

Abstract
The 90 kDa heat shock proteins (Hsp90) are molecular chaperones that are responsible for the folding and/or trafficking of ~400 client proteins, many of which are directly associated with cancer progression. Consequently, inhibition of Hsp90 can exhibit similar activity as combination therapy as multiple signaling nodes can be targeted simultaneously. In fact, seventeen small-molecule inhibitors that bind the Hsp90 N-terminus entered clinical trials for the treatment of cancer, all of which exhibited pan-inhibitory activity against all four Hsp90 isoforms. Unfortunately, most demonstrated undesired effects alongside induction of the pro-survival heat shock response. As a result, isoform-selective inhibitors have been sought to overcome these detriments. Described herein is a structure-based approach to design Hsp90β-selective inhibitors along with preliminary SAR. In the end, compound 5 was shown to manifest ~370-fold selectivity for Hsp90β versus Hsp90α, and induced the degradation of select Hsp90β-dependent clients. These data support the development of Hsp90β-selective inhibitors as a new paradigm to overcome the detriments associated with pan-inhibition of Hsp90.
Keywords: Hsp90, Hsp90β, Hsp90β-selective inhibitors, Cancer therapy, Protein folding
Graphical Abstract

The high identity shared among the various Hsp90 isoforms (>85%) within the N-terminal ATP-binding pocket has made the development of isoform selective inhibitors very challenging. Recently, we reported the first Hsp90β-selective inhibitor, which induced the degradation of Hsp90β-dependent clients without induction of Hsp90 levels. Structure-activity relationships for the first Hsp90β-selective inhibitor are disclosed herein and led to the identification of compound 5, which exhibited ~370-fold selectivity for Hsp90β over Hsp90α.
Introduction
The 90 kDa heat shock proteins (Hsp90) are molecular chaperones that are responsible for regulation of the cellular proteome and are conserved across almost all living organisms.[1–4] In eukaryotic cells and under normal physiological conditions, Hsp90 levels represent 1–2% of the total cellular protein, however during stress, these levels can increase to 4–6%.[5–8] Hsp90 plays the role of a molecular chaperone for ~400 client protein substrates by helping them maintain or achieve their three-dimensional conformation via activation, trafficking and/or degradation.[4,9] Many of these client proteins contribute to the growth, proliferation, metastasis and/or angiogenesis of cancer cells.[10] Furthermore, Hsp90 clients are associated with all ten hallmarks of cancer, and consequently, Hsp90 inhibitors can exhibit properties similar to multi-drug combination therapy, since multiple therapeutic pathways can be targeted simultaneously.[9,10]
Mammalian cells contain two cytosolic Hsp90 isoforms; inducible Hsp90α and constitutively expressed Hsp90β. In addition, the endoplasmic reticulum (ER) localized 94-kDa glucose-regulated protein (Grp94) and mitochondrial tumor necrosis factor receptor-associated protein 1 (TRAP1) are also Hsp90 isoforms.[11–13] Although Hsp90 isoforms share more than 85% structural identity within the N-terminal ATP binding pocket (21 out of 29 residues are totally conserved and the remaining 8 share high similarity), their function and client proteins vary.[14–16]
Seventeen pan-inhibitors of Hsp90[17–20] underwent clinical evaluation and unfortunately, most demonstrated deleterious side effects, such as cardiotoxicity, ocular toxicity, and/or dose-limiting toxicities.[17,21–23] Such issues raised the question of whether isoform-selective inhibition of Hsp90 can overcome these detriments? Unfortunately, the development of isoform-selective inhibitors is particularly challenging due to the high identity and homology shared among all four isoforms, particularly within their N-terminal ATP-binding pockets. Despite the high identity shared among the isoforms, recent studies identified several scaffolds that manifest selective binding for Grp94, which is the least homologous of all the paralogs.[24–26]
Unlike the other isoforms, Hsp90α and Hsp90β share ~95% identity within the N-terminal nucleotide-binding pocket, differing by only two amino acid residues (Hsp90α contains Ser52 and Ile91 in lieu of Ala52 and Leu91 for Hsp90β), which makes the development Hsp90α- or Hsp90β-selective inhibitors a daunting task.[27,28] Based on careful analysis of the co-crystal structures of radicicol bound to both Hsp90α and Hsp90β, a structure-based approach to design Hsp90β-selective inhibitors was pursued. In fact, we recently reported the first small molecule Hsp90β-selective inhibitor KUNB31 (Figure 1), which selectively induced the degradation of Hsp90β-dependent clients without the induction of Hsp90 levels.[27] In addition, KUNB31 was shown to manifest anti-proliferative activity at low micromolar concentrations against select cancer cell lines. Herein, we report the synthesis, evaluation, and preliminary SAR studies for KUNB31 analogs as Hsp90β-selective inhibitors.
Figure 1.

Structure of the known Hsp90 N-terminal inhibitor KUNB31, which showed >50 fold Hsp90β-selectivity.
Results and Discussion
Design
The design of Hsp90β-selective inhibitors was initiated by comparing the amino acids (Hsp90β contains Ala52 and Leu91 in lieu of Ser52 and Ile91 in Hsp90α) that differentiate the binding sites and the three water molecules that reside within the bottom pocket of each isoform. The co-crystal structure of Hsp90β bound to the pan-inhibitor radicicol revealed Thr184 and Asp93 to form hydrogen-bond interactions with radicicol’s carbonyl and 4-phenol (Figure 2b and 2c) via three conserved water molecules. However, these water molecules play a different role in each isoform due to the replacement of Ser52 in Hsp90α with Ala52 in Hsp90β. This subtle difference between the co-crystal structures of Hsp90α and Hsp90β suggests that an appendage at the 3- or 4-position of the resorcinol ring may allow access to a channel that resides in Hsp90β, but not in Hsp90α. Based on this hypothesis and the evaluation of initial compounds, KUNB31 was proposed to fit into the Hsp90β ATP-binding pocket, but did not appear to bind Hsp90α based on computational studies.[27] Therefore, it was proposed that the 2-phenol and amide moiety of KUNB31 would mimic the 2-phenol and lactone moiety present in the natural product, radicicol.
Figure 2.

Water-mediated network of hydrogen bonds. a Structures of the known Hsp90 N-terminal inhibitors radicicol and Fluoro-KUNB31 (numbers are given for clarity). b Modeling of radicicol into the N-terminal ATP-binding site of Hsp90α (PDB code: 2XAB). c Modeling of radicicol into the N-terminal ATP-binding site of Hsp90β (PDB code: 1UYM). d Co-crystal structure of Fluoro-KUNB31 bound to Hsp90β.[27]
The 5-isopropyl appendage was designed to introduce hydrophobic interactions with a hydrophobic pocket and most importantly, the isoxazole ring serves as a bicyclic analog, wherein the 3- and 4-positions of the resorcinol moiety were combined to introduce favorable interactions with the Hsp90β-isoform. KUNB31 was found to manifest an apparent Kd of 180 nM against Hsp90β, while exhibiting ~50-fold selectivity over Hsp90α and Grp94. The co-crystal structure of Hsp90β and the fluorinated analog, fluoro-KUNB31, revealed the molecule to bind similar to that predicted by computational studies (Fig 2a and 2d). Notably, the 5-isopropyl substituent established hydrophobic interactions with Phe138 and Val181of Hsp90β, whereas the isoxazole ring displaced both conserved water molecules A and B. Based on the co-crystal structure, it was envisioned that other heterocycles or substituted ring systems could serve as surrogates for the isoxazole ring and help to establish structure-activity relationships and the role played by each atom within the isoxazole ring. In addition, the proposed molecules can establish hydrogen-bonding interactions and elucidate the role played by each heteroatom with water molecules A and B (Fig 2b).
Chemistry
Synthesis of the proposed analogs started via the preparation of oxygen- and nitrogen-containing heterocycles to replace the isoxazole moiety present in KUNB31. The oxygen-containing furan derivative 5, was prepared in seven steps from the commercially available resorcinol derivative, 5-bromo-2,4-dihydroxybenzoic acid (1), which was readily converted into methyl ester 2 via Fischer esterification (Scheme 1). Selective alkylation of the 4-phenol was achieved using 2-bromoacetaldehyde diethyl acetal at 100°C in the presence of potassium carbonate, followed by exposure to Amberlyst-15 in toluene to yield the bicyclic benzofuran, 3.[29] Reactivity of the 4-phenol over the 2-hydroxyl results from the 2-phenol’s participation in an intra-molecular hydrogen bond with the neighboring carbonyl. Suzuki coupling of 3 with Molander’s potassium isopropenyltrifluoroborate,[30] followed by hydrogenation under Wilkinson’s conditions gave the 5-isopropylbenzofuran, 4. Finally, 4 was converted into amide 5 upon saponification of the methyl ester, followed by coupling of the resulting acid with isoindoline in the presence of HATU and DIPEA (Scheme 1).
Scheme 1.

Synthesis of benzofuran 5.
The nitrogen-containing indole, indazole, benzotriazole and benzimidazole derivatives were prepared from aniline 8, which was derived from commercially available methyl 4-amino-2-methoxybenzoate (6, Scheme 2). Selective iodination at the 5-position of aniline 6 was achieved with N-iodosuccinamide to yield iodoaniline 7. The isopropyl moiety was then installed via a Suzuki coupling reaction with Molendar’s isopropenyltrifluoroborate, followed by hydrogenation of the resulting olefin to afford 8.
Scheme 2.

Synthesis of aniline 8.
In order to generate the carbon-carbon bond at the 3-position, aniline 8 was converted into iodoaniline 9, which served as a common intermediate for the preparation of both indole 11 and indazole 13 (Scheme 3). A Sonogashira coupling reaction between intermediate 9 and trimethylsilyl acetylene gave the corresponding ortho-alkynyl aniline 10, which was then subjected to copper-mediated cyclization and concomitant TMS elimination to afford indole 11.[31] Iodoaniline 9 could also be converted into the corresponding ortho-toluidine intermediate 12, via a Suzuki coupling reaction with trimethyl boroxine. Toluidine 12 was subsequently converted into the diazonium salt, which upon treatment with potassium acetate in the presence of 18-crown-6 afforded indazole 13.[32]
Scheme 3.

Synthesis of indole 11 and indazole 13.
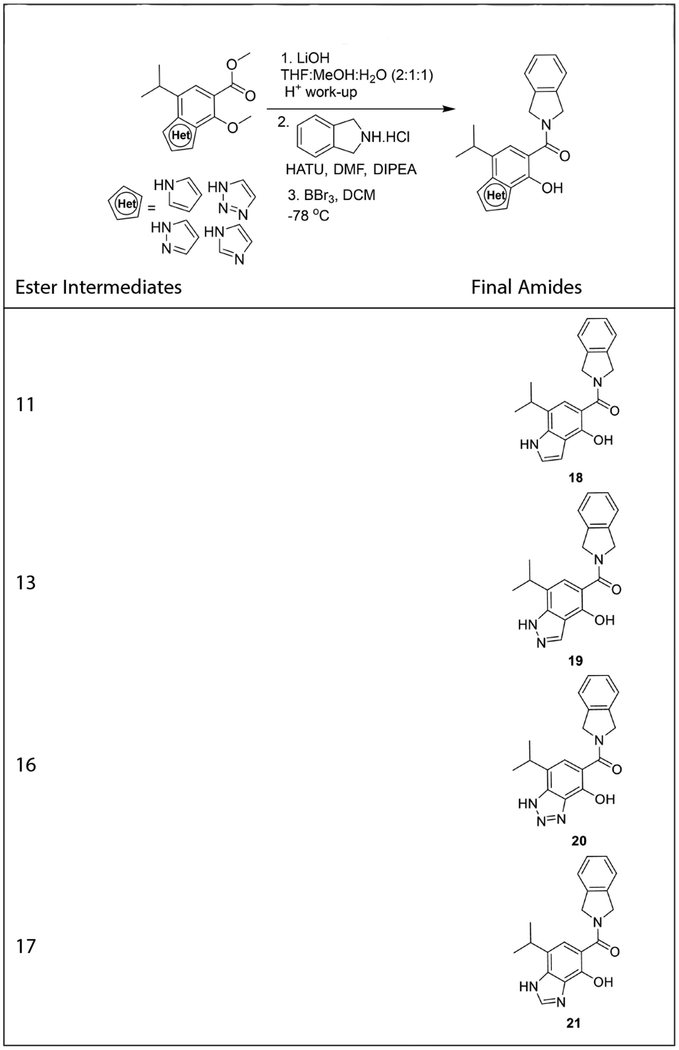
The benzotriazole and benzimidazole derivatives were synthesized via the conversion of intermediate 8 into nitroaniline, 14 (Scheme 4). Nitration was performed on acetylated 8 using ammonium nitrate in the presence of trifluoroacetic anhydride to provide the corresponding ortho-nitro acetanilide, which upon de-acetylation gave 14.[33,34] Reduction of the nitro group gave the requisite aniline, 15.[34] Treatment of 15 with iso-amyl nitrite in the presence of acetic acid gave the corresponding bicyclic benzotriazole 16,[35] whereas heating the same diamine in formic acid along with triethyl orthoformate at 120°C (in a sealed tube) afforded benzimidazole 17.[36] All ester intermediates (11, 13, 16 and 17) were subjected to hydrolysis, followed by coupling of the resulting carboxylic acids with isoindoline in the presence of HATU and DIPEA to afford the desired amides (Table 1). Cleavage of the methyl ethers gave the corresponding phenols 18, 19, 20, and 21, respectively.
Scheme 4.

Synthesis of benzotriazole 16 and benzimidazole 17.
Table 1.
Conversion of methyl ester into amide.
|
In order to examine the hydrogen bond interactions manifested by the 3- and 4-positions of the bicyclic ring system, a series of indanone derivatives was prepared. Commercially available 4-hydroxy indane 22 was converted into the corresponding 2-methoxy-5-bromo-1-benzoate (25) in 5 steps (Scheme 5).
Scheme 5.

Synthesis of indanone analogs 29a and 29b.
Hydroxyl-directed ortho-formylation of 22 using paraformaldehyde in the presence of magnesium chloride and trimethylamine, followed by methylation of the phenol and Pinnick oxidation of the resulting aldehyde gave 2-methoxy benzoic acid (23), which was isolated as methyl ester 24. Selective bromination at the 5-position provided access to the indanone. Benzylic oxidation of 25 with chromium trioxide gave two separable regio-isomeric ketones, 26a and 26b, in a 3:1 ratio, respectively. Structural assignments of 26a and 26b were based on 1H and 13C NMR spectral data. In particular, the H6 proton of 26a was shifted downfield (δ 8.14 ppm) due to the deshielding nature of the carbonyl at the para position, as compared to H6 of 26b (δ 7.80 ppm), which is located meta to the ketone. Similarly, the 13C NMR signal for the β-CH2 of 26b (δ 22.2 ppm) shifted upfield due to the shielding effect of the ortho-methoxy group, as compared to the β-CH2 of 26a (δ 27.3 ppm). Both 26a and 26b were subjected to a Suzuki coupling reaction with Molendar’s isopropenyltrifluoroborate and subsequent hydrogenation of the resulting olefin to give 27a and 27b, respectively. Hydrolysis of 27a and 27b and coupling of the requisite carboxylic acid with isoindoline in the presence of HATU and DIPEA gave the corresponding amides, 28a and 28b, which underwent demethylation to afford phenols, 29a and 29b, respectively.
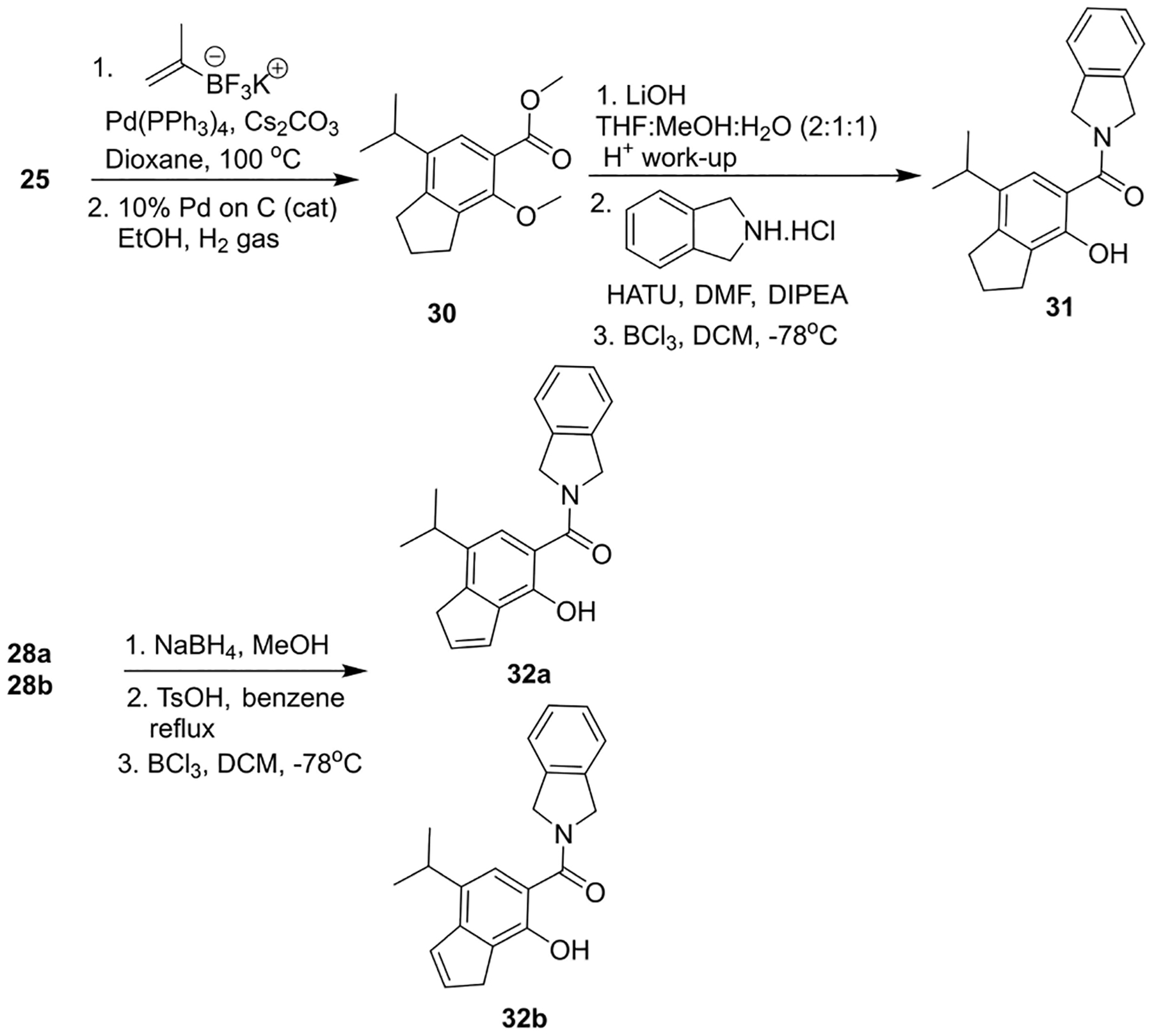
The bicyclic indane and indene scaffolds were prepared to determine the role played by the heteroatoms as compared to 29a and 29b. Indane 31 was prepared via intermediate 25 by installation of the isopropyl group into the 5-position, followed by hydrolysis of the ester, an amide coupling reaction with isoindoline, and finally, demethylation (Scheme 6). Isomeric ketones, 28a and 28b, were reduced to the corresponding alcohols before an acid-mediated dehydration reaction afforded the olefinic products, which underwent dealkylation to furnish isomers 32a and 32b.
Scheme 6.
Synthesis of indane 31 and indene analogs 32a and 32b.
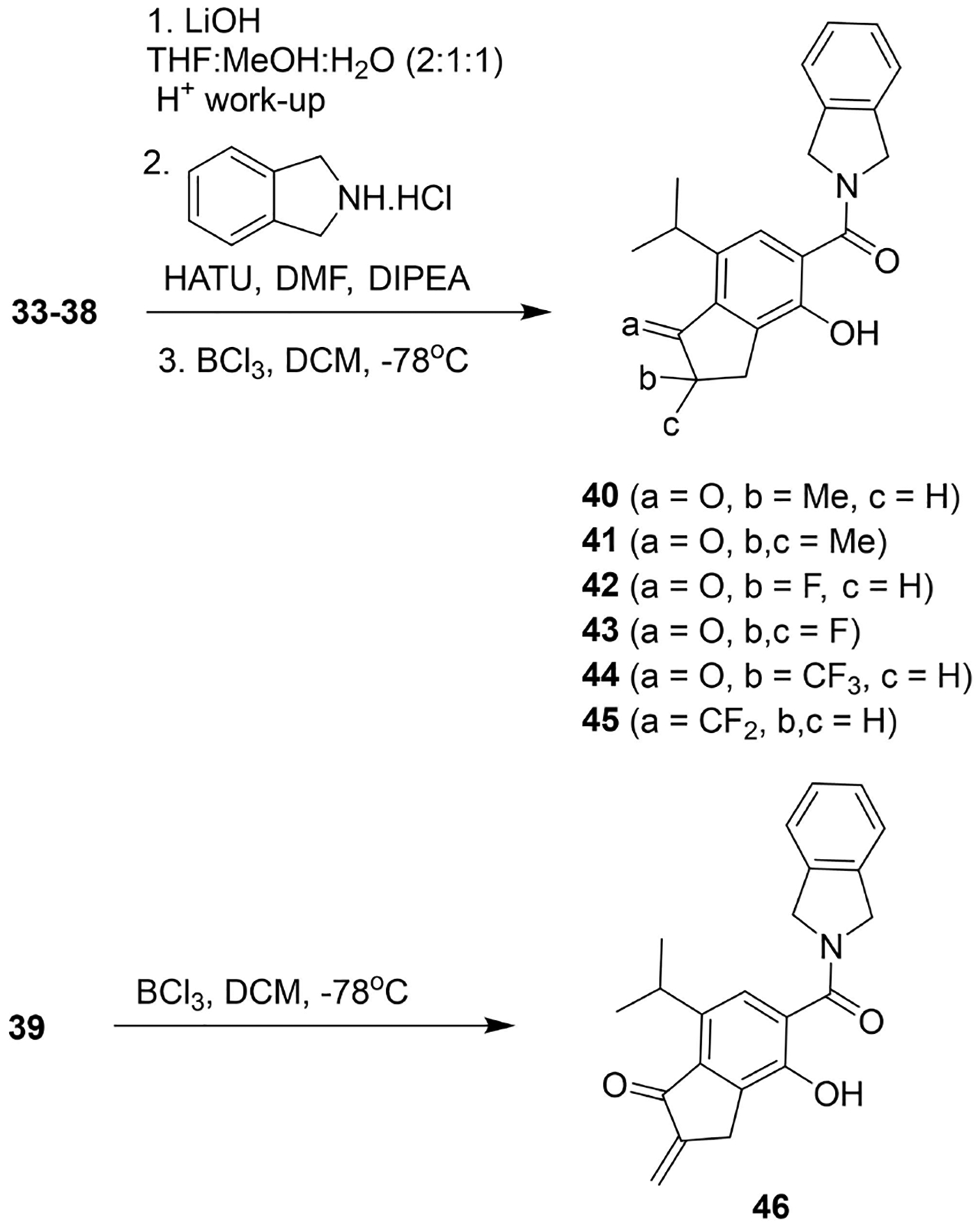
Utilizing scaffolds 27b and 28b, substituted indanone scaffolds were prepared by placing substituents onto the α- and β-positions, as well as by transforming the ketone into an electronically similar fluoro or difluoro alkene (Scheme 7). α-Methyl and α,α-dimethyl derivatives (33 and 34) were prepared via generation of the α-carbanion of 27b, followed by alkylation with iodomethane.[37] Exposure of 27b to Selectfluor gave the corresponding fluoro-derivative, 35.[38] Whereas generation of the tertiarybutylsilyl enol ether of 35 followed by treatment with Selectfluor produced the difluoro-derivative, 36.[39] Finally, the enol acetate of 27b was treated with a cationic trifluoromethylating reagent to give the α-trifluormethylated indanone, 37.[40] The difluoro analog 38, resulted from a reaction between 27b and (difluoromethylene)phosphonium ylide, which was generated in situ from CF3TMS and PPh3 in the presence of LiI.[41] α-Methylene 39 was prepared from intermediate 28b via an iron (III)-catalyzed homologation reaction (Scheme 7).[42] Compounds 33–38 were subjected to hydrolysis and the resulting carboxylic acids converted into the corresponding amides, which upon demethylation gave phenols 40–45 (Scheme 8). Similarly, phenol 46 was realized after demethylation of 39. Indenone 50 was prepared from intermediate 27b and was converted into the corresponding benzyl-protected phenol 47, followed by hydrolysis of the ester and subsequent amide coupling with isoindoline to give 48. The ketone of 48 was converted into the vinyl triflate, which underwent Saegusa oxidation conditions to produce the α, β-unsaturated ketone, 49. Hydrogenolysis of 49 afforded indanone 50 (Scheme 9). Oxime 51 and methyl oxime 52 were prepared from 29b under similar conditions enlisting hydroxylamine and methoxylamine hydrochloride, respectively (Scheme 10).
Scheme 7.

Synthesis of substituted indanones 33–39.
Scheme 8.
Synthesis of analogs 40–46.
Scheme 9.

Synthesis of α,β-unsaturated analog 50.
Scheme 10.

Synthesis of oxime 50 and methyl oxime 52.
Two tetralone derivatives were prepared to examine the effect of ring expansion at the 3- and 4-positions of the resorcinol moiety (Scheme 11). Bromination of commercially available 5-methoxy-1-tetralone (53) using N-bromosuccinimide gave the corresponding bromine derivative 54. The isopropenyl group was introduced via a Suzuki coupling reaction with potassium isopropenyl trifluoroborate followed by reduction of the olefin to afford 55. Formylation of 55 via a Duff reaction gave aldehyde 56, which underwent oxidation with chromium trioxide to produce a mixture of regio-isomeric keto acids, 57a and 57b, in a 3:1 ratio, respectively. Treatment of the acids with HATU and isoindoline afforded the separable and isomeric amides, 58a and 58b. Finally, Lewis-acid catalyzed demethylation of 58a and 58b yielded the corresponding phenols, 59a and 59b, respectively.
Scheme 11.

Synthesis of tetralone analogs 59a & 59b.
Structure-activity relationships
The binding affinity of KUNB31 analogs for Hsp90α and Hsp90β was determined via fluorescence polarization (FP) assays (entry 1, Table 2). The co-crystal structure of fluoro-KUNB31 bound to Hsp90β revealed the isoxazole of KUNB31 to displace two conserved water molecules (A and B) upon binding to Hsp90β and to provide hydrogen bonding interactions with Ala52 and Leu91.[27]
Table 2.
IC50 values of heterocyclic derivatives against Hsp90α and Hsp90β determined using fluorescence polarization (FP) assay.
| Entry | Compound | HSP90β [IC50 in μM] | HSP90α [IC50 in μM] | Selectivity |
|---|---|---|---|---|
| 1 | KUNB31 [a] | 0.18 ± 0.01 | 9.55 ± 1.08 | > 50 |
| 2 | 5 | 0.27 ± 0.09 | > 100 | > 370 |
| 3 | 18 | > 100 | > 100 | NA |
| 4 | 19 | > 100 | > 100 | NA |
| 5 | 20 | > 100 | > 100 | NA |
| 6 | 21 | 0.45 ± 0.15 | 2.22 ± 0.19 | 4.9 |
| 7 | 29a | > 100 | > 100 | NA |
| 8 | 29 b | 0.15 ± 0.04 | 0.79 ± 0.14 | >5 |
| 9 | 31 | 5.23 ± 0.13 | > 100 | > 19.1 |
| 10 | 32a | 8.29 ± 0.17 | > 100 | >12 |
| 11 | 32b | 1.86 ± 0.11 | > 100 | > 53 |
| 12 | 40 | 8.83 ± 1.48 | > 100 | >11 |
| 13 | 41 | > 100 | > 100 | NA |
| 14 | 42 | 0.34 ± 0.01 | 2.42 ± 0.07 | 7.1 |
| 15 | 43 | 18.19 ± 1.3 | > 100 | >5 |
| 16 | 44 | 19.59 ± 2.2 | > 100 | >5 |
| 17 | 45 | > 100 | > 100 | NA |
| 18 | 46 | > 100 | > 100 | NA |
| 19 | 50 | 6.75 ± 0.01 | 43.13 ± 11.5 | >5 |
| 20 | 51 | > 100 | > 100 | NA |
| 21 | 52 | > 100 | > 100 | NA |
| 22 | 59a | > 100 | > 100 | NA |
| 23 | 59 b | > 100 | > 100 | NA |
KUNB31 IC50 values were taken from Ref. [27].
Replacement of the isoxazole ring with a furan (entry 2) appears to produce similar activity, but albeit a lower affinity for Hsp90α. The data also suggests that a hydrogen bond acceptor at the 4-position of the resorcinol moiety is important for Hsp90β affinity. Benzofuran analog 5 exhibited a IC50 of 0.27 μM against Hsp90β, while exhibiting no measurable affinity for Hsp90α (IC50 >100). Introduction of the nitrogen atom into the 3- and/or 4-positions of the resorcinol ring appears detrimental to both affinity and selectivity (entry 5). The binding affinity of the indanone analogs was also investigated, which included a ketone at the 4-position to extend the hydrogen bond acceptor. The inclusion of a hydrogen bond acceptor at the 4-position of the resorcinol ring improved the binding affinity for other analogs, however indanone 29b exhibited strong affinity for both isoforms, and consequently low selectivity. In contrast, 29a did not exhibit affinity for either isoform (entries 7 and 8).
The importance of a hydrogen bond acceptor at the 4-position is further supported by the poor binding affinity manifested by indane derivative 31. Indene 32b manifested better affinity and selectivity for Hsp90β as compared to 32a. Surprisingly, substituted indanone 40 did not display better affinity for Hsp90β, but did exhibit greater affinity than the dimethyl product 41, which was not tolerated. Similarly, introduction of fluorine or trifluoromethyl at the α-position (42 and 44) was tolerable but the difluoro compound (43) displayed both poor affinity and selectivity. Fluoro-alkene 45 and the Michael acceptor 46 did not display good affinity nor selectivity for either isoform, and 50 exhibited slightly better affinity for Hsp90β than Hsp90α (entries 16–19).
In vitro evaluation
The anti-proliferative activity manifested by these compounds was evaluated against human HT-29 (colorectal adenocarcinoma), NCI–H1299 (human non-small cell lung cancer), and UM-UC3 (human bladder cell carcinoma) cell lines (Table 3). Compound 5 manifested an IC50 of 18.84 ± 2.60 μM, 17.54 ± 0.42 μM, and 71.63 ± 3.73 μM against HT29, NCI-H1299, and UM-UC3 cancer cell lines, respectively. Although compounds 5, 29b, and 50 were found to exhibit activity against various cancer cell lines, other compounds were relatively inactive or exhibited weak cellular activity against the cell lines tested. In contrast, compound 50 exhibited low micromolar activity, which appears to be independent of Hsp90β inhibition.
Table 3.
IC50 values of heterocyclic derivatives against HT29, NCI H1299 and UM-UC3 cell lines.
| Entry | Compound | HT29[a] [IC50 in μM] | NCI—H1299[b] [IC50 in μM] | UM-UC3[c] [IC50 in μM] |
|---|---|---|---|---|
| 1 | KUNB31 [d] | 3.7 ± 0.34 | Not tested | 3.01 ± 0.56 |
| 2 | 5 | 18.84 ± 2.6 | 17.54 ± 0.42 | 71.63 ± 3.73 |
| 3 | 18 | 61.95 ± 1.42 | > 100 | > 100 |
| 4 | 19 | > 100 | > 100 | > 100 |
| 5 | 20 | > 100 | 74.99 ± 21.4 | > 100 |
| 6 | 21 | 93.74 ± 2.67 | 59.96 ± 10.5 | 52.95 ± 3.32 |
| 7 | 29 a | 63.54 ± 1.69 | 67.62 ± 4.13 | > 100 |
| 8 | 29 b | 21.72 ± 2.60 | 10.02 ± 3.28 | 55.55 ± 1.34 |
| 9 | 31 | 59.33 ± 0.18 | 58.10 ± 0.34 | 57.4 ± 13.68 |
| 10 | 32 a | 51.79 ± 2.18 | 46.13 ± 1.11 | 51.3 ± 1.83 |
| 11 | 32 b | 47.35 ± 2.94 | 41.39 ± 1.20 | 59.4 ± 4.8 |
| 12 | 40 | 60.25 ± 0.63 | 59.04 ± 1.78 | 70.4 ± 0.77 |
| 13 | 41 | 54.33 ± 1.32 | 51.31 ± 0.15 | 51.9 ± 1.27 |
| 14 | 42 | 58.29 ± 1.51 | 20.6 ± 0.28 | 69.9 ± 0.77 |
| 15 | 43 | > 100 | > 100 | > 100 |
| 16 | 44 | 75.29 ± 0.66 | 57.76 ± 8.01 | 59.65 ± 0.21 |
| 17 | 45 | Not tested | Not tested | Not tested |
| 18 | 46 | 66.52 ± 1.35 | 68.2 ± 1.20 | > 100 |
| 19 | 50 | 2.33 ± 0.48 | 1.53 ± 0.16 | 1.18 ± 0.1 |
| 20 | 51 | 51.17 ± 2.23 | 33.73 ± 1.78 | 51.65 ± 3.32 |
| 21 | 52 | 51.17 ± 2.23 | 85.95 ± 7.7 | 78.25 ± 15.7 |
| 22 | 59 a | 51.17 ± 2.23 | 83.33 ± 1.31 | > 100 |
| 23 | 59 b | Not tested | Not tested | Not tested |
colon cancer cell line
non-small cell lung carcinoma cell line
bladder cancer cell line.
KUNB31 IC50 values were taken from Ref. [27].
Western blot analyses were then performed on HT-29 cell lysates treated with compound 5 to determine the effect of Hsp90β inhibition on cancer cells. Compound 5 was chosen as it manifested the greatest selectivity towards Hsp90β as compared to Hsp90α (>370 fold). After a 24 h incubation, compound 5 induced the degradation of CDK4 and Akt, which are both known Hsp90β-dependent client proteins in a dose dependent manner.[27] In contrast, the protein expression levels of survivin, which is an Hsp90α-dependent client protein, remained unaffected until higher concentrations of compound 5 were reached, which provides evidence of isoform-selective inhibition in the cellular environment. Total levels of Hsp90 client proteins Erk5 and Her-2 also decreased at higher concentrations of compound 5 against the HT-29 cell line. The levels of Hsp70 appear to be induced upon treatment with compound 5, which is consistent with earlier studies (Figure 3).
Figure 3.

Western blot analysis of client proteins with compound 5 in HT-29 cell line. DMSO and 500 nM Geldanamycin (GDA) were used as negative and positive controls, respectively.
Conclusion
Most of the Hsp90 pan-inhibitors that have undergone clinical evaluation displayed adverse events, the mechanisms for which remained poorly understood.[17–19,21,22] However, it was proposed that inhibition of an individual isoform could provide a novel opportunity to minimize the toxicities associated with pan-inhibition, as a more defined set of clients can be modulated. Consequently, efforts have focused on the development of isoform-selective inhibitors of Hsp90 to treat a variety of diseases. Previously, the first small molecule Hsp90β-selective inhibitor (KUNB31) was disclosed and selectively induced the degradation of Hsp90β-dependent clients without the induction of Hsp90 levels.[27] Based on the KUNB31 structure, we synthesized, evaluated, and performed preliminary SAR studies on KUNB31 analogs as Hsp90β-selective inhibitors. Compound 5 was shown to manifest ~370-fold selectivity for Hsp90β versus Hsp90α. Furthermore, Western blot analysis confirmed that the Hsp90β selective inhibitor 5 induced the degradation of Hsp90β-dependent clients, while exhibiting minimal or no effect on Hsp90α-dependent clients. Further optimization of compound 5 will likely lead to the development of more selective and potent compounds, which can be used to elucidate the role played by Hsp90β in cancer and other diseases.
Experimental Section
General Information
All reactions were performed in oven-dried glassware under an argon atmosphere unless otherwise stated. Commercially available anhydrous solvents and reagents were utilized during synthesis. Flash column chromatography was performed using silica gel (40–63 μm particle size). The 1H spectra were recorded on a Brucker instrument at 500, and 400 MHz frequencies and 13 C NMR were recorded at 126, and 101 MHz frequencies. Data are reported as p=pentet, q=quartet, t=triplet, d=doublet, s=singlet, bs=broad singlet, m=multiplet, coupling constant(s) in Hz. High resolution mass spectral data were obtained on a time-of-flight mass spectrometer, and analysis was performed using electrospray ionization. The final products were determined to be ≥ 95% purity using an Agilent technologies Infinity II instrument with a Poroshell 120 EC-C18 2.7 μm (4.6×100 mm) column using a solvent gradient of solvent A (H2O with 0.1% trifluoroacetic acid) and solvent B (acetonitrile with 0.1% trifluoroacetic acid). Thin-layer chromatography was performed with TLC silica gel 60F254 plates purchased from Millipore Sigma and visualized by UV light.
Methyl 5-bromo-4-(2,2-dimethoxyethoxy)-2-hydroxybenzoate (3):
Anhydrous potassium carbonate (K2CO3) (2.1 g, 15 mmol, 1.5 equiv.) was added to a solution of 2 (2.5 g, 10 mmol) in DMF (20 mL) and vigorously stirred. Bromoacetaldehyde diethyl acetal (10.0 g, 7.6 mL, 51 mmol 5.1 equiv.) was added dropwise over 5 min. The resulting suspension heated at 100°C for 2 h under inert Argon atmosphere. After the reaction mixture was cooled to rt, the suspension was poured into 5% aqueous solution of potassium hydrogen sulfate (KHSO4 aq, 200 mL) at 0°C, and then extracted with EtOAc (3×100 mL). The combined organic layers were washed with brine, dried over anhydrous sodium sulfate (Na2SO4), and concentrated in vacuo. The light brown oily residue was used in the next step without further purification. The crude brown oil was dissolved in toluene (50 mL) and Amberlyst-15 (310 mg) was added to the solution. The reaction mixture was stirred at reflux for 4 h and equipped with Dean-Stark apparatus to remove water. After cooling to rt, the resulting suspension was filtered, and the residue washed with toluene (80 mL). The combined filtrate was concentrated in vacuo and the residue was purified by silica gel column chromatography (EtOAc/hexane; 1:9 to afford product 3 as off-white solid (1.2 g, Yield 44%, over 2 steps): 1H NMR (400 MHz, CDCl3): δ (ppm) 11.40 (s, 1H), 7.94 (s, 1H), 7.64 (d, J=7.6 Hz, 1H), 7.05 (d, J=7.1 Hz, 1H), 3.98 (s, 3H); 13C NMR (101 MHz, CDCl3): δ (ppm) 170.5, 157.0, 156.4, 145.2, 128.3, 118.5, 107.8, 106.1, 94.6, 52.7. MS (ESI) m/z 271 (M + 1), 273 (M + 3).
Methyl 4-hydroxy-7-isopropylbenzofuran-5-carboxylate (4):
A solution of 3 (500 mg, 1.84 mmol) in 1,4-dioxane (10 mL) and water (0.5 mL) was treated with cesium carbonate (1.80 mg, 13.0 mmol, 3 equiv.) followed by potassium isopropenyltrifluoroborate (409 mg, 2.77 mmol, 1.5 equiv.) in a reaction vessel. The reaction mixture was thoroughly degassed by purging with Ar for about 15 min and thereafter tetrakis(triphenylphosphine)palladium (0) (213 mg, 0.2 mmol, 0.1 equiv.) was added under inert Argon atmosphere. The reaction vessel was then sealed with a Teflon cap and heated to 100°C for 12 h. The reaction mixture was cooled to rt, and filtered through a small pad of celite, and then washed with ethyl acetate a few times. Solvent was removed to afford a dark brown oily residue. 1H NMR spectra of the crude residue showed the desirable signals and so the crude residue was used in the next step without further purification. In a round bottom flask, the crude residue from the previous step was dissolved in absolute EtOH (20 ml) and charged with Wilkinson’s catalyst (340 mg, 0.368 mmol, 0.2 equiv.) while stirring. The flask was 1st evacuated by vacuum and then filled with H2 gas from a balloon. This process was repeated for a few times and then finally left the reaction mixture in presence of H2 under a balloon pressure for 12 h. After the H2 balloon was removed, the reaction mixture was filtered through a pad of celite and eluted with EtOAc (50 mL). The eluent was concentrated, and the residue purified by flash chromatography (SiO2, 1:9 ethyl acetate/hexanes) to afford pure compound 4 as off-white solid (200 mg, Yield 46%, over 2 steps). 1H NMR (400 MHz, CDCl3): δ (ppm) 11.32 (s, 1H), 7.68–7.45 (m, 2H), 6.97 (d, J=2.2 Hz, 1H), 3.96 (s, 3H), 3.42–3.16 (m, 1H), 1.36 (d, J=7.0 Hz, 6H). 13C NMR (101 MHz, CDCl3): δ (ppm) 171.5, 158.0, 156.0, 144.1, 125.0, 122.0, 117.0, 105.9, 105.0, 52.3, 28.5, 22.7. MS (ESI) m/z 235 (M + 1).
(4-Hydroxy-7-isopropylbenzofuran-5-yl)(isoindolin-2-yl) methanone (5):
Methyl ester 4 (54 mg, 0.23 mmol) was dissolved in mixed solvent consisting of THF:MeOH:H2O (2:1:1, 10 ml) followed by solid LiOH (17 mg, 0.69 mmol) was added to the solution in portions. The reaction mixture was stirred at room temperature for 18 h. The solvent was concentrated, and the residue was dissolved in cold aqueous HCl solution (1 N) to adjust the pH around 2. The aqueous phase was then extracted with EtOAc (3 × 20 ml) and combined organic layers were dried (Na2SO4) and concentrated to afford crude carboxylic acid residue as off-white solid, which was used in the next step without further purification. The carboxylic acid was dissolved in Dry DMF (5 mL) and then HATU (130 mg, 0.23 mmol) DIPEA (0.1 mL, 0.68 mmol) were added to this solution. The reaction mixture was stirred at room temperature for 10 min. before isoindoline. HCl salt (53 mg, 1.22 mmol) was added to the same and continue stirring for 16 h at room temperature. The reaction mixture was then poured on crushed ice, which gave a light-yellow solid, which was collected via filtration and washed with cold water. The crude solid was then purified by column chromatography (silica gel 120 – 200 mesh, gradient 2–10% EtoAc/n-Hexane) to afford pure product 5 (44 mg, Yield 60%, over 2 steps) as a pale-yellow solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 11.65 (s, 1H), 7.58 (d, J=2.2 Hz, 1H), 7.39 (s, 1H), 7.31 (s, 4H), 6.98 (d, J=2.2 Hz, 1H), 5.13 (s, 4H), 3.51–3.23 (m, 1H), 1.41 (d, J=6.9 Hz, 6H); 13C NMR (101 MHz, CDCl3): δ (ppm) 172.1, 156.1, 154.4, 144.1, 128.0, 123.4, 122.8, 120.9, 117.7, 110.0, 105.1, 54.4, 28.8, 22.9; HRMS (ESI) m/z [M+H]+ calculated for C20H20NO3 322.1434, found 322.1437.
Methyl 4-amino-5-iodo-2-methoxybenzoate (7):
A solution of methyl 4-amino-2-methoxybenzoate (15 g, 0.083 mol) in dry acetonitrile (500 ml) was treated with N-iodosuccinimide (27.85 g, 0.123 mol) at 0°C. The reaction mixture was stirred at room temperature for 3 h. The mixture was concentrated and then diluted with EtOAc, the organic layer was washed with sat. aq. NaHCO3, dried over anhydrous Na2SO4 and concentrated under reduced pressure to afford the product 7 (25 g, Yield 97%) as yellowish brown amorphous solid, which was used in the next step without further purification. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.16 (s, 1H), 6.27 (s, 1H), 4.52 (br. s, 2H), 3.82 (s, 3H), 3.81 (s, 3H); 13C NMR (101 MHz, CDCl3): δ (ppm) 164.8, 161.7, 151.6, 142.8, 111.2, 97.3, 71.6, 56.1, 51.7; MS (ESI) m/z 308 (M + 1).
Methyl 4-amino-5-isopropyl-2-methoxybenzoate (8):
Cesium carbonate (10.61 g, 97.71 mmol), potassium isopropenyltrifluoroborate (5.78 g, 39.08 mmol) and water (20 mL) were added sequentially to an 1,4-dioxane (200 ml) solution of 7 (10 g, 32.57 mmol) in a sealed tube. The reaction mixture was degassed by bubbling argon for about 10 min. Tetrakis(triphenylphosphine)palladium(0) (3.76 g, 3.27 mmol) was then added under the same inert atmosphere. The reaction tube was sealed with a cap lined with a Teflon tape and heated at 120°C for 12 h. The reaction mixture was cooled to rt and filtered through a small pad of celite (eluted with ethyl acetate 200 mL). Solvent was removed and the residue was purified by flash chromatography (silica gel, gradient elution 5–15% EtOAc/hexanes) to afford Methyl 4-amino-2-methoxy-5-(prop-1-en-2-yl)benzoate (6.4 g, Yield 89%) as an off-white amorphous solid.1H NMR (400 MHz, CDCl3): δ (ppm) 7.57 (s, 1H), 6.23 (s, 1H), 5.24 (s, 1H), 4.98 (s, 1H), 3.79 (s, 3H), 3.76 (s, 3H), 1.99 (s, 3H); MS (ESI) m/z 222 (M + 1).
A hydrogen pressure vessel was filled with ethanolic (100 ml) solution of Methyl 4-amino-2-methoxy-5-(prop-1-en-2-yl)benzoate (6 g, 27.14 mmol). Palladium on carbon (10%, 800 mg) and molecular sieves (10 g) were added to this solution. The reaction mixture was degassed and purged with hydrogen gas before it was set to stir for 16 h under a hydrogen pressure (300 psi). The reaction mixture was filtered through a pad of celite and eluted with EtOAc. The eluent was concentrated, and the residue purified by flash chromatography (silica gel, 2–10% ethyl acetate/hexanes) to afford pure compound 34 (5.2 g, Yield 93%) as an off-white solid. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 7.52 (s, 1H), 6.35 (s, 1H), 5.84 (s, 2H), 3.76 (S, 3H), 3.73 (s, 3H), 2.99–2.90 (m, 1H), 1.19 (s, 3H), 1.17 (s, 3H); MS (ESI) m/z 224 (M + 1).
Methyl 4-amino-3-iodo-5-isopropyl-2-methoxybenzoate (9):
A solution of methyl 4-amino-2-methoxybenzoate 8 (5 g, 22.3 mmol) in dry acetonitrile (100 mL) was treated with N-iodosuccinimide (5.02 g, 26.7 mmol) at 0°C. The resulting mixture was stirred at room temperature for 6 h. The mixture was concentrated and partitioned between EtOAc (200 mL) and sat. aq. NaHCO3 solution (100 mL). The organic phase was washed with sat. aq. NaHCO3 solution (2 × 100 mL) and brine (100 mL), dried over anhydrous Na2SO4 and concentrated under reduced pressure, the residue was purified by column chromatography (silica gel, 100–200 mesh, 2–10% EtOAc/hexane) to afford the pure product 9 (25 g, Yield: 97%) as a pale yellow solid.1H NMR (400 MHz, CDCl3) δ (ppm) 7.64 (s, 1H), 3.82 (s, 3H), 3.77 (s, 3H), 2.81–2.70 (m, 1H), 1.22 (s, 3H), 1.20 (s, 3H); MS (ESI) m/z 350 (M + 1).
Methyl 4-amino-5-isopropyl-2-methoxy-3-((trimethylsilyl)ethynyl) benzoate (10):
A solution of compound 9 (2 g, 5.73 mmol) in dry Et3N (30 mL) in a sealed tube was treated with ethynyl-trimethylsilane (1.58 mL, 11.46 mmol), PdCl2(PPh3)2 (80 mg, 0.11 mmol) and CuI (5 mg, 0.028 mmol). The tube was sealed with a cap and lined with a Teflon tape. The mixture was heated at reflux for 4 h. The solvent was removed under reduced pressure and the residue was filtered through Celite using EtOAc (100 mL) as solvent. The solvent was removed, and the crude residue was absorbed on silica and purified by column chromatography (silica gel, 2–15% EtOAc/Hexane) to afford pure compound 10 (1.6 g, Yield: 87%) as an off-white solid.1H NMR (400 MHz, CDCl3): δ (ppm) 7.75 (s, 1H), 4.07 (s, 3H), 3.98 (s, 3H), 2.97–2.85 (m, 1H), 1.39 (s, 3H), 1.38 (s, 3H), 0.41 (s, 9H); MS (ESI) m/z 320 (M + 1).
Methyl 7-isopropyl-4-methoxy-1H-indole-5-carboxylate (11):
A mixture of compound 10 (1.5 g, 4.7 mmol), copper(I) iodide (895 mg, 9.4 mmol), and DMF (20 mL) was stirred under argon at 110°C for 4 h. The reaction mixture was diluted with diethyl ether (50 mL) and filtered through Celite. The residue was rinsed with another portion of diethyl ether (50 mL) and the combined filtrates were washed with water (50 mL) and then brine (50 mL). The organic layer was separated, dried over Na2SO4, filtered, and concentrated. The crude residue was purified by column chromatography (silica gel, eluting with 10–20% EtOAc/Hexane) to afford pure product 11 (750 mg, Yield: 65%) as colourless amorphous solid.1H NMR (400 MHz, CDCl3): δ (ppm) 8.36 (bs, 1H), 7.47 (s, 1H), 7.15–7.11 (m, 1H), 6.71–6.67 (m, 1H), 4.00 (s, 3H), 3.85 (s, 3H), 3.16–3.05 (m, 1H), 1.32 (s, 3H), 1.30 (s, 3H); MS (ESI) m/z 248 (M + 1).
Methyl 4-amino-5-isopropyl-2-methoxy-3-methylbenzoate (12):
A solution of compound 9 (1 g, 2.86 mmol) in 1,4-dioxane (15 mL) was treated with trimethyl boroxine (0.8 mL, 5.72 mmol), cesium carbonate (1.86 g, 5.72 mmol) and 1,1’-bis(diphenylphosphino) ferrocene-palladium(II) dichloride dichloromethane complex (116 mg, 0.14 mmol). The reaction mixture was degassed by bubbling Ar and then heated in a microwave reactor at 130°C. for 1.5 h. The mixture was partitioned between water (30 mL) and ethyl acetate (2 × 60 mL). The combined organics were dried over sodium sulphate, filtered and evaporated to dryness. The residue was purified by column chromatography on silica gel eluting with an ethyl acetate/hexane (10–20%) gradient to afford compound 12 (620 mg, Yield: 91%) as an off-white solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.55 (s, 1H), 3.80 (s, 3H), 3.70 (s, 3H), 2.81–2.73 (m, 1H), 2.06 (s, 3H), 1.21 (s, 3H), 1.19 (s, 3H); MS (ESI) m/z 238 (M + 1).
Methyl 7-isopropyl-4-methoxy-1H-indazole-5-carboxylate (13):
A suspension of compound 12 (500 mg, 2.10 mmol) in water (3 mL) was cooled to 0°C in an ice bath and then treated with HBF4 solution (48% Wt. in H2O, 4 mL, 21.0 mmol). After 5 min NaNO2 (180 mg, 2.52 mmol) dissolved in water (5 mL) was added dropwise and stirred for 1 h at 0°C. The reaction gave a yellow solid precipitate, which was filtered and dried to afford product (400 mg), which was immediately used to the next step. A stirred solution of 6-isopropyl-3-methoxy-4-(methoxycarbonyl)-2-methylbenzenediazonium tetrafluoroborate (400 mg, 1.19 mmol) in chloroform (10 mL) was treated with potassium acetate (350 mg, 3.57 mmol) and 18-crown-ether (20 mg, 0.04 mmol). The reaction mixture was stirred at room temperature for 12 h. the reaction mixture was portioned between CHCl3 (50 mL) and water (20 mL). The combined organic layers were dried over sodium sulphate and evaporated to dryness. The residue was purified by column chromatography on silica gel eluting with an ethyl acetate/hexane (10–20%) gradient to afford compound 13 (150 mg, Yield: 91% for 2 steps) as an off-white solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.28 (s, 1H), 7.63 (s, 1H), 4.15 (s, 3H), 3.87 (s, 3H), 3.28–3.17 (m, 1H), 1.35 (s, 3H), 1.33 (s, 3H); MS (ESI) m/z 249 (M + 1).
Methyl 4-acetamido-5-isopropyl-2-methoxy-3-nitrobenzoate (14):
A solution of 8 (2.0 g, 9.0 mmol) in dry DCM (30 ml) under Argon atmosphere was treated with trimethylamine (3.7 ml, 27.0 mmol) at 0°C followed by acetic anhydride (1.7 ml, 18.0 mmol). The reaction mixture was warmed to room temperature and stirred for additional 3 h. After quenching the reaction mixture with saturated NaHCO3 solution, DCM layer was separated, and aqueous layer was extracted with DCM. The combined organic layers were dried over Na2SO4 and then concentrated under reduced pressure. The crude residue was purified by flash chromatography (silica gel, 2–10% ethyl acetate/hexanes) to afford pure Methyl 4-acetamido-5-isopropyl-2-methoxybenzoate (1.9 g, Yield 80%) as yellow solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.74 (s, 1H), 7.48 (s, 1H), 3.85 (s, 6H), 3.01–2.80 (m, 1H), 2.21 (s, 3H), 1.21 (d, J=6.8 Hz, 6H); 13C NMR (101 MHz, CDCl3): δ (ppm) 168.9, 166.6, 158.2, 139.4, 129.3, 115.7, 106.4, 56.2, 52.0, 27.3, 24.9, 23.0; MS (ESI) m/z 265 (M + 1).
A solution of Methyl 4-acetamido-5-isopropyl-2-methoxybenzoate (1.3 g, 4.9 mmol) in dry DCM (20 ml) was treated with TFAA (4.1 ml, 29 mmol), followed by solid amonium nitrate (780 mg, 9.8 mmol). The brown color reaction mixture was allowed to stir for 30 min at room temperature before it was quenched with cold water. The recation mixture was diluted with DCM and then organic layer was separated. After extracting the aqueous layer with DCM a few times, the organic layers were combined and washed with saturated NaHCO3 solution, followed by brine. Combined organic layer was then dried ovet Na2SO4 and concentrated under reduced pressure to afford light brown solid, which was used in the next step without further purification.
The crude light brown solid was dissolved in MeOH (20 ml) and cooled to 0°C. Conc. H2SO4 (0.8 ml, 15 mmol) was added to it slowly dropwise with stirring. After the addition was completed, the reaction mixture was brought to RT and then refluxed for 16 h. After cooling the reaction mixture to RT, the solvent was evaporated and the oily residue was dissolved into EtOAc. The organic layer was washed with cold saturated solution of NaHCO3, followed by water, dried over Na2SO4 and concentrated to a brown solid, which was purified by flash chromatography (silica gel, 2–5% EtOAc/hexanes) to afford pure 14 (530 mg, Yield 45% over 2 steps) as brown solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 6.98 (d, J=1.2 Hz, 1H), 4.23 (s, 3H), 3.91 (s, 3H), 3.19 (m, J=6.9, 1.2 Hz, 1H), 1.26 (d, J=6.8 Hz, 6H).
Methyl 7-isopropyl-4-methoxy-1H-benzo[d][1,2,3]triazole-5-carboxylate (16):
Nitro compound 14 (100 mg, 0.373 mmol) was dissolved into MeOH (10 ml) and the resulting solution was degassed by purging Argon for 10 min. Pd/C was added followed by trimethylamine (0.5 ml, 4 mmol). After the reaction mixture was stirred under 1 atmosphere pressure of H2 gas filled balloon for 6 h, it was filtered through a pad of Celite and rinsed with MeOH. The filtrate was concentrated to a brown viscus liquid (15), which was dissolved into AcOH (5 ml). To this solution was dropwise added i-amylnitrite (0.1 ml, 0.8 mmol) via a micro syringe at room temperature under Ar. The reaction mixture was stirred at the same temperature for 0.5 h and then diluted with water and EtOAc. The organic layer was separated, and the aqueous layer was extracted with EtOAc. The combined organic layer was washed with cold saturated NaHCO3 solution, followed by water, dried over Na2SO4 and concentrated under reduced pressure to a brown gum, which was purified by flash chromatography (silica gel, 5–15% EtOAc/hexanes) to afford pure 16 (55 mg, Yield 54% over 2 steps) as off-white solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.72 (s, 1H), 4.54 (s, 3H), 3.97 (s, 3H), 3.44 (m, 1H), 1.35 (d, J=6.9 Hz, 6H); 13C NMR (101 MHz, CDCl3): δ (ppm) 166.7, 160.5, 157.3, 152.9, 139.0, 128.6, 120.6, 61.7, 52.7, 29.3, 22.1. HRMS (ESI) m/z [M+H]+ calculated for C12H16N3O3 250.1189, found 250.1186.
Methyl 7-isopropyl-4-methoxy-1H-benzo[d]imidazole-5-carboxy-late (17):
Nitro compound 14 (100 mg, 0.373 mmol) was dissolved into MeOH (10 ml) and the resulting solution was degassed by purging Ar for 10 min. Pd/C was added followed by trimethylamine (0.5 ml, 4 mmol). After the reaction mixture was stirred under 1 atmosphere pressure of H2 gas filled balloon for 6 h, it was filtered through a pad of Celite and rinsed with MeOH. The filtrate was concentrated to a brown viscus liquid (15), which was dissolved into trietheoxyetahne (2 ml) and then formic acid (0.05 ml, 1.3 mmol) was added to it. The reaction mixture was placed in a seal tube and heated to 120°C for 48 h. Solvent was evaporated off and the residue was absorbed on silica for purification by flash chromatography (silica gel, 30–35% EtOAc/hexanes) to afford pure 17 (70 mg, Yield 76% over two steps) as white solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.16 (s, 1H), 7.62 (s, 1H), 4.13 (s, 3H), 3.95 (s, 3H), 3.43 (m, 1H), 1.35 (d, J=6.9 Hz, 6H). HRMS (ESI) m/z [M+H]+ calculated for C13H17N2O3 249.1247, found 249.1233.
General procedure for ester hydrolysis followed by amidification:
Methyl ester (11, 13,16 and 17, 1 mmol) was dissolved in 10 mL of THF: MeOH: H2O (2:1:1). Solid lithium hydroxide (3 mmol) was added to this solution in portions with stirring. The reaction mixture, which was initially a suspension eventually became a clear solution with continuous stirring. The stirring continued at ambient temperature until the SM was fully consumed (monitored by TLC). The solvent was evaporated under reduced pressure and the residue was acidified with 1 N HCl (until pH ~2) to obtain an off-white solid, which was extracted with EtOAc. The organic layer was dried (Na2SO4) and concentrated to afford the corresponding carboxylic acid, which was taken to the next step without further purification.
The carboxylic acid (1 mmol) was dissolved in anhydrous DMF (5 mL) and HATU (1.5 mmol) was added followed by DIPEA (3 mol). The reaction mixture was allowed to stir at rt for 10 min. before isoindoline. HCl salt (1.5 mmol) was added to the reaction mixture and stirred for another18 h. After the complete consumption of SM (monitored by TLC) the reaction mixture was quenched with the addition of crushed ice, which gave a yellow to brown solid precipitate. The solid was separated by filtration and re-dissolved in EtOAc, washed with brine, dried (Na2SO4) and concentrated to afford an off-white residue, which was purified by column chromatography (silica gel 120 – 200 mesh, gradient 2–15% EtOAc/n-Hexane) to offered corresponding pure amide.
Isoindolin-2-yl(7-isopropyl-4-methoxy-1H-indol-5-yl)methanone:
1H NMR (400 MHz, DMSO-d6): δ (ppm) 11.30 (s, 1H), 7.43–7.20 (m, 5H), 6.82 (s, 1H), 6.63 (s, 1H), 4.84 (s, 2H), 4.54 (s, 2H), 3.94 (s, 3H), 3.82–3.77 (m, 1H), 1.29 (s, 3H), 1.28 (s, 3H); MS (ESI) m/z 335 (M + 1).
Isoindolin-2-yl(7-isopropyl-4-methoxy-1H-indazol-5-yl) methanone:
1H NMR (400 MHz, CDCl3): δ (ppm) 8.22 (s, 1H), 7.92 (s, 1H), 7.33–7.13 (m, 3H), 7.09–7.00 (m, 2H), 5.24 (s, 2H), 4.96 (s, 2H), 4.10 (s, 3H), 3.27–3.18 (m, 1H), 1.29 (s, 6H); MS (ESI) m/z 336 (M + 1).
Isoindolin-2-yl(4-isopropyl-7-methoxy-1H-benzo[d][1,2,3]triazol-6-yl)methanone:
1H NMR (400 MHz, CDCl3): δ (ppm) 15.35 (s, 1H), 7.45–6.80 (m, 5H), 5.04 (s, 2H), 4.69 (m, 2H), 4.40 (s, 3H), 2.99 (s, 1H), 0.92 (s, 6H); 13C NMR (101 MHz, CDCl3): δ (ppm) 171.29, 169.22, 165.94, 162.86, 136.52, 136.14, 136.13, 127.71, 127.55, 123.03, 61.44, 52.38, 38.71, 36.67, 29.64, 22.48. HRMS (ESI) m/z [M+H]+ calculated for C19H21N4O2 337.1661, found 337.1659.
Isoindolin-2-yl(7-isopropyl-4-methoxy-1H-benzo[d]imidazol-5-yl) methanone:
1H NMR (400 MHz, MeOH-d4): δ (ppm) 8.13–7.95 (m, 1H), 7.43–6.93 (m, 5H), 5.01 (s, 2H), 4.69 (s, 2H), 4.19 (s, 3H), 3.37 (m, 1H), 1.36 (m, 6H). HRMS (ESI) m/z [M+H]+ calculated for C20H22N3O2 336.1705, found 336.1706.
General procedure for de-methylation/Synthesis of compounds (18–21):
The appropriate amide (1 mmol) was dissolved in dry DCM (5 mL) and cooled to −78°C before BCl3 solution (3 mmol, 1 M sol. in DCM) was added to it dropwise via a syringe. The reaction was continued at −78°C for 2 h. and the progress of the reaction was monitored by TLC (50% EtoAc/n-Hexane). After completion of the reaction cold water was added to quench the reaction and gradually warmed up to rt. The organic layer was separated, and the aqueous layer was extracted with DCM a few times. The combined organic layers were dried (Na2SO4) and concentrated under reduced pressure. The crude residue was purified by column chromatography (silica gel 120 – 200 mesh, gradient 10–15% EtoAc/n-Hexane) to afford corresponding products (18–21) as pale-yellow solid.
(4-hydroxy-7-isopropyl-1H-indol-5-yl)(isoindolin-2-yl)methanone (18):
1H NMR (400 MHz, CDCl3): δ (ppm) 11.84 (s, 1H), 8.34 (s, 1H), 7.24–7.11 (m, 4H), 7.07 (s, 1H), 6.74 (s, 1H), 5.08 (s, 4H), 3.20–3.07 (m, 1H), 1.31 (s, 6H); HRMS (ESI+), m/z [M+Na+] calculated for C20H20N2O2Na 343.3816; found 343.1836.
(4-Hydroxy-7-isopropyl-1H-indazol-5-yl)(isoindolin-2-yl) methanone:
1H NMR (400 MHz, CDCl3): δ (ppm) 12.37 (s, 1H), 8.25 (s, 1H), 7.38 (s, 1H), 7.30–7.21 (m, 4H), 7.19 (s, 1H), 5.06 (s, 4H), 3.23–3.14 (m, 1H), 1.38 (s, 3H), 1.36 (s, 3H); HRMS (ESI+), m/z [M+Na+] calculated for C19H19N3O2Na 344.3746; found 344.3425.
(7-Hydroxy-4-isopropyl-1H-benzo[d][1,2,3]triazol-6-yl)(isoindolin-2-yl)methanone:
1H NMR (400 MHz, CDCl3): δ (ppm) 7.37–6.94 (m, 7H), 5.05 (s, 2H), 4.66 (s, 2H), 4.41 (s, 3H), 2.98 (s, 1H), 1.08–0.70 (m, 6H); 13C NMR (126 MHz, CDCl3) δ (ppm) 169.61, 144.70, 136.04, 135.74, 127.96, 127.78, 123.10, 122.67, 122.57, 119.67, 61.65, 53.61, 52.70, 29.87, 22.41. HRMS (ESI) m/z [M+H]+ calculated for C18H19N4O2 323.1504, found 323.1502.
(4-Hydroxy-7-isopropyl-1H-benzo[d]imidazol-5-yl)(isoindolin-2-yl) methanone:
1H NMR (400 MHz, DMSO-d6): δ (ppm) 9.29 (s, 1H), 7.46–7.03 (m, 6H), 4.88 (s, 2H), 4.74 (s, 2H), 3.40 (hept, J=7.2 Hz, 1H), 1.31 (d, J=6.8 Hz, 6H). 13C NMR (101 MHz, DMSO-d6): δ (ppm) 167.9, 141.1, 137.8, 129.4, 128.7, 126.6, 125.8, 123.4, 120.9, 119.7, 28.6, 23.2, 21.5. HRMS (ESI) m/z [M+H]+ calculated for C19H20N3O2 322.1548, found 322.1550.
4-Methoxy-2,3-dihydro-1H-indene-5-carboxylic acid (23):
Compound 23 was synthesized by a series of sequential steps as shown in the Scheme 5. The synthesis begun from the formylation of Indan-4-ol using previously reported procedure (ref 1) to provide 4-hydroxy-2,3-dihydro-1H-indene-5-carbaldehyde, which was methylated by dissolving the aldehyde (20 g, 0.12 mol) in dry Acetone (200 mL) added potassium carbonate (34 g, 0.25 mol) and Methyl Iodide (31 mL, 0.49 mol). To the reaction mixture was heated to 70–75°C. for 6 h. The solvent was evaporated and to the residue added water (200 mL) and Ethyl acetate (500 mL). The organic layer was separated, the aqueous layer was extracted with Ethyl acetate (2×100 mL) and the combined organic layers dried over anhydrous Sodium sulphate and evaporated at reduced pressure to give 4-methoxy-2,3-dihydro-1H-indene-5-carbaldehyde (20 g, Yield: 93%) as an off-white solid. 1H NMR (500 MHz, CDCl3): δ (ppm) 10.30 (s, 1H), 7.59 (d, J=7.7 Hz, 1H), 6.98 (d, J=7.7 Hz, 1H), 3.87 (s, 3H), 2.96 (t, J=7.4 Hz, 2H), 2.87 (t, J=7.4 Hz, 2H), 2.12–2.02 (m, 2H); MS (ESI) m/z 177 (M + 1).
To a suspension of 4-methoxy-2,3-dihydro-1H-indene-5-carbaldehyde (18 g, 0.10 mol) in acetonitrile (200 mL) is added a solution of sodium chlorite (23 g, 0.26 mol) and sodium dihydrogen phosphate (61 g, 0.51 mol) in water (30 mL). To the solution is added 2-methyl-2-butene (32 mL, 31 mol). The resulting homogeneous solution is stirred at room temperature for 2 h. After completion of the reaction, the solvent was evaporated, and the residue is diluted with water and acidified with 1 M hydrochloric acid to pH=1 and extracted with ethyl acetate (3×200 mL). The combined organic layers are extracted with 1 M sodium hydroxide solution which is further acidified with 6 M hydrochloric acid and extracted with ethyl acetate (3×200 ml). The organic layer is washed with brine (200 mL), dried over sodium sulfate and concentrated to yield the acid 23 (19 g, Yield: 88%) as pale-yellow solid, which was used to the next step without any further purification; MS (ESI) m/z 191 (M − 1).
Methyl 4-methoxy-2,3-dihydro-1H-indene-5-carboxylate (24):
Compound 23 (18 g, 94 mmol) was dissolved in dry Acetonitrile (360 mL) and potassium carbonate (26 g, 190 mmol) and Methyl Iodide (12 mL, 190 mmol) were added. To the reaction mixture was heated to 70–75°C. for 6 h. The solvent was evaporated and to the residue added water (200 mL) and Ethyl acetate (500 mL). The organic layer was separated, the aqueous layer was extracted with Ethyl acetate (2× 100 mL) and the combined organic layers dried over Sodium sulphate and evaporated at reduced pressure to afford product 24 (18.2 g, Yield: 95%) as an off-white solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.56 (d, J=7.7 Hz, 1H), 6.93 (d, J=7.7 Hz, 1H), 3.82 (s, 3H), 3.79 (s, 3H), 2.93–2.84 (m, 4H), 2.09–1.98 (m, 2H); 13C NMR (101 MHz, CDCl3): δ (ppm) 167.1, 156.7, 152.0, 137.6, 130.3, 121.9, 119.8, 61.0, 52.1, 33.5, 29.9, 25.4; MS (ESI) m/z 207 (M + 1).
Methyl 7-bromo-4-methoxy-2,3-dihydro-1H-indene-5-carboxylate (25):
Compound 24 (15 g, 72.7 mmol) was dissolved in Dry acetonitrile (300 mL) and NBS (16 g, 87.2 mmol) was added. The reaction mixture was stirred at room temperature for 12 h, the solvent was evaporated on rotavapor at reduced pressure, the obtained crude product was dissolved in EtoAc (300 mL) and washed with brine (100 mL) and H2O (100 mL), the organic layer was separated and dried over anhydrous Na2SO4 and concentrated on rotavapor. The product was purified by column chromatography (silica gel 120 – 200 mesh, gradient 2–15% EtoAc/n-Hexane) to afford pure product 25 (17 g, Yield: 82%) as yellow solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.72 (s, 1H), 3.82 (s, 3H), 3.77 (s, 3H), 3.01 (t, J=7.5 Hz, 2H), 2.89 (t, J=7.5 Hz, 2H), 2.11–2.01 (m, 2H); 13C NMR (101 MHz, CDCl3): δ (ppm) 178.1, 165.7, 155.9, 139.5, 132.9, 124.0, 113.7, 61.2, 52.4, 35.2, 31.3, 24.2; MS (ESI) m/z 285 (M + 1), 287 (M + 3).
Methyl 7-bromo-4-methoxy-3-oxo-2,3-dihydro-1H-indene-5-carboxylate (26a) & Methyl 7-bromo-4-methoxy-1-oxo-2,3-dihydro-1H-indene-5-carboxylate (26b):
To a stirred solution of compound 18 (3.6 g, 12.6 mmol) in acetic acid (20 mL) was added Chromium trioxide (3.38 g, 37.8 mmol) dissolved in a mixture of acetic acid (16 mL) and water (5 mL). The reaction mixture was heated to 70–75°C for 48 h. The solvent was evaporated and to the residue added water (20 mL) and Ethyl acetate (50 mL). The organic layer was separated, the aqueous layer was extracted with Ethyl acetate (2× 30 mL) and the combined organic layers dried over anhydrous Sodium sulphate and evaporated at reduced pressure, the residue was purified by column chromatography (Silica gel 100–200 mesh, hexane/EtOAc 5–30%) to afford 26a and 26b (2.2 g and 760 mg, Yield 82%) as a 3:1 isomeric mixture as an amorphous solid. Isomer-I (26b): 1H NMR (400 MHz, CDCl3): δ (ppm) 7.80 (s, 1H), 3.96 (s, 3H), 3.94 (s, 3H), 3.14–3.10 (m, 2H), 2.79–2.76 (m, 2H); 13C NMR (101 MHz, CDCl3): δ (ppm) 203.3, 165.0, 156.8, 151.6, 138.1, 135.2, 129.9, 112.7, 61.8, 53.0, 37.1, 22.2; MS (ESI) m/z 300 (M + 1), 302 (M + 3); Isomer-II (26a): 1H NMR (400 MHz, CDCl3) δ (ppm) 8.14 (s, 1H), 4.04 (s, 3H), 3.93 (s, 3H), 3.08–3.03 (m, 2H), 2.79–2.74 (m, 2H); 13C NMR (101 MHz, CDCl3): δ (ppm) 202.7, 165.1, 160.6, 157.8, 139.9, 136.0, 132.0, 126.2, 115.5, 63.4, 53.1, 37.0, 27.3; MS (ESI) m/z 300 (M + 1), 302 (M + 3).
Methyl 7-isopropyl-4-methoxy-3-oxo-2,3-dihydro-1H-indene-5-carboxylate (27a):
In an oven dried sealed tube was charged with 26a (1.13 g, 4.34 mmol), cesium carbonate (3.69 g, 13.0 mmol), potassium isopropenyltrifluoroborate (671 mg, 5.21 mmol) and 1,4-dioxane (20 mL), water (2 mL) closed with septum. The reaction mixture degassed with an argon for about 30 min to eliminate the oxygen. Tetrakis(triphenylphosphine)palladium (0) (501 mg, 0.43 mmol) was added under argon condition. The tube was sealed with a cap lined with a Teflon tape. The tube was heated at 100°C for 12 h, cooled to rt, and filtered through a small pad of celite (elution with ethyl acetate 50 mL). Solvent was distilled of at reduced pressure and the residue purified by flash chromatography (silica gel 100–200 mesh, 1:3 ethyl acetate/hexanes) to afford methyl 4-methoxy-3-oxo-7-(prop-1-en-2-yl)-2,3-dihydro-1H-indene-5-carboxylate (950 mg, Yield 84%) as a colorless solid.1H NMR (400 MHz, CDCl3): δ (ppm) 7.44 (s, 1H), 5.14 (s, 1H), 4.88 (s, 1H), 3.88 (s, 6H), 3.08 (t, J=6.1 Hz, 2H), 2.65 (t, J=6.0 Hz, 2H), 2.03 (s, 3H);); MS (ESI) m/z 261 (M + 1).
Palladium on carbon (10%, 100 mg) and molecular sieves were added to a solution of methyl 4-methoxy-3-oxo-7-(prop-1-en-2-yl)-2,3-dihydro-1H-indene-5-carboxylate (900 mg, 3.44 mmol) in ethanol (50 mL). The suspension was stirred for 16 h under a hydrogen atmosphere (balloon pressure) before it was filtered through a pad of celite and eluted with EtOAc (50 mL). The eluent was concentrated, and the residue purified by flash chromatography (silica gel, 1:9 ethyl acetate/hexanes) to afford pure compound 27a (820 mg, Yield 92%) as an off-white solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.57 (s, 1H), 4.08–4.00 (m, 1H), 3.90 (S, 3H), 3.84 (s, 3H), 3.04 (t, J=5.9 Hz, 2H), 2.64 (t, J=5.9 Hz, 2H), 1.17 (s, 3H), 1.15 (s, 3H); MS (ESI) m/z 263 (M + 1).
6-(Isoindoline-2-carbonyl)-4-isopropyl-7-methoxy-2,3-dihydro-1H-inden-1-one (28a):
Compound 27a (800 mg, 3.44 mmol) was dissolved in 10 mL of THF:MeOH:H2O (2:1:1) added Lithium hydroxide (219 mg, 9.16 mmol). The suspension was stirred for 1 h at room temperature. The solvent was concentrated, and the residue was dissolved in Ice cold water (10 mL) and acidified with Con. HCl until pH ~2, the obtained solid was filtered and dried to afford 7-isopropyl-4-methoxy-3-oxo-2,3-dihydro-1H-indene-5-carboxylic acid (650 mg, Yield: 93%) as an off-white solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.94 (s, 1H), 4.11–4.01 (m, 1H), 3.99 (S, 3H), 3.11 (t, J=5.9 Hz, 2H), 2.71 (t, J=5.9 Hz, 2H), 1.19 (s, 3H), 1.17 (s, 3H); MS (ESI) m/z 249 (M + 1).
The carboxylic acid (110 mg, 0.443 mmol) was dissolved in Dry DMF (5 mL), HATU (202 mg, 0.531 mmol) and DIPEA (0.27 mL, 1.32 mmol) were added. The reaction mixture was stirred at room temperature for 10 min. followed by the Isoindoline (69 mg, 0.443 mmol) and continue the stirring for 5 h at rt. After completion of the reaction the solvent was evaporated on rotavapor at reduced pressure, the crude obtained was dissolved in EtoAc (50 mL) and washed with brine (20 mL) and H2O (20 mL), the organic layer was separated and dried over anhydrous Na2SO4 and concentrated on rotavapor. The crude product was purified by column chromatography (silica gel 120 – 200 mesh, gradient 2–15% EtoAc/n-Hexane) to offered pure product 28a (130 mg, Yield 82%) as yellow solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.38–7.25 (m, 4H), 7.14 (d, J=7.4 Hz, 1H), 5.03 (s, 2H), 4.63 (bs, 2H), 4.17–4.09 (m, 1H), 3.90 (s, 3H), 3.14–3.09 (m, 2H), 2.74–2.70 (m, 2H), 1.23 (s, 3H), 1.22 (s, 3H); 13C NMR (100 MHz, CDCl3): δ (ppm) 206.8, 168.0, 151.1, 148.4, 145.9, 136.2, 135.9, 135.4, 134.9, 127.8, 127.6, 124.0, 123.1, 122.6, 61.2, 53.5, 52.1, 36.9, 26.8, 23.1, 22.2; MS (ESI) m/z 350 (M + 1).
7-hydroxy-6-(isoindoline-2-carbonyl)-4-isopropyl-2,3-dihydro-1H-inden-1-one (29a):
Compound 28a (100 mg, 0.28 mmol) was dissolved in Dry DCM (5 mL), BCl3 (1 M sol. in DCM, 0.57 mL, 0.84 mmol) was added at −78°C. The reaction was continued at −78°C for 2 h. the progress of the reaction was monitored by TLC (50% EtoAc/n-Hexane). After completion of the reaction the solvent was evaporated on rotavapor at reduced pressure, the crude obtained was dissolved in EtoAc (20 mL) and washed with brine (10 mL) and H2O (10 mL), the organic layer was separated and dried over anhydrous Na2SO4 and concentrated on rotavapor. The crude product was purified by column chromatography (silica gel 120 – 200 mesh, gradient 2–15% EtoAc/n-Hexane) to afford pure product 29a (25 mg, Yield 26%) as pale-yellow solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 10.35 (s, 1H), 7.41(s, 1H), 7.37–7.11 (m, 4H), 5.02 (s, 4H), 4.10–4.00 (m, 1H), 3.02–2.94 (m, 2H), 2.67–2.60 (m, 2H), 1.20 (s, 6H); 13C NMR (101 MHz, CDCl3): δ (ppm) 207.9, 170.5, 154.7, 145.0, 139.1, 136.75, 135.7, 130.5, 128.2, 124.4, 124.1, 123.6, 122.9, 121.0, 55.9, 53.4, 37.3, 26.7, 23.6, 22.0; HRMS (ESI+), m/z [M+ Na+] calculated for C21H21NO3Na 358.3916; found 358.1496.
Methyl 7-isopropyl-4-methoxy-1-oxo-2,3-dihydro-1H-indene-5-carboxylate (27b):
In an oven dry sealed tube was charged with 26b (1.13 g, 4.34 mmol), cesium carbonate (3.69 g, 13.0 mmol), potassium isopropenyltrifluoroborate (671 mg, 5.21 mmol) and 1,4-dioxane (20 mL), water (2 mL) closed with septum. The reaction mixture was purged with an argon for about 30 min to eliminate the oxygen. Tetrakis(triphenylphosphine)palladium (0) (501 mg, 0.43 mmol) was added under nitrogen condition. The tube was sealed with a cap lined with a Teflon tape. The tube was heated at −100°C for 12 h, cooled to rt, and filtered through a small pad of celite (elution with ethyl acetate). Solvent was removed and the residue purified by flash chromatography (silica gel, 1:3 ethyl acetate/hexanes) to afford methyl 4-methoxy-1-oxo-7-(prop-1-en-2-yl)-2,3-dihydro-1H-indene-5-carboxylate (950 mg, Yield 84%) as a colorless solid.1H NMR (400 MHz, CDCl3): δ (ppm) 7.48 (s, 1H), 5.13 (s, 1H), 4.82 (s, 1H), 3.93 (s, 3H), 3.83 (s, 3H), 3.08 (t, J=6.1 Hz, 2H), 2.65 (t, J=6.0 Hz, 2H), 2.03 (s, 3H); MS (ESI) m/z 261 (M + 1).
Palladium on carbon (10%, 100 mg) and molecular sieves were added to an ethanolic (50 mL) solution of methyl 4-methoxy-1-oxo-7-(prop-1-en-2-yl)-2,3-dihydro-1H-indene-5-carboxylate (1 g, 3.83 mmol). The suspension was stirred for 16 h under a hydrogen atmosphere (balloon pressure) before it was filtered through a pad of celite and eluted with EtOAc (50 mL). The eluent was concentrated, and the residue purified by flash chromatography (silica gel, 1:9 ethyl acetate/hexanes) to afford pure compound 27b (910 mg, Yield 92%) as an off-white solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.80 (s, 1H), 3.93 (S, 3H), 3.84 (s, 3H), 3.05–2.98 (m, 3H), 2.68–2.63 (m, 2H), 1.22 (s, 3H), 1.20 (s, 3H); MS (ESI) m/z 263 (M + 1).
5-(Isoindoline-2-carbonyl)-7-isopropyl-4-methoxy-2,3-dihydro-1H-inden-1-one (28b):
To a suspension of Compound 27b (900 mg, 3.44 mmol) in 10 mL of THF:MeOH:H2O (2:1:1) Lithium hydroxide (222 mg, 1.03 mmol) was added. The suspension was stirred for 1 h at room temperature. The solvent was concentrated, and the residue was dissolved in Ice cold water (10 mL) and acidified with Con. HCl until pH ~2, the obtained solid was filtered and dried to afford 7-isopropyl-4-methoxy-1-oxo-2,3-dihydro-1H-indene-5-carboxylic acid (680 mg, Yield: 93%) as an off-white solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 8.23 (s, 1H), 4.17 (s, 3H), 3.11–3.07 (m, 2H), 3.06–2.99 (m, 1H), 2.75–2.70 (m, 2H), 1.23 (s, 6H); MS (ESI) m/z 249 (M + 1).
The carboxylic acid (110 mg, 0.443 mmol) was dissolved in Dry DMF (5 mL), HATU (202 mg, 0.531 mmol) and DIPEA (0.27 mL, 1.32 mmol) were added. The reaction mixture was stirred at room temperature for 10 min. Isoindoline (69 mg, 0.443 mmol) was added and continue the stirring for 5 h at rt. After completion of the reaction the solvent was evaporated on rotavapor at reduced pressure, the crude obtained was dissolved in EtoAc (50 mL) and washed with brine (20 mL) and H2O (20 mL), the organic layer was separated and dried over anhydrous Na2SO4 and concentrated on rotavapor. The crude product was purified by column chromatography (silica gel 120 – 200 mesh, gradient 2–15% EtoAc/n-Hexane) to offered pure product 28b (130 mg, Yield 82%) as yellow solid. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 7.54 (s, 1H), 7.40–7.24 (m, 4H), 4.85 (s, 2H), 4.50 (s, 2H), 3.83 (s, 3H), 3.15–3.03 (m, 3H), 2.70–2.63 (m, 2H), 1.25 (s, 3H), 1.23 (s, 3H); MS (ESI) m/z 350 (M + 1).
4-Hydroxy-5-(isoindoline-2-carbonyl)-7-isopropyl-2,3-dihydro-1H-inden-1-one (29b):
Compound 28b (100 mg, 0.28 mmol) was dissolved in Dry DCM (5 mL) and BCl3 (1 M sol. in DCM, 0.57 mL, 0.84 mmol) was added at −78°C. The reaction was continued at −78°C for 2 h. the progress of the reaction was monitored by TLC (50% EtoAc/n-Hexane). After completion of the reaction the solvent was evaporated on rotavapor at reduced pressure, the crude obtained was dissolved in EtoAc (20 mL) and washed with brine (10 mL) and H2O (10 mL), the organic layer was separated and dried over anhydrous Na2SO4 and concentrated on rotavapor. The crude product was purified by column chromatography (silica gel 120 – 200 mesh, gradient 2–15% EtoAc/n-Hexane) to afford pure product 30 (28 mg, Yield 27%) as a pale-yellow solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 9.53 (s, 1H), 7.47 (s, 1H), 7.37–.02 (m, 4H), 4.96 (s, 2H), 4.73 (s, 2H), 3.08–2.94 (m, 3H), 2.75–2.70 (m, 2H), 1.20 (s, 3H), 1.18 (s, 3H); 13C NMR (101 MHz, CDCl3): δ (ppm) 210.3, 167.3, 154.5, 151.8, 137.94, 136.8, 136.4, 133.3, 127.9, 123.2, 122.8, 122.6, 53.3, 52.0, 36.2, 29.5, 24.7, 23.2; HRMS (ESI+), m/z [M+Na+] calculated for C21H21NO3Na 358.3916; found 358.1464.
Methyl 7-isopropyl-4-methoxy-2,3-dihydro-1H-indene-5-carboxy-late (30):
Compound 30 was synthesized by a procedure similar to compound 27a (1.2 g, Yield: 85%) as an off-white solid 1H NMR (400 MHz, CDCl3): δ (ppm) 7.52 (s, 1H), 3.89 (s, 3H), 3.82 (s, 3H), 3.03–2.84 (m, 5H), 2.17–1.98 (m, 2H), 1.22 (d, J=7.0 Hz, 6H). 13C NMR (101 MHz, CDCl3): δ (ppm) 167.4, 154.4, 149.3, 139.9, 137.5, 126.5, 122.3, 61.0, 52.1, 31.8, 30.7, 30.0, 25.0, 23.1; MS (ESI) m/z 249 (M + 1).
Isoindolin-2-yl(7-isopropyl-4-methoxy-2,3-dihydro-1H-inden-5-yl) methanone:
Followed the similar procedure described for the synthesis of compound 28a (0.2 g, Yield: 75%) as an off-white solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.40–7.09 (m, 4H), 5.30 (s, 1H), 5.02 (s, 2H), 4.68 (s, 2H), 3.83 (s, 3H), 3.07–2.85 (m, 5H), 2.20–2.07 (m, 2H), 1.24 (d, J=7.0 Hz, 6H). 13C NMR (101 MHz, CDCl3): δ (ppm) 169.5, 150.4, 145.7, 140.3, 137.0, 136.5, 135.8, 128.5, 127.69, 122.7, 61.01, 53.7, 52.2, 31.5, 30.8, 25.1, 23.1. HRMS (ESI) m/z [M + 1]+ calculated for C21H24NO2 322.1803, found 322.1802.
(4-hydroxy-7-isopropyl-2,3-dihydro-1H-inden-5-yl)(isoindolin-2-yl)methanone (31):
Compound 31 was synthesized by a similar procedure described for 29a (0.030 g, Yield: 62%) as a pale yellow solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 10.62 (s, 1H), 7.29 (d, J=22.6 Hz, 5H), 5.08 (s, 4H), 2.98 (dt, J=21.9, 7.2 Hz, 5H), 2.12 (p, J=7.5 Hz, 2H), 1.27 (d, J=6.9 Hz, 6H). 13C NMR (101 MHz, CDCl3): δ (ppm) 171.7, 154.2, 148.4, 134.5, 136.1, 131.7, 127.9, 122.8, 122.4, 115.2, 32.05, 30.58, 29.53, 24.77, 23.52. HRMS (ESI) m/z [M+H]+ calculated for C21H24NO2 322.1803, found 322.1802.
Isoindolin-2-yl(7-isopropyl-4-methoxy-1H-inden-5-yl)methanone:
Followed the similar procedure described for compound 28a (0.12 g, Yield: 76%) as an off-white solid.1H NMR (400 MHz, CDCl3): δ (ppm) 7.32–6.90 (m, 6H), 6.64–6.48 (m, 1H), 4.94 (s, 2H), 4.58 (s, 2H), 3.85 (s, 3H), 3.45 (t, J=2.0 Hz, 2H), 3.22–3.03 (m, 1H), 1.21 (d, J=6.9 Hz, 6H); 13C NMR (101 MHz, CDCl3): δ (ppm) 169.7, 150.2, 146.2, 137.0, 136.6, 135.0, 133., 130.14, 127.8, 123.4, 123.2, 122.7, 60.9, 53.8, 37.6, 30.6, 23.6. MS (ESI) m/z 334 (M + 1).
(4-Hydroxy-7-isopropyl-1H-inden-5-yl)(isoindolin-2-yl)methanone (32a):
Compound 32a was synthesized by a similar procedure described for 29a (0.042 g, Yield: 69%) as a pale yellow solid. 1H NMR (400 MHz, Benzene-d6) δ (ppm) 11.71 (s, 1H), 6.94 (m, 4H), 6.75 (m, 2H), 6.00 (dt, J=5.6, 2.0 Hz, 1H), 4.49 (m, 4H), 2.86 (t, J=2.0 Hz, 2H), 2.73–2.47 (m, 1H), 0.96 (d, J=6.9 Hz, 6H); 13C NMR (101 MHz, CDCl3): δ (ppm) δ 171.9, 153.7, 148.2, 137.5, 131.9, 130.4, 129.9, 128.4, 127.9, 124.5, 123.5, 113.8, 50.3, 37.2, 30.5, 23.9; HRMS (ESI) m/z [M+H]+ calculated for C21H22NO2 320.1635, found 320.1645.
Isoindolin-2-yl(4-isopropyl-7-methoxy-1H-inden-6-yl)methanone:
Followed the similar procedure described for compound 28a (0.110 g, Yield: 73%) as a pale yellow solid. 1H NMR (400 MHz, Benzene-d6) δ 7.28 (s, 1H), 7.07 (t, J=1.9 Hz, 1H), 7.04–6.80 (m, 3H), 6.68–6.53 (m, 1H), 6.30 (dt, J=5.6, 2.0 Hz, 1H), 4.98 (s, 2H), 4.47 (s, 2H), 3.74 (s, 3H), 3.09 (t, J=2.0 Hz, 2H), 2.85 (hept, J=6.9 Hz, 1H), 1.13 (d, J=6.9 Hz, 6H). 13C NMR (101 MHz, Benzene-d6) δ 168.3, 148.4, 144.0, 139.5, 137.1, 136.6, 133.5, 130.3, 128.8, 122.9, 122.4, 121.7, 61.9, 53.3, 52.11, 37.9, 30.7, 22.7; MS (ESI) m/z 334 (M + 1).
(7-hydroxy-4-isopropyl-1H-inden-6-yl)(isoindolin-2-yl)methanone (32b):
Compound 32a was synthesized by a similar procedure described for 29a (0.34 g, Yield: 63%) as a pale yellow solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.37 (s, 1H), 7.21 (d, J=19.1 Hz, 4H), 6.97 (dt, J=5.6, 1.9 Hz, 1H), 6.68 (dt, J=5.6, 1.9 Hz, 1H), 5.05 (s, 4H), 3.41 (q, J=2.8, 2.0 Hz, 2H), 3.25–3.11 (m, 1H), 1.27 (d, J=6.9 Hz, 6H); 13C NMR (101 MHz, CDCl3): δ (ppm) 171.9, 153.7, 148.1, 137.5, 131.9, 130.4, 129.9, 127.9, 123.5, 113.8, 37.2, 30.5, 23.9; HRMS (ESI) m/z [M+H]+ calculated for C21H22NO2 320.1635, found 320.1645.
Methyl 7-isopropyl-4-methoxy-2-methyl-1-oxo-2,3-dihydro-1H-indene-5-carboxylate (33):
1 M LiN(SiMe3)2 solution in THF (0.8 mL, 0.8 mmol) was added to a solution of indanone derivative 27b (200 mg, 0.762 mmol) in THF (5 mL) and DMI (2.5 mL) at −40°C and the reaction mixture was stirred for 1 h at the same temperature. MeI (0.1 mL, 2.29 mmol) was added to the mixture at −40°C. After stirring at rt for 16 h, the reaction was quenched with 10% NH4Cl aq. (10 mL). THF was removed in vacuo, the mixture was extracted with EtOAc (20 mL×2). The combined organic layer was washed with H2O (20 mL) and brine (20 mL), dried (Na2SO4) and concentrated to afford an off-white residue, which was purified by column chromatography (silica gel 120 – 200 mesh, gradient 2–5% EtOAc/n-Hexane) to afford pure 33 (150 mg, 71%).[43] 1H NMR (400 MHz, CDCl3): δ (ppm) 7.63 (s, 1H), 4.15–4.04 (m, 1H), 3.96 (s, 3H), 3.91 (s, 3H), 3.45–3.33 (m, 1H), 2.76–2.62 (m, 2H), 1.31 (d, J=7.3 Hz, 3H), 1.25–1.21 (2Xd, J=6.9 Hz, 6H); 13C NMR (101 MHz, CDCl3): δ (ppm) 209.6, 166.9, 154.9, 147.7, 145.3, 136.8, 129.2, 127.4, 61.7, 52.7, 42.9, 31.4, 27.1, 23.3, 16.50; MS (ESI) m/z 276 (M + 1).
5-(isoindoline-2-carbonyl)-7-isopropyl-4-methoxy-2-methyl-2,3-dihydro-1H-inden-1-one:
Followed the similar procedure described for compound 28a (0.13 g, Yield:69%) an amorphous solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.41–7.22 (m, 4H), 7.16 (d, J=7.4 Hz, 1H), 5.03 (s, 2H), 4.65 (s, 2H), 4.20–4.05 (m, 1H), 3.41 (m, 1H), 2.77–2.61 (m, 2H), 1.34 (d, J=7.1 Hz, 3H), 1.26–1.22 (2Xd, J=6.9 Hz, 6H); 13C NMR (101 MHz, CDCl3): δ (ppm) 209.35, 168.22, 146.61, 146.22, 136.45, 136.16, 135.15, 134.96, 128.01, 127.81, 124.20, 123.25, 122.78, 61.41, 53.67, 52.33, 42.64, 31.53, 27.16, 23.31, 23.29, 16.53; MS (ESI) m/z 364 (M + 1).
4-Hydroxy-5-(isoindoline-2-carbonyl)-7-isopropyl-2-methyl-2,3-di-hydro-1H-inden-1-one (40):
Compound 40 was synthesized by a similar procedure described for 29a (0.028 g, Yield: 48%) as a yellow solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 10.39 (s, 1H), 7.49 (s, 1H), 7.35 7.26 (m, 5H), 5.09 (s, 4H), 4.12 (m, 1H), 3.46–3.20 (m, 1H), 2.79–2.49 (m, 2H), 1.41–1.14 (m, 9H); 13C NMR (101 MHz, CDCl3): δ (ppm) 210.4, 170.6, 154.7, 154.2, 143.2, 139.3, 136.1, 130.7, 123.7, 122.7, 121.0, 54.5, 42.8, 31.0, 26.9, 23.7, 23.6, 16.6; MS (ESI) m/z 350 (M + 1).
Methyl 7-isopropyl-4-methoxy-2,2-dimethyl-1-oxo-2,3-dihydro-1H-indene-5-carboxylate (34):
1 M LiN(SiMe3)2 solution in THF (2.4 mL, 2.4 mmol) was added to a solution of indanone derivative 27b (200 mg, 0.762 mmol) in THF (5 mL) and DMI (2.5 mL) at −40°C and the reaction mixture was stirred for 1 h at the same temperature. MeI (0.2 mL, 3.81 mmol) was added to the mixture at −40°C. After stirring at rt for 16 h, the reaction was quenched with 10% NH4Cl aq. (10 mL). THF was removed in vacuo, the mixture was extracted with EtOAc (20 mL×2). The combined organic layer was washed with H2O (20 mL) and brine (20 mL), dried (Na2SO4) and concentrated to afford an off-white residue, which was purified by column chromatography (silica gel 120 – 200 mesh, gradient 2–5% EtOAc/n-Hexane) to afford pure 34 (130 mg, 59%).1H NMR (400 MHz, CDCl3): δ (ppm) 7.63 (s, 1H), 4.13–4.02 (m, 1H), 3.95 (s, 3H), 3.89 (s, 3H), 2.96 (s, 2H), 1.24 – 1.21 (2X s, 12H); 13C NMR (101 MHz, CDCl3): δ (ppm) 211.51, 166.92, 155.00, 146.36, 145.75, 135.78, 129.28, 127.33, 61.61, 52.72, 46.11, 39.23, 27.10, 25.61, 23.27. MS (ESI) m/z 291 (M + 1).
5-(isoindoline-2-carbonyl)-7-isopropyl-4-methoxy-2,2-dimethyl-2,3-dihydro-1H-inden-1-one:
Followed the similar procedure described for compound 28a (0.11 g, Yield:69%) as an off-white solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.42–7.08 (m, 5H), 5.03 (s, 2H), 4.65 (s, 2H), 4.12 (m, 1H), 3.89 (s, 3H), 2.98 (s, 2H), 1.32–1.14 (m, 9H); 13C NMR (101 MHz, CDCl3) δ (ppm) 211.36, 168.30, 146.70, 145.36, 136.49, 136.20, 128.05, 127.85, 124.26, 123.29, 122.82, 61.39, 53.72, 52.36, 45.84, 39.41, 27.26, 25.71, 23.33. MS (ESI) m/z 378 (M + 1).
4-hydroxy-5-(isoindoline-2-carbonyl)-7-isopropyl-2,2-dimethyl-2,3-dihydro-1H-inden-1-one (41):
Compound 41 was synthesized by a similar procedure described for 29a (0.034 g, Yield: 43%) as yellow solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 10.36 (s, 1H), 7.50 (s, 1H), 7.29 (d, J=24.8 Hz, 4H), 5.09 (d, J=4.9 Hz, 4H), 4.18–3.98 (m, 1H), 2.93–2.91 (m, 2H), 1.31–1.20 (m, 12H); 13C NMR (101 MHz, CDCl3): δ (ppm) 212.3, 170.6, 154.7, 141.8, 139.7, 135.1, 134.6, 128.1, 125.9, 123.7, 121.1, 46.0, 38.8, 38.7, 26.9, 26.8, 25.7, 25.6, 23.7, 23.4. MS (ESI) m/z 364 (M + 1).
Methyl 2-fluoro-7-isopropyl-4-methoxy-1-oxo-2,3-dihydro-1H-indene-5-carboxylate (35):
Solid Selectfluor (297 mg, 0.838 mmol) was added to a stirring solution of indanone derivative 27b (200 mg, 0.762 mmol) in MeOH (5 mL) followed by concentrated H2SO4 (0,05 ml) and the mixture was heated to 50 °C for 16 h under Ar. After the reaction mixture was cooled off, the solid was filtered out and solution was evaporated to dryness. The dry residue was then re-dissolved in EtOAc and washed with water followed by brine, dried (Na2SO4) and concentrated to afford an off-white residue, which was purified by column chromatography (silica gel 120 – 200 mesh, gradient 2–5% EtOAc/n-Hexane) to afford pure 35 as off-white solid (140 mg, 66%).[38] 1H NMR (400 MHz, CDCl3): δ (ppm) 7.60 (s, 1H), 5.15 (ddd, J=50.9, 8.1, 4.6 Hz, 1H), 3.93–3.86 (m, 1H), 3.89 (s, 3H), 3.84 (s, 3H), 3.59 (ddd, J=17.3, 7.7, 6.6 Hz, 1H), 3.05 (dddd, J=23.0, 17.3, 4.5, 1.0 Hz, 1H), 1.17 (2X d, J=8.0 Hz, 6H); 13C NMR (101 MHz, CDCl3): δ (ppm) 199.9 (d, J=15.2), 166.2, 154.7, 145.8 (d, J=2.0 Hz), 143.6 (d, J=6.1 Hz), 134.0, 130.5, 128.0, 90.5 (d, J=190.9 Hz), 61.72, 52.71, 29.8 (d, J=22.2), 27.40, 22.9 (J=45.5 Hz); 19F NMR (376 MHz, Chloroform-d) δ 192.32; MS (ESI) m/z 381 (M + 1).
2-fluoro-5-(isoindoline-2-carbonyl)-7-isopropyl-4-methoxy-2,3-di-hydro-1H-inden-1-one:
Followed the similar procedure described for compound 28a (0.12 g, Yield:72%) as a pale-yellow solid. 1H NMR (400 MHz, CDCl3) δ (ppm) 7.46–7.23 (m, 4H), 7.16 (s, 1H), 5.38–5.12 (m, 1H), 5.03 (s, 2H), 4.64 (s, 2H), 4.02 (h, J=6.9 Hz, 1H), 3.90 (s, 3H), 3.67 (dt, J=17.3, 7.5 Hz, 1H), 3.26–3.06 (m, 1H), 1.32–1.14 (m, 6H);13C NMR (101 MHz, CDCl3) δ (ppm) 199.8 (d, J=15.1), 167.57, 151.2 (d, J=2 Hz), 146.90, 142.7 (d, J=6.0 Hz), 136.6, 136.1 (d, J=23.2 Hz), 132.4, 128.1 (J=22.0 Hz), 125.19, 123.0 (d, J=48.0 Hz), 90.5 (d, J=191.9 Hz), 61.58, 53.60, 52.36, 30.1 (d, J=22.2 Hz), 27.7, 23.3, 22.9;HRMS (ESI) m/z [M+H]+ calculated for C22H23FNO3 368.1659, found 368.1656.
2-fluoro-4-hydroxy-5-(isoindoline-2-carbonyl)-7-isopropyl-2,3-di-hydro-1H-inden-1-one (42):
Compound 42 was synthesized by a similar procedure described for 29a (0.026 g, Yield: 39%). 1H NMR (400 MHz, CDCl3) δ (ppm) 10.43 (s, 1H), 7.51 (s, 1H), 7.32–7.25 (m, 4H), 5.26–5.11 (m, 1H), 5.17–5.04 (m, 4H), 3.96 (q, J=6.9 Hz, 1H), 3.60 (dt, J=17.4, 7.5 Hz, 1H), 3.12–2.96 (m, 1H), 1.25 (2Xd, J=6.9 Hz, 6H); 13C NMR (101 MHz, CDCl3) δ (ppm) 200.8, 170.0, 154.7, 139.9, 139.1, 136.1, 133.5, 128.2, 124.5, 122.9, 122.4, 90.9 (d, J=191.9 Hz), 77.4, 29.6 (d, J=20.2 Hz), 27.4, 23.5 (d, J=100.6 Hz).19F NMR (376 MHz, CDCl3) δ 192.32. MS (ESI) m/z 354 (M + 1).
Methyl 2,2-difluoro-7-isopropyl-4-methoxy-1-oxo-2,3-dihydro-1H-indene-5-carboxylate (36):
Compound 27 (140 mg, 0.5 mmol) and triethyl amine (0.4 ml, 303 mg, 3.0 mmol) were dissolved in methylene chloride (4 mL). The solution was cooled to 0°C, and tert-butyl-dimethylsilyl trifluorosulfonate (0.2 mL, 0.7 mmol) was added. The reaction was allowed to warm to ambient temperature, stirred 16 h, and then diluted with ethyl ether. The organic solution was washed with saturated NaHCO3 solution, followed by 1 N HCl, and finally by brine before it was dried (Na2SO4). The solvent was removed under vacuum to give the silyl enol ether which was used in the next step without further purification and/or characterization. The silyl enol ether was dissolved in acetonitrile (5 mL), and Selectfluor (0.194 mg, 0.55 mmol) was added. The reaction was stirred at ambient temperature for 5 h. The solvent was then evaporated under vacuum, and the residue taken in methylene chloride and filtered.[44] The filtrate was washed with water, dried (Na2SO4) and concentrated to afford an off-white residue, which was purified by column chromatography (silica gel 120 – 200 mesh, gradient 2–5% EtOAc/n-Hexane) to afford pure 36 as light-yellow solid (100 mg, 67%).1H NMR (400 MHz, CDCl3): δ (ppm) 7.71–7.58 (m, 1H), 3.94–3.88 (m, 1H), 3.91 (s, 3H), 3.84 (s, 3H), 3.53–3.33 (m, 2H), 1.19 (d, J=6.9 Hz, 6H); 13C NMR (101 MHz, CDCl3): δ (ppm) 206.2, 165.9, 154.6, 146.9, 141.9, 132.3, 131.3, 128.5, 116.2 (t, J=256.5 Hz), 61.8, 52.9, 33.9 (t, J=26.3 Hz), 27.7, 22.8; 19F NMR (376 MHz, CDCl3): δ (ppm) – 110.32. MS (ESI) m/z 299 (M + 1).
2,2-difluoro-5-(isoindoline-2-carbonyl)-7-isopropyl-4-methoxy-2,3-dihydro-1H-inden-1-one:
Followed the similar procedure described for compound 28a (0.12 g, Yield: 64%) as a pale-yellow solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.34–7.00 (m, 5H), 4.92 (s, 2H), 4.54 (s, 2H), 3.94–3.83 (m, 1H), 3.80 (s, 3H), 3.43 (t, J=12.8 Hz, 2H), 1.15 (d, J=6.9 Hz, 6H); 13C NMR (101 MHz, CDCl3): δ (ppm) 191.7, 191.4 (t, J=27.0), 167.2, 151.2, 147.94, 141.1 (t, J=6.0 Hz), 137.3, 136.1, 130.8 (t, J=3.0 Hz), 128.2, 125.8, 123.3, 116.4 (t, J=254.0 Hz), 61.6, 53.6, 52.5, 34.10 (t, J=26.3 Hz), 28.00, 22.97; 19F NMR (376 MHz, CDCl3): δ (ppm) 110.3. MS (ESI) m/z 386 (M + 1).
2,2-difluoro-4-hydroxy-5-(isoindoline-2-carbonyl)-7-isopropyl-2,3-dihydro-1H-inden-1-one (43):
Compound 43 was synthesized by a similar procedure described for 29a (0.019 g, Yield: 28%) as yellow solid. 1H NMR (400 MHz, CDCl3) δ (ppm) 8.03 (s, 1H), 7.75–7.30 (m, 4H), 5.10 (q, J=8.1 Hz, 4H), 3.74–3.55 (m, 1H), 3.56–3.36 (m, 2H), 1.39–1.16 (m, 6H);13C NMR (101 MHz, CDCl3) δ (ppm) 169.7, 159.9, 146.2, 140.9, 137.1, 134.4, 131.1, 128.8, 124.9, 124.5 (m), 123.8, 50.57, 33.6 (m), 33. 27.8 (m), 23.2 (m) [ketone carbonyl signal is obscure]; 19F NMR (376 MHz, CDCl3) δ (ppm) 110.47;HRMS (ESI) m/z [M+H]+ calculated for C21H19F2NO3 372.1407, found 372.1405.
Methyl 7-isopropyl-4-methoxy-1-oxo-2-(trifluoromethyl)-2,3-dihydro-1H-indene-5-carboxylate (37):
ketone 27 (300 mg, 1.14 mmol) and TsOH•H2O (294 mg, 1.72 mmol) were added in acetic anhydride (5 mL) in a sealed tube with screw cap. The mixture was heated at 120°C for 48 h and then cooled to room temperature. The mixture was then diluted with Et2O (30 mL), washed with saturated NaHCO3 (aq, 10 mL), dried (Na2SO4), concentrated under reduced pressure to afford a light brown residue, which was used in the next step without further purification or characterization.
CF3SO2Na (711.6 mg, 4.56 mmol), Cu(OTf)2 (62 mg, 0.171 mmol), enol acetates (267 mg, 1,14 mmol) were added in MeCN (5 mL) in sequence. After the reaction mixture was cooled to 0°C, an aqueous solution of TBHP (0.8 mL, 5.70 mmol, 70% solution by weight) was added to the reaction mixture with stirring. After stirring for 2 h, the mixture was concentrated under reduced pressure and then purified by column chromatography (silica gel 120 – 200 mesh, gradient 2–5% EtOAc/n-Hexane) to afford pure 37 as light-yellow solid (100 mg, 27%).[40] 1H NMR (400 MHz, CDCl3): δ (ppm) 7.68 (s, 1H), 4.08–3.98 (m, 1H), 3.97 (s, 3H), 3.93 (s, 3H), 3.50–3.33 (m, 2H), 3.23 (m, 1H), 1.24 (2Xd, J=2.9 Hz, 6H); 13C NMR (101 MHz, Chloroform-d) δ 196.9, 166.3, 154.7, 146.5, 145.9, 129.9, 128.1, 126.2, 123.3, 61.7, 52.7, 50.30 (q, J=27.3), 27.1, 24.1, 23.00, 22.9; 19F NMR (376 MHz, CDCl3): δ (ppm) 67.93; HRMS (ESI) m/z [M+H]+ calculated for C16H18F3O4 331.1156, found 331.1151.
5-(Isoindoline-2-carbonyl)-7-isopropyl-4-methoxy-2-(trifluoromethyl)-2,3-dihydro-1H-inden-1-one:
Followed the similar procedure described for compound 28a (0.11 g, Yield: 65%) as pale-yellow solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.37–7.06 (m, 5H), 4.98 (s, 2H), 4.59 (s, 2H), 4.08–3.94 (m, 1H), 3.85 (d, J=16.5 Hz, 3H), 3.38 (m, 2H), 3.20 (m, 1H), 1.18 (dd, J=7.0, 3.7 Hz, 6H); 13C NMR (101 MHz, CDCl3): δ (ppm) 196.7, 167.7, 151.2, 146.9, 145.6, 136.2, 135.9, 128.1, 127.9, 125.3, 123.3, 122.83, 61.5, 53.6, 52.3, 50.25 (q, J=27.3), 27.3, 23.14.; 19F NMR (376 MHz, CDCl3) δ (ppm): −67.87; HRMS (ESI) m/z [M+H]+ calculated for C23H23F3NO3 418.1632, found 418.1625.
4-Hydroxy-5-(isoindoline-2-carbonyl)-7-isopropyl-2-(trifluoromethyl)-2,3-dihydro-1H-inden-1-one (44):
Compound 44 was synthesized by a similar procedure described for 29a (XX g, Yield: XX %). 1H NMR (400 MHz, CDCl3) δ (ppm): 10.52 (s, 1H), 7.65–7.21 (m, 5H), 5.10 (s, 4H), 4.05 (m, 1H), 3.52–3.28 (m, 2H), 3.30–3.12 (m, 1H), 1.27 (t, J=6.8 Hz, 6H); 13C NMR (101 MHz, CDCl3) δ (ppm): 197.6, 169.8, 154.5, 141.8, 139.7, 135.2, 130.8, 128.1, 126.3, 124.3, 123.5, 121.6, 55.5, 53.2, 50.29 (q, J=27.3), 26.8, 23.3; 19F NMR (376 MHz, CDCl3) δ (ppm): 67.92; HRMS (ESI) m/z [M+Na]+ calculated for C22H20F3NNaO3 426.128454, found 426.128749.
Methyl 1-(difluoromethylene)-7-isopropyl-4-methoxy-2,3-dihydro-1H-indene-5-carboxylate (38):
To an oven dry microwave vial (50 mL, Biotage) charged with triphenylphosphine (2.25 g, 8.57 mmol), lithium iodide (765 mg, 5.71 mmol) and compound 27b (750 mg, 2.85 mmol) under inert atmosphere. To this vial, anhydrous toluene (17 mL) and anhydrous DMF (3 mL) were injected. Finally, TMSCF3 (1.06 mL, 7.14 mmol) was added through a syringe. The resulting mixture was heated at 45°C for 40 h. As the reaction proceeded, the reaction mixture also turned from pale orange to dark brown. The reaction vial was cooled down to room temperature and quenched with water (30 mL) and extracted with EtOAc (3×50 mL).The combined organic layer was dried over anhydrous Na2SO4, filtered and dried on a rotary evaporator. The residue was purified by column chromatography (silica gel, eluted with 3–30% ethyl acetate/hexane gradient) to afford the pure product 47 (470 mg, Yield: 54%) as an off-white amorphous solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.48 (s, 1H), 3.75 (s, 3H), 3.66 (s, 3H), 3.02–2.94 (s, 1H), 2.83–2.79 (m, 2H), 2.67–2.60 (m, 2H), 1.07 (d, J=6.7 Hz, 6H); 19F NMR (400 MHz, CDCl3): δ (ppm) −79.72, −79.59, −85.72, −85.6; MS (ESI) m/z 297 (M + 1).
(1-(difluoromethylene)-7-isopropyl-4-methoxy-2,3-dihydro-1H-inden-5-yl)(isoindolin-2-yl)methanone:
Followed the similar procedure described for compound 28a to offered pure product (120 mg, Yield: 75%) as a pale-yellow solid. 1H NMR (400 MHz, DMSO-d6): δ (ppm) 7.42–7.28 (m, 4H), 7.08 (s, 1H), 5.00 (s, 2H), 4.67 (s, 2H), 3.65 (s, 3H), 3.26–3.18 (m, 1H), 3.07–3.00 (m, 2H), 2.45–2.32 (m, 2H), 1.18 (d, J=6.7 Hz, 6H); 13C NMR (100 MHz, CDCl3): δ (ppm) 169.5, 150.2, 143.2, 137.9, 136.8, 136.4, 127.9, 124.6, 123.5, 123.2, 122.8, 61.1, 53.9, 52.3, 50.2, 30.3, 29.4, 28.4, 25.3, 23.3; MS (ESI) m/z 384 (M + 1).
(1-(difluoromethylene)-4-hydroxy-7-isopropyl-2,3-dihydro-1H-inden-5-yl)(isoindolin-2-yl)methanone (45):
Compound 45 was synthesized by a similar procedure described for 29a to afford pure product 45 (25 mg, Yield 24%) as a pale yellow solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.35–7.24 (m, 4H), 7.08 (s, 1H), 5.00 (s, 2H), 4.67 (s, 2H), 3.26–3.18 (m, 1H), 3.07–3.00 (m, 2H), 3.45–2.32 (m, 2H), 1.18 (m, 6H); 13C NMR (101 MHz, CDCl3): δ (ppm) 169.3, 150.5, 142.1, 141.7, 136.9, 136.8, 136.5, 130.2, 127.8, 123.5, 122.8, 61.1, 53.8, 52.3, 49.3, 30.6, 30.4, 29.7, 24.2, 23.5; HRMS (ESI+), m/z [M+ Na+] calculated for C22H21F2NO2Na 392.1426; found 392.1348.
5-(isoindoline-2-carbonyl)-7-isopropyl-4-methoxy-2-methylene-2,3-dihydro-1H-inden-1-one (39):
Synthesized using literature procedure[41] to afford pure product 39 (35 mg, Yield 42%) as a pale-yellow solid. 1H NMR (400 MHz, CDCl3) δ 7.36–7.16 (m, 4H), 7.08 (d, J=7.4 Hz, 1H), 6.27 (td, J=2.3, 0.9 Hz, 1H), 5.59 (q, J=1.6 Hz, 1H), 4.96 (s, 2H), 4.56 (s, 2H), 4.17 (m, 1H), 3.84 (s, 3H), 1.27–1.09 (d, J=6.9 Hz, 6H).13C NMR (101 MHz, CDCl3) δ 193.4, 168.0, 151.0, 146.8, 143.1, 136.3, 136.0, 135.2, 127.9, 127.7, 124.3, 123.1, 122.6, 119.4, 61.4, 53.5, 52.2, 28.6, 27.2, 23.1; MS (ESI) m/z 362 (M + 1).
4-hydroxy-5-(isoindoline-2-carbonyl)-7-isopropyl-2-methylene-2,3-dihydro-1H-inden-1-one (46):
Compound 46 was synthesized by a similar procedure described for 29a to afford pure product 46 (34 mg, Yield 28%) as yellow solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.47 (s, 1H), 7.19 (s, 4H), 6.27 (m, 1H), 5.69–5.51 (m, 1H), 5.13–4.92 (m, 4H), 4.25–4.07 (m, 1H), 1.29 (d, J=6.9 Hz, 6H).13C NMR (101 MHz, CDCl3): δ (ppm) 194.1, 170.3, 154.5, 143.5, 139.7, 130.7, 128.4, 123.6, 124.2, 120.9, 120.0, 119.9, 119.6, 50.2, 28.2, 26.8, 23.4; MS (ESI) m/z 348 (M + 1).
Methyl 4-(benzyloxy)-7-isopropyl-1-oxo-2,3-dihydro-1H-indene-5-carboxylate (47):
Compound 47 was synthesized by two steps, demethylation of 27b by a similar procedure described for 29a to afford phenolic product as an off white solid (330 mg, Yield: 71%). 1H NMR (400 MHz, CDCl3): δ (ppm) 10.69 (s, 1H), 7.64 (s, 1H), 4.05–3.85 (m, 4H), 2.99–2.93 (m, 2H), 2.66–2.60 (m, 2H), 1.23 (d, J=6.8 Hz, 6H); 13C NMR (101 MHz, CDCl3): δ (ppm) 207.6, 170.4, 156.9, 144.7, 139.3, 138.8, 125.1, 115.2, 52.6, 37.1, 26.4, 23.2, 21.5; MS (ESI) m/z 249 (M + 1).
The phenolic compound (0.5 g, 2.01 mmol) was dissolved in dry acetonitrile (18 mL), benzyl bromide (0.287 mL, 2.42 mmol) and K2CO3 (0.362 g, 2.62 mmol) were added. The reaction mixture was heated to reflux for 12 h. Cooled to rt and the solvent was evaporated at reduced pressure, the crude residue was purified by column chromatography (silica gel 100–200 mesh, eluted with 3–30% ethyl acetate/hexane gradient) to afford the pure product 47 (585 mg, Yield: 85%) as an off-white amorphous solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.61 (s, 1H), 7.41–7.23 (m, 5H), 4.97 (s, 2H), 4.10–3.98 (m, 1H), 3.82 (s, 3H), 2.96–2.91 (m, 2H), 2.61–2.54 (m, 2H), 1.18 (d, J=6.8 Hz, 6H); 13C NMR (101 MHz, CDCl3): δ (ppm) 207.0, 166.6, 153.6, 149.9, 145.2, 137.2, 137.0, 129.4, 128.5, 128.3, 128.1, 127.3, 76.2, 52.5, 37.1, 26.7, 23.1, 22.4; MS (ESI) m/z 339 (M + 1).
4-(benzyloxy)-5-(isoindoline-2-carbonyl)-7-isopropyl-2,3-dihydro-1H-inden-1-one (48):
Followed the similar procedure described for compound 28a to offered pure product 48 (600 mg, Yield: 92%) as a pale-yellow solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.38–7.22 (m, 8H), 7.11 (d, J=7.4 Hz, 1H), 5.07 (s, 2H), 5.00 (s, 2H), 4.61 (s, 2H), 4.21–4.10 (m, 1H), 3.02 (s, 2H), 2.68 (t, J=6.2 Hz, 2H) 1.24 (d, J=6.8 Hz, 6H); 13C NMR (101 MHz, CDCl3): δ (ppm) 206.9, 167.7, 150.4, 149.3, 146.6, 137.0, 136.4, 136.1, 135.9, 135.6, 128.7, 128.5, 128.2, 128.0, 127.8, 124.3, 123.2, 122.8, 76.4, 53.7, 52.4, 37.2, 27.1, 23.3, 22.6; MS (ESI) m/z 426 (M + 1).
4-(benzyloxy)-5-(isoindoline-2-carbonyl)-7-isopropyl-1H-inden-1-one (49):
Compound 48 (0.5 g, 1.18 mmol) in dry benzene (10 mL) at 0 ○C under Argon was added successively Et3N (0.328 mL, 2.35 mmol) and TMSOTf (0.319 mL, 1.76 mmol). The mixture was warmed to room temperature and stirred for 10 min. The reaction mixture was again cooled to 0 ○C and diluted with ether (20 mL) and NaHCO3 solution (10 mL). The layers were separated, and the aqueous phase was extracted with ether (3×10 mL). The combined organic extracts were washed with brine (20 mL), then dried on anhydrous Na2SO4, filtered, and evaporated under reduced pressure to afford the silyl enol ether as a light-yellow oil (580 mg). The silyl enol ether was dissolved in dry DCM (10 mL) and added dropwise to an aluminum foil-wrapped flask containing a suspension of Pd(OAc)2 (262 mg, 1.0 mmol) in dry acetonitrile (5 mL) under Ar. The mixture was stirred at room temperature for 1 hr, then filtered through a short aluminum foil-wrapped column of silica gel (100 mL, DCM). The filtrate was evaporated under reduced pressure, and the residue was purified by column chromatography (aluminum foil-wrapped column, silica gel 100–200 mesh; 0–40% hexane/EtOAc) to afford the pure Product 49 (360 mg, Yield: 72%) as yellow solid.[45] 1H NMR (400 MHz, CDCl3): δ (ppm) 7.41 (d, J=6.1 Hz, 1H), 7.28–7.14 (m, 8H), 7.06 (d, J=7.3 Hz, 1H), 5.72 (d, J=6.1 Hz, 1H), 4.98 (s, 2H), 4.89 (s, 2H), 4.58 (s, 2H), 3.97–3.88 (m, 1H), 1.14 (d, J=6.8 Hz, 6H); MS (ESI) m/z 424 (M + 1).
4-hydroxy-5-(isoindoline-2-carbonyl)-7-isopropyl-1H-inden-1-one (50):
Borantrichloride.methyl sulfide complex (237 mg, 1.32 mmol) was added to a solution of compound 49 (280 mg, 0.66 mmol) in dry DCM (10 mL) at −10°C. The reaction mixture was slowly warmed to room temperature and stirred for about 1 h. The reaction was quenched with water (10 mL) and the organic layer was separated and dried over anhydrous Na2SO4, filtered and evaporated at reduced pressure. The crude obtained was purified by column chromatography (silica gel 100–200 mesh, 10–30% Hexane/EtOAc) to afford the pure Product 50 (143 mg, Yield: 62%) as yellow solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.40 (s, 1H), 7.33–7.15 (m, 3H), 7.03 (d, J=7.3 Hz, 1H), 5.19 (d, J=6.2 Hz, 1H), 5.15 (d, J=6.2 Hz, 1H), 4.88 (s, 2H), 4.56 (s, 2H), 3.21–3.06 (m, 1H), 1.14 (d, J=6.8 Hz, 6H); 13C NMR (101 MHz, CDCl3): δ (ppm) 202.2, 167.4, 150.4, 150.4, 146.9, 146.2, 137.0, 136.4, 136.0, 135.6, 134.0, 128.5, 128.1, 127.7, 123.0, 122.6, 53.3, 52.3, 48.4, 26.7, 23.2, 23.0; HRMS (ESI+), m/z [M+H+] calculated for C21H20NO3 334.1438; found 334.1429.
(4-hydroxy-1-(hydroxyimino)-7-isopropyl-2,3-dihydro-1H-inden-5-yl)(isoindolin-2-yl)methanone (51):
Hydroxylamine hydrochloride (16 mg, 0.22 mmol), pyridine (24 mg, 0.3 mmol) were added to a solution of compound 29b (50 mg, 0.15 mmol) in ethanol (1 mL). The reaction mixture was heated to reflux for about 2 h. The reaction mixture was cooled, and the solvent was evaporated on rotavapor, the crude was re dissolved in EtoAc (5 mL) and washed with water (2×5 mL) the organic layer was separated and dried over anhydrous Na2SO4, filtered and evaporated at reduced pressure to afford the pure Product 51 (40 mg, Yield: 89%) as an off white solid no further purification required. 1H NMR (500 MHz, DMSO-d6): δ (ppm) 11.06 (s, 1H), 9.49 (s, 1H), 7.55–7.10 (m, 5H), 4.85 (s, 2H), 4.69 (s, 2H), 4.05–3.89 (m, 1H), 2.96 (s, 4H), 1.16 (s, 6H); 13C NMR (126 MHz, CDCl3): δ (ppm) 170.9, 164.1, 154.1, 137.6, 137.1, 136.3, 127.6, 123.5, 122.4, 116.8, 62.2, 28.4, 26.5, 24.9, 23.2; HRMS (ESI+), m/z [M+H+] calculated for C21H23N2O3 351.1703; found 351.1704.
(4-hydroxy-7-isopropyl-1-(methoxyimino)-2,3-dihydro-1H-inden-5-yl)(isoindolin-2-yl)methanone (52):
O-Methylhydroxylamine hydrochloride (19 mg, 0.22 mmol), pyridine (24 mg, 0.3 mmol) were added to a solution of compound 29b (50 mg, 0.15 mmol) in ethanol (1 mL). The reaction mixture was heated to reflux for about 2 h. The reaction mixture was cooled, and the solvent was evaporated on rotavapor, the crude was re dissolved in EtoAc (5 mL) and washed with water (2×5 mL) the organic layer was separated and dried over anhydrous Na2SO4, filtered and evaporated at reduced pressure to afford the pure Product 52 (44 mg, Yield: 90%) as an off white solid no further purification required. 1H NMR (500 MHz, CDCl3): δ (ppm) 10.61 (s, 1H), 7.42 (s, 1H), 7.38–7.27 (m, 4H), 5.10 (s, 4H), 4.08–3.95 (m, 4H), 3.00–2.87 (m, 4H), 1.27 (d, J=6.8 Hz, 6H); 13C NMR (126 MHz, DMSO-d6): δ (ppm) 168.4, 164.1, 148.5, 148.3, 137.2, 136.4, 135.8, 127.8, 127.7, 124.9, 123.7, 123.3, 53.4, 52.2, 28.1, 26.4, 25.2, 23.6; HRMS (ESI+), m/z [M+H]+ calculated for C22H25N2O3 365.1860; found 365.1859.
8-bromo-5-methoxy-3,4-dihydronaphthalen-1(2H)-one (54):
5-methoxy-1-tetralone (5 g, 28 mmol) was dissolved in Dry CH3CN (160 mL), and NBS (6 g, 33.7 mmol) was added. After 48 h, the solvent was evaporated on rotavapor at reduced pressure, the crude obtained was dissolved in EtoAc (300 mL) and washed with brine (100 mL) and H2O (100 mL), the organic layer was separated and dried over anhydrous Na2SO4 and concentrated on rotavapor. The residue was purified by column chromatography (silica gel 120 – 200 mesh, gradient 2–15% EtoAc/n-Hexane) to give 2 (6.2 g, Yield 96%) as a yellow solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.52 (dt, J=8.7, 0.7 Hz, 1H), 6.84 (d, J=8.7 Hz, 1H), 3.86 (s, 3H), 2.91 (t, J=6.2 Hz, 2H), 2.68 (t, J=6.7 Hz, 2H), 2.14–2.07 (m, 2H). MS (EI): m/z 254 (M + 1, 100), 256 (M + 3, 96).
5-methoxy-8-(prop-1-en-2-yl)-3,4-dihydronaphthalen-1(2H)-one:
In an oven dry sealed tube was charged with 2 (5.0 g, 19.68 mmol, 1 equiv.), cesium carbonate (19.23 g, 59.0 mmol, 3 equiv.), potassium isopropenyltrifluoroborate (3.49 g, 23.6 mmol, 1.2 equiv.) and tetrahydrofuran (60 mL), water (6 mL) closed with septum. The reaction mixture was purged with an argon for about 15 min to eliminate the oxygen. Tetrakis(triphenylphosphine)palladium (0) (2.27 g, 1.96 mmol, 0.1 equiv.) was added under nitrogen condition. The tube was sealed with a cap lined with a Teflon tape. The tube was heated at 100°C for 12 h, cooled to rt, and filtered through a small pad of celite (elution with ethyl acetate). Solvent was removed and the residue purified by flash chromatography (SiO2, 1:3 ethyl acetate/hexanes) to afford product (3.3 g, Yield 78%) as an off-white solid.1H NMR (400 MHz, CDCl3): δ (ppm) 6.96 (d, J=8.3 Hz, 1H), 6.88 (d, J=8.3 Hz, 1H), 4.94 (s, 1H), 4.66 (s, 1H), 3.79 (s, 3H), 2.83 (t, J=6.2 Hz, 2H), 2.55 (t, J=6.7 Hz, 2H), 2.05–1.99 (m, 2H), 1.93 (s, 3H);MS (ESI) m/z 217 (M + 1).
5-isopropyl-8-methoxy-1,2,3,4-tetrahydronaphthalene (55):
A hydrogen pressure vessel was filled with ethanolic (100 ml) solution of 5-methoxy-8-(prop-1-en-2-yl)-3,4-dihydronaphthalen-1(2H)-one (3.2 g, 14.7 mmol) in ethyl acetate (100 mL). Palladium on carbon (10%, 300 mg) was added. The suspension was stirred for 16 h under a hydrogen atmosphere (balloon pressure) before it was filtered through a pad of celite and eluted with EtOAc (50 mL). The eluent was concentrated, and the residue purified by flash chromatography (SiO2, 1:9 ethyl acetate/hexanes) to afford pure compound 55 (2.4 g, Yield 84%) as an off-white solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 6.99 (d, J=8.4 Hz, 1H), 6.61 (d, J=8.4 Hz, 1H), 3.71 (s, 3H), 3.06–2.95 (m, 1H), 2.70–2.57 (m, 4H), 1.74–1.63 (m, 4H), 1.13 (s, 3H), 1.11 (s, 3H); MS (ESI) m/z 205 (M + 1).
4-isopropyl-1-methoxy-5,6,7,8-tetrahydronaphthalene-2-carbaldehyde (56):
Compound 55 (2 g, 9.8 mmol) was dissolved in trifluoroacetic acid (20 mL) and Hexamethylenetetramine (4.12 g, 29.4 mmol) was added. The reaction mixture was heated to 90°C for 12 h. The solvent was removed under reduced pressure, the residue was dissolved in Ethyl acetate (100 mL) washed with water (50 mL×2) the organic layer was separated and dried over anhydrous Na2SO4 and concentrated on rotavapor. The crude product was purified by column chromatography (silica gel 120 – 200 mesh, gradient 2–15% EtoAc/n-Hexane) to afford compound 5 (1.6 g, Yield 70%) as an off-white solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 10.26 (s, 1H), 7.51 (s, 1H), 3.79 (s, 3H), 3.10–2.98 (m, 1H), 2.76–2.69 (m, 4H), 1.81–1.66 (m, 4H), 1.15 (s, 3H), 1.13 (s, 3H); MS (ESI) m/z 233 (M + 1).
4-isopropyl-1-methoxy-5-oxo-5,6,7,8-tetrahydronaphthalene-2-carboxylic acid (57a) & 4-isopropyl-1-methoxy-8-oxo-5,6,7,8-tetrahydronaphthalene-2-carboxylic acid (57b):
Compound 56 (1.5 g, 6.46 mmol) was dissolved in acetic acid (10 mL) and chromium trioxide (1.93 g, 19.3 mmol) solution (dissolved in 10 mL of acetic acid) was added drop wise for about 15 minutes. The reaction mixture was heated to 60°C for 10 h. The solvent was evaporated on rotavapor at reduced pressure, the crude obtained was dissolved in EtoAc (50 mL) and washed with brine (20 mL) and H2O (20 mL), the organic layer was separated and dried over anhydrous Na2SO4 and concentrated on rotavapor. The crude product was purified by column chromatography (silica gel 120 – 200 mesh, gradient 2:1–5% EtoAc/n-Hexane) to offered a mixture (1:3) of Isomeric products 57a and 57b (240 mg and 730 mg, Yield 58%) Isomer -I (57a): 1H NMR (400 MHz, CDCl3): δ (ppm) 8.18 (s, 1H), 7.19 (s, 1H), 3.92 (s, 3H), 3.15–3.16 (m, 1H), 2.95 (t, J=6.3 Hz, 2H), 2.62 (t, J=6.3 Hz, 2H), 2.11–2.04 (m, 2H), 1.21 (s, 3H), 1.19 (s, 3H);MS (ESI) m/z 263 (M + 1); Isomer-II (57b): 1H NMR (400 MHz, CDCl3): δ (ppm) 7.94 (s, 1H), 3.85 (s, 3H), 2.90 (t, J=6.3 Hz, 2H), 2.64 (t, J=6.3 Hz, 2H), 2.08–1.99 (m, 3H), 1.18 (s, 3H), 1.17 (s, 3H);MS (ESI) m/z 263 (M + 1)
7-(isoindoline-2-carbonyl)-5-isopropyl-8-methoxy-3,4-dihydronaphthalen-1(2H)-one (58a):
Compound 58a was synthesized by a similar procedure described for 28a to afford pure product (109 mg, Yield 75%) as yellow solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.29–7.15 (m, 4H), 7.03 (d, J=7.2 Hz, 1H), 4.93 (s, 2H), 4.78 (bs, 2H), 3.78 (s, 3H), 3.15–3.05 (m, 1H), 2.90 (t, J=6.3 Hz, 2H), 2.60 (t, J=6.3 Hz, 2H), 2.09–2.01 (m, 2H), 1.18 (s, 3H), 1.16 (s, 3H); MS (ESI) m/z 369 (M + 1).
8-hydroxy-7-(isoindoline-2-carbonyl)-5-isopropyl-3,4-dihydronaphthalen-1(2H)-one (59a):
Compound 59a was synthesized by a similar procedure described for 29a to afford pure product (23 mg, Yield 27%) as pale-yellow solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.61–7.50 (m, 2H), 7.03–6.87 (m, 3H), 5.01 (s, 4H), 4.05–3.95 (m, 1H), 2.94–2.83 (m, 2H), 2.66–2.56 (m, 2H), 2.12–1.99 (m, 2H), 1.19 (s, 6H); HRMS (ESI+), m/z [M+Na+] calculated for C22H23NO3Na 372.4279; found 372.4128.
6-(isoindoline-2-carbonyl)-8-isopropyl-5-methoxy-3,4-dihydronaphthalen-1(2H)-one (58b):
Compound 58b was synthesized by a similar procedure described for 28a to afford pure product (109 mg, Yield 75%) as yellow solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.23–7.09 (m, 4H), 7.10 (d, J=7.2 Hz, 1H), 4.87 (s, 2H), 4.48 (bs, 2H), 3.66 (s, 3H), 2.88–2.78 (m, 2H), 2.54 (t, J=6.3 Hz, 2H), 2.01–1.91 (m, 3H), 1.08 (s, 3H), 1.06 (s, 3H); MS (ESI) m/z 369 (M + 1).
5-hydroxy-6-(isoindoline-2-carbonyl)-8-isopropyl-3,4-dihydronaphthalen-1(2H)-one (59b):
Compound 58b was synthesized by a similar procedure described for 29b to afford pure product (19 mg, Yield 25%) as pale-yellow solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 12.99 (s, 1H), 7.30–7.16 (m, 4H), 7.07 (d, J=7.2 Hz, 1H), 4.97 (s, 2H), 4.69 (s, 2H), 3.12–3.01 (m, 1H), 2.91 (t, J=6.3 Hz, 2H), 2.65 (t, J=6.5 Hz, 2H), 2.12–2.03 (m, 2H), 1.16 (s, 3H), 1.14 (s, 3H); 13C NMR (101 MHz, CDCl3): δ (ppm) HRMS (ESI+), m/z [M+Na+] calculated for C22H23NO3Na 372.4279; found 372.4128.
Florescence polarization
The assay was performed as described previously.[27] Briefly, the assay was performed in a 96-well format in black, flat-bottom plates (Santa Cruz Biotechnology) with a final volume of 100μL. Twenty-five microliters of assay buffer (20mM HEPES, pH 7.3, 50mM KCl, 5mM MgCl2, 20mM Na2MoO4, 2mM DTT, 0.1mg/mL BGG, and 0.01% NP-40) containing 6nM FITC-GDA (fluorescent tracer, stock in DMSO and diluted in assay buffer) and 50μL of assay buffer containing 10nM of either Hsp90α or Hsp90β were added to each well. Compounds were tested in triplicate (1% DMSO final concentration). For each plate, wells containing buffer only (background), tracer in buffer only (low polarization control), and protein and tracer in buffer with 1% DMSO (high polarization control) were included. Plates were incubated at 4°C with rocking for 24h. Polarization values (in mP units) were measured at 37°C with an excitation filter at 485nm and an emission filter at 528 nm. Polarization values were correlated to % tracer bound and compound concentrations. The IC50 values were calculated by GraphPad Prism (La Jolla, CA) and/or Microsoft excel software and reported as mean ± SD.
Cell viability assay
HT29, UM-UC3 and NCI-H1299 cells were seeded in 96-well plates at a cell density of 2000 cells/well. Compounds or vehicle were administered at the desired concentrations (1% DMSO final concentration) and incubated for 72h. DMSO was used as a negative control for cell viability. Values of the viability of compound-treated cells were expressed as a percentage of that of DMSO-treated cells. Luminescent signals were measured by the Cell Titer-Glo luminescent cell viability assay (Promega, Madison, WI) according to the manufacturer’s instructions on a BioTek Synergy 4 plate reader (BioTek Instruments, Winooski, VT). The experiments were performed in triplicate, and the data were analyzed. The IC50 values were calculated by GraphPad Prism (La Jolla, CA) and/or Microsoft excel software and reported as mean ±SD.
Western Blot Analysis
HT29 cells were grown in RPMI containing 1% Pen-Strep (VWR, K952–100 ML) and 10% FBS+ (Atlas Biologicals, F-0500-D), and plated in a 10 cm plate (VWR, 10861–696). At ~70% confluence, cells were treated with GDA at 500 nM and compound 5 at 5, 10, 20, 30, 40, and 50 μM for 24 h. The cell extracts were harvested in 1X RIPA buffer (Cell Signaling Technology, Inc.; Danvers, MA), containing protease and phosphatase inhibitors (Roche). Lysates were centrifuged at 12,000g for 25min at 4°C. Protein concentrations were determined by a BCA protein assay (Thermo Scientific, Rockford, IL). The samples were then boiled in sample loading buffer and equal amounts of samples were resolved on a 10% SDS-PAGE gel and then transferred to a polyvinylidene fluoride membrane (PVDF) and immunoblotted with the corresponding specific antibodies. Membranes were incubated with an appropriate horseradish peroxidase-labeled secondary antibody, developed with a chemiluminescent substrate, and visualized using Biorad Chemidoc imaging system.
Supplementary Material
Acknowledgements
The authors would like to acknowledge financial support of this work from the National Institutes of Health (CA213566).
Footnotes
Supporting information for this article is available on the WWW under https://doi.org/10.1002/chem.202102574
Conflict of Interest
The authors declare no conflict of interest.
References
- [1].Ritossa F Experientia 1962, 18, 571–573. [Google Scholar]
- [2].Gupta RS, Mol. Bio. Evol 1995, 12, 1063–1073. [DOI] [PubMed] [Google Scholar]
- [3].Stechmann A, Cavalier-Smith T, J. Mol. Evol 2003, 57, 408–419. [DOI] [PubMed] [Google Scholar]
- [4].Jarosz DF, Lindquist S, Taipale M, Nat. Rev. Mol. Cell Biol 2010, 11, 515–528. [DOI] [PubMed] [Google Scholar]
- [5].Whitesell L, Lindquist SL, Nat. Rev. Cancer 2005, 5, 761–772. [DOI] [PubMed] [Google Scholar]
- [6].Picard D, Cell. Mol. Life Sci 2002, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Chiosis G, Neckers L, ACS Chem. Biol 2006, 1, 279–284. [DOI] [PubMed] [Google Scholar]
- [8].Stravopodis DJ, Margaritis LH, Voutsinas GE, Curr. Med. Chem 2007, 14, 3122–3138. [DOI] [PubMed] [Google Scholar]
- [9].Krukenberg KA, Street TO, Lavery LA, Agard DA, Q. Rev. Biophys 2011, 44, 229–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Miyata Y, Nakamoto H, Neckers L, Curr. Pharm. Des 2013, 19, 347–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Sreedhar AS, Kalmár E, Csermely P, Shen Y, FEBS Lett. 2004, 562, 11–15. [DOI] [PubMed] [Google Scholar]
- [12].Chen B, Piel WH, Gui L, Bruford E, Monteiro A, Genomics. 2005, 86, 627–637. [DOI] [PubMed] [Google Scholar]
- [13].Garg G, Khandelwal A, Blagg BSJ, Adv. Cancer Res 2016, 129, 51–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Roe SM, O’Brien R, Prodromou C, Piper PW, Pearl LH, Ladbury JE, Panaretou B, The EMBO. J 1998, 17, 4829–4836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Prince TL, Kijima T, Tatokoro M, Lee S, Tsutsumi S, Yim K, Rivas C, Alarcon S, Schwartz H, Khamit-Kush K, Scroggins BT, Beebe K, Trepel JB, Neckers L, PLoS One 2015, 10, e0141786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Zubrienė A, Gutkowska M, Matulienė J, Chaleckis R, Michailovienė V, Voroncova A, Venclovas C, Zylicz A, Zylicz M, Matulis D, Biophys. Chem 2010, 152, 153–163. [DOI] [PubMed] [Google Scholar]
- [17].Bhat R, Tummalapalli SR, Rotella DP, J. Med. Chem 2014, 57, 8718–8728. [DOI] [PubMed] [Google Scholar]
- [18].Barrott JJ, Haystead TAJ, FEBS J. 2013, 280, 1381–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Jhaveri K, Taldone T, Modi S, Chiosis G, Biochim. Biophys. Acta 2012, 1823, 742–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Khandelwal A, Crowley VM, Blagg BSJ, Med. Res. Rev 2016, 36, 92–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Neckers L, Workman P, Clin. Cancer Res 2012, 18, 64–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Garcia-Carbonero R, Carnero A, Paz-Ares L, Lancet Oncol. 2013, 14, 358. [DOI] [PubMed] [Google Scholar]
- [23].Powers MV, Workman P, FEBS Lett. 2007, 581, 3758–3769. [DOI] [PubMed] [Google Scholar]
- [24].Patel PD, Yan P, Seidler PM, Patel HJ, Sun W, Yang C, Que NS, Taldone T, Finotti P, Stephani RA, Gewirth DT, Chiosis G, Nat. Chem. Biol 2013, 9, 677–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Duerfeldt AS, Peterson LB, Maynard JC, Ng CL, Eletto D, Ostrovsky O, Shinogle HE, Moore DS, Argon Y, Nicchitta CV, Blagg BSJ, J. Am. Chem. Soc 2012, 134, 9796–9804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Crowley VM, Khandelwal A, Mishra S, Stothert AR, Huard DJE, Zhao J, Muth A, Duerfeldt AS, Kizziah JL, Lieberman RL, Dickey CA, Blagg BSJ, J. Med. Chem 2016, 59, 3471–3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Khandelwal A, Kent CN, Balch M, Peng S, Mishra SJ, Deng J, Day VW, Liu W, Subramanian C, Cohen M, Holzbeierlein JM, Matts R, Blagg BSJ, Nat. Commun 2018, 9, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Mishra SJ, Liu W, Beebe K, Banerjee M, Kent CN, Munthali V, Koren J, Taylor JA, Neckers LM, Holzbeierlein J, Blagg BSJ, J. Med. Chem 2021, 64, 1545–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Shill MC, Das AK, Itou T, Karmakar S, Mukherjee PK, Mizuguchi H, Kashiwada Y, Fukui H, Nemoto H, Bioorg. Med. Chem 2015, 23, 6869–6874. [DOI] [PubMed] [Google Scholar]
- [30].Molander GA, Sandrock DL, Curr. Opin. Drug Discov. Devel 2009, 12, 811–823. [PMC free article] [PubMed] [Google Scholar]
- [31].Ezquerra J, Pedregal C, Lamas C, Barluenga J, Pérez M, García-Martín MA, González JM, J. Org. Chem 1996, 61, 5804–5812. [Google Scholar]
- [32].Crestey F, Collot V, Stiebing S, Rault S, Tetrahedron 2006, 62, 7772–7775. [Google Scholar]
- [33].Botvay A, Máthé Á, Pöppl L, Polymer 1999, 40, 4965–4970; [Google Scholar]; Máthé, Pöppl L, Polymer 1999, 40, 4965–4970. [Google Scholar]
- [34].Renneberg D, Dervan PB. J. Am. Chem. Soc 2003, 125, 5707–5716. [DOI] [PubMed] [Google Scholar]
- [35].Bolgunas S, Clark DA, Hanna WS, Mauvais PA, Pember SO, J. Med. Chem 2006, 49, 4762–4766. [DOI] [PubMed] [Google Scholar]
- [36].Chiu C, Chiu H, Lee D, Lu T, RSC Adv. 2018, 8, 26407–26415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Mahapatra T, Jana N, Nanda S, Tetrahedron: Asymmetry 2008, 19, 1224–1232. [Google Scholar]
- [38].Liu J, Chan J, Bryant CM, Duspara PA, Lee EE, Powell D, Yang H, Liu Z, Walpole C, Roberts E, Batey RA, Tetrahedron Lett. 2012, 53, 2971–2975. [Google Scholar]
- [39].Pigeon X, Bergeron M, Barabé F, Dubé P, Frost HN, Paquin J, Angew. Chem. Int. Ed 2010, 49, 1123–1127. [DOI] [PubMed] [Google Scholar]
- [40].Lu Y, Li Y, Zhang R, Jin K, Duan C, J. Fluorine Chem 2014, 161, 128–133. [Google Scholar]
- [41].Krishnamoorthy S, Kothandaraman J, Saldana J, Prakash GKS, Eur. J. Org. Chem 2016, 2016, 4965–4969. [Google Scholar]
- [42].Li Y, Lou S, Zhou Q, Zhu L, Zhu L, Li L, Eur. J. Org. Chem 2015, 2015, 3044–3047. [Google Scholar]
- [43].Takeda T, Terada M, J. Am. Chem. Soc 2013, 135, 15306–15309. [DOI] [PubMed] [Google Scholar]
- [44].Leanna RM, Pat US. Appl. Publ., US.20080287676 A1, 20 Nov 2008. [Google Scholar]
- [45].Hauser FM, Zhou M, Sun Y, Synth. Commun 2002, 31, 77–80. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.