

ABSTRACT
Glioblastoma (GBM) is an aggressive primary brain tumor with unique immunity predominated by myeloid cells. GBM cells have been implicated to evade immune attack through hijacking myeloid-affiliated transcriptional programs to establish an immunosuppressive microenvironment. However, molecular features of immune-evading GBM cells in heterogeneous GBMs and their interactions with immune cells remain unclear. Herein, we employed single-cell RNA sequencing (scRNA-seq) and bulk RNA-seq data to develop an in silico method for delineating GBM immune signature and identifying new molecular subsets for immunotherapy. We identified a new GBM cell subset, termed TC-6, that harbored immune-invading signature and actively interacted with tumor-associated macrophages (TAMs) to orchestrate an immune-suppressive niche. Proinflammatory transcriptional factors STAT1, STAT2, IRF1, IRF2, IRF3, and IRF7 were identified as the core regulons defining TC-6 subsets. Further immune transcriptome analyses revealed three immune subtypes (C1, C2, and C3). C3 subtype GBMs were enriched with TC-6 cells and immunosuppressive TAMs, and exhibited an immunomodulatory signature that associated with reduced efficacy of anti-PD-1 treatment. Interferon-related DNA damage resistance signaling was upregulated in C3 GBMs, predicting shortened survival of GBM patients who received chemo-radiation treatment. Treatment of OSI-930 as a molecular agent targeting c-kit and VEGFR2 tyrosine kinases may compromise the immunomodulatory signature of C3 GBMs and synergize with chemo-radiation therapy. We further developed a simplified 11-gene set for defining C3 GBMs. Our work identified TC-6 subset as an immune-evading hub that creates an immunomodulatory signature of C3 GBMs, gaining insights into the heterogeneity of GBM immune microenvironment and holding promise for optimized anti-GBM immunotherapy.
KEYWORDS: glioblastoma, molecular subtyping, single-cell RNA sequencing, prognosis
Introduction
Glioblastoma (GBM) is the most lethal and prevalent malignant brain tumor with marked cellular and molecular heterogenicity. GBM exhibits a dismal response to standard-of-care therapeutics including maximal safe surgical resection, radiation and chemo-treatment.1 Genetic profiling of GBM has identified distinct transcriptional subtypes (termed proneural/classical/mesenchymal subtypes), reflecting the distinct states of GBM tumor cells.2,3 Whether and how GBM heterogenicity is driven by the intrinsic evolution of tumor cells or the extrinsic microenvironmental cues remains largely unknown. A recent work by Gangoso etal.4 reveals that GBM cells employ myeloid transcriptional circuits to establish a tumor-supportive, immunosuppressive microenvironment. However, molecular features of immune-evading GBM cells in heterogeneous GBMs and their interactions with immune cells remain unclear.
Immune checkpoint blockade (ICB) treatment reshapes the scope of anti-tumortherapeutics.5 In GBMs, although preclinical results of ICB treatment are promising,6,7 clinical trials are under evaluation with discrepant responsiveness.8,9 Genetic changes of PTEN and MAPK signaling molecules, as well as the activation of WNT-β-catenin pathway in GBM cells, have been proposed as intrinsic immune evasion mechanisms.8,10 Adaptive changes of tumor microenvironment, including induction of PD-1 expression and sustained immune activation, also contribute to neoadjuvant anti-PD-1 treatment inefficacy.11,12 Likewise, a growing number of data imply that sustained interferon(IFN) signaling arose from PD-1 blockade is a major driving force of anti-tumor adaptive immune resistance.13,14 In clinical settings, comprehensive analyses of the interactions between tumor-infiltrating immune cells and tumor cells could predict immunotherapy efficiency.4,15 However, whether there are GBM cell populations that could drive immune evasion and create a specific immune signature associated with ICB ineffectiveness remains largely unknown.
Herein, we employed single-cell RNA-seqencing (scRNA-seq) and bulk RNA-seq data to develop an in silico method for delineating GBM immune signature, and identified a unique tumor subset (TC-6) as a hub for creating an immunomodulatory signature of human GBMs. TC-6 actively interacts with tumor microenvironmental (TME) cells and acquires TAM-affiliated transcriptional programs to elicit immunosuppression. To delineate specific transcriptional programs, we performed single-cell regulatory network inference and clustering (SCENIC) based on the scRNA-seq data to determine the core gene regulatory networks for immune subtyping. Three immunological subtypes were identified with distinct genomic alterations, immunotherapeutic response, and patient survival. Our work provides insights into the transcriptional modules in GBMs, with potential value for developing and optimizing therapeutic strategies against this lethal tumor.
Materials and methods
Human GBM cohorts
The Cancer Genome Atlas (TCGA) GBM gene expression data (Affymetrix Human Genome U133A microarray platform, n = 529) and the corresponding patient clinical information data were from the UCSC data portal (https://xenabrowser.net). Quantile normalization was performed on gene expression data using the limma R package. RNA sequencing (RNA-seq) raw data of primary GBMs (n = 154) and recurrent GBMs (n = 5) were obtained from the TCGA portal (http://tcga-data.nci.nih.gov/tcga/) and were normalized using Deseq2 R package. GBM gene expression profiles of Chinese patients were retrieved from the Chinese Glioma Genome Atlas (CGGA) (http://www.cgga.org.cn/). The batch effect between mRNAseq-325 series and mRNAseq-693 series was corrected using the “ComBat” function of sva R package. RNA-seq data of recurrent GBM patients treated with adjuvant, post-surgical anti-PD-1 blockade were retrieved from GEO portal (GSE121810, https://www.ncbi.nlm.nih.gov/geo/).
Enrichment analysis
Gene-set enrichment analysis (GSEA) was performed using GSEA software (v4.0.1) (https://www.gsea-msigdb.org/gsea). Enrichment results with a criterion of p-value < 0.05 and false discovery rate (FDR) < 0.25 were considered as significant. Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses were performed using the clusterProfiler R package. The files of oncogenic tumor biology were interpreted and visualized using the enrichplot R package. Single-sample GSEA (ssGSEA) algorithm was employed for analyzing the enrichment scores usingGSVA package. Gene sets were derived from the Broad Institute’s Molecular Signatures Database (MsigDB) or literature resources.
Immune signature analysis
CIBERSORT platform (https://cibersort.stanford.edu/) was used to estimate immune cell proportions in GBM dataset based on the LM22 reference profile. xCell (https://xcell.ucsf.edu/) gene signature-based method was used for calculating the proportions of Th1 and Th2 cells. Computing gene set scores for representative immune signatures (immune checkpoint, immunosuppression modulators for TC-6, immune response to tumor cells, et al.) were performed through ssGSEA algorithm. Immune-related indices and an IFN-related DNA damage signature have been defined in previous studies.16,17 Immune-related functional modules used for GSEA analyses were summarized in Supplementary Table 1.
SynergySeq
SynergySeq platform (https://schurerlab.shinyapps.io/synergyseq/) was used for identifying compounds associated with inflammatory signature and with potential to combine with chemo-radiotherapy.18 The LINCS compounds displaying high reversal scores of disease signature and high dissimilarity of cytotoxic therapeutics (chemoagent temozolomide (TMZ) and radiation) were considered as potential molecular candidates for combined treatment. The candidates with signature discordance score ≥ 0.5 and TMZ orthogonality score < 0.02 were selected for in silico compound library screening.
Single-cell RNA sequencing data analyses
GBM scRNA-seq dataset was downloaded from Sequence Read Archive (SRA, PRJNA579593). The CellRanger software (version 2.1.0) was performed to process single-cell data to demultiplex, align reads against the GRCh38 human reference genome, and generate feature-barcode matrices. Raw matrix was preprocessed using computational methods deposited in the Seurat R package. Quality control was performed to further ensure that only high-quality single-cell data was processed further, and cells with fewer than 4000 genes/cell and fewer than 8000 UMIs/cell were removed, as well were cells with greater than 20% of their transcriptome represented in mitochondrial transcripts. We applied the Canonical Correlation Analysis (CCA) algorithm to obtain the batch-corrected expression matrix to remove the batch effects from different donors. To cluster single cells by their expression profiles, we chose 20 principal components and used an unsupervised graph-based clustering algorithm Louvain (resolutions: 0.15). Cell type annotations were based on the expression of cellular entity gene marker visualized on the t-SNE plot. Gene signatures for different cell subsets were identified as log2 fold-change value > 0.5 and expression percent > 50% in all qualified cells (Supplementary Table 2). SCENIC (https://aertslab.org/) protocol was used for single-cell regulatory network inference.19 The inference of regulons was performed as follows: (1) Identification of co-expression modules between transcription factors and the potential targets based on the gene-expression matrix through GENIE3; (2) Screening for the significantly enriched motif of the correct upstream regulators to identify truly functional regulons through cis-regulatory motif analysis from RcisTarget; (3) Calculation of the activity score of each regulon using AUCell algorithm. The regulon-activity matrix for each cell was used for dimensionality reduction for visualization via the t-SNE technique. For cell-type specific regulons identification, calcRSS function was performed based on regulon specificity score. For trajectory inference, we used the spliced and unspliced transcript reads to calculate the RNA velocity using velocity.Rscript with the Cell Ranger output. All the tumor cell subsets were analyzed and the evolution-based trajectory was projected onto the UMAP plot.
Cell-cell communication inference
The intracellular interactions through receptor-ligand complexes were analyzed through CellphoneDB (www.cellphonedb.org).20 The average expression level of ligands and receptors in each cell type was generated and presented as a null distribution of each ligand-receptor pair. The likelihood of cell-specificity of a given receptor-ligand complex was determined.
GBM immunological subtyping
Genes in the metagenes (n = 112) were used for GBM immunological subtyping using R package(parameter: pItem = 0.8, pFeature = 1, reps = 1000) with Euclidean clustering method. The best cluster number (k) was determined using Consensus Cumulative Distribution Function (CDF) Plot. The t-SNE-based approach was employed for subtype assignment validation using the Rtsne R package. The molecular similarity between the predicted subtypes from the TCGA dataset and those from the CGGA dataset was compared using Subclass Mapping on the platform of GenePattern.
Generation of the C3 gene classifier
Prediction Analysis for Microarrays (PAM)21 was employed for identifying subset-associated genes using the nearest shrunken centroids through the PAMR package. GBM samples from the TCGA database were randomly stratified into the training and validation cohorts with a ratio of 2:1. The PAM fits the model for 100 different threshold values, 10-fold cross-validations were used to choose the optimal threshold value for centroid shrinkage. The accuracy of the 11-gene classifier was calculated and validated through external GBM datasets.
Genomic mutation analysis
The mutation annotation format (MAF) file with aggregated mutation information of GBM cases was obtained from the TCGA portal. Candidate somatic single nucleotide variations were identified using Mutect algorithms. The mutational summary and landscape plots were performed by the maftools R package. The “mafCompare” function was applied to compare two different cohorts to identify differentially mutated genes, and results were visualized using forest plots. The “clinicalenrichment” function was used for various groupwise and pairwise comparisons to identify enriched mutations and copy number alterations (CNAs) in each subtype. The “plotEnrichmentResults” function was used to plot these results. MutSigCV1.41 on the GenePattern platform was used to identify the mutated genes highly relevant to cancer (q-value <0.25).
Immunohistochemical (IHC) and immunofluorescent (IF) staining
Human GBM specimens were from the Biobank of Southwest Hospital, Army Medical University, with informed consent from patients or their guardians. The usage of human specimens was approved by the Ethics Committees of Southwest Hospital and was in accordance with the principles of the Helsinki Declaration. IHC and IF staining were performed on formalin-fixed, paraffin-embedded GBM sections as previously described.22 Primary antibodies used in this study were listed in Supplementary Table 3. Dako REAL EnVision Detection System was used for visualization following the manufacturer’s protocol. IHC staining was evaluated independently by two neuropathologists blinded to C3 status of each tumor. The expression level of each molecule was semi-quantitatively scored based on the staining area and staining intensity of positive tumor cells as previously described.23 The proportion of positive tumor cells (staining area) was scored as follows: 0 (no positive tumor cells), 1 (<5% positive tumor cells), 2 (5%–19% positive tumor cells), 3 (20%–50% positive tumor cells), 4 (>50% positive tumor cells). The staining intensity was evaluated according to the following criteria: 0 (no staining), 1 (weak staining = light yellow), 2 (moderate staining = yellow brown), and 3 (strong staining = brown). The staining index (SI) was calculated as staining intensity score × staining area score for each field of microscope.
RNA-sequencing (RNA-seq)
The RNA of GBM specimens from Southwest Hospital was extracted using TRIzol (Invitrogen). The cDNA libraries were constructed according to the TruSeqRNA Sample Preparation v2 protocol, and sequenced using an Illumina HiSeq X sequencer in paired-end sequencing mode (2 × 150 bp reads). The HISAT alignment tool was used to align all sequencing raw reads to the human genome (hg38) reference sequence. Only uniquely and properly mapped read pairs were used for further analysis. Cufflinks program was used to assemble individual transcripts from reads aligned to the human genome.
Statistical analyses
Statistical analyses were performed using R software (3.6.3) or SPSS (19.0 Inc, Chicago, IL). Kruskal–Wallis H test was used for comparisons of variables with abnormal distribution among three independent groups. The Mann–Whitney U test was used for the comparison between two independent groups when the dependent variable was continuous but was not normally distributed. The Chi-Square test was used for comparison of categorical variables. Kaplan–Meier survival curve, Log-rank test and multivariate Cox regression analyses were used for survival analyses. The forest plot was used for the visualization of covariate effects using the forest model R package. P< 0.05 or 0.01 was considered statistical significance.
Results
Single-cell RNA sequencing identifies a unique tumor cell subset orchestrating immunosuppression
To delineate GBM immune TME feature at the single-cell level, we used scRNA-seq data from 5 cases of IDH wild-type human GBMs (SRA, PRJNA579593) and performed unsupervised graph-based clustering on 36789 cells. Sixteen subsets were identified and were annotated based on CNAs and canonical cell markers (Figure 1a). Glioma cell lineages were characterized by the gain of chromosome 7 and the loss of chromosome 10.24 Normalization of CNA profiles revealed chromosomal aberrations in each cell and 6 tumor cell subsets (termed TC-1 to TC-6) were identified in all the samples analyzed (Supplementary Figure S1a-b). TC-1 to TC-5 subsets recapitulated the features of neural progenitor-like (NPC-like), oligodendrocyte-progenitor-like (OPC-like), astrocyte-like (AC-like), and mesenchymal-like (MES-like) GBM cells identified in previous studies,2 whereas TC-6 did not match the feature of abovementioned subsets. Of note, TC-6 showed high expression of EGFR and IFN-stimulated genes (CXCL10, STAT1, ISG15, IFI44L) (Figure 1b). Functional enrichment analyses of hallmark collection indicated that TC-6 was associated with signatures involving IFN response, epithelial-mesenchymal transition, hypoxia, and inflammatory response (Figure 1c). To address the interplay between these tumor cell subsets and immune cells, we performed an unbiased ligand-receptor interaction analysis among these TC subsets and niche cells (Supplementary Figure S2a, Figure 1d). In comparison with other TC subsets, TC-6 subset actively interacted with TAMs (TAM-1, -2) through predicted pathways involving TAM recruitment and phenotype reprogramming (CX3CR1-CX3CL1 axis; CSF1-CSF1R axis), growth factor-initiated tumor growth (MIF-EGFR axis, Supplementary Figure S2b) and “Do not Eat Me” immune surveillance (LILRB1-HLA-F axis).25,26 TC-6 subset was associated with abundant inflammatory stimuli (IL1β, IL1RN, MIF and Oncostatin-M) mediating the TAM reprogramming (Figure 1e). Further analyses of TCGA GBM cohort indicated that TAM signature was positively correlated with TC-6 signature (Figure 1f). In silico cellular interaction analyses also suggest TC-6 subset may interact with tumor-infiltrating T cells in GBMs to regulate T cell recruitment (CXCR3-CXCL10 axis; SPN-ICAM1 axis), expansion and cytotoxic activity (CD94:NKG2A-HLA-E axis and TIGIT-NECTIN2 axis).27–29 Intriguingly, in comparison with other tumor cell subsets, TC-6 exhibited an upregulation of immune checkpoints PD-L1, IDO1, NT5E (CD73), and LGALS9 for immune evasion30–32 (Figure 1g). RNA velocity estimation indicated that TC-6 was the precursor cells of other GBM cell populations (Supplementary Figure S3a). Altogether, these data suggest TC-6 subset as a unique tumor cell subset that could actively interact with TAMs and T cells to create an immunosuppressive microenvironment.
Figure 1.
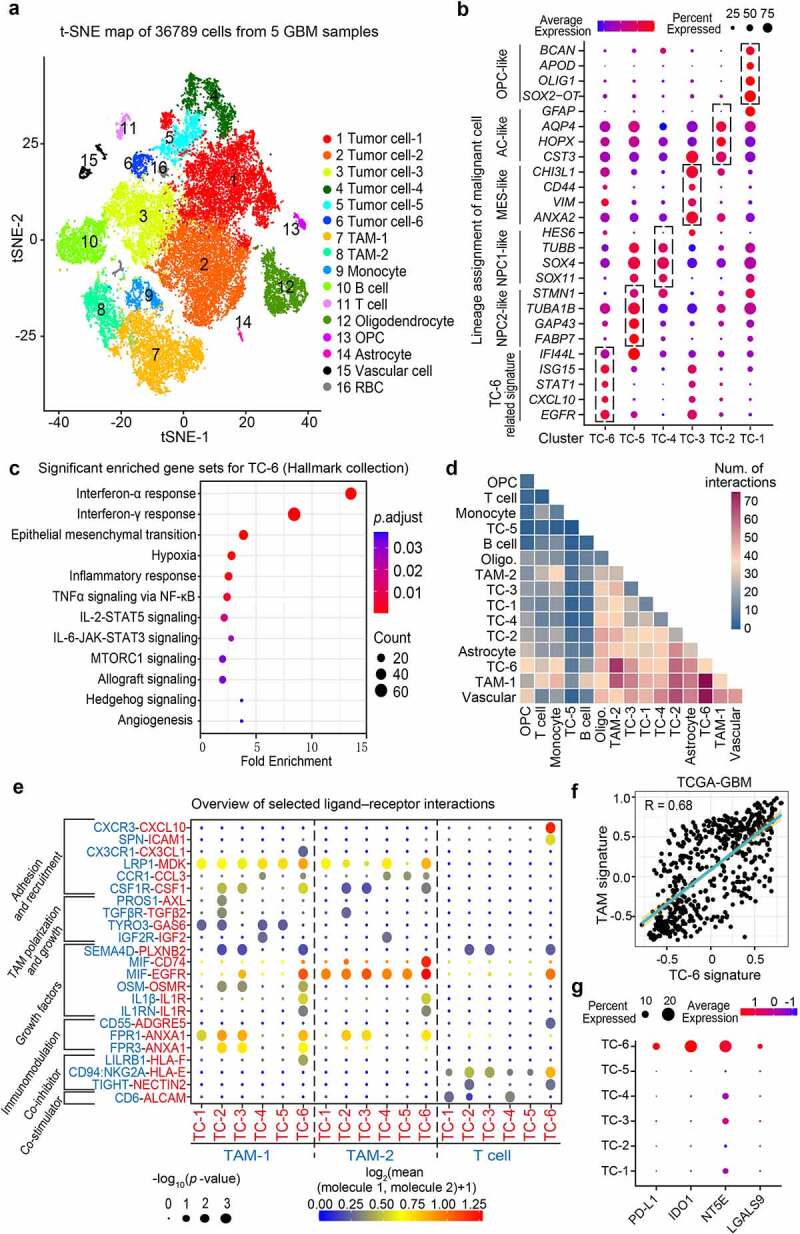
Single-cell RNA sequencing identifies a novel tumor cell subset for orchestrating immunosuppression. (a) t-SNE plot showing 16 major cell types using the single-cell RNA sequencing data from 5 cases of GBMs. TAM, Tumor-associated macrophages; RBC, Red blood cell; OPC, Oligodendrocyte progenitor cell. (b) Dot plots showing the distinct cell state markers in the indicated tumor cell subsets (TC-1, -2, -3, -4, -5, and -6). Dot size was proportional to the fraction of cells expressing specific genes. Color intensity corresponds to the relative expression of specific genes. TC, Tumor cell; NPC, Neural progenitor cell; AC, Astrocyte; MES, Mesenchymal. (c) Enrichment analyses of the differential expressed genes in TC-6 versus other subsets (log2 fold-change > 0.5, min.pct > 0.10). The bubble size indicates the number of genes in each term, and different colors correspond to different adjusted p-values. (d) Heatmap showing the total number of interactions between cell types inferred from whole clusters of GBMs using CellphoneDB. Oligo., Oligodendrocyte. (e) Overview of representative ligand–receptor interactions between tumor cells and immune cells. P-values are indicated by circle size, the average expression of interacting molecules in cluster 1 and 2 are indicated by colors. (f) Correlation plot between TC-6 subset and TAM subset in TCGA GBM cohort, correlation coefficient was listed on the top. (g) Dot plots showing the expression of known immune checkpoints (PD-L1, IDO1, NT5E, and LGALS9) across the TC subsets.
Identification of core regulons coordinating the TC-6 phenotype
To identify transcriptional regulatory networks determining the state of each cell subset, we applied single-cell regulatory network inference and clustering (SCENIC) to evaluate the activity of the gene regulatory networks (GRNs, termed regulons). Eighty regulons were recognized across different cell subsets (Figure 2a). To evaluate the performance of SCENIC, annotations inferred from the expression profiling were mapped to the t-SNE clustering of regulon activity matrix (Supplementary Figure S3b). Cell entity clustering was similar to those defined by transcriptome profiling. Among the 88 regulons, IRF1, IRF2, IRF3, IRF7, STAT1, and STAT2 were identified as specific regulators for TC-6 based on the Regulon Specificity Score (RSS) (Figure 2b).33 TC-6 also showed high gene set activities in these regulons (Figure 2c) that may mediate unique immune transcriptional programs.
Figure 2.
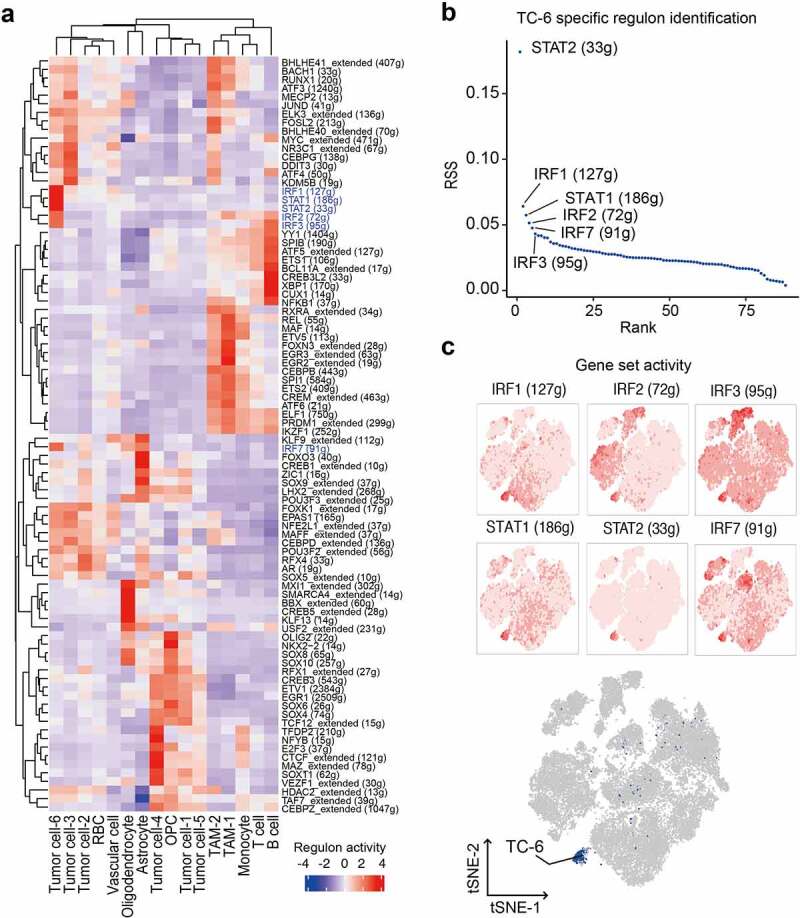
Identification of core regulons delineating the TC-6 phenotype. (a) Heatmap showing the normalized activity score of representative regulons across different subsets of GBM tissues. (b) Identification of TC-6 subset specific regulators based on the Regulon Specificity Score (RSS, y-axis). (c) Activities of IRF1, IRF2, IRF3, IRF7, STAT1 and STAT2 regulons on t-SNE clustering, cells were colored according to the corresponding activity score of regulon (upper panels). Dark blue dots representative of TC-6 in the t-SNE single-cell map were shown in the bottom panel.
Novel GBM subtyping based on TC-6 core regulons
We next investigated whether the core regulons of TC-6 could be used to define GBM immune subtypes. The workflow was shown in Figure 3a. The top 30 genes with high Genie3weight were screened and 112 candidate genes were identified as metagenes (Supplementary Table 4). TCGA GBM dataset was clustered based on the metagenes using consensus clustering. The results showed that three-subtype-clustering was the most optimized clustering method (Figure 3b, Supplementary Figure S4a, b), and was consistent with the clustering of spectral patterns through t-SNE dimensionality reduction (Figure 3c). The rationality and reproducibility of the three-subtype-clustering method were validated through the CGGA GBM cohort (Supplementary Figure S4c,d). Most metagenes associated with TC-6 subtype were upregulated in C3 subtype (Supplementary Figure S4e), and TC6 signature score was significantly higher in C3 subtype of GBMs as compared with C1 or C2 subtype of GBMs (Supplementary Figure S4f), indicating C3 subtype of GBMs are enriched with TC-6 subset. To determine the prognostic significance of immunological subtypes, we employed TCGA and CGGA GBM cohorts and found that C3 subtype GBMs displayed shortened survival relative to non-C3 subtype GBMs (Figure 3d,e).
Figure 3.

Novel GBM subtyping based on TC-6 core regulons. (a) The workflow of TC-6 regulons-based subtyping of human GBMs. (b) Consensus clustering based on metagenes from core regulons identified three subgroups. (c) t-SNE analysis of the three subtypes. (d-e) Kaplan–Meier survival analyses of overall survival in GBMs with C3 and non-C3 subtypes using the TCGA mRNA cohort (d) and CGGA RNA-seq cohort (e). **, p < 0.01; *, p < 0.05.
IFN-related DNA damage resistance signature informs therapeutic resistance to chemo/radio-therapy in C3-subtype GBMs
We further delineate the molecular features and clinical characteristics of the three subtypes. C3 subtype of GBMs was associated with wild-type IDH1, non-glioma-CpG Island methylator phenotype (non-G-CIMP), and classical subtype features of glioma samples (Figure 4a)1 . ssGSEA analysis revealed distinct functions of C1-C3 subtypes. Intriguingly, C3 subtype was associated with IFN-related DNA damage resistance (IRDS), cytosolic DNA sensing pathway, and inflammatory response (Figure 4a). Meanwhile, C1 subtype was enriched with G2/M checkpoints and Notch signaling gene signature. C2 subtype was associated with pathways regulating hypoxia, angiogenesis, and glycolysis. To address how C3 molecular feature underlies worse clinical outcomes of GBM patients who received chemo/radiation, we analyzed IRDS gene set associated with chemo/radio-resistance.16,34 GSEA analysis revealed that IRDS was significantly enriched in C3 subtype relative to non-C3 subtypes (Figure 4b). Twenty-four of 44 IRDS genes were among the top 10% genes differentially expressed in C3 versus non-C3 subtype (Figure 4b). Consistently, the majority of the C3-associated leading-edge genes in IRDS were upregulated in recurrent GBMs relative to the paired primary GBMs (Figure 4b). Besides, C3 GBMs overexpressed another geneset associated with chemo/radio-resistance (Figure 4c). To investigate whether targeting C3 signature could reverse the resistance to chemoagent TMZ and radiation thus improve therapeutic efficacy, we used the SynergySeq platform and screened for the potential agents targeting C3-subtype GBMs. Orthogonal plot analyses identified potential candidate agents that targeted C3 signature and could combine with TMZ/radiation (Figure 4d, Supplementary Table 5).2 OSI-930, a brain-penetrable molecular inhibitor of c-kit, VEGFR2 and CSF1 R,35 was identified as a potential C3-targeting agent. We also identified EX-527, a SIRT1 inhibitor exhibiting anti-tumor effects on glioma cells,36 as a promising chemo-agent for C2-subtype GBMs (Supplementary Table 6).
Figure 4.
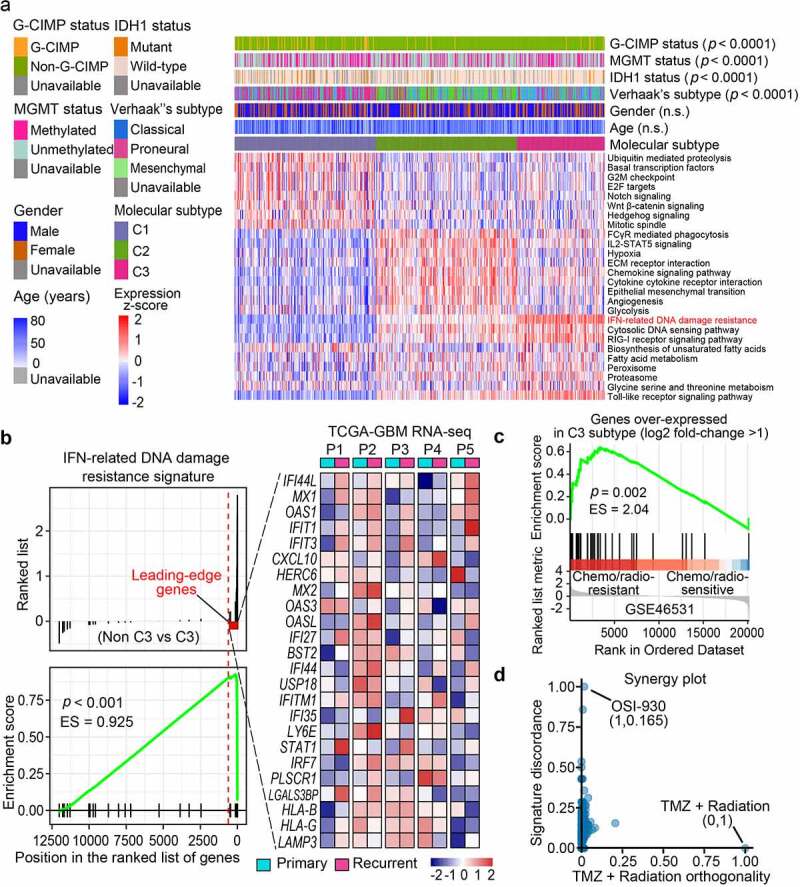
IFN-related DNA damage resistance signature informs therapeutic resistance to chemo/radio-therapy treatment in C3-subtype GBMs. (a)3 The landscape of clinical and molecular characteristics among different subtypes (C1, C2, C3). The normalized ssGSEA score for functional gene sets was plotted through a heatmap. G-CIMP, Glioma-CpG island methylator phenotype; MGMT, O6-methylguanine-DNA methyltransferase; ECM, Extracellular matrix. (b) GSEA plot of the enrichment of IFN-related DNA damage resistance signature in the C3 subtype relative to the non-C3 subtypes (C1 and C2). Leading-edge genes were labeled as the red dotted line (left panel). Heatmap in the right panel showed the expression of these leading-edge genes in five pairs of primary-recurrent GBM samples from the TCGA database. (c) GSEA plot of C3 over-expressed geneset (log2 fold-change >1) in temozolomide (TMZ)/radiation-resistant group relative to the TMZ/radiation-sensitive group. (d) Rankings of the library of integrated network-based cellular signatures (LINCS) compounds based on their concordance with TMZ/radiation therapy and discordance with C3 signature. The x-axis value suggests the concordant degree with TMZ/radiation and the y-axis value suggests the discordance with C3 signature.
Genomic landscape of the three immunological subtypes
To determine whether the three immunological subtypes were associated with genomic changes of GBM cells,3 we evaluated the distribution of somatic gene alterations among the three subtypes using MutSig algorithms. As shown in Supplementary Figure S5a–c,4 gene mutations associated with GBM progression (EGFR, PTEN, TP53, PDGFRA, NF1, RB1, PIK3CA, and IDH1) varied across C1-C3 subtypes. Of note, C3 subtype had the highest genomic alteration (mutation and regional DNA amplification) rate of EGFR and PIK3CA (70%, 16%) relative to C1 (45%, 6%) and C2 (52%, 12%) subtypes. HYDIN mutation, a recently identified cancer-associated antigen involving adaptive immune responses,37 was associated with poor prognosis of GBM patients and showed higher mutation frequency in C3 subtype GBMs relative to C1 and C2 subtypes (Supplementary Figure S5b–d). EGFR amplification was also significantly higher in C3 (Supplementary Figure S5e). We determined the protein level and kinase phosphorylation of EGFR across all three subtypes using TCGA datasets. As expected, C3 subtype GBMs exhibited a higher expression of EGFR and phosphorylated EGFR relative to C1 and C2 subtypes (Supplementary Figure S5f), which may sustain cell survival and compromise chemo-radiation-induced cytotoxicity.
Distinct immunological characteristics of the immunological subtypes
We further profiled the immune milieu and found that C1-C3 subsets manifested distinct immune signatures (Figure 5a) and different immune cell fractions (Supplementary Figure S6a–c). C1 subtype was defined as a lymphocyte-dominant subtype, harboring increased Th1 lymphocytes (Figure 5a) and CD8+ T lymphocytes (Supplementary Figure S6a–c). High indel-derived neoantigens and immune response to tumor cells in C1 subtype informed increased probability of immune responsiveness to immune checkpoint inhibitors (Figure 5a). C2 subtype was defined as a myeloid-predominant subtype, showing an increased fraction of monocytes, macrophages and neutrophils (Figure 5a, Supplementary Figure S6a–c). C3 subtype was featured by the enrichment of polarized macrophages (i.e., tumor-associated macrophages) and NK cells (Supplementary Figure S6a–c), reduced indel neoantigens and cancertestis antigens, increased expressions of immune checkpoint molecules and immunosuppressive modulators overexpressed in TC-6 (Figure 5a). Correlation analyses of TCGA GBM cohort revealed an association between immunosuppressive TAM signature and TC-2, -3 and -6 signature (Supplementary Figure S7a,b). The immunological characteristics of C1-C3 subtype GBMs were summarized in Figure 5b.
Figure 5.
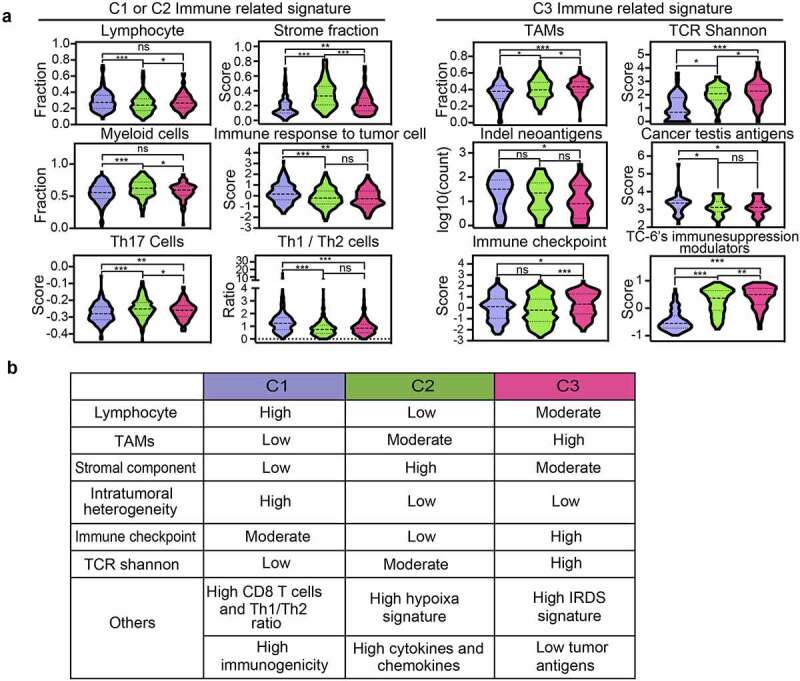
Immunological and clinical characteristics of GBMs with distinct immunological subtypes. (a) Scoring or fraction of immune cell components and indices in C1, C2 and C3 subtypes. (b) The key immune signatures among C1, C2 and C3 subtypes. IFN, Interferon; TCR, T cell receptor; Tfh, T follicular helper.
C3 subtype GBMs manifest reduced efficacy to anti-PD-1 therapy
Recent clinical trials of neoadjuvant anti-PD-1 treatment in GBMs showed a modest effect in improving patient survival.7,8 To determine whether the efficacy of anti-PD-1 treatment is associated with C1-C3 immunological subtypes, we investigated a GBM cohort (GSE121810) treated with anti-PD-112 and assigned the GBM cases to immunological subtypes. As expected, C3-subtype GBMs showed a shortened overall survival (median: 87.5 days) relative to C2-subtype GBMs (median: 236.9 days) or C1-subtype GBMs (median: 329.0 days). Kaplan–Meier survival analyses confirmed that C3-subtype GBMs exhibited worst patient outcome relative to C1- or C2-subtype GBMs (Figure 6a; C3 vs. C1: p = 0.0067; C3 vs. C2: p = 0.002; C2 vs. C1: p = 0.1875). C3 subtype GBMs were enriched for pathways regulating IFN-γ response, T cell exhaustion, TGF-β signaling and TC-6-associated immunosuppressive modulators (Figure 6b,c), thus informing a reduced efficacy to anti-PD-1 therapy.
Figure 6.
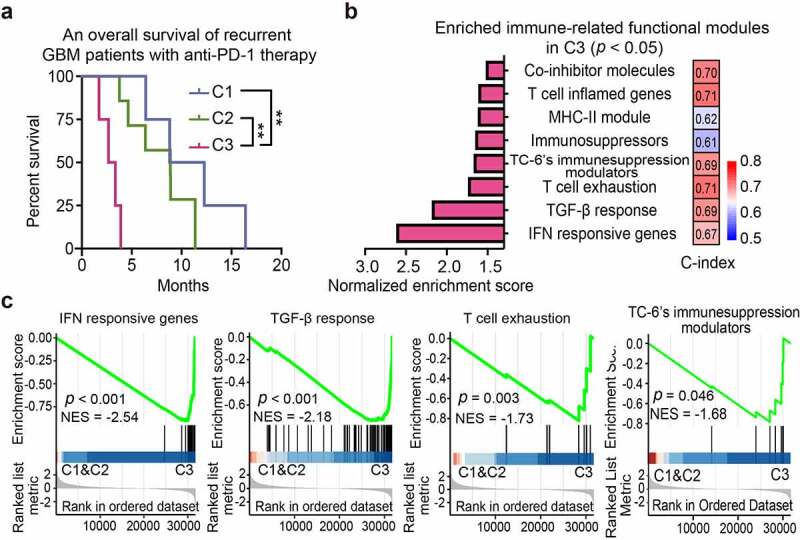
C3 subtype predicts poor immunotherapeutic response. (a) Kaplan–Meier analyses of patient overall survival of anti-PD-1-treated GBMs with different subtypes (GSE121810). (b) GSEA analysis of enriched immune modules with statistical significance in C3 subtype relative to the non-C3 subtypes. The column plot (left panel) showed the normalized enrichment score of each module. Concordance index (C-index) was presented for the indicated characteristic immune module (right panel). Predictive power (high to low) was indicated as bar color (red to blue). (c) GSEA plots of the representative enriched functional modules in C3 subtype. NES, Normalized enrichment score.
Development of an 11-gene C3 classifier for GBM molecular diagnosis
To simplify a biomarker set defining C3 GBMs for molecular diagnosis, we used the PAM method to develop a multigene classifier. TCGA GBMs were randomly stratified into training/testing cohorts at the ratio of 2:1, and 10-fold cross-validation was used to compute the cross-validation error (Supplementary Figure S8a). An 11-gene classifier was identified as the best optimal set, showing the best sensitivity (0.978), accuracy (0.989), specificity (0.992) and the highest discriminant performance using the testing cohort (Figure 7a, Supplementary Figure S8b,c).5 The efficiency of the 11-gene C3 GBM classifier was confirmed by testing cohort, external TCGA RNA-seq testing dataset and CGGA RNA-seq testing dataset (Figure 7b,c). The immune functional modules associated with IFN-γ response, antigen processing and presentation were enriched in C3-subtype GBMs using the 11-gene-classifier (Supplementary Figure S8d). C3-subtype GBMs identified through the 11-gene classifier showed worse outcomes relative to the non-C3 GBMs using the TCGA and CGGA GBM cohorts (Figure 7d,e). Multivariate Cox regression analysis confirmed that the 11-gene C3 classifier was a promising and independent biomarker set for predicting GBM patient survival (Supplementary Figure S9a). Intriguingly, most of the classifier genes were upregulated in TC-6 subsets with good consistency, suggesting that TC-6 subset is a determinant for defining C3 immune signature (Supplementary Figure S9b). We analyzed bulk RNA-seq data from six cases of human GBMs and found a consistent upregulation of the 11 classifier genes in C3 subtype GBMs (Figure 7f). Moreover, the TC-6 and IFN-related DNA damage resistance signatures were significantly enriched in C3-subtype GBMs relative to non-C3-subtype GBMs (Figure 7g). IHC staining confirmed upregulated expression of 11 classifier genes (Figure 8a,b), TAM-1 marker (CD163), TAM-2 markers (CTSD and GPNMB) and EGFR (Supplementary Figure S10a,b) in C3-subtype GBMs relative to non-C3-subtype GBMs. These data indicate that the 11-gene classifier is sufficient for defining C3-subtype GBMs with potential value for molecular diagnosis and prognostic determination6.
Figure 7.
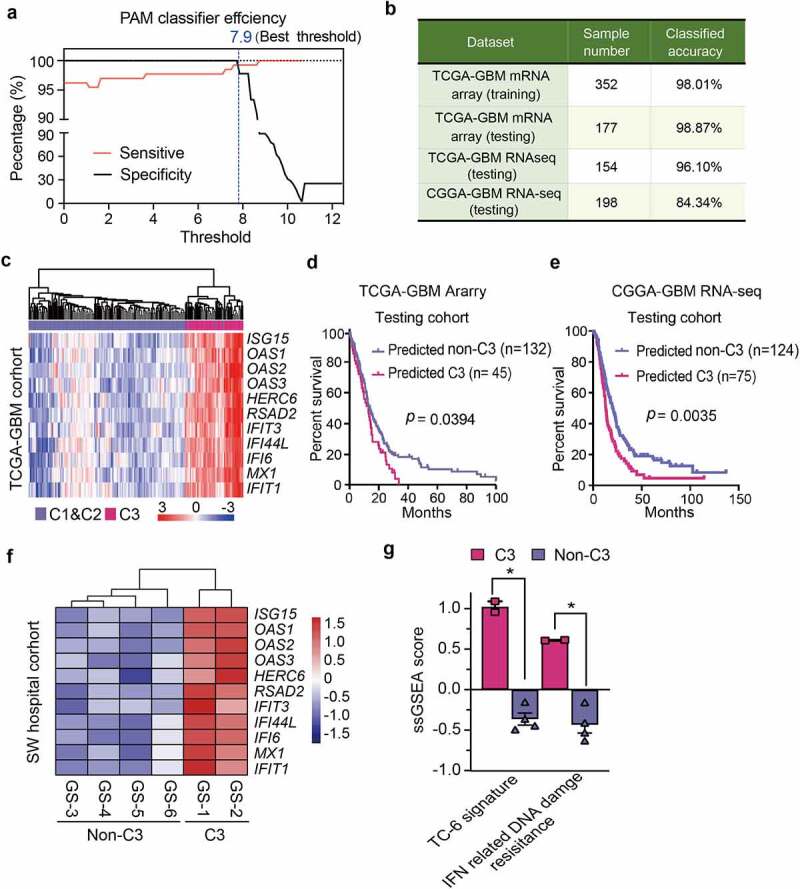
Development of an 11-gene C3 classifier for GBM molecular diagnosis. (a) Plots of sensitivity (red) and specificity (black) of the shrinkage parameter (thresholds) computed by cross-validation. The value (7.9, indicaded in blue line) yielded a subset of 11 genes with the most optimized efficiency. (b) Percentage of classified samples using the 11-gene classifier for C3 subtype in the indicated GBM datasets. The TCGA mRNA (training set) was employed as the predictor model. TCGA mRNA dataset, TCGA RNA-seq dataset, CGGA RNA-seq dataset were used as the testing sets. (c) Unsupervised hierarchical clustering of the TCGA mRNA testing set based on the 11-gene classifier. Metagenes-based subtypes are indicated with different colors at the top column. (d-e) Kaplan–Meier analyses of GBM patients from the TCGA dataset (d) and CGGA dataset (e) based on the 11-gene classifier of C3 subtype. (f) Unsupervised hierarchical clustering of six human GBM samples of RNA-seq data based on the 11-gene classifier of C3 subtype. (g) Enrichment score of TC-6 signature and IFN-related DNA damage resistance in human GBMs with non-C3 and C3 subtypes. The data was presented as the mean ± SEM. **, p < 0.01; *, p < 0.05.
Figure 8.
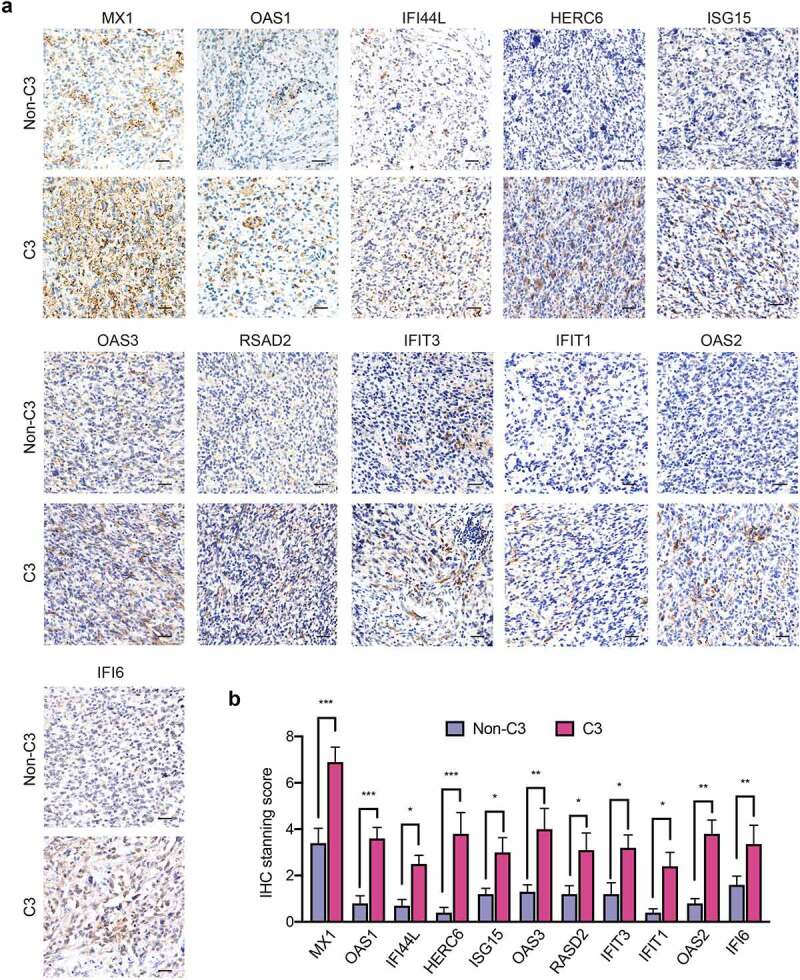
IHC staining of 11 classifier genes in C3-subtype GBMs relative to non-C3-subtypes. (a) Representative IHC images showing the expressions of 11 classifier genes in human GBMs with C3 and non-C3 subtypes. Scale bar: 50 μm. (b) Semi-quantification of IHC staining of 11 classifier genes in human GBMs with C3 and non-C3 subtypes. ***, p < 0.001; **, p< 0.01; *, p < 0.05.
Discussion
The limited understandings of GBM tumor cell heterogeneity and the complex interaction with immune components impede the development of effective immune therapeutics. Herein, we employed scRNA-seq and bulk RNA-seq data to develop an in silico method for delineating GBM immune signature. We identified TC-6 subset as a hub for creating an immunomodulatory signature of C3 subtype GBMs, holding promise for advancing molecular diagnosis of GBM immune feature and subsequent immunotherapy development. We speculate that the generation of TC-6 subset is not elicited via intrinsic genetic evolution, but through immunoediting reprogramming by immune cells and niche factors such as IFN. A recent work by Gangoso et al.4 indicated that IFN-γ treatment induced IRF8 expression in GBM cells to contribute to immune evasion. In agreement with this, our results showed that IRF1, IRF2, IRF3, IRF7, STAT1, and STAT2 downstream of IFN-γ signaling were highly activated in TC-6 subset to launch a myeloid-affiliated transcriptional program. While the sources of IFN-γ in GBMs warrant further exploration, emerging data have demonstrated that tumor-infiltrating macrophages especially the proinflammatory macrophages and brain-residing microglia secreted abundant IFN-γ.38 We speculate that IFN-γ signaling may represent a sustaining circuit that enables the retrospective interaction between TC-6 cells and tumor-infiltrating myeloid cells, thus rendering tumor cells undergo myeloid-affiliated transcriptional reconfiguration and creating an immunosuppressive environment in C3 subtype GBMs. Apart from immune cells, our in silico analyses also suggest a possible interplay between TC-6 and vascular cells through EFGR and VEGF signaling. Vascular components are important participants of GBM immunity. Besides, glioma-vascular interactions facilitate vasculogenic mimicry, vessel co-option mediated invasion, and glioma stem cell stemness.39–41 Future studies are warranted to investigate whether and how TC-6 could interact with vascular cells to reshape immune cell recruitment and functions.
GBMs are typically T cell-deprived immune cold tumors that may not respond to ICB therapy. The molecular signatures related to unfavorable ICB response in GBMs remain largely unknown. The enriched IFN signature in C3 subtype GBMs prompts us to investigate its association with anti-PD-1 efficiency. IFN signaling has been implicated in orchestrating antitumor responses, whereas conflicting data exist regarding the role of IFN signaling in restraining ICB treatment.42,43 Recent evidence supports that induction of IFN-γ at the low tumor burden stage could promote apoptosis of tumor-cytotoxic CD8+ T cells, resulting in a compromised long-term response of memory T cells and impaired ICB therapeutic efficacy.44 Besides, tumors may occupy a natural defensive mechanism from immune response.45,46 IFN-γ could also exert an immune-suppressive function by inducing the expression of immune inhibitory factors like PD-L1/2, IDO1 and TIM3 to antagonize innate and adaptive immune responses in solid tumors.10,13,45,47 Our work identified upregulated PD-L1 and IDO1 in TC-6 subsets, which might be attributed to sustained inflammatory stimuli like IFNs in C3 subtype GBM microenvironment. Delineating immune features in GBMs received neoadjuvant anti-PD-1 are warranted in future works to confirm our in silico findings of the role of C3 signature in predicting anti-PD-1 response.
Previous Transcriptomic analyses revealed proneural, classical, and mesenchymal subtypes as the major GBM molecular clusters.48 One of the major limitations of this molecular classification system is the limited involvement of environmental cues, especially the immune-competent signature. Our classification scheme integrates immune cell entity and tumor-immune cell interaction analyses to make a supplement to Verhaak’s GBM subtyping to delineate GBM molecular features. We also integrate tumor genomic alternations such as IDH1, NF1 and EGFRvIII in our analyses, as genomic mutation patterns are fundamental for inflammatory factor production, peripheral immune cell recruitment and activation.8,49,50 EGFR amplification and mutation were frequently observed in C3 subtype GBMs and were predominately presented in TC-6 cells. Previous studies have demonstrated that EGFR activation impaired anti-tumoral immunity in lung carcinoma and melanoma.51–53 GBM shares many of the molecular drivers used by other solid tumors, but possesses a unique immune environment that renders immunotherapy failure.46 It is worthwhile to investigate the unique mutational patterns in C3 subtype GBMs and their association with immune reprogramming.
In conclusion, we developed a novel, reliable, and feasible subtype classification system for delineating GBM immune signatures. A simplified 11-gene classifier defining C3-subtype GBMs shows the potential value for prognostic determination and anti-PD-1 efficacy prediction. Our findings of a unique GBM cell subset employing myeloid transcriptional circuits to create an immunomodulatory signature delineate the interplays of GBM cell subsets and infiltrating myeloid cells, with potential value for GBM molecular diagnosis and immunotherapy development.
Supplementary Material
Acknowledgments
All authors would like to thank the specimen donors and Biobank of Southwest Hospital for providing paraffin sections.
Funding Statement
This research was supported by the National Natural Science Foundation of China under Grants [Nos. 31991172 and 81922056] and Science and Technology Innovation Project of Chongqing Science and Technology Commission, China [No. cstc2021yszx-jcyj0006].
Footnotes
In the revised Figure 3, “10-gene classifier” has been replaced by “11-gene classifier” in panel a.
In the revised Supplementary tables, “LINCS IID” has been replaced by “LINCS ID” in Table S5 and Table S6.
In the revised Figure 4, “CIMP” has been replaced by “G-CIMP” in the top left corner of panel a.
The label of y-axis has been added in the revised Figure S5c.
In the revised Figure S8b and c, “C1&C2” has been replaced by“Non-C3”.
In the revised Figure 7, “C1&C2” has been replaced by“Non-C3”in panel d and panel e.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
References
- 1.Furnari FB, Fenton T, Bachoo RM, Mukasa A, Stommel JM, Stegh A, Hahn WC, Ligon KL, Louis DN, Brennan C, et al. Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev. 2007;21(21):2683–15. doi: 10.1101/gad.1596707. [DOI] [PubMed] [Google Scholar]
- 2.Neftel C, Laffy J, Filbin MG, Hara T, Shore ME, Rahme GJ, Richman AR, Silverbush D, Shaw ML, Hebert CM, et al. An integrative model of cellular states, plasticity, and genetics for glioblastoma. Cell. 2019;178(4):835–849 e21. doi: 10.1016/j.cell.2019.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang Q, Hu B, Hu X, Kim H, Squatrito M, Scarpace L, deCarvalho AC, Lyu S, Li P, Li Y, et al. Tumor evolution of glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell. 2017;32(1):42–56 e6. doi: 10.1016/j.ccell.2017.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gangoso E, Southgate B, Bradley L, Rus S, Galvez-Cancino F, McGivern N, Güç E, Kapourani C-A, Byron A, Ferguson KM, et al. Glioblastomas acquire myeloid-affiliated transcriptional programs via epigenetic immunoediting to elicit immune evasion. Cell. 2021;184(9):2454–2470 e26. doi: 10.1016/j.cell.2021.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nishino M, Ramaiya NH, Hatabu H, Hodi FS.. Monitoring immune-checkpoint blockade: response evaluation and biomarker development. Nat Rev Clin Oncol. 2017;14(11):655–668. doi: 10.1038/nrclinonc.2017.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zeng J, See AP, Phallen J, Jackson CM, Belcaid Z, Ruzevick J, Durham N, Meyer C, Harris TJ, Albesiano E, et al. Anti-PD-1 blockade and stereotactic radiation produce long-term survival in mice with intracranial gliomas. Int J Radiat Oncol Biol Phys. 2013;86(2):343–349. doi: 10.1016/j.ijrobp.2012.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McGranahan T, Therkelsen KE, Ahmad S, Nagpal S. Current state of immunotherapy for treatment of glioblastoma. Curr Treat Options Oncol. 2019;20(3):24. doi: 10.1007/s11864-019-0619-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhao J, Chen AX, Gartrell RD, Silverman AM, Aparicio L, Chu T, Bordbar D, Shan D, Samanamud J, Mahajan A, et al. Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat Med. 2019;25(3):462–469. doi: 10.1038/s41591-019-0349-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jackson CM, Lim M. Immunotherapy for glioblastoma: playing Chess, not checkers. Clin Cancer Res. 2018;24(17):4059–4061. doi: 10.1158/1078-0432.CCR-18-0491. [DOI] [PubMed] [Google Scholar]
- 10.Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. 2017;168(4):707–723. doi: 10.1016/j.cell.2017.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ito H, Nakashima H, Chiocca EA. Molecular responses to immune checkpoint blockade in glioblastoma. Nat Med. 2019;25(3):359–361. doi: 10.1038/s41591-019-0385-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cloughesy TF, Mochizuki AY, Orpilla JR, Hugo W, Lee AH, Davidson TB, Wang AC, Ellingson BM, Rytlewski JA, Sanders CM, et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat Med. 2019;25(3):477–486. doi: 10.1038/s41591-018-0337-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jacquelot N, Yamazaki T, Roberti MP, Duong CPM, Andrews MC, Verlingue L, Ferrere G, Becharef S, Vétizou M, Daillère R, et al. Sustained Type I interferon signaling as a mechanism of resistance to PD-1 blockade. Cell Res. 2019;29(10):846–861. doi: 10.1038/s41422-019-0224-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Benci JL, Xu B, Qiu Y, Wu TJ, Dada H, Twyman-Saint Victor C, Cucolo L, Lee DSM, Pauken KE, Huang AC, et al. Tumor interferon signaling regulates a multigenic resistance program to immune checkpoint blockade. Cell. 2016;167(6):1540–1554 e12. doi: 10.1016/j.cell.2016.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang L, Sfakianos JP, Beaumont KG, Akturk G, Horowitz A, Sebra RP, Farkas AM, Gnjatic S, Hake A, Izadmehr S, et al. Myeloid cell-associated resistance to PD-1/PD-L1 blockade in urothelial cancer revealed through bulk and single-cell RNA sequencing. Clin Cancer Res. 2021;27(15):4287-4300 . doi: 10.1158/1078-0432.CCR-20-4574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Khodarev NN, Beckett M, Labay E, Darga T, Roizman B, Weichselbaum RR. STAT1 is overexpressed in tumors selected for radioresistance and confers protection from radiation in transduced sensitive cells. Proc Natl Acad Sci U S A. 2004;101(6):1714–1719. doi: 10.1073/pnas.0308102100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Ou Yang T-H, Porta-Pardo E, Gao GF, Plaisier CL, Eddy JA, et al. The immune landscape of cancer. Immunity. 2018;48(4):812–830 e14. doi: 10.1016/j.immuni.2018.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stathias V, Jermakowicz AM, Maloof ME, Forlin M, Walters W, Suter RK, Durante MA, Williams SL, Harbour JW, Volmar C-H, et al. Drug and disease signature integration identifies synergistic combinations in glioblastoma. Nat Commun. 2018;9(1):5315. doi: 10.1038/s41467-018-07659-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aibar S, Gonzalez-Blas CB, Moerman T, Huynh-Thu VA, Imrichova H, Hulselmans G, Rambow F, Marine J-C, Geurts P, Aerts J, et al. SCENIC: single-cell regulatory network inference and clustering. Nat Methods. 2017;14(11):1083–1086. doi: 10.1038/nmeth.4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Efremova M, Vento-Tormo M, Teichmann SA, Vento-Tormo R. CellPhoneDB: inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat Protoc. 2020;15(4):1484–1506. doi: 10.1038/s41596-020-0292-x. [DOI] [PubMed] [Google Scholar]
- 21.Dabney AR. Classification of microarrays to nearest centroids. Bioinformatics. 2005;21(22):4148–4154. doi: 10.1093/bioinformatics/bti681. [DOI] [PubMed] [Google Scholar]
- 22.Shi Y, Guryanova OA, Zhou W, Liu C, Huang Z, Fang X, Wang X, Chen C, Wu Q, He Z, et al. Ibrutinib inactivates BMX-STAT3 in glioma stem cells to impair malignant growth and radioresistance. Sci Transl Med. 2018;10(443). doi: 10.1126/scitranslmed.aah6816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang Y, Liu Y, Yao X, Ping Y, Jiang T, Liu Q, Xu S, Huang J, Mou H, Gong W, et al. Annexin 1 released by necrotic human glioblastoma cells stimulates tumor cell growth through the formyl peptide receptor 1. Am J Pathol. 2011;179(3):1504–1512. doi: 10.1016/j.ajpath.2011.05.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama S, Zheng S, Chakravarty D, Sanborn J, Berman S, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155(2):462–477. doi: 10.1016/j.cell.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barkal AA, Weiskopf K, Kao KS, Gordon SR, Rosental B, Yiu YY, George BM, Markovic M, Ring NG, Tsai JM, et al. Engagement of MHC class I by the inhibitory receptor LILRB1 suppresses macrophages and is a target of cancer immunotherapy. Nat Immunol. 2018;19(1):76–84. doi: 10.1038/s41590-017-0004-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhu Y, Yang J, Xu D, Gao X-M, Zhang Z, Hsu JL, Li C-W, Lim S-O, Sheng -Y-Y, Zhang Y, et al. Disruption of tumour-associated macrophage trafficking by the osteopontin-induced colony-stimulating factor-1 signalling sensitises hepatocellular carcinoma to anti-PD-L1 blockade. Gut. 2019;68(9):1653–1666. doi: 10.1136/gutjnl-2019-318419. [DOI] [PubMed] [Google Scholar]
- 27.Harjunpaa H, Guillerey C. TIGIT as an emerging immune checkpoint. Clin Exp Immunol. 2020;200(2):108–119. doi: 10.1111/cei.13407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Abd Hamid M, Wang RZ, Yao X, Fan P, Li X, Chang X-M, Feng Y, Jones S, Maldonado-Perez D, Waugh C, et al. Enriched HLA-E and CD94/NKG2A interaction limits antitumor CD8+ tumor-infiltrating T lymphocyte responses. Cancer Immunol Res. 2019;7(8):1293–1306. doi: 10.1158/2326-6066.CIR-18-0885. [DOI] [PubMed] [Google Scholar]
- 29.Wang W, Chong WP, Li C, Chen Z, Wu S, Zhou H, Wan Y, Chen W, Gery I, Liu Y, et al. Type I interferon therapy limits CNS autoimmunity by inhibiting CXCR3-mediated trafficking of pathogenic effector T cells. Cell Rep. 2019;28(2):486–497 e4. doi: 10.1016/j.celrep.2019.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goswami S, Walle T, Cornish AE, Basu S, Anandhan S, Fernandez I, Vence L, Blando J, Zhao H, Yadav SS, et al. Immune profiling of human tumors identifies CD73 as a combinatorial target in glioblastoma. Nat Med. 2020;26(1):39–46. doi: 10.1038/s41591-019-0694-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li F, Zhang R, Li S, Liu J. IDO1: an important immunotherapy target in cancer treatment. Int Immunopharmacol. 2017;47:70–77. doi: 10.1016/j.intimp.2017.03.024. [DOI] [PubMed] [Google Scholar]
- 32.Yang R, Sun L, Li C-F, Wang Y-H, Yao J, Li H, Yan M, Chang W-C, Hsu J-M, Cha J-H, et al. Galectin-9 interacts with PD-1 and TIM-3 to regulate T cell death and is a target for cancer immunotherapy. Nat Commun. 2021;12(1):832. doi: 10.1038/s41467-021-21099-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Suo S, Zhu Q, Saadatpour A, Fei L, Guo G, Yuan G-C. Revealing the critical regulators of cell identity in the mouse cell atlas. Cell Rep. 2018;25(6):1436–1445 e3. doi: 10.1016/j.celrep.2018.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weichselbaum RR, Ishwaran H, Yoon T, Nuyten DSA, Baker SW, Khodarev N, Su AW, Shaikh AY, Roach P, Kreike B, et al. An interferon-related gene signature for DNA damage resistance is a predictive marker for chemotherapy and radiation for breast cancer. Proc Natl Acad Sci U S A. 2008;105(47):18490–18495. doi: 10.1073/pnas.0809242105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garton AJ, Crew AP, Franklin M, Cooke AR, Wynne GM, Castaldo L, Kahler J, Winski SL, Franks A, Brown EN, et al. OSI-930: a novel selective inhibitor of Kit and kinase insert domain receptor tyrosine kinases with antitumor activity in mouse xenograft models. Cancer Res. 2006;66(2):1015–1024. doi: 10.1158/0008-5472.CAN-05-2873. [DOI] [PubMed] [Google Scholar]
- 36.Wang T, Li X, Sun SL. EX527, a Sirt-1 inhibitor, induces apoptosis in glioma via activating the p53 signaling pathway. Anticancer Drugs. 2020;31(1):19–26. doi: 10.1097/CAD.0000000000000824. [DOI] [PubMed] [Google Scholar]
- 37.Laske K, Shebzukhov YV, Grosse-Hovest L, Kuprash DV, Khlgatian SV, Koroleva EP, Sazykin AY, Penkov DN, Belousov PV, Stevanović S, et al. Alternative variants of human HYDIN are novel cancer-associated antigens recognized by adaptive immunity. Cancer Immunol Res. 2013;1(3):190–200. doi: 10.1158/2326-6066.CIR-13-0079. [DOI] [PubMed] [Google Scholar]
- 38.Hambardzumyan D, Gutmann DH, Kettenmann H. The role of microglia and macrophages in glioma maintenance and progression. Nat Neurosci. 2016;19(1):20–27. doi: 10.1038/nn.4185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Griveau A, Seano G, Shelton SJ, Kupp R, Jahangiri A, Obernier K, Krishnan S, Lindberg OR, Yuen TJ, Tien A-C, et al. A glial signature and Wnt7 signaling regulate glioma-vascular interactions and tumor microenvironment. Cancer Cell. 2018;33(5):874–889 e7. doi: 10.1016/j.ccell.2018.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhu TS, Costello MA, Talsma CE, Flack CG, Crowley JG, Hamm LL, He X, Hervey-Jumper SL, Heth JA, Muraszko KM, et al. Endothelial cells create a stem cell niche in glioblastoma by providing NOTCH ligands that nurture self-renewal of cancer stem-like cells. Cancer Res. 2011;71(18):6061–6072. doi: 10.1158/0008-5472.CAN-10-4269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hardee ME, Zagzag D. Mechanisms of glioma-associated neovascularization. Am J Pathol. 2012;181(4):1126–1141. doi: 10.1016/j.ajpath.2012.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brockwell NK, Owen KL, Zanker D, Spurling A, Rautela J, Duivenvoorden HM, Baschuk N, Caramia F, Loi S, Darcy PK, et al. Neoadjuvant interferons: critical for effective PD-1-based immunotherapy in TNBC. Cancer Immunol Res. 2017;5(10):871–884. doi: 10.1158/2326-6066.CIR-17-0150. [DOI] [PubMed] [Google Scholar]
- 43.Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 2015;27(4):450–461. doi: 10.1016/j.ccell.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pai CS, Huang JT, Lu X, Simons DM, Park C, Chang A, Tamaki W, Liu E, Roybal KT, Seagal J, et al. Clonal deletion of tumor-specific T cells by interferon-gamma confers therapeutic resistance to combination immune checkpoint blockade. Immunity. 2019;50(2):477–492 e8. doi: 10.1016/j.immuni.2019.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, Gandhi L, Redig AJ, Rodig SJ, Asahina H, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun . 2016;10501. doi: 10.1038/ncomms10501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jackson CM, Choi J, Lim M. Mechanisms of immunotherapy resistance: lessons from glioblastoma. Nat Immunol. 2019;20(9):1100–1109. doi: 10.1038/s41590-019-0433-y. [DOI] [PubMed] [Google Scholar]
- 47.Bartok O, Pataskar A, Nagel R, Laos M, Goldfarb E, Hayoun D, Levy R, Körner P-R, Kreuger IZM, Champagne J, et al. Anti-tumour immunity induces aberrant peptide presentation in melanoma. Nature. 2020;590(4845):332–337. doi: 10.1038/s41586-020-03054-1. [DOI] [PubMed] [Google Scholar]
- 48.Wang Q, Hu B, Hu X, Kim H, Squatrito M, Scarpace L, deCarvalho AC, Lyu S, Li P, Li Y, et al. Tumor evolution of glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell. 2018;33(1):152. doi: 10.1016/j.ccell.2017.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang XC, Wang J, Shao GG, Wang Q, Qu X, Wang B, Moy C, Fan Y, Albertyn Z, Huang X, et al. Comprehensive genomic and immunological characterization of Chinese non-small cell lung cancer patients. Nat Commun. 2019;10(1):1772. doi: 10.1038/s41467-019-09762-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, Berent-Maoz B, Pang J, Chmielowski B, Cherry G, et al. Genomic and transcriptomic features of response to Anti-PD-1 therapy in metastatic melanoma. Cell. 2016;165(1):35–44. doi: 10.1016/j.cell.2016.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Akbay EA, Koyama S, Carretero J, Altabef A, Tchaicha JH, Christensen CL, Mikse OR, Cherniack AD, Beauchamp EM, Pugh TJ, et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov. 2013;3(12):1355–1363. doi: 10.1158/2159-8290.CD-13-0310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen N, Fang W, Zhan J, Hong S, Tang Y, Kang S, Zhang Y, He X, Zhou T, Qin T, et al. Upregulation of PD-L1 by EGFR activation mediates the immune escape in EGFR-driven NSCLC: implication for optional immune targeted therapy for NSCLC patients with EGFR mutation. J Thorac Oncol. 2015;10(6):910–923. doi: 10.1097/JTO.0000000000000500. [DOI] [PubMed] [Google Scholar]
- 53.Zaiss DM, van Loosdregt J, Gorlani A, Bekker CJ, Gröne A, Sibilia M, Van bergen en henegouwen PP, Roovers R, Coffer P, Sijts AAM. Amphiregulin enhances regulatory T cell-suppressive function via the epidermal growth factor receptor. Immunity. 2013;38(2):275–284. doi: 10.1016/j.immuni.2012.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.