

Abstract
Micheliolide (MCL) is a naturally occurring sesquiterpene lactone that selectively targets leukemic stem cells (LSCs), which persist after conventional chemotherapy for myeloid leukemias, leading to disease relapse. To overcome modest MCL cytotoxicity, analogs with ≈two–threefold greater cytotoxicity against LSCs are synthesized via late-stage chemoenzymatic C—H functionalization. To enhance bone marrow delivery, MCL analogs are entrapped within bone-targeted polymeric nanoparticles (NPs). Robust drug loading capacities of up to 20% (mg drug mg−1 NP) are obtained, with release dominated by analog hydrophobicity. NPs loaded with a hydrolytically stable analog are tested in a leukemic mouse model. Median survival improved by 13% and bone marrow LSCs are decreased 34-fold following NPMCL treatments versus controls. Additionally, selective leukemic cell and LSC cytotoxicity of the treatment versus normal hematopoietic cells is observed. Overall, these studies demonstrate that MCL-based antileukemic agents combined with bone-targeted NPs offer a promising strategy for eradicating LSCs.
Keywords: drug delivery, leukemia, micheliolide, small molecule drugs, targeted delivery, late-stage C-H functionalization
1. Introduction
Chemotherapy resistant leukemic stem cells (LSCs), which reside in the bone marrow microenvironment (BMME), contribute to disease relapse in blast crisis chronic myeloid leukemia (bcCML) and acute myeloid leukemia (AML). Compounded by an aberrant accumulation of inflammatory mediators, the BMME becomes dysfunctional and no longer supports normal hematopoiesis and skeletal remodeling, which ultimately results in patient mortality.[1–3] LSCs possess similar characteristics as hematopoietic stem cells (HSCs), including self-renewing potential and multipotential differentiation,[4] complicating eradication of LSCs while sparing HSCs using conventional chemotherapeutic agents.
A highly sought-after approach in leukemia treatment is the selective elimination of LSCs.[4] Recent studies showed that plant-derived sesquiterpene lactone (SQL) natural products such as parthenolide (PTL) and micheliolide (MCL) can kill LSCs selectively over normal hematopoietic cells.[5–8] PTL and MCL share a reactive α-methylene-γ-lactone moiety, which is critical for antileukemic activity and mediates the covalent modification of various target proteins in leukemia cells, including nuclear factor kappa B for PTL[9,10] and pyruvate kinase M2 and other proteins for MCL.[11] Induction of oxidative stress in leukemia cells and LSCs has also been linked to the antileukemic activity of PTL and MCL.[5,12] A C13-dimethylamino adduct pro-drug of MCL (DMAMCL) was recently shown to improve survival in a xenograft model of human AML[8,13] and showed promise for the treatment of gliomas (Trial ID: ACTRN12616000228482) and other malignancies.[14–16] Despite this progress, MCL has modest activity against LSCs in vitro (LC50 ≈ 15 μM) and improvement of this antileukemic activity has been hampered by poor accessibility to modifications that do not abrogate its anticancer activity (e.g., C13 and C4-OH).[8,13,17] In addition, as for other cytotoxic compounds, systemic delivery of this compound has risks associated with side-effects and/or off-target toxicity.[18]
Delivery systems offer many advantages to the delivery of small molecule drugs such as MCL. Drug delivery systems (DDSs) can enhance drug solubility via encapsulation, adsorption, or covalent attachment and can protect drugs from serum degradation.[19] Furthermore, DDS physiochemical characteristics (i.e., size, surface charge, surface chemistry) can be altered to improve drug circulation time.[20] However, achieving tissue selectivity remains a challenge.[18] In this study, late-stage chemoenzymatic C—H functionalization using engineered cytochrome P450 enzymes[7,21,22] was applied to develop MCL analogs with enhanced cytotoxicity against acute myeloid leukemia (AML) and biphenotypic B myelomonocytic leukemia cells. To enable targeted delivery to the bone marrow, these novel MCL analogs were combined with amphiphilic poly(styrene-alt-maleic anhydride)-b-poly(styrene) (PSMA-b-PS)-based nanoparticles (NPs)[23–25] functionalized with a tartrate-resistant acid phosphatase (TRAP) binding peptide (TBP) useful for bone-specific drug delivery.[23,26] Herein, we report that TBP-NPs exhibited efficient loading and release of the MCL analogs, protected hydrolytically susceptible MCL analogs from hydrolysis in serum, and enabled bone-targeted delivery of an improved triazolefunctionalized MCL analog, resulting in a reduction of the burden of leukemia stem cells in a murine model of blast-crisis chronic myeloid leukemia.
2. Results
2.1. Micheliolide Analogs Show Improved In Vitro Toxicity against Leukemia Cells
Previous studies discovered that the plant-derived SQL MCL has promising cytotoxic activity (LC50: 20–50 μM) against various cancer cells, including AML cells.[8,14–16] Attempts to further develop MCL have involved functionalization of the hydroxyl group at C4 position of MCL or modification of the reactive α-methylene-γ-lactone moiety, which resulted in either no change or loss in antileukemic activity.[8,17] To overcome these challenges, we recently developed two engineered cytochrome P450 enzymes useful for regio- and stereoselective hydroxylation of aliphatic positions C2 and C14 in MCL.[12] Importantly, chemoenzymatic modification of these sites yielded MCL analogs with improved antileukemic activity.[12] Building upon these findings, a panel of C2- and C14-functionalized MCL analogs were selected and further developed for incorporation into bone-targeted NPs (TBP-NPs (Figure 1). Specifically, a panel of arylester analogs was prepared in view of the beneficial effect of fluorinated aryl groups installed at positions C2 and C14 on MCL anticancer activity.[12] Furthermore, two carbamate analogs of MCL-13 and MCL-14, namely MCL-38 and MCL-39, were designed, in which the p-CF3-aryl group is appended to the MCL scaffold via a carbamate linkage. Carbamates are widely explored in medicinal chemistry due to greater hydrolytic and enzymatic stability compared to esters.[27] Finally, a triazole analog of MCL-19, termed MCL-64, was designed to replace the ester group with a bioisosteric[28] and non-hydrolyzable triazole linkage (Figure 1). Collectively, this panel of MCL analogs was chosen to study the effects of a) substitution position b) lipophilicity, and c) linkage chemistry (ester vs carbamate vs triazole) on antileukemic activity, serum stability, and analog loading capacity (LC) and release properties from the NPs.
Figure 1.
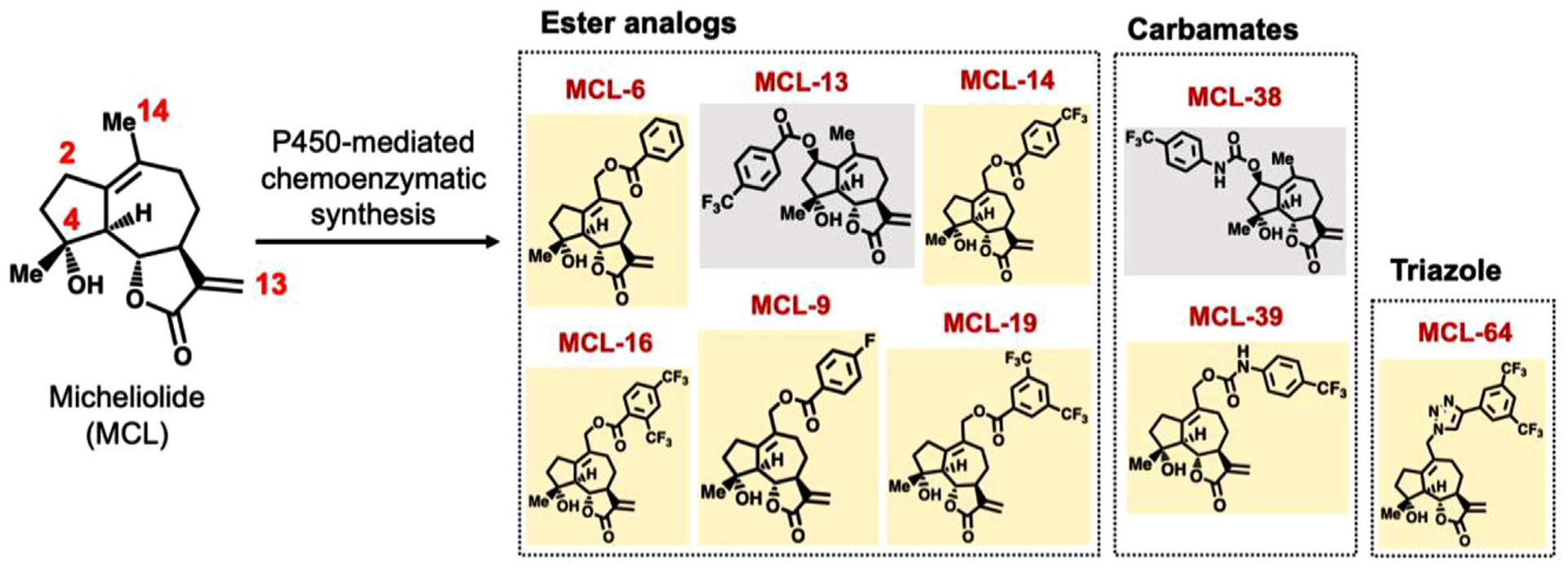
Structure of MCL analogs using P450-mediated chemoenzymatic synthesis.
The antileukemic activity of MCL analogs was assessed in cell viability assays using M9-ENL1 (pro-B cell acute lymphoblastic leukemia) and MV-411 cells (biphenotypic B-myelomonocytic leukemia) (Table 1). The ester analogs showed up to tenfold improvement in cytotoxicity of M9-ENL1 cells, with a half-maximal lethal concentration (LC50) of 1.8–5.1 μM compared to 15.4 μM for MCL. Notably, the novel carbamate analogs exhibited comparable (MCL-38 vs MCL-13) or improved (MCL-39 vs MCL-14) antileukemic activity (LC50 = 3.2–3.6 μm) compared to the ester analogs. In addition, the triazole-based analog, MCL-64, showed favorable cytotoxicity (LC50: 2.2 μM), which corresponded to a twofold improvement over the ester analog, MCL-19. A similar trend was observed using MV-411 cells, in which the MCL analogs showed two to sixfold greater antileukemic activity compared to the parent compound (LC50 = 1.0–1.9 μM vs 5.6 μM; Table 1), thus highlighting the beneficial effect of the C2- or C14-substitutions on the activity of MCL. Further characterization revealed a insignificant (or minor) relationship between LC50 and lipophilicity as defined by CLogP (R2 = 0.38) (Figure S1, Supporting Information). MCL-64 emerged as one of the most effective compounds, with LC50 of 1.2 μM against MV411 cells (Table 1). Importantly, these studies defined the novel ester and carbamate analogs MCL-19 and MCL-39, as well as the triazole analog, MCL-64, as potent antileukemic analogs (Table 1) for further assessment of stability.
Table 1.
LC50 of MCL and its analogs against M9-ENL1 and MV411 cells.
| Class | Compound | CLogP1 | LC50 [μM] M9-ENL1 | LC50 [μM]2 MV-411 |
|---|---|---|---|---|
| Parent | MCL | 1.7 | 15.4 ± 1.1 | 5.6 ± 2.8 |
| Esters | MCL-6 | 2.8 | 20 ± 3 | 1.4 ± 0.3** |
| MCL-9 | 2.9 | 4.8 ± 1 | 1.9 ± 0.1** | |
| MCL-13 | 4.0 | 1.8 ± 1 | 1.8 ± 0.7** | |
| MCL-14 | 3.7 | 10 ± 2 | 1.0 ± 0.1** | |
| MCL-16 | 4.5 | 5.2 ± 1 | 1.0 ± 0.4** | |
| MCL-19 | 4.5 | 4.1 ± 1.1 | 1.7 ± 0.9** | |
| Carbamates | MCL-38 | 3.6 | 4.3 ± 1 | 2.9 ± 0.6 |
| MCL-39 | 3.2 | 3.3 ± 1 | 2.9 ± 1.4 | |
| Triazole | MCL-64 | 4.0 | 2.2 ± 0.1 | 1.2 ± 0.5** |
CLogP represents an estimation of the logarithm of partition coefficient [n-Octanol/water] as determined by ChemDraw;
Half-maximal lethal concentration. Data represents mean ± std (n = 3).
p < 0.01 represents significant differences between MCL analogs and MCL using one-way ANOVA and Dunnett’s multiple comparisons test used to compare MCL analogs to MCL.
Selected C14-substituted MCL analogs were tested for chemical and hydrolytic stability. Analogs were incubated in phosphate-buffered saline (PBS) and blood serum at 50% v/v and 100% v/v for 24-h, followed by HPLC analysis (Figure 2). All analogs were stable in PBS over 24 h (Figure 2). However, nearly complete hydrolysis (>80–90%) of the ester analog, MCL-19, was observed after a 24 h incubation in both 50% and 100% serum (Figure 2A). As anticipated, the carbamate analog MCL-39 showed improved hydrolytic stability compared to the ester analog, with 25–50% compound remaining intact in 50% and 100% serum after 24 h (Figure 2B). In contrast, the triazole analog, MCL-64, showed excellent stability in blood serum with no degradation observed (Figure 2C). Overall, P450 chemoenzymatic modifications enabled the synthesis of analogs with labile ester linkages, stable carbamate linkages, and nonhydrolyzable triazole linkages with improved cytotoxicity against leukemic cells for NP loading.
Figure 2.
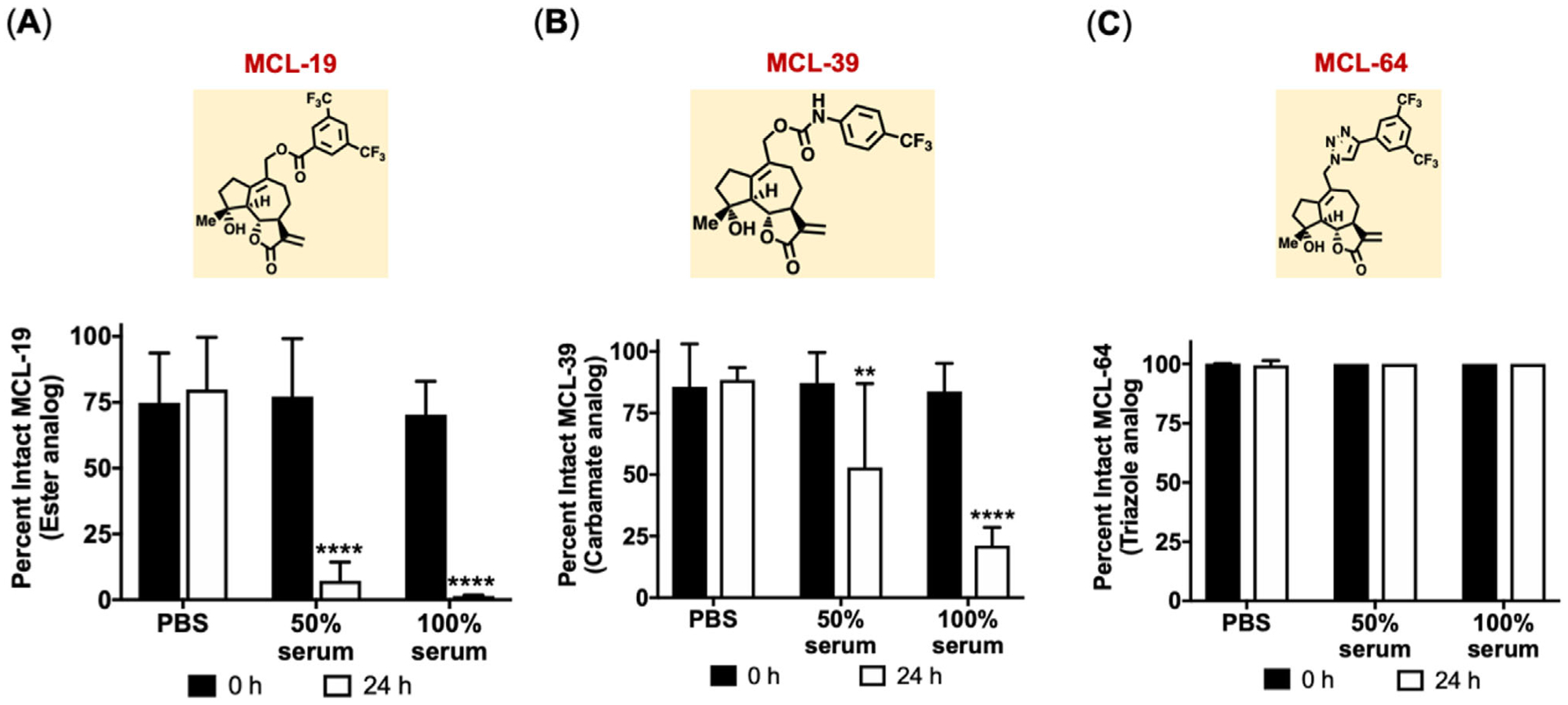
Stability of ester, carbamate, and triazole MCL analogs in blood serum. Percent intact drug for MCL analogs after incubating with PBS, 50% and 100% mouse blood serum. A) MCL-19 (ester), B) MCL-39 (carbamate), and C) MCL-64 (triazole). Data represents mean ± SD (n = 3). **p < 0.01 and ****p < 0.0001 represents significance between 0 and 24 h time points for each incubation condition using two-way ANOVA followed by Sidak’s multiple comparisons.
2.2. Effect of MCL Modifications on Loading and Release from PSMA-b-PS Nanoparticles
Poly(styrene-alt-maleic anhydride)-b-poly(styrene) (PSMA-b-PS)-based NPs are versatile DDS with the ability to load and controllably release a variety of small-molecule compounds.[23–26] Furthermore, upon the introduction of TRAP-TBP, PSMA-b-PS NPs (TBP-NP) exhibit preferential bone accumulation versus untargeted or scrambled peptide targeted NP controls.[23] PSMA-b-PS based diblock copolymers were synthesized via one-step reversible addition-fragmentation chain transfer (RAFT) polymerizations, as previously described.[23–26,29] Diblock copolymers had overall molecular weights of 71.7 kDa (20.7 kDa 1st block, 51 kDa 2nd block or PSMA100-b-PS494) and a polydispersity index of 1.05, indicating well-controlled reactions. Diblock copolymers self-assembled into NPs were monodispersed with sizes 85 ± 1 nm with an overall surface charge of −36 ± 1 mV prior to drug loading.
P450-mediated chemoenzymatic modifications increased the lipophilicity of MCL, which provided a unique opportunity to test the relationship between compound CLogP and LC. NPs exhibited robust loading of MCL at ≈14% LC (Figure 3A). In comparison, LCs of the ester, triazole, and carbamate MCL analogs were between 0.4–10%, ≈ 16%, and ≈ 6–23%, respectively (Figure 3A). There was no relationship between compound CLogP and LC (R2 = 0.023%), indicating drug-core interactions were more complex than described by hydrophobicity alone (Figure S3, Supporting Information). C2 site-modified MCL analogs (−13 and −38) demonstrated significantly lower loading (0.4 ± 0.5%, and 6 ± 5% for ester and carbamate analogs) compared to their C14 site-modified counterparts (Figure 3A). Notably, MCL analogs (−38 and −39), which contained the same type of modification at different positions loaded at ≈ 6% and ≈ 23%, respectively, further highlighting the impact of positional changes on loading (Figure 3A). Drug loading resulted in 32% to 54% increases in NP size compared to unloaded NPs with no detectable change in surface charge compared to unloaded NP controls (Figure 3B). Further characterization of loaded NPs showed inverse linear relationships between LC and size (R2 = 0.65) (Figure S3, Supporting Information), suggesting more compact PS cores as a consequence of greater π-π stacking.[30]
Figure 3.
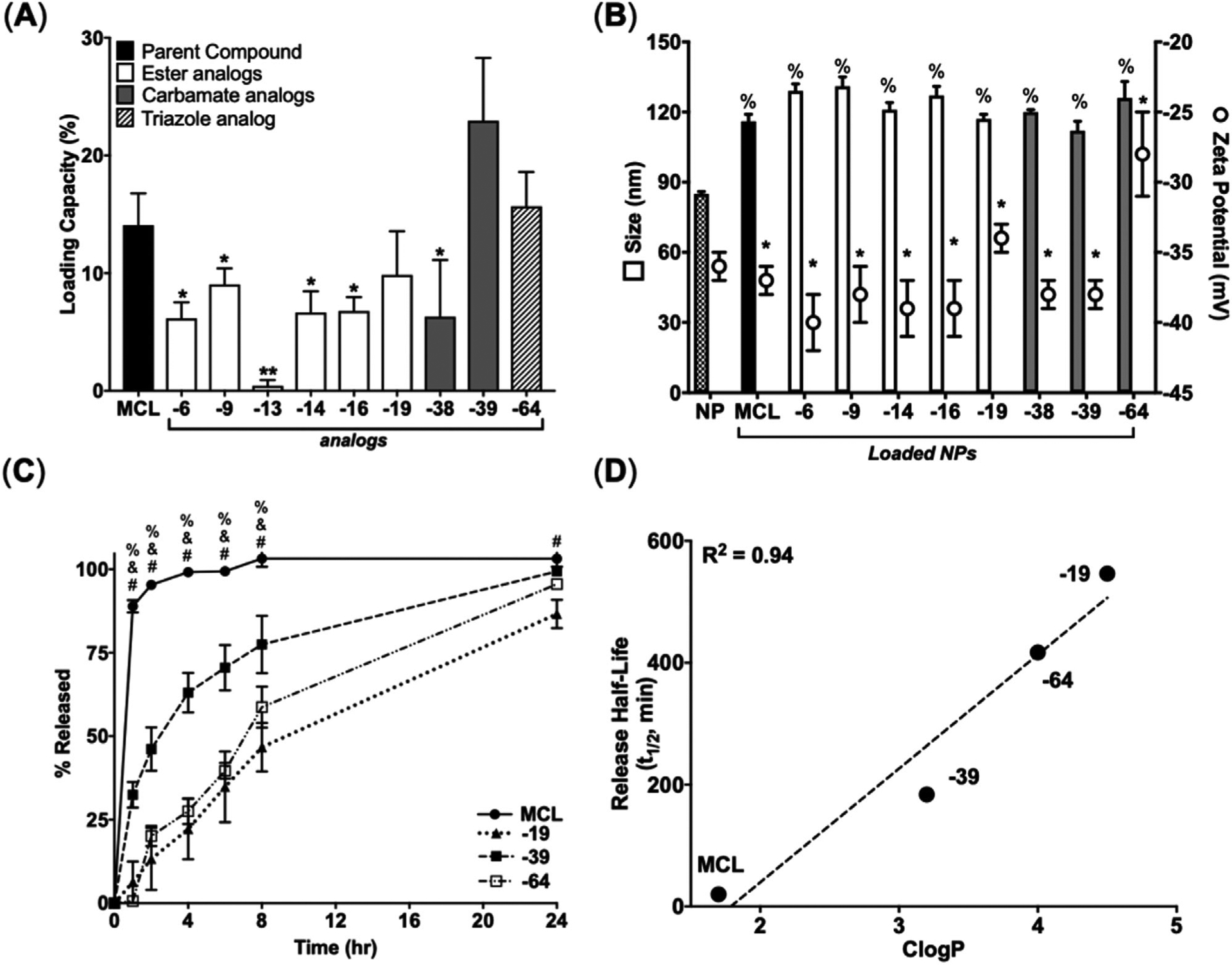
Structural modifications of MCL impact loading in PSMA-b-PS NPs and hydrophobicity impact release from PSMA-b-PS NPs. A) LC for MCL and MCL analogs after loading. Data represent mean ± SD. (n = 3). *p < 0.05 and **p < 0.01 represents statistical differences between MCL and MCL analogs using unpaired two-tailed t-tests. B) Physiochemical properties of loaded NPs. *p < 0.05 represents statistical differences between MCL and MCL analogs for size and % p < 0.05 shows statistical differences comparing MCL and MCL analogs for zeta potential using one-way ANOVA followed by Dunnett’s multiple comparisons test. C) Cumulative release of MCL and MCL analogs from NPs. Data represent mean ± SD. (n = 3). %, & and # represents statistical differences between MCL and MCL-39, MCL-64, and MCL-19, respectively, at each time point using two-way ANOVA followed by Dunnett’s multiple comparisons test. D) Linear correlation between release half-life of MCL and selected MCL analogs plotted against their calculated partition coefficient (CLogP) as determined by ChemDraw.
Given excellent NP loading characteristics, improved cytotoxicity, and increased hydrophobicity, the ester (−19), carbamate (−39), and triazole (−64) analogs were further investigated in NP release studies. MCL was released rapidly from NPs with a half-life of 0.33 h while carbamate (−39), triazole (−64), and ester (−19) analogs had release half-lives of 3, 7, and 9 h, respectively (Figure 3C). Interestingly, linear regression between release half-lives and CLogP revealed that drug hydrophobicity predicts release half-lives (R2 = 0.95) (Figure 3D). Overall, loading studies suggested that drug-core interactions favored modifications at the C14 position of MCL and had a greater impact on LC of MCL analogs than hydrophobicity while hydrophobicity impacted release more than positional modifications.
2.3. Nanoparticles Impart Serum Stability to Ester MCL Analogs
To test whether NP loading could protect the ester analog (MCL-19) from degradation, stability in the presence of 50% serum, a physiologically relevant concentration, was investigated over time with free MCL-19 used as a control (Figure 4A). After 8 h of incubation, free MCL-19 showed near-complete hydrolysis to MCL-OH with only 7 ± 2% of the intact compound while the NP encapsulated MCL-19 showed that 31 ± 15% of active MCL-19 remained (Figure 4A). Free MCL-19 in serum degraded rapidly with a half-life of 18 min (Figure 4A). NP loading of MCL-19 extended the degradation half-life of the drug to 6 h (Figure 4A). Interestingly, the degradation rate for NP-loaded MCL-19 was determined to be 5.3 h−1 and the release rate of MCL-19 from NPs from 0–8 h was 5.7 h−1 (Figures 2C and 4A). The close agreement between degradation and release rates for MCL-19 suggests that degradation occurs immediately upon release from NPs. As expected, the serum stability of the carbamate analogs, MCL-39 and MCL-64, was unaffected by serum incubation (Figure 4B,C). Previous studies using PTL showed that NP encapsulation abrogated cytotoxicity due to rapid and robust uptake of NPs, thus limiting the drug exofacial thiol binding mechanism of cytotoxicity.[24] After incubating MV411 cells with NP-loaded MCL analogs, a similar phenomenon was observed with generally reduced cytotoxicity of fourfold for MCL, twofold for MCL-19, 0.8-fold for MCL-39, while cytotoxicity was unchanged for MCL-64 (Table 2). Notably, previous studies[24] show that unloaded NPs exhibit no cytotoxicity to MV411 cells across a broad range of concentrations, well above the ranges used here.
Figure 4.
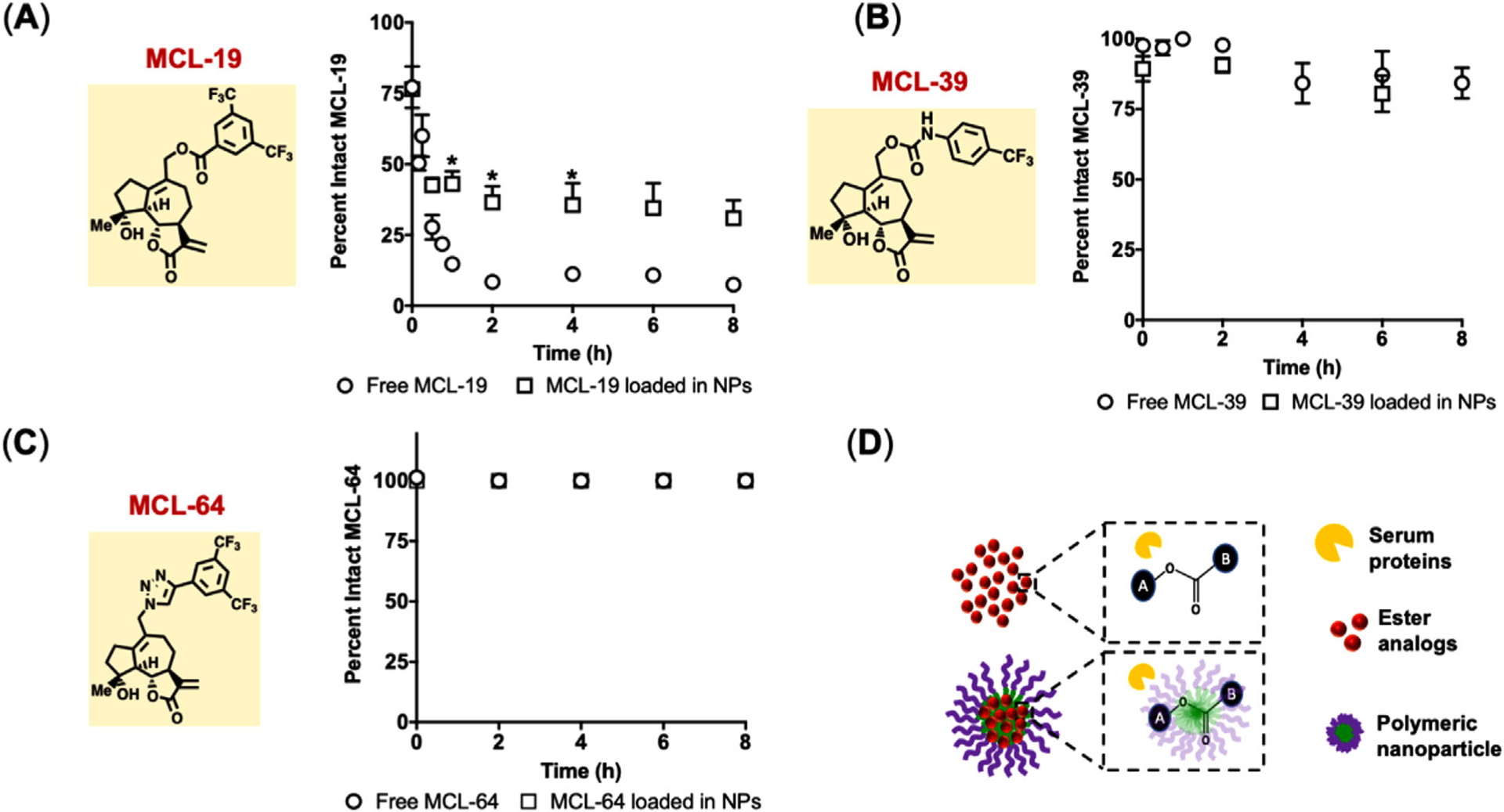
Degradable ester bonds are partially protected from degradation via NP encapsulation. Percent intact drug after incubation in 50% mouse serum. A) MCL-19, B) MCL-39, and C) MCL-64. Data represents mean ± SEM (n = 3–9). *p < 0.05 represents statistical differences between free drug and NP loaded drug at each time point using two-way ANOVA followed by Sidak’s multiple comparisons test. D) Schematic of serum proteins interacting with ester analogs in free and in NP form.
Table 2.
LC50 of loaded NPs against MV411 cells.
| Loaded NP | LC50 [μM] | Δ vs free |
|---|---|---|
| MCL | 23.7 ± 2.6 | fourfold |
| MCL-19 | 3.2 ± 0.5 | twofold |
| MCL-39 | 2.2 ± 0.6 | 0.8-fold |
| MCL-64 | 1.7 ± 0.1 | onefold |
Data represents mean ± std (n = 3).
2.4. Triazole Analog (MCL-64)-Loaded Bone-Targeted NPs Reduce LSC Burden In Vivo
MCL-64 was selected for in vivo delivery using a bcCML model[1] due to > fourfold enhancement in toxicity compared to MCL, unchanged LC and release over ≈24 h from NPs, and excellent serum stability. Due to release kinetics over 24 h and 10% TBP-NP accumulation at 24 h (Figure S7, Supporting Information), efficacy studies adapted a treatment regimen whereby injections (0.4 mg MCL-64 as free drug and loaded within NP) were performed 2 d after leukemia induction and every other day for up to 6 doses (for overall doses of 2.4 mg MCL-64 (117 mg kg−1) as free drug and in NP with constant NP dose of 150 mg kg−1) (Figure 5A).[26] TBP-NPMCL-64 treatment resulted in a median survival rate of 21.5 d versus 19 d for free drug (Figure 5B).
Figure 5.
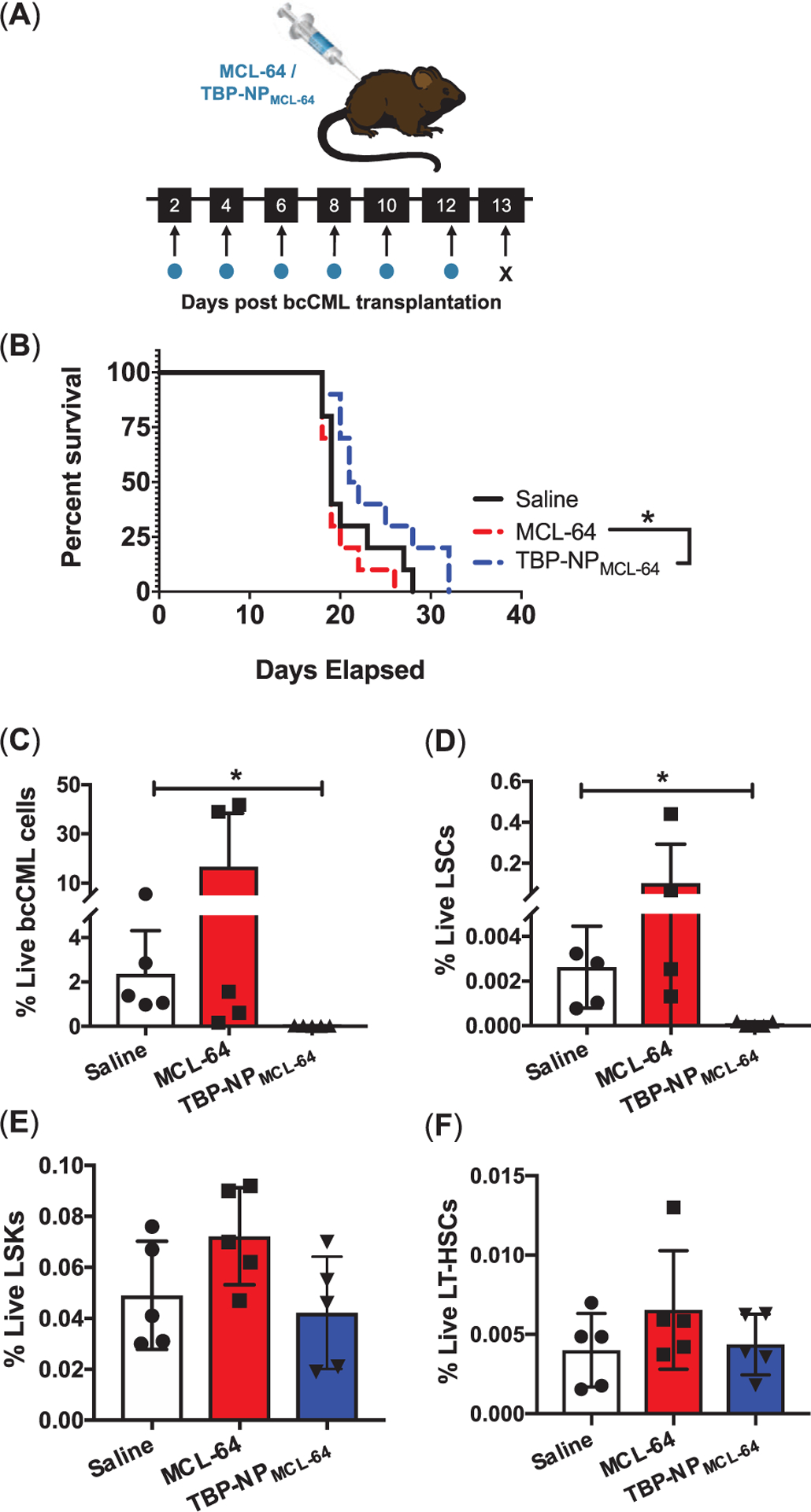
A decrease in marrow LSCs is observed via TBP-NP mediated delivery of MCL-64. A) Schematic and treatment for free MCL-64 and TBP-NP loaded MCL-64, B) bcCML mouse survival after treating with saline, free MCL-64, and TBP-NP loaded MCL-64. Data represents n = 10 per group. *p < 0.05 represents statistical analysis comparing free MCL-64 to MCL-64 in TBP-NPs performed using log-rank (Mantel-cox) test. C) GFP+ bcCML cells, D) LSC cells, E) LSK cells, and F) LT-HSCs in the marrow after free MCL-64 and TBP-NP loaded MCL-64 treatment. Data represents mean ± SD (n = 4–5). *p < 0.05 represents statistical differences comparisons using unpaired t-tests.
Leukemia cells in the bcCML model are GFP+, which allow for discrimination between malignant and non-malignant cells. NP treatments significantly decreased total GFP+ bcCML cells by 121-fold compared to control bcCML mice in the marrow (Figure 5C). TBP-NPMCL-64 treatment decreased LSC populations twofold from 0.06% to 0.03% (Figure 5D). To test the effect of MCL-64 treatment on normal hematopoiesis, lineage−, sca-1+, c-kit+ (LSKs), and long-term hematopoietic stem cells (LT-HSCs), defined as GFP−/LSK/Flt3−/CD48−/CD150+ (Figure S4, Supporting Information), were analyzed in the bone marrow using flow cytometry. Treatment with free drug and TBP-NP drug had no effect on LSKs and LT-HSCs, suggesting that treatment had minimal off-target effects in the bone marrow (Figure 5E,F). Notably, studies repeated using a separate batch of bcCML cells and a lower dose of MCL-64 (free and in TBP-NPs) resulted in similar findings of improved survival, decreased LSCs, and negligible effects on normal hematopoiesis (Figure S5, Supporting Information). Furthermore, TBP-NPs without drugs did not change bcCML, LSC, LSK, and LT-HSC populations (Figure S6, Supporting Information). Altogether, these data suggest that MCL-64 delivery is selective towards LSC and bcCML cells while sparing non-leukemic hematopoietic cells in the marrow.
3. Discussion
Despite significant improvements in antileukemic drug discovery, clinical trials fail, in part, due to poor aqueous solubility of drugs, leading to rapid systemic clearance, poor target versus off-target tissue accumulation, and dose-limiting toxicities.[31] This is especially true in AML and bcCML, where the 5-year survival rate has remained stagnant at < 35% for the past 30 years. Disease relapse can be attributed to persistent LSCs.[32–34] Therefore, designing drugs that selectively eliminate LSCs is critical to improving patient survival. In this study, novel stereo- and regioselective modifications were performed to enhance the toxicity of the naturally occurring drug, MCL, against chemotherapy-evading LSCs and bone-targeted NP (TBP-NP) mediated delivery was adapted. Drug-core interactions that enabled loading were dependent on the site of modification, while NP release correlated with drug hydrophobicity. Furthermore, the triazole analog MCL-64 was found to exhibit excellent serum stability, while ester and carbamate analogs had variable serum-mediated degradation that was circumvented by NP loading. TBP-NPs loaded with MCL-64 significantly reduced LSCs and bcCML cells, while free MCL-64 showed no significant impact. TBP-NPs loaded with MCL-64 improved survival and minimally affected non-leukemic hematopoietic marrow cells, suggesting selectivity towards LSCs and bcCML cells. Overall, this study highlighted that a promising compound may only be successful in vivo once it overcomes the barriers associated with delivery, including poor aqueous solubility and tissue targeting.
Predicting delivery system drug LC and release is challenging due to multifold parameters, such as hydrophobicity, electrostatic interaction, dipole-dipole interaction, hydrogen bonding, drug size, and the thermodynamics of mixing that impact loading.[35–37] Loading experiments using PSMA-b-PS NPs and the SQL, PTL, showed that hydrophobic core characteristics were necessary to achieve robust PTL loading.[25] In this study, the MCL scaffold remained unchanged while the position and type of drug modification were altered. Positional modifications impacted LC of the carbamate analogs (MCL-38 (C2) and MCL-39 (C14)) and ester analogs MCL-13 (C2) and MCL-6 (C14). Since the composition of NPs in these studies was kept constant, it is unlikely that the core thermal properties alone contributed to differences in LC-based upon positional modifications. Also, LC did not correlate with hydrophobicity. Though solubility of the drugs was not directly investigated to enable Flory–Huggins interaction parameter calculation, drug-polymer miscibility is likely to be impacted by positional modifications.[38] Additionally, at the C14 position, an extra carbon group extends the carbamate group away from the parent compound, increasing flexibility, which may contribute to greater π-stacking with the PS cores and slower release rates. Indeed, simulations of drug release from polymeric NPs suggested that enthalpic interactions, rather than physical barriers, dominated drug release from NPs.[39] The correlation of 0.95 obtained for the CLogP and release half-life suggested stronger π-π stacking between highly hydrophobic drugs and the PS core of the NPs. Historical analyses of compounds loaded within PSMA-b-PS NPs[23–25] show a strong correlation of 0.99 when release rate and CLogP of loaded compounds were plotted (data not shown). A few outliers exist, including drugs with amine groups (i.e., doxorubicin, 3-amino-6-(4-((4-methylpiperazin-1-yl)sulfonyl)phenyl)-N-(pyridin-3-yl)pyrazine-2-carboxamide (AR28)) that showed slower release rates due to electrostatic interactions with carboxylates of PSMA corona blocks.[23,25] Based on the release results, it is postulated that the drug release rate from PS can be controlled based on drug hydrophobicity and/or charge.
Drug candidates are often limited by unstable bonds easily cleaved via hydrolysis or serum protein esterase activity. Therefore, DDSs can serve as a protective barrier to degradation. In this study, the ester-bearing MCL analog, MCL-19, was partially protected from degradation upon NP loading. This data is corroborated in other DDSs. For example, the serum hydrolysis half-life of Camptothecin was improved from minutes to hours when entrapped within a variety of polymeric and solid-lipid NPs.[40–43] Nevertheless, polymeric micelle NPs are dynamic structures, whereby diblock polymer chains exchange constantly. Additionally, exchange can be exacerbated by protein adsorption to NPs in vitro or in vivo.[35] Therefore, it is likely that dynamic exchange of diblocks, which allows for some protein and/or water accessibility to the entrapped drug, underpins incomplete protection observed for MCL-19.[35]
Active targeting using ligands can be used to improve tissue-specific drug biodistribution. The targeted NP system (TBP-NP) used here has been previously shown to increase bone accumulation twofold versus untargeted NP.[23] In the bcCML model, ≈10% of TBP-NP accumulated at the bone. TBP-NP mediated delivery of MCL-64 resulted in significant reductions in LSCs compared to free drug, which is attributed to improved tissue-specific delivery via NP targeting. Our findings of improved survival using MCL-64 are not as dramatic compared to studies using the water-soluble analog, dimethylamino-MCL (DMAMCL).[8,13] However, the different types of disease, and the use of an immune-competent syngeneic model, complicate the ability to compare results, as the bcCML model used here progresses more rapidly than the xenograft model used previously.[8,13] Improving the survival of bcCML mice therefore demonstrates the potency of TBP-NPMCL-64 and further highlights the importance of retaining drug at the target site. Survival studies also suggest that TBP-NP delivery of MCL-64 increases cytotoxicity to LSCs and abrogates systemic toxicity of MCL-64 due to decreased off-target accumulation.
Despite the observed decreases in bcCML and LSC burden after TBP-NP, the lack of a prolonged therapeutic effect underscores the complex interplay between the BMME and leukemic cells. Previous efforts to treat bcCML with TBP-NPs delivering a potent chemokine receptor inhibitor revealed partial restoration of the BMME and a modest reduction of bcCML.[26] Others show that combinatory approaches are needed to achieve therapeutic benefits in aggressive leukemia models.[44,45] To drive long-term survival in the bcCML model, MCL-64 can be loaded together with BMME modifying drugs (i.e., chemokine receptor inhibitors) and conventional therapies (i.e., tyrosine kinase inhibitors, anthracyclines, etc.) in future studies.
The findings from this study have broad implications for developing new therapeutic agents with applications beyond leukemia. PSMA-b-PS NPs offer opportunities to increase drug solubility and stability, as detailed here. The bone targeting platform improves drug circulation time to achieve therapeutically relevant doses of the drug in the BMME, which is important for reducing deleterious off-target drug effects. A limitation of the drug loading study is that the repertoire of compounds tested did not provide enough breadth to definitively assess and predict structure-activity relationships. While the definitive parameters required for drug loading were not established, the ability to regio-selectively modify a parent compound and measure LC was informative and these techniques may be adaptable for other DDSs. Based on these studies and others, compounds released from PSMA-b-PS NPs can be predicted based on CLogP, but correlations that predict drug loading behavior were elusive. For future iterations of this work, high throughput screening methods[46] may be useful to predict compound loading within NP cores.
4. Conclusion
Integrated drug development and delivery approach were utilized to demonstrate the antileukemic potency of novel MCL derivatives. Specifically, P450 enzymatic modifications were used to develop a subset of MCL derivatives with enhanced antileukemic activity and unique properties for NP loading and delivery. Due to poor aqueous solubility and tissue-specific targeting in vivo, bone-targeted PSMA-b-PS NPs were used to encapsulate and deliver selected derivatives. LC was impacted by positional modification while the release was affected by derivative hydrophobicity. Triazole and carbamate MCL derivatives exhibited excellent serum stability while ester derivatives were readily degraded. Nevertheless, NP entrapment mitigated degradation. Significant reductions in LSC burden were only observed in leukemic mice treated using the lead derivative, MCL-64 delivered via bone-targeted NPs compared with drug alone and untreated mice. Additionally, leukemic mice treated with free MCL-64 succumbed to disease by day 19 while bone-targeted NPMCL-64 survived. The results of this study stress the importance of drug delivery during the process of drug discovery and contribute significant findings regarding the development of LSC-specific inhibitors.
5. Experimental Section
Materials:
Chemicals used in this study were purchased from Sigma-Aldrich, Fisher Scientific, and Alfa Aesar unless otherwise noted. Distilled/deionized water (ddH2O, resistivity = 18 MΩ) and spectroscopic grade solvents were used for all studies. Mice used to establish the bcCML model (6–8-week-old male C57BL/6 (CD45.1)) were purchased from the breeding core facility at the Wilmot Cancer Institute and studies were conducted with prior approval from the University of Rochester School of Medicine and Dentistry, University Committee for Animal Resources (UCAR).
Synthesis of Ester and Carbamate MCL Analogs:
The MCL analogs were synthesized via chemoenzymatic synthesis using two engineered variants of cytochrome P450BM3 (CYP102A1) from Bacillus megaterium for the site-selective hydroxylation of MCL to give 2(R)-hydroxy-MCL (P450 variant VF10) or 14-hydroxy-MCL (P450 variant V-H10(47C,87I)).[12] Briefly, large scale enzymatic hydroxylation reactions were carried using 200 mg MCL dissolved at a final concentration of 2.5 mM in 320 mL potassium phosphate buffer (50 mM, pH 8.0) containing DMSO at 2% (v/v). To obtain 2(R)-hydroxy-MCL, P450 variant VF10 (final concentration: 2.5 μM, 0.1 mol%) was added to the solution followed by the addition of a NADPH regeneration system containing: PTDH (2 μM), NADP+ (150 μM), and sodium phosphite (50 mM). The reaction mixture was stirred at 80 rpm for 12 h at room temperature and the crude product was extracted with dichloromethane (6 × 90 mL). The collected organic layers were dried over Na2SO4, concentrated under vacuum, and purified by flash chromatography on silica gel (40% n-hexane/ 60% ethyl acetate) to afford 2(R)-hydroxy-MCL as a white solid (175 mg, 82% yield): 1H NMR (500 MHz, CDCl3) d 1.32 (s, 3H), 1.34–1.43 (m, 1H), 1.91–1.95 (m, 4H), 2.13 (d, 1H, J = 13.8 Hz), 2.21–2.30 (m, 2H), 2.34 (dd, 2H, J = 14.5, 27.1 Hz), 2.72 (t, 1H, J = 9.7 Hz), 3.02 (d, 1H, J = 10.3 Hz), 3.73 (t, 1H, J = 10.1 Hz), 4.64 (t, 1H, J = 5.8 Hz), 5.30 (s, 1H), 5.50 (d, 1H, J = 1.9 Hz), and 6.22 (d, 1H, J = 2.7 Hz); 13C NMR (126 MHz, CDCl3): d 24.07, 24.61, 25.86, 35.59, 48.59, 49.88, 58.52, 71.91, 77.93, 83.65, 119.60, 136.25, 138.22, 138.76, and 169.45; MS (ESI) calculated for C15H20O4 m/z: 264.32; found: 287.2 [M+Na]+, 551.2 [2M+Na]+. To prepare 14-hydroxy-MCL, a similar procedure was applied using P450 variant V-H10(47C,87I) (final concentration: 2.5 μM, 0.1 mol%) in 1 M phosphate buffer (pH 7.0) to afford 14-hydroxy-MCL as a white solid (140 mg, 66% yield). 1H NMR (500 MHz, CDCl3) δ = 1.27–1.36 (m, 4H), 1.74–1.80 (m, 1H), 1.84 (dd, 1H, J = 7.8, 16.5 Hz), 2.13–2.21 (m, 3H), 2.26–2.36 (m, 2H), 2.52 (d, 1H, J = 8.4 Hz), 2.56 (d, 1H, J = 8.2 Hz), 2.61–2.73 (m, 2H), 2.80 (d, 1H, J = 10.5 Hz), 3.81 (t, 1H, J = 10.3 Hz), 4.08 (s, 1H), 5.51 (d, 1H, J = 2.9 Hz), and 6.21 (d, 1H, J = 3.2 Hz); 13C NMR (126 MHz, CDCl3) δ = 22.80, 25.85, 28.97, 30.35, 38.30, 50.08, 58.80, 65.53, 80.04, 84.06, 119.67, 134.32, 136.75, 138.71, and 169.55; MS (ESI) calculated for C15H20O4 m/z: 264.32; found: 287.2 [M+Na]+, 551.2 [2M+Na]+. The ester analogs MCL-6, MCL-9 MCL-13, MCL-14, MCL-16, and MCL-19 were prepared via esterification of 2(R)-hydroxy-MCL or 14-hydroxy-MCL as described elsewhere.[12] The carbamate analogs, MCL-38 and MCL-39 were prepared using the following procedure. To a solution of 2(R)-hydroxy-MCL (12 mg, 0.05 mmol), and 4-trifluoro-phenyl isocyanate (8 μL, 0.07 mmol) in anhydrous DCM (2 mL) under argon atmosphere, DBTDL (dibuthyltin dilaurate) (20% mol) was added and the reaction was stirred at room temperature for 12-h. Upon completion of the reaction as determined by TLC, a saturated solution of NaHCO3 was added and the aqueous phase was extracted with DCM (3x). The collected organic layers were dried over Na2SO4, concentrated in vacuo and the crude product was purified by flash chromatography on silica gel (eluting mixture EtOAc 50% in n-Hexane) to obtain MCL-38 as a white solid (14 mg, 90% yield).1H NMR (500 MHz, CDCl3): d 1.29 (s, 3H), 1.45–1.51 (m, 1H), 1.77 (s, 3H), 2.01 (dd, 1H, J1 = 13.0 Hz, J2 = 4.5 Hz), 2.16 (dd, 1H, J1 = 14.0 Hz, J2 = 2.5 Hz), 2.35 (s, 2H), 2.51 (dd, 1H, J1 = 13.0 Hz, J2 = 5.0 Hz), 2.73 (t, 1H, J = 9.0 Hz), 3.02 (d, 1H, J = 10.0 Hz), 3.84 (t, 1H, J = 10.0 Hz), 5.55 (d, 1H, J = 3.0 Hz), 5.65 (t, 1H, J = 7.5 Hz), 6.26 (d, 1H, J = 3.5 Hz), 6.86 (s, 1H, NH), 7.50 (d, 2H, J = 8.5 Hz), and 7.56 (d, 2H, J = 8.5 Hz); 13C NMR (126 MHz, CDCl3): d 24.1, 24.2, 25.7, 35.7, 45.7, 48.5, 58.2, 74.5, 83.8, 116.1, 118.0 (2C), 120.3, 123.0, 126.3 (2C), 130.8, 133.6, 138.2, 139.3, 140.9, 152.6, and 169.2; 19F NMR (376 MHz, CDCl3):0.37; MS (ESI) calcd for C23H24F3NO5 m/z: 451.44; found: 475.4 [M+Na]+, 925.6 [2M+Na]+. The same procedure was carried out starting from 14-hydroxy-MCL to obtain MCL-59 as a white solid (29 mg, 93% yield). 1H NMR (500 MHz, CDCl3): d 1.32–1.39 (m, 4H), 1.83–1.90 (m, 2H), 215–2.25 (m, 2H), 2.36–2.41 (m, 1H), 2.52 (d, 1H, J = 16.0 Hz), 2.63–2.72 (m, 2H), 2.83 (d, 1H, J = 10.5 Hz), 3.83 (t, 1H, J = 10.0 Hz), 4.64 (s, 2H), 5.51 (d, 1H, J = 2.5 Hz), 6.22 (d, 1H, J = 3.0 Hz), 6.94 (s, 1H, NH), 7.50 (d, 2H, J = 8.5 Hz), and 7.55 (d, 2H, J = 8.5 Hz); 13C NMR (126 MHz, CDCl3): d 22.8, 25.6, 29.9, 30.9, 38.2, 49.9, 58.9, 68.2, 80.1, 83.6, 118.0 (2C), 119.8, 123.0, 125.3 (J = 138.5 Hz), 126.3 (2C), 129.7, 138.4, 140.5, 140.9, 153.1, and 169.4; 19F NMR (376 MHz, CDCl3): 0.36; MS (ESI) calcd for C23H24F3NO5 m/z: 451.44; found: 475.4 [M+Na]+, 925.6 [2M+Na]+. NMR spectra are included in the Supporting Information section (Figure S8, Supporting Information).
Synthesis of MCL-64:
MCL-64 was synthesized via conversion of 14-hydroxy-MCL to 14-azido-MCL, followed by copper catalyzed azide/alkyne cycloaddition (CuAAC) reaction. To a cooled solution of 14-hydroxy-MCL in dry toluene (0 °C, under argon) was added diphenyl-phosphoryl azide (DPPA, 2 equiv) and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU, 2 equiv). The reaction was gradually warmed to room temperature and stirred for six hours. The crude reaction mixture was directly loaded onto silica gel and purified by flash chromatography (gradient eluent 20%–40% EtOAc in hexanes) to obtain 14-azido-MCL as a light-yellow oil (80% yield). 1H NMR (500 MHz, CDCl3): d 1.30–1.39 (m, 4H), 1.84–1.90 (m, 2H), 2.17 (d, 1H, J = 8.5 Hz), 2.28–2.39 (m, 2H), 2.47 (d, 1H, J = 12.5 Hz), 2.51–2.57 (m, 1H), 2.73 (t, 1H, J = 7.5 Hz), 2.86 (d, 1H, J = 8.0 Hz), 3.82–3.89 (m, 2H), 4.21 (q, 1H, J = 8.0 Hz), 5.51 (d, 1H, J = 3.0 Hz), and 6.23 (d, 1H, J = 3.0 Hz); 13C NMR (126 MHz, CDCl3): d 22.6, 25.5, 29.7, 32.1, 38.1, 49.8, 56.1, 58.8, 80.0, 83.6, 119.8, 129.3, 138.3, 140.1, and 169.3; MS (ESI) calculated for C15H19N3O4 m/z: 289.34; found: 290.2 [M+H]+, 312.2 [M+Na]+. 14-azido-MCL was dissolved in a solution of 1:1 DCM/H2O and purged under argon. The mixture was vigorously stirred followed by the addition of 1-ethynyl-3,5-bis(trifluoromethyl)benzene (1.2 equiv), copper sulfate pentahydrate (1.5 equiv) and sodium ascorbate (5.5 equiv) at room temperature. The resulting orange mixture was stirred vigorously for <1-h followed by extraction with DCM (3x). The combined organic layers were dried over Na2SO4, concentrated in vacuo and purified by flash chromatography on silica gel (gradient eluting mixture 20–50% EtOAc in hexanes) to obtain the MCL-64 as a clear oil (60% yield). 1H NMR (500 MHz, CDCl3): d 1.27 (m, 1H), 1.39 (s, 3H), 1.89–1.97 (m, 2H), 2.03–2.14 (m, 3H), 2.35 (d, 1H, J = 16.5 Hz), 2.50–2.58 (m, 1H), 2.67–2.77 (m, 2H), 2.92 (d, 1H, J = 10.5 Hz), 3.85 (t, 1H, J = 10.5 Hz), 5.01 (s, 2H), 5.48 (d, 1H, J = 3.0 Hz), 6.21 (d, 1H, J = 3.5 Hz), 7.82 (d, 2H, J = 6.2 Hz), and 8.27 (s, 2H); 13C NMR (126 MHz, CDCl3): d 23.0, 25.0, 30.1, 31.0, 38.2, 49.8, 55.4, 59.2, 80.1, 83.3, 120.1, 121.8, 125.7, 128.5, 132.3, 132.5, 132.6, 138.1, 141.7, 145.6, and 169.1; MS (ESI) calculated for C25H23F6N3O3 m/z: 527.16; found: 528.3 [M+H]+. NMR spectra are included in the Supporting Information section (Figure S8, Supporting Information).
MTT Cell Viability Studies with M9-ENL-1 Cells:
M9-ENL1 cells were maintained in a 37 °C humidified incubator with 5% CO2. Cells were plated at a density of 106 cells mL−1 in αMEM culture media (Invitrogen) supplemented with 5% human plasma, 20% FBS, the cytokines SCF, IL-3, IL-7, and FLT3 ligand (Peprotech), and penicillin/ streptomycin. Prior to testing, the compounds were diluted into a complete cell culture medium and the media was supplemented with sterile DMSO (10% of final volume). Compounds were then diluted into the culture media, at the desired concentration, to yield a final DMSO volume (v/v) of 1% with a total of 6 replicates per dose (n = 6). Following a 24-h incubation period, 20 μL of a 5 mg mL−1 Thiazolyl Blue Tetrazolium Bromide solution was added directly to the culture media. After incubation at 37 °C for 3 h, the plates were centrifuged (4000 rpm, 5 min), the media was removed, and 100 μL of DMSO was added to solubilize the formazan product. The resulting OD was measured at 550 nm using a multi-well plate reader (Tecan). The values were normalized against the wells containing the DMSO vehicle (1% DMSO) and the resultant values were fitted to a non-linear regression plot; [Inhibitor] versus normalized response with a variable slope, using GraphPad Version 7.03.
Diblock Synthesis, Characterization, and NP Self-Assembly:
Amphiphilic diblock copolymers consisting of maleic anhydride (MA) and styrene (STY) monomers were synthesized via one-step RAFT polymerization (Schematic S1, Supporting Information).[23–26,29] Excess distilled STY (99% ACS grade) was polymerized with MA, which was recrystallized from chloroform, using a 4:1 [STY]:[MA] ratio in the presence of 4-cyano-4-dodecylsulfanyltrithiocarbonyl sulfanyl pentanoic acid (DCT) chain transfer agent (CTA) and 100:1 [MA]/[CTA]. Radical initiator, 2,2’–azo-bis (isobutylnitrile) (AIBN) recrystallized from methanol was added at a ratio of 2:1 [CTA]:[Initiator] in dioxane (50% w/w) and the reaction was purged on ice for 45 mins and placed in a 60 °C oil bath.[23–26,29] At 72-h, the polymerization reaction was halted by air exposure and solubilized in acetone prior to precipitating in petroleum ether and drying under vacuum at room temperature. Synthesized polymers were characterized via gel permeation chromatography (GPC) using a Shimadzu system equipped with a differential refractometry (Shimadzu RID-27 6 10A), a light scattering detector (Wyatt Technology DAWN TREOS), a solvent pump (Shimadzu LC-1 20AD), and a column oven set at 60 °C (Shimadzu CTO-20A). Analysis was performed using a 3-μm linear gel column (Tosoh TSK-Gel 3 Super HM-N, 6.0 mm ID × 15 cm) in series with a 3-μm guard column (Tosoh Biosciences) with a mobile phase consisting of spectroscopic grade DMF/0.05 M LiCl, a flow rate of 0.35 mL min−1, and polymer solvated at 1 mg mL−1 in the mobile phase. Refractive index increment (dn/dc) of PSMA-b-PS polymers determined experimentally to be 0.142 mL g−1 was used to calculate molecular weights and polydispersity indices (PDIs).[23–26,29] MA conversion was analyzed via 1H NMR (Bruker 300 MHz) by preparing pre- and post-polymerization solutions (before precipitation) in deuterated dimethyl sulfoxide (DMSO).
To form NPs, diblock copolymers (200 mg) were dissolved in dimethylformamide (DMF, 30 mL) and ddH2O (30 mL) was added using a syringe pump set at 24.4 μL min−1.[23–26,29] Samples were dialyzed against ddH2O for 3 days (MWCO 6–8 kDa) and filtered using 0.2 μM cellulose acetate filters. Lyophilization was used to determine gravimetric NP concentrations and stored at 4 °C in ddH2O. Dynamic light scattering (DLS, Malvern Instrument, Worcestershire, UK) was used to measure the size and zeta potential of NPs at 0.2 mg mL−1 concentrations in PBS, pH 7.4.
MCL Analogs Loading and Characterization:
MCL analogs were dissolved in chloroform at 3 mg mL−1. NPs in water were added to stirring solutions of each drug using 1:3 drug:NP weight ratios. Drug-NP solutions were stirred overnight uncovered in a chemical fume hood to allow evaporation of chloroform. Aggregated and insoluble drug was separated by 2 rounds of centrifugation at 4000 rpm and centrifugal filtration at 2000 rpm for 10 min (100 000 MWCO Amicon Ultra-15 centrifugal filter device (Millipore)) was used to separate any unloaded drug from the drug-NP solution. The final concentrate was reconstituted in ddH2O.
Drug loading was quantified using high-performance liquid chromatography (HPLC) with a mobile phase consisting of A) HPLC grade water and B) as HPLC grade methanol for MCL-6, MCL-13, MCL-14, MCL-16, MCL-9, MCL-38, and MCL-64. For MCL-19 and MCL-39, HPLC mobile phases were as follows A) HPLC grade water + 0.1% formic acid, B) acetonitrile + 0.05% formic acid. HPLC analysis was performed on a Kromasil C18 column (50 mm × 4.6 mm, 5 μm particle size, 100 Å pore size). Drug elution was monitored at 210 nm (Shimadzu). Flow conditions were set at 0.5 mL min−1 with a gradient elution (0–3 min 5% B, 3–9 min 70% B, and 9–10 min 5% B) for MCL-6, MCL-13, MCL-14, MCL-16, MCL-9, MCL-38, and MCL-64 and with a gradient elution (0–2 min 95% B, 2–7 min 35%–95% B, 7–11 min 95% B, and 11–12 min 5% B) for MCL-19 and MCL-39. Standards and samples were prepared for each drug molecule in methanol (0, 3.125, 6.25, 12.5, 25, and 50 mg mL−1) and filtered using a 0.22 μm polytetrafluoroethylene (PTFE) filter prior to analysis on HPLC. Drug LC was calculated as mg drug loaded/mg NPs × 100. In a subset of experiments, acetone was used as an organic solvent following the same approaches outlined above (Figure S2, Supporting Information).
Determination of LC50 of Selected MCL Analogs Using MV411 Cells:
Human bi-phenotypic B myelomonocytic leukemia MV411 cells (ATCC, CRL-9591) maintained at 1–5 × 106 cells mL−1 in Iscove’s modified Dulbecco Media (IMDM) were supplemented with 10% v/v heat-inactivated fetal bovine serum (FBS) and 1% v/v penicillin-streptomycin (PS) at 37 °C in 5% CO2 prior to experiments. Cells were plated at 500 000 cells/well (≈260 000 cells cm−2) and various concentrations of free drug dissolved in DMSO or loaded NP in PBS were added to wells and incubated at 37 °C in 5% CO2 for 24 h. Cell viability was determined using alamarBlue (Invitrogen, Cat #: DAL1025). Fluorescence was detected using Cytation 5 (BioTek Instruments, Ex/Em = 530/590). Wells containing only media plus Alamar blue reagent was used for background subtractions. Data were normalized to untreated wells and LC50 was measured by plotting Log (concentration) versus normalized values and using the built-in nonlinear regression fit function in GraphPad Prism 7.
Drug Release from PSMA-b-PS Based NPs:
Drug release was performed using dialysis tubing with MWCO of 6–8 kDa and phosphate buffer saline, pH 7.4 set to 37 °C to emulate physiological conditions. At t = 0,1, 2, 4, 6, 8, 24, 48, and 72 h, 100 μL aliquots were collected, and the sink condition was replaced at t = 2, 8, 24 and 48 h. Samples were analyzed after adding 100 μL of methanol to each sample via HPLC as previously described.
Serum Stability of MCL Analogs:
To assess the stability of MCL analogs, free compounds (5 μg) were added to PBS (pH 7.4), 100% mouse serum (Life Technologies, Cat #: 10 401), or 1:1 [100% mouse serum]/[PBS] in room temperature. Additionally, drug-loaded NPs in ddH2O was added to mouse serum (1:1/[100% serum]). Samples were extracted twice with ethyl acetate (300 μL) by centrifugation at 11 000 rpm for 2 mins. Combined supernatants were dried under a nitrogen stream, dissolved in methanol (200 μL), filtered using 0.22 μm PTFE filters, and then analyzed via HPLC as described previously.
Preparation and Characterization of Targeted NPs and Treatment of bcCML Mice:
To generate TRAP TBP (sequence: TPLSYLKGLVTVG), microwave-assisted solid-phase peptide synthesis (CEM Corp, Liberty1 synthesizer) was used.[23] Briefly, Fluorenylmethyloxycarbonyl chloride (FMOC)-protected amino acids (AAPPTec and Peptides International) were coupled with an activator mix of 0.5 M O-(benzotriazole-1-yl)-N, N, N’, N’-tetramethyluronium hexafluorophosphate (HBTU) in DMF and an activator base mix of 2 M N, N–Diisopropylethylamine (DIEA) in 1-methyl-2-pyrrolidinone (NMP) with deprotection of individual amino acids achieved using 5% piperazine in DMF. Peptides were cleaved from Fmoc-Gly-Wang resin (Millipore, MA) using 92.5% trifluoroacetic acid (TFA), 2.5% H2O, 2.5% 3, 6-dioxa-1,8-ocatanedithiol (DODT), and 2.5% triisopropylsilane (TIPS) for 2-h, precipitated in ice-cold diethyl ether, and dried in vacuum. Molecular weights were validated using matrix-assisted laser desorption ionization time-of-light mass spectrometry (MALDI-TOF) (Brüker Autoflex III) and equal volumes of TBP (10 mg mL−1 30:70 [v/v] acetonitrile: 0.1% TFA in H2O) and 10 mg mL−1 a-cyano-4-hydroxycinnamic acid.
Carbodiimide chemistry using 1-ethyl-3-(3-dimethylamino)propyl carbodiimide (EDC, Thermo Fisher) ([EDC]/[polymer] = 10:1), 5 mM hydroxysulfosuccinimide (sulfo-NHS, ThermoFisher), and TBP ([TBP]/[polymer] = 10:1 in 0.1 M sodium phosphate buffer (pH 7.4)) was used to conjugate TBP to NPs overnight.[23] Samples were dialyzed against ddH2O for 72 h (MWCO 6–8 kDa) and peptide content was determined using O-Phthaldialdehyde (OPA, Thermo Fisher, Ex/EM = 360 nm/455 nm) and NP tracking analysis. (NTA, Nanosight NS300). Drug loading was achieved by using 4:1 [chloroform]/[acetone]. HPLC was used to access LC as mentioned previously.
All murine experiments were performed under protocols approved by the institutional animal care and use committee (IACUC) (University of Rochester, Rochester, New York, USA). The bcCML model was established via infection with MSCV-BCR/ABL-IRES-green fluorescent protein (GFP) and Nup98/HoxA9-yellow fluorescent protein (YFP) vectors into lineage-negative, sca1+, c-kit negative (LSK) cells enriched via FACs sorting (Figure S4, Supporting Information).[1] Briefly, bcCML cells (2 × 104 in 2% FBS in PBS) were tail-vein injected into sub-lethally irradiated (6 Gy, 137Cs radiation source (GAMMACELL-40) primary recipients. Non-irradiated secondary recipients were then tail vein injected with 2 × 105 splenic cells from primary recipients to establish the model. Free MCL-64 was initially dissolved in DMSO and then diluted in sterile saline immediately prior to injection and MCL-64 loaded TBP-NPs dissolved in PBS, pH 7.4 (0.15 mg MCL-64 and 0.4 mg, 150 mg kg−1 NPs) were intraperitoneally injected starting at day 2 twice daily and thereafter for a total of 6 doses. Control treatments included a saline vehicle. At day 13, mice were sacrificed via CO2 and long bones flushed with PBS using a 25-gauge needle to retrieve bone marrow cells for flow cytometry.[1] Briefly, bone marrow cells were resuspended in 1 mg mL−1 collagenase type IV, 2 mg mL−1 dispase, and 10 U mL−1 DNase in Hank’s Balanced Salt Solution (HBSS). Red blood cells (RBCs) were lysed for 5 min at room temperature using RBC lysis buffer (156 mM NH4Cl, 127 μM EDTA, and 12 mM NaHCO3). Samples were resuspended in 100 μL 2% FBS in PBS with 1 μL of each antibody and incubated at 4°C for 25 min. Following incubation, cells were washed with 1 mL of 2% FBS in PBS. Cells were then resuspended in 2% FBS in PBS with 0.1 μg mL−1 of 4’,6-Diamidino-2-phenylindole dihydrochloride (DAPI) and 1 × 106 events were collected via a LSR-II Fortessa (BD Biosciences). Data were analyzed via FlowJo version 13 9.6.5 (Tree Star) with fluorescence minus one (FMO) controls used for gating. Survival studies were performed using the aforementioned procedures and monitoring mice over time.
Biodistribution of NPs in bcCML Mice:
TBP-NPs were loaded with IR780 using a solvent exchange.[23,25] Briefly, TBP-NPs were added to the stirring solution of IR780 in acetone. IR780 loaded TBP-NPs were dialyzed against ddH2O for 3 days and ddH2O was refreshed every 4–8 h. bcCML mice were injected at D7 with fluorescent TBP-NPs, and in vivo live imaging (IVIS) was performed 24 h later after mice were sacrificed and tissues were collected. Untreated bcCML controls were used for background subtraction of the fluorescent signal and the percent total signal was calculated based on the total radiance efficiency (TRE) of each tissue per the TRE of each mouse.
Statistical Analysis:
All statistical tests were performed using GraphPad Prism 7.0 software. One-way analysis of variance (ANOVA) with Dunnett’s multiple comparisons test or two-way ANOVA with Sidak’s multiple comparisons test was used in experiments consisting of two or more groups and unpaired student’s t-test were used for comparisons between 2 groups. Kaplan–Meier curves were analyzed with the log-rank (Mantel-Cox) test. Statistical significance was indicated as *p < 0.05, **p < 0.01, and ****p < 0.0001. For in vivo experiments, the number of mice per condition was calculated based on a power analysis with β = 0.80. All data shown are expressed as mean ± standard deviation (SD) or standard error mean (SEM).
Supplementary Material
Acknowledgements
General: The authors gratefully thank Dr. James McGrath (University of Rochester) for equipment use. Funding: This work was supported by the National Science Foundation Career Award (CBET1450987 (D.S.W.B)), National Institutes of Health (NIH) P30 AR069655, R01 AR064200, R01 AR056696 (D.S.W.B), F31 CA228391 (M.A.F), R01 GM098628 (R.F.), University of Rochester CTSA award number UL1 TR002001 (D.S.W.B. and B.F.), University Research Award (D.S.W.B., R.F., and B.J.F.), and Drug Discovery Grant (D.S.W.B).
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Conflict of Interest
The MCL analogs and TRAP binding peptides described here are part of patent applications, WO2019040335A1 and WO2014152451, respectively, filed by the University of Rochester.
Contributor Information
Marian A. Ackun-Farmmer, University of Rochester, Department of Biomedical Engineering, 308 Robert B. Goergen Hall, Box 270168, Rochester, NY 14627, USA University of Rochester Medical Center, Center for Musculoskeletal Research, 308 Robert B. Goergen Hall, Box 270168, Rochester, NY 14627, USA.
Hanan Alwaseem, University of Rochester, Department of Chemistry, 418 Hutchison Hall, RC Box 270216, Rochester, NY 14627-0216, USA.
Michele Counts, University of Rochester, Department of Biomedical Engineering, 308 Robert B. Goergen Hall, Box 270168, Rochester, NY 14627, USA.
Benjamin J. Frisch, University of Rochester Medical Center, Department of Pathology and Laboratory Medicine, 601 Elmwood Ave, Box 704, Rochester, NY 14642, USA University of Rochester, Department of Biomedical Engineering, 308 Robert B. Goergen Hall, Box 270168, Rochester, NY 14627, USA; University of Rochester Medical Center, Center for Musculoskeletal Research, 308 Robert B. Goergen Hall, Box 270168, Rochester, NY 14627, USA; University of Rochester, James P. Wilmot Cancer Institute, School of Medicine and Dentistry, 601 Elmwood Ave, Rochester, NY 14642, USA.
Rudi Fasan, University of Rochester, Department of Chemistry, 418 Hutchison Hall, RC Box 270216, Rochester, NY 14627-0216, USA.
Danielle S. W. Benoit, University of Rochester Medical Center. Department of Orthopaedics. 308 Robert B. Goergen Hall, Box 270168, Rochester, NY 14627, USA University of Rochester, Department of Biomedical Engineering, 308 Robert B. Goergen Hall, Box 270168, Rochester, NY 14627, USA; University of Rochester Medical Center, Center for Musculoskeletal Research, 308 Robert B. Goergen Hall, Box 270168, Rochester, NY 14627, USA; University of Rochester, James P. Wilmot Cancer Institute, School of Medicine and Dentistry, 601 Elmwood Ave, Rochester, NY 14642, USA; University of Rochester, Materials Science Program, 308 Robert B. Goergen Hall, Box 270168, Rochester, NY 14627, USA; University of Rochester, Department of Chemical Engineering, 308 Robert B. Goergen Hall, Box 270168, Rochester, NY 14627, USA.
Data Availability Statement
Data reported in this manuscript can be requested from the corresponding authors.
References
- [1].Frisch BJ, Ashton JM, Xing L, Becker MW, Jordan CT, Calvi LM, Blood 2012, 119, 540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Bowers M, Zhang B, Ho Y, Agarwal P, Chen C-C, Bhatia R, Blood 2015, 125, 2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Krevvata M, Silva BC, Manavalan JS, Galan-Diez M, Kode A, Matthews BG, Park D, Zhang CA, Galili N, Nickolas TL, Dempster DW, Dougall W, Teruya-Feldstein J, Economides AN, Kalajzic I, Raza A, Berman E, Mukherjee S, Bhagat G, Kousteni S, Blood 2014, 124, 2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Pei S, Jordan CT, Best Pract. Res., Clin. Haematol 2012, 25, 415. [DOI] [PubMed] [Google Scholar]
- [5].Guzman ML, Rossi RM, Li X, Corbett C, Hassane DC, Bushnell T, Carroll M, Sullivan E, Neelakantan S, Crooks PA, Jordan CT, Blood 2006, 108, 237. [Google Scholar]
- [6].Kolev JN, O’dwyer KM, Jordan CT, Fasan R, ACS Chem. Biol 2014, 9, 164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Tyagi V, Alwaseem H, O’dwyer KM, Ponder J, Li QY, Jordan CT, Fasan R, Bioorg. Med. Chem 2016, 24, 3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Zhang Q, Lu Y, Ding Y, Zhai J, Ji Q, Ma W, Yang M, Fan H, Long J, Tong Z, Shi Y, Jia Y, Han B, Zhang W, Qiu C, Ma X, Li Q, Shi Q, Zhang H, Li D, Zhang J, Lin J, Li L-Y, Gao Y, Chen Y, J. Med. Chem 2012, 55, 8757. [DOI] [PubMed] [Google Scholar]
- [9].Hehner SP, Heinrich M, Bork PM, Vogt M, Ratter F, Lehmann V, Schulze-Osthoff K, Dröge W, Schmitz ML, J. Biol. Chem 1998, 273, 1288. [DOI] [PubMed] [Google Scholar]
- [10].Neelakantan S, Nasim S, Guzman ML, Jordan CT, Crooks PA, Bioorg. Med. Chem. Lett 2009, 19, 4346. [DOI] [PubMed] [Google Scholar]
- [11].Li J, Li S, Guo J, Li Q, Long J, Ma C, Ding Y, Yan C, Li L, Wu Z, Zhu H, Li KK, Wen L, Zhang Q, Xue Q, Zhao C, Liu N, Ivanov I, Luo M, Xi R, Long H, Wang PG, Chen Y, J. Med. Chem 2018, 61, 4155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Alwaseem H, Giovani S, Crotti M, Welle K, Jordan CT, Ghaem-maghami S, Fasan R, ACS Cent. Sci 2021, 7, 841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Ji Q, Ding Y-H, Sun Y, Zhang Y, Gao H-E, Song H-N, Yang M, Liu X-L, Zhang Z-X, Li Y-H, Gao Y-D, Oncotarget 2016, 7, 65012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].An Y, Guo W, Li L, Xu C, Yang D, Wang S, Lu Y, Zhang Q, Zhai J, Fan H, Qiu C, Qi J, Chen Y, Yuan S, PLoS One 2015, 10, e0116202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Yu L, Chen W, Tang Q, Ji K-Y, Cancer Manage. Res 2019, 11, 9203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Jia Y, Zhang C, Zhou L, Shi Y, Tong Z, Xu H, OncoTargets Ther. 2015, 8, 2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ma W-W, Shi Q-Q, Ding Y-H, Long J, Zhang Q, Chen Y, Molecules 2013, 18, 5980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Bertrand N, Wu J, Xu X, Kamaly N, Farokhzad OC, Adv. Drug Delivery Rev 2014, 66, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Narvekar M, Xue HY, Eoh JY, Wong HL, AAPS PharmSciTech 2014, 15, 822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Yoo J-W, Chambers E, Mitragotri S, Curr. Pharm. Des 2010, 16, 2298. [DOI] [PubMed] [Google Scholar]
- [21].Alwaseem H, Frisch BJ, Fasan R, Bioorg. Med. Chem 2018, 26, 1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zhang K, Shafer BM, Demars MD, Stern HA, Fasan R, J. Am. Chem. Soc 2012, 134, 18695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Wang Y, Newman MR, Ackun-Farmmer M, Baranello MP, Sheu T-J, Puzas JE, Benoit DSW, ACS Nano 2017, 11, 9445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Baranello MP, Bauer L, Jordan CT, Benoit DSW, Cell Mol. Bioeng 2015, 8, 455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Baranello MP, Bauer L, Benoit DSW, Biomacromolecules 2014, 15, 2629. [DOI] [PubMed] [Google Scholar]
- [26].Ackun-Farmmer MA, Soto CA, Lesch ML, Byun D, Yang L, Calvi LM, Benoit DSW, Frisch BJ, FASEB J. 2021, 35, e21402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ghosh AK, Brindisi M, J. Med. Chem 2015, 58, 2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Dick A, Cocklin S, Pharmaceuticals 2020, 13, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Ackun-Farmmer MA, Alatise KL, Cross G, Benoit DSW, Adv. Biosyst 2020, 4, 2000172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Gong J, Huo M, Zhou J, Zhang Y, Peng X, Yu D, Zhang H, Li J, Int. J. Pharm 2009, 376, 161. [DOI] [PubMed] [Google Scholar]
- [31].Benoit DSW, Overby CT, Sims KR Jr, Ackun-Farmmer MA, in Biomaterials Science, 4th ed. (Eds. Wagner WR, Sakiyama-Elbert SE, Zhang G, Yaszemski MJ), Academic Press, Elsevier, United States: 2020, pp. 1237–1266. [Google Scholar]
- [32].Ghiaur G, Wroblewski M, Loges S, Semin. Hematol 2015, 52, 200. [DOI] [PubMed] [Google Scholar]
- [33].Behrmann L, Wellbrock J, Fiedler W, Front. Oncol 2018, 8, 444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Zhou H-S, Carter BZ, Andreeff M, Cancer Biol. Med 2016, 13, 248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Kim S, Shi Y, Kim JY, Park K, Cheng J-X, Expert Opin. Drug Delivery 2010, 7, 49. [DOI] [PubMed] [Google Scholar]
- [36].Letchford K, Liggins R, Burt H, J. Pharm. Sci 2008, 97, 1179. [DOI] [PubMed] [Google Scholar]
- [37].Liu J, Xiao Y, Allen C, J. Pharm. Sci 2004, 93, 132. [DOI] [PubMed] [Google Scholar]
- [38].Van Der Sman RGM, Food Hydrocolloids 2019, 96, 396. [Google Scholar]
- [39].Buxton GA, Clarke N, Soft Matter 2007, 3, 1513. [DOI] [PubMed] [Google Scholar]
- [40].Opanasopit P, Yokoyama M, Watanabe M, Kawano K, Maitani Y, Okano T, J. Controlled Release 2005, 104, 313. [DOI] [PubMed] [Google Scholar]
- [41].Singer JW, Bhatt R, Tulinsky J, Buhler KR, Heasley E, Klein P, De Vries P, J. Controlled Release 2001, 74, 243. [DOI] [PubMed] [Google Scholar]
- [42].Martins S, Tho I, Reimold I, Fricker G, Souto E, Ferreira D, Brandl M, Int. J. Pharm 2012, 439, 49. [DOI] [PubMed] [Google Scholar]
- [43].Zunino F, Dallavalle S, Laccabue D, Beretta G, Merlini L, Pratesi G, Curr. Pharm. Des 2002, 8, 2505. [DOI] [PubMed] [Google Scholar]
- [44].Mu C-F, Xiong Y, Bai X, Sheng Y-J, Cui J, Mol. Pharmaceutics 2017, 14, 274. [DOI] [PubMed] [Google Scholar]
- [45].Pan XQ, Zheng X, Shi G, Wang H, Ratnam M, Lee RJ, Blood 2002, 100, 594. [DOI] [PubMed] [Google Scholar]
- [46].Shah M, Agrawal Y, Pharm. Dev. Technol 2013, 18, 582. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data reported in this manuscript can be requested from the corresponding authors.