

Abstract
Introduction
Our previous case–control study demonstrated that a high level of intestinal alkaline phosphatase (IAP), an endotoxin-detoxifying anti-inflammatory enzyme secreted by villus-associated enterocytes and excreted with stool, plays a protective role against type 2 diabetes mellitus (T2DM) irrespective of obesity. In the current study, we investigated the long-term effect of IAP deficiency (IAPD) on the pathogenesis of T2DM.
Research design and methods
A healthy cohort of participants without diabetes (30–60 years old), comprising 188 without IAPD (IAP level: ≥65 U/g stool) and 386 with IAPD (IAP level: <65 U/g stool), were followed up for 5 years. We measured stool IAP (STAP) and fasting plasma glucose, and calculated the risk ratio (RR) using log-binomial regression model.
Results
T2DM incidence rates were 8.0%, 11.7%, 25.6%, and 33.3% in participants with ‘persistent no IAPD’ (IAP level: always ≥65 U/g stool), ‘remittent IAPD’ (IAP level: increased from <65 U/g stool to ≥65 U/g stool), ‘persistent IAPD’ (IAP level: always <65 U/g stool), and ‘incident IAPD’ (IAP level: decreased from ≥65 U/g stool to <65 U/g stool), respectively. Compared with ‘persistent no IAPD’ the risk of developing T2DM with ‘incident IAPD’ was 270% higher (RR: 3.69 (95% CI 1.76 to 7.71), χ2 p<0.001). With ‘persistent IAPD’ the risk was 230% higher (RR: 3.27 (95% CI 1.64 to 6.50), p<0.001). ‘Remittent IAPD’ showed insignificant risk (RR: 2.24 (95% CI 0.99 to 5.11), p=0.0541). Sensitivity analyses of persistent IAP levels revealed that, compared with participants of the highest persistent IAP pentile (always >115 U/g stool), the rate of increase of fasting glycemia was double and the risk of developing T2DM was 1280% higher (RR: 13.80 (95% CI 1.87 to 101.3), p=0.0099) in participants of the lowest persistent IAP pentile (always <15 U/g stool). A diabetes pathogenesis model is presented.
Conclusions
IAPD increases the risk of developing T2DM, and regular STAP tests would predict individual vulnerability to T2DM. Oral IAP supplementation might prevent T2DM.
Keywords: diabetes mellitus, type 2
Significance of this study.
What is already known about this subject?
The deficiency of intestinal alkaline phosphatase (IAP), an anti-inflammatory gut enzyme secreted by intestinal enterocytes and excreted with stool, is directly associated with type 2 diabetes mellitus (T2DM), and a high level of IAP plays a protective role against T2DM irrespective of obesity.
IAP deficiency (IAPD) is also directly associated with type 1 diabetes mellitus.
What are the new findings?
This 5-year prospective cohort study shows that IAPD increases the rate of fasting plasma glucose.
IAPD increases the risk of developing T2DM.
Remission of IAPD prevents the development of T2DM.
How might these results change the focus of research or clinical practice?
Regular stool IAP tests would diagnose vulnerability to T2DM, and prevention of IAPD would prevent T2DM.
Intervention of IAPD by oral IAP supplementation might prevent T2DM.
Eradication of the worldwide pandemic of T2DM is possible by early diagnosis of IAPD-associated vulnerability to T2DM followed by pre-emptive preventive measures.
Introduction
Type 2 diabetes mellitus (T2DM), a chronic metabolic disease manifested with hyperglycemia and insulin resistance, is a major global health problem with devastating consequences in terms of morbidity, mortality, and healthcare costs. T2DM affected approximately 463 million adults worldwide (9.3% of world population) in 2019 and is projected to affect approximately 578 million (10.2%) in 2030 and 700 million (10.8%) in 2045 (see online supplemental appendix A on T2DM statistics).1–3 The pathogenesis of T2DM is poorly understood. Various factors have been postulated to be involved in the development of T2DM, notably genetic polymorphism, ethnicity, metabolic syndrome, obesity, diets, infection, dysbiosis, autoimmunity, drugs, stress, and pregnancy.4–11 Low-grade systemic inflammation has been implicated to play a pivotal role in the pathogenesis of T2DM.8–11 Death of Gram-negative bacteria results in fragmentation of the cell wall, releasing its lipopolysaccharide (LPS), an endotoxin that causes inflammation. Intestinal luminal LPS is usually excreted with stool; however, under certain circumstances, such as increased gut permeability and increased intake of a high-fat diet, alcohol, and fructose, LPS may translocate to blood circulation, causing endotoxemia.12 Recently, ‘metabolic endotoxemia’, defined as persistently increased levels of LPS in blood, has been shown to induce low-grade systemic inflammation, leading to insulin resistance, hyperglycemia (T2DM), dyslipidemia, and fatty liver in mice.12 13 We have previously shown that LPS is detoxified by intestinal alkaline phosphatase (IAP), a gut enzyme secreted by villus-associated enterocytes and partially excreted with stool.7 12 IAP functions as an anti-inflammatory enzyme by detoxifying LPS and other bacterial toxins through dephosphorylation (see online supplemental appendix B for IAP functions).7 12 14 15 We have previously demonstrated that mice deficient in IAP (IAP knockout, Akp−/−) develop metabolic syndrome (hyperglycemia, dyslipidemia, and fatty liver) that can be prevented by oral IAP supplementation.12 Our subsequent case–control human study identified that the average level of IAP was approximately 65 U/g stool in the healthy population,7 and accordingly an IAP level of ≥65 U/g stool is considered high (normal) and protective against T2DM. In contrast, the average IAP level detected in patients with T2DM was only 35 U/g stool.7 Obese people with normal IAP level did not develop T2DM. The study established that IAP deficiency (IAPD), defined as <65 U of IAP per gram of stool, is directly associated with T2DM.7 Based on the above-mentioned coherent animal and human studies,7 12 we hypothesized that IAPD might be an independent risk factor of T2DM in humans. Thus, we conducted a 5-year prospective cohort study and here we report that IAPD plays a pivotal role in the development of T2DM. We also provide a model defining the pathway of IAPD-mediated pathogenesis of diabetes.
bmjdrc-2021-002643supp001.pdf (533KB, pdf)
Methods
Study design and participants
We followed up the health status of a healthy cohort of 671 participants without diabetes (30–60 years old) of our previous case–control study7 for 5 years. IAPD was considered as the major exposure variable and incidence of T2DM as the outcome variable, where baseline physical and biochemical profiles, including body mass index (BMI), fasting plasma glucose (FPG), and IAP values of each participant, were taken into account for outcome measurement (figure 1; see online supplemental appendix C for details).
Figure 1.
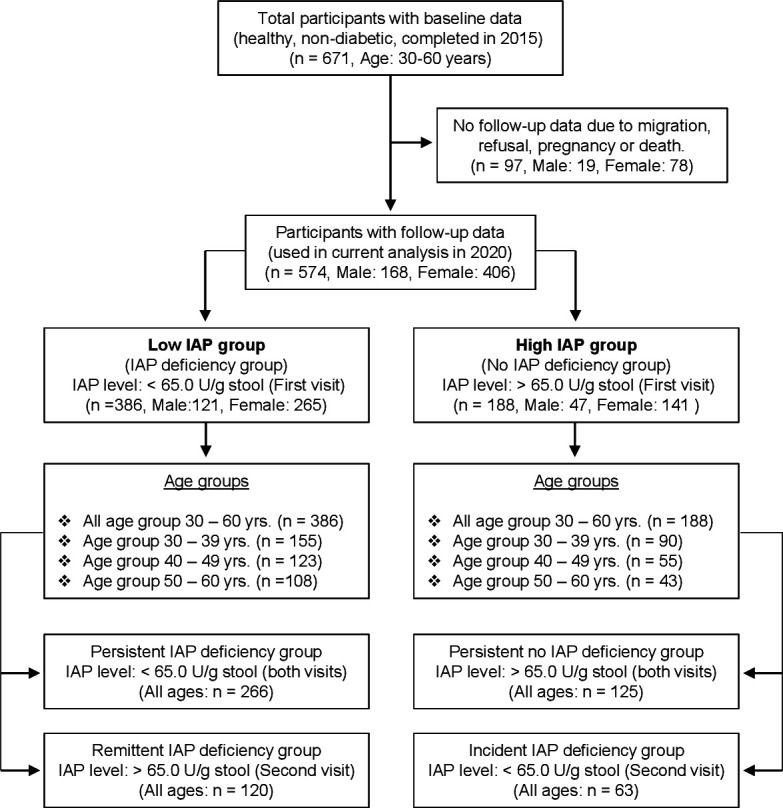
Flow chart of a 5-year prospective cohort study defining the correlation between deficiency of intestinal alkaline phosphatase (IAP) and development of type 2 diabetes mellitus (T2DM). The participants were selected from the control healthy population without diabetes of a previous case–control study (case: patients with T2DM; control: healthy participants) that established that IAP deficiency (IAPD) is directly associated with T2DM, and the average IAP level in the healthy population without diabetes was approximately 65.0 U/g stool.7 The participants, 30–60 years old, were stratified into two groups based on baseline IAP levels (low IAP group (IAPD group): <65.0 U/g stool; high IAP group (no IAPD group): ≥65.0 U/g stool). The follow-up data of 97 participants were not available due to migration, refusal, pregnancy, or death of participants. Four participants in their 50s died of myocardial infarction. It was observed that IAP levels in some individuals remained persistently stable (within group-specific limit) and in other persons IAP levels temporally changed (increased or decreased) during the 5-year period. Remittent IAPD is defined as a condition when the second visit IAP value of a person is more than the respective persistent IAP value (transition from <65 U/g stool to ≥65.0 U/g stool). Incident IAPD is defined as a condition when the second visit IAP value of a person is less than the respective persistent IAP value (transition from ≥65 U/g stool to <65.0 U/g stool). There was no statistically significant difference between low and high IAP groups in baseline characteristics except age (43.2±8.8 years vs 40.2±8.9 years, respectively, p<0.05). In a specific age group there was no statistically significant difference in age between ‘persistent IAPD group’ and ‘remittent IAPD group’ as well as between ‘persistent no IAPD group’ and ‘incident IAPD group’.
Each participant consented to participate in the study and signed an informed consent form approved by the National Research Ethics Committee (see Ethics approval).
Laboratory procedures
We measured blood pressure and body mass index (BMI), and serum cholesterol, high-density lipoproteins (HDL), low-density lipoproteins (LDL), triglycerides, creatinine, and alanine aminotransferase (ALT). The T2DM status of each participant was confirmed by measuring fasting plasma glucose (FPG) (at least 10 hours) concentration and/or hemoglobin A1c (HbA1c) concentration. An FPG level of ≥7.0 mmol/L (126 mg/dL) or HbA1c level ≥6.5% was considered diagnostic for T2DM (see online supplemental appendix C for details).1 Stool alkaline phosphatase (STAP) assay was performed following the procedures as previously described (see online supplemental appendix C for details).7 In brief, a specific amount of stool was dissolved in a specific amount of stool dilution buffer (eg, 100 mg stool to 5 mL stool dilution buffer), homogenized and centrifuged, and supernatant containing STAP was collected and followed by measuring STAP concentration by an automatic chemistry analyzer. STAP assays were performed by laboratory technologists who were blinded to the diagnoses of participants. Because 80% of STAP is IAP and 20% of STAP is of bacterial origin,7 IAP is synonymously used for STAP in this article.
Statistical analysis
Statistical analysis was performed using the SAS V.9.4 software. χ2 test was used to examine the association between IAPD group and T2DM status. The correlation between IAP and different risk factors of T2DM was evaluated by Pearson’s correlation coefficient. Adjusted generalized linear model of regression analysis was used to determine adjusted mean difference in IAP between T2DM groups (T2DM vs non-T2DM). Log-binomial regression analysis was used to calculate the unadjusted and adjusted risk ratios (RR) (see online supplemental appendix C for details). We also performed repeated measure analysis of variance (ANOVA) to examine the association of the IAP level with T2DM. A p value of <0.05 was considered statistically significant.
Results
Both baseline and follow-up data were available for 574 participants and were thus included in the study, and 97 participants were excluded because of unavailability of follow-up data due to migration, refusal, pregnancy, and death (figure 1). There was no significant difference between the included and excluded groups in terms of physical and biochemical parameters (online supplemental table S1). The characteristics of all included participants are shown in online supplemental table S2.
Based on the follow-up data, we observed that 20% of the participants developed T2DM in 5 years. Confirming our previous results,7 we found that a high IAP level plays a protective role against T2DM (figure 2A). The percentile distribution shows that IAP levels are low in patients with T2DM at all percentile points compared with healthy participants without diabetes (controls) (figure 2B). We also found that obese people with high IAP do not develop T2DM (figure 2C).
Figure 2.
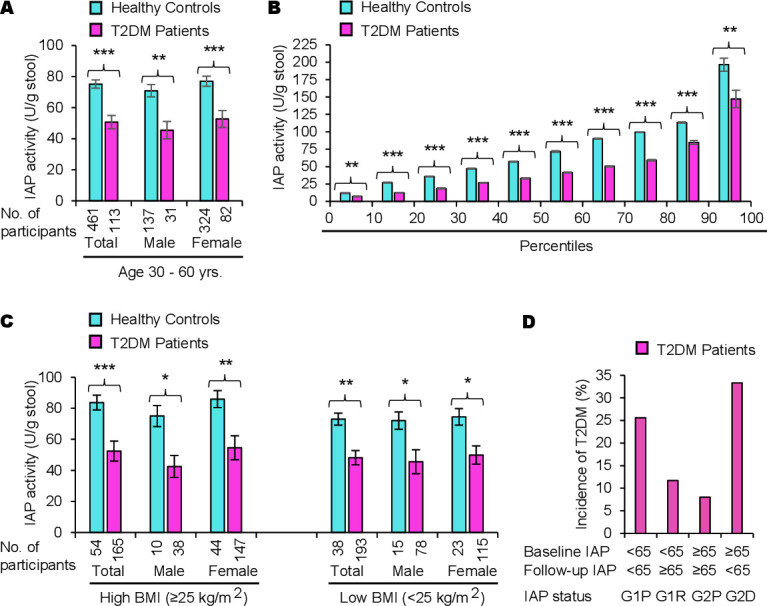
A high level of intestinal alkaline phosphatase (IAP) plays a protective role against type 2 diabetes mellitus (T2DM) irrespective of obesity. (A) Intestinal alkaline phosphatase deficiency (IAPD) is associated with T2DM. Participants, aged 30–60 years old, were screened for T2DM (see figure 1) and diagnosed as having T2DM or no T2DM (healthy controls). Data are summarized as mean (average)±SEM. Statistical significance of the difference between two groups was tested using unpaired two-tailed Student’s t-test. *p<0.05, **p<0.01, ***p<0.001. The post-hoc statistical power analyses revealed the powers for respective total, male, and female groups to be 100%, 100%, and 100%, respectively, which validated the adequacy of power (conventionally, >80% power at α=0.05) for respective sample sizes. Percentage loss of IAP in patients with T2DM compared with healthy controls: total, 32.6%; male, 35.8%; female, 31.6%. The average IAP level is 8.2% less in healthy control men compared with healthy control women; however, the difference is not significant (p=0.27561). (B) Percentile distributions showing IAP levels are low in patients with diabetes at all percentile points compared with healthy controls. Individual IAP values from each group (healthy controls or patients with T2DM) were organized from the lowest to the highest and then the average IAP value within each 10th percentile was calculated (n=11 within each 10th percentile for patients with T2DM and n=46 within each 10th percentile for healthy controls). The average values (mean±SEM) for corresponding percentiles are plotted. Note: Only the values in the first and last 10th percentile divisions will be greatly affected if an ‘outlier’ (a few extremely low or high values, compared with the most other values, affecting the mean value) is present. The values within the 10th and 90th percentiles are real, not affected by outliers. (C) A high level of IAP protects from T2DM irrespective of obesity. Patients with T2DM as well as healthy control participants were categorized into two groups, one group with high body mass index (BMI ≥25.0 kg/m2, includes obese and overweight persons) and the other group with low BMI (<25.0 kg/m2). Note: Persons with high BMI and high levels of IAP do not develop T2DM. There was no significant difference in IAP levels in healthy controls of high BMI and low BMI groups as well as in patients with T2DM of high and low BMI groups. (D) IAPD is associated with a higher incidence of T2DM. The percentage of incidence was calculated for the 5 years (see table 3). Note: Participants with ‘persistent IAPD’ (G1P) and ‘incident IAPD’ (G2D) have much higher incidence of T2DM compared with participants with ‘persistent no IAPD’ (G2P) and ‘remittent IAPD’ (G1R).
The baseline characteristics of participants grouped by IAPD status are shown in online supplemental table S3 and the follow-up characteristics of the IAPD groups are shown in table 1.
Table 1.
Follow-up characteristics of participants grouped by status of IAPD from 2015 to 2020
| Follow-up characteristics | Persistent no IAPD | Incident IAPD | Persistent IAPD | Remittent IAPD |
| IAP level (U/g stool) (for defining IAPD) | Baseline: ≥65.0 Follow-up: ≥65.0 |
Baseline: ≥65.0 Follow-up:<65.0 |
Baseline: <65.0 Follow-up: <65.0 |
Baseline: <65.0 Follow-up: ≥65.0 |
| Number of participants (N=574) | 125 (male 31; female 94) | 63 (male 16; female 47) | 266 (male 80; female 186) | 120 (male 41; female 79) |
| Age group (years) at baseline | 30–60 | 30–60 | 30–60 | 30–60 |
| Average age (years) at baseline | 40.2±8.9 | 42.1±10.1 | 43.2±8.8* | 39.8±9.1 |
| Height (m) at baseline | 1.5±0.1 | 1.5±0.2 | 1.5±0.2 | 1.5±0.2 |
| Weight (kg) | 60.3±10.8 | 57.5±9.3 | 59.2±8.8 | 61.4±11.0 |
| BMI (kg/m2) | 26.0±5.3 | 25.6±4.7 | 25.3±3.8 | 25.8±4.4 |
| Systolic BP (mm Hg) | 128.9±17.1 | 133.3±20.4 | 131.6±16.0 | 127.5±16.5 |
| Diastolic BP (mm Hg) | 79.1±9.2 | 81.2±10.2 | 79.8±8.4 | 79.7±8.9 |
| Creatinine (mg/dL) | 0.89±0.21 | 0.97±0.41 | 0.92±0.22 | 0.89±0.29 |
| Cholesterol (mg/dL) | 178.0±32.7 | 189.5±39.0 | 178.2±31.9 | 181.5±36.9 |
| HDL (mg/dL) | 42.4±7.7 | 43.5±9.7 | 42.1±8.8 | 43.3±8.6 |
| LDL (mg/dL) | 103.9±27.3 | 112.5±31.8 | 102.9±26.4 | 106.5±32.1 |
| Triglycerides (mg/dL) | 160.9±68.2 | 166.2±64.1 | 164.5±67.7 | 155.7±63.4 |
| ALT (U/L) | 38.3±16.4 | 33.3±21.7 | 38.4±18.5 | 34.7±16.3 |
| FPG (mmol/L) | 5.6±1.4 | 6.7±3.4* | 6.1±1.7* | 5.8±1.7 |
Data are summarized as mean (average)±SD for each variable.
Statistical significance of the difference between two respective groups was examined using unpaired two-tailed Student’s t-test (reference group: persistent no IAPD).
*p<0.01.
ALT, alanine aminotransferase; BMI, body mass index; BP, blood pressure; FPG, fasting plasma glucose; HDL, high-density lipoproteins; IAP, intestinal alkaline phosphatase; IAPD, IAP deficiency; LDL, low-density lipoproteins.
Validating our previous data,7 Pearson’s correlation coefficient analysis showed no correlation between IAP level and age, gender, FPG, BMI, ALT, serum creatinine, blood pressure, or lipid level (online supplemental table S4). A generalized linear regression model predicted a strong association of IAP with T2DM, confirming our previous observation (table 2).
Table 2.
Generalized linear regression model showing an association of intestinal alkaline phosphatase with T2DM
| Source | DF | Type III SS | Mean square | F value | Pr > F |
| T2DM status | 1 | 46 343.23514 | 46 343.23514 | 18.11 | <0.0001 |
| Sex | 1 | 0.99577 | 0.99577 | 0.00 | 0.9843 |
| Age | 1 | 5658.29100 | 5658.29100 | 2.21 | 0.1376 |
| BMI at visit 1 | 1 | 5252.19590 | 5252.19590 | 2.05 | 0.1525 |
| BMI at visit 2 | 1 | 245.95209 | 245.95209 | 0.10 | 0.7566 |
| Creatinine at visit 1 | 1 | 525.10855 | 525.10855 | 0.21 | 0.6507 |
| Creatinine at visit 2 | 1 | 4891.03284 | 4891.03284 | 1.91 | 0.1674 |
| Cholesterol at visit 1 | 1 | 1627.98235 | 1627.98235 | 0.64 | 0.4254 |
| Cholesterol at visit 2 | 1 | 6104.75510 | 6104.75510 | 2.39 | 0.1230 |
| HDL at visit 1 | 1 | 3563.31536 | 3563.31536 | 1.39 | 0.2385 |
| HDL at visit 2 | 1 | 11 898.08018 | 11 898.08018 | 4.65 | 0.0315 |
| LDL at visit 1 | 1 | 2424.42424 | 2424.42424 | 0.95 | 0.3308 |
| LDL at visit 2 | 1 | 6897.93373 | 6897.93373 | 2.70 | 0.1012 |
| TG at visit 1 | 1 | 2420.23875 | 2420.23875 | 0.95 | 0.3312 |
| TG at visit 2 | 1 | 9132.44536 | 9132.44536 | 3.57 | 0.0594 |
| ALT at visit 1 | 1 | 3584.14371 | 3584.14371 | 1.40 | 0.2371 |
| ALT at visit 2 | 1 | 117.00346 | 117.00346 | 0.05 | 0.8307 |
| SBP at visit 1 | 1 | 556.47151 | 556.47151 | 0.22 | 0.6411 |
| SBP at visit 2 | 1 | 12 008.77300 | 12 008.77300 | 4.69 | 0.0307 |
| DBP at visit 1 | 1 | 16.35648 | 16.35648 | 0.01 | 0.9363 |
| DBP at visit 2 | 1 | 2099.57910 | 2099.57910 | 0.82 | 0.3654 |
ALT, alanine aminotransferase; BMI, body mass index; DBP, diastolic blood pressure; DF, degrees of freedom; F, F statistic; HDL, high-density lipoproteins; LDL, low-density lipoproteins; Pr, probability; SBP, systolic blood pressure; SS, sum of squares; T2DM, type 2 diabetes mellitus; TG, triglycerides.
A repeated measure ANOVA also showed a very strong association of IAP with T2DM (online supplemental table S5).
We assessed unadjusted relative risks of developing T2DM in different IAPD groups (figure 3). Compared with the ‘persistent no IAPD’ group, we observed that the ‘incident IAPD’ and ‘persistent IAPD’ groups had significantly higher risk of developing T2DM, especially approximately 7.0-fold increased risk in younger men (30–39 years old) with ‘incident IAPD’. In contrast, like ‘persistent no IAPD’, ‘remittent IAPD’ was associated with less risk of developing T2DM.
Figure 3.

Association between intestinal alkaline phosphatase deficiency (IAPD) and incidence of type 2 diabetes mellitus (T2DM) stratified by gender and age. A prospective cohort of healthy participants without diabetes (n=574, 30–60 years old) were followed up for 5 years. Based on baseline and follow-up IAP values, the participants were classified as having ‘persistent no IAPD’ (intestinal alkaline phosphatase (IAP) level: always ≥65 U/g stool), ‘persistent IAPD’ (IAP level: always <65 U/g stool), ‘incident IAPD’ (IAP level: decreased from ≥65 U/g stool to <65 U/g stool), or ‘remittent IAPD’ (IAP level: increased from <65 U/g U/g stool to ≥65 U/g stool). Post-hoc statistical power analyses revealed adequacy of power (conventionally, >80% power at α=0.05) for different comparative groups (‘persistent no IAPD’ vs ‘incident IAPD’: 98.6%; ‘persistent no IAPD’ vs ‘persistent IAPD’: 99.4%; ‘persistent no IAPD’ vs ‘remittent IAPD’: 90.0%), validating the adequacy of sample size for each group as calculated by an online program (http://clincalc.com/Stats/Power.aspx). Data are unadjusted relative risk (RR) and 95% CI.
Table 3 shows the incidence rates of T2DM, which were 8.0%, 11.7%, 25.6%, and 33.3% in participants with ‘persistent no IAPD’, ‘remittent IAPD’, ‘persistent IAPD’, and ‘incident IAPD’, respectively (also see figure 2D).
Table 3.
Intestinal alkaline phosphatase deficiency (IAPD) is associated with increased risk of developing type 2 diabetes mellitus (T2DM)
| IAPD status | Total participants | T2DM n (%) |
Relative risk (95% CI) | ||||
| Model 1* | Model 2† | Model 3‡ | Model 4§ | Model 5¶ | |||
| Persistent no IAPD | 125 | 10 (8.0) | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| Incident IAPD | 63 | 21 (33.3) | 4.17 (2.09 to 8.30)*** |
3.78 (1.89 to 7.54)*** |
4.31 (2.00 to 9.27)*** |
4.17 (1.93 to 9.00)*** |
3.69 (1.76 to 7.71)*** |
| Persistent IAPD | 266 | 68 (25.6) | 3.20 (1.70 to 5.99)*** |
3.01 (1.60 to 5.65)*** |
3.33 (1.65 to 6.71)*** |
3.27 (1.62 to 6.61)*** |
3.27 (1.64 to 6.50)*** |
| Remittent IAPD | 120 | 14 (11.7) | 1.46 (0.67 to 3.16) |
1.49 (0.69 to 3.21) |
2.00 (0.88 to 4.56) |
2.05 (0.89 to 4.68) |
2.24 (0.99 to 5.11) |
***p<0.001.
*Model 1: unadjusted model.
†Model 2: adjusted for age and sex at baseline (visit 1).
‡Model 3: adjusted for variables in model 2 plus body mass index, systolic blood pressure, and diastolic blood pressure at baseline (visit 1).
§Model 4: adjusted for variables in model 3 plus creatinine, cholesterol, high-density lipoproteins (HDL), low-density lipoproteins (LDL), triglycerides, and alanine aminotransferase at baseline (visit 1).
¶Model 5: adjusted for variables in model 4 plus fasting plasma glucose at baseline (visit 1).
The data indicated that compared with the ‘persistent no IAPD’ group, the incidence rate was more than 4.0-fold higher in the ‘incident IAPD’ group. Table 3 also shows the relative risks of developing T2DM in different IAPD groups after adjusting for various confounding factors, such as age, gender, BMI, systolic blood pressure, diastolic blood pressure, cholesterol, HDL, LDL, triglycerides, creatinine, ALT, and FPG. Compared with ‘persistent no IAPD’, ‘incident IAPD’ was associated with 3.7-fold increased risk of developing T2DM (RR: 3.69 (95% CI 1.76 to 7.71), p<0.001). Similarly, ‘persistent IAPD’ was associated with 3.3-fold increased risk (RR: 3.27 (95% CI 1.64 to 6.50), p<0.001). In contrast, the relative risk of developing T2DM in the ‘remittent IAPD’ group was insignificant (RR: 2.24 (95% CI 0.99 to 5.11), p=0.0541).
To delineate the sensitivity of different persistent levels of IAP on developing T2DM, we calculated T2DM incidence rates, relative risks, and rates of increase of FPG for five persistent levels of IAP (table 4).
Table 4.
The lower the persistent IAP level the higher the rate of increase of fasting glycemia and relative risk of developing T2DM
| Pentile | Persistent IAP level (U/g stool) |
Participants, n (T2DM, %) |
Relative risk (95% CI) | First visit FPG level, mmol/L (mg/dL) | Second visit FPG level, mmol/L (mg/dL) | Percentage increase of FPG level |
| Pentile 1 | 0–15.0 | 30 (14, 46.7) | 13.8 (1.87 to 101.3)** | 4.4±0.7 (79.2±12.6) | 6.9±2.5*** (124.2±45.0) | 34.1*** |
| Pentile 2 | 15.1–33.0 | 38 (8, 21.1) | 6.9 (0.91 to 52.60) | 4.5±0.8 (81.0±14.4) | 6.1±1.0*** (109.8±18.0) | 25.9* |
| Pentile 3 | 33.1–55.0 | 39 (9, 23.1) | 7.0 (0.93 to 52.57) | 4.3±0.8 (77.4±14.4) | 6.0±1.1*** (108.0±19.8) | 25.2** |
| Pentile 4 | 55.1–115.0 | 58 (8, 13.8) | 4.8 (0.63 to 36.71) | 4.3±0.8 (77.4±14.4) | 5.6±1.0*** (100.8±18.0) | 23.9* |
| Pentile 5 | >115.0 | 34 (1, 2.9) | 1.0 | 4.5±0.7 (81.0±12.6) | 5.5±0.7*** (99.0±12.6) | 17.3 |
All participants, 30–60 years old, were stratified into pentiles based on baseline IAP levels, and then participants with persistent IAP level (during follow-up visit) were identified.
Data are summarized as mean (average)±SD for each variable.
Pentile 5 was the reference pentile for calculating the statistical significance of difference in the percentage of increase of FPG levels (Student’s t-test) as well as evaluating age-adjusted and gender-adjusted relative risk between two pentiles.
*p<0.05, **p<0.01, ***p<0.001.
FPG, fasting plasma glucose; IAP, intestinal alkaline phosphatase; T2DM, type 2 diabetes mellitus.
Compared with participants with the highest levels of persistent IAP (always >115 U/g stool, pentile 5), participants with the lowest level of persistent IAP (always ≤15.0 U/g stool, pentile 1) had approximately 16.0-fold higher incidence rate of T2DM (2.9% vs 46.7%, respectively). As expected, the age-adjusted and gender-adjusted relative risk also increased by 13.80-fold in pentile 1 (RR: 13.80 (95% CI 1.87 to 101.3), p=0.0099). A graphical presentation of percentage of risk of T2DM associated with each pentile is shown in online supplemental figure S1. Table 4 also shows that, compared with pentile 5, the rate of increase in glycemia was double in pentile 1 (17.4% vs 34.1%, respectively, p<0.001). The rate of increase in FPG was not dependent on age (online supplemental table S6).
Discussion
This observational prospective cohort study shows that IAPD is an independent risk factor of T2DM. In the context of confounding factors affecting IAP levels, confirming our previous observations,7 Pearson’s correlation coefficient analysis showed that age, gender, and various physical and biochemical risk factors do not have any significant effect on IAP levels (online supplemental table S4). The participants were on an unrestricted diet, and it is expected that if the sample sizes of related groups are high the diet should not affect the outcome of the study because both groups would be similarly affected by diet. The sample sizes of related groups in this study were large enough to achieve more than 90% post-hoc statistical power (figure 3) and hence we believe it is unlikely that diet had any effect on the outcome of study. The participants were from diverse socioeconomic background and therefore we think that socioeconomic conditions did not impact the outcome of the study. Obesity has been implicated in the pathogenesis of T2DM;1–3 however, we found that obese people with a high level of IAP do not develop T2DM (figure 2C), indicating a critical role of IAP in preventing T2DM even in the presence of obesity.
This study established IAPD as an independent risk factor of T2DM, and adjusted relative risk analyses demonstrated that various confounding factors did not have any effect on the outcome of the study (table 3). Regarding the cause and consequence relationship between IAPD and T2DM, at the beginning of study all participants were without diabetes and 386 participants had IAPD and therefore IAPD cannot be a consequence of T2DM; instead T2DM is the consequence of IAPD. In the context of IAPD being a causal factor of T2DM, we anticipate that a case–control interventional study with oral supplementation of IAP will be required. Based on our previous study, we proposed that ‘temporal IAP profiling’, defined as regular monitoring of stool IAP, should be able to diagnose if a person has ‘incipient (latent) metabolic syndrome’, including ‘incipient T2DM’.7 The current study also established that ‘temporal IAP profiling (regular STAP tests)’ will identify an individual vulnerable to develop T2DM.
We have previously shown that oral IAP supplementation prevents T2DM in mice,12 and the current human study shows that ‘remittent IAPD’ prevents T2DM (table 3). Based on these studies, we anticipate that oral IAP supplementation might prevent T2DM in humans.
We strongly believe that IAPD as an independent risk factor plays a pivotal role in the pathogenesis of T2DM in humans based on the following findings of our previous and current studies: (1) IAPD (IAP gene knockout) causes diabetes (hyperglycemia) in mice; (2) chronic inhibition of intestinal luminal IAP activity in mice by oral supplementation of phenylalanine, an inhibitor of IAP, in drinking water causes diabetes; (3) chronic oral supplementation of IAP prevents diabetes in IAP knockout mice; (4) IAPD is directly associated with T2DM in humans; (5) overall incidence rate of T2DM is approximately 4.2-fold higher in humans with IAPD; (6) overall relative risk of developing T2DM is increased by approximately 3.7-fold in humans with IAPD; (7) the lower the persistent IAP level the higher (approximately 16.1-fold) the incidence rate of T2DM in humans; (8) the lower the persistent IAP level the higher (approximately 13.8-fold) the relative risk of developing T2DM in humans; (9) the lower the persistent IAP level the higher (approximately twofold) the rate of increase of fasting glycemia in humans; and (10) remission of IAPD prevents T2DM in humans, as shown in this study and in Malo7 and Kaliannan et al.12 It is to note that an association between high serum alkaline phosphatase (liver/bone/kidney alkaline phosphatase) and new-onset T2DM has been reported in a population with hypertension.16 Serum alkaline phosphatase is a proinflammatory marker and we think increased serum alkaline phosphatase might indicate IAPD-induced systemic inflammation in this patient population. Based on this study and other published data, we developed a model depicting the molecular mechanisms of IAPD-mediated diabetes pathogenesis (figure 4). Brief description and related references on the steps of the diabetes pathogenesis pathway are provided in the following sections.
Figure 4.
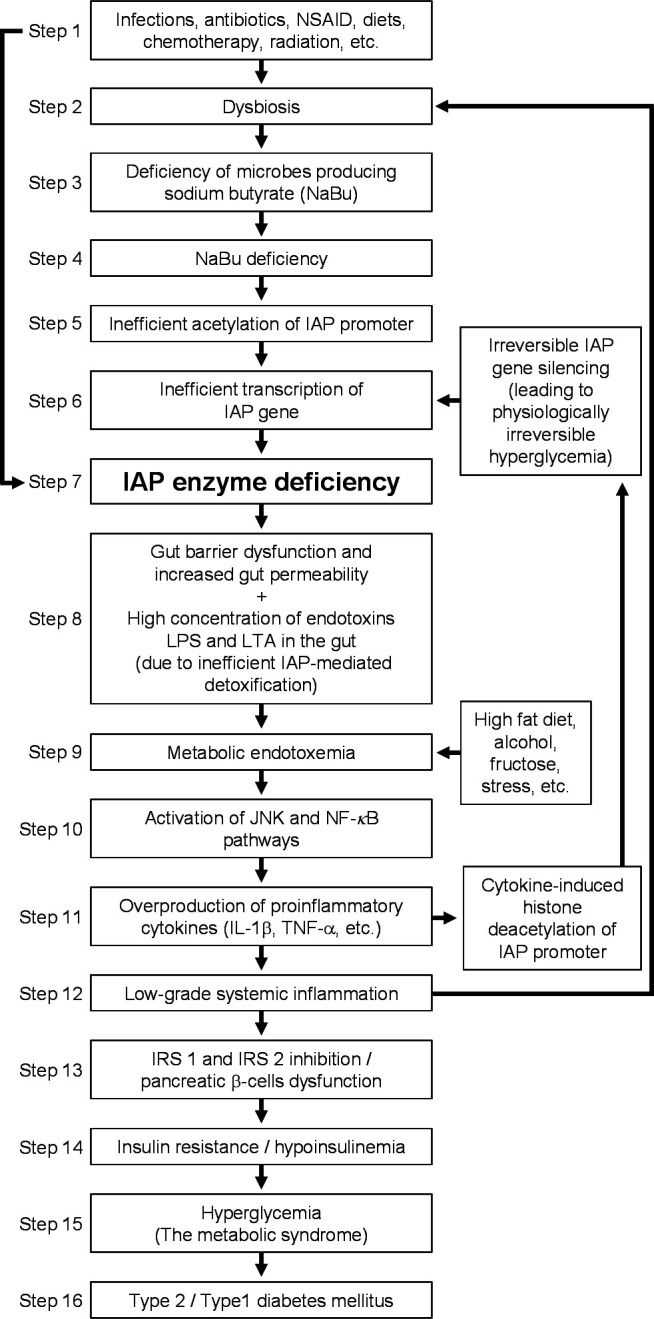
The pathway of diabetes pathogenesis. IAP, intestinal alkaline phosphatase; IL-1β, interleukin-1β; IRS, insulin receptor substrate; JNK, c-Jun N-terminal kinases; LPS, lipopolysaccharides; LTA, lipoteichoic acids; NF-kB, nuclear factor-kB; NSAID, non-steroidal anti-inflammatory drugs; TNF-α, tumor necrosis factor-α.
Steps 1 and 2: induction of dysbiosis
Dysbiosis is defined as an imbalance in type and number of normal microbes, and numerous diseases have been linked to dysbiosis including T2DM, type 1 diabetes mellitus (T1DM), cancer, coronary artery disease, and AIDS.17–19 The homeostasis of normal intestinal microbiota could be disrupted by various factors, leading to dysbiosis such as diets, infections, antibiotics, chemotherapy, and radiation.20–24 It is anticipated that some dysbiotic factors might reduce the number of bacteria that produce sodium butyrate (NaBu) (see steps 3 and 4).
Steps 3 and 4: sodium butyrate deficiency
Butyric acid, a short-chain fatty acid, is produced by fermentation of dietary fibers by the gut microbiota, and the deficiency of sodium butyrate (NaBu, sodium salt of butyric acid) is associated with specific types of dysbioses. Lassenius et al25 showed that NaBu deficiency as well as IAPD are associated with T1DM. In a metagenome-wide association study of gut microbiota in T2DM, Qin et al26 demonstrated a decrease of some species of universal butyrate-producing bacteria.
Steps 5–7: IAP enzyme deficiency
Histone deacetylases (HDACs) inhibit the transcription of a gene, and an HDAC inhibitor, such as NaBu, increases transcription. We have previously shown that, in human colon cancer HT-29 cells, NaBu transcriptionally activates the IAP gene by H3 histone hyperacetylation.27 28 It has been shown that NaBu upregulates the IAP gene in pig intestine explants.29 Based on the fact that specific types of dysbioses can decrease butyrate production (steps 3 and 4), it is expected that under such a dysbiotic condition the transcription of IAP gene is reduced or silenced, resulting in IAP enzyme deficiency. It is anticipated that factors described in step 1 can also directly affect intestinal physiology, leading to inhibition of IAP activity and/or gene expression.
Steps 8 and 9: development of metabolic endotoxemia
Metabolic endotoxemia is defined as a twofold to threefold persistent increase of bacterial endotoxin LPS in blood circulation compared with normal levels.7 12 13 30 Previously, we have shown that IAP dephosphorylates and thus detoxifies various proinflammatory factors, including LPS, lipoteichoic acids, cytosine-phosphate-guanosine DNA, ATP, uridine diphosphate, and flagellin.7 12 We have shown that IAPD leads to endotoxemia.12 It has also been demonstrated that endotoxemia is precipitated by alcohol, fructose, and stress.31
Steps 10–12: chronic systemic inflammation and irreversible IAP gene silencing
We and other laboratories have shown that metabolic endotoxemia in mice precipitates low-grade systemic inflammation as evidenced by increased serum levels of proinflammatory cytokines tumor necrosis factor-alpha (TNF-α) and interleukin 1beta (IL-1b).12 13 Also, human volunteers receiving endotoxin as intravenous bolus injection or infusion showed increased levels of TNF-α, IL-6, and IL-1b.32 33
In the presence of NaBu, we have previously shown that proinflammatory cytokines TNF-α and IL-1β cannot inhibit the expression of IAP in HT-29 cells. However, in the absence of NaBu, these cytokines inhibit the expression of IAP gene, and post-treatment with NaBu cannot reverse this IAP silencing, which indicates that proinflammatory cytokines in the absence of NaBu probably permanently silence the IAP gene.34 It is to point out that inflammation can also cause dysbiosis, further promoting IAPD.17
Steps 13 and 14: insulin resistance and hypoinsulinemia
Insulin resistance, defined as the impaired ability of circulating insulin to regulate normal cellular glucose uptake, occurs due to defective insulin-mediated signal transduction.35 Insulin binds to its receptor and activates its kinase domain, which is followed by activation of the insulin receptor substrate family (IRS 1–6), the scaffolding proteins required to mediate signal transduction.35 Gene knockout studies revealed that mice deficient in IRS-1 develop insulin resistance, whereas mice deficient in IRS-2 develop insulin resistance and also beta cell apoptosis, leading to hypoinsulinemia.36 37 The proinflammatory cytokines TNF-α and IL-1β inhibit the activity of IRS-1 and IRS-2 proteins and, as expected, lead to the development of insulin resistance and T2DM.38 39
Steps 15 and 16: development of the metabolic syndrome and T2DM
As discussed above, IAPD leads to metabolic endotoxemia, which precipitates low-grade systemic inflammation, and the resulting increased levels of proinflammatory cytokines TNF-α and IL-1β inhibit IRS proteins, leading to insulin resistance, hypoinsulinemia, hyperglycemia, and T2DM. Further, it is well known that chronic inflammation inflicts damages to pancreatic beta cells, vascular endothelial cells, and hepatocytes, and dysfunction of these cells precipitates metabolic syndrome, characterized by hyperglycemia, hypertension, and dyslipidemia.12 Metabolic syndrome ultimately leads to the development of T2DM, heart disease, hypertension, and other metabolic diseases.40 The model depicted here exemplifies the IAPD-mediated pathogenesis pathway of diabetes.
Conclusions
IAPD increases the risk of diabetes. Regular monitoring of stool alkaline phosphatase (STAP tests) would identify individual vulnerability to diabetes. Oral IAP supplementation might prevent diabetes.
Acknowledgments
We are grateful to Professors Syed S Islam, MBBS, PhD (Epidemiology, West Virginia University), Abu Abdullah, MBBS, MPH, PhD (Epidemiology, Boston University), and Abdullah Baqui, MBBS, MPH, DrPH (Johns Hopkins University), for their critical reviews of the manuscript and statistical analyses. We dedicate the article to Professor Syed S Islam, who recently died.
Footnotes
Contributors: Study concept and theory: MSM; research design: MSM, AKAK, JM, TS, SI, KK, MAH, SR, MFP; participant recruitment: MSM, JM, MMHR, GB, SAT, SKB, TS; data acquisition: MSM, JM, MMHR, GB, SAT, SKB, TS; data analyses and interpretation: MSM, AKAK, MJA, JM, SKB, TS, SI, KK, MAH, SR, MFP; statistical analyses: MSM, MJA, AKAK, JM, SKB, TS, SI, SR, KK, MAH, MFP; drafting of the manuscript: MSM, MJA, SI, AKAK, KK, MAH, SR, MFP; critical review of the manuscript for important intellectual content: all authors; obtained funding: MSM; approval of the manuscript: all authors; study supervision: MSM. MSM is the guarantor of this work.
Funding: The study was funded by grants (to MSM) from the Ministry of Education, the People’s Republic of Bangladesh (Dhaka, Bangladesh; grant number: LS2019937) and the Bangladesh Medical Research Council (Dhaka, Bangladesh; grant number BMRC/HPNSP-Grant/2019-2020/569(1-60)). The study was further supported by generous personal donations from Dr Mohammed Salehuddin, MD (Tuscaloosa, Alabama, USA) and Dr ABM Haroon, MBBS (Samorita Hospital, Dhaka, Bangladesh). The funding sources (Ministry of Education, the People’s Republic of Bangladesh, Dhaka, Bangladesh, and the Bangladesh Medical Research Council, Dhaka, Bangladesh) did not play any role in the development of theory and study design. The funding sources were also not involved in the collection, analyses, and interpretation of data, or in writing and submission of the manuscript. The corresponding author had full access to all the data in the study and took the decision for publication of the data.
Competing interests: None declared.
Provenance and peer review: Not commissioned; externally peer reviewed.
Supplemental material: This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information. All data generated or analyzed during this study are included in the published article. No applicable resources were generated or analyzed during the current study.
Ethics statements
Patient consent for publication
Not required.
Ethics approval
This study involves human participants and was reviewed and approved by the Bangladesh Medical Research Council, Government of the People's Republic of Bangladesh, Dhaka, Bangladesh (BMRC registration number: 150 02 10 2018; ref: BMRC/NREC/2016-2019/28). Participants gave informed consent to participate in the study before taking part.
References
- 1.American Diabetes Association . 2. Classification and Diagnosis of Diabetes: Standards of Medical Care in Diabetes-2018. Diabetes Care 2018;41:S13–27. 10.2337/dc18-S002 [DOI] [PubMed] [Google Scholar]
- 2.Saeedi P, Petersohn I, Salpea P, et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res Clin Pract 2019;157:107843. 10.1016/j.diabres.2019.107843 [DOI] [PubMed] [Google Scholar]
- 3.Cho NH, Shaw JE, Karuranga S, et al. IDF diabetes atlas: global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res Clin Pract 2018;138:271–81. 10.1016/j.diabres.2018.02.023 [DOI] [PubMed] [Google Scholar]
- 4.Zierath JR. Major Advances and Discoveries in Diabetes - 2019 in Review. Curr Diab Rep 2019;19:118. 10.1007/s11892-019-1255-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Skyler JS, Bakris GL, Bonifacio E, et al. Differentiation of diabetes by pathophysiology, natural history, and prognosis. Diabetes 2017;66:241–55. 10.2337/db16-0806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kahn SE, Cooper ME, Del Prato S. Pathophysiology and treatment of type 2 diabetes: perspectives on the past, present, and future. Lancet 2014;383:1068–83. 10.1016/S0140-6736(13)62154-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Malo MS. A high level of intestinal alkaline phosphatase is protective against type 2 diabetes mellitus irrespective of obesity. EBioMedicine 2015;2:2016–23. 10.1016/j.ebiom.2015.11.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Velloso LA, Eizirik DL, Cnop M. Type 2 diabetes mellitus--an autoimmune disease? Nat Rev Endocrinol 2013;9:750–5. 10.1038/nrendo.2013.131 [DOI] [PubMed] [Google Scholar]
- 9.Franks PW, McCarthy MI. Exposing the exposures responsible for type 2 diabetes and obesity. Science 2016;354:69–73. 10.1126/science.aaf5094 [DOI] [PubMed] [Google Scholar]
- 10.Masters SL, Latz E, O'Neill LAJ. The inflammasome in atherosclerosis and type 2 diabetes. Sci Transl Med 2011;3:81ps17. 10.1126/scitranslmed.3001902 [DOI] [PubMed] [Google Scholar]
- 11.Stumvoll M, Goldstein BJ, van Haeften TW. Type 2 diabetes: principles of pathogenesis and therapy. Lancet 2005;365:1333–46. 10.1016/S0140-6736(05)61032-X [DOI] [PubMed] [Google Scholar]
- 12.Kaliannan K, Hamarneh SR, Economopoulos KP, et al. Intestinal alkaline phosphatase prevents metabolic syndrome in mice. Proc Natl Acad Sci U S A 2013;110:7003–8. 10.1073/pnas.1220180110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cani PD, Amar J, Iglesias MA, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007;56:1761–72. 10.2337/db06-1491 [DOI] [PubMed] [Google Scholar]
- 14.Lallès J-P. Recent advances in intestinal alkaline phosphatase, inflammation, and nutrition. Nutr Rev 2019;77:710–24. 10.1093/nutrit/nuz015 [DOI] [PubMed] [Google Scholar]
- 15.Buchet R, Millán JL, Magne D. Multisystemic functions of alkaline phosphatases. Methods Mol Biol 2013;1053:27–51. 10.1007/978-1-62703-562-0_3 [DOI] [PubMed] [Google Scholar]
- 16.Zhang Y, Zhou C, Li J, et al. Serum alkaline phosphatase levels and the risk of new-onset diabetes in hypertensive adults. Cardiovasc Diabetol 2020;19:186. 10.1186/s12933-020-01161-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DeGruttola AK, Low D, Mizoguchi A, et al. Current understanding of dysbiosis in disease in human and animal models. Inflamm Bowel Dis 2016;22:1137–50. 10.1097/MIB.0000000000000750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carding S, Verbeke K, Vipond DT, et al. Dysbiosis of the gut microbiota in disease. Microb Ecol Health Dis 2015;26:26191. 10.3402/mehd.v26.26191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Malo MS, Alam SN, Mostafa G, et al. Intestinal alkaline phosphatase preserves the normal homeostasis of gut microbiota. Gut 2010;59:1476–84. 10.1136/gut.2010.211706 [DOI] [PubMed] [Google Scholar]
- 20.Brown K, DeCoffe D, Molcan E, et al. Diet-Induced dysbiosis of the intestinal microbiota and the effects on immunity and disease. Nutrients 2012;4:1095–119. 10.3390/nu4081095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Engen PA, Green SJ, Voigt RM, et al. The gastrointestinal microbiome: alcohol effects on the composition of intestinal microbiota. Alcohol Res 2015;37:223–36. [PMC free article] [PubMed] [Google Scholar]
- 22.Syer SD, Wallace JL, Environmental WJL. Environmental and NSAID-enteropathy: dysbiosis as a common factor. Curr Gastroenterol Rep 2014;16:377. 10.1007/s11894-014-0377-1 [DOI] [PubMed] [Google Scholar]
- 23.Montassier E, Gastinne T, Vangay P, et al. Chemotherapy-driven dysbiosis in the intestinal microbiome. Aliment Pharmacol Ther 2015;42:515–28. 10.1111/apt.13302 [DOI] [PubMed] [Google Scholar]
- 24.Stecher B. The roles of inflammation, nutrient availability and the commensal microbiota in enteric pathogen infection. Microbiol Spectr 2015;3. 10.1128/microbiolspec.MBP-0008-2014 [DOI] [PubMed] [Google Scholar]
- 25.Lassenius MI, Fogarty CL, Blaut M, et al. Intestinal alkaline phosphatase at the crossroad of intestinal health and disease - a putative role in type 1 diabetes. J Intern Med 2017;281:586–600. 10.1111/joim.12607 [DOI] [PubMed] [Google Scholar]
- 26.Qin J, Li Y, Cai Z, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012;490:55–60. 10.1038/nature11450 [DOI] [PubMed] [Google Scholar]
- 27.Hinnebusch BF, Henderson JW, Siddique A, et al. Transcriptional activation of the enterocyte differentiation marker intestinal alkaline phosphatase is associated with changes in the acetylation state of histone H3 at a specific site within its promoter region in vitro. J Gastrointest Surg 2003;7:237–45. discussion 44-5. 10.1016/S1091-255X(02)00140-3 [DOI] [PubMed] [Google Scholar]
- 28.Shin J, Carr A, Corner GA, et al. The intestinal epithelial cell differentiation marker intestinal alkaline phosphatase (ALPi) is selectively induced by histone deacetylase inhibitors (HDACi) in colon cancer cells in a Kruppel-like factor 5 (KLF5)-dependent manner. J Biol Chem 2014;289:25306–16. 10.1074/jbc.M114.557546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Melo ADB, Silveira H, Bortoluzzi C, et al. Intestinal alkaline phosphatase and sodium butyrate may be beneficial in attenuating LPS-induced intestinal inflammation. Genet Mol Res 2016;15:gmr15048875. 10.4238/gmr15048875 [DOI] [PubMed] [Google Scholar]
- 30.Erridge C, Attina T, Spickett CM, et al. A high-fat meal induces low-grade endotoxemia: evidence of a novel mechanism of postprandial inflammation. Am J Clin Nutr 2007;86:1286–92. 10.1093/ajcn/86.5.1286 [DOI] [PubMed] [Google Scholar]
- 31.Hamarneh SR, Kim B-M, Kaliannan K, et al. Intestinal alkaline phosphatase attenuates alcohol-induced hepatosteatosis in mice. Dig Dis Sci 2017;62:2021–34. 10.1007/s10620-017-4576-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Andreasen AS, Krabbe KS, Krogh-Madsen R, et al. Human endotoxemia as a model of systemic inflammation. Curr Med Chem 2008;15:1697–705. 10.2174/092986708784872393 [DOI] [PubMed] [Google Scholar]
- 33.Fullerton JN, Segre E, De Maeyer RPH, et al. Intravenous endotoxin challenge in healthy humans: an experimental platform to investigate and modulate systemic inflammation. J Vis Exp 2016. 10.3791/53913. [Epub ahead of print: 16 05 2016]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Malo MS, Biswas S, Abedrapo MA, et al. The pro-inflammatory cytokines, IL-1beta and TNF-alpha, inhibit intestinal alkaline phosphatase gene expression. DNA Cell Biol 2006;25:684–95. 10.1089/dna.2006.25.684 [DOI] [PubMed] [Google Scholar]
- 35.Haeusler RA, McGraw TE, Accili D. Biochemical and cellular properties of insulin receptor signalling. Nat Rev Mol Cell Biol 2018;19:31–44. 10.1038/nrm.2017.89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Araki E, Lipes MA, Patti ME, et al. Alternative pathway of insulin signalling in mice with targeted disruption of the IRS-1 gene. Nature 1994;372:186–90. 10.1038/372186a0 [DOI] [PubMed] [Google Scholar]
- 37.Withers DJ, Gutierrez JS, Towery H, et al. Disruption of IRS-2 causes type 2 diabetes in mice. Nature 1998;391:900–4. 10.1038/36116 [DOI] [PubMed] [Google Scholar]
- 38.Bouzakri K, Ribaux P, Halban PA. Silencing mitogen-activated protein 4 kinase 4 (MAP4K4) protects beta cells from tumor necrosis factor-alpha-induced decrease of IRS-2 and inhibition of glucose-stimulated insulin secretion. J Biol Chem 2009;284:27892–8. 10.1074/jbc.M109.048058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ballak DB, Stienstra R, Tack CJ, et al. Il-1 family members in the pathogenesis and treatment of metabolic disease: focus on adipose tissue inflammation and insulin resistance. Cytokine 2015;75:280–90. 10.1016/j.cyto.2015.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Saklayen MG. The global epidemic of the metabolic syndrome. Curr Hypertens Rep 2018;20:12. 10.1007/s11906-018-0812-z [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
bmjdrc-2021-002643supp001.pdf (533KB, pdf)
Data Availability Statement
All data relevant to the study are included in the article or uploaded as supplementary information. All data generated or analyzed during this study are included in the published article. No applicable resources were generated or analyzed during the current study.