

Abstract
Metabolic disorders (MDs), including type-1 and 2 diabetes (T2DM) and chronic obesity, are among the faster growing diseases globally and are a primary risk factor for Alzheimer’s disease (AD). The term “type-3 diabetes” has been proposed for AD due to the interrelated cellular, metabolic, and immune features shared by diabetes, insulin resistance (IR), and the cognitive impairment and neurodegeneration found in AD. Patients with MDs and/or AD commonly exhibit altered glucose homeostasis and IR; systemic chronic inflammation encompassing all of the periphery, blood-brain barrier (BBB), and central nervous system (CNS); pathological vascular remodeling; and increased BBB permeability which allows transfusion of neurotoxic molecules from the blood to the brain. This review summarizes the components of the BBB, mechanisms through which MDs alter BBB permeability via immune and metabolic pathways, the contribution of BBB dysfunction to the manifestation and progression of AD, and current avenues of therapeutic research which address BBB permeability. In addition, issues with the translational applicability of current animal models of AD regarding BBB dysfunction and proposals for future directions of research that address the relationship between MDs, BBB dysfunction, and AD are discussed.
Article Summary:
This review summarizes the components of the blood-brain barrier (BBB), mechanisms through which metabolic disorders alter BBB permeability via immune and metabolic pathways, the contribution of BBB dysfunction to the manifestation and progression of Alzheimer’s disease, and current avenues of therapeutic research which address BBB permeability
The incidence of metabolic disorders, such as obesity and diabetes, is increasing globally with a corresponding increase in AD. Diabetes is defined by hyperglycemia caused by impaired insulin secretion (type-1), resistance to insulin (type-2), or both1. The number of individuals with diabetes worldwide has quadrupled since 1980, making diabetes the 4th most common cause of death from non-communicable disease. Obesity rates in the United states are three times the global average, with 39.8% of adults having a body-mass index of 30 or greater2 and globally since 1975, the number of individuals classified as obese has tripled to 650 million adults, or 13% of the population3. Obesity is also associated with IR, as well as dyslipidemia, and hypertension4. At the same time, in the United States AD is the sixth most common cause of death. There are 5.8 million patients currently diagnosed with AD in the US, a number that may grow to over 13 million people by 2050 as the baby-boomer generation ages5. AD is characterized by functional cognitive impairment coupled with increased deposition of beta amyloid protein (Aβ) and aggregation of hyperphosphorylated tau protein6. Unfortunately, shared causal mechanisms between MDs and AD lead to significant comorbidity, with some researchers referring to AD as “type-3 diabetes”7–9 due to the commonalities. Understanding the metabolic, immunological, and vascular complications of MDs and AD is an essential component of developing and implementing interventions aimed at reducing the number of individuals who develop AD, identifying those in the early stage of the disease, slowing or stopping its progression, and alleviating symptoms such as cognitive impairment. Many of these commonalities occur at the BBB, the selectively permeable series of membranes and cells that protect the CNS.
Role of the BBB
The BBB regulates the transport of ions and molecules (including nutrients) to and from the brain. The barrier regulates adequate supply of oxygen, glucose, and trace elements while excluding toxic plasma components and leukocytes which could damage neural tissue if not excluded10. The endothelial layer that comprises the barrier has four primary components: endothelial cells, mural cells, cells of the basal lamina, and glia. The endothelial cells of the barrier are highly polarized and held together in tight junctions that exclude large and hydrophilic molecules and regulate passage of smaller molecules and ions. Tight junctions are composed of integral proteins such as occludins, claudins, zonula occludens protein-1, and alpha and beta-catenin which form the tight junctions or adhere the junctions to the cytoarchitecture11. Importantly, the BBB endothelial layer protects the CNS from peripheral immune cells and other immune factors which would cause inflammation and damage within the brain12. The inner/abluminal membrane contains two types of mural cells: pericytes and vascular smooth muscle cells. Pericytes support remodeling and tone of the approximately 600–700km (in an adult human) of cerebral vasculature, forming tendrils that ensheath the capillary wall13. Vascular smooth muscle cells surround arteries and contract or expand to control blood flow14 and hence provide oxygen. Constant oxygen supply is vital as hypoxia damages neurons within minutes10. Two layers of basal lamina surround the abluminal surface and provide a structural framework: an endothelial membrane that is secreted by endothelial cells and a parenchymal membrane secreted by astrocytes15. The basal lamina is selectively permeable to, and regulates concentrations of, growth factors, hormones, and essential nutrients16,17. Both layers function to maintain barrier integrity, and the parenchymal layer further connects astrocyte processes to the membrane15. Among glial cells functioning at the BBB, astrocytes are particularly important. Astrocytes regulate blood flow, ionic concentration (including potassium and calcium), and glucose transport from periphery facing endothelial cells to neurons18,19. Under healthy conditions the components of the BBB work together to maintain homeostasis and efficiently regulate influx and efflux of essential materials in the CNS. However, MDs can impede healthy functioning and compromise the BBB.
Metabolic disorders: disruptions of the BBB relevant to AD
MDs impair molecular transport, vascular structure, and tight junction integrity20–23. Chronic obesity and T2DM damage the tight junctions of the barrier and increase permeability to larger peripheral blood products22,23. These effects are particularly robust in the hippocampus24; this is potentially important because cognitive and metabolic processes specifically in the hippocampus are also markedly impaired by IR, so that hippocampal cognitive dysfunction is a key symptom of T2DM and obesity25,26. In patients with T2DM, which is defined by systemic insulin resistance, specifically hippocampal damage is evident early in the development of the disease and persists throughout disease progression27–29. Insulin resistance within the brain and specifically within the hippocampus causes cognitive impairment: in particular, insulin regulates glucose metabolism in the hippocampus (as well as in the periphery)30,31 and is an essential component of hippocampal memory including regulation of local glucose metabolism via the insulin sensitive glucose transporter 432–34. Rats perform better on spatial memory tasks after intrahippocampal administration of insulin and performance is impaired by blocking intrahippocampal insulin signaling25,35. IR is manifest in the hippocampus of individuals with MDs and AD and is correlated with cognitive impairment36; moreover, IR is apparent in hippocampal and cortical cells derived from AD patients postmortem even without a prior MD diagnosis37. Hence, MDs appear likely to have effects on the BBB that correlate with, but may precede, cognitive and brain-metabolic impacts.
Consistent with this, BBB dysfunction is apparent in IR mice before any cognitive effects of a high-fat diet (HFD)38. Diet-induced obese (DIO) mice exhibit early BBB dysfunction at the hippocampus and hypothalamus due to increased production of reactive oxygen species, causing cellular stress and a decrease in the tight junction proteins claudin-5 and zonula occludens-139,40. Other rodent models of T2DM, such as Zucker diabetic fatty rats, natively show the same decrease in occludins and claudin-5, most significantly in the hippocampus23. Tight junction disruption at the hippocampus corresponds with increased hippocampal expression of the proinflammatory signaling molecules interferon-gamma (IFN-γ) and interleukin-1beta (IL-1β)41. Further, hyperglycemia, as manifest in T2DM, increases production of protein kinase C (PKC) causing a reduction of bioavailable nitric oxide (NO) and a thickening of basement membranes, respectively impairing vasorelaxation and inducing vasoconstriction42,43. Alterations in glucose transport across the BBB is evident in the early phases of AD and may be another link between pathological metabolic changes and AD development and progression. The primary glucose transporter at the brain endothelium, glucose transporter-1 (GLUT-1) is reduced in AD patients44–46 as is GLUT-3, an important neuronal glucose transporter47. GLUT-1 deficient transgenic mice overexpressing human APP Swedish mutation exhibit increased amyloidosis, reduced cerebral vascularization and blood-flow, and decreased GLUT-1 expression, despite showing no changes in blood glucose levels48. Hypoglycemia, a common side-effect of insulin treatment, decreases claudin-5 expression and disrupts the functioning of zona occluden-149, and even among elderly patients with T2DM, the number of hypoglycemic events is associated with increased risk of dementia50. Dyslipidemia, a component of central obesity, contributes to a chronic inflammatory response51 and, among patients with AD, is associated with significantly increased incidence of BBB dysfunction52. In both diabetes and obesity, metabolic and immune changes at the BBB contribute to an inflammatory cascade that increases the risk of cognitive impairment via impaired molecular transport and vascular changes53.
Chronically elevated inflammation damages the cerebral vasculature and is a key factor linking metabolic disorders, BBB degeneration, and AD (figure 1)54,55. Inflammation is an immune mechanism that is a key component of the body’s adaptive response to pathogens and injury56, but severe or chronic inflammation can lead to pathology57,58. T2DM causes endothelial cells at the BBB to increase expression of receptor for advanced glycation end-products (RAGE) which activates the proinflammatory nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB) pathway and leads to further increased production of pro-inflammatory cytokines interleukin-6 (Il-6) and tumor-necrosis factor alpha (TNF-α)53,55,59 causing an impaired endothelial vasodilatory response42. BBB inflammation induces vascular remodeling leading to hypoxia and increases secretion of hypoxia-induced factor 1a (HIF1a)60 which enhances Aβ toxicity and continues to increase the production of inflammatory cytokines locally61. Over time, these vascular impairments lead to chronic cerebral hypoperfusion and upregulated expression of RAGE, leading to increased Aβ deposition in the brain and further neuroinflammation via the NF-κB pathway62. Hypoperfusion also causes pericytes to detach from the basement membrane, increasing vascular permeability to large, and potentially neurotoxic, molecules from the periphery63. The resulting reduction in pericyte coverage correlates with cortical and hippocampal capillary and BBB dysfunction and occurs secondarily to decreases in cerebral blood-flow, indicating that hypoxia and hypoperfusion instigate the change63. Hypoperfusion, inflammation, and reduced pericyte coverage at the hippocampus contributes to a loss of hippocampal volume in conjunction with progressive cognitive impairment in both T2DM and AD24,64. AD is associated with cerebral, and specifically hippocampal, hypometabolism65,66; hence, reduction in supply of glucose and/or oxygen subsequent to hypoperfusion is likely to be a major causal link between MDs and AD. Through the process of inflammation and hypoxia induced vascular damage, MDs cause chronic damage to the BBB and increase AD risk via hypometabolism and increased concentration of Aβ.
Figure 1:
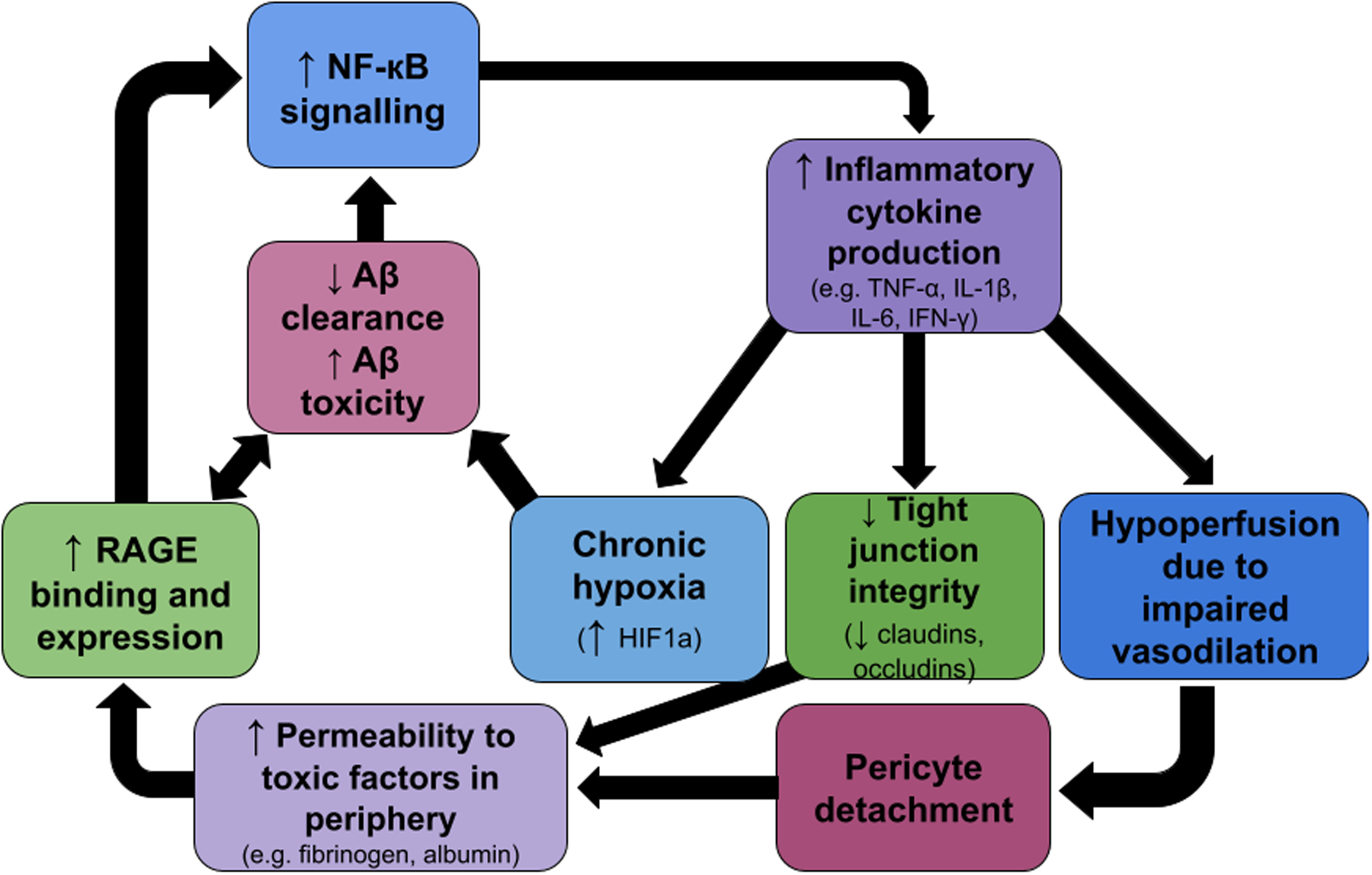
A simplified schematic representing some of the cascading positive feedback loops connecting inflammatory responses, vascular damage, BBB changes, and increased amyloid toxicity.
Contribution of BBB breakdown to AD progression
BBB breakdown is a critical component of AD pathology (reviewed in table 1). A low level of BBB breakdown in medial temporal regions is a normal aspect of aging; during the etiology of AD, however, this breakdown increases (initially prior to cognitive impairment) with the extent of barrier dysfunction predictive of the magnitude of cognitive impairment67–73. Nation et al (2019) found that increased permeability is not correlated with an increase in CSF amyloid or tau in patients with early cognitive impairment69, suggesting the possibility of multiple converging mechanisms and specifically that BBB breakdown may be a causal factor for AD independent of abnormal central protein factors. A similar result is shown in transgenic mice expressing the AD risk factor gene apolipoprotein 4 (APOE4) and 5 AD risk factor transgenes altering amyloid precursor protein (APP) and presenillin-1 (PS1) (APOE4;5xFAD). These mice display increased BBB permeability along with reduced cerebral blood-flow and pericyte coverage in the cortex and hippocampus74. APOE4;5xFAD mice and AD patients also display increased concentrations of fibrinogen in the brain, a key clotting factor in the periphery which incites neuroinflammation and demyelination upon infiltration to the CNS74–78. The BBB impairment shown in APOE4;5xFAD mice is due, at least in part, to activity in the inflammatory cyclophilin A/matrix metalloproteinase 9 (CyA/MMP9) pathway, as pharmacological inhibition of the pathway attenuated neuronal loss and improved cognitive performance74, again suggesting that proinflammatory cascades contribute significantly to barrier dysfunction. As in the proinflammatory cascade observed in MDs, BBB breakdown throughout AD progression is due in part to the impact of increased Aβ on the brain endothelium67–69,71,73. One of the first neural structures to be adversely impacted by AD is the hippocampus which is both a target of the early impact of MDs and a site of early BBB permeability70–73. Dysregulated molecular transport at the BBB increases AD pathology risk: hyperinsulinemia, as seen in MDs, damages the vasculature of the BBB while decreasing clearance of toxic Aβ42 oligomers and increasing permeability of the barrier to peripheral Aβ4279. Neuronal Aβ accumulation is a hallmark of AD pathology, and Aβ oligomers specifically are neurotoxic and cause a pronounced inflammatory response80,81. Simultaneously, increased expression of vasoconstrictive endothillin-1 and Aβ accumulation in vessel walls causes a thinning of neuronal capillaries and a reduction in total vascularization in the brains of patients with AD and T2DM causing chronic hypoperfusion82 and DIO mice display a similar vascular insufficiency and are at increased risk for transient ischemic strokes causing acute hypoperfusion83. These factors incite a feed-forward mechanism in BBB dysfunction whereby metabolic dysregulation and inflammation cause hypoperfusion and impair Aβ clearance; these in turn induce vascular changes, worsen cerebral hypoperfusion, further degrade the barrier, and increase inflammation61. Aβ efflux is dependent on both concentration and active transport via RAGE and low density lipoprotein receptor-1 (LRP-1) and estimates for total Aβ clearance across the BBB range from 25–85% of total efflux84–86. Therefore, any reduction in Aβ transport may contribute to AD. This is supported by observed reductions in LRP-1 in both AD patients and APOE knockout mice84 and by impairment in Aβ clearance in mice injected with radiolabeled Aβ1–40 and an anti-LRP-1 antibody85. In vitro models using human AD cultured neurons exposed to Aβ found that barrier permeability increased along with increased levels of reactive oxygen species (ROS), MMP2, and IFN-y as well as decreased expression of the tight junction-regulating proteins Claudin-1,5, and VE-cadherin87. Although the effect of Aβ to increase permeability of the BBB has been shown several times, work analyzing cognitive impairment and barrier integrity in patients with AD and several other neurodegenerative disorders found that it may be a secondary effect in many cases: barrier permeability was more strongly associated with diabetes and damage to the brain microvasculature than amyloid burden or genetic risk factors88. It is likely that several overlapping dysfunctions cause BBB breakdown in patients with both MDs and (pre-)AD. Overall, converging evidence indicates that BBB breakdown may be a critical component in the progression from MDs to AD.
Table 1:
summary of cited literature regarding metabolic dysfunction at the BBB and AD and the role of BBB dysfunction in AD.
| Metabolic dysfunction at the BBB | Mooradian (1997)21 | Diabetes is associated with pathological changes to BBB microvasculature |
| Rhea et al. (2017)22 | Review: obesity decreases expression of TJ proteins, thins cerebrovasculature, and increases neuroinflammation | |
| Chang et al. (2014)39 | HFD disrupts BBB function and increases neuronal apoptosis | |
| Salameh et al. (2019)40 | DIO disrupts hippocampal and hypothalamic BBB | |
| Roy et al. (2010)43 | Diabetes causes thickening of vascular basement membrane | |
| Sajja et al. (2014)59 | Hypoglycemia reduces claudin-5 and disrupts zona occluden-1 function | |
| Serlin et al. (2011)53 | Diabetes induces vascular damage, impairs glucose transport, and increases RAGE signaling | |
| Yamagishi and Imaizumi (2005)55 | Hyperglycemia increases productions of advanced glycation end products and reactive oxygen species, resulting in vascular | |
| Li et al. (2016)83 | BBB disruption is more severe in obese mice following ischemic stroke | |
| Metabolic dysfunction in AD | Allen et al. (2004)20 | Meta-analysis: T2DM is associated with increased risk and faster progression of AD |
| Roberts et al. (2014)24 | Mid-life diabetes is associated with loss of brain volume and increased cognitive impairment | |
| McNay et al. (2010)25, McNay & Recknagel (2011)26 | Central insulin resistance impairs spatial working memory | |
| Bruel et al. (2011)27 | T2DM is associated with reduction in HPC volume in obese adolescents | |
| den Heijer et al. (2003)28 | T2DM is associated with reduction in HPC volume | |
| Gold et al. (2007)29 | T2DM associated with reduced HPC volume and impaired cognitive performance in middle aged | |
| McNay and Pearson-Leary (2020)33 | Reduced glucose transport associated with cognitive impairment in rats | |
| Biessels & Reagan (2015)36 | IR is evident in brains of patients with T2DM and AD | |
| Talbot et al. (2012)37 | Reduced insulin signaling evident in the HPC in AD patients without T2DM | |
| Hardigan et al. (2016)42 | Diabetes induces cerebrovascular dysfunction resulting in cognitive impairment | |
| Mosconi et al. (2010)66, (2013)65 | Decreased brain glucose utilization occurs early in AD disease progression and correlates with severity | |
| Swaminathan et al. (2018)79 | Peripheral insulin administration alters the clearance of Aβ40 and Aβ42 | |
| Small et al. (1995)98 | APOE4 carriers display reduced parietal metabolism | |
| BBB dysfunction in AD | Rhea et al. (2020)91 | Review of the impact of APOE4 and insulin on the BBB |
| Takechi et al. (2017)38 | Cognitive decline is preceded by BBB dysfunction in IR mice | |
| Horwood and Davies (1994)44, Kalaria and Harik (1989)46, Mooriadian (1997)21, Simpson et al. (1994)47 | BBB glucose transport reduced in AD | |
| Bowman et al. (2018)52 | AD patients with dyslipidemia exhibit increased BBB permeability | |
| Chakraborty et al. (2017)61 | Review: vascular disturbance impairs BBB function and increased AD risk | |
| Yang et al. (2020)62 | Cerebral hypoperfusion increases neuroinflammation, Aβ accumulation, and BBB permeability | |
| Liu et al. (2019)63 | Chronic hypoperfusion reduces BBB integrity and decreases pericyte coverage | |
| Montagne et al. (2015)67 | BBB breakdown is evident in early AD and correlates with degree of cognitive impairment | |
| Montagne et al. (2020)68 | BBB breakdown in APOE4 carriers precedes and predicts cognitive decline independent of Aβ or tau | |
| Nation et al. (2019)69 | BBB breakdown is evident in individuals with early mild cognitive impairment independent of Aβ or tau | |
| Montagne et al. (2021)74, Bell et al. (2012)93, Halliday et al. (2016)94 | APOE4 accelerated BBB dysfunction is dependent on the Cyclophilin A/MMP9 pathway and independent of Aβ | |
| Cortes-Canteli et al. (2010)75 | Fibrinogen clots colocalize with Aβ, and fibrinogen depletion improves cognitive performance | |
| Hultman et al. (2013)76 | APOE4 exacerbates Aβ associated fibrinogen deposition | |
| Miners et al. (2018)77 | Decreased BBB integrity and pericyte marker PDGFRβ is associated with increased fibrinogen deposition and Aβ accumulation in the brain. | |
| Ryu and McLarnon (2009)78 | Co-administration of Aβ42 and fibrinogen cause pronounced neuroinflammation which can be reduced by blocking microglial activation | |
| Bailey et al. (2004)82 | Review of the contributions of microvascular pathology to AD | |
| Nelson et al. (2017)85, Shibata et al. (2000)84 | Aβ influx/efflux is dependent on BBB RAGE and LRP1/2 | |
| Zlokovic (2013)92 | Review of the cerebrovascular effects of APOE4 | |
| Main et al. (2018)95 | BBB regeneration is impaired by APOE4 following traumatic brain injury | |
| Alata et al. (2015)96 | APOE4 mice display decreased cerebral vascularization and glucose transport across the BBB | |
| Zipser et al. (2007)100 | Prothrombin levels in the cortex correlate with Braak stage |
Abbreviations: Aβ, beta amyloid protein; AD, Alzheimer’s disease; APOE, apolipoprotein E; BBB, blood–brain barrier; DIO, diet-induced obese; HFD, high-fat diet; HPC, hematopoietic progenitor cell; IR, insulin resistance; LRP, low density lipoprotein receptor; MMP, metalloproteinase; PDFGR, platelet-derived growth factor receptor; RAGE, receptor for advanced glycation end-products; T2DM, type 2 diabetes; TJ, tight junction.
Genotype can also contribute to BBB degradation relevant to AD. Carriers of the APOE4 isoform are at increased risk for the development of AD partially due to alterations at the BBB. Although APOE4 and low-density lipoproteins do not cross the BBB89,90, trans-BBB transport of insulin91 and Aβ92 is partially regulated by APOE4 and is impaired in APOE4 carriers. APOE4 carriers have increased activity in the proinflammatory CyA/MMP9 pathway in pericytes and endothelial cells, leading to apoptosis, increased barrier permeability, and impaired barrier repair when compared to APOE3 carriers68,93–95. APOE4 mice displayed a 29% reduction in glucose transport and a 41.3% increase in RAGE expression at the BBB96. Decreased brain glucose metabolism is a hallmark symptom of AD65,66 which is pronounced in positron emission tomography scans of APOE4 carriers prior to symptoms of cognitive impairment97,98, during mild cognitive impairment (MCI)99, and after progression to AD65, indicating a relationship between APOE4 genotype, cerebral metabolic dysfunction, and AD risk. In cognitively unimpaired subjects, APOE4 carriers exhibit increased permeability of the BBB at the hippocampus and parahippocampal gyrus which is not explained by elevated Aβ or tau68. Within AD patients, APOE4 status predicts increased accumulation of the peripheral clotting factor prothrombin, an indicator of BBB leakage and vascular damage, in both prefrontal cortex and CSF100. The complexity of the interrelation between metabolic factors, immune functioning, and APOE genotype limits the utility of approaches which account only for one aspect and is likely a significant reason why effective treatment strategies for AD have been elusive to-date.
Therapeutic targets: insulin resistance and the BBB.
The BBB is both a critical target for, and a significant barrier to, the development of therapeutic agents for the treatment of AD101. To date, therapeutics targeting amyloid and tau have failed to slow progression and/or reduce symptoms of AD102. Hence, novel therapies targeting other aspects of the disease including aberrant inflammation, hypometabolism, and BBB permeability are required. Drugs currently approved to treat metabolic dysfunction have promise to improve BBB function and reduce the pace of cognitive impairment. The glucagon-like peptide-1 (GLP-1) analogue liraglutide crosses the BBB103 and improves cognition in animal models104,105 and an exploratory trial of the GLP-1 analog dulaglutide found potential for slowing cognitive decline in T2DM patients106. Central delivery of insulin is another treatment for brain metabolic dysfunction that shows promise in treating AD. Insulin transport into the brain is reduced in both patients with obesity and AD107,108. In DIO mice, exogenous insulin increased efflux and decreased influx of Aβ across the BBB, thereby reducing Aβ accumulation, and improved cognitive performance109. However, as insulin influx to the brain is saturable, the amount of insulin that can be transported across the BBB is inherently limited110. To overcome this limitation, insulin may be administered intranasally, thereby bypassing the BBB and entering the brain alongside the olfactory and trigeminal neural pathways111,112. By administering insulin intranasally rather than peripherally, therapeutically relevant doses may be delivered directly to the CNS and avoid potential side effects of large doses of insulin administered to the periphery113. In mice overexpressing APP and PS1, intranasal insulin reduced Aβ plaque formation, reduced Aβ production, improved insulin signaling, and alleviated cognitive impairments114. In both wild-type and senescence-prone accelerated mice (a model of accelerated and sporadic AD), intranasal insulin is detected in the brain within 5 minutes of administration and remains increased for up to 60 minutes post-administration115. In clinical trials, patients with MCI to moderate AD symptoms receiving a daily dose of intranasal insulin over the course of 4 months improved cognitive functioning and decreased hyper-phosphorylated Tau and Aβ42 ratios116.
There are, however, caveats to the use of insulin in restoring brain metabolism, improving cognition, and decreasing Aβ load. These issues include sex differences in the BBB’s response to inflammation117,118 and the impact of APOE genotype on BBB integrity as discussed above. One study found that intranasal insulin improved cognition in APOE4 negative males but worsened cognition in APOE4 negative females119 while a subsequent study in the same lab found that four months of intranasal insulin improved cognition for APOE4 carriers but worsened cognition for non-APOE4 carriers120. It is possible, therefore, that intranasal insulin may only be an efficacious therapy for a subset of AD patients at most. Exogenous insulin may also be less effective in the context of MDs. DIO rats required an order of magnitude higher dose of intrahippocampal insulin than control and diet-resistant rats in order to display cognitive enhancement121. Chronic systemic insulin therapy can also result in recurrent hypoglycemia which, when severe enough to require hospitalization or emergency intervention, is itself associated with an increased risk of dementias, both AD and vascular122. Moreover, insulin and Aβ compete for a common breakdown pathway123,124, so that hyperinsulinemia is associated with impaired degradation of Aβ125 and chronic insulin therapy at clinically useful doses may potentially lead to an unwelcome increase in brain amyloid burden. There is convincing evidence that some level of insulin is produced in the brain, rather than being transported from the periphery34; speculatively, a further potential therapeutic approach would be to support and/or increase such production.
Targeting the neuroinflammatory response is a viable, if complex, alternative avenue of therapeutic development which has the potential to attenuate damage to the BBB and retard development of AD. Administration of non-steroidal anti-inflammatory drugs (NSAIDs), a once-promising avenue of treatment126, has unfortunately been unsuccessful. A randomized clinical trial (RCT) administering 220mg of Naproxen or placebo twice daily for two years concluded that Naproxen was ineffective at slowing AD symptom progression and was associated with adverse health events127. Similar results have been found with other NSAIDs128. The lack of clinical efficacy and increase in adverse events may be due to NSAIDs’ common affinity for inhibition of cyclooxygenase 1/prostaglandin G/H 1 (COX-1) which has numerous positive homeostatic functions besides the inflammatory response, and lower affinity for COX-2, which functions much more exclusively as a proinflammatory regulator128. These results indicate that more specific modulation of the immune system may be required. Treatments involving administration of exogenous anti-inflammatory cytokines and their agonists, or antagonists to pro-inflammatory cytokine activity, are currently being investigated. The regulatory/anti-inflammatory cytokine interleukin-10 (IL-10) regulates microglia morphology and the production of IL-6/TNF-α: upregulations of IL-10 attenuates BBB impairment and improves cognition129. Targeting RAGE is another viable avenue of therapeutic intervention currently under development which may reduce inflammation, improve vascular function, and slow cognitive decline in T2DM and AD. The RAGE inhibitor FPS-AM1 passes through the BBB and reduces microglial activation, oxidative stress, and proinflammatory cytokine signalling130, while the RAGE inhibitor Azeliragon slows cognitive deterioration in patients with mild to moderate AD131. As RAGE is a binding target for Aβ, administration of soluble RAGE binds to Aβ in the periphery and prevents RAGE mediated translocation across the BBB132
In clinical trials, resveratrol, a naturally occurring polyphenol found naturally in grapes, has been shown to be well tolerated, improve cognitive outcomes, and preserve BBB integrity while upregulating IL-10 gene expression and concentrations133. Resveratrol also attenuates BBB permeability due to HFD39. Longitudinal epidemiological studies and animal models also indicate that caffeine protects against BBB degenerations and decreases risk of AD development, likely through reducing the effects of oxidative stress and increasing insulin sensitivity134,135. Caffeine also protects against HFD-induced reduction of brain-derived neurotrophic factor (BDNF) and cognitive impairment136. Preliminary animal models have also indicated a benefit from the use of interferon-β1a (IFNβ1a), a regulatory cytokine commonly used to treat multiple sclerosis and a potent inhibitor of IL-6, IL-1β, TNF-α, and IFN-γ137. Dietary interventions aimed at controlling the specific inflammatory factors which impair BBB integrity therefore have potential to delay or prevent AD by preserving normal molecular transport and protecting against vascular injury.
Lifestyle factors should also be addressed with the goal of reducing risk or severity of AD. Regular exercise improves insulin sensitivity, cognitive performance, and endothelial function and decreases inflammatory reactivity at the BBB138,139, and while the exact mechanism of action is not known, the improvement likely involves increased expression of BDNF which promotes neuronal survival and synaptic integrity and stabilizes the vasculature140. In longitudinal studies and RCTs involving chronically obese individuals, taking part in a voluntary weight-loss intervention (diet, exercise, or bariatric surgery) was associated with significant improvement in memory and attention141. Following a weight-loss intervention, neuroinflammation and IR decreased while BBB transport of insulin and leptin increased22. This suggests that treatment for chronic obesity may also reduce risk for the development of AD via improvement of BBB functioning. From a dietary perspective, a procognitive and anti-inflammatory diet such as The Mediterranean-Dietary Approach to Systolic Hypertension (DASH) Intervention for Neurodegenerative Delay (MIND) prevents cognitive decline142 and is associated with a reduce AD risk143. Patients with T2DM adhering to the Mediterranean diet show improved glycemic control144 and reduced high-density lipoprotein and overall adiposity145 and patients with cardiovascular disease on the Mediterranean diet or DASH show reduced biomarkers of inflammation146,147. This suggests that the neuroprotective effect of the MIND diet occurs via beneficial metabolic and immune changes148. Preservation of BBB integrity through non-pharmacological or lifestyle interventions will likely delay or prevent the onset of AD.
Importance of accurate models and early identification
In evaluating models for AD research, attention should be paid to the function of the BBB to ensure translational accuracy. For instance, a comparison of double transgenic APP/PS1 with control B6/SJL mice did not find any difference in barrier permeability to albumin, insulin, or Aβ40149. Another study using APP/PS2, human Tau, and APOE4 knock-in mice lines failed to detect significant alteration in BBB permeability to radiolabeled tracers and albumin150. These findings may suggest that the models do not accurately recapitulate the impact of AD in humans10,68,151. It is also possible that the failure to observe differences is due to methodological issues, as brain perfusion prior to tissue collection may remove non-immune antibodies from the brain and radiolabeled tracers may lack the sensitivity and resolution to detect BBB changes in the context of neurodegenerative disease152. Administration of intranasal insulin increased glucose metabolism in the hippocampus only in wild-type, but not APP/PS1 mice153 indicating that APP/PS1 mice may have altered metabolism in a non-translationally relevant manner. Taken together, this may indicate a limitation of the transgenic models themselves. A potential explanation is that the various transgenic mouse lines do not capture the long-term immunological and metabolic alterations leading to a chronic inflammatory state and vascular changes seen in most AD patients. Due to these inconsistencies, it may be beneficial to concentrate on animal models of AD which include the metabolic and immunological risk antecedents, such as HFD-induced obesity and T2DM.
Patients with MDs exhibit peripheral and CNS IR, chronic systemic inflammation in the CNS, BBB, and periphery, and marked impairment of the cerebrovasculature. Cerebrovascular impairment causes hypoxia and hypoperfusion leading to pericyte loss, damage to tight-junctions, and a further exacerbated inflammatory response. These factors combine to increase AD risk by increasing permeability of the BBB to cytotoxic peripheral blood products, impairing transport of insulin into and Aβ out of the brain, and damaging regions such as the hippocampus and prefrontal cortex. In order to identify patients with MDs who are at risk for developing AD early enough for the progression to be slowed by non-pharmacological lifestyle interventions, health screenings should include rigorous assessment for cognitive impairments154. Given the cascade nature of the inflammatory response, BBB breakdown, and amyloid accumulation in AD, early identification of at-risk individuals before the onset of severe symptoms is an important step in slowing disease progression. Awareness of the interrelation between metabolic, immune, and vascular functioning may also open new treatment modalities in the future.
Acknowledgements:
This work was funded by NIH R01AG050598. The authors would like to thank Angelina Tassone for her help producing figure 1.
Data Availability Statement:
Data sharing is not applicable to this article as no new data were created or analyzed in the current study.
References
- 1.Kerner W, Brückel J, German Diabetes Association. Definition, classification and diagnosis of diabetes mellitus. Exp Clin Endocrinol Diabetes Off J Ger Soc Endocrinol Ger Diabetes Assoc 2014; 122: 384–6. [DOI] [PubMed] [Google Scholar]
- 2.Hales C Prevalence of obesity among adults and youth: United States, 2015–2016. NCHS data brief, no 288. 2017; published online June 6. https://www.cdc.gov/nchs/products/databriefs/db288.htm (accessed May 6, 2020). [PubMed] [Google Scholar]
- 3.World Health Organization. Noncommunicable diseases country profiles 2018. WHO. 2018. http://www.who.int/nmh/publications/ncd-profiles-2018/en/ (accessed May 6, 2020). [Google Scholar]
- 4.Engin A The Definition and Prevalence of Obesity and Metabolic Syndrome. Adv Exp Med Biol 2017; 960: 1–17. [DOI] [PubMed] [Google Scholar]
- 5.2020 Alzheimer’s disease facts and figures. Alzheimers Dement J Alzheimers Assoc 2020; published online March 10. DOI: 10.1002/alz.12068. [DOI] [Google Scholar]
- 6.Soria Lopez JA, González HM, Léger GC. Alzheimer’s disease. Handb Clin Neurol 2019; 167: 231–55. [DOI] [PubMed] [Google Scholar]
- 7.de la Monte SM, Wands JR. Alzheimer’s Disease is Type 3 Diabetes—Evidence Reviewed. J Diabetes Sci Technol 2008; 2: 1101–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kandimalla R, Thirumala V, Reddy PH. Is Alzheimer’s disease a Type 3 Diabetes? A critical appraisal. Biochim Biophys Acta Mol Basis Dis 2017; 1863: 1078–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Steen E, Terry BM, J. Rivera E, et al. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease – is this type 3 diabetes? J Alzheimers Dis 2005; 7: 63–80. [DOI] [PubMed] [Google Scholar]
- 10.Zlokovic BV. The Blood-Brain Barrier in Health and Chronic Neurodegenerative Disorders. Neuron 2008; 57: 178–201. [DOI] [PubMed] [Google Scholar]
- 11.Ueno M Molecular Anatomy of the Brain Endothelial Barrier: An Overview of the Distributional Features. Curr Med Chem 2007; 14: 1199–206. [DOI] [PubMed] [Google Scholar]
- 12.Muldoon LL, Alvarez JI, Begley DJ, et al. Immunologic privilege in the central nervous system and the blood-brain barrier. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab 2013; 33: 13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Winkler EA, Bell RD, Zlokovic BV. Central nervous system pericytes in health and disease. Nat Neurosci 2011; 14: 1398–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aird WC. Phenotypic Heterogeneity of the Endothelium: II. Representative Vascular Beds. Circ Res 2007; 100: 174–90. [DOI] [PubMed] [Google Scholar]
- 15.Del Zoppo GJ, Milner R, Mabuchi T, Hung S, Wang X, Koziol JA. Vascular matrix adhesion and the blood-brain barrier. Biochem Soc Trans 2006; 34: 1261–6. [DOI] [PubMed] [Google Scholar]
- 16.Arends F, Lieleg O. Biophysical Properties of the Basal Lamina: A Highly Selective Extracellular Matrix. In: Travascio F, ed. Composition and Function of the Extracellular Matrix in the Human Body. InTech, 2016. DOI: 10.5772/62519. [DOI] [Google Scholar]
- 17.Thomsen MS, Routhe LJ, Moos T. The vascular basement membrane in the healthy and pathological brain. J Cereb Blood Flow Metab 2017; 37: 3300–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cabezas R, à vila M, Gonzalez J, et al. Astrocytic modulation of blood brain barrier: perspectives on Parkinson’s disease. Front Cell Neurosci 2014; 8. DOI: 10.3389/fncel.2014.00211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Daneman R The blood-brain barrier in health and disease. Ann Neurol 2012; 72: 648–72. [DOI] [PubMed] [Google Scholar]
- 20.Allen KV, Frier BM, Strachan MWJ. The relationship between type 2 diabetes and cognitive dysfunction: longitudinal studies and their methodological limitations. Eur J Pharmacol 2004; 490: 169–75. [DOI] [PubMed] [Google Scholar]
- 21.Mooradian AD. Central nervous system complications of diabetes mellitus — a perspective from the blood–brain barrier. Brain Res Rev 1997; 23: 210–8. [DOI] [PubMed] [Google Scholar]
- 22.Rhea EM, Salameh TS, Logsdon AF, Hanson AJ, Erickson MA, Banks WA. Blood-Brain Barriers in Obesity. AAPS J 2017; 19: 921–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yoo DY, Yim HS, Jung HY, et al. Chronic type 2 diabetes reduces the integrity of the blood-brain barrier by reducing tight junction proteins in the hippocampus. J Vet Med Sci 2016; 78: 957–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roberts RO, Knopman DS, Przybelski SA, et al. Association of type 2 diabetes with brain atrophy and cognitive impairment. Neurology 2014; 82: 1132–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McNay EC, Ong CT, McCrimmon RJ, Cresswell J, Bogan JS, Sherwin RS. Hippocampal memory processes are modulated by insulin and high-fat-induced insulin resistance. Neurobiol Learn Mem 2010; 93: 546–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McNay EC, Recknagel AK. Reprint of: ‘Brain insulin signaling: A key component of cognitive processes and a potential basis for cognitive impairment in type 2 diabetes’. Neurobiol Learn Mem 2011; 96: 517–28. [DOI] [PubMed] [Google Scholar]
- 27.Bruehl H, Sweat V, Tirsi A, Shah B, Convit A. Obese Adolescents with Type 2 Diabetes Mellitus Have Hippocampal and Frontal Lobe Volume Reductions. Neurosci Med 2011; 2: 34–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.den Heijer T, Vermeer SE, van Dijk EJ, et al. Type 2 diabetes and atrophy of medial temporal lobe structures on brain MRI. Diabetologia 2003; 46: 1604–10. [DOI] [PubMed] [Google Scholar]
- 29.Gold SM, Dziobek I, Sweat V, et al. Hippocampal damage and memory impairments as possible early brain complications of type 2 diabetes. Diabetologia 2007; 50: 711–9. [DOI] [PubMed] [Google Scholar]
- 30.Lundqvist MH, Almby K, Abrahamsson N, Eriksson JW. Is the Brain a Key Player in Glucose Regulation and Development of Type 2 Diabetes? Front Physiol 2019; 10: 457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Petersen MC, Shulman GI. Mechanisms of Insulin Action and Insulin Resistance. Physiol Rev 2018; 98: 2133–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McNay EC. Your Brain on Insulin: From Heresy to Dogma. Perspect Psychol Sci 2014; 9: 88–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McNay EC, Pearson-Leary J. GluT4: A central player in hippocampal memory and brain insulin resistance. Exp Neurol 2020; 323: 113076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McNay EC, Recknagel AK. Brain insulin signaling: A key component of cognitive processes and a potential basis for cognitive impairment in type 2 diabetes. Neurobiol Learn Mem 2011; 96: 432–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gladding JM, Abbott KN, Antoniadis CP, Stuart A, Begg DP. The Effect of Intrahippocampal Insulin Infusion on Spatial Cognitive Function and Markers of Neuroinflammation in Diet-induced Obesity. Front Endocrinol 2018; 9. DOI: 10.3389/fendo.2018.00752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Biessels GJ, Reagan LP. Hippocampal insulin resistance and cognitive dysfunction. Nat Rev Neurosci 2015; 16: 660–71. [DOI] [PubMed] [Google Scholar]
- 37.Talbot K, Wang H-Y, Kazi H, et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J Clin Invest 2012; 122: 1316–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Takechi R, Lam V, Brook E, et al. Blood-Brain Barrier Dysfunction Precedes Cognitive Decline and Neurodegeneration in Diabetic Insulin Resistant Mouse Model: An Implication for Causal Link. Front Aging Neurosci 2017; 9: 399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chang H-C, Tai Y-T, Cherng Y-G, et al. Resveratrol Attenuates High-Fat Diet-Induced Disruption of the Blood–Brain Barrier and Protects Brain Neurons from Apoptotic Insults. J Agric Food Chem 2014; 62: 3466–75. [DOI] [PubMed] [Google Scholar]
- 40.Salameh TS, Mortell WG, Logsdon AF, Butterfield DA, Banks WA. Disruption of the hippocampal and hypothalamic blood–brain barrier in a diet-induced obese model of type II diabetes: prevention and treatment by the mitochondrial carbonic anhydrase inhibitor, topiramate. Fluids Barriers CNS 2019; 16: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hwang IK, Choi JH, Nam SM, et al. Activation of microglia and induction of pro-inflammatory cytokines in the hippocampus of type 2 diabetic rats. Neurol Res 2014; 36: 824–32. [DOI] [PubMed] [Google Scholar]
- 42.Hardigan T, Ward R, Ergul A. Cerebrovascular complications of diabetes: focus on cognitive dysfunction. Clin Sci 2016; 130: 1807–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Roy S, Ha J, Trudeau K, Beglova E. Vascular basement membrane thickening in diabetic retinopathy. Curr Eye Res 2010; 35: 1045–56. [DOI] [PubMed] [Google Scholar]
- 44.Horwood N, Davies DC. Immunolabelling of hippocampal microvessel glucose transporter protein is reduced in Alzheimer’s disease. Virchows Arch Int J Pathol 1994; 425: 69–72. [DOI] [PubMed] [Google Scholar]
- 45.Mooradian AD, Chung HC, Shah GN. GLUT-1 expression in the cerebra of patients with Alzheimer’s disease. Neurobiol Aging 1997; 18: 469–74. [DOI] [PubMed] [Google Scholar]
- 46.Kalaria RN, Harik SI. Reduced glucose transporter at the blood-brain barrier and in cerebral cortex in Alzheimer disease. J Neurochem 1989; 53: 1083–8. [DOI] [PubMed] [Google Scholar]
- 47.Simpson IA, Chundu KR, Davies-Hill T, Honer WG, Davies P. Decreased concentrations of GLUT1 and GLUT3 glucose transporters in the brains of patients with Alzheimer’s disease. Ann Neurol 1994; 35: 546–51. [DOI] [PubMed] [Google Scholar]
- 48.Winkler EA, Nishida Y, Sagare AP, et al. GLUT1 reductions exacerbate Alzheimer’s disease vasculo-neuronal dysfunction and degeneration. Nat Neurosci 2015; 18: 521–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sajja RK, Prasad S, Cucullo L. Impact of altered glycaemia on blood-brain barrier endothelium: an in vitro study using the hCMEC/D3 cell line. Fluids Barriers CNS 2014; 11: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chin SO, Rhee SY, Chon S, et al. Hypoglycemia is associated with dementia in elderly patients with type 2 diabetes mellitus: An analysis based on the Korea National Diabetes Program Cohort. Diabetes Res Clin Pract 2016; 122: 54–61. [DOI] [PubMed] [Google Scholar]
- 51.Reitz C. Dyslipidemia and the risk of Alzheimer’s disease. Curr Atheroscler Rep 2013; 15: 307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bowman GL, Kaye JA, Quinn JF. Dyslipidemia and blood-brain barrier integrity in Alzheimer’s disease. Curr Gerontol Geriatr Res 2012; 2012: 184042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Serlin Y, Levy J, Shalev H. Vascular pathology and blood-brain barrier disruption in cognitive and psychiatric complications of type 2 diabetes mellitus. Cardiovasc Psychiatry Neurol 2011; 2011: 609202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huber JD, Egleton RD, Davis TP. Molecular physiology and pathophysiology of tight junctions in the blood–brain barrier. Trends Neurosci 2001; 24: 719–25. [DOI] [PubMed] [Google Scholar]
- 55.Yamagishi S, Imaizumi T. Diabetic Vascular Complications: Pathophysiology, Biochemical Basis and Potential Therapeutic Strategy. Curr Pharm Des 2005; 11: 2279–99. [DOI] [PubMed] [Google Scholar]
- 56.Taams LS. Inflammation and immune resolution: Editotial. Clin Exp Immunol 2018; 193: 1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.DiSabato DJ, Quan N, Godbout JP. Neuroinflammation: the devil is in the details. J Neurochem 2016; 139: 136–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Heneka MT, Kummer MP, Latz E. Innate immune activation in neurodegenerative disease. Nat Rev Immunol 2014; 14: 463–77. [DOI] [PubMed] [Google Scholar]
- 59.Schmidt AM, Weidman E, Lalla E, et al. Advanced glycation endproducts (AGEs) induce oxidant stress in the gingiva: a potential mechanism underlying accelerated periodontal disease associated with diabetes. J Periodontal Res 1996; 31: 508–15. [DOI] [PubMed] [Google Scholar]
- 60.Shen Y, Gu J, Liu Z, et al. Inhibition of HIF-1α Reduced Blood Brain Barrier Damage by Regulating MMP-2 and VEGF During Acute Cerebral Ischemia. Front Cell Neurosci 2018; 12. DOI: 10.3389/fncel.2018.00288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chakraborty A, de Wit NM, van der Flier WM, de Vries HE. The blood brain barrier in Alzheimer’s disease. Vascul Pharmacol 2017; 89: 12–8. [DOI] [PubMed] [Google Scholar]
- 62.Yang H, Wang W, Jia L, et al. The Effect of Chronic Cerebral Hypoperfusion on Blood-Brain Barrier Permeability in a Transgenic Alzheimer’s Disease Mouse Model (PS1V97L). J Alzheimers Dis 2020; 74: 261–75. [DOI] [PubMed] [Google Scholar]
- 63.Liu Q, Radwanski R, Babadjouni R, et al. Experimental chronic cerebral hypoperfusion results in decreased pericyte coverage and increased blood-brain barrier permeability in the corpus callosum. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab 2019; 39: 240–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.McRae-McKee K, Evans S, Hadjichrysanthou C, Wong MM, de Wolf F, Anderson RM. Combining hippocampal volume metrics to better understand Alzheimer’s disease progression in at-risk individuals. Sci Rep 2019; 9: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mosconi L Glucose metabolism in normal aging and Alzheimer’s disease: Methodological and physiological considerations for PET studies. Clin Transl Imaging 2013; 1. DOI: 10.1007/s40336-013-0026-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mosconi L, Berti V, Glodzik L, Pupi A, De Santi S, de Leon MJ. Pre-clinical detection of Alzheimer’s disease using FDG-PET, with or without amyloid imaging. J Alzheimers Dis JAD 2010; 20: 843–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Montagne A, Barnes SR, Sweeney MD, et al. Blood-Brain Barrier Breakdown in the Aging Human Hippocampus. Neuron 2015; 85: 296–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Montagne A, Nation DA, Sagare AP, et al. APOE4 leads to blood–brain barrier dysfunction predicting cognitive decline. Nature 2020; 581: 71–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nation DA, Sweeney MD, Montagne A, et al. Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat Med 2019; 25: 270–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Convit A, de Leon MJ, Golomb J, et al. Hippocampal atrophy in early Alzheimer’s Disease: Anatomic specificity and validation. Psychiatr Q 1993; 64: 371–87. [DOI] [PubMed] [Google Scholar]
- 71.Bobinski M, Wegiel J, Wisniewski HM, et al. Atrophy of Hippocampal Formation Subdivisions Correlates with Stage and Duration of Alzheimer Disease. Dement Geriatr Cogn Disord 1995; 6: 205–10. [DOI] [PubMed] [Google Scholar]
- 72.Geula C Abnormalities of neural circuitry in Alzheimer’s disease: Hippocampus and cortical cholinergic innervation. Neurology 1998; 51: S18–29. [DOI] [PubMed] [Google Scholar]
- 73.Jaroudi W, Garami J, Garrido S, Hornberger M, Keri S, Moustafa AA. Factors underlying cognitive decline in old age and Alzheimer’s disease: the role of the hippocampus. Rev Neurosci 2017; 28. DOI: 10.1515/revneuro-2016-0086. [DOI] [PubMed] [Google Scholar]
- 74.Montagne A, Nikolakopoulou AM, Huuskonen MT, et al. APOE4 accelerates advanced-stage vascular and neurodegenerative disorder in old Alzheimer’s mice via cyclophilin A independently of amyloid-β. Nat Aging 2021; 1: 506–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cortes-Canteli M, Paul J, Norris EH, et al. Fibrinogen and β-Amyloid Association Alters Thrombosis and Fibrinolysis: A Possible Contributing Factor to Alzheimer’s Disease. Neuron 2010; 66: 695–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hultman K, Strickland S, Norris EH. The APOE ε4/ε4 Genotype Potentiates Vascular Fibrinogen Deposition in Amyloid-Laden Vessels in the Brains of Alzheimer’s Disease Patients. J Cereb Blood Flow Metab 2013; 33: 1251–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Miners JS, Schulz I, Love S. Differing associations between Aβ accumulation, hypoperfusion, blood–brain barrier dysfunction and loss of PDGFRB pericyte marker in the precuneus and parietal white matter in Alzheimer’s disease. J Cereb Blood Flow Metab 2018; 38: 103–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ryu JK, McLarnon JG. A leaky blood-brain barrier, fibrinogen infiltration and microglial reactivity in inflamed Alzheimer’s disease brain. J Cell Mol Med 2009; 13: 2911–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Swaminathan SK, Ahlschwede KM, Sarma V, et al. Insulin differentially affects the distribution kinetics of amyloid beta 40 and 42 in plasma and brain. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab 2018; 38: 904–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Forloni G, Balducci C. Alzheimer’s Disease, Oligomers, and Inflammation. J Alzheimers Dis JAD 2018; 62: 1261–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jongbloed W, Bruggink KA, Kester MI, et al. Amyloid-β oligomers relate to cognitive decline in Alzheimer’s disease. J Alzheimers Dis JAD 2015; 45: 35–43. [DOI] [PubMed] [Google Scholar]
- 82.Bailey TL, Rivara CB, Rocher AB, Hof PR. The nature and effects of cortical microvascular pathology in aging and Alzheimer’s disease. Neurol Res 2004; 26: 573–8. [DOI] [PubMed] [Google Scholar]
- 83.Li C, Jiang Z, Lu W, Arrick D, McCarter K, Sun H. Effect of obesity on early blood–brain barrier disruption following transient focal cerebral ischemia. Obes Sci Pract 2016; 2: 58–68. [Google Scholar]
- 84.Shibata M, Yamada S, Kumar SR, et al. Clearance of Alzheimer’s amyloid-ss(1–40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J Clin Invest 2000; 106: 1489–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Nelson AR, Sagare AP, Zlokovic BV. Role of clusterin in the brain vascular clearance of amyloid-β. Proc Natl Acad Sci U S A 2017; 114: 8681–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Roberts KF, Elbert DL, Kasten TP, et al. Amyloid-β efflux from the central nervous system into the plasma: Brain Efflux of Amyloid-β. Ann Neurol 2014; 76: 837–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Shin Y, Choi SH, Kim E, et al. Blood–Brain Barrier Dysfunction in a 3D In Vitro Model of Alzheimer’s Disease. Adv Sci 2019; 6: 1900962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Janelidze S, Hertze J, Nägga K, et al. Increased blood-brain barrier permeability is associated with dementia and diabetes but not amyloid pathology or APOE genotype. Neurobiol Aging 2017; 51: 104–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mahley RW. Central Nervous System Lipoproteins. Arterioscler Thromb Vasc Biol 2016; 36: 1305–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wang H, Eckel RH. What are Lipoproteins doing in the Brain? Trends Endocrinol Metab TEM 2014; 25: 8–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rhea EM, Raber J, Banks WA. ApoE and cerebral insulin: Trafficking, receptors, and resistance. Neurobiol Dis 2020; 137: 104755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zlokovic BV. Cerebrovascular Effects of Apolipoprotein E: Implications for Alzheimer Disease. JAMA Neurol 2013; 70: 440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bell RD, Winkler EA, Singh I, et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature 2012; 485: 512–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Halliday MR, Rege SV, Ma Q, et al. Accelerated pericyte degeneration and blood–brain barrier breakdown in apolipoprotein E4 carriers with Alzheimer’s disease. J Cereb Blood Flow Metab 2016; 36: 216–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Main BS, Villapol S, Sloley SS, et al. Apolipoprotein E4 impairs spontaneous blood brain barrier repair following traumatic brain injury. Mol Neurodegener 2018; 13: 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Alata W, Ye Y, St-Amour I, Vandal M, Calon F. Human apolipoprotein E ɛ4 expression impairs cerebral vascularization and blood–brain barrier function in mice. J Cereb Blood Flow Metab 2015; 35: 86–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Reiman EM, Caselli RJ, Yun LS, et al. Preclinical evidence of Alzheimer’s disease in persons homozygous for the epsilon 4 allele for apolipoprotein E. N Engl J Med 1996; 334: 752–8. [DOI] [PubMed] [Google Scholar]
- 98.Small GW, Mazziotta JC, Collins MT, et al. Apolipoprotein E type 4 allele and cerebral glucose metabolism in relatives at risk for familial Alzheimer disease. JAMA 1995; 273: 942–7. [PubMed] [Google Scholar]
- 99.Liu M, Chen X, Paranjpe M, et al. Quantitative imaging APOE4 effects on brain tau deposition and glucose metabolism in mild cognitive impairment patients with [18F]AV1451 and [18F]FDG PET. J Nucl Med 2018; 59: 80–80. [Google Scholar]
- 100.Zipser BD, Johanson CE, Gonzalez L, et al. Microvascular injury and blood–brain barrier leakage in Alzheimer’s disease. Neurobiol Aging 2007; 28: 977–86. [DOI] [PubMed] [Google Scholar]
- 101.Harilal S, Jose J, Parambi DJG, et al. Revisiting the Blood-brain barrier: A hard nut to crack in the transportation of drug Molecules. Brain Res Bull 2020; published online April 18. DOI: 10.1016/j.brainresbull.2020.03.018. [DOI] [PubMed] [Google Scholar]
- 102.Sigurdsson EM. Alzheimer’s therapy development: A few points to consider. In: Progress in Molecular Biology and Translational Science. Elsevier, 2019: 205–17. [DOI] [PubMed] [Google Scholar]
- 103.Femminella GD, Frangou E, Love SB, et al. Evaluating the effects of the novel GLP-1 analogue liraglutide in Alzheimer’s disease: study protocol for a randomised controlled trial (ELAD study). Trials 2019; 20: 191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Batista AF, Forny-Germano L, Clarke JR, et al. The diabetes drug liraglutide reverses cognitive impairment in mice and attenuates insulin receptor and synaptic pathology in a non-human primate model of Alzheimer’s disease. J Pathol 2018; 245: 85–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Vargas-Soria M, Carranza-Naval MJ, del Marco A, Garcia-Alloza M. Role of liraglutide in Alzheimer’s disease pathology. Alzheimers Res Ther 2021; 13: 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Cukierman-Yaffe T, Gerstein HC, Colhoun HM, et al. Effect of dulaglutide on cognitive impairment in type 2 diabetes: an exploratory analysis of the REWIND trial. Lancet Neurol 2020; 19: 582–90. [DOI] [PubMed] [Google Scholar]
- 107.Craft S, Peskind E, Schwartz MW, Schellenberg GD, Raskind M, Porte D. Cerebrospinal fluid and plasma insulin levels in Alzheimer’s disease: Relationship to severity of dementia and apolipoprotein E genotype. Neurology 1998; 50: 164–8. [DOI] [PubMed] [Google Scholar]
- 108.Urayama A, Banks WA. Starvation and Triglycerides Reverse the Obesity-Induced Impairment of Insulin Transport at the Blood-Brain Barrier. Endocrinology 2008; 149: 3592–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Vandal M, White PJ, Tremblay C, et al. Insulin Reverses the High-Fat Diet-Induced Increase in Brain A and Improves Memory in an Animal Model of Alzheimer Disease. Diabetes 2014; 63: 4291–301. [DOI] [PubMed] [Google Scholar]
- 110.Banks WA, Owen JB, Erickson MA. Insulin in the brain: There and back again. Pharmacol Ther 2012; 136: 82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hanson LR, Frey WH. Intranasal delivery bypasses the blood-brain barrier to target therapeutic agents to the central nervous system and treat neurodegenerative disease. BMC Neurosci 2008; 9: S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kellar D, Craft S. Brain insulin resistance in Alzheimer’s disease and related disorders: mechanisms and therapeutic approaches. Lancet Neurol 2020; 19: 758–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chapman CD, Schiöth HB, Grillo CA, Benedict C. Intranasal insulin in Alzheimer’s disease: Food for thought. Neuropharmacology 2018; 136: 196–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Mao Y-F, Guo Z, Zheng T, et al. Intranasal insulin alleviates cognitive deficits and amyloid pathology in young adult APPswe/PS1dE9 mice. Aging Cell 2016; 15: 893–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Rhea EM, Humann SR, Nirkhe S, Farr SA, Morley JE, Banks WA. Intranasal Insulin Transport is Preserved in Aged SAMP8 Mice and is Altered by Albumin and Insulin Receptor Inhibition. J Alzheimers Dis JAD 2017; 57: 241–52. [DOI] [PubMed] [Google Scholar]
- 116.Craft S, Claxton A, Baker LD, et al. Effects of Regular and Long-Acting Insulin on Cognition and Alzheimer’s Disease Biomarkers: A Pilot Clinical Trial. J Alzheimers Dis JAD 2017; 57: 1325–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Erickson MA, Liang WS, Fernandez EG, Bullock KM, Thysell JA, Banks WA. Genetics and sex influence peripheral and central innate immune responses and blood-brain barrier integrity. PLOS ONE 2018; 13: e0205769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Torres L, Bynoe MS. Influence of gender on blood brain barrier permeability and adenosine receptor signaling. FASEB J 2017; 31: 1042.3–1042.3. [Google Scholar]
- 119.Claxton A, Baker LD, Wilkinson CW, et al. Sex and ApoE Genotype Differences in Treatment Response to Two Doses of Intranasal Insulin in Adults with Mild Cognitive Impairment or Alzheimer’s Disease. J Alzheimers Dis 2013; 35: 789–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Claxton A, Baker LD, Hanson A, et al. Long-Acting Intranasal Insulin Detemir Improves Cognition for Adults with Mild Cognitive Impairment or Early-Stage Alzheimer’s Disease Dementia. J Alzheimers Dis 2015; 44: 897–906. [DOI] [PubMed] [Google Scholar]
- 121.McNay EC, Ong CT, McCrimmon RJ, Cresswell J, Bogan JS, Sherwin RS. Hippocampal memory processes are modulated by insulin and high-fat-induced insulin resistance. Neurobiol Learn Mem 2010; 93: 546–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Whitmer RA. Hypoglycemic Episodes and Risk of Dementia in Older Patients With Type 2 Diabetes Mellitus. JAMA 2009; 301: 1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Mullins RJ, Diehl TC, Chia CW, Kapogiannis D. Insulin Resistance as a Link between Amyloid-Beta and Tau Pathologies in Alzheimer’s Disease. Front Aging Neurosci 2017; 9. DOI: 10.3389/fnagi.2017.00118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Rad SK, Arya A, Karimian H, et al. Mechanism involved in insulin resistance via accumulation of β-amyloid and neurofibrillary tangles: link between type 2 diabetes and Alzheimer’s disease. Drug Des Devel Ther 2018; 12: 3999–4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Schilling MA. Unraveling Alzheimer’s: Making Sense of the Relationship between Diabetes and Alzheimer’s Disease1. J Alzheimers Dis 2016; 51: 961–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Josa M, Urizar JP, Rapado J, et al. Pharmacokinetic/pharmacodynamic modeling of antipyretic and anti-inflammatory effects of naproxen in the rat. J Pharmacol Exp Ther 2001; 297: 198–205. [PubMed] [Google Scholar]
- 127.Meyer P-F, Tremblay-Mercier J, Leoutsakos J, et al. INTREPAD: A randomized trial of naproxen to slow progress of presymptomatic Alzheimer disease. Neurology 2019; 92: e2070–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Walker D, Lue L-F. Anti-inflammatory and Immune Therapy for Alzheimers Disease: Current Status and Future Directions. Curr Neuropharmacol 2007; 5: 232–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Lin R, Chen F, Wen S, Teng T, Pan Y, Huang H. Interleukin-10 attenuates impairment of the blood-brain barrier in a severe acute pancreatitis rat model. J Inflamm Lond Engl 2018; 15: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Deane R, Singh I, Sagare AP, et al. A multimodal RAGE-specific inhibitor reduces amyloid β-mediated brain disorder in a mouse model of Alzheimer disease. J Clin Invest 2012; 122: 1377–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Burstein AH, Sabbagh M, Andrews R, Valcarce C, Dunn I, Altstiel L. Development of Azeliragon, an Oral Small Molecule Antagonist of the Receptor for Advanced Glycation Endproducts, for the Potential Slowing of Loss of Cognition in Mild Alzheimer’s Disease. J Prev Alzheimers Dis 2018; 5: 149–54. [DOI] [PubMed] [Google Scholar]
- 132.Deane R, Du Yan S, Submamaryan RK, et al. RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat Med 2003; 9: 907–13. [DOI] [PubMed] [Google Scholar]
- 133.Sawda C, Moussa C, Turner RS. Resveratrol for Alzheimer’s disease: Alzheimer’s disease. Ann N Y Acad Sci 2017; 1403: 142–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Chen X, Ghribi O, Geiger JD. Caffeine Protects Against Disruptions of the Blood-Brain Barrier in Animal Models of Alzheimer’s and Parkinson’s Diseases. J Alzheimers Dis 2010; 20: S127–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Eskelinen MH, Kivipelto M. Caffeine as a Protective Factor in Dementia and Alzheimer’s Disease. J Alzheimers Dis 2010; 20: S167–74. [DOI] [PubMed] [Google Scholar]
- 136.Moy GA, McNay EC. Caffeine prevents weight gain and cognitive impairment caused by a high-fat diet while elevating hippocampal BDNF. Physiol Behav 2013; 109: 69–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Mudò G, Frinchi M, Nuzzo D, et al. Anti-inflammatory and cognitive effects of interferon-β1a (IFNβ1a) in a rat model of Alzheimer’s disease. J Neuroinflammation 2019; 16: 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Cohrdes C, Bretschneider J. Can social support and physical activity buffer cognitive impairment in individuals with depressive symptoms? Results from a representative sample of young to older adults. J Affect Disord 2018; 239: 102–6. [DOI] [PubMed] [Google Scholar]
- 139.Małkiewicz MA, Szarmach A, Sabisz A, Cubała WJ, Szurowska E, Winklewski PJ. Blood-brain barrier permeability and physical exercise. J Neuroinflammation 2019; 16. DOI: 10.1186/s12974-019-1403-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Wang R, Holsinger RMD. Exercise-induced brain-derived neurotrophic factor expression: Therapeutic implications for Alzheimer’s dementia. Ageing Res Rev 2018; 48: 109–21. [DOI] [PubMed] [Google Scholar]
- 141.Veronese N, Facchini S, Stubbs B, et al. Weight loss is associated with improvements in cognitive function among overweight and obese people: A systematic review and meta-analysis. Neurosci Biobehav Rev 2017; 72: 87–94. [DOI] [PubMed] [Google Scholar]
- 142.Morris MC, Tangney CC, Wang Y, et al. MIND diet slows cognitive decline with aging. Alzheimers Dement 2015; 11: 1015–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.McGrattan AM, McGuinness B, McKinley MC, et al. Diet and Inflammation in Cognitive Ageing and Alzheimer’s Disease. Curr Nutr Rep 2019; 8: 53–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Mattei J, Bigornia SJ, Sotos-Prieto M, Scott T, Gao X, Tucker KL. The Mediterranean Diet and 2-Year Change in Cognitive Function by Status of Type 2 Diabetes and Glycemic Control. Diabetes Care 2019; published online May 23. DOI: 10.2337/dc19-0130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Mohammadpour S, Ghorbaninejad P, Janbozorgi N, Shab-Bidar S. Associations between adherence to MIND diet and metabolic syndrome and general and abdominal obesity: a cross-sectional study. Diabetol Metab Syndr 2020; 12: 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Casas R, Sacanella E, Urpí-Sardà M, et al. Long-Term Immunomodulatory Effects of a Mediterranean Diet in Adults at High Risk of Cardiovascular Disease in the PREvención con DIeta MEDiterránea (PREDIMED) Randomized Controlled Trial. J Nutr 2016; 146: 1684–93. [DOI] [PubMed] [Google Scholar]
- 147.Soltani S, Chitsazi MJ, Salehi-Abargouei A. The effect of dietary approaches to stop hypertension (DASH) on serum inflammatory markers: A systematic review and meta-analysis of randomized trials. Clin Nutr Edinb Scotl 2018; 37: 542–50. [DOI] [PubMed] [Google Scholar]
- 148.Nelson AR, Sweeney MD, Sagare AP, Zlokovic BV. Neurovascular dysfunction and neurodegeneration in dementia and Alzheimer’s disease. Biochim Biophys Acta BBA - Mol Basis Dis 2016; 1862: 887–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Poduslo JF, Curran GL, Wengenack TM, Malester B, Duff K. Permeability of Proteins at the Blood–Brain Barrier in the Normal Adult Mouse and Double Transgenic Mouse Model of Alzheimer’s Disease. Neurobiol Dis 2001; 8: 555–67. [DOI] [PubMed] [Google Scholar]
- 150.Bien-Ly N, Boswell CA, Jeet S, et al. Lack of Widespread BBB Disruption in Alzheimer’s Disease Models: Focus on Therapeutic Antibodies. Neuron 2015; 88: 289–97. [DOI] [PubMed] [Google Scholar]
- 151.Sweeney MD, Sagare AP, Zlokovic BV. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat Rev Neurol 2018; 14: 133–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Montagne A, Zhao Z, Zlokovic BV. Alzheimer’s disease: A matter of blood–brain barrier dysfunction? J Exp Med 2017; 214: 3151–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Gabbouj S, Natunen T, Koivisto H, et al. Intranasal insulin activates Akt2 signaling pathway in the hippocampus of wild-type but not in APP/PS1 Alzheimer model mice. Neurobiol Aging 2019; 75: 98–108. [DOI] [PubMed] [Google Scholar]
- 154.Biessels GJ, Whitmer RA. Cognitive dysfunction in diabetes: how to implement emerging guidelines. Diabetologia 2019; published online Aug 16. DOI: 10.1007/s00125-019-04977-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in the current study.