

ABSTRACT
As the most seriously controlled mycotoxin produced by Aspergillus spp. and Penicillium spp., ochratoxin A (OTA) results in various toxicological effects and widely contaminates agro-products. Biological detoxification is the highest priority regarding OTA in food and feed industry, but currently available detoxification enzymes have relatively low effectiveness in terms of time and cost. Here we show a superefficient enzyme, ADH3, identified from Stenotrophomonas acidaminiphila that has a strong ability to transform OTA into nontoxic ochratoxin-α by acting as an amidohydrolase. Recombinant ADH3 (1.2 μg/mL) completely degrades 50 μg/L OTA within 90 s, while the other most efficient OTA hydrolases available take several hours. The kinetic constant showed that rADH3 (Kcat/Km) catalytic efficiency was 56.7 to 35,000 times higher than those of previous hydrolases rAfOTase, rOTase, and commercial carboxypeptidase A (CPA). Protein structure-based assay suggested that ADH3 has a preference for hydrophobic residues to form a larger hydrophobic area than other detoxifying enzymes at the cavity of the catalytic sites, and this structure allows OTA easier access to the catalytic sites. In addition, ADH3 shows considerable temperature adaptability to exert hydrolytic function at the temperature down to 0°C or up to 70°C. Collectively, we report a superefficient OTA detoxifying enzyme with promising potential for industrial applications.
IMPORTANCE Ochratoxin A (OTA) can result in various toxicological effects and widely contaminates agro-products and feedstuffs. OTA detoxifications by microbial strains and bio-enzymes are significant to food safety. Although previous studies showed OTA could be transformed through several pathways, the ochratoxin-α pathway is recognized as the most effective one. However, the most currently available enzymes are not efficient enough. Here, a superefficient hydrolase, ADH3, which can completely transform 50 μg/L OTA into ochratoxin-α within 90 s was screened and characterized. The hydrolase ADH3 shows considerable temperature adaptability (0 to 70°C) to exert the hydrolytic function. Findings of this study supplied an efficient OTA detoxifying enzyme and predicted the superefficient degradation mechanism, laying a foundation for future industrial applications.
KEYWORDS: ochratoxin A, detoxifying enzyme, catalytic mechanism, food safety, mycotoxin pollution
INTRODUCTION
Ochratoxin A (OTA) easily contaminates a variety of agro-products, such as cereals, fruits, and cocoa and coffee beans, and consequently causes unqualified and harmful processed foods and feedstuffs (1, 2). As the most seriously controlled mycotoxin, OTA shows teratogenic, potential carcinogenic (group IIB carcinogen), and mutagenic effects, and poses a serious threat to human health (1, 3–6). The development of detoxification methods with high efficiency receives great interest in food and agriculture areas (7–9). As shown in Fig. 1, OTA is mainly degraded (or transformed) through four metabolic pathways including (i) peptidic bond cleavage to produce ochratoxin-α (OTα) and l-β-phenylalanine; (ii) dechlorinating from the isocoumarin ring to produce ochratoxin-B (OTB) and further degraded to ochratoxin β (OTβ); (iii) hydroxylation at isocoumarin ring to produce ochratoxin hydroquinone (OTHQ); and (iv) opening the lactone ring to produce lactone-opened ochratoxin A (OP-OTA) (10, 11). OTα is much less toxic metabolite compared to the parent chemical OTA, followed by OTB and OP-OTA (12, 13), and hereby transformation of OTA to OTα is recognized as one of the most effective pathway for OTA detoxification.
FIG 1.

Metabolic pathways of ochratoxin A degradation.
Among the detoxification methods, biodetoxification using microbial strains and bioenzymes has become the hot spot of the field, due to the serial merits including environmental-friendly, safety and high efficiency (14–19). Some identified hydrolases, such as carboxypeptidase A (CPA) and carboxypeptidase Y (CPY) have been commercially obtained on OTA detoxification, but the efficiencies were relatively low (20–23). As far as we know, the most efficient commercial hydrolase CPA (67 μg/mL protein) takes 1 h to degrade less than 10% of 50 μg/L substrate OTA, showing low efficiency and high costs and emergently needing new hydrolase with high efficiency (23, 24).
In our previous study, a difunctional strain of Stenotrophomonas acidaminiphila CW117 which shows efficiently degradation activities on aflatoxin B1 and OTA was isolated by enrichment method, and aflatoxin degradation was characterized (25). In this study, OTA degradation is further investigated, and OTA hydrolases are screened and focused for its efficient activity.
RESULTS
Ochratoxin A is degraded and transformed into ochratoxin α by strain CW117.
In a previous study, aflatoxin B1 degradation by strain CW117 was characterized (25), but the difunctional strain equally degrades OTA efficiently. As illustrated in Fig. 2A, the strain CW117 completely degraded 50 μg/L OTA within 60 h. Further investigation found that cells suspension and cell lysate in phosphate-buffered saline (PBS, pH 7.2) showed efficient degrading activity on OTA, but the cell-free supernatant from the culture of strain CW117 had no degradation ability (Fig. 2B). At the same cell density, fresh cells completely degraded 50 μg/L OTA in about 6 h while the cell lysate took 12 h (Fig. 2C). The results indicated that the degradation agent from CW117 is intracellular.
FIG 2.

Degradation characteristics of S. acidaminiphila CW117 on OTA. (A) Degradation dynamic of the strain CW117. (B) Degradation assays on the culture supernatant, cells suspension, and cell lysate. (C) Degradation dynamics of cells suspension and cell lysates. (D) HPLC analysis of OTA degradation by cell-suspension of the CW117 (the chromatograms from top to bottom: panel 1, OTA and OTα standards; panel 2, the OTA chromatogram at 0 h; panel 3, the degradation result at 3 h; panel 4, the degradation result at 6 h. (E) OTA degradation and OTα production by cells suspension and cell lysates.
The degradation process of OTA by strain CW117 and the degradation product were monitored by high-performance liquid chromatography (HPLC) analysis. The treatment of OTA by cell suspension or cell lysate produced a new chromatographic peak with the same retention time of OTα (Fig. 2D). With the incubation time extension, the chromatographic peak of OTA was decreased gradually, which was accompanied with the increase of the new product. After the incubation with cell suspension of strain CW117 for 6 h, OTA peak was completely disappeared. By liquid chromatography-tandem mass spectrometry (MS/MS), the new product derived from OTA degradation by strain CW117 produced [M+H+] at m/z 257 as precursor ion in MS spectrum, and product ions at m/z 167 and 210.9 in MS/MS spectrum, which are consistent with OTα mass spectrometry (Fig. S1) (10). The degradation dynamics analysis showed that at each time point of degradation process, the molar equivalent of OTα production was equal to the OTA degradation, indicating that the degraded OTA was completely transformed into OTα (Fig. 1E). The product OTα was not decreased, indicating that OTα cannot be degraded further by the strain.
Effects of environmental factors (or as enzymatic characterization) on the ability of the strain to degrade OTA were investigated (Fig. 3A to D). The optimal temperature for OTA degradation was 20°C to 40°C. The function to degrade OTA was destroyed and decreased to 5% of original activity by the temperature up to 60°C. However, low temperature showed a little effect on the ability to degrade OTA. Even the incubation temperature down to 0°C, the function was decreased only by 17%. The optimal pH for the OTA degradation of strain CW117 was 6 to 8, while pH 5 or 10 significantly inhibited the function. Cu2+ or Zn2+ ions or 1% SDS significantly destroyed the degrading ability of strain CW117 on OTA. Other factors including Mg2+, Ca2+, Fe3+, metal-chelators (EDTA and EGTA), and proteinase K showed moderate effects to inhibit the degrading function. The enzymatic characterization indicated that the OTA degradation agent should be allocated to active proteins.
FIG 3.
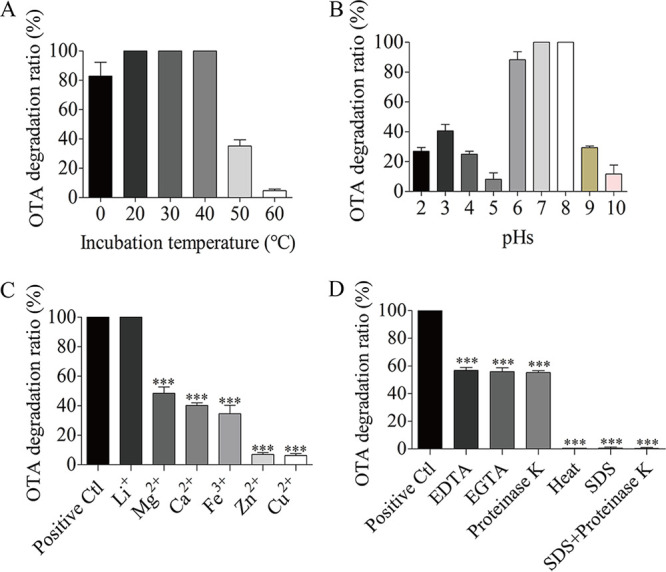
Enzymatic characterization on OTA degradation of S. acidaminiphila CW117. (A) The temperature evaluation and optimal temperature. (B) The pH evaluation and optimal pH. (C) The metal ions effect on degradation activity. (D) The effects of metal-chelator and protein denaturant on degradation activity.
Identification of superefficient OTA detoxification enzyme ADH3 from strain CW117.
The results above indicated that the degradation agent of CW117 as intracellular enzyme(s) to hydrolyze the amide bond of the chemical and produce non-toxic chemical OTα (10, 12, 26). Degradation of OTA to OTα is attributed to amide bond cleavage in OTA structure (10). A complete genome analysis of CW117 was performed to prospect potential candidates including peptidase, amidase, amidohydrolase and carboxypeptidase that possible to hydrolyze amido bond, and thus to degrade OTA. By the CW117 genome (GenBank accession no. CP062156.1) analyses, 53 hydrolase genes (Table S4) were screened and they are overexpressed in Escherichia coli for testing OTA degradation. As a result, only hydrolase of ADH3 (Table S4, H7691_12935) showed efficient OTA degradation activity, and thus selected for further analysis. Based on the result of multiple sequences alignments (Fig. S5, Fig. 4A) and OTA degradation tests, ADH3 which encoded by the gene adh3 was identified as the amido hydrolase in strain CW117 for OTA degradation. ADH3 was characterized as amidohydrolase and composed of 427 amino acid residues with a predicted molecular weight of 45.6 kDa and isoelectronic point of 6.9. The recombinant protein rADH3 (Fig. S6) showed strong ability to degrade OTA. With the concentration of 1.2 μg/mL protein, rADH3 completely degraded OTA of 0.1 μM in 90 s. The degraded product was OTα as produced by strain CW117 (the MS/MS spectrum was identical to Fig. S1).
FIG 4.

Characterization of detoxification enzyme ADH3 and mainstream commercial enzyme CPA on OTA degradation. (A) The amino acid sequence of ADH3 (the red and amplified residues are the active sites). (B) The purified recombinant ADH3 overexpressed by E. coli BL21. (C) The effects of metal-chelator and protein denaturant on degradation activity. (D) The metal ion effect on degradation activity. (E) The temperature evaluation and optimal temperature. (F) The pH evaluation and optimal pH.
As reported above in cell suspension or cell lysate prepared from strain CW117, rADH3 was susceptible for some environmental factors including protein denaturants (1% SDS) and proteinase K (Fig. 4B to F). However, ADH3 showed higher temperature adaptability to exert the degradation function than its host strain CW117 and the commercial detoxification enzyme CPA, it showed the considerable activity even the temperature down to 0°C or up to 70°C (Fig. 4E). Several previously identified OTA detoxification enzymes including OTase, CP, PJ_1540, and AfOTase (24, 26–28) were overexpressed in E. coli (Table 1), and the recombinant proteins were purified in this study to compare the abilities of OTA detoxification with ADH3 (Fig. 5, Fig. 6). The kinetic study showed that Km value of rADH3 was 0.000039 mM, and the Kcat/Km value was 303937.85 s−1.mM−1, while the Kcat/Km values of other OTA detoxification enzymes were from 0.55 to 5363.25 s−1.mM−1 (Table 2). Compared with the mainstream commercial detoxification enzyme CPA identified from bovine pancreas with a Kcat/Km value of 8.7561 s−1.mM−1, the current rADH3 increased the hydrolytic efficiency of OTA by almost 35,000 times, showing an extremely high detoxification ability.
TABLE 1.
OTA detoxification enzymes identified from other microbial strains with known gene sequences
| Detoxification enzyme | Expression primersa | GenBank accession no. or locus tag (reference) |
|---|---|---|
| OTase | F:CCGGAATTCATGGTCCGCCGAATTGC R:CCGCTCGAGCTACAGAAAAGGATTACGTGC |
KJ854920.1 (4) |
| CP | F:CCGGAATTCATGAACATCACGAAATGGAAAC R:CCGCTCGAGTTAAAACCAGCCTGTTACCG |
KP161493.1 (3) |
| PJ_1540 | F:CCGGAATTCATGACACACAAAAGCGCTC R:CCGCTCGAGTTAGAACAAGTTGCTAAAGAAC |
PJ15_1540 (5) |
| AfOTase | F:CCGGAATTCCAAGCCAGTAACCCCAT R:CCGCTCGAGCTACGGTTTTTTGTGATCC |
BVZ28_02750 (6) |
The bold and underlined sequences in primers indicate the restriction sites for endonucleases EcoRI and XhoI in forward (F) and reverse (R) primers, respectively.
FIG 5.
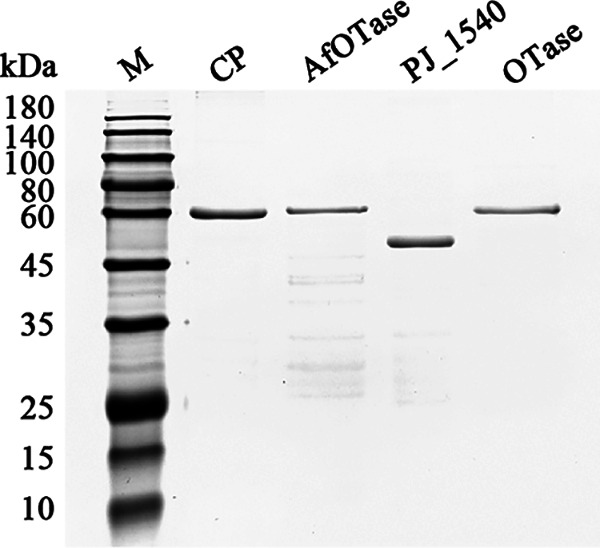
SDS-PAGE analysis of heterologous expressed rOTase, rCP, rPJ_1540, and rAfOTase by E. coli. Recombinant protein AfOTase was expressed by vector pGEX-4T-1 and E. coli BL21, recombinant proteins CP and OTase were expressed by vector pET32a and E. coli BL21(DE3), and recombinant protein PJ_1540 was expressed by vector pET28a and E. coli BL21(DE3). His-tag for pET32a is 15 kDa, His-tag for pET28a is 4 kDa, and GST-tag for pGEX-4T-1 is 26 kDa. The recombinant proteins with His-tag was purified by nickel-nitrilotriacetic acid agarose resin (Ni–NTA, Qiagen, CA, USA), and recombinant protein with GST-tag purified by GSTrap FF (GE Healthcare, Piscataway, NJ, USA) according to manufacturer's instructions.
FIG 6.
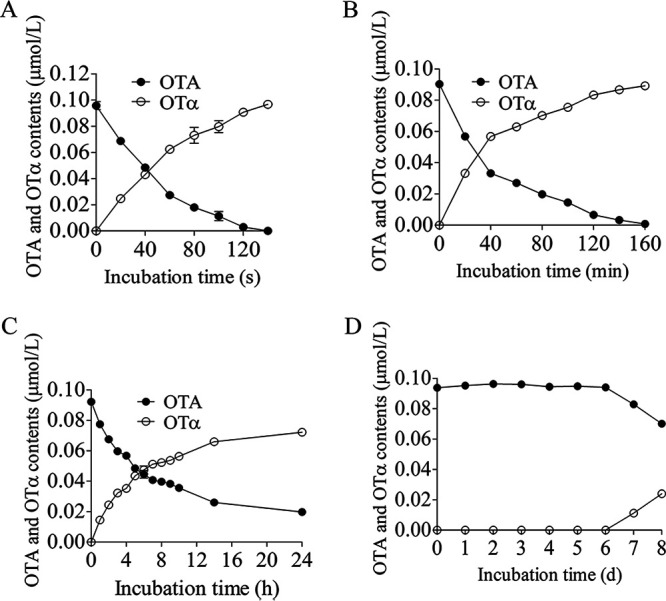
The dynamics comparison of OTA degradation and OTα production by detoxification enzymes ADH3, OTase, and commercial enzymes. (A) The dynamics of OTA degradation and OTα production by 0.8 μg/mL rADH3 protein. (B) The dynamics of OTA degradation and OTα production by 15 μg/mL OTase protein. (C) The dynamics of OTA degradation and OTα production by 15 μg/mL CPA (carboxypeptidase A) protein (D). The dynamics of OTA degradation and OTα production by 15 μg/mL CPY (carboxypeptidase Y) protein.
TABLE 2.
Steady-state kinetic constants of the OTA detoxification enzymes with known polypeptide sequencea
| Enzyme | Identity with ADH3 (E-value) |
Vmax (mmol.min−1.mg−1) |
Km (mM) | Kcat (s−1) |
Kcat/Km (s−1.mM−1) |
|---|---|---|---|---|---|
| ADH3 | - | 173.00 | 0.000039 | 11.97 | 303937.85 |
| CPA | - | 0.1118 | 0.00734 | 0.06428 | 8.7561 |
| CPY | - | 0.00006 | 0.02037 | 0.00006 | 0.00302 |
| OTase | 32.7% (1e-47) | 1.034 | 0.00082 | 1.19 | 1444.71 |
| CP | 44.4% (0.24) | 0.00074 | 0.00055 | 0.00079 | 1.43 |
| PJ_1540 | 28.6% (1.4) | 0.00068 | 0.00091 | 0.00050 | 0.55 |
| AfOTase | 44.4% (0.38) | 11.19 | 0.0025 | 13.24 | 5363.25 |
The detoxification enzymes ADH3, OTase, CP, PJ_1540, and AfOTase were expressed and purified in our laboratory. The commercial detoxification enzyme CPA (Catalogue no. C9268) and CPY (Catalogue no. C3888) were obtained from Sigma–Aldrich (Shanghai, China). The ADH3 is originally from S. acidaminiphila; CPA is originally from bovine pancreas; CPY is originally from yeast; OTase is originally from A. niger (24); CP, is originally from B. amyloliquefaciens (27, 54); PJ_1540 is originally from Acinetobacter sp. (28); AfOTase is originally from A. faecalis (26).
ADH3’s unique structure contributes to its superefficient OTA detoxification activity.
Based on the homology model of ADH3 (Fig. 7A), we selected 10 candidate amino acid residues located at the catalytic active center of ADH3 for point mutation analysis. Those ADH3 mutants were recombinantly expressed in E. coli BL21 to compare OTA degradation abilities. Activity validation results suggested that four residues (83H, 85H, 271H, and 344D) on ADH3, which form quaternary catalytic center, were important for OTA degradation (Fig. 7B). Point mutations on those four residues resulted in complete function loss (Fig. 7A and B). As listed in Table 2, the OTA detoxification activity of ADH3 is ∼210 and ∼56 times stronger than that of OTase and AfOTase, respectively. Solvent accessibility (SAS) assay revealed that the catalytic center on ADH3 has wider hydrophobic areas and larger open areas than that on OTase, and thus facilitates substrate OTA to reach catalytic center (Fig. 7C). Compared with ADH3, the catalytic center of OTase was located deep in the pore area, and it sank ∼5Å. Furthermore, we analyzed the root mean squared deviation (RMSD) and radius of gyration (Rg) of quaternary catalytic sites in those three OTA detoxification enzymes using molecular dynamics (MD) simulation. Among them, ADH3 quaternary catalytic site has the highest RMSD (Fig. 7D) and Rg (Fig. 7E), suggesting the most actively catalytic site.
FIG 7.

Comparison of the catalytic center structures of different OTA degrading enzymes. (A, B) Activity analysis linked point mutation experiments demonstrated that four amino acid residues (83H, 85H, 271H, and 344D) that constitute the quaternary catalytic center are critical to OTA degradation activity of ADH3. (C) Solvent accessibility (SAS) results indicated that the catalytic site of ADH3 was more hydrophobic than that in Otase. (D, E) Molecular dynamic analysis of the four residues of quaternary catalytic site of AHD3, possessed the highest root mean squared deviation (RMSD, D) and radius of gyration (Rg, E).
DISCUSSION
In this study, OTA degradation activity of S. acidaminiphila CW117 to convert OTA into OTα through peptidic bond cleavage was validated and characterized. The key factor responsible for OTA detoxification in the strain of CW117 was identified and characterized as an amidohydrolase ADH3, which showed superefficient OTA-transformation ability. At time of this submission, only three types of OTA hydrolases with known gene (or protein) sequences were published, there are carboxypeptidase (i.e., CPA, CPY, CP, PJ_1540), amidase (i.e., OTase), and N-acyl-L-amino acid amidohydrolase (AfOTase) (21–24, 26, 28). Sequence alignment indicates that ADH3 was the OTA hydrolase of superfamily amidohydrolase, but the catalytic efficiency is much efficient than AfOTase, and the two hydrolases shows only 10.99% amino acid sequences similarity from each other, which indicate that ADH3 should be a novel amidohydrolase (26). Compared with the first commercial OTA detoxifying enzyme CPA, which takes several hours to degrade 50 μg/L OTA on the concentration of 67 μg/mL protein (23, 24), ADH3 takes less than 90 s with only 1.2 μg/mL protein, suggesting much lower costs for industry application (Fig. S6E).
ADH3’s function to degrade OTA was compared with the most efficient OTA detoxification enzyme OTase identified from Aspergillus niger (24), the first commercial detoxification enzyme CPA (21–23), and CPY (16, 24). As illustrated in Fig. 6, 0.8 μg/mL ADH3 took 2.3 min to degrade 0.1 μM OTA (Fig. 6A), 15 μg/mL OTase took 160 min to degrade 0.09 μM OTA (Fig. 6B), 15 μg/mL CPA took 24 h to degrade only 78.5% of 0.09 μM OTA (Fig. 6C), and 15 μg/mL CPY needed 8 days to degrade only 25.3% of 0.093 μM (Fig. 6D). In the study of Dobritzsch et al., the apparent degradation rates of OTase and CPA were compared (24), but other OTA hydrolases did not compare directly. Here, we further compared apparent degradation rate of ADH3 with OTase and two other commercial hydrolases, and found ADH3 showed the distinguished OTA hydrolytic activity. In addition, the results of apparent degradation rates determined in different laboratories might be variable due to different experimental conditions (e.g., buffer solution, pH, temperature), and the kinetic constants are generally recommended to enzymatic characterization. Among the identified OTA hydrolases, CPA was only hydrolase characterized for its kinetic constants (i.e., Kcat, Km), but other hydrolases were not tested (23). After we determined the seven identified OTA hydrolases (ADH3, CP, CPA, CPY, OTase, AfOTase, PJ_1540), the results of kinetic study were consistent to apparent degradation rates which equally suggested that ADH3 was the distinguished OTA hydrolases (Table 2). For example, the Kcat/Km value of rADH3 was 56.7, 210, and 35,000 times higher than that of rAfOTase, rOTase, and CPA, respectively (Table 2). All the data indicate that the activity of ADH3 was much higher than that of other enzymes.
Based on the protein structures and catalytic mechanism previous, the ADH3 structure was modeled from known crystal structures 2QS8 (29), 6SLF (30), and 4C5Y (24), and the superefficient catalytic mechanism was predicted. The quaternary catalytic site formed by the residues (83H, 85H, 271H, and 344D) is key to ADH3 activity as revealed by point-mutagenesis (Fig. 7A). The conservation of catalytic residues suggests that substrate hydrolysis follows a mechanism identical to that proposed for other OTA hydrolysis enzymes (24, 31, 32). Cleavage might be facilitated by the polarization of the carbonyl bond by coordination to the three His residues (83H, 85H, and 271H), which also stabilize the developing oxyanion. Protonation of the nascent amino group by 344D leads to cleavage of the carbon-nitrogen bond (Fig. 7A and B). A structure-based assay revealed that ADH3 has a preference for hydrophobic residues to form a larger hydrophobic area at the cavity of the catalytic sites and thus gives the substrate OTA easier access to catalytic sites (Fig. 7C). Twenty-nanosecond MD simulation revealed that ADH3 catalytic site has high RMSD and Rg (Fig. 7D and E). The active thermodynamic properties of ADH3 catalytic sites likely account for its superefficient catalytic efficiency. In addition, the catalytic center of most amidohydrolases is the catalytic triad (33, 34), but for ADH3 is catalytic quaternary, which may be another reason for its superefficient catalytic activity. Combined with molecular dynamics analysis of this unique catalytic site, the catalytic site structure of ADH3 might provide a model for future amidohydrolases activity modification.
Conclusions.
In this study, an amidohydrolase ADH3 with a strong ability to transform OTA into nontoxic product ochratoxin-α was discovered. The rADH3 catalytic efficiency (Kcat/Km) was 56.7 to 35,000 times higher than those of previous published detoxifying enzymes rAfOTase, rOTase, and commercial enzyme CPA. The catalytic mechanism prediction suggested that ADH3 forms a larger hydrophobic area than other detoxifying enzymes at the cavity of the catalytic sites, and this structure gives the OTA easier access to catalytic sites. In addition, ADH3 shows considerable temperature adaptability (0 to 70°C) to exert the hydrolytic function. Collectively, detoxifying enzyme ADH3 showed promising potential for industrial applications.
MATERIALS AND METHODS
Ochratoxin A degradation by Stenotrophomonas acidaminiphila CW117.
Fresh culture of strain CW117 (1.0 mL) was inoculated into 100 mL nutrient broth (NB, Difco) containing about 50 μg/L OTA, and agitated at 160 revolutions per minute (rpm) and 37°C. The E. coli DH5α was used as the negative control. During degradation tests, 1.0 mL of the culture was taken at time points of 0, 1st, 3rd, 6th, 9th, 12th, 24th, 36th, 48th, and 60th h, and OTA residues were cleaned by OchraTest™ column (OchraTest™, Vicam, Milford, MA, USA) and analyzed by HPLC following a previously described method (10). During HPLC analysis, OTA was separated by an XBridgeTM C18 column (4.6 × 250 mm, 5 μm; Waters, USA), and detected by the fluorescence detector 2475 (Waters, USA) with excitation wavelength of 340 nm and emission wavelength of 460 nm. The mobile phase consisted of acetonitrile, water, and glacial acetic acid by the volume ratio of 48:51:10 and the flow rate was 1.0 mL/min, with the injection volume of 10 μL.
The degradation agents (i.e., culture supernatant, cell lysate, and cell suspension) of strain CW117 were prepared by procedures similar to those described by Cai et al. (25). Strain CW117 was grown in NB at 37°C for 48 h. Bacterial cells were collected by centrifugation at 8,000 × g at 4°C for 5 min; by centrifugation, the pellet and culture supernatant were separated. Then, the culture supernatant was passed through a sterile filter (0.22 μm, Millipore, Billerica, MA, USA) to produce the cell-free culture supernatant. The cell suspension was prepared by resuspending bacterial cells in PBS (pH 7.2) after the cell pellet washed twice with distilled water. The cell lysate was prepared by disintegrating the cell suspension with an ultrasonicator (Qsonica Q700, Newtown, CT, USA), then centrifuged at 10,000 × g for 10 min, and passed through the supernatant with a sterile filter (0.22 μm, Millipore, Billerica, MA, USA). After degradation agents prepared, OTA degradation activity was tested. Briefly, 2.0 mL degradation agent (culture supernatant, cell suspension, or cell lysate) was mixed with the same volume of OTA standard (diluted with PBS, pH 7.2) at a final concentration of about 50 μg/L, and incubated at 37°C for 12 h, and the OTA residues were analyzed by HPLC as above. Degradation dynamics of cell suspension (i.e., cells) or cell lysate was determined by 10.0 mL degradation agent mixed with 10 mL of OTA standard as above, and the OTA residues were analyzed at time points 0, 1st, 3rd, 6th, 9th, 12th, and 24th h. The sterile NB was selected as the control for degradation tests.
Identification and quantification of degradation products.
During the sample analysis on OTA residues in degradation tests, one degradation product that affined to OchraTest column (Vicam, Milford, MA, USA) was observed and collected by the immunoaffinity (i.e., OchraTest) column according to the manufacturer’s instructions. The degradation product was further analyzed with OTA and OTα standards by HPLC and LC-MS/MS according to the method of Wei et al. (10). The dynamics of OTA degradation and OTα production by cell suspension (i.e., cells) and cell lysate were performed in 20 mL degradation mixture as above, and the degradation samples were collected at 0, 1st, 3rd, 6th, 9th, and 12th h for OTA and OTα determination by HPLC. The sterile NB was selected as the control for the degradation tests.
OTA degradation characterization on S. acidaminiphila CW117.
Unless otherwise indicated, OTA solution in the following tests was prepared by diluting OTA standard with PBS (pH 7.2). The 1.0 mL cell suspension (PBS, pH 7.2) was mixed with the same volume of OTA solution (final concentration 40 μg/L), and incubated at 0, 20, 30, 40, 50, and 60°C for 24 h, respectively, and then OTA residues were analyzed. For pH evaluation, the collected cell pellet were resuspended in buffers with different pH values (pH 2.0 to 5.0, glycine-HCl buffer; pH 6.0 to 7.0, phosphate buffer; pH 8.0 to 9.0, Tris-HCl buffer; pH 10.0 to 12.0, glycine-NaOH buffer). After that, 1.0 mL prepared cell suspension was mixed with 1.0 mL OTA solution that prepared by the same pH buffer (final concentration 40 μg/L), and incubated at 37°C for 24 h, then the OTA residues were analyzed. For metal ions test, the collected cell pellet was suspended in PBS (pH 7.2) containing 0.02 mol/L each metal ion. The 1.0 mL prepared cells suspension mixed with the same volume of OTA solution (final concentration 40 μg/L) and incubated at 37°C for 6 h, and the OTA residues were analyzed. For metal-chelator and protein denaturant tests, the collected cell pellet was resuspended in PBS (pH 7.2) which contained 0.2 mol/L metal-chelator (EDTA or EGTA), 1.0 mg/mL proteinase K, 1% SDS, or 1.0 mg/mL proteinase K plus 1% SDS, and the prepared cell suspensions were incubated at 37°C for 6 h before the OTA degradation tests. After pretreatment, 1.0 mL cell suspension with different treatments was mixed with the same volume of OTA solution (final concentration 40 μg/L) and incubated at 37°C for 12 h, and then the OTA residues were analyzed. The PBS with corresponding metal-chelator or denaturant was selected as negative control and untreated bacterial cells were selected as positive control.
Complete genome analysis.
S. acidaminiphila CW117 was cultivated in NB with 160 rpm shaking for 48 h. Genomic DNA was extracted by using the DNA isolation kit (Qiagen, Shenzhen, China) and purified by phenol:chloroform:isoamyl alcohol (25:24:1, vol/vol/vol) extraction. Genome sequencing was performed by Guangdong MAGIGENE Biotechnology Co., Ltd (Guangzhou, China) using the PacBio Sequel II and Novaseq 6000 sequencer. The reads from PacBio Sequel were assembled using SMRT Link v5.1.0, and the reads contained data from Novaseq and PacBio Sequel were assembled using Unicycle v0.4.8. Putative genes were identified using Glimmer 3.02 (35) or Prodigal (36). Noncoding RNAs of tRNA and rRNA were identified using tRNAscan-SE (version 1.3.1), rRNAmmer (version 1.2), and the sRNA was identified using Rfam database and screened by cmsearch. Interspersed repeats and tandem repeats were analyzed by RepeatMasker (version open-4.0.5) and Tandem repeats finder (version 4.07b). Prophage was identified by PHAST, and Genomic Islands (GIs) were predicted by IslandPath-DIOMB. Annotation was performed with BLAST search against databases (37), including the National Center for Biotechnology Information (NCBI) nonredundant proteins (NR) (38), clusters of orthologous groups of proteins (COG) (39), the Kyoto Encyclopedia of Genes and Genomes (KEGG) (40), Gene Ontology (GO) (41), Swiss-prot (42), Pfam (43), Carbohydrate-Active Enzymes Database (CAZy) (44), pathogen host interactions (PHI) (45), virulence factors of pathogenic bacteria (VFDB) (46), and Antibiotic Resistance Genes Database (ARDB) (47). After gene annotation, amido bond hydrolases, which are considered as the possible OTA detoxification genes, were screened for following protein expression and OTA degradation assay in vitro.
Gene cloning, protein expression, and purification.
The candidate genes were amplified by using CW117 genomic DNA and each gene primer pair (Text S1, Table S4) with Primerstar max DNA polymerase (TaKaRa, Dalian, China). The purified PCR product (e.g., adh3 fragment) and expression plasmid pGEX-4T-1 were digested by BamHI (or other corresponding endonuclease) and XhoI (or other corresponding endonuclease) at 37°C overnight, respectively. The digest mixture contained 1.0 μL each endonuclease (e.g., XhoI and BamHI), 1 μg of purified PCR fragment (or 1 μg pGEX-4T-1), and 5 μL of 10× H buffer (TaKaRa, Dalian, China), and supplemented distilled water up to 50 μL. The purified digested fragment and digested expression plasmid were ligated by solution I DNA ligase for 1 h at 16°C. The recombinant plasmid (e.g., pGEX-4T-1/adh3) was constructed by ligation solution consisting of 2 μL digested pGEX-4T-1, 4 μL digested PCR fragment, and 6 μL ligase. Thereafter, recombinant plasmid (5 μL) was transformed to E. coli Trans1-T1 and identified by PCR sequencing. The positive clone was enriched by Luria-Bertani (LB) broth contained 50 μg/mL ampicillin (AmpR), and the recombinant plasmid was extracted by Plasmid MiniPrep Kit (Axygen, NY, USA) according to manufacturer's instructions. The recombinant plasmid (e.g., pGEX-4T-1/adh3) was transformed to E. coli BL21 by heated shock and the transformant was incubated in AmpR LB broth at appropriate temperature (from 16°C to 37°C for different genes) with agitation of 160 rpm. When the culture optical density (OD600) reached about 0.6, 0.1 mM Isopropyl β-D-1-thiogalactopyranoside (IPTG) was added for induction, and followed by additional 4 h to 6 h incubation (48).
Additionally, the genes of OTA detoxification enzymes OTase, CP, PJ_1540, and AfOTase identified from other microbial strains with known polypeptide sequences were commercially synthesized by Sangon Biotech (Shanghai, China). Protein expressions of the synthesized genes were performed using the vector pGEX-4T-1 (for gene afotase), pET32a (for genes cp and otase), and pET28a (for gene pj_1540), the restriction endonucleases EcoRI and XhoI, and the primer pairs as Table 1, under previously described conditions (24, 26–28).
After protein expression, bacterial cells were disintegrated by an ultrasonicator and centrifuged at 10,000 × g for 10 min, and the crude enzyme for the expressed gene was produced from the supernatant by passing through a sterile filter (0.22 μm, Millipore, Billerica, MA, USA). OTA degradation activity of each recombinant protein was firstly evaluated by incubating the crude enzyme with OTA solution (PBS, pH 7.2), and the crude enzyme with OTA degradation activity was selected for further purification. The active recombinant proteins (i.e., ADH3, OTase, CP, PJ_1540, and AfOTase) were purified by affinity chromatography with GSTrap FF (GE Healthcare, Piscataway, NJ, USA) or nickel-nitrilotriacetic acid agarose resin (Ni–NTA, Qiagen, CA) according to manufacturer's instructions. The purity of recombinant protein was examined by SDS-PAGE (Bio-Rad, Hercules, CA, USA), and the protein concentration was evaluated by spectrophotometrically method with bovine serum albumin (BSA) as a standard (49).
OTA degradation characterization of rADH3.
OTA degradation by rADH3 was performed in 1 mL degradation mixture that contained 1.2 μg rADH3 protein and 50 ng OTA in PBS (pH 7.2), at 37°C for 90 s, and then the catalytic reaction was stopped by 3.0 mL acetonitrile. OTA residue was determined by HPLC. As a parallel test, 1.2 μg denatured rADH3 in PBS degradation mixture (pH 7.2) was used as the negative control. Moreover, the OTA degradation product by rADH3 was collected using the same procedures of degradation product from the strain CW117, and the analytical conditions of LC-MS/MS identification were consistent as above. The dynamics of OTA degradation and OTα production by rADH3 were performed in 5 mL PBS degradation mixture (pH 7.2) which contained 4.0 μg rADH3 protein (0.8 μg/mL) and 0.5 nM OTA (i.e., 0.1 μmol/L), and the degradation samples were collected at 0 s to 140 s with the interval of 20 s for OTA and OTα determination by HPLC. Here, 4.0 μg denatured rADH3 was selected for the negative control as parallel tests. Comparatively, the dynamics of OTA degradation and OTα production by rOTase and commercial enzymes of CPA (Catalogue no. C9268) and CPY (Catalogue no. C3888) from Sigma–Aldrich were evaluated by the same procedure, but the protein content for each commercial enzyme or rOTase was 75 μg (i.e., 15 μg/mL) and incubation times were 160 min for rOTase, 24 h for CPA, and 8 days for CPY.
The pH range and optimal pH of rADH3 were evaluated by adding 1.0 μg rADH3 protein to 1 mL OTA solution (final concentration 50 μg/L) that prepared by different pH buffers (pH 2.0 to 5.0, glycine-HCl buffer; pH 6.0 to 7.0, phosphate buffer; pH 8.0 to 9.0, Tris-HCl buffer; pH 10.0 to 11.0, glycine-NaOH buffer), and incubated at 37°C for 2 min. The temperature range and optimal temperature of rADH3 were evaluated by adding 1.0 μg rADH3 protein to 1 mL OTA solution (final concentration 50 μg/L, PBS, pH 7.2), and incubated at 0 to 100°C with 10°C as the interval for 2 min. In SDS and proteinase tests, the rADH3 was firstly pretreated by final concentration of 1% SDS, 1 mg/mL proteinase K, or 1% SDS plus 1 mg/mL proteinase K, at 37°C for 6 h, respectively. After that, the degradation activity of the pretreated rADH3 was determined as above at 37°C for 2 min. The PBS with corresponding denaturant or untreated rADH3 was selected as negative and positive control. In metal-chelator tests, ADH3 was first pretreated by 0.1 mol/L EDTA (EDTA) or ethylene glycol-bis(beta-aminoethyl ether)-N-tetraacetic acid (EGTA) at 37°C for 6 h. After that, the degradation activity of the pretreated rADH3 was determined as above at 37°C for 2 min. The PBS with corresponding metal-chelator or untreated rADH3 was selected as negative and positive control. The metal ions effect on ADH3 was evaluated by adding 1.0 μg rADH3 protein to 1.0 mL OTA solution (final concentration 50 μg/L) that contained 0.01 mol/L metal ion (i.e., Li+, Ca2+, Mg2+, Fe2+, Zn2+, or Cu2+), and the degradation tests were performed at 37°C for 2 min. The OTA degradation mixture without any metal ion was selected the control. The characterization on mainstream commercial enzyme CPA was evaluated by the same procedure, but the protein content for CPA was 15 μg/mL and incubation time was 48 h.
Kinetic constants determination.
During the kinetic constants determination, enzymatic reactions were carried out under the optimal conditions of 1 min for ADH3, 20 min for OTase and AfOTase, 1 h for CPA, 36 h for CP, 80 h for PJ_1540, and 136 h for CPY at appropriate substrate concentrations (i.e., 30-to 90 μg/L OTA), respectively. The degradation product OTα was cleaned using the OchraTest immunoaffinity column (Vicam, Milford, MA, USA) and determined by HPLC following the method as above. The reaction rate versus the substrate concentration was plotted, then the kinetic constants of Kcat, Km (including Kcat/Km), and Vmax were calculated as Stander et al. by a nonlinear regression of the Michaelis-Menten equation with GraphPad PRISM version 5.0 (GraphPad Software, La Jolla, CA) (23).
Enzymes modeling and MD simulation.
The homology models of ADH3, AfOTase, and OTase were modeled from known crystal structures 2QS8 (29), 6SLF (30), and 4C5Y (24), respectively. A short-time (10 ps) MD simulation was performed to obtain the optimized structure model for SAS assay. MD simulation of enzymes was performed to evaluate the stability and conformational changes of these enzymes. All MD simulations were performed using Amber99SB-ILDN force field (50) using GROMACS 5.1 package (51), and running on high-performance Linux system (National Supercomputing Center in Shenzhen). During MD simulations, all the systems were solvated using TIP3P (52) water model in a periodic box, followed by addition Na+ or Cl– ions to neutralize the systems. Before MD simulations, energy minimization, NVT (canonical ensemble), and NPT (isothermal-isobaric ensemble) equilibration were performed. Finally, MD simulations were run for 20 ns time scale under constant temperature (300 K) and pressure (1 atm). The catalytic sites predicted from MD simulations were substituted with glycine using the point mutation method (53), respectively. Each gene mutant was expressed using the vector pGEX-4T-1 and E. coli BL21, and OTA degradation activity of mutated protein was determined as ADH3.
ACKNOWLEDGMENT
This study was supported by the National Natural Science Foundation of China (32172319, 31671949), National Key R&D Program of China (2017YFC1600902), and Anhui Provincial Key Research and Development Plan (202004h07020007).
We declare no conflict of interest.
Footnotes
Supplemental material is available online only.
Contributor Information
Ren Lai, Email: rlai@mail.kiz.ac.cn.
Yu Zhou, Email: microbes@ahau.edu.cn.
Hideaki Nojiri, University of Tokyo.
REFERENCES
- 1.Stoev SD. 2013. Food safety and increasing hazard of mycotoxin occurrence in foods and feeds. Crit Rev Food Sci Nutr 53:887–901. 10.1080/10408398.2011.571800. [DOI] [PubMed] [Google Scholar]
- 2.Zhang J, Zhu L, Chen H, Li M, Zhu X, Gao Q, Wang D, Zhang Y. 2016. A polyketide synthase encoded by the gene An15g07920 is involved in the biosynthesis of Ochratoxin A in Aspergillus niger. J Agric Food Chem 64:9680–9688. 10.1021/acs.jafc.6b03907. [DOI] [PubMed] [Google Scholar]
- 3.Hou L, Gan F, Zhou X, Zhou Y, Qian G, Liu Z, Huang K. 2018. Immunotoxicity of ochratoxin A and aflatoxin B1 in combination is associated with the nuclear factor kappa B signaling pathway in 3D4/21cells. Chemosphere 199:718–727. 10.1016/j.chemosphere.2018.02.009. [DOI] [PubMed] [Google Scholar]
- 4.Marin-Kuan M, Nestler S, Verguet C, Bezencon C, Piguet D, Mansourian R, Holzwarth J, Grigorov M, Delatour T, Mantle P, Cavin C, Schilter B. 2006. A toxicogenomics approach to identify new plausible epigenetic mechanisms of ochratoxin a carcinogenicity in rat. Toxicol Sci 89:120–134. 10.1093/toxsci/kfj017. [DOI] [PubMed] [Google Scholar]
- 5.Ostry V, Malir F, Toman J, Grosse Y. 2017. Mycotoxins as human carcinogens—the IARC Monographs classification. Mycotoxin Res 33:65–73. 10.1007/s12550-016-0265-7. [DOI] [PubMed] [Google Scholar]
- 6.Zepnik H, Pahler A, Schauer U, Dekant W. 2001. Ochratoxin A-induced tumor formation: is there a role of reactive ochratoxin A metabolites? Toxicol Sci 59:59–67. 10.1093/toxsci/59.1.59. [DOI] [PubMed] [Google Scholar]
- 7.Loi M, Fanelli F, Liuzzi VC, Logrieco AF, Mule G. 2017. Mycotoxin biotransformation by native and commercial enzymes: present and future perspectives. Toxins (Basel) 9:111. 10.3390/toxins9040111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Taylor MC, Jackson CJ, Tattersall DB, French N, Peat TS, Newman J, Briggs LJ, Lapalikar GV, Campbell PM, Scott C, Russell RJ, Oakeshott JG. 2010. Identification and characterization of two families of F420 H2-dependent reductases from Mycobacteria that catalyse aflatoxin degradation. Mol Microbiol 78:561–575. 10.1111/j.1365-2958.2010.07356.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li J, Huang J, Jin Y, Wu C, Shen D, Zhang S, Zhou R. 2018. Mechanism and kinetics of degrading aflatoxin B1 by salt tolerant Candida versatilis CGMCC 3790. J Hazard Mater 359:382–387. 10.1016/j.jhazmat.2018.05.053. [DOI] [PubMed] [Google Scholar]
- 10.Wei W, Qian Y, Wu Y, Chen Y, Peng C, Luo M, Xu J, Zhou Y. 2020. Detoxification of ochratoxin A by Lysobacter sp. CW239 and characteristics of a novel degrading gene carboxypeptidase cp4. Environ Pollut 258:113677. 10.1016/j.envpol.2019.113677. [DOI] [PubMed] [Google Scholar]
- 11.Wu Q, Dohnal V, Huang L, Kuca K, Wang X, Chen G, Yuan Z. 2011. Metabolic pathways of ochratoxin A. Curr Drug Metab 12:1–10. 10.2174/138920011794520026. [DOI] [PubMed] [Google Scholar]
- 12.Haq M, Gonzalez N, Mintz K, Jaja-Chimedza A, De Jesus CL, Lydon C, Welch A, Berry JP. 2016. Teratogenicity of ochratoxin A and the degradation product, ochratoxin alpha, in the zebrafish (Danio rerio) embryo model of vertebrate development. Toxins (Basel)) 8:40. 10.3390/toxins8020040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heussner AH, Bingle LE. 2015. Comparative Ochratoxin toxicity: a review of the available data. Toxins (Basel)) 7:4253–4282. 10.3390/toxins7104253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen W, Li C, Zhang B, Zhou Z, Shen Y, Liao X, Yang J, Wang Y, Li X, Li Y, Shen XL. 2018. Advances in biodetoxification of Ochratoxin A: a review of the past five decades. Front Microbiol 9:1386. 10.3389/fmicb.2018.01386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abrunhosa L, Ines A, Rodrigues AI, Guimaraes A, Pereira VL, Parpot P, Mendes-Faia A, Venancio A. 2014. Biodegradation of ochratoxin A by Pediococcus parvulus isolated from Douro wines. Int J Food Microbiol 188:45–52. 10.1016/j.ijfoodmicro.2014.07.019. [DOI] [PubMed] [Google Scholar]
- 16.Abrunhosa L, Paterson RR, Venancio A. 2010. Biodegradation of ochratoxin A for food and feed decontamination. Toxins (Basel) 2:1078–1099. 10.3390/toxins2051078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang JQ, Fang X, Tian X, Chen P, Lin JL, Guo XX, Li JX, Fan Z, Song WM, Chen FY, Ahati R, Wang LJ, Zhao Q, Martin C, Chen XY. 2020. Aromatization of natural products by a specialized detoxification enzyme. Nat Chem Biol 16:250–256. 10.1038/s41589-019-0446-8. [DOI] [PubMed] [Google Scholar]
- 18.Rodriguez H, Reveron I, Doria F, Costantini A, De Las Rivas B, Muňoz R, Garcia-Moruno E. 2011. Degradation of Ochratoxin A by brevibacterium species. J Agric Food Chem 59:10755–10760. 10.1021/jf203061p. [DOI] [PubMed] [Google Scholar]
- 19.Zhang HH, Wang Y, Zhao C, Wang J, Zhang XL. 2017. Biodegradation of ochratoxin A by Alcaligenes faecalis isolated from soil. J Appl Microbiol 123:661–668. 10.1111/jam.13537. [DOI] [PubMed] [Google Scholar]
- 20.Pitout MJ, Nel W. 1969. The inhibitory effect of ochratoxin A on bovine carboxypeptidase A in vitro. Biochem Pharmacol 18:1837–1843. 10.1016/0006-2952(69)90279-2. [DOI] [PubMed] [Google Scholar]
- 21.Stander MA, Bornscheuer UT, Henke E, Steyn PS. 2000. Screening of commercial hydrolases for the degradation of ochratoxin A. J Agric Food Chem 48:5736–5739. 10.1021/jf000413j. [DOI] [PubMed] [Google Scholar]
- 22.Abrunhosa L, Santos L, Venâncio A. 2006. Degradation of ochratoxin A by proteases and by a crude enzyme of Aspergillus niger. Food Biotechnol 20:231–242. 10.1080/08905430600904369. [DOI] [Google Scholar]
- 23.Stander MA, Steyn PS, van Der Westhuizen FH, Payne BE. 2001. A kinetic study into the hydrolysis of the ochratoxins and analogues by carboxypeptidase A. Chem Res Toxicol 14:302–304. 10.1021/tx000221i. [DOI] [PubMed] [Google Scholar]
- 24.Dobritzsch D, Wang H, Schneider G, Yu S. 2014. Structural and functional characterization of ochratoxinase, a novel mycotoxin-degrading enzyme. Biochem J 462:441–452. 10.1042/BJ20140382. [DOI] [PubMed] [Google Scholar]
- 25.Cai M, Qian Y, Chen N, Ling T, Wang J, Jiang H, Wang X, Qi K, Zhou Y. 2020. Detoxification of aflatoxin B1 by Stenotrophomonas sp. CW117 and characterization the thermophilic degradation process. Environ Pollut 261:114178. 10.1016/j.envpol.2020.114178. [DOI] [PubMed] [Google Scholar]
- 26.Zhang H, Zhang Y, Yin T, Wang J, Zhang X. 2019. Heterologous expression and characterization of a novel ochratoxin A degrading enzyme, N-acyl-L-amino acid amidohydrolase, from alcaligenes faecalis. Toxins (Basel) 11:518. 10.3390/toxins11090518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chang X, Wu Z, Wu S, Dai Y, Sun C. 2015. Degradation of ochratoxin A by Bacillus amyloliquefaciens ASAG1. Food Addit Contam Part A Chem Anal Control Expo Risk Assess 32:564–571. 10.1080/19440049.2014.991948. [DOI] [PubMed] [Google Scholar]
- 28.Liuzzi VC, Fanelli F, Tristezza M, Haidukowski M, Picardi E, Manzari C, Lionetti C, Grieco F, Logrieco AF, Thon MR, Pesole G, Mule G. 2016. Transcriptional analysis of Acinetobacter sp. neg1 capable of degrading ochratoxin A. Front Microbiol 7:2162. 10.3389/fmicb.2016.02162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xiang DF, Xu C, Kumaran D, Brown AC, Sauder JM, Burley SK, Swaminathan S, Raushel FM. 2009. Functional annotation of two new carboxypeptidases from the amidohydrolase superfamily of enzymes. Biochemistry 48:4567–4576. 10.1021/bi900453u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Natsch A, Emter R. 2020. The specific biochemistry of human axilla odour formation viewed in an evolutionary context. Philos Trans R Soc Lond B Biol Sci 375:20190269. 10.1098/rstb.2019.0269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Seibert CM, Raushel FM. 2005. Structural and catalytic diversity within the amidohydrolase superfamily. Biochemistry 44:6383–6391. 10.1021/bi047326v. [DOI] [PubMed] [Google Scholar]
- 32.Thoden JB, Phillips GN, Jr, Neal TM, Raushel FM, Holden HM. 2001. Molecular structure of dihydroorotase: a paradigm for catalysis through the use of a binuclear metal center. Biochemistry 40:6989–6997. 10.1021/bi010682i. [DOI] [PubMed] [Google Scholar]
- 33.Weber BW, Kimani SW, Varsani A, Cowan DA, Hunter R, Venter GA, Gumbart JC, Sewell BT. 2013. The mechanism of the amidases: mutating the glutamate adjacent to the catalytic triad inactivates the enzyme due to substrate mispositioning. J Biol Chem 288:28514–28523. 10.1074/jbc.M113.503284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ribeiro AJM, Tyzack JD, Borkakoti N, Holliday GL, Thornton JM. 2020. A global analysis of function and conservation of catalytic residues in enzymes. J Biol Chem 295:314–324. 10.1074/jbc.REV119.006289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Delcher AL, Bratke KA, Powers EC, Salzberg SL. 2007. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 23:673–679. 10.1093/bioinformatics/btm009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, Hauser LJ. 2010. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402. 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pruitt KD, Tatusova T, Maglott DR. 2007. NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res 35:D61–D65. 10.1093/nar/gkl842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tatusov RL, Galperin MY, Natale DA, Koonin EV. 2000. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res 28:33–36. 10.1093/nar/28.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kanehisa M, Goto S, Kawashima S, Okuno Y, Hattori M. 2004. The KEGG resource for deciphering the genome. Nucleic Acids Res 32:D277–D280. 10.1093/nar/gkh063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. 2000. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25:25–29. 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boutet E, Lieberherr D, Tognolli M, Schneider M, Bairoch A. 2007. UniProtKB/Swiss-Prot. Methods Mol Biol 406:89–112. 10.1007/978-1-59745-535-0_4. [DOI] [PubMed] [Google Scholar]
- 43.El-Gebali S, Mistry J, Bateman A, Eddy SR, Luciani A, Potter SC, Qureshi M, Richardson LJ, Salazar GA, Smart A, Sonnhammer ELL, Hirsh L, Paladin L, Piovesan D, Tosatto SCE, Finn RD. 2019. The Pfam protein families database in 2019. Nucleic Acids Res 47:D427–D432. 10.1093/nar/gky995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM, Henrissat B. 2014. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res 42:D490–D495. 10.1093/nar/gkt1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Urban M, Cuzick A, Seager J, Wood V, Rutherford K, Venkatesh SY, De Silva N, Martinez MC, Pedro H, Yates AD, Hassani-Pak K, Hammond-Kosack KE. 2020. PHI-base: the pathogen-host interactions database. Nucleic Acids Res 48:D613–D620. 10.1093/nar/gkz904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu B, Zheng D, Jin Q, Chen L, Yang J. 2019. VFDB 2019: a comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res 47:D687–D692. 10.1093/nar/gky1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu B, Pop M. 2009. ARDB–antibiotic resistance genes database. Nucleic Acids Res 37:D443–D447. 10.1093/nar/gkn656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yoshida S, Hiraga K, Takehana T, Taniguchi I, Yamaji H, Maeda Y, Toyohara K, Miyamoto K, Kimura Y, Oda K. 2016. A bacterium that degrades and assimilates poly(ethylene terephthalate). Science 351:1196–1199. 10.1126/science.aad6359. [DOI] [PubMed] [Google Scholar]
- 49.Pace CN, Vajdos F, Fee L, Grimsley G, Gray T. 1995. How to measure and predict the molar absorption coefficient of a protein. Protein Sci 4:2411–2423. 10.1002/pro.5560041120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lindorff-Larsen K, Piana S, Palmo K, Maragakis P, Klepeis JL, Dror RO, Shaw DE. 2010. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins: Structure, Function, and Bioinformatics 78:1950–1958. 10.1002/prot.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, Lindahl E. 2015. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2:19–25. 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- 52.Mahoney MW, Jorgensen WL. 2000. A five-site model for liquid water and the reproduction of the density anomaly by rigid, nonpolarizable potential functions. J Chem Phys 112:8910–8922. 10.1063/1.481505. [DOI] [Google Scholar]
- 53.Han Y, Li B, Yin TT, Xu C, Ombati R, Luo L, Xia Y, Xu L, Zheng J, Zhang Y, Yang F, Wang GD, Yang S, Lai R. 2018. Molecular mechanism of the tree shrew's insensitivity to spiciness. PLoS Biol 16:e2004921. 10.1371/journal.pbio.2004921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Azam MS, Yu D, Liu N, Wu A. 2019. Degrading ochratoxin A and zearalenone mycotoxins using a multifunctional recombinant enzyme. Toxins (Basel) 11:301. 10.3390/toxins11050301. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Text S1, Tables S1 to S4, Fig. S1 to S6. Download AEM.01964-21-s0001.pdf, PDF file, 1.3 MB (1.3MB, pdf)