

Abstract
Heart failure is the one of the leading causes of death in the United States. Heart failure is a complex syndrome caused by numerous diseases, including severe myocardial infarction (MI). MI occurs after an occlusion of a cardiac artery causing downstream ischemia. MI is followed by cardiac remodeling involving extensive remodeling and fibrosis, which, if the original insult is severe or prolonged, can ultimately progress into heart failure. There is no “cure” for heart failure because therapies to regenerate dead tissue are not yet available. Previous studies have shown that in both post-MI and post-ischemia-reperfusion (I/R) models of heart failure, administration of cortical bone stem cell (CBSC) treatment leads to a reduction in scar size and improved cardiac function. Our first study investigated the ability of mouse CBSC-derived exosomes (mCBSC-dEXO) to recapitulate mouse CBSCs (mCBSC) therapeutic effects in a 24-h post-I/R model. This study showed that injection of mCBSCs and mCBSC-dEXOs into the ischemic region of an infarct had a protective effect against I/R injury. mCBSC-dEXOs recapitulated the effects of CBSC treatment post-I/R, indicating exosomes are partly responsible for CBSC’s beneficial effects. To examine if exosomes decrease fibrotic activation, adult rat ventricular fibroblasts (ARVFs) and adult human cardiac fibroblasts (NHCFs) were treated with transforming growth factor β (TGFβ) to activate fibrotic signaling before treatment with mCBSC- and human CBSC (hCBSC)-dEXOs. hCBSC-dEXOs caused a 100-fold decrease in human fibroblast activation. To further understand the signaling mechanisms regulating the protective decrease in fibrosis, we performed RNA sequencing on the NHCFs after hCBSC-dEXO treatment. The group treated with both TGFβ and exosomes showed a decrease in small nucleolar RNA (snoRNA), known to be involved with ribosome stability.
NEW & NOTEWORTHY Our work is noteworthy due to the identification of factors within stem cell-derived exosomes (dEXOs) that alter fibroblast activation through the hereto-unknown mechanism of decreasing small nucleolar RNA (snoRNA) signaling within cardiac fibroblasts. The study also shows that the injection of stem cells or a stem-cell-derived exosome therapy at the onset of reperfusion elicits cardioprotection, emphasizing the importance of early treatment in the post-ischemia-reperfusion (I/R) wounded heart.
Listen to this article’s corresponding podcast at https://ajpheart.podbean.com/e/cortical-bone-stem-cells-effects-on-cardiac-wound-healing/.
Keywords: cardiac fibroblast, cortical bone stem cell, exosome, fibroblast activation, ribosome stability
INTRODUCTION
As of 2019, cardiovascular disease (CVD) is the largest contributor to mortality in the United States. The CVD umbrella contains numerous diseases such as hypertension, coronary artery disease (CAD), myocardial infarction (MI), and congenital heart failure (CHF). Each culminates in a decline in heart function leading to heart failure (HF) for which the final therapeutic option is heart transplantation, leading to a significant need for curative or preventative therapeutics (1).
Myocardial infarctions (MI) occur when blood flow is blocked within one of the vessels that would otherwise supply cellular nutrients to the heart, causing a cascade of tissue death downstream of the occluded area, resulting in an infarcted region of cardiac tissue (2). The immediate response to a myocardial infarction is referred to as the inflammatory phase of wound healing. This response is rapid, occurring at onset of the ischemic period and lasting for one to 3 days (3). During this phase, the cardiac tissue is hypoxia and undergoes alterations to the mechanical stretch of cardiac muscle (4). For this short period of time, the cardiac extracellular matrix (ECM) is composed of a highly dynamic matrix consisting of fibrin and fibronectin (4). Matrix metalloproteinase (MMP) activity and expression levels rise, as well as NF-κB and Toll-like receptor signaling (5). With or without reperfusion, the post-MI wound healing process is complex, ultimately leading to the formation of scar tissue at the site of the infarct, through a process termed fibrosis (4). At the level of the ECM, there is an increase in the synthesis of collagens (in particular collagen-III) and laminin. There is also an increase in the synthesis of adhesion proteins and matricellular proteins (6). The signaling molecules that are involved with these changes involved decreased expression of inflammatory mediators such as angiotensin-II, endothelin-1, fibroblast growth factor, platelet-derived growth factor (PDGF), tumor growth factor β1 (TGFβ1), tumor growth factor β2 (TGFβ2), and interleukin-10 (7). Over time, the scarred myocardium, which is necessary for wall stabilization, can lead to diastolic filling defects, decreased myocardial contractility, and impaired electrical conductivity (8). These changes lead to decreases in the heart’s pumping function and can progress to heart failure.
In a clinical setting, rescue of a patient undergoing an MI involves opening of the occluded vessel allowing reperfusion of the oxygen-starved downstream tissue (9). Although this rescues portions of the damaged heart and can save a patient’s life, reperfusion leads to a separate disease phenotype called ischemia-reperfusion injury (I/R), which causes myocyte death by both necrosis and apoptosis (10). Thus, research into therapeutics that can alleviate tissue death after I/R is an essential field.
Recent studies have shown that specific stem-cell therapies can lead to improved post-MI or -I/R cardiac function and decreases in scar size (11). Unfortunately, these are not curative therapeutics due to mitigating factors including low engraftment rate of stem cells, a potential for hostile immune response in the treated animal, tumorigenicity (12, 13) (increasing the danger for off-target tumor growth) (14, 15), and failure of stem cells to regenerate new tissue. Fortunately, the beneficial effects of stem cells have been shown to be regulated via paracrine signaling factors, one of which is the release of stem cell-derived exosomes (dEXO) into the surrounding tissue (12, 13, 16–19). Furthermore, studies have shown that the long-lasting effects of dEXOs is due to the delivery of miRNA and mRNA (17, 20) to target cells, leading to alterations in cell signaling within recipient cells. In the heart, cardiomyocyte-derived exosomes contain miRNAs that cause apoptotic signaling cascades in neighboring cardiomyocytes (21), as well as miRNAs that promote fibroblast proliferation (22).
Our group has previously shown that bone-derived stem cell, termed a cortical bone stem cell (CBSC), when administered directly into the post-MI border zone (in both mouse and pig models) causes to a significant decrease in scar size and improved heart structure and function (23). In both animal models, CBSC treatment reduced myocardial scar size, increased angiogenesis within the border zone of the infarct, and improved cardiac function (24, 25). The mechanism by which CBSCs instigate a protective effect on the post-I/R cardiac remodeling process is the major question in this study. Herein, we explore the hypothesis that the release of exosomes from CBSCs leads to alterations in fibroblast signaling and reduces post-I/R scar size.
METHODS
In Vivo Experiments
Murine myocardial I/R injury.
Animal procedures were approved by the Temple University School of Medicine Institutional Animal Care and Use Committee. Male C57BL/6J (Jackson Stock No. 000664) mice (13 wk old; Jackson Laboratories) were subjected to baseline echocardiography (echo) 1 wk before surgery to confirm normal cardiac function. Male mice were used for these studies.
Before surgery, mice were anesthetized with ketamine (50 mg/kg) and xylazine (8 mg/kg) and received injections of 0.2 mL saline and heparin (200 U/kg). Mice were intubated with a PE-60 tubing and ventilated (Harvard Apparatus Mini-Vent Model 845) with 100% oxygen. A 3-cm incision was made along the skin, and a sternotomy was performed between ribs 2 and 3 using a thermal cautery unit (Geiger). The left coronary artery (LCA) was ligated, and a 3-mm piece of PE-10 tubing was placed between the suture and the heart on top of the LCA. After 45 m of ischemia, the suture and PE-10 tubing were removed to allow the heart to fully reperfuse. Reperfusion was visually confirmed. Immediately after the heart began to reperfuse, four 10 µL intramuscular injections [1× phosphate-buffered saline (PBS), CBSCs, CBSC-dEXOs] were given around the infarct border zone. The chest wall and skin incisions were closed. Following the completion of surgery, mice were placed in a recovery cage that was heated and received subcutaneous injections of 0.3 mL sterile saline and buprenorphine (0.1 mg/kg). Reperfusion occurred for 24 h. Following reperfusion, mice were anesthetized and intubated as previously described. Blood was collected via the inferior vena cava (IVC) for cardiac troponin measurements post-I/R. The left carotid artery was cannulated with a catheter containing 2% Evan’s blue dye (EBD). After cannulation, the LCA was religated and EBD (1 mL) was perfused through the heart. The heart was rapidly excised, washed with 1× PBS, and cut into five 1-mm cross-sectional slices by a McIlwin Tissue Chopper. Heart slices were incubated at 37°C with 1% 2-3-5-triphenyltetrazolium chloride (TTC) to differentiate between the regions of infarction and area at risk (AAR). The individual heart slices were imaged, weighed, quantified, and labeled as the left ventricle (LV), AAR, and infarcted regions. All measurements were performed in a blinded manner.
In Vitro Experiments
CBSC isolation and culture.
Mouse and human CBSCs were isolated as previously described (24, 25). Institutional Review Board (IRB) approval for human CBSC isolation: Temple University, IRB No. 23594: Human Bone Samples from Orthopedic Surgery to Identify Novel Stem Cell Population. CBSCs are cultured in a proprietary CBSC growth media, licensed to the authors by MyocardTherapeutics, LLC.
Exosome isolation.
To isolate exosomes from stem cells, mCBSCs (passage 10) and hCBSCs (passage 22) were cultured in 10 cm2 dishes for 2 days and allowed to grow to ∼90% confluency. Both cell types, mCBSCs and hCBSCs, were then passaged and replated in forty 150 cm2 flasks in normal CBSC growth media for 24 h at a density of 200,000 cells/flask. Following this initial 24-h growth period, the normal CBSC growth media was removed and replaced with low-exosome CBSC growth media (normal CBSC growth media with added System Biosciences EXO-FBS-250A-1). Both cell types were cultured in the low-exosome growth media, which causes secretion of exosomes into the supernatant. After 4 days, the supernatant containing exosomes was collected and filtered through a 0.22-µM media sterilizer filter (Corning, 430517). The filtered supernatant was concentrated in 100,000 media concentrator conicals via centrifugation at >1,000 g for 30 min. After centrifugation, the concentrate was collected and pipetted into 25 mL glass ultracentrifugation conicals. To remove any cellular debris, the concentrate was ultracentrifuged at 20,000 g for 30 min. Following the removal of cellular debris, the media was collected and slowly layered on top of 4 mL of sucrose gradient (pH = 7.4). The media + sucrose gradient was ultracentrifuged at 100,000 g for 1 h to pull the exosomes into the sucrose gradient. Following the ultracentrifugation, the media above the exosome layer was removed and 1× PBS was added to the conical at the equal volume of the previously removed media. The remaining solution within the conicals was then mixed to dissipate the gradient effect of the remaining sucrose. The conicals were then ultracentrifuged again at 100,000 g for 1 h to precipitate out the exosomes into a pellet. The exosome protein concentration was measured via Bradford Concentration Assay (BCA), and the exosome particle size and concentration were measured via Nanosight assay.
qPCR.
Adult mouse aortic fibroblasts (MAAFs) were provided by Dr. Raj Kishore and cultured in six-well plates in fibroblast growth medium. Cells were plated in six-well plates at ∼40,000 cells/well. Cells were treated with either 72 h CM from mCBSCs, 72 h CM from MAAFs, 2.5 × 108 particles of mCBSC-dEXOs, and base growth medium for 72 h, at which point, the cells were lysed and RNA was collected with Qiagen RNEasy RNA Isolation kit. RNA was quantified using Nanodrop, and then cDNA was constructed using (Qiagen RNeasy Micro Kit, Cat. No. 74004). qPCR was run using SYBR green (Qiagen, 204057). Primers for fibrosis and angiogenesis-related genes were purchased from Thermo Fisher Scientific, as listed in Table 1.
Table 1.
PCR primer sequences
| Oligo Name | Sequence (5′ to 3′) |
|---|---|
| mMMP2 | |
| Forward | CCCCATGTGTCTTCCCCTTC |
| Reverse | GTGTAGATCGGGGCCATCAG |
| mCol1A1 | |
| Forward | TTCTCCTGGCAAAGACGGAC |
| Reverse | CCATCGGTCATGCTCTCTCC |
| mCol3A1 | |
| Forward | GAGGAATGGGTGGCTATCCG |
| Reverse | TTGCGTCCATCAAAGCCTCT |
| mVEGFA | |
| Forward | CCCACGTCAGAGAGCAACAT |
| Reverse | GGAATTAGACAGCAGCGGCA |
| GAPDH | |
| Forward | CCTGGAGAAACCTGCCAAGTATG |
| Reverse | AGAGTGGGAGTTGCTGTTGAAGTC |
Fibroblast activation assay.
Primary ARVFs were isolated via the “chop” method (26). Alternatively, human cardiac fibroblasts (Lonza CC2904) were purchased and used for the human cardiac fibroblast activation assay. P1 fibroblasts were plated at ∼1,800 cells/well in a 96-well plate and grown overnight in fibroblast growth media. Twenty-four hours later, the media was changed to ∼1% FBS media and the cells were starved overnight. The following day, cells received treatments of 1x, 10x, 100x, and 1,000x mCBSC or hCBSC-dEXOs ± TGFβ (where x = 2,000 exosome particles/cell treated). After 72 h, the cells were fixed in 4% paraformaldehyde and then stained with antibodies for αSMA (Sigma A5228, 1:2,000), fibronectin (Abcam ab1943954, 1:1,000), and DAPI (EMD Millipore 268298-10MG) and secondary antibodies (Thermo Fisher Scientific A28175, goat anti-mouse-488, 1:400, A27040, goat anti-rabbit-647, 1:400, and 62249, Hoechst 33342, 1:2,000). The cells were then imaged with CellInsight microscope CX7, and the mean fluorescent intensity measured via CellInsight software. Antibodies were validated by comparing αSMA and fibronectin negative fibroblasts to positive myofibroblasts.
Long RNA sequencing.
Primary human ventricular cardiac fibroblasts (NHCFs) were purchased from Lonza (Lonza CC2904) and cultured in Lonza fibroblast medium (Lonza CC-4526) plus fetal bovine serum (FBS; Gibco 10439024), fibroblast growth factor (Peprotech AF-100-18B), insulin-transferrin-selenium (ITS; Lonza Biowhittaker 17838Z), and Penicillin streptomycin glutamine (PSG; Gibco 10378016). Passage 1 cells were cultured in 10 cm dishes and then passaged and plated at 200,000 cells per well in six-well plates. Cells were allowed to adhere and grow for 24 h in Lonza + 10% FBS media and then were serum starved overnight in Lonza + 1% FBS. Following the overnight serum starvation, cell medium was aspirated and replaced with Lonza + 1% FBS media and cells were treated with hCBSC-dEXOs ± 10 ng/mL TGFβ (Invivogen rcyc-htgfb1). Following 72 h of incubation at 37°C, cells were lysed and RNA was isolated using the Qiagen miRNeasy isolation kit (Qiagen RNeasy Micro Kit 74004). PolyA mRNAs were enriched via poly-T RNA purification beads using the TrueSeq stranded mRNA library prep kit. Hiseq rapid SR cluster kit was used to amplify enriched mRNAs before multiplexing and reading via the HiSeq rapid SBS kit. Single-end 75-bp fragments were read at a depth of ∼40 M reads per sample. Sequencing was performed on Illumina HiSeq2500 sequencer using Illumina sequencing kits. RNA transcripts were aligned to the human genome (hg38) using HISAT2 v2.1.0 before quantification using HTSeq v0.11.2. DESeq2 v1.22.2 was used to perform differential expression analysis between the following groups: 1) normal human ventricular cardiac fibroblasts (NHCFs) treated with vehicle and NHCFs treated with TGFβ, 2) NHCFs treated with vehicle and NHCFs treated with hCBSCs, and 3) NHCFs treated with TGFβ and NHCFs treated with TGFβ and hCBSCs. Significantly differentially expressed genes met the criteria of a fold change (FC) ≥|1.5| and false discovery rate (FDR) of ≤0.05. Heatmaps were created using R v3.5.3 package pHeatmap, v1.0.12. Gene ontology (GO) analysis was performed using R package clusterProfiler v3.10.1 and visualized using Prism 9. EXOSeq data submission to the GEO repository is in process (RNASeq GEO ID: GSE174490).
The hg38 genome contains annotations for the full transcripts of mRNA, miRNA, and snoRNA allowing analysis of all transcript types discussed in this paper. Analysis of each transcript type followed the same sequence of events as described above.
Exosome RNA sequencing.
To isolate exosomes from stem cells for sequencing, mCBSCs (passage 10) and hCBSCs (passage 22) were cultured in 10 cm2 dishes for 2 days and allowed to grow to ∼90% confluency. Both cell types, mCBSCs and hCBSCs, were then passaged and replated in forty 150-cm2 flasks in normal CBSC growth media for 24 h at a density of 200,000 cells/flask.
Transcripts per kilobase million (TPM) was calculated for each RNA transcript as follows:
Reads per kilobase (RPK) = Transcript counts/length of transcript in kilobase
Per million scaling factor = Sum of all RPKs per sample/1,000,000
TPM = Individual RPK/ million scaling factors
Transcripts with a TPM greater than or equal to 100 were selected for further analysis. Gene ontology (GO) analysis was performed using R package clusterProfiler, v3.10.1 and visualized using Prism 9.
miRNA target prediction and GO analysis.
miRDB was used to predict targets for each miRNA identified in the EXOSeq as significantly enriched (TPM ≥ 100). Human and mouse miRNAs were queried against miRDB’s species-specific databases. All targets with a score of 100 were selected for further analysis. For miRNAs without target scores of 100, the single highest-ranking target was chosen for further analysis. GO analysis was performed using R package clusterProfiler, v3.10.1.
Statistical Analysis
Data are represented as means ± SD. Two-sided testing was used for all statistical tests. A P value of ≤0.05 was used to determine significance for all statistical tests. The distributions of all continuous variables were tested for normality assumptions using GraphPad Prism. Comparisons for data with a single measurement were performed using the unpaired t test or the Mann–Whitney U-test depending on the distribution of the data. One sample t tests were used for the experimental data in Fig. 3, given the low n number of samples in the vehicle comparison group. Comparisons for data with repeated measurements were performed using one-way ANOVA, followed by Tukey’s multiple comparisons test using the GraphPad Prism software. All statistics analysis was performed with GraphPad Prism (Version 8.0d, GraphPad, La Jolla, CA).
Figure 3.

mCBSC-dEXOs decrease activation of profibrotic genes in murine adult aortic fibroblasts (MAAFs). A–D: qPCR quantification of MAAFs fibroblasts following 72 h treatment with base growth medium, MAAF-CM, mCBSC-CM, and mCBSC-dEXOs (2.5 × 108 particles/cell treated). All data are normalized to GAPDH. A: mCBSC exosome treatment leads to 0.5-fold decrease in Col1A1 expression in murine endothelial fibroblasts (MEF). B: Col3A1 is reduced by 0.5 fold by MEF-CM treatment, to 0.25 fold by mCBSC-CM treatment, and to 0.20 fold by mCBSC exosome treatment. C: MMP2 is increased in MAAFs treatment with MAAF CM, but remains consistent between the base growth medium and the mCBSC-CM and mCBSC-dEXO treatments. D: VEGFA is increased in the MAAF-CM-treated MAAFs but remains unchanged in the MAAFs following mCBSC-CM and mCBSC-dEXO treatment (base growth medium: n = 2; MAAF-CM: n = 3; mCBSC-CM: n = 3; mCBSC-dEXO: n = 3; one-sample t tests were performed). *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001. CBSCs, cortical bone stem cells; MAAF-CM, MAAF conditioned medium; mCBSC, mouse CBSCs; mCBSC-CM, mCBSC conditioned medium; mCBSC-dEXO, mouse CBSC-derived exosomes; MMP2, Matrix metalloproteinase 2.
RESULTS
Isolation of Human and Mouse Cortical Bone Stem Cell dEXOs (hCBSC-dEXO; mCBSC-dEXO)
CBSCs were isolated from both mouse and human primary bone cells as described previously (24, 25). Exosomes were isolated from both mCBSCs and hCBSCs by collecting the cell culture media and ultracentrifuging, as described and illustrated in Fig. 1A. Briefly, the CBSCs are cultured in exosome-free CBSC growth media for 2 days to reach confluency. The supernatant, now considered a conditioned media (CM) containing the full CBSC secretome, is collected before a series of ultracentrifugation steps to isolate and precipitate exosomes. After isolation, the appropriate morphological phenotypes of mCBSCs (Fig. 1B) and hCBSCs (Fig. 1E) were visually confirmed. Subsequently, vesicle collection was visually identified (Fig. 1, C and F) before confirmation of concentrated isolation of specific particle sizes (Fig. 1, D and G) via Nanosight imaging.
Figure 1.
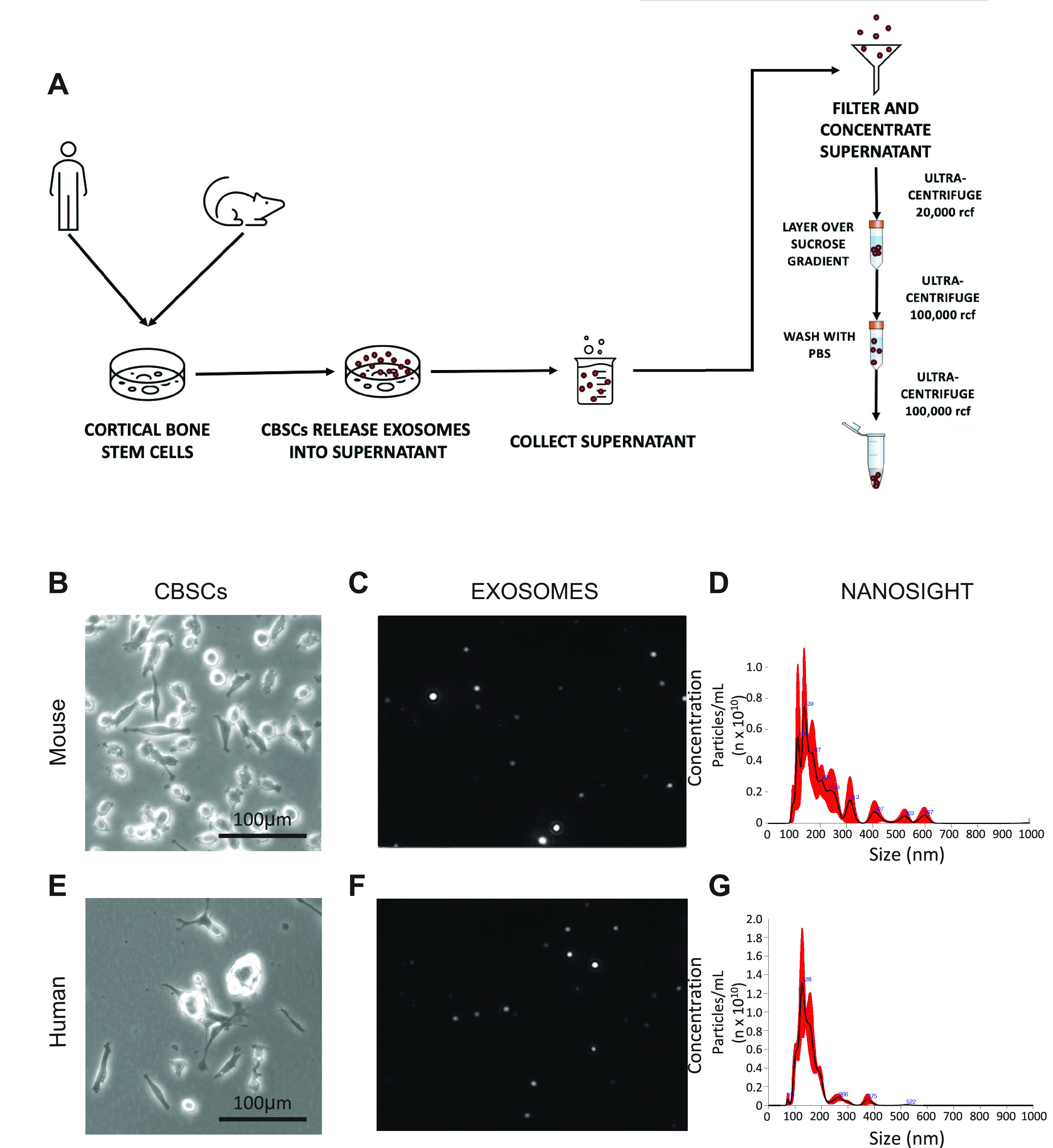
Isolation of human and mouse cortical bone stem-cell dEXOs. A: illustration of hCBSC and mCBSC-dEXO as described in methods. B: ×10 magnification of mCBSCs in vitro. C: representative Nanosight still of mCBSC-derived particles. D: Nanosight quantification of mCBSCs size and concentration. E: ×10 magnification of p22 hCBSCs in vitro. F: representative Nanosight still of hCBSC-derived particles. G: Nanosight quantification of hCBSCs size and concentration.
mCBSCs and mCBSC-dEXOs Reduce Infarct Size in a Murine Model of I/R
Previous CBSC studies in our laboratory have shown that CBSC treatment causes a decrease in fibrosis in the post-MI mouse heart and post-I/R pig heart (23–25, 27, 28). Although we have not seen the CBSCs reduce the immediate ischemic injury in our past studies, other laboratories investigating cardiosphere-derived cells (CDC) observed cardioprotection in rats and pigs when administering CDC-dEXOs post-IR (20). Additionally, one of the prevailing theories in the stem cell field implicates exosome release as a method of paracrine signaling, conferring the cells protective mechanism (29). These data led to our design of a murine in vivo experiment with two goals. First, to understand if CBSCs are protective because they reduce fibrosis over time or because they reduce wound size leading to decreased long-term fibrosis due to a smaller initial injury. Second, given studies showing secreted vesicles from stem cells exhibiting the protective effects of stem cells, are CBSC dEXOs part of the mechanism by which stem cells are protective?
To examine these questions, we performed a 24-h I/R murine study using mCBSCs and mCBSC-dEXOs injected immediately on reperfusion. To begin, mice were subjected to echocardiography, to confirm normal baseline cardiac function, 1 wk before surgery. Before the myocardial infarction, the mice all showed cardiac ejection fraction >65%. Mice first underwent 45 m of ischemia after which the ligation was removed and mice were immediately injected with saline, 100,000 mCBSCs, or 2.5 × 108 particles of mCBSC-dEXOs. There was no difference in AAR between the three groups, but both mCBSC- and mCBSC-dEXO-treated mice had statistically significant reductions in infarct size (Fig. 2B, quantified Fig. 2C) as compared with vehicle-treated mice. These data indicate that the injection of the mCBSC-dEXOs and mCBSCs confers cardioprotection when administered in the border zone immediately following reperfusion.
Figure 2.
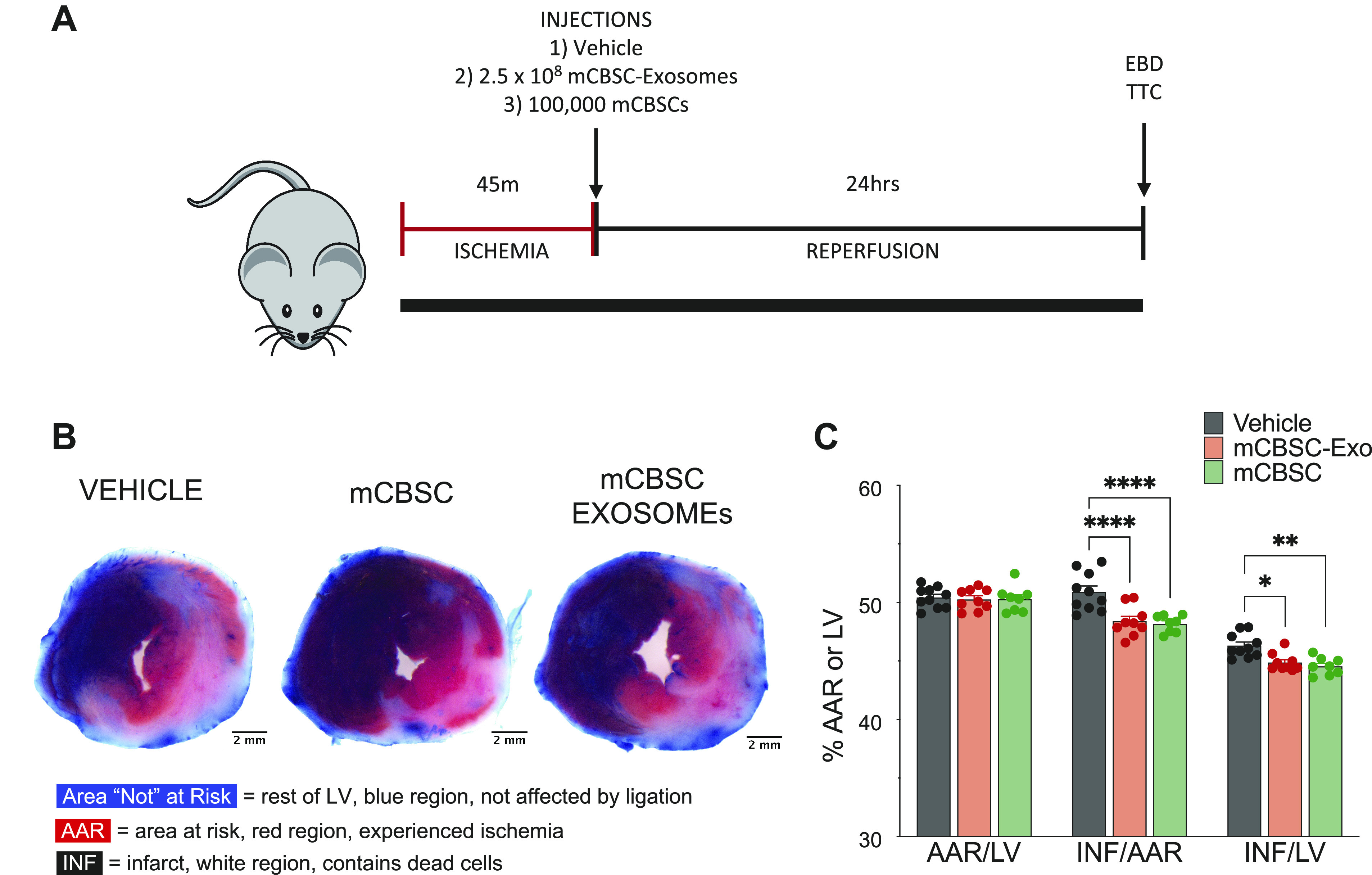
CBSCs and CBSC-dEXOs reduce infarct size in a murine model of I/R. A: illustration of 24-h myocardial I/R injury study design as described in methods. B: representative images of 1 mm cross sections of hearts following EBD and TTC staining. C: quantification AAR (red) vs. LV (blue) area, indicating consistency of injury in each animal and treatment. Quantification of infarct size (white) per AAR and infarct size per LV indicating a significant decrease in infarct size in the mCBSC-treated mice and mCBSC-dEXO-treated mice (vehicle: n = 10; mCBSC-Exo: n = 9; mCBSC-treated mice: n = 8; all males; two-way ANOVA tests were performed). *P ≤ 0.05; **P ≤ 0.01; ****P ≤ 0.0001. AAR, area at risk; CBSCs, cortical bone stem cells; CBSC-dEXOs, CBSC-derived exosomes; EBD, Evan’s blue dye; I/R, ischemia-reperfusion; LV, left ventricle; mCBSC, mouse CBSCs; mCBSC-dEXO, mouse CBSC-derived exosomes; TTC, 2-3-5-triphenyltetrazolium chloride.
Interestingly, both mCBSCs and their dEXOs exhibited a similar level of cardioprotection in the post-I/R heart, suggesting that CBSC-dEXOs may be responsible for the therapeutic effects of CBSCs. This led to our desire to understand if mCBSC-dEXOs could recapitulate the signaling effects of CBSCs on wound healing.
mCBSC-dEXOs Decrease Activation of Profibrotic Genes in Murine Adult Aortic Fibroblasts
There are several ways cells have been shown to signal through paracrine mechanisms. Cells release into media their full secretome, containing chemokines, cytokines, vesicles, and exosomes (30). To examine which paracrine signaling factors contribute to the mechanism by which CBSCs aid in wound healing and reduce fibrotic signaling, we examined conditioned media, containing all of these paracrine factors, and CBSC-dEXOs, to see whether the CBSC-dEXOs could induce the same signaling effects as the full secretome of CBSCs.
For 72 h, murine adult aortic fibroblasts (MAAFs) were treated with post-72 h MAAF-CM, mCBSC-CM, mCBSC-dEXOs, and base growth media as a vehicle treatment and RNA was isolated for qPCR analysis. Expression of profibrotic genes Collagen Type I, and III, MMP2, and VEGFA were decreased in fibroblasts after treatment with mCBCS-CM and mCBSC-dEXO. The decrease in expression of profibrotic genes was greater in the mCBSC-dEXO group than the mCBSC group for Col1A1 (Fig. 3A), Col3A1 (Fig. 3B), and MMP2 (Fig. 3C), though there was no statistical difference between the effect of mCBSC-CM and mCBSC-dEXOs on MAAF production of VEGFA (Fig. 3D). The vehicle-treated cells have an n of 2, indicating that more rigorous examination of these effects will be warranted in future studies. Still, these results together suggest that mCBSC-dEXOs are capable of recapitulating the antifibrotic effects of mCBSCs alone.
mCBSC-dEXOs Reduce ARVF Activation
Primary ARVFs were cultured in 96-well plates in normal serum media at 1,800 cells per well. After 24 h, the media was changed to a low-serum media overnight. In the morning, the media was replaced with low-serum media ± TGFβ (50 ng/mL) and the cells were treated with increasing concentrations of mCBSC-dEXOs, from 2,000 exosome particles per cell treated up to 2,000,000 exosome particles per cell treated (Supplemental Fig. S1A; https://doi.org/10.6084/m9.figshare.15101379.v1). After 72 h, the cells were fixed, stained, and imaged for αSMA and fibronectin (Fig. 4A) . There was a dose-dependent reduction of αSMA expression in the ARVFs (Fig. 4B), supporting the idea that mCBSC-dEXOs reduce fibroblast activation. There was a smaller but still significant reduction in fibronectin (Fig. 4C), implying there is less extracellular matrix (ECM) deposition by the ARVFs in response to mCBSC-dEXO treatment.
Figure 4.
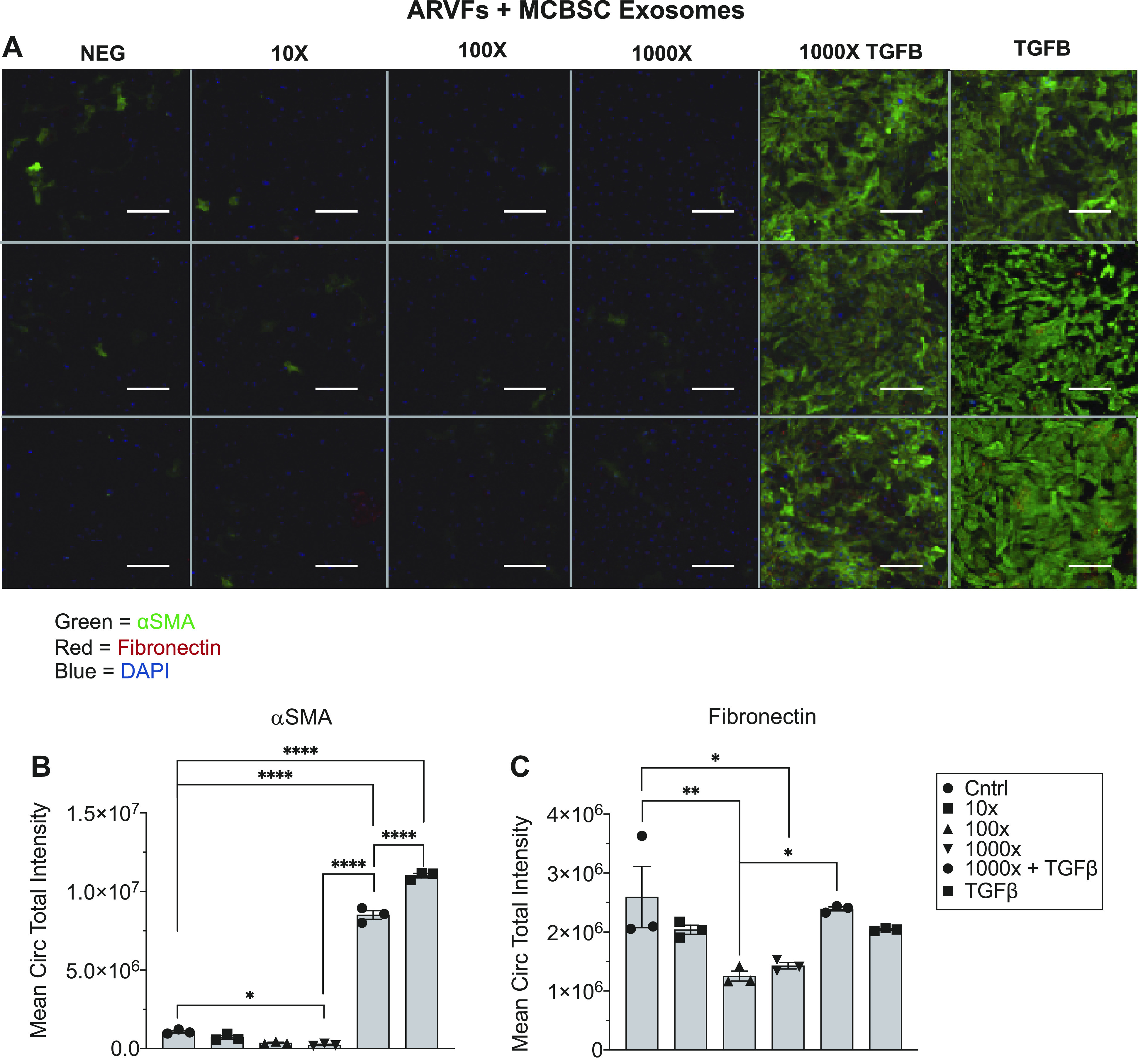
mCBSC-dEXOs treatment of ARVF reduces fibrotic activation marker expression in the presence of TGFβ. A: representative image of plate of ARVFs treated with mCBSC-dEXOs. B: quantification of αSMA expression. C: quantification of fibronectin expression (n = 3 for all groups; ordinary one-way ANOVA tests were performed, followed by Tukey’s multiple comparison test). *P ≤ 0.05; **P ≤ 0.01; ****P ≤ 0.0001. (Scale bar = 100 μM). ARVFs, adult rat ventricular fibroblasts; CBSCs, cortical bone stem cells; mCBSC-dEXO, mouse CBSC-derived exosomes; TGFβ, transforming growth factor β.
hCBSC-dEXOs Reduce ARVF Activation
To examine if human exosomes had the same inhibitory potential, hCBSC-dEXOs were used to treat ARVFs as described above (Supplemental Fig. S1B). A dose-dependent reduction in αSMA expression (Fig. 5A, quantified Fig. 5B) and fibronectin (Fig. 5A, quantified Fig. 5C) was observed. These data support the idea that there is still less ECM deposition by the ARVFs following hCBSC-dEXO treatment, even though the exosomes are from a different species.
Figure 5.
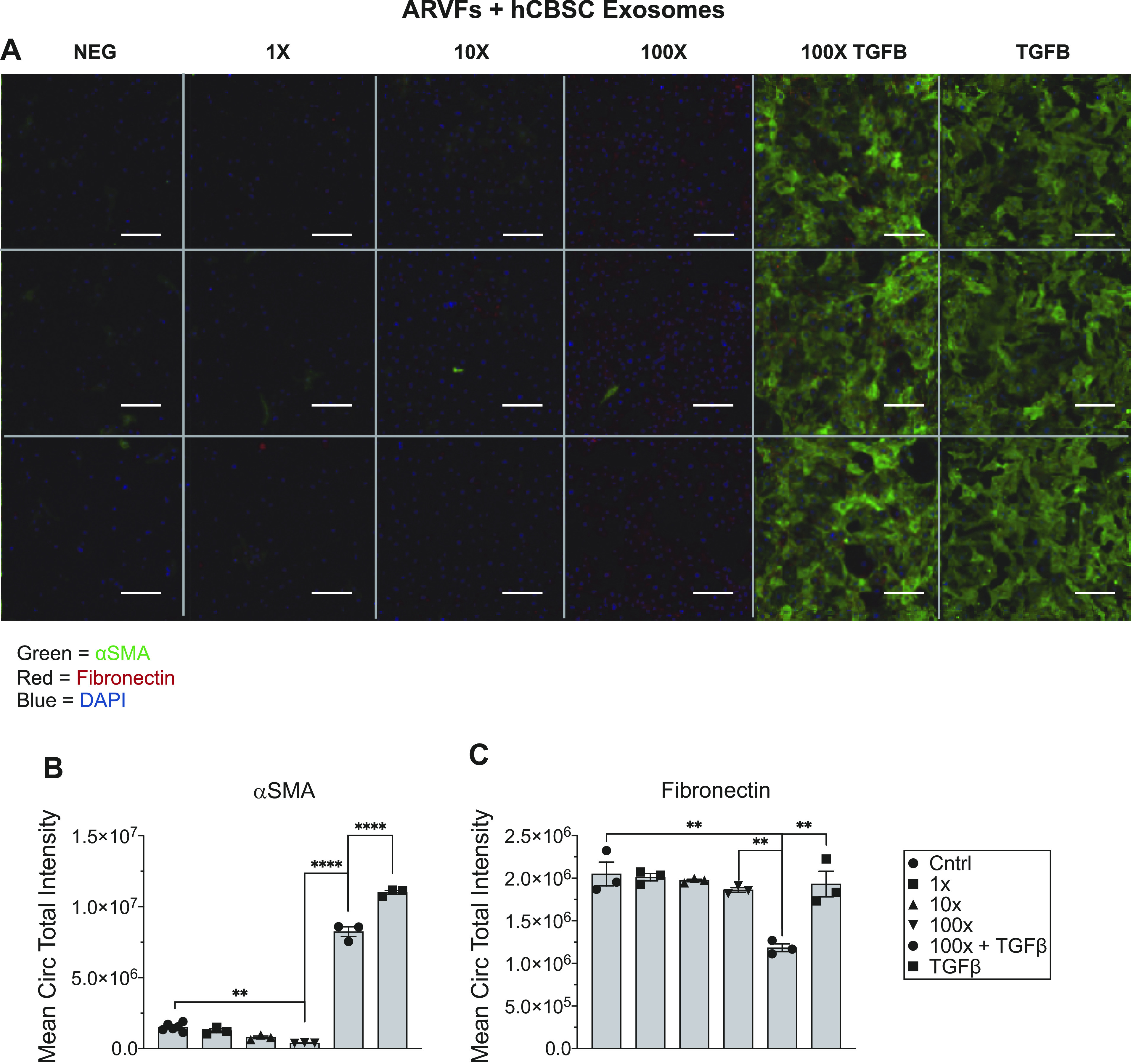
hCBSC-dEXOs treatment of ARVF reduces fibrotic activation marker expression in the presence of TGFβ. A: representative image of plate of ARVFs treated with hCBSC-dEXOs. B: quantification of αSMA expression (Control n = 6, for all other groups n = 3). C: quantification of fibronectin expression (n = 3 in all groups; ordinary one-way ANOVA tests were performed, followed by Tukey’s multiple comparison test). **P ≤ 0.01; ****P ≤ 0.0001. (Scale bar = 100 μM). ARVFs, adult rat ventricular fibroblasts; CBSCs, cortical bone stem cells; hCBSC-dEXOs, human CBSC-derived exosomes; TGFβ, transforming growth factor β.
hCBSC-dEXOs Reduce NHCF Activation
Next, primary NHCFs were cultured and treated with hCBSCs, as described above and illustrated in Supplemental Fig. S1C. In the NHCF + hCBSC-dEXOs condition, αSMA expression was reduced to a level similar to nonactivated fibroblasts, even post-TGFβ activation (Fig. 6A, quantified Fig. 6B). Fibronectin was also reduced in a dose-dependent fashion following hCBSC-dEXO treatment (Fig. 6A, quantified Fig. 6C). These finding support the idea that hCBSC-dEXOs have a significant antifibrotic effect by reducing fibroblast activation to myofibroblasts. Additionally, hCBSC-dEXOs were found to have a more profound inhibitory effect on fibrotic activation in NHCFs as compared with ARVFs, implying there is species-specific signaling occurring. One potential mechanism explaining how exosomes alter the phenotype of recipient cells is that exosomes contain miRNA that modify gene expression. To explore this idea, we investigated the RNA content of mouse and human CBSC-dEXOs.
Figure 6.
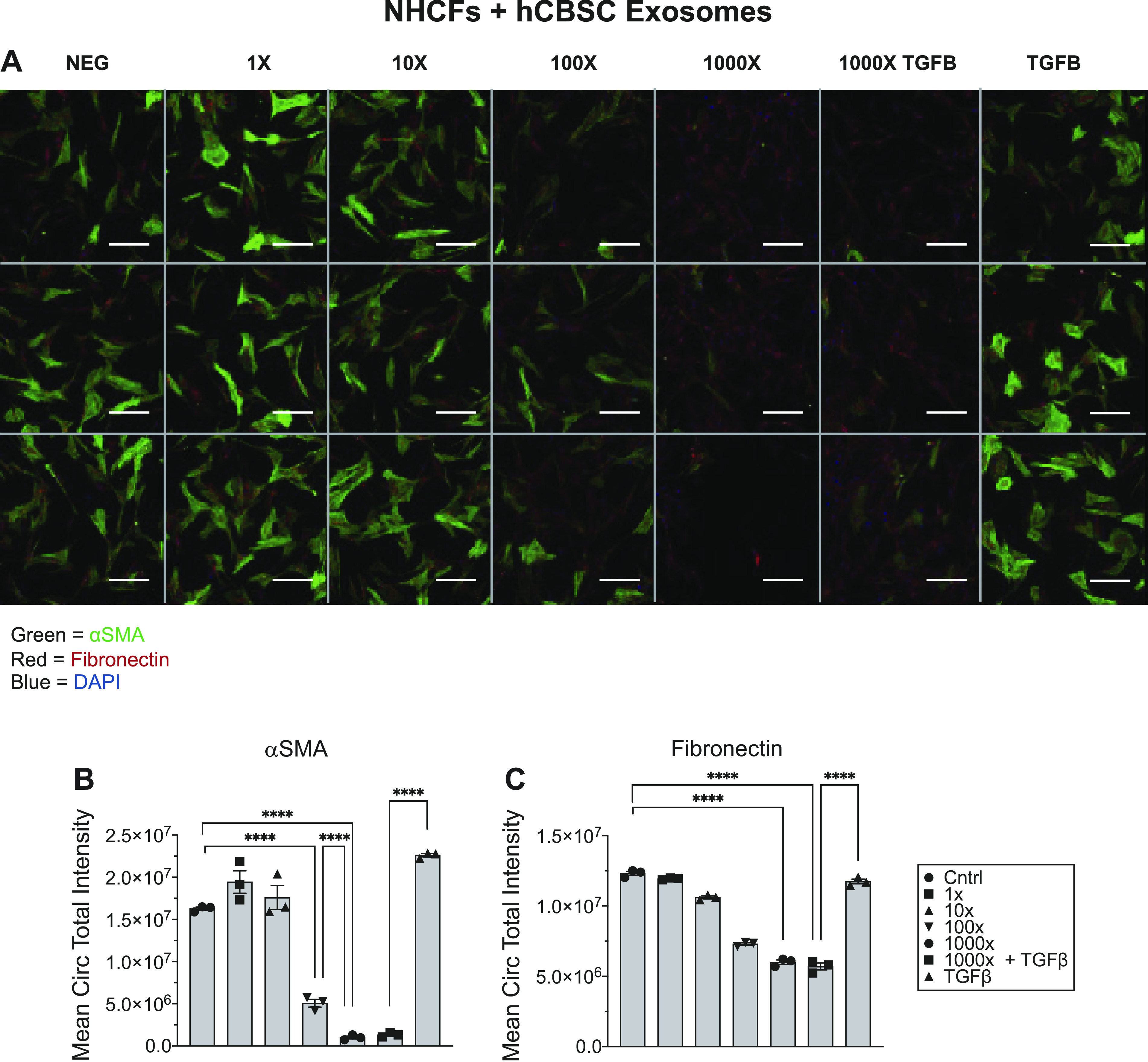
hCBSC-dEXOs treatment of NHCFs reduces fibrotic activation marker expression in the presence of TGFβ. A: representative image of plate of NHCFs treated with hCBSC-dEXOs. B: quantification of αSMA expression. C: quantification of fibronectin expression (n = 3 for each group; ordinary one-way ANOVA tests were performed, followed by Tukey’s multiple comparison test). ****P ≤ 0.0001. (Scale bar = 100 μM). CBSCs, cortical bone stem cells; hCBSC-dEXOs, human CBSC-derived exosomes; NHCFs, normal human ventricular cardiac fibroblasts; TGFβ, transforming growth factor β.
Comparison of Mouse CBSC-dEXOs with Human CBSC-dEXOs
To further explore both the mechanisms by which exosomes alter recipient cell phenotypes and the species-specific effect, we performed RNASeq on mCBSC-dEXOs and hCBSC-dEXOs to understand their respective RNA makeup. In terms of exosomal RNA content, the two species contain similar, but not identical, RNA components.
The mCBSC-dEXO RNA content consisted of 40% miRNA, 20% rRNA, 18% tRNA, 10% piRNA, 8% rfam, and 3% tRNA-like sequences (Fig. 7A). mCBSC-dEXO RNA with a TPM of greater than or equal to 100 was subsetted for gene ontological analysis of cellular components (Fig. 7C), molecular functions (Fig. 7E), and biological processes (Fig. 7G). GO indicated that these are involved in regulating gene expression, as evidenced by enrichment of the biological processes “positive regulation of gene expression” and “catabolic processes” (Fig. 7G), as well as enrichment of cellular component “ribonucleoprotein complex” (Fig. 7E).
Figure 7.
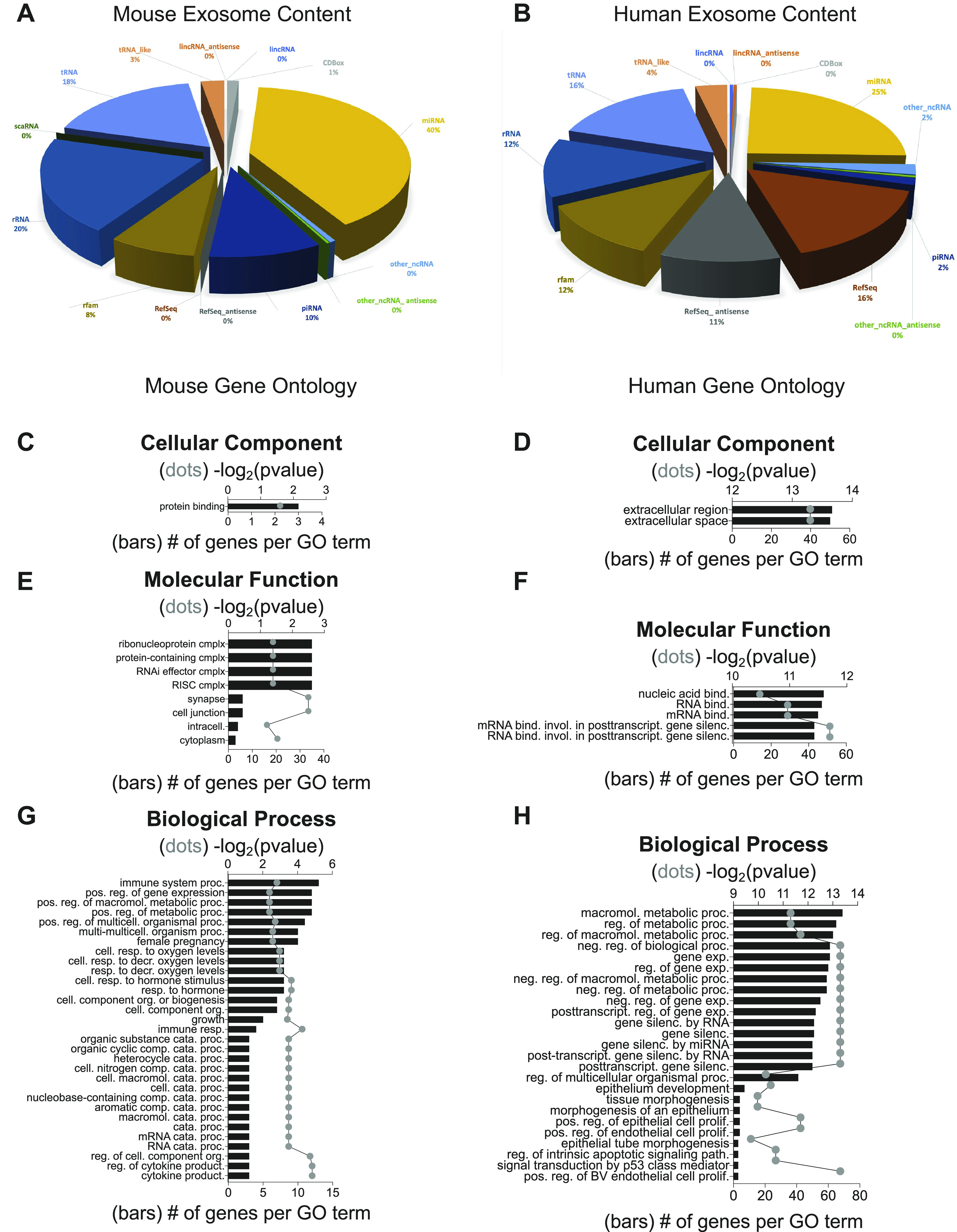
Exosomal RNA content from mCBSC-dEXO and hCBSC-dEXO differs in both RNA species percentages and gene ontology (GO) enrichment. Small RNASeq analysis of RNA content from mCBSC-dEXOs and hCBSC-dEXOs. A: pie chart of RNA species quantities in mCBSC-dEXOs. B: pie chart of RNA species quantities in hCBSC-dEXOs. Gene ontology for mCBSC-dEXO cellular component (C), hCBSC-dEXO cellular component (D), mCBSC-dEXO molecular function (E), hCBSC-dEXO molecular function (F), mCBSC-dEXO biological process (G), and hCBSC-dEXO biological process (n = 4 for each group; H). CBSCs, cortical bone stem cells; hCBSC-dEXO, human CBSC-derived exosome; mCBSC-dEXO, mouse CBSC-derived exosomes.
To understand how exactly the mouse exosome content could be driving phenotypic changes, we began by examining the targets of the miRNA contained within the mCBSC-dEXOs using miRDB prediction methods. Although numerous targets did appear (Supplemental Table S1; https://doi.org/10.6084/m9.figshare.15101415.v1), GO analysis did not result in significant enrichment, possibly due to the predictive nature of our search, which likely includes predicted targets which are not targets in vitro/vivo.
Next, we examine the human CBSC-dEXO RNA content and found it consisted of 25% miRNA, 16% refSeq RNA, 16% tRNA, 12% rRNA, 12% rfam, 11% RefSeq antisense sequence, 4% tRNA-like sequences, 2% piRNA, and 2% other_ncRNAs (Fig. 7B). We again subsetted the hCBSC-dEXO RNA to those with a TPM of greater than or equal to 100 and performed gene ontological analysis of cellular components (Fig. 7D), molecular function (Fig. 7F), and biological processes (Fig. 7H). Human exosomes contained RNA that primarily altered regulation of metabolic processes. Specifically, hCBSC-dEXO contained RNA important in regulating the molecular functions of “posttranscriptional gene silencing” and “RNA binding” (Fig. 7F) as well as numerous biological processes such as “negative regulation of gene expression” and “gene silencing” (Fig. 7H). The changes in the regulatory landscape could be driven by the miRNA and snoRNA found within the hCBSC-dEXOs, which are then delivered to recipient cells.
Again, to examine exosome signaling via its miRNA content, we found the miRNA targets using miRDB. Similarly, numerous targets appeared (Supplemental Table S2; https://doi.org/10.6084/m9.figshare.15101427.v1), but GO analysis did not result in significant enrichment. The dEXO target search did reveal that many of the predicted targets of the miRNA found within the exosomes would lead to negative regulation of the acute inflammatory response, which would be a direction for future investigations in vitro.
hCBSC-dEXOs Cause Downregulation of miRNA and snoRNA Related to Ribosome Stability and Protein Translation in Human CFs
To understand the interplay between CBSC-dEXOs signaling content and their recipient cells, we examined the RNA of NHCFs via RNASeq. RNASeq was performed on normal human cardiac fibroblasts (NHCFs) + vehicle (VEH), NHCFs + TGFβ, NHCFs + hCBSC-dEXOs (NHCF + EXO), and NHCFs + TGFβ + hCBSC-dEXOs (NHCF + TGFβ+EXO) to examine the effect of CBSC-dEXO signaling on fibroblast RNA content in each condition.
First, we determined the effects of TGFβ on fibroblast gene expression. Principal component analysis (PCA) showed distinct separation between the NHCF + VEH and the NHCF + TGFβ group indicating a change in the transcriptional profiles (Supplemental Fig. S2A; https://doi.org/10.6084/m9.figshare.15101385.v1). Volcano plot visualization shows there are 656 significantly downregulated genes and 479 upregulated genes in the TGFβ condition, as defined by a FC ≥ |1.5| and an FDR ≤ 0.05 (Supplemental Fig. S2C). A heatmap of the top 100 genes showed that genes such as TMEM26, PHOSPH1, and many SNORD family genes, involved in stabilizing protein translation, were upregulated, whereas FOXF2 and CDC25, along with many other genes associated with cell growth regulation, were downregulated (Supplemental Fig. S2B). Profibrotic genes examined in Fig. 3, Col1A1, MMP2, and VEGFA had a significant positive in fold change in the TGFβ condition. Gene Ontology (GO) analysis revealed increases in molecular functions associated with fibroblast activation such as “cytokine activation” and “receptor binding,” “growth factor activation,” and “hormone activation” (Supplemental Fig. S2D). Cellular component GO analysis revealed associations with changes in the “membrane” and “complex of collagen trimers” (Supplemental Fig. S2E) whereas enriched Biological Processes included the expected activation of the immune system as evidenced by “complement activation” and of the fibroblast gene program as evidenced by transcription factor “SMAD protein signaling transduction,” “RNA processing,” “protein processing,” and “protein maturation.”
Treatment of NHCFs with hCBSC exosomes barely changed the transcriptional landscape of NHCFs in the absence of pathological signaling (TGFβ). There was little separation between the NHCF + VEH group and the NHCF + EXO group as evidenced by the limited variance in the PCA plot (Supplemental Fig. S3A; https://doi.org/10.6084/m9.figshare.15101412.v2). This was supported by the single significantly upregulated gene, ribonuclease inhibitor 1 (RNH1), in the NHCF + EXO-treated condition (Supplemental Fig. S3B).
Given the protective effect of exosomes on fibroblast markers, we determined if deleterious transcriptional changes induced by TGFβ were altered by exosome treatment. There were significant differences between the NHCF + TGFβ and NHCF + TGFβ + EXO groups indicating distinct transcriptional profiles (Fig. 8A). Plotting of genes with a FC ≥ |1.5| and an FDR ≤ 0.05 led to the identification of 42 genes, which were downregulated postexosome treated, and 1 gene, which was significantly upregulated (Fig. 8C). Heatmap of the 43 different genes shows upregulation of tubulin in exosome-treated group and downregulation of a variety of snoRNA genes that are important in regulating ribosome stability and important for protein translation (Fig. 8B). Within the downregulated group was miR7641-2, which targets CXCL2, an important cytokine signaling protein, suggesting decreased inflammatory signaling. Overall, these results suggest that the addition of hCBSC-dEXOs into the fibrotic environment lowers global protein translation levels. This can lead to a decrease in the translation of fibroblast genes important in the transition from fibroblast to myofibroblast. Analysis of GO molecular functions significantly altered between these groups indicates that “cyclic compound binding” is altered, which further suggests that the binding of cellular components to RNA is changed by the hCBSC-dEXO treatment (Fig. 8D). Additionally, biological processes involved in “metabolic processes,” “RNA,” “ribonucleic complex subunit organization,” “mRNA processing,” and “mRNA splicing” were altered, suggesting a change in the transcriptional and translational machinery in the cell (Fig. 8E). This was further confirmed by the changes in the cellular component GO category related to the “nuclear lumen,” “RNP complex,” “spliceosomal snRNP complex,” and “small nuclear RNP complex” (Fig. 8F).
Figure 8.
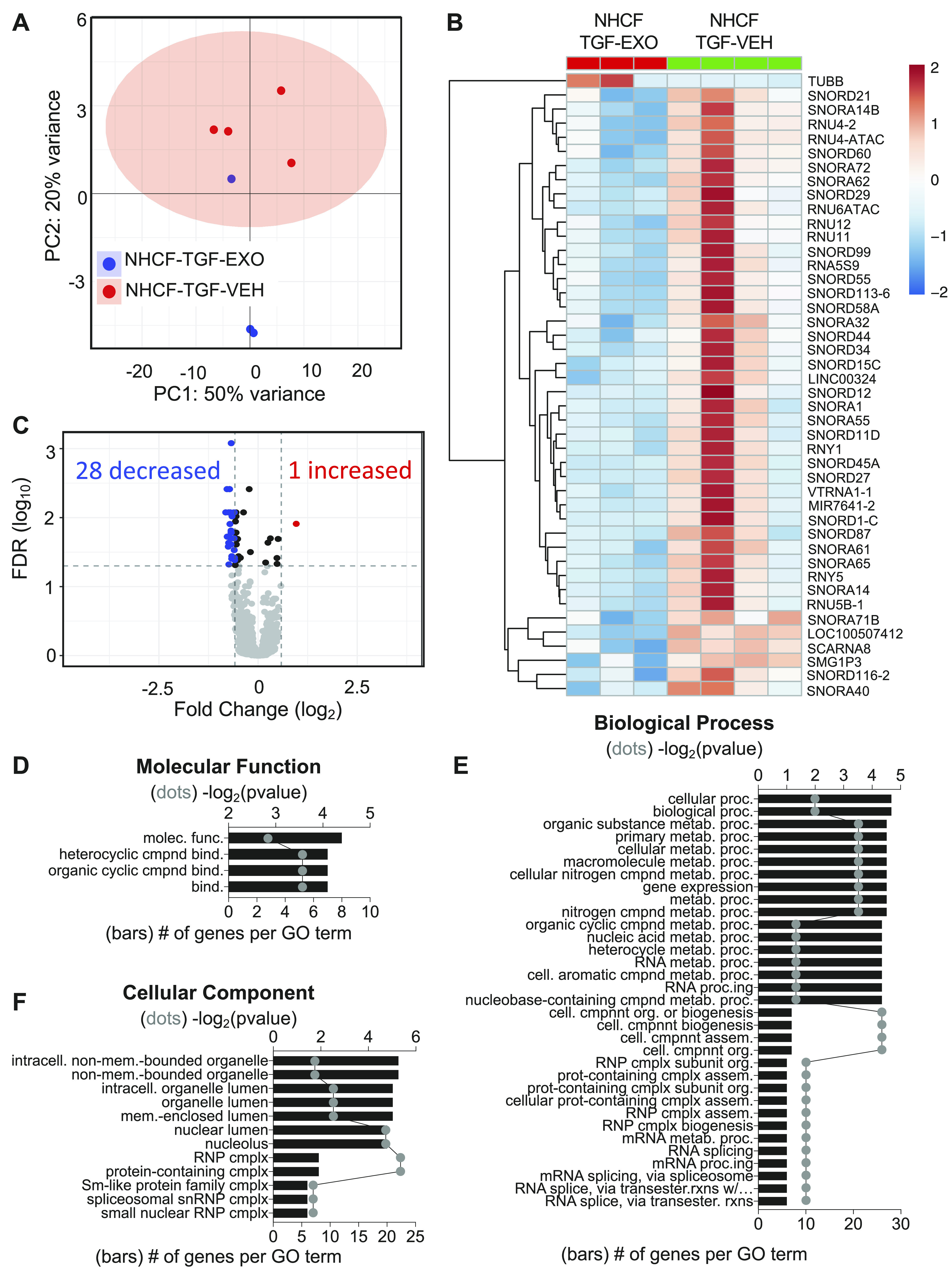
hCBSC-dEXO treatment of activated NHCFs leads to downregulation in miRNA and snoRNA related to ribosome stability and protein translation. Differential expression and gene ontology analysis of NHCFs activated with TGFβ to induce fibrosis and treated with either VEH or hCBSC exosomes. A: principal component analysis shows two distinct transcriptional profiles between NHCF-TGF-VEH and NHCF-TGFβ-EXO. B: heatmap of the 1 significantly increased transcript and 20 significantly deceased transcripts. C: volcano plot analysis showing transcripts with a FC ≥ |1.5| and FDR ≤ 0.05. Gene ontology analysis indicating the molecular function (D), biological process (E), and cellular components of genes (F) that are significantly differentially expressed (n = 4 for TGF-VEH; n = 3 for TGF-EXO). CBSCs, cortical bone stem cells; FC, fold change; FDR, false discovery rate; hCBSC-dEXO, human CBSC-derived exosome; NHCFs, normal human ventricular cardiac fibroblasts; snoRNA, small nucleolar RNA; TGFβ, transforming growth factor β; VEH, vehicle.
DISCUSSION
Exosome research in the context of post-MI injury has rapidly expanded in the past 10 years. This is, in part, due to the modest therapeutic benefits following stem cell treatment and a lack of understanding into the full mechanism of stem cells beneficial effects. Thankfully, studies such as Ling Gao et al. more clearly elucidated the paracrine mechanism by which stem cells are cardioprotective. Specifically, they found that human-induced pluripotent stem cell (hIPSC)-dEXOs were able to recapitulate the beneficial effect of hiPSC-cardiomyocytes and hiPSC-endothelial on cardiac hypertrophy (31). They posited that the cells and dEXOs therapeutic effect was due to the exosomal miRNAs, which they proved by recapitulating the beneficial effect using synthetic mimics of the most abundant exosomal miRNAs. This advancement aided in the fields shift of focus to understanding the exosomal miRNA signaling mechanisms preventing disease.
Our laboratory identified a novel bone-derived stem cell, CBSCs, which we found improved cardiac output and decreased scar size without providing cardioprotection, in both mouse and pig MI models. Specifically, initial infarct size 24 h post-MI was unaltered by CBSC treatment in both the mouse and pig models. Importantly, there were long-term alterations in scar size and improvements in cardiac function following treatment with CBSCs in both species, suggesting a possible antifibrotic effect (24, 25).
Experimentally, our studies were not originally designed ideally for study of cardioprotection. The initial mouse study of mCBSCs used a permanent ligation model of mouse MI, due to increased model consistency over I/R, but provides a massive insult, obscuring nuanced cardioprotective signaling mechanisms (24). The pig study, however, did examine CBSCs effect on cardiac I/R, which, while more clinically relevant, was limited by the necessity for myocardial mapping, a process which leads to 60 m of reperfusion and the reperfusion injury signaling cascade, before injection of swine CBSCs (25). Given the long-term protective effects of CBSCs on heart function, we wanted to more fully understand the CBSCs signaling mechanisms. We designed a mouse study using the more clinically relevant I/R injury, which would allow for immediate injection of mCBSCs and mCBSC-dEXOs on reperfusion and could reveal possible cardioprotective effects of either mCBSCs or mCBSC-dEXOs. We saw that injection of both mCBSCs and mCBSC-dEXOs immediately on reperfusion led to cardioprotection, emphasizing the importance of early intervention in the clinical treatment of myocardial infarctions with ischemia-reperfusion injury.
There is a need for further study beyond the scope of this investigation to more deeply explore the mechanisms of what precisely is occurring to confer cardioprotection. We would, in future studies, inspect the levels of mortality following CBSC and CBSC-derived exosome treatment, as well as investigate cellular markers of injury, and look at long-term effects of CBSC and CBSC-derived exosome treatment on cardiac function and cardiac scar size. These more detailed investigations would help to derive the answer to the mechanism of cardioprotection within this study. Furthermore, we would also add in an additional treatment group, which would be one of exosome-free CBSC-conditioned medium. This group would allow us to be more certain that the exosomes are the responsible player in all of the extracellular milieu released by CBSCs.
These data, and the Ling Gao et al. study (31), led us to posit that CBSCs release paracrine factors, specifically exosomes, which alter cardiac signaling. We found that addition of CBSC-dEXOs to TGFβ-activated fibroblasts caused a reduction in αSMA and fibronectin, two essential fibrotic markers. Additionally, we found that intraspecies exosome treatments led to the most significant protection against fibrosis, as indicated by the NHCF + TGFβ + hCBSC-dEXO condition (Fig. 3, G–I), an important observation for designing future human clinical treatments. Our past pig study saw a reduction in fibrosis when comparing CBSCs to saline in an I/R model, with scar size at 3 mo post-I/R reduced to 8.5% of left ventricular volume in the CBSC-treated animals, as opposed to 16.1% of LV volume in the vehicle-treated animals (25).
We then inspected the commonalities and differences between mouse and human CBSC-dEXOs. We found that both mouse and humans CBSC-dEXOs contain miRNA, rRNA, and tRNA important in gene silencing, translational stability, and translation efficiency, respectively, suggesting exosomes function via alterations to both the transcriptional and translation landscape of target cells. Interestingly, there are species-specific differences. mCBSC-dEXOs contained more piRNA, important in silencing of transposable elements (Fig. 7A and Supplemental Table S1), whereas hCBSCs contained >10% of RefSeq genes (Fig. 7B and Supplemental Table S2), important in metabolic function and regulation of transcription.
Despite their differences, the predominant RNA contained in mouse and human CBSC-dEXOs is miRNAs. The importance of miRNAs as paracrine signaling factors from CBSCs is highlighted by Kraus et al.’s study identifying CBSC-derived miR-18a as the driver of decreased myofibroblast activation (32). Although miR-18a was found in both mCBSC-dEXOs and hCBSC-dEXOs in this study, it was not found to be subsequently upregulated in the NCHFs treated with hCSBC-dEXOs. We did find numerous miRNAs that were upregulated in hCBSC-dEXOs, such as miR-378a, let-7a-3, and miR-31 (Supplemental Table S2), which alter translation and fibrotic signaling. Studies using synthetic mimetics of said miRNA postfibroblast activation are key to understanding the role of these miRNA in alleviating fibrotic signaling.
We then examined how exosomal content alters the signaling of target cells using RNASeq. We found that GO biological processes that were enriched in the hCBSC-dEXOs, such as “macromolecular metabolic processes” and “gene expression” (Fig. 7H), were also enriched in the NHCFs treated with TGFB and hCBSC-dEXOs (Fig. 8E), suggesting a link between exosomal content uptake and changes in the target cells biological processes. The majority of the genes altered by exosome treatment in activated fibroblasts were snoRNA, which are involved in ribosome stabilization and influencing ribosomal RNA processing (33) by binding to rRNA and altering RNA stability (34) leading to increased levels of protein translation (35). Specific snoRNA function is dependent on where snoRNAs bind to rRNA. Binding within the C/D box of rRNA leads primarily to increased 2′O-ribose-methyl group additions. Alternatively, binding of snoRNAs to the H/ACA box leads to increased pseudouridylation (33). Although the effect of snoRNA on rRNA conformation is not fully understood, currently conformational changes caused by snoRNA are believed to enhance ribosomal efficiency (34), suggesting a decrease in snoRNAs can led to ribosome destabilization and alterations to translation. We examined genes associated with translation (data not shown due to nonsignificance) and found no significant difference in expression. This suggests that the reduction in snoRNA genes is not altering global translation but is instead causing targeted changes to the translational landscape. Further studies into the associations of specific snoRNA with translation of specific genes are necessary to confirm this hypothesis.
Additional ncRNA transcripts associated with gene regulation were downregulated in the TGFB + EXO treated condition, specifically, ro-associated noncoding RNAs (RNYs), small nuclear RNA (RNUs), LINC00324, LOC100507412, and miR-7641-2. RNYs are proposed to be important in small RNA quality control and regulation of cellular stress response and proliferation, though the exact mechanism is unknown (36). Although RNUs are important in the splicing of introns and alternative gene splicing (37), LINC00324 has been studied in gastric cancer, in which knockdown of this lncRNA inhibits proliferation, migration, and invasion of cancer cells (38). LOC100507412 is an uncharacterized long ncRNA, thus necessitating further study. miR7641 is known to suppress CXCL1 at both translational and posttranslational levels in human mesenchymal stem cells (MSC) (39) but was previously unidentified in CFs. In MSCs, miR-7641-2 downregulates CXCL2, a gene involved in neutrophil recruitment. Given we did not see upregulation of CXCL2 in NHCFs, further study into the targets of miR-7641-2 in NHCFs is required. Overall, the genes altered by hCSBC-dEXO treatment are primarily from newly identified families of ncRNAs, whose function is related to mRNA and translation regulation. Given the nascence of study pertaining to these RNA families, further experimentation is required, but study has the potential for creation of novel therapeutic strategies.
In this work, we investigated the effect of exosomes specifically on cardiac fibroblasts. We acknowledge, however, that in the complex environment of the postmyocardial infarction heart, the exosomes most likely may not target strictly to cardiac fibroblasts but could also bind to and deposit their contents into cardiac myocytes or cardiac endothelial cells, necessitating study into endogenous cardiac exosomal targeting to examine the uptake of CBSC-dEXOs into specific cardiac cell subtypes. Furthermore, we acknowledge that there may be alternative endogenous exosome release that is altered by the injection of CBSC-derived exosomes, which deserves additional investigation. A limitation of this study that must be acknowledged is the study of solely exogenous exosome injection, as well as that this study is limited to the investigation of CBSC and CBSC-derived exosomes’ effects on male mice. We also observed a difference in infarct size in this study as compared with our previous studies. We hypothesize that it is the change in study design, with the immediate injection of exosomes and CBSCs, which allows for the reduced infarct size. Our previous studies were designed in such a way that there was either a permanent infarct model, which was too overwhelming to find the small amount of cardioprotection as seen here, or required ∼60 min of reperfusion before the injection of therapy. We believe it is the immediate injection of therapeutics in the more sensitive model of I/R that led to the successful cardioprotective ability of both the exosomes and the CBSCs.
In summary, we investigated the ability of CBSC-dEXOs to recapitulate the antifibrotic abilities of CBSCs both in vitro and in vivo, and found that mCBSC-dEXOs and hCBSC-dEXOs exerted the same antifibrotic and therapeutic effects as their respective species of CBSCs. We discovered that hCBSC-dEXOs are specifically antifibrotic in their ability to reduce αSMA activation and fibronectin deposition even in the presence of a strong fibroblast activation signal and that this activity is possibly due to CBSC-derived exosomal snoRNA content reducing ribosomal stability within the treated cells. snoRNA-driven alteration in protein translation within the proinflammatory environment of the post-MI/R heart may reduce fibroblast activation. Furthermore, we investigated whether mCBSCs and mCBSC-dEXOs could provide cardioprotection in a murine I/R model in which treatment was administered immediately on reperfusion and found that both mCBSCs and mCBSC-dEXO postreperfusion led to decreased infarct size and cardioprotection. mCBSC-dEXO-treated animals exhibited the same level of cardioprotection as the mCBSC-treated animals, implying that mCBSC-dEXOs may be the main driver of mCBSC-mediated cardioprotection. Our findings led to the hypothesis that mCBSC-dEXOs induce alterations to cardiac remodeling via a decrease in fibroblast activation, leading to long-term reductions in cardiac scar, and therefore improvements in cardiac repair and function. Our findings on the cardioprotective effect of mCBSC-dEXOs, in particular, warrant a longer term post-I/R study.
SUPPLEMENTAL DATA
Supplemental Fig. S1: https://doi.org/10.6084/m9.figshare.15101379.v1.
Supplemental Fig. S2: https://doi.org/10.6084/m9.figshare.15101385.v1.
Supplemental Fig. S3: https://doi.org/10.6084/m9.figshare.15101412.v2.
Supplemental Table S1: https://doi.org/10.6084/m9.figshare.15101415.v1.
Supplemental Table S2: https://doi.org/10.6084/m9.figshare.15101427.v1.
GRANTS
This work was supported by American Heart Association Grant 19PRE34380532 (to G.J.S.) and National Heart, Lung, and Blood Institute Grants 5P01HL134608-04 (to R.K.) and 5R01HL139960-03 (to S.R.H.).
DISCLOSURES
S. R. Houser is a named inventor on intellectual property filings that are related to the cortical bone stem cells used in this study. In addition, S. R. Houser is a cofounder and scientific advisor and holds equity in MyocardTherapeutics, LLC, a biotech startup that will license S. R. Houser’s cortical bone cell technology from Temple University for commercial development and clinical trials. MyocardTherapeutics, LLC, has not funded any aspect of this research.
AUTHOR CONTRIBUTIONS
G.J.S., S.M., R.K., T.A.M., J.W.E., S.R.H., and K.A.K. conceived and designed research; G.J.S., R.B., A.N.H., A.L.H., Y.Y., H.K., D.E., and J.J. performed experiments; G.J.S., E.K.M., A.N.H., and A.L.H. analyzed data; G.J.S., S.M., S.R.H., and E.K.M. interpreted results of experiments; G.J.S. prepared figures; G.J.S. drafted manuscript; G.J.S., S.R.H., E.K.M., and A.N.H. edited and revised manuscript; G.J.S., R.B., S.M., R.K., T.A.M., J.W.E., S.R.H., E.K.M., A.N.H., A.L.H., Y.Y., K.A.K., H.K., D.E., and J.J. approved final version of manuscript.
REFERENCES
- 1.Buckberg GD, Nanda NC, Nguyen C, Kocica MJ. What is the heart? Anatomy, function, pathophysiology, and misconceptions. J Cardiovasc Dev Dis 5: 33, 2018. doi: 10.3390/jcdd5020033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tham YK, Bernardo BC, Ooi JYY, Weeks KL, McMullen JR. Pathophysiology of cardiac hypertrophy and heart failure: signaling pathways and novel therapeutic targets. Arch Toxicol 89: 1401–1438, 2015. p doi: 10.1007/s00204-015-1477-x. [DOI] [PubMed] [Google Scholar]
- 3.Lüscher TF. The sooner, the better: anti-inflammation in acute myocardial infarction. Eur Heart J 41: 4100–4102, 2020. doi: 10.1093/eurheartj/ehaa752. [DOI] [PubMed] [Google Scholar]
- 4.Frangogiannis NG. The inflammatory response in myocardial injury, repair, and remodelling. Nat Rev Cardiol 11: 255–265, 2014. doi: 10.1038/nrcardio.2014.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eckle T, Eltzschig HK. Toll-like receptor signaling during myocardial ischemia. Anesthesiology 114: 490–492, 2011. doi: 10.1097/ALN.0b013e31820a4d78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schnee JM, Hsueh WA. Angiotensin II, adhesion, and cardiac fibrosis. Cardiovasc Res 46: 264–268, 2000. doi: 10.1016/S0008-6363(00)00044-4. [DOI] [PubMed] [Google Scholar]
- 7.Kerner T, Ahlers O, Reschreiter H, Bührer C, Möckel M, Gerlach H. Adhesion molecules in different treatments of acute myocardial infarction. Crit Care 5: 145–150, 2001. doi: 10.1186/cc1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Talman V, Ruskoaho H. Cardiac fibrosis in myocardial infarction—from repair and remodeling to regeneration. Cell Tissue Res 365: 563–581, 2016. doi: 10.1007/s00441-016-2431-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Frangogiannis NG, Smith CW, Entman ML. The inflammatory response in myocardial infarction. Cardiovasc Res 53: 31–47, 2002. doi: 10.1016/S0008-6363(01)00434-5. [DOI] [PubMed] [Google Scholar]
- 10.van Empel VPM, Bertrand ATA, Hofstra L, Crijns HJ, Doevendans PA, De Windt LJ. Myocyte apoptosis in heart failure. Cardiovasc Res 67: 21–29, 2005. doi: 10.1016/j.cardiores.2005.04.012. [DOI] [PubMed] [Google Scholar]
- 11.Orlic D, Kajstura J, Chimenti S, Jakoniuk I, Anderson SM, Li B, Pickel J, McKay R, Nadal-Ginard B, Bodine DM, Leri A, Anversa P. Bone marrow cells regenerate infarcted myocardium. Nature 410: 701–705, 2001. doi: 10.1038/35070587. [DOI] [PubMed] [Google Scholar]
- 12.Abdelwahid E, Kalvelyte A, Stulpinas A, de Carvalho KAT, Guarita-Souza LC, Foldes G. Stem cell death and survival in heart regeneration and repair. Apoptosis 21: 252–268, 2016. doi: 10.1007/s10495-015-1203-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Duelen R, Sampaolesi M. Stem cell technology in cardiac regeneration: a pluripotent stem cell promise. EBioMedicine 16: 30–40, 2017. doi: 10.1016/j.ebiom.2017.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abou-Saleh H, Zouein FA, El-Yazbi A, Sanoudou D, Raynaud C, Rao C, Pintus G, Dehaini H, Eid AH. The march of pluripotent stem cells in cardiovascular regenerative medicine. Stem cell Res Ther 9: 201, 2018. doi: 10.1186/s13287-018-0947-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goradel NH, Hour FG-, Negahdari B, Malekshahi ZV, Hashemzehi M, Masoudifar A, Mirzaei H. Stem cell therapy: a new therapeutic option for cardiovascular diseases. J Cell Biochem 119: 95–104, 2018. doi: 10.1002/jcb.26169. [DOI] [PubMed] [Google Scholar]
- 16.Lemcke H, Voronina N, Steinhoff G, David R. Recent progress in stem cell modification for cardiac regeneration. Stem cells Int 2018: 1909346–1909346, 2018. doi: 10.1155/2018/1909346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kishore R, Khan M. More than tiny sacks: stem cell exosomes as cell-free modality for cardiac repair. Circ Res 118: 330–343, 2016. doi: 10.1161/CIRCRESAHA.115.307654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yuan Y, Du W, Liu J, Ma W, Zhang L, Du Z, Cai B. Stem cell-derived exosome in cardiovascular diseases: macro roles of micro particles. Front Pharmacol 9: 547–547, 2018. doi: 10.3389/fphar.2018.00547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Prathipati P, Nandi SS, Mishra PK. Stem cell-derived exosomes, autophagy, extracellular matrix turnover, and miRNAs in cardiac regeneration during stem cell therapy. Stem cell Rev Rep 13: 79–91, 2017. doi: 10.1007/s12015-016-9696-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gallet R, Dawkins J, Valle J, Simsolo E, de Couto G, Middleton R, Tseliou E, Luthringer D, Kreke M, Smith RR, Marbán L, Ghaleh B, Marbán E. Exosomes secreted by cardiosphere-derived cells reduce scarring, attenuate adverse remodelling, and improve function in acute and chronic porcine myocardial infarction. Eur Heart J 38: 201–211, 2017. doi: 10.1093/eurheartj/ehw240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang J, Wang F, Sun X, Chu X, Jiang R, Wang Y, Pang L. Myocardial infarction cardiomyocytes-derived exosomal miR-328-3p promote apoptosis via Caspase signaling. Am J Transl Res 13: 2365–2378, 2021. [PMC free article] [PubMed] [Google Scholar]
- 22.Yang J, Yu X, Xue F, Li Y, Liu W, Zhang S. Exosomes derived from cardiomyocytes promote cardiac fibrosis via myocyte-fibroblast cross-talk. Am J Transl Res 10: 4350–4366, 2018. [PMC free article] [PubMed] [Google Scholar]
- 23.Mohsin S, Troupes CD, Starosta T, Sharp TE, Agra EJ, Smith S, Duran JM, Zalavadia N, Zhou Y, Kubo H, Berretta RM, Houser SR. Unique features of cortical bone stem cells associated with repair of the injured heart. Circ Res 117: 1024–1033, 2015. [Erratum in Circ Res 127(11): e271, 2020]. doi: 10.1161/CIRCRESAHA.115.307362. [DOI] [PubMed] [Google Scholar]
- 24.Duran JM, Makarewich CA, Sharp TE, Starosta T, Zhu F, Hoffman NE, Chiba Y, Madesh M, Berretta RM, Kubo H, Houser SR. Bone-derived stem cells repair the heart after myocardial infarction through transdifferentiation and paracrine signaling mechanisms. Circ Res 113: 539–552, 2013. doi: 10.1161/CIRCRESAHA.113.301202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sharp TE, Schena GJ, Hobby AR, Starosta T, Berretta RM, Wallner M, 3rd, Borghetti G, Gross P, Yu D, Johnson J, Feldsott E, Trappanese DM, Toib A, Rabinowitz JE, George JC, Kubo H, Mohsin S, Houser SR. Cortical bone stem cell therapy preserves cardiac structure and function after myocardial infarction. Circ Res 121: 1263–1278, 2017. doi: 10.1161/CIRCRESAHA.117.311174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stratton MS, Bagchi RA, Felisbino MB, Hirsch RA, Smith HE, Riching AS, Enyart BY, Koch KA, Cavasin MA, Alexanian M, Song K, Qi J, Lemieux ME, Srivastava D, Lam MPY, Haldar SM, Lin CY, McKinsey TA. Dynamic chromatin targeting of BRD4 stimulates cardiac fibroblast activation. Circ Res 125: 662–677, 2019. doi: 10.1161/CIRCRESAHA.119.315125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hobby ARH, Sharp TE, Berretta RM, Borghetti G, Feldsott E, Mohsin S, Houser SR. Cortical bone-derived stem cell therapy reduces apoptosis after myocardial infarction. Am J Physiol Heart Circ Physiol 317: H820–H829, 2019. doi: 10.1152/ajpheart.00144.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mohsin S, Houser SR. Cortical bone derived stem cells for cardiac wound healing. Korean Circ J 49: 314–325, 2019. doi: 10.4070/kcj.2018.0437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Khan M, Nickoloff E, Abramova T, Johnson J, Verma SK, Krishnamurthy P, Mackie AR, Vaughan E, Garikipati VN, Benedict C, Ramirez V, Lambers E, Ito A, Gao E, Misener S, Luongo T, Elrod J, Qin G, Houser SR, Koch WJ, Kishore R. Embryonic stem cell-derived exosomes promote endogenous repair mechanisms and enhance cardiac function following myocardial infarction. Circ Res 117: 52–64, 2015. doi: 10.1161/CIRCRESAHA.117.305990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Park S-R, Kim J-W, Jun H-S, Roh JY, Lee H-Y, Hong I-S. Stem cell secretome and its effect on cellular mechanisms relevant to wound healing. Mol Ther 26: 606–617, 2018. doi: 10.1016/j.ymthe.2017.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gao L, Wang L, Wei Y, Krishnamurthy P, Walcott GP, Menasché P, Zhang J. Exosomes secreted by hiPSC-derived cardiac cells improve recovery from myocardial infarction in swine. Sci Transl Med 12: eaay1318, 2020. doi: 10.1126/scitranslmed.aay1318. [DOI] [PubMed] [Google Scholar]
- 32.Kraus L, Ma L, Yang Y, Nguyen F, Hoy RC, Okuno T, Khan M, Mohsin S. Cortical bone derived stem cells modulate cardiac fibroblast response via miR-18a in the heart after injury. Front Cell Dev Biol 8: 494–494, 2020. doi: 10.3389/fcell.2020.00494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kiss T, Fayet E, Jády BE, Richard P, Weber M. Biogenesis and intranuclear trafficking of human box C/D and H/ACA RNPs. Cold Spring Harb Symp Quant Biol 71: 407–417, 2006. doi: 10.1101/sqb.2006.71.025. [DOI] [PubMed] [Google Scholar]
- 34.Bachellerie J-P, Cavaillé J, Hüttenhofer A. The expanding snoRNA world. Biochimie 84: 775–790, 2002. p doi: 10.1016/S0300-9084(02)01402-5. [DOI] [PubMed] [Google Scholar]
- 35.Dieci G, Preti M, Montanini B. Eukaryotic snoRNAs: a paradigm for gene expression flexibility. Genomics 94: 83–88, 2009. doi: 10.1016/j.ygeno.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 36.Köhn M, Pazaitis N, Hüttelmaier S. Why YRNAs? About versatile RNAs and their functions. Biomolecules 3: 143–156, 2013. doi: 10.3390/biom3010143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Valadkhan S, Gunawardane LS. Role of small nuclear RNAs in eukaryotic gene expression. Essays Biochem 54: 79–90, 2013. doi: 10.1042/bse0540079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang S, Cheng Y, Yang P, Qin G. Silencing of long noncoding RNA LINC00324 interacts with MicroRNA-3200-5p to attenuate the tumorigenesis of gastric cancer via regulating BCAT1. Gastroenterol Res Pract 2020: 4159298, 2020. doi: 10.1155/2020/4159298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yoo JK, Jung HY, Kim C-H, Son WS, Kim JK. miR-7641 modulates the expression of CXCL1 during endothelial differentiation derived from human embryonic stem cells. Arch Pharm Res 36: 353–358, 2013. doi: 10.1007/s12272-013-0067-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Fig. S1: https://doi.org/10.6084/m9.figshare.15101379.v1.
Supplemental Fig. S2: https://doi.org/10.6084/m9.figshare.15101385.v1.
Supplemental Fig. S3: https://doi.org/10.6084/m9.figshare.15101412.v2.
Supplemental Table S1: https://doi.org/10.6084/m9.figshare.15101415.v1.
Supplemental Table S2: https://doi.org/10.6084/m9.figshare.15101427.v1.