

Abstract
The aging population is at a higher risk for age-related diseases and infections. This observation could be due to immunosenescence: the decline in immune efficacy of both the innate and the adaptive immune systems. Age-related immune decline also links to the concept of ‘inflamm-aging,’ whereby aging is accompanied by sterile chronic inflammation. Along with a decline in immune function, aging is accompanied by a widespread of ‘omics’ remodeling. Transcriptional landscape changes linked to key pathways of immune function have been identified across studies, such as macrophages having decreased expression of genes associated to phagocytosis, a major function of macrophages. Therefore, a key mechanism underlying innate immune cell dysfunction during aging may stem from dysregulation of youthful genomic networks. In this review, we discuss both molecular and cellular phenotypes of innate immune cells that contribute to age-related inflammation.
Keywords: inflammaging, macrophages, neutrophil, immunosenescence, inflammation
Introduction
The human population is aging, which has led to the rise in prevalence of many so-called age-related diseases. Not only is the aging population much more susceptible to age-related diseases, they are also more susceptible to infections. For example, elderly individuals are at a higher risk of developing severe COVID-19 or complications from influenza infections [1,2]. This increased chance of infection can be due to the decline of the function of the immune system, a phenomenon called ‘immunosenescence’ [3]. Age-related changes in the function of the immune system are also accompanied by a chronic sterile inflammation, a mechanism dubbed ‘inflamm-aging,’ which is thought to promote age-related disease and functional decline [4]. Inflamm-aging is associated with many different factors, most typically encompassing increases in pro-inflammatory cytokines tumor necrosis factor alpha [TNFa], interleukin 1 beta [IL1b] and interleukin 6 [IL6] [5]. Although these cytokines are involved in normal immune function, persistent levels of pro-inflammatory cytokines can lead to tissue damage, contributing to increased prevalence of chronic diseases (e.g. Alzheimer’s disease (AD), cancer, etc.) [6].
An important source of inflammatory signals in aged organisms is thought to be the accumulation of senescent cells across tissues [5,7]. Indeed, accumulating evidence has shown that senescent cells are characterized by a senescence-associated secretory phenotype [8–10], which includes a panoply of pro-inflammatory cytokines, proteases, growth factors and metabolites [10,11]. The impact of senescent cells on age-related inflammation, and their potential role as a target for pro-longevity therapies (i.e. senolytic drugs) have been extensively reviewed elsewhere [12–15], but remain somewhat uncertain. Aberrant immune activation with age could also stem from increased permeability of the intestinal mucosa (i.e. ‘leaky gut’) [16,17] related to changes to the gut microbiota [18,19], which may directly contribute to increased systemic inflammation. Age-related increase in genomic instability may itself also drive aspects of inflammaging. Indeed, re-activation of LINE-1 transposable elements during aging and in senescent cells has been proposed to drive an interferon response, thus contributing to sterile inflammation [20–22]. In addition, chronic DNA-damage signaling itself, for instance in aged lymphocytes, may also render them more activation-prone through innate receptors even in the absence of infection [23].
Generally, overall immune dysfunction is a hallmark of aging, with functional decline impacting both innate immunity and adaptative immunity [3,24,25]. Age-related deficiencies in hematopoietic stem cells [HSCs], which reside in the bone marrow and differentiate throughout life to give rise to mature cells of the innate and adaptive immune systems, are thought to underlie at least partly dysfunction of mature immune cells [26]. For instance, aged HSCs from mice show large epigenomic and transcriptomic differences compared to youthful HSCs which are likely to adversely impact their differentiation capacity [27,28]. In addition, aged HSCs tend to produce higher numbers of innate cells from the myeloid lineage, a phenomenon called ‘myeloid bias’ [26,28,29]. Interestingly, a recent study using heterochronic bone marrow transplant has shown that at least part of the inflammatory milieu of aged animals directly stems from the activity of bone marrow hematopoietic progenitors [30]. Conversely, HSC function can be directly remodeled by inflammatory signals, such as those observed during normal aging [31]. Downstream of HSCs and of special relevance to ‘inflamm-aging’ are cells of the innate immune system, which constitute the first line of defense against pathogens. Key cell populations of the innate immune system include monocytes, macrophages, neutrophils, natural killer cells and dendritic cells (DCs) [32].
Importantly, a key mechanism underlying innate immune cell dysfunction during aging may stem from dysregulation of youthful genomic networks. Indeed, aging is accompanied by widespread remodeling of transcriptional landscapes across tissues and cell types (reviewed in [33]). In addition, age-related inflammatory signatures at the transcriptional levels have been observed across species and tissues, suggesting that such ‘omic’ remodeling is a conserved aging response [34,35].
In this review, we will focus on how innate immune cells act as key contributors to age-related inflammation (Figure 1). We will discuss both molecular and cellular phenotypes which have been described in the aging innate immune system, and how they could relate to the phenomenon of inflamm-aging and immunosenescence.
Figure 1 .
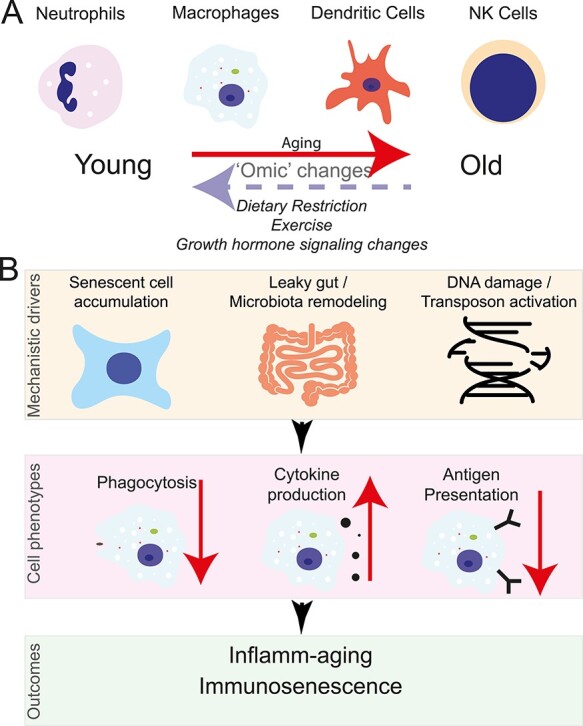
Functional genomics insights into inflamm-aging and immunosenescence through the lens of the innate immune system. (A) Aging results in a widespread of molecular changes to terminally differentiated immune cells. These changes can be partially rescued through different interventions. (B) Possible contributors to age-related changes in macrophage function that will ultimately result in immunosenescence and inflamm-aging.
Macrophages
Macrophages are a central hub or ‘one stop shop’ in the adult innate immune system. Macrophages accomplish a variety of key tasks, such as phagocytosis, cytokine production, antigen presentation and assist in wound healing [36]. Far from being a homogenous cell type, macrophages have various embryonic primordium origins (i.e. fetal liver/yolk sac versus bone marrow stem cells), broadly corresponding to macrophages classified as ‘tissue resident’ (as old as the individual; e.g. microglia, peritoneal macrophages) versus ‘de novo’ (continuously produced throughput life; e.g. monocyte-derived) [37]. These different populations of macrophages show clear epigenomic and transcriptional differences (at least in mouse) [38–41], which are partially shaped by exposure to niche signals [38,39]. Such molecular differences can lead to differential abilities of macrophages subtypes to respond to challenges, or sense pathogen-associated molecular patterns [PAMPs].
Based on the differences between location and the effects the microenvironment has on gene expression, many differentially expressed genes are important for specialized functions [40,41]. Driving differences in gene expression, tissue-resident macrophage populations have different transcription factors [TFs] that specifically regulate their gene expression [38,39,42]. For instance in mice, Gata6 is highly expressed in peritoneal macrophages, Bhlhe40 in large peritoneal macrophages, Spic in splenic macrophages, Sall1 in microglia and PPARg in alveolar macrophages, which may drive both specific basal states and age-related changes [42,43]. Disruption of Gata6 in the myeloid lineage leads to defects in mouse peritoneal macrophage homeostatic proliferation and in inflammatory response [44]. Even though tissue-resident macrophages each have a specific TF that regulates their phenotype, PU.1 is thought to regulate many TFs associated to tissue-resident macrophages [38,39,42]. Intriguingly, decreased levels of PU.1 in microglia associate to delayed onset of Alzheimer’s disease in humans, suggesting that differences in tissue-macrophage phenotypes may underlie differences in aging trajectories [45,46]. Further studies will need to be conducted to understand how these TFs are regulated with aging, and how they may generally influence immunosenescence and inflamm-aging.
Aging transcriptome landscape
Accumulating evidence has revealed that de novo and tissue-resident macrophage populations from both rat and mice maintain their specificities throughout life despite undergoing widespread remodeling of their transcriptional landscapes with aging (Table 1). For example, in a study comparing the impact of organismal aging on 2 populations of tissue-resident macrophages from mice (i.e. adipose versus splenic macrophages), Camell et al. [47] showed that both macrophage types maintain their transcriptional specificities throughout life, although they are both affected by aging. Interestingly, age-related differentially expressed genes were quite different between the two types of macrophages, suggesting that there are macrophage population specific responses to aging [47]. Although each transcriptomic study of aging macrophages from both rat and mice identified clear specificities to each type of macrophage (even showing differences between the same macrophage type across studies), a number of recurrent misregulated functional categories stand out, including upregulation of cytokine pathways and downregulation of phagosome/endosome-related pathways (Table 1). However, variability in the reported age-related remodeling of macrophages may stem from true biological differences (e.g. niche-effects on macrophages, or intrinsic differences linked to embryonic origin). In contrast, it is also possible that these differences stem from technical differences (e.g. microarray versus RNA-seq, bioinformatic pipelines), from differences in key biological covariates (e.g. ethnicity/genetic background, circadian time), or, merely from how young and old age groups are defined (e.g. young mice defined as young as 2 months or as old as 6 months old, old mice defined as young as 12 months or as old as 30 months old). Harmonized studies or meta-analyses taking into account these variables will be key to understand general principles of age-related macrophage transcriptional remodeling.
Table 1.
Age-related changes in the transcriptional landscape of different populations of macrophages
| Population | Species | Strain | Ages | Sex | Upregulated categories (selected) |
Downregulated categories (selected) |
References |
|---|---|---|---|---|---|---|---|
| Aorta macrophages | R. norvegicus | Sprague–Dawley | 5 m versus 27 m | B | M1-proinflammatory macrophage signature | M2-macrophage gene signature | [89] |
| Adipose tissue macrophages | M. musculus | C57BL/6 J | 3 m versus 24 m | M | Cellular ketone Cellular aldehyde Lipid Fatty acid derivative |
[47] | |
| M. musculus | C57BL/6 J | 2 m versus 20 m | M | Phagocytosis C-type lectin receptors |
[127] | ||
| R. norvegicus | Sprague–Dawley | 5 m versus 27 m | B | M1-proinflammatory macrophage signature | M2-macrophage gene signature | [89] | |
| Alveolar macrophages | M. musculus | C57BL/6 | 2–4 m versus 22–24 m | NA | Inflammatory pathways Vascular endothelial growth factor signaling |
Metaphase checkpoint Initiation of mitosis Spindle assembly Chromosome separation |
[56] |
| M. musculus | C57BL/6 J | 3 m versus 23 m | NA | Immune and inflammatory responses Fibrosis |
[128] | ||
| Bone callus macrophages | M. musculus | C57BL/6 J | 3 m versus 24 m | NA | Negative regulation of protein processing Cell chemotaxis Complement receptor signaling pathway |
Antigen processing and presentation Regulation of immune response Cytokine activity Phagosome en0docytosis |
[129] |
| Bone marrow macrophages (in vivo) | M. musculus | C57BL/6 J | 2-4 m versus 20-30 m | M | Innate immune response Inflammatory response Cytokine-cytokine receptor Toll-like receptor NFkB signaling pathways |
[51] | |
| H. sapiens | NA | <50 years versus > 50 years | NA | Regulation of immune system process Defense response |
[51] | ||
| Kidney macrophages | M. musculus | C57BL/6 JN | 1 m versus 3 m versus 18 m versus 21 m versus, 24 m versus 30 m | B | M1-proinflammatory macrophage signature | M2-macrophage gene signature | [130] |
| Microglia | M. musculus | C57BL/6 J | 4 m versus 22 m | F | Immune cell adhesion Chemotaxis Cytokine signaling Anti-microbial effector responses Interferon responses |
Endocytosis/phagocytosis Cytoskeletal reorganization Antigen processing and presentation |
[131] |
| M. musculus | C57BL/6 J | 3 m versus 22 m | M | Immune system process Response to virus Interferon signaling |
[132] | ||
| M. musculus | C57BL/6 J | 3 m versus 12 m versus 18 m versus 24 m | M | Lipid biosynthetic process Regulation of signal transduction Intracellular protein transport Positive regulation of endocytosis Microglial cell activation |
[133] | ||
| M. musculus | C57BL/6 N | 2 m versus 6 m versus 9 m versus 12 m | M | Inflammatory response Innate immune response Cell chemotaxis MHC protein complex Cytokine receptor binding |
[134] | ||
| M. musculus | C57BL/6 JN | 1 m versus 3 m versus 18 m versus 21 m versus, 24 m versus 30 m | B | Major histocompatibility complex (MHC) class I genes Interferon responsive or regulatory genes |
[130] | ||
| Nerve macrophage | M. musculus | C57BL/6 | 3 m versus 15 m | F | Organismal death Organization of cytoskeleton Microtubule dynamics |
Pro-inflammatory cytokines Retinoic acid receptor Activation Estrogen receptor signaling Peroxisome proliferator-activated receptors signaling |
[135] |
| Skeletal muscle macrophages | M. musculus | C57BL/6 J | 4 m versus 20-24 m | NA | Response to stress Response to interferon beta Negative regulation of macromolecule biosynthesis Negative regulation of cell adhesion |
Defense response Response to LPS Cellular homeostasis Regulation of cytokine secretion |
[136] |
| Skin macrophages | R. norvegicus | Sprague–Dawley | 5 m versus 27 m | B | M1-proinflammatory macrophage signature | M2-macrophage gene signature | [89] |
| Thiogallate-induced peritoneal exudate macrophages | M. musculus | C57BL/6 J | 3 m versus 18 m | F | Cholesterol biosynthesis Response to stress Response to stimulus |
[137] | |
F: Female; M: Male; B: both sexes; NA: sex not specified in the article; m: months.
Age-related functional decline
Although there was some variability among studies, pathways related to endosome or phagosome biology were found to be generally downregulated with age across transcriptomic studies. Indeed, macrophages are primarily phagocytes, and multiple studies across species have observed defects in this core process with organismal aging (Table 2). Studies evaluating phagocytic capacities of various populations of macrophages from both rat and mice with aging, including peritoneal macrophages, Kupffer cells, alveolar macrophages, bone marrow macrophages and bone marrow-derived macrophages [BMDMs] (i.e. differentiated in vitro from bone marrow progenitors and monocytes), again show contrasting results, consistent with the notion that different macrophage populations may have specific aging trajectories (Table 2). After an infection or in response to tissue damage, macrophages are required to clear up apoptotic cells, senescent cells and cell debris (through a process called ‘efferocytosis’) [48], as well as neutrophil extracellular traps (NETs) [49]. Intriguingly, two distinct populations of mouse macrophages have been shown to be defective in efferocytosis with aging [50,51]. The impact of decreased efferocytotic capacity occurs in normal tissue homeostasis, but also in the ability to resolve inflammation. For instance, in a peritonitis mouse model, efferocytosis of apoptotic neutrophils by peritoneal macrophages was shown to decrease with age, leading to an improper resolution of inflammation [50]. Importantly, a decreased ability to clear senescent cells through efferocytosis could also indirectly drive increased inflammation due to an increasing burden of cells producing SASP factors [52,53]. Thus, overall age-related loss of phagocytic and efferocytic capacity of macrophages may lead to increased susceptibility to infections and/or defects in tissue homeostasis [54–56], ultimately leading to immune dysfunction and increased inflammation.
Table 2.
Age-related changes to phagocytosis of different populations of macrophages
| Population | Species | Strain | Ages | Sex | Phagocytosis cargo | Age-related change | Reference |
|---|---|---|---|---|---|---|---|
| Alveolar macrophages | R. norvegicus | F-344xBN | 6 m versus 12 m versus 18 m | NA | Opsonized K. pneumoniae 43,816 | increase | [138] |
| M. musculus | C57BL/6 | 2-4 m versus 16 m versus 22–24 m versus 33 m | NA | Alexa Fluor 488 beads | decrease | [56] | |
| Bone marrow-derived macrophages (in vitro differentiation) | M. musculus | C57BL/6 | 2–4 m versus 18–22 m | F | S. pneumoniae | decrease | [18] |
| M. musculus | C57BL/6 | 2–3 m versus 15–20 m | M | Fluorescent particles | no difference | [139] | |
| Bone marrow macrophages (in vivo) | M. musculus | C57BL/6 J | 2–4 m versus 20–30 m | M | Senescent neutrophils | decrease | [51] |
| Kupffer Cells | R. norvegicus | F344 | ~1 m versus 12 m versus 24–25 m | M | Polystyrene/sucrose microsphere | increase | [140] |
| Microglia | M. musculus | C57Bl/6 J | 2–3 m versus 20–22 m | M | Fluoresbrite Yellow Green Carboxylate microsphere (1 μm) | decrease | [141] |
| Peritoneal Macrophages | M. musculus | BALB/c | 3 m versus 15 m | B | Latex beads (1.09 μm) | decrease | [142] |
| M. musculus | BALB/c | 3–4 m versus 20–23 m | B | Latex beads (1.09 μm) | decrease | [143] | |
| M. musculus | BALB/c | 3 m versus 15 m | M | Opsonized Candida albicans Latex beads Latex beads (1.09 μm) |
No change | [144] | |
| M. musculus | C57BL/6 | 2–4 m versus 18–22 m | F | S. pneumoniae | decrease | [18] | |
| M. musculus | C57BL/6 | 2–3 m versus 15–20 m | M | Fluorescent particles | decrease | [139] | |
| M. musculus | BALB/c | 2 m versus 20 m | M | Apoptotic neutrophils | decrease | [50] | |
| M. musculus | CF-1 | 3 m versus 22 m | M | Carbon uptake | decrease | [145] | |
| Thiogallate-induced peritoneal exudate macrophages | M. musculus | B6C3-F1 B6.Gld |
2 m versus 24 m | M | Apoptotic TAMRA/SE-labeled apoptotic Jurkat T cells | decrease | [146] |
F: Female; M: Male; B: both sexes; NA: sex not specified in the article; m: months.
When macrophages are exposed to specific stimuli in vitro, they are thought to adopt specific genomic programs by ‘polarization’ towards so-called M1 or M2 phenotype, which secrete different cytokines/chemokines [57–59]. The picture is more complex in vivo, where macrophage activation phenotypes are more spread along a spectrum [60]. Although this dichotomy corresponds to a more nuanced spectrum in vivo, it has been used to determine whether macrophages are more pro-inflammatory or pro-tissue remodeling. With aging, macrophages can become skewed towards one end of the polarization spectrum, and this skewing will create a microenvironment that can be detrimental to surrounding cell types. In the liver and adipose tissue, macrophages tend to skew towards an M1 phenotype, while bone marrow, spleen, lymph nodes, lung and muscle seem to skew more towards an M2 phenotype [61]. For example, mouse Kupffer cells and adipose tissue macrophages were reported to skew towards a Cd38+ M1 phenotype with aging, leading to increased inflammation and consumption of nicotinamide adenine dinucleotide [NAD], with detrimental impact on tissue homeostasis [62]. Typically, monocytes recruited to a site of infection tend to be M1-like, while tissue-resident macrophages tend to be more M2-like [63]. However, aging has been associated to increased skewing of both resident and non-resident macrophages to M1-like phenotypes [64,65]. Differential skewing of macrophages with aging may underlie the recurrent upregulation of pro-inflammatory cytokine-related pathways, which have also been observed at the transcriptional level (Table 1).
Toll-like receptors [TLR] are important for recognition of PAMPs, and thus to trigger cellular activation in response to TLR-associated signals [66]. Studies have shown aging leads to a dysfunctional response to PAMP stimuli, notably resulting in misregulation of cytokine production [67–73]. Macrophages also have the ability to present antigens to lymphocytes, and antigen presentation capacity by macrophages has been reported to decline with age [74]. However, the mechanisms behind this decrease are unclear, as different macrophage populations show opposite trends in regulation of antigen-presenting complex genes (e.g. MHC I and MHC II) and proteins. For instance, MHCII mRNA levels increase with age in rat microglia and mouse bone marrow macrophages [51,75]. In contrast, BMDMs from young and old mice showed decreased expression of MHC class II protein and mRNA [76]. Whether due to increased or decreased production of MHC, dysfunction in the antigen presentation machinery is likely to impact the resolution of inflammation and promote a more inflammatory state.
Similar to transcriptional differences, age-related functional changes in macrophage function are not homogeneous in the literature. There are a few possible explanations behind the apparent inconsistencies One of the possibilities that could explain the differences observed is that the innate immune system can be modulated in response to sex-steroids, including estrogens [77]. Although this has not yet been studied systematically throughout aging, populations of macrophages (e.g. microglia, have been shown to be extremely sex-dimorphic in young animals [78–80]). Since most studies have thus far been conducted in only one sex (Tables 1 and 2), it is likely that differences between studies may partially result from differences in the biological sex of the used animals. Furthermore, contrasting results on phagocytic capacity may also be due to differences in the cargo used to perform the assay, including differential impacts of aging on ‘neutral’ cargo phagocytosis (e.g. latex beads), TLR-mediated phagocytosis (e.g. Zymosan, bacteria), or opsonic versus non-opsonic phagocytosis (Table 2). Finally, another variable that can contribute to the differences observed between the different tissue-resident macrophages is the impact of their niche microenvironment, which shapes many of their phenotypes [38,39], and may be itself driving differences in aging trajectories. Therefore, experimental design choices and differences between studies will greatly impact the final observations.
Neutrophils
Neutrophils or ‘polymorphonuclear cells’ are the most abundant leukocyte among human blood cells, composing 50–70% of white blood cells throughout life [81]. Neutrophils are short-lived cells, with an estimated cellular lifespan ranging from only hours to days upon terminal differentiation, in a process that has been dubbed ‘Neutrophil aging,’ distinct from organismal aging [82–84]. Thus, they are continually generated in the bone marrow and released into circulation to contribute to overall immune surveillance [83,85]. Key processes that neutrophils can undergo upon activation by microbial signals include secretion of antimicrobial granules and release of ‘NETs’ [83,86]. Although neutrophils are essential for immune surveillance as a ‘first line-of-defense,’ they can also contribute to and aggravate inflammatory disease [83,85]. Indeed, emerging evidence suggests that neutrophils play important roles in chronic inflammation [87].
Aging transcriptomic landscape
Although changes in gene expression regulation throughout lifespan have been reported across many tissues and cell types [33,34], how organismal aging (rather than ‘daily’ cellular aging) affects neutrophils is still largely unknown. However, despite their continued turnover, the transcriptional landscape of primary mouse bone-marrow neutrophils seems to be influenced by organismal aging [88], although age-related transcriptional changes are dwarfed by large sex-differences. Differentially expressed genes of neutrophils showed an upregulation of genes associated with immune signaling and autophagy accompanied by the downregulation of chromatin-associated genes [88]. Thus, the upregulated of autoimmunity as well as downregulation of chromatin genes could lead to the mis-regulation of NETs upon microbial signals. Interestingly, a single-cell atlas of rat aging organs showed increased infiltration of neutrophils in adipose tissue, liver and kidney [89]. In addition, neutrophils showed large gene expression differences with aging across tissues in that dataset, although the functional analysis of these differentially expressed genes was not discussed [89]. Although the broader relevance of these findings to other populations of neutrophils, to neutrophils from humans or to neutrophils exposed to priming signals will be required, changes in neutrophil transcriptomes could lead to functional defects with aging.
Age-related functional decline
Although they have yet not been studied as extensively as macrophages, evidence suggests that neutrophils from aged organisms are dysfunctional on many levels [90–95]. Thus, age-related neutrophil dysfunction likely contributes to overall immune dysfunction, with changes in ‘primed’ NETosis [91,92], chemotaxis [93], intracellular granule secretion [93,96], phagocytosis [94,97] and pathogen killing [91,94,95]. Interestingly, neutrophils can be primed by TNFa for increased NETosis [92], and circulating TNFa levels are known to increase with aging [18]. Notably, NETosis capacity with aging has only been reported in a primed state (i.e. TNFa pre-incubation of human circulating blood monocytes [92] or thioglycolate-elicitation of mouse peritoneal neutrophils [91], which is known to mimic TNFa priming [98]), and the ‘naïve’ unprimed state has not yet been investigated. It will be important to determine whether aged naïve neutrophils have increased NETosis capacity, as the aged milieu may itself prime them for unscheduled activation and promote further acceleration of the inflammatory response.
Importantly, age-related dysfunction of macrophages may itself lead to modulation of the pool of neutrophils, by allowing them to survive longer in an impaired state. Indeed, bone-marrow macrophages are responsible for clearing senescent neutrophils by efferocytosis [99], and this process is defective in the bone marrows of aged animals [51]. Since senescent neutrophils are functionally distinct from fresh neutrophils in terms of anti-microbial properties and inflammatory recruitment [100], changes in the efficiency and timing of senescent neutrophil clearing may drive aspects of observed defects in neutrophils from aged organisms.
Dendritic cells
DCs represent the primary antigen-presenting cells of the innate immune system, and are notably responsible for initiating a primary T-lymphocyte response to non-self antigens [101]. They play a major role among innate immune cells as the major link between the innate immune and adaptive immune responses [101]. DCs are, in other words, the surveillance cells of innate immunity, monitoring the presence of antigens, from pathogens or tumors [101]. Generally, DCs can be divided into conventional DCs (cDCs) and plasmacytoid DCs (pDCs) [102]. They are found across tissues, and play key roles in the communication with cells from the adaptive arm of the immune system (e.g. in the thymus or lymphoid organs).
Aging transcriptomic landscape
Upon age-associated thymic involution, gene set-enrichment analysis revealed that mouse thymic DCs show a moderate but significant increase in proinflammatory gene expression programs [103]. However, for both splenic and thymic DCs in mice, transcriptomic studies identified relatively few age-related transcriptional differences in mice [103,104].
A recent study of human peripheral blood cells captured ‘omic’ changes in circulating DCs with human aging, including at the single cell RNA-seq level [105]. DCs from aged individuals showed upregulation of interferon-stimulated genes and pro-inflammatory cytokine, consistent with an increased inflammatory phenotype of DCs with human aging [105]. Overrepresented functional categories linked to upregulation with aging included apoptosis and interferon gamma signaling [105]. Interestingly, genes involved in DC antigen presentation (e.g. CLEC12A, TXNIP), or self-tolerance (e.g. MALAT1, AHR) significantly decreased with aging in circulating DCs [105], consistent with decreased antigen-presenting ability. However, with age, MHCII and costimulatory molecules were downregulated across DCs subtypes [106]. Thus, based on transcriptional profiles, aged human DCs seem to acquire a pro-inflammatory phenotype while losing antigen-presenting ability.
Age-related functional decline
While the numbers and phenotype of DC subsets are broadly unaffected in older subjects, their abilities to migrate and process antigens are thought to be significantly compromised [107]. Accumulating studies have shown age-related changes in cDC and pDC function and often compare the two types. Aged cDCs have a phenotype similar to those from younger subjects, while pDCs seem to acquire decreased TLR 7 and 9 expression [107–109]. Furthermore, cytokine secretion in aged-cDCs shows systematically higher TNF-α and IL-6 secretion and lower activation by PAMPs (e.g. Pam3Cys, flagellin, poly IC, lipoteioic acid) [107]. In contrast, pDCs generally show an age-related decrease in IFN-α production [107,110,111]. The mechanisms delineating cDC–pDC interactions in the aging process will deserve further exploration. DCs have been reported to have a decreased ability to prime CD4+ T cells in older subjects [112]. However, whether or not the issue is due to dysfunctions in (i) antigen presentation, (ii) antigen presentation response or (iii) both remains unclear [112]. Tissue-specific responses may also play a role in shaping DC phenotypes with aging. For instance, while the population of cDC in the lymph nodes and spleen of aged mice remained stable, lung DCs increased in numbers with age [104]. Additionally, reconstitution of cDCs by bone marrow precursors was higher in aged mice compared to young mice [104]. In vitro experiments found that both young and aged DC in general were similarly capable of both direct and cross presentation of antigens [104].
Natural killer cells
Natural killer (NK) cells are cytotoxic innate lymphocytes that also play a crucial part in the innate immune system. They are activated by interferons or cytokines to defend against viruses and potentially tumors [113]. Upon activation, they can then specifically target infected or oncogenic cells for death.
Aging transcriptomic landscape
Although NK cells have not been as extensively studied with organism aging, emerging evidence suggests that they undergo significant changes which are consistent with immune dysfunction [105,114]. Through a study of peripheral blood cells in humans throughout aging, Zheng et al. [105] captured ‘omic’ changes in NK cells with human aging at the single cell level. Indeed, they observed age-associated increases in circulating NK cells, together with a reprogrammed immune landscape with age [105]. For example, scRNA-seq analysis of NK cell-status allowed them to distinguish circulating NKs in three distinct immune states: the CD16low CD56bright subset [NK1], the CD16high CD56dim CD57− low-cytotoxic subset [NK2] and the CD16high CD56dim CD57+ late subset [NK3] [105]. Aged healthy adults (>60 years) had decreased numbers of NK1 but expanded NK2 and NK3 compartments compared to their young adult counterparts, suggesting genomic reprogramming of these cells with aging [105]. Interestingly, aged healthy adults exhibited higher expression genes related to apoptotic responses to lipopolysaccharide, apoptotic signaling and lower virus defense responses [105]. Together, with aging, NK cells seem to exhibit a heightened inflammatory state and show impediments in antiviral response and activity [105]. However, further studies of purified NK cells with aging will be needed to understand the factors driving observed dysfunction.
Age-related functional decline
Much of NK cell activity relies on DC activation, as both types of innate cells work in reciprocity across both innate and adaptive immunity in virus control and tumor immunology [115]. Defects of DCs in aged C57BL/6 mice cascades into failure to properly activate NK cells with aging, thus leading to decreased ability to clear tumor cells [115]. NK cells have been shown to demonstrate tissue-specific immune responses associated with age-related changes. For example, in older adults, the cytotoxicity of the CD56low NK population significantly decreases, which is thought to result from age-related defects in perforin mobilization to the immune synapse [116–118]. It is interesting to note however, that the CD56low NK cell population expands as the CD56hi NK cell population [responsible for cytokine production] dwindles during the aging process [118]. To note, these phenomena have been mostly studied with aging in the periodontal region [119]. Overall, the impact of aging on functional phenotypes of NK cells is only starting to be understood and will require further investigation.
Conclusions and perspectives
Accumulating evidence has shown that innate immune cells exhibit remodeling of their transcriptional programs, which is likely to drive aspects of age-related dysfunction in many of their key phenotypes (Figure 1). Immune modulation is a leading candidate in understanding the aging process. Interestingly, naked mole rats, an extremely long-lived rodent model (~30 years compared to ~3 years for laboratory mice) have a unique immune system that is distinct from the mice model [120]. Naked mole rats seem to have a greater emphasis on innate immunity than laboratory mice [120], consistent with the notion that the tuning of innate immunity is key for healthy longevity. Although general themes of age-related changes in innate immune cells ‘omic’ landscapes are consistent with the notion of inflamm-aging, studies have shown relatively little overlap thus far (Tables 1 and 2), which warrants a systematic study including all necessary covariates to understand the impact of aging on the innate immune system.
Accumulating studies have shown that specific interventions (e.g. exercise, dietary restriction, etc.) can exert pro-health and pro-longevity effects. A key question is thus how these pro-longevity interventions might modulate inflamm-aging phenotypes. Interestingly, a recent study which profiled organs at the single cell level in aging rats and in response to dietary restriction [DR] found that DR can rescue dysfunction in macrophage polarization and reverse immune cell infiltration in various organs [89]. Furthermore, DR was associated to downregulation of pathways related to immune response, inflammation and response to stimuli (e.g. LPS, interleukins), and to upregulation of pathways related to regeneration, response to growth factors and extracellular matrix [89]. Whereas aging tends to lead to a pro-inflammatory ‘M1’-like phenotype of adipose tissue macrophages [121,122], a recent study showed that exercise is able to reverse this age-related pro-inflammatory skewing [123]. To note, exercise may both suppress the infiltration of M1-like macrophages and promote reprogramming to a more ‘M2’ phenotype [124]. Both exercise and DR can modulate transduction from nutrient signaling pathways, thus promoting healthy longevity. It will be key to understand how they may influence the development of inflamm-aging and immunosenescence phenotypes.
Finally, although discussed studies have reported age-related changes in innate immune cell processes, there is still little known about how these changes are influenced by biological sex. Indeed, both the adult mammalian immune system [80,125] and the aging process [126] are sex-dimorphic, suggesting that the study of inflamm-aging should be stratified as a function of sex. Indeed, a pioneer study has revealed that peripheral immune cells are differentially regulated with aging in men and women [125]. Thus, to understand the impact of immune decline on aging and to permit the development of broadly applicable therapeutic strategies, it will be key to systematically include sex a biological variable of interest in future functional genomic studies of inflamm-aging.
Key Points
Immune decline is a hallmark of aging.
Aging associates with a state of chronic sterile inflammation.
Innate immune cells undergo widespread molecular and functional remodeling with aging.
Acknowledgements
We thank members of the Benayoun lab for helpful discussions and feedback on the manuscript.
Ryan J. Lu is a PhD student from the Biology of Aging program at the Leonard Davis School of Gerontology at the University of Southern California. His research focuses on sex-dimorphism in mechanisms driving age-related dysfunction in macrophages and neutrophils.
Emily K. Wang is a student in the Master of Science in Gerontology program at the Leonard Davis School of Gerontology at the University of Southern California. Her research focuses on identifying conserved sex-dimorphic signatures across macrophage populations.
Bérénice A. Benayoun is an assistant professor of Gerontology at the Leonard Davis School of Gerontology at the University of Southern California. Her research focuses on leveraging big data methods to uncover new regulatory mechanisms driving the aging process in vertebrate species.
Funding
R.L. is supported by a Diana Jacobs Kalman/AFAR Scholarships for Research in the Biology of Aging. B.A.B. is supported by NIA grants R00 AG049934 and R21 AG063739, Pew Biomedical Scholar award #00034120, an innovator grant from the Rose Hills Foundation, a collaborative grant from the Simon Foundation, a junior scholar award from the Global Consortium for Reproductive Longevity and Equality, a Glenn and AFAR Junior Faculty Award and the Kathleen Gilmore Biology of Aging Research Award.
References
- 1. Thompson WW, Shay DK, Weintraub E, et al. Influenza-associated hospitalizations in the United States. JAMA 2004;292:1333–1340. [DOI] [PubMed] [Google Scholar]
- 2. Perrotta F, Corbi G, Mazzeo G, et al. COVID-19 and the elderly: insights into pathogenesis and clinical decision-making. Aging Clin Exp Res 2020;32:1599–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Aiello A, Farzaneh F, Candore G, et al. Immunosenescence and its hallmarks: how to oppose aging strategically? A review of potential options for therapeutic intervention. Front Immunol 2019;10:2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Franceschi C, Bonafe M, Valensin S, et al. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann N Y Acad Sci 2000;908:244–254. [DOI] [PubMed] [Google Scholar]
- 5. Franceschi C, Campisi J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J Gerontol A Biol Sci Med Sci 2014;69(Suppl 1):S4–S9. [DOI] [PubMed] [Google Scholar]
- 6. Michaud M, Balardy L, Moulis G, et al. Proinflammatory cytokines, aging, and age-related diseases. J Am Med Dir Assoc 2013;14:877–882. [DOI] [PubMed] [Google Scholar]
- 7. Gorgoulis V, Adams PD, Alimonti A, et al. Cellular senescence: defining a path forward. Cell 2019;179:813–827. [DOI] [PubMed] [Google Scholar]
- 8. Coppe JP, Patil CK, Rodier F, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol 2008;6:2853–2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wiley CD, Liu S, Limbad C, et al. SILAC analysis reveals increased secretion of hemostasis-related factors by senescent cells. Cell Rep 2019;28:3329–3337 e3325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Basisty N, Kale A, Jeon OH, et al. A proteomic atlas of senescence-associated secretomes for aging biomarker development. PLoS Biol 2020;18:e3000599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. James EL, Michalek RD, Pitiyage GN, et al. Senescent human fibroblasts show increased glycolysis and redox homeostasis with extracellular metabolomes that overlap with those of irreparable DNA damage, aging, and disease. J Proteome Res 2015;14:1854–1871. [DOI] [PubMed] [Google Scholar]
- 12. Coppe JP, Desprez PY, Krtolica A, et al. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol 2010;5:99–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Campisi J, Robert L. Cell senescence: role in aging and age-related diseases. Interdiscip Top Gerontol 2014;39:45–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Partridge L, Fuentealba M, Kennedy BK. The quest to slow ageing through drug discovery. Nat Rev Drug Discov 2020;19:513–532. [DOI] [PubMed] [Google Scholar]
- 15. Kang C. Senolytics and senostatics: a two-pronged approach to target cellular senescence for delaying aging and age-related diseases. Mol Cells 2019;42:821–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Annaert P, Brouwers J, Bijnens A, et al. Ex vivo permeability experiments in excised rat intestinal tissue and in vitro solubility measurements in aspirated human intestinal fluids support age-dependent oral drug absorption. Eur J Pharm Sci 2010;39:15–22. [DOI] [PubMed] [Google Scholar]
- 17. Hollander D, Tarnawski H. Aging-associated increase in intestinal absorption of macromolecules. Gerontology 1985;31:133–137. [DOI] [PubMed] [Google Scholar]
- 18. Thevaranjan N, Puchta A, Schulz C, et al. Age-associated microbial dysbiosis promotes intestinal permeability, systemic inflammation, and macrophage dysfunction. Cell Host Microbe 2017;21:455–466 e454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kim M, Benayoun BA. The microbiome: an emerging key player in aging and longevity. Transl Med Aging 2020;4:103–116. [PMC free article] [PubMed] [Google Scholar]
- 20. Van Meter M, Kashyap M, Rezazadeh S, et al. SIRT6 represses LINE1 retrotransposons by ribosylating KAP1 but this repression fails with stress and age. Nat Commun 2014;5:5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. De Cecco M, Ito T, Petrashen AP, et al. L1 drives IFN in senescent cells and promotes age-associated inflammation. Nature 2019;566:73–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Simon M, Van Meter M, Ablaeva J, et al. LINE1 Derepression in aged wild-type and SIRT6-deficient mice drives inflammation. Cell Metab 2019;29:871–885 e875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cavanagh MM, Weyand CM, Goronzy JJ. Chronic inflammation and aging: DNA damage tips the balance. Curr Opin Immunol 2012;24:488–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lopez-Otin C, Blasco MA, Partridge L, et al. The hallmarks of aging. Cell 2013;153:1194–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kennedy BK, Berger SL, Brunet A, et al. Geroscience: linking aging to chronic disease. Cell 2014;159:709–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. SanMiguel JM, Young K, Trowbridge JJ. Hand in hand: intrinsic and extrinsic drivers of aging and clonal hematopoiesis. Exp Hematol 2020;91:1–9. doi: 10.1016/j.exphem.2020.09.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sun D, Luo M, Jeong M, et al. Epigenomic profiling of young and aged HSCs reveals concerted changes during aging that reinforce self-renewal. Cell Stem Cell 2014;14:673–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Khokhar ES, Borikar S, Eudy E, et al. Aging-associated decrease in the histone acetyltransferase KAT6B is linked to altered hematopoietic stem cell differentiation. Exp Hematol 2020;82:43–52 e44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Elias HK, Bryder D, Park CY. Molecular mechanisms underlying lineage bias in aging hematopoiesis. Semin Hematol 2017;54:4–11. [DOI] [PubMed] [Google Scholar]
- 30. Das MM, Godoy M, Chen S, et al. Young bone marrow transplantation preserves learning and memory in old mice. Commun Biol 2019;2:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hormaechea-Agulla D, Le DT, King KY. Common sources of inflammation and their impact on hematopoietic stem cell biology. Curr Stem Cell Rep 2020;1–12. https://pubmed.ncbi.nlm.nih.gov/32837857/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hoebe K, Janssen E, Beutler B. The interface between innate and adaptive immunity. Nat Immunol 2004;5:971–974. [DOI] [PubMed] [Google Scholar]
- 33. Lai RW, Lu R, Danthi PS, et al. Multi-level remodeling of transcriptional landscapes in aging and longevity. BMB Rep 2019;52:86–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Benayoun BA, Pollina EA, Singh PP, et al. Remodeling of epigenome and transcriptome landscapes with aging in mice reveals widespread induction of inflammatory responses. Genome Res 2019;29:697–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Barth E, Srivastava A, Stojiljkovic M, et al. Conserved aging-related signatures of senescence and inflammation in different tissues and species. Aging (Albany NY) 2019;11:8556–8572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Linehan E, Fitzgerald DC. Ageing and the immune system: focus on macrophages. Eur J Microbiol Immunol (Bp) 2015;5:14–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Epelman S, Lavine KJ, Beaudin AE, et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity 2014;40:91–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lavin Y, Winter D, Blecher-Gonen R, et al. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell 2014;159:1312–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gosselin D, Link VM, Romanoski CE, et al. Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell 2014;159:1327–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gautier EL, Shay T, Miller J, et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat Immunol 2012;13:1118–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hickman SE, Kingery ND, Ohsumi TK, et al. The microglial sensome revealed by direct RNA sequencing. Nat Neurosci 2013;16:1896–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chen S, Yang J, Wei Y, et al. Epigenetic regulation of macrophages: from homeostasis maintenance to host defense. Cell Mol Immunol 2020;17:36–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jarjour NN, Schwarzkopf EA, Bradstreet TR, et al. Bhlhe40 mediates tissue-specific control of macrophage proliferation in homeostasis and type 2 immunity. Nat Immunol 2019;20:687–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rosas M, Davies LC, Giles PJ, et al. The transcription factor Gata6 links tissue macrophage phenotype and proliferative renewal. Science 2014;344:645–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rustenhoven J, Smith AM, Smyth LC, et al. PU.1 regulates Alzheimer's disease-associated genes in primary human microglia. Mol Neurodegener 2018;13:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Huang KL, Marcora E, Pimenova AA, et al. A common haplotype lowers PU.1 expression in myeloid cells and delays onset of Alzheimer's disease. Nat Neurosci 2017;20:1052–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Camell CD, Sander J, Spadaro O, et al. Inflammasome-driven catecholamine catabolism in macrophages blunts lipolysis during ageing. Nature 2017;550:119–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dalli J, Serhan CN. Pro-resolving mediators in regulating and conferring macrophage function. Front Immunol 2017;8:1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Farrera C, Fadeel B. Macrophage clearance of neutrophil extracellular traps is a silent process. J Immunol 2013;191:2647–2656. [DOI] [PubMed] [Google Scholar]
- 50. Arnardottir HH, Dalli J, Colas RA, et al. Aging delays resolution of acute inflammation in mice: reprogramming the host response with novel nano-proresolving medicines. J Immunol 2014;193:4235–4244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Frisch BJ, Hoffman CM, Latchney SE, et al. Aged marrow macrophages expand platelet-biased hematopoietic stem cells via interleukin-1B. JCI Insight 2019;5:e124213. doi: 10.1172/jci.insight.124213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Watanabe S, Kawamoto S, Ohtani N, et al. Impact of senescence-associated secretory phenotype and its potential as a therapeutic target for senescence-associated diseases. Cancer Sci 2017;108:563–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kale A, Sharma A, Stolzing A, et al. Role of immune cells in the removal of deleterious senescent cells. Immun Ageing 2020;17:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Belchamber KBR, Donnelly LE. Macrophage dysfunction in respiratory disease. Results Probl Cell Differ 2017;62:299–313. [DOI] [PubMed] [Google Scholar]
- 55. Bachiller S, Jimenez-Ferrer I, Paulus A, et al. Microglia in neurological diseases: a road map to brain-disease dependent-inflammatory response. Front Cell Neurosci 2018;12:488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wong CK, Smith CA, Sakamoto K, et al. Aging impairs alveolar macrophage phagocytosis and increases influenza-induced mortality in mice. J Immunol 2017;199:1060–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Orecchioni M, Ghosheh Y, Pramod AB, et al. Macrophage polarization: different gene signatures in M1(LPS+) vs. classically and M2(LPS-) vs. alternatively activated macrophages. Front Immunol 2019;10:1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Roszer T. Understanding the mysterious M2 macrophage through activation markers and effector mechanisms. Mediators Inflamm 2015;2015:816460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Duluc D, Delneste Y, Tan F, et al. Tumor-associated leukemia inhibitory factor and IL-6 skew monocyte differentiation into tumor-associated macrophage-like cells. Blood 2007;110:4319–4330. [DOI] [PubMed] [Google Scholar]
- 60. Ginhoux F, Schultze JL, Murray PJ, et al. New insights into the multidimensional concept of macrophage ontogeny, activation and function. Nat Immunol 2016;17:34–40. [DOI] [PubMed] [Google Scholar]
- 61. Jackaman C, Tomay F, Duong L, et al. Aging and cancer: the role of macrophages and neutrophils. Ageing Res Rev 2017;36:105–116. [DOI] [PubMed] [Google Scholar]
- 62. Covarrubias AJ, Kale A, Perrone R, et al. Senescent cells promote tissue NAD(+) decline during ageing via the activation of CD38(+) macrophages. Nat Metab 2020;2:1265–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Goldberg EL, Dixit VD. Drivers of age-related inflammation and strategies for healthspan extension. Immunol Rev 2015;265:63–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Becker L, Nguyen L, Gill J, et al. Age-dependent shift in macrophage polarisation causes inflammation-mediated degeneration of enteric nervous system. Gut 2018;67:827–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Gibon E, Loi F, Cordova LA, et al. Aging affects bone marrow macrophage polarization: relevance to bone healing. Regen Eng Transl Med 2016;2:98–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol 2003;21:335–376. [DOI] [PubMed] [Google Scholar]
- 67. Boehmer ED, Goral J, Faunce DE, et al. Age-dependent decrease in toll-like receptor 4-mediated proinflammatory cytokine production and mitogen-activated protein kinase expression. J Leukoc Biol 2004;75:342–349. [DOI] [PubMed] [Google Scholar]
- 68. Boehmer ED, Meehan MJ, Cutro BT, et al. Aging negatively skews macrophage TLR2- and TLR4-mediated pro-inflammatory responses without affecting the IL-2-stimulated pathway. Mech Ageing Dev 2005;126:1305–1313. [DOI] [PubMed] [Google Scholar]
- 69. Chelvarajan RL, Collins SM, Van Willigen JM, et al. The unresponsiveness of aged mice to polysaccharide antigens is a result of a defect in macrophage function. J Leukoc Biol 2005;77:503–512. [DOI] [PubMed] [Google Scholar]
- 70. Fallah MP, Chelvarajan RL, Garvy BA, et al. Role of phosphoinositide 3-kinase-Akt signaling pathway in the age-related cytokine dysregulation in splenic macrophages stimulated via TLR-2 or TLR-4 receptors. Mech Ageing Dev 2011;132:274–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Mahbub S, Deburghgraeve CR, Kovacs EJ. Advanced age impairs macrophage polarization. J Interferon Cytokine Res 2012;32:18–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Renshaw M, Rockwell J, Engleman C, et al. Cutting edge: impaired toll-like receptor expression and function in aging. J Immunol 2002;169:4697–4701. [DOI] [PubMed] [Google Scholar]
- 73. van Duin D, Mohanty S, Thomas V, et al. Age-associated defect in human TLR-1/2 function. J Immunol 2007;178:970–975. [DOI] [PubMed] [Google Scholar]
- 74. Plowden J, Renshaw-Hoelscher M, Gangappa S, et al. Impaired antigen-induced CD8+ T cell clonal expansion in aging is due to defects in antigen presenting cell function. Cell Immunol 2004;229:86–92. [DOI] [PubMed] [Google Scholar]
- 75. Frank MG, Barrientos RM, Biedenkapp JC, et al. mRNA up-regulation of MHC II and pivotal pro-inflammatory genes in normal brain aging. Neurobiol Aging 2006;27:717–722. [DOI] [PubMed] [Google Scholar]
- 76. Herrero C, Sebastian C, Marques L, et al. Immunosenescence of macrophages: reduced MHC class II gene expression. Exp Gerontol 2002;37:389–394. [DOI] [PubMed] [Google Scholar]
- 77. Kovats S. Estrogen receptors regulate innate immune cells and signaling pathways. Cell Immunol 2015;294:63–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Guneykaya D, Ivanov A, Hernandez DP, et al. Transcriptional and translational differences of microglia from male and female brains. Cell Rep 2018;24:2773–2783 e2776. [DOI] [PubMed] [Google Scholar]
- 79. Villa A, Gelosa P, Castiglioni L, et al. Sex-specific features of microglia from adult mice. Cell Rep 2018;23:3501–3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Gal-Oz ST, Maier B, Yoshida H, et al. ImmGen report: sexual dimorphism in the immune system transcriptome. Nat Commun 2019;10:4295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Nah EH, Kim S, Cho S, et al. Complete blood count reference intervals and patterns of changes across pediatric, adult, and geriatric ages in Korea. Ann Lab Med 2018;38:503–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Zhang D, Chen G, Manwani D, et al. Neutrophil ageing is regulated by the microbiome. Nature 2015;525:528–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Shah B, Burg N, Pillinger MH. Chapter 11 - Neutrophils. In: Firestein GS, Budd RC, Gabriel SE et al. (eds). Kelley and Firestein's Textbook of Rheumatology 10th edn. Amsterdam, Netherlands: Elsevier, 2017, 169–188.e163. [Google Scholar]
- 84. Ballesteros I, Rubio-Ponce A, Genua M, et al. Co-option of neutrophil fates by tissue environments. Cell 2020;183:1282–1297 e1218. [DOI] [PubMed] [Google Scholar]
- 85. Furze RC, Rankin SM. Neutrophil mobilization and clearance in the bone marrow. Immunology 2008;125:281–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Sollberger G, Tilley DO, Zychlinsky A. Neutrophil extracellular traps: the biology of chromatin externalization. Dev Cell 2018;44:542–553. [DOI] [PubMed] [Google Scholar]
- 87. Soehnlein O, Steffens S, Hidalgo A, et al. Neutrophils as protagonists and targets in chronic inflammation. Nat Rev Immunol 2017;17:248–261. [DOI] [PubMed] [Google Scholar]
- 88. Lu R, Taylor S, Contrepois K, et al. Multi-omic profiling of primary mouse neutrophils reveals a pattern of sex and age-related functional regulation. bioRxiv 2020; 2020.2007.2006.190595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Ma S, Sun S, Geng L, et al. Caloric restriction reprograms the single-cell transcriptional landscape of Rattus Norvegicus aging. Cell 2020;180:984–1001 e1022. [DOI] [PubMed] [Google Scholar]
- 90. Tseng CW, Liu GY. Expanding roles of neutrophils in aging hosts. Curr Opin Immunol 2014;29:43–48. [DOI] [PubMed] [Google Scholar]
- 91. Tseng CW, Kyme PA, Arruda A, et al. Innate immune dysfunctions in aged mice facilitate the systemic dissemination of methicillin-resistant S. aureus. PloS one 2012;7:e41454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Hazeldine J, Harris P, Chapple IL, et al. Impaired neutrophil extracellular trap formation: a novel defect in the innate immune system of aged individuals. Aging Cell 2014;13:690–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Sapey E, Greenwood H, Walton G, et al. Phosphoinositide 3-kinase inhibition restores neutrophil accuracy in the elderly: toward targeted treatments for immunosenescence. Blood 2014;123:239–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Wenisch C, Patruta S, Daxbock F, et al. Effect of age on human neutrophil function. J Leukoc Biol 2000;67:40–45. [DOI] [PubMed] [Google Scholar]
- 95. Simell B, Vuorela A, Ekstrom N, et al. Aging reduces the functionality of anti-pneumococcal antibodies and the killing of Streptococcus pneumoniae by neutrophil phagocytosis. Vaccine 2011;29:1929–1934. [DOI] [PubMed] [Google Scholar]
- 96. McLaughlin B, O'Malley K, Cotter TG. Age-related differences in granulocyte chemotaxis and degranulation. Clin Sci (Lond) 1986;70:59–62. [DOI] [PubMed] [Google Scholar]
- 97. Butcher SK, Chahal H, Nayak L, et al. Senescence in innate immune responses: reduced neutrophil phagocytic capacity and CD16 expression in elderly humans. J Leukoc Biol 2001;70:881–886. [PubMed] [Google Scholar]
- 98. Itou T, Collins LV, Thorén FB, et al. Changes in activation states of murine polymorphonuclear leukocytes (PMN) during inflammation: a comparison of bone marrow and peritoneal exudate PMN. Clin Vaccine Immunol 2006;13:575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Casanova-Acebes M, Pitaval C, Weiss LA, et al. Rhythmic modulation of the hematopoietic niche through neutrophil clearance. Cell 2013;153:1025–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Adrover JM, Del Fresno C, Crainiciuc G, et al. A neutrophil timer coordinates immune defense and vascular protection. Immunity 2019;51:966–967. [DOI] [PubMed] [Google Scholar]
- 101. Clark GJ, Angel N, Kato M, et al. The role of dendritic cells in the innate immune system. Microbes Infect 2000;2:257–272. [DOI] [PubMed] [Google Scholar]
- 102. Gigley JP, Khan IA. Plasmacytoid DC from aged mice down-regulate CD8 T cell responses by inhibiting cDC maturation after Encephalitozoon cuniculi infection. PloS one 2011;6:e20838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Ki S, Park D, Selden HJ, et al. Global transcriptional profiling reveals distinct functions of thymic stromal subsets and age-related changes during thymic involution. Cell Rep 2014;9:402–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Tan SY, Cavanagh LL, d'Advigor W, et al. Phenotype and functions of conventional dendritic cells are not compromised in aged mice. Immunol Cell Biol 2012;90:722–732. [DOI] [PubMed] [Google Scholar]
- 105. Zheng Y, Liu X, Le W, et al. A human circulating immune cell landscape in aging and COVID-19. Protein Cell 2020;11:740–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Wong C, Goldstein DR. Impact of aging on antigen presentation cell function of dendritic cells. Curr Opin Immunol 2013;25:535–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Agrawal A, Agrawal S, Gupta S. Role of dendritic cells in inflammation and loss of tolerance in the elderly. Front Immunol 2017;8:896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Zacca ER, Crespo MI, Acland RP, et al. Aging impairs the ability of conventional dendritic cells to cross-prime CD8+ T cells upon stimulation with a TLR7 ligand. PloS one 2015;10:e0140672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Panda A, Qian F, Mohanty S, et al. Age-associated decrease in TLR function in primary human dendritic cells predicts influenza vaccine response. J Immunol 2010;184:2518–2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Stout-Delgado HW, Yang X, Walker WE, et al. Aging impairs IFN regulatory factor 7 up-regulation in plasmacytoid dendritic cells during TLR9 activation. J Immunol 2008;181:6747–6756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Jing Y, Shaheen E, Drake RR, et al. Aging is associated with a numerical and functional decline in plasmacytoid dendritic cells, whereas myeloid dendritic cells are relatively unaltered in human peripheral blood. Hum Immunol 2009;70:777–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Fulop T, Larbi A, Dupuis G, et al. Immunosenescence and inflamm-aging as two sides of the same coin: friends or foes? Front Immunol 2017;8:1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Larbi A, Franceschi C, Mazzatti D, et al. Aging of the immune system as a prognostic factor for human longevity. Physiology (Bethesda) 2008;23:64–74. [DOI] [PubMed] [Google Scholar]
- 114. Pinti M, Appay V, Campisi J, et al. Aging of the immune system: focus on inflammation and vaccination. Eur J Immunol 2016;46:2286–2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Guo Z, Tilburgs T, Wong B, et al. Dysfunction of dendritic cells in aged C57BL/6 mice leads to failure of natural killer cell activation and of tumor eradication. Proc Natl Acad Sci U S A 2014;111:14199–14204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Caligiuri MA. Human natural killer cells. Blood 2008;112:461–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Hazeldine J, Hampson P, Lord JM. Reduced release and binding of perforin at the immunological synapse underlies the age-related decline in natural killer cell cytotoxicity. Aging Cell 2012;11:751–759. [DOI] [PubMed] [Google Scholar]
- 118. Shaw AC, Goldstein DR, Montgomery RR. Age-dependent dysregulation of innate immunity. Nat Rev Immunol 2013;13:875–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Ebersole JL, Graves CL, Gonzalez OA, et al. Aging, inflammation, immunity and periodontal disease. Periodontol 2000 2016;72:54–75. [DOI] [PubMed] [Google Scholar]
- 120. Hilton HG, Rubinstein ND, Janki P, et al. Single-cell transcriptomics of the naked mole-rat reveals unexpected features of mammalian immunity. PLoS Biol 2019;17:e3000528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Heilbronn LK, Campbell LV. Adipose tissue macrophages, low grade inflammation and insulin resistance in human obesity. Curr Pharm Des 2008;14:1225–1230. [DOI] [PubMed] [Google Scholar]
- 122. Ni Y, Ni L, Zhuge F, et al. Adipose tissue macrophage phenotypes and characteristics: the key to insulin resistance in obesity and metabolic disorders. Obesity (Silver Spring) 2020;28:225–234. [DOI] [PubMed] [Google Scholar]
- 123. Baek KW, Lee DI, Jeong MJ, et al. Effects of lifelong spontaneous exercise on the M1/M2 macrophage polarization ratio and gene expression in adipose tissue of super-aged mice. Exp Gerontol 2020;141:111091. [DOI] [PubMed] [Google Scholar]
- 124. Kawanishi N, Yano H, Yokogawa Y, et al. Exercise training inhibits inflammation in adipose tissue via both suppression of macrophage infiltration and acceleration of phenotypic switching from M1 to M2 macrophages in high-fat-diet-induced obese mice. Exerc Immunol Rev 2010;16:105–118. [PubMed] [Google Scholar]
- 125. Marquez EJ, Chung CH, Marches R, et al. Sexual-dimorphism in human immune system aging. Nat Commun 2020;11:751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Sampathkumar NK, Bravo JI, Chen Y, et al. Widespread sex dimorphism in aging and age-related diseases. Hum Genet 2020;139:333–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Hall BM, Gleiberman AS, Strom E, et al. Immune checkpoint protein VSIG4 as a biomarker of aging in murine adipose tissue. Aging Cell 2020;19:e13219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Angelidis I, Simon LM, Fernandez IE, et al. An atlas of the aging lung mapped by single cell transcriptomics and deep tissue proteomics. Nat Commun 2019;10:963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Clark D, Brazina S, Yang F, et al. Age-related changes to macrophages are detrimental to fracture healing in mice. Aging Cell 2020;19:e13112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Tabula Muris Consortium . A single-cell transcriptomic atlas characterizes ageing tissues in the mouse. Nature 2020;583:590–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Grabert K, Michoel T, Karavolos MH, et al. Microglial brain region-dependent diversity and selective regional sensitivities to aging. Nat Neurosci 2016;19:504–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Deczkowska A, Matcovitch-Natan O, Tsitsou-Kampeli A, et al. Mef2C restrains microglial inflammatory response and is lost in brain ageing in an IFN-I-dependent manner. Nat Commun 2017;8:717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Ma W, Cojocaru R, Gotoh N, et al. Gene expression changes in aging retinal microglia: relationship to microglial support functions and regulation of activation. Neurobiol Aging 2013;34:2310–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Pan J, Ma N, Yu B, et al. Transcriptomic profiling of microglia and astrocytes throughout aging. J Neuroinflammation 2020;17:97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Stratton JA, Eaton S, Rosin NL, et al. Macrophages and associated ligands in the aged injured nerve: a defective dynamic that contributes to reduced axonal regrowth. Front Aging Neurosci 2020;12:174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Runyan CE, Welch LC, Lecuona E, et al. Impaired phagocytic function in CX3CR1(+) tissue-resident skeletal muscle macrophages prevents muscle recovery after influenza a virus-induced pneumonia in old mice. Aging Cell 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Lin JB, Sene A, Santeford A, et al. Oxysterol signatures distinguish age-related macular degeneration from physiologic aging. EBioMedicine 2018;32:9–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Mancuso P, McNish RW, Peters-Golden M, et al. Evaluation of phagocytosis and arachidonate metabolism by alveolar macrophages and recruited neutrophils from F344xBN rats of different ages. Mech Ageing Dev 2001;122:1899–1913. [DOI] [PubMed] [Google Scholar]
- 139. Linehan E, Dombrowski Y, Snoddy R, et al. Aging impairs peritoneal but not bone marrow-derived macrophage phagocytosis. Aging Cell 2014;13:699–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Hilmer SN, Cogger VC, Le Couteur DG. Basal activity of Kupffer cells increases with old age. J Gerontol A Biol Sci Med Sci 2007;62:973–978. [DOI] [PubMed] [Google Scholar]
- 141. Ritzel RM, Patel AR, Pan S, et al. Age- and location-related changes in microglial function. Neurobiol Aging 2015;36:2153–2163. [DOI] [PubMed] [Google Scholar]
- 142. Ferrandez MD, De la Fuente M. Effects of age, sex and physical exercise on the phagocytic process of murine peritoneal macrophages. Acta Physiol Scand 1999;166:47–53. [DOI] [PubMed] [Google Scholar]
- 143. De La Fuente M. Changes in the macrophage function with aging. Comp Biochem Physiol A Comp Physiol 1985;81:935–938. [DOI] [PubMed] [Google Scholar]
- 144. Ortega E, Forner MA, Barriga C, et al. Effect of age and of swimming-induced stress on the phagocytic capacity of peritoneal macrophages from mice. Mech Ageing Dev 1993;70:53–63. [DOI] [PubMed] [Google Scholar]
- 145. Videla LA, Tapia G, Fernandez V. Influence of aging on Kupffer cell respiratory activity in relation to particle phagocytosis and oxidative stress parameters in mouse liver. Redox Rep 2001;6:155–159. [DOI] [PubMed] [Google Scholar]
- 146. Aprahamian T, Takemura Y, Goukassian D, et al. Ageing is associated with diminished apoptotic cell clearance in vivo. Clin Exp Immunol 2008;152:448–455. [DOI] [PMC free article] [PubMed] [Google Scholar]