study, two classes of methods comparisons were evaluated in order to yield accurate simulations at minimum possible computational cost, which we will refer to as computational efficiency. The first class was fundamental MD algorithmic optimizations, explained in detail in Section 2.2, which were applied to all simulations to improve computational efficiency. The second class, which is a primary focus of this work, is the identification, development, and evaluation of methods in detail that can improve the computational efficiency of MD simulation of the polymerization and hydration steps of the process. The methods used for each step of the simulation workflow, error reduction approaches, analysis of the pore morphology and topology, and the computational environment are described in turn in the sections that follow.
2.2. Molecular Dynamics Fundamentals
All MD simulations were performed using the Large-scale Atomic/Molecular Massively Parallel Simulator (LAMMPS, https://lammps.sandia.gov), an open-source MD simulator [23]. We utilized a potential energy function based upon the CHARMM and AMBER frameworks [24, 25].
The force field we utilized includes bonded interactions comprised of bonds, bond angles, dihedral torsion angles, and improper dihedral angles. The non-bonded components of the force field were defined by Lennard-Jones (LJ) and Coulombic interactions. This simple representation of interatomic forces provides sufficient accuracy for simulating organic molecules in aqueous environments, while remaining computationally inexpensive relative to more complex force fields [24, 25].
We utilized the Antechamber function in AMBER to assign General Amber Force Field (GAFF) interaction parameters and LJ parameters. We used AM1-BCC to assign partial charges to each atom [26]. Molecular structures of constituent molecules, including m-phenylenediamine (MPD), trimesoyl chloride (TMC), hexane, water, and hydroxide were equilibrated using assigned parameters. All interaction parameters and charges used are listed in the Supplementary Materials Section S1.
A velocity-Verlet integrator was used to perform time integration of the potential energy equation [27]. Both the canonical (NVT) ensemble and isothermal-isobaric (NPT) ensemble were utilized to conserve the number of atoms (N), domain temperature (T), and either the domain volume (V) or domain pressure (P) [28]. A Nose-Hoover style thermostat or barostat was used to conserve temperature and pressure, respectively.
To improve the computational efficiency, the SHAKE and RATTLE algorithms were used to constrain the lengths and angles of covalent hydrogen bonds to their equilibrium values to reduce the computational cost of evaluating those interactions and enable longer time steps. [29, 30]. Short-range LJ and Coulombic interactions were computed for atom pairs separated by fewer than 10 angstroms (Å) [31], which was large enough to give results consistent with larger cutoff distances while improving efficiency of computing pair-wise forces. Coulombic interactions between atoms separated by more than 10 Å were calculated via the particle-particle particle-mesh method (PPPM) [32, 33].
2.3. Polymerization
To simulate polymerization of monomers that comprise typical commercial crosslinked aromatic polyamide TFC active layers, we built upon a polymerization method first introduced by Harder et al. [13]. In this method, MPD and TMC were randomly packed at a stoichiometric ratio of 3:2 into a cubic, periodic domain. Changes to this ratio are known to affect properties and performance of active layers [34], which is easily modified using this method. Two alternative approaches were evaluated to assess the effect on the resultant polymerized structure and the computational cost: a solvent flooded approach, and a solvent-free approach. This is the first comparison of polymerization methods of this kind to determine the extent to which this simulation choice affects the structure and properties of the simulated membranes and the simulation cost.
For the more realistic solvent-flooded approach, the MPD and TMC packed domain was flooded with hexane molecules, as first described by Zhang et al. [19]. Hexane was used as a solvent because MPD partitions into the organic phase during interfacial polymerization, where it reacts with TMC [35]. Hexane flooding was omitted for the solvent-free approach, thereby reducing the atom count and resulting in less computationally costly simulations.
After random packing and hexane flooding, if used, atomic positions were relaxed via minimization of potential energy. Using a 10 fs time step and a steepest descent approach, relaxation was carried out until the relative change in potential energy between sequential time steps was less than 10−5. After relaxation, polymerization was simulated in an NVT ensemble at 300 K, with a time step of 2 fs.
REACTER, a parallel algorithm implemented within LAMMPS for performing predefined reactions in atomistic MD solutions, was utilized to perform the polymerization of MPD and TMC via formation of amide bonds between amine groups located on MPD molecules and acyl chloride groups located on TMC molecules [36]. During polymerization, nitrogen atoms of MPD amine groups and carbon atoms of TMC acyl chloride groups that passed within 5 Å of each other were reacted to form an amide bond. Concurrently with the formation of the amide bond, the chlorine atom from the TMC and hydrogen atom from MPD were removed. Polymerization was run for at least 750,000 fs, or until no new reactions occurred, and crosslinking degrees were determined based on the fraction of the maximum number of theoretical amide bonds that formed. Achievement of steady state always took fewer than 750,000 fs due to the pre-mixed state of the domain and the relatively high initial concentrations of MPD and TMC. After polymerization of MPD and TMC, hexane was removed from the system if it was originally present, and the remaining acyl chloride groups were hydrolyzed into carboxylic acid groups. To perform the hydrolysis, the simulation domain was flooded with free hydroxides and the system was then relaxed via steepest descent with a 10 fs time step and a 10−5 relative potential energy tolerance stopping criterion. Oxygen atoms from free hydroxides and carbon atoms from acyl chloride groups were then reacted if they passed within 5 Å of each other. As they reacted, byproduct chlorine atoms were removed. Hydrolysis continued until all acyl chloride groups were hydrolyzed. After hydrolysis, any unreacted hydroxides were removed, leaving only the crosslinked aromatic polyamide.
2.4. Equilibration
While the polyamide matrix condensed during polymerization, the density of the final matrix did not reach experimentally determined active layer dry densities in the short time scales attainable via the MD polymerization approach used. Therefore, the matrix was equilibrated via compression at elevated temperatures to achieve experimentally validated densities, and then relaxed at ambient temperatures and pressures to ensure stability of the resultant structures. Utilizing elevated temperatures and compression, equilibration simulations only took 1.5 ns.
The equilibration procedure consisted of a three-step compression loop, modified from a method to compress simulations of glassy polymers to experimental densities [37]. All steps used a time step of 2 fs, and each simulation step was run for 50,000 fs. The first step used an NVT ensemble at 1,000 K to allow the polyamide to rapidly change conformations. The second step was an NVT ensemble at 300 K to return the temperature to typical conditions. The third step was an NPT ensemble at 300 K and an increasing sequence of pressure steps—1, 10, 100, and 1,000 bar—to compress the polymer. The compression loops were continued until the relative change in polyamide density between loops was less than 0.01 g/cm3. After compression, the polymer was relaxed via an NPT ensemble at 300 K and 1 bar for 500,000 fs to equilibrate the system under target conditions.
In order to use these domains as active layers with a feed (−z) and permeate (+z) face, one dimension must be non-periodic. We therefore utilized an unwrapping method to delete a 2 Å thick x-y planar slice of the polyamide domain to break the periodic condition in the z direction [19]. Dry domains were equilibrated after unwrapping via NPT at 1 bar and 300 K for 106 fs (1 nanosecond, ns) to ensure stability of unwrapped domains.
2.5. Hydration
After equilibration, the polyamide matrix was exposed on two faces to reservoirs of water molecules, thereby hydrating the domain as water molecules diffused into the domain. We evaluated alternative approaches to speed the computationally costly hydration process by pre-inserting water molecules into pores that may eventually be occupied by water after hydration. The targeted cavity sizes were based upon the well-established existence of so-called network and aggregate pores within crosslinked aromatic polyamide; network and aggregate pores have radii of 2.1–2.4 Å and 3.5–4.5 Å, respectively [38]. While random insertion methods have been used to hydrate polyamide simulations before, this is the first comparison of hydration simulation approaches to determine their capability to speed up hydration and evaluate whether they affect polyamide properties (including steady state water content) and performance.
Three alternative hydration methods were evaluated: a control without water molecule insertion, insertion into aggregate pores, and insertion into aggregate and network pores. After the water insertion step, all alternatives were exposed to identical cubes of pure water equilibrated via NPT at 1 bar and 300 K for 100,000 fs; exposure occurred at the −z and +z faces of the polyamide domain.
To hold the domain volume constant while applying feed and permeate pressures, graphene planes were used as pistons to enforce the confining pressure while using an NVT ensemble [9]. Graphene pistons consisted of a rigid plane of hexagonal close-packed carbon atoms with a lattice constant of 2.46 Å. The graphene pistons were restricted such that only forces acting on the piston through the center of mass of the plane in the z direction were non-zero, thereby preventing rotation and shear movements of the piston. A compensating force was added to the graphene pistons to pressurize the feed and permeate reservoirs to 1 bar during hydration to simulate realistic active layer hydration. Hydration simulations were carried out in an NVT ensemble at 300 K with a time step of 2 fs.
Previous MD simulations of polyamide hydration via diffusion were carried out until the change in water concentration in the polyamide matrix was zero between successive time steps [19]. To assess the effects of hydration simulation alternatives, we simulated hydration for a minimum of 300 ns to observe fluctuations and ensure steady state.
To calculate water concentrations and pore-size distributions of hydrated domains, snapshots of the system state, including positions and identifying information of all atoms, were saved every 100,000 time steps (0.2 ns). In all domains, the polyamide-water interface near the domain boundaries was ill-defined and rough. To remove edge effects of this interface on water concentration and pore-size distributions, we evaluated pore-size distributions and water concentration within centered, truncated regions of the domain that were as large as possible without including the polyamide-water interface. The truncated regions were selected to be the portion of the domain where the slope of the water concentration profile was zero.
A rolling time average was used to smooth temporal variations in water concentrations and pore-size distributions resulting from the small size of the spatial domains and normal temporal variations typical of molecular methods. Time averages utilized an averaging window of 50 ns. Thus, all water concentrations and pore-size distributions reported for hydrated domains are time-averaged results from truncated domains. Pore-size distributions for dry domains were collected using compressed and equilibrated cubic periodic domains prior to hydration. Probability density functions (PDFs) of pore diameters within a polyamide domain were used to describe the pore-size distribution.
Time to steady state hydration was calculated as the time at which the slope of the time-averaged water concentration during hydration was zero. Due to entropic fluctuations, the water concentration value oscillated about a steady state value. In the event that these oscillations resulted in multiple points of zero slope, the first occurrence was chosen as the time to steady state.
2.6. Operation
After hydration, polyamide domains were immobilized so that a pressure difference between the feed and permeate sides of the polyamide could be established, while not restricting local entropic fluctuations of the polyamide. Domains were immobilized by tethering a random selection of atoms within a 1 Å slice of the domain located 10 Å from the permeate face. Nitrogen or carbon atoms found in amide, amine, or carboxyl groups were selected randomly to prevent over-constraint of hydrogen atoms or full immobilization of aromatic rings. The tethering was achieved by flexibly “bonding” the atoms in place. Fewer than 5% of the atoms in the 1 Å slice were required to be tethered in order to prevent movement of the matrix in the z direction.
To create a pressure difference between the feed and permeate reservoirs, a force was applied to the feed graphene piston until the pressure in the feed reservoir was 72.3 bar, while the permeate reservoir was maintained at 1 bar. Operation was simulated using an NVT ensemble at 300 K with a time step of 2 fs.
To calculate the water permeability for comparison to real active layers, we computed the number of water molecules exiting the polyamide domain during operation simulation over each time increment to determine the volumetric flux of water Jw using
| (1) |
where MW is the molar mass of water, ρw is the density of water at 300 K and 1 bar, NA is the Avogadro constant, Axy is the cross-sectional area of the polyamide domain, Np is the number of permeated water molecules, and t is permeation time. Snapshots of water molecule locations were collected every 0.2 ns, and to smooth temporal variations ΔNP was time-averaged over 1 ns. Operation simulations were carried out for 25 ns, and Jw was calculated over the final 20 ns of operation.
Based upon the flux of water through the polyamide, the water permeability Kw was calculated as
| (2) |
where Pf is the pressure in the feed reservoir, Pp is the pressure in the permeate reservoir, and L is the thickness (z-direction) of the polyamide domain.
2.7. Error Reduction
Two methods were evaluated to reduce the stochastic error in our MD simulations, growing the number of realizations per alternative for a fixed-size domain and growing the size of the domain. This is the first reported comparison between ensemble realization size scaling and domain size scaling performed for atomistic MD simulations of polyamide domains, hydrated or otherwise. Increasing the number of realizations per ensemble was accessible only to the smallest domain sizes we evaluated due to the significant increases in computational cost with domain size. The control polymerization ensemble consisted of 20 unique random packings of MPD and TMC. We then duplicated these packings, and flooded the duplicates with hexane molecules to create an ensemble of 20 realizations for the more realistic polymerization alternative. For hydration, each alternative had an ensemble of three unique random packings of MPD and TMC. When ensembles were used, results were averaged over the set of realizations to determine membrane properties and performance. Table 1 shows the number of realizations used for each simulation alternative.
Table 1:
Number of realizations per ensemble used to evaluate MD simulation step approaches.
| Domain Scale | 1x | 8x | 64x | |
|---|---|---|---|---|
| MD simulation step | Polymerization | Hydration | Hydration | Hydration |
| Ensemble size | 20 | 3 | 1 | 1 |
The second method evaluated to reduce simulation errors was to increase the size of the simulated domain. Four domain sizes were utilized, a baseline domain (1x), an 8-times larger (8x) by atom count, as well as a 64-times larger (64x) and a 512-times larger (512x) domain. As domain volume scales with atom count, these domains were also effectively 8-times, 64-times, and 512-times larger by volume. The 512x domain was only polymerized without hexane present, compressed, and equilibrated, not used in either hydration or operation simulations. Table 2 contains the total number of molecules and atoms, and the size of the truncated domain used at each scale and simulation step. The size of the truncated region that was analyzed scaled with domain thickness.
Table 2:
Domain molecule counts, atom counts and length scales.
| Domain Scale | 1x | 8x | 64x | 512x | |
|---|---|---|---|---|---|
| MPD molecules | 300 | 2,400 | 19,200 | 153,600 | |
| TMC molecules | 200 | 1,600 | 12,800 | 102,400 | |
|
| |||||
| Polymerization step alternative | NH | H | NH | NH | NH |
| Hexane molecules | - | 1,000 | - | - | - |
| Atoms during polymerization | 8,400 | 28,400 | 67,200 | 537,600 | 4,300,800 |
| x-y planar area (Å2) | 442 | 442 | 882 | 1762 | 3522 |
|
| |||||
| Number water molecules | 3,600 | 14,400 | 57,600 | - | |
| Atoms during hydration and operation | 19,200 | 110,400 | 710,400 | - | |
| Truncated domain thickness (Å) | 30 | 70 | 150 | - | |
Polymerization step alternatives H and NH refer to polymerization with and without hexane present, respectively.
Typical domain sizes used in the MD literature are comparable with our 1x domains, and the largest (by volume) previously reported in the literature is on the order of our 8x domain [39]. The scale of the 64x domain is an order of magnitude larger than that domain, and within the range of the intrinsic polyamide thickness (10–30 nm), measured experimentally [40, 41].
2.8. Pore Morphology and Topology
At the molecular scale, crosslinked aromatic polyamide can be considered a porous medium, where the pore space is typically referred to as the free volume. From here forward, we will refer to the free volume as the pore space of the polyamide. Of primary concern when determining property-performance relationships of a porous medium is the geometric characterization of the pore space. We examined three approaches to characterize the pore morphology and topology: pore-size distribution, medial axis analysis, and Minkowski functionals. This is the first time that the concepts of medial axis analysis and Minkowski functionals are used in the context of polyamide molecular morphology.
To determine the pore-size distribution within the polyamide domain, we utilized a Monte Carlo cavity energetic sizing algorithm (CESA) method in which a candidate point for a cavity was randomly selected within the domain. That point was then moved to the nearest local minimum in the LJ potential field via steepest descent. The cavity surrounding the point was then expanded until the net LJ interactions with neighbor atoms was zero [42, 43]. We utilized the Free Volume Toolkit (https://bitbucket.org/frank_willmore/vacuumms) to perform the CESA sizing using truncated snapshots of polyamide atom locations collected every 200,000 fs during hydration [44]. Interested readers are directed to the previous publications for more details on the CESA method [42–45].
Identification of the pathways through the polyamide pore space that traverse the domain from feed to permeate faces is an integral step in evaluating the characteristics of those pathways that control polyamide properties and performance [46, 47]. To do so, we introduce in the context of polyamide molecular morphology the concept of medial axis analysis. The medial axis of the pore space describes the central axes of pathways through the polyamide pore space [46, 48]. To evaluate the medial axis, we discretized the domains into a 3D binary voxel map. We performed a grid independence study to determine a voxel size of 0.05 Å generated results that were independent of domain resolution, see Supplementary Materials Section S2. Domain voxels were then divided into two sets, those voxels that make up the pore space of the domain Vf, and those voxels that make up the volume occupied by the atoms, Va. Using the atomic LJ potential field, voxels within the repulsive LJ region surrounding an atom were said to be in Va. As the domain was a binary map, all other voxels were therefore part of Vf. The medial axis was then identified via a 3D voxel thinning of Vf, implemented in PoreSpy [48–50].
Pore-size distributions generated using the CESA method and medial axis analysis provided useful measures of the pore space but did not fully describe the pore-space morphology and topology. To address this shortcoming, we also implemented Minkowski functional analysis of the polyamide pore space to provide integral measures of the known invariant pore-space characteristics.
The Minkowski functionals are a unique and complete set of the invariant characteristics of a 3D object of positive reach, including the volume, surface area, the integral of the mean bounding surface curvature, and the integral of the Gaussian curvature of the bounding surface [51–53]. The Gaussian curvature can be related to the Euler characteristic, a numerical description of topology, using the Gauss-Bonnet theorem [53, 54]. Critical to the importance of Minkowski functionals is Hadwiger’s theorem, which states that any property of a convex morphological body that is additive, motion invariant and conditionally continuous can be written as a linear combination of the Minkowski functionals [51, 53, 55]. Such properties are of primary concern in the development of mechanistic property-performance relationships of polyamide active layers that fully account for the effects of molecular morphology and topology [56, 57]. Therefore, Minkowski functionals can be used as comprehensive quantitative descriptors of the polyamide film morphology and topology.
Minkowski functionals alone are not capable of defining the accessibility of the pore space to particles of a given size, i.e. no information about the distribution of pore sizes within a pore space is generated by Minkowski functionals [58]. Minkowski functionals were therefore evaluated for pore-space opening maps. Opening maps were generated by identifying the portions of pore space accessible to spheres of increasing size (also termed structuring elements). A description of the opening map generation procedure, technically described as morphological erosion and dilation of the pore space, and application examples are available in the literature [e.g., 59–61]. The Minkowski functionals of the opening maps for each structuring element size can then be evaluated to yield morphological description of the pore space for the full range of molecule sizes capable of permeating the polyamide domain [58].
To evaluate the Minkowski functionals, we discretized the domains into a 3D binary voxel map using a resolution of 0.05 Å. Domain voxels were then divided into two sets, using the same masking procedure described for medial axis analysis. We then utilized QuantIm (https://www.quantim.ufz.de) to compute the morphological opening and Minkowski functionals for the pore space Vf [58]. The opening map was generated for a range of sphere sizes spanning from a single point to the maximally sized sphere that could fit in at least one location within the discretized representation of the pore space.
2.9. Computational Environment
High-performance computing (HPC) resources were used to perform this work. The machines utilized were Expanse at the San Diego Supercomputing Center and Bridges-2 at the Pittsburgh Supercomputing Center. We utilized the standard compute nodes on each machine, which both consist of two AMD EPYC 7742 CPUs, containing 64 cores per CPU, yielding 128 cores per node. LAMMPS supports parallelized, distributed memory computation utilizing openMPI, and for shared memory uses OpenMP threading.
C++ code was compiled using g++ 10.2.0 and C code was compiled using gcc 10.2.0; the −O2 optimization flag was utilized to improve performance. Optimal simulation speeds were achieved utilizing a combination of MPI and threading via OpenMP: 32 MPI processes were assigned to each node utilized, and 4 OpenMP threads were used per MPI process—yielding one thread for each of the 128 cores present on a node. Based upon scaling results of the simulations, the number of nodes was varied with the size of the domain to ensure efficient use of the HPC resources. One node was used for all simulations of the 1x domain, two nodes for the 8x domain, and three nodes for the 64x domain. Four nodes were used to simulate polymerization and equilibrations of the 512x domain.
3. Results
3.1. Overview
The aim of this work is to advance methods for molecular-scale simulation of polyamide membrane synthesis, hydration, and operational performance that increase computational efficiency and provide a basis to improve insights derived from simulation. The sections that follow present results in turn on each of these aspects of the work.
3.2. Polymerization
Figure 1 depicts the different steps and elements of the polymerization process. The process starts with the packing of MPD and TMC (Figure 1(a)) into a cubic periodic domain with or without hexane (Figure 1(b)). An oligomer representative of the reaction of MPD and TMC is presented in Figure 1(a). As polymerization proceeds, MPD and TMC condense slightly, and when there are no more monomers to react, unreacted acyl chlorides are hydrolyzed (Figure 1(c)). Finally, the polymerized and hydrolyzed polyamide is compressed and equilibrated to experimentally validated densities (Figure 1(d)).
Figure 1:
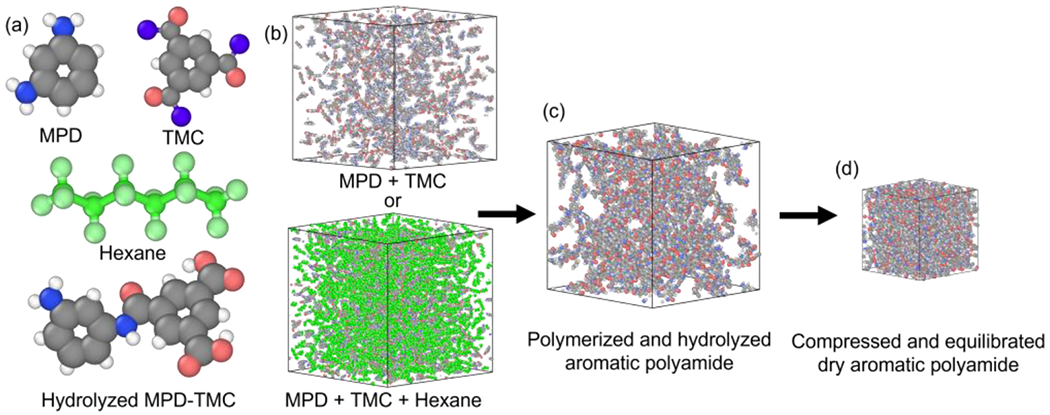
Polymerization and equilibration of membrane structures: (a) molecular structures of MPD monomer, TMC monomer, and hexane molecule, and hydrolyzed MPD-TMC oligomer, where white, gray, blue, pink, and purple represent hydrogen, carbon, nitrogen, oxygen, and chlorine, respectively while carbon and hydrogen of hexane molecules are represented in green; (b) example cubic periodic domain duplicates packed with MPD and TMC shown on top, and additionally flooded with hexane on bottom; (c) polymerized and hydrolyzed polyamide matrix; and (d) final compressed and equilibrated polyamide domain.
Details of the polymerization simulations for the 1x domain are given in Table 2. Simulation update rates, described by the ratio between time simulated to real time used to perform that simulation, were 32.01 ns/day and 39.03 ns/day for the cases with and without hexane, respectively. Thus, the update rate was improved by 22% by excluding hexane from the simulations.
After equilibration and unwrapping of the domain to form the −z and +z faces, both polymerization alternatives (i.e., with and without hexane present) were hydrated without pre-insertion of water molecules. Figure 2 shows the ensemble (20 realizations) and time-averaged concentration of water for both polymerization approaches. The shaded region around each line represents two times the standard deviation of the time average, which was used as a measure of the uncertainty in the ensemble and time-averaged values depicted by the trend lines. Table 3 summarizes results for the effect of polymerization conditions on the ensemble averaged (20 replicates) values of membrane properties commonly discussed in relation to membrane performance, and water permeability calculated during operation simulation. The crosslinking degree and dry density of simulated films were approximately 0.90 and 1.26 g/cm3 in accordance with experimentally measured values. The average water concentration and water permeability at equilibrium were in the 0.12–0.15 g/cm3 and 0.16–0.17 x 10−6 Lm−1hr−1bar−1 ranges, respectively.
Figure 2:
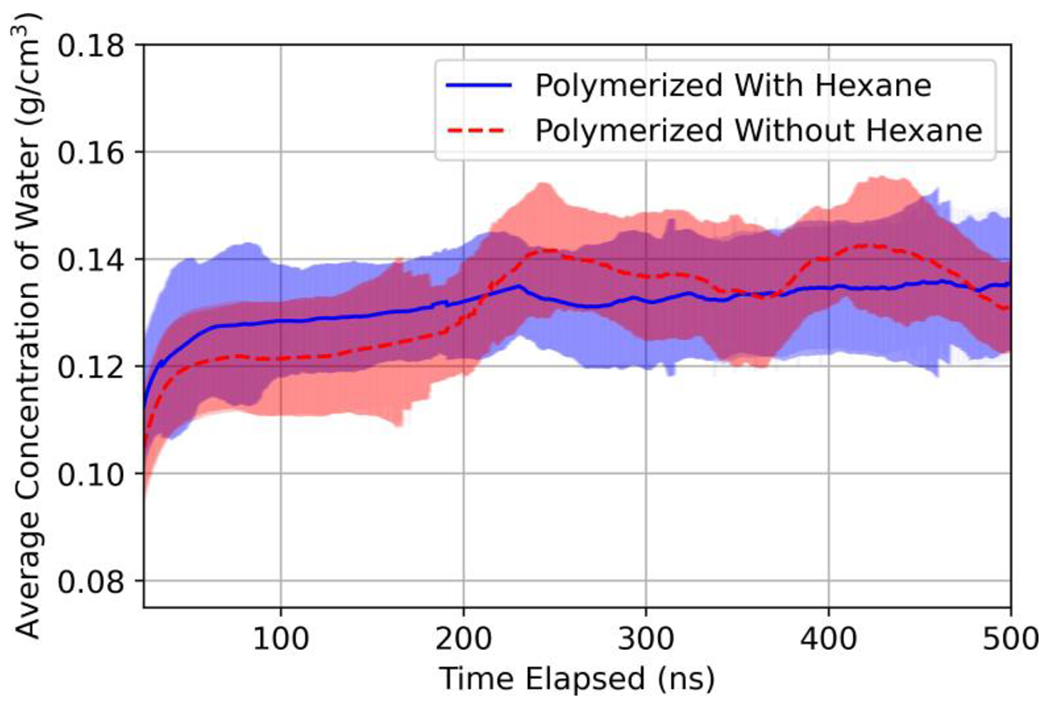
Ensemble averaged concentration of water molecules (g/cm3) during hydration of 1x domain polymerization alternatives. Shaded regions represent two times the standard deviation of the averaged values.
Table 3:
Effect of polymerization simulation approach on 1x domain properties and performance.
| Polymerization Alternative | With hexane | Without hexane |
|---|---|---|
| Crosslinking degree, DC (−) | 0.90±0.02 | 0.90±0.01 |
| Dry density, ρd (g/cm3) | 1.26±0.01 | 1.26±0.01 |
| Thickness, L (Å) | 36±5 | 36±4 |
| Water concentration, ρw (g/cm3) | 0.135±0.013 | 0.131±0.009 |
| Time to steady state hydration, tSS (ns) | 83±6 | 85±7 |
| Water permeability, Kw x 10−6(Lm−1hr−1bar−1) | 0.17±0.03 | 0.16±0.02 |
3.3. Hydration
Figure 3 depicts the different insertion approaches used prior to hydration. To observe the long-time-scale behavior of the three hydration approaches (i.e., no water insertion, insertion into aggregate pores, insertion into network and aggregate pores), we simulated each approach over 800 ns for the lx domain polymerized in the presence and absence of hexane.
Figure 3:
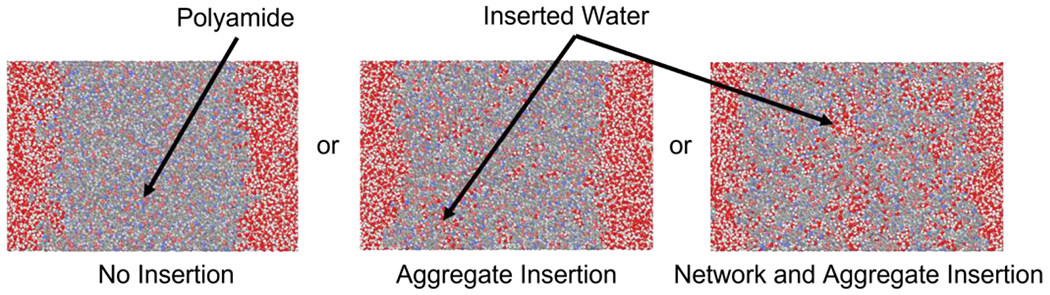
Water insertion method alternatives performed for an 8x domain, where white, gray, blue, and pink, represent hydrogen, carbon, nitrogen, and oxygen. Red represents oxygen in water.
Figure 4(a) and 4(b) show the ensemble (3 realizations) and time-averaged water concentration within the polyamide domain for the hexane and no hexane simulations, respectively. Table 4 contains the ensemble averaged data for all hydration step alternatives evaluated at the 1x domain scale. The water concentration at steady state for the cases of no insertion and insertion into aggregate pores was in the 0.112–0.147 g/cm3 range, and significantly greater for the case of insertion into aggregate and network pores, at 0.146–0.202 g/cm3. Similarly, the time to steady-state hydration was in the range of 73–78 ns for no insertion and aggregate pore insertion cases, and significantly greater in the range of 221–277 ns for the case of insertion in aggregate and network pores.
Figure 4:
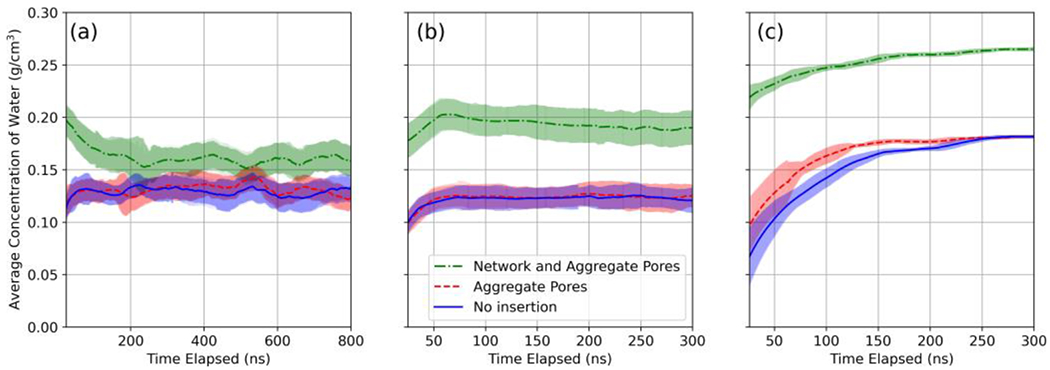
Ensemble averaged concentration of water molecules (g/cm3) during hydration of: 1x domains (a) polymerized with hexane; (b) polymerized without hexane; (c) 8x domains polymerized without hexane. Shaded regions represent two times the standard deviation of the averaged values denoted by the trend lines.
Table 4:
Effect of hydration simulation approach on 1x domain properties and performance.
| Polymerization Alternative | With Hexane | Without Hexane | ||||
|---|---|---|---|---|---|---|
| Crosslinking Degree, DC (−) | 0.89±0.04 | 0.89±0.04 | 0.89±0.04 | 0.91±0.03 | 0.91±0.03 | 0.91±0.03 |
| Dry density, ρd (g/cm3) | 1.26±0.02 | 1.26±0.02 | 1.26±0.02 | 1.26±0.03 | 1.26±0.03 | 1.26±0.03 |
| Insertion Method | No | A | N + A | No | A | N + A |
| Thickness, L (Å) | 36±5 | 33±6 | 33±5 | 36±4 | 34±3 | 34±5 |
| Water concentration, ρw (g/cm3) | 0.135±0.012 | 0.124±0.012 | 0.164±0.018 | 0.122±0.008 | 0.125±0.008 | 0.191±0.011 |
| Time to steady state hydration, tSS (ns) | 73±6 | 78±5 | 221±15 | 75±6 | 75±7 | 277±13 |
| Water permeability, Kw x 10−6 (Lm−1hr−1bar−1) | 0.17±0.03 | 0.18±0.03 | 0.73±0.14 | 0.16±0.02 | 0.16±0.02 | 0.55±0.11 |
Insertion methods No, N, and A refer to no insertion, network insertion, and aggregate insertion, respectively.
We also evaluated the time-averaged water concentration within 8x domains polymerized without hexane present (Figure 4(c)). Final average water concentrations for no insertion and insertion into aggregate pores were identical at 0.180 g/cm3, while insertion into network and aggregate pores resulted in 0.265 g/cm3.
3.4. Error Reduction
Two methods were evaluated to reduce stochastic MD simulation error, ensemble averaging methods and increasing the simulated domain size. Figure 5 shows polyamide water concentration of the 1x, 8x, and 64x domains polymerized without hexane present. To show how ensemble averaging can reduce error, we present three ensemble alternatives for the 1x domain (i.e., one aggregate insertion realization, three aggregate insertion realizations, and 23 no insertion and aggregate insertion realizations), two ensemble alternatives for the 8x domain (i.e., one aggregate insertion realization, one no insertion and one aggregate insertion realization) for the 8x domain, and one 64x domain and hydrated with aggregate insertion. The properties of the 8x and 64x polyamide domains are summarized in Table 5. Results show that the water concentration at equilibrium in 1x, 8x, and 64x domains was approximately 0.125 g/cm3, 0.180 g/cm3, and 0.240 g/cm3, respectively, which for the 64x domain are all on the order of experimentally reported values (ρw =0.19–0.31 g/cm3) [62]. Further, water permeabilities for all domains, 0.16–1.484 x 10−6 Lm−1hr−1bar−1, were comparable to water permeabilities calculated from in-lab experiment (0.1–1.1 × 10−6 Lm−1hr−1bar−1) [62,63].
Figure 5:
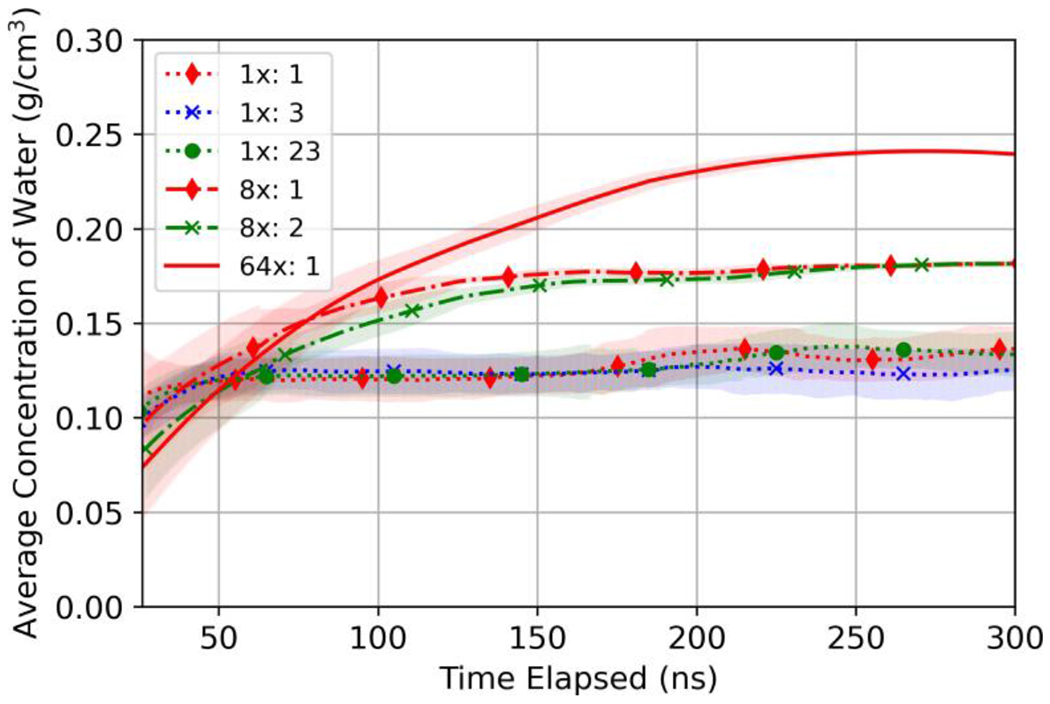
Ensemble averaged concentration of water molecules (g/cm3) during hydration of each domain scale polymerized without hexane present. Numbers next to domain scale in the legend denote the number of realizations in the ensemble. Shaded regions represent two times the standard deviation of the rolling average.
Table 5:
Polyamide domain properties as a function of system size.
| Domain Scale | 8x | 64x | ||
|---|---|---|---|---|
| Crosslinking degree, DC (−) | 0.89 | 0.89 | 0.89 | 0.89 |
| Dry density, ρd (g/cm3) | 1.27 | 1.27 | 1.27 | 1.27 |
| Insertion method | No | A | N + A | A |
| Thickness, L (Å) | 78 | 79 | 79 | 140 |
| Water concentration, ρw (g/cm3) | 0.180±0.005 | 0.180±0.004 | 0.265±0.001 | 0.240±0.000 |
| Time to steady state hydration, tSS (ns) | 262 | 146 | 290 | 283 |
| Water permeability, Kw x 10−6 (Lm−1hr−1bar−1) | 0.446 | 0.435 | 0.821 | 1.484 |
Insertion methods No, N, and A refer to no insertion, network insertion, and aggregate insertion, respectively.
3.5. Pore Morphology and Topology
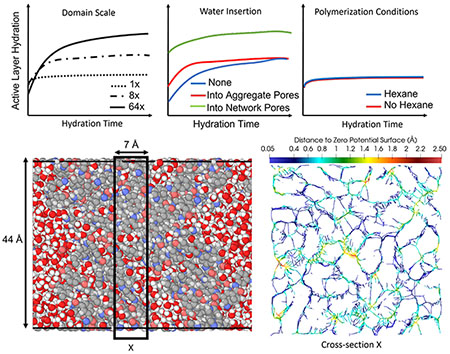
Figure 5 shows the pore-size distributions for dry, equilibrated domains across all sizes evaluated. All pore-size distributions are unimodal with larger domains having distributions shifted to larger cavity diameters compared with smaller domains.
Pore-size distributions were also evaluated for the three hydration approaches for 1x domains polymerized in the presence of hexane. Pore-size distributions are displayed for the initial (after insertion of water molecules) and final (at steady state). The corresponding initial and final ensemble results are presented in Figure 7 for 1x domains polymerized in the presence (a) and absence of hexane (b), and 8x domains polymerized in the absence of hexane (c). Results show that hydration shifted the pore sizes to bimodal distributions (Figure 7) for all hydration approaches compared with the unimodal distributions observed for dry domains (Figure 6).
Figure 7:
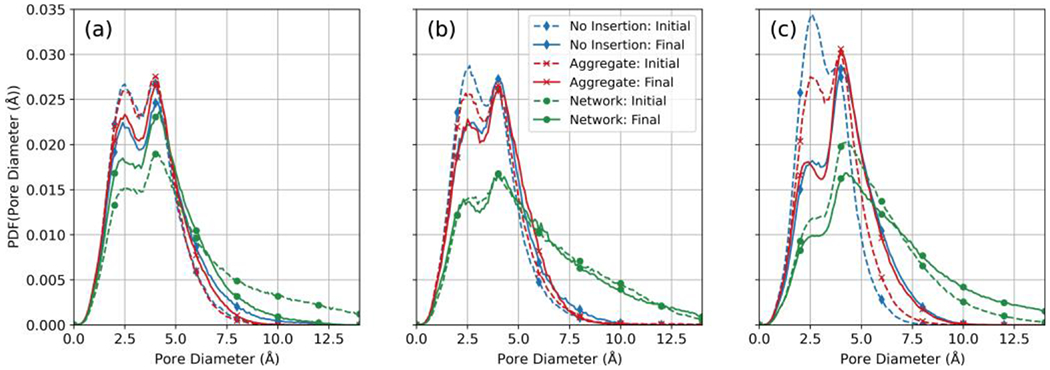
Initial and final ensemble (3 realizations) averaged pore-diameter-size distributions during hydration of: (a) 1x domains polymerized with hexane; (b) 1x domains polymerized without hexane; (c) 8x domains polymerized without hexane.
Figure 6:
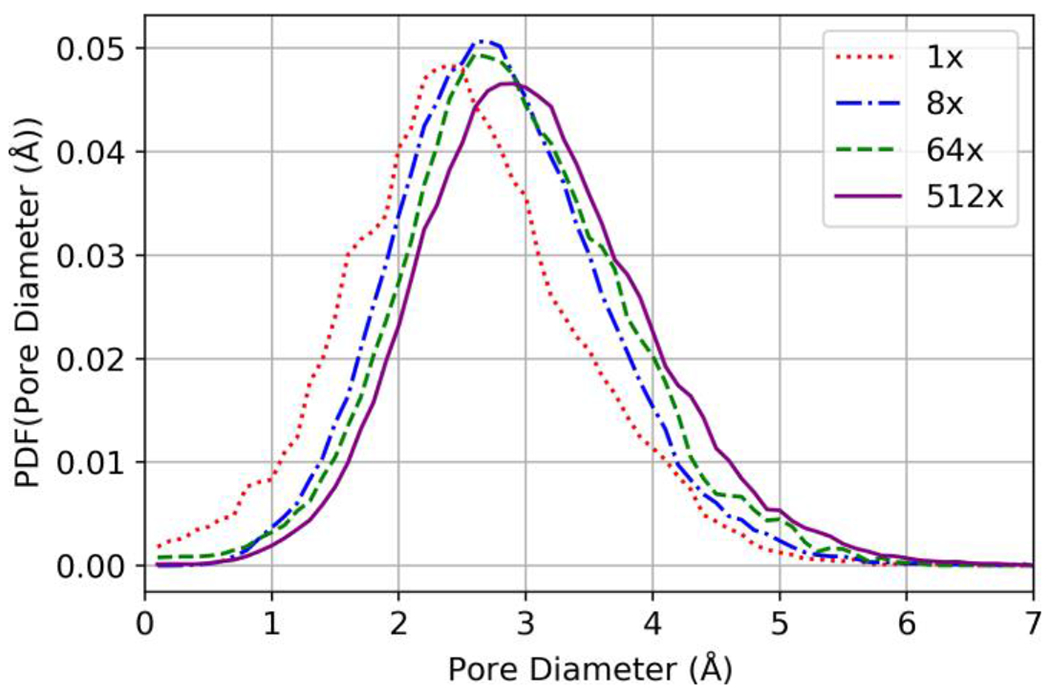
Pore-size distributions of dry, compressed and equilibrated polyamide domains across all domain scales.
Figure 8 illustrates a comparison across domain sizes of the initial and final pore-size distributions observed during hydration after insertion of water into aggregate pores. Results show a shift toward larger pore sizes and a reduction in the amplitude of the first peak of the bimodal distribution after hydration with a larger domain.
Figure 8:
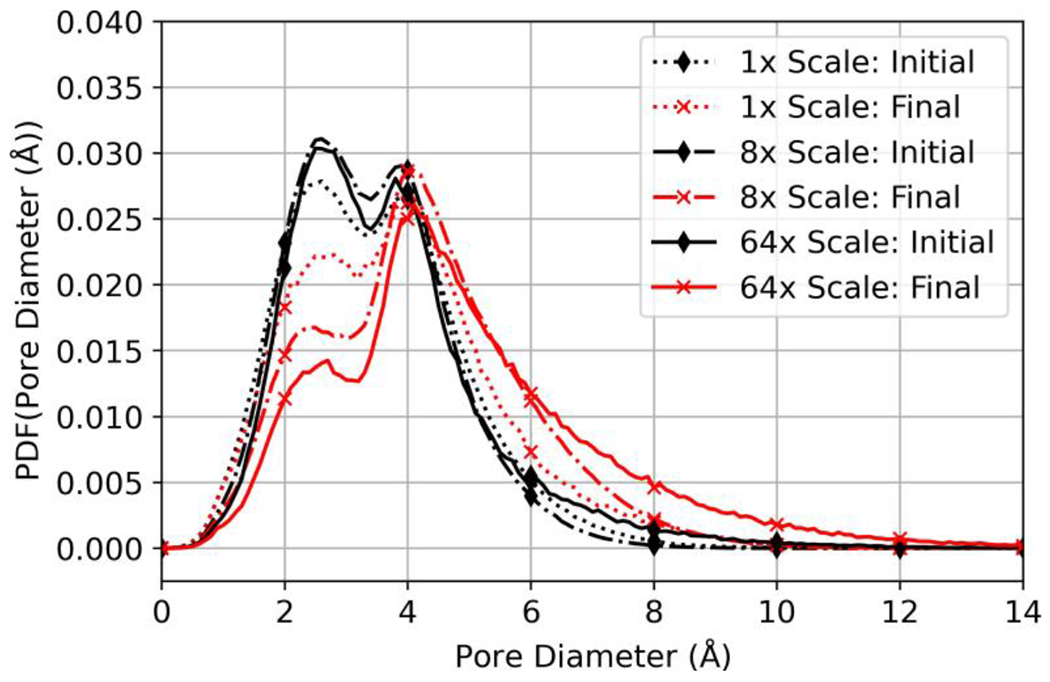
Initial and final ensemble averaged pore-diameter-size distributions during hydration of each domain scale polymerized without hexane present.
Figure 9 shows the medial axis of the polyamide pore space located within a 7 Å thick region of a hydrated 1x domain. The maximum sized cavity in this slice was approximately 5 Å in diameter.
Figure 9:
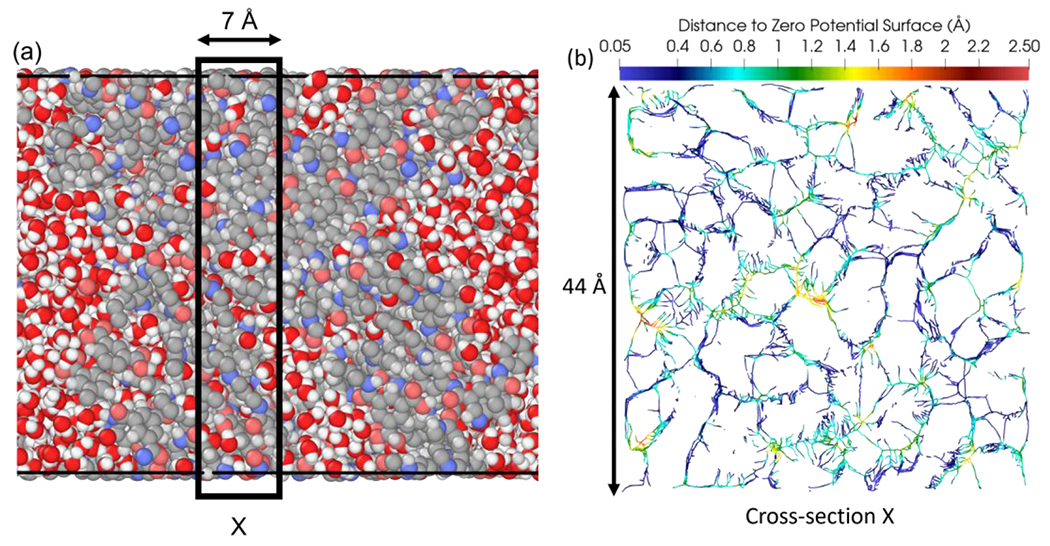
Hydrated 1x domain (a) where white, gray, blue, and pink represent hydrogen, carbon, nitrogen, and oxygen of the polyamide respectively, and red and white represent oxygen and hydrogen of water, boxed region represents thickness of cross-section; (b) medial axis representation of the polyamide pore space within a 7 Å cross section, where color scale represents distance from the medial axis to the surface of the volume occupied by atoms, as defined by a net zero potential energy surface.
Figure 10 illustrates the Minkowski functionals evaluated for an opening map of the pore space of a 1x domain polymerized without hexane in a dry and hydrated state. All Minkowski functionals were normalized by the domain volume. The voxel size was 0.053 Å3. Results show that Minkowski functionals differ for dry and hydrated states, where porosity is increased, the slope of the surface area density profile is reduced, and the peaks of the mean curvature density and Euler characteristic densities are reduced as the domains are hydrated. The size of the maximum accessible structuring element was 4.2 Å and 9 Å for the dry and hydrated states, respectively.
Figure 10:
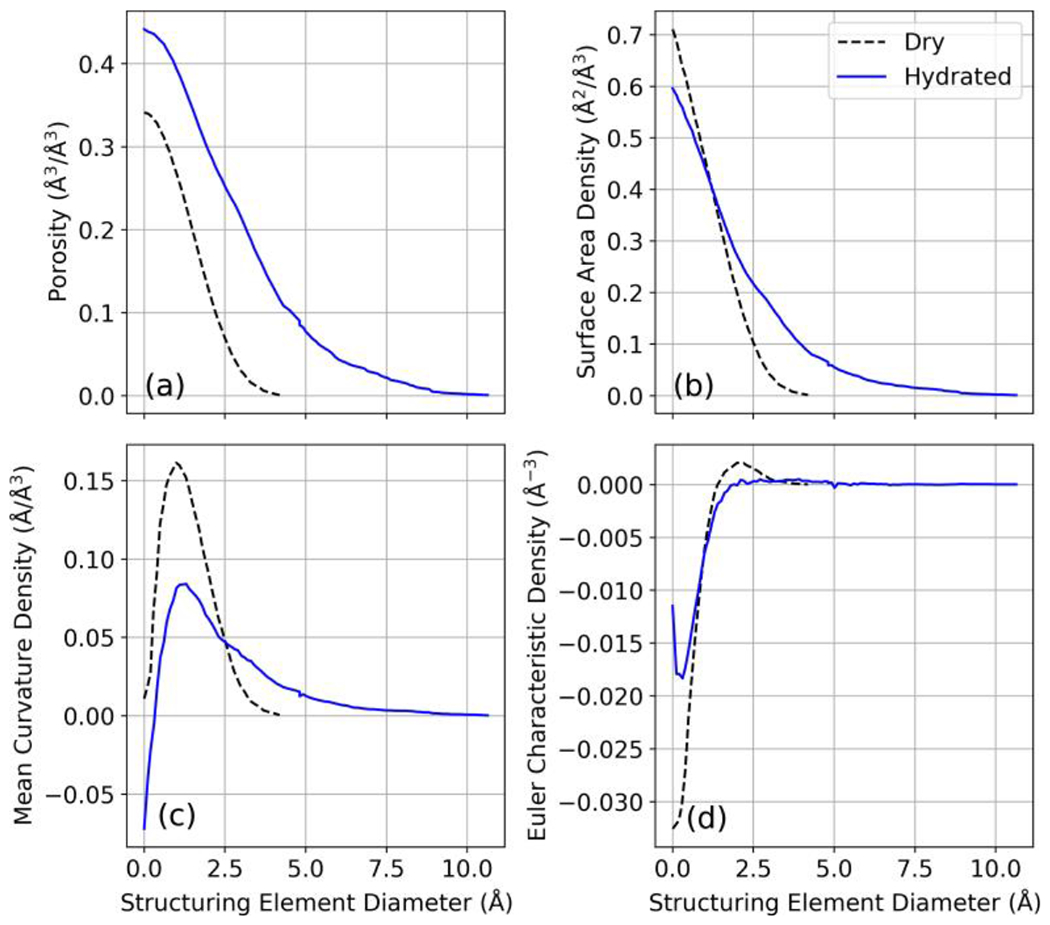
Minkowski functionals of dry and hydrated 1x domain pore-space opening map: (a) pore-volume density, i.e. porosity; (b) surface-area density; (c) mean surface-curvature density; and (d) Euler characteristic density.
4. Discussion
Development of realistic and computationally efficient polymerization methods is necessary to enable simulation of large and therefore more representative polyamide domains. Polymerization in a model solvent like hexane improves the realism of the simulation conditions but incurs a significant computational burden. This burden increases as the number of atoms N increases, as MD simulations have optimal complexity O(N log N) per time step when using fast Fourier Transform methods like PPPM, as was done in this study [64].
In evaluating the effect of a more computationally efficient polymerization step on polyamide hydration and performance, we found that simulating polymerization without hexane present increased the simulation rate by 22%. As shown in Table 3, the crosslinking degrees, dry densities, steady-state water concentration, time to steady state, and water permeability for both polymerization step alternatives were all within two standard deviations of each other. Therefore, simplifying the polymerization step by removing hexane did not result in a change in domain properties or performance. Accordingly, all larger scale domains were polymerized without hexane present.
Achievement of steady state during hydration requires more simulation time than all other simulation steps, regardless of domain scale. This is because without pre-hydrating the polymer, hydration must occur via diffusion, the time scale of which scales with distance squared. As the time step is fixed in MD simulations, this longer simulation time translates directly to a computational burden for hydration that scales with the square of the distance for a diffusive process.
Examining the ensembles of 1x domains used to evaluate hydration alternatives, the no insertion and aggregate insertion resulted in overlapping bounds computed as two standard deviations about the mean for all measures reported in Table 4 and for the water concentration profiles shown in Figures 4(a) and 4(b). This was likely due to the small size of the domain combined with relative infrequency of larger scale pores in the dry polyamide, as seen by the similar initial pore-size distributions of the 1x domains shown in Figures 7(a–b). Final pore-size distributions of the no insertion and aggregate insertion methods were also similar (Figures 7(a–b)).
The network and aggregate insertion method applied to the 1x domain resulted in elevated water concentrations (Figure 4(a–b)), time to steady state, and water permeability (Table 4). The pore-size distributions also exhibited increased frequency of larger scale pores not found in the other hydration alternatives (Figure 7(a–b)). In the case of the 1x domain polymerized with hexane present, the network and aggregate insertion ensemble did dehydrate to approach the no insertion and aggregate insertion alternatives but did not converge to the values of alternatives over the relatively long times simulated (Figure 7(a)).
Comparing the hydration alternatives for the 8x domain scale however, we found a difference (116 ns) in the time to steady state between the no insertion and the aggregate insertion cases, while final water concentration and water permeability were preserved, as shown in Figure 4(c) and Table 5. The aggregate insertion method reduced the time to steady state from 262 ns for the no insertion case to 146 ns. Comparing the time to steady state for the corresponding alternatives of the 1x domain, 75 ns, the scaling was effectively reduced from a factor of approximately 3.5 for the no insertion case to approximately 2 for aggregate insertion.
Figure 7(c) shows a marked difference in the amplitude of first peak between the no insertion and aggregate insertion methods for the initial pore-size distributions of the 8x domains. This difference is consistent with the change in the pore-size distribution after hydration and the reduced time observed to steady state for the aggregate insertion case compared to the no insertion case. The final distributions for the no insertion and aggregate insertion hydration alternatives were similar. At the 8x scale, the network and aggregate insertion method led to altered pore-size distributions and increased time to steady state, water concentrations, and water permeability (Figure 4(c) and Table 5).
Because aggregate insertion was such an effective method for reducing time to steady state, while not altering final results, we hydrated the 64x domain using aggregate insertion to enable analysis of this large domain. Time to steady state hydration for the 64x domain was 283 ns (Table 5), comparing this to the time for 1x and 8x domains, 75 ns and 146 ns, respectively, it is apparent that aggregate insertion results in a linear scaling of time to steady state with domain thickness. This is an improvement over typical diffusive scaling (i.e., thickness squared) as described previously.
Because our MD workflow contained a stochastic component, we used multiple realizations of the 1x domain to quantify uncertainty and time-averaging to smooth entropic fluctuations. Figure 5 shows the effect of utilizing ensembles of increasing size for the 1x and 8x domains. Ensemble averaging of the 1x domain served to smooth temporal variations and the mean value trend line changed by more than two standard deviations, which can be observed at 250 ns by comparing the results between 3 and 23 realizations.
In addition to ensemble averaging, we also evaluated increasing the domain scale as a method to reduce the stochastic error of results. Increasing the domain scale decreased the variance more than increasing number of realizations per ensemble over the conditions simulated in this work, as exemplified by the reduction in the averaging bounds shown in Figure 5.
Importantly, increasing the scale of the domain revealed that results for MD simulations of polyamide were dependent on domain size for the scales evaluated. Steady-state water concentration increased with domain scale, as seen in Figure 5. Furthermore, the dry pore-size distributions in Figure 6 and the initial and final hydrated pore-size distributions shown in Figure 8, show that larger scale pores become more frequent as the domain scale is increased.
Comparing results to experimental observations, the 64x domain had water concentrations in closest agreement with commercial TFC active layers (ρw = 0.19 – 0.31 g/cm3) [62, 63]. Accordingly, increasing the domain scale of future MD simulations is critical for producing realistic property-performance relationships of polyamide. The need for large-scale simulations makes the development of techniques for improving the computational efficiency a useful endeavor. Methods such as coarse-graining may be required to enable more efficient simulation of large domains [18, 65, 66]. Furthermore, MD implementations utilizing graphics processing units (GPUs) or hybrid CPU-GPU architectures have recently proven effective methods for improving the computational efficiency of evaluating pair-wise interactions [32, 33, 67].
All polyamide domains showed the existence of a bimodal distribution for pore sizes during hydration (Figure 8), one centered at the diameter of a cavity capable of existing between close packed aromatic rings (2.5 Å) [39, 68, 69], and another characteristic of network pores, having diameters distributed about 4 Å [38]. For the no insertion and aggregate insertion approaches, hydration resulted in a decrease in the peak centered at 2.5 Å, while the frequency of pores with diameters > 4 Å increased.
Monte Carlo based pore-size distributions are a frequently used tool to analyze the pore space of molecular polyamide domains, and both the dry and hydrated pore-size distributions collected for this study are consistent with those previously collected in literature [13, 14, 19, 21, 39, 70]. However, pore-size distributions generated via Monte Carlo methods do not fully describe the pore morphology and topology.
Figure 9 shows the medial axis of the pore space within a 1x domain. In its current state, this mapping of the medial axis shows the “skeleton” of the pore space. By identifying portions of this medial axis that connect the feed and permeate faces, one can reveal the pathways through the pore space available to a molecule. Ultimately, it is the characteristics of these available pathways that will determine the properties and performance of a polyamide active layer.
In order to highlight a method to evaluate those characteristics, we implemented Minkowski functional analysis of the whole pore space. Figure 10 shows that evaluations of Minkowski functionals for polyamide pore space opening maps are a promising method for characterizing the molecular pore space. The size of the maximum accessible structuring element was 4.2 Å and 9 Å for the dry and hydrated states, respectively. During hydration, the domain porosity increased from 0.34 to 0.43, and porosity increased for all opening maps created with comparable structuring element sizes, indicating a swelling of the pore space due to hydration. The slope of the surface area density curve was reduced after hydration for opening maps created with comparable structuring element sizes, and the initial surface area density decreased from 0.71 Å−1 to 0.58 Å−1. The maximum mean curvature density decreased during hydration from 0.16 Å−2 to 0.08 Å−2. Similarly, the maximum Euler characteristic density decreased from 0.002 Å−3 to 4 x 10−4 Å−3.
Future work will utilize both medial axis analysis to identify the available pathways and Minkowski functional analysis to describe the accessibility and characteristics of those pathways, to determine the function of molecular structure in mechanistic property-performance relationships of polyamide active layers.
5. Conclusions
We investigated methods to: (1) improve computational efficiency; (2) reduce error; and (3) evaluate pore space (molecular free volume) morphology and topology in MD simulations of polyamide. We present the following conclusions based on the results and discussion presented previously.
Presence of hexane during polymerization simulations did not affect observed properties or performance of the final polyamide domains. By polymerizing without hexane, the simulation update rate improved by 22%.
Inserting water molecules directly into aggregate pores of 883 Å3 polyamide domains prior to hydration reduced the time to steady-state hydration.
Medial axis analysis of the polyamide pore space elucidated the pathways available for molecules to travel through the polymer.
Minkowski functional methods were used to quantify characteristics of the polyamide pore space that are useful for developing property-performance relationships that account for the effects of molecular structure.
Polyamide properties and performance were dependent on domain size. Larger sized domains had increased steady-state water concentrations and increased frequency of larger scale pores.
The largest scale simulation of polyamide (1763 Å3) showed water concentrations that were in close agreement with experimentally observed water concentrations of hydrated commercial TFC active layers.
Large-scale domains provide computational challenges that motivate future consideration of coarse graining and multiscale methods.
Supplementary Material
Highlights.
Molecular dynamics simulations of polyamide synthesis and hydration were performed.
More computationally efficient, but less realistic synthesis did not affect results.
Water insertion into large polymer pores sped hydration without affecting results.
The morphology and topology of the molecular potential field was analyzed.
Simulation results depended on scale; larger scales approached experimental values.
6. Acknowledgements
This work was supported by the US National Science Foundation (NSF) Graduate Research Fellowship Program Grant No. DGE-1650115, the National Institute of Environmental Health Sciences Superfund Basic Research Program (P42-ES031007), the University of North Carolina at Chapel Hill (UNC) Creativity Hubs Research Initiative, and the Caroline H. and Thomas S. Royster Fellowship at UNC. This work used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by NSF grant number ACI-1548562, specifically the resources Bridges-2 at the Pittsburgh Supercomputing Center and Expanse at the San Diego Supercomputing Center, through allocation CTS200041.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Additional force field parameters, including atomic partial charges, Lennard-Jones parameters, and bonded interaction constants. Minkowski functional grid independence study results.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].Shannon MA, Bohn PW, Elimelech M, Georgiadis JG, Mariñas BJ, Mayes AM, Science and technology for water purification in the coming decades, Nature 452 (7185) (2008) 301–310. doi: 10.1038/nature06599. URL http://www.nature.com/articles/nature06599 [DOI] [PubMed] [Google Scholar]
- [2].Elimelech M, Phillip WA, The Future of Seawater Desalination: Energy, Technology, and the Environment, Science 333 (6043) (2011) 712–717. doi: 10.1126/science.1200488. URL https://www.sciencemag.org/lookup/doi/10.1126/science.1200488 [DOI] [PubMed] [Google Scholar]
- [3].Petersen RJ, Composite reverse osmosis and nanofiltration membranes, Journal of Membrane Science 83 (1) (1993) 81–150. doi: 10.1016/0376-7388(93)80014-0. URL https://linkinghub.elsevier.com/retrieve/pii/0376738893800140 [DOI] [Google Scholar]
- [4].Lee KP, Arnot TC, Mattia D, A review of reverse osmosis membrane materials for desalination—Development to date and future potential, Journal of Membrane Science 370 (1-2) (2011) 1–22. doi: 10.1016/j.memsci.2010.12.036. URL https://linkinghub.elsevier.com/retrieve/pii/S0376738810010045 [DOI] [Google Scholar]
- [5].Geise GM, Park HB, Sagle AC, Freeman BD, McGrath JE, Water permeability and water/salt selectivity tradeoff in polymers for desalination, Journal of Membrane Science 369 (1-2) (2011) 130–138. doi: 10.1016/j.memsci.2010.11.054. URL https://linkinghub.elsevier.com/retrieve/pii/S0376738810009233 [DOI] [Google Scholar]
- [6].Geise GM, Paul DR, Freeman BD, Fundamental water and salt transport properties of polymeric materials, Progress in Polymer Science 39(1) (2014) 1–42. doi: 10.1016/j.progpolymsci.2013.07.001. URL https://linkinghub.elsevier.com/retrieve/pii/S0079670013000804 [DOI] [Google Scholar]
- [7].Wang J, Dlamini DS, Mishra AK, Pendergast MTM, Wong MC, Mamba BB, Freger V, Verliefde AR, Hoek EM, A critical review of transport through osmotic membranes, Journal of Membrane Science 454 (2014) 516–537. doi: 10.1016/j.memsci.2013.12.034. URL https://linkinghub.elsevier.com/retrieve/pii/S0376738813009873 [DOI] [Google Scholar]
- [8].Ebro H, Kim YM, Kim JH, Molecular dynamics simulations in membrane-based watertreatment processes: A systematic overview, Journal of Membrane Science 438 (2013) 112–125. doi: 10.1016/j.memsci.2013.03.027. URL https://linkinghub.elsevier.com/retrieve/pii/S0376738813002226 [DOI] [Google Scholar]
- [9].Ridgway HF, Orbell J, Gray S, Molecular simulations of polyamide membrane materials used in desalination and water reuse applications: Recent developments and future prospects, Journal of Membrane Science 524 (2017) 436–448. doi: 10.1016/j.memsci.2016.11.061. URL https://linkinghub.elsevier.com/retrieve/pii/S0376738816318798 [DOI] [Google Scholar]
- [10].Lonsdale HK, Merten U, Riley RL, Transport properties of cellulose acetate osmotic membranes, Journal of Applied Polymer Science 9 (4) (1965) 1341–1362. doi: 10.1002/app,1965.070090413. URL [DOI] [Google Scholar]
- [11].Wijmans J, Baker R, The solution-di ffusion model: a review, Journal of Membrane Science 107 (1-2) (1995) 1–21. doi: 10.1016/0376-7388(95)00102-I. URL https://linkinghub.elsevier.com/retrieve/pii/037673889500102I [DOI] [Google Scholar]
- [12].Paul D, Reformulation of the solution-di ffusion theory of reverse osmosis, Journal of Membrane Science 241 (2) (2004) 371–386. doi: 10.1016/j.memsci.2004.05.026. URL https://linkinghub.elsevier.com/retrieve/pii/S0376738804004120 [DOI] [Google Scholar]
- [13].Harder E, Walters DE, Bodnar YD, Faibish RS, Roux B, Molecular Dynamics Study of a Polymeric Reverse Osmosis Membrane, The Journal of Physical Chemistry B 113 (30) (2009) 10177–10182. doi: 10.1021/jp902715f. URL 10.1021/jp902715f [DOI] [PubMed] [Google Scholar]
- [14].Ding M, Szymczyk A, Goujon F, Soldera A, Ghoufi A, Structure and dynamics of water confined in a polyamide reverse-osmosis membrane: A molecular-simulation study, Journal of Membrane Science 458 (2014) 236–244. doi: 10.1016/j.memsci.2014.01.054. URL https://linkinghub.elsevier.com/retrieve/pii/S0376738814000775 [DOI] [Google Scholar]
- [15].Kolev V, Freger V, Hydration, porosity and water dynamics in the polyamide layer of reverse osmosis membranes: A molecular dynamics study, Polymer 55 (6) (2014) 1420–1426. doi: 10.1016/j.polymer.2013.12.045. URL https://www.sciencedirect.com/science/article/pii/S0032386113011610 [DOI] [Google Scholar]
- [16].Hughes ZE, Gale JD, A computational investigation of the properties of a reverse osmosis membrane, J. Mater. Chem 20 (2010) 7788–7799. doi: 10.1039/C0JM01545H. URL 10.1039/C0JM01545H [DOI] [Google Scholar]
- [17].Wei W, Liu J, Jiang J, Atomistic Simulation Study of Polyarylate/Zeolitic-Imidazolate Framework Mixed-Matrix Membranes for Water Desalination, ACS Applied Nano Materials 3 (10) (2020) 10022–10031. doi: 10.1021/acsanm.0c02004. URL 10.1021/acsanm.0c02004 [DOI] [Google Scholar]
- [18].Muscatello J, M üller E, Mostofi A, Sutton A, Multiscale molecular simulations of the formation and structure of polyamide membranes created by interfacial polymerization, Journal of Membrane Science 527 (2017) 180–190. doi: 10.1016/j.memsci.2016.11.024. URL https://linkinghub.elsevier.com/retrieve/pii/S0376738816314909 [DOI] [Google Scholar]
- [19].Zhang H, Wu MS, Zhou K, Law AW-K, Molecular Insights into the Composition–Structure–Property Relationships of Polyamide Thin Films for Reverse Osmosis Desalination, Environmental Science & Technology 53 (11) (2019) 6374–6382. doi: 10.1021/acs.est.9b02214. URL 10.1021/acs.est.9b02214 [DOI] [PubMed] [Google Scholar]
- [20].Gao W, She F, Zhang J, Dum ée LF, He L, Hodgson PD, Kong L, Understanding water and ion transport behaviour and permeability through poly(amide) thin film composite membrane, Journal of Membrane Science 487 (2015) 32–39. doi: 10.1016/j.memsci.2015.03.052. URL https://linkinghub.elsevier.com/retrieve/pii/S0376738815002409 [DOI] [Google Scholar]
- [21].Shen M, Keten S, Lueptow RM, Dynamics of water and solute transport in polymeric reverse osmosis membranes via molecular dynamics simulations, Journal of Membrane Science 506 (2016) 95–108. doi: 10.1016/j.memsci.2016.01.051. URL https://linkinghub.elsevier.com/retrieve/pii/S0376738816300527 [DOI] [Google Scholar]
- [22].Kolev V, Freger V, Molecular Dynamics Investigation of Ion Sorption and Permeation in Desalination Membranes, The Journal of Physical Chemistry B 119 (44) (2015) 14168–14179. doi: 10.1021/acs.jpcb.5b06566. URL 10.1021/acs.jpcb.5b06566 [DOI] [PubMed] [Google Scholar]
- [23].Plimpton S, Fast Parallel Algorithms for Short-Range Molecular Dynamics, Journal of Computational Physics 117 (1) (1995) 1–19. doi: 10.1006/jcph.1995.1039. URL https://linkinghub.elsevier.com/retrieve/pii/S002199918571039X [DOI] [Google Scholar]
- [24].Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA, A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules, Journal of the American Chemical Society 117 (19) (1995) 5179–5197. doi: 10.1021/ja955032e. URL 10.1021/ja955032e [DOI] [Google Scholar]
- [25].MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FTK, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiórkiewicz-Kuczera J, Yin D, Karplus M, All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins †, The Journal of Physical Chemistry B 102 (18) (1998) 3586–3616. doi: 10.1021/jp973084f. URL 10.1021/jp973084f [DOI] [PubMed] [Google Scholar]
- [26].Jakalian A, Jack DB, Bayly CI, Fast, e fficient generation of high-quality atomic charges, am1-bcc model: II. parameterization and validation, Journal of Computational Chemistry 23 (16) (2002) 1623–1641. arXiv: 10.1002/jcc.10128, doi: 10.1002/jcc.10128. URL 10.1002/jcc.10128 [DOI] [PubMed] [Google Scholar]
- [27].Tuckerman ME, Alejandre J, L ópez-Rendón R, Jochim AL, Martyna GJ, A Liouville-operator derived measure-preserving integrator for molecular dynamics simulations in the isothermal–isobaric ensemble, Journal of Physics A: Mathematical and General 39 (19) (2006) 5629–5651. doi: 10.1088/0305-4470/39/19/S18. URL 10.1088/0305-4470/39/19/S18 [DOI] [Google Scholar]
- [28].Shinoda W, Shiga M, Mikami M, Rapid estimation of elastic constants by molecular dynamics simulation under constant stress, Physical Review B 69 (13) (2004) 134103. doi: 10.1103/PhysRevB.69.134103. URL 10.1103/PhysRevB.69.134103 [DOI] [Google Scholar]
- [29].Ryckaert J-P, Ciccotti G, Berendsen HJ, Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes, Journal of Computational Physics 23 (3) (1977) 327–341. doi: 10.1016/0021-9991(77)90098-5. URL https://linkinghub.elsevier.com/retrieve/pii/0021999177900985 [DOI] [Google Scholar]
- [30].Andersen HC, Rattle: A “velocity” version of the shake algorithm for molecular dynamics calculations, Journal of Computational Physics 52 (1) (1983) 24–34. doi: 10.1016/0021-9991(83)90014-1. URL https://linkinghub.elsevier.com/retrieve/pii/0021999183900141 [DOI] [Google Scholar]
- [31].Price DJ, Brooks CL, A modified TIP3P water potential for simulation with Ewald summation, The Journal of Chemical Physics 121 (20) (2004) 10096–10103. doi: 10.1063/1.1808117. URL [DOI] [PubMed] [Google Scholar]
- [32].Brown WM, Wang P, Plimpton SJ, Tharrington AN, Implementing molecular dynamics on hybrid high performance computers – short range forces, Computer Physics Communications 182 (4) (2011) 898–911. doi: 10.1016/j.cpc.2010.12.021. URL https://linkinghub.elsevier.com/retrieve/pii/S0010465510005102 [DOI] [Google Scholar]
- [33].Brown WM, Kohlmeyer A, Plimpton SJ, Tharrington AN, Implementing molecular dynamics on hybrid high performance computers – Particle–particle particle-mesh, Computer Physics Communications 183 (3) (2012) 449–459. doi: 10.1016/j.cpc.2011.10.012. URL https://linkinghub.elsevier.com/retrieve/pii/S0010465511003444 [DOI] [Google Scholar]
- [34].Khorshidi B, Thundat T, Fleck BA, Sadrzadeh M, Thin film composite polyamide membranes: parametric study on the influence of synthesis conditions, RSC Advances 5 (68) (2015) 54985–54997. doi: 10.1039/C5RA08317F. URL http://xlink.rsc.org/?DOI=C5RA08317F [DOI] [Google Scholar]
- [35].Freger V, Kinetics of Film Formation by Interfacial Polycondensation, Langmuir 21 (5) (2005) 1884–1894. doi: 10.1021/la048085v. URL 10.1021/la048085v [DOI] [PubMed] [Google Scholar]
- [36].Gissinger JR, Jensen BD, Wise KE, REACTER: A Heuristic Method for Reactive Molecular Dynamics, Macromolecules 53 (22) (2020) 9953–9961. doi: 10.1021/acs.macromol.0c02012. URL [DOI] [Google Scholar]
- [37].Abbott LJ, Hart KE, Colina CM, Polymatic: a generalized simulated polymerization algorithm for amorphous polymers, Theoretical Chemistry Accounts 132 (3) (2013) 1334. doi: 10.1007/s00214-013-1334-z. URL 10.1007/s00214-013-1334-z [DOI] [Google Scholar]
- [38].Cahill DG, Freger V, Kwak S-Y, Microscopy and Microanalysis of Reverse-Osmosis and Nanofiltration Membranes, MRS Bulletin 33 (1) (2008) 27–32. doi: 10.1557/mrs2008.11. URL 10.1557/mrs2008.11. [DOI] [Google Scholar]
- [39].Wei T, Zhang L, Zhao H, Ma H, Sajib MSJ, Jiang H, Murad S, Aromatic Polyamide Reverse-Osmosis Membrane: An Atomistic Molecular Dynamics Simulation, The Journal of Physical Chemistry B 120 (39) (2016) 10311–10318. doi: 10.1021/acs.jpcb.6b06560. URL 10.1021/acs.jpcb.6b06560 [DOI] [PubMed] [Google Scholar]
- [40].Song X, Gan B, Qi S, Guo H, Tang CY, Zhou Y, Gao C, Intrinsic Nanoscale Structure of Thin Film Composite Polyamide Membranes: Connectivity, Defects, and Structure–Property Correlation, Environmental Science & Technology 54 (6) (2020) 3559–3569. doi: 10.1021/acs.est.9b05892. URL 10.1021/acs.est.9b05892 [DOI] [PubMed] [Google Scholar]
- [41].Peng LE, Yao Z, Yang Z, Guo H, Tang CY, Dissecting the Role of Substrate on the Morphology and Separation Properties of Thin Film Composite Polyamide Membranes: Seeing Is Believing, Environmental Science & Technology 54 (11) (2020) 6978–6986. doi: 10.1021/acs.est.0c01427. URL 10.1021/acs.est.0c01427 [DOI] [PubMed] [Google Scholar]
- [42].in ‘t Veld PJ, Stone MT, Truskett TM, Sanchez IC, Liquid Structure via Cavity Size Distributions, The Journal of Physical Chemistry B 104 (50) (2000) 12028–12034. doi: 10.1021/jp001934c. URL 10.1021/jp001934c [DOI] [Google Scholar]
- [43].Willmore FT, Wang X, Sanchez IC, Free volume properties of model fluids and polymers: Shape and connectivity, Journal of Polymer Science Part B: Polymer Physics 44 (9) (2006) 1385–1393. doi: 10.1002/polb.20793. URL 10.1002/polb.20793 [DOI] [Google Scholar]
- [44].Willmore FT, A toolkit for the analysis and visualization of free volume in materials, in: Proceedings of the 1st Conference of the Extreme Science and Engineering Discovery Environment on Bridging from the extreme to the campus and beyond-XSEDE ’12, ACM Press, Chicago, Illinois, 2012, p. 1. doi: 10.1145/2335755.2335826. URL http://dl.acm.org/citation.cfm?doid=2335755.2335826 [DOI] [Google Scholar]
- [45].Jiang Y, Willmore FT, Sanders D, Smith ZP, Ribeiro CP, Doherty CM, Thornton A, Hill AJ, Freeman BD, Sanchez IC, Cavity size, sorption and transport characteristics of thermally rearranged (TR) polymers, Polymer 52 (10) (2011) 2244–2254. doi: 10.1016/j.polymer.2011.02.035. URL https://linkinghub.elsevier.com/retrieve/pii/S0032386111001558 [DOI] [Google Scholar]
- [46].Lindquist WB, Lee S-M, Coker DA, Jones KW, Spanne P, Medial axis analysis of void structure in three-dimensional tomographic images of porous media, Journal of Geophysical Research: Solid Earth 101 (B4) (1996) 8297–8310. doi: 10.1029/95JB03039. URL 10.1029/95JB03039 [DOI] [Google Scholar]
- [47].Lindquist W, Venkatarangan A, Investigating 3D geometry of porous media from high resolution images, Physics and Chemistry of the Earth, Part A: Solid Earth and Geodesy 24 (7) (1999) 593–599. doi: 10.1016/S1464-1895(99)00085-X. URL https://linkinghub.elsevier.com/retrieve/pii/S146418959900085X [DOI] [Google Scholar]
- [48].Lee T, Kashyap R, Chu C, Building Skeleton Models via 3-D Medial Surface Axis Thinning Algorithms, CVGIP: Graphical Models and Image Processing 56 (6) (1994) 462–478. doi: 10.1006/cgip.1994.1042. URL https://linkinghub.elsevier.com/retrieve/pii/S104996528471042X [DOI] [Google Scholar]
- [49].van der Walt S, Schönberger JL, Nunez-Iglesias J, Boulogne F, Warner JD, Yager N, Gouillart E, Yu T, scikit-image: image processing in Python, PeerJ 2 (2014) e453. doi: 10.7717/peeij.453. URL https://peerj.com/articles/453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Gostick J, Khan Z, Tranter T, Kok M, Agnaou M, Sadeghi M, Jervis R, PoreSpy: A Python Toolkit for Quantitative Analysis of Porous Media Images, Journal of Open Source Software 4 (37) (2019) 1296. doi: 10.21105/joss.01296. URL 10.21105/joss.01296 [DOI] [Google Scholar]
- [51].Hadwiger H, Vorlesungen über inhalt, Oberfläche und isoperimetrie, Vol. 93, Springer-Verlag, 1957. [Google Scholar]
- [52].Klain DA, Rota G-C, others, Introduction to geometric probability, Cambridge University Press, 1997. [Google Scholar]
- [53].Ohser J, Mucklich F, Statistical analysis of microstructures in materials science, John Wiley & Sons, 2000. [Google Scholar]
- [54].Hilfer R, Review on Scale Dependent Characterization of the Microstructure of Porous Media, Transport in Porous Media 46 (2) (2002) 373–390. doi: 10.1023/A:1015014302642. URL 10.1023/A:1015014302642 [DOI] [Google Scholar]
- [55].Klain DA, A short proof of Hadwiger’s characterization theorem, Mathematika 42 (2) (1995) 329–339, publisher: London Mathematical Society. [Google Scholar]
- [56].Armstrong RT, McClure JE, Robins V, Liu Z, Arns CH, Schlüter S, Berg S, Porous Media Characterization Using Minkowski Functionals: Theories, Applications and Future Directions, Transport in Porous Media 130(1) (2019) 305–335. doi: 10.1007/sll242-018-1201-4. URL 10.1007/sll242-018-1201-4 [DOI] [Google Scholar]
- [57].Miller CT, Bruning K, Talbot CL, McClure JE, Gray WG, Nonhysteretic Capillary Pressure in Two-Fluid Porous Medium Systems: Definition, Evaluation, Validation, and Dynamics, Water Resources Research 55 (8) (2019) 6825–6849. doi: 10.1029/2018WR024586. URL 10.1029/2018WR024586 [DOI] [Google Scholar]
- [58].Vogel H-J, Weller U, Schlüter S, Quantification of soil structure based on Minkowski functions, Computers & Geosciences 36 (10) (2010) 1236–1245. doi: 10.1016/j.cageo.2010.03.007. URL https://linkinghub.elsevier.com/retrieve/pii/S009830041000169X [DOI] [Google Scholar]
- [59].Matheron G, Random sets and integral geometry, Wiley, New York, 1975, type: Book. [Google Scholar]
- [60].Serra J, Image analysis and mathematical morphology, Academic Press, London, 1982. [Google Scholar]
- [61].Hilpert M, Miller CT, Pore-morphology-based simulation of drainage in totally wetting porous media, Advances in Water Resources 24 (3-4) (2001) 243–255. doi: 10.1016/S0309-1708(00)00056-7. URL https://linkinghub.elsevier.com/retrieve/pii/S0309170800000567 [DOI] [Google Scholar]
- [62].Lin L, Lopez R, Ramon GZ, Coronell O, Investigating the void structure of the polyamide active layers of thin-film composite membranes, Journal of Membrane Science 497 (2016) 365–376. doi: 10.1016/j.memsci.2015.09.020. URL https://linkinghub.elsevier.com/retrieve/pii/S0376738815301836 [DOI] [Google Scholar]
- [63].Lin L, Weigand TM, Farthing MW, Jutaporn P, Miller CT, Coronell O, Relative importance of geometrical and intrinsic water transport properties of active layers in the water permeability of polyamide thin-film composite membranes, Journal of Membrane Science 564 (2018) 935–944. doi: 10.1016/j.memsci.2018.08.002. URL https://linkinghub.elsevier.com/retrieve/pii/S0376738817317519 [DOI] [Google Scholar]
- [64].Sagui C, Darden T, Multigrid methods for classical molecular dynamics simulations of biomolecules, The Journal of Chemical Physics 114 (15) (2001) 6578–6591. doi: 10.1063/1.1352646. URL 10.1063/1.1352646 [DOI] [Google Scholar]
- [65].Oizerovich-Honig R, Raim V, Srebnik S, Simulation of Thin Film Membranes Formed by Interfacial Polymerization, Langmuir 26 (1) (2010) 299–306. doi: 10.1021/la9024684. URL 10.1021/la9024684 [DOI] [PubMed] [Google Scholar]
- [66].Riniker S, Allison JR, van Gunsteren WF, On developing coarse-grained models for biomolecular simulation: a review, Physical Chemistry Chemical Physics 14 (36) (2012) 12423. doi: 10.1039/c2cp40934h. URL http://xlink.rsc.org/?DOI=c2cp40934h [DOI] [PubMed] [Google Scholar]
- [67].Brown WM, Yamada M, Implementing molecular dynamics on hybrid high performance computers—Three-body potentials, Computer Physics Communications 184 (12) (2013) 2785–2793. doi: 10.1016/j.cpc.2013.08.002. URL https://linkinghub.elsevier.com/retrieve/pii/S0010465513002634 [DOI] [Google Scholar]
- [68].Singh P, Ray P, Xie Z, Hoang M, Synchrotron SAXS to probe crosslinked network of polyamide ‘reverse osmosis’ and ‘nanofiltration’ membranes, Journal of Membrane Science 421-422 (2012) 51–59. doi: 10.1016/j.memsci.2012.06.029. URL https://linkinghub.elsevier.com/retrieve/pii/S0376738812004887 [DOI] [Google Scholar]
- [69].Fu Q, Verma N, Ma H, Medellin-Rodriguez FJ, Li R, Fukuto M, Stafford CM, Hsiao BS, Ocko BM, Molecular Structure of Aromatic Reverse Osmosis Polyamide Barrier Layers, ACS Macro Letters 8 (4) (2019) 352–356. doi: 10.1021/acsmacrolett.9b00077. URL 10.1021/acsmacrolett.9b00077 [DOI] [PubMed] [Google Scholar]
- [70].Araki T, Cruz-Silva R, Tejima S, Takeuchi K, Hayashi T, Inukai S, Noguchi T, Tanioka A, Kawaguchi T, Terrones M, Endo M, Molecular Dynamics Study of Carbon Nanotubes/Polyamide Reverse Osmosis Membranes: Polymerization, Structure, and Hydration, ACS Applied Materials & Interfaces 7 (44) (2015) 24566–24575. doi: 10.1021/acsami.5b06248. URL 10.1021/acsami.5b06248 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.