

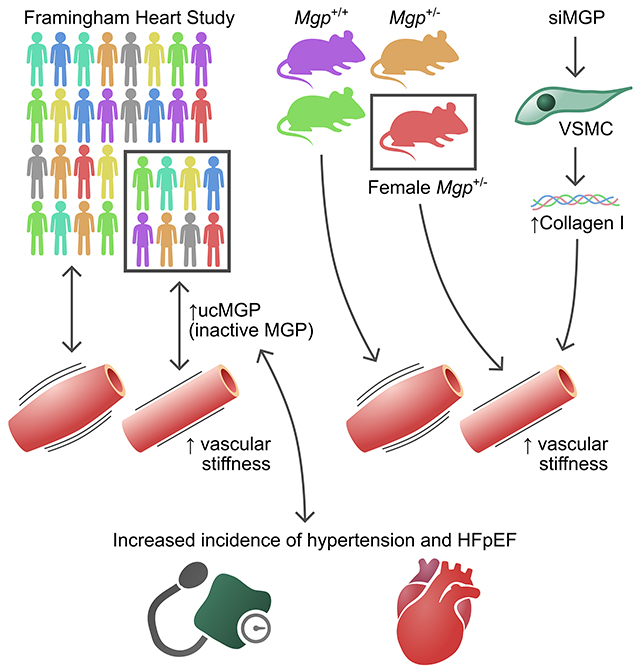
Abstract
OBJECTIVE:
Arterial stiffness is a risk factor for cardiovascular disease, including heart failure with preserved ejection fraction (HFpEF). Matrix Gla protein (MGP) is implicated in vascular calcification in animal models and circulating levels of the uncarboxylated form of MGP (ucMGP) are associated with cardiovascular disease-related and all-cause mortality in human studies. However, the role of MGP in arterial stiffness is uncertain.
APPROACH AND RESULTS:
We examined the association of ucMGP levels with vascular calcification, arterial stiffness including carotid-femoral pulse wave velocity (PWV), and incident heart failure in community-dwelling adults from the Framingham Heart Study. To further investigate the link between MGP and arterial stiffness, we compared aortic PWV in age- and sex-matched young (4-month-old) and aged (10-month-old) wild-type and Mgp+/− mice. Among 7066 adults, we observed significant associations between levels of ucMGP and measures of arterial stiffness, including PWV and pulse pressure. Longitudinal analyses demonstrated an association between baseline ucMGP levels and future increases in systolic blood pressure and incident HFpEF. Aortic PWV was increased in older, but not young, female Mgp+/− mice compared to wild-type mice, and this augmentation in PWV was associated with increased aortic elastin fiber fragmentation and collagen accumulation.
CONCLUSIONS:
This translational study demonstrates an association between ucMGP levels and arterial stiffness and future HFpEF in a large observational study, findings that are substantiated by experimental studies showing that mice with Mgp heterozygosity develop arterial stiffness. Taken together, these complementary study designs suggest a potential role of therapeutically targeting MGP in HFpEF.
Keywords: arterial stiffness, heart failure, matrix GLA protein
GRAPHICAL ABSTRACT:
A graphical abstract is available for this article.
Introduction
Among patients with heart failure (HF), more than half have HF with preserved rather than reduced ejection fraction (HFpEF; HFrEF).1 Despite the high prevalence, mortality, and economic burden of HFpEF, no specific treatments for this disorder have been identified.2 Hypertension is a well-established risk factor for HFpEF, and recent data suggest that increased central blood pressure and large vessel stiffness influence ventricular-arterial coupling, and are integral to the pathophysiology of HFpEF.3, 4 Greater arterial stiffness increases left ventricular (LV) pulsatile afterload and induces LV hypertrophy and diastolic dysfunction, both hallmarks of cardiac remodeling in HFpEF.5 Stiffening of the large elastic arteries is associated with increased risk of systolic hypertension and HF as well as coronary artery disease, stroke, and atrial fibrillation.6 Despite this, there is currently no approved treatment to prevent aortic stiffness. Carotid-femoral pulse wave velocity (PWV), which is the gold standard measure of aortic stiffness, approximately doubles between the ages of 30 and 70 years in humans.7 Given the rapidly growing elderly population,8 there is a great unmet clinical need to determine the molecular mechanisms leading to increased arterial stiffness.
Matrix Gla protein (MGP) is a vitamin K-dependent extracellular matrix protein that inhibits vascular calcification. Mice that lack MGP develop spontaneous arterial calcification at an early age.9, 10 Human polymorphisms in the MGP gene have been associated with both the presence and progression of coronary artery calcification11 and with incident myocardial infarction,12 although the results of these association studies have not always been replicated.13, 14 The biological activity of MGP depends on γ-glutamylcarboxylase, an enzyme that converts inactive, uncarboxylated MGP (ucMGP) to active, carboxylated MGP (cMGP).15 In addition, MGP contains phosphorylated serine residues that may also regulate its activity.15 Vitamin K serves as an essential cofactor for MGP carboxylation and cMGP is the active form of the protein that confers protection against vascular calcification.16 Many studies have shown an important association between increased ucMGP levels and cardiovascular disease, chronic kidney disease, and mortality.17, 18
Higher levels of ucMGP have also been associated with increased vascular stiffness in select patient samples.19, 20 Arterial stiffness reflects the reduced ability of an artery to expand and contract in response to changes in blood pressure. Arterial stiffness increases with advancing age and is associated with a higher incidence of hypertension, adverse myocardial remodeling with diastolic dysfunction, and HFpEF.21 Although higher levels of ucMGP are associated with arterial stiffness, a causal link between ucMGP and vascular compliance remains to be determined and the relationship of ucMGP levels with new-onset hypertension and HFpEF is unknown.
In this study, we examined the association of plasma ucMGP levels with arterial stiffness in a large community-based sample. In the context of the known close association between arterial stiffness and heart failure risk, we also examined the association between ucMGP with adverse LV remodeling and the development of hypertension and HFpEF. We also studied mice with heterozygous deletion of the Mgp gene to determine effects on the closely related trait of aortic pulse wave velocity. Taken together, our human and mouse studies sought to implicate an important role of MGP in increased arterial stiffness as a precursor to HFpEF.
Materials and Methods
The data supporting the study findings will be made available on reasonable request. FHS data are made publicly available and can be accessed through the National Institutes of Health database of genotypes and phenotypes (https://www.ncbi.nlm.nih.gov/gap/). Please see the Major Resources Table in the Supplemental Materials.
Study sample
We studied participants in the Framingham Heart Study, a prospective longitudinal community-based observational cohort study. Participants who attended the following baseline examinations were included: Offspring cohort exam 7 (1998-2001, n=3539) and Third Generation exam 1 (2002-2005, n=4095).22, 23 Individuals with prior heart failure (n=51), end-stage renal disease (estimated glomerular filtration rate, eGFR<30 ml/min/1.73m2, n=25), prevalent atrial fibrillation (n=140), or missing biomarker data (n=152) were excluded. No participants were taking warfarin, which could alter MGP status. In addition, individuals missing information concerning key clinical covariates in primary analyses (n=200) were excluded, leaving 7066 individuals for analysis. For each outcome of interest, we further excluded individuals who were missing ascertainment of baseline disease status or the outcome (Supplemental Table 4). The study was approved by the appropriate institutional review boards and all participants provided written informed consent.
Clinical, biomarker, and imaging assessment
Participants underwent a comprehensive medical history, anthropometric assessment, and physical examination. Blood pressure was measured in a seated position after at least 5 minutes of rest, and hypertension was defined as a systolic blood pressure (SBP) of at least 140 mmHg, diastolic blood pressure (DBP) of at least 90 mmHg, or current use of antihypertensive medication. Plasma samples were obtained, and immediately processed and stored at −80°C until assayed. Total ucMGP was assayed using a modified sandwich enzyme-linked immunosorbent assay approach, multiplexed on a Luminex xMAP platform (Sigma-Aldrich).24 The assay had a detection range of 119-9110 pg/mL, with inter-assay and intra-assay coefficients of variation of 17.8% and 10.8%, respectively. Participants underwent evaluation of vascular function using applanation tonometry and assessment of brachial artery flow-mediated vasodilation, as well as standard transthoracic echocardiography at the time of baseline (Third Generation) or subsequent examination (Offspring cohort exam 8, 2005-2008) as previously described.25 A subset of 3,339 individuals underwent multidetector computed tomography with evaluation of coronary, thoracic aortic, and abdominal aortic calcium measurements.26
Genome-wide genotyping and imputation
Genome-wide genotyping was performed using the Affymetrix 500K mapping arrays and 50K supplemental Human Gene Focused arrays (Affymetrix, Inc., Santa Clara, CA) and the Illumina Human Exome BeadChip v.1.0 (Exome Chip; Illumina, Inc., San Diego, CA). Imputation was performed using the 1000 Genomes Project Reference Panel using MACH.27, 28 Results were filtered for imputation quality ratio <0.8 and minor allele frequency (MAF) <0.01, and a genomic locus was defined using linkage disequilibrium r2<0.1. We determined if top SNPs had in silico associations with expression quantitative trait loci (eQTLs) and relevant clinical phenotypes.29
Clinical outcome ascertainment
We followed participants for longitudinal changes in BP and incident hypertension between the baseline and follow-up examinations (Offspring exam 8, 2005-2008; Third Generation exam 2, 2008-2011). Changes in SBP, DBP, and pulse pressure were defined as the change between the baseline and follow-up examinations. Participants were also followed every two years for medical updates. Medical records were reviewed and incident HF outcomes were adjudicated by a three-physician panel. Medical records were also reviewed for assessment of LV function at or around the time of the first HF incident. Each incident HF event was categorized as HFpEF (LVEF≥50%), HFrEF (LVEF<50%), or unclassified (no LV function assessment available at or around the time of HF event).30
Statistical analysis of Framingham cohort data
Baseline clinical characteristics were summarized for each of the two cohorts. ucMGP concentrations were natural log-transformed due to a right-skewed distribution. Clinical correlates of ucMGP were examined using stepwise linear regression, forcing age and sex into the model, and using P=0.10 for entry and P=0.05 for retention in the model. We used linear regression to examine the association between the ucMGP level and measures of cardiovascular function, including arterial stiffness, endothelial function, as well as cardiac structure and function. Analyses were first adjusted for age and sex, and then additionally adjusted for systolic blood pressure, anti-hypertensive drug treatment, diabetes mellitus, body-mass index, smoking status, total and high-density lipoprotein cholesterol, and prevalent myocardial infarction. Vascular and echocardiographic strain analyses were also adjusted for heart rate. PWV was transformed as the negative inverse due to skewness (−1000/PWV) as previously described.25 CT-based analyses examined continuous calcium scores after adding 1 unit and log-transformation. In cross-sectional analyses, we adjusted for multiple hypothesis testing using a Bonferroni-corrected P-value threshold of P < 0.01 (5 primary traits: coronary artery calcium, PWV, left atrial diameter, left ventricular mass index).
We examined the association between the level of ucMGP and changes in BP and incident hypertension at baseline and follow-up examinations using linear and logistic regression, respectively. For continuous blood pressure analyses, we imputed blood pressure for participants taking anti-hypertensive medications by adding 5 mmHg to the diastolic and 10 mmHg to the systolic blood pressure, as previously described.31 Models were first adjusted for age and sex, and further adjusted for baseline blood pressure, body mass index, diabetes, smoking, total and high-density lipoprotein cholesterol, and prevalent myocardial infarction. Multivariable Cox regression models were used to examine the association between ucMGP level and incident HFpEF or HFrEF. Models accounted for multiple competing risks (death, other subtype of HF, and unclassified HF). We tested the proportional hazards assumption and found it was not violated. We also performed sex-stratified analyses and tested for ucMGP*sex interactions. All analyses were conducted using SAS version 9.4 software (SAS Institute).
A genome-wide association study of MGP concentrations was performed using an additive genetic model in a linear mixed effects model (“LMEKIN” function, Kinship Package, R). We defined cis variants within 1 Mb of the MGP gene. The threshold for genome-wide significance was set at p<5x10−8 with suggestive associations at p<1x10−6. We performed Mendelian Randomization testing using two cis-SNP variants (P<10−5, LD r2<0.1) as instrumental variables in order to examine whether MGP was causal for arterial stiffness as ascertained by PWV.32
Mouse Echocardiography
All experiments involving mice were approved by the Partners Subcommittee on Research Animal Care (Protocol #2008N000169). C57Bl/6 wild-type and MGP+/− heterozygous mice were used in this study.9, 33 Heterozygous mice were chosen for this study, since MGP−/− homozygous mice invariably succumb to severe aortic calcification with aortic rupture at ~6 weeks of age.9 Mice were anesthetized with continuous flow ~1.5% isoflurane gas and placed in a supine position for imaging. Mice were subsequently prepared for imaging by connecting ECG electrodes to limbs and using a chemical hair remover (Nair, Carter-Horner, Mississauga, ON, Canada) on the skin over the area to be imaged.34 Mouse echocardiography was performed using an ultrasound biomicroscope (Vevo770, VisualSonics, Toronto, ON, Canada).35 Imaging of the abdominal aorta was conducted using the RMV 704 scanner probe with a central frequency of 40 MHz, a focal length of 6 mm, and a field of view of 10 mm x 10 mm, as previously described.35 Briefly, using the M-mode setting, the velocity of blood flow in the center of the aortic vessel was measured at the proximal and distal ends of the field of view. The distances between the foot of the flow waveform and the R-wave of the ECG (arrival times) and the distance between the two points of measurement in the aorta were determined post-acquisition using the Vevo770 software and averaged over 5-10 cardiac cycles. PWV was calculated by dividing the distance between the proximal and distal locations (in millimetres) by the difference in the proximal and distal arrival times of the waveforms (in milliseconds), as previously described.35, 36
Mouse Cardiac Magnetic Resonance
C57Bl/6 wild-type and MGP+/− heterozygote mice were anesthetized with continuous flow ~2% isoflurane gas in a chamber and then transferred to a face mask and placed in a headfirst prone position for MRI scanning. Mice were subsequently prepared for imaging by connecting ECG electrodes to limbs, placing a body temperature probe, and placing a respiratory pillow under their abdomen. Mouse cardiac magnetic resonance (CMR) was performed on a small bore 4.7T scanner (Biospec 47/40 USR, Bruker, Billerica, MA, USA). As previously described,37, 38 2D time resolved phase contrast imaging (98x98um2, 1mm slice thickness, Repetition Time [TR]/Echo Time [TE] = 5.3ms/1.6ms, five 1ms trigger delays with interleaving for view sharing temporal resolution of 1ms, four averages) of the descending aorta was conducted at two single perpendicular slices 1 cm apart in encoding for the through-plane flow at VENC=170 cm/sec. PWV was calculated from slice separation (1cm) divided by the time difference of the upslope from the velocity time curve using the multi-point transit time method39 of the pulse traveling from one slice to the other. Conventional myocardial balanced steady-state free precession imaging (TR = 8ms, TE = 2.5ms, 128x128um2, 1mm slice thickness, 20 phases)40 was performed to measure cardiac function. Reference myocardial T1 mapping (single echo non-selective inversion recovery gradient echo, TR = 3008ms, TE =2.4ms, TI = 150, 300, 500, 900, 1500, and 3000ms) was performed to yield native T1 values.
Aortic Immunofluorescence and Histology
Aortas were fixed in formalin (10%) for 24 h prior to paraffinization and sectioning (6-μm). Staining with Verhoeff-Van Gieson and Masson’s Trichrome (Thermo Scientific, MI, USA) were performed for assessment of elastin integrity and collagen deposition, respectively. Two-dimensional images were analyzed using image J software. Aortas were also embedded and cryopreserved in optimal cutting-temperature medium (Sakura Tissue-Tek, Zoeterwoude, Netherlands), and 6-µm sections were prepared. To detect specific collagens in aortas, frozen tissue sections were fixed in cold 100% methanol and incubated with primary antibodies specific for COL1 (Abcam, #ab34710) and COL3 (Abcam, #ab6310). The location of F-Actin and nuclei were identified by staining with F-actin (ActinGreenTM 488 ReadyProbes, ThermoFisher Scientific, USA) and 4’,6-diamidino-2-phenylindole (DAPI), respectively.
Separate histological sections from the same mice were prepared for ex vivo alizarin red staining to access calcium content, as previously described.41, 42 Briefly, sections were deparaffinized and stained with 2% Alizarin red (pH 4.2) for 5 min at room temperature and rinsed repeatedly with distilled water. Images of whole aortas were captured with a low magnification microscope (magnification, ×10; Nikon Eclipse 80i microscope) in a darkened background.
siRNA-mediated Knockdown of MGP in Vascular Smooth Muscle Cells
siRNA directed against human and mouse MGP (siMGP) and scrambled control siRNA (siSC) were obtained from Dharmacon (SMARTpool, Thermo Scientific). HASMCs (Cell Applications #355-75a) and MOVAS (ATCC CRL-2797) were transfected with siRNA using Lipofectamine RNAiMAX reagent, as described by the manufacturer (Life Technologies).
Measurement of gene expression by quantitative RT-PCR
Total RNA from aortas and cells were extracted by the phenol/guanidinium method. Reverse transcription was performed using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA). A Mastercycler ep Realplex (Eppendorf, Hamburg, Germany) was used for real-time amplification and quantification of transcripts. Relative expression of target transcripts were normalized to levels of 18S ribosomal RNA, determined using the relative CT method. Taqman® gene expression assays were used to quantify mRNA levels encoding MGP, RUNX2, COL1A1, and COL1A2.
Near-infrared imaging and quantification of aortic calcification
To quantify aortic calcification, age-matched wild-type and Mgp+/− mice, as well as Mgp−/− mice as a positive control, were injected via the tail vein with OsteoSense-680 (PerkinElmer, 2 µl/g each) 24 hours before euthanasia, as described previously.33 Aortas were isolated and analyzed ex vivo by fluorescence reflectance imaging using an Odyssey Imaging System (LI-COR Biotechnology, Lincoln, NE) and software version 3.0.16.43, 44
Statistical analysis
Statistical analyses for the murine experiments were performed using Graph Pad Prism 8.0 (GraphPad Software, La Jolla, CA) and Stata 13.0 (StataCorp LLC). The Shapiro-Wilk test was used to determine the normality of each continuous variable, and all such variables were found to be normally distributed. Data are reported as mean ± SEM, unless otherwise indicated. For more than 2 group comparisons of continuous variables, two-tailed 1-way analysis of variance (ANOVA) with Sidak’s post-hoc testing was employed. A two-sided P<0.05 was considered to be statistically significant.
Results
Baseline characteristics of the 7066 participants (mean age 49 years, 54% women) are presented in Supplemental Table 1. Higher ucMGP levels were associated with older age and male sex. In age- and sex-adjusted analyses, higher ucMGP concentrations were also associated with other cardiovascular risk factors including higher blood pressure, higher body mass index, hyperlipidemia, and lower eGFR (Supplemental Table 2). In addition, levels of ucMGP were higher among those with previous myocardial infarction.
Among a subset of 3,339 individuals with ascertainment of vascular calcification, 44% had coronary, 23% had thoracic, and 53% had abdominal aortic calcification as defined by a CT-based calcium score of >0. We found no significant associations between the levels of ucMGP and coronary, thoracic aortic, or abdominal aortic calcium deposition (P>0.05 for all, Table 1). In contrast, ucMGP levels were associated with measures of arterial stiffness, including carotid-femoral PWV and central pulse pressure (Table 1). A 1-unit higher log-transformed ucMGP was associated with a 0.26 standard deviation higher PWV (negative inverse transformed) in age- and sex-adjusted analyses (s.e. 0.02, P<0.0001). After adjusting for other clinical covariates, the ucMGP levels remained independently associated with PWV (beta 0.08, s.e. 0.02, P<0.0001). Higher levels of ucMGP were also associated with decreased brachial flow-mediated vasodilation (age- and sex-adjusted beta −0.08, s.e. 0.03, P<0.0001), which is a marker of endothelial dysfunction; however, this association was not significant in the multivariable-adjusted model (P=0.39).
Table 1.
Association of ucMGP and cardiovascular structure and function
| Age- and sex-adjusted | Multivariable-adjusted | |||||
|---|---|---|---|---|---|---|
|
|
||||||
| Beta | s.e. | P-value | Beta | s.e. | P-value | |
| Cardiac CT measures | ||||||
| Coronary artery calcium | 0.007 | 0.015 | 0.668 | −0.014 | 0.016 | 0.378 |
| Thoracic aortic calcium | −0.008 | 0.012 | 0.509 | −0.002 | 0.012 | 0.866 |
| Abdominal aortic calcium | 0.015 | 0.014 | 0.271 | −0.010 | 0.014 | 0.480 |
|
| ||||||
| Vascular function measures | ||||||
| Carotid femoral pulse wave velocity* | 0.122 | 0.009 | <0.001 | 0.040 | 0.008 | <0.001 |
| Aortic augmentation index* | −0.015 | 0.012 | 0.214 | −0.001 | 0.012 | 0.922 |
| Central pulse pressure (carotid)* | 0.086 | 0.014 | <0.001 | 0.018 | 0.012 | 0.144 |
| Brachial flow-mediated vasodilation* | −0.040 | 0.012 | 0.001 | −0.011 | 0.013 | 0.386 |
|
| ||||||
| Echocardiographic measures | ||||||
| Left atrial diameter | 0.109 | 0.011 | <0.001 | −0.003 | 0.011 | 0.771 |
| LV end-diastolic dimension | 0.063 | 0.012 | <0.001 | 0.007 | 0.012 | 0.545 |
| Relative wall thickness | 0.050 | 0.013 | <0.001 | −0.007 | 0.014 | 0.604 |
| LV mass | 0.081 | 0.011 | <0.001 | −0.021 | 0.010 | 0.034 |
| LV fractional shortening | 0.006 | 0.013 | 0.649 | −0.004 | 0.014 | 0.790 |
| LV global longitudinal strain* | 0.134 | 0.017 | <0.001 | 0.029 | 0.016 | 0.074 |
| LV circumferential strain* | 0.035 | 0.018 | 0.047 | 0.034 | 0.018 | 0.059 |
| LV radial strain* | −0.019 | 0.018 | 0.293 | −0.020 | 0.019 | 0.294 |
| LV transverse strain* | −0.042 | 0.018 | 0.019 | −0.001 | 0.019 | 0.941 |
Multivariable-adjusted model, adjusted for age, sex, systolic blood pressure, hypertension treatment, diabetes mellitus, body-mass index, smoking, total and HDL cholesterol, and prevalent myocardial infarction. LV mass analyses adjusted for height and weight in place of BMI.
Also adjusted for heart rate
Beta = SD unit difference in response variable per 1-SD unit difference in log-transformed ucMGP
Higher levels of ucMGP were also associated with echocardiographic measures of cardiac structure and function, including increased left atrial diameter and relative wall thickness as well as worse LV global longitudinal strain (P≤0.0002 for all, Table 1). In multivariable-adjusted analyses, ucMGP remained associated with worse global longitudinal strain (P=0.005); however this relationship was attenuated after additionally adjusting for heart rate (P=0.07).
In longitudinal multivariable analyses, we observed that higher baseline ucMGP levels were associated with future increases in systolic but not diastolic blood pressure, resulting in a widening in pulse pressure, adjusting for other clinical variables (Table 2). Using multivariable analyses, 1-unit higher baseline log-transformed ucMGP level was associated with a future 1.7 mmHg increase in pulse pressure (s.e. 0.34, P<0.0001). Baseline ucMGP levels were associated with the development of hypertension: 1-unit higher baseline ucMGP level was associated with a two-fold higher odds of future hypertension (age- and sex-adjusted OR 2.04, 95% CI 1.67-2.49, P<0.0001). This association was attenuated after multivariable adjustment (OR 1.23, 95% CI 0.99-1.54, P=0.07). We selected covariates a priori based on the fact that they were known to be traditional clinical risk factors associated with the development of heart failure and hypertension in prior studies based in Framingham Heart Study participants. Further adjustment for eGFR did not result in substantive differences. It is of note that the associations of ucMGP with central pulse pressure, brachial flow-mediated vasodilation, and LV relative wall thickness were attenuated when adjusting for systolic blood pressure, thus supporting a direct impact of ucMGP on vascular function.
Table 2.
Association of ucMGP and longitudinal cardiovascular outcomes
| Age- and sex-adjusted | Multivariable-adjusted | |||||
|---|---|---|---|---|---|---|
|
| ||||||
| Change in blood pressure | beta | s.e. | P-value | beta | s.e. | P-value |
| Systolic blood pressure | −0.01 | 0.01 | 0.43 | 0.05 | 0.01 | <0.001 |
| Diastolic blood pressure | −0.06 | 0.01 | <.0001 | −0.01 | 0.01 | 0.62 |
| Pulse pressure | 0.03 | 0.01 | 0.04 | 0.06 | 0.01 | <0.001 |
| Future hypertension | OR | 95% CI | P-value | OR | 95% CI | P-value |
| Incident hypertension | 1.41 | (1.28 - 1.55) | <0.001 | 1.11 | (0.99 - 1.23) | 0.07 |
| Future Heart Failure | HR | 95% CI | P-value | HR | 95% CI | P-value |
| Incident heart failure | 1.52 | (1.28 - 1.80) | <0.001 | 1.34 | (1.13 - 1.59) | 0.001 |
| Incident HFpEF | 1.74 | (1.37 - 2.21) | <0.001 | 1.51 | (1.18 - 1.94) | 0.001 |
| Incident HFrEF | 1.32 | (1.00 - 1.73) | 0.048 | 1.24 | (0.95 - 1.62) | 0.12 |
Multivariable-adjusted model, adjusted for age, sex, systolic blood pressure, hypertension treatment, diabetes mellitus, body-mass index, smoking, total and HDL cholesterol, and prevalent myocardial infarction. Blood pressure and hypertension analyses simultaneously adjusted for systolic and diastolic blood pressure. In addition, heart failure analyses were adjusted for baseline LV hypertrophy and left bundle branch block.
Beta = 1-SD unit change per 1-SD unit difference in log-transformed ucMGP
OR = Odds Ratio per 1-SD unit difference in log-transformed ucMGP
HR = Hazard Ratio per 1-SD unit difference in log-transformed ucMGP
Over a median follow-up period of 12 years, 197 participants developed incident heart failure, with 101 classified as HFpEF and 77 as HFrEF. Higher baseline ucMGP levels were associated with increased risk of incident overall HF (multivariable-adjusted hazard ratio (HR) 1.84, 95% CI 1.28-2.63, P=0.001). Of note, ucMGP levels were specifically associated with incident HFpEF with no significant association with HFrEF (HFpEF: HR 2.37, 95% CI 1.40-4.00, P=0.001; HFrEF: HR 1.57, 95% CI 0.89-2.74, P=0.12).
We found 2 genome-wide significant loci on chromosome 12 associated with circulating ucMGP levels (Supplemental Table 3): rs1800801 in the 5’ untranslated region of the MGP gene itself (P<10−100), and rs4764124, which is located within an intron of the SMCO3 gene [single-pass membrane protein with coiled-coil domains 3] and is near the MGP locus (P=6.9x10−10). There were two missense variants in linkage disequilibrium with rs1800801: rs4236 (MGP, P=1.7x10−38) and rs11276 (ART4 [ADP-ribosyltransferase 4], P=5.5x10−38), both predicted to be tolerated, benign variants,45, 46 and two synonymous SNPs rs1861698 (ART4, P<10−100) and rs3088189 (ART4, P=6.0x10−38) that were also associated with circulating ucMGP levels. We found two cis-protein quantitative trait loci (cis-pQTLs, defined as loci associated with ucMGP levels with P<1x10−5, located within 1 Mb of the MGP gene, pruned to linkage disequilibrium r2<0.1) and used these as instrumental variables for ucMGP to test associations with PWV using two-sample Mendelian randomization. One of the two cis-pQTLs was suggestive of a causal relationship of MGP levels to PWV (MR test P=0.04).
We searched our top MGP locus rs1800801 against published databases of gene expression and known clinical phenotypes.29 We found that rs1800801 was associated with MGP expression in various tissues (nerve, P=1.0x10−11; lung, P=1.1x10−4; thyroid P=1.9x10−6) as well as mRNA expression of other genes. In vascular beds, rs1800801 was associated with expression of SMCO3 (aorta, P=1.4x10−5; coronary, P=1.1x10−4), C12orf60 (tibial artery, P=5.8x10−6, aorta P=7.0x10−8, coronary P=8.7x10−4), ERP27 (aorta, P=1.6x10−17; coronary P=2.3x10−9), and ARHGDIB (tibial artery, P=4.7x10−5; aorta P=3.7x10−6). Missense variants in MGP were also associated with lumbar bone mineral density (lowest P=1.9x10−4).
Functional data indicates that MGP is a potent inhibitor of vascular calcification.9, 16 However, in our study, higher ucMGP levels were associated with increased arterial stiffness, increased cardiovascular disease, and adverse outcomes presumably because of counterregulatory or compensatory mechanisms related to MGP expression and/or possibly because ucMGP is not adequately modified by vitamin K-dependent gamma carboxylase to form cMGP, the biologically active protein.17 To determine whether MGP levels are causally related to vascular stiffness, we compared aortic PWV in age- and sex-matched young and old Mgp+/− and wild-type mice. All female mice were nulliparous. It was not possible to study Mgp−/− mice because these mice develop spontaneous arterial calcification by two weeks of age and die of aortic rupture by ~6 weeks of age.9 In contrast to Mgp−/− mice, heterozygous Mgp+/− mice do not develop appreciable arterial calcification even at 12 months of age, as measured by Osteosense near-infrared fluorescence (Supplemental Figure 1a). Furthermore, there was no detectable difference in calcium content in aortic sections from Mgp+/− mice, compared to wild-type mice, as evidenced by Alizarin red staining (Supplemental Figure 1b). In 4-month-old mice (Figure 1a), the aortic PWV assessed by echocardiography was similar in Mgp+/− and wild-type mice, with no apparent sex differences (mean ±SEM; Female: 1.8 ± 0.2 m/s vs. 2.3 ± 0.3, P=0.38; Male: 2.2 ± 0.2 vs 2.2 ± 0.4 m/s, P=0.99). However, at 10 months of age (Figure 1b), female Mgp+/− mice had a higher aortic PWV compared to female wild-type mice (7.6 ± 0.5 vs 2.5 ± 0.2 m/s, P<0.0001). There was no difference between the aortic PWV of 10-month-old male Mgp+/− mice and wild-type mice (3.6 ± 0.7 vs 3.5 ± 0.5 m/s, P>0.99). These results indicate that Mgp heterozygosity in older female mice causes increased arterial stiffness.
Figure 1: Arterial stiffness is increased in 10-month-old female Mgp+/− mice.
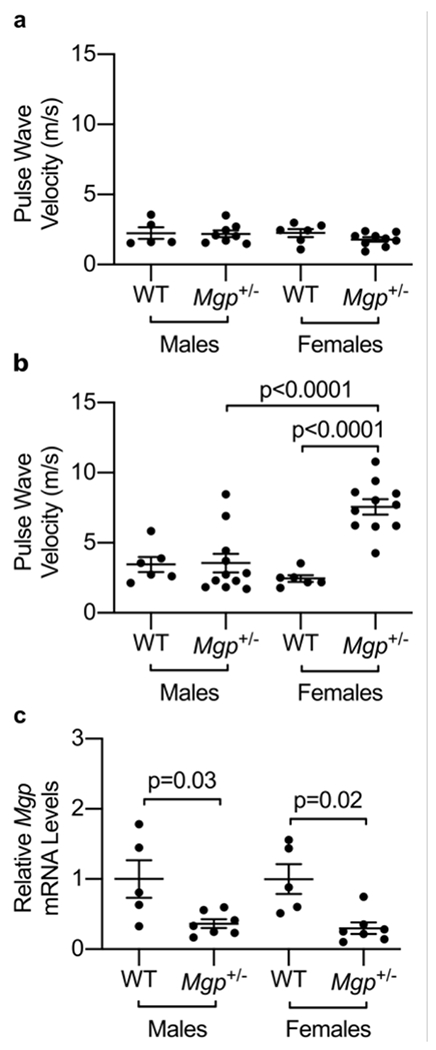
(a) No difference was observed in pulse wave velocity (PWV) between female and male wild-type (WT) and Mgp+/− mice at 4 months of age. (b) 10-month-old female Mgp+/− mice had increased PWV compared to age-matched WT female mice, but no difference between male Mgp+/− and WT mice was observed. PWV was higher in 10-month-old female Mgp+/− mice than their male counterparts. (c) Mgp heterozygosity is associated with a >50% decrease in aortic expression of Mgp mRNA. mRNA expression levels of Mgp were significantly lower in aged Mgp+/− female and male mice (n=7 in each group) compared to age-matched wild-type mice (n=5 in each group). No difference in aortic Mgp mRNA levels was detected between Mgp+/− female and male mice. Statistical comparisons were made with a one-way ANOVA with Sidak’s post-hoc testing.
To determine whether the difference in aortic stiffness observed in older female versus male Mgp+/− mice is due to sex-specific differences in Mgp expression levels, aortic Mgp mRNA levels were measured by qPCR (Figure 1c). Mgp expression levels were decreased by >60% in aortas isolated from aged female and male Mgp+/− mice when compared to age-matched wild-type female and male mice. These results suggest that the sex-specific differences in the effects of Mgp heterozygosity on the development of vascular stiffness are not associated with sex-specific differences in aortic Mgp mRNA expression levels.
To further support our echocardiographic findings, we assessed PWV and myocardial T1 mapping using mouse cardiac magnetic resonance (CMR). As shown in Supplemental Figure 2a–b, PWV was increased in female Mgp+/− mice (7.6 ± 0.6 m/s) compared to male Mgp+/− mice (2.7 ± 0.1 m/s, P<0.0001) and female wild-type (2.3 ± 0.1 m/s, P<0.0001). Further, T1-weighted signals (a marker of LV fibrosis) were higher in the left ventricles of female Mgp+/− mice (1337 ± 7.0 ms) relative to female wild-type mice (Supplemental Figure 2c–d, 1313 ± 4.7 ms, P=0.045). However, female Mgp+/− mice did not exhibit different LVEF or LV end-systolic and end-diastolic volumes (Supplemental Figure 3) compared to male Mgp+/− mice or wild-type mice. These findings indicate that Mgp heterozygosity in older female mice results in increased aortic stiffness and is associated with a higher degree of cardiac fibrosis compared to wild-type mice.
Aortic stiffness is primarily dependent on changes in the matrix composition of the vessel’s medial layer with elastin fiber disruption and increased collagen deposition. In particular, collagen I plays a crucial role in arterial stiffening, the expression of which is induced by Runt-related transcription factor 2 (Runx2).47 We therefore sought to investigate the pathologic consequences underlying the role of MGP heterozygosity in arterial stiffness. First, we determined the histologic changes associated with Mgp heterozygosity. We focused on female mice, given the fact we only demonstrated increased stiffness in female Mgp heterozygote mice. While the aortas of 10-month-old wild-type mice were histologically normal, the aortas of 10-month-old Mgp+/− mice had elastin fiber fragmentation (Figure 2a) and collagen accumulation (by 34.1 ± 7.0%) in the medial layer (Figure 2b). A substantial induction of Collagen I (COLI) protein expression was observed in the aortic media from Mgp+/− mice via immunofluorescence (Figure 2c). However, no increase in COL3 was observed in the aortas from Mgp heterozygote mice. To further study the role of MGP on the regulation of collagen expression, we treated MOVAS (a mouse aortic smooth muscle cell line, Figure 2d) and human aortic smooth muscle cells (HuASMCs, Figure 2e) with an siRNA targeting MGP. Silencing MGP (~70% in MOVAS and 95% in HuASMCs) increased the expression of RUNX2 (~30% in MOVAS and >2-fold in HuASMCs). In addition, and consistent with the immunofluorescent results, knockdown of MGP resulted in an increase in Col1a1 and Col1a2 mRNA levels in mouse aortic SMCs (Figure 2d). Similarly, in human aortic SMCs, knockdown of MGP increased COL1A2 by >40% but did not significantly affect COL1A1 expression. These findings indicate that MGP heterozygosity is associated with increased aortic collagen I protein expression and that, similarly, reduced MGP levels in vascular smooth muscle cells result in increased expression of RUNX2 and collagen I.
Figure 2: Mgp heterozygosity is associated with increased medial collagen accumulation and elastin disruption and reduced MGP expression in vascular smooth muscle cells increases collagen I expression.
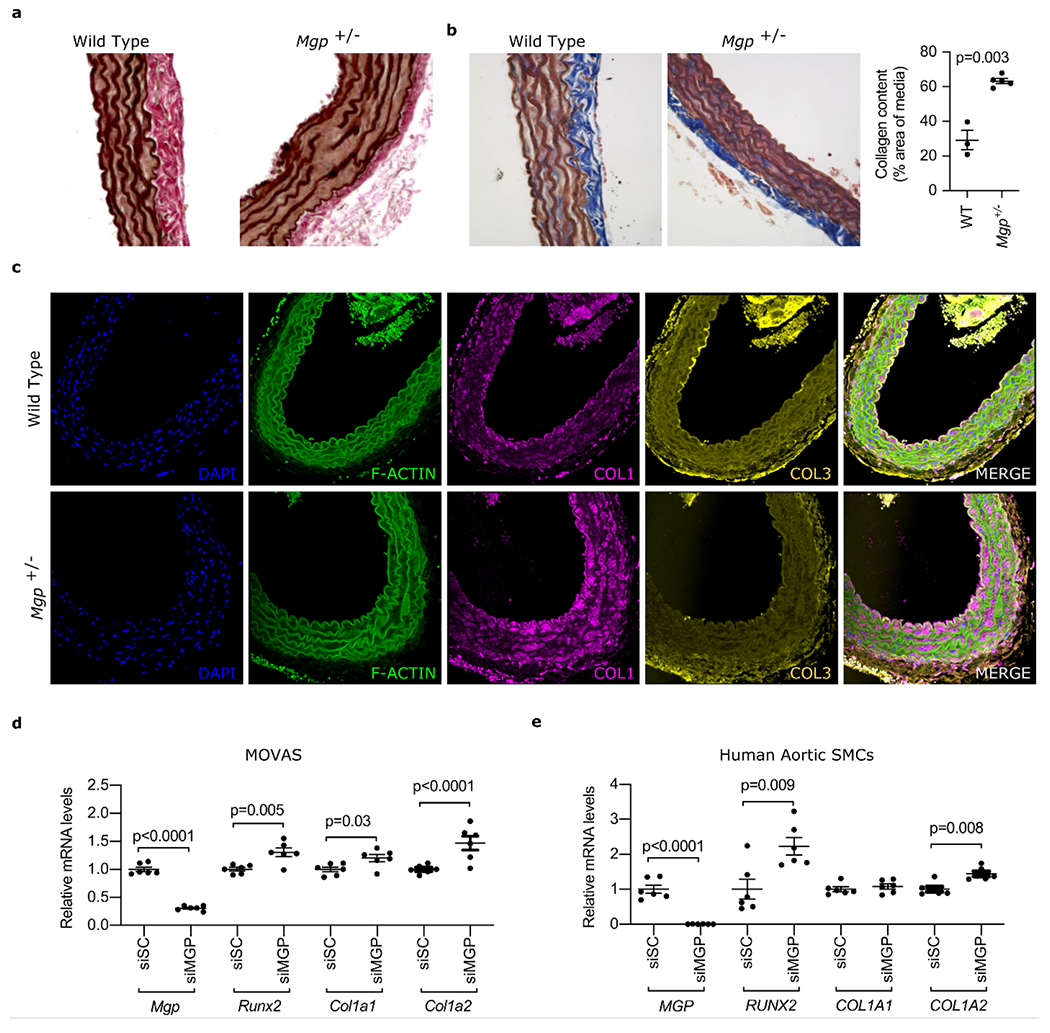
(a) to (c) show sections of aortas from 10-month-old female wild-type (WT) and Mgp+/− mice. (a) Verhoeff–Van Gieson (VVG) staining showing disrupted elastin fiber integrity in the aortas of female Mgp+/− mice compared to wild-type mice. (b) Masson’s Trichrome staining demonstrating increased collagen (blue staining) in the medial layer of aortas from female Mgp+/− mice compared to wild-type mice (quantification shown in the right panel, Student’s t-test). (c) Aortas isolated from WT and Mgp+/− mice were stained for F-Actin (green), COL1 (pink), COL3 (gold) and DNA (blue, DAPI). Increased levels of COL1, but not COL3, protein were seen in aortas from female Mgp+/− mice compared to wild-type mice. (d) Treatment of MOVAS (n=6 biologically independent samples in each group) with siMGP (resulting in >70% knockdown of Mgp mRNA) caused an increase in Runx2, Col1a1, and Col1a2 expression (Student’s t-test). (e) Treatment of human aortic smooth muscle cells (n=6 biologically independent samples in each group) with siMGP (resulting in >90% knockdown of MGP mRNA) increased RUNX2 and COL1A2 mRNA levels (Student’s t test).
Discussion
We examined the role of MGP in the development of arterial stiffness as a precursor to HFpEF using complementary studies in a large human community-based cohort and a mouse model of MGP heterozygosity. Our findings were as follows: first, increased levels of ucMGP were associated with increased vascular stiffness as measured by aortic PWV and were associated with measures of cardiac function including LV strain. Second, circulating ucMGP levels portended the future development of pulse pressure widening and HFpEF in a large cohort. To determine if MGP was causally related to vascular stiffness, we took two approaches. We used SNPs associated with ucMGP levels as instrumental variables and found that Mendelian randomization analysis implicated ucMGP as causal for PWV. We also ascertained aortic PWV in MGP heterozygote mice. Compared to matched wild-type mice, female MGP+/− mice had a three-fold increase in aortic PWV with concomitant cardiac fibrosis. Taken together, these observations are consistent with a direct role for MGP in regulating the development of vascular stiffness. Our findings also suggest that MGP-related vascular effects may contribute to the risk of HFpEF.
Although our mouse models indicate a protective benefit of MGP in arterial stiffness, higher ucMGP levels were associated with increased arterial stiffness, increased cardiovascular disease, and adverse outcomes presumably because (1) circulating MGP levels are upregulated secondarily and compensatory to a primary insult and/or (2) ucMGP is not adequately modified by vitamin K-dependent gamma carboxylase to form cMGP, the biologically active protein. Our prior work in patients with calciphylaxis and End-Stage Renal Disease suggest that, while ucMGP levels are elevated in states of vascular disease, the ratio of cMGP:ucMGP is lower in the disease state.17 Similarly, in the calcification disorder pseudoxanthoma elasticum, defective carboxylation of MGP is observed and the ratio of cMGP:ucMGP is reduced.48, 49 Furthermore, in patients with chronic heart failure, while levels of ucMGP were higher with worsening HF severity, the ratio of cMGP to ucMGP was lower in more severe states of HF.50 Therefore, reduced activation of MGP to the carboxylated form appears to be an important factor in cardiovascular disorders. Similarly, our in vitro and in vivo findings support a protective role of active MGP in arterial stiffness. We therefore hypothesize that arterial stiffness is manifested by inadequate or insufficient modification to the active (carboxylated) form of MGP.
There are different species of MGP in the circulation and, to become fully active, MGP must both be carboxylated and phosphorylated.51 Both ucMGP and dp-ucMGP (dephosphorylated ucMGP) represent fractions of MGP that are inactive.15 Furthermore, both ucMGP and dp-ucMGP have been associated with cardiovascular disease, however some differences have been noted in these associations.17, 51–55 For instance, ucMGP has been associated with cardiovascular disease in patients with known CAD but not in diabetics, whereas dp-ucMGP is associated with risk in diabetics.56 One potential explanation for this is that ucMGP, which consists of phosphorylated MGP species, may behave differently than dp-ucMGP at a biologic level in tissues, thus differentially affecting circulating levels. A limitation of our investigation is that whereas ucMGP associations with arterial stiffness, hypertension, and HFpEF were determined, dp-ucMGP was not measured.
While the primary focus of MGP has been on its role in the development of vascular calcification, a few previous studies have suggested a potential association with arterial stiffness. 19, 20, 57–60 Recent studies demonstrated that aortic PWV independently correlated with dp-ucMGP in European populations (~1000 individuals).19, 20, 58, 60 In addition, separate smaller studies (<200 individuals) have found a strong association between dp-ucMGP and CF-PWV in both hypertensive and diabetic individuals, further supporting a role for MGP with arterial stiffness.57, 59 We extend these findings to show an association of circulating ucMGP levels with arterial stiffness in >7000 individuals. Whilst a previous study found an association between dp-ucMGP and worse outcomes in patients with chronic heart failure, ours is the first study to show such an association with future incident HFpEF.50 It has been suggested that abnormal ventricular-vascular coupling, caused by increased arterial stiffness, may contribute to the pathophysiology of HFpEF.50
MGP has an established protective role against vascular calcification through inhibition of bone morphogenetic protein (BMP) signaling and direct binding and inhibition of calcium phosphate crystal growth in the extracellular matrix.33, 61 In human studies, both polymorphisms in the MGP gene and increased circulating MGP levels have been associated with coronary artery calcification and cardiovascular events.11, 62 Furthermore, mice that lack both alleles of MGP develop spontaneous arterial calcification at an early age.9 For this reason, previous studies have suggested that MGP influences vascular stiffness through its role in preventing calcification.20, 57 It was interesting, therefore, that ucMGP levels were associated with vascular stiffness in the absence of an association with detectable vascular calcification. However, our study is limited in that the human and mouse imaging techniques may not be able to detect microcalcifications.63, 64 Therefore, we cannot exclude the possibility that microcalcification is the mechanism by which MGP heterozygosity increases aortic stiffness. Our findings are substantiated by Mendelian randomization results showing that ucMGP genetic proxies are associated with PWV in humans. Importantly, ucMGP requires activation by a vitamin K-dependent carboxylase, therefore making the MGP pathway modifiable with vitamin K therapy. Indeed, recent studies have demonstrated that Vitamin K2 (VK2) treatment is protective against age-related aortic stiffening in 243 healthy adults, but it will be important to study the effects of vitamin K therapy in larger cohorts with vascular disease.65 We further demonstrated that 10-month-old female, but not male, MGP+/− mice developed increased aortic stiffness, in the context of ~50% lower aortic MGP mRNA levels, compared to wild-type mice. It was interesting that only female MGP+/− mice developed increased aortic stiffness, especially given recent findings that VK2 treatment prevented age-related aortic stiffness increases only in post-menopausal women.65 These findings allude to a possible female-dominant role for MGP in the development of vascular stiffness. Taken together, our results suggest that MGP heterozygosity is a cause for arterial stiffness and that, in humans, arterial stiffness is associated with increased ucMGP levels, the inactive form of the protein.
The exact mechanisms that link ucMGP with arterial stiffness, particularly prior to detectable arterial macrocalcification, remain unclear. We show here a direct effect of MGP deficiency on aortic collagen expression in mice, pointing to a possible mechanistic link between MGP and arterial stiffness. Arterial compliance is regulated, in large part, by the relative contribution of the scaffolding proteins: collagen and elastin. Previous studies have reported a prominent role for type I collagen metabolism in the development of arterial stiffness.66, 67 Similarly, our data points to an influence of MGP on type I collagen accumulation. Furthermore, MGP may influence stiffness through its inhibition of Runx2 expression, which was previously found to induce arterial stiffness in a diabetic mouse model.47 Interestingly, whilst the in vitro data points to a mechanistic link between MGP deficiency and increased arterial stiffness through the increased production of collagen, reduced levels of Mgp in male aortas did not manifest in increased PWV. This possibly relates to sex-specific levels of complexity in the in vivo model that remain unknown. In supportive clinical evidence, VK2 treatment lowered arterial stiffness only in women, which may suggest that relative MGP deficiency may have a greater influence on arterial stiffness in females.65 Future studies will be needed to understand the interplay between MGP and sex-specific hormones and/or gene expression and how this relates to arterial stiffening.
Beyond its association with arterial stiffness, we show that ucMGP is associated with LV systolic mechanics, as well as the future development of hypertension and HFpEF. Hypertension is a well-established risk factor for HFpEF, and recent data suggest that central blood pressure and large vessel stiffness in particular appear integral to the pathophysiology of HFpEF.3, 68 Arterial stiffness increases LV pulsatile afterload and induces LV hypertrophy and diastolic dysfunction, both hallmarks of cardiac remodeling in HFpEF.3, 5 Further, concomitant vascular and ventricular stiffening results in abnormal “coupling”, adversely affecting cardiovascular function.69 Our findings substantiate an important role of ucMGP in this process.
We also explored genetic determinants of ucMGP levels using GWAS. Not surprisingly, we found an association between circulating ucMGP levels and rs1800801 near the MGP gene (P<10−100). This MGP gene variant has previously been associated with vascular calcification and atherosclerotic disease.70 Previous studies have demonstrated that this SNP has a crucial role on the promoter activity of the MGP gene.12 We then used SNPs associated with ucMGP levels as instrumental variables and found that Mendelian randomization analysis implicated ucMGP as causal for PWV, strengthening our observational findings of ucMGP and arterial stiffness.
There are limitations in our study that deserve mention. First, this study focuses on the measurement of ucMGP and not the active, carboxylated form of MGP (cMGP). Antibodies specific to cMGP are not available at this time, as they once were previously.17 Thus, we are unable to comment on the relative ratio of active:inactive MGP. Second, we cannot exclude residual confounding or establish causal relationships with our cohort study by virtue of its observational design. Third, our cohort study included middle-aged and older adults who were largely of European ancestry; the generalizability of our human data to other ages and races/ethnicities is unknown. Fourth, both Mendelian Randomization analysis and a murine model were used to provide evidence of a causal link between MGP and arterial stiffness. However, only female Mgp+/− mice developed increased aortic PWV and not male Mgp+/− mice. Sex-specific differences were not observed in the human cohort. Future investigation is needed to understand the sex-specific findings of Mgp+/− mice.
In summary, we identified novel associations between ucMGP and increased risk of large artery stiffening, elevated systolic blood pressure, pulse pressure widening, and future HFpEF. These observational findings are reinforced by Mendelian randomization analyses showing a putative causal association with PWV, as well as functional studies using Mgp+/− mice, that provide further evidence for a mechanistic role for MGP in the protection against large artery stiffening with aging. Our findings support the novel hypothesis that MGP regulates arterial stiffness.
Supplementary Material
Highlights Section:
Higher levels of uncarboxylated matrix GLA protein (ucMGP) were associated with increased vascular stiffness as measured by aortic pulse wave velocity and were associated with measures of cardiac function including LV strain.
ucMGP levels predicted the future development of hypertension and HFpEF.
Mendelian randomization analysis implicated ucMGP as causally linked to pulse wave velocity.
MGP heterozygosity in mice was discovered to be a novel model of arterial stiffness.
MGP regulates collagen synthesis by vascular smooth muscle cells.
Acknowledgements
R. Malhotra and J.E. Ho conceived the study, acquired, and analyzed the data, and wrote the article. C.J. Nicholson designed and conducted experiments, acquired, and analyzed data, and wrote the article. C. Slocum, H.H. Sigurslid, C.L.L. Cardenas, R. Li, S.L. Boerboom, and C. Nguyen designed and conducted experiments and edited the manuscript. D. Wang, V. Bhambhani, Y. Chen, S. Hwang and C. Yao performed the clinical study, analyzed data, and edited the manuscript. F. Ichinose, D.B. Bloch, M.E. Lindsay, G.D. Lewis, J.R. Aragam, U. Hoffman, G.F. Mitchell, N.M. Hamburg, R.S. Vasan, E.J. Benjamin, M.G. Larson, W.M. Zapol, S. Cheng, J.D. Roh, C.J. O’Donnell, D. Levy provided assistance in designing the study and edited the manuscript.
Sources of Funding
This work was partially supported by the National Heart, Lung and Blood Institute’s Framingham Heart Study (Contracts N01-HC-25195 and HHSN268201500001I) and by the Division of Intramural Research (D.L.) of the National Heart, Lung and Blood Institute. Dr. Malhotra is supported by NHLBI grants R01 HL142809 and R01 HL159514, the Wild Family Foundation, the American Heart Association (18TPA34230025), and a Massachusetts General Hospital Hassenfeld Scholar Award. Dr. Roh is supported by NIA grant K76AG064328. Dr. Ho is supported by NIH grants R01 HL134893, R01 HL140224, and K24 HL153669. Dr. Cheng is supported by R01-HL134168, R01-HL131532, R01-HL143227, and R01-HL142983. Dr. Benjamin was supported by NIH grants R01HL128914; 2R01 HL092577; 1R01 HL141434 01A1; 1R01HL60040; 1R01HL70100, and American Heart Association, 18SFRN34110082. The views expressed in this manuscript are those of the authors and do not necessarily represent the views of the National Heart, Lung, and Blood Institute; the National Institutes of Health; or the U.S. Department of Health and Human Services.
Nonstandard Abbreviations and Acronyms
- BMI
Body mass index
- BP
Blood Pressure
- CI
Confidence interval
- cMGP
carboxylated Matrix GLA Protein
- CMR
Cardiac Magnetic Resonance
- COL1
Collagen 1
- COL1A1
Collagen Type I Alpha 1 Chain
- COL1A2
Collagen Type I Alpha 2 Chain
- COL3
Collagen 3
- CT
Computed Tomography
- DAPI
4’,6-diamidino-2-phenylindole
- DBP
Diastolic Blood Pressure
- dp-ucMGP
de-phosphorylated Uncarboxylated Matrix GLA Protein
- ECG
Electrocardiogram
- eGFR
estimated glomerular filtration rate
- eQTL
expression quantitative trait loci
- GWAS
Genome-wide Association Study
- HASMC
Human Aortic Smooth Muscle Cell
- HDL
High-density lipoprotein
- HF
Heart Failure
- HFpEF
Heart Failure with preserved Ejection Fraction
- HFrEF
Heart Failure with reduced Ejection Fraction
- LV
Left ventricular
- MGP
Matrix GLA Protein
- MOVAS
Murine Aortic Vascular Smooth Muscle
- PWV
Pulse Wave Velocity
- RT-PCR
Real-Time Polymerase Chain Reaction
- RUNX2
Runt-related transcription Factor 2
- SBP
Systolic Blood Pressure
- S.E.
Standard error
- SEM
Standard error of the mean
- TE
Echo Time
- TR
Repetition Time
- ucMGP
uncarboxylated Matrix GLA Protein
- VK2
Vitamin K2
Footnotes
Disclosures
Dr. Malhotra receives consulting fees from MyoKardia (now BMS) and Third Pole, has a sponsored research agreement with Bayer Pharmaceuticals and Amgen, and is a co-founder of Patch Inc.
References
- 1.Oktay AA, Rich JD, Shah SJ. The emerging epidemic of heart failure with preserved ejection fraction. Current heart failure reports. 2013;10:401–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE Jr., Drazner MH, et al. 2013 accf/aha guideline for the management of heart failure: Executive summary: A report of the american college of cardiology foundation/american heart association task force on practice guidelines. Circulation. 2013;128:1810–1852 [DOI] [PubMed] [Google Scholar]
- 3.Weber T, Chirinos JA. Pulsatile arterial haemodynamics in heart failure. Eur Heart J. 2018;39:3847–3854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weber T, Wassertheurer S, O’Rourke MF, Haiden A, Zweiker R, Rammer M, et al. Pulsatile hemodynamics in patients with exertional dyspnea: Potentially of value in the diagnostic evaluation of suspected heart failure with preserved ejection fraction. J Am Coll Cardiol. 2013;61:1874–1883 [DOI] [PubMed] [Google Scholar]
- 5.Chirinos JA. Arterial stiffness: Basic concepts and measurement techniques. J Cardiovasc Transl Res. 2012;5:243–255 [DOI] [PubMed] [Google Scholar]
- 6.Sutton-Tyrrell K, Najjar SS, Boudreau RM, Venkitachalam L, Kupelian V, Simonsick EM, et al. Elevated aortic pulse wave velocity, a marker of arterial stiffness, predicts cardiovascular events in well-functioning older adults. Circulation. 2005;111:3384–3390 [DOI] [PubMed] [Google Scholar]
- 7.Determinants of pulse wave velocity in healthy people and in the presence of cardiovascular risk factors: ‘Establishing normal and reference values’. Eur Heart J. 2010;31:2338–2350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.North BJ, Sinclair DA. The intersection between aging and cardiovascular disease. Circ Res. 2012;110:1097–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Luo G, Ducy P, McKee MD, Pinero GJ, Loyer E, Behringer RR, et al. Spontaneous calcification of arteries and cartilage in mice lacking matrix gla protein. Nature. 1997;386:78–81 [DOI] [PubMed] [Google Scholar]
- 10.Malhotra R, Mauer AC, Lino Cardenas CL, Guo X, Yao J, Zhang X, et al. Hdac9 is implicated in atherosclerotic aortic calcification and affects vascular smooth muscle cell phenotype. Nat Genet. 2019;51:1580–1587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cassidy-Bushrow AE, Bielak LF, Levin AM, Sheedy PF 2nd, Turner ST, Boerwinkle E, et al. Matrix gla protein gene polymorphism is associated with increased coronary artery calcification progression. Arteriosclerosis, thrombosis, and vascular biology. 2013;33:645–651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Herrmann SM, Whatling C, Brand E, Nicaud V, Gariepy J, Simon A, et al. Polymorphisms of the human matrix gla protein (mgp) gene, vascular calcification, and myocardial infarction. Arteriosclerosis, thrombosis, and vascular biology. 2000;20:2386–2393 [DOI] [PubMed] [Google Scholar]
- 13.Roustazadeh A, Najafi M, Amirfarhangi A, Nourmohammadi I. No association between mgp rs1800802 polymorphism and stenosis of the coronary artery. Ann Saudi Med. 2013;33:149–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang Y, Chen J, Zhang Y, Yu W, Zhang C, Gong L, et al. Common genetic variants of mgp are associated with calcification on the arterial wall but not with calcification present in the atherosclerotic plaques. Circ Cardiovasc Genet. 2013;6:271–278 [DOI] [PubMed] [Google Scholar]
- 15.Schurgers LJ, Spronk HM, Skepper JN, Hackeng TM, Shanahan CM, Vermeer C, et al. Post-translational modifications regulate matrix gla protein function: Importance for inhibition of vascular smooth muscle cell calcification. J Thromb Haemost. 2007;5:2503–2511 [DOI] [PubMed] [Google Scholar]
- 16.Yao Y, Shahbazian A, Bostrom KI. Proline and gamma-carboxylated glutamate residues in matrix gla protein are critical for binding of bone morphogenetic protein-4. Circ Res. 2008;102:1065–1074 [DOI] [PubMed] [Google Scholar]
- 17.Nigwekar SU, Bloch DB, Nazarian RM, Vermeer C, Booth SL, Xu D, et al. Vitamin k-dependent carboxylation of matrix gla protein influences the risk of calciphylaxis. J Am Soc Nephrol. 2017;28:1717–1722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thamratnopkoon S, Susantitaphong P, Tumkosit M, Katavetin P, Tiranathanagul K, Praditpornsilpa K, et al. Correlations of plasma desphosphorylated uncarboxylated matrix gla protein with vascular calcification and vascular stiffness in chronic kidney disease. Nephron. 2017;135:167–172 [DOI] [PubMed] [Google Scholar]
- 19.Pivin E, Ponte B, Pruijm M, Ackermann D, Guessous I, Ehret G, et al. Inactive matrix gla-protein is associated with arterial stiffness in an adult population-based study. Hypertension. 2015;66:85–92 [DOI] [PubMed] [Google Scholar]
- 20.Mayer O Jr., Seidlerova J, Wohlfahrt P, Filipovsky J, Vanek J, Cifkova R, et al. Desphospho-uncarboxylated matrix gla protein is associated with increased aortic stiffness in a general population. J Hum Hypertens. 2016;30:418–423 [DOI] [PubMed] [Google Scholar]
- 21.Shirwany NA, Zou MH. Arterial stiffness: A brief review. Acta Pharmacol Sin. 2010;31:1267–1276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kannel WB, Feinleib M, McNamara PM, Garrison RJ, Castelli WP. An investigation of coronary heart disease in families. The framingham offspring study. American journal of epidemiology. 1979;110:281–290 [DOI] [PubMed] [Google Scholar]
- 23.Splansky GL, Corey D, Yang Q, Atwood LD, Cupples LA, Benjamin EJ, et al. The third generation cohort of the national heart, lung, and blood institute’s framingham heart study: Design, recruitment, and initial examination. American journal of epidemiology. 2007;165:1328–1335 [DOI] [PubMed] [Google Scholar]
- 24.Ho JE, Lyass A, Courchesne P, Chen G, Liu C, Yin X, et al. Protein biomarkers of cardiovascular disease and mortality in the community. J Am Heart Assoc. 2018;7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mitchell GF, Hwang SJ, Vasan RS, Larson MG, Pencina MJ, Hamburg NM, et al. Arterial stiffness and cardiovascular events: The framingham heart study. Circulation. 2010;121:505–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hoffmann U, Massaro JM, D’Agostino RB Sr., Kathiresan S, Fox CS, O’Donnell CJ. Cardiovascular event prediction and risk reclassification by coronary, aortic, and valvular calcification in the framingham heart study. J Am Heart Assoc. 2016;5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Genomes Project C, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, et al. A global reference for human genetic variation. Nature. 2015;526:68–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huan T, Rong J, Liu C, Zhang X, Tanriverdi K, Joehanes R, et al. Genome-wide identification of microrna expression quantitative trait loci. Nat Commun. 2015;6:6601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Staley JR, Blackshaw J, Kamat MA, Ellis S, Surendran P, Sun BB, et al. Phenoscanner: A database of human genotype-phenotype associations. Bioinformatics. 2016;32:3207–3209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ho JE, Enserro D, Brouwers FP, Kizer JR, Shah SJ, Psaty BM, et al. Predicting heart failure with preserved and reduced ejection fraction: The international collaboration on heart failure subtypes. Circ Heart Fail. 2016;9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cui JS, Hopper JL, Harrap SB. Antihypertensive treatments obscure familial contributions to blood pressure variation. Hypertension. 2003;41:207–210 [DOI] [PubMed] [Google Scholar]
- 32.Mitchell GF, Verwoert GC, Tarasov KV, Isaacs A, Smith AV, Yasmin, et al. Common genetic variation in the 3’-bcl11b gene desert is associated with carotid-femoral pulse wave velocity and excess cardiovascular disease risk: The aortagen consortium. Circ Cardiovasc Genet. 2012;5:81–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Malhotra R, Burke MF, Martyn T, Shakartzi HR, Thayer TE, O’Rourke C, et al. Inhibition of bone morphogenetic protein signal transduction prevents the medial vascular calcification associated with matrix gla protein deficiency. PLoS One. 2015;10:e0117098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Williams R, Needles A, Cherin E, Zhou YQ, Henkelman RM, Adamson SL, et al. Noninvasive ultrasonic measurement of regional and local pulse-wave velocity in mice. Ultrasound Med Biol. 2007;33:1368–1375 [DOI] [PubMed] [Google Scholar]
- 35.Nicholson CJ, Seta F, Lee S, Morgan KG. Microrna-203 mimics age-related aortic smooth muscle dysfunction of cytoskeletal pathways. J Cell Mol Med. 2017;21:81–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hartley CJ, Taffet GE, Michael LH, Pham TT, Entman ML. Noninvasive determination of pulse-wave velocity in mice. Am J Physiol. 1997;273:H494–500 [DOI] [PubMed] [Google Scholar]
- 37.Herold V, Parczyk M, Morchel P, Ziener CH, Klug G, Bauer WR, et al. In vivo measurement of local aortic pulse-wave velocity in mice with mr microscopy at 17.6 tesla. Magn Reson Med. 2009;61:1293–1299 [DOI] [PubMed] [Google Scholar]
- 38.Parczyk M, Herold V, Klug G, Bauer WR, Rommel E, Jakob PM. Regional in vivo transit time measurements of aortic pulse wave velocity in mice with high-field cmr at 17.6 tesla. J Cardiovasc Magn Reson. 2010;12:72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fielden SW, Fornwalt BK, Jerosch-Herold M, Eisner RL, Stillman AE, Oshinski JN. A new method for the determination of aortic pulse wave velocity using cross-correlation on 2d pcmr velocity data. J Magn Reson Imaging. 2008;27:1382–1387 [DOI] [PubMed] [Google Scholar]
- 40.Hiba B, Richard N, Janier M, Croisille P. Cardiac and respiratory double self-gated cine mri in the mouse at 7 t. Magn Reson Med. 2006;55:506–513 [DOI] [PubMed] [Google Scholar]
- 41.Huesa C, Millan JL, van ‘t Hof RJ, MacRae VE. A new method for the quantification of aortic calcification by three-dimensional micro-computed tomography. Int J Mol Med. 2013;32:1047–1050 [DOI] [PubMed] [Google Scholar]
- 42.O’Rourke C, Shelton G, Hutcheson JD, Burke MF, Martyn T, Thayer TE, et al. Calcification of vascular smooth muscle cells and imaging of aortic calcification and inflammation. J Vis Exp. 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Derwall M, Malhotra R, Lai CS, Beppu Y, Aikawa E, Seehra JS, et al. Inhibition of bone morphogenetic protein signaling reduces vascular calcification and atherosclerosis. Arteriosclerosis, thrombosis, and vascular biology. 2012;32:613–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Malhotra R, Wunderer F, Barnes HJ, Bagchi A, Buswell MD, O’Rourke CD, et al. Hepcidin deficiency protects against atherosclerosis. Arteriosclerosis, thrombosis, and vascular biology. 2019;39:178–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vaser R, Adusumalli S, Leng SN, Sikic M, Ng PC. Sift missense predictions for genomes. Nat Protoc. 2016;11:1–9 [DOI] [PubMed] [Google Scholar]
- 47.Raaz U, Schellinger IN, Chernogubova E, Warnecke C, Kayama Y, Penov K, et al. Transcription factor runx2 promotes aortic fibrosis and stiffness in type 2 diabetes mellitus. Circ Res. 2015;117:513–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li Q, Jiang Q, Schurgers LJ, Uitto J. Pseudoxanthoma elasticum: Reduced gamma-glutamyl carboxylation of matrix gla protein in a mouse model (abcc6−/−). Biochem Biophys Res Commun. 2007;364:208–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vanakker OM, Martin L, Schurgers LJ, Quaglino D, Costrop L, Vermeer C, et al. Low serum vitamin k in pxe results in defective carboxylation of mineralization inhibitors similar to the ggcx mutations in the pxe-like syndrome. Lab Invest. 2010;90:895–905 [DOI] [PubMed] [Google Scholar]
- 50.Ueland T, Dahl CP, Gullestad L, Aakhus S, Broch K, Skardal R, et al. Circulating levels of non-phosphorylated undercarboxylated matrix gla protein are associated with disease severity in patients with chronic heart failure. Clin Sci (Lond). 2011;121:119–127 [DOI] [PubMed] [Google Scholar]
- 51.Roumeliotis S, Dounousi E, Eleftheriadis T, Liakopoulos V. Association of the inactive circulating matrix gla protein with vitamin k intake, calcification, mortality, and cardiovascular disease: A review. Int J Mol Sci. 2019;20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cranenburg EC, Brandenburg VM, Vermeer C, Stenger M, Muhlenbruch G, Mahnken AH, et al. Uncarboxylated matrix gla protein (ucmgp) is associated with coronary artery calcification in haemodialysis patients. Thromb Haemost. 2009;101:359–366 [PubMed] [Google Scholar]
- 53.Delanaye P, Krzesinski JM, Warling X, Moonen M, Smelten N, Medart L, et al. Dephosphorylated-uncarboxylated matrix gla protein concentration is predictive of vitamin k status and is correlated with vascular calcification in a cohort of hemodialysis patients. BMC Nephrol. 2014;15:145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schurgers LJ, Barreto DV, Barreto FC, Liabeuf S, Renard C, Magdeleyns EJ, et al. The circulating inactive form of matrix gla protein is a surrogate marker for vascular calcification in chronic kidney disease: A preliminary report. Clin J Am Soc Nephrol. 2010;5:568–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schurgers LJ, Teunissen KJ, Knapen MH, Kwaijtaal M, van Diest R, Appels A, et al. Novel conformation-specific antibodies against matrix gamma-carboxyglutamic acid (gla) protein: Undercarboxylated matrix gla protein as marker for vascular calcification. Arteriosclerosis, thrombosis, and vascular biology. 2005;25:1629–1633 [DOI] [PubMed] [Google Scholar]
- 56.Dalmeijer GW, van der Schouw YT, Magdeleyns EJ, Vermeer C, Verschuren WM, Boer JM, et al. Matrix gla protein species and risk of cardiovascular events in type 2 diabetic patients. Diabetes Care. 2013;36:3766–3771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chirinos JA, Sardana M, Syed AA, Koppula MR, Varakantam S, Vasim I, et al. Aldosterone, inactive matrix gla-protein, and large artery stiffness in hypertension. J Am Soc Hypertens. 2018;12:681–689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hashmath Z, Lee J, Gaddam S, Ansari B, Oldland G, Javaid K, et al. Vitamin k status, warfarin use, and arterial stiffness in heart failure. Hypertension. 2019;73:364–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sardana M, Vasim I, Varakantam S, Kewan U, Tariq A, Koppula MR, et al. Inactive matrix gla-protein and arterial stiffness in type 2 diabetes mellitus. Am J Hypertens. 2017;30:196–201 [DOI] [PubMed] [Google Scholar]
- 60.Wei FF, Thijs L, Cauwenberghs N, Yang WY, Zhang ZY, Yu CG, et al. Central hemodynamics in relation to circulating desphospho-uncarboxylated matrix gla protein: A population study. J Am Heart Assoc. 2019;8:e011960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schurgers LJ, Uitto J, Reutelingsperger CP. Vitamin k-dependent carboxylation of matrix gla-protein: A crucial switch to control ectopic mineralization. Trends in molecular medicine. 2013;19:217–226 [DOI] [PubMed] [Google Scholar]
- 62.Liu Y-P, Gu Y-M, Thijs L, Knapen MHJ, Salvi E, Citterio L, et al. Inactive matrix gla protein is causally related to adverse health outcomes: A mendelian randomization study in a flemish population. Hypertension. 2015;65:463–470 [DOI] [PubMed] [Google Scholar]
- 63.Relucenti M, Heyn R, Petruzziello L, Pugliese G, Taurino M, Familiari G. Detecting microcalcifications in atherosclerotic plaques by a simple trichromic staining method for epoxy embedded carotid endarterectomies. Eur J Histochem. 2010;54:e33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang Y, Osborne MT, Tung B, Li M, Li Y. Imaging cardiovascular calcification. J Am Heart Assoc. 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.CVaH Vik. Effect of menaquinone-7 (vitamin k2) on vascular elasticity in healthy subjects: Results from a one-year study. Vascular Diseases and Therapeutics. 2020;5:1–4 [Google Scholar]
- 66.Ishikawa J, Kario K, Matsui Y, Shibasaki S, Morinari M, Kaneda R, et al. Collagen metabolism in extracellular matrix may be involved in arterial stiffness in older hypertensive patients with left ventricular hypertrophy. Hypertens Res. 2005;28:995–1001 [DOI] [PubMed] [Google Scholar]
- 67.McNulty M, Mahmud A, Spiers P, Feely J. Collagen type-i degradation is related to arterial stiffness in hypertensive and normotensive subjects. J Hum Hypertens. 2006;20:867–873 [DOI] [PubMed] [Google Scholar]
- 68.Reddy YNV, Andersen MJ, Obokata M, Koepp KE, Kane GC, Melenovsky V, et al. Arterial stiffening with exercise in patients with heart failure and preserved ejection fraction. J Am Coll Cardiol. 2017;70:136–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Borlaug BA, Kass DA. Ventricular-vascular interaction in heart failure. Heart Fail Clin. 2008;4:23–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sheng K, Zhang P, Lin W, Cheng J, Li J, Chen J. Association of matrix gla protein gene (rs1800801, rs1800802, rs4236) polymorphism with vascular calcification and atherosclerotic disease: A meta-analysis. Sci Rep. 2017;7:8713. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.