

Abstract
Glioblastoma and other high-grade gliomas (HGGs) are the most common and deadly primary brain tumors. Due to recent advances in immunotherapy and improved clinical outcomes in other disease sites, the study of immunotherapy in HGG has increased significantly. Herein, we summarize and evaluate existing evidence and ongoing clinical trials investigating the use of immunotherapy in the treatment of HGG, including therapeutic vaccination, immune checkpoint inhibition, adoptive lymphocyte transfer, and combinatorial approaches utilizing radiation and multiple modalities of immunotherapy. Special attention is given to the mechanisms by which radiation may improve immunogenicity in HGG, why this motivates the study of radiation in combination with immunotherapy, and how to determine optimal dosing and scheduling of radiation. Though larger randomized controlled trials have not consistently shown improvements in clinical outcomes, this area of research is still in its early stages and a number of important lessons can be taken away from the studies that have been completed to date. Many studies found a subset of patients who experienced durable responses, and analysis of their immune cells and tumor cells can be used to identify biomarkers that predict therapeutic response, as well as additional glioma-specific targets that can enhance therapeutic efficacy in a challenging tumor type.
Keywords: Glioblastoma (GBM), vaccine, adoptive transfer, immune checkpoint inhibitor (ICI), radiotherapy
Introduction
Primary brain tumors are a heterogeneous group of cancers with an estimated 23,820 new cases and 17,760 deaths in the United States in 2019 (1). Glioblastoma (GBM), a type of high-grade glioma (HGG), is the most common primary brain tumor, accounting for about 80% of cases, affecting patients of all ages. Standard treatment for GBM includes maximal safe resection followed by chemoradiation [radiotherapy with daily temozolomide (TMZ)] and 6–12 cycles of adjuvant TMZ, with or without alternating electric field therapy (2-4). Despite advances in all three modalities, clinical outcomes after standard treatment remain poor with a median survival in the range of 15 months and 5-year overall survival (OS) of less than 10% (5,6). Unfortunately, with a median progression-free survival (PFS) of approximately 7 months after initial treatment, patients overwhelmingly develop recurrent disease, portending a median survival of 1–4.5 months from the time of progression (7). Various second-line treatments including additional surgical resection, alternating electric field therapy, bevacizumab (anti-VEGF), and chemotherapy are used, but none have consistently demonstrated prolonged survival after recurrence (8-13). In recent years, immune checkpoint inhibitors (ICIs) have demonstrated efficacy in multiple malignancies, including melanoma (14-16), renal cell carcinoma (17), non-small cell lung cancer (NSCLC) (18,19), and small cell lung cancer (20). In response to the clear need for improved treatments, and the promising results seen in other settings, the investigation of immunotherapy for the treatment of primary brain tumors has increased significantly. In this review, we will discuss the unique immunologic characteristics of the central nervous system (CNS), as well as the existing and emerging evidence regarding the use of immunotherapy in HGG. We will then focus our discussion on combinatorial therapies with an emphasis on the use of immunotherapy in conjunction with radiation in HGG. Clinical trials were identified based on the most advanced phase of research and most recent publication for all types of immunotherapy in HGG. We present the following article in accordance with the Narrative Review reporting checklist (available at http://dx.doi.org/10.21037/tcr-20-1933).
Immune reactions in the CNS
The CNS has traditionally been considered an immune-privileged site. This assumption was based, in part, on observations that allogeneic tissue grafts placed in the brains of animal models could actively grow (21). This phenomenon was attributed to the presence of the blood-brain barrier (BBB) and the absence of a lymphatic drainage system or specialized antigen presenting cells (APCs) (22-24). However, this assumption has been challenged by a number of studies that have instead shown an immune-specialized capacity of the CNS (25). Microglia have been identified as resident APCs in the CNS given their phenotypical and functional similarities to professional APCs, namely dendritic cells (DCs) and macrophages (26,27). Pathways of lymphatic drainage have been identified along cranial nerve sheaths (28), with up to 47% of colony-stimulating factor (CSF) draining to cervical lymph nodes observed in one study of radio-labeled albumin injected into the brains of rabbits (29), with confirmation in more modern studies (30,31). The BBB, while regulating ion concentrations and preventing the passive transport of macromolecules (32), does not uniformly prevent the passage of immune cells. Activated cytotoxic T cells (CTLs) specific to CNS antigens cross the BBB (33). These findings suggest that tumor-specific antigens (TSAs) could be presented by microglia within the CNS or by professional APCs within cervical lymph nodes and initiate a T cell-mediated response against tumor cells. A summary of immune mechanisms and therapeutic targets is shown in Figure 1.
Figure 1.
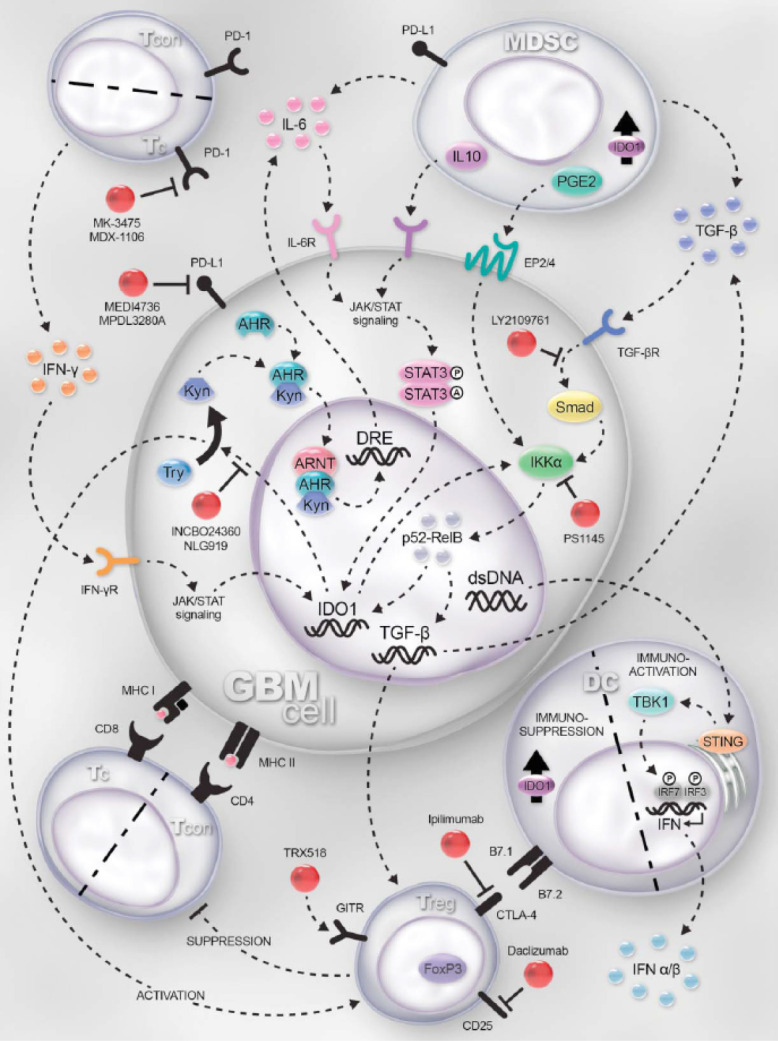
Mechanisms of immune reactions and therapeutic targets in glioblastoma (GBM). GBM cells, tumor-resident dendritic cells (DCs) and myeloid-derived suppressor cells (MDSC) express indoleamine-2,3-dioxygenase 1 (IDO1). IDO1 expression is regulated by the JAK/STAT and NF-κB pathways, which is induced by IFN-γ and TGF-β-receptor activation, respectively. IDO1 is a cytoplasmic enzyme that metabolizes tryptophan (Trp) to kynurenine (Kyn). Within the GBM cell, Kyn complexes with the aryl hydrocarbon receptor (Ahr), cytoplasmically, facilitating the nuclear translocation and further docking with aryl hydrocarbon receptor nuclear translocator (ARNT) to transcriptionally regulate IL-6, acting as an autocrine loop that amplifies and sustains IDO1 expression. Simultaneously, extracellular Kyn suppresses T effector responses while activating regulatory T cell (Treg; CD4+CD25+FoxP3+) function through a presumably overlapping mechanism. IDO1 directly activates NF-kB signaling which maintains and/or upregulates TGF-β expression. Increased TGF-β levels upregulate CTLA-4 and GITR expression by Treg. CTLA-4 interacts with B7.1 (CD80) and B7.2 (CD86) on DC, resulting in the induction of IDO1 (in DC) and commensurate downregulation of antigen presentation to T cells. Both GBM and MDSC express TGF-b, which synergizes with PD-L1 to suppress the T cell effector response via interaction with PD-1. Moreover, interleukin-10 (IL-10)- and prostaglandin E2 (PGE2)-expressing MDSC act on their cognate receptors expressed by GBM to ramify JAK/STAT and NK-κB-mediated signaling. DNA released by dead/dying GBM cells is phagocytized by resident DC to activate the STING pathway leading to type 1 interferon (α/β) expression, supporting increased effectiveness of anti-GBM immunity. PD-1 is highly expressed by tumor-infiltrating cytotoxic T cells and PD-L1 is upregulated on cancer/stromal cells in response to T-cell-secreted IFN-γ. Blocking the interaction of PD-1-expressing T cells with PD-L1 leads to increased effector function and enhanced GBM immunity. Targets for immunomodulation are shown in red. Note: Although IDO1 expression and signaling are shown in GBM cells, shared signaling patterns are presumed to be present in DC and MDSC as well. TCON: conventional CD4+FoxP3− T cell; TREG: regulatory CD4+FoxP3+ T cell; TC: cytotoxic CD8+ T cell; INCBO24360/NLG919: inhibitors of IDO1; PS1145: inhibitor of the NF-κB pathway; TRX518: humanized monoclonal agonistic antibody for GITR; Ipilimumab: humanized monoclonal antibody for CTLA-4; LY2109761: TGF-β receptor kinase inhibitor; MK-3475/MDX-1106: humanized monoclonal antibodies to PD-1; MEDI4736/MPDL3280A: humanized monoclonal antibodies to PD-L1; Anti-Gr1: mSC-depleting antibody; Daclizumab: humanized anti-CD25 (IL-2Ra); STING: stimulator of interferon genes; TBK1: TANK-binding kinase 1; IRF3/7: interferon regulatory factor 3/7; STAT3: signal transducer and activator of transcription 3. Reprinted from Binder et al. (25) (https://www.tandfonline.com/doi/full/10.1080/2162402X.2015.1082027?scroll=top&needAccess=true) without changes under the terms of the Creative Commons Attribution License.
The critical question now is not whether tumor-directed immune reactions occur in the CNS, but rather how the immune specializations within the brain can hinder or enhance a tumor-specific immune response. We will discuss the history and current state of clinical research of immunotherapy in HGG, as well as how the lessons learned from these studies are directing ongoing clinical trials and future research.
Therapeutic vaccination
Therapeutic vaccination is intended to generate a tumor- and patient-specific immune response (34). This is achieved by inoculating the patient with TSAs or tumor-associated antigens (TAAs). These antigens are ingested by DCs and other professional APCs and presented to naïve CD8+ and CD4+ T cells via major histocompatibility complex (MHC)-I and MHC-II, respectively. In vivo or co-administered stimulatory agents lead these T cells to mature into antigen-specific CTLs (CD8+), which initiate a cell-mediated response, and helper T cells (CD4+ Th1 and Th2 cells), which initiate an antibody-mediated response (35). Ideally, this cascade leads to an acquired tumor-directed immune response that correlates to clinically significant disease control. While meant to achieve the same end result, vaccines come in various forms, including peptide vaccines, heat shock protein (HSP) vaccines, and DC vaccines, each of which are forms of active immunotherapy, in which the immune system is activated to target cancer cells (36,37). On the other hand, viral, or oncolytic, vaccines are a form of gene therapy, in which a tumor-associated gene is modified to create a tumor-targeted virus (36). Completed and ongoing clinical trials investigating these therapies in adult HGG are summarized in Table 1.
Table 1. Therapeutic vaccination clinical trials.
| Trial number | Abbreviated trial name | Target | Study design | Population | Intervention/arms | Control | Number of analyzed patients | Survival, median months (95% CI) | Additional outcomes | Trial status | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Peptide vaccines | |||||||||||
| NCT00643097 | ACTIVATE | EGFRvIII | Phase II, single arm | Adults, new GBM s/p GTR and chemoRT | Rindopepimut vaccine every 2 weeks ×3 then monthly concomitant with standard adjuvant TMZ, until progression or death | Matched cohort (more KPS 80) | Experimental: n=18; Matched cohort: n=17 | PFS: 14.2 (9.9–17.6) vs. 6.3 (4.1–9.0); OS: 26.0 (21.0–47.7) vs. 15.0 (11.4–19.8) | – | Published | (38) |
| NCT00643097 | ACT II | EGFRvIII | Phase II, 2 arms | Adults, new GBM s/p GTR and chemoRT | Rindopepimut vaccine every 2 weeks ×3 then monthly until progression or death and after the 3rd vaccine: Arm I: standard dose TMZ (200 mg/m2 on days 1–5); Arm II: dose intensified TMZ (100 mg/m2 on days 1–21) | Matched cohort (same as above) | Arm I: 12; Arm II: 10; matched cohort: same as above | PFS (Arms I and II): 15.2 (11.0–18.5); OS (Arms I and II): 23.6 (18.5–33.1) | – | Published | (39) |
| NCT00458601 | ACT III | EGFRvIII | Phase II, single arm | Adults, new GBM s/p GTR and chemoRT | Rindopepimut vaccine every 2 weeks ×3 then monthly, concomitant with standard adjuvant TMZ until intolerance or progression | None | 65 | PFS: 9.2 (7.4–11.3); OS: 21.8 (17.9, 26.5) | – | Published | (40) |
| NCT01480479 | ACT IV | EGFRvIII | Phase III, randomized, double-blind, placebo-controlled | Adults, new GBM s/p GTR and chemoRT | Rindopepimut vaccine every 2 weeks ×2 then monthly, concomitant with standard adjuvant TMZ until intolerance or progression | Placebo vaccine | 405 | PFS: 8.0 (7.1–8.5) vs. 7.4 (6.0–8.7); OS: 20.1 (18.5–22.1) vs. 20.0 (18.1–21.9) | – | Published | (41) |
| NCT01498328 | ReACT | EGFRvIII | Phase II, randomized, double-blind, placebo-controlled | Adults, relapsed GBM, bevacizumab-naïve | Rindopepimut vaccine every 2 weeks ×3 then monthly with bevacizumab given every 2 weeks | Placebo vaccine | 73 | PFS6: 28% vs. 16% (P=0.12); PFS: HR 0.72 (0.43–1.21, P=0.22); OS: HR 0.53 (0.32–0.88, P=0.01) | – | Published | (42) |
| NCT02454634 | NOA-16 | IDH1R132H | Phase I, single arm | Adults, new HGG s/p chemoRT | IDH1R132H peptide vaccine every 4 weeks ×8 with topical imiquimod, concomitant with standard adjuvant TMZ | None | 33 | 32-week PFS: 87.5% | 2 serious AEs, 1 probably related; 93.3% CTL and/or humoral response | Completed, abstract only | (43) |
| NCT02193347 | RESIST | IDH1R132H | Phase I, single arm | Adults, recurrent grade II gliomas | IDH1R132H peptide vaccine every 2 weeks ×3, followed by re-resection, followed by maintenance vaccine with TMZ | None | 24 enrolled | N/A | Primary: toxicity; secondary: immunogenicity | Active, not recruiting | N/A |
| NCT01250470 | SurVaxM Peptide Vaccine | Survivin | Phase I, single arm | Adults, recurrent HGG, HLA-A*02(+) or HLA-A*03(+) | SurVaxM vaccine every 2 weeks ×4 | None | 9 | PFS: 4.1; OS: 20.2 | 1 G3 AE, not related to vaccine | Completed | (44) |
| Multi-peptide vaccines | |||||||||||
| NCT01222221 | Cancer Research UK IMA950-101 | 11 GBM TAAs | Phase I, 2 arms | Adults, new GBM, HLA-A*02(+) | IMA950/GM-CSF vaccine injected 11 times over 24 weeks: Arm I: started 7–14 days prior to chemoRT; Arm II: started 7 days after chemoRT, concomitant with standard adjuvant TMZ | None | 45: Arm I: 22; Arm II: 23 | PFS6: 74.4%; PFS9: 30.8%; OS: 15.3 months | 2 dose-limiting grade 3 AEs; OS for ISR vs. no ISR: 26.7 vs. 13.2 (HR 0.33, P=0.0001) | Published | (45) |
| NCT01920191 | IMA950 Multi-peptide Vaccine with Poly-ICLC | 11 GBM TAAs | Phase I/II, single arm | Adults, new HGG, HLA-A*02(+) | IMA950/poly-ICLC vaccine injected 9 or 11 (protocol revision) times over 24 weeks starting 7 days concomitant with standard adjuvant TMZ | None | GBM: 16; Grade III astrocytoma: 3 | For GBM pts: PFS6: 81%; PFS9: 63%; OS: 19 months (17.3–27.9) | – | Published | (46) |
| UMIN000001243 | ITK-1 Personalized Peptide Vaccine | 4 of 14 GBM TAAs based on 4 highest IgG titers | Phase I, single arm | Adults, recurrent GBM, HLA-A*24(+) | ITK-1 (14 peptide candidates, 4 chosen per highest IgG titers for each) vaccine every week ×6 | None | 12 | PFS6 16.7%; PFS: 2.3 (1.7–3.5); OS: 10.6 (8.0–12.5) | – | Published | (47) |
| N/A | ITK-1 Personalized Peptide Vaccine vs. Placebo | 4 of 14 GBM TAAs based on 4 highest IgG titers | Phase III, randomized, double-blind, placebo-controlled | Adults, recurrent GBM, HLA-A*24(+) | ITK-1 (12 peptide candidates, 4 chosen per highest IgG titers for each) vaccine every week ×12 | Placebo vaccine | 88: Experimental: 58; Control: 30 | OS: 8.4 (6.6–10.6) vs. 8.0 (4.8–12.9) | Unfavorable OS in experimental group associated with SART2-93 peptide selection, ≥70 years old, >70 kg body weight, and PS 3; OS for pts without SART2-93 and age <70 years old: 9.6 (7.3–12.0) vs. 4.7 (3.7 vs. 6.8), HR 0.49, P=0.031 | Published | (48) |
| NCT02149225 | GAPVAC-101 | 5–10 unmutated TAAs and 1–2 mutated TAAs based on tumor mutation/transcriptome analysis | Phase I, single arm | Adults, new GBM, HLA-A*02(+) or HLA-A*24(+) | APVAC1 (5–10 synthetic unmutated antigens), followed by APVAC2 (1–2 synthetic neoantigens), concomitant with standard adjuvant TMZ. Both vaccines personalized based patient's tumor mutation/transcriptome analyses | None | 15 | PFS: 14.2; OS: 29.0 | – | Published | (49) |
| UMIN000000002 | An Autologous Tumor Vaccine with RT | Autologous tumor peptides | Phase I/IIa, single arm | Adults, new GBM | Autologous formalin-fixed tumor vaccine every week ×3 starting on week 4 of radiation without no TMZ | None | 22 | PFS: 7.6 (4.3–13.6); OS: 19.8 (13.8–31.3) | – | Published | (50) |
| UMIN000001426 | An Autologous Tumor Vaccine with Adjuvant TMZ | Autologous tumor peptides | Phase I/IIa, single arm | Adults, new GBM | Autologous formalin-fixed tumor vaccine every week ×3 starting on first day of standard adjuvant TMZ | None | 24 | PFS: 8.2 (CI N/R); 33% progression free at 24 months, associated with diameter of DTH (delayed-type hypersensitivity) response; OS: 22.2 (2.7–41.7) | – | Published | (51) |
| NCT01903330 | Bevacizumab +/− Gliovac (ERC1671) | Autologous tumor peptides and pooled allogeneic peptides | Phase II, randomized, double-blind, placebo-controlled | Adults, recurrent GBM, bevacizumab-naïve | Gliovac (ERC1671, autologous and pooled allogenic tumor peptides), GM-CSF, cyclophosphamide, and bevacizumab in 4-week cycles until intolerance or progression | Placebo and bevacizumab | 84 expected | Interim analysis (n=9): PFS: 7.3 vs. 5.4; OS: 12.1 vs. 7.6 | Primary: toxicity; secondary: PFS, OS, immunogenicity | Recruiting, interim results published | (52) |
| Heat shock protein vaccines | |||||||||||
| NCT00293423 | HSPPC-96 Vaccine (phase I) | HSPPC-96 | Phase I, single arm | Adults, recurrent GBM | Re-resection followed by autologous HSPPC-96 vaccine every 2 weeks (n=6) or every week ×4 then every 2 weeks (n=6) until intolerance or progression | None | 12 | OS: 10.1 | No severe AEs | Published | (53) |
| NCT00293423 | HSPPC-96 Vaccine (phase II) | HSPPC-96 | Phase II, single arm | Adults, recurrent GBM | Re-resection followed by autologous HSPPC-96 vaccine every 2 weeks (n=6) or every week ×4 then every 2 weeks (n=6) until intolerance or progression (median 6 doses) | None | 41 | PFS: 4.4 (3.2–5.5); OS: 9.8 (8.0–11.6) | 1 serious AE related to vaccine | Published | (54) |
| NCT01814813 | Bevacizumab +/− HSPPC-96 Vaccine | HSPPC-96 | Phase II, randomized | Adults, recurrent GBM | Re-resection followed by: Arm I: HSPPC-96 vaccine and concomitant bevacizumab for 12 2-week cycles or until progression; Arm II: HSPPC-96 vaccine, bevacizumab added if progression, for 12 2-week cycles or until 2nd progression | Arm III: bevacizumab alone until progression | 90 | PFS (P<0.01), OS (P=0.16): Arm I: 3.7 (2.9–5.4), 6.6 (5.4–10.4); Arm II: 2.5 (2.0–3.5), 9.2 (5.7–11.6); Arm III: 5.3 (3.7–8.0), 10.7 (8.8–17.2) | – | Active, not recruiting, unpublished data | (55) |
| NCT00905060 | HeatShock | HSPPC-96 | Phase II, single arm | Adults, new GBM s/p ≥90% resection and chemoRT | HSPPC-96 vaccine weekly ×4, then monthly concomitant with standard adjuvant TMZ, until depletion of vaccine or progression | None | 46 | PFS: 17.8 (11.3–21.6); OS: 23.8 (19.8–30.2) | – | Completed, abstract only | (56) |
| NCT03650257 | Adjuvant TMZ +/− HSPPC-96 Vaccine | HSPPC-96 | Phase II, randomized | Adults, new GBM s/p ≥80% resection and chemoRT | Starting 2 weeks after chemoRT, HSPPC-96 vaccine weekly ×4, then in 2 weeks, then in 3 weeks concomitant with standard adjuvant TMZ | SOC treatment | 150 expected | N/A | Primary: OS; secondary: PFS, immunogenicity, AEs | Recruiting | N/A |
| Dendritic cell vaccines—peptide loaded | |||||||||||
| N/A | EGFRvIII-targeted DC Vaccine | EGFRvIII | Phase I, single arm, 3+3 | Adults, new GBM s/p resection and chemoRT | After standard chemoRT, DC vaccine every 2 weeks ×3, dose escalated in groups of 3 patients | Historical control | 12 | OS: 22.8 (17.5–29.0) vs. 15.6 (historical) | – | Published | (57) |
| N/A | ICT-107 | 6 GBM TAAs | Phase I, single arm | Adults, new or recurrent GBM or brainstem glioma, HLA-A*01(+) and/or HLA-A*02(+) | ICT-107 DC vaccine every 2 weeks ×3 after standard chemoRT (new) or re-resection (recurrent) and prior to standard adjuvant TMZ (new) | None | New GBM: 16; Recurrent GBM: 3; Brainstem: 1 | New GBM (n=16); PFS: 16.9 (8.9–49.8); OS: 38.4 (25.9–40.7) | – | Published | (58) |
| NCT01280552 | Adjuvant TMZ +/− ICT-107 | 6 GBM TAAs | Phase II, randomized, double-blind, placebo-controlled | Adults, new GBM, HLA-A*01(+) and/or HLA-A*02(+) | ICT-107 DC vaccine weekly ×4 prior to standard adjuvant TMZ, then concomitant with TMZ at month 1, 3, 6, and every 6 months thereafter | Placebo control (unpulsed DCs) | Experimental: 81; Control: 43 | PFS: 11.2 (8.2–13.1) vs. 9.0 (5.5–10.3), HR=0.57, P=0.011; OS: 17.0 (13.7–20.6) vs. 15.0 (12.3–23.1), HR=0.87, P=0.58 | – | Published | (59) |
| Dendritic cell vaccines—tumor lysate loaded | |||||||||||
| NCT00068510 | Autologous Tumor Lysate DC Vaccine (phase I) | Tumor lysate (autologous) | Phase I, single arm | Adults, new HGG s/p resection and chemoRT | Before standard adjuvant TMZ, DC vaccine every 2 weeks ×3, then concomitant with TMZ every 3 months until depletion of vaccine or progression | Phase I cohort of peptide-loaded DC vaccine (NCT00612001) | 28 | PFS: 18.1 vs. 9.6; OS: 34.4 vs. 14.5 | – | Published | (60) |
| EY-DOH-MD #0910072504 | Autologous Tumor Lysate DC Vaccine (phase I/II) | Tumor lysate (autologous) | Phase I/II, single arm | Adults, new or recurrent HGG | After resection and standard RT (new, use of TMZ not reported) or re-resection (recurrent), DC vaccine given weekly ×4, every 2 weeks ×2, every month ×4 | Matched cohort | New GBM: 8; Recurrent GBM: 8; Recurrent WHO III: 1 | New and recurrent GBM (n=16): OS: 17.1 vs. 12.5 | – | Published | (61) |
| N/A | ChemoRT +/− Autologous Tumor Lysate DC Vaccine | Tumor lysate (autologous) | Phase II, randomized | Adults, new GBM s/p resection and chemoRT | Concomitant with chemoRT, DC vaccine given 10 times over 6 months | SOC treatment | Experimental: n=18; SOC control: n=16 | PFS: 8.5 vs. 8.0 (P=0.075); OS: 31.9 vs. 15.0 (P<0.002) | – | Published | (62) |
| NCT01006044 | Autologous Tumor Lysate DC Vaccine (phase II) | Tumor lysate (autologous) | Phase II | Adults, new GBM s/p resection with <1 cc residual | DC vaccine given prior to chemoRT ×1, then concomitant with standard adjuvant TMZ, monthly ×3, bimonthly ×4, quarterly until depletion of vaccine | None | 31 | PFS: 12.7 (7–16); OS: 23.4 (16–33.1) | – | Published | (63) |
| NCT00576446 | Resection with Gliadel Wafers followed by Autologous Tumor Lysate DC Vaccine | Tumor lysate (autologous) | Phase I, single arm | Adults, new or recurrent HGG | Resection with Gliadel Wafer placement, then DC vaccine given every 2 weeks ×3 (sequencing with standard therapies N/R) | None | New GBM: 8; New WHO III: 3; Recurrent GBM: 15; Recurrent WHO III: 2 | PFS, OS: New GBM: 4.8 (1.2–25.5), 27.7 (10.5–39.1); recurrent GBM: 1.9 (0.6–3.6), 10.9 (6.3–21.3) | – | Published | (64) |
| NCT01213407 | Adjuvant TMZ +/− Audencel | Tumor lysate (autologous) | Phase II, randomized | Adults, new GBM, s/p resection (≥ 70%) and chemoRT | After chemoRT, Audencel (DC vaccine) given weekly ×4, then monthly concomitant with standard adjuvant TMZ | SOC treatment | Experimental: 34; SOC control: 42 | PFS: 6.6 (4.5–9.1) vs. 6.9 (5.8–9.3), P=0.83; OS: 18.3 (14.2–21.8) vs. 18.4 (11.3–22.1), HR=0.99, P=0.89 | – | Published | (65) |
| NCT00045968 | Adjuvant TMZ +/− DCVax®-L | Tumor lysate (autologous) | Phase III, randomized, double-blind, placebo-controlled | Adults, new GBM s/p resection and chemoRT | Concomitant with standard adjuvant TMZ, DCVax-L every 10 days ×3, then months 2, 4, 8, then every 6 months thereafter, crossover permitted (90% received DCVax-L) | Placebo vaccine | Experimental: 232; Control: 99 | Entire cohort: OS: 23.1 (21.2–25.4) | – | Active, not recruiting, interim results published | (66) |
| NCT01808820 | Autologous Tumor Lysate DC Vaccine with Imiquimod | Tumor lysate (autologous) | Phase I, single arm | Subjects 13 y or older, recurrent HGG, s/p re-resection (≤2 cc residual) | DC vaccine/imiquimod given weekly ×4 | None | 20 expected | N/A | Primary: toxicity; secondary: PFS, OS, immunogenicity | Active, not recruiting | N/A |
| NCT01204684 | Autologous Tumor Lysate DC Vaccine +/− Imiquimod or Poly-ICLC | Tumor lysate (autologous) | Phase II, randomized | Adults, new or recurrent HGG | Arm I: DC vaccine with placebo cream or injection | Arm I | 60 expected | N/A | Primary: most effective combination; secondary: PFS, OS | Active, not recruiting | N/A |
| Arm II: DC vaccine with imiquimod cream | |||||||||||
| Arm III: DC vaccine with poly-ICLC (sequencing with standard therapies N/R) | |||||||||||
| NCT01957956 | Allogenic Tumor Lysate DC Vaccine (New GBM) | Tumor lysate (allogeneic) | Early Phase I | Adults, new GBM | DC Vaccine starting on second cycle of standard adjuvant TMZ, every 4 weeks ×11 or until intolerance or progression | None | 21 expected | N/A | Primary: toxicity; secondary: OS, PFS, ORR, time to response, duration of response | Active, not recruiting | N/A |
| NCT03360708 | Allogenic Tumor Lysate DC Vaccine (recurrent GBM) | Tumor lysate (allogeneic) | Early phase I | Adults, recurrent GBM | DC vaccine every 3 weeks ×13 or until intolerance or progression | None | 20 expected | N/A | Primary: toxicity; secondary: OS, PFS, ORR, time to response, duration of response | Recruiting | N/A |
| NCT02010606 | Allogenic Tumor Lysate DC Vaccine from a GBM Stem-like Cell Line | Tumor lysate (allogeneic) | Phase I, single arm | Adults, new or recurrent GBM | Starting after chemoRT (new), DC vaccine given weekly ×4, then every 8 weeks concomitant with standard adjuvant TMZ, until depletion of vaccine or progression | None | 39 enrolled | N/A | Primary: toxicity; secondary: OS, PFS, QoL, ORR, immunogenicity | Active, not recruiting | N/A |
| Dendritic cell vaccines—mRNA loaded | |||||||||||
| NCT00846456 | GSC Antigen mRNA DC Vaccine | Autologous GSC antigens | Phase I/II, single arm | Adults, new GBM s/p resection (≤5 cc residual) | After chemoRT, DC vaccine twice in first week, weekly ×3, then every 2 weeks concomitant with standard adjuvant TMZ | Matched cohort (n=10) | 7 | PFS: 22.8 vs. 7.8, P=0.0018; OS: 25.0 vs. 19.2, P=0.11 | – | Completed, interim results published | (67) |
| NCT02649582 | ADDIT-GLIO | WT1 | Phase I/II, single arm | Adults, new GBM | One week after chemoRT, DC vaccine given weekly ×3, then monthly concomitant with standard adjuvant TMZ, for up to 12 cycles | None | 20 expected | N/A | Primary: OS; secondary: feasibility, toxicity, immunogenicity, ORR, QoL | Recruiting | N/A |
| NCT02709616 | PERCELLVAC | Autologous TAAs | Phase I, single arm | Adults, new GBM | After chemoRT, DC vaccine every 2 weeks concomitant with standard adjuvant TMZ | None | 10 expected | N/A | Primary: toxicity; secondary: OS, PFS, immunogenicity | Active, not recruiting | N/A |
| Dendritic cell vaccines—glioma stem cell loaded | |||||||||||
| NCT01567202 | ChemoRT +/− Autologous GSC DC Vaccine | Autologous GSC antigens | Phase II, randomized, double-blind, placebo-controlled | Adults, new or recurrent GBM s/p ≥95% resection | 2 weeks after resection, DC vaccine weekly ×3, prior to and/or concomitant with standard chemoRT (new GBM) | Placebo vaccine | Experimental: 22; Placebo control: 21 | PFS: 7.7 vs. 6.9, P=0.75; OS: 13.7 vs. 10.7, P=0.05 | – | Published | (68) |
| NCT01171469 | Allogeneic GSC DC Vaccine | Allogeneic GSC antigens | Phase I, single arm | All ages, recurrent HGG or medulloblastoma | DC vaccine/imiquimod every 2 weeks ×4, then every 4 weeks ×10 | None | 8 enrolled | N/A | Primary: MTD; secondary: time to progression | Completed, not published | N/A |
| Dendritic cell vaccines—viral antigen loaded | |||||||||||
| NCT00639639 | CMV pp65 DC vaccine | pp65 | Phase I, single arm | Adults, new GBM s/p >90% resection | DC vaccine given every 2 weeks ×3, then every 4 weeks ×3–9 or until progression, concomitant with adjuvant dose intensified TMZ | Matched cohort (n=23) | 11 | PFS: 25.3 (11.0–not reached) vs. 8.0 (6.2–10.8), P=0.0001; OS: 41.1 (21.6–not reached) vs. 19.2 (14.3–21.3), P=0.0001 | – | Published | (69) |
| NCT03927222 | I-ATTAC | pp65 | Phase II, single arm | Adults, new GBM, MGMT unmethylated, CMV(+) | DC vaccine every 2 weeks ×3, then monthly ×7, concomitant with adjuvant dose intensified TMZ | None | 48 expected | N/A | Primary: OS; secondary: PFS, toxicity, immunogenicity | Recruiting | N/A |
| NCT02465268 | ATTAC-II | pp65 | Phase II, randomized, double-blind, placebo-controlled | Adults, new GBM | DC vaccine every 2 weeks ×3, then monthly ×7, concomitant with adjuvant TMZ | Placebo vaccine | 120 expected | N/A | Primary: OS; secondary: PFS, immunogenicity | Recruiting | N/A |
| Viral (oncolytic) vaccines | |||||||||||
| NCT01491893 | Recombinant Poliovirus (PVSRIPO, phase I) | CD155 | Phase I, single arm | Adults, recurrent GBM | PVSRIPO vaccine intratumoral injection ×1 | Matched cohort (n=104) | 61 | OS: 12.5 (9.9–15.2) vs. 11.3 (9.8–12.5) | 1 DLT: grade 4 intracranial hemorrhage; 1 grade 5 seizure attributed to PVSRIPO; 19% had grade 3+ AEs | Published | (70) |
| NCT02986178 | PVSRIPO (phase II) | CD155 | Phase II, single arm | Adults, recurrent GBM | PVSRIPO vaccine intratumoral injection ×1 | None | 122 expected | N/A | Primary: ORR; secondary: OS, PFS, safety | Recruiting | N/A |
| NCT00805376 | DNX-2401 Oncolytic Adenovirus | E1A mutant | Phase I, 2 arms | Adults, recurrent HGG | Arm I: DNX-2401 vaccine intratumoral injection ×1 | None | Arm I: 25; Arm II: 12 | OS: 13.0 | No DLTs; no serious AEs | Published | (71) |
| Arm II: DNX-2401 vaccine intratumoral injection ×1, then resection 14 days later with resection cavity vaccine injection | |||||||||||
| NCT01470794 | Toca 511 (phase I) | Glioma-selective | Phase I, single arm | Adults, recurrent HGG | Re-resection with resection cavity injection of Toca 511 vaccine and oral 5-FC | None | 53 (23 in phase III eligible subgroup, based on population and dose received) | OS: 11.9 (10.7–15.1) | – | Published | (72) |
| NCT02414165 | Toca 5 Trial (phase II/III) | Glioma-selective | Phase II/III, randomized | Adults, recurrent anaplastic astrocytoma or GBM | Re-resection with resection cavity injection of Toca 511 vaccine and oral 5-FC | SOC treatment (lomustine, TMZ, or bevacizumab) | 403 | OS: 11.1 vs. 12.2, HR 1.06, P=0.6154 | – | Completed, abstract only | (73) |
EGFRvIII, epidermal growth factor variant III; GBM, glioblastoma; s/p, status post; GTR, gross total resection; chemoRT, chemoradiation; KPS, Karnofsky performance status; TMZ, temozolomide; n, number of patients; PFS, progression free survival; OS, overall survival; PFS6, 6 month progression free survival; HGG, high grade glioma (i.e., WHO Grade III or IV glioma); AE, adverse event; CTL, cytotoxic T lymphocyte; G3, grade 3; TAA, tumor-associated antigen; GM-CSF, granulocyte-macrophage colony-stimulating factor; PFS9, 9 month progression free survival; ISR, injection site reaction; Poly-ICLC, polyinosinic-polycytidylic acid-poly-l-lysine carboxymethylcellulose; RT, radiation; HSPPC-96, heat shock protein peptide complex 96; SOC, standard of care; DC, dendritic cell; N/R, not reported; ORR, objective response rate; QoL, quality of life; GSC, glioma stem cell; WT1, Wilms tumor 1; MTD, maximum tolerated dose; CMV, cytomegalovirus; pp65, phosphoprotein 65; MGMT, O6-methylguanine DNA methyltransferase; DLT, dose limiting toxicity; 5-FC, 5-fluorocytosine.
Peptide vaccines: active immunotherapy
Peptide vaccines are non-cell-based vaccines by which tumor antigen is directly inoculated, to be ingested by APCs and presented to T cells. These antigens are often prepared in combination with carrier proteins and immunostimulatory adjuvants, such as granulocyte-macrophage colony-stimulating factor (GM-CSF) and polyinosinic-polycytidylic acid-poly-L-lysine carboxymethylcellulose (poly-ICLC) (74). The ideal peptide is one that has a high affinity for MHC-I, thereby increasing the probability of being cross-presented to stimulate CD8+ T cells. A number of such glioma TAAs have been identified, including gp100, AIM-2, TRP-2, and MAGE-1 (58). However, the most studied peptide in HGG is epidermal growth factor receptor variant III (EGFRvIII), a cell surface protein with a tumor-specific epitope expressed by approximately one third of GBMs (75,76). This protein is seen in several other epithelial tumors, but not on normal tissue (77). Its immunogenicity and efficacy as a therapeutic vaccine has been studied in a series of phase II and III trials. In the ACTIVATE phase II, multicenter trial, 18 adults with newly diagnosed EGFRvIII positive GBM who underwent a gross total resection and chemoradiation were given rindopepimut (also known as CDX-110), an EGFRvIII-targeted peptide vaccine, with standard adjuvant TMZ and found to have significantly better outcomes compared to a matched cohort (38). They reported a median PFS of 14.2 months (95% CI, 9.9–17.6) vs. 6.3 months (95% CI, 4.1–9.0) in the matched cohort and median OS of 26.0 months (95% CI, 21.0–47.7) vs. 15.0 months (95% CI, 11.4–19.8). Interestingly, for recurrent tumors that were re-resected (n=11), 82% no longer exhibited EGFRvIII expression, suggesting selective eradication of EGFRvIII positive cells versus downregulation of EGFRvIII as an adaptive immune escape mechanism.
In the follow-up phase II trial, ACT II, patients meeting the same inclusion criteria were either given standard dose TMZ (n=12) or dose-intensified TMZ (n=10) (39). Despite more severe and sustained lymphopenia in the dose-intensified group, these patients paradoxically exhibited greater magnitudes of antibody- and cell-mediated immune responses. Again, the median PFS and OS for the entire cohort were favorable (15.2 and 23.6 months, respectively) compared to the same matched cohort described above. The ACT III study (also single-arm, phase II, multicenter) was designed to confirm these results and did so, with a cohort of 65 patients achieving a median OS of 21.8 months (40). However, in the anticipated randomized, double-blind, placebo-controlled, phase III trial, ACT IV, interim analysis showed no OS benefit (median 20.1 vs. 20.0 months, P=0.93), and the trial was closed early (41). The use of rindopepimut was also investigated in the recurrent setting in the ReACT randomized, phase II trial, where bevacizumab was given with or without rindopepimut to 73 patients. The investigators found a trend toward improved PFS at 6 months (28% vs. 16%, P=0.12), the same median PFS of 3.7 months between groups, and, notably, a significant improvement in OS [hazard ratio (HR) 0.53, 95% confidence interval (CI), 0.32–0.88] (42). The authors concluded that efforts to validate the potential benefits of rindopepimut in larger trials were warranted.
Early clinical investigations of a vaccine targeting an IDH type 1 mutant peptide, IDH1R132H, are underway. This point mutation in IDH-1 is a common driver in glioma tumor development (78-80), and an immunogenic MHC-II-binding epitope has recently been identified and confirmed in murine models (81). The clinical trials utilizing an IDH1R132H peptide vaccine [(43), NCT03893903, NCT02193347], as well as the survivin-targeted vaccine, SurVaxM (44), are summarized in Table 1.
To optimize the likelihood of immunogenicity and tumor-directed response, multi-peptide vaccines target a number of tumor antigens commonly seen in HGG. Utilized in several clinical trials, IMA950 is a multi-peptide vaccine containing 11 TAAs, which are expressed on the majority of GBMs via human leukocyte antigen (HLA) receptors (82). Cancer Research UK IMA950-101, a phase I, two-arm, first-in-human study of IMA950 included 45 adults with newly diagnosed GBM who were HLA-A*02 positive (45). Eleven injections were given over 24 weeks starting either before (arm I) or after (arm II) standard chemoradiation. Overall, there were two grade 3 dose-limiting toxicities, 90% developed a TAA-specific response, and 50% developed a response to multiple TAAs. PFS was 74.4% at 6 months and 30.8% at 9 months. Median OS was 15.3 months, but, interestingly, patients who had an injection-site reaction (58% overall) had prolonged OS (26.7 vs. 13.2 months, P=0.0001). In subsequent studies, investigators attempted to optimize the immunogenicity of the vaccine by use of the adjuvant poly-ICLC, which had previously shown efficacy in a murine model (83), and human studies with other peptide vaccines (84,85). In one phase I/II study of newly diagnosed WHO grade III (n=3) and IV (n=16) glioma patients, the first 6 patients received IMA950 intradermally and poly-ICLC intramuscularly (IM) (46). Then the protocol was changed to optimize vaccine formulation by mixing IMA950 and poly-ICLC, which was injected subcutaneously (SC, n=7) or IM (n=6). The initial formulation only led to single peptide CD8+ T cell responses, while the mixed formulation elicited multipeptide CD8+ and CD4+ T cell responses. Median OS for the GBM patients was 19 months. IMA950/poly-ICLC mixed and administered SC is being further investigated in a phase I/II, randomized trial with or without pembrolizumab (NCT03665545).
While the prior mentioned studies used the same synthetic or allogeneic peptide(s) for all patients, several studies have investigated the use of personalized multi-peptide vaccines that include antigens for which the patient has the highest IgG titers (47-49). Others have created personalized vaccines by using antigen collected from the patient’s resected tumor (50,51). These, and other ongoing studies, are summarized in Table 1. This approach has been used more frequently in DC vaccination, which is discussed in further detail below.
HSP vaccines: active immunotherapy
HSPs normally regulate chaperone proteins and facilitate proper protein folding (86), but in the setting of GBM, they bind multiple TAAs, forming heat shock protein peptide complexes (HSPPCs) (87). Like other molecules, they are ingested, processed, and presented by APCs, making them a potential vector for multi-peptide TAA recognition (88). Furthermore, HSPPCs bind macrophages and stimulate the production of proinflammatory cytokines (89), and also bind immature DCs prompting them to mature (90), thereby contributing to both innate and adaptive immune responses. Thus, HSPPCs are attractive for use in therapeutic vaccinations due to their immunostimulatory capacities and the range of antigens they carry. They also provide a personalized therapy as they are harvested from the patient’s tumor. The most commonly used HSPPC in clinical trials is HSPPC-96. In a phase I, single arm trial, an autologous HSPPC-96 vaccine was given to 12 patients with recurrent GBM, and 11 exhibited an immune response with a median OS of 10.8 months, compared to 3.7 months in the non-responder, with no severe adverse events (53). In a follow-up phase II trial in 41 patients, survival results were similar with a median OS of 9.8 months (95% CI, 8.0–11.6) and PFS of 4.4 months (95% CI, 3.2–5.5) (54). The same group initiated a phase II trial of 90 patients with recurrent GBM randomized to HSPPC-96 vaccine with concomitant bevacizumab, HSPPC-96 with addition of bevacizumab and continued vaccine administration at progression, or bevacizumab alone. Though the study is still active, the investigators have released data showing no PFS or OS benefit [Parney I, 2019, unpublished data (55)]. The HSPPC-96 vaccine is also being studied in the treatment of newly diagnosed GBM. One phase II, single arm trial of 46 patients, published in abstract only, achieved an impressive median PFS of 17.9 months (95% CI, 11.3–21.6) and OS of 23.8 months (95% CI, 19.8–30.2) (56). A larger randomized phase II study is currently underway (NCT03650257).
DC vaccines: active immunotherapy
Unlike peptide and HSP vaccines, DC vaccines are cell-based vaccines produced by harvesting the patient’s DCs, culturing and priming the cells ex vivo, then reinoculating the patient with them. DCs are the most powerful stimulators of cell-mediated immune response, so priming them ex vivo should optimize the likelihood of obtaining such a response. DCs can be “pulsed” or “loaded” with tumor peptides, products of lysed autologous tumor cells, tumor-derived mRNA, glioma stem cells (GSCs), and viral antigens that have been associated with HGG (36). Numerous early stage clinical trials to assess the safety and efficacy of DC vaccines in HGG have been published since the 1990’s, and are summarized in other reviews (91,92). Notably, the rate of adverse events, and most significantly the rate of adverse autoimmune reactions, has been much lower than those seen with ICIs (91,93), so only pertinent reports of severe toxicities are mentioned here. We will focus our review of DC vaccines on trials that have made OS comparisons, as well as recently published and ongoing studies, all of which are summarized in Table 1.
Overall, the summarized studies of DC vaccines show promising survival results, with median OS for newly diagnosed GBM ranging from 17.0 to 41.1 months and PFS from 6.6 up to 25.3 months. While survival times are expectedly shorter for studies in recurrent GBM, with median OS ranging from 10.9 to 15.3 months and PFS from 1.9 to 6.3 months, these compare very favorably to historical controls. Unfortunately, improved survival has not been uniformly seen in the subset of these studies that are randomized. For example, in single arm, phase I trial, ICT-107, a DC vaccine loaded with 6 GBM TAAs, was used to treat 20 adults with newly diagnosed or recurrent GBM or brainstem gliomas that were HLA-A*01 and/or HLA-A*02 positive (58). Among the 16 newly diagnosed GBM patients, median OS was an impressive 40.1 months. A follow-up study was designed as a phase II, randomized, double-blind, placebo-controlled trial, which included only new diagnoses of GBM that were, again, HLA-A*01 and/or HLA-A*02 positive (59). Patients were randomized to receive either the ICT-107 DC vaccine (n=81), or a placebo unpulsed DC vaccine (n=43), prior to and concomitant with standard adjuvant TMZ. Unfortunately, there was no significant difference between OS (17.0 vs. 15.0 months, HR 0.87, P=0.58), with values resembling historical controls. PFS was, however, significantly improved in the experimental arm (11.2 vs. 9.0 months, HR 0.57, P=0.011) but the absolute benefit was modest. In another larger randomized, phase II trial, Audencel, a tumor lysate-pulsed DC vaccine, was given after completion of standard chemoradiation (n=34), but there was no significant improvement in OS (18.3 vs. 18.4 months, HR 0.99, P=0.89) or PFS (6.6 vs. 6.9 months, P=0.83) compared to standard adjuvant TMZ (n=42) (65). However, the randomized studies are not uniformly negative. In another randomized, phase II trial of a tumor lysate DC vaccine, patients in the experimental arm received the vaccine 10 times over the course of 6 months following standard chemoradiation and experienced a markedly improved OS of 31.9 months as compared to 15.0 months in the standard of care (SOC) control arm (P<0.002) (62). A modest but significant OS benefit was also seen in a study of a GSC loaded DC vaccine (68). This phase II, randomized, double-blind, placebo-controlled study included both newly diagnosed and recurrent GBM patients who underwent a near total resection and received either 3 injections of the GSC vaccine (n=22) or a placebo vaccine (n=21). Median OS was 13.7 months in the experimental arm and 10.7 months in the placebo control arm (P=0.05). When controlling for IDH1 and TERT promoter status, B7-H4 expression, and new or recurrent disease, the experimental arm had more significantly prolonged OS (HR 2.5, P=0.02) and a nonsignificant trend toward improved PFS (HR 1.37, P=0.37).
The reasons for the varied results of these studies are not clear but may be related to appropriate patient selection. For example, in the study reported by Wen et al., patients positive for either HLA-A*01 or HLA-A*02 were included, but the HLA-A*02 positive patients exhibited a higher rate of immune response which correlated to a higher rate of OS compared to non-responders (59). Furthermore, in this study, 33% of the control arm patients had a detectable immune response to one or more TAAs contained in the experimental vaccine, perhaps suggesting that the placebo unpulsed DC vaccine was stimulating an immune response to in vivo TAAs. A follow-up phase III trial is being conducted now, including only HLA-A*02 positive patients with a less active placebo, though it is currently suspended due to financial issues (NCT02546102). Another notable ongoing study is the phase III trial investigating DCVax-L, a tumor lysate DC vaccine, given to patients with newly diagnosed GBM concomitant with standard adjuvant TMZ, with crossover allowed following recurrence (NCT00045968). An interim analysis reported an impressive median OS of 23.1 months (95% CI, 21.2–25.4) for the entire cohort (n=331), but the primary endpoint of PFS has not yet been assessed (66).
Viral vaccines: gene therapy
Viral, or oncolytic, vaccines are used in the treatment of malignant disease when a specific virus or viral gene product has been associated with the tumor. In GBM, the cytomegalovirus (CMV) antigen pp65 has been associated with GBM (94) and studied in a number of viral antigen-loaded DC vaccine studies (see Table 1). Several studies have also investigated the direct inoculation of various modified nonpathogenic viruses, including PVSRIPO [recombinant polio-rhinovirus (70)], DNX-2401 [a tumor-selective oncolytic adenovirus (71)], and Toca 511 [a gamma-retrovirus (72)], with interesting results. In each of these studies, the virus is injected directly into the tumor and/or resection cavity. In the case of PVSRIPO, the virus selectively infects tumor cells due to their overexpression of CD155. APCs are also infected but chronically and sublethally, leading to constitutive activation with a proinflammatory response that supports a T cell mediated tumor response. In a single arm, phase I trial published in the New England Journal of Medicine, 61 patients with recurrent GBM received an intratumoral injection of PVSRIPO with one dose-limiting toxicity (grade 4 intracranial hemorrhage), one grade 5 toxicity (seizure), and 19% rate of grade 3 or higher adverse events (70). Median OS was 12.5 months (95% CI, 9.9–15.2), which was not significantly different from a matched cohort, 11.3 months (95% CI, 9.8–12.5). However, OS in the experimental group reached a plateau of 21% (95% CI, 11–33%) at 24 months, and this was sustained at 36 months, leading the investigators to open a larger phase II trial (NCT02986178). Both of the other studies also had a subset of patients who achieved a sustained response. In the DNX-2401 trial, 20% of patients (5 of 25) survived more than 3 years after the injection, and 3 of the 5 patients had more than a 95% reduction in their tumor volume (71). In the Toca 511 trial, patients with recurrent HGG underwent re-resection with injection of the vector followed by oral 5-fluorocytosine (5-FC). The vector encodes cytosine deaminase, which converts 5-FC to 5-fluorouracil (5-FU) within tumor cells. Within the study of 53 patients, 23 were deemed the “phase III eligible subgroup” based on their uniformity of patient and treatment characteristics. Of the subgroup, 21.7% (5 of 23) had a complete response and remain alive and in response 33.9 to 52.2 months after Toca 511 administration (72). The follow-up phase III trial enrolled 403 patients, and, unfortunately, interim analysis recently showed that the primary endpoint of OS was not met (11.1 vs. 12.2 months, HR 1.06, P=0.6154) (73). The investigators did note that pre-planned subgroup analyses showed compelling results, but these data are not yet published, and as with preceding studies, may show subsets of patients with durable responses. Interestingly, a similar rate of about 20% of patients in all three trials had a sustained response. This may suggest a common immunologic feature of the tumor or microenvironment that potentiates the therapy. Comparison of biomarkers may elucidate this phenomenon.
Immune checkpoint inhibition: passive immunotherapy
In the normally functioning immune system, immune cells express checkpoint proteins, such as CTLA-4 and PD-1, which, when bound by their ligands, such as PD-L1, attenuate immune cells to prevent normal tissue damage. In the setting of malignant disease, tumor cells escape the immune system in part by overexpressing inhibitory proteins like PD-L1 (95,96). ICIs are monoclonal antibodies that block CTLA-4, PD-1, or PD-L1 to prevent interactions between these proteins, thereby circumventing T cell attenuation and tumor cell immune escape. As opposed to active immunotherapy, ICIs are a type of passive immunotherapy, in which an immune effector molecule is introduced to the patient’s body (37). These therapies have significantly improved survival in a number of solid tumors (14-20), which has generated clinical research in a broad range of disease sites, including primary brain tumors (see Table 2). The most commonly used ICIs in clinical practice for other solid tumors, and under investigation for use in HGG, are ipilimumab (anti-CTLA-4), nivolumab (anti-PD-1), pembrolizumab (anti-PD-1), atezolizumab (anti-PD-L1), and durvalumab (anti-PD-L1).
Table 2. Immune checkpoint inhibition clinical trials.
| Trial number | Abbreviated trial name | Target | Study design | Population | Intervention/Arms | Control | Number of analyzed patients | Survival, median months (95% CI) | Additional outcomes | Trial status | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Basket trials of immune checkpoint inhibitors | |||||||||||
| NCT01375842 | PCD4989g | PD-L1 | Phase Ia, single arm | Adults, recurrent GBM | Atezolizumab given every 3 weeks until progression or intolerance | None | 16 | PFS: 1.2 (0.7–10.7); OS: 4.2 (range, 1.2–18.8+); 3 pts with long-term survival (range, 16.0–18.8+) | PR: 1 (6.3%); SD: 3 (18.8%); AEs: 3 G3, 1 serious AE leading to interruption | Published | (97) |
| NCT02054806 | Keynote-028 | PD-1 | Phase I, single arm | Adults, recurrent GBM, bevacizumab-naïve | Pembrolizumab every 2 weeks until progression, intolerance, or 24 mo |
None | 26 | PFS: 2.8 (1.9–9.1); OS: 14.4 (10.3–not reached) | PR: 1 (3.8%); SD: 12 (46.2%), median duration 39.4 weeks (range, 7.1+–85.9+); G3–4 AEs: 4 | Active, not recruiting, abstract only | (98) |
| NCT02335918 | Varlilumab (anti-CD27) and nivolumab | PD-1, CD27 | Phase II | Adults, recurrent GBM, bevacizumab-naïve | Varlilumab and nivolumab every 2 weeks ×16 | None | 22 | OS: 9.7 (6.7–14.8), 8 pts survived >12 mo (range: 13.7–23+); PR: 2; SD: 9 | Serious AEs: 2; DLT: 0 | Completed, abstract only | (99) |
| NCT02526017 | Cabiralizumab (anti-CSF-1R) and nivolumab | PD-1, CSF-1R | Phase 1a/1b, 2 arms | Adults, recurrent GBM | Arm I: Cabiralizumab; Arm II: Cabiralizumab and nivolumab | None | 295 expected (entire cohort) | N/A | Primary: toxicity, ORR; secondary: OS, PFS, immunogenicity, biomarkers | Active, not recruiting | N/A |
| Single agent immune checkpoint inhibitors | |||||||||||
| NCT02550249 | Neoadjuvant and adjuvant nivolumab | PD-1 | Phase II, single arm | Adults, new or recurrent GBM | Nivolumab 2 weeks prior to resection ×1, then every 2 weeks (starting after chemoRT, concomitant with adjuvant TMZ for newly diagnosed) until progression or intolerance | None | Newly diagnosed: 3; recurrent: 27 | PFS: 4.1 (2.8–5.5); OS: 7.3 (5.4–7.9) | G3–4 AE: 1 | Published | (100) |
| NCT02852655 | Adjuvant +/− neoadjuvant pembrolizumab | PD-1 | Phase II, randomized | Adults, recurrent GBM | Arm I: Pembrolizumab 2 weeks prior to resection ×1, then every 3 weeks until progression or intolerance; Arm II: After resection, every 3 weeks until progression or intolerance | None | Arm I: 16; Arm II: 19 | PFS: 3.3 vs. 2.4 (HR 0.43, P=0.03); OS: 13.7 vs. 7.5 (HR 0.39, P=0.04) | G3–4 AE: 10 (67%) vs. 7 (47%), P=0.46; AEs leading to treatment discontinuation: 2 vs. 0 | Published | (101) |
| NCT02017717 | CheckMate-143 | PD-1 | Phase III, randomized | Adults, recurrent GBM | Nivolumab every 2 weeks until progression or intolerance | Bevacizumab every 2 weeks until progression or intolerance | Experimental: 184; Control: 185 | PFS: 1.5 vs. 3.5; OS: 9.8 vs. 10.0 | G3–4 AE: 18% vs. 15%; AEs leading to treatment discontinuation: 10% vs. 15% | Active, not recruiting, abstract only | (102) |
| NCT02617589 | CheckMate-498 | PD-1 | Phase III, randomized | Adults, new GBM, MGMT unmethylated | Nivolumab every 2 weeks concomitant with standard radiation without TMZ, then every 4 weeks after radiation until progression or intolerance | SOC treatment | 550 expected | N/A | Primary: OS; secondary: PFS | Active, not recruiting, unpublished data | (103) |
| NCT02667587 | CheckMate-548 | PD-1 | Phase III, randomized, double-blind, placebo-controlled | Adults, new GBM, MGMT methylated | Nivolumab concomitant with standard chemoRT | Placebo infusion | 693 expected | N/A | Primary: OS, PFS | Active, not recruiting, unpublished data | (104) |
| NCT02337686 | Pembrolizumab and re-resection | PD-1 | Phase II, single arm | Adults, recurrent GBM | Pembrolizumab ×1–2 prior to resection, then afterwards until progression or intolerance | Matched SOC cohort | Experimental: 15; matched cohort: 10 | PFS: 7 [4–16]; OS: not reached | – | Active, not recruiting, abstract only | (105) |
| NCT02530502 | Pembrolizumab with chemoRT | PD-1 | Phase I, single arm | Adults, new GBM, s/p resection | Pembrolizumab every 3 weeks, concomitant with standard chemoRT | None | 4 enrolled | N/A | Primary: DLT; secondary: toxicity, immunogenicity, biomarkers | Active, not recruiting | N/A |
| NCT04047706 | Nivolumab and IDO1 inhibition (BMS-986205) with RT or ChemoRT | PD-1, IDO1 | Phase I, 2 arms | Adults, new GBM | Arm I: nivolumab every 2 weeks ×3 and IDO1 inhibitor daily concomitant with standard chemoRT and with adjuvant TMZ; Arm II: nivolumab and IDO1 inhibitor with radiation alone and adjuvantly | None | 30 expected | N/A | Primary: toxicity; secondary: OS, PFS, ORR | Recruiting | N/A |
| NCT02336165 | Durvalumab with RT (new) or with/without bevacizumab (recurrent) | PD-L1 | Phase II, 5 arms | Adults, new MGMT unmethylated, or recurrent GBM | Durvalumab (MEDI4736) every 2 weeks and: Arm I (newly diagnosed): concomitant radiation without TMZ; Arm II–IV (bevacizumab-naïve): 3 dose levels of bevacizumab including none; Arm V (bevacizumab-refractory): continued bevacizumab | None | 159 enrolled; Arm V: 22 | Arm V: PFS: range, 0.2–5.6, 11 pts (50%) with PFS ≥1.8 mo; OS: range, 0.2–11.9, 8 pts (36%) with OS ≥5.1 mo | Primary: OS, PFS; secondary: toxicity, QoL, biomarkers | Active, not recruiting, abstract only (re: Arm V) | (106) |
| NCT02337491 | Pembrolizumab +/− bevacizumab | PD-1 | Phase II, randomized | Adults, recurrent GBM | Arm I: pembrolizumab every 3 weeks and bevacizumab every 2 weeks until progression or intolerance; Arm II: pembrolizumab every 3 weeks until progression or intolerance | None | Arm I: 50; Arm II: 30 | PFS: 4.1 (2.8–5.5) vs. 1.4 (1.4–2.7); OS: 8.8 (7.7–14.2) vs. 10.3 (8.5–12.5) | – | Completed, abstract and unpublished data | (107,108) |
| NCT02311582 | Pembrolizumab +/− MRI-guided laser ablation | PD-1 | Phase I/II, 2 arms | Adults, recurrent GBM | Phase II: Arm I: MLA, then pembrolizumab every 3 weeks; Arm II: pembrolizumab every 3 weeks | None | 58 expected | N/A | Primary: PFS; secondary: OS, immunogenicity, biomarkers | Recruiting | N/A |
| NCT03718767 | Nivolumab for IDH-mutated glioma with hypermutator phenotype | PD-1 | Phase II, single arm | Adults, recurrent glioma, IDH-mutated with tumor specific mutational load | Nivolumab every 2 weeks ×8, then every 4 weeks ×12 | None | 95 expected | N/A | Primary: PFS; secondary: OS, QoL | Recruiting | N/A |
| NCT03047473 | The SEJ Study | PD-L1 | Phase II, single arm | Adults, new GBM s/p chemoRT | Avelumab every 2 weeks with standard adjuvant TMZ | None | 30 expected | Interim analysis (n=8): PFS: 11.9; CR: 2 (25%); PR: 1 (12.5%); SD: 1 (12.5%); AEs requiring treatment break: 2 | Primary: toxicity; secondary: OS, PFS, ORR, biomarkers | Active, not recruiting, abstract only | (109) |
| Dual immune checkpoint inhibitors | |||||||||||
| NCT02017717 | CheckMate-143 | PD-1, CTLA-4 | Phase I, randomized | Adults, recurrent GBM | Arm I: nivolumab 3 mg/kg every 2 weeks; Arm II: nivolumab 1 mg/kg and ipilimumab 3 mg/kg every 3 weeks ×4, then nivolumab 3 mg/kg every 2 weeks; Arm III: nivolumab 3 mg/kg every and ipilimumab 1 mg/kg every 3 weeks ×4, then nivolumab 3 mg/kg every 2 weeks (nonrandomized) | None | Arm I: 10; Arm II: 10; Arm III: 20 | PFS, OS: Arm I: 1.9 (1.3–4.6), 10.4 (4.1–22.8); Arm II: 1.5 (0.5–2.8), 9.2 (3.9–12.7); Arm III: 2.1 (1.4–2.8), 7.3 (4.7–12.9) | PR: 1 in Arm I, 2 in Arm III; SD (≥12 weeks): 2 in Arm I, 2 in Arm II, 4 in Arm III; AEs leading to discontinuation: 1 in Arm I, 3 in Arm II, 4 in Arm III | Published | (110) |
| NCT02311920 | Ipilimumab and/or Nivolumab | PD-1, CTLA-4 | Phase I, randomized | Adults, new GBM s/p chemoRT | Adjuvant TMZ and: Arm I: ipilimumab every 4 weeks ×4, then every 3 months ×4; Arm II: nivolumab every 2 weeks ×8, then every 2 weeks ×24; Arm III: ipilimumab every 4 weeks ×4 and nivolumab every 2 weeks ×32 | None | 32 enrolled | N/A | Primary: toxicity; secondary: OS, biomarkers | Active, not recruiting | N/A |
| NCT02658981 | Adult Brain Tumor Consortium 1501 | PD-1, LAG-3, CD137 | Phase I, 4 arms | Adults, recurrent GBM | Arm I: anti-LAG-3 (BMS-986016) every 2 weeks; Arm II: anti-CD137 (urelumab, BMS-663513) every 3 weeks; Arm III: anti-LAG-3 and nivolumab every 2 weeks; Arm IV: anti-CD137 every 4 weeks and nivolumab every 2 weeks (arm closed 10/16/18) | None | 100 expected | Interim analysis (n=44): OS: Arm I: 8; Arm II: 14; Arm III: 7; Arm IV: N/R | Primary: toxicity; secondary: OS, PFS, ORR | Recruiting, interim analysis published (abstract only) | (111) |
| NCT02794883 | Tremelimumab (anti-CTLA-4) and/or Durvalumab | PD-L1, CTLA-4 | Phase II, 3 arms | Adults, recurrent HGG | Re-resection after first cycle of: Arm I: tremelimumab every 4 weeks; Arm II: durvalumab every 2 weeks; Arm III: tremelimumab every 4 weeks and durvalumab every 2 weeks | None | 36 enrolled | N/A | Primary: immunogenicity; secondary: toxicity, OS, PFS, biomarkers | Active, not recruiting | N/A |
| NCT03707457 | Nivolumab and anti-GITR, IDO1 Inhibitor, or Ipilimumab | PD-1, GITR, IDO1, CTLA-4 | Phase I, 3 arms | Adults, recurrent GBM | Arm I: nivolumab and anti-GITR every 4 weeks; Arm II: nivolumab every 4 weeks and IDO1 inhibitor daily; Arm III: nivolumab and ipilimumab every 3 weeks ×4, then nivolumab every 4 weeks | None | 30 expected | N/A | Primary: toxicity; secondary: OS, PFS, immunogenicity, biomarkers | Active, not recruiting | N/A |
| NCT03233152 | GlitIpNi | PD-1, CTLA-4 | Phase I, single arm | Adults, recurrent GBM | Nivolumab ×1 24 hours prior to re-resection with ipilimumab injection into resection cavity, then nivolumab every 2 weeks ×5 | None | 6 expected | N/A | Primary: toxicity, OS, PFS | Recruiting | N/A |
PD-L1, programmed death ligand 1; GBM, glioblastoma; PFS, progression-free survival; OS, overall survival; PR, partial response; SD, stable disease; AE, adverse event; PD-1, programmed death-1; G3–4, grade 3–4; DLT, dose limiting toxicity; CSF-1R, colony-stimulating factor 1 receptor; ORR, objective response rate; chemoRT, chemoradiation; TMZ, temozolomide; HR, hazard ratio; MGMT, O6-methylguanine DNA methyltransferase; SOC, standard of care; RT, radiation; IDO1, indoleamine-2,3-dioxygenase 1; QoL, quality of life; MLA, MRI-guided laser ablation; CTLA-4, cytotoxic T-lymphocyte-associated protein 4; LAG-3, lymphocyte-activation gene 3; n, number of patients; HGG, high grade glioma (i.e., WHO Grade III or IV glioma); GITR, glucocorticoid-induced TNFR family related gene.
Early data on the safety and efficacy of ICIs for use in recurrent GBM comes from several basket trials. The phase Ia PCD4989g study included 16 adults with recurrent GBM who received atezolizumab every 3 weeks until disease progression or intolerance (97). Median PFS (1.2 months, 95% CI, 0.7–10.7) and OS (4.2 months, 95% CI, 1.2–18.8+) were comparable to historical controls, but 3 patients (18.8%) experienced long-term survival of 16.0+ months. The investigators reported three grade 3 toxicities, with one serious adverse event leading to a treatment interruption. Recurrent bevacizumab-naïve GBM patients were also included in the Keynote-028 basket trial of pembrolizumab (NCT02054806). The analysis of 26 patients at a median follow-up of 14 months, published in abstract form only, showed a more favorable PFS of 2.8 months (95% CI, 1.9–9.1) and OS of 14.4 months (95% CI, 10.3–not reached) (98). Furthermore, while one patient had a partial response (PR), 12 patients (46.2%) had stable disease (SD) with a median duration of 9.1 months (range, 1.6+–9.8+). Safety data was comparable to the PCD4989g study, with report of four grade 3–4 toxicities, none of which required treatment interruption.
The presence of durable responders and acceptable safety outcomes led to the development of additional glioma-specific clinical trials, the results of which have been mixed but generally underwhelming. A recent meta-analysis was done on all studies of GBM that have been published in full manuscript form and report on efficacy and survival outcomes after treatment with ICIs (112). This included four retrospective studies (113-116), two phase I studies (97,110), and two phase II studies (100,101), encompassing 203 patients (predominantly with recurrent GBM) treated with atezolizumab, nivolumab, ipilimumab, and pembrolizumab as monotherapy or in combination with each other or bevacizumab. The median PFS and OS were 2.1 and 7.3 months, respectively, which is favorable compared to historical controls, but not as impressive as the results of vaccine therapy trials, which report median OS on the range of about 10–11 months in patients with recurrent GBM (47,53,54,70).
Moreover, three large randomized trials have reported disappointing results on interim analyses. In the CheckMate-143 trial, adults with recurrent GBM received either nivolumab (n=184) or SOC bevacizumab (n=185) with no significant differences in PFS (1.5 vs. 3.5 months) or OS (9.8 vs. 10.0 months) (102). In the CheckMate-498 trial, adults with newly diagnosed O-6-methylguanine-DNA methyltransferase (MGMT) unmethylated GBM were randomized to receive nivolumab every 2 weeks concomitant with radiation (without TMZ) and adjuvant nivolumab every 4 weeks, or SOC chemoradiation with TMZ, with an expected 550 patients (NCT02617589). In a press release on their website, Bristol-Myers Squibb stated that the experimental treatment “failed to prolong overall survival” compared to the control arm [Bristol-Myers Squibb, 2019, unpublished data (103)]. The CheckMate-548 phase III, randomized, double-blind, placebo-controlled study of standard chemoradiation with TMZ, with or without concomitant nivolumab, for an expected 693 patients with newly diagnosed MGMT-methylated GBM, has also failed to meet its primary endpoint of PFS, while the OS endpoint has yet to mature [Bristol-Myers Squibb, 2019, unpublished data (104)].
Various hypotheses have been proposed to explain the lackluster outcomes of these trials, including low mutational burden of gliomas, low prevalence of PD-1 expressing CD8+ T cells coupled with low PD-L1 expression of glioma tumor cells, and immunosuppressive characteristics of the glioma tumor microenvironment (TME). Tumor mutational “burden” or “load” (TML) is a quantification of the rate of nonsynonymous mutations (i.e., those tumor DNA mutations causing an alteration to the transcribed amino acid sequence). TML is correlated to the number of neoantigens transcribed, which is in turn correlated to the likelihood of a tumor-specific immune response and response to ICIs (117-119). High mutational burden is seen often in some solid tumor histologies, such as melanoma and NSCLC, but this level of burden is seen in <10% of GBM (120,121). A subset of patients with GBM have been identified as having a hypermutator phenotype, which is associated with mismatch repair (MMR) deficiency (121,122). While the prevalence of MMR deficiency in GBM is low (<5%) (121,123), a phase II trial is currently open to investigate the efficacy of nivolumab in patients with recurrent gliomas with the hypermutator phenotype (NCT03718767). Regarding checkpoint proteins, higher PD-1 and PD-L1 expression has been shown to predict response to ICIs in other cancers (124,125). Several studies have found low rates of PD-1 expression on tumor infiltrating leukocytes (TILs) in glioma specimens (~34%) with even lower rates of PD-L1 expression on tumor cells (~7%) (121,126), suggesting that these inhibitory pathways may play a less significant role in glioma immune escape compared to other cancers. Several ongoing studies are investigating the use of novel ICIs targeting checkpoint proteins that may be more pertinent to immune escape in gliomas, such as CD137 and LAG-3 [NCT02658981, (111)], GITR (NCT03707457), and CD27 [NCT02335918, (99)]. Many ongoing studies are also evaluating for more reliable biomarkers to predict a response to therapy. For example, in the Adult Brain Tumor Consortium 1501 trial, one patient had a complete response and, prior to immunotherapy, was found to have higher T cell receptor (TCR) clonality and peripheral blood with more CD137+CD8+ T cells and fewer Foxp3+CD137+CD4+ T cells than the other 5 patients analyzed (111). The glioma TME also seems to play a large role in suppressing tumor-specific immune responses. These immune modulating mechanisms are varied and complex, including cytokine expression that favors regulatory T cells (Tregs, which are CTLA-4+ and secrete immunosuppressive cytokines TGF-β and IL-10) and recruits immunosuppressive macrophages (via CSF-1), as well as expression of pro-apoptotic cell surface proteins (e.g., CD95 and CD70) (127). Indoleamine-2,3-dioxygenase 1 (IDO1), expressed in high levels in GBM specimens, causes inhibition and apoptosis of CTLs as well as amplification of Tregs (128). Several ongoing studies are utilizing nivolumab in combination with inhibitors of immune suppressors, including an IDO1 inhibitor (NCT03707457), cabiralizumab (a monoclonal anti-CSF-1R antibody, NCT02526017), and galunisertib (a TGF-β receptor I inhibitor, NCT02423343).
Another approach being studied in HGG is the use of dual immune checkpoint blockade, based on its efficacy in other tumor types (129-131). In the phase I exploratory study of CheckMate-143, patients were randomized to receive nivolumab or combined nivolumab/ipilimumab at various doses (110). The investigators found similar efficacy between arms, but in the dual ICI arms there were more adverse events leading to treatment discontinuation, which was the basis for using nivolumab alone in the phase III study. However, toxicity was acceptable overall, and a number of studies are open to further investigate the efficacy and safety of dual ICIs in newly diagnosed GBM and recurrent HGG (see Table 2).
Of note, the safety of ICIs has generally been considered acceptable in the published trials of their use in gliomas. However, in comparison to vaccine therapies, which have very rare adverse events and dose limiting toxicities, ICIs can cause severe immune-related toxicities in some patients. Thus, their relative benefit must be considered in relation to the potential for toxicity. While these toxicities are felt to be acceptable in the treatment of other malignancies, these disease sites have a more clearly defined benefit that has yet to be shown in HGG.
Adoptive lymphocyte transfer (ALT): passive immunotherapy
As another form of passive immunotherapy, ALT aims to provide an exogenous tumor directed T cell population. This population may be developed from tumor-specific T cells, either CTLs harvested from peripheral blood or TILs from tumor specimens, which are expanded ex vivo before being (re)introduced into the patient following chemotherapy preconditioning (132). Alternatively, normal T cells can be harvested from peripheral blood and modified to express tumor-specific TCRs or chimeric antigen receptors (CARs) (133,134). TCR T cells are developed by identifying TCR genes in tumor-reactive T cells, then isolating and transferring these genes to normal T cells to activate them against a TAA. On the other hand, CAR T cells are developed by engineering a receptor composed of the variable regions of an antibody specific for a TAA, linked to intracellular signaling proteins and co-stimulatory molecules. TCR T cells can only recognize intracellular antigens by processes of MHC expression and co-stimulation, both of which are often downregulated by tumor cells. On the other hand, CAR T cells can recognize peptides or cell surface components (including carbohydrates and glycolipids) in an MHC-independent manner without a need for co-stimulation, giving them a broader range than TCR T cells, more tumor infiltrative capacity than monoclonal antibodies, and fewer barriers to activating an anti-tumor immune response than normal T cells (135). There are adoptive cell therapies that utilize other cellular populations, such as natural killer cells, lymphokine-activated killer cells, and gamma-delta cells, but these are used less often in current clinical research. Because of the distinct advantages of CAR T cells, the majority of adoptive cell research utilizes this approach and will be the focus of this section, with additional studies summarized in Table 3.
Table 3. Adoptive lymphocyte transfer clinical trials.
| Trial number | Abbreviated trial name | Target | Study design | Population | Intervention/arms | Control | Number of analyzed patients | Survival, median months (95% CI) | Additional outcomes | Trial status | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|
| CAR T cells | |||||||||||
| NCT02209376 | EGFRvIII CAR T cells | EGFRvIII | Phase I, single arm | Adults, recurrent GBM, EGFRvIII(+) | CAR T cells infused ×1 | None | 10 | OS: 8.2 | AEs: 3 (neurologic, possibly related) | Published | (136) |
| NCT01454596 | EGFRvIII CAR T cells with Chemo and IL-2 | EGFRvIII | Phase I, dose escalation | Adults, recurrent GBM, EGFRvIII(+) | Conditioning chemotherapy (flu/cy), then CAR T cells with IL-2 ×1 | None | 18 | PFS: 1.3 (IQR, 1.1–1.9); OS: 6.9 (IQR, 2.8–10.0) | 2 treatment related adverse events (hypoxia), including 1 death (highest dose level) | Published | (137) |
| NCT03170141 | EGFRvIII CAR T cells with Chemo | EGFRvIII | Phase I, single arm | Adults, recurrent GBM, EGFRvIII(+) | Conditioning chemotherapy (flu/cy), then CAR T cells ×1 | None | 20 expected | N/A | Primary: toxicity; secondary: OS, PFS, ORR, immunogenicity | Enrolling by invitation | N/A |
| NCT02664363 | ExCeL | EGFRvIII | Phase I, single arm | Adults, new GBM, EGFRvIII(+) | Up to 3 cycles of standard adjuvant TMZ, followed by CAR T cells ×1, followed by additional adjuvant TMZ | None | 3 | N/A | Primary: MTD; secondary: toxicity | Terminated (study funding ended) | N/A |
| NCT01109095 | HERT-GBM | HER2 | Phase I, single arm | Adults (n=10) and children (n=7), recurrent GBM, HER2(+) | CAR T cells ×1, up to 6 times if objective response | None | 17 | PFS: 3.5; OS: 11.1 (4.1–27.2); PR: 1; SD: 7 | Serious AEs: 0 | Published | (138) |
| NCT02442297 | iCAR | HER2 | Phase I, 2 arms | Adults and children, recurrent GBM, HER2(+) | CAR T cells ×3 at various dose levels; Arm I: high risk (51–100% HER2 positive); Arm II: standard risk (1–50%) | None | 28 expected | N/A | Primary: toxicity; secondary: ORR | Recruiting | N/A |
| NCT00730613 | IL13Rα2 CAR T cells (single arm) | IL13Rα2 | Phase I, single arm | Adults, recurrent GBM | Resection and catheter placement, followed by intracavitary or intratumoral CAR T cell infusion, up to 12 times | None | 3 | OS: 10.3 (range, 8.6–13.9) | G3 AE: 2 (headache, transient neurologic symptoms) | Published | (139) |
| NCT02208362 | IL13Rα2 CAR T cells (5 arms) | IL13Rα2 | Phase I, 5 arms | Adults and children 12+ years old, recurrent HGG, IL13Rα2(+) | CAR T cell infusion weekly ×3, then repeated at least 1 week later until progression or depletion of product: Arm I: intratumoral; Arm II: intracavitary; Arm III: intraventricular; Arm IV: intratumoral and intraventricular; Arm V: intratumoral and intraventricular vaccine therapy | None | 92 expected | Case report: pt in arm IV had CR of all intracranial and spinal tumors, maintained for 7.5 mo, no G3+ toxicities | Primary: toxicity; secondary: OS, PFS, ORR, immunogenicity, QoL | Recruiting, case report published | (140) |
| NCT02575261 | SOC treatment +/− EphA2 CAR T cells | EphA2 | Phase I/II, randomized | Adults, new or recurrent GBM, EphA2(+) | CAR T cell infusion | SOC treatment | 60 enrolled | N/A | Primary: immunogenicity; secondary: ORR, PFS, toxicity | Completed, not published | N/A |
| CTLs | |||||||||||
| ACTRN12609000338268 | CMV-specific T cells | CMV peptides | Phase I, single arm | Adults, recurrent GBM, CMV(+) serology | Activated T cell infusions every 4 weeks ×3 or until depletion, concomitant chemotherapy or bevacizumab allowed (n=9) | None | 11 | PFS: 8.1 (range, 3.6–58.7); OS: 13.3 (range, 4.4–79.9+) | Serious AEs: 0 | Published | (141) |
| NCT02060955 | ALECSAT vs. bevacizumab and irinotecan | Tumor antigens (to activate CD8+, CD4+ T cells, NK cells) | Phase II, randomized | Adults, recurrent GBM | ALECSAT infusion weeks 4, 9, 14, 26, 46, crossover permitted | Bevacizumab and irinotecan every 2 weeks | Experimental: 15; Control: 10 (175 planned) | PFS: 1.0 vs. 5.4, HR 0.16, P<0.001; OS: 5.0 vs. 6.8, HR 0.45, P=0.19 | – | Terminated, abstract only | (142) |
| NCT02799238 | ChemoRT +/− ALECSAT | Tumor antigens (to activate CD8+, CD4+ T cells, NK cells) | Phase II, randomized | Adults, new GBM | ALECSAT infusion starting concomitantly with chemoRT, every 4 weeks ×3, then every 3 months | SOC treatment | 62 expected | N/A | Primary: PFS; secondary: OS, ORR, toxicity, QoL | Active, not recruiting | N/A |
| TILs | |||||||||||
| NCT03347097 | Autologous TILs vs. PD1-TILs | Tumor antigens | Phase I, 2 arms | Adults, new GBM | Starting 10 days after standard chemoRT, infused twice every 30 days without adjuvant TMZ: Arm I: TIL infusion; Arm II: PD-1-TIL infusion (transgenic modified TILs expressing PD-1) | None | 40 expected | N/A | Primary: toxicity; secondary: OS, PFS, ORR | Recruiting | N/A |
| TCR T cells | |||||||||||
| NCT01082926 | Intratumoral GRm13Z40-2 T cells | IL-13-zetakine, HyTK | Phase I, single arm | Adults, recurrent HGG | Intratumoral GRm13Z40-2 CTL infusion on days 1 and 3 and IL-2 on days 2–5, weekly ×2 | None | 6 enrolled | N/A | Primary: toxicity | Completed, not published | N/A |
| Gamma-delta T cells | |||||||||||
| NCT04165941 | Intratumoral γδ T cells | Tumor antigens | Phase I, single arm | Adults, new GBM | Intratumoral γδ T cells 1–3 times with standard adjuvant TMZ | None | 12 expected | N/A | Primary: MTD; secondary: OS, PFS, immunogenicity | Recruiting | N/A |
CAR, chimeric antigen receptor; EGFRvIII, epidermal growth factor variant III; GBM, glioblastoma; OS, overall survival; AE, adverse event; IL-2, interleukin-2; Flu/cy, fludarabine/cyclophosphamide; PFS, progression free survival; IQR, interquartile range; ORR, objective response rate; TMZ, temozolomide; MTD, maximum tolerated dose; HER2, human epidermal growth factor receptor 2; PR, partial response; SD, stable disease; IL13Rα2, interleukin 13 receptor subunit alpha 1; G3, grade 3; HGG, high grade glioma (i.e., WHO Grade III or IV glioma); Pt, patient; QoL, quality of life; SOC, standard of care; CTL, cytotoxic T lymphocyte; EphA2, EPH receptor A2; CMV, cytomegalovirus; n, number of patients; NK, natural killer; HR, hazard ratio; ChemoRT, chemoradiation; TIL, tumor infiltrating lymphocyte; PD-1, programmed death-1; TCR, T cell receptor; IL-13, interleukin 13.
The majority of CAR T cell preclinical and clinical trials utilize EGFRvIII, HER2, IL13Rα2, or EphA2 as targets. Two phase I studies using EGFRvIII CAR T cells in adults with recurrent EGFRvIII positive GBM have been published to date. In a first-in-human trial of 10 patients who received a single infusion of CAR T cells without preconditioning chemotherapy, the authors reported 3 neurological adverse events that were possibly related to treatment, but may have been related to the disease itself (136). The median OS for this cohort was 8.2 months. Notably, one patient had SD 18 months later at the time of publication. Interestingly, the investigators obtained post-treatment tissue from 7 patients, five of which exhibited less TAA expression with increased expression of inhibitory cell surface proteins and Tregs compared to pre-treatment specimens, indicating adaptive changes in the local TME. The second published phase I trial included 18 patients who received preconditioning chemotherapy followed by EGFRvIII CAR T cell infusion with IL-2 in a dose-escalated design (137). There were 2 treatment-related adverse events, including one death at the highest CAR T cell dose level. Similar to the prior study, median survival outcomes were unimpressive with a PFS of 1.3 months (IQR, 1.1–1.9) and OS of 6.9 months (IQR, 2.8–10.0), but one patient achieved a progression-free interval of 12.5 months, two patients lived more than 12 months, and a third was alive at the time of analysis 59 months later. An additional phase I EGFRvIII CAR T cell trial in recurrent GBM is open (NCT03170141), and another, in newly diagnosed GBM that infused CAR T cells during standard maintenance TMZ, was terminated after enrolling 3 patients, results not yet reported (NCT02664363).
HER2 is expressed on up to 80% of GBM tumors, and, like EGFRvIII, is not expressed in normal neurons or glia (143). In the phase I, single arm HERT-GBM study, 17 patients (10 adults and 7 children) with recurrent GBM were treated with HER2-targeted CAR T cells infused at least once, and up to 6 times if an objective response was achieved (138). Eight patients experienced clinical benefit with one PR lasting 9 months and 7 with SD lasting 2–29 months. Three patients were alive without disease at 24–29 months. Median OS was 11.1 months (95% CI, 4.1–27.2) from the time of progression and 24.5 months (95% CI, 17.2–34.6) from the time of initial diagnosis. The iCAR trial is a phase I follow-up study from the same group of investigators that separates patients into “high risk” and “standard risk” groups based on the degree of HER2 positivity of the tumor (51–100% or 1–50%, respectively, NCT02442297).
Another recently published first-in-human phase I CAR T cell trial investigated the use of IL13Rα2 as a target for adults with recurrent GBM (139). IL-13 receptor α2 is overexpressed by 45–75% of GBM tumors, not significantly expressed on normal brain tissue, and, when bound to its ligand IL-13, releases the immunosuppressive TGF-β, making it an attractive therapeutic target (144-146). In this trial, 3 patients underwent resection of recurrent tumor with placement of a catheter, followed by intracavitary or intratumoral CAR T cell infusion up to 12 times. Two patients had grade 3 adverse events, one headache and another with transient neurologic symptoms. One patient’s tumor was analyzed before and after CAR T cell therapy and was noted to have less IL13Rα2 expression, presenting the same question observed in earlier vaccine trials: does this represent selective eradication of antigen positive tumor cells, or downregulation of antigen presentation as a mechanism of adaptive immune escape? Surprisingly, the patient with the lowest pre-treatment expression of IL13Rα2 was one of 2 who had an objective response and a favorable OS of 10.3 months, compared to 8.6 and 13.9 months in the other two patients. A larger follow-up study is now recruiting patients 12 years or older with recurrent HGG and allocating to 5 arms with various modes of delivery, including intratumoral, intracavitary, intraventricular, or a combination (NCT02575261). The investigators published a case report on one study participant with recurrent multifocal GBM who received 6 intracavitary and 10 intraventricular infusions with a complete response of all intracranial and spinal tumors, maintained 7.5 months later with no grade 3 or higher adverse events (140).
Similar to the use of ICIs, the existing clinical trials utilizing ALT have shown impressive durable responses in subsets of patients, though the majority of study patients experience a disease course similar to those receiving standard care. One limitation of ALT is that the patient must have a tumor that expresses the target of interest. In the initial trial of EGFRvIII CAR T cells, the authors reported that the tumors of 369 patients were evaluated for possible study entry, but only 79 (21%) were EGFRvIII positive (136). The heterogeneity of glioma tumors also poses a challenge. As seen in the trial of IL13Rα2 CAR T cells, there may only be selective killing of tumor cells that express the target antigen, leaving the remaining tumor cells free to divide and repopulate. Researchers are developing T cells loaded with multiple CARs (147) or bispecific CARs (148) in attempts to overcome this issue. Alternatively, the lack of antigen positive tumor cells after ALT may represent adaptive immune escape, in which case a combined modality approach may be more efficacious, a topic that is discussed further below.
In terms of safety, the two most concerning ALT-related toxicities that have been seen in the treatment of other malignancies are cytokine release syndrome and on-target/off-tumor effects (149). Cytokine release syndrome occurs as a result of diffuse triggering of T cells leading to overproduction of proinflammatory cytokines, manifesting as a combination of fever, hypotension, hypoxia, and tachycardia. On-target/off-tumor effects refer to cross reactivity between tumor cells and normal tissue cells that also express the target antigen. This was observed in the early trials of ALT in melanoma, in which there was damage to melanocytes of the skin, eye, and cochlea (150). These effects have not been observed in the published clinical trials of ALT in HGG. With the exception of a treatment-related death in the dose escalation study of EGFRvIII CAR T cells, the observed toxicities have been infrequent and transient.
Radiation and immunotherapy
The term abscopal effect was first used in 1953 to describe the phenomenon of radiation’s effect on decreasing the size of tumors “at a distance from the irradiated volume” (151). This distant effect on non-radiated tumors has since been shown to be immune-mediated in preclinical studies and case reports (152-154). It is hypothesized that radiation’s direct effects of tumor cell killing initiate a cascade of proinflammatory cytokine release and ingestion of dead cells by APCs, which in turn release immunostimulatory cytokines and present TAAs to CD8+ T cells, which differentiate into CTLs to produce a tumor-specific immune response, a process termed immunogenic cell death (155,156). Apart from direct cytotoxic effects of DNA damage, radiation may increase the immunogenicity of tumor cells by various mechanisms. Non-lethal DNA mutations caused by radiation may lead to the transcription of neoantigens, increasing mutational burden and likelihood of immune recognition (157). Double-stranded DNA fragments that enter the cytoplasm indirectly activate stimulator of interferon genes (STING), which leads to the production of IFN-I, which is involved in the maturation of glioma-directed DCs (158,159). In radiation-induced cell death, damage-associated molecular patterns (DAMPs), such as high-mobility-group box 1 (HMGB1) (160), are released and cause increased expression of costimulatory molecules on DCs, which in turn activate CTLs (161). Microglia also express DAMP receptors which, when bound, activate these cells to enhance antigen presentation (162). Radiation also increases MHC-I expression, thereby increasing the recognition of TAAs and neoantigens by CD8+ T cells (163). Given the role of radiation in SOC treatment for gliomas, and its apparent role in increasing tumor immunogenicity, its combination with immune therapies may have a synergistic effect. This has in fact been seen in a number of preclinical trials in which radiation delivered in combination with blockade of PD-1, CTLA-4, and other checkpoint proteins leads to improved tumor response and survival (164-166).
When considering the addition of immunotherapy to SOC treatment, the appropriate sequencing and timing of therapies is unknown. Most of the patients with newly diagnosed HGG included in the studies discussed in the previous sections (see Tables 1-3) underwent standard chemoradiation with neoadjuvant, concurrent, and/or adjuvant experimental immunotherapy. These studies are categorized by timing of immunotherapy relative to radiation in Table 4. One of these studies compared neoadjuvant and concurrent immunotherapy to adjuvant immunotherapy (45). Namely, newly diagnosed GBM patients in the Cancer Research UK IMA950-101 trial received a multi-peptide vaccine starting 7–14 days prior to chemoradiation that was continued concurrently (arm I, n=22) or started 7 days after completion of chemoradiation (arm II, n=23). Overall, there was a trend toward more TAA responses per patient in arm II (mean, 1.6 in arm I vs. 2.2 in arm II, P=0.295). Furthermore, when patients were assessed for any vaccine response at three different timepoints relative to the first vaccination, patients in arm I had a higher rate of response at timepoint 1 (83% vs. 57%), but a lower rate at timepoints 2 (7% vs. 21%) and 3 (10% vs. 21%). In other words, patients in arm I had a more robust response prior to chemoradiation that was not maintained during the course of treatment, whereas patients in arm II had a response that was less robust upfront but was more durable. These findings suggest chemoradiation may limit induction and maintenance of a tumor-specific immune response, possibly due to chemotherapy- and radiation-induced lymphodepletion; on the other hand, chemotherapy-induced lymphodepletion in the adjuvant setting may cause a subdued induction response followed by lymphoid recovery allowing for expansion and maintenance of the response. While clinical outcomes were the same between groups, the study was not randomized in design or powered to detect survival differences. The vast majority of vaccine therapy trials in newly diagnosed gliomas listed in Table 4 deliver the experimental therapy adjuvantly, whereas most of the ICI trials utilize concurrent delivery. These studies may provide clues as to how sequencing and timing can be optimized for various types of therapeutics, but larger randomized trials are needed to improve our understanding of these details. Particularly, the potential advantages of initiating immunotherapy prior to radiation are unknown due to a paucity of studies designed in this fashion (see Table 4).
Table 4. Clinical Trials of Immunotherapy with Conventionally Fractionated Radiation, Categorized by Timing of Immunotherapy.
| Trial number | Abbreviated trial name | Target | Additional details | Reference |
|---|---|---|---|---|
| Neoadjuvant and adjuvant | ||||
| NCT02550249 | Neoadjuvant and adjuvant nivolumab | PD-1 | Table 2 | (100) |
| NCT01006044 | Autologous tumor lysate DC vaccine (phase II) | Tumor lysate (autologous) | Table 1 | (63) |
| Adjuvant | ||||
| NCT03047473 | The SEJ Study | PD-L1 | Table 2 | (109) |
| NCT02311920 | Ipilimumab and/or nivolumab | PD-1, CTLA-4 | Table 2 | N/A |
| NCT03347097 | Autologous TILs vs. PD1-TILs | Tumor antigens | Table 3 | N/A |
| NCT04165941 | Intratumoral γδ T Cells | Tumor antigens | Table 3 | N/A |
| NCT00643097 | ACTIVATE | EGFRvIII | Table 1 | (38) |
| NCT00643097 | ACT II | EGFRvIII | Table 1 | (39) |
| NCT00458601 | ACT III | EGFRvIII | Table 1 | (40) |
| NCT01480479 | ACT IV | EGFRvIII | Table 1 | (41) |
| NCT02454634 | NOA-16 | IDH1R132H | Table 1 | (43) |
| NCT01920191 | IMA950 multi-peptide vaccine with poly-ICLC | 11 GBM TAAs | Table 1 | (46) |
| NCT02149225 | GAPVAC-101 | 5–10 unmutated TAAs and 1–2 mutated TAAs based on tumor mutation/transcriptome analysis | Table 1 | (49) |
| UMIN000001426 | An autologous tumor vaccine with adjuvant TMZ | Autologous tumor peptides | Table 1 | (51) |
| NCT00905060 | HeatShock | HSPPC-96 | Table 1 | (56) |
| NCT03650257 | Adjuvant TMZ +/− HSPPC-96 vaccine | HSPPC-96 | Table 1 | N/A |
| N/A | EGFRvIII-targeted DC vaccine | EGFRvIII | Table 1 | (57) |
| N/A | ICT-107 | 6 GBM TAAs | Table 1 | (58) |
| NCT01280552 | Adjuvant TMZ +/− ICT-107 | 6 GBM TAAs | Table 1 | (59) |
| NCT00068510 | Autologous tumor lysate DC vaccine (phase I) | Tumor lysate (autologous) | Table 1 | (60) |
| EY-DOH-MD #0910072504 | Autologous tumor lysate DC vaccine (phase I/II) | Tumor lysate (autologous) | Table 1 | (61) |
| N/A | ChemoRT +/− autologous tumor lysate DC vaccine | Tumor lysate (autologous) | Table 1 | (62) |
| NCT01213407 | Adjuvant TMZ +/− Audencel | Tumor lysate (autologous) | Table 1 | (65) |
| NCT00045968 | Adjuvant TMZ +/− DCVax®-L | Tumor lysate (autologous) | Table 1 | (66) |
| NCT01957956 | Allogenic tumor lysate DC vaccine (new GBM) | Tumor lysate (allogeneic) | Table 1 | N/A |
| NCT02010606 | Allogenic tumor lysate DC vaccine from a GBM stem-like cell line | Tumor lysate (allogeneic) | Table 1 | N/A |
| NCT00846456 | GSC antigen mRNA DC vaccine | Autologous GSC antigens | Table 1 | (67) |
| NCT02649582 | ADDIT-GLIO | WT1 | Table 1 | N/A |
| NCT02709616 | PERCELLVAC | Autologous TAAs | Table 1 | N/A |
| NCT00639639 | CMV pp65 DC vaccine | pp65 | Table 1 | (69) |
| NCT03927222 | I-ATTAC | pp65 | Table 1 | N/A |
| NCT02465268 | ATTAC-II | pp65 | Table 1 | N/A |
| Concurrent | ||||
| NCT02617589 | CheckMate-498 | PD-1 | Table 2 | (103) |
| NCT02667587 | CheckMate-548 | PD-1 | Table 2 | (104) |
| NCT02530502 | Pembrolizumab with chemoRT | PD-1 | Table 2 | N/A |
| NCT02336165 | Durvalumab with RT (new) or with/without bevacizumab (recurrent) | PD-L1 | Table 2 | (106) |
| NCT04047706 | Nivolumab and IDO1 inhibition (BMS-986205) with RT or ChemoRT | PD-1, IDO1 | Table 2 | N/A |
| NCT02799238 | ChemoRT +/− ALECSAT | Tumor antigens (to activate CD8+, CD4+ T cells, NK cells) | Table 3 | N/A |
| UMIN000000002 | An autologous tumor vaccine with RT | Autologous tumor peptides | Table 1 | (50) |
| NCT01567202 | ChemoRT +/− autologous GSC DC vaccine | Autologous GSC antigens | Table 1 | (68) |
| Other | ||||
| NCT01222221 | Cancer Research UK IMA950-101 | 11 GBM TAAs | Table 1 | (45) |
PD-1, programmed death-1; DC, dendritic cell; PD-L1, programmed death ligand 1; CTLA-4, cytotoxic T-lymphocyte-associated protein 4; TIL, tumor infiltrating lymphocyte; EGFRvIII, epidermal growth factor variant III; Poly-ICLC, polyinosinic-polycytidylic acid-poly-l-lysine carboxymethylcellulose; GBM, glioblastoma; TAA, tumor associated antigen; TMZ, temozolomide; HSPPC-96, heat shock protein peptide complex 96; ChemoRT, chemoradiation; GSC, glioma stem cell; WT1, Wilms tumor 1; CMV, cytomegalovirus; pp65, phosphoprotein 65; RT, radiation; IDO1, indoleamine-2,3-dioxygenase 1; NK, natural killer.
While the prior discussion focused on the use of conventionally-fractionated radiation therapy [CFRT, using smaller doses of 1.8–2.0 Gray (Gy) per fraction daily over 6 or more weeks], some have hypothesized that fractionated stereotactic radiosurgery (fSRS, using doses of 6–8 Gy per fraction over 3–5 treatments) may be more effective in generating an immune response. Such “ablative” doses may lead to increased tumor antigen release and, in preclinical studies, have shown favorable changes to the TME (167,168). Though the use of fSRS has not proven beneficial in the treatment of GBM (169), its use in combination with immunotherapy has not been well studied. A number of clinical trials are underway to investigate the benefit of fSRS and ICIs (and one study using EGFRvIII CAR T cells) in recurrent glioma (Table 5). One phase I, single arm study published interim results on 23 patients treated with fSRS to a dose of 30 Gy in 5 fractions with bevacizumab and concurrent dose-escalated pembrolizumab with 53% of patients experiencing a complete or PR lasting at least 6 months, and 6- and 12-month OS rates of 94% and 64%, respectively (170). There was one grade 3 treatment-related toxicity of elevated liver transaminases. As an unstudied domain, these upcoming trials are an exciting source of data to potentially support new and meaningful treatment approaches.
Table 5. Clinical trials of immunotherapy with fractionated stereotactic radiosurgery.
| Trial number | Abbreviated trial name | Target | Study design | Population | Intervention/arms | Control | Number of analyzed patients | Survival, median months (95% CI) | Additional outcomes | Trial status | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|
| NCT02313272 | Pembrolizumab with fSRS and bevacizumab | PD-1 | Phase I, single arm, 3+3 | Adults, recurrent HGG | fSRS (30 Gy/5 fx) with concomitant dose-escalated pembrolizumab every 3 weeks and bevacizumab every 2 weeks | None | 32 enrolled; 23 analyzed | 6-mo OS: 94%; 12-mo OS: 64%; CR or PR ≥6 mo: 53% | G3+ AE: 1 (elevated transaminases) | Active, not recruiting, abstract only | (170) |
| NCT02866747 | STERIMGLI | PD-L1 | Phase I/II, randomized | Adults, recurrent GBM | fSRS (24 Gy/3 fx) with durvalumab starting day of 3rd fx, then every 4 weeks | fSRS (24 Gy/3 fx) alone | 62 expected | Phase I results: G3+ AE: 1 (vestibular neuritis) | Primary: DLT, ORR; secondary: OS, PFS, QoL, toxicity | Suspended (Interim analysis), abstract only (phase I) | (171) |
| NCT02968940 | Avelumab with fSRS | PD-L1 | Phase II, single arm | Adults, WHO II or III IDH-mutant glioma transformed to GBM | fSRS (30 Gy/5 fx) with avelumab every 2 weeks | None | 43 expected | N/A | Primary: toxicity, PFS; secondary: OS, ORR | Active, not recruiting | N/A |
| NCT03961971 | Anti-Tim-3 (MBG453) and spartalizumab (anti-PD-1) with fSRS | Tim-3, PD-1 | Phase I, single arm | Adults, recurrent GBM | Anti-Tim-3 and spartalizumab ×1, then fSRS, then anti-Tim-3 and spartalizumab every 4 weeks until progression or intolerance | None | 15 expected | N/A | Primary: toxicity; secondary: OS, PFS, ORR | Active, not recruiting | N/A |
| NCT02829931 | Nivolumab with fSRS | PD-1 | Phase I, single arm | Adults, recurrent HGG | fSRS (30 Gy/5 fx) then nivolumab every 2 weeks ×8, then nivolumab every 4 weeks ×4 | None | 26 expected | N/A | Primary: toxicity; secondary: OS, ORR, biomarkers | Recruiting | (172) |
| NCT03743662 | Nivolumab with fSRS and bevacizumab | PD-1 | Phase II, 2 arms | Adults, recurrent GBM, MGMT-methylated | Arm I: nivolumab every 2 weeks ×2, then fSRS (30 Gy/5 fx) with bevacizumab every 2 weeks ×2, then nivolumab every 2 weeks until progression or intolerance; Arm II: Addition of surgery after first 2 cycles of nivolumab, prior to fSRS | None | 94 expected | N/A | Primary: OS; secondary: PFS, ORR | Recruiting | N/A |
| NCT04225039 | Anti-GITR (INCAGN01876) and anti-PD-1 (INCMGA00012) with fSRS | GITR, PD-1 | Phase II, 3 arms | Adults, recurrent GBM | Arm I: anti-GITR and anti-PD-1 ×1, then fSRS (24 Gy/3 fx), then anti-PD-1 every 4 weeks and anti-GITR every 2 weeks until progression or intolerance; Arm II: Arm I with surgery following fSRS; Arm III: Arm I with surgery and no RT | None | 32 expected | N/A | Primary: ORR; secondary: OS, PFS, toxicity | Not yet recruiting | N/A |
| NCT03283631 | INTERCEPT | EGFRvIII | Phase I, single arm | Adults, recurrent GBM, EGFRvIII(+) | fSRS followed by same-day intratumoral infusion of CAR T cells | None | 24 expected | N/A | Primary: MTD; secondary: OS, PFS, immunogenicity | Recruiting | N/A |
fSRS, fractionated stereotactic radiosurgery; PD-1, programmed death-1; HGG, high grade glioma (i.e., WHO Grade III or IV glioma); Gy, Gray; Fx, fractions; OS, overall survival; CR, complete response; PR, partial response; G3, grade 3; AE, adverse event; PD-L1, programmed death ligand 1; GBM, glioblastoma; DLT, dose limiting toxicity; ORR, objective response rate; PFS, progression free survival; QoL, quality of life; MGMT, O6-methylguanine DNA methyltransferase; GITR, glucocorticoid-induced TNFR family related gene; EGFRvIII, epidermal growth factor variant III; CAR, chimeric antigen receptor; MTD, maximum tolerated dose.
Of note, while it appears radiation may augment tumor-specific immune responses, concerns have been raised regarding the potential immunosuppressive effects of standard treatment for HGG. Patients treated with radiation and TMZ (and, in many cases, glucocorticoids) often have significant reductions in CD4+ T cell counts that are long-lasting and have been associated with early death due to tumor progression (173). While chemotherapy-induced lymphodepletion is a well-understood phenomenon, radiation-induced lymphodepletion may impede immune-mediate tumor control due to direct damage to lymphocytes, though these mechanisms are not well understood (37). One published phase I/IIa study of 22 patients with newly diagnosed GBM received an autologous tumor peptide vaccine concomitant with and after standard radiation without TMZ with favorable PFS and OS outcomes of 7.6 months (95% CI, 4.3–13.6) and 19.8 months (95% CI, 13.8–31.3), respectively (50). However, the unpublished results of the large CheckMate-498 trial, which compared SOC treatment to concurrent and adjuvant nivolumab without TMZ, showed no survival benefit [Bristol-Myers Squibb, 2019, unpublished data (103)]. Additional studies of various immunotherapy modalities with or without TMZ are needed to further characterize the effects of chemotherapy on tumor-directed immune responses and clinical outcomes. Several ongoing studies are omitting concurrent or adjuvant TMZ, including one phase I trial of nivolumab and IDO1 inhibition in newly diagnosed GBM that includes separate treatment arms with or without TMZ (NCT04047706). Details regarding chemotherapy use in these studies are summarized in Table 4.
Combination immunotherapies
One of the predominant approaches in recently opened clinical trials for newly diagnosed and recurrent HGG is combination immunotherapy, i.e., utilizing a combination of therapeutic vaccination, ICI, and ALT. Each individual therapy stimulates or augments the immune system by a different mechanism: ICIs overcome local immunosuppression of the TME and T cell anergy, while vaccines and ALT overcome nonimmunogenic properties of the tumor and augment the limited specificity and size of the pre-existing T cell population, respectively. Preclinical studies have, indeed, shown improved immune and tumor responses with the use of an ICI in conjunction with a vaccine or ALT (174-177). Moreover, hypotheses of improved responses to combination therapy are also derived from observations of some of the early single-modality clinical trials discussed above. For example, O’Rourke et al. observed that after administration of EGFRvIII CAR T cells to patients with recurrent GBM, resection specimens expressed higher levels of inhibitor cell surface proteins, including PD-L1, with similar levels of PD-1 expression on TILs (136). Thus, the addition of an ICI that blocks interactions between PD-1 and PD-L1 would presumably limit this mechanism of adaptive immune escape. This hypothesis is the basis of an ongoing phase I, single arm trial of patients with newly diagnosed MGMT-unmethylated GBM, utilizing short-course radiation (40 Gy in 15 fractions) without TMZ followed by combined EGFRvIII CAR T cell and pembrolizumab infusions every 3 weeks for 3 and 4 cycles, respectively [NCT03726515, (178)].
In another single-modality trial, Schuessler et al. reported the results of a phase I, single arm trial of 11 adults with recurrent GBM and CMV-positive serology, who received autologous CMV-specific T cell infusions (141). Notably, one patient who enrolled after a second recurrence was still alive more than 6 years later at the time of publication. The investigators observed that patients with less durable control (i.e., PFS <100 days) had significant differences in gene expression compared to durable responders, including higher expression of CTLA-4, again implying the potential benefit of combined treatment with an ICI. Moreover, one subject underwent subsequent resection, and analysis of TILs showed that only approximately one third of antigen-specific T cells were polyfunctional. Polyfunctional T cells (i.e., those that express multiple pro-inflammatory cytokines and cytotoxic markers), have been associated with prolonged survival in melanoma patients treated with DC vaccination (179), and Reap et al. hypothesized that the addition of pp65 mRNA-loaded DCs to CMV-specific T cell therapy would generate more polyfunctional T cells (180). In their phase I study, 17 patients with newly diagnosed GBM were randomized to receive activated T cells once and either the DC vaccine or placebo every 2 weeks for three cycles, concomitant with standard adjuvant TMZ. They did in fact find that the presence of polyfunctional T cells (positive for IFN-γ, TNF-α, and CCL3) was correlated with OS (R =0.7371, P=0.0369) in patients that received combination therapy.
Limited data exist on clinical outcomes of combination immunotherapy in HGG at this time, but current reports include a phase I trial published in 2000 (181) and unpublished data from the AVeRT study [Archer G, 2020, unpublished data (182)]. In the former, a phase I, single arm trial reported by Sloan et al., 19 adults with recurrent HGG (2 with anaplastic astrocytoma, 1 with gliosarcoma, and 16 with GBM) underwent resection followed by administration of an autologous whole tumor cell vaccine (with GM-CSF) and anti-CD3 T cell therapy with a favorable median OS of 12 months (range, 6–28) (181). In the phase I AVeRT study, 6 adults with recurrent HGG were randomized to receive 4 cycles of biweekly nivolumab with (n=3) or without (n=3) three additional biweekly cycles of nivolumab in combination with a pp65 mRNA-loaded DC vaccine. Patients in both arms then underwent re-resection followed by biweekly nivolumab until progression and 5 cycles of monthly DC vaccine infusions. The 3 patients who received both nivolumab and the DC vaccine prior to resection had PFS and OS of 6.3 months (4.7–10.7) and 15.3 months (4.73–not reached), respectively, compared to 4.3 months (2.1–5.3) and 8.0 months (5.7–8.3) in the patients who received nivolumab only prior to resection [Archer G, 2020, unpublished data (182)]. To date, the number of HGG patients treated with combination immunotherapy is small, but these early data and the scientific basis of their possible synergy are encouraging. Additional ongoing studies are summarized in Table 6.
Table 6. Combination immunotherapy clinical trials.
| Trial number | Abbreviated trial name | Target | Study design | Population | Intervention/arms | Control | Number of analyzed patients | Survival, median months (95% CI) | Additional outcomes | Trial status | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Vaccine + ICI | |||||||||||
| NCT04013672 | SurVaxM peptide vaccine and pembrolizumab | Survivin, PD-1 | Phase II, 2 arms | Adults, recurrent GBM | SurVaxM/GM-CSF/Montanide ISA 51 every 2 weeks times 4 then every 3 months, with pembrolizumab every 3 weeks, until progression or intolerance; Arm I: anti-PD-1 therapy naïve; Arm II: failed previous anti-PD-1 therapy | None | 51 expected | N/A | Primary: PFS; secondary: toxicity | Recruiting | N/A |
| NCT03893903 | AMPLIFY-NEOVAC | IDH1R132H, PD-L1 | Phase I, 3 arms, randomized | Adults, recurrent HGG excluding 1p/19q co-deletion and nuclear ATRX loss | Experimental drug every 2 weeks ×3, followed by re-resection, followed by maintenance experimental drug: Arm I: IDH1R132H peptide vaccine; Arm II: IDH1R132H peptide vaccine with avelumab; Arm III: avelumab | None | 48 expected | N/A | Primary: RLT; secondary: immunogenicity, AEs, ORR, OS, PFS | Recruiting | N/A |
| NCT03665545 | IMA950-106 | 11 GBM TAAs, PD-1 | Phase I/II, 2 arms, randomized | Adults, recurrent GBM, HLA-A*02(+) | Arm I: IMA950/poly-ICLC vaccine alone; Arm II: IMA950/poly-ICLC vaccine with pembrolizumab | None | 24 expected | N/A | Primary: toxicity; secondary: PFS, OS, QoL, immunogenicity | Recruiting | N/A |
| NCT03018288 | Pembrolizumab +/− HSPPC-96 vaccine | PD-1, HSPPC-96 | Phase II, randomized, double-blind | Adults, new GBM | Arm I: pembrolizumab every 3 weeks ×3 concomitant with standard chemoRT with TMZ; Arm II: Arm I with addition of HSPPC-96 vaccine post-chemoradiation weekly ×4, then every 3 weeks ×6 | Arm III: Arm II with placebo vaccine | 14 enrolled | N/A | Primary: OS | Suspended (root cause analysis being conducted) | N/A |
| NCT04201873 | Autologous tumor lysate DC vaccine +/− pembrolizumab | Tumor lysate (autologous), PD-1 | Phase I, randomized | Adults, recurrent GBM | Pembrolizumab ×1 2 weeks prior to re-resection, then every 3 weeks after resection until progression or intolerance. DC vaccine/poly-ICLC after resection every 2 weeks ×3 | Placebo pembrolizumab with experimental DC vaccine/poly-ICLC on same schedule | 40 expected | N/A | Primary: toxicity, immunogenicity; secondary: OS, PFS, biomarkers | Recruiting | N/A |
| NCT02529072 | AVeRT | pp65, PD-1 | Phase I, randomized | Adults, recurrent HGG | Arm I: nivolumab every 2 weeks ×4, then re-resection, then biweekly nivolumab until progression and monthly CMV pp65 DC vaccine ×5; Arm II: Nivolumab every 2 weeks ×4, then nivolumab and CMV pp65 DC vaccine every 2 weeks ×3, then re-resection, then biweekly nivolumab until progression and monthly CMV pp65 DC vaccine ×5 | None | Arm I: 3; Arm II: 3 | PFS, OS: Arm I: 4.3 (2.1–5.3), 8.0 (5.7–8.3); Arm II: 6.3 (4.7–10.7), 15.3 (4.73–not reached) | – | Completed, unpublished data | (182) |
| NCT02798406 | DNX-2401 oncolytic adenovirus and pembrolizumab | E1A mutant, PD-1 | Phase II, single arm | Adults, recurrent GBM | DNX-2401 vaccine intratumoral injection ×1, then pembrolizumab every 3 weeks for 2 years or until progression or intolerance | None | 49 enrolled | N/A | Primary: ORR; secondary: OS, PFS | Active, not recruiting | N/A |
| Vaccine + other | |||||||||||
| NCT02366728 | ELEVATE | pp65, IL-2R | Phase II, randomized, double-blind, placebo-controlled | Adults, new GBM s/p resection with <1 cm residual in maximal dimension | Arm I: placebo DC vaccine; Arm II: DC vaccine; Arm III: DC vaccine with basiliximab (anti-IL-2R) | Arm I: placebo vaccine | 100 expected | N/A | Primary: OS, immunogenicity; secondary: PFS | Active, not recruiting | N/A |
| Vaccine + ALT | |||||||||||
| N/A | Autologous whole tumor vaccine and anti-CD3 T cells | Tumor antigens, CD3 | Phase I, single arm | Adults, recurrent HGG | Re-resection followed by vaccine/GM-CSF ×2, followed by activated T cells ×1 | None | 19 | OS: 12 mo (range, 6–28) | – | Published | (181) |
| NCT00693095 | CMV-specific T cells +/− CMV pp65 DC vaccine | pp65 | Phase I, randomized | Adults, new GBM | Arm I: activated T cells ×1 and DC vaccine every 2 weeks ×3 concomitant with standard adjuvant TMZ; Arm II: Arm I with saline placebo instead of DC vaccine | None | Arm I: 9; Arm II: 8 | PFS and OS N/R; polyfunctional T cells correlated with OS (R =0.7371, P=0.0369) in Arm I | G3+ AEs: 0 | Published | (180) |
| ALT + ICI | |||||||||||
| NCT03726515 | EGFRvIII CAR T cells and pembrolizumab | EGFRvIII, PD-1 | Phase I, single arm | Adults, new GBM, MGMT-unmethylated, EGFRvIII(+) | 2–3 weeks after short-course RT (40 Gy/15 fx) without TMZ, CAR T cells and pembrolizumab given every 3 weeks ×3 and ×4, respectively | None | 7 expected | N/A | Primary: toxicity; secondary: OS, PFS, ORR | Active, not recruiting | (178) |
| NCT04003649 | IL13Rα2 CAR T cells and nivolumab +/− ipilimumab | IL13Rα2, PD-1, CTLA-4 | Phase I, randomized | Adults, recurrent GBM, IL13Rα2(+) | Re-resection with intraventricular catheter placement, then: Arm I: ipilimumab and nivolumab ×1, then two weeks later, nivolumab and intraventricular CAR T cells ×4 or more; Arm II: nivolumab and intraventricular CAR T cells ×4 or more | None | 60 expected | N/A | Primary: toxicity, feasibility; secondary: OS, PFS, ORR, immunogenicity, biomarkers, QoL | Recruiting | N/A |
| ICI + other | |||||||||||
| NCT02423343 | Galunisertib (anti-TGFβRI) and nivolumab | PD-1, TGFβRI | Phase Ib, single arm | Adults, recurrent GBM | Galunisertib for 14 days every 4 weeks ×4 and nivolumab every 2 weeks ×8 | None | 75 enrolled (entire cohort) | N/A | Primary: MTD | Active, not recruiting | N/A |
| NCT04160494 | D2C7 (EGFR-targeted immunotoxin) and atezolizumab | EGFRwt/EGFRvIII, PD-L1 | Phase I, single arm | Adults, recurrent GBM | Intratumoral D2C7 ×1, then atezolizumab ×1 2 weeks later, then possible re-resection 2 weeks later, then atezolizumab every 3 weeks for 2 years or until progression or intolerance | None | 18 expected | N/A | Primary: toxicity | Recruiting | N/A |
ICI, immune checkpoint inhibitor; PD-1, programmed death-1; GBM, glioblastoma; GM-CSF, granulocyte-macrophage colony-stimulating factor; PFS, progression-free survival; PD-L1, programmed death ligand 1; HGG, high grade glioma (i.e., WHO Grade III or IV glioma); ATRX, alpha-thalassemia/mental retardation, X-linked; RLT, regime limiting toxicity; AE, adverse event; ORR, objective response rate; OS, overall survival; TAA, tumor associated antigen; Poly-ICLC, polyinosinic-polycytidylic acid-poly-l-lysine carboxymethylcellulose; QoL, quality of life; HSPPC-96, heat shock protein peptide complex 96; ChemoRT, chemoradiation; TMZ, temozolomide; DC, dendritic cell; pp65, phosphoprotein 65; CMV, cytomegalovirus; E1A, early region 1A; IL-2R, interleukin 2 receptor; N/R, not reported; G3, grade 3; MGMT, O6-methylguanine DNA methyltransferase; EGFRvIII, epidermal growth factor variant III; RT, radiation; Gy, Gray; fx, fractions; CAR, chimeric antigen receptor; IL13Rα2, interleukin 13 receptor subunit alpha 1; CTLA-4, cytotoxic T-lymphocyte-associated protein 4; TGFβRI, TGFβ receptor I; MTD, maximum tolerated dose; EGFRwt, epidermal growth factor wild type.
Conclusions
This discussion has attempted to comprehensively review the existing data and ongoing studies of immunotherapy and radiation in HGG, including therapeutic vaccination, immune checkpoint inhibition, and ALT. Though larger randomized controlled trials have not consistently shown improvements in clinical outcomes, this area of research is still in its early stages and a number of important lessons can be taken away from the studies that have been completed to date. Many studies found a subset of patients who experienced durable responses, and analysis of their immune cells and tumor cells can be used to identify biomarkers that predict therapeutic response, as well as additional glioma-specific targets that can enhance therapeutic efficacy. Given the rationale for potential synergy with radiation and combination immunotherapies, future research should focus on these approaches. Identification of biomarkers to predict the efficacy of radiation combined with immunotherapy is of great importance in appropriately selecting patients for more aggressive treatment regimens. This specific area of combinatorial therapy is only in its infancy, and the results of ongoing trials are anticipated to push us further along the path to achieving long-term survival for patients with GBM and other HGGs.
Acknowledgments
Funding: None.
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Footnotes
Provenance and Peer Review: This article was commissioned by the Guest Editors (Dr. Arya Amini and Dr. Tyler Robin) for the series “Synergy in Action: Novel Approaches to Combining Radiation Therapy and Immunotherapy” published in Translational Cancer Research. The article has undergone external peer review.
Reporting Checklist: The authors have completed the Narrative Review reporting checklist. Available at http://dx.doi.org/10.21037/tcr-20-1933
Peer Review File: Available at http://dx.doi.org/10.21037/tcr-20-1933
Conflicts of Interest: All authors have completed the ICMJE uniform disclosure form (available at http://dx.doi.org/10.21037/tcr-20-1933). The series “Synergy in Action: Novel Approaches to Combining Radiation Therapy and Immunotherapy” was commissioned by the editorial office without any funding or sponsorship. DEN reports personal fees from DNAtrix, outside the submitted work. DRO reports grants from American Heart Association, grants from American Cancer Society, grants from Medtronic, grants from Agios, outside the submitted work. CGR reports grants from Takeda, personal fees from Genentech, and personal fees from AstraZeneca, outside the submitted work. The authors have no other conflicts of interest to declare.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin 2019;69:7-34. 10.3322/caac.21551 [DOI] [PubMed] [Google Scholar]
- 2.National Comprehensive Cancer Network. Central Nervous System Cancers (Version 1.2020). Available online: https://www.nccn.org/professionals/physician_gls/pdf/cns.pdf. Accessed March 18, 2020.
- 3.Stupp R, Mason WP, Van Den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 2005;352:987-96. 10.1056/NEJMoa043330 [DOI] [PubMed] [Google Scholar]
- 4.Stupp R, Taillibert S, Kanner A, et al. Effect of tumor-treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma: a randomized clinical trial. JAMA 2017;318:2306-16. 10.1001/jama.2017.18718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Krex D, Klink B, Hartmann C, et al. Long-term survival with glioblastoma multiforme. Brain 2007;130:2596-606. 10.1093/brain/awm204 [DOI] [PubMed] [Google Scholar]
- 6.Stupp R, Hegi ME, Mason WP, et al. European Organisation for Research and Treatment of Cancer Brain Tumour and Radiation Oncology Groups; National Cancer Institute of Canada Clinical Trials Group. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol 2009;10:459-66. 10.1016/S1470-2045(09)70025-7 [DOI] [PubMed] [Google Scholar]
- 7.Park JK, Hodges T, Arko L, et al. Scale to predict survival after surgery for recurrent glioblastoma multiforme. J Clin Oncol 2010;28:3838-43. 10.1200/JCO.2010.30.0582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Taal W, Oosterkamp HM, Walenkamp AM, et al. Single-agent bevacizumab or lomustine versus a combination of bevacizumab plus lomustine in patients with recurrent glioblastoma (BELOB trial): a randomised controlled phase 2 trial. Lancet Oncol 2014;15:943-53. 10.1016/S1470-2045(14)70314-6 [DOI] [PubMed] [Google Scholar]
- 9.Wick W, Puduvalli VK, Chamberlain MC, et al. Phase III study of enzastaurin compared with lomustine in the treatment of recurrent intracranial glioblastoma. J Clin Oncol 2010;28:1168-74. 10.1200/JCO.2009.23.2595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brandes AA, Tosoni A, Amista P, et al. How effective is BCNU in recurrent glioblastoma in the modern era? A phase II trial. Neurology 2004;63:1281-4. 10.1212/01.WNL.0000140495.33615.CA [DOI] [PubMed] [Google Scholar]
- 11.Stupp R, Wong ET, Kanner AA, et al. NovoTTF-100A versus physician's choice chemotherapy in recurrent glioblastoma: a randomised phase III trial of a novel treatment modality. Eur J Cancer 2012;48:2192-202. 10.1016/j.ejca.2012.04.011 [DOI] [PubMed] [Google Scholar]
- 12.Friedman HS, Prados MD, Wen PY, et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol 2009;27:4733-40. 10.1200/JCO.2008.19.8721 [DOI] [PubMed] [Google Scholar]
- 13.Quant EC, Norden AD, Drappatz J, et al. Role of a second chemotherapy in recurrent malignant glioma patients who progress on bevacizumab. Neuro Oncol 2009;11:550-5. 10.1215/15228517-2009-006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hodi FS, O'Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010;363:711-23. 10.1056/NEJMoa1003466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med 2015;372:320-30. 10.1056/NEJMoa1412082 [DOI] [PubMed] [Google Scholar]
- 16.Robert C, Schachter J, Long GV, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med 2015;372:2521-32. 10.1056/NEJMoa1503093 [DOI] [PubMed] [Google Scholar]
- 17.Motzer RJ, Escudier B, McDermott DF, et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med 2015;373:1803-13. 10.1056/NEJMoa1510665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brahmer J, Reckamp KL, Baas P, et al. Nivolumab versus docetaxel in advanced squamous-cell non–small-cell lung cancer. N Engl J Med 2015;373:123-35. 10.1056/NEJMoa1504627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reck M, Rodríguez-Abreu D, Robinson AG, et al. Pembrolizumab versus chemotherapy for PD-L1–positive non–small-cell lung cancer. N Engl J Med 2016;375:1823-33. 10.1056/NEJMoa1606774 [DOI] [PubMed] [Google Scholar]
- 20.Horn L, Mansfield AS, Szczęsna A, et al. First-line atezolizumab plus chemotherapy in extensive-stage small-cell lung cancer. N Engl J Med 2018;379:2220-9. 10.1056/NEJMoa1809064 [DOI] [PubMed] [Google Scholar]
- 21.Murphy JB, Sturm E. Conditions determining the transplantability of tissues in the brain. J Exp Med 1923;38:183-97. 10.1084/jem.38.2.183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cancilla PA, DeBault LE. Neutral amino acid transport properties of cerebral endothelial cells in vitro. J Neuropathol Exp Neurol 1983;42:191-9. 10.1097/00005072-198303000-00008 [DOI] [PubMed] [Google Scholar]
- 23.Fabry Z, Raine CS, Hart MN. Nervous tissue as an immune compartment: the dialect of the immune response in the CNS. Immunol Today 1994;15:218-24. 10.1016/0167-5699(94)90247-X [DOI] [PubMed] [Google Scholar]
- 24.Theele DP, Streit WJ. A chronicle of microglial ontogeny. Glia 1993;7:5-8. 10.1002/glia.440070104 [DOI] [PubMed] [Google Scholar]
- 25.Binder DC, Davis AA, Wainwright DA. Immunotherapy for cancer in the central nervous system: current and future directions. Oncoimmunology 2015;5:e1082027. 10.1080/2162402X.2015.1082027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lowe J, MacLennan KA, Powe DG, et al. Microglial cells in human brain have phenotypic characteristics related to possible function as dendritic antigen presenting cells. J Pathol 1989;159:143-9. 10.1002/path.1711590209 [DOI] [PubMed] [Google Scholar]
- 27.Ulvestad E, Williams K, Bjerkvig R, et al. Human microglial cells have phenotypic and functional characteristics in common with both macrophages and dendritic antigen-presenting cells. J Leukoc Biol 1994;56:732-40. 10.1002/jlb.56.6.732 [DOI] [PubMed] [Google Scholar]
- 28.Harling-Berg C, Knopf PM, Merriam J, et al. Role of cervical lymph nodes in the systemic humoral immune response to human serum albumin microinfused into rat cerebrospinal fluid. J Neuroimmunol 1989;25:185-93. 10.1016/0165-5728(89)90136-7 [DOI] [PubMed] [Google Scholar]
- 29.Yamada S, DePasquale M, Patlak CS, et al. Albumin outflow into deep cervical lymph from different regions of rabbit brain. Am J Physiol 1991;261:H1197-204. [DOI] [PubMed] [Google Scholar]
- 30.Aspelund A, Antila S, Proulx ST, et al. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J Exp Med 2015;212:991-9. 10.1084/jem.20142290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Louveau A, Herz J, Alme MN, et al. CNS lymphatic drainage and neuroinflammation are regulated by meningeal lymphatic vasculature. Nat Neurosci 2018;21:1380-91. 10.1038/s41593-018-0227-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wolburg H, Noell S, Mack A, et al. Brain endothelial cells and the glio-vascular complex. Cell Tissue Res 2009;335:75-96. 10.1007/s00441-008-0658-9 [DOI] [PubMed] [Google Scholar]
- 33.Galea I, Bernardes-Silva M, Forse PA, et al. An antigen-specific pathway for CD8 T cells across the blood-brain barrier. J Exp Med 2007;204:2023-30. 10.1084/jem.20070064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tanaka S, Louis DN, Curry WT, et al. Diagnostic and therapeutic avenues for glioblastoma: no longer a dead end? Nat Rev Clin Oncol 2013;10:14-26. 10.1038/nrclinonc.2012.204 [DOI] [PubMed] [Google Scholar]
- 35.Liao W, Lin JX, Leonard WJ. IL-2 family cytokines: new insights into the complex roles of IL-2 as a broad regulator of T helper cell differentiation. Curr Opin Immunol 2011;23:598-604. 10.1016/j.coi.2011.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oh T, Sayegh ET, Fakurnejad S, et al. Vaccine therapies in malignant glioma. Curr Neurol Neurosci Rep 2015;15:508 10.1007/s11910-014-0508-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reznik E, Smith AW, Taube S, et al. Radiation and immunotherapy in high-grade gliomas. Am J Clin Oncol 2018;41:197-212. 10.1097/COC.0000000000000406 [DOI] [PubMed] [Google Scholar]
- 38.Sampson JH, Heimberger AB, Archer GE, et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J Clin Oncol 2010;28:4722-9. 10.1200/JCO.2010.28.6963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sampson JH, Aldape KD, Archer GE, et al. Greater chemotherapy-induced lymphopenia enhances tumor-specific immune responses that eliminate EGFRvIII-expressing tumor cells in patients with glioblastoma. Neuro Oncol 2011;13:324-33. 10.1093/neuonc/noq157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schuster J, Lai RK, Recht LD, et al. A phase II, multicenter trial of rindopepimut (CDX-110) in newly diagnosed glioblastoma: the ACT III study. Neuro Oncol 2015;17:854-61. 10.1093/neuonc/nou348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weller M, Butowski N, Tran DD, et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): a randomised, double-blind, international phase 3 trial. Lancet Oncol 2017;18:1373-85. 10.1016/S1470-2045(17)30517-X [DOI] [PubMed] [Google Scholar]
- 42.Reardon DA, Desjardins A, Vredenburgh JJ, et al. Rindopepimut with Bevacizumab for Patients with Relapsed EGFRvIII-Expressing Glioblastoma (ReACT): Results of a Double-Blind Randomized Phase II Trial. Clin Cancer Res 2020;26:1586-94. 10.1158/1078-0432.CCR-18-1140 [DOI] [PubMed] [Google Scholar]
- 43.Platten M, Schilling D, Bunse L, et al. A mutation-specific peptide vaccine targeting IDH1R132H in patients with newly diagnosed malignant astrocytomas: A first-in-man multicenter phase I clinical trial of the German Neurooncology Working Group (NOA-16). J Clin Oncol 2018;36:2001. 10.1200/JCO.2018.36.15_suppl.2001 [DOI] [Google Scholar]
- 44.Fenstermaker RA, Ciesielski MJ, Qiu J, et al. Clinical study of a survivin long peptide vaccine (SurVaxM) in patients with recurrent malignant glioma. Cancer Immunol Immunother 2016;65:1339-52. 10.1007/s00262-016-1890-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rampling R, Peoples S, Mulholland PJ, et al. A cancer research UK first time in human phase I trial of IMA950 (novel multipeptide therapeutic vaccine) in patients with newly diagnosed glioblastoma. Clin Cancer Res 2016;22:4776-85. 10.1158/1078-0432.CCR-16-0506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Migliorini D, Dutoit V, Allard M, et al. Phase I/II trial testing safety and immunogenicity of the multipeptide IMA950/poly-ICLC vaccine in newly diagnosed adult malignant astrocytoma patients. Neuro Oncol 2019;21:923-33. 10.1093/neuonc/noz040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Terasaki M, Shibui S, Narita Y, et al. Phase I trial of a personalized peptide vaccine for patients positive for human leukocyte antigen-A24 with recurrent or progressive glioblastoma multiforme. J Clin Oncol 2011;29:337-44. 10.1200/JCO.2010.29.7499 [DOI] [PubMed] [Google Scholar]
- 48.Narita Y, Arakawa Y, Yamasaki F, et al. A randomized, double-blind, phase III trial of personalized peptide vaccination for recurrent glioblastoma. Neuro Oncol 2019;21:348-59. 10.1093/neuonc/noy200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hilf N, Kuttruff-Coqui S, Frenzel K, et al. Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature 2019;565:240-5. 10.1038/s41586-018-0810-y [DOI] [PubMed] [Google Scholar]
- 50.Muragaki Y, Maruyama T, Iseki H, et al. Phase I/IIa trial of autologous formalin-fixed tumor vaccine concomitant with fractionated radiotherapy for newly diagnosed glioblastoma. J Neurosurg 2011;115:248-55. 10.3171/2011.4.JNS10377 [DOI] [PubMed] [Google Scholar]
- 51.Ishikawa E, Muragaki Y, Yamamoto T, et al. Phase I/IIa trial of fractionated radiotherapy, temozolomide, and autologous formalin-fixed tumor vaccine for newly diagnosed glioblastoma. J Neurosurg 2014;121:543-53. 10.3171/2014.5.JNS132392 [DOI] [PubMed] [Google Scholar]
- 52.Bota DA, Chung J, Dandekar M, et al. Phase II study of ERC1671 plus bevacizumab versus bevacizumab plus placebo in recurrent glioblastoma: interim results and correlations with CD4+ T-lymphocyte counts. CNS Oncol 2018;7:CNS22. 10.2217/cns-2018-0009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Crane CA, Han SJ, Ahn B, et al. Individual patient-specific immunity against high-grade glioma after vaccination with autologous tumor derived peptides bound to the 96 KD chaperone protein. Clin Cancer Res 2013;19:205-14. 10.1158/1078-0432.CCR-11-3358 [DOI] [PubMed] [Google Scholar]
- 54.Bloch O, Crane CA, Fuks Y, et al. Heat-shock protein peptide complex–96 vaccination for recurrent glioblastoma: a phase II, single-arm trial. Neuro Oncol 2014;16:274-9. 10.1093/neuonc/not203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Parney I. Unpublished data. 2019. Available online: https://clinicaltrials.gov/ct2/show/results/NCT01814813. Accessed March 20, 2020.
- 56.Bloch O, Raizer JJ, Lim M, et al. Newly diagnosed glioblastoma patients treated with an autologous heat shock protein peptide vaccine: PD-L1 expression and response to therapy. J Clin Oncol 2015;33:2011-. 10.1200/jco.2015.33.15_suppl.2011 [DOI] [Google Scholar]
- 57.Sampson JH, Archer GE, Mitchell DA, et al. An epidermal growth factor receptor variant III-targeted vaccine is safe and immunogenic in patients with glioblastoma multiforme. Mol Cancer Ther 2009;8:2773-9. 10.1158/1535-7163.MCT-09-0124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Phuphanich S, Wheeler CJ, Rudnick JD, et al. Phase I trial of a multi-epitope-pulsed dendritic cell vaccine for patients with newly diagnosed glioblastoma. Cancer Immunol Immunother 2013;62:125-35. 10.1007/s00262-012-1319-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wen PY, Reardon DA, Armstrong TS, et al. A randomized double-blind placebo-controlled phase II trial of dendritic cell vaccine ICT-107 in newly diagnosed patients with glioblastoma. Clin Cancer Res 2019;25:5799-807. 10.1158/1078-0432.CCR-19-0261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Prins RM, Wang X, Soto H, et al. Comparison of glioma-associated antigen peptide-loaded versus autologous tumor lysate-loaded dendritic cell vaccination in malignant glioma patients. J Immunother 2013;36:152-7. 10.1097/CJI.0b013e3182811ae4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chang CN, Huang YC, Yang DM, et al. A phase I/II clinical trial investigating the adverse and therapeutic effects of a postoperative autologous dendritic cell tumor vaccine in patients with malignant glioma. J Clin Neurosci 2011;18:1048-54. 10.1016/j.jocn.2010.11.034 [DOI] [PubMed] [Google Scholar]
- 62.Cho DY, Yang WK, Lee HC, et al. Adjuvant immunotherapy with whole-cell lysate dendritic cells vaccine for glioblastoma multiforme: a phase II clinical trial. World Neurosurg 2012;77:736-44. 10.1016/j.wneu.2011.08.020 [DOI] [PubMed] [Google Scholar]
- 63.Inogés S, Tejada S, de Cerio AL, et al. A phase II trial of autologous dendritic cell vaccination and radiochemotherapy following fluorescence-guided surgery in newly diagnosed glioblastoma patients. J Transl Med 2017;15:104. 10.1186/s12967-017-1202-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rudnick JD, Sarmiento JM, Uy B, et al. A phase I trial of surgical resection with Gliadel Wafer placement followed by vaccination with dendritic cells pulsed with tumor lysate for patients with malignant glioma. J Clin Neurosci 2020;74:187-93. 10.1016/j.jocn.2020.03.006 [DOI] [PubMed] [Google Scholar]
- 65.Buchroithner J, Erhart F, Pichler J, et al. Audencel Immunotherapy Based on Dendritic Cells Has No Effect on Overall and Progression-Free Survival in Newly Diagnosed Glioblastoma: A Phase II Randomized Trial. Cancers (Basel) 2018;10:372. 10.3390/cancers10100372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liau LM, Ashkan K, Tran DD, et al. First results on survival from a large Phase 3 clinical trial of an autologous dendritic cell vaccine in newly diagnosed glioblastoma. J Transl Med 2018;16:142. 10.1186/s12967-018-1507-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vik-Mo EO, Nyakas M, Mikkelsen BV, et al. Therapeutic vaccination against autologous cancer stem cells with mRNA-transfected dendritic cells in patients with glioblastoma. Cancer Immunol Immunother 2013;62:1499-509. 10.1007/s00262-013-1453-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yao Y, Luo F, Tang C, et al. Molecular subgroups and B7-H4 expression levels predict responses to dendritic cell vaccines in glioblastoma: an exploratory randomized phase II clinical trial. Cancer Immunol Immunother 2018;67:1777-88. 10.1007/s00262-018-2232-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Batich KA, Reap EA, Archer GE, et al. Long-term survival in glioblastoma with cytomegalovirus pp65-targeted vaccination. Clin Cancer Res 2017;23:1898-909. 10.1158/1078-0432.CCR-16-2057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Desjardins A, Gromeier M, Herndon JE, et al. Recurrent Glioblastoma Treated with Recombinant Poliovirus. N Engl J Med 2018;379:150-61. 10.1056/NEJMoa1716435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lang FF, Conrad C, Gomez-Manzano C, et al. Phase I Study of DNX-2401 (Delta-24-RGD) Oncolytic Adenovirus: Replication and Immunotherapeutic Effects in Recurrent Malignant Glioma. J Clin Oncol 2018;36:1419-27. 10.1200/JCO.2017.75.8219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cloughesy TF, Landolfi J, Vogelbaum MA, et al. Durable complete responses in some recurrent high-grade glioma patients treated with Toca 511 + Toca FC. Neuro Oncol 2018;20:1383-92. 10.1093/neuonc/noy075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cloughesy T, Petrecca K, Walbert T, et al. LTBK-08. TOCA 511 & TOCA FC versus standard of care in patients with recurrent high grade glioma. Neuro Oncol 2019;21:vi284. 10.1093/neuonc/noz219.1199 [DOI] [Google Scholar]
- 74.Butowski N. Immunostimulants for malignant gliomas. Neurosurg Clin N Am 2010;21:53-65. 10.1016/j.nec.2009.08.007 [DOI] [PubMed] [Google Scholar]
- 75.Heimberger AB, Hlatky R, Suki D, et al. Prognostic effect of epidermal growth factor receptor and EGFRvIII in glioblastoma multiforme patients. Clin Cancer Res 2005;11:1462-6. 10.1158/1078-0432.CCR-04-1737 [DOI] [PubMed] [Google Scholar]
- 76.Wong AJ, Ruppert JM, Bigner SH, et al. Structural alterations of the epidermal growth factor receptor gene in human gliomas. Proc Natl Acad Sci U S A 1992;89:2965-9. 10.1073/pnas.89.7.2965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Humphrey PA, Wong AJ, Vogelstein B, et al. Anti-synthetic peptide antibody reacting at the fusion junction of deletion-mutant epidermal growth factor receptors in human glioblastoma. Proc Natl Acad Sci U S A 1990;87:4207-11. 10.1073/pnas.87.11.4207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Balss J, Meyer J, Mueller W, et al. Analysis of the IDH1 codon 132 mutation in brain tumors. Acta neuropathologica 2008;116:597-602. 10.1007/s00401-008-0455-2 [DOI] [PubMed] [Google Scholar]
- 79.Bardella C, Al-Dalahmah O, Krell D, et al. Expression of Idh1R132H in the murine subventricular zone stem cell niche recapitulates features of early gliomagenesis. Cancer Cell 2016;30:578-94. 10.1016/j.ccell.2016.08.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yan H, Parsons DW, Jin G, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med 2009;360:765-73. 10.1056/NEJMoa0808710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schumacher T, Bunse L, Pusch S, et al. A vaccine targeting mutant IDH1 induces antitumour immunity. Nature 2014;512:324-7. 10.1038/nature13387 [DOI] [PubMed] [Google Scholar]
- 82.Dutoit V, Herold-Mende C, Hilf N, et al. Exploiting the glioblastoma peptidome to discover novel tumour-associated antigens for immunotherapy. Brain 2012;135:1042-54. 10.1093/brain/aws042 [DOI] [PubMed] [Google Scholar]
- 83.Zhu X, Nishimura F, Sasaki K, et al. Toll like receptor-3 ligand poly-ICLC promotes the efficacy of peripheral vaccinations with tumor antigen-derived peptide epitopes in murine CNS tumor models. J Transl Med 2007;5:10. 10.1186/1479-5876-5-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Okada H, Butterfield LH, Hamilton RL, et al. Induction of robust type-I CD8+ T-cell responses in WHO grade 2 low-grade glioma patients receiving peptide-based vaccines in combination with poly-ICLC. Clin Cancer Res 2015;21:286-94. 10.1158/1078-0432.CCR-14-1790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Okada H, Kalinski P, Ueda R, et al. Induction of CD8+ T-cell responses against novel glioma-associated antigen peptides and clinical activity by vaccinations with α-type 1 polarized dendritic cells and polyinosinic-polycytidylic acid stabilized by lysine and carboxymethylcellulose in patients with recurrent malignant glioma. J Clin Oncol 2011;29:330-6. 10.1200/JCO.2010.30.7744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Agashe VR, Hartl FU. editors. Roles of molecular chaperones in cytoplasmic protein folding. Seminars in cell & developmental biology, Elsevier, 2000. [DOI] [PubMed] [Google Scholar]
- 87.Graner MW, Bigner DD. Chaperone proteins and brain tumors: potential targets and possible therapeutics. Neuro Oncol 2005;7:260-78. 10.1215/S1152851704001188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Nishikawa M, Takemoto S, Takakura Y. Heat shock protein derivatives for delivery of antigens to antigen presenting cells. Int J Pharm 2008;354:23-7. 10.1016/j.ijpharm.2007.09.030 [DOI] [PubMed] [Google Scholar]
- 89.Asea A, Kraeft SK, Kurt-Jones EA, et al. HSP70 stimulates cytokine production through a CD14-dependant pathway, demonstrating its dual role as a chaperone and cytokine. Nat Med 2000;6:435-42. 10.1038/74697 [DOI] [PubMed] [Google Scholar]
- 90.Panjwani NN, Popova L, Srivastava PK. Heat shock proteins gp96 and hsp70 activate the release of nitric oxide by APCs. J Immunol 2002;168:2997-3003. 10.4049/jimmunol.168.6.2997 [DOI] [PubMed] [Google Scholar]
- 91.Anguille S, Smits EL, Lion E, et al. Clinical use of dendritic cells for cancer therapy. Lancet Oncol 2014;15:e257-67. 10.1016/S1470-2045(13)70585-0 [DOI] [PubMed] [Google Scholar]
- 92.Srinivasan VM, Ferguson SD, Lee S, et al. Tumor vaccines for malignant gliomas. Neurotherapeutics 2017;14:345-57. 10.1007/s13311-017-0522-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Draube A, Klein-González N, Mattheus S, et al. Dendritic cell based tumor vaccination in prostate and renal cell cancer: a systematic review and meta-analysis. PLoS One 2011;6:e18801. 10.1371/journal.pone.0018801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cobbs CS, Harkins L, Samanta M, et al. Human cytomegalovirus infection and expression in human malignant glioma. Cancer Res 2002;62:3347-50. [PubMed] [Google Scholar]
- 95.Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996;271:1734-6. 10.1126/science.271.5256.1734 [DOI] [PubMed] [Google Scholar]
- 96.Töpfer K, Kempe S, Müller N, et al. Tumor evasion from T cell surveillance. J Biomed Biotechnol 2011;2011:918471. 10.1155/2011/918471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lukas RV, Rodon J, Becker K, et al. Clinical activity and safety of atezolizumab in patients with recurrent glioblastoma. J Neurooncol 2018;140:317-28. 10.1007/s11060-018-2955-9 [DOI] [PubMed] [Google Scholar]
- 98.Reardon DA, Kim TM, Frenel JS, et al. ATIM-35. Results of the phase IB KEYNOTE-028 multi-cohort trial of pembrolizumab monotherapy in patients with recurrent PD-L1-positive glioblastoma multiforme (GBM). Neuro Oncol 2016;18:vi25-6. 10.1093/neuonc/now212.100 [DOI] [Google Scholar]
- 99.Reardon D, Kaley T, Iwamoto F, et al. ATIM-23. Anti-CD27 agonist antibody varlilumab in combination with nivolumab for recurrent glioblastoma (rGBM): phase 2 clinical trial results. Neuro Oncol 2018;20:vi6. 10.1093/neuonc/noy148.018 [DOI] [Google Scholar]
- 100.Schalper KA, Rodriguez-Ruiz ME, Diez-Valle R, et al. Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma. Nat Med 2019;25:470-6. 10.1038/s41591-018-0339-5 [DOI] [PubMed] [Google Scholar]
- 101.Cloughesy TF, Mochizuki AY, Orpilla JR, et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat Med 2019;25:477-86. 10.1038/s41591-018-0337-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Reardon DA, Omuro A, Brandes AA, et al. OS10.3 Randomized Phase 3 Study Evaluating the Efficacy and Safety of Nivolumab vs Bevacizumab in Patients With Recurrent Glioblastoma: CheckMate 143. Neuro Oncol 2017;19:iii21. 10.1093/neuonc/nox036.071 [DOI] [Google Scholar]
- 103.Bristol-Myers Squibb. Unpublished data. 2019. Available online: https://news.bms.com/press-release/corporatefinancial-news/bristol-myers-squibb-announces-phase-3-checkmate-498-study-did. Accessed April 2, 2020.
- 104.Bristol-Myers Squibb. Unpublished data. 2019. Available online: https://news.bms.com/press-release/corporatefinancial-news/bristol-myers-squibb-provides-update-phase-3-opdivo-nivolumab-. Accessed April 3, 2020.
- 105.De Groot JF, Penas-Prado M, Mandel JJ, et al. Window-of-opportunity clinical trial of a PD-1 inhibitor in patients with recurrent glioblastoma. J Clin Oncol 2018;36:abstr 2008.
- 106.Reardon D, Kaley T, Dietrich J, et al. Atim-12. Phase 2 study to evaluate the clinical efficacy and safety of Medi4736 (Durvalumab [Dur]) in patients with Bevacizumab (Bev)-refractory recurrent Glioblastoma (Gbm). Neuro Oncol 2017;19:vi28. 10.1093/neuonc/nox168.108 [DOI] [Google Scholar]
- 107.Reardon DA, Nayak L, Peters KB, et al. Phase II study of pembrolizumab or pembrolizumab plus bevacizumab for recurrent glioblastoma (rGBM) patients. J Clin Oncol 2018;36:abstr 2006.
- 108.Reardon D. Unpublished data. 2019. Available online: https://clinicaltrials.gov/ct2/show/results/NCT02337491. Accessed April 7, 2020.
- 109.Jacques FH, Nicholas G, Lorimer I, et al. Avelumab in newly diagnosed glioblastoma multiforme: The SEJ study. J Clin Oncol 2019. doi: . 10.1200/JCO.2019.37.15_suppl.e13571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Omuro A, Vlahovic G, Lim M, et al. Nivolumab with or without ipilimumab in patients with recurrent glioblastoma: results from exploratory phase I cohorts of CheckMate 143. Neuro Oncol 2018;20:674-86. 10.1093/neuonc/nox208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lim M, Ye X, Piotrowski AF, et al. Updated phase I trial of anti-LAG-3 or anti-CD137 alone and in combination with anti-PD-1 in patients with recurrent GBM. J Clin Oncol 2019;37:abstr 2017.
- 112.Brahm CG, van Linde ME, Enting RH, et al. The Current Status of Immune Checkpoint Inhibitors in Neuro-Oncology: A Systematic Review. Cancers 2020;12:586. 10.3390/cancers12030586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Carter T, Shaw H, Cohn-Brown D, et al. Ipilimumab and Bevacizumab in Glioblastoma. Clin Oncol (R Coll Radiol) 2016;28:622-6. 10.1016/j.clon.2016.04.042 [DOI] [PubMed] [Google Scholar]
- 114.Blumenthal DT, Yalon M, Vainer GW, et al. Pembrolizumab: first experience with recurrent primary central nervous system (CNS) tumors. J Neurooncol 2016;129:453-60. 10.1007/s11060-016-2190-1 [DOI] [PubMed] [Google Scholar]
- 115.Chamberlain MC, Kim BT. Nivolumab for patients with recurrent glioblastoma progressing on bevacizumab: a retrospective case series. J Neurooncol 2017;133:561-9. 10.1007/s11060-017-2466-0 [DOI] [PubMed] [Google Scholar]
- 116.Mantica M, Pritchard A, Lieberman F, et al. Retrospective study of nivolumab for patients with recurrent high grade gliomas. J Neurooncol 2018;139:625-31. 10.1007/s11060-018-2907-4 [DOI] [PubMed] [Google Scholar]
- 117.Gubin MM, Zhang X, Schuster H, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 2014;515:577-81. 10.1038/nature13988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Rizvi NA, Hellmann MD, Snyder A, et al. Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Science 2015;348:124-8. 10.1126/science.aaa1348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Snyder A, Makarov V, Merghoub T, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med 2014;371:2189-99. 10.1056/NEJMoa1406498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Colli LM, Machiela MJ, Myers TA, et al. Burden of nonsynonymous mutations among TCGA cancers and candidate immune checkpoint inhibitor responses. Cancer Res 2016;76:3767-72. 10.1158/0008-5472.CAN-16-0170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hodges TR, Ott M, Xiu J, et al. Mutational burden, immune checkpoint expression, and mismatch repair in glioma: implications for immune checkpoint immunotherapy. Neuro Oncol 2017;19:1047-57. 10.1093/neuonc/nox026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Bouffet E, Larouche V, Campbell BB, et al. Immune checkpoint inhibition for hypermutant glioblastoma multiforme resulting from germline biallelic mismatch repair deficiency. J Clin Oncol 2016;34:2206-11. 10.1200/JCO.2016.66.6552 [DOI] [PubMed] [Google Scholar]
- 123.Eckert A, Kloor M, Giersch A, et al. Microsatellite instability in pediatric and adult high-grade gliomas. Brain Pathol 2007;17:146-50. 10.1111/j.1750-3639.2007.00049.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ansell SM, Lesokhin AM, Borrello I, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin's lymphoma. N Engl J Med 2015;372:311-9. 10.1056/NEJMoa1411087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Herbst RS, Soria JC, Kowanetz M, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014;515:563-7. 10.1038/nature14011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Garber ST, Hashimoto Y, Weathers SP, et al. Immune checkpoint blockade as a potential therapeutic target: surveying CNS malignancies. Neuro Oncol 2016;18:1357-66. 10.1093/neuonc/now132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Nduom EK, Weller M, Heimberger AB. Immunosuppressive mechanisms in glioblastoma. Neuro Oncol 2015;17:vii9-14. 10.1093/neuonc/nov151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zhai L, Lauing KL, Chang AL, et al. The role of IDO in brain tumor immunotherapy. J Neurooncol 2015;123:395-403. 10.1007/s11060-014-1687-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hellmann MD, Ciuleanu TE, Pluzanski A, et al. Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. N Engl J Med 2018;378:2093-104. 10.1056/NEJMoa1801946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Motzer RJ, Tannir NM, McDermott DF, et al. Nivolumab plus ipilimumab versus sunitinib in advanced renal-cell carcinoma. N Engl J Med 2018;378:1277-90. 10.1056/NEJMoa1712126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Wolchok JD, Chiarion-Sileni V, Gonzalez R, et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med 2017;377:1345-56. 10.1056/NEJMoa1709684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015;348:62-8. 10.1126/science.aaa4967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.June CH, O’Connor RS, Kawalekar OU, et al. CAR T cell immunotherapy for human cancer. Science 2018;359:1361-5. 10.1126/science.aar6711 [DOI] [PubMed] [Google Scholar]
- 134.Park TS, Rosenberg SA, Morgan RA. Treating cancer with genetically engineered T cells. Trends Biotechnol 2011;29:550-7. 10.1016/j.tibtech.2011.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Bielamowicz K, Khawja S, Ahmed N. Adoptive cell therapies for glioblastoma. Front Oncol 2013;3:275. 10.3389/fonc.2013.00275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.O’Rourke DM, Nasrallah MP, Desai A, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med 2017;9:eaaa0984. 10.1126/scitranslmed.aaa0984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Goff SL, Morgan RA, Yang JC, et al. Pilot Trial of Adoptive Transfer of Chimeric Antigen Receptor–transduced T Cells Targeting EGFRvIII in Patients With Glioblastoma. J Immunother 2019;42:126-35. 10.1097/CJI.0000000000000260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Ahmed N, Brawley V, Hegde M, et al. HER2-Specific Chimeric Antigen Receptor-Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA Oncol 2017;3:1094-101. 10.1001/jamaoncol.2017.0184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Brown CE, Badie B, Barish ME, et al. Bioactivity and Safety of IL13Rα2-Redirected Chimeric Antigen Receptor CD8+ T Cells in Patients with Recurrent Glioblastoma. Clin Cancer Res 2015;21:4062-72. 10.1158/1078-0432.CCR-15-0428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Brown CE, Alizadeh D, Starr R, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med 2016;375:2561-9. 10.1056/NEJMoa1610497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Schuessler A, Smith C, Beagley L, et al. Autologous T-cell Therapy for Cytomegalovirus as a Consolidative Treatment for Recurrent Glioblastoma. Cancer Res 2014;74:3466-76. 10.1158/0008-5472.CAN-14-0296 [DOI] [PubMed] [Google Scholar]
- 142.Haslund C, Muhic A, Lukacova S, et al. OS2.7 An open-labelled, randomized phase II study in patients with recurrent Glioblastoma Multiforme comparing progression free survival of ALECSAT (Autologous lymphoid effector cells specific against tumour-cells) versus Bevacizumab/Irinotecan. Neuro Oncol 2016;18:iv6. 10.1093/neuonc/now188.017 [DOI] [Google Scholar]
- 143.Liu G, Ying H, Zeng G, et al. HER-2, gp100, and MAGE-1 are expressed in human glioblastoma and recognized by cytotoxic T cells. Cancer Res 2004;64:4980-6. 10.1158/0008-5472.CAN-03-3504 [DOI] [PubMed] [Google Scholar]
- 144.Debinski W, Gibo DM, Hulet SW, et al. Receptor for Interleukin 13 Is a Marker and Therapeutic Target for Human High-Grade Gliomas. Clin Cancer Res 1999;5:985-90. [PubMed] [Google Scholar]
- 145.Fichtner-Feigl S, Strober W, Kawakami K, et al. IL-13 signaling through the IL-13α2 receptor is involved in induction of TGF-β1 production and fibrosis. Nat Med 2006;12:99-106. 10.1038/nm1332 [DOI] [PubMed] [Google Scholar]
- 146.Jarboe JS, Johnson KR, Choi Y, et al. Expression of interleukin-13 receptor α2 in glioblastoma multiforme: implications for targeted therapies. Cancer Res 2007;67:7983-6. 10.1158/0008-5472.CAN-07-1493 [DOI] [PubMed] [Google Scholar]
- 147.Hegde M, Corder A, Chow KK, et al. Combinational targeting offsets antigen escape and enhances effector functions of adoptively transferred T cells in glioblastoma. Mol Ther 2013;21:2087-101. 10.1038/mt.2013.185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Grada Z, Hegde M, Byrd T, et al. TanCAR: A Novel Bispecific Chimeric Antigen Receptor for Cancer Immunotherapy. Mol Ther Nucleic Acids 2013;2:e105. 10.1038/mtna.2013.32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Minagawa K, Zhou X, Mineishi S, et al. Seatbelts in CAR therapy: how safe are CARS? Pharmaceuticals 2015;8:230-49. 10.3390/ph8020230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Johnson LA, Morgan RA, Dudley ME, et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood 2009;114:535-46. 10.1182/blood-2009-03-211714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Mole RH. Whole body irradiation; radiobiology or medicine? Br J Radiol 1953;26:234-41. 10.1259/0007-1285-26-305-234 [DOI] [PubMed] [Google Scholar]
- 152.Demaria S, Ng B, Devitt ML, et al. Ionizing radiation inhibition of distant untreated tumors (abscopal effect) is immune mediated. Int J Radiat Oncol Biol Phys 2004;58:862-70. 10.1016/j.ijrobp.2003.09.012 [DOI] [PubMed] [Google Scholar]
- 153.Postow MA, Callahan MK, Barker CA, et al. Immunologic correlates of the abscopal effect in a patient with melanoma. N Engl J Med 2012;366:925-31. 10.1056/NEJMoa1112824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Stamell EF, Wolchok JD, Gnjatic S, et al. The abscopal effect associated with a systemic anti-melanoma immune response. Int J Radiat Oncol Biol Phys 2013;85:293-5. 10.1016/j.ijrobp.2012.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Fonteneau JF, Larsson M, Bhardwaj N. Interactions between dead cells and dendritic cells in the induction of antiviral CTL responses. Curr Opin Immunol 2002;14:471-7. 10.1016/S0952-7915(02)00358-8 [DOI] [PubMed] [Google Scholar]
- 156.Zhang B, Bowerman NA, Salama JK, et al. Induced sensitization of tumor stroma leads to eradication of established cancer by T cells. J Exp Med 2007;204:49-55. 10.1084/jem.20062056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.McGranahan N, Furness AJ, Rosenthal R, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016;351:1463-9. 10.1126/science.aaf1490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Deng L, Liang H, Xu M, et al. STING-dependent cytosolic DNA sensing promotes radiation-induced type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity 2014;41:843-52. 10.1016/j.immuni.2014.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Ohkuri T, Ghosh A, Kosaka A, et al. STING contributes to antiglioma immunity via triggering type I IFN signals in the tumor microenvironment. Cancer Immunol Res 2014;2:1199-208. 10.1158/2326-6066.CIR-14-0099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Apetoh L, Ghiringhelli F, Tesniere A, et al. Toll-like receptor 4–dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med 2007;13:1050-9. 10.1038/nm1622 [DOI] [PubMed] [Google Scholar]
- 161.Vanpouille-Box C, Alard A, Aryankalayil MJ, et al. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat Commun 2017;8:15618. 10.1038/ncomms15618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Savage CD, Lopez-Castejon G, Denes A, et al. NLRP3-Inflammasome Activating DAMPs Stimulate an Inflammatory Response in Glia in the Absence of Priming Which Contributes to Brain Inflammation after Injury. Front Immunol 2012;3:288. 10.3389/fimmu.2012.00288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Reits EA, Hodge JW, Herberts CA, et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J Exp Med 2006;203:1259-71. 10.1084/jem.20052494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Zeng J, See AP, Phallen J, et al. Anti-PD-1 blockade and stereotactic radiation produce long-term survival in mice with intracranial gliomas. Int J Radiat Oncol Biol Phys 2013;86:343-9. 10.1016/j.ijrobp.2012.12.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Belcaid Z, Phallen JA, Zeng J, et al. Focal radiation therapy combined with 4-1BB activation and CTLA-4 blockade yields long-term survival and a protective antigen-specific memory response in a murine glioma model. PloS One 2014;9:e101764. 10.1371/journal.pone.0101764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Kim JE, Patel MA, Mangraviti A, et al. Combination Therapy with Anti-PD-1, Anti-TIM-3, and Focal Radiation Results in Regression of Murine Gliomas. Clin Cancer Res 2017;23:124-36. 10.1158/1078-0432.CCR-15-1535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Filatenkov A, Baker J, Mueller AMS, et al. Ablative Tumor Radiation Can Change the Tumor Immune Cell Microenvironment to Induce Durable Complete Remissions. Clin Cancer Res 2015;21:3727-39. 10.1158/1078-0432.CCR-14-2824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Dovedi SJ, Cheadle EJ, Popple AL, et al. Fractionated radiation therapy stimulates antitumor immunity mediated by both resident and infiltrating polyclonal T-cell populations when combined with PD-1 blockade. Clin Cancer Res 2017;23:5514-26. 10.1158/1078-0432.CCR-16-1673 [DOI] [PubMed] [Google Scholar]
- 169.Tsao MN, Mehta MP, Whelan TJ, et al. The American Society for Therapeutic Radiology and Oncology (ASTRO) evidence-based review of the role of radiosurgery for malignant glioma. Int J Radiat Oncol Biol Phys 2005;63:47-55. 10.1016/j.ijrobp.2005.05.024 [DOI] [PubMed] [Google Scholar]
- 170.Sahebjam S, Forsyth P, Arrington J, et al. ATIM-18. A PHASE I trial of hypofractionated stereotactic irradiation (HFSRT) with pembrolizumab and bevacizumab in patients with recurrent high grade glioma (NCT02313272). Neuro Oncol 2017;19:vi30. 10.1093/neuonc/nox168.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Pouessel D, Mervoyer A, Larrieu-Ciron D, et al. Hypofractionnated stereotactic radiotherapy and anti-PDL1 durvalumab combination in recurrent glioblastoma: Results of the phase I part of the phase I/II STERIMGLI trial. J Clin Oncol 2018;36:abstr 2046.
- 172.Sahebjam S, Forsyth PAJ, Arrington J, et al. Nivolumab combined with hypofractionated stereotactic irradiation (HFSRT) for patients with recurrent high grade gliomas: A phase I trial (NCT02829931). J Clin Oncol 2017. doi: . 10.1200/JCO.2017.35.15_suppl.TPS2084 [DOI] [Google Scholar]
- 173.Grossman SA, Ye X, Lesser G, et al. Immunosuppression in Patients with High-Grade Gliomas Treated with Radiation and Temozolomide. Clin Cancer Res 2011;17:5473-80. 10.1158/1078-0432.CCR-11-0774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Antonios JP, Soto H, Everson RG, et al. PD-1 blockade enhances the vaccination-induced immune response in glioma. JCI Insight 2016;1:e87059. 10.1172/jci.insight.87059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Field CS, Hunn MK, Ferguson PM, et al. Blocking CTLA-4 while priming with a whole cell vaccine reshapes the oligoclonal T cell infiltrate and eradicates tumors in an orthotopic glioma model. Oncoimmunology 2017;7:e1376154. 10.1080/2162402X.2017.1376154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.John LB, Devaud C, Duong CPM, et al. Anti-PD-1 Antibody Therapy Potently Enhances the Eradication of Established Tumors By Gene-Modified T Cells. Clin Cancer Res 2013;19:5636-46. 10.1158/1078-0432.CCR-13-0458 [DOI] [PubMed] [Google Scholar]
- 177.Yin Y, Boesteanu AC, Binder ZA, et al. Checkpoint Blockade Reverses Anergy in IL-13Rα2 Humanized scFv-Based CAR T Cells to Treat Murine and Canine Gliomas. Mol Ther Oncolytics 2018;11:20-38. 10.1016/j.omto.2018.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Bagley S, Desai A, Binder Z, et al. RBTT-12. A phase I study of EGFRvIII-directed CAR T cells combined with PD-1 inhibition in patients with newly, diagnosed, MGMT-unmethylated glioblastoma: trial in progress. Neuro Oncol 2019;21:vi221. 10.1093/neuonc/noz175.923 [DOI] [Google Scholar]
- 179.Wimmers F, Aarntzen EHJG, Duiveman-deBoer T, et al. Long-lasting multifunctional CD8+ T cell responses in end-stage melanoma patients can be induced by dendritic cell vaccination. OncoImmunology 2015;5:e1067745. 10.1080/2162402X.2015.1067745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Reap EA, Suryadevara CM, Batich KA, et al. Dendritic Cells Enhance Polyfunctionality of Adoptively Transferred T Cells That Target Cytomegalovirus in Glioblastoma. Cancer Res 2018;78:256-64. 10.1158/0008-5472.CAN-17-0469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Sloan AE, Dansey R, Zamorano L, et al. Adoptive immunotherapy in patients with recurrent malignant glioma: preliminary results of using autologous whole-tumor vaccine plus granulocyte-macrophage colony-stimulating factor and adoptive transfer of anti-CD3-activated lymphocytes. Neurosurg Focus 2000;9:e9. 10.3171/foc.2000.9.6.10 [DOI] [PubMed] [Google Scholar]
- 182.Archer G. Unpublished data. 2020. Available online: https://clinicaltrials.gov/ct2/show/results/NCT02529072. Accessed March 20, 2020.