

Abstract
Although plasmin inhibitors could be used in multiple disorders, their use has been restricted to preventing blood loss in hemostatic dysregulation because of poor efficacy and adverse effects of current agents. We reasoned that a new class of direct inhibitors that offer better efficacy, selectivity, and safety could be discovered by exploiting allosterism in plasmin, a protease homologous to other allosteric serine proteases. We report on the synthesis, biological activity, and mechanism of action of a group of small molecules, called non-saccharide glycosaminoglycan mimetics (NSGMs), as direct allosteric plasmin inhibitors. Our results show that distinct NSGMs selectively inhibit human full-length plasmin. The molecule inhibited clot lysis, alluding to its promise as an allosteric regulator of plasmin. We show that direct allosteric inhibition of plasmin could led to new antifibrinolytic agent(s) that may exhibit better efficacy, potency, selectivity, and safety in comparison to current therapy.
Graphical Abstract
INTRODUCTION
Plasmin, a member of the superfamily of serine proteases, cleaves cross-linked fibrin present in blood clots and serves as the sole mediator of natural fibrinolysis. Homeostatic hemostasis, the process of controlled clot formation, relies on the action of plasmin to limit excessive clot deposition. Such homeostasis may become dysregulated due to natural attenuation of fibrinogenesis, e.g., hemophilia,1 or over-activation of fibrinolysis, e.g., some states of disseminated intravascular coagulation, chronic liver disease, and leukemia.2-4 Hemostasis may also become dysregulated due to incidental abnormalities, e.g., major cardiac surgeries5 or chemotherapy-induced thrombocytopenia in cancer.6 In either case of such natural or man-made hemostatic dysregulation, bleeding complications are common, which can be alleviated by reducing plasmin biosynthesis or activity.
Plasmin can also be generated at cell surfaces by activation of plasminogen, which binds to its receptors that are abundantly expressed on cells.7 Localized generation of plasmin is important for a range of processes, of which some are disease producing such as chronic inflammation and tumor meta-stasis.8,9 Plasmin inhibition in these cases could be potentially beneficial as observed in the case of colitis,10 postischemic neutrophil migration,11 and metastasis of prostate carcinoma cells.12
Despite the diverse indications in which reduction of plasmin activity would be advantageous, only two agents have been approved for clinical use. Tranexamic acid and ε-aminocaproic acid, two lysine analogs that compete with fibrin to bind to lysine-binding sites in the kringle domains of plasminogen and thereby reduce activation of the zymogen to plasmin.8 Although the two agents have significantly advanced antifibrinolytic therapy, their indirect mechanism involving competition of much smaller inhibitors with macromolecular fibrin and multiple sites of engagement between plasminogen and fibrin reduces their efficacy of inhibition. Additionally, the agents induce adverse effects on the central GABA receptor,13 especially for tranexamic acid that carries the risk of seizures and renal dysfunction.14
Aprotinin, a Kunitz-type protein that competitively inhibits plasmin, is currently not available in the U.S. due to significant mortality and morbidity associated with its use.15,16 It a rather nonspecific protein that targets multiple serine proteases, e.g., trypsin, kallikrein, elastase, factor D, and others,8 which could be problematic. Yet, its inhibition of several serine proteases of the coagulation and inflammation systems was also an advantage, which led Canada and Europe to approve its use for a limited number of hyperfibrinolytic conditions.
Structurally, plasmin is a two-chain protein composed of an N-terminal heavy chain of 561 residues that form five kringle domains and a C-terminal light chain of 230 residues that form the catalytic domain.17 The catalytic domain displays high structural homology to trypsin and its family members18 (Figure 1), although there is one interesting difference. Plasmin lacks the 95–100 group of residues (chymotrypsinogen numbering), which form a β-hairpin in most other serine proteases resulting in a more open active site region.19 This contributes to its broader substrate specificity than most serine proteases,20 yet has offered opportunities of developing orthosteric inhibitors that exhibit high selectivity. Considerable effort has been expended on developing active site inhibitors to date and has been fairly successful.8,9,21-28 Of particular note is a recent work that reveals selective subnanomolar cyclic plasmin active-site inhibitors.26-28
Figure 1.

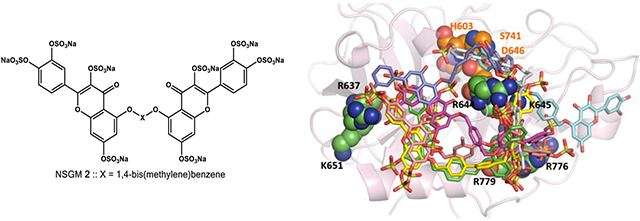
Electrostatic potential surface maps of anion-binding exosites of serine proteases. These exosites are targeted by sulfated nonsaccharide glycosaminoglycan mimetics (NSGMs). The maps show extensive positive charge density for all four proteases (plasmin (3UIR) (A), thrombin (1XMN) (B), factor Xa (2GD4) (C), and factor XIa (1ZHM) (D)) but with significant structural differences, which come in handy in developing selective NSGM inhibitors. The electrostatic potential surface was calculated using APBS tool. Electropositive (+1) surface is coded blue, while electronegative (−1) surface is in red.
We reasoned that an alternative approach of allosterism would be worth investigating because allosteric sites of serine proteases are significantly less conserved compared to their active sites.29-31 This approach may yield selective inhibitors of plasmin and would in addition introduce a new class of antifibrinolytics that may bear future potential. A priori, allosteric networks should exist in plasmin because of its three-dimensional similarity to other serine proteases that are known to display allosterism. In fact, plasmin contains a region of strongly electropositive charge density in the manner of thrombin, factor Xa, and factor XIa, each of which is known to bind to heparin (Figure 1).32-35 Work performed in the 1980s has suggested that heparin directly interacts with plasmin and allosterically modulates its catalytic activity.36 We have also earlier shown that a group of synthetic oligomeric mimetics of heparin (Mr ≈ 3000–5000) directly bind and allosterically inhibit human plasmin.37 Thus, we posited that appropriate sulfated small molecules, referred to as non-saccharide glycosaminoglycan mimetics (NSGMs), should be possible to discover that allosterically inhibit plasmin.
NSGMs are a new class of molecules that afford major advantages in drug discovery and/or chemical biology.38 Structurally, NSGMs possess multiple sulfate groups on an aromatic backbone. This endows them with simultaneous ionic and hydrophobic properties that introduce unique capabilities to recognize proteins. NSGMs tend to be highly water-soluble, readily synthesizable, and generally nontoxic to cells.38,39 Thus, NSGMs as direct, allosteric inhibitors of plasmin are likely to promise higher therapeutic efficacy than that achieved with current antifibrinolytics and possess other advantages necessary in clinically successful agents.
To discover potent allosteric sulfated diflavonoids, we studied a focused library of 15 analogs based on NSGMs designed earlier.40 Herein, we report on the synthesis, biological activity, and mechanism of action of these analogs. The results show that a specific sulfated diflavonoid 2 displays excellent potency (IC50 ≈ 6.3 μM) and affinity (KD ≈ 0.7 μM) for human full-length plasmin. The molecule utilizes direct, reversible, allosteric mechanism of action and is at least 22-fold more selective for plasmin in comparison thrombin, factor Xa, and factor XIa. Agent 2 inhibited clot lysis with an IC50 of 8.8 μM, which alludes to its promise as the first lead allosteric regulator of plasmin. We expect that the avenue of direct allosteric inhibition of plasmin will eventually lead to antifibrinolytic agents that are more efficacious, potent, selective, and safe than current approved drugs.
RESULTS AND DISCUSSION
Rationale behind Developing a Focused Library of Sulfated Diflavonoid NSGMs.
Our previous work on sulfated NSGMs inhibiting serine proteases has established that these unique molecules typically exhibit allosteric mechanism of action with the dual property of high selectivity and efficacy.40-44 For example, a group of sulfated diflavonoids were earlier found to inhibit plasmin with more than 5-fold selectivity over the closely related thrombin and factor Xa and display variable efficacies in the range of 38–100%.40 In that work, structure–activity studies had revealed that a four-atom linker connecting two quercetin moieties, each with sulfate groups at positions 3, 7, 3′, and 4′, led to a plasmin inhibition. Yet, the potency (IC50 ≥ 75 μM) and selectivity (5-fold) of the best sulfated diflavonoid were moderate. Thus, in this study, we developed a focused library of 15 sulfated diflavonoids analogs. The site of coupling each flavonoid moiety was maintained constant, while variations were introduced in the length and nature of linker as well as the number of sulfate groups on each flavonoid monomer (Scheme 1 and Table 1). Briefly, the molecules belong to three categories: (a) the octasulfated quercetin-based homodimers (five molecules); (b) the tetrasulfated apigenin-based homodimers (nine molecules); and (c) the hexasulfated quercetin-apigenin heterodimers (one molecule). The nature of the linker was either nonaromatic including ethylene (7), propylene (8), butylene (9), and unsaturated butylene (10) or aromatic including p,p-xylene (2, 4, 11, 13, 16), m,m-xylene (5 and 14), 4,4′-bis(methylene)-1,1′-biphenyl (3 and 12), and 1,3-bis(methylene)pyridine (6 and 15). The number of sulfate groups varied from 8 (1–6) to 6 (16) to 4 (7–15). All NSGM analogs possess sulfate groups at positions 7 and 4′ of their monomers. However, octasulfated NSGM analogs (1–6) have additional four sulfate groups at positions 3 and 3′ of their monomers, whereas hexasulfated NSGM analog (16) possesses an additional pair of sulfate groups at positions 3 and 3′ of one of the monomers. The tetrasulfated NSGM analogs (7–15) possess no additional sulfate groups.
Scheme 1. Chemical Synthesis of Sulfated NSGMsa.
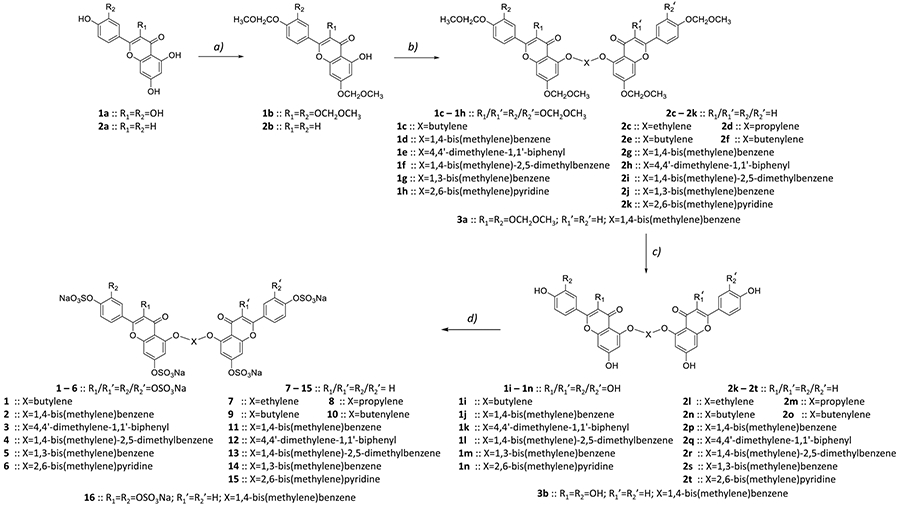
a(a) MOM-Cl (2 or 4 equiv), DIPEA, DCM, rt, 12 h, 50–60%; (b) K2CO3 (2 equiv), dibromo linker derivative (1 equiv), DMF, rt, 12 h, 30–50%; (c) TMS-Br, CH2Cl2, −30, 1 h, then 0°C, 12–24 h, 70–90%; (d) SO3/Me3N (6 equiv/−OH), Et3N (10 equiv/−OH), CH3CN, microwave, 90–100 °C, 4–8 h, 60–90%.
Table 1.
In Vitro Inhibition of Human Plasmin by NSGMsa
| ||||||
|---|---|---|---|---|---|---|
| Inhibitor | R1/R1` | R2/R2` | X | IC50 (μM) | HS | Δ Y (%) |
| 1 | OSO3Na | OSO3Na |
|
76 ± 12b | 1.0 ± 0.2 | 72 ± 9 |
| 2 | OSO3Na | OSO3Na |
|
6.3 ± 0.4 | 0.7 ± 0.1 | 93 ± 4 |
| 3 | OSO3Na | OSO3Na |
|
37 ± 7 | 0.8 ± 0.2 | 80 ± 11 |
| 4 | OSO3Na | OSO3Na |
|
6.5 ± 0.7 | 0.7 ± 0.1 | 104 ± 7 |
| 5 | OSO3Na | OSO3Na |
|
73 ± 11 | 0.6 ± 0.1 | 107 ± 7 |
| 6 | OSO3Na | OSO3Na |
|
231 ± 27 | 0.8 ± 0.2 | 90 ± 5 |
| 7 | H | H |
|
71 ± 2 | 2.4 ± 0.3 | 79 ± 3 |
| 8 | H | H |
|
>1000 | ND c | ND |
| 9 | H | H |
|
282 ± 17 | 1.8 ± 0.3 | 90 ± 8 |
| 10 | H | H |
|
121 ± 18 | 0.9 ± 0.2 | 90 ± 11 |
| 11 | H | H |
|
34 ± 1 | 2.4 ± 0.3 | 89 ± 2 |
| 12 | H | H |
|
47 ± 2 | 1.1 ± 0.1 | 79 ± 3 |
| 13 | H | H |
|
20 ± 1 | 2.0 ± 0.1 | 94 ± 2 |
| 14 | H | H |
|
34 ± 2 | 2.3 ± 0.5 | 86 ± 4 |
| 15 | H | H |
|
42 ± 3 | 1.2 ± 0.2 | 63 ± 4 |
| 16 | H/OSO3Na | H/OSO3Na |
|
14 ± 1 | 0.9 ± 0.1 | 109 ± 6 |
The IC50, HS, and ΔY values were obtained following nonlinear regression analysis of direct inhibition of human plasmin in appropriate Tris-HCl buffers of pH 7.4 at 37 °C. Inhibition was monitored spectrophotometrically. See Experimental Part for details.
Errors represent ±1 SE.
Not determined.
Synthesis of Sulfated NSGM Analogs.
The synthesis of NSGMs was achieved in four steps involving selective protection flavonoid monomers, dimerization, deprotection, and persulfation (Scheme 1). The conditions employed in each of these steps were slightly modified versions of our earlier methods39,40 and described in detail in the Experimental Part. Briefly, quercetin (1a) or apigenin (2a) was treated with appropriate equivalents of methoxy-O-methyl (MOM) chloride at room temperature under basic conditions to yield MOM-protected intermediate 1b or 2b, respectively. The phenolic group at position 5 remained free in this condition due to the intramolecular H-bond with the adjacent carbonyl group. These intermediates were then made to react with 1 equiv of the desired dibromo linker in the presence of K2CO3 (Scheme 1; step b) at room temperature to yield the corresponding protected dimers 1c–h or 2c–k in yields of 30–50%. Likewise, for synthesizing heterodimer 3a two consecutive SN2 reactions were used with two different substrates (i.e., protected quercetin and protected apigenin). MOM deprotection was then performed using bromotrimethylsilane at −30 °C (1 h) and 0 °C (12–24 h), giving polyphenolic precursors 1i–n, 2k–t, and 3b (Scheme 1; step c) in quantitative yields. The final step for the generation of each sulfated diflavonoid 2–16 was microwave-assisted chemical sulfation in CH3CN using SO3/Me3N as the sulfating agent and Et3N as a base, as described in our earlier studies.39,40,45 The reaction was run for 4–8 h at 90–100 °C and resulted in per-sulfated products in quantitative yields (Scheme 1, step d). The structural identities of all 15 NSGMs were confirmed using 1H and 13C NMR spectroscopies and UPLC–MS. The purity of each product was >95% (see Experimental Part).
Plasmin Inhibition Potential of Sulfated NSGMs.
Inhibition of human plasmin by the 15 NSGMs 2–16 and a polyphenol precursor 1j was measured using Spectrozyme PL hydrolysis assay at pH 7.4 and 37 °C, as described earlier.40 This assay utilizes the direct relationship between the initial rate of substrate hydrolysis and catalytic activity of a protease. The fractional decrease in initial rate of hydrolysis in the presence of an inhibitor is analyzed using a dose–response eq 1 to calculate the potency (IC50, HS) and efficacy (Y0, YM) parameters (see Experimental Part). Figure 2 shows the semilog inhibition curves observed for representative NSGMs. The range of inhibitory potency was found to be broad (6.3 μM to >1 mM), while the efficacy for most inhibitors was high (>80%).
Figure 2.
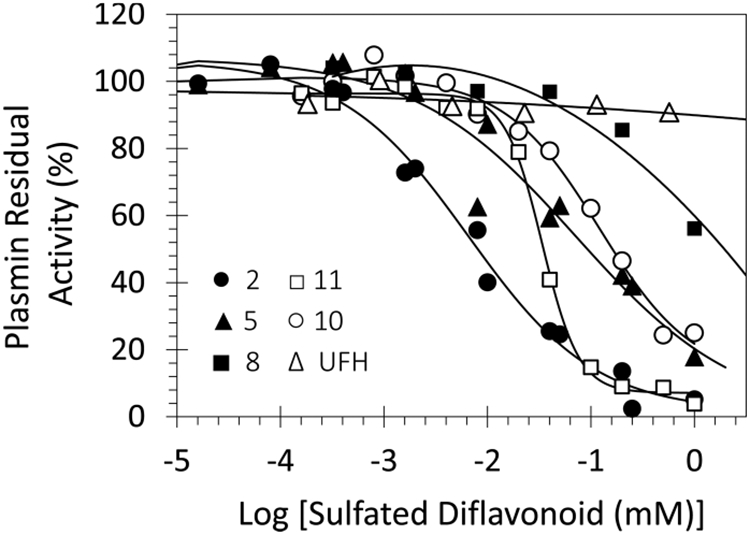
Representative profiles of direct inhibition of human plasmin by sulfated NSGMs and UFH. The inhibition of plasmin was measured spectrophotometrically through a chromogenic substrate hydrolysis assay at pH 7.4 and 37 °C. Solid lines represent the sigmoidal fits to the data to obtain IC50, HS, YM, and Y0 using eq 1, as described in the Experimental Part.
Although quercetin- and apigenin-based NSGMs (i.e., 1–6 versus 7–15, respectively) differ from each other in the number of sulfate groups (8 versus 4, respectively), their range of IC50 values was rather similar (6.3–231 μM versus 20–282 μM, respectively). Quercetin analogs 2 and 4 were the most potent molecules with IC50 values of 6.3 and 6.5 μM. In contrast, apigenin analog 13 was the most potent (IC50 ≈ 20.0 ± 0.5 μM, Table 1). It is worth mentioning here that among the linkers screened, the most detrimental linker was propylene (NSGM 8, IC50 > 1 mM; see Table 1), which was much less tolerated than ethylene (e.g., 7, IC50 = 71 μM) as well as butylene (e.g., 9, IC50 = 282 μM). This is an unusual observation and appears to indicate different binding geometries for NSGMs that differ in only one methylenic group (i.e., 7 (or 9) versus 8). This is also a major departure from the observation with allosteric inhibitors of human thrombin, which seem to prefer much longer linkers,41 suggesting an altered basis of recognition despite considerable homology between the two proteases.
For both quercetin analogs and apigenin analogs, p,p-xylenic linkers (i.e., 2 and 4 or 11 and 13) are the most potent anti-plasmin molecules. In contrast, longer aromatic linkers (e.g., 3 and 13) or m,m-xylenic linkers (e.g., 5 and 14) or heterocyclic linkers (e.g., 6 and 15) were less potent (see Table 1). Also, aromatic linkers in general appear to exhibit better inhibition potency than aliphatic linkers for both series of NSGMs (Table 1 and ref 37). This implies that a particular three-dimensional and distance relationship between the two monomeric moieties is important for plasmin recognition.
Comparing analogs in the apigenin series with their counterparts in the quercetin series reveals three interesting trends. For some linkers, a quercetin to apigenin change is associated with 3- to 5-fold decrease in plasmin inhibition potency (e.g., 2 vs 11; 4 vs 13). For others, there is a 2- to 6-fold increase in potency (e.g., 5 vs 14 and 6 vs 15), while for still others, there is no significant change (e.g., 3 vs 12). This implies that for preferred linkers, higher sulfation density is better, while for less preferred linkers, sulfation does not guarantee better inhibition potential. This supports the idea that appropriate NSGM scaffold and structure are critical for developing clinically relevant inhibitors of serine proteases.41-44
To evaluate the value of sulfation, we measured plasmin inhibition by polyphenol 1j, which is the nonsulfated precursor of NSGM 2. The polyphenol precursor inhibited human plasmin with a potency of 116 μM, which is about 18.3-fold less than that observed for 2, indicating the role of sulfate groups in achieving potent plasmin inhibition. However, this result is very surprising. No small nonsulfated precursor of NSGMs has been found to inhibit serine proteases studied to date, e.g., thrombin or factor XIa as yet,41-44 which implies that absence of sulfate groups on NSGMs negates their ability to recognize a serine protease. In contrast, 1j retains moderate plasmin inhibition potential.
Kinetics of Plasmin Inhibition by NSGM 2 and Its Precursor 1j.
To understand the basis for NSGM’s inhibitory potential, the kinetics of Spectrozyme PL hydrolysis by plasmin was measured at pH 7.4 and 37 °C in the presence of the inhibitor 2 and its polyphenolic precursor 1j. As expected, the initial velocity varied in a hyperbolic manner with increasing concentration of the substrate at all concentrations of inhibitor 2 and polyphenol 1j (Figure 3). The KM for Spectrozyme PL in the absence of inhibitor 2 was found to be 0.050 mM (Table 2), which decreased ~ 12.5-fold upon increasing the inhibitor’s concentration to 200 μM (0.004 mM). At the same time, the VMAX decreased 4-fold from 42.2 mAU/min to 10.4 mAU/min for the same range of inhibitor concentration. For inhibitor 1j, the KM for Spectrozyme PL increased ~ 13.5-fold, while the VMAX remained essentially unchanged (50.6–51.3 mAU/min) as a function of inhibitor concentration reaching 400 μM.
Figure 3.
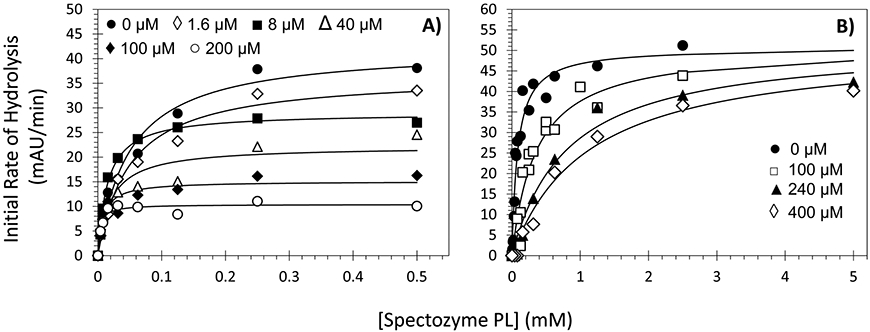
Michaelis–Menten kinetics of human plasmin in the presence of NSGM 2 (A) or its polyphenolic precursor 1j (B). The initial rate of hydrolysis at various substrate concentrations was measured spectrophotometrically in pH 7.4 buffer at 37 °C. Solid lines represent the nonlinear regressional fits to the data using the eq 2 to yield KM and VMAX.
Table 2.
Michaelis–Menten Kinetics of Human Plasmin in the Presence of NSGM 2 or Its Precursor 1ja
| [inhibitor] (μM) | KM b (mM) | VMAX b (mAU/min) | |
|---|---|---|---|
| 2 | 0 | 0.050 ± 0.001 | 42.2 ± 2.3 |
| 1.6 | 0.050 ± 0.001 | 36.3 ± 2.0 | |
| 8 | 0.020 ± 0.001 | 29.0 ± 0.6 | |
| 40 | 0.020 ± 0.007 | 22.1 ± 1.9 | |
| 100 | 0.008 ± 0.003 | 15.1 ± 1.0 | |
| 200 | 0.004 ± 0.001 | 10.4 ± 0.5 | |
| 1j | 0 | 0.083 ± 0.014 | 50.6 ± 2.4 |
| 100 | 0.357 ± 0.082 | 51.0 ± 4.6 | |
| 240 | 0.807 ± 0.180 | 51.7 ± 3.9 | |
| 400 | 1.122 ± 0.233 | 51.3 ± 4.0 |
KM and VMAX values of Spectrozyme PL substrate hydrolysis by human plasmin were measured under physiologic conditions. mAU indicates milliabsorbance units.
Error represents ±1 SE.
The two inhibitors (2 and 1j) are thus fundamentally different with respect to their mechanism of plasmin inhibition. While the affinity of the substrate increased in the presence of NSGM 2, it decreased in the presence of precursor 1j. Simultaneously, NSGM 2 brought about a substantial decrease in the rate of hydrolysis, whereas 1j did not change this rate. This implies that NSGM 2 induces uncompetitive inhibition, while inhibitor 1j works in a competitive manner. Alternatively, sulfated NSGM 2 is an allosteric inhibitor of human plasmin, whereas the nonsulfated, polyphenolic inhibitor 1j is an orthosteric inhibitor.
Selectivity of NSGM 2 and Its Precursor 1j against Related Serine Proteases.
The rationale behind targeting an allosteric site on human plasmin was to achieve inhibition selectivity over closely related serine proteases. To assess this feature, proteolysis of appropriate small peptide-based chromogenic substrates of a variety of homologous serine proteases was measured (Figure 4, see Experimental Part for details). The IC50 values of inhibitor 2 against thrombin were 156 μM, suggesting a plasmin selectivity of 22-fold (Table 3). Likewise, 2 displayed selective indices of 70-, 26-, >277-, >138-, >61-, and 27-fold against factors Xa, XIa, IXa, XIIa, trypsin, and chymotrypsin (Table 3). Thus, the allosteric inhibitor 2 displays excellent selectivity against a number of coagulation serine proteases. In contrast, its polyphenolic precursor 1j displays plasmin selectivity of only approximately 2-fold against thrombin, factor Xa, and factor XIa, which is not very useful. This implies that unsulfated precursor 1j does not differentiate these proteases.
Figure 4.
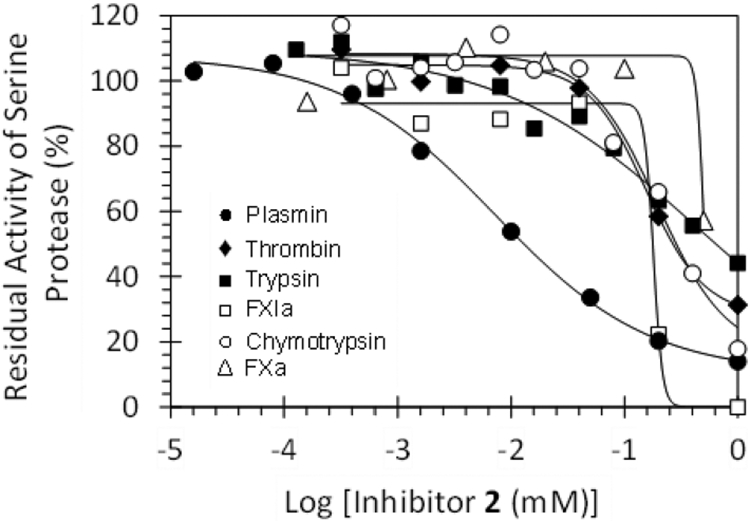
Direct inhibition of serine proteases by NSGM 2. The inhibition of plasmin (●), thrombin (♦), trypsin (■), factor XIa (□), chymotrypsin (○), and factor Xa (△) by inhibitor 2 was studied using chromogenic substrate hydrolysis assay at pH 7.4 and 37 °C, as described in Experimental Part. Solid lines represent the sigmoidal dose–response fits (eq 1) to the data to obtain the values of IC50, ΔY, and HS.
Table 3.
Selectivity of NSGM 2 and Its Precursor 1j against a Panel of Related Serine Proteasesa
| inhibitor | enzyme | IC50 b (μM) | ΔY b,c (%) | SId |
|---|---|---|---|---|
| 2 | plasmin | 7.2 ± 0.6 | 98 ± 5 | |
| thrombin | 156 ± 14 | 77 ± 8 | 22 | |
| FXa | ~505 | ND | 70 | |
| FXIa | 180 ± 20 | 93 ± 11 | 26 | |
| FIXa | >2000 | ND | >277 | |
| FXIIa | >1000 | ND | >138 | |
| trypsin | >440 | ND | >61 | |
| chymotrypsin | 196 ± 39 | 92 ± 9 | 27 | |
| 1j | plasmin | 116 ± 9 | 111 ± 8 | |
| thrombin | 267 ± 28 | 91 ± 11 | 2 | |
| FXa | 212 ± 30 | 104 ± 17 | 2 | |
| FXIa | 208 ± 32 | 109 ± 16 | 2 |
Direct inhibition of human enzymes was measured in appropriate Tris-HCl buffers of pH 7.4 at 37 °C. Inhibition was monitored spectrophotometrically. See Experimental Part for details.
Selectivity index.
Errors represent ±1 SE.
Not determined.
Thermodynamic Affinity of Sulfated Diflavonoids for Human Plasmin.
Although the IC50 values of the sulfated NSGMs have been rigorously defined, their thermodynamic affinity (KD) remained undefined. In general, the affinities of sulfated saccharide and nonsaccharide ligands for their targets, such as thrombin, factor XIa, plasmin, and antithrombin, have been measured using intrinsic37,44,46-48 as well as extrinsic44-49 fluorescence probes. For example, heparins induce a 30–40% increase in the intrinsic tryptophan fluorescence of antithrombin,46 while sucrose octasulfate decreases the intrinsic fluorescence of thrombin by 5–10%.47 Among the nonsaccharide ligands, sulfated tetrahydroisoquinolines,48 low molecular weight lignins,37 and sulfated pentagalloyl glucosides44 induced a decrease in the intrinsic fluorescence of antithrombin, plasmin, and factor XIa, while sulfated quinazolin-4(3H)-ones induced a 50–90% increase in the fluorescence of DEGR-factor XIa.49 Thus, we used the intrinsic tryptophan fluorescence to probe the interaction of NSGMs being studied here with plasmin and measure their KD.
A saturating decrease of ~100% in the intrinsic fluorescence of plasmin was measured for NSGM 2 at pH 7.4 and 37 °C, which could be fitted using the standard quadratic binding eq 3 to calculate a KD of 700 ± 100 nM (Figure 5 and Supporting Information Table S1). Such potent inhibition of a serine protease by a small molecule NSGM inhibitor has not been observed earlier. Inhibitors 4, 10, and 13 induced a 90%, 101%, and 108% loss in the fluorescence of the intrinsic tryptophan, which implied affinity values of 1.0 ± 0.1 μM, 3.6 ± 0.6 μM, and 1.9 ± 0.2 μM, respectively. These affinities are also very good and show that this group of NSGMs displays excellent affinity for human plasmin.
Figure 5.
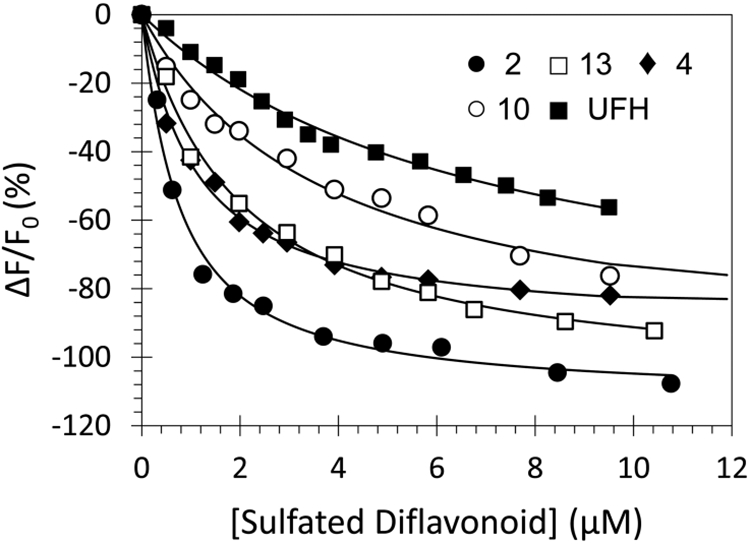
Spectrofluorometric measurement of the affinity of human plasmin for NSGMs 2 (●), 4 (♦), 10 (○), 13 (□), and UFH (■) at pH 7.4 and 37 °C using the intrinsic tryptophan fluorescence (λEM = 348 nm, λEX = 280 nm). Solid lines represent the nonlinear regressional fits using quadratic eq 3 to derive KD and ΔFMAX. See details in the Experimental Part.
Yet, the measured KD and IC50 values for these inhibitors are different (e.g., 0.7 μM versus 6.3 μM for 2). Although a priori KD does not have to be equal to be always equal to KI for allosteric inhibitors, NSGMs developed to date for thrombin and factor XIa have displayed essentially equivalent KD and IC50.41-44,49 This has been the case because the NSGMs developed to date have displayed essentially ideal non-competitive inhibition mechanism. In contrast, the NSGMs developed here for plasmin display uncompetitive inhibition mechanism. In this mechanism, the inhibitor binds to both the free enzyme (E) and the enzyme–substrate complex (E:S) with significantly different affinities (e.g., >5-fold), which forms the basis for observed KD to be less than the IC50.
To further probe whether NSGMs can possess the capacity to differentially recognize free plasmin and plasmin–substrate complex, we measured the affinity of NSGMs to bind to dansyl-EGR-chloromethylketone-plasmin. In this plasmin, the active site His has been irreversibly modified with chloromethylketone, which is expected to mimic the acyl enzyme intermediate formed in the E:S complex. The hypothesis was that if the conformation of allosteric site in the free enzyme is different from that in the E:S complex (or alternatively, active-site blocked plasmin), then affinities of NSGMs for the two forms of plasmin (active, free enzyme versus active-site blocked enzyme) would be different. Once again, the affinity of NSGMs 2 and 13 for active-site-blocked plasmin was measured using change in intrinsic fluorescence (see Figure S1). The KD values calculated using the quadratic binding equation were 4.4 ± 0.4 μM and 5.4 ± 0.3 μM for 2 and 13, respectively. These values, especially for NSGM 2, are significantly different from the affinity of 700 ± 100 nM measured for the complex of 2 with active plasmin but very similar to the observed IC50 of 6.3 μM. The results are in line with the expectation from mechanistic basis of different conformational states of the free E and E:S complex and imply that NSGMs bind to plasmin at a site other than the active site, or allosteric site.
In contrast to NSGMs, unfractionated heparin (UFH), which is more highly sulfated than NSGMs, was found to moderately bind to plasmin with a KD of 6.7 ± 0.8 μM (Figure 5 and Supporting Information Table S1). More importantly, UFH did not directly inhibit human plasmin at the highest concentration tested of 570 μM at pH 7.4 and 37 °C (Figure 2). Thus, heparins are not able to induce appropriate conformational change in free plasmin to effect inhibition. This suggests that the NSGM backbone is critical. These results highlight the significance of the aromatic scaffold of sulfated NSGMs recognition as well as inhibition of human plasmin.
NSGM 2 Inhibition of Plasmin Is Reversible.
To assess whether the interaction of sulfated analog 2 with plasmin is ionic, we measured the IC50 of plasmin inhibition by molecule 2 at pH 7.4 and 37 °C in the presence of varying levels of NaCl. As the salt concentration decreased from 200 to 0 mM, the IC50 decreased more than 6-fold from 17.1 μM to 2.8 μM (Figure 6A and Supporting Information Table S2). This suggests that inhibition of plasmin by NSGM 2 is most probably driven by electrostatic interactions between sulfate groups and their counterparts Arg and Lys on plasmin. Therefore, we hypothesized that inhibitor 2’s activity may be reversed by protamine, which is a clinically used arginine-rich polypeptide that counteracts the anticoagulant activity of UFH.50 Human plasmin was first treated with high concentration of inhibitor 2 (200 μM) and the recovery of plasmin activity by protamine studied spectrophotometrically through hydrolysis of Spectrozyme PL at pH 7.4 and 37 °C (Figure 6D). The efficacy and the effective concentration of protamine to restore 50% of enzyme activity (EC50) were calculated and found to be ~100% and 19.9 ± 0.8 mg/mL, respectively. Thus, this class of allosteric inhibitors, in particular NSGM 2, interacts with plasmin through ionic interactions and its inhibition is rapidly and fully reversed by protamine.
Figure 6.
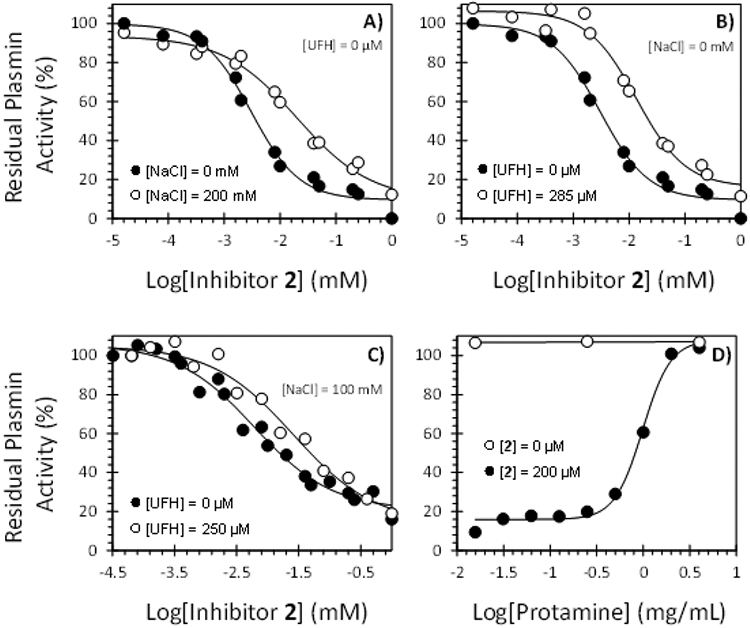
Inhibition of human plasmin by NSGM 2 as a function of NaCl and/or UFH: (A) with NaCl and without UFH; (B) without NaCl and with UFH; (C) with both NaCl and UFH. Inhibition was studied using the chromogenic substrate hydrolysis assay at pH 7.4 and 37 °C. (D) Profile of recovery of plasmin activity in the presence of increasing concentrations of protamine (0–4 mg/mL) in the presence and absence of NSGM 2 (200 μM). Solid lines represent the sigmoidal fits to the data to obtain IC50 (or EC50), HS, YM, and Y0 using eq 1 or 4, as described in the Experimental Part.
Sulfated NSGM 2 Competes with Heparin.
UFH is a variably sulfated, highly heterogeneous and polydisperse mixture of linear sulfated glycosaminoglycan (GAG) chains. Heparin has long been clinically used as an anticoagulant by enhancing the inactivation of coagulation proteases, particularly thrombin.51 In addition to this effect, heparin also affects the fibrinolytic system. In this connection, the heparin–plasmin interaction has received significant attention, particularly to explore routes to novel antifibrinolytic agents.8,9 Yet, the heparin-binding site(s) on plasmin is(are) yet to be identified. Most probably, the site(s) is(are) located in plasmin’s catalytic domain, although the kringle domains, or both, could also be heparin’s site of action. Several studies have exploited these putative heparin-binding site(s) to introduce heparin mimetics as anti-plasmin agents including low molecular weight lignins,37 chemically modified dextran sulfate derivatives,52 and sulfated polyvinyl alcohol–acrylate copolymers.53
To assess whether NSGM 2 is a heparin mimetic, we studied its plasmin inhibition profile in the presence of UFH. The apparent IC50 values of plasmin inhibition were measured at varying levels of UFH at pH 7.4 and 37 °C (Table 4). In the absence of any added NaCl (I = 0.05), the IC50 of plasmin inhibition increased from 2.8 to 10.4 μM as the concentration of UFH changed from 0 to 285 μM (Figure 6B). In the presence of 100 mM NaCl, the inhibition potency changed from 6.1 μM at 0 mM UFH to 25.9 μM at 250 μM UFH (Figure 6C). Both these results indicate that NSGM 2 progressively becomes a weaker allosteric inhibitor as the concentration of UFH increases. Alternatively, the results suggest some form of competitive effect between UFH and NSGM 2, although it is difficult to ascertain whether the competition arises from direct competitive binding or due to indirect conformational changes arising from simultaneous engagement of both ligands by plasmin. Nevertheless, allosteric inhibitor 2 appears to function as a heparin mimetic and most probably inhibits human plasmin by binding to or in the vicinity of a putative anion-binding site(s) on plasmin.
Table 4.
Inhibition of Human Plasmin by NSGM 2 in the Presence of UFHa
| [NaCl] (mM) | [UFH] (μM) | IC50 b (μM) | HS b | ΔY b (%) |
|---|---|---|---|---|
| 0 | 0 | 2.8 ± 0.3 | 1.0 ± 0.3 | 90 ± 7 |
| 15 | 3.6 ± 0.3 | 1.3 ± 0.3 | 82 ± 5 | |
| 50 | 4.6 ± 0.7 | 0.8 ± 0.1 | 93 ± 13 | |
| 285 | 10.4 ± 1.2 | 1.0 ± 0.2 | 93 ± 7 | |
| 100 | 0 | 6.1 ± 0.6 | 0.7 ± 0.1 | 85 ± 5 |
| 50 | 14.1 ± 1.4 | 1.0 ± 0.2 | 81 ± 6 | |
| 250 | 25.9 ± 4.6 | 0.7 ± 0.1 | 91 ± 11 |
The IC50, HS, and ΔY values were obtained following nonlinear regression analysis of direct inhibition of human plasmin in appropriate Tris-HCl buffers of pH 7.4 at 37 °C. Inhibition was monitored spectrophotometrically.
Errors represent ±1 SE.
Structure–Activity Relationship Studies Further Support Allosteric Binding.
If NSGMs do bind in the heparin-binding site of plasmin, we reasoned that the structure–activity relationship (SAR) of this set of NSGMs could be computationally analyzed. We performed GOLD-based docking and scoring study of a group of seven NSGMs, including 2, 3, 4, 8, 10, and 11 and the nonsulfated polyphenolic precursor 1j, following our earlier methodology of GAGs and GAG mimetics docking to proteins.54-57 The molecules were docked onto the putative heparin-binding site of plasmin using an unbiased search algorithm of 300 GA runs. The best 20 docked solutions for each NSGM were analyzed for the consistency of binding, and the pose corresponding to the highest GOLDSCORE was selected for analyses of interactions at an atomic level.
Figure 7A shows the best pose of each NSGM overlaid on each other, while individual poses for selected other NSGMs are presented in panels C–F. At a qualitative level, the study showed that higher potency NSGMs interacted consistently with the site formed by R637, R644, and R779. Yet, there were differences in the hydrogen bonding interactions made by each inhibitor, which should theoretically lead to different IC50 values. Lower potency NSGMs appeared to interact with other residues in the vicinity of the above three. At a quantitative level, Figure 7B shows the relationship between potency and GOLDSCORE. The plot shows that the inhibition potency correlates well with GOLDSCORE (R2 = 0.92). This correlation is arguably the best observed so far for NSGM–protein interactions. This implies that the quantitative model could be used for designing advanced analogs of 2 in an iterative manner. Finally, the docking study confirms the results of the Michaelis studies (see above) that the unsulfated inhibitor 1j (cyan colored) is an active site ligand. In fact, 1j does not interact with the allosteric site. The primary reason for this is the lack of sulfate groups, which are critical for targeting the heparin-binding allosteric site.
Figure 7.

GOLD-based docking and scoring study of NSGMs binding to human plasmin. (A) Overlay of the best docked pose for NSGMs 1j (cyan), 2 (magenta), 3 (yellow), 4 (pink), 8 (white), 10 (blue), and 11 (green). (B) Relationship between −log IC50 (in M units) and GOLDSCORE. (C─F) Hydrogen bond interactions by inhibitors 1j, 2, 4, and 8 are shown. The key interacting residues are shown as spheres, and the inhibitors are shown as sticks.
NSGM 2 Inhibits Clot Lysis.
A key experiment to advance the concept that NSGM 2 could be a promising lead molecule is to evaluate its potency in inhibiting plasmin-mediated clot fibrinolysis. Thus, an in vitro study was performed in a 96-well platform. A fibrin-rich clot was first generated by inducing fibrinogenolysis using human thrombin and factor XIIIa in 20 mM Tris-HCl, pH 7.4, containing 10 mM CaCl2 at 37 °C. The resulting clot was allowed to stabilize for 15 min at 37 °C, following which NSGM 2 (or vehicle) at desired concentrations was added and allowed to equilibrate. Finally, clot lysis was initiated by addition of human plasmin and monitored by measuring absorbance at 405 nm as a function of time. Figure 8A shows the relative change in A405 at various time intervals in the presence of inhibitor 2 at 0–666 μM concentrations. Plasmin-mediated hydrolysis of the fibrin-rich clot results in a decrease in the relative A405 over time. Under the conditions studied, the clot was completely lysed in 290 min in the absence of NSGM 2. However, a significant inhibition/delay in clot lysis was observed in the presence of 2 in a dose-dependent manner. Plotting the rate of clot lysis (i.e., fibrinolysis) over 50–200 min versus the corresponding concentrations of inhibitor 2 shows a semilog sigmoidal relationship, which could be analyzed using dose–response eq 1 to calculate the IC50 of fibrinolysis inhibition. An apparent IC50 of 8.8 ± 1.0 μM and efficacy of 69.1 ± 6.9% (Figure 8B) were calculated using this nonlinear regressional analysis. This IC50 is similar to that determined using the small peptide substrate (Spectrozyme PL). Overall, the allosteric inhibitor 2 was found to inhibit plasmin-mediated fibrinolysis, which is key for further development of such molecules as promising lead entities.
Figure 8.
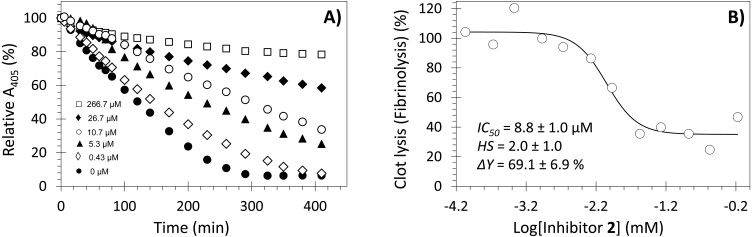
Effect of various concentrations of NSGM 2 on clot lysis (fibrinolysis) over time (A) and the dose dependent inhibition of clot lysis by inhibitor 2 (B). The effect was determined at pH 7.4 and 37 °C by measuring the UV absorbance of each well containing different concentration of 2 at 405 nm over 410 min. The rate of fibrinolysis over the time period of 50–200 min in the presence of NSGM 2 was transformed into % fibrinolysis using the rate in the absence of NSGM 2. Experiments were performed at pH 7.4 and 37 °C, as described in the Experimental Part. Solid lines represent the sigmoidal dose–response fits (eq 1) to the data to obtain the values of IC50, ΔY, and HS.
CONCLUSION AND SIGNIFICANCE
The fundamental hypothesis behind this work was that human plasmin should be possible to allosterically inhibit considering its high homology with other serine proteases, especially thrombin and factor XIa, which have been targeted by NSGMs of different structural features.37,40-43,48,49 The coagulation proteases possess one or more allosteric anion-binding site(s) that are recognized by heparin and related GAGs.33,34,58-60 Yet, these exosites are not structurally identical because their Arg/Lys residues are distinct in terms of the number, location, and orientation.32-35 More interestingly, the exosites differ in terms of their hydrophobic subsites or surrounding regions,32-35 which likely serve as recognition elements for NSGMs.41-44,49 This concept is further supported by plasmin inhibitors studied here.
This work shows that sulfated NSGMs can modulate plasmin’s catalytic activity with moderate-to-high potency by interacting with at least one anion-binding site. A specific NSGM 2 was found to inhibit human plasmin with at least 12-fold better potency than previous report.37 Also, the molecule displayed good antifibrinolysis potential. Further work will be needed to assess its advanced pharmacological applicability before advancing its clinical relevance. This would include its effect on blood loss in animal models such as liver laceration model and tail bleeding assays as well as its ex vivo effects on human blood in thromboelastographic experiments. At the present time, the NSGM 2 represents the best and most exciting lead toward the first allosteric regulator of human plasmin.
The value of this work also lies in the state of antifibrinolytics in use today. Only two agents, i.e., tranexamic acid and ε-aminocaproic acid, are in clinical use. Both of these suffer from severe lack of efficacy8,61,62 and adverse consequences. Aprotinin may be introduced in the U.S., but there are dangers associated with its use.8,15,16 The pace of discovering new direct plasmin inhibitors is increasingly becoming feverish, especially with the introduction of an amidinobenzylamide derivative, which inhibits plasmin as well as urokinase.63 Some of the inhibitors in the category of orthosteric inhibitors display nanomolar potencies.26-28 This high potency bodes well for further clinical studies. However, potency alone does not guarantee success. The ratio of potency to toxicity of a molecule is better measure of clinical success. In this respect, NSGMs are likely to offer more promise because of their high water solubility. In fact, literature reports show that highly sulfated small molecules possess little cytotoxicity.39,64 Thus, the concept of NSGM-mediated allosterism as an avenue for discovery of antifibrinolytics is novel, radical, and likely to be potentially rewarding.
There are other positive characteristics of NSGMs, in general, and 2, in particular. First, NSGM 2 inhibition of plasmin was instantaneously and fully reversed by protamine. Although the studies performed here related to in vitro settings, they allude to the possibility of in vivo reversal. Second, considering that these molecules are highly sulfated and will likely not penetrate the blood–brain barrier or placental barrier, we expect minimal central nervous system or fetal toxicities. This is important because both tranexamic acid and ε-aminocaproic acid appear to reach the brain. Third, NSGMs are readily synthesizable.38,45 This aspect is typically not appreciated but is extremely important because newer antifibrinolytics have to be inexpensive in order to be clinically viable. Yet, despite these positives, much further work is needed.
Overall, our work attempts to show that allosteric inhibition of plasmin is a viable approach for discovery of antifibrinolytics. We have discovered a homogeneous sulfated NSGM as the first promising lead anti-plasmin agent. NSGMs target the heparin-binding sites on plasmin. Further studies in animal models are needed to assess the clinical potential of NSGMs as antifibrinolytic agents. This work does not address their possible use as antimetastatic agents in treatment of cancer, where possibilities are equally promising.
EXPERIMENTAL PART
Chemicals, Reagents, Enzymes, and Substrates.
All anhydrous organic solvents were purchased from Sigma-Aldrich (Milwaukee, WI) or Fisher (Pittsburgh, PA) and used as such. Other solvents used were reagent grade and used as purchased. Analytical TLC was performed using UNIPLATE silica gel GHLF 250 μm precoated plates (ANALTECH, Newark, DE). Silica gel (200–400 mesh, 60 Å), fibrinogen, and UFH were from Sigma-Aldrich. Chemical reactions sensitive to air or moisture were carried out under nitrogen atmosphere in oven-dried glassware. Reagent solutions, unless otherwise noted, were handled under a nitrogen atmosphere using syringe techniques. Flash chromatography was performed using Teledyne ISCO (Lincoln, NE). Combiflash RF was performed using disposable normal silica cartridges of 30–50 μm particle size, 230–400 mesh size, and 60 Å pore size. The flow rate of the mobile phase was in the range of 18–35 mL/min, and mobile phase gradients of ethyl acetate/hexanes and CH2Cl2/CH3OH were used to elute compounds. Human plasmin, thrombin, factor Xa, and factor XIa were obtained from Haematologic Technologies (Essex Junction, VT). Stock solutions of serine proteases (plasmin, thrombin, factor Xa, and factor XIa) were prepared in 20 mM Tris-HCl buffer, pH 7.4, containing 100 mM NaCl, 2.5 mM CaCl2, 0.1% PEG8000, and 0.02% Tween80. Chromogenic substrates of thrombin (Spectrozyme TH), plasmin (Spectrozyme PL), and factor Xa (Spectrozyme FXa) were obtained from Sekisui Diagnostics (Lexington, MA). The chromogenic substrate of factor XIa (Chromogenix S-2366) was from DiaPharma (West Chester, OH).
Chemical Characterization.
1H and 13C NMR were recorded on Bruker-400 MHz spectrometer in either CD3OD, CDCl3, acetone-d6, D2O, or DMSO-d6. Signals, in part per million (ppm), are relative either to the residual peak of the solvent or to the internal standard of TMS. The NMR data are reported as chemical shift (ppm), integration, multiplicity of signal (s = singlet, d = doublet, t = triplet, q = quartet, dd = doublet of doublet, m= multiplet), and coupling constants (Hz). ESI-MS of compounds were recorded using Waters Acquity TQD MS spectrometer in positive or negative ion mode. Samples were dissolved in methanol and infused at a rate of approximately 20 μL/min. Ionization conditions were optimized for each intermediate and final persulfated product to maximize the ionization of the parent ion. Final persulfated NSGMs 2–16 were obtained in high overall yields and had >95% purity. Representative 1H NMR, 13C NMR, MS, and UPLC profiles of the final products are presented in Supporting Information. The compounds studied here as inhibitors of plasmin do not exhibit pan assay interference (PAINS), and this aspect is described in more detail in the Supporting Information available online.
General Procedure for Flavonoid Monomers Protection by Methoxymethyl (MOM) Chloride.
The quercetin and apigenin molecules were protected using MOM-Cl, as reported earlier.39,40 Briefly, to a solution of quercetin or apigenin (1 equiv) in DCM, N,N-diisopropylethylamine (DIPEA) (4–8 equiv) and MOM chloride (4 equiv for quercetin and 2 equiv for apigenin) were added under nitrogen atmosphere. After vigorous stirring at 0 °C for 1 h, the reaction mixture was allowed to warm to room temperature over 2 h and the stirring was maintained for 12 h. The resulting mixture was diluted with water (100 mL), extracted with ethyl acetate (200 mL), and then the organic layer was dried over Na2SO4, concentrated under reduced pressure, and purified by flash column chromatography to afford the tetraprotected quercetin 1b as a yellow solid in yields of 50–55% and the diprotected apigenin 2b as a cream solid in yields of 52–60%. Spectral characteristics of purified protected flavonoid monomers are as follows.
2-(3,4-Bis(methoxymethoxy)phenyl)-5-hydroxy-3,7-bis-(methoxymethoxy)-4H-chromen-4-one (1b).
1H NMR (400 MHz, CDCl3) δ 12.47 (s, 1H), 7.84 (d, J = 2.1 Hz, 1H), 7.64 (dd, J = 8.7, 2.1 Hz, 1H), 7.25–7.11 (m, 1H), 6.54 (d, J = 2.2 Hz, 1H), 6.39 (d, J = 2.1 Hz, 1H), 5.24 (d, J = 7.4 Hz, 4H), 5.17 (s, 2H), 5.11 (s, 2H), 3.47 (d, J = 4.6 Hz, 6H), 3.42 (s, 3H), 3.17 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 178.60, 162.94, 161.91, 156.67, 156.49, 149.65, 146.60, 135.63, 124.42, 123.92, 117.74, 115.66, 106.63, 99.70, 97.81, 95.64, 95.10, 94.22, 94.14, 57.72, 56.41, 56.35. MS (ESI) calculated for C23H26O11 [(M + H)]+, m/z 478.15, found for [(M + H)]+, m/z 478.907.
5-Hydroxy-7-(methoxymethoxy)-2-(4-(methoxymethoxy)-phenyl)-4H-chromen-4-one (2b).
1H NMR (400 MHz, CDCl3) δ 12.70 (s, 1H), 7.81–7.77 (m, 2H), 7.13–7.08 (m, 2H), 6.61 (d, J = 2.2 Hz, 1H), 6.54 (s, 1H), 6.43 (d, J = 2.2 Hz, 1H), 5.20 (d, J = 3.0 Hz, 4H), 3.46 (d, J = 1.2 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ 182.48, 164.01, 163.00, 162.15, 160.27, 157.61, 127.99, 124.69, 116.61, 104.72, 100.10, 94.37, 94.30, 56.34, 56.21. MS (ESI) calculated for C19H18O7 [(M + Na)]+, m/z 359.11, found for [(M + H)]+, m/z 359.171.
General Procedure for Flavonoid Dimerization by SN2 Reaction.
The quercetin- and apigenin-based monomers were coupled under basic conditions using different dibromo linkers as reported earlier.39,40 Briefly, to a solution of tetraprotected quercetin or dibrotected apigenin (1 equiv) in N,N-dimethylformamide (DMF) was added K2CO3 (2.5 equiv), and the mixture was stirred for 2 min. This was followed by addition of appropriate dibromo linker (0.5 equiv) and vigorous stirring for 12 h at room temperature. After the reaction completion as indicated by TLC, the reaction mixture was diluted with a mixture of ethyl acetate/H2O (50 mL; 1:1 mixture). The organic layer was separated, and the aqueous phase was further extracted with ethyl acetate (2 × 25 mL). The organic layer was then washed with saturated NaCl solution (25 mL). The three organic layers were combined, dried over anhydrous Na2SO4, and concentrated under reduced pressure to afford the crude intermediates 1c–3a which were further purified using flash chromatography on silica gel (70–85% ethyl acetate in hexanes). The pure intermediates were obtained as white to cream solids in yields of 30–50%. Spectral characteristics of intermediate 1c matched earlier reports.39,40 Spectral characteristics of the new purified intermediates are as follows.
5,5′-((1,4-Phenylenebis(methylene))bis(oxy))bis(2-(3,4-bis-(methoxymethoxy)phenyl)-3,7-bis(methoxymethoxy)-4H-chromen-4-one) (1d).
1H NMR (400 MHz, CDCl3) δ 7.97 (s, 1H), 7.86 (s, 2H), 7.62 (dd, J = 29.3, 8.9 Hz, 5H), 7.40 (t, J = 7.8 Hz, 1H), 7.16 (s, 1H), 6.65 (d, J = 2.4 Hz, 2H), 6.44 (s, 2H), 5.38–5.10 (m, 20H), 3.49 (d, J = 5.2 Hz, 12H), 3.43 (s, 6H), 3.19 (s, 6H). 13C NMR (100 MHz, CDCl3) δ 173.63, 161.28, 159.75, 158.56, 153.34, 149.19, 146.58, 137.96, 135.89, 127.03, 125.02, 123.71, 117.85, 115.78, 110.47, 98.64, 97.81, 95.88, 95.73, 95.18, 94.39, 77.32, 77.00, 76.68, 70.73, 57.65, 57.60, 56.41, 56.33. MS (ESI) calculated for C54H58O22 [(M + K)]+, m/z 1097.44, found for [(M + K)]+, m/z 1097.373.
5,5′-(([1,1′-Biphenyl]-4,4′-diylbis(methylene))bis(oxy))bis(2-(3,4-bis(methoxymethoxy)phenyl)-3,7-bis(methoxymethoxy)-4H-chromen-4-one) (1e).
1H NMR (400 MHz, CDCl3) δ 7.84 (d, J = 2.1 Hz, 2H), 7.68–7.51 (m, 11H), 7.20 (d, J = 4.6 Hz, 1H), 6.64 (d, J = 2.2 Hz, 2H), 6.44 (d, J = 2.2 Hz, 2H), 5.26–5.22 (m, 12H), 5.17 (s, 4H), 5.15 (s, 4H), 3.47 (d, J = 6.1 Hz, 12H), 3.41 (s, 6H), 3.17 (s, 6H). 13C NMR (100 MHz, CDCl3) δ 173.61, 161.26, 159.82, 158.58, 153.36, 149.22, 146.61, 140.29, 137.98, 135.49, 127.41, 127.35, 127.31, 127.29, 127.27, 125.04, 123.72, 117.92, 115.84, 110.56, 98.70, 97.82, 95.98, 95.76, 95.21, 94.40, 70.77, 57.59, 56.41, 56.39, 56.31. MS (ESI) calculated for C60H62O22 [(M + K)]+, m/z 1173.47, found for [(M + K)]+, m/z 1173.046.
5,5′-(((2,5-Dimethyl-1,4-phenylene)bis(methylene))bis-(oxy))bis(2-(3,4-bis(methoxymethoxy)phenyl)-3,7-bis-(methoxymethoxy)-4H-chromen-4-one) (1f).
1H NMR (400 MHz, CDCl3) δ 7.87 (d, J = 2.1 Hz, 2H), 7.68 (d, J = 2.1 Hz, 1H), 7.66 (d, J = 2.1 Hz, 1H), 7.49 (s, 2H), 7.23 (d, J = 2.7 Hz, 2H), 6.67 (d, J = 2.2 Hz, 2H), 6.46 (d, J = 2.2 Hz, 2H), 5.26 (d, J = 4.3 Hz, 8H), 5.24–5.03 (m, 12H), 3.51 (d, J = 4.8 Hz, 12H), 3.46 (s, 6H), 3.20 (s, 6H), 2.36 (s, 6H). 13C NMR (100 MHz, CDCl3) δ 173.47, 161.28, 159.94, 158.59, 153.22, 149.18, 146.60, 137.93, 133.59, 133.34, 129.86, 125.07, 123.67, 117.90, 115.84, 110.52, 98.50, 97.77, 95.82, 95.75, 95.21, 94.42, 69.43, 57.56, 56.40, 56.31, 18.60. MS (ESI) calculated for C56H62O22 [(M + K)]+, m/z 1125.34, found for [(M + K)]+, m/z 1125.026.
5,5′-((1,3-Phenylenebis(methylene))bis(oxy))bis(2-(3,4-bis-(methoxymethoxy)phenyl)-3,7-bis(methoxymethoxy)-4H-chromen-4-one) (1g).
1H NMR (400 MHz, CDCl3) δ 7.87 (d, J = 2.1 Hz, 2H), 7.70–7.55 (m, 5H), 7.42 (t, J = 7.6 Hz, 1H), 7.23 (d, J = 1.9 Hz, 2H), 6.66 (d, J = 2.1 Hz, 2H), 6.45 (d, J = 2.2 Hz, 2H), 5.26 (d, J = 4.0 Hz, 12H), 5.20 (s, 4H), 5.17 (s, 4H), 3.50 (d, J = 5.3 Hz, 12H), 3.44 (s, 6H), 3.20 (s, 6H). 13C NMR (100 MHz, CDCl3) δ 173.64, 162.49, 161.29, 159.74, 158.55, 153.36, 149.17, 146.55, 137.92, 136.71, 129.21, 126.15, 124.98, 124.57, 123.70, 117.80, 115.74, 110.43, 98.62, 97.79, 95.86, 95.70, 95.16, 94.36, 70.80, 57.60, 56.42, 56.34, 56.32. MS (ESI) calculated for C54H58O22 [(M + K)]+, m/z 1097.44, found for [(M + K)]+, m/z 1097.337.
5,5′-((Pyridine-2,6-diylbis(methylene))bis(oxy))bis(2-(3,4-bis(methoxymethoxy)phenyl)-3,7-bis(methoxymethoxy)-4H-chromen-4-one) (1h).
1H NMR (400 MHz, CDCl3) δ 8.89–8.80 (m, 2H), 8.53–8.47 (m, 1H), 7.84 (d, J = 2.1 Hz, 2H), 7.66 (d, J = 2.1 Hz, 1H), 7.64 (d, J = 2.1 Hz, 1H), 7.19 (d, J = 2.0 Hz, 2H), 6.75 (d, J = 2.0 Hz, 2H), 6.62 (d, J = 2.1 Hz, 2H), 5.70 (s, 4H), 5.26–5.22 (m, 8H), 5.21 (s, 4H), 5.15 (s, 4H), 3.49 (s, 6H), 3.47 (s, 6H), 3.45 (s, 6H), 3.15 (s, 6H). 13C NMR (100 MHz, CDCl3) δ 173.72, 161.59, 158.57, 157.78, 154.25, 149.44, 146.65, 137.88, 124.70, 123.82, 117.93, 115.83, 110.02, 99.20, 97.80, 96.84, 95.75, 95.19, 94.32, 66.12, 57.68, 56.57, 56.36, 56.33. MS (ESI) calculated for C53H57NO22 [(M + H)]+, m/z 1060.34, found for [(M + H)]+, m/z 1060.072.
5,5′-(Ethane-1,2-diylbis(oxy))bis(7-(methoxymethoxy)-2-(4-(methoxymethoxy)phenyl)-4H-chromen-4-one) (2c).
1H NMR (400 MHz, CDCl3) δ 7.85–7.76 (m, 5H), 7.38–6.82 (m, 5H), 6.78 (s, 2H), 6.65 (s, 2H), 5.18 (s, 8H), 4.54 (s, 4H), 3.80–3.26 (m, 12H). 13C NMR (100 MHz, CDCl3) δ 177.50, 162.49, 159.84, 159.52, 128.08, 116.57, 100.23, 96.63, 94.47, 94.25, 68.32, 56.58, 56.26. MS (ESI) calculated for C40H38O14 [(M + Na)]+, m/z 765.13, found for [(M + Na)]+, m/z 765.393.
5,5′-(Propane-1,3-diylbis(oxy))bis(7-(methoxymethoxy)-2-(4-(methoxymethoxy)phenyl)-4H-chromen-4-one) (2d).
1H NMR (400 MHz, CDCl3) δ 7.76–7.63 (m, 4H), 7.11–6.97 (m, 4H), 6.63 (d, J = 2.2 Hz, 2H), 6.50 (d, J = 2.2 Hz, 2H), 6.43 (s, 2H), 5.18–5.13 (m, 8H), 4.38 (t, J = 5.7 Hz, 4H), 3.42 (t, J = 1.7 Hz, 12H), 2.53–2.36 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 177.39, 161.47, 160.45, 160.40, 159.59, 159.38, 127.55, 125.11, 116.43, 110.04, 107.89, 98.43, 95.51, 94.27, 65.72, 56.38, 56.17. MS (ESI) calculated for C41H40O14 [(M + Na)]+, m/z 779.14, found for [(M + Na)]+, m/z 779.400.
5,5′-(Butane-1,4-diylbis(oxy))bis(7-(methoxymethoxy)-2-(4-(methoxymethoxy)phenyl)-4H-chromen-4-one) (2e).
1H NMR (400 MHz, CDCl3) δ 7.73 (d, J = 8.7 Hz, 4H), 7.06 (d, J = 8.7 Hz, 4H), 6.59 (d, J = 2.1 Hz, 2H), 6.49–6.36 (m, 4H), 5.17 (d, J = 7.6 Hz, 8H), 4.28–4.10 (m, 4H), 3.43 (d, J = 2.6 Hz, 12H), 2.25–2.17 (m, 4H). 13C NMR (100 MHz, CDCl3) δ 177.27, 161.39, 160.39, 160.35, 159.61, 159.37, 127.64, 127.53, 125.16, 116.45, 109.97, 107.91, 98.03, 95.43, 94.35, 94.30, 69.12, 56.38, 56.16, 25.84. MS (ESI) calculated for C42H42O14 [(M + Na)]+, m/z 793.16, found for [(M + Na)]+, m/z 793.442.
(E)-5,5′-(But-2-ene-1,4-diylbis(oxy))bis(7-(methoxymethoxy)-2-(4-(methoxymethoxy)phenyl)-4H-chromen-4-one) (2f).
1H NMR (400 MHz, CDCl3) δ 7.77 (d, J = 8.9 Hz, 4H), 7.09 (d, J = 8.9 Hz, 4H), 6.72 (d, J = 2.2 Hz, 2H), 6.52 (s, 2H), 6.48–6.42 (m, 4H), 5.20 (d, J = 6.4 Hz, 8H), 4.76–4.65 (m, 4H), 3.46 (d, J = 9.1 Hz, 12H). 13C NMR (100 MHz, CDCl3) δ 177.38, 161.42, 159.81, 159.66, 159.52, 127.61, 126.47, 125.06, 116.46, 107.95, 99.99, 95.86, 94.43, 94.28, 68.88, 56.44, 56.17. MS (ESI) calculated for C42H40O14 [(M + Na)]+, m/z 791.14, found for [(M + Na)]+, m/z 791.150.
5,5′-((1,4-Phenylenebis(methylene))bis(oxy))bis(7-(methoxymethoxy)-2-(4-(methoxymethoxy)phenyl)-4H-chromen-4-one) (2g).
1H NMR (400 MHz, CDCl3) δ 7.79–7.72 (m, 4H), 7.58 (s, 4H), 7.11–7.03 (m, 4H), 6.70 (d, J = 2.2 Hz, 2H), 6.52 (s, 2H), 6.44 (d, J = 2.2 Hz, 2H), 5.23–5.13 (m, 12H), 3.43 (s, 12H). 13C NMR (100 MHz, CDCl3) δ 177.36, 161.32, 160.65, 159.65, 159.50, 135.91, 127.62, 126.94, 125.04, 116.54, 116.44, 110.37, 107.96, 99.02, 96.11, 94.40, 94.25, 70.71, 56.43, 56.20. MS (ESI) calculated for C46H42O14 [(M + Na)]+, m/z 841.16, found for [(M + Na)]+, m/z 841.574.
5,5′-(([1,1′-Biphenyl]-4,4′-diylbis(methylene))bis(oxy))bis(7-(methoxymethoxy)-2-(4-(methoxymethoxy)phenyl)-4H-chromen-4-one) (2h).
1H NMR (400 MHz, CDCl3) δ 7.91–7.79 (m, 5H), 7.64–7.51 (m, 7H), 7.16–7.04 (m, 6H), 6.89–6.73 (m, 2H), 6.60–6.46 (m, 2H), 5.32–5.10 (m, 12H), 3.49–3.43 (m, 12H). 13C NMR (100 MHz, CDCl3) δ 177.59, 159.78, 159.68, 140.30, 135.16, 128.19, 127.29, 127.27, 116.59, 99.42, 96.15, 94.52, 94.25, 70.85, 56.55, 56.27. MS (ESI) calculated for C52H46O14 [(M + Na)]+, m/z 917.19, found for [(M + Na)]+, m/z 917.522.
5,5′-(((2,5-Dimethyl-1,4-phenylene)bis(methylene))bis-(oxy))bis(7-(methoxymethoxy)-2-(4-(methoxymethoxy)-phenyl)-4H-chromen-4-one) (2i).
1H NMR (400 MHz, CDCl3) δ 7.96–7.65 (m, 4H), 7.57 (s, 2H), 7.20–7.01 (m, 6H), 6.81 (d, J = 2.1 Hz, 2H), 6.55 (d, J = 2.2 Hz, 2H), 5.32–5.02 (m, 12H), 3.55–3.31 (m, 12H), 2.36 (s, 6H). 13C NMR (100 MHz, CDCl3) δ 177.43, 159.86, 159.64, 133.44, 133.06, 129.48, 127.87, 116.52, 98.97, 95.98, 94.50, 94.25, 69.31, 56.52, 56.23, 18.60. MS (ESI) calculated for C48H46O14 [(M + Na)]+, m/z 869.19, found for [(M + Na)]+, m/z 869.488.
5,5′-((1,3-Phenylenebis(methylene))bis(oxy))bis(7-(methoxymethoxy)-2-(4-(methoxymethoxy)phenyl)-4H-chromen-4-one) (2j).
1H NMR (400 MHz, CDCl3) δ 7.94–7.80 (m, 4H), 7.75 (s, 1H), 7.60–7.48 (m, 2H), 7.44–7.35 (m, 1H), 7.24 (s, 2H), 7.14–6.99 (m, 4H), 6.79 (d, J = 2.1 Hz, 2H), 6.64–6.51 (m, 2H), 5.31–5.14 (m, 12H), 3.55–3.31 (m, 12H). 13C NMR (100 MHz, CDCl3) δ 177.60, 162.43, 160.29, 159.74, 159.65, 136.48, 129.10, 128.12, 126.10, 124.65, 116.57, 99.32, 96.09, 94.51, 94.25, 70.89, 56.55, 56.26. MS (ESI) calculated for C52H46O14 [(M + Na)]+, m/z 841.16, found for [(M + Na)]+, m/z 841.510.
5,5′-((Pyridine-2,6-diylbis(methylene))bis(oxy))bis(7-(methoxymethoxy)-2-(4-(methoxymethoxy)phenyl)-4H-chromen-4-one) (2k).
1H NMR (400 MHz, CDCl3) δ 8.73 (s, 2H), 8.38 (s, 1H), 7.92–7.57 (m, 4H), 7.17–6.98 (m, 4H), 6.82 (d, J = 2.1 Hz, 2H), 6.63 (d, J = 2.2 Hz, 2H), 6.55 (s, 2H), 5.62 (s, 4H), 5.40–5.01 (m, 8H), 3.63–3.25 (m, 12H). 13C NMR (100 MHz, CDCl3) δ 177.43, 161.77, 161.63, 159.99, 159.52, 157.70, 151.80, 130.89, 127.80, 124.58, 116.56, 109.77, 107.56, 99.79, 97.13, 94.38, 94.26, 66.22, 56.60, 56.23. MS (ESI) calculated for C45H41O14 [(M + Na)]+, m/z 842.15, found for [(M + Na)]+, m/z 842.48.
2-(3,4-Bis(methoxymethoxy)phenyl)-3,7-bis(methoxymethoxy)-5-((4-(((7-(methoxymethoxy)-2-(4-(methoxymethoxy)phenyl)-4-oxo-4H-chromen-5-yl)oxy)methyl)benzyl)-oxy)-4H-chromen-4-one (3a).
1H NMR (400 MHz, CDCl3) δ 7.88–7.81 (m, 3H), 7.66 (dd, J = 8.7, 2.1 Hz, 1H), 7.61–7.55 (m, 3H), 7.25–7.16 (m, 2H), 7.14–7.05 (m, 3H), 6.78 (d, J = 2.2 Hz, 1H), 6.65 (d, J = 2.2 Hz, 1H), 6.51 (d, J = 2.2 Hz, 1H), 6.44 (d, J = 2.3 Hz, 1H), 5.28–5.13 (m, 16H), 3.54–3.39 (m, 16H), 3.19 (s, 2H). 13C NMR (100 MHz, CDCl3) δ 177.50, 173.63, 161.30, 159.74, 158.55, 153.35, 149.19, 146.58, 137.95, 127.88, 127.03, 127.00, 125.01, 123.71, 117.85, 116.52, 115.78, 110.44, 98.64, 97.82, 96.10, 95.87, 95.73, 95.18, 94.39, 94.24, 84.54, 77.32, 77.00, 76.68, 70.71, 57.64, 57.60, 56.51, 56.42, 56.32, 56.24. MS (ESI) calculated for C50H50O18, [(M + Na)]+, m/z 961.29, found for [(M + Na)]+, m/z 961.45.
General Procedure for Preparation of Polyphenolic Flavonoid Dimers by MOM Deprotection.
The MOM groups were completely deprotected by bromotrimethylsilane (TMS-Br). Briefly, to a solution of MOM-protected molecule in dry CH2Cl2, TMS-Br (6 equiv per MOM group) was added. The reaction mixture was stirred at −30 °C for an hour and then at 0 °C for 12–24 h. The deprotection was monitored using UPLC–MS until completion. After that, ethyl acetate (25 mL) was added to precipitate the polyphenol product from the reaction mixture. The precipitate was filtered, washed with excess ethyl acetate to remove the excess TMS-Br, and dried to obtain pure polyphenols 1i–3b as yellow to orange solid which were used in subsequent reactions without further purification. Spectral characteristics of intermediate 1i matched earlier reports.39,40 Spectral characteristics of the resulting new polyphenols are as follows.
5,5′-((1,4-Phenylenebis(methylene))bis(oxy))bis(2-(3,4-dihydroxyphenyl)-3,7-dihydroxy-4H-chromen-4-one) (1j).
1H NMR (400 MHz, DMSO-d6) δ 10.73 (s, 2H), 9.34 (bs, 1H), 8.81 (s, 1H), 7.73 (s, 4H), 7.67 (d, J = 2.2 Hz, 2H), 7.51 (dd, J = 8.4, 2.2 Hz, 2H), 6.89 (d, J = 8.5 Hz, 2H), 6.54–6.46 (m, 4H), 5.25 (s, 4H). 13C NMR (100 MHz, DMSO-d6) δ 171.07, 162.36, 159.20, 157.89, 146.94, 145.03, 142.02, 137.24, 136.08, 126.54, 125.47, 122.28, 119.12, 115.58, 114.53, 105.45, 97.13, 94.86, 69.49. MS (ESI) calculated for C36H26O14 [(M + H)]+, m/z 707.13, found for [(M + H)]+, m/z 706.892.
5,5′-(([1,1′-Biphenyl]-4,4′-diylbis(methylene))bis(oxy))bis(2-(3,4-dihydroxyphenyl)-3,7-dihydroxy-4H-chromen-4-one) (1k).
1H NMR (400 MHz, DMSO-d6) δ 10.84 (s, 4H), 9.22 (bs, 2H), 7.77 (q, J = 8.5 Hz, 9H), 7.67 (d, J = 2.2 Hz, 2H), 7.54–7.52 (m, 1H), 7.50 (d, J = 2.2 Hz, 1H), 6.89 (d, J = 8.4 Hz, 3H), 6.51 (s, 4H), 5.29 (s, 4H). 13C NMR (100 MHz, DMSO-d6) δ 171.07, 162.35, 159.20, 157.89, 146.93, 145.02, 142.01, 139.06, 137.26, 136.17, 127.21, 126.51, 122.29, 119.13, 115.57, 115.04, 114.52, 105.48, 97.17, 69.41. MS (ESI) calculated for C44H40O14 [(M + H)]+, m/z 783.16, found for [(M + H)]+, m/z 783.113.
5,5′-(((2,5-Dimethyl-1,4-phenylene)bis(methylene))bis-(oxy))bis(2-(3,4-dihydroxyphenyl)-3,7-dihydroxy-4H-chromen-4-one) (1l).
1H NMR (400 MHz, DMSO-d6) δ 10.79 (bs, 2H), 7.63 (d, J = 2.2 Hz, 2H), 7.57 (s, 2H), 7.48 (dd, J = 8.5, 2.2 Hz, 2H), 6.86 (d, J = 8.5 Hz, 2H), 6.53 (d, J = 2.1 Hz, 2H), 6.48 (d, J = 2.0 Hz, 2H), 5.14 (s, 4H), 2.37 (s, 6H). 13C NMR (100 MHz, DMSO-d6) δ 170.94, 162.34, 159.38, 157.91, 146.91, 145.02, 141.91, 137.14, 133.99, 133.23, 129.79, 122.28, 119.10, 115.57, 114.50, 105.42, 97.07, 94.78, 68.56. MS (ESI) calculated for C40H30O14 [(M + H)]+, m/z 735.17, found for [(M + H)]+, m/z 734.960.
5,5′-((1,3-Phenylenebis(methylene))bis(oxy))bis(2-(3,4-dihydroxyphenyl)-3,7-dihydroxy-4H-chromen-4-one) (1m).
1H NMR (400 MHz, DMSO-d6) δ 10.81 (bs, 6H), 7.65 (dd, J = 8.8, 2.2 Hz, 5H), 7.55–7.47 (m, 3H), 6.87 (dd, J = 8.5, 3.0 Hz, 2H), 6.49 (t, J = 1.9 Hz, 2H), 6.40 (d, J = 2.0 Hz, 1H), 6.17 (d, J = 2.0 Hz, 1H), 5.23 (s, 4H). 13C NMR (100 MHz, DMSO-d6) δ 175.79, 171.04, 163.84, 162.35, 160.67, 159.22, 157.88, 156.09, 147.66, 146.92, 145.02, 137.24, 136.85, 135.68, 122.28, 121.91, 119.94, 119.14, 115.58, 115.03, 114.50, 105.46, 102.98, 98.15, 93.32, 69.82. MS (ESI) calculated for C36H26O14 [(M + H)]+, m/z 707.13, found for [(M + H)]+, m/z 706.960.
5,5′-((Pyridine-2,6-diylbis(methylene))bis(oxy))bis(2-(3,4-dihydroxyphenyl)-3,7-dihydroxy-4H-chromen-4-one) (1n).
1H NMR (400 MHz, DMSO-d6) δ 10.77 (bs, 2H), 8.28–8.12 (m, 3H), 7.68 (d, J = 2.2 Hz, 2H), 7.53 (dd, J = 8.5, 2.2 Hz, 2H), 6.90 (d, J = 8.5 Hz, 2H), 6.62–6.45 (m, 4H), 5.40 (s, 4H). 13C NMR (100 MHz, DMSO-d6) δ 171.19, 162.46, 158.58, 157.81, 155.47, 147.03, 145.04, 142.42, 137.30, 122.20, 120.52, 119.23, 115.59, 114.58, 105.42, 97.46, 95.30, 69.88. MS (ESI) calculated for C37H25NO14 [(M + H)]+, m/z 708.14, found for [(M + H)]+, m/z 707.920.
5,5′-(Ethane-1,2-diylbis(oxy))bis(7-hydroxy-2-(4-hydroxyphenyl)-4H-chromen-4-one) (2l).
1H NMR (400 MHz, DMSO-d6) δ 10.67 (s, 2H), 10.16 (s, 2H), 7.84 (d, J = 8.8 Hz, 4H), 6.97–6.79 (m, 4H), 6.59–6.53 (m, 4H), 6.49 (s, 2H), 4.38 (s, 4H). 13C NMR (100 MHz, DMSO-d6) δ 175.50, 162.27, 160.39, 159.94, 159.50, 158.98, 127.96, 127.76, 125.49, 121.47, 115.81, 107.68, 105.93, 95.95, 68.01. MS (ESI) calculated for C32H22O10 [(M + H)]+, m/z 566.12, found for [(M + H)]+, m/z 567.103.
5,5′-(Propane-1,3-diylbis(oxy))bis(7-hydroxy-2-(4-hydroxyphenyl)-4H-chromen-4-one) (2m).
1H NMR (400 MHz, DMSO-d6) δ 11.52 (s, 2H), 7.93 (d, J = 8.1 Hz, 2H), 7.83 (dd, J = 8.8, 2.8 Hz, 2H), 6.92 (dd, J = 12.1, 8.4 Hz, 5H), 6.77–6.39 (m, 5H), 4.33–4.23 (m, 4H), 2.31–2.18 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 175.66, 162.36, 160.37, 159.88, 159.80, 158.99, 137.62, 131.37, 128.02, 127.72, 125.46, 121.48, 116.59, 115.80, 107.24, 105.93, 97.11, 95.18, 64.88, 20.74. MS (ESI) calculated for C33H24O10 [(M + H)]+, m/z 581.14, found for [(M + H)]+, m/z 581.273.
5,5′-(Butane-1,4-diylbis(oxy))bis(7-hydroxy-2-(4-hydroxyphenyl)-4H-chromen-4-one) (2n).
1H NMR (400 MHz, DMSO-d6) δ 11.35 (s, 2H), 10.24 (bs, 2H), 8.29–7.69 (m, 5H), 7.01–6.87 (m, 4H), 6.75–6.22 (m, 5H), 4.80–3.87 (m, 4H), 2.44–1.61 (m, 4H). 13C NMR (100 MHz, DMSO-d6) δ 175.50, 162.30, 160.34, 159.91, 158.94, 145.52, 137.69, 128.04, 127.69, 127.60, 125.46, 121.53, 116.59, 115.80, 107.26, 105.94, 95.06, 60.03, 48.57, 20.74. MS (ESI) calculated for C34H26O10 [(M + H)]+, m/z 595.15, found for [(M + H)]+, m/z 595.952.
(E)-5,5′-(But-2-ene-1,4-diylbis(oxy))bis(7-hydroxy-2-(4-hydroxyphenyl)-4H-chromen-4-one) (2o).
1H NMR (400 MHz, DMSO-d6) δ 10.61 (s, 2H), 10.09 (s, 2H), 7.76 (d, J = 8.8 Hz, 4H), 6.82 (d, J = 8.7 Hz, 4H), 6.45 (d, J = 2.1 Hz, 2H), 6.41 (s, 2H), 6.33 (d, J = 2.2 Hz, 2H), 6.28 (s, 2H), 4.58 (s, 4H). 13C NMR (100 MHz, DMSO-d6) δ 175.27, 162.26, 160.63, 160.35, 159.84, 159.17, 154.90, 127.84, 127.72, 121.51, 121.22, 115.95, 115.81, 108.35, 107.37, 106.00, 105.80, 88.69, 68.18. MS (ESI) calculated for C34H24O10 [(M + H)]+, m/z 593.14, found for [(M + H)]+, m/z 593.032.
5,5′-((1,4-Phenylenebis(methylene))bis(oxy))bis(7-hydroxy-2-(4-hydroxyphenyl)-4H-chromen-4-one) (2p).
1H NMR (400 MHz, DMSO-d6) δ 11.48 (s, 2H), 10.32 (s, 2H), 8.17–7.79 (m, 5H), 7.64 (s, 3H), 7.01–6.84 (m, 5H), 6.66 (d, J = 8.4 Hz, 3H), 6.58–6.40 (m, 2H), 5.21 (s, 4H). 13C NMR (100 MHz, DMSO-d6) δ 175.30, 160.65, 159.90, 159.13, 158.00, 154.93, 135.90, 127.85, 127.73, 126.88, 126.81, 121.23, 116.09, 115.96, 115.82, 108.57, 105.82, 98.00, 88.96, 69.77. MS (ESI) calculated for C38H26O10 [(M + H)]+, m/z 643.15, found for [(M + H)]+, m/z 642.978.
5,5′-(([1,1′-Biphenyl]-4,4′-diylbis(methylene))bis(oxy))bis(7-hydroxy-2-(4-hydroxyphenyl)-4H-chromen-4-one) (2q).
1H NMR (400 MHz, DMSO-d6) δ 11.47 (s, 2H), 10.28 (bs, 2H), 7.94 (d, J = 8.8 Hz, 2H), 7.84 (d, J = 8.8 Hz, 2H), 7.78–7.58 (m, 8H), 7.44–7.23 (m, 2H), 7.12–6.83 (m, 4H), 6.77–6.59 (m, 2H), 6.59–6.41 (m, 2H), 5.24 (s, 4H). 13C NMR (100 MHz, DMSO-d6) δ 175.31, 160.66, 160.38, 159.91, 159.14, 158.01, 154.94, 128.61, 128.50, 127.86, 127.74, 127.51, 127.42, 127.00, 126.97, 126.56, 126.43, 126.30, 121.23, 116.10, 115.96, 115.83, 108.57, 105.84, 88.96, 69.66. MS (ESI) calculated for C44H30O10 [(M + H)]+, m/z 719.19, found for [(M + H)]+, m/z 719.021.
5,5′-(((2,5-Dimethyl-1,4-phenylene)bis(methylene))bis-(oxy))bis(7-hydroxy-2-(4-hydroxyphenyl)-4H-chromen-4-one) (2r).
1H NMR (400 MHz, DMSO-d6) δ 11.46 (s, 2H), 10.30 (bs, 2H), 7.93 (d, J = 8.8 Hz, 3H), 7.87–7.76 (m, 1H), 7.68–7.55 (m, 2H), 7.00–6.86 (m, 5H), 6.71 (s, 2H), 6.65 (s, 1H), 6.56–6.50 (m, 2H), 5.12 (s, 4H), 2.33 (s, 6H). 13C NMR (100 MHz, DMSO-d6) δ 175.27, 160.64, 159.86, 159.14, 158.09, 154.94, 133.69, 132.89, 129.49, 127.84, 127.71, 121.26, 115.95, 115.82, 108.51, 105.83, 97.74, 88.79, 68.49, 18.13. MS (ESI) calculated for C40H30O10 [(M + H)]+, m/z 671.18, found for [(M + H)]+, m/z 670.831.
5,5′-((1,3-Phenylenebis(methylene))bis(oxy))bis(7-hydroxy-2-(4-hydroxyphenyl)-4H-chromen-4-one) (2s).
1H NMR (400 MHz, DMSO-d6) δ 10.71 (bs, 4H), 7.96–7.91 (m, 1H), 7.87–7.82 (m, 3H), 7.77 (s, 1H), 7.62 (dd, J = 7.6, 1.6 Hz, 1H), 7.54–7.35 (m, 2H), 6.92 (dd, J = 8.8, 6.8 Hz, 4H), 6.78 (s, 1H), 6.56 (d, J = 2.1 Hz, 1H), 6.53 (s, 1H), 6.50–6.48 (m, 2H), 6.20 (d, J = 2.1 Hz, 1H), 5.22 (s, 4H). 13C NMR (100 MHz, DMSO-d6) δ 181.69, 175.59, 164.09, 162.26, 161.42, 161.13, 160.39, 159.95, 159.29, 159.00, 157.28, 136.93, 128.42, 127.84, 127.74, 126.06, 121.17, 115.94, 115.83, 107.56, 105.98, 102.83, 95.61, 93.93, 69.85. MS (ESI) calculated for C38H26O10 [(M + H)]+, m/z 643.16, found for [(M + H)]+, m/z 643.148.
5,5′-((Pyridine-2,6-diylbis(methylene))bis(oxy))bis(7-hydroxy-2-(4-hydroxyphenyl)-4H-chromen-4-one) (2t).
1H NMR (400 MHz, DMSO-d6) δ 10.83 (s, 2H), 10.23 (s, 2H), 8.11–8.01 (m, 3H), 7.86 (d, J = 8.8 Hz, 4H), 6.93 (d, J = 8.8 Hz, 4H), 6.61 (d, J = 2.0 Hz, 2H), 6.56 (s, 2H), 6.54–6.43 (m, 2H), 5.28 (s, 4H). 13C NMR (100 MHz, DMSO-d6) δ 175.72, 162.39, 160.46, 160.15, 158.98, 158.84, 155.80, 138.48, 127.78, 121.44, 120.28, 115.84, 107.44, 106.01, 98.04, 95.88, 70.29. MS (ESI) calculated for C37H25NO10 [(M + H)]+, m/z 644.15, found for [(M + H)]+, m/z 643.922.
2-(3,4-Dihydroxyphenyl)-3,7-dihydroxy-5-((4-(((7-hydroxy-2-(4-hydroxyphenyl)-4-oxo-4H-chromen-5-yl)oxy)methyl)-benzyl)oxy)-4H-chromen-4-one (2b).
1H NMR (400 MHz, DMSO-d6) δ 10.66 (s, 3H), 7.85–7.76 (m, 2H), 7.70 (d, J = 8.2 Hz, 2H), 7.64–7.58 (m, 2H), 7.55–7.37 (m, 2H), 6.91–6.77 (m, 3H), 6.56–6.35 (m, 5H), 5.21–5.14 (m, 4H). 13C NMR (100 MHz, DMSO-d6) δ 175.65, 162.29, 160.42, 159.26, 159.00, 157.87, 146.90, 145.02, 137.34, 136.10, 128.42, 127.77, 126.57, 126.47, 126.36, 122.32, 121.47, 115.94, 115.83, 115.57, 107.51, 105.99, 105.45, 95.53, 69.47. MS (ESI) calculated for C38H26O12 [(M + H)]+, m/z 675.15, found for [(M + Na)]+, m/z 675.23.
General Procedure for Chemical Sulfation of Polyphenols.
Sulfation of polyphenols was performed using microwave-assisted chemical protocol, as described earlier.39,40,45 Briefly, to a stirred solution of polyphenol in anhydrous CH3CN (~3 mL) at room temperature, Et3N (10 equiv/−OH group) and SO3/Me3N complex (6 equiv/−OH) were added. The reaction vessel was sealed and microwaved (CEM Discover, Cary, NC) for 4–8 h at 90 °C. The reaction mixture was cooled and concentrated in vacuo at temperature of <30 °C. The reaction mixture was then purified on Combiflash RF system using CH2Cl2/CH3OH mobile system (6:4) to obtain the persulfated molecules. The fractions containing the desired molecule were pooled together, concentrated in vacuo, and reloaded onto a SP Sephadex C-25 column for sodium exchange. Desired fractions containing sodium salts of the persulfated molecules were pooled, concentrated in vacuo, and lyophilized to obtain a fluffy white powder. All sulfation reactions were quantitative with >65% yield. Spectral characteristics of sulfated diflavonoid 1 matched earlier reports.39,40 Spectral characteristics of the new final persulfated products are as follows.
Sodium 4-(5-((4-(((2-(3,4-Bis(sulfonatooxy)phenyl)-4-oxo-3,7-bis(sulfonatooxy)-4H-chromen-5-yl)oxy)methyl)benzyl)-oxy)-4-oxo-3,7-bis(sulfonatooxy)-4H-chromen-2-yl)-1,2-phenylene Bis(sulfate) (2).
1H NMR (400 MHz, DMSO-d6) δ 8.15 (d, J = 2.3 Hz, 2H), 8.09 (d, J = 2.3 Hz, 1H), 8.08 (d, J = 2.3 Hz, 1H), 7.76 (s, 4H), 7.65 (d, J = 8.9 Hz, 2H), 7.11 (d, J = 2.0 Hz, 2H), 6.82 (d, J = 2.2 Hz, 2H), 5.25 (s, 4H). 13C NMR (100 MHz, DMSO-d6) δ 172.98, 158.73, 158.34, 157.10, 153.49, 146.53, 142.95, 135.72, 135.32, 126.82, 124.50, 123.58, 119.89.03, 118.80, 109.56, 100.72, 99.38, 70.15. MS (ESI) calculated for C38H18Na8O38S8 [(M − 2Na)/2]2−, m/z 737.8314, found for [(M − 2Na)/2]2−, m/z 738.2780.
Sodium 4-(5-((4′-(((2-(3,4-Bis(sulfonatooxy)phenyl)-4-oxo-3,7-bis(sulfonatooxy)-4H-chromen-5-yl)oxy)methyl)-[1,1′-bi-phenyl]-4-yl)methoxy)-4-oxo-3,7-bis(sulfonatooxy)-4H-chromen-2-yl)-1,2-phenylene Bis(sulfate) (3).
1H NMR (400 MHz, DMSO-d6) δ 8.13 (dd, J = 4.6, 2.3 Hz, 2H), 8.10–8.03 (m, 2H), 7.76 (s, 8H), 7.64 (dd, J = 8.9, 3.7 Hz, 2H), 7.07 (dd, J = 3.8, 2.0 Hz, 2H), 6.79 (d, J = 2.3 Hz, 2H), 5.26 (s, 4H). 13C NMR (100 MHz, DMSO-d6) δ 172.47, 158.61, 158.10, 157.02, 153.12, 146.35, 142.89, 139.19, 135.93, 135.29, 127.52, 126.65, 123.87, 119.98, 118.80, 109.80, 100.83, 69.86. MS (ESI) calculated for C44H22Na8O38S8 [(M − 2Na)/2]2−, m/z 775.8471, found for [(M − 2Na)/2]2−, m/z 776.6650
Sodium 4-(5-((4-(((2-(3,4-Bis(sulfonatooxy)phenyl)-4-oxo-3,7-bis(sulfonatooxy)-4H-chromen-5-yl)oxy)methyl)-2,5-dimethylbenzyl)oxy)-4-oxo-3,7-bis(sulfonatooxy)-4H-chromen-2-yl)-1,2-phenylene Bis(sulfate) (4).
1H NMR (400 MHz, DMSO-d6) δ 8.14 (d, J = 2.4 Hz, 2H), 8.05 (dd, J = 8.9, 2.4 Hz, 2H), 7.63 (d, J = 8.9 Hz, 2H), 7.55 (s, 2H), 7.11 (d, J = 2.0 Hz, 2H), 6.83 (d, J = 2.1 Hz, 2H), 5.14 (s, 4H), 2.38 (s, 6H). 13C NMR (100 MHz, DMSO-d6) δ 172.68, 158.91, 158.18, 157.07, 153.22, 146.42, 142.90, 135.19, 133.85, 133.62, 130.20, 124.48, 123.80, 120.05, 118.84, 109.68, 100.63, 99.25, 69.10, 18.30. MS (ESI) calculated for C40H22Na8O38S8 [(M − 2Na)/2]2−, m/z 751.8471, found for [(M − 2Na)/2]2−, m/z 751.7020.
Sodium 4-(5-((3-(((2-(3,4-Bis(sulfonatooxy)phenyl)-4-oxo-3,7-bis(sulfonatooxy)-4H-chromen-5-yl)oxy)methyl)benzyl)-oxy)-4-oxo-3,7-bis(sulfonatooxy)-4H-chromen-2-yl)-1,2-phenylene Bis(sulfate) (5).
1H NMR (400 MHz, DMSO-d6) δ 8.13 (dd, J = 6.3, 2.4 Hz, 2H), 8.03–7.97 (m, 2H), 7.67–7.40 (m, 6H), 7.17 (d, J = 2.0 Hz, 2H), 6.76 (d, J = 2.1 Hz, 2H), 5.28 (s, 4H). 13C NMR (100 MHz, DMSO-d6) δ 172.75, 158.71, 158.17, 157.05, 153.27, 146.42, 142.91, 136.74, 135.28, 128.64, 126.35, 124.52, 123.76, 119.99, 118.83, 109.69, 100.84, 99.30, 70.28. MS (ESI) calculated for C38H18Na8O38S8 [(M − 2Na)/2]2−, m/z 737.8314, found for [(M − 2Na)/2]2−, m/z 737.6929.
Sodium 4-(5-((6-(((2-(3,4-Bis(sulfonatooxy)phenyl)-4-oxo-3,7-bis(sulfonatooxy)-4H-chromen-5-yl)oxy)methyl)pyridin-2-yl)methoxy)-4-oxo-3,7-bis(sulfonatooxy)-4H-chromen-2-yl)-1,2-phenylene Bis(sulfate) (6).
1H NMR (400 MHz, DMSO-d6) δ 8.15–8.05 (m, 7H), 7.65 (d, J = 8.9 Hz, 2H), 7.15 (d, J = 2.0 Hz, 2H), 6.78 (d, J = 2.2 Hz, 2H), 5.31 (s, 4H). 13C NMR (100 MHz, DMSO-d6) δ 172.62, 158.19, 157.04, 155.89, 153.35, 146.41, 142.89, 135.29, 124.48, 123.80, 120.27, 119.95, 118.74, 109.61, 99.43, 70.76. MS (ESI) calculated for C37H17NNa8O38S8 [(M − 2Na)/2]2−, m/z 738.3290, found for [(M − 2Na)/2]2−, m/z 738.7981.
Sodium 4-Oxo-5-(2-((4-oxo-7-(sulfonatooxy)-2-(4-(sulfonatooxy)phenyl)-4H-chromen-5-yl)oxy)ethoxy)-2-(4-(sulfonatooxy)phenyl)-4H-chromen-7-yl Sulfate (7).
1H NMR (400 MHz, DMSO-d6) δ 8.06–8.00 (m, 5H), 7.47 (s, 2H), 7.41–7.20 (m, 5H), 6.92–6.66 (m, 2H), 4.41 (s, 4H). 13C NMR (100 MHz, DMSO-d6) δ 175.78, 160.06, 157.54, 156.67, 155.59, 154.17, 127.21, 124. 68, 120.13, 110.67, 107.01, 101.69, 93.82, 67.92. MS (ESI) calculated for C32H18Na4O22S4 [(M −2Na)/2]2−, m/z 463.9484, found for [(M −2Na)/2]2−, m/z 464.0431.
Sodium 4-Oxo-5-(3-((4-oxo-7-(sulfonatooxy)-2-(4-(sulfonatooxy)phenyl)-4H-chromen-5-yl)oxy)propoxy)-2-(4-(sulfonatooxy)phenyl)-4H-chromen-7-yl Sulfate (8).
1H NMR (400 MHz, DMSO-d6) δ 7.97–7.94 (m, 4H), 7.32–7.30 (m, 4H), 7.17 (d, J = 2.2 Hz, 2H), 6.71–6.70 (m, 4H), 4.35–4.28 (m, 4H), 2.30–2.23 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 175.87, 159.82, 158.99, 158.17, 158.08, 156.26, 127.02, 125.09, 120.10, 109.54, 107.33, 100.23, 99.50, 65.27 MS (ESI) calculated for C33H20Na4O22S4 [(M−2Na)/2]2−, m/z 470.9562, found for [(M −2Na)/2]2−, m/z 471.1272.
Sodium 4-Oxo-5-(4-((4-oxo-7-(sulfonatooxy)-2-(4-(sulfonatooxy)phenyl)-4H-chromen-5-yl)oxy)butoxy)-2-(4-(sulfonatooxy)phenyl)-4H-chromen-7-yl Sulfate (9).
1H NMR (400 MHz, DMSO-d6) δ 7.97–7.94 (m, 4H), 7.33 (dd, J = 8.9, 1.2 Hz, 4H), 7.17 (d, J = 1.9 Hz, 2H), 6.73 (t, J = 1.7 Hz, 2H), 6.66 (d, J = 0.9 Hz, 2H), 4.22–4.03 (m, 4H), 2.09–1.99 (m, 4H). 13C NMR (100 MHz, DMSO-d6) δ 175.80, 175.49, 159.80, 159.71, 159.15, 158.17, 158.11, 157.98, 156.54, 156.30, 155.49, 154.15, 127.1, 127.00, 125.11, 124.86, 120.15, 110.55, 109.54, 107.31, 107.12, 101.10, 100.51, 99.45, 93.05, 68.65, 25.52, 25.31, 25.13. MS (ESI) calculated for C34H22Na4O22S4 [(M − 2Na)/2]2−, m/z 477.9641, found for [(M − 2Na)/2]2−, m/z 478.2439.
Sodium (E)-4-Oxo-5-((4-((4-oxo-7-(sulfonatooxy)-2-(4-(sulfonatooxy)phenyl)-4H-chromen-5-yl)oxy)but-2-en-1-yl)-oxy)-2-(4-(sulfonatooxy)phenyl)-4H-chromen-7-yl Sulfate (10).
1H NMR (400 MHz, DMSO-d6) δ 7.99–7.93 (m, 4H), 7.35–7.31 (m, 4H), 7.21 (d, J = 2.0 Hz, 2H), 6.72 (d, J = 2.2 Hz, 2H), 6.68 (s, 2H), 6.40 (s, 2H), 4.69 (s, 4H). 13C NMR (100 MHz, DMSO-d6) δ 175.74, 159.88, 158.57, 158.20, 158.13, 156.36, 127.00, 126.89, 125.03, 120.11, 109.54, 107.30, 99.63, 68.34. MS (ESI) calculated for C34H20Na4O22S4 [(M − 2Na)/2]2−, m/z 476.9562, found for [(M − 2Na)/2]2−, m/z 477.1390.
Sodium 4-Oxo-5-((4-(((4-oxo-7-(sulfonatooxy)-2-(4-(sulfonatooxy)phenyl)-4H-chromen-5-yl)oxy)methyl)benzyl)-oxy)-2-(4-(sulfonatooxy)phenyl)-4H-chromen-7-yl Sulfate (11).
1H NMR (400 MHz, DMSO-d6) δ 7.92–7.79 (m, 4H), 7.59 (s, 4H), 7.28–7.21 (m, 4H), 7.13 (d, J = 2.1 Hz, 2H), 6.71 (d, J = 2.2 Hz, 2H), 6.61 (s, 2H), 5.14 (s, 4H). 13C NMR (100 MHz, DMSO-d6) δ 175.80, 159.99, 158.54, 158.23, 158.12, 156.39, 136.08, 127.03, 126.82, 125.05, 120.14, 109.77, 107.33, 101.09, 99.89, 69.91. MS (ESI) calculated for C38H22Na4O22S4 [(M − 2Na)/2]2−, m/z 501.9641, found for [(M − 2Na)/2]−, m/z 502.2589.
Sodium 4-Oxo-5-((4′-(((4-oxo-7-(sulfonatooxy)-2-(4-(sulfonatooxy)phenyl)-4H-chromen-5-yl)oxy)methyl)-[1,1′-bi-phenyl]-4-yl)methoxy)-2-(4-(sulfonatooxy)phenyl)-4H-chromen-7-yl Sulfate (12).
1H NMR (400 MHz, DMSO-d6) δ 8.00–7.87 (m, 4H), 7.84–7.58 (m, 8H), 7.34 (d, J = 8.9 Hz, 4H), 7.23 (d, J = 2.1 Hz, 2H), 6.82 (d, J = 2.2 Hz, 2H), 6.72 (s, 2H), 5.27 (s, 4H). 13C NMR (100 MHz, DMSO-d6) δ 175.81, 160.00. 158.53, 158.23, 158.12, 156.39, 139.14, 136.04, 127.50, 127.05, 126.58, 125.04, 120.14, 109.76, 107.32, 101.07, 99.91, 69.75. MS (ESI) calculated for C40H26Na4O22S4 [(M − 2Na)/2]2−, m/z 540.07, found for [(M − 2Na)/2]−, m/z 539.761.
Sodium 5-((2,5-Dimethyl-4-(((4-oxo-7-(sulfonatooxy)-2-(4-(sulfonatooxy)phenyl)-4H-chromen-5-yl)oxy)methyl)benzyl)-oxy)-4-oxo-2-(4-(sulfonatooxy)phenyl)-4H-chromen-7-yl Sulfate (13).
1H NMR (400 MHz, DMSO-d6) δ 7.99 (d, J = 8.9 Hz, 4H), 7.69 (s, 2H), 7.34 (d, J = 8.9 Hz, 4H), 7.25 (d, J = 2.1 Hz, 2H), 6.87 (d, J = 2.2 Hz, 2H), 6.74 (s, 2H), 5.14 (s, 4H), 2.37 (s, 6H). 13C NMR (100 MHz, DMSO-d6) δ 175.77, 159.90, 158.66, 158.23, 158.13, 156.34, 133.87, 132.96, 129.61, 127.01, 125.08, 120.13, 109.69, 107.32, 100.86, 99.73, 68.72, 18.16. MS (ESI) calculated for C40H26Na4O22S4 [(M − 2Na)/2]2−, m/z 515.9797, found for [(M − 2Na)/2]2−, m/z 515.8131.
Sodium 4-Oxo-5-((3-(((4-oxo-7-(sulfonatooxy)-2-(4-(sulfonatooxy)phenyl)-4H-chromen-5-yl)oxy)methyl)benzyl)-oxy)-2-(4-(sulfonatooxy)phenyl)-4H-chromen-7-yl Sulfate (14).
1H NMR (400 MHz, DMSO-d6) δ 7.98–7.83 (m, 4H), 7.71 (s, 1H), 7.61 (dd, J = 7.6, 1.6 Hz, 2H), 7.41 (t, J = 7.6 Hz, 1H), 7.26 (d, J = 8.9 Hz, 4H), 7.23 (d, J = 2.1 Hz, 2H), 6.79 (d, J = 2.2 Hz, 2H), 6.71 (s, 2H), 5.23 (s, 4H). 13C NMR (100 MHz, DMSO-d6) δ 175.82, 159.97, 158.54, 158.22, 158.12, 156.38, 136.84, 128.37, 127.03, 126.15, 125.31, 125.05, 120.13, 109.76, 107.32, 101.10, 99.90, 70.09. MS (ESI) calculated for C38H22Na4O22S4 [(M − 2Na)/2]2−, m/z 501.9641, found for [(M − 2Na)/2]2−, m/z 502.2365.
Sodium 4-Oxo-5-((6-(((4-oxo-7-(sulfonatooxy)-2-(4-(sulfonatooxy)phenyl)-4H-chromen-5-yl)oxy)methyl)pyridin-2-yl)methoxy)-2-(4-(sulfonatooxy)phenyl)-4H-chromen-7-yl Sulfate (15).
1H NMR (400 MHz, DMSO-d6) δ 8.08–7.92 (m, 7H), 7.36 (d, J = 8.9 Hz, 4H), 7.29 (d, J = 2.0 Hz, 2H), 6.80 (d, J = 2.2 Hz, 2H), 6.75 (s, 2H), 5.29 (s, 4H). 13C NMR (100 MHz, DMSO-d6) δ 175.94, 160.15, 158.23, 158.20, 158.08, 156.42, 156.09, 137.84, 127.08, 124.99, 120.15, 119.98, 109.58, 107.33, 100.81, 99.95, 99.49, 70.65. MS (ESI) calculated for C37H21NNa4O22S4 [(M − 2Na)/2]2−, m/z 502.4617, found for [(M − 2Na)/2]2−, m/z 502.3524.
Sodium 4-(4-Oxo-5-((4-(((4-oxo-7-(sulfonatooxy)-2-(4-(sulfonatooxy)phenyl)-4H-chromen-5-yl)oxy)methyl)benzyl)-oxy)-3,7-bis(sulfonatooxy)-4H-chromen-2-yl)-1,2-phenylene Bis(sulfate) (16).
1H NMR (400 MHz, DMSO-d6) δ 8.07 (d, J = 2.4 Hz, 1H), 7.99 (dd, J = 8.9, 2.3 Hz, 1H), 7.96–7.79 (m, 2H), 7.66–7.49 (m, 5H), 7.27 (dd, J = 9.0, 2.2 Hz, 2H), 7.22–7.09 (m, 1H), 7.01 (d, J = 2.0 Hz, 1H), 6.79–6.54 (m, 3H), 5.16 (d, J = 5.5 Hz, 4H). 13C NMR (100 MHz, DMSO-d6) δ 176.35, 173.11, 160.43, 159.10, 158.71, 158.56, 157.48, 156.82, 153.60, 146.83, 143.33, 136.51, 135.76, 127.56, 127.45, 125.56, 124.28, 120.63, 120.40, 119.19, 110.22, 107.84, 101.55, 100.36, 99.72, 70.43. MS (ESI) calculated for C38H20Na6O30S6 [(M − 2Na)/2]2−, m/z 619.8977, found for [(M − 2Na)/2]2−, m/z 620.4034.
UPLC–MS Analysis of Sulfated Diflavonoids.
Analysis of sulfated NSGMs was performed using a Waters Acquity H-class UPLC system equipped with a photodiode array detector and triple quadrupole mass spectrometer. A reversed-phase Waters BEH C18 column of particle size 1.7 μm and 2.1 mm × 50 mm dimensions at 30 ± 2 °C was employed. Solvent A consisted of 25 mM n-hexylamine in water containing 0.1% (v/v) formic acid, while solvent B consisted of 25 mM n-hexylamine in acetonitrile–water mixture (3:1 v/v) containing 0.1% (v/v) formic acid. A linear gradient of 3% solvent B per min over 20 min (initial solvent B proportion was 20% v/v) at a flow rate of 500 μL/min was used. The sample was monitored for absorbance in the range of 190–400 nm and then directly introduced into the mass spectrometer with ESI-MS detection. The following parameters were used: capillary voltage, 4 kV; cone voltage, 20 V; desolvation temperature, 350 °C; nitrogen gas flow, 650 L/h. Multiple mass scans were collected in the range of 250–1500 amu within 0.25 s and averaged to enhance the signal-to-noise ratio.
Direct Inhibition of Serine Proteases.
Direct inhibition of human plasmin was measured using a chromogenic substrate hydrolysis assay on a microplate reader (FlexStation III, Molecular Devices, Sunnyvale, CA, USA), as reported earlier.40,44 Briefly, to each well of a 96-well microplate containing 85 μL of 20 mM Tris-HCl buffer, pH 7.4, containing 100 mM NaCl, 2.5 mM CaCl2, 0.1% PEG8000, and 0.02% Tween80 at 37 °C was added 5 μL of potential plasmin inhibitor (0–20 mM aqueous solution) or vehicle alone and 5 μL of enzyme (stock of 500 nM, effective concentration in the well is ~25 nM). After 5 min of incubation, 5 μL of 1 mM Spectrozyme PL was rapidly added and the residual enzyme activity was measured from the initial rate of increase in A405. Relative residual enzyme activity (Y) as a function of the concentration of sulfated molecule was fitted using logistic eq 1 to obtain the potency (IC50), efficacy (ΔY,) and Hill slope (HS) of inhibition.
| (1) |
In this equation, Y is the ratio of residual plasmin activity in the presence of inhibitor to that in its absence (fractional residual activity). YM and Y0 are the maximum and minimum possible values of the fractional residual protease activity. The difference between these two values is used to evaluate the inhibitor’s efficacy to reduce the residual enzyme activity under the assay condition. IC50 is the concentration of the inhibitor that results in 50% inhibition of enzyme activity, and it is used to evaluate the inhibitor’s potency.
Direct inhibition of thrombin, factor IXa, factor Xa, factor XIa, factor XIIa, trypsin, and chymotrypsin by inhibitor 2 was measured using a chromogenic substrate hydrolysis assay on a microplate reader (FlexStation III, Molecular Devices), as reported earlier.41,42 Briefly, to each well of a 96-well microplate containing 85–185 μL of 20 mM Tris-HCl buffer, pH 7.4, containing 100 mM NaCl, 2.5 mM CaCl2, 0.1% PEG8000, and 0.02% Tween80 at either 25 °C (thrombin) or 37 °C (factor IXa, factor Xa, factor XIa, factor XIIa, trypsin, and chymotrypsin) or 20 mM Tris-HCl buffer, pH 7.4, containing 100 mM NaCl, 2.5 mM CaCl2, 0.1% PEG8000, 0.02% Tween80, and 33% ethylene glycol at 37 37 °C (factor IXa) was added 5 μL of inhibitor 2 (0–20 mM) or vehicle and 5 μL of the enzyme. The final concentrations of the enzymes were 6 nM (thrombin), 250 nM (factor IXa), 1.09 nM (factor Xa), 0.75 nM (factor XIa), 10 nM (factor XIIa), 0.145 μg/mL (trypsin), and 0.5 mg/mL (chymotrypsin). After 5 min of incubation, 5 μL of 1.0 mM Spectrozyme TH, 10.0 mM Spectrozyme FIXa, 2.5 mM Spectrozyme FXa, 6.9 mM S-2366, 5.0 mM Spectrozyme FXIIa, 3.2 mM SS-2222, 5.0 mM Spectrozyme Cty was rapidly added and the residual enzyme activity was measured from the initial rate of increase in absorbance at 405 nm. Relative residual enzyme activity (Y, activity in the presence of inhibitor to that in its absence) as a function of inhibitor 2 concentrations was fitted using logistic eq 1 to obtain the IC50, ΔY, and HS of inhibition. Similar selectivity studies were performed for polyphenolic precursor 1j.
Michaelis–Menten Kinetics of Spectrozyme PL Hydrolysis.
The initial rate of Spectrozyme PL hydrolysis by human plasmin (~25 nM) was monitored from the linear increase in absorbance at 405 nm corresponding to less than 10% consumption of the substrate. The initial rate was measured as a function of various concentrations of the substrate (0–2 mM) in the presence of fixed concentration of inhibitor 2 (0–200 μM) or inhibitor 1j (0–400 μM) in 20 mM Tris-HCl buffer, pH 7.4, containing 100 mM NaCl, 2.5 mM CaCl2, 0.1% PEG8000, and 0.02% Tween80 at 37 °C. The data were fitted by Michaelis–Menten eq 2 to determine KM and VMAX:
| (2) |
Fluorescence-Based Binding Affinity.
Fluorescence experiments were performed using a QM4 spectrofluorometer (Photon Technology International, Birmingham, NJ) in 20 mM Tris-HCl buffer, pH 7.4, containing 100 mM NaCl, 2.5 mM CaCl2, and 0.1% PEG8000 at 37 °C. The affinities of human plasmin for inhibitors 2, 4, 10, and 13 were measured using the change in the intrinsic tryptophan fluorescence (λEM = 348 nm, λEX = 280 nm) at varying concentrations of the inhibitors. The titrations were performed by adding aliquots of 250 μM aqueous solution of inhibitor 2, 4, 10, or 13 to 200 μL solution of plasmin (160 nM) and monitoring the fluorescence intensity at the appropriate λEM. The excitation and emission slits were set to 1.0 mm. The observed change in fluorescence (ΔF) relative to initial fluorescence (F0) was fitted using eq 3 to obtain the dissociation constant (KD) and the maximal change in fluorescence (ΔFMAX) at saturation. Each measurement was performed three times. Likewise, UFH binding affinity was measured in similar fashion. In this equation, L indicates the sulfated inhibitor or UFH.
| (3) |
Preparation of Active Site Blocked Plasmin.
The active site of human plasmin was blocked using 1,5-dansyl-EGR-cmk using a previously described method.65 Briefly, human plasmin (25 μM) was treated with 1,5-dansyl-EGR-cmk (1 mM) in phosphate buffered saline (PBS) at 37 °C for 2 h. At the end of the incubation, the activity of the treated enzyme was determined using the chromogenic substrate hydrolysis to confirm active site blockage. Excess 1,5-dansyl-EGR-cmk was removed by overnight dialysis against PBS.
Fluorescence-Based Binding Affinity of 2 and 13 for Active Site Blocked Plasmin.
Affinity of compounds 2 and 13 for active site blocked plasmin was determined using the changes in intrinsic tryptophan fluorescence (λEM = 348 nm, λEX = 280 nm) at varying inhibitor concentrations. The buffer used was a 20 mM Tris-HCl buffer, pH 7.4, containing 100 mM NaCl, 2.5 mM CaCl2, and 0.1% PEG8000 at 37 °C. The titrations were performed as with free plasmin but with a concentration of active-site-blocked plasmin of 500 nM. The data so obtained were fitted using eq 3 to obtain the dissociation constant (KD) and the maximal change in fluorescence (ΔFMAX) at saturation. Each measurement was performed three times.
Protamine Reversibility of Plasmin Inhibition.
To assess the in vitro reversibility of plasmin inhibition, the activity profiles were measured in increasing concentrations of protamine sulfate in the presence of inhibitor 2 (200 μM) at pH 7.4 and 37 °C. Generally, each well of the 96-well microplate contained 86 μL of 20 mM Tris-HCl buffer of pH 7.4 containing 100 mM NaCl, 2.5 mM CaCl2, 0.02% Tweeen80, and 0.1% PEG8000 to which 1 μL of inhibitor 2 (20 mM) or vehicle, 3 μL of plasmin (stock of 1.6 μM), and 5 μL of protamine sulfate (0–80 mg/mL; effective concentrations were 0–4 mg/mL) were sequentially added. After a 5 min incubation, 5 μL of Spectrozyme PL (1 mM) was rapidly added and the restored plasmin activity was measured from the initial rate of increase in absorbance at 405 nm over the time period of 20–120 s. Stock of protamine sulfate was serially diluted to give nine different aliquots in the wells. Relative restored plasmin activity at each concentration of protamine was calculated from the ratio of plasmin activity in the presence and absence of the reversing agent. Equation 4 was used to fit the dose dependence of restored protease activity to obtain the effective concentration of reversing agent required to restore 50% of enzyme activity at specific inhibitor concentration (EC50) and the efficacy (ΔY) of reversing process.
| (4) |
Effect of NSGM 2 on Plasmin-Mediated Clot Lysis.
The method followed here was similar to that reported earlier37 with a few modifications. This in vitro experiment was conducted in 96-well platform in pH 7.4 buffer at 37 °C. The clot was generated by the addition of 80 μL of a solution of thrombin (2.5 μg/mL) in 20 mM Tris-HCl containing 10 mM CaCl2 to 60 μL of a solution of fibrinogen (10 mg/mL) and factor XIIIa (2 μg/mL) in 20 mM Tris-HCl containing 10 mM CaCl2. The resulting clot was stabilized for 15 min at 37 °C. The absorbance was then measured at 405 nm and was about 0.91 across all 16 wells. Following this, 5 μL of inhibitor 2 (or vehicle) (effective concentrations in the well were 0–666 μM) was added to each well resulting in no significant change in the A405 of the corresponding wells. Finally, 5 μL of human plasmin (stock of 1.6 μM) in 20 mM Tris-HCl buffer of pH 7.4 containing 2.5 mM CaCl2 and 100 mM NaCl was added to each well and the A405 of each well was recorded over more than 400 min at various time intervals. By semilog plotting the rate of clot lysis (fibrinolysis) over the time period of 50–200 min versus the corresponding concentrations of inhibitor 2 using the logistic eq 1, the IC50 of fibrinolysis inhibition by this inhibitor and its efficacy were determined.
Molecular Modeling Studies.
The 3D coordinates of plasmin were extracted from Protein Data Bank (PDB code 3UIR). Molecular modeling studies was performed using protein preparation tool in Tripos Sybyl-X version 2.2 (https://www.certara.com/software/molecular-modeling-and-simulation/sybyl-x-suite/). Hydrogen atoms were added and minimized keeping all heavy atoms as aggregates. Potential site of binding was identified by analyzing the proteins electrostatic potential (Figure 1A) using APBS tool in PyMOL Molecular Graphics System, version 1.5.0.4 (Schrödinger, New York, NY). The potential binding site included residues R637, R644, K645, K651, R776, and R779, and all residues within 18 Å of centroid were identified as part of the binding site. Inhibitors 1j, 2, 3, 4, 8, 10, and 11 were modeled in Sybyl and docked into the structure using GOLD, version 5.2,66 as described earlier.54 For each inhibitor, 300 genetic algorithm run, 100 000 iterations were employed in which the early termination option was disabled. Each docked pose was scored using GOLDSCORE, and all poses were retained.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by Grants HL107152, HL090586, and HL128639 from the National Institutes of Health to U.R.D. R.A.A.-H. acknowledges the Donald and Nancy Abraham Postdoctoral Research Fellowship from the VCU School of Pharmacy and a Postdoctoral Research Grant from the VCU Office of Research and Innovation. We also thank Brianna Mackie and Dr. Akul Mehta of VCU for their technical support in characterization efforts, and Dr. Faik Musayev for help with protein purification. We thank the computational facilities provided by National Center for Research Resources (award number S10RR027411).
ABBREVIATIONS USED
- GAG
glycosaminoglycan
- NSGM
non-saccharide glycosaminoglycan mimetic
- SAR
structure–activity relationship
- UFH
unfractionated heparin
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.6b01474.
Data corresponding to the thermodynamic studies of NSGM–plasmin interaction; salt dependence studies of NSGM–plasmin interaction; and spectral profiles (NMR, UPLC, ESI-MS) of NSGMs (PDF)
Molecular formula strings and some data (CSV)
The authors declare no competing financial interest.
REFERENCES
- (1).Wolberg AS Plasma and cellular contributions to fibrin network formation, structure and stability. Haemophilia 2010, 16, 7–12. [DOI] [PubMed] [Google Scholar]
- (2).Leebeek FW; Rijken DC The fibrinolytic status in liver diseases. Semin. Thromb. Hemostasis 2015, 41, 474–480. [DOI] [PubMed] [Google Scholar]
- (3).Wada H; Matsumoto T; Yamashita Y Diagnosis and treatment of disseminated intravascular coagulation (DIC) according to four DIC guidelines. J. Intensive Care 2014, 2, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Stein E; McMahon B; Kwaan H; Altman JK; Frankfurt O; Tallman MS The coagulopathy of acute promyelocytic leukaemia revisited. Best Pract. Res. Clin. Haematol 2009, 22, 153–163. [DOI] [PubMed] [Google Scholar]
- (5).Ranucci M Hemostatic and thrombotic issues in cardiac surgery. Semin. Thromb. Hemostasis 2015, 41, 84–90. [DOI] [PubMed] [Google Scholar]
- (6).Kuter DJ Managing thrombocytopenia associated with cancer chemotherapy. Oncology (Williston Park) 2015, 29, 282–294. [PubMed] [Google Scholar]
- (7).Castellino FJ; Ploplis VA Structure and function of the plasminogen/plasmin system. Thromb. Haemostasis 2005, 93, 647–654. [DOI] [PubMed] [Google Scholar]
- (8).Al-Horani RA; Desai UR Recent advances on plasmin inhibitors for the treatment of fibrinolysis-related disorders. Med. Res. Rev 2014, 34, 1168–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Swedberg JE; Harris JM Natural and engineered plasmin inhibitors: Applications and design strategies. ChemBioChem 2012, 13, 336–348. [DOI] [PubMed] [Google Scholar]
- (10).Munakata S; Tashiro Y; Nishida C; Sato A; Komiyama H; Shimazu H; Dhahri D; Salama Y; Eiamboonsert S; Takeda K; Yagita H; Tsuda Y; Okada Y; Nakauchi H; Sakamoto K; Heissig B; Hattori K Inhibition of plasmin protects against colitis in mice by suppressing matrix metalloproteinase 9-mediated cytokine release from myeloid cells. Gastroenterology 2015, 148, 565–578. [DOI] [PubMed] [Google Scholar]
- (11).Reichel CA; Lerchenberger M; Uhl B; Rehberg M; Berberich N; Zahler S; Wymann MP; Krombach F Plasmin inhibitors prevent leukocyte accumulation and remodeling events in the postischemic microvasculature. PLoS One 2011, 6, e17229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Deryugina EI; Quigley JP Cell surface remodeling by plasmin: a new function for an old enzyme. J. Biomed. Biotechnol 2012, 2012, 564259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Lecker I; Wang DS; Romaschin AD; Peterson M; Mazer CD; Orser BA Tranexamic acid concentrations associated with human seizures inhibit glycine receptors. J. Clin. Invest 2012, 122, 4654–4666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Dhir A Antifibrinolytics in cardiac surgery. Ann. Card. Anaesth 2013, 16, 117–125. [DOI] [PubMed] [Google Scholar]
- (15).Mangano DT; Tudor IC; Dietzel C The risk associated with aprotinin in cardiac surgery. N. Engl. J. Med 2006, 354, 353–365. [DOI] [PubMed] [Google Scholar]
- (16).Schneeweiss S; Seeger JD; Landon J; Walker AM Aprotinin during coronary-artery bypass grafting and risk of death. N. Engl. J. Med 2008, 358, 771–783. [DOI] [PubMed] [Google Scholar]
- (17).Wang X; Lin X; Loy JA; Tang J; Zhang XC Crystal structure of the catalytic domain of human plasmin complexed with streptokinase. Science 1998, 281, 1662–1665. [DOI] [PubMed] [Google Scholar]
- (18).Perona JJ; Craik CS Structural basis of substrate specificity in the serine proteases. Protein Sci. 1995, 4, 337–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Parry MA; Fernandez-Catalan C; Bergner A; Huber R; Hopfner KP; Schlott B; Gührs KH; Bode W The ternary microplasmin-staphylokinase-microplasmin complex is a proteinase-cofactor-substrate complex in action. Nat. Struct. Biol 1998, 5, 917–923. [DOI] [PubMed] [Google Scholar]
- (20).Hervio LS; Coombs GS; Bergstrom RC; Trivedi K; Corey DR; Madison EL Negative selectivity and the evolution of protease cascades: the specificity of plasmin for peptide and protein substrates. Chem. Biol 2000, 7, 443–453. [DOI] [PubMed] [Google Scholar]
- (21).Hidaka K; Gohda K; Teno N; Wanaka K; Tsuda Y Active site-directed plasmin inhibitors: Extension on the P2 residue. Bioorg. Med. Chem 2016, 24, 545–553. [DOI] [PubMed] [Google Scholar]
- (22).Cheng L; Pettersen D; Ohlsson B; Schell P; Karle M; Evertsson E; Pahlén S; Jonforsen M; Plowright AT; Boström J; Fex T; Thelin A; Hilgendorf C; Xue Y; Wahlund G; Lindberg W; Larsson LO; Gustafsson D Discovery of the fibrinolysis inhibitor AZD6564, acting via interference of a protein-protein Interaction. ACS Med. Chem. Lett 2014, 5, 538–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Teno N; Gohda K; Wanaka K; Tsuda Y; Akagawa M; Akiduki E; Araki M; Masuda A; Otsubo T; Yamashita Y Novel type of plasmin inhibitors: providing insight into P4 moiety and alternative scaffold to pyrrolopyrimidine. Bioorg. Med. Chem 2015, 23, 3696–3704. [DOI] [PubMed] [Google Scholar]
- (24).Teno N; Gohda K; Yamashita Y; Otsubo T; Yamaguchi M; Wanaka K; Tsuda Y Plasmin inhibitors with hydrophobic amino acid-based linker between hydantoin moiety and benzimidazole scaffold enhance inhibitory activity. Bioorg. Med. Chem. Lett 2016, 26, 2259–2261. [DOI] [PubMed] [Google Scholar]
- (25).Teno N; Gohda K; Wanaka K; Tsuda Y; Sueda T; Yamashita Y; Otsubo T Pyrrolo-pyrimidine-inhibitors with hydantoin moiety as spacer can explore P4/S4 interaction on plasmin. Bioorg. Med. Chem 2014, 22, 2339–2352. [DOI] [PubMed] [Google Scholar]
- (26).Saupe SM; Steinmetzer T A new strategy for the development of highly potent and selective plasmin inhibitors. J. Med. Chem 2012, 55, 1171–1180. [DOI] [PubMed] [Google Scholar]
- (27).Saupe SM; Leubner S; Betz M; Klebe G; Steinmetzer T Development of new cyclic plasmin inhibitors with excellent potency and selectivity. J. Med. Chem 2013, 56, 820–831. [DOI] [PubMed] [Google Scholar]
- (28).Hinkes S; Wuttke A; Saupe SM; Ivanova T; Wagner S; Knörlein A; Heine A; Klebe G; Steinmetzer T Optimization of cyclic plasmin inhibitors: From benzamidines to benzylamines. J. Med. Chem 2016, 59, 6370–6386. [DOI] [PubMed] [Google Scholar]
- (29).Merdanovic M; Mönig T; Ehrmann M; Kaiser M Diversity of allosteric regulation in proteases. ACS Chem. Biol 2013, 8, 19–26. [DOI] [PubMed] [Google Scholar]
- (30).Christopoulos A Allosteric binding sites on cell-surface receptors: novel targets for drug discovery. Nat. Rev. Drug Discovery 2002, 1, 198–210. [DOI] [PubMed] [Google Scholar]
- (31).Hauske P; Ottmann C; Meltzer M; Ehrmann M; Kaiser M Allosteric regulation of proteases. ChemBioChem 2008, 9, 2920–2928. [DOI] [PubMed] [Google Scholar]
- (32).Millers EK; Johnson LA; Birrell GW; Masci PP; Lavin MF; de Jersey J; Guddat LW The structure of human microplasmin in complex with textilinin-1, an aprotinin-like inhibitor from the Australian brown snake. PLoS One 2013, 8, e54104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Carter WJ; Cama E; Huntington JA Crystal structure of thrombin bound to heparin. J. Biol. Chem 2005, 280, 2745–2749. [DOI] [PubMed] [Google Scholar]
- (34).Johnson DJ; Li W; Adams TE; Huntington JA Antithrombin-S195A factor Xa-heparin structure reveals the allosteric mechanism of antithrombin activation. EMBO J. 2006, 25, 2029–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Jin L; Pandey P; Babine RE; Weaver DT; Abdel-Meguid SS; Strickler JE Mutation of surface residues to promote crystallization of activated factor XI as a complex with benzamidine: an essential step for the iterative structure-based design of factor XI inhibitors. Acta Crystallogr., Sect. D: Biol. Crystallogr 2005, 61, 1418–1425. [DOI] [PubMed] [Google Scholar]
- (36).Yomtova VM; Stambolieva NA; Blagoev BM Kinetic study of the effect of heparin on the amidase activity of trypsin, plasmin and urokinase. Thromb. Haemostasis 1983, 49, 199–203. [PubMed] [Google Scholar]
- (37).Henry BL; Abdel Aziz M; Zhou Q; Desai UR Sulfated, low-molecular-weight lignins are potent inhibitors of plasmin, in addition to thrombin and factor Xa: novel opportunity for controlling complex pathologies. Thromb. Haemostasis 2010, 103, 507–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Desai UR The promise of sulfated synthetic small molecules as modulators of glycosaminoglycan function. Future Med. Chem 2013, 5, 1363–1366. [DOI] [PubMed] [Google Scholar]
- (39).Patel NJ; Karuturi R; Al-Horani RA; Baranwal S; Patel J; Desai UR; Patel BB Synthetic, non-saccharide, glycosaminoglycan mimetics selectively target colon cancer stem cells. ACS Chem. Biol 2014, 9, 1826–1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Al-Horani RA; Karuturi R; White DT; Desai UR Plasmin regulation through allosteric, sulfated, small molecules. Molecules 2015, 20, 608–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Verespy S III; Mehta AY; Afosah D; Al-Horani RA; Desai UR Allosteric partial inhibition of monomeric proteases. Sulfated coumarins induce regulation, not just inhibition, of thrombin. Sci. Rep 2016, 6, 24043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Sidhu PS; Abdel Aziz MH; Sarkar A; Mehta AY; Zhou Q; Desai UR Designing allosteric regulators of thrombin. Exosite 2 features multiple subsites that can be targeted by sulfated small molecules for inducing inhibition. J. Med. Chem 2013, 56, 5059–5070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Abdel Aziz MH; Sidhu PS; Liang A; Kim JY; Mosier PD; Zhou Q; Farrell DH; Desai UR Designing allosteric regulators of thrombin. Monosulfated benzofuran dimers selectively interact with Arg173 of exosite 2 to induce inhibition. J. Med. Chem 2012, 55, 6888–6897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Al-Horani RA; Desai UR Designing allosteric inhibitors of factor XIa. Lessons from the interactions of sulfated pentagalloyl glucopyranosides. J. Med. Chem 2014, 57, 4805–4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Al-Horani RA; Karuturi R; Verespy S 3rd; Desai UR Synthesis of glycosaminoglycan mimetics through sulfation of polyphenols. Methods Mol. Biol 2015, 1229, 49–67. [DOI] [PubMed] [Google Scholar]
- (46).Desai UR; Petitou M; Björk I; Olson ST Mechanism of heparin activation of antithrombin. Role of individual residues of the pentasaccharide activating sequence in the recognition of native and activated states of antithrombin. J. Biol. Chem 1998, 273, 7478–7487. [DOI] [PubMed] [Google Scholar]
- (47).Desai BJ; Boothello R; Mehta AY; Scarsdale JN; Wright HT; Desai UR Interaction of thrombin with sucrose octasulfate. Biochemistry 2011, 50, 6973–6982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Al-Horani RA; Liang A; Desai UR Designing nonsaccharide allosteric activators of antithrombin for accelerated inhibition of factor Xa. J. Med. Chem 2011, 54, 6125–6138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Karuturi R; Al-Horani RA; Mehta SC; Gailani D; Desai UR Discovery of allosteric modulators of factor XIa by targeting hydrophobic domains adjacent to its heparin-binding site. J. Med. Chem 2013, 56, 2415–2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Hartman SK; Teruya J Practice guidelines for reversal of new and old anticoagulants. DM, Dis.-Mon 2012, 58, 448–461. [DOI] [PubMed] [Google Scholar]
- (51).Henry BL; Desai UR Anticoagulants: Drug discovery and development. In Burger’s Medicinal Chemistry, 7th ed.; Rotella D, Abraham DJ, Eds.; John Wiley and Sons: New York, 2010; pp 365–408. [Google Scholar]
- (52).Ledoux D; Papy-Garcia D; Escartin Q; Sagot MA; Cao Y; Barritault D; Courtois J; Hornebeck W; Caruelle JP Human plasmin enzymatic activity is inhibited by chemically modified dextrans. J. Biol. Chem 2000, 275, 29383–29390. [DOI] [PubMed] [Google Scholar]
- (53).Vörös G; Kolev K; Csomor K; Machovich R Inhibition of plasmin activity by sulfated polyvinylalcohol-acrylate copolymers. Thromb. Res 2000, 100, 353–361. [DOI] [PubMed] [Google Scholar]
- (54).Sankaranarayanan NV; Desai UR Toward a robust computational screening strategy for identifying glycosaminoglycan sequences that display high specificity for target proteins. Glycobiology 2014, 24, 1323–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Argade MD; Mehta AY; Sarkar A; Desai UR Allosteric inhibition of human factor XIa: discovery of monosulfated benzofurans as a class of promising inhibitors. J. Med. Chem 2014, 57, 3559–3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Abdel Aziz MH; Sidhu PS; Liang A; Kim JY; Mosier PD; Zhou Q; Farrell DH; Desai UR Designing allosteric regulators of thrombin. Monosulfated benzofuran dimers selectively interact with Arg173 of exosite 2 to induce inhibition. J. Med. Chem 2012, 55, 6888–6897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Al-Horani RA; Liang A; Desai UR Designing nonsaccharide, allosteric activators of antithrombin for accelerated inhibition of factor Xa. J. Med. Chem 2011, 54, 6125–6138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Monteiro RQ Targeting exosites on blood coagulation proteases. An. Acad. Bras. Cienc 2005, 77, 275–280. [DOI] [PubMed] [Google Scholar]
- (59).Zhao M; Abdel-Razek T; Sun MF; Gailani D Characterization of a heparin-binding site on the heavy chain of factor XI. J. Biol. Chem 1998, 273, 31153–31159. [DOI] [PubMed] [Google Scholar]
- (60).Badellino KO; Walsh PN Localization of a heparin binding site in the catalytic domain of FXIa. Biochemistry 2001, 40, 7569–7580. [DOI] [PubMed] [Google Scholar]
- (61).Royston D Blood-sparing drugs: Aprotinin, tranexamic acid, and epsilon-aminocaproic acid. Int. Anesthesiol. Clin 1995, 33, 155–179. [PubMed] [Google Scholar]
- (62).Makhija N; Sarupria A; Kumar Choudhary S; Das S; Lakshmy R; Kiran U Comparison of epsilon aminocaproic acid and tranexamic acid in thoracic aortic surgery: Clinical efficacy and safety. J. Cardiothorac. Vasc. Anesth 2013, 27, 1201–1207. [DOI] [PubMed] [Google Scholar]
- (63).Schweinitz A; Steinmetzer T; Banke IJ; Arlt MJ; Stürzebecher A; Schuster O; Geissler A; Giersiefen H; Zeslawska E; Jacob U; Krüger A; Stürzebecher J Design of novel and selective inhibitors of urokinase-type plasminogen activator with improved pharmacokinetic properties for use as antimetastatic agents. J. Biol. Chem 2004, 279, 33613–33622. [DOI] [PubMed] [Google Scholar]
- (64).Correia-da-Silva M; Sousa E; Pinto MM Emerging sulfated flavonoids and other polyphenols as drugs: nature as an inspiration. Med. Res. Rev 2014, 34, 223–279. [DOI] [PubMed] [Google Scholar]
- (65).Bøtkær KA; Byszuk AA; Andersen LM; Christensen A; Andreasen PA; Blouse GE Nonproteolytic induction of catalytic activity into the single-chain form of urokinase-type plasminogen activator by dipeptides. Biochemistry 2009, 48, 9606–9617. [DOI] [PubMed] [Google Scholar]
- (66).Jones G; Willett P; Glen RC; Leach AR; Taylor R Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol 1997, 267, 727–748. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.